Clinical Genetics
• Explore how recent advances in genetics have changed the field of health care.
• Discuss the essential competencies in genetics and genomics for all nurses.
• Describe expanded roles for nurses in genetics and genetic counseling.
• Discuss key findings of the Human Genome Project.
• Describe the different types of genetic testing.
• Identify genetic disorders commonly tested for in maternity and women’s health nursing.
• Explore the possible benefits and risks of pharmacogenomics.
• Discuss the current status of gene therapy.
• Examine the ethical, legal, and social implications of the Human Genome Project.
• Explain the key concepts of basic human genetics.
• Discuss the education and counseling needs of individuals and families who undergo genetic testing.
• Explore the availability of genetic testing for individuals and families from diverse backgrounds.
• Describe the role of genomics in cancer.
• Identify genetics resources for nurses and other health care professionals.

Recent advances in molecular biology and genomics have revolutionized the field of health care by providing the tools needed to determine the hereditary component of many diseases, as well as improve our ability to predict susceptibility to disease, onset and progression of disease, and response to medications (Feero, Guttmacher, & Collins, 2008; Ginsburg & Willard, 2009, Guttmacher, McGuire, Ponder, & Stefansson, 2010). This increase in genetic knowledge has resulted in a gradual shift from genetics to genomics. Genetics is the study of individual genes and their effect on relatively rare single gene disorders, whereas genomics is the study of all the genes in the human genome together, including their interactions with each other, the environment, and the influence of other psychosocial factors and cultural factors. Genes are basic physical units of inheritance that are passed from parents to offspring and contain the information needed to specify traits. The genome is the entire set of genetic instructions found in each cell. For these and other definitions of genetic terms, visit the Talking Glossary of Genetic Terms (www.genome.gov/Glossary).
With growing public interest in personalized genomic information (information about much or all of a person’s genome), increasing development of practice guidelines, mounting commercial pressures, and ever-increasing opportunities for individuals, families, and communities to participate in the direction and design of their genomic health care, genetic services are rapidly becoming an integral part of routine health care (Guttmacher et al., 2010). Moreover, many individuals and families have participated in direct-to-consumer genetic testing (testing marketed directly to consumers through television, print advertisements, and websites for companies such as DNA Direct [www.dnadirect.com/web], 23 and Me [www.23andme.com], and DeCODEme [www.decodeme.com]). Although much of the information provided by direct-to-consumer testing companies is recreational (ancestry information, information about type of ear wax, and bitter taste perception), some of it is health related and could be interpreted as diagnosis (Evans & Green, 2009). Because of this, direct-to-consumer testing that is provided without the involvement of competent health care professionals may be not only unhelpful but also harmful (Guttmacher et al.; McGuire & Burke, 2010).
Due to their frontline position in the health care system and their long-standing history of providing holistic family-centered care, nurses are likely to be among the first health care professionals to whom individuals and families turn with questions about genetic risk and susceptibility and to seek guidance regarding the complexities of genetic testing and interpretation. Nowhere is this more apparent than in the area of maternity and women’s health care (Dolan, Biermann, & Damus, 2007). A growing number of maternity and women’s health nurses are offering and interpreting genetic tests. Although most of these tests are being used to determine a client’s risk of having a child affected by a genetic condition such as Down syndrome, cystic fibrosis (CF), or sickle cell disease, the number of tests being used to determine the presence of, or susceptibility to, adult-onset disorders (e.g., hereditary colorectal cancer, hereditary breast and ovarian cancer, and Huntington’s disease [HD]) continues to rise. Additionally, nurses working in maternity and women’s health are caring for an increasing number of individuals and families who are dealing with complex ethical, legal, and social issues associated with genetic testing and the experience of living with someone who has a genetic condition (Hamilton, 2009; Sparbel & Williams, 2009; Van Riper, 2007; Van Riper & Gallo, 2006).
Nursing Expertise in Genetics and Genomics
Essential Competencies in Genetics and Genomics for All Nurses
Nearly 50 organizations, including the Association of Women’s Health, Obstetric and Neonatal Nurses and the National Association of Neonatal Nurses, have endorsed the Essential Nursing Competencies and Curriculum Guidelines for Genetics and Genomics. The guidelines were developed by an independent panel of nurse leaders from clinical, research, and academic settings and published by the American Nurses Association and the National Human Genome Research Institute (NHGRI) of the National Institutes of Health (Jenkins & Calzone, 2007). According to the guidelines, all nurses need to have minimal competencies in genetics and genomics regardless of their academic preparation, practice setting, or specialty. Some of the competencies most relevant to nurses in the area of maternity and woman’s health include:
• Constructs a pedigree from collected family history information using standardized symbols and terminology.
• Develops a nursing care plan that incorporates genetic and genomic assessment information.
• Recognizes when one’s own attitudes and values related to genetics and genomic science may affect care provided to clients.
• Provides clients with credible, accurate, appropriate, and current genetic and genomic information, resources, services, and/or technologies that facilitate decision making.
• Demonstrates in practice the importance of tailoring genetic and genomic information and services to clients based on their culture, religion, knowledge level, literacy, and preferred language.
• Assesses client’s knowledge, perceptions, and responses to genetic and genomic information.
• Facilitates referrals for specialized genetic and genomic services for clients as needed.
Expanded Roles for Maternity and Women’s Health Nurses
• Expanded roles for nurses with expertise in genetics and genomics are developing in many areas of maternity and women’s health nursing. These areas include but are not limited to:
• Prenatal screening and testing (Dolan et al., 2007; Hamilton, 2009; Sparbel & Williams, 2009)
• Neonatal genetic screening and testing (Kenner, Lewis, Pressler, & Little, 2008)
• Palliative care for infants with life-threatening genetic conditions and their families (Shaw, 2008; Wirth, 2009)
• The identification and care of individuals with genetic conditions and their families (Gallo, Knafl, & Angst, 2009; Lynch, Snyder, & Lynch, 2009; Ranweiler, 2009; Schiefelbein & Cheeseman, 2009; Snyder, Lynch, & Lynch, 2009; Van Riper, 2007)
• The care of women with genetic conditions who require specialized care during pregnancy, such as women with congenital heart disease (Khairy, Ouyang, Fernandes, Lee-Parritz, Economy, & Landzberg, 2006), cystic fibrosis (McMullen, Pasta, Frederick, Konstan, Morgan, Schechter, et al., 2006), and Factor V Leiden (Horne & McCloskey, 2006; Weinstein, 2009)
The Oncology Nursing Society (ONS) (www.ons.org) has taken an active role in providing oncology nurses with the education and resources they need to integrate genetics and genomics into all phases of care for individuals and families affected by chance. ONS offers online updates, regional classes, and position statements related to genetics and genomics (Hamilton, 2009).
Human Genome Project and Implications for Clinical Practice
Two key findings from the Human Genome Project were that (1) all human beings are 99.9% identical at the deoxyribonucleic acid (DNA) level, and (2) there are probably about 20,500 genes in the human genome. The finding that human beings are 99.9% identical at the DNA level should help discourage the use of science as a justification for drawing biologically precise racial boundaries around certain groups of people (Collins, 2004). Originally scientists had estimated that there were 50,000 to 140,000 genes in the human genome. It had been assumed that the main reason that humans are more evolved and more highly sophisticated than other species is that they have more genes. A new explanation for human complexity, given the relatively small number of genes, is that humans are more efficient with their genes. Humans are able to do much more with their genes than are other species. Instead of producing only one protein per gene, most human genes produce at least three proteins.
Importance of Family History
Completion of the Human Genome Project and the resultant identification of the inherited causes for many diseases has created a renewed interest in family history (Clarke, 2009; De Sevo, 2009; Dolan et al., 2007; Dolan & Moore, 2007; Feero et al., 2008; Ginsburg & Willard, 2009; Hinton, 2008; Soloman, Jack, & Feero, 2008; Wattendorf & Hadley, 2005). Although it is easy to be impressed by the almost 1900 genetic tests currently available, family history will most likely continue to be the single most cost-effective piece of genetic information. Rich and colleagues (2004) described family history as the most important tool for diagnosis and risk assessment in health care genetics and a critical tool in the use of predictive testing in primary care. Solomon and colleagues argued that a complete three-generation family history that includes ethnicity information concerning both sides of family is the best genetic “test” applicable to preconception care. When nurses and other clinicians conduct a family history, they can gain not only valuable information about the structure of the family and diseases that affect various individuals in the family, but also a rich understanding of family relationships, social context, occupations, lifestyle, and health habits. In addition, the process of collecting this information often facilitates the development of a relationship between the client/family and the clinician. In 2004, the U.S. Department of Health and Human Services launched the Family History Initiative by designating Thanksgiving Day as National Family History Day. The U.S. Surgeon General encouraged families to use their family gatherings as a time to talk about and collect important family health history. A number of family history tools are available free of charge online. One of the most widely used is the My Family Health Portrait (https://familyhistory.hhs.gov). Another recently developed tool is the family health history tool, Does it run in the family? that was developed by the Genetic Alliance (www.doesitruninthefamily.org).
Gene Identification and Testing
Initial efforts to sequence and analyze the human genome have proven invaluable in the identification of genes involved in disease and in the development of genetic tests. Hundreds of genes involved in diseases such as breast cancer, colorectal cancer, Alzheimer’s disease, and CF have been identified. The number of commercially available genetic tests continues to increase and can be found on GeneTests, a publicly funded genetics information resource for clinicians (www.ncbi.nlm.nih.gov/sites/GeneTests/?db=GeneTests).
Genetic testing involves the analysis of human DNA, ribonucleic acid (RNA), chromosomes (threadlike packages of genes and other DNA in the nucleus of a cell), or proteins to detect abnormalities related to an inherited condition. Genetic tests can be used to examine directly the DNA and RNA that make up a gene (direct or molecular testing), look at markers that are coinherited with a gene that causes a genetic condition (linkage analysis), examine the protein products of genes (biochemical testing), or examine chromosomes (cytogenetic testing). Cytogenetic analysis of malignant tissue has become a mainstay of oncology.
Most of the genetic tests now offered in clinical practice are tests for single-gene disorders in clients with clinical symptoms or who have a family history of a genetic disease. Some of these genetic tests are prenatal tests or tests used to identify the genetic status of a pregnancy at risk for a genetic condition. Current prenatal testing options include maternal serum screening (a blood test used to see if a pregnant woman is at increased risk for carrying a fetus with a neural tube defect or chromosomal abnormalities such as Down syndrome, trisomy 18 and trisomy 13), fetal ultrasound or sonogram (an imaging technique using high-frequency sound waves to produce images of the fetus inside the uterus), and invasive procedures (chorionic villus sampling and amniocentesis) (see Chapter 26 for discussion of these tests). Other tests are carrier screening tests used to identify individuals who have a gene mutation for a genetic condition but do not show symptoms of the condition because it is an autosomal recessive condition (e.g., CF, sickle cell disease, and Tay-Sachs disease). Another type of genetic testing is predictive testing, which is used to clarify the genetic status of asymptomatic family members. The two types of predictive testing are presymptomatic and predispositional. Mutation analysis for HD, a neurodegenerative disorder, is an example of presymptomatic testing. If the gene mutation for HD is present, symptoms of HD are certain to appear if the individual lives long enough. Testing for a BRCA1 gene mutation to determine breast cancer susceptibility is an example of predispositional testing. Predispositional testing differs from presymptomatic testing in that a positive result (indicating that a BRCA1 mutation is present) does not indicate a 100% risk of developing the condition (breast cancer).
In addition to using genetic tests to test for single-gene disorders in clients with clinical symptoms or who have a family history of a genetic disease, genetic tests are being used for population-based screening. For example, newborn screening for phenylketonuria (PKU) and other inborn errors of metabolism (IEMs) has been going on in the United States and many other countries for decades (Guttmacher et al., 2010; Kenner et al., 2008). Initially, state-mandated newborn screening in the United States was concerned with only a few conditions. With the advent of tandem mass spectrometry, the number of conditions tested for during newborn screening grew rapidly. Currently, most states use blood spots collected from newborns to test for at least 30 different metabolic and genetic diseases. The four conditions most commonly tested for are PKU, congenital hypothyroidism, galactosemia, and sickle cell disease. A complete list of conditions tested for in each state is available on the National Newborn Screening and Genetics Resource website.
Another type of population-based screening is carrier screening for single-gene disorders such as CF, sickle cell disease, and Tay-Sachs disease either preconceptually or prenatally. In 2001, the American College of Obstetricians and Gynecologists and the American College of Medical Genetics (ACOG & ACMG) began recommending that clinicians offer carrier screening for CF to individuals with a family history of CF, reproductive partners of individuals who have CF, and couples in whom one or both partners are Caucasian and are planning a pregnancy or seeking prenatal care (ACOG & ACMG, 2001). One outcome of this broader CF carrier screening is that more and more individuals are being informed they have a CF mutation. Unfortunately, the correlation between genotype (an individual’s collection of genes) and phenotype (an individual’s observable traits) is poor for many of the more than 1400 CF mutations identified to date. That is, whereas some CF mutations are associated with significant health problems (poor growth, greasy stools, and chronic respiratory problems) others are not. Because of this, the significance of many CF mutations is uncertain. As a result, nurses and other health care professionals are increasingly being asked to communicate results with uncertain significance to individuals and families during the preconception and prenatal period (Dolan et al., 2007). In 2008, the NHGRI held a workshop to discuss lessons learned and new opportunities for population-based carrier screening (www.genome.gov/27026048). One of the main conclusions from this workshop was that a more coherent and systematic approach is needed for the introduction of new tests into population-based screening programs.
Pharmacogenomics
One of the most promising clinical applications of the Human Genome Project has been pharmacogenomic testing (the use of genetic information to guide a client’s drug therapy) (Ginsburg & Willard, 2009). Associations between genetic variation and drug effect have been observed for a number of commonly used drugs, including warfarin, an anticoagulant commonly used to reduce the risk of thromboembolic events in clients with a history of deep vein thrombosis, pulmonary embolism, myocardial infarction, or atrial fibrillation (Lanfear & McCleod, 2007; Meckley, Gudgeon, Anderson, Williams, & Veenstra, 2010). Warfarin is a drug with a narrow therapeutic index; it can result in serious bleeding with supratherapeutic doses and thromboembolic events with subtherapeutic doses). Because of this and the fact that there is a great deal of inter- and intraclient dose variation, warfarin is one of the most common causes of serious adverse drug reactions. Fortunately there is mounting evidence that genotype-guided warfarin dosing may not only help reduce the serious adverse drug reactions commonly associated with warfarin, but increase dosing accuracy, shorten the time to dose stabilization, and help identify individuals who may require more frequent monitoring. In August 2007, the U.S. Food and Drug Administration (FDA) approved updated labeling for warfarin. The updated labeling acknowledges that individuals with variations in their CYP2C9 and VKORC1 genes may require a lower initial dose of warfarin. However, there are not enough clinical data yet to recommend that this type of testing be mandatory.
Pharmacogenomic testing can also be used to target therapies. Trastuzumab (Herceptin), a monoclonal antibody that specifically targets HER2/neu overexpressing breast tumors, is an example of a drug for which an obligatory genetic test has been developed (Ginsburg & Willard, 2009). The purpose of this obligatory genetic test is to identify the subset of women with breast cancer who overexpress HER2/neu. Women who overexpress HER2/neu are most likely the only breast cancer clients who will benefit from taking trastuzumab (www.herceptin.com/index.jsp).
Gene Therapy
In the early 1990s, a great deal of optimism was felt about the possibility of using gene therapy to correct a long list of inherited diseases. Generally, gene therapy involves inserting a healthy copy of the defective gene into the somatic cells (any cell of the body except sperm and egg cells) of the affected individual. Although the early optimism about gene therapy was probably never fully justified, gene therapy has now moved from preclinical to clinical studies for many diseases ranging from hemophilia and other single gene disorders to complex disorders such as cancer, human immunodeficiency virus, and cardiovascular disorders (Gillet, Macadangdang, Fathke, Gottesman, & Kimchi-Sarfaty, 2009). Early hype, failures, and tragic events, such as the death of Jessie Gelsinger (an 18-year-old male with an X-linked genetic liver disease who was the first person publicly identified as having died in a gene therapy clinical trial) have now largely been replaced by stepwise progress in carefully developed, scientifically precise clinical trails (Gillet et al.; Kohn & Candotti, 2009). Major challenges to gene therapy include figuring out how to target the right gene to the right location in the right cells, expressing the transferred gene at the right time, and minimizing adverse reactions.
Ethical, Legal, and Social Implications
Before the beginning of the Human Genome Project, widespread concern about misuse of the information gained through genetics research resulted in 5% of the Human Genome Project budget being designated for the study of the ethical, legal, and social implications (ELSIs) of human genome research. Two large ELSI programs were created to identify, analyze, and address the ELSIs of human genome research at the same time that the basic science issues were being studied. During the past decade, issues of high priority for these programs have been privacy and fairness in the use and interpretation of genetic information; clinical integration of new genetics technologies; issues surrounding genetics research, such as possible discrimination and stigmatization; and education for professionals and the general public about genetics, genetics health care, and ELSI of human genome research. Both ELSI programs have excellent websites that include large amounts of educational information, as well as links to other informative sites (www.genome.gov/10001618; www.ornl.gov/sci/techresources/Human_Genome/elsi/elsi.shtml).
The major risk associated with genetic testing concerns what happens with the information gained through testing: it may result in increased anxiety and altered family relationships; it may be difficult to keep confidential; and it may result in discrimination and stigmatization. More important, there is still a large gap between the ability to test for a genetic condition and the ability to treat the same condition. In addition, informed consent is difficult to ensure when some of the outcomes, benefits, and risks of genetic testing remain unknown. Also many of the tests being used are as yet imperfect—few have a 100% detection rate. Individuals and families who receive false-positive results (the test results falsely indicate that a person or fetus is affected by a genetic condition) may terminate an unaffected pregnancy or undergo unwarranted extreme measures such as bilateral prophylactic mastectomy. Individuals and families who receive false-negative results (the test results indicate that a person or fetus is not affected by a genetic condition when in fact the person or fetus is affected) may fail to follow surveillance strategies designed to improve their health outcomes because they have been falsely reassured that they are not at increased risk for a specific condition.
Factors Influencing the Decision to Undergo Genetic Testing
The decision to undergo genetic testing is seldom autonomous and based solely on the needs and preferences of the individual being tested. Instead, it is often a decision based on feelings of responsibility and commitment to others (Van Riper, 2005; Van Riper & McKinnon, 2004). For example, a woman who is receiving treatment for breast cancer may undergo BRCA1/BRCA2 mutation testing not because she wants to find out if she carries a BRCA1 or BRCA2 mutation, but because her two unaffected sisters have asked her to be tested, and she feels a sense of responsibility and commitment to them. A female airline pilot with a family history of HD, who has no desire to find out if she has the gene mutation associated with HD, may undergo mutation analysis for HD because she feels she has an obligation to her family, her employer, and the people who fly with her.
Decisions about genetic testing are shaped, and in many instances constrained, by factors such as social norms, where care is received, and socioeconomic status. Most pregnant women in the United States now have at least one ultrasound examination, many undergo some type of multiple-marker screening, and a growing number undergo other types of prenatal testing. The range of prenatal testing options available to a pregnant woman and her family may vary significantly, based on where the woman receives prenatal care and her socioeconomic status. Certain types of prenatal testing may not be available in smaller communities and rural settings (e.g., chorionic villus sampling and fluorescent in situ hybridization [FISH] analysis). In addition, certain types of genetic testing may not be offered in conservative medical communities (e.g., preimplantation diagnosis). Some types of genetic testing are expensive and typically not covered by health insurance. Because of this, these tests may be available only to a relatively small number of individuals and families: those who can afford to pay for them.
Cultural and ethnic differences also have a significant effect on decisions about genetic testing. When prenatal diagnosis was first introduced, the principal constituency was a self-selected group of Caucasian, well-informed, middle- to upper-class women. Today the widespread use of genetic testing has introduced prenatal testing to new groups of women, women who had not previously considered genetics services. The fact that many of the women undergoing prenatal testing may not share mainstream U.S. views about the role of medicine and prenatal care, the meaning of disability, or how to respond to scientific risks and uncertainties further amplifies the complexity of ethical issues associated with prenatal testing.
The genetic testing experience raises fundamental questions about the mutual obligations of kin. Are individuals morally obligated to alert extended family members about inherited health risks? Conversely, do extended family members have a moral obligation to participate in research designed to determine genetic risk when unwanted information about them may be generated in the process? Another important question that must be considered is “Whose gene is it?” This question is likely to stimulate a great deal of debate, especially in the area of preimplantation genetic testing.
Clinical Genetics
Human development is a complicated process that depends on the systematic unraveling of instructions found in the genetic material of the egg and the sperm. Development from conception to birth of a normal, healthy baby occurs without incident in most cases; occasionally, however, some anomaly in the genetic code of the embryo creates a birth defect or disorder.
Genes and Chromosomes
The hereditary material carried in the nucleus of each of the somatic cells determines an individual’s characteristics. This material, called deoxyribonucleic acid (DNA), forms threadlike strands known as chromosomes. Each chromosome is composed of the many smaller segments of DNA referred to as genes. Genes, or combinations of genes, contain coded information that determines an individual’s unique characteristics. The code is found in the specific linear order of the molecules that combine to form the strands of DNA. Genes control both the types of proteins that are made and the rate at which they are produced. Genes never act in isolation; they always interact with other genes and the environment.
All normal human somatic cells contain 46 chromosomes arranged as 23 pairs of homologous (matched) chromosomes; one chromosome of each pair is inherited from each parent. There are 22 pairs of autosomes that control most traits in the body, and one pair of sex chromosomes. Whereas the Y chromosome is primarily concerned with sex determination, the X chromosome contains genes that are involved in much more than sex determination. The larger female chromosome is called the X; the smaller male chromosome is the Y. Generally the presence of a Y chromosome causes an embryo to develop as a male; in the absence of a Y chromosome, the individual develops as a female. Thus in a normal female, the homologous pair of sex chromosomes are XX, and in a normal male, the homologous pair are XY.
Homologous chromosomes (except the X and Y chromosomes in males) have the same number and arrangement of genes. In other words, if one chromosome has a gene for hair color, its partner chromosome also will have a gene for hair color, and these hair-color genes will have the same loci or be located in the same place on the two chromosomes. Although both genes code for hair color, they may not code for the same hair color. Genes at corresponding loci on homologous chromosomes that code for different forms or variations of the same trait are called alleles. An individual having two copies of the same allele for a given trait is said to be homozygous for that trait; with two different alleles, the person is heterozygous for the trait.
The term genotype typically is used to refer to the genetic makeup of an individual when discussing a specific gene pair, but at times genotype is used to refer to an individual’s entire genetic makeup or all the genes that the individual can pass on to future generations. Phenotype refers to the observable expression of an individual’s genotype, such as physical features, a biochemical or molecular trait, and even a psychologic trait. A trait or disorder is considered dominant if it is expressed or phenotypically apparent when only one copy of an allele associated with the trait is present. It is considered recessive if it is expressed only when two copies of the alleles associated with the trait are present.
As more is learned about genetics and genomics, the concepts of dominance and recessivity have become more complex, especially in X-linked disorders. For example, traits considered to be recessive may be expressed even when only one copy of a gene located on the X chromosome is present. This occurs frequently in males because males have only one X chromosome; thus they have only one copy of the gene located on the X chromosome. Whichever gene is present on the one X chromosome determines which trait is expressed. Females, conversely, have two X chromosomes, so they have two copies of the genes located on the X chromosome. However, in any female somatic cell, only one X chromosome is functioning (otherwise, there would be inequality in gene dosage between males and females). This process, known as X-inactivation or the Lyon hypothesis, is generally a random occurrence. That is, there is a fifty-fifty chance as to whether the maternal X or the paternal X is inactivated. Occasionally the percentage of cells that have the X with an abnormal or mutant gene is very high. This helps explain why hemophilia, an X-linked recessive disorder, can clinically manifest itself in a female known to be a heterozygous carrier (a female who has only one copy of the gene mutation). It also helps explain why traditional methods of carrier detection are less effective for X-linked recessive disorders; the possible range for enzyme activity values can vary greatly, depending on which X chromosome is inactivated.
Chromosomal Abnormalities
Chromosomal abnormalities are a major cause of reproductive loss, congenital problems, and gynecologic disorders; the incidence is approximately 0.6% in newborns, 6% in stillbirths, and 60% in spontaneous abortions (Martin, 2008). Errors resulting in chromosomal abnormalities can occur during mitosis (cell division occurring in somatic cells that results in two identical daughter cells containing a diploid number of chromosomes) or meiosis (division of a sex cell into two and four haploid cells). These errors can occur in either the autosomes or the sex chromosomes. Even without the presence of obvious structural malformations, small deviations in chromosomes can cause problems in fetal development.
The pictorial analysis of the number, form, and size of an individual’s chromosomes is known as a karyotype. Cells from any nucleated, replicating body tissue (not red blood cells, nerves, or muscles) can be used. The most commonly used tissues are white blood cells and fetal cells in amniotic fluid. The cells are grown in a culture and arrested when they are in metaphase (during metaphase, the chromosomes are condensed and visible with a light microscope), and then the cells are dropped onto a slide. This breaks the cell membranes and spreads the chromosomes, making them easier to visualize. Next the cells are stained with special stains (e.g., Giemsa stain) that create striping or “banding” patterns. These patterns aid in the analysis because they are consistent from person to person. Once the chromosome spreads are photographed or scanned by a computer, they are cut out and arranged in a specific numeric order according to their length and shape. The chromosomes are numbered from largest to smallest, 1 to 22, and the sex chromosomes are designated by the letter X or Y. Each chromosome is divided into two “arms” designated by p (short arm) and q (long arm). A female karyotype is designated as 46, XX and a male karyotype is designated as 46, XY. Figure 3-1 illustrates the chromosomes in a body cell.
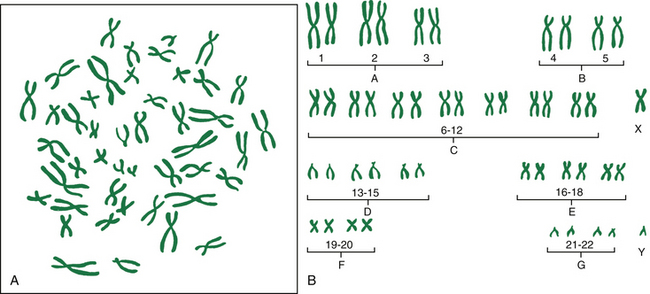
FIG. 3-1 Chromosomes during cell division. A, Example of photomicrograph. B, Chromosomes arranged in karyotype; female and male sex-determining chromosomes.
Autosomal Abnormalities
Autosomal abnormalities involve differences in the number or structure of autosome chromosomes (pairs 1 through 22). They result from unequal distribution of genetic material during gamete (egg and sperm) formation.
Abnormalities of Chromosome Number: A euploid cell is a cell with the correct or normal number of chromosomes within the cell. Since most gametes are haploid (1N, 23 chromosomes) and most somatic cells are diploid (2N, 46 chromosomes), they are both considered euploid cells. Deviations from the correct number of chromosomes per cell can be one of two types: (1) polyploidy, in which the deviation is an exact multiple of the haploid number of chromosomes or one chromosome set (23 chromosomes); or (2) aneuploidy, in which the numerical deviation is not an exact multiple of the haploid set. A triploid (3N) cell is an example of a polyploidy. It has 69 chromosomes. A tetraploid (4N) cell, also an example of a polyploidy, has 92 chromosomes.
Aneuploidy is the most commonly identified chromosome abnormality in humans and the leading genetic cause of mental retardation. A monosomy is the product of the union between a normal gamete and a gamete that is missing a chromosome. Monosomic individuals only have 45 chromosomes in each of their cells. The product of the union of a normal gamete with a gamete containing an extra chromosome is a trisomy. The most common autosomal aneuploid conditions involve trisomies. Trisomic individuals have 47 chromosomes in most or all their cells.
The vast majority of trisomies occur during oogenesis (the process by which a premeiotic female germ cell divides into a mature egg) and the incidence of these types of chromosomal errors increases exponentially with advancing maternal age (Hassold & Hunt, 2009; Hunt & Hassold, 2008). Although variation exists among trisomies with regard to the parent and stage of origin of the extra chromosome, most trisomies are maternal meiosis I (MI) errors. This means that most trisomies are caused by nondisjunction during the first meiotic division. The first meiotic division involves the segregation of homologous or similar chromosomes. One pair of chromosomes fails to separate. One resulting cell contains both chromosomes, and the other contains none. The fact that most trisomies are maternal MI errors is not that surprising, because maternal MI occurs over a long time span. It is initiated in precursor cells during fetal development, but it is not completed until the time those cells undergo ovulation after menarche.
The most common trisomal abnormality is Down syndrome (DS). Approximately one in every 733 newborns has DS and it is estimated that there are more than 400,000 individuals with DS living in the United States (Canfield, Honein, Yuskiv, Xing, Mai, Collins, et al., 2006). Ninety-five percent of individuals with DS have trisomy 21 or an extra chromosome 21 (47, XX +21, female with DS; or 47, XY +21, male with DS). Another type of DS, translocation, occurs when extra chromosome 21 material is present in every cell of the individual but it is attached to another chromosome. In the third type of DS, mosaicism, extra chromosome 21 material is found in some but not all of the cells.
Although the clinical presentation of DS is complex and variable (Ranweiler, 2009), all individuals with DS have some level of mental retardation. Common characteristics seen in individuals with Down syndrome are as follows:
• Oblique palpebral fissures or an upward slant to the eyes
• Epicanthal folds or small skin folds on the inner corners of the eyes
• Small, white, crescent-shaped spots on the irises called Brushfield spots
• A flat facial profile that usually includes a somewhat depressed nasal bridge and a small nose
• Enlargement of the tongue in relationship to size of the mouth
• Small ears, which may be abnormally shaped or abnormally rotated
• Short, broad hands with a fifth finger that has one flexion crease instead of two
• A single deep crease across the center of the palm, often referred to as a simian crease
• Excessive space between large and second toe
• Hyperflexibility, an excessive ability to extend the joints
Some individuals with DS have all of these characteristics, but others have only a few. Figure 3-2 is a picture of an infant with DS who has some of the characteristics commonly associated with that syndrome (see Nursing Care Plan).
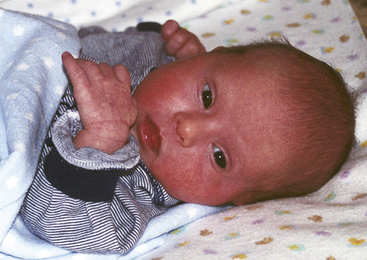
FIG. 3-2 Infant with Down syndrome. Note upward slant to eyes, flat nasal bridge, slightly protruding tongue, and mottled skin. (Courtesy Thomas and Christie Coghill, Clayton, NC.)
Congenital abnormalities and diseases found in individuals with DS are the same as those that occur in the general population, but individuals with DS are affected more often and more severely by specific abnormalities and diseases than are typically developing individuals. For example, congenital heart disease occurs in 40% to 50% of individuals with DS, whereas the overall incidence of congenital heart disease in the general populations is around 0.8%. Leukemia occurs in 1 of every 150 children with DS. This is 20 times higher than in the general population. Other health problems commonly seen in individuals with DS include vision and hearing defects, sleep apnea, thyroid disease, atlantoaxial instability, and gastrointestinal abnormalities. Individuals with DS have a higher mortality rate from infectious disease than do individuals who do not have DS (see Chapter 36).
Although the risk of having a child with DS increases with maternal age (incidence is approximately 1 in 1200 for a 25-year-old woman; 1 in 350 for a 35-year-old woman; and 1 in 30 for a 45-year-old woman), children with Down syndrome can be born to mothers of any age (National Down Syndrome Society, 2010). Eighty percent of children with Down syndrome are born to mothers younger than 35 years. The risk of a mother having a second child with Down syndrome is about 1% when the cause of the Down syndrome is trisomy 21.
During the past 30 years fundamental changes have occurred in the care of individuals with DS. These changes, which underscore the importance of the family and emphasize the need for health promotion and health protection activities, have resulted in individuals with DS living longer and enjoying an improved
 NURSING CARE PLAN
NURSING CARE PLAN
The Family Living with a Child with Down Syndrome
Interrupted family processes related to birth of a neonate with Down syndrome
The parents will verbalize accurate information about Down syndrome, including implications for future pregnancies.
Nursing Interventions/Rationales
• Assess knowledge base of parents regarding the clinical signs and symptoms of Down syndrome to correct any misconceptions and establish basis for teaching plan.
• Provide information throughout the genetic evaluation regarding risk status and clinical signs and symptoms of Down syndrome to give parents a realistic picture of neonate’s defects and assist with decision making for future pregnancies.
• Use therapeutic communication during discussions with the parents to provide opportunity for expression of concern.
• Refer to support groups, social services, or counseling to assist family with cohesive actions and decision making.
• Refer to child development specialist to provide family with realistic expectations regarding cognitive and behavioral differences of child with Down syndrome.
Situational low self-esteem related to diagnosis of Down syndrome as evidenced by parents’ statements of guilt and shame
The parents will express an increased number of positive statements regarding their neonate with Down syndrome.
Nursing Interventions/Rationales
• Assist parents to list strengths and coping strategies that have been helpful in past situations to use appropriate strategies during this situational crisis.
• Encourage expression of feelings using therapeutic communication to provide clarification and emotional support.
• Clarify and provide information regarding Down syndrome to decrease feelings of guilt and gradually increase feelings of positive self-esteem.
• Refer for further counseling as needed to provide more in-depth and ongoing support.
Risk for impaired parenting related to birth of neonate with Down syndrome
Nursing Interventions/Rationales
• Assist parents to see and describe normal aspects of infant to promote bonding.
• Encourage and assist with breastfeeding if that is parents’ choice of feeding method to facilitate closeness with infant and provide benefits of breast milk.
• Assure parents that information regarding the neonate will remain confidential to assist the parents to maintain some situational control and allow for time to work through their feelings.
• Discuss and role-play with parents ways of informing family and friends of infant’s diagnosis and prognosis to promote positive aspects of infant and decrease potential isolation from social interactions.
• Provide anticipatory guidance about what to expect as infant develops to assist family to facilitate optimum development of their infant.
Spiritual distress related to situational crisis of child born with Down syndrome
Parents will seek appropriate support persons (family members, priest, minister, rabbi) for assistance.
Nursing Interventions/Rationales
• Listen for cues indicative of parents’ feelings (“Why did God do this to us?”) to identify messages indicating spiritual distress.
• Acknowledge parents’ spiritual concerns and encourage expression of feelings to help build a therapeutic relationship.
• Facilitate visits from clergy and provide privacy during visits to demonstrate respect for parents’ relationship with clergy.
• Encourage parents to discuss concerns with clergy to use expert spiritual care resources to help the parents.
• Facilitate interaction with family members and other support persons to encourage expressions of concern and seek comfort.
Social isolation related to full-time caretaking responsibilities for a neonate with Down syndrome
Nursing Interventions/Rationales
• Provide opportunity for parents to express feelings about caring for a neonate with Down syndrome to facilitate effective communication and trust.
• Discuss with parents their expectations about caring for the neonate to identify potential areas of concern.
• Assist parents to identify potential caregiving resources to permit parents to return to a routine at home.
• Identify appropriate referrals for home care to provide continuity of care.
quality of life. The life expectancy for individuals with DS has increased from 9 years in 1929 to at least 50 to 60 years. For decades it was assumed that living in a family that includes an individual with DS was a negative experience. Findings from more recent studies do not provide support for this notion (Cuskelly, Hauser-Cram, & Van Riper, 2009; Van Riper, 2007). Many families living with DS have described the experience as positive and growth producing (see also Chapter 36).
Other autosomal trisomies that maternity nurses may see in practice are trisomy 18 and trisomy 13. Trisomy 18 (Edward syndrome) is more common than trisomy 13 (Patau syndrome); it occurs in about 1 out of every 3000 live births versus 1 out of every 10,000 live births for trisomy 13. Infants with trisomy 18 may exhibit more than 130 different anomalies, but some of the major phenotypic features and medical complications are small for gestational age or low birth weight; craniofacial abnormalities including cleft lip and/or palate, small mouth, and small jaw; weak cry; feeding difficulties; cardiac malformations; central nervous system manifestations including hypertonia, seizures, and apnea; and extremity malformations such as small fingernails and toenails, clenched fist with index finger overlapping the third finger, and rocker-bottom feet (Shaw, 2008).
As with trisomy 18, infants with trisomy 13 have numerous abnormalities, the most common of which are central nervous system anomalies, visual abnormalities, microcephaly (small head), absent nasal bridge, cleft lip and palate, holoprosencephaly (one large eyelike structure in the center of the face due to fusion of the developing eyes), capillary hemangiomas, cardiac defects, extremity deformities including polydactyly (extra fingers or toes), renal abnormalities, and genital abnormalities (Wirth, 2009).
Infants with trisomy 18 and trisomy 13 are usually severely to profoundly retarded. Although both conditions have a poor prognosis, with the vast majority of affected infants dying before they reach their first birthday, a growing number of infants with these trisomies are living longer and a small number are actually living into their 20s and 30s.
Nondisjunction also can occur during mitosis. If this occurs early in development when cell lines are forming, the individual has a mixture of cells, some with a normal number of chromosomes and others either missing a chromosome or containing an extra chromosome. This condition is known as mosaicism. The most common form of mosaicism in autosomes is mosaic Down syndrome.
Depending on when the nondisjunction occurs during development, different body tissues will have different numbers of chromosomes. The clinical characteristics of DS may be mild or with varying degrees of severity, depending on the number and location of the abnormal cells. An individual with mosaic DS may have normal intelligence. Mosaicism of both trisomy 18 and trisomy 13 has been reported. Both situations usually lead to a partial clinical expression of the phenotype. Infants who have mosaic trisomy 18 or trisomy 13 usually have a longer life span than infants with these disorders who are not mosaic.
Abnormalities of Chromosome Structure
Structural abnormalities can occur in any chromosome. Types of structural abnormalities include translocation, duplication, deletion, microdeletion, and inversion. Translocation results when there is an exchange of chromosomal material between two chromosomes. Exposure to certain drugs, viruses, and radiation can cause translocations, but often they arise for no apparent reason. The two major types of translocation are reciprocal and robertsonian. Reciprocal translocations are the most common. In a reciprocal translocation, either the parts of the two chromosomes are exchanged equally (balanced translocation) or a part of a chromosome is transferred to a different chromosome, creating an unbalanced translocation because there is extra chromosomal material—extra of one chromosome but correct amount or deficient amount of the other chromosome. In a balanced translocation, the individual is phenotypically normal because there is no extra chromosome material; it is just rearranged. In an unbalanced translocation, the individual will be both genotypically and phenotypically abnormal.
In a robertsonian translocation, the short arms (p arms) of two different acrocentric chromosomes (chromosomes with very short p arms) break, leaving sticky ends that then cause the two long arms (q arms) to stick together. This forms a new, large chromosome that is made of the two long arms. The individual with a balanced robertsonian translocation has 45 chromosomes. Because the short arm of acrocentric chromosomes contains genes for ribosomal RNA and these genes are represented elsewhere, the individual usually does not show any symptoms.
Deletions result in the loss of chromosomal material and partial monosomy for the chromosome involved. Loss of chromosomal material at the end of a chromosome is referred to as a terminal deletion. In contrast, loss of chromosomal material anywhere else in the chromosome is called an interstitial deletion. The resulting clinical phenotype of either a terminal or an interstitial deletion depends on how much of the chromosome has been lost and the number and function of the genes contained in the missing segment. Microdeletions are deletions too small to be detected by standard cytogenetic techniques. These deletions can be identified with FISH analysis. FISH technology uses a single-stranded piece of DNA with a fluorescent label that adheres to its complementary piece of DNA in the chromosome being investigated.
Whenever a portion of a chromosome is deleted from one chromosome and added to another, the gamete produced can have either extra copies of genes or too few copies. The clinical effects produced can be mild or severe, depending on the amount of genetic material involved. Two of the more common conditions are the deletion of the short arm of chromosome 5 (cri du chat syndrome) and the deletion of the long arm of chromosome 18. Cri du chat syndrome, so named after the typical mewing cry of the affected infant, causes severe mental retardation with microcephaly and unusual facial appearance. Deletion of the long arm of chromosome 18 causes severe psychomotor retardation with multiple organ malformations. Velocardiofacial syndrome, characterized by cardiac and craniofacial abnormalities, is an example of a microdeletion. In this syndrome, a very small piece of the long arm of chromosome 22 is missing. Microdeletions in the Y chromosome have been found in men with infertility problems.
Inversions are deviations in which a portion of the chromosome has been rearranged in reverse order. Few birth defects have been attributed to the presence of inversions, but it is suspected that inversions may be responsible for problems with infertility and miscarriages. Some inversions can be detected prenatally. Inversions do not appear to occur randomly; more than 40% of all inversions involve chromosome 9 (Lashley, 2005).
Sex Chromosome Abnormalities
Several sex chromosome abnormalities are caused by nondisjunction during gametogenesis in either parent. The most common deviation in females is Turner syndrome or monosomy X (45, X). The affected female is missing an X chromosome. She usually exhibits juvenile external genitalia with undeveloped ovaries. She is short and often has webbing of the neck, a low hairline in the back, low-set ears, and lymphedema of her hands and feet. Intelligence may be impaired. Most affected embryos miscarry spontaneously. In most cases of Turner syndrome, it is the paternal X or Y that is lost.
The most common deviation in males is Klinefelter syndrome, or trisomy XXY. The affected male has an extra X chromosome and exhibits poorly developed secondary sexual characteristics and small testes. He is infertile, usually tall, and may be slow to learn (www.genetic.org). Males who have mosaic Klinefelter syndrome may be fertile.
Patterns of Genetic Transmission
Heritable characteristics are those that can be passed on to offspring. The patterns by which genetic material is transmitted to the next generation are affected by the number of genes involved in the expression of the trait. Many phenotypic characteristics result from two or more genes on different chromosomes acting together (referred to as multifactorial inheritance); others are controlled by a single gene (referred to as unifactorial inheritance). Specialists in genetics (e.g., geneticists, genetic counselors, and nurses with advanced expertise in genetics and genomics) predict the probability of the presence of an abnormal gene from the known occurrence of the trait in the individual’s family and the known patterns by which the trait is inherited.
Multifactorial Inheritance
Most common congenital malformations result from multifactorial inheritance, a combination of genetic and environmental factors. Examples are cleft lip, cleft palate, congenital heart disease, neural tube defects, and pyloric stenosis. Each malformation can range from mild to severe, depending on the number of genes for the defect present or the amount of environmental influence. A neural tube defect can range from spina bifida, a bony defect in the lumbar region of the vertebrae with little or no neurologic impairment, to anencephaly, absence of brain development, which is always fatal. Some malformations occur more often in one sex. For example, pyloric stenosis and cleft lip are more common in males, and cleft palate is more common in females.
Unifactorial Inheritance
If a single gene controls a particular trait or disorder, its pattern of inheritance is referred to as unifactorial mendelian, or single-gene inheritance. The number of single-gene disorders far exceeds the number of chromosomal abnormalities. Potential patterns of inheritance for single-gene disorders include autosomal dominant, autosomal recessive, and X-linked dominant and recessive modes of inheritance.
Autosomal Dominant Inheritance: Autosomal dominant inheritance disorders are those in which only one copy of a variant allele is needed for phenotypic expression. The variant allele may be a result of a mutation, a spontaneous and permanent change in the normal gene structure, in which case the disorder occurs for the first time in the family. Usually an affected individual comes from multiple generations having the disorder. An affected parent who is heterozygous for the trait has a 50% chance of passing the variant allele to each offspring (Fig. 3-3, B and C). There is a vertical pattern of inheritance (there is no skipping of generations; if an individual has an autosomal dominant disorder such as HD, so must one of the parents). Males and females are equally affected.
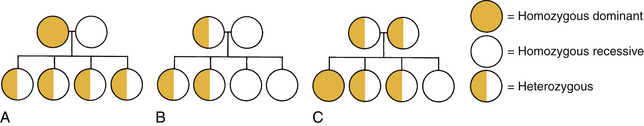
FIG. 3-3 Possible offspring in three types of matings. A, Homozygous-dominant parent and homozygous-recessive parent: children all heterozygous, displaying dominant trait. B, Heterozygous parent and homozygous recessive parent: children 50% heterozygous, displaying dominant trait; 50% homozygous, displaying recessive trait. C, Both parents heterozygous: children 25% homozygous, displaying dominant trait; 25% homozygous, displaying recessive trait; 50% heterozygous, displaying dominant trait.
Autosomal dominant disorders are not always expressed with the same severity of symptoms. For example, a woman who has an autosomal dominant disorder may show few symptoms and may not become aware of her diagnosis until after she gives birth to a severely affected child. Predicting whether an offspring will have a minor or severe abnormality is not possible. Examples of autosomal dominant disorders are HD, Marfan syndrome, neurofibromatosis, myotonic dystrophy, Stickler syndrome, Treacher Collins syndrome, and achondroplasia (dwarfism).
Neurofibromatosis (NF) is a progressive disorder of the nervous system that causes tumors to form on nerves anywhere in the body. NF affects all races, all ethnic groups, and both sexes equally. Half of the cases of NF result from a spontaneous genetic mutation, whereas the other half of cases are inherited in an autosomal dominant manner. Individuals with NF due to a spontaneous mutation have a 50% chance of transmitting the variant allele to the next generation with each pregnancy. Two genetically distinct forms of NF include NF1, the most common type, with an incidence of 1 in 4000 and NF2, with an incidence of 1 in 40,000 (Clarke, 2009). The most distinctive features of NF1 are multiple neurofibromas (benign, soft tumors), freckles in the axilla or groin, and patches of skin pigmentation called café-au-lait spots. Generally, symptoms of NF1 are mild and affected individuals are able to live healthy, productive lives. Individuals with NF2 typically develop bilateral vestibular schwannomas (tumors on the eighth cranial nerves, the hearing and balance nerves) that often cause pressure damage to nearby nerves, which may result in headaches, facial pain, and facial numbness. Other symptoms typically experienced by individuals with NF2 include tinnitus (ringing noise in the ear) and poor balance. Some individuals with NF2 experience hearing loss in their teen years.
No treatment for NF is available, other than the surgical removal of the tumors. Once removed, the tumors may grow back.
Factor V Leiden (FVL) is the most common inherited risk factor for primary and recurrent venous thromboembolisms (Moll, 2006). It is an autosomal dominant disorder that markedly increases an individual’s risk for deep vein thrombosis (blood clots in the large veins of the legs) and pulmonary emboli (blood clots that travel through the bloodstream and become embedded in the lungs), especially if the individual is a woman who (1) uses oral contraceptives, (2) is pregnant, or (3) is on hormone replacement therapy during menopause. FVL is due to a mutation in the Factor V gene, which leads to activated protein C (APC) resistance. If a woman is heterozygous (has inherited one copy of the FVL mutation), she has a four- to eightfold increase in her chance of developing venous blood clots, but her risk increases by a factor as high as 75 to 80 if she is homozygous (has inherited two copies of the FVL mutation) (National Institutes of Health, 2010). Women who carry the FVL mutation should not take oral contraceptives. In addition, if they become pregnant and they have a history of blood clots, it is recommended that they receive prophylactic anticoagulation (with low-molecular-weight heparin) during their pregnancy and for 6 weeks postpartum (Marik & Plante, 2008).
FVL can be accurately detected with genetic testing, but the most cost-effective way to screen for FVL is taking a careful individual and family history. Women with a personal or close family history of venous blood clots, pulmonary emboli, early onset and recurrent preeclampsia, recurrent fetal growth restriction, recurrent pregnancy loss and stillbirth, or placental abruption should be screened for FVL.
Autosomal Recessive Inheritance: Autosomal recessive inheritance disorders are those in which both genes of a pair are forms associated with the disorder to be expressed. Heterozygous individuals have only one variant allele and are unaffected clinically because their normal gene (wild-type allele) overshadows the variant allele. They are known as carriers of the recessive trait. Because these recessive traits are inherited by generations of the same family, an increased incidence of the disorder occurs in consanguineous matings (closely related parents). For the trait to be expressed, two carriers must each contribute a variant allele to the offspring (see Fig. 3-3, C). The chance of the trait occurring in each child is 25%. A clinically normal offspring may be a carrier of the gene. Autosomal recessive disorders have a horizontal pattern of inheritance, rather than the vertical pattern seen with autosomal dominant disorders. That is, autosomal recessive disorders are usually observed in one or more siblings, but not in earlier generations. Males and females are equally affected. Most recessive disorders tend to have severe clinical manifestations, and affected offspring may not reproduce. If they do, all their offspring will at least be carriers for the disorder. Most inborns errors of metabolism (IEMs), such as PKU, galactosemia, maple syrup urine disease, Tay-Sachs disease, sickle cell anemia, and CF, are autosomal recessive inherited disorders.
Inborn Errors of Metabolism: More than 350 inborn errors of metabolism have been recognized (Jorde, Carey, & Bamshad, 2010). Individually, IEMs are relatively rare, but collectively, they are common (1 in 5000 live births). Archibald Garrod first used the term “inborn errors of metabolism” in 1908 when he described variants of metabolism. Garrod recognized that IEMs illustrate our “chemical individualities.” As noted previously, most IEMs are inherited in an autosomal recessive pattern. IEMs occur when a gene mutation reduces the efficiency of encoded enzymes to a level at which normal metabolism cannot occur. Defective enzyme action interrupts the normal series of chemical reactions from the affected point onward. The result may be an accumulation of a damaging product, such as phenylalanine in PKU, or the absence of a necessary product, such as the lack of melanin in albinism caused by lack of tyrosinase. Diagnostic and carrier testing is available for a growing number of IEMs. In addition, many states have started screening for specific IEMs as part of their expanded newborn screening programs using tandem mass spectrometry. However, many of the deaths caused by IEMs are due to enzyme variants not currently screened for in many of the newborn screening programs (Jorde et al.).
Phenylketonuria is a relatively uncommon autosomal recessive disorder. A deficiency in the liver enzyme phenylalanine hydroxylase results in failure to metabolize the amino acid phenylalanine, allowing its metabolites to accumulate in the blood. The incidence of this disorder is 1 in every 10,000 to 20,000 births. The highest incidence is found in Caucasians (from northern Europe and the United States). It is rarely seen in Jewish, African, or Japanese populations. Screening for PKU is routinely performed as part of state-mandated newborn screening in the United States.
Tay-Sachs disease is a lipid-storage disease that occurs more commonly in Ashkenazi Jews and French-Canadians from Quebec (Lashley, 2005). It results from a deficiency in hexosaminidase. Until age 4 to 6 months, infants with Tay-Sachs disease appear normal; their facial features are considered very beautiful. Then the clinical symptoms appear: apathy and regression in motor and social development and decreased vision. Death occurs between ages 3 and 4 years. No treatment exists for Tay-Sachs disease.
Infantile Krabbe’s disease or globoid-cell leukodystrophy is a lysosomal storage disorder characterized by failure of the process of myelination in the central and peripheral nervous systems, rapidly progressive neurologic deterioration, and death (often before the age of 2 years). Findings from a study by Escolar and colleagues (2005) suggest that transplantation of umbilical-cord blood from unrelated donors in newborns with infantile Krabbe’s disease favorably altered the natural history of this disease. Infants who underwent transplantation before the development of symptoms demonstrated continued gains in developmental skills and progressive central myelination, and most had age-appropriate cognitive function and receptive language skills. However, if transplantation occurred after the onset of symptoms, there was minimal neurologic improvement. These findings have implications for decisions regarding the addition of screening for lysosomal storage disorders to existing newborn screening programs.
X-linked Dominant Inheritance: X-linked dominant inheritance mimics autosomal dominant inheritance, except that male-to-male transmission cannot occur unless the father has Klinefelter syndrome due to XY disomy (Simpson & Elias, 2003). X-linked dominant inheritance disorders occur in males and heterozygous females, but because of X inactivation, affected females are usually less severely affected than affected males and they are more likely to transmit the abnormal gene (variant allele) to their offspring (Lashley, 2005). Heterozygous females (females who have one wild-type allele and one variant allele) have a 50% chance of transmitting the abnormal gene (variant allele) to each offspring. The variant allele is often lethal in affected males because, unlike affected females, they have no normal gene (wild-type allele). Mating of an affected male and an unaffected female is uncommon as a result of the tendency for the variant allele to be lethal in affected males. Relatively few X-linked dominant disorders have been identified. Two examples are vitamin D–resistant rickets and Rett syndrome.
X-linked Recessive Inheritance: Abnormal genes for X-linked recessive inheritance disorders are carried on the X chromosome. Females may be heterozygous or homozygous for traits carried on the X chromosome because they have two X chromosomes. Males are hemizygous because they have only one X chromosome, which carries genes with no alleles on the Y chromosome. Therefore X-linked recessive disorders are most commonly manifested in the male, with the abnormal gene on his single X chromosome. Hemophilia, color blindness, and Duchenne muscular dystrophy are X-linked recessive disorders.
A man with an X-linked recessive disorder receives the disease-associated allele from his carrier mother on her affected X chromosome. Female carriers (those heterozygous for the trait) have a 50% probability of transmitting the disease-associated allele to each offspring. An affected man can pass the disease-associated allele to his daughters, but not to his sons. The daughters will be carriers of the trait if they receive a normal gene on the X chromosome from their mother. They will be affected only if they receive a disease-associated allele on the X chromosome from both their mother and their father.
Fragile X syndrome (FXS), the most common inherited form of cognitive impairment, is an X-linked disorder that has a complex pattern of inheritance. FXS is caused by a trinucleotide repeat expansion (CGG) at a “fragile site” on the long arm of the X chromosome. Most people have 5 to 40 CGG repeats. Individuals with FXS have more than 200 CGG repeats. The abnormally expanded CGG segment inactivates or silences the FMR1 (fragile X mental retardation) gene, which prevents the gene from producing a protein called fragile X mental retardation protein. Loss of this protein leads to the characteristic physical features (large ears, long face, prominent forehead, protruding ears, hypermobile joints, and macroorchidism or increased testicular volume in postpubertal males) and behavior problems (poor eye contact, hyperactivity, social avoidance, repetitive speech, and self-injurious behavior) associated with FXS (Jorde et al., 2010; Schneider, Hagerman, & Hessl, 2009). Males and females can be affected by FXS, but because males have only one X chromosome, a CGG repeat expansion on one X is likely to affect males more severely than females. Also, the degree of cognitive impairment tends to be milder and more variable in females than in males. Unlike DS, FXS is not generally detectable through a physical examination at birth. Delays and behavioral abnormalities gradually become apparent during the first 2 years of life, but ultimately the diagnosis of FXS can be verified only through DNA testing (Sherman, Pletcher, & Driscoll, 2005).
Individuals with more than 55 but fewer than 200 CGG repeats are said to be permutation carriers. These individuals were originally thought to be unaffected, but recent research has shown that about 20% of adult carrier females may develop premature ovarian failure (cessation of menses before 40 years of age). Elderly male permutation carriers may manifest fragile X-associated tremor/ataxia syndrome (FXTAS), which consists of parkinsonism, intention tremors, autonomic dysfunction, peripheral neuropathy, weakness in the legs, cognitive decline, and cerebellar ataxia (www.nfx.org).
Cancer Genomics
Gene Mutations That Can Lead to Cancer
There are three main ways that people acquire gene mutations that can lead to cancer. The first is from the environment. Known factors in the environment that cause cancer are ultraviolet (UV) light (skin cancer) and tobacco smoke (lung cancer). The second way that people acquire mutations is by chance. Normal metabolic processes can generate chemicals that damage DNA. Third, people inherit mutations from their parents; hereditary mutations are thought to be a major factor in about 5% to 10% of all cancers.
The two main types of genes that have been recognized as playing a critical role in the development of cancer are oncogenes and tumor suppressor genes (American Cancer Society, 2010). Oncogenes are mutated forms of proto-oncogenes. The main functions of proto-oncogenes are to encourage and promote normal growth and development. When proto-oncogenes mutate to become carcinogenic oncogenes, the result is excessive cell multiplication. The activation of oncogenes has been compared to a jammed accelerator in a car. Most mutations of proto-oncogenes are acquired mutations, such as mutations in the KIT gene which are thought to cause most cases of gastrointestinal stromal tumor (GIST). This type of cancer can be treated with drugs that target the KIT gene, such as imatinib (Gleevec). Two examples of inherited mutations of proto-oncogenes are ERBB2, located on chromosome 13, and KRAS2, located on chromosome 12. ERBB2 is involved in breast, ovarian, lung, gastric, and salivary gland cancers. KRAS2 is involved in breast, pancreatic, thyroid, colorectal, bladder, and lung cancers, as well as acute myeloid leukemia.
Tumor suppressor genes normally function to inhibit or “put the brakes on” the cell growth and division cycle. They function to prevent the development of tumors. Mutations in tumor suppressor genes cause the cell to ignore one or more of the components of the network of inhibitory signals, removing the brakes from the cell cycle. This results in a higher rate of uncontrolled growth: cancer. Acquired mutations of the TP53 gene appear in a wide range of cancers, including lung, colorectal, and breast cancer. Examples of inherited tumor suppressor genes include APC, located on chromosome 5 and involved with familial adenomatous polyposis of the colon (FAP); BRCA1, located on chromosome 17 and associated with hereditary breast cancer and ovarian cancer; and RB1, found on chromosome 13 and involved with familial retinoblastoma.
Hereditary Breast and Ovarian Cancer
Breast cancer is a common disease and a central concern in women’s health. In the United States, breast cancer is the most common form of cancer for women and the second most common cause of death. Hereditary mutations are considered to be a key factor in approximately 5% to 10% of all breast and ovarian cancers. Another 15% to 20% of female breast cancers occur in women who have a family history of breast and ovarian cancer but do not carry a mutation in one of the genes that are known to be strongly associated with breast and ovarian cancer susceptibility.
BRCA1 and BRCA2 mutations account for approximately 70% to 85% of hereditary breast and ovarian cancer (HBOC). These mutations are inherited in an autosomal dominant pattern; thus each offspring of an individual found to carry a BRCA mutation has a 50% chance of inheriting the same mutation. Ashkenazi Jews are 10 times more likely to have BRCA1 and/or BRCA2 mutations than are the general population. According to estimates of lifetime risk, approximately 12% of women in the general population will develop breast cancer sometime during their lifetime, compared to about 60% of women who have inherited a deleterious mutation in their BRAC1 or BRCA2 gene (National Cancer Institute, 2010). Another way of saying this is that a woman with a deleterious BRCA1 or BRCA2 mutation is about five times more likely to develop breast cancer than a woman who does not carry a deleterious BRCA1 or BRCA2 mutation. Even though only about 6% of the men who carry a BRCA mutation develop breast cancer, men who carry a BRCA mutation have a 50% chance of passing the mutation on to their offspring. As far as lifetime risk estimates for ovarian cancer, about 1.4% of women in the general population will be diagnosed with ovarian cancer during their lifetime, compared with 15% to 40% of women who have a deleterious BRCA 1 or BRCA2 mutation. Carriers of BRCA1 mutations may also be at increased risk for colon cancer, though this has been disputed. Carriers of BRCA2 mutations may be at increased risk for pancreatic, prostate, gallbladder and bile duct, and stomach cancers, hematologic malignancies, and malignant melanoma.
Genetic testing for HBOC has been commercially available in the United States since 1995. The cost for testing (current in 2010) is approximately $3,000 for full-sequence BRCA1 and BRCA2 testing, and $350 for an analysis of relatives of an individual with an identified mutation. Women newly diagnosed with breast cancer are increasingly being asked to consider undergoing BRCA1 and BRCA2 testing before they make decisions about their treatment options. This request for testing is occurring because a number of studies have shown that a woman’s short-term risk of developing a second breast cancer is substantially affected by whether she carries a BRCA1 or BRCA2 mutation, and prophylactic surgery has been found to decrease the risk of breast and ovarian cancer by more than 90% (Hartmann, Degnim, & Schaid, 2004; McDonnell, Schaid, Myers, Grant, Donohue, Woods, et al., 2001; Rebbeck, Friebel, Lynch, Neuhausen, van’t Veer, Garber, et al., 2004). The main advantage to offering BRCA1 and BRCA2 testing before the onset of treatment is that it gives women who are found to carry a deleterious mutation the option of choosing risk-reduction surgery concurrent with therapeutic surgical treatment.
Colon Cancer
Colon cancer is the third leading cause of cancer-related death in women. According to estimates from the American Cancer Society (2010), 102,900 new cases of colon cancer and 39,670 new cases of rectal cancer were diagnosed in 2009 and there were 51,370 deaths due to colorectal cancer. Only 10% of colon cancer cases are likely to involve a mutation in one of several predisposing genes. Two examples of predisposing genes are mutations in the APC tumor suppressor gene and mutations in a mismatch repair gene. Mutations in the APC tumor suppressor gene have been associated with FAP, a rare, autosomal dominant syndrome that accounts for about 1% of all colon cancer. It is typically diagnosed clinically. Affected individuals have 100 to 1000 polyps in their colon by the time they are 20 to 30 years old. Genetic testing is greater than 80% sensitive. Identification of high risk individuals guides surveillance strategies and the timing of a prophylactic colectomy. Low risk individuals can stop the increased surveillance.
Hereditary nonpolyposis colon cancer (HNPCC) results from mutations in one of many mismatch repair (MMR) genes. Mutations in MSH2 and MLH1 account for 50% to 60% of HNPCC. Families at high risk for HNPCC often have three or more relatives with colorectal cancer; colorectal cancer present in at least two generations; and a diagnosis of colorectal cancer before age 50 years in at least one case. HNPCC is characterized by an increased risk of colon cancer and other cancers that include cancers of the ovary, endometrium, stomach, small intestine, upper urinary tract, hepatobiliary tract, brain, and skin. The lifetime risk of colon cancer for individuals with HNPCC is approximately 80%. The majority of these cancers occur in the proximal colon. Genetic tests are available to test for MSH2 and MLH1. Testing should be done first on the affected family member. At-risk clients should be offered a prophylactic colectomy. Women may be offered a total abdominal hysterectomy with a salpingo-oophorectomy to decrease cancer risk. If colon cancer develops, a total colectomy is recommended.
Genetic Counseling
Genetic counseling is a service that grew out of a need for professionals who could provide genetics information, education, and support to individuals and families with ongoing or potential genetic health concerns. In 2010 there were 31 accredited genetic counseling programs in the United States and international genetic counseling programs in 15 different countries. At the same time, there were 2448 American Board of Genetic Counseling (ABGC)–certified genetic counselors. The National Society of Genetic Counselors was formed in 1979 and the International Society of Nurses in Genetics (ISONG) was formed in 1988. The number of nursing programs offering courses in genetics and genomics is growing rapidly. A small number of graduate nursing programs offer advanced practice courses and/or specialty options in genetics and genomics. Examples of these are the University of California San Francisco School of Nursing, the University of Pittsburgh School of Nursing, and the University of Iowa College of Nursing.
Definition of Genetic Counseling
In 1975 an ad hoc committee of the American Society of Human Genetics developed a formal definition of genetic counseling. According to this definition:
Genetic counseling is a communication process that deals with the human problems associated with the occurrence or risk of occurrence of a genetic disorder in a family. This process involves an attempt by one or more appropriately trained person to help the individual or family to (1) comprehend the medical facts including the diagnosis, probable course of the disorder, and the available management; (2) appreciate the way heredity contributes to the disorder and the risk of recurrence in specified relatives; (3) understand the alternatives for dealing with the risk of recurrence; (4) choose a course of action that seems to them appropriate in view of their risk, their family goals, and their ethical and religious standards and act in accordance with that decision; and (5) make the best possible adjustment to the disorder in an affected family member and/or to the risk of recurrence of that disorder.
Access and Referral to Genetic Counseling
Genetic counseling is typically provided by a team of genetics specialists that includes clinical geneticists (physicians with an MD or DO), medical geneticists with a PhD, genetics fellows, genetics counselors, and, in a growing number of cases, advanced practice genetics nurse specialists. Cytogeneticists, biochemical geneticists, and molecular geneticists support the clinical genetics team by providing laboratory expertise that helps with the diagnosis and management of individuals and families affected by genetic conditions.
Until recently, most individuals and families interested in receiving genetic counseling went to regional genetics centers or major medical centers. Genetic counseling also was provided in outreach or satellite genetics clinics, public health clinics, and some community hospitals. Now that genetics is entering the mainstream of health care, genetic counseling is being offered in a wide variety of other settings. These include, but are not limited to, managed health care organizations, commercial facilities, and private practices. A number of specialized groups provide genetics education and counseling for individuals and families affected by specific genetic disorders, such as DS, CF, diabetes, muscular dystrophy, HD, and cancer. Genetic counseling also is offered over the Internet.
Individuals and families seek out, or are referred for, genetic counseling for a wide variety of reasons and at all stages of their lives. Some seek preconception or prenatal information; others are referred after the birth of a child with a birth defect or a suspected genetic condition; still others seek information because they have a family history of a genetic condition. Regardless of the setting or the individual and family’s stage of life, genetic counseling should be offered and available to all individuals and families who have questions about genetics and their health. However, there is currently a shortage of appropriately trained genetics professionals who can provide genetic counseling. This means that many individuals and families will not be offered genetic counseling when they undergo genetic testing. Moreover, some of the genetics education and counseling that is provided will be inadequate.
It may take years before a sufficient number of health care professionals feel comfortable and are proficient in providing genetic counseling (Burke & Kirk, 2006; Cashion, 2009). Until then it is imperative that all health care professionals become familiar with existing genetics resources, such as the Centers for Disease Control and Prevention, the Genetic Alliance, the National Coalition for Health Professional Education in Genetics, Genetics Education Program for Nurses at Cincinnati Children’s Hospital Medical Center, NHGRI Education, and others. Some of these resources may be in health care professionals’ own communities, but others are regional, national, and international.
Estimation of Risk
Most families with a history of genetic disease want an answer to the following question: What is the chance that our future children will have this disease? Because the answer to this question may have profound implications for individual family members and the family as a whole, health care professionals must be able to answer this question as accurately as they can in a timely manner. In some cases, estimation of risk is rather straightforward; in other cases, it is complicated. Because of this, health care professionals should be prepared to refer families with a history of genetic disease to genetics professionals if they are at all unsure. Again, the answer to this question can have profound implications for individual family members and the family as a whole, so health care professionals must do their best to ensure that the question is answered accurately.
If a couple has not yet had children, but they are known to be at risk for having children with a genetic disease, they will be given an occurrence risk. Once the mating of a couple has produced one or more children with a genetic disease, the couple will be given a recurrence risk. Both occurrence and recurrence risks are determined by the mode of inheritance for the genetic disease in question. For genetic diseases caused by a factor that segregates during cell division (genes and chromosomes), risk can be estimated with a high degree of accuracy by application of mendelian principles.
In an autosomal dominant disorder, both the occurrence and recurrence risk is 50%, or one in two, when one parent is affected and the other is not. The recurrence risk for autosomal recessive disorders is 25%, or one in four, if both parents are carriers (they each have one recessive disease gene and one normal gene). Occasionally an individual homozygous for a recessive disease gene mates with an individual who is a carrier of the same recessive gene. In this case, the recurrence risk is 50%, or one in two. If two individuals affected by an autosomal recessive disorder mate, all of their children will be affected (See Fig. 3-3). For X-linked disorders, recurrence risk is related to the sex of the child. Translocation disorders have a high risk of recurrence.
A number of autosomal disorders display fairly complex patterns of inheritance, making estimation of risk somewhat difficult. For example, if a child is born with a genetic disease and there has been no history of the disease in the family, the disease may have been caused by a new mutation (this is more likely if the disease in question is an autosomal dominant disorder, such as achondroplasia). If the child’s genetic disease has been caused by a new mutation, the recurrence risk for the parents’ subsequent children is low (1% to 2%), but it is not as low as that for the general population. Offspring of the affected child may have a substantially elevated occurrence risk.
The risk of recurrence for multifactorial conditions can be estimated empirically. An empiric risk is based not on genetics theory but rather on experience and observation of the disorder in other families. Recurrence risks are determined by applying the frequency of a similar disorder in other families to the case under consideration.
An important concept to be emphasized to individuals and families during a genetic counseling session is that each pregnancy is an independent event. For example, in monogenic disorders in which the risk factor is one in four that the child will be affected, the risk remains the same no matter how many affected children are already in the family. Families may maintain the erroneous assumption that the presence of one affected child ensures that the next three will be free of the disorder. However, “chance has no memory.” The risk is one in four for each pregnancy. Conversely, in a family with a child who has a disorder with multifactorial causes, the risk increases with each subsequent child born with the disorder.
Interpretation of Risk
The guiding principle for genetics counselors has traditionally been nondirectiveness. According to the principle of nondirectiveness, the individual who is providing genetic counseling respects the right of the individual or family being counseled to make autonomous decisions. Counselors using a nondirective approach avoid making recommendations and try to communicate genetics information in an unbiased manner. The first step in providing nondirective counseling is becoming aware of one’s own values and beliefs. Another important step is recognizing how those values and beliefs can influence or interfere with the communication of genetics information.
If the individual who is providing genetic counseling has difficulty being nonjudgmental and objective, he or she may either intentionally or unintentionally influence the decision-making process. Individuals and families also may pressure the counselor to make decisions for them with questions such as, “What would you do if you were me?” Families and individuals need education, guidance, and support throughout the counseling process. They should be given the facts and possible consequences, as well as all of the assistance they need in problem solving, but the final decision regarding a course of action must be their own.
Multiple Roles for Nurses in Genetics
Nurses play many roles in genetics. Some nurses play a key role in the identification of families in need of genetic counseling, and they collaborate with other health care professionals to make referrals to specialists in genetics. Other nurses take a more active role in genetic counseling. For example, these nurses might provide appropriate genetics information before, during, and after the initial genetic counseling session; construct family pedigrees of three or more generations; clarify the genetics information that family members receive during counseling sessions or from other sources such as the public library, the Internet, or support groups; help families manage the ongoing challenges associated with living with a genetic disorder; make referrals to support groups and national organizations; and provide long-term follow-up of families affected by genetic conditions.
Probably the most important of all nursing functions is to provide emotional support during all aspects of the counseling process. Feelings that are generated under the real or imagined threat posed by a genetic disorder are as varied as the people being counseled. Responses may include a variety of stress reactions, such as apathy, denial, anger, hostility, fear, embarrassment, grief, and loss of self-esteem. Guilt and self-blame are universal reactions. Many look on the disorder as a stigma, especially if the disorder is visible to others. Old wives’ tales, superstitions, and long-held misconceptions may influence a family’s reaction to a genetic disorder (Clinical Reasoning).
 CLINICAL REASONING
CLINICAL REASONING
Counseling About Genetic Risk
Sylvia confides in you that several infants have been born into her family with serious anomalies. From previous conversations you know that she is opposed to abortion and would never consider having one. Sylvia is currently 6 weeks pregnant, and her physician has urged her to have chorionic villus sampling (CVS). Sylvia asks you for information about CVS and the implications of a finding that her fetus has serious anomalies.
1. Evidence—Is there sufficient evidence to provide Sylvia with answers to her questions about CVS?
2. Assumptions—What assumptions can be made about CVS and what can be discovered using this technology?
a. The risks of this technology
b. The benefits of this technology
c. The fit between Sylvia’s beliefs and the implications of a positive diagnosis
d. Sylvia’s acceptance of the birth of a child with an anomaly
3. What implications and priorities for nursing can be made at this time?
Future Promise of Genetics
Overall, the Human Genome Project and other sequencing efforts have been a huge success. Our understanding of the human genome, as well as other genomes, has grown exponentially during the past decade. The increased availability of genetic testing and other genetics services gives individuals and families unprecedented opportunities to learn whether they have heightened risk for certain diseases or the potential to transmit gene mutations to their offspring. Awareness of genetic risk also can facilitate informed health care decisions and, in some cases, can promote risk reduction behaviors that have the potential to reduce morbidity and mortality. Ultimately it is hoped that advances in molecular biology and genomics will make it possible to offer diagnostic, preventive, and treatment options not only for genetic diseases but also for common diseases such as cancer, atherosclerosis, diabetes, and Alzheimer’s disease.
Recent advances made possible through the Human Genome Project have been remarkable, but our ability to offer treatment options, even for single-gene disorders, remains limited. Progress in the acquisition of genetics information and the development of genetics technology continues to outpace the development of therapeutic interventions. For most genetic conditions, therapeutic interventions are nonexistent or disappointingly limited. Consequently the most useful means of reducing the incidence of genetic disorders now is preventing transmission. Only three reproductive options exist for individuals at risk for transmitting a genetic disorder: the avoidance of pregnancy; genetic diagnosis during an ongoing pregnancy; and prevention of transmission of an altered gene or genes through preimplantation genetics. For many families, none of these options is viewed as acceptable.
Dialogue among pregnant women, expectant families, health care professionals, and disability advocates concerning prenatal testing for DS and other genetic disorders is urgently needed. Clinical and technical information must be complemented by social understanding of the experience of disability in contemporary society. The picture of life with a disability should be more balanced than that currently portrayed. It is critical that the voices of individuals and families living with disabilities be heard.
Nurses are in an ideal position to help individuals and families maximize the benefits of the genetics revolution, but first, nurses need (1) a working knowledge of human genetics, (2) an awareness of recent advances in genetics and genomics, and (3) an understanding of the potential effects of genomic discoveries on individual and family well-being. More research is needed concerning the family experience of genetic testing. Nurses must understand why individuals and families decide to undergo genetic testing. Nurses also need to be aware of how individuals and families define and manage ethical, legal, and social issues that emerge during the genetic testing experience.
 COMMUNITY ACTIVITY
COMMUNITY ACTIVITY
• Select a hereditary disorder such as cystic fibrosis, muscular dystrophy, hemophilia, Tay-Sachs disease, or sickle cell anemia. Visit the website of the national organization. Locate accredited care centers that are in your community. Do the centers offer preconception counseling?
• Visit the Genetic Alliance website at www.geneticalliance.org. Select a disorder and go to the disease information search link. Review the client information sections about clinical description, insurance issues, research and treatment.
• Share your findings with your classmates in a clinical conference.
KEY POINTS
• Recent advances in molecular biology and genomics have revolutionized the field of health care by providing the tools needed to determine the hereditary component of many diseases.
• Increasingly, nurses from all specialty areas, as well as all practice settings, are expected to have competencies in genetics and genomics.
• The major force behind the genetics revolution has been the Human Genome Project.
• All humans are 99.9% identical at the DNA level.
• Approximately 20,000 to 25,000 genes are found in the human genome.
• Most of the genetic tests being offered in clinical practice are tests for single-gene disorders.
• Pharmacogenomics will probably be the most immediate clinical application of the Human Genome Project.
• The decision to undergo genetic testing is often based on feelings of responsibility and commitment to others.
• Genes are the basic units of heredity responsible for all human characteristics. They comprise 23 pairs of chromosomes: 22 pairs of autosomes and 1 pair of sex chromosomes.
• Chromosomal abnormalities occur in autosomes and sex chromosomes.
• Multifactorial inheritance includes genetic and environmental contributions.
• Advances in genetics have complex ethical, legal, and social implications.
 Audio Chapter Summaries Access an audio summary of these Key Points on
Audio Chapter Summaries Access an audio summary of these Key Points on 