Hemolytic Disorders and Congenital Anomalies
• Differentiate between physiologic and pathologic jaundice in the newborn.
• Develop a nursing plan of care for the prevention, identification, and management of the neonate with hyperbilirubinemia.
• Compare Rh and ABO incompatibility and the implications for neonatal outcomes.
• Explain nursing management to prevent the pathologic consequences of hyperbilirubinemia.
• Review prenatal diagnosis of neonatal disorders.
• Present assessment strategies during the postnatal period to aid in diagnosis of congenital disorders.
• Describe each congenital disorder presented in this chapter and identify the priority of nursing care for each.
• Describe preoperative and postoperative nursing care of the newborn.
• Develop a nursing care plan for parents of a newborn with a defect or disorder.

Many physiologic alterations occur in infants during the newborn period. The nurse must be alert to any deviations from normal, ranging from an obvious congenital anomaly, such as myelomeningocele, to a less obvious deviation such as a congenital heart defect that may not be symptomatic at birth. The nurse must possess the assessment skills necessary to detect any deviation from normal, as well as the knowledge base necessary to participate in the skilled care needed for affected infants. The nurse also must be cognizant of the special needs of the family with a child who is born with or acquires an abnormal condition.
The complications that affect newborns can stem from three basic problems: (1) problems relating to gestational age or intrauterine growth that does not follow normal patterns, such as a preterm birth; (2) acquired problems resulting from maternal or newborn physiologic factors, such as ABO incompatibility or respiratory distress; and (3) physical problems, such as congenital anomalies. This chapter focuses on acquired problems and congenital anomalies in the neonate.
Hyperbilirubinemia
Hyperbilirubinemia is a condition in which the total serum bilirubin level in the blood is increased. Values are abnormal based on gestational age, days of life, and the baby’s general physical condition. Hyperbilirubinemia is characterized by a yellow discoloration of the skin, mucous membranes, sclera, and various organs. This discoloration is referred to as jaundice, or icterus. Jaundice is caused primarily by the accumulation in the skin of unconjugated bilirubin, a breakdown product of hemoglobin formed after its release from hemolyzed red blood cells (RBCs). Jaundice first appears in the infant’s face and head and progresses downward toward the toes. It dissipates in the reverse order. Physiologic jaundice, discussed in Chapters 23 and 24, is the most common abnormal finding in newborns and is usually benign. The challenge in the care of neonates with hyperbilirubinemia is to distinguish physiologic jaundice from a serious clinical pathologic condition.
Physiologic Jaundice
Physiologic jaundice occurs in about 60% of healthy term newborns and almost all preterm infants (Watson, 2009). It typically arises more than 24 hours after birth. In both Caucasian and African-American infants, it is manifested by a progressive increase in the unconjugated bilirubin level in cord blood from 2 mg/dl to a mean peak of 5 to 6 mg/dl between 60 and 72 hours of age. In Asian and Native-American infants, the level may increase to 10 to 14 mg/dl between 72 and 120 hours of age. Resolution in Caucasian and African-American newborns is evidenced by a rapid decline in the unconjugated bilirubin level to 2 mg/dl by 5 days after birth; in Asian and Native-American infants, this takes 7 to 10 days.
Physiologic jaundice is more common in preterm infants in whom the serum bilirubin level typically reaches a mean peak of 10 to 12 mg/dl by the fifth or sixth day of life. It takes longer for the maximal concentration to be reached in preterm than in full-term infants because of the preterm infant’s immature liver function and slower metabolic processes.
Pathologic Jaundice
Pathologic jaundice is the result of an increased level of total serum bilirubin that if left untreated can result in acute bilirubin encephalopathy or kernicterus. Although the terms are often used interchangeably, acute bilirubin encephalopathy describes the acute central nervous system manifestations seen in the first weeks after birth, whereas the term kernicterus is used to describe the chronic and permanent results of bilirubin toxicity (American Academy of Pediatrics [AAP], Subcommittee on Hyperbilirubinemia, 2004). Kernicterus, though rare, still occurs. Adherence to guidelines by the American Academy of Pediatrics (AAP Subcommittee on Hyperbilirubinemia) and a clinical position statement by the Association of Women’s Health, Obstetric and Neonatal Nurses (AWHONN, 2005) should result in kernicterus becoming largely preventable. The following findings support a diagnosis of pathologic jaundice and, if encountered in an infant, warrant further investigation (Bradshaw, 2010; Watson, 2009):
• Serum bilirubin concentrations of greater than 4 mg/dl in cord blood
• Clinical jaundice evident within 24 hours of birth
• Total serum bilirubin levels increasing by more than 5 mg/dl in 24 hours or increasing at a rate of 0.5 mg/dl or more over a 4- to 8-hour period
• A serum bilirubin level in a term newborn that exceeds 15 mg/dl at any time or clinical jaundice lasting more than 10 days
• A serum bilirubin level in a preterm newborn that exceeds 10 mg/dl at any time
• Any case of visible jaundice that persists for more than 10 days of life in a term infant or 21 days in a preterm infant, unless the infant is receiving breast milk
AAP guidelines (2004) and the clinical position statement by AWHONN (2005) note the need for universal screening to identify elevated bilirubin level in newborns to facilitate the prevention of acute bilirubin encephalopathy and kernicterus. The use of hour-specific serum bilirubin levels to predict newborns at risk for rapidly rising levels is an official recommendation by the AAP Subcommittee on Hyperbilirubinemia (2004) for the monitoring of healthy neonates at 35 weeks of gestation or more before discharge from the hospital. Use of a nomogram helps determine which newborns might need further evaluation after discharge. In many institutions the nomogram is used to determine the infant’s risk for development of hyperbilirubinemia requiring medical treatment or closer screening (see Fig. 24-8). Risk factors recognized to place infants in the high risk category include gestational age less than 38 weeks, breastfeeding, previous sibling with significant jaundice, and jaundice appearing before discharge. It is recommended that healthy infants (35 weeks of gestation or greater) receive follow-up care and assessment of bilirubin within 3 days of discharge if discharged at less than 24 hours and a risk assessment with tools such as the hour-specific nomogram (see box 36-1). Likewise, newborns discharged at 24 to 47.9 hours should receive follow-up evaluation within 4 days (96 hours), and those discharged between 48 and 72 hours should receive follow-up within 5 days (AAP Subcommittee on Hyperbilirubinemia, 2004). Other key elements of the recommendations of the AAP Subcommittee include promoting successful breastfeeding; recognizing that visual estimation of the degree of jaundice, particularly in darkly pigmented infants, can lead to errors; and providing parents with written and verbal information about newborn jaundice.
Many potential causes of pathologic hyperbilirubinemia are found in neonates (Box 36-2). The most common are hemolytic disorders of the newborn.
Hemolytic Disease of the Newborn
Hemolytic diseases of the newborn occur most often if the blood groups of the mother and baby are different. The most common are ABO and Rh factor incompatibilities.
The four major blood groups in the ABO system are A, B, AB, and O. People with type A blood have A antigen; those with type B have B antigen; those with type AB have both A and B antigens; and those with type O have no antigens. In turn, people with type A blood have plasma antibodies to type B blood; those with type B blood have antibodies to type A blood; those with type AB blood have no antibodies; and those with type O blood have antibodies to types A and B blood. If a person is administered or exposed to an incompatible blood type, he or she will form antibodies against the antigen in that blood, with agglutination, or clumping, occurring as the antibodies in the plasma mix with the antigens of the different blood group.
The Rh factor, a genetically determined factor present on RBCs, can be a major source of incompatibility. Of the several forms of the Rh antigen, the D antigen is the most significant because it causes the most antibody production in a person who is Rh negative. A person who has the Rh factor is considered Rh positive; a person without it is considered Rh negative. For example, a mother who has A-negative blood has the A antigen, plasma antibodies to the B antigen, and no Rh factor on her RBCs.
Hemolytic disorders occur when maternal antibodies are present naturally or form in response to an antigen from the fetal blood crossing the placenta and entering the maternal circulation. The maternal antibodies of the immunoglobulin G (IgG) class in turn cross the placenta, causing hemolysis of the fetal RBCs, resulting in hyperbilirubinemia and jaundice.
Rh Incompatibility
Rh incompatibility, or isoimmunization, occurs when an Rh-negative mother has an Rh-positive fetus who inherits the dominant Rh-positive gene from the father. If the mother is Rh negative and the father is Rh positive and homozygous for the Rh factor, all the offspring will be Rh positive. If the father is heterozygous for the factor, there is a 50% chance that each infant born of the union will be Rh positive and a 50% chance that each will be born Rh negative. An Rh-negative fetus is in no danger because it has the same Rh factor as the mother. An Rh-negative fetus with an Rh-positive mother also is in no danger. Only the Rh-positive fetus of an Rh-negative mother is at risk. From 10% to 15% of all Caucasian couples and about 5% of African-American couples have Rh incompatibility. It is rare in Asian couples due to the high incidence of ABO incompatibility, which is protective against Rh isoimmunization because of rapid destruction of the fetal RBCs which prevents Rh antigen exposure and maternal antibody production (Diehl-Jones & Askin, 2010). The incidence of Rh sensitization and resulting hemolytic disease of the newborn has decreased dramatically since the development of Rho(D) immunoglobulin in 1968. New treatment modalities, including early detection and fetal blood transfusions, have improved the outcome of affected fetuses.
The pathogenesis of Rh incompatibility is as follows. Hematopoiesis (the formation, production, and maintenance of blood cells) in the fetus is well established by the ninth week of gestation (Blackburn, 2007; Diehl-Jones & Askin, 2010). When fetal RBCs that contain the Rh antigen pass through the placenta into the maternal circulation absent the Rh antigen, the maternal immune system produces antibodies against the foreign fetal antigens. The process of antibody formation is called maternal sensitization. Sensitization can occur during pregnancy, birth, miscarriage or induced abortion, amniocentesis, external cephalic version, or trauma. Usually women become sensitized in their first pregnancy with an Rh-positive fetus but do not produce enough antibodies to cause lysis (destruction) of fetal blood cells. During subsequent pregnancies, antibodies form in response to repeated contact with the antigen from the fetal blood, and lysis of fetal RBCs results. The overall incidence of isoimmunization in Rh-negative mothers who are at risk is less than 10% and only 5% of mothers with isoimmunization have babies with hemolytic disease (Stoll, 2007). Multiple gestation, placental abruption, placenta previa, manual removal of the placenta, and cesarean birth increase the incidence of transplacental hemorrhage and the risk of isoimmunization.
Severe Rh incompatibility results in marked fetal hemolytic anemia because the fetal erythrocytes are destroyed by maternal Rh-positive antibodies. Although the placenta usually clears the bilirubin resulting from the RBC breakdown, in extreme cases fetal bilirubin levels increase. This results in fetal jaundice, also known as icterus gravis.
The fetus compensates for the anemia by producing large numbers of immature erythrocytes to replace those hemolyzed, thus the name for this condition: erythroblastosis fetalis. In hydrops fetalis, the most severe form of this disease, the fetus has marked anemia, cardiac decompensation, cardiomegaly, and hepatosplenomegaly. Hypoxia results from the severe anemia. In addition, because of the decreased intravascular oncotic pressure involved, fluid leaks out of the intravascular space. This results in generalized edema, as well as effusions into the peritoneal (ascites), pericardial, and pleural (hydrothorax) spaces. The placenta is often edematous, which, along with the edematous fetus, can cause the uterus to rupture.
Intrauterine or early neonatal death can occur as a result of hydrops fetalis, although intrauterine transfusions and early birth of the fetus can help to avert this. Intrauterine transfusion involves the infusion of Rh-negative, type O blood into the umbilical vein. The frequency of intrauterine transfusions varies according to institution and fetal hydropic status, but it can be as often as every 2 weeks until the fetus reaches pulmonary maturity at approximately 37 to 38 weeks of gestation. Studies of the use of intrauterine transfusions have demonstrated a high survival rate and low risk of disabilities in the surviving infant (Gruslin & Moore, 2006; Moise, 2010).
ABO Incompatibility
ABO incompatibility is the most common cause of hemolytic disease in the newborn, although the anemia that results is usually mild. ABO maternal blood group incompatibility occurs in approximately 20% of infants; however, only about 5% have clinical manifestations (Cunningham, Leveno, Bloom, Hauth, Rouse, & Spong, 2010). It occurs if the fetal blood type is A, B, or AB, and the maternal type is O. It occurs rarely in infants with type B blood born to mothers with type A blood. The incompatibility arises because naturally occurring anti-A and anti-B antibodies are transferred across the placenta to the fetus. Unlike the situation that pertains to Rh incompatibility, firstborn infants can be affected because mothers with type O blood already have anti-A and anti-B antibodies in their blood. Such a newborn can have a weakly positive direct Coombs’ test (also referred to as a direct antiglobulin test [DAT]). The cord bilirubin level usually is less than 4 mg/dl, and any resulting hyperbilirubinemia usually can be treated with phototherapy. Exchange transfusions are required only occasionally. Although ABO incompatibility is a common cause of hyperbilirubinemia, it rarely precipitates significant anemia resulting from the hemolysis of RBCs.
Other Causes of Hemolytic Jaundice
It is not within the scope of this text to discuss the many potential causes of hemolytic jaundice in childhood. However, among African-Americans and persons of Mediterranean heritage there is a high incidence of glucose-6-phosphate dehydrogenase deficiency (G6PD) (Wilkins, 2010). Because it is a sex-linked disease, male offspring are affected more often than females. A deficiency of a red blood cell enzyme in combination with exposure to an oxidant stressor (such as sepsis) results in hemolysis and a decreased RBC life (Diehl-Jones & Askin, 2010). The increase in the destruction of RBCs overwhelms the immature neonatal liver’s ability to conjugate the indirect bilirubin. Treatment is the same as for any newborn with rapidly rising serum bilirubin levels. Other metabolic and inherited conditions that increase hemolysis and can cause jaundice in the infant include galactosemia, Crigler-Najjar disease, and hypothyroidism (Blackburn, 2007).
Acute Bilirubin Encephalopathy
The goal of the care given the infant with hyperbilirubinemia is the prevention of acute bilirubin encephalopathy, which is caused by the deposition of bilirubin in the brain, especially within the basal ganglia, the cerebellum, and the hippocampus. Normally bilirubin does not cause harm because it is bound to albumin and carried to the liver to undergo conjugation. However, once albumin binding sites are saturated, the bilirubin circulates as unconjugated (indirect) bilirubin, which is highly lipid soluble and capable of crossing the blood-brain barrier. Circulation of unconjugated bilirubin can reach toxic levels and become deposited in the basal ganglia, resulting in the yellowish staining of the brain tissue and the necrosis of neurons.
Acute bilirubin encephalopathy, which can develop in newborns with no apparent signs of clinical jaundice, is directly related to the total serum bilirubin level, although these levels alone do not predict the risk of brain injury. In a term infant, a serum bilirubin level of 25 mg/dl is considered the upper limit, beyond which the risk for acute bilirubin encephalopathy increases. However, the condition can occur at much lower levels in premature infants or infants with other complications. In low birth weight preterm infants, a peak total serum bilirubin level as low as 6.5 mg/dl has been associated with the development of acute bilirubin encephalopathy (National Institute of Child Health and Human Development [NICHD], 1985; Watson, 2009).
Some of the perinatal events that increase the likelihood of acute bilirubin encephalopathy, even at these lower bilirubin levels, include hypoxia, asphyxia, acidosis, hypothermia, hypoglycemia, sepsis, treatment with certain medications, and hypoalbuminemia. In essence, any condition that interferes with the conjugation of bilirubin or competes for albumin-binding sites increases the risk that unconjugated bilirubin can pass through the blood-brain barrier and cause damage to the central nervous system.
Acute bilirubin encephalopathy is associated with acute and long-term signs of neurologic damage (AAP Subcommittee on Hyperbilirubinemia, 2004; Bradshaw, 2010; Volpe, 2008). The clinical manifestations typically appear between 2 and 6 days after birth and go through several phases as the disease progresses. During the first phase the newborn is hypotonic and lethargic and shows a poor suck and depressed or absent Moro reflex. These more subtle signs are followed by the appearance of a high-pitched cry, opisthotonos (severe muscle spasm that causes the back to arch acutely), spasticity, hyperreflexia, and fever. If allowed to progress to the third phase, the neonate will demonstrate a shrill cry, apnea, deep stupor to coma, seizures, and hearing and visual disturbances.
About half of the affected infants survive, although they often have permanent sequelae including extrapyramidal movement disorders (especially dystonia and athetosis), gaze abnormalities (especially an upward gaze), auditory disturbances (especially sensorineural hearing loss), intellectual deficits (rarely in the mentally retarded range), and enamel dysplasia of the deciduous teeth. Movement abnormalities and auditory disturbances are almost always present.
Kernicterus is the irreversible, chronic sequela of bilirubin toxicity (Bradshaw, 2010; Volpe, 2008; Watson, 2009). Over the first year of life a baby demonstrates the characteristic findings of hypotonia, active deep tendon reflexes, persistent tonic neck reflex, and difficulty meeting developmental milestones. Characteristics of a fully developed encephalopathy include the permanent sequelae identified above. Treatment for kernicterus is supportive.
Care Management
It is important to determine the blood type and Rh factor of the pregnant woman prenatally. Early identification of the Rh-negative woman must occur, and care must be taken to prevent sensitization. The nurse must obtain a thorough history to assess for events that could have caused her to develop antibodies to the Rh factor. Such events include (1) previous pregnancy with an Rh-positive fetus; (2) transfusion with Rh-positive blood, which causes immediate sensitization; (3) miscarriage or induced abortion after 8 or more weeks of gestation; (4) amniocentesis performed for any reason; (5) premature separation of the placenta; and (6) trauma.
Because hematopoiesis is well developed in the fetus by the ninth week of gestation, a woman who has had a miscarriage or induced abortion after this time or has previously given birth to a child may have been inoculated with fetal blood at the time of placental separation. During amniocentesis, the needle can cause localized damage to the single layer of cells that separates the maternal and fetal circulation in the placenta, thereby allowing fetal RBCs to enter the maternal circulation.
If any of these events has occurred, the nurse checks the woman’s record to determine whether she has received Rho(D) immunoglobulin, such as RhoGAM (WinGAM), which is a commercial preparation of passive antibodies against the Rh factor (see the Medication Guide, p. 499). This injection of anti-Rh antibodies destroys any fetal RBCs in the maternal circulation and blocks the maternal antibody production. RhoGAM is 90% effective in preventing sensitization. It is recommended that it be given to an Rh-negative mother at 28 weeks of gestation; within 72 hours after delivery; after an invasive procedure such as amniocentesis, chorionic villus sampling (CVS), or percutaneous umbilical blood sampling (PUBS); and any time there is a risk of fetal-maternal hemorrhage. It is also recommended after induced abortion and ectopic pregnancy (Box 36-3). (Diehl-Jones & Askin, 2010; Moise, 2010; Wong, DeSandre, Sibley, & Stevenson, 2006)
At the first prenatal visit of an Rh-negative woman with a fetus who may be Rh positive, an indirect Coombs’ test should be done to determine whether she has antibodies to the Rh antigen. In this test the maternal blood serum is mixed with Rh-positive RBCs. If the Rh-positive RBCs agglutinate or clump, this indicates that maternal antibodies are present or that the mother has been sensitized. The dilution of the specimen of blood at which clumping occurs determines the titer, or level, of maternal antibodies. This titer indicates the degree of maternal sensitization. A level of 1:8 rarely results in fetal jeopardy. If the titer reaches 1:16, amniocentesis is performed to determine optical density (ΔOD) of amniotic fluid to estimate fetal hemolytic process (see Chapter 26). Rising bilirubin levels can indicate the need for an intrauterine transfusion. Genetic testing allows early identification of paternal zygosity at the RhD gene locus, thus allowing earlier detection of the potential for isoimmunization and precluding further maternal or fetal testing (Moise, 2010).
The indirect Coombs’ test is repeated at 28 weeks. If the result remains negative, indicating that sensitization has not occurred, the woman is given an intramuscular injection of Rho(D) immunoglobulin. If the test result is positive, showing that sensitization has occurred, it is then repeated every 4 to 6 weeks to monitor the maternal antibody titer as just described.
Prevention of hyperbilirubinemia is the primary prenatal focus of care. The implementation of interventions focused on the care of the woman whose fetus is at risk for hyperbilirubinemia is essential to prevent problems in the newborn. Prenatal control of diabetes mellitus, prevention of maternal infection, avoidance of drugs such as diazepam and salicylates near the time of birth, and prevention of preterm birth reduce the risk.
The fetus and maternal antibody titers are monitored prenatally. Several methods are used to detect fetal anemia: amniocentesis to measure ΔOD 450, PUBS or cordocentesis, and middle cerebral artery peak systolic velocity (MCA-PSV). Although ultrasound-guided cordocentesis is the gold standard for detecting fetal anemia, MCA-PSV is being used increasingly as an accurate, noninvasive means to detect fetal anemia. This study is done using Doppler ultrasound (Moise, 2010).
If amniocentesis reveals that the ΔOD is high or the MCA-PSV is increased, cordocentesis is performed to assess fetal hematocrit. If the hematocrit is less than 30%, intrauterine transfusion is indicated (Moise, 2010). Intrauterine transfusion can be done every 1 to 2 weeks between 26 and 32 weeks. If the endangered fetus is at more than 32 weeks of gestation, a preterm birth may be indicated, usually by cesarean.
Postpartum interventions focus on preventing sensitization in the mother, if it has not occurred already, and treating any complications in the neonate resulting from the hemolysis of RBCs (see the Nursing Care Plan). The unsensitized Rh-negative mother whose baby is Rh positive should receive Rho(D) immunoglobulin within 72 hours of birth to prevent her from producing antibodies to the fetal blood cells that entered her bloodstream during the birth. One dose accommodates approximately 30 ml of fetal RBCs (Diehl-Jones & Askin, 2010).
At birth the neonate’s cord blood is sent to the laboratory to determine the infant’s blood type and Rh status. A direct Coombs’ test is performed on this cord blood to determine whether there are maternal antibodies in the fetal blood. If antibodies are present, the titer, indicating the degree of maternal sensitization, is measured. If the titer is 1:64, an exchange transfusion is indicated. In addition, the prevention of or prompt therapy for perinatal asphyxia, acidosis, cold stress, sepsis, and hypoglycemia will decrease the newborn’s risk for severe hemolytic disease and the susceptibility to kernicterus. Early feeding is initiated to stimulate stooling and thus facilitate the removal of bilirubin. See Chapter 24 for a discussion of phototherapy.
Exchange transfusions are needed infrequently because of the improved recognition of neonates at risk of hemolytic disease that can result from isoimmunization. Other factors must always be considered as well, particularly the clinical condition of the infant, because it is a procedure with potential complications. Guidelines for the initiation of exchange transfusion in relation to serum bilirubin levels in infants of more than 35 weeks of gestation can be found in the 2004 AAP Clinical Practice Guideline (AAP Subcommittee on Hyperbilirubinemia, 2004).
Exchange transfusion is accomplished by alternately removing a small amount of the infant’s blood and replacing it with an equal amount of donor blood. Exchange transfusion replaces the RBCs that would otherwise be hemolyzed by circulating maternal antibodies, removes the antibodies responsible for hemolysis, and corrects the anemia caused by hemolysis of the infant’s sensitized RBCs. It also reduces the serum bilirubin level in infants who have severe hyperbilirubinemia from any cause. If the infant has Rh incompatibility, type O Rh-negative blood is used for transfusion so the maternal antibodies still present in the infant do not hemolyze the transfused blood. Depending on the infant’s size, maturity, and condition, amounts of 5 to 20 ml of the infant’s blood are removed at one time and replaced with donor blood. The double volume or two-volume exchange replaces approximately 170 ml/kg of body weight, or 87% of the infant’s total blood volume. The procedure requires approximately 1 hour. After the procedure, phototherapy is continued and the bilirubin levels are monitored every 4 hours (Martin & Cloherty, 2008; Watson, 2009; Wong et al., 2006).
The infant is monitored closely during and after the procedure, including the heart rate and rhythm, respirations, blood pressure, temperature, and perfusion. Preservatives in donor blood lower the infant’s serum calcium and magnesium levels. It is not uncommon for calcium gluconate to be given during the exchange transfusion if symptoms of hypocalcemia become evident. Symptoms of hypocalcemia include jitteriness, irritability,
 NURSING CARE PLAN
NURSING CARE PLAN
The Infant with Hyperbilirubinemia
Risk for injury related to hemolytic disease and treatment effects
Bilirubin levels decrease with treatment; no evidence exists of harmful effects from phototherapy (e.g., no eye irritation, dehydration, temperature instability, or skin breakdown); and no complications occur from exchange transfusions.
Nursing Interventions/Rationales
• Initiate early feedings to enhance excretion of bilirubin in stools.
• Observe skin and mucous membranes for signs of jaundice, indicative of increasing bilirubin levels; monitor total serum bilirubin levels to determine rate of increase and treatment response.
• Note time of jaundice onset to help distinguish physiologic from other causes of jaundice.
• Observe for signs of hypoxia, hypothermia, hypoglycemia, and metabolic acidosis, which occur as a result of hyperbilirubinemia and increase the risk of brain damage.
• Initiate phototherapy per physician’s order to decrease bilirubin levels.
• During phototherapy, shield infant’s eyes to prevent damage to corneas and retinas; keep infant nude and change positions frequently for maximal body surface exposure; cleanse skin frequently to prevent irritation; maintain adequate fluid intake to prevent dehydration; monitor body temperature to prevent hyperthermia.
• Before exchange transfusion, keep infant on nothing-by-mouth (NPO) status (2 to 4 hours) to prevent aspiration; check donor blood for compatibility to prevent transfusion reaction; have resuscitation equipment (oxygen, Ambu bag, endotracheal tubes, laryngoscope) at bedside in preparation for emergency action.
• Assist physician with exchange transfusion procedure; track amounts of blood withdrawn and transfused to maintain balanced blood volume; maintain body temperature to avoid hypothermia and cold stress; monitor vital signs and observe for signs of hypocalcemia, hypomagnesemia.
• After transfusion, continue to monitor vital signs, signs of hypocalcemia and hypomagnesemia; monitor for hypoglycemia; check umbilical cord for bleeding.
Interrupted breastfeeding related to discharge of mother and continued hospitalization of infant
Mother states desire to continue breastfeeding as much as possible and expresses and stores milk when she cannot come to the hospital to nurse the infant.
Nursing Interventions/Rationales
• Encourage the mother to come to the hospital to nurse infant as often as feasible for her to maintain milk supply and encourage bonding.
• Provide private and comfortable space for nursing that is available 24 hours per day to support the mother in breastfeeding.
• Instruct the mother in methods to express and store breast milk to ensure a safe and adequate milk supply.
• Provide written educational materials and audiovisual aids to demonstrate proper techniques for expressing and storing milk and to allow the mother to learn at her own pace.
• Recommend use of a breast pump and pump according to recommended guidelines (e.g., pump a minimum of five times a day; pump a minimum of 100 minutes a day; pump long enough to soften breasts) to provide maximum stimulation for milk production.
• Reassure the mother that infant’s nutritional needs will be met through expressed milk or other methods to allay anxiety.
• Review the daily routine of the mother to advise her on ways to incorporate pumping into her daily schedule.
• Provide information about breastfeeding support groups to help the mother obtain emotional support from other breastfeeding mothers.
Parental role conflict related to separation from hospitalized infant and interruptions of family life due to travel to hospital to see infant.
Parents will share responsibilities for child care and home maintenance; parents will visit infant in hospital as often as feasible.
Nursing Interventions/Rationales
• Suggest to parents that they mutually establish a routine for child care for siblings and home maintenance to enhance communication and provide necessary home activities.
• Encourage parents to seek assistance and support from friends, relatives, and community groups such as their church to meet their needs and ensure that a minimal level of appropriate functioning is maintained.
• Encourage parents to travel to the hospital as often as is desirable and feasible to promote breastfeeding and bonding with infant.
Deficient knowledge related to administration of home phototherapy
Nursing Interventions/Rationales
• Explore family’s willingness to try home phototherapy to evaluate feasibility of home therapy option.
• Explore family’s understanding of jaundice and proposed therapy to establish baseline for teaching.
• Teach family with demonstration–return demonstration, allowing for several practice sessions, and supplement with written materials with pictorial representations to ensure safe and optimal results.
• Include the following in your instructions: placement of lamp or fiberoptic unit; proper eye care and patching; proper skin care; proper positioning under lamp; provision of increased fluid intake; monitoring of time under lamp; monitoring of temperature and skin, eyes, feeding patterns, stooling and voiding patterns; observation for complications to ensure that the family understands and implements safe and effective use of phototherapy.
• Stress importance of obtaining the prescribed bilirubin tests on schedule as a way of tracking success of therapy.
• Give parents a contact number if they have any questions while carrying out therapy to offer ongoing support and increase parent comfort.
convulsions, tachycardia, and electrocardiogram changes. The nurse monitors the neonate for hypoglycemia during the several hours after the exchange because the high glucose content of the preservatives can stimulate insulin secretion. These high risk neonates typically have dextrose support through an intravenous route (Martin & Cloherty, 2008; Watson, 2009).
Planning for rehabilitative measures is necessary if kernicterus occurs. The family will need the services of many community resources to care for the affected child. An interdisciplinary approach that includes social services must be taken.
Congenital Anomalies
A congenital anomaly is a defect that is present at birth and can be caused by genetic or environmental factors, or both. It is defined as a physical, metabolic, anatomic, or behavioral deviation from the normal pattern of development (Moore & Persaud, 2007). Congenital defects are reported to occur in 2% to 3% of all live births (Bay, Steele, & Davis, 2007), but this number increases to approximately 6% by 5 years, when more anomalies are diagnosed. In addition, the incidence of congenital malformations in fetuses that are aborted is higher than that in infants who are born alive, thus adding to the overall incidence. Major congenital defects are the leading cause of death in infants younger than 1 year of age in the United States, accounting for 20.4% of infant deaths (Heron, Sutton, Xu, Ventura, Strobino, & Guyer, 2010). Although the incidences of other causes of neonatal mortality have decreased, the death rate associated with most congenital anomalies has essentially remained stable since 1932.
The desired and expected outcome of every wanted pregnancy is a normal, functioning infant with a good intellectual potential. Fulfillment of this hope depends on numerous hereditary and environmental factors. Probably all human characteristics have a genetic component, including those that produce symptoms or physical abnormalities that impair the fitness of the person. Some disorders or diseases occur through the influence of a single gene or the combined action of many genes inherited from the parents; others result from the action of the intrauterine environment. Many defects appear to occur as the result of multifactorial inheritance, the interaction of multiple genes with environmental factors that affect the embryonic development of the affected system. Examples of these include neural tube defects, congenital heart defects, developmental dysplasia of the hip, and cleft lip or palate. In about half of all cases of congenital anomalies, there is no identifiable cause. A chromosomal abnormality or gene alteration is responsible for approximately 40% of anomalies (Manning, 2009). Evidence indicates that maternal obesity is significantly linked to spina bifida, cardiac defects, diaphragmatic hernia, hypospadias, omphalocele, anorectal atresia, and limb reductions (Cunningham et al., 2010; Waller, Shaw, Rasmussen, Hobbs, Canfield, Siega-Riz, et al., 2007),
Ways of detecting and preventing some of the congenital anomalies are being improved continually, as are surgical techniques for the care of the fetus and newborn with certain anomalies. Promoting the availability of these services to populations at risk challenges community health care systems. An interdisciplinary team approach is vital for providing holistic care: the surgical treatment, rehabilitation, and education of the child, as well as psychosocial and financial assistance for the parents. Parental disappointment and disillusion add to the complexity of the nursing care needed for these infants.
The most common congenital anomalies that cause serious problems in the neonate are congenital heart disease, neural tube defects, cleft lip or palate, clubfoot, and developmental dysplasia of the hip. These are thought to result from the interaction of multiple genetic and environmental factors. Minor anomalies are less apparent but are important to identify because they can be a part of a characteristic pattern of malformations. That is, they can point to the presence of a more serious major anomaly and aid in its diagnosis. The presence of a minor anomaly indicates the need for further evaluation of the neonate for other anomalies. Minor malformations are more common in areas of the body that have variable features, such as the face and distal extremities. Some of the most common minor malformations include the lack of a helical fold of the pinna, low-set ears, alterations in hair pattern or texture, absent philtrum or a hairy patch or birthmark over the vertebral column (Hudgins & Cassidy, 2006).
Cardiovascular System Anomalies
During fetal development, cell division and differentiation of the organs and tissues of a particular body system sometimes occur rapidly. During these critical periods particular body systems are more susceptible to environmental influences than they are later in gestation. For example, the critical period for the cardiovascular system is from week 3 of embryonic development to week 8, when many women are not aware that they are pregnant.
Congenital cardiac anomalies, with an overall prevalence of approximately 81 per 10,000 births, are the most common of all congenital malformations (Bernstein, 2007; Reller, Strickland, Riehle-Colarusso, Mahle, & Correa, 2009). Congenital heart defects (CHDs) are anatomic abnormalities in the heart that are present at birth, although they may not be diagnosed immediately. Congenital heart disease occurs more frequently in preterm infants. There is an increased likelihood of cardiac disease in neonates that are small for gestational age (SGA), as well as in those with congenital infections.
Ventricular septal defect, the most common type of heart defect with increased pulmonary blood flow (acyanotic lesion) has a prevalence of 27.5 per 10,000 births, and accounts for about 30% to 35% of all congenital heart defects. Tetralogy of Fallot has an incidence of 4.7 per 10,000 births, and is the most common cardiac defect with decreased pulmonary blood flow (cyanotic lesion) (Bernstein, 2007; Reller et al., 2009). Congenital heart defects are often associated with other extracardiac defects such as renal agenesis, omphalocele, tracheoesophageal fistula, and diaphragmatic hernias. Congenital heart disease is the leading cause of death in children with congenital defects (Bernstein, 2007).
The etiology of CHDs is multifactorial, chromosomal and/or genetic, and due to environmental teratogens (Sadowski, 2010). This is important information for parents because they often feel guilty thinking that they have done something to cause the defect. The etiology is also important when it comes to counseling the family regarding recurrence risks for future pregnancies. If the parent or a sibling is diagnosed with a CHD, the risk is increased 3- to 4-fold, and if two first-order relatives have a CHD, the recurrence risk is increased 10-fold (Park, 2008). Maternal factors that are known to be associated with a higher incidence of CHDs include the following (Hartas, Tsounias & Gupta-Malhotra, 2009):
• Ingestion of folic acid antagonists, progesterone, estrogen, lithium, warfarin (Coumadin), or anticonvulsants such as phenytoin
• Use of the acne medication isotretinoin (Accutane)
• Metabolic disorders such as diabetes mellitus and phenylketonuria
Maternal smoking is associated with a greater risk of congenital heart disease. Infants born to women who are heavy smokers (≥25 cigarettes per day) have more than twice the risk of having a congenital septal defect (Malik, Cleves, Honein, Romitti, Botto, Yang, et al., 2008).
Maternal obesity can increase the risk of congenital heart anomalies. An above normal body mass index has been associated with an increased risk for all congenital heart defects, and specifically conotruncal defects, tetralogy of Fallot, total anomalous pulmonary venous return, hypoplastic left heart syndrome, right ventricular outflow tract defects, and septal defects (Gilboa, Correa, Botto, Rasmussen, Waller, Hobbs, et al., 2010).
Genetic factors are implicated in the pathogenesis of CHDs. It now appears that a much greater percentage of CHDs than believed to be true in the past can be attributed to single gene mutations. This recognition is based on animal models, studies of human familial patterns of inheritance, and the finding of cardiovascular defects as a part of syndromes exhibiting mendelian patterns of inheritance. An example of this is the identification of chromosome 22q11 deletions in infants with DiGeorge syndrome. This syndrome affects an estimated 1 in 4000 live births, making this one of the most frequent genetic disorders with congenital heart defects. The gene or genes located in the chromosomal region involved with DiGeorge syndrome appear to play a major role in the development of cardiovascular defects, particularly truncus arteriosus, absent pulmonary valve syndrome, tetralogy of Fallot, pulmonary atresia, and other abnormalities involving the aortic arch. Congenital heart disease is found in more than 50% of children with trisomy 21 (Down syndrome), 90% of neonates with trisomy 18 (Gilbert syndrome), and 40% of neonates with Turner syndrome (Bernstein, 2007).
Although traditionally a CHD has been classified as either cyanotic or acyanotic, a classification that categorizes cardiac defects physiologically is now considered more descriptive. The first of the four categories in this classification includes defects that result in increased pulmonary blood flow. Examples of the CHDs in this category are defects that eventually manifest with congestive heart failure such as atrial and ventricular septal defects and patent ductus arteriosus. The second category includes defects that involve decreased pulmonary blood flow that produce cyanosis. The most common example of this type of defect is tetralogy of Fallot. The third category includes those defects that cause obstruction to blood flow out of the heart. Pulmonary stenosis is an example of an obstruction to the flow of blood out of the right side of the heart (ventricle) that causes cyanosis. Coarctation of the aorta and subaortic stenosis are examples of obstructions to the flow of blood out of the left side of the heart that can result in pulmonary venous congestion that leads to a left-sided congestive heart failure. The fourth category comprises those complex cardiac anomalies that involve blood flow composed of a mixed amount of oxygen saturated and oxygen desaturated blood in the heart or great vessels. These include such defects as transposition of the great vessels and total anomalous venous return (Table 36-1 and Fig. 36-1).
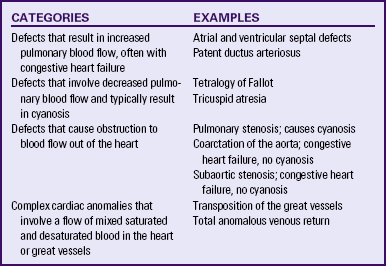
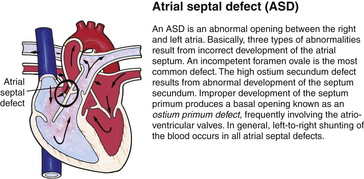
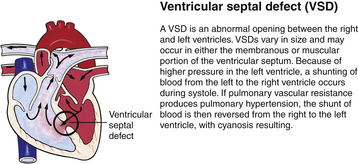
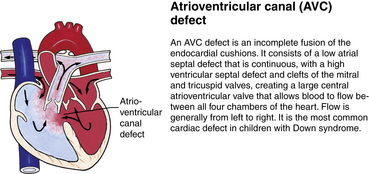
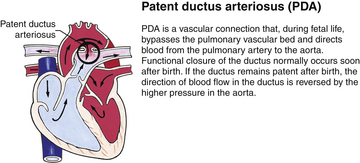
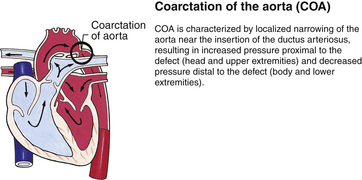
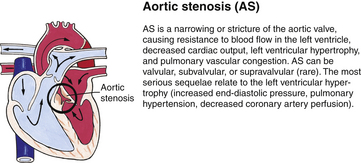
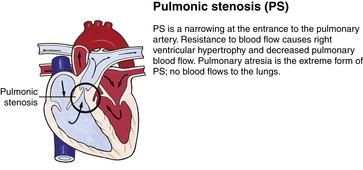
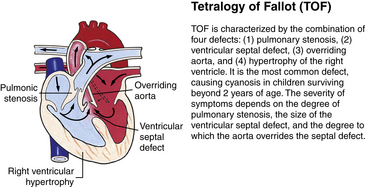
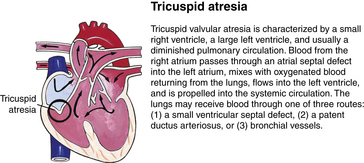
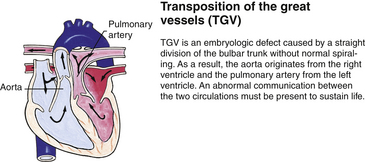
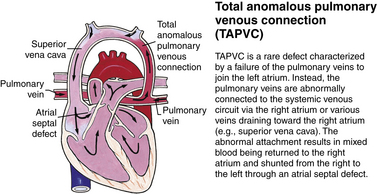
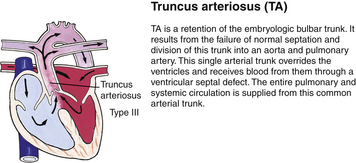
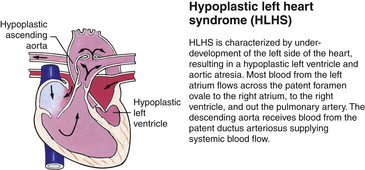
FIG. 36-1 Congenital heart abnormalities. (Modified from Hockenberry, M., & Wilson, D. [2011]. Wong’s nursing care of infants and children [9th ed.]. St. Louis: Mosby.)
Severe CHDs often are evident immediately after birth, especially defects that cause cyanosis such as transposition of the great vessels. Infants with these anomalies are transferred directly to neonatal intensive care units, preferably those that are equipped to diagnose and medically/surgically treat these types of cardiac emergencies. Even though the structural or functional anomalies are always present at birth, the affected newborns can be asymptomatic because the defect is too small to interfere with sufficient blood flow to the lung for oxygenation or does not interfere with delivery of oxygenated blood to the tissues. With growth and maturation, the defect can become apparent as the infant or child is exposed to stresses such as growth demands or infection.
If symptoms are present at birth, they can be obvious with the first cry, which can be weak and muffled or loud and breathless. Affected newborns can exhibit cyanosis that is not relieved when given supplemental oxygen, with the cyanosis increasing whenever the child cries or is in the supine position. The bluish gray, dusky color of cyanotic infants can be mild, moderate, or severe. Other infants can be acyanotic and pale, with or without mottling on exertion, which includes crying, feeding, or stooling.
The affected newborn’s activity level varies from restlessness to lethargy, and possibly unresponsiveness, except to pain. Persistent bradycardia (a resting heart rate of less than 80 beats/min) or tachycardia (a rate exceeding 160 beats/min) can be noted. The cardiac rhythm can be abnormal, and various murmurs may be heard. Signs of congestive heart failure and decreased tissue perfusion can also become evident (Hartas et al., 2009; Sadowski, 2010).
Because the cardiac and respiratory systems function together, cardiac disease can be manifested by respiratory signs and symptoms. The nurse assesses the respiratory rate while the newborn is in a resting state. Tachypnea, a respiratory rate greater than 60 breaths/min without dyspnea is typically a subtle clue that the baby possibly has a cardiac malformation. Increased respiratory depth or hyperpnea is often noted when the neonate has a cardiac lesion that is obstructing blood flow to the lung. Signs that can indicate the development of congestive heart failure are feeding difficulties and increasing respiratory distress, especially tachypnea such that the baby has to stop feeding to breathe.
A major role of the nurse is to assess infants for abnormal findings, which must be reported immediately. Newborns exhibiting these symptoms require prompt diagnosis and appropriate therapy in a neonatal or pediatric intensive care unit. Interventions include administering oxygen as ordered, administering cardiotonic and other medications such as diuretics that rid the body of accumulated fluid, decreasing the workload of the heart by maintaining a thermoneutral environment, feeding with the gavage method if necessary, and preventing crying if this precipitates cyanosis. Various diagnostic tests such as echocardiography and cardiac catheterization are performed to obtain specific information about the defect and the need for surgical intervention. Significant improvements in diagnosis, medical management, and surgical treatment of CHDs have caused the death rate to decrease significantly, with the result that more of these children are reaching adulthood (Hartas et al., 2009).
Central Nervous System Anomalies
Most congenital anomalies of the central nervous system (CNS) result from defects in the closure of the neural tube during fetal development. Although the cause of neural tube defects (NTDs) is unknown, they are thought to stem from the interaction of many genes that can be influenced by factors in the fetal environment. Environmental influences such as maternal treatment with anticonvulsants (e.g., valproic acid or carbamazepine), treatment with methotrexate (a chemotherapeutic medication), folic acid deficiency, and maternal diabetes have been implicated (Zupancic, 2008). Excessive maternal body heat exposure during the early first trimester, significant febrile illness, and lower socioeconomic status can increase the risk of an NTD (Sterk, 2010).
Maternal folic acid deficiency has a direct bearing on failure of the neural tube to close. Therefore, as a preventive measure, folic acid supplementation (0.4 mg/day) is recommended for women of childbearing age.
The incidence of NTD is about 1 per 1000 live births. However, the incidence varies according to race, geographic area, socioeconomic status, and ethnicity (Zupancic, 2008). Prevalence estimates for the two most common NTDs indicate that spina bifida occurs in approximately 18 of every 100,000 live births and anencephaly occurs in approximately 11 of every 100,000 live births (Mathews, 2008). Approximately 95% of all NTDs occur in families that have no history of NTDs. Primary NTDs have an increased recurrence rate in subsequent pregnancies although secondary NTDs do not. Primary NTDs are the result of failure of the anterior neuropore to close or a disruption of an already closed neural tube between 18 and 25 days of gestation (Moore & Persaud, 2007; Volpe, 2008). Myelomeningocele, encephalocele, and anencephaly are examples of primary NTDs. Secondary neural tube defects account for 5% of NTDs. They are due to disruption in development of the lower sacral or coccygeal segments during secondary neurulation between 26 days and 8 weeks of gestation (Moore & Persaud; Volpe). Examples of secondary NTDs are meningocele, and sacral agenesis/dysgenesis.
Although an NTD is usually an isolated defect, it can occur with some chromosomal abnormalities and syndromes and with other defects such as cleft palate, ventricular septal defect, tracheoesophageal fistula, diaphragmatic hernia, imperforate anus, and renal anomalies. Some NTDs are diagnosed prenatally with fetal ultrasonography and the finding of elevated levels of alpha-fetoprotein in the amniotic fluid and maternal serum. Increased use of prenatal diagnostic techniques and termination of pregnancies have also had an effect on the overall incidence of birth of infants with NTDs.
Encephalocele and Anencephaly
Encephalocele and anencephaly are abnormalities resulting from failure of the anterior end of the neural tube to close. An encephalocele is a herniation of the brain and meninges through a skull defect. Treatment consists of surgical repair and shunting to relieve hydrocephalus, unless a major brain malformation is present. Most of these infants will have some degree of cognitive deficit. Anencephaly is the absence of both cerebral hemispheres and of the overlying skull. This condition is incompatible with life; many of the infants are stillborn or die within a few days of birth. Comfort measures are provided until the infant eventually dies of respiratory failure.
Spina Bifida
Spina bifida, the most common defect of the CNS, results from failure of the neural tube to close at some point. The two categories of spina bifida are spina bifida occulta and spina bifida manifesta. Spina bifida occulta, the milder form, is a malformation in which the posterior portion of the laminas fails to close, but the spinal cord or meninges do not herniate or protrude through the defect. Skin typically covers the opening in the spinal cord. There can also be a bulge under the skin where the ends of the spinal cord terminate in fatty tissue. Often a birthmark or a hairy patch is present above the defect.
Spina bifida manifesta occurs predominantly in the lumbar or lumbosacral regions and includes meningocele and myelomeningocele. A meningocele is a herniation of the meninges at the site of the defect in the vertebral column, and is typically covered by skin. The baby’s neurologic function tends to be normal unless other abnormalities are present. A myelomeningocele is a herniation of the meninges and spinal cord at the site of the defect, with or without skin or vertebral covering (Fig. 36-2). The sac can tear easily, allowing cerebrospinal fluid (CSF) to leak out and providing an entry for infectious agents into the CNS. Because the nerves are involved, there are motor and sensory deficits below the lesion. Eighty percent of the lesions occur in the lumbar area and 95% of affected neonates will have hydrocephalus, typically due to an Arnold-Chiari malformation (Lynam & Verklan, 2010; Volpe, 2008). An Arnold-Chiari malformation results from the improper development and downward displacement of the hindbrain into the cervical spinal canal, which blocks the flow of CSF from communicating with the spinal column and results in the development of hydrocephalus. The long-term prognosis in an affected infant can be determined to a large extent at birth, with the degree of neurologic dysfunction related to the level of the lesion, as the level determines the nerves involved. Prenatal diagnosis makes possible a scheduled cesarean birth, allowing for more careful delivery of the infant’s back to try to prevent rupture of the meningeal sac.
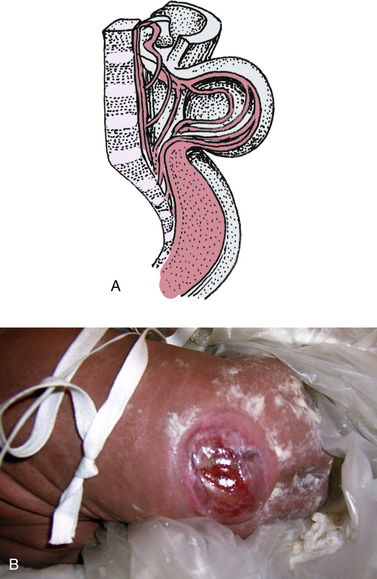
FIG. 36-2 A, Myelomeningocele. Note absence of vertebral arches. B, Myelomeningocele (spina bifida). (Courtesy Cheryl Briggs, RNC, Annapolis, MD.)
A major preoperative nursing intervention for a neonate with a myelomeningocele is to protect the protruding sac from injury, rupture, and resultant risk of CNS infection. Such infants should be positioned in a prone-kneeling position and the knees protected from skin breakdown. The sac should be covered with a sterile, moist, nonadherent dressing, and cared for using sterile technique. A drape should be placed over the buttocks below the lesion and secured using the drape’s adhesive to keep the lesion free of meconium or stool. Neurosurgery and urology consultations should occur immediately after the neonate is admitted. A thorough physical examination will be done to evaluate the level of the injury, sensory involvement, sphincter control (anal wink), and measurement of the frontal-occipital circumference to assess for hydrocephalus (Lynam & Verklan, 2010; Volpe, 2008). Intake and output are recorded to document the number and character of the voidings and stools as well as the leakage of urine and stool. Nursing assessment includes observations of movement or absence of movement as well as the quality of movements of the lower extremities.
Nurses provide support and information to parents as they begin to learn to cope with an infant who has immediate needs for intensive care and who probably will have long-term needs as well. Based on the type and position of the lesion, along with the neonate’s clinical condition, early surgical repair is advocated to preserve cognitive function, decrease the risk of sepsis, and improve prognosis for ambulation (Sterk, 2010). Surgical shunt procedures to prevent increasing hydrocephalus may be needed, such as a ventriculoperitoneal shunt.
 NURSING ALERT
NURSING ALERT
Infants with myelomeningocele are at increased risk for developing latex sensitivity, so they must not come into direct or secondary contact with any products or equipment containing latex.
Maternal-fetal surgery to repair a fetal myelomeningocele is a promising new development in maternal-fetal intervention. The rationale for this intervention is based on direct and indirect evidence that some of the neural damage resulting from the myelomeningocele is acquired in the later part of gestation, caused by amniotic fluid exposure or trauma to the exposed neural elements. The improvement in hydrocephalus and the Arnold-Chiari II malformation can decrease the need for a ventriculoperitoneal shunt. There does not appear to be any improvement in neurologic function, including problems with continence. The term birth of an infant with myelomeningocele is a viable alternative to fetal repair. Numerous other significant issues have been addressed in the consideration of maternal-fetal surgery for this and other nonlethal anomalies (Hirose & Farmer, 2009).
Hydrocephalus
Hydrocephalus is a condition in which there is excess CSF in the ventricles of the brain due to overproduction (rare) or a decrease in reabsorption. The most common etiology for the neonate is excess ventricular CSF due to aqueductal flow obstruction. The obstruction prevents the CSF from leaving the head and flowing into the spinal column. Other etiologies include congenital viral infections, myelomeningocele with Arnold-Chiari malformation, and congenital masses or tumors (Lynam & Verklan, 2010; Volpe, 2008).
The neonate presents with an increasing frontal-occipital circumference because the head circumference is increasing at an abnormal rate as a result of the increase in CSF pressure (Fig. 36-3). The sutures are widened and the fontanels are full or bulging and tense. Classic signs of increasing intracranial pressure (vomiting, lethargy, and irritability) can be attributed to feeding intolerance. It is thought that “setting sun eyes,” (eyes that are rotated downward) indicate permanent damage to the brain tissue. Serial head ultrasound is used to evaluate the evolving condition. Neurosurgery and genetics services are consulted to evaluate for the placement of a ventriculoperitoneal shunt versus placement of a reservoir and to assess for the presence of congenital anomalies (Lynam & Verklan, 2010; Volpe, 2008).

FIG. 36-3 Mother providing kangaroo care to preterm twins; the one on the right has hydrocephalus. The characteristic appearance is an enlarged head, thinning of the scalp, distended scalp veins, and a full fontanel. (Courtesy Cheryl Briggs, RNC, Annapolis, MD.)
The nurse provides support to the family by teaching and involving them in their baby’s care as much as possible. Minimal stimulation protocols that promote the use of limited handling, dim lighting, and attention to the baby’s cues will help keep the baby calm. The head needs to be positioned carefully, repositioned at least once every 4 hours, and attention given such that the head is not positioned on the shunt side postoperatively. Gel-filled pillows can provide some comfort for the neonate. The frontal-occipital circumference needs to be measured serially, and depending on the rate of increase, every 4 hours to every 24 hours. Neurologic assessments and observation for signs of increasing intracranial pressure should be done every 4 to 8 hours, depending on the baby’s clinical condition.
The baby should be fed in a semi-reclining position with the head well-supported. The method, amount, and frequency of feeding depend on the infant’s tolerance and energy level. The nurse should be alert to the possibility of emesis, a frequent occurrence in the presence of increased intracranial pressure, and should maintain aspiration precautions. Nonnutritive sucking, touching, and cuddling needs should be met.
In addition to serial intracranial ultrasonography, the diagnosis is also made using computed tomography and magnetic resonance imaging (MRI); antenatal diagnosis can be made by fetal ultrasonography. The surgical correction of hydrocephalus involves the placement of a shunt that goes from the ventricles of the brain usually to the peritoneum to allow the drainage of excess CSF. Damaged or destroyed brain tissue cannot be restored. The long-term prognosis in affected infants depends on the presence and extent of such tissue damage, along with the cause of the hydrocephalus, the presence of concurrent neurologic problems, and the long-term success of the shunt procedure.
Parent teaching regarding the shunt should be done both pre- and postoperatively. Providing handouts or if possible, seeing an infant with a shunt already in place can be helpful. Parents also need to be taught signs of a blocked shunt and signs of infection such as increasing intracranial pressure and changes in the baby’s feeding patterns.
Microcephaly
Microcephaly refers to a head circumference that measures two or more standard deviations below the mean for age and sex (Lynam & Verklan, 2010). Brain growth is usually restricted and thus mental retardation is common. Maternal risk factors include congenital viral infections, chromosomal disorders, and malnutrition. Fetal and neonatal factors include inflammation, birth trauma, and sequelae of hypoxic-ischemic encephalopathy. If the result of an in utero insult, the baby is typically born with a small head and brain, which gives the forehead a backward sloping appearance. Diagnostic evaluations include a complete maternal history, evaluation of the events surrounding the birth, computed tomography (CT) or MRI to evaluate brain volume, and a neurologic assessment. Typically genetics and infectious disease consults are also obtained. Although neurologic deficits are not present at birth, developmental delays will become evident. The baby’s outcome and prognosis depend on the severity of the microcephaly. Treatment for the infant is supportive. The parents will need support and education to help them learn to care for a child with cognitive impairment and developmental delays.
Respiratory System Anomalies
Screening for congenital anomalies of the respiratory system is necessary even in infants who are apparently normal at birth. Respiratory distress at birth or shortly thereafter can be the result of lung immaturity or anomalous development. Congenital laryngeal web and bilateral choanal atresia are readily apparent at birth. Respiratory distress caused by diaphragmatic hernia and tracheoesophageal fistula can appear immediately or be delayed, depending on the severity of the defect.
Laryngeal Web and Choanal Atresia
A laryngeal web, which is uncommon, results from the incomplete separation of the two sides of the larynx and is most often between the vocal cords. This is a surgical emergency such that perforation of the web using an endotracheal tube can be lifesaving. Choanal atresia (Fig. 36-4) is the most common congenital anomaly of the nose. The posterior nares can be blocked by a bony or soft-tissue obstruction. The obstruction can be unilateral or bilateral. The baby can display symptoms of respiratory distress and cyanosis or pallor that are relieved whenever the baby is crying. Inability to pass a suction catheter through the nose into the pharynx is highly suggestive of the diagnosis. Securing an oral airway into the posterior pharynx and maintaining the baby in the prone position will provide a patent airway and time to evaluate for the presence of other abnormalities. Definitive therapy involves creating a patency through the bony or soft tissue obstruction, and use of serial obturators to dilate the new airway passages (Pappas & Walker, 2010; Ringer & Hansen, 2008).
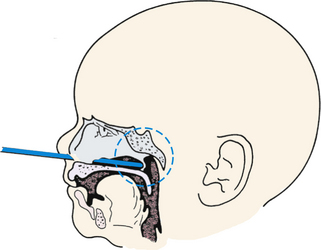
FIG. 36-4 Choanal atresia. Posterior nares are obstructed by membrane or bone either bilaterally or unilaterally. Infant becomes cyanotic at rest. With crying, newborn’s color improves. Nasal discharge is present. Snorting respirations often are observed with increased respiratory effort. Newborn may be unable to breathe and eat at the same time. Diagnosis is made by noting inability to pass small feeding tube through one or both nares. (Used with permission of Ross Products Division, Abbott Laboratories, Inc., Columbus, OH 43216. From Clinical Education Aid #6, Copyright 1963, Ross Products Division, Abbott Laboratories, Inc.)
Congenital Diaphragmatic Hernia
Congenital diaphragmatic hernia (CDH) results from a defect in the formation of the diaphragm, allowing the abdominal organs to be displaced into the thoracic cavity. It occurs in approximately 1 in 3000 to 4000 live births and despite improvements in care management, the mortality rate remains high, at about 20% to 30% (Tsao & Lally, 2008). The etiology is unknown; however, it is not uncommon to find concomitant chromosomal anomalies, especially trisomies 12, 18, and 21, as well as abnormalities of the gastrointestinal, genitourinary, and central nervous systems. Intestinal malrotation is also associated with CDH. Approximately 80% of the hernias occur on the left side in the posterior diaphragm known as the foramen of Bochdalek (Holder, Klaassens, Tibboel, de Klein, Lee, & Scott, 2007; Ringer & Hansen, 2008). Poorer outcomes are associated with right-sided or bilateral defects (Brownlee, Howatson, Davis, & Sabharwal, 2009).
A small defect may not be detected on an early prenatal ultrasound. Fetuses with CDH tend to have polyhydramnios, which typically prompts the obstetrician to obtain an ultrasound. A major advantage of diagnosing CDH prenatally is that it allows the baby to be born in a tertiary hospital that is equipped to manage all the cardiorespiratory problems associated with the defect and to rapidly stabilize the neonate’s precarious condition. Hernias can be repaired by fetal surgery in some research institutions; however, intrauterine surgical correction of CDH has met with poor neonatal outcomes in many cases. Severe defects can be managed with the baby being born by cesarean using the EXIT (EX-utero intrapartum treatment) procedure in which the neonate is transitioned immediately from placental support to extracorporeal membrane oxygenation (ECMO) (Ringer & Hansen, 2008).
Depending on the size of the defect and how much developed lung tissue is present, the baby may not be in any significant distress at birth. A small defect can distress the baby when feeding such that symptoms of respiratory distress (tachypnea, pallor, mottled, cyanosis) become evident. Babies with a large defect will have significant distress at birth because the viscera present in the thoracic cavity during embryonic life prevented the normal development of the lung (Fig. 36-5). Characteristic symptoms include respiratory distress, cyanosis, heart sounds shifted to the right, and low blood pressure. There will be increasing distress as the bowels fill with air. The abdomen is scaphoid shaped and the chest appears barrel shaped due to the abdominal contents being in the chest. Diagnosis can be made on the basis of the x-ray finding of loops of intestine in the thoracic cavity and the absence of intestine in the abdominal cavity. The severity of the clinical picture is considered to be a neonatal emergency (Bradshaw, 2010; Ringer & Hansen, 2008).
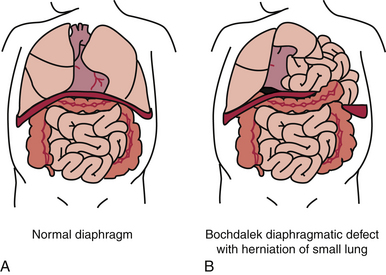
FIG. 36-5 A, Normal diaphragm separating the abdominal and thoracic cavities. B, Diaphragmatic hernia with a small lung and abdominal contents in the thoracic cavity. (From Erlich P., & Coran, A. [2007]. Diaphragmatic hernia. In R. Kliegman, R. Behrman, H. Jenson, & B. Stanton [Eds.], Nelson textbook of pediatrics [18th ed.]. Philadelphia: Saunders.)
Surgical repair is performed as soon as the infant is stable. Preoperative nursing interventions include participating in the stabilization of the infant’s cardiopulmonary condition until surgical repair can be done. Inhaled nitrous oxide (NO) has been used in many centers with moderate success to treat the accompanying persistent pulmonary hypertension (Askin & Diehl-Jones, 2010). Gastric contents are aspirated and suction applied to decompress the gastrointestinal tract and prevent further cardiothoracic compromise. Oxygen therapy, mechanical ventilation, and the correction of acidosis are necessary in infants with large defects. Pulmonary hypertension can occur as a result of the lung hypoplasia. ECMO or high-frequency oscillatory ventilation can be used in infants with severe circulatory and respiratory complications (see Chapter 37) (Bradshaw, 2010; Migliazza, Bellan, Alberti, Auriemma, Burgio, Locatelli, & Colombo, 2007).
The prognosis depends largely on the degree of fetal pulmonary development and the success of surgical diaphragmatic closure, but the prognosis in severe cases is often poor. As a rule, finding the liver in the thorax is associated with the worst prognosis. Approximately 20% of infants with CDH die before they are able to have surgical repair. It is unclear whether the timing of the surgery or the condition of the infant at the time of repair is more important to improved outcomes (Bianco-Batlles, Mohamed, & Hammad, 2010). Overall survival rates have improved with the advent of inhaled NO, improved management of high-frequency ventilation, and ECMO. Of those who survive, some have chronic lung disease with long-term oxygen dependency, feeding difficulties, and gastroesophageal reflux (GER).
Gastrointestinal System Anomalies
Anomalies in the gastrointestinal (GI) system can occur anywhere along the GI tract, from the mouth to the anus. Some anomalies, such as cleft lip, omphalocele, and gastroschisis, are apparent at birth. Others, including cleft palate, esophageal atresia, pyloric stenosis, intestinal obstructions, and imperforate anus, become apparent as the infant is further assessed or becomes symptomatic.
Cleft Lip and Palate
Facial clefts are among the most common congenital anomalies. Cleft lip or palate is a congenital midline fissure, or opening, in the lip or palate resulting from failure of the primary palate to fuse (Fig. 36-6). One or both deformities can occur, and nasal deformity can be present. Multiple genetic and, to a lesser extent, environmental factors (e.g., maternal infection, maternal smoking, radiation exposure, alcohol ingestion, and treatment with medications such as corticosteroids, lithium, retinoids, and phenytoin) appear to be involved in their development. Approximately one third of cases of cleft lip or palate are associated with a major anomaly such as Pierre Robin syndrome (Manning, 2009).
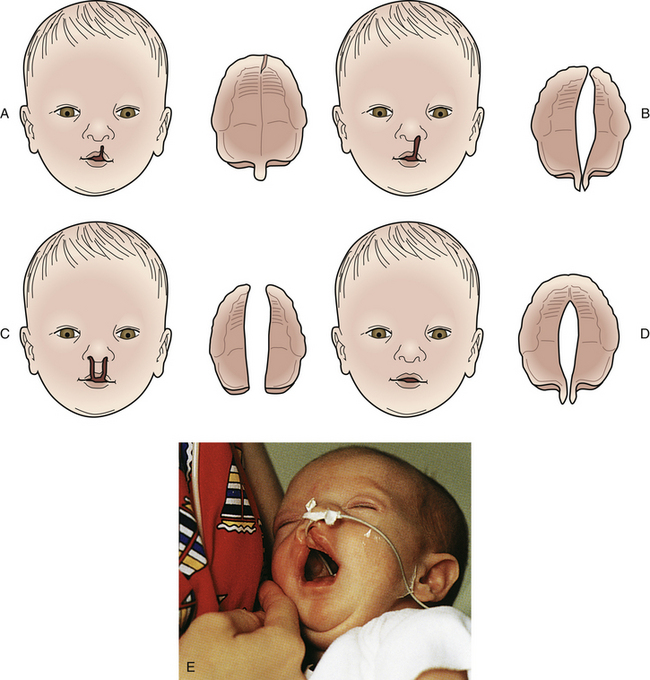
FIG. 36-6 Variations in clefts of lip and palate at birth. A, Notch in vermilion border. B, Unilateral cleft lip and cleft palate. C, Bilateral cleft lip and cleft palate. D, Cleft palate. E, Infant with complete unilateral cleft lip. Note the feeding tube. (A-D, From Hockenberry, M., & Wilson, D. [2011]. Wong’s nursing care of infants and children [9th ed.]. St. Louis: Mosby. E, From Dickason, E., Silverman, B., & Kaplan, J. [1998]. Maternal-infant nursing care [3rd ed.]. St. Louis: Mosby.)
Cleft lip with or without cleft palate occurs in approximately 10.48 per 10,000 live births. Cleft palate alone occurs in 6.39 per 10,000 live births (Centers for Disease Control and Prevention [CDC], 2006).
The defect can range from a simple notch in the lip to complete separation of the lip that extends to the floor of the nose. The treatment for a cleft lip is surgical repair, which usually is done between ages 6 and 12 weeks, if the infant is healthy and free of infection. Advances in surgical techniques have made it possible for some infants, particularly those with unilateral cleft lip, to have a near-normal appearance. The results of the repair depend on the severity of the defect, with more severe bilateral cleft lip requiring surgical repair done in stages (Thigpen, 2007).
Anomalies of the palate often occur in association with cleft lip. Cleft palate alone is more common in female infants and occurs more frequently as a constituent of certain syndromes. This defect can range from a cleft in the uvula to a complete cleft of the hard and soft palates that can be unilateral, bilateral, or midline. Feeding is difficult because the cleft lip renders the newborn unable to maintain a seal around a nipple; the cleft palate renders the infant unable to form a vacuum to maintain suction when feeding. In addition, the inability to suck and swallow normally allows milk to pool in the nasopharynx, which increases the likelihood of aspiration. Furthermore, as the infant attempts to suck, milk often comes out through the cleft and the nares. Although the degree of difficulty depends on the size of the cleft, feeding problems are greater in infants with a cleft palate than in those with a cleft lip. Breastfeeding can be successful in some infants, particularly if the infant has a cleft lip alone. Bottle feeding can be successful in some infants. There are special nipples, bottles, and appliances available to aid in feeding (Fig. 36-7). In general, parents of infants with these defects need education, support, and encouragement as they learn to feed their baby. This can help minimize anxiety and frustration while promoting competence and confidence in providing infant care. The type of surgical repair to close the cleft palate is based on the degree of the cleft and, if severe, can necessitate repair done in stages. The cleft lip is usually repaired when the neonate reaches approximately 3600-4000 grams, and the palate is repaired at 1 to 2 years of age (Sterk, 2010). Early repair helps avert some of the speech problems that may occur in people with palate defects. Long-term care is often necessary for children with a cleft palate and involves the combined efforts of a health care team that includes acute care and community nurses; social workers; ear, nose, and throat and plastic surgeons; speech therapists; and orthodontists.
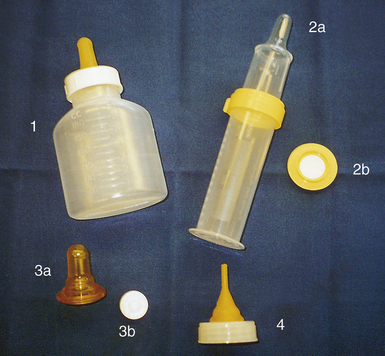
FIG. 36-7 Aids in feeding infants with cleft lip and palate 1, Mead Johnson bottle and nipple for cleft palate. Cleft palate nipple system (2a) with valve (2b) to regulate flow. Haberman feeder (3a) with disk (3b) to control flow of milk. 4, Ross cleft palate assembly. Nipple can be trimmed to accommodate palate size. (Courtesy Shannon Perry, Phoenix, AZ.)
Parents ofinfants with a cleft lip or palate need much support, particularly in the case of a cleft lip, because this is both a cosmetic and functional defect. Recognizing that this may interfere with normal parent-infant bonding in the neonatal period, the nurse must assess for this and intervene appropriately.
Esophageal Atresia and Tracheoesophageal Fistula
Esophageal atresia (EA) and tracheoesophageal fistula (TEF), the most life-threatening anomalies of the esophagus, typically occur together, although they can occur singly. The prevalence of EA and TEF is approximately 2.37 per 10,000 live births (CDC, 2006). Esophageal atresia is a congenital anomaly in which the esophagus ends in a blind pouch, thus failing to form a continuous passageway to the stomach. TEF is an abnormal connection between the esophagus and the trachea. The most common variant is the combination of a proximal EA, in which the esophagus ends in a blind pouch, with a distal TEF, in which the lower esophagus exits the stomach and is connected to the trachea by a fistula, rather than forming a continuous tube to the upper esophagus (Fig. 36-8). Variations of the anomalies are possible, depending on the presence or absence of a TEF, the site of the fistula, and the location and degree of the esophageal obstruction.
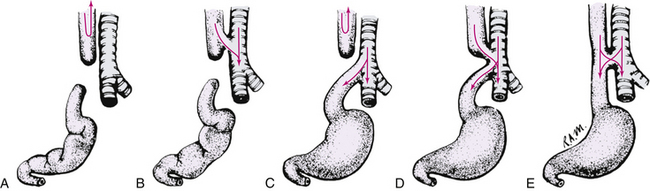
FIG. 36-8 Congenital atresia of esophagus and tracheoesophageal fistula. A, Upper and lower segments of esophagus end in blind sac, occurring in 5% to 8% of such infants. B, Upper segment of esophagus ends in atresia and connects to trachea by fistulous tract, occurring rarely. C, Upper segment of esophagus ends in blind pouch; lower segment connects with trachea by small fistulous tract, occurring in 80% to 95% of such infants. D, Both segments of esophagus connect by fistulous tracts to trachea, occurring in less than 1% of such infants. Infant may aspirate with first feeding. E, Esophagus is continuous but connects by fistulous tract to trachea; known as H-type. (From Hockenberry, M., & Wilson, D. [2011]. Wong’s nursing care of infants and children [9th ed.]. St. Louis: Mosby.)
The presence of a midline defect such as EA or TEF is often accompanied by another significant embryonic defect such as a cardiac anomaly, cleft lip and/or palate, or vertebral, genitourinary, or abdominal wall defect (Lovvorn, Glenn, Pacetti, & Carter, 2011). The affected baby who is preterm and/or small for gestational age typically has other malformations such that a good outcome is not expected.
The defect can be diagnosed prenatally. A small or absent stomach can be detected during a fetal ultrasound. There is usually a history of polyhydramnios because the fetus is unable to swallow the amniotic fluid. At birth, the clinical presentation depends on the type of anomaly that is present. Infants with the life-threatening anomaly EA with TEF show significant respiratory difficulty immediately after birth. Esophageal atresia with or without TEF results in excessive oral secretions, drooling, and feeding intolerance. When fed, the infant may swallow, but then cough and gag and return the fluid through the nose and mouth. Respiratory distress can result from aspiration or from the acute gastric distention produced by the TEF. Choking, coughing, and cyanosis occur after even a small amount of fluid is taken by mouth.
NURSING ALERT
Any infant with excessive oral secretions and respiratory distress should not be fed orally until a physician is consulted.
Nursing interventions are supportive until surgery is performed. The infant with EA and TEF should be kept in a supine position with the head of the bed elevated about 30 degrees to facilitate respiratory efforts and prevent reflux and aspiration of gastric contents. Antireflux and antacid medication may be given to minimize gastroesophageal reflux and prevent acid-induced pneumonitis. An orogastric tube (Replogle tube) is placed in the proximal esophageal pouch and attached to low continuous suction to remove secretions and decrease the possibility of aspiration (Lovvorn et al., 2011). The infant requires close observation and intervention to maintain a patent airway. Other supportive measures include thermoregulation, maintaining fluid and electrolyte balance intravenously as well as acid-base balance, and prevention of any further complications as a result of an associated defect. Surgical correction, done in one stage if possible, consists of ligating the fistula and anastomosing the two segments of the esophagus. The chances for survival in those infants in a good risk category exceed 95% depending on the presence of associated defects and the infant’s birth weight. Preterm infants with cardiac or chromosomal anomalies have the highest mortality rate. Many infants with EA and TEF will have postoperative issues related to feeding difficulties such as GER and esophageal strictures requiring periodic dilation (Bradshaw, 2010; Magnuson, Parry, & Chwals, 2006).
Omphalocele and Gastroschisis
Omphalocele and gastroschisis are two of the more common congenital defects of the abdominal wall. Omphalocele occurs in approximately 2 of every 10,000 live births, whereas the prevalence of gastroschisis is about 3.7 in 10,000 live births (CDC, 2006). An omphalocele is a covered defect of the umbilical ring into which varying amounts of the abdominal organs can herniate (Fig. 36-9, A). The peritoneal sac covering the defect can rupture during or after birth. Many of the infants born with an omphalocele are preterm and nearly 50% have an underlying chromosomal abnormality, usually trisomies 12, 18, or 21. Congenital heart defects are often associated with omphalocele (Lovvorn et al., 2011).
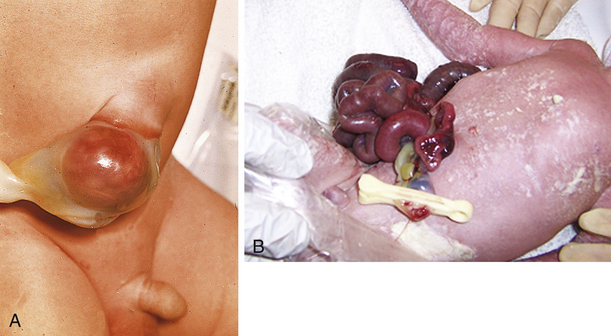
FIG. 36-9 A, Omphalocele. B, Gastroschisis of bowel and stomach. (A, From O’Doherty, N. [1986]. Neonatology: Micro atlas of the newborn. Nutley, NJ: Hoffmann-La Roche; B, courtesy Cheryl Briggs, RNC, Annapolis, MD.)
Gastroschisis is the herniation of the bowel through a defect in the abdominal wall to the right of the umbilical cord (see Fig. 36-9, B). No membrane covers the contents as it does with an omphalocele. Gastroschisis is 3 to 4 times more common than omphalocele and is not usually associated with other major congenital anomalies or syndromes. Intestinal atresia can occur with gastroschisis (Lovvorn et al., 2011).
The preoperative nursing care is similar for infants with either defect. Exposure of the viscera causes problems with thermoregulation and fluid and electrolyte balance. Immediately after birth, the neonate’s torso should be placed in an impermeable, clear plastic bowel bag to decrease insensible water losses, maintain thermoregulation, and prevent contamination of the exposed viscera (Bradshaw, 2010). It is essential that the nurse assess the exposed viscera frequently to detect any changes in perfusion to the exposed abdominal contents. The baby should be placed in a side-lying position and the viscera supported with a blanket roll to prevent vascular compromise to a torqued intestine. Prior to surgery, the exposed viscera should be kept covered with sterile moistened saline gauze and plastic wrap. Gastric decompression with a Replogle tube (a special type of gastric tube) connected to low intermittent wall suction is also necessary to prevent aspiration pneumonia and to allow as much bowel as possible to be placed into the abdomen during surgery. Antibiotics, fluid and electrolyte replacement, and thermoregulation are needed for physiologic support (Lovvorn et al., 2011).
Surgery is usually performed soon after birth. If complete closure is impossible because of the small size of the defect and the large amount of viscera to be replaced, a Silastic silo or patch (Dow Corning, Midland, MI) is placed. This protects the contents as they are gradually placed back into the abdominal cavity and minimizes symptoms of respiratory distress as the increasing intraabdominal pressure pushes against the diaphragm. The defect is closed surgically after the exposed visceral contents have been reduced; this reduction process usually takes 7 to 10 days. The prognosis depends on the size of the defect and the presence of associated anomalies. It is generally expected that there will be complications related to intestinal dysfunction, such as feeding difficulties, dysmotility, or short-gut syndrome if a substantial amount of the intestine had to be removed.
Parental support is essential because the infant has an obvious disfiguring anomaly that can be shocking and repulsive in appearance. Depending on the size of the defect, the infant can also be critically ill before surgery. The nurse must be aware of the effect this may have on parental bonding and intervene appropriately as the parents cope with this crisis.
Gastrointestinal Obstruction
Congenital intestinal obstruction can occur anywhere in the GI tract and takes one of the following forms: atresia, which is a complete obliteration of the passage; partial obstruction, in which the symptoms can vary in severity and sometimes not be detected in the neonatal period; or malrotation of the intestine, which leads to twisting of the intestine (volvulus) and obstruction. Esophageal atresia, discussed previously, is a type of GI obstruction. Duodenal atresia, midgut malrotation and volvulus, jejunoileal atresia, necrotizing enterocolitis, and meconium ileus are the most common causes of neonatal intestinal obstruction. Meconium ileus is an obstruction caused by impacted meconium; over 90% of infants who are born with with meconium ileus have cystic fibrosis, a life-threatening chronic illness (Lovvorn et al., 2011).
Signs of neonatal intestinal obstruction occur early. It can be suspected in pregnant women with polyhydramnios. The neonate with an intestinal obstruction displays the following cardinal signs: bilious vomiting, abdominal distention, and failure to pass normal amounts of meconium in the first 24 hours. High intestinal obstruction is characterized by vomiting, even if the infant is not being fed orally. Distention usually indicates a low obstruction, with vomiting occurring later. Abdominal distention can elevate the diaphragm, which can cause respiratory difficulties.
Nursing care is aimed at supporting the infant until surgical intervention can be carried out to eliminate the obstruction. Oral feedings are withheld, an orogastric tube is placed to low intermittent wall suction, and intravenous therapy is initiated to provide needed fluid and electrolytes. In infants with an intestinal obstruction, surgery consists of resecting the obstructed area of bowel and anastomosing the nonaffected bowel, or creating an ostomy and allowing the bowel to rest. In recent years the survival rate for these infants has risen to 90% to 95% as a result of improved medical and nursing management, as well as a better understanding of the total problem.
Imperforate Anus
Imperforate anus is a term used to describe a wide range of congenital disorders involving the anus and rectum and genitourinary system (Fig. 36-10). These anomalies are relatively common, with an incidence of approximately 1 in 5000 live births (Bradshaw, 2010; Lovvorn et al., 2011). Occurring more in male than in female infants, they result from the failure of the urogenital sinus and cloaca to differentiate during weeks 7 and 8 of gestational life (Blackburn, 2007). Such infants have no anal opening, and commonly there is also a fistula from the rectum to the perineum or genitourinary system (Fig. 36-11). Types of anorectal malformations include the typical cloaca in females, which involves the vagina, colon, and urethra forming a single common passage in the perineum. Others include the low rectovaginal fistula (female) and rectourethral bulbar fistula (male). Imperforate anus can occur in isolation or in combination with other congenital defects such as esophageal atresia, duodenal atresia, renal anomalies, or vertebral anomalies. Extensive surgical repair is often required in stages for the more complex types of anorectal malformations. Infants with high imperforate anus typically have bowel incontinence even after surgical repair. In some cases the anomaly involves stenotic areas, or a thin translucent membrane can cover the anal opening. Treatment for such a membrane is anoplasty followed by daily dilation, which parents learn to do. The preoperative nursing care is similar to that described for other GI obstructions. Imperforate anus is often associated with other anomalies, with nearly half having additional genitourinary anomalies, which can complicate long-term care. Outcome is excellent with low imperforate anus (Bradshaw, 2010).
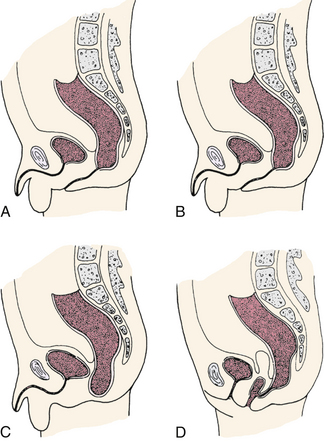
FIG. 36-10 Types of imperforate anus. Anal sphincter muscle may be present and intact. A, High lesion opening onto perineum through narrow fistulous tract. B, High lesion ending in fistulous tract to urinary tract. C, Low lesion in bowel passes through puborectal muscle. D, High lesion ending in fistulous tract to vagina.
Musculoskeletal System Anomalies
The two most common musculoskeletal system anomalies seen in neonates are developmental dysplasia of the hip and congenital clubfoot. Both of these conditions must be detected and treated early for successful correction.
Developmental Dysplasia of the Hip
The broad term developmental dysplasia of the hip (DDH) describes a spectrum of disorders related to abnormal development of one or all of the components of the hip joint that can develop at any time during fetal life, infancy, or childhood. A change in terminology from congenital hip dysplasia (CHD) and congenital dislocation of the hip to DDH more properly reflects a variety of hip abnormalities in which there is a shallow acetabulum, subluxation, or dislocation.
The incidence of DDH in the United States is 1.5 per 1000 live births (Butler, 2007). The etiology is unknown, and believed to be multifactorial. Certain factors such as sex, birth order, family history, intrauterine position, birth type, joint laxity, and postnatal positioning are believed to affect the risk of DDH. Predisposing factors associated with DDH can be divided into three broad categories: (1) physiologic factors, which includes maternal hormone secretion and intrauterine positioning; (2) mechanical factors, which involves breech presentation, multiple fetus, oligohydramnios, and large infant size; other mechanical factors may include continued maintenance of the hips in adduction and extension that will in time cause a dislocation; and (3) genetic factors, which entail a higher incidence of DDH in siblings of affected infants, and an even greater incidence of recurrence if a sibling and one parent were affected.
Figure 36-12 illustrates the three degrees of DDH, which are described as follows.
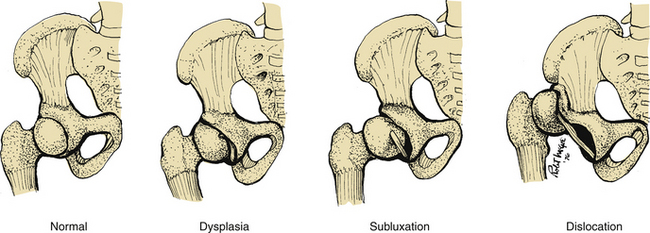
• Acetabular dysplasia (or preluxation)—mildest form of DDH in which there is neither subluxation nor dislocation. There is a delay in acetabular development evidenced by osseous hypoplasia of the acetabular roof that is oblique and shallow, although the cartilaginous roof is comparatively intact. The femoral head remains in the acetabulum.
• Subluxation—accounts for the largest percentage of DDH. Subluxation implies incomplete dislocation of the hip and is sometimes regarded as an intermediate state in the development from primary dysplasia to complete dislocation. The femoral head remains in contact with the acetabulum, but a stretched capsule and ligamentum teres cause the head of the femur to be partially displaced. Pressure on the cartilaginous roof inhibits ossification and produces a flattened socket.
Dislocation—The femoral head loses contact with the acetabulum and is displaced posteriorly and superiorly over the fibrocartilaginous rim. The ligamentum teres is elongated and taut. DDH is often not detected at the initial examination after birth; thus all infants should be carefully monitored for hip dysplasia at follow-up visits throughout the first year of life. In the newborn period dysplasia usually appears as hip joint laxity rather than as outright dislocation. Subluxation and the tendency to dislocate can be demonstrated by the Ortolani or Barlow tests. The Ortolani and Barlow tests are most reliable from birth to 2 or 3 months of age (see Fig. 23-11). Other signs of DDH are shortening of the limb on the affected side (Galeazzi sign, Allis sign), asymmetric thigh and gluteal folds, and broadening of the perineum (in bilateral dislocation).
NURSING ALERT
The Ortolani and Barlow tests must be performed by an experienced clinician to prevent fracture or other damage to the hip.
Treatment is begun as soon as the condition is recognized because early intervention is more favorable to the restoration of normal bony architecture and function. The longer treatment is delayed, the more severe the deformity, the more difficult the treatment, and the less favorable the prognosis. The treatment varies with the age of the child and the extent of the dysplasia. The goal of treatment is to obtain and maintain a safe, congruent position of the hip joint to promote normal hip joint development and ambulation.
The hip joint is maintained by dynamic splinting in a safe position with the proximal femur centered in the acetabulum in an attitude of flexion and abduction. Of the numerous devices available, the Pavlik harness is the most widely used, and with time, motion, and gravity, the hip works into a more abducted, reduced position (www.pavlikharness.com). The harness is worn continuously until the hip is proved stable on clinical and radiographic examination, typically around 3 months. It has an 80% success rate for the treatment of classic DDH. If not effective, traction, casting, and even surgery can be necessary to stabilize the hip (Krasser, 2008).
NURSING ALERT
The former practice of double- or triple-diapering for DDH is not recommended because it promotes hip extension, thus worsening proper hip development.
In addition to the major intervention of assessing and helping identify the disorder, another key nursing intervention is teaching the parents about the care of the infant as he/she
 CLINICAL REASONING
CLINICAL REASONING
Developmental Dysplasia of the Hip (DDH)
Anna, a 4.3-kg Caucasian neonate, is admitted to the newborn nursery after a cesarean birth for breech presentation. Anna appears to be a healthy newborn and is being examined by the pediatric nurse practitioner (PNP). Anna’s vital signs are within the normal range and her physical examination is normal except for findings related to her left hip. The assessment findings were followed by an ultrasound, and it was determined that Anna has DDH.
1. Evidence—Are there any predisposing factors that would alert the PNP to suspect DDH? Is there evidence to determine her diagnosis and preferred treatment?
2. Assumptions—What assumptions can be made about the following items?
a. Physical assessment techniques that will be done to assess for DDH
b. The immediate plan of care for Anna after a diagnosis of DDH
c. The ability for Anna’s mother to breastfeed her daughter
d. The effect of having an infant with a congenital disorder
3. What implications and priorities for nursing care can be made at this time?
will remain in the harness continuously during the treatment. Because the harness is worn during a time of maximal growth, it is necessary for the parents to adjust the infant’s care to accommodate the infant’s changing needs. The baby can develop a brachial plexus palsy due to increased tension of the shoulder harness if the harness is not modified according to the neonate’s growth. Thorough and ongoing follow-up care is necessary, as is psychosocial support for the family (Butler, 2007).
Clubfoot
Congenital clubfoot is a deformity of the foot and ankle that includes forefoot adduction, midfoot supination, hindfoot varus, and ankle equinus. Deformities of the foot and ankle are described according to the position of the ankle and foot. The more common positions involve the following variations:
• Talipes varus—An inversion or a bending inward
• Talipes valgus—An eversion or bending outward
• Talipes equinus—Plantar flexion in which the toes are lower than the heel
• Talipes calcaneus—Dorsiflexion, in which the toes are higher than the heel
Most cases of clubfoot are a combination of these positions. The most frequently occurring type is the composite deformity talipes equinovarus (TEV). In this abnormality the foot appears C-shaped, pointing downward and inward; the ankle is inverted; and the Achilles tendon is shortened. The foot appears small, wide, and stiff, and the lower leg appears small because of hypoplasia of the calf muscles. Unless treated, further stiffening occurs, and bony changes will result. Unilateral clubfoot is somewhat more common than bilateral clubfoot and can occur as an isolated defect or in association with other disorders or syndromes, such as chromosomal aberrations, arthrogryposis (a generalized immobility of the joint), cerebral palsy, or spina bifida.
Clubfoot is one of the most common congenital anomalies, occurring in approximately 1.5 per 1000 live births, with two times more male than female infants affected. The etiology is thought to be multifactorial and can involve a genetic predisposition, chromosomal anomalies, abnormalities of the uterine environment, and neuromuscular pathologies (Butler, 2007; Sterk, 2010).
Serial casting is begun shortly after birth, before discharge from the nursery. Successive casts allow for gradual stretching of skin and tight structures on the medial side of the foot. Manipulation and casting are repeated frequently (every week) to accommodate the rapid growth of early infancy. The extremity or extremities are often casted or splinted until maximum correction is achieved, usually within 8 to 12 weeks. If needed, surgical correction is done before the infant begins to walk.
Because these infants are often placed in a cast before discharge, the nurse must teach parents necessary care, including how to protect the cast and assess the toes for neurovascular compromise. This is particularly important because of the potential for the infant to outgrow the cast. As is true with the birth of any child with an anomaly, the nurse should be supportive of the parents as they learn the ways to meet the infant’s normal needs, as well as those brought about by the infant’s physical problem.
Polydactyly
Occasionally an infant is born with extra digits on the hands or feet. In some instances, polydactyly is hereditary. If there is little or no bone involvement, the extra digit is tied with silk suture soon after birth. The finger or toe falls off within a few days, leaving a small scar. When there is bone involvement, surgical repair is indicated.
Genitourinary System Anomalies
Anomalies involving the genitourinary system can be distressing to parents because they can be readily apparent and, in the case of some conditions, because of the concern about sexuality and reproductive functioning. These anomalies range from obvious anomalies of the external genitalia, such as hypospadias, to those involving internal organs that are not obvious but can cause damage to the urinary tract. An example of the latter is an obstruction in the urinary tract that can cause hydronephrosis, which is the abnormal collection of urine in the renal pelvis, that can eventually destroy the kidney.
Hypospadias and Epispadias
Hypospadias constitutes a range of penile anomalies associated with an abnormally located urinary meatus. The meatus can open below the glans penis or anywhere along the ventral surface of the penis, the scrotum, or the perineum. It is the most common anomaly of the penis, affecting approximately 88.7 in 100,000 male infants (Osterman, Martin, & Menacker, 2009). Hypospadias is classified according to the location of the meatus and the presence or absence of chordee, which is a ventral curvature of the penis (Figs. 36-13 and 36-14). The cause is unknown, although it is thought to be of multifactorial inheritance.
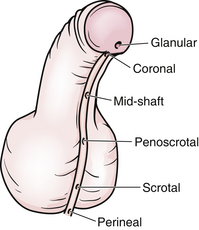
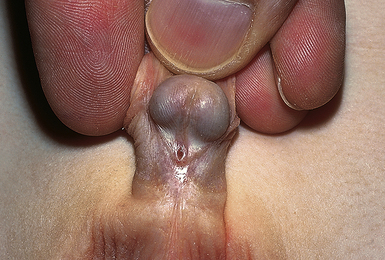
FIG. 36-14 Hypospadias. (Courtesy H. Gil Rushton, MD, Children’s National Medical Center, Washington, DC.)
Mild cases of hypospadias are often repaired for cosmetic reasons and involve a single surgical procedure. In more severe cases, several surgeries are required to reconstruct the urethral opening and correct the chordee, thereby straightening the penis. The goals are to improve the appearance of the genitalia, make it possible for the child to urinate in a standing position, and have a sexually adequate organ. These infants are not circumcised because the foreskin can be needed during surgical repair. Repair is done early, between 6 and 12 months of age, so that the child’s body image is not impaired (Botwinski, 2010).
Epispadias, a rare anomaly, results from failure of urethral canalization. About half of the affected infants are males who have a widened pubic symphysis and a broad spadelike penis with the urethral opening on the dorsal surface. In females there is a wide urethra and a bifid clitoris. Severity ranges from a mild anomaly to a severe one that is associated with exstrophy of the bladder. Surgical correction is necessary, and affected male infants should not be circumcised.
Exstrophy of the Bladder
The most common bladder anomaly is exstrophy (Fig. 36-15), which often occurs in conjunction with epispadias. It is rare, occurring in approximately 1 in 35,000 to 40,000 live births, and males are affected twice as often as females (Elder, 2007). It results from the abnormal development of the bladder, abdominal wall, and pubic symphysis that causes the bladder, the urethra, and the ureteral orifices to be exposed. The bladder is visible in the suprapubic area as a red mass with numerous folds, with urine draining from it onto the infant’s skin. Immediately after birth the exposed bladder should be covered with a sterile, nonadherent dressing to protect its delicate surface until closure can be performed. It is recommended that reconstructive surgery be started in the neonatal period, such that the bladder is closed within 48 hours. Parents will need detailed instruction along with support and encouragement as they deal with caring for an infant who has such an obvious defect. Repair is completed before school age, if possible, although some children never attain normal voiding patterns and later may be considered for surgery for urinary diversion.
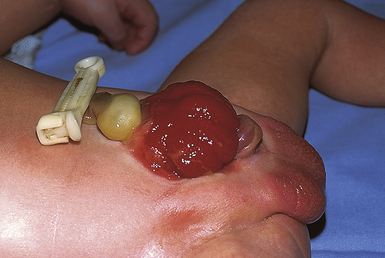
Ambiguous Genitalia
The nurse is often the one to discover ambiguous genitalia in the newborn (Fig. 36-16) during a physical assessment. Erroneous or abnormal sexual differentiation can be a genetic aberration, such as congenital adrenal hypoplasia, which can be life threatening because it involves the deficiency of all adrenal cortical hormones. Other possible causes of sexual ambiguity include chromosomal abnormalities, defective sex hormone synthesis in male infants, and the placental transfer of masculinizing agents to female fetuses. Sex assignment should be based on data gathered from the following sources: maternal and family history, including the ingestion of steroids during pregnancy, and relatives with ambiguous genitalia or who died during the neonatal period; physical examination; chromosomal analysis (results are available in 2 to 3 days); endoscopy, ultrasonography, and radiographic contrast studies; biochemical tests, such as analysis of urinary steroid excretion, which helps detect several of the adrenal cortical syndromes; and, in some instances, laparotomy or gonad biopsy
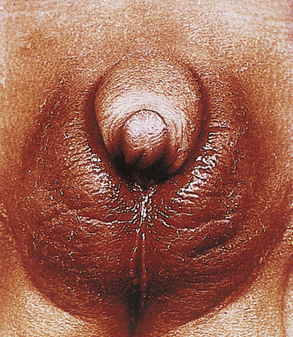
FIG. 36-16 Ambiguous external genitals (i.e., structure can be enlarged clitoral hood and clitoris or malformed penis). (Courtesy Edward S. Tank, MD, Division of Urology, Oregon Health Science University, Portland.)
Assessment and management of a newborn with ambiguous genitalia requires urgency and sensitivity. Therapeutic intervention, including any counseling and surgery, should be started as soon as possible. Care is best managed by a multidisciplinary team consisting of the primary physician, pediatric endocrinologist, geneticist, surgeon, social worker, and nurses. An infant born with ambiguous genitalia should not receive a sex assignment until diagnostic testing provides enough information for a well-informed decision (Chi, Lee, & Neely, 2008).
An appropriate gender assignment should be based on the following: age at presentation, potential for mature sexual function, potential fertility, and the long-term psychologic and intellectual effect on the child and family. Parents need much support as they learn to deal with this very challenging situation.
Care Management
Refined testing procedures are available to monitor fetal development. Prenatal diagnostic techniques such as amniocentesis, ultrasonography, alpha-fetoprotein measurements, chorionic villus sampling, PUBS, fetal nuchal translucency (FNT) screening, and gene probes contribute information to the database (see Chapter 26). Although they are a valuable adjunct to prenatal care, these tests cannot identify all congenital disorders. Furthermore, ethical issues surround such testing, and the nurse must be prepared to support the family’s decision regarding these tests. If a disorder is detected and the family decides to proceed with the pregnancy, the advantage is that appropriate care can be made available for the infant immediately at birth.
The nurse reviews the maternal history and medical information in the prenatal record for risk factors associated with congenital disorders. These factors include various medical, surgical, and social conditions and their treatments (see Chapter 30); maternal infection (see Chapter 7); maternal endocrine and metabolic disorders (see Chapter 29); and infection and drug dependence in the newborn (see Chapter 35).
Perinatal Diagnosis
Many congenital anomalies require intervention soon after birth. By careful observations in the birth room or nursery, the nurse can identify most of these conditions. An excessive amount of amniotic fluid, polyhydramnios, is commonly associated with congenital anomalies in the newborn, and such infants should be examined closely at the earliest possible time.
Oligohydramnios, an insufficient amount of amniotic fluid, is associated primarily with anomalies of the urinary tract that prevent normal micturition in utero. It is most often associated with renal agenesis or dysplasia and obstructive lesions in the lower urinary tract. Anomalies of the ears are associated with renal abnormalities. Bilateral renal agenesis, resulting in oligohydramnios, commonly manifests as Potter syndrome, which is characterized by atypical facial appearance consisting of a flat nose, recessed chin, epicanthal folds, and low-set abnormal ears; limb abnormalities; pulmonary hypoplasia; and fetal growth restriction. These conditions can be diagnosed prenatally.
Postnatal Diagnosis
Apgar scoring and a brief assessment are completed for all neonates after birth. Any deviations from normal are reported to the primary health care provider immediately. A thorough assessment of all body systems follows to identify anomalies and determine the best management plan for the neonate and the family.
Some infants have multiple congenital anomalies. A recognized pattern of malformations is referred to as a syndrome. The most common affecting about 14 of every 10,000 births (CDC, 2006), is Down syndrome, (Fig. 36-17), with the diagnosis confirmed early in the neonatal period.
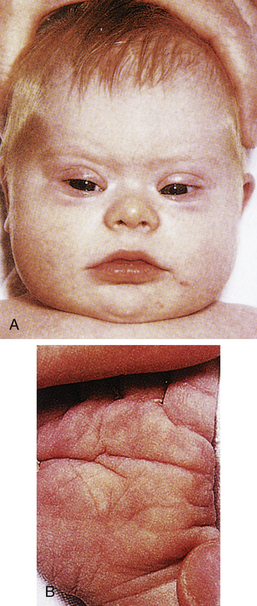
Genetic Diagnosis
Diagnostic procedures for the detection of genetic disorders are performed after birth at any time from the postnatal period through adulthood. Many tests exist for various disorders; only the most frequently used tests are discussed here.
Newborn Screening
The most widespread use of postnatal testing for genetic disease is the routine screening of newborns for inborn errors of metabolism such as phenylketonuria (PKU), galactosemia, hemoglobinopathy (sickle cell disease and thalassemias), and hypothyroidism; these are the minimum mandatory newborn screening tests in most states in the United States. Newborn screening testing in Canada varies by province.
An inborn error of metabolism (IEM) is the term applied to a large group of disorders caused by a metabolic defect that results from the absence of or change in a protein, usually an enzyme, and mediated by the action of a certain gene. These defects can involve any substrate produced from protein, carbohydrate, or fat metabolism. Inborn errors of metabolism are recessive disorders, so a person must receive a defective gene from each parent. The parents usually are unaffected because their normal dominant gene directs the synthesis of sufficient protein to meet their metabolic needs under normal circumstances.
With the advent of new biochemical techniques, it is possible to detect the abnormal gene responsible for causing an increasing number of these disorders early in the neonatal period so appropriate therapies to prevent further morbidity can be implemented. Deoxyribonucleic acid (DNA) sequencing along with mutational analysis, molecular diagnosis, and enzyme assays can be used to identify carriers, confirm the diagnosis, and play a role in genetic counseling with the family (Schiefelbein & Cheeseman, 2009; Sterk, 2010).
Phenylketonuria is an amino acid disorder that results from a deficiency of the enzyme phenylalanine dehydrogenase (see Chapter 3). This deficiency can cause elevated levels of phenylalanine that result in CNS damage. Severe effects of PKU are rare because of early recognition through newborn screening (Blackburn, 2007). The test for PKU is not reliable, however, until the newborn has ingested adequate amounts of breast milk or formula.
NURSING ALERT
The nurse must document the initial ingestion of milk and perform the test for PKU at least 24 hours after that time.
If the infant has PKU, treatment includes a diet low in protein plus the addition of a special amino acid–containing formula that does not contain phenylalanine. Despite compliance with treatment, many affected children have some intellectual impairment. Successful management and outcome are largely dependent on early identification of the condition, modifying the diet, and compliance with the treatment regimen throughout the entire life (Sterk, 2010).
Galactosemia, caused by a deficiency of the enzyme galactose 1-phosphate uridyltransferase, results in the inability to convert galactose to glucose. Galactosemia can be detected by measuring the blood levels of galactose in the urine of newborns suspected of having the disease who have ingested formula containing galactose. Early symptoms are vomiting, weight loss, persistent jaundice, and CNS symptoms, including poor feeding, drowsiness, and seizures. Escherichia coli sepsis occurs in a large number of affected infants. If the disorder goes untreated, the galactose levels will continue to increase, and the affected infant will show failure to thrive, developmental delay, cataracts, jaundice, hepatomegaly, and cirrhosis of the liver, with death possibly occurring in the first month of life. Therapy consists of eliminating galactose from the diet and beginning a lactose-free diet. The condition precludes breastfeeding because lactose is present in breast milk (Sterk, 2010).
Congenital hypothyroidism results from a deficiency of thyroid hormones, and can be permanent (requires treatment for life) or transient (spontaneously resolves). All states in the United States routinely screen for hypothyroidism by measuring thyroxine (T4) in a drop of blood obtained from a heelstick at 2 to 5 days of age. At this time the normally expected increase in T4 is lacking in newborns with hypothyroidism. It is more often included as part of the newborn screen done in the first 24 to 48 hours or before discharge. Neonatal screening consists of an initial filter-paper blood-spot thyroxine (T4) measurement followed by measurement of thyroid-stimulating hormone (TSH) in specimens with low T4 values. Early screening can have false-positive results. Treatment is thyroid replacement. In the newborn, the results of thyroid function studies are elevated in comparison with values in older children; therefore, it is important to document the timing of the tests. In preterm and sick full-term infants, thyroid function levels are usually lower than in the healthy full-term infant. A repeat T4 and TSH can be elevated after 30 weeks (corrected age) in newborns born before that time and after resolution of the acute illness in the sick full-term infant.
If the baby is untreated, symptoms usually appear after 6 weeks and include bradycardia; hypothermia; hypotension; hyporeflexia; abdominal distention; umbilical hernia; coarse, dry hair; thick, dry skin that feels cold; anemia; widely patent cranial sutures; and retarded bone age beginning at birth. The most disabling problem, however, is delayed development of the nervous system, leading to severe developmental delay. Once identified, treatment is started immediately using synthetic T4 (L-thyroxine) as a thyroid replacement (Sterk, 2010).
Cytogenetic Studies
Abnormalities can occur in either the autosomes or the sex chromosomes. Chromosomal disorders often can be diagnosed on the basis of the clinical manifestations alone. However, an infant may have a clinical appearance that is only suggestive of a problem. Cytogenetic studies must be done to confirm or rule out a suspected diagnosis. Newer techniques in molecular cytogenetic analysis make possible a more precise identification of risk for having a fetus affected with a genetic defect such as phenylketonuria.
Disorders in the number or structure of chromosomes can be diagnosed by a karyotype (see Fig. 3-1). A karyotype is a photographic enlargement of the chromosomes arranged by their numbered pairs.
Abnormalities of the sex chromosomes make up about half of all the chromosomal abnormalities occurring in the newborn. The most common test for sex chromosome abnormalities is the buccal smear, using cells scraped from the mucosa inside the mouth. When prepared and stained, these show the number of inactive X chromosomes, also known as an X-chromatin mass or a Barr body. Each cell, whether male or female, has one genetically active X chromosome. Therefore, a normal female has one active X chromosome and one Barr body, which is on the inactive X chromosome. A normal male has no Barr bodies because he has only one genetically active X chromosome.
Dermatoglyphics
Dermatoglyphics is the study of the patterns formed by the ridges in the skin on the digits, palms, and soles. These patterns, formed early in development, are strongly correlated with the effects of chromosomes. The addition or deletion of genetic material produces alterations in the loops, swirls, and arches of the finger and toe prints, in the palm lines, and in the flexion creases on the palms of the hands and soles of the feet. Characteristic dermatoglyphic patterns have been noted for almost all the chromosomal abnormalities, including trisomies 13, 18, and 21.
An infant with Down syndrome (trisomy 21) can have a single palmar crease(Fig. 36-17), a single flexion crease of the fifth digit, and an open-field pattern on the ball of the foot (Matthews & Robin, 2011). The characteristic dermatoglyphic feature in a child with Turner syndrome is the large size of the dermal patterns on the fingers and toes. Certain fingerprint patterns also may be found in those people who have cardiac valvular problems later in life. Asymmetry of palmar ridges has been reported in congenital anomalies such as cleft lip and palate and congenital vertebral anomaly.
Newborn Care
A collaborative health team approach that includes specialists and community service representatives is needed in the care of infants with some disorders. Surgical intervention in
 NURSING PROCESS
NURSING PROCESS
The Newborn with a Congenital Anomaly
The initial physical examination of the newborn (see Chapter 24) can reveal obvious congenital anomalies, although some anomalies are not evident until the infant is a few hours or days old. Ongoing assessment is important to identify the presence of anomalies.
Expected outcomes can apply to the infant and to the parents and family. Expected outcomes for the infant include that he or she will:
• Maintain adequate physiologic functioning (airway, breathing, circulation)
Expected outcomes for the parents and family include that they will:
• Demonstrate normal grief responses.
• Perceive the infant as a family member.
• Verbalize understanding of the anomaly, management, alternative courses of action, community resources, prognosis, and care needed after hospital discharge.
• Demonstrate ability to focus on new knowledge and skills needed to care for infant.
the neonatal period can be necessary for the infant requiring either immediate correction or a palliative procedure to relieve the symptoms of the anomaly until definitive correction can be done. There is a higher morbidity and mortality rate in neonates than in older children or adults undergoing similar procedures. However, despite these problems unique to neonates, advances in surgical techniques, anesthesia, and the nursing care given in intensive care nurseries have been responsible for decreasing the risk of surgery in neonates.
The health care team must be highly skilled to meet the needs of these high risk infants. In addition to stabilization of the infant’s condition (oxygenation and perfusion of tissues), other preoperative interventions, such as orogastric tube placement for abdominal decompression, attention to thermoregulation and pain management, and the maintenance of fluid and electrolyte balance, are implemented to manage specific problems.
Parents and Family Support
While the infant is receiving optimal care, the parents have needs that must be met as they deal with the crisis of having an infant with an abnormal condition. The nurse carefully assesses their reactions, which are likely to be those typical of a grief response. Facilitating their understanding of the information given them about their infant’s condition is a vital nursing intervention. A newly diagnosed disorder often implies the need for the implementation of a therapeutic regimen. For example, the disorder may be an inborn error of metabolism, such as PKU, which requires consistent and rigid adherence to a diet. The family may need help with securing the required formula and receiving counseling from the clinical dietitian. The nurse stresses the importance of maintaining the diet, keeping an adequate supply of special preparations, and avoiding the use of unauthorized substitutions.
Referral to appropriate agencies is another essential component of the follow-up management, and the nurse should make the parents aware of all possible sources of aid, including pertinent literature, parent groups, and national organizations. Many organizations and foundations, such as the Cystic Fibrosis Foundation and the Muscular Dystrophy Association, provide services and counseling for families of affected children. There are also numerous parent groups the family can join. There they can share experiences and receive mutual support in coping with problems similar to those of other group members. Nurses should be familiar with the services available in their community that provide assistance and education to families with these special problems.
A major nursing function is providing emotional support to the family during all aspects of the care of the infant born with a defect or disorder. The feelings stemming from the real or imagined threat posed by a congenital anomaly are as varied as the people being counseled. Responses may include apathy, denial, anger, hostility, fear, embarrassment, grief, and loss of self-esteem (see Chapter 38).
Parents benefit from seeing before-and-after pictures of other babies born with the same defect. Coupled with other verbal and nonverbal supportive care, this visual reassurance may be effective in allaying their concerns.
Families need much information, guidance, and support as they make decisions regarding the care of their infants. Once they have been given the facts and possible consequences and all the assistance they need in problem solving, the final decision regarding a course of action must be their own. It is then incumbent on health care providers to support the family’s decision.
Nurses frequently encounter children with genetic diseases and families in which there is a risk that a disorder can be transmitted to or occur in an offspring. It is a responsibility of nurses to be alert to situations in which persons could benefit from a genetic evaluation and counseling, to be aware of the local genetic resources, to aid the family in finding services, and to offer support and care for children and families affected by genetic conditions. Local genetic clinics can be located through several sites, such as GeneTests (www.genetests.org), a publicly funded medical genetics information resource developed for physicians and other health care providers, which is available at no cost to all interested persons. Another resource is the National Society of Genetic Counselors (www.nsgc.org), which lists genetic counselors by states in the United States.
 COMMUNITY ACTIVITY
COMMUNITY ACTIVITY
• Visit the American Pregnancy Association website (www.americanpregnancy.org) and go to the birth defects and disorders link. Select a birth defect such as cleft lip and palate, congenital heart defects, or spina bifida. Review the information regarding neonatal effects, risk factors, diagnosis and resources. At the resources link, visit the website of the national organization. Locate a clinic that specializes in the care and treatment of the birth defect in your community.
• Visit the American Academy of Pediatrics website (www.aap.org). Go to the health topics link and search for jaundice. Review the questions and answers about jaundice for parents and the management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation clinical practice guideline for health care providers.
KEY POINTS
• Hyperbilirubinemia is caused by a variety of factors, including maternal-fetal Rh and ABO incompatibility.
• Erythroblastosis fetalis leads to anemia, edema, and the cytotoxic effects of unconjugated bilirubin.
• The injection of Rho(D) immunoglobulin in Rh-negative and Coombs’ test–negative women provides passive immunity and minimizes the possibility of isoimmunization.
• Neonatal exchange transfusion with type O, Rh-negative RBCs serves to treat anemia and acidosis and to remove bilirubin, maternal antibodies, and fetal RBCs that are beginning to hemolyze.
• Major congenital defects are the leading cause of death in term neonates.
• The most common major congenital anomalies that cause serious problems in the neonate are congenital heart disease, neural tube defects, cleft lip or palate, and developmental dysplasia of the hip.
• Minor anomalies can be part of a characteristic pattern of malformations.
• Current technology permits the prenatal diagnosis of many congenital anomalies and disorders.
• The most widespread use of postnatal testing for genetic disease is the routine screening of newborns for inborn errors of metabolism.
• The curative and rehabilitative problems of an infant with a congenital disorder are often complex and require a multidisciplinary approach to care.
• Parents often need special instruction (e.g., meeting nutrition requirements, cast care, or home phototherapy) before they take a high risk infant home.
• The supportive care given to the parents of infants with an abnormal condition must begin at birth, or at the time of diagnosis and continue for years.
 Audio Chapter Summaries Access an audio summary of the Key Points on
Audio Chapter Summaries Access an audio summary of the Key Points on 