Chapter 13 Urogenital System
The urogenital system is divided functionally into the urinary system and the genital system. Embryologically, these systems are closely associated with one another, especially during their early stages of development.
The urogenital system develops from the intermediate mesenchyme derived from the dorsal wall of the embryo (Fig. 13-1A and B). During folding of the embryo in the horizontal plane (see Chapter 6), the intermediate mesenchyme (embryonic connective tissue in the mesoderm) is carried ventrally and loses its connection with the somites (Fig. 13-1C and D). A longitudinal elevation of the mesenchyme—the urogenital ridge—forms on each side of the dorsal aorta (Fig. 13-1F). The part of the urogenital ridge that gives rise to the urinary system is the nephrogenic cord (Fig. 13-1C to F); the part that gives rise to the genital system is the gonadal ridge (see Fig. 13-18C).
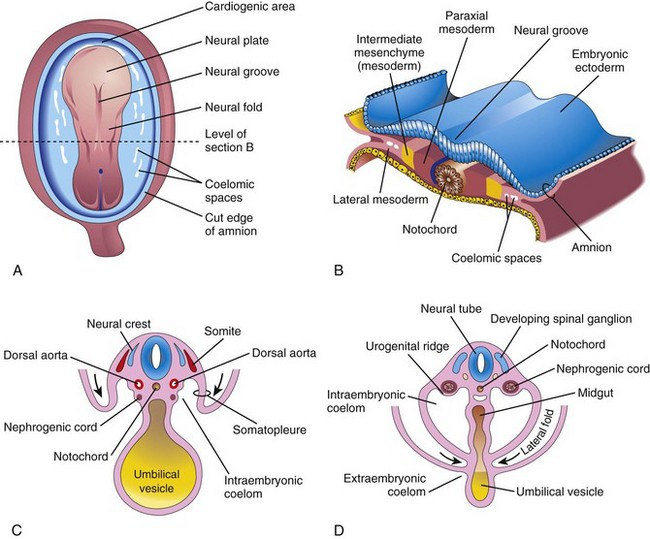
Figure 13–1 A, Dorsal view of an embryo during the third week (approximately 18 days). B, Transverse section of the embryo, showing the position of the intermediate mesenchyme before lateral folding occurs. C, Transverse section of the embryo after the commencement of folding. D, Transverse section of the embryo, showing the lateral folds meeting each other ventrally.
Development of Urinary System
The urinary system begins to develop before the genital system; it consists of the kidneys, ureters, urinary bladder, and urethra.
Development of Kidneys and Ureters 
Three sets of excretory organs, or kidneys, develop in human embryos. The first set—the pronephroi—is rudimentary and never functions. The second set—the mesonephroi—is well developed and functions for a brief period. The third set—the metanephroi—becomes the permanent kidneys.
Pronephroi
The transitory pronephroi appear early in the fourth week of development. They are represented by a few cell clusters in the neck region (Fig. 13-2A). The pronephric ducts run caudally and open into the cloaca (Fig. 13-2B). The pronephroi degenerate but most of the pronephric ducts persist and are used by the next set of kidneys.
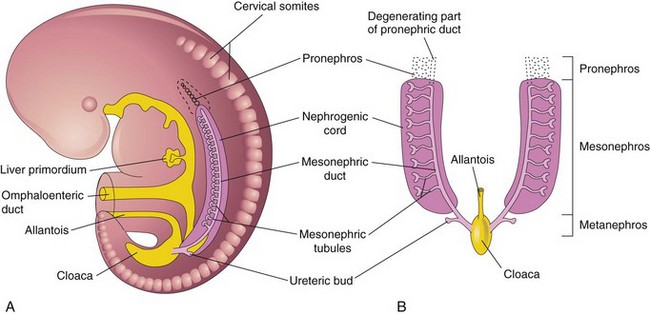
Mesonephroi
The large mesonephroi appear late in the fourth week caudal to the pronephroi (Fig. 13-2) and function as interim kidneys until the permanent kidneys develop (Fig. 13-3). The mesonephroi consist of glomeruli and mesonephric tubules (Fig. 13-3C to F). The tubules open into the mesonephric ducts, originally the pronephric ducts. The mesonephric ducts open into the cloaca. The mesonephroi degenerate toward the end of the first trimester; however, their tubules become the efferent ductules of the testes, and the mesonephric ducts have several adult derivatives in the male (Table 13-1).
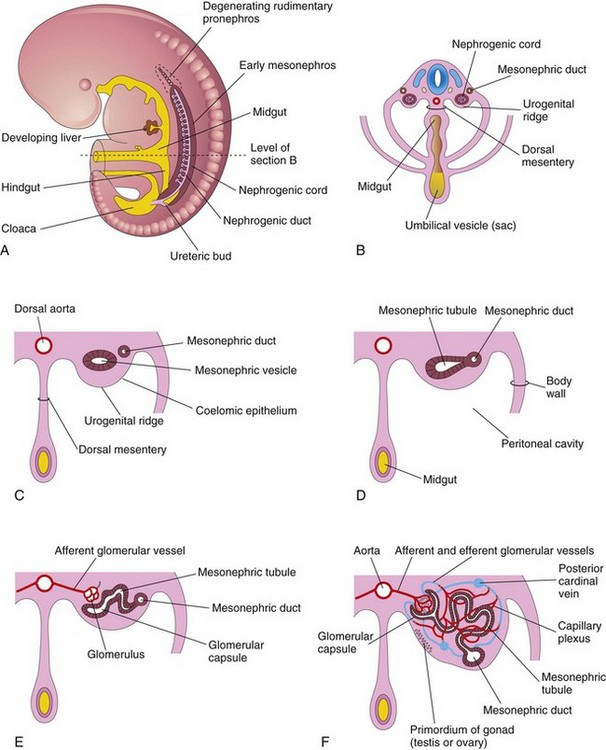
Figure 13–3 A, Lateral view of a 5-week embryo showing the extent of the mesonephros and the primordium of the metanephros—permanent kidney. B, Transverse section of the embryo showing the nephrogenic cords from which the mesonephric tubules develop. C to F, Transverse sections showing successive stages in the development of a mesonephric tubule between the 5th and 11th weeks. Note in C that the mesenchymal cell cluster in the nephrogenic cord has developed a lumen, thereby forming a mesonephric vesicle. The vesicle soon becomes an S-shaped mesonephric tubule and extends laterally to join the mesonephric duct. The expanded medial end of the mesonephric tubule is invaginated by blood vessels to form a glomerular capsule. The cluster of capillaries projecting into this capsule is the glomerulus.
Table 13–1 Adult Derivatives and Vestigial Remains of Embryonic Urogenital Structures*
| MALE | EMBRYONIC STRUCTURE | FEMALE |
|---|---|---|
| Testis | Indifferent gonad | Ovary |
| Seminiferous tubules | Cortex | Ovarian follicles |
| Rete testis | Medulla | Rete ovarii |
| Gubernaculum | Gubernaculum | Ovarian ligament |
| Round ligament of the uterus | ||
| Efferent ductules of testis | Mesonephric tubules | Epoophoron |
| Paradidymis | Paroophoron | |
| Appendix of epididymis | Mesonephric duct | Appendix vesiculosa |
| Duct of epididymis | Duct of epoophoron | |
| Ductus deferens | Longitudinal duct, Gartner duct | |
| Ureter, pelvis, calices, and collecting tubules | Ureter, pelvis, calices, and collecting tubules | |
| Ejaculatory duct and seminal gland | ||
| Appendix of testis | Paramesonephric duct | Hydatid (of Morgagni) |
| Uterine tube | ||
| Uterus | ||
| Urinary bladder | Urogenital sinus | Urinary bladder |
| Urethra (except navicular fossa) | Urethra | |
| Prostatic utricle | Vagina | |
| Prostate | Urethral and paraurethral glands | |
| Bulbourethral glands | Greater vestibular glands | |
| Seminal colliculus | Sinus tubercle | Hymen |
| Penis | Phallus | Clitoris |
| Glans of penis | Glans of clitoris | |
| Corpora cavernosa of penis | Corpora cavernosa of clitoris | |
| Corpus spongiosum of penis | Bulb of vestibule | |
| Ventral aspect of penis | Urogenital folds | Labia minora |
| Scrotum | Labioscrotal swellings | Labia majora |
Metanephroi
The metanephroi—the primordia of the permanent kidneys—begin to develop early in the fifth week and start to function approximately 4 weeks later. Urine formation continues throughout fetal life. The urine is excreted into the amniotic cavity and mixes with the amniotic fluid. The permanent kidneys develop from two sources of mesodermal origin (Fig. 13-4A):
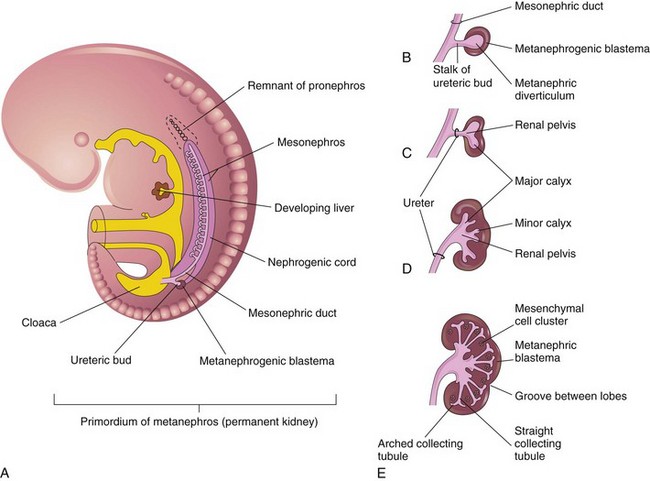
Figure 13–4 Development of the metanephros, the primordium of the permanent kidney. A, Lateral view of a 5-week embryo, showing the primordium of the metanephros. B to E, Successive stages in the development of the ureteric bud (fifth to eighth weeks). Observe the development of the ureter, the renal pelvis, the calices, and the collecting tubules.
The ureteric bud is an outgrowth from the mesonephric duct, near its entrance into the cloaca, and is the primordium of the ureter, renal pelvis, calices, and collecting tubules (Fig. 13-4B to E). The elongating bud penetrates the metanephrogenic blastema—a mass of cells derived from the nephrogenic cord—that forms the nephrons (Fig. 13-4B). The stalk of the ureteric bud becomes the ureter, and the cranial part of the diverticulum undergoes repetitive branching. The branches form the collecting tubules (Figs. 13-4C to E and 13-5).
The straight collecting tubules undergo repeated branching, forming successive generations of collecting tubules. The first four generations of tubules enlarge and coalesce to form the major calices (Fig. 13-4C to E); the second four generations coalesce to form the minor calices. The end of each arched collecting tubule induces clusters of mesenchymal cells in the metanephrogenic blastema to form small metanephric vesicles (Fig. 13-5A). These vesicles elongate and become the renal tubules (Fig. 13-5B and C). The proximal ends of these tubules are invaginated by glomeruli. The renal corpuscle (glomerulus and glomerular capsule) and its proximal convoluted tubule, nephron loop (of Henle), and distal convoluted tubule constitute a nephron (Fig. 13-5D). Each distal convoluted tubule contacts an arched collecting tubule. The tubules become confluent, forming a uriniferous tubule.
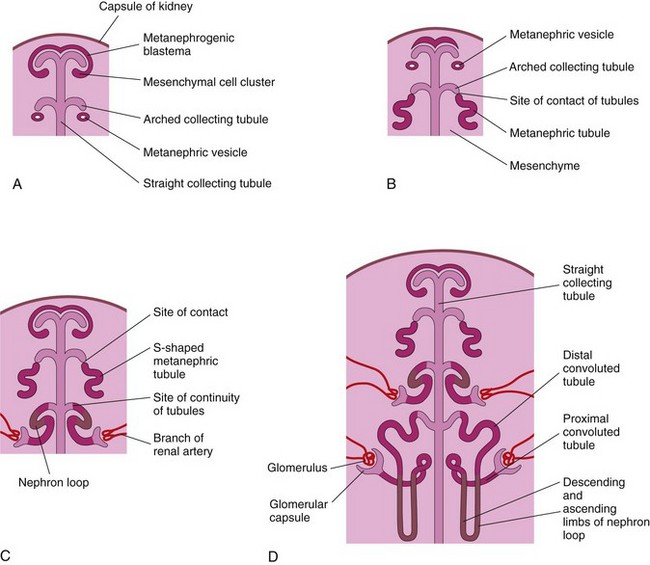
Figure 13–5 Illustrations of stages in nephrogenesis—the development of nephrons. A, Nephrogenesis commences approximately at the beginning of the eighth week. B and C, Note that the metanephric tubules, the primordia of the nephrons, become continuous with the collecting tubules to form uriniferous tubules. D, The number of nephrons more than doubles from 20 weeks to 38 weeks. Observe that nephrons are derived from the metanephric mass of the mesoderm and that the collecting tubules are derived from the metanephric diverticulum.
Branching of the metanephric diverticulum depends on an inductive signal from the metanephric mesoderm—differentiation of the nephrons depends on induction by the collecting tubules. The molecular aspects of the reciprocal interactions between the metanephric mesenchyme and the collecting tubules are shown in Figure 13-6.
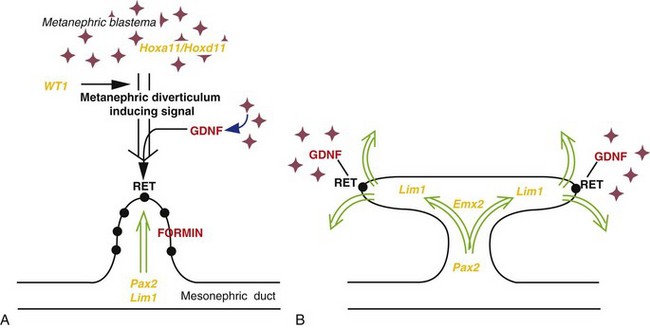
Figure 13–6 Molecular control of kidney development. A, Formation of the metanephric diverticulum requires inductive signals derived from the metanephric blastema under the control of transcription factors (yellow text), such as WT1, and signaling molecules (red text), including GDNF and its epithelial receptor, RET. Normal metanephric diverticulum response to these inductive signals is under the control of transcription factors, such as Pax2, Pax8, Lim1, and the FORMIN gene. B, Branching of the metanephric diverticulum is initiated and maintained by interaction with the mesenchyme under the regulation of genes such as Emx2 and specified expression of GDNF and RET at the tips of the invading metanephric diverticulum.
(From Piscione TD, Rosenblum ND: The malformed kidney: disruption of the glomerular and tubular development. Clin Genet 56:342, 1999.)
The fetal kidneys are subdivided into lobes. The lobulation usually disappears during infancy as the nephrons increase and grow. At term, nephron formation is complete—each kidney containing approximately 2 million nephrons. Functional maturation of the kidneys occurs after birth.
Positional Changes of Kidneys
The metanephric kidneys lie close to each other in the pelvis (Fig. 13-7A). As the abdomen and pelvis grow, the kidneys gradually come to lie in the abdomen and move farther apart (Fig. 13-7B and C). The caudal part of the embryo grows away from the kidneys so that the kidneys occupy progressively higher cranial levels. As the kidneys change their positions “ascend,” they rotate medially almost 90 degrees. By the ninth week, the kidneys come in contact with the suprarenal glands as they attain their adult position (Fig. 13-7C and D).
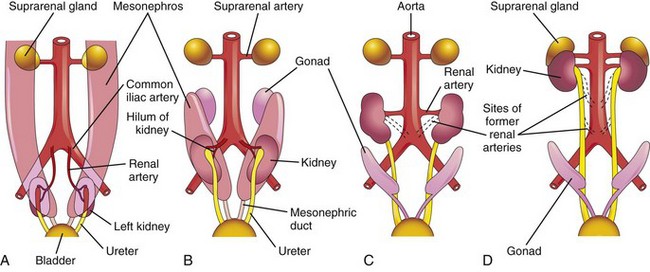
Figure 13–7 Ventral views of the abdominopelvic region of embryos and fetuses (sixth to ninth weeks) showing medial rotation and “ascent” of the kidneys from the pelvis to the abdomen. A and B, Observe also the decrease in size of the mesonephroi. C and D, Note that as the kidneys “ascend,” they are supplied by arteries at successively higher levels, and that the hilum of the kidney (where the vessels and nerves enter) is eventually directed anteromedially.
Changes in Blood Supply of Kidneys
Initially, the renal arteries are branches of the common iliac arteries (Fig. 13-7A and B). Later, the kidneys receive their blood supply from the distal end of the aorta (Fig. 13-7C). The kidneys receive their most cranial arterial branches from the abdominal aorta which become the renal arteries. Normally, the caudal primordial branches undergo involution and disappear.
Accessory Renal Arteries
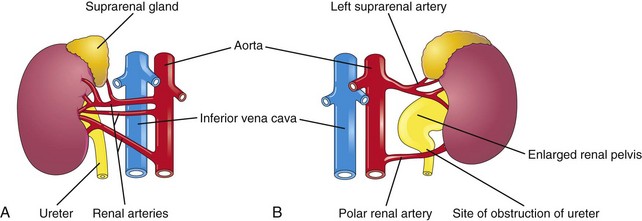
The common variations in the blood supply to the kidneys reflect the manner in which the blood supply continually changes during embryonic and early fetal life (Fig. 13-7). Approximately 25% of adult kidneys have accessory (supernumerary) renal arteries, usually arising from the aorta, superior or inferior to the main renal artery (Fig. 13-8A and B). An accessory artery to the inferior pole (polar renal artery) may cross anterior to the ureter and obstruct it, causing hydronephrosis, or distention of the pelvis and calices with urine (Fig. 13-8B). Renal arteries are end arteries; consequently, if an accessory artery is damaged or ligated, the part of the kidney supplied by it will become ischemic. Accessory arteries are approximately twice as common as accessory veins.
Congenital Anomalies of Kidneys and Ureters
Unilateral renal agenesis occurs in approximately 1 in 1000 newborn infants (Fig. 13-9A). Boys are affected more often than girls, and the left kidney is usually the one that is absent. The other kidney usually undergoes compensatory hypertrophy and performs the function of the missing kidney.
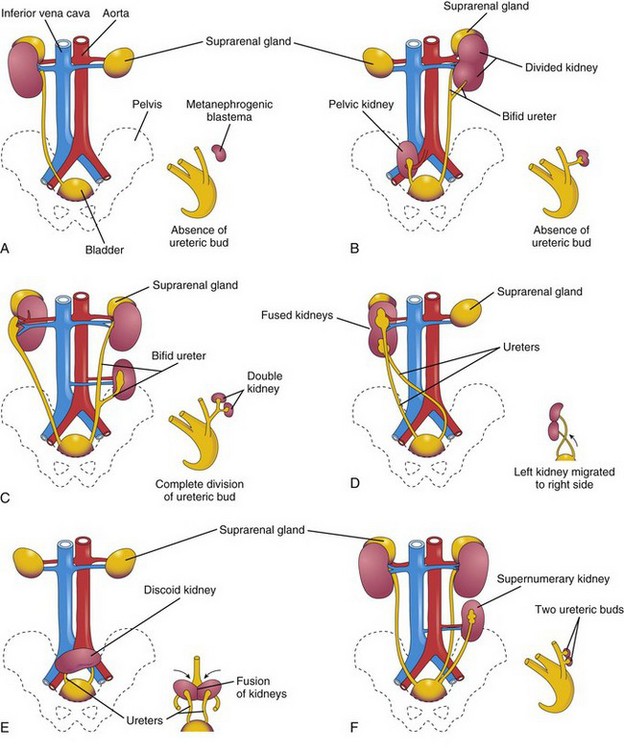
Figure 13–9 Various anomalies of the urinary system. The small sketch at the lower right of each illustration shows the probable embryologic basis of the anomaly. A, Unilateral renal agenesis. B, On the right side of the fetus, a pelvic kidney; on the left side of the fetus, a divided kidney with a bifid ureter. C, On the right side of the fetus, malrotation of the kidney; on the left side of fetus, a bifid ureter and a supernumerary kidney. D, Crossed fused renal ectopia. The left kidney has crossed over and fused with the right kidney. E, Discoid kidney resulting from fusion of the kidneys while they were in the pelvis. F, Supernumerary left kidney resulting from the development of two metanephric diverticula.
Bilateral renal agenesis is associated with oligohydramnios (small amount of amniotic fluid) because little or no urine is excreted into the amniotic cavity. This condition occurs in approximately 1 in 3000 births, is three times more common in males, and is incompatible with postnatal life. These infants also have pulmonary hypoplasia. Failure of the metanephric diverticulum to penetrate the metanephric blastema results in absence of renal development because no nephrons are induced by the collecting tubules to develop from the metanephric blastema.
Malrotation of Kidneys
If the kidney does not rotate, the hilum faces anteriorly (embryonic position) (Figs. 13-7 and 13-9C). If the hilum faces posteriorly, then rotation has progressed too far; if it faces laterally, medial rotation has occurred. Abnormal rotation of the kidneys is often associated with ectopic kidneys.
Ectopic Kidneys
One or both kidneys may be in an abnormal position (Fig. 13-9B and E). Most ectopic kidneys are located in the pelvis, but some lie in the inferior part of the abdomen. Pelvic kidneys and other forms of ectopia result from failure of the kidneys to “ascend.”
Fusion Anomalies
Crossed Fused Ectopia
Sometimes a kidney crosses to the other side, resulting in crossed renal ectopia, with or without fusion. An unusual kidney abnormality is unilateral fused kidneys (Fig. 13-9D). In such cases, the developing kidneys fuse while they are in the pelvis, and one kidney moves to its normal position, carrying the other one with it.
Horseshoe Kidney
In approximately 1 in 500 persons, the poles of the kidneys are fused (usually the inferior poles) (Fig. 13-10). Normal ascent of the fused kidneys is prevented because they are caught by the root of the inferior mesenteric artery. The function of these kidneys is preserved and each has a normal ureter and blood supply.
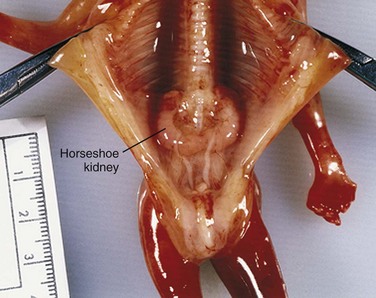
Figure 13–10 Horseshoe kidney in a female fetus (13 weeks). This anomaly resulted from fusion of the inferior poles of the kidneys while they were in the pelvis.
(Courtesy of Dr. D.K. Kalousek, Department of Pathology, University of British Columbia, Children’s Hospital, Vancouver, British Columbia, Canada.)
Duplications of Urinary Tract
Duplications of the abdominal part of the ureter and the renal pelvis are common, but a kidney in excess of the normal number (supernumerary kidney) is rare (Fig. 13-9C and F). These duplicates result from division of a metanephric diverticulum. Incomplete division of the ureteric primordium results in a divided kidney with a bifid ureter (Fig. 13-9B). Complete division results in a double kidney with a bifid ureter or with separate ureters (Fig. 13-11). A supernumerary kidney with its own ureter probably results from the formation of two metanephric diverticula.
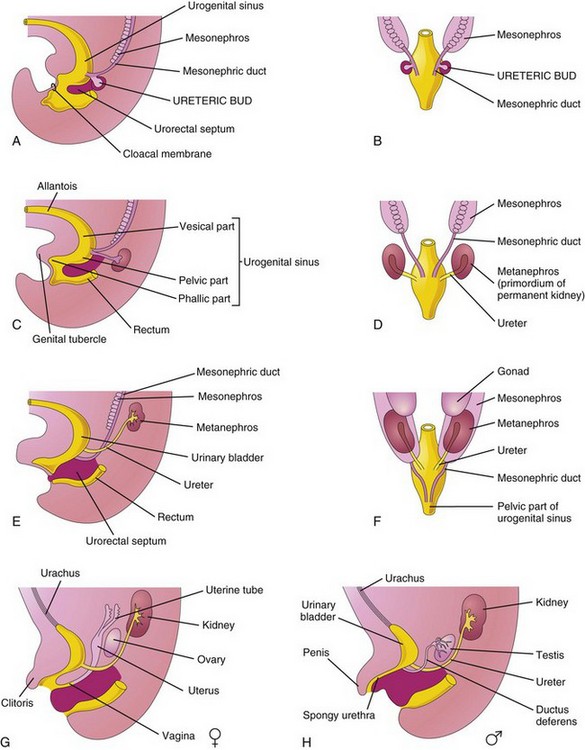
Figure 13–11 Division of the cloaca into the urogenital sinus and rectum; absorption of the mesonephric ducts; development of the urinary bladder, urethra, and urachus; and changes in the location of the ureters. A, Lateral view of the caudal half of a 5-week embryo. B, D, and F, Dorsal views. C, E, G, and H, Lateral views. The stages shown in G and H are reached by the 12th week of development.
Development of Urinary Bladder
Division of the cloaca by the urorectal septum into a dorsal rectum and a ventral urogenital sinus is described in Chapter 12. For descriptive purposes, the urogenital sinus is divided into three parts (Fig. 13-11A and C):
• A cranial vesical part that forms most of the bladder and is continuous with the allantois
• A middle pelvic part that becomes the urethra in the neck of the bladder, the prostatic part of the urethra in males, and the entire urethra in females
• A caudal phallic part that grows toward the genital tubercle—the primordium of the penis or the clitoris
Initially, the bladder is continuous with the allantois (Fig. 13-11C). The allantois soon constricts and becomes a thick, fibrous cord, the urachus (Fig. 13-11G). In adults, the urachus is represented by the median umbilical ligament. As the bladder enlarges, distal parts of the mesonephric ducts are incorporated into its dorsal wall (Fig. 13-11B to H) and contribute to the formation of the connective tissue in the trigone of the bladder. The epithelium of the entire bladder is derived from the endoderm of the urogenital sinus. The other layers of the bladder wall develop from the adjacent splanchnic mesenchyme. As the mesonephric ducts are absorbed, the ureters come to open separately into the urinary bladder (Fig. 13-11C to H). In males, the orifices of the mesonephric ducts move close together and enter the prostatic part of the urethra as the caudal ends of these ducts become the ejaculatory ducts. In females, the distal ends of the mesonephric ducts degenerate.
Ectopic Ureter
In males, an ectopic ureter may open into the neck of the bladder, the prostatic part of the urethra, the ductus deferens, the prostatic utricle, or the seminal gland. In females, an ectopic ureter may enter the neck of the bladder, the urethra, the vagina, or the vestibule of the vagina. An ectopic ureter results when the ureter is carried caudally with the mesonephric duct and is incorporated into the caudal portion of the vesical part of the urogenital sinus.
Urachal Anomalies
A remnant of the lumen usually persists in the inferior part of the urachus in infants. In approximately 50% of cases, the lumen is continuous with the cavity of the bladder. Remnants of the epithelial lining of the urachus may give rise to urachal cysts (Fig. 13-12A). The patent inferior end of the urachus may dilate to form a urachal sinus that opens into the bladder. The lumen in the superior part of the urachus may also remain patent and form a urachal sinus that opens at the umbilicus (Fig. 13-12B). Very rarely, the entire urachus remains patent and forms a urachal fistula that allows urine to escape from its umbilical orifice (Fig. 13-12C).
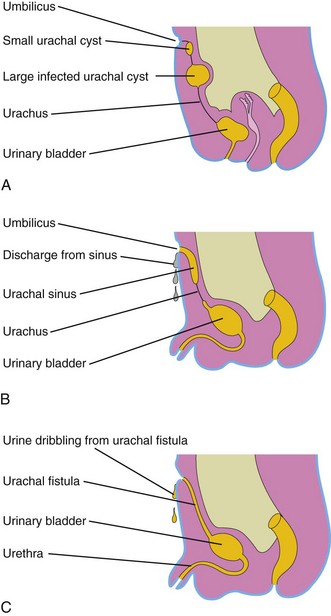
Figure 13–12 Urachal anomalies. A, Urachal cysts. The most common site for these cysts is in the superior end of the urachus, just inferior to the umbilicus. B, Two types of urachal sinuses are shown: one that opens into the bladder and one that opens at the umbilicus. C, Patent urachus or urachal fistula connecting the bladder and umbilicus.
Exstrophy of Bladder
Exstrophy of the bladder is a severe anomaly that occurs in approximately 1 in 10,000 to 1 in 40,000 births, predominantly affecting males (Fig. 13-13). Exposure and protrusion of the mucosal surface of the posterior wall of the bladder characterize this congenital anomaly. The trigone of the bladder and the ureteric orifices are exposed, and urine dribbles intermittently from the everted bladder.
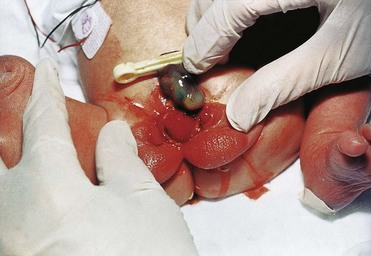
Figure 13–13 A male infant with exstrophy of the bladder. Because of defective closure of the inferior part of the anterior abdominal wall and the anterior wall of the bladder, the urinary bladder appears as an everted, bulging mass inferior to the umbilicus.
(Courtesy of A.E. Chudley, MD, Department of Pediatrics and Child Health, University of Manitoba, Children’s Hospital, Winnipeg, Manitoba, Canada.)
Epispadias, in which the urethra opens on the dorsum of the penis, and wide separation of the pubic bones are associated with complete exstrophy of the bladder. In some cases, the penis is divided into two parts, and the scrotum is bifid (split). Exstrophy of the bladder is believed to be caused by failure of the mesenchymal cells to migrate between the ectoderm and the endoderm of the infra-abdominal wall (cloacal membrane) during the fourth week (Fig. 13-14B and C). As a result, no muscle or connective tissue forms in the abdominal wall over the urinary bladder. Rupture of the fragile cloacal membrane results in wide communication between the exterior and the mucous membrane of the bladder. Rupture of the membrane before division of the cloaca by the urorectal septum leads to exstrophy of the cloaca, resulting in exposure of both the bladder and the hindgut.
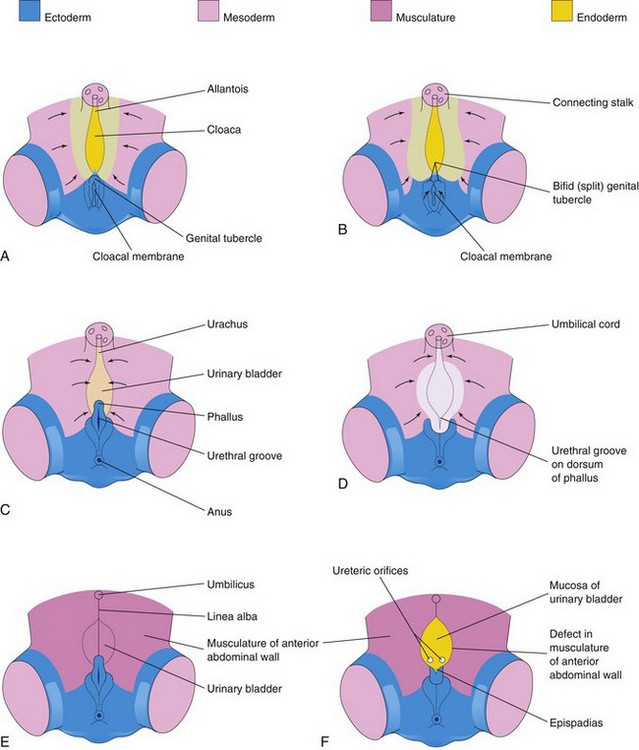
Figure 13–14 A, C, and E, Normal stages in the development of the infraumbilical abdominal wall and the penis during the fourth to eighth weeks. Note that mesoderm and (later) muscle reinforce the ectoderm of the developing anterior abdominal wall. B, D, and F, Probable stages in the development of exstrophy of the bladder and epispadias. In B and D, note that the mesenchyme (embryonic connective tissue) does not extend into the anterior abdominal wall anterior to the urinary bladder. Also note that the genital tubercle is located in a more caudal position than usual, and that the urethral groove has formed on the dorsal surface of the penis. In F, the surface ectoderm and the anterior wall of the bladder have ruptured, resulting in exposure of the posterior wall of the bladder. Note that the musculature of the anterior abdominal wall is present on each side of the defect.
(Adapted from Patten BM, Barry A: The genesis of exstrophy of the bladder and epispadias. Am J Anat 90:35, 1952.)
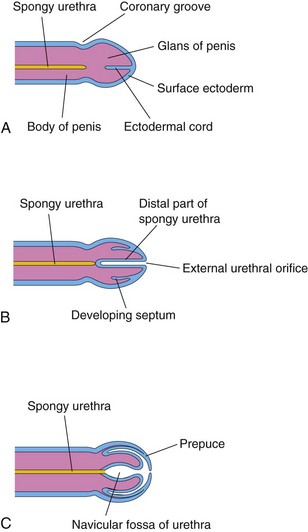
Development of Urethra
The epithelium of most of the male urethra and the entire female urethra is derived from the endoderm of the urogenital sinus (Figs. 13-11 and 13-15). The distal part of the urethra in the glans of the penis is derived from a solid cord of ectodermal cells that grows from the tip of the glans penis to meet the part of the spongy urethra derived from the phallic part of the urogenital sinus (Fig. 13-15A to C). The ectodermal cord canalizes and joins the rest of the spongy urethra; consequently, the epithelium of the terminal part of the urethra is derived from the surface ectoderm. The connective tissue and the smooth muscle of the urethra in both sexes are derived from the splanchnic mesenchyme.
Development of Suprarenal Glands
The cortex of the suprarenal (adrenal) gland develops from the mesenchyme lining the posterior abdominal wall, whereas the medulla differentiates from the adjacent sympathetic ganglion derived from the neural crest cells (Fig. 13-16A and B). These cells differentiate into the secretory cells of the suprarenal medulla. The cortex is evident during the sixth week as an aggregation of mesenchymal cells seen bilaterally between the root of the dorsal mesentery and the developing gonad (see Fig. 13-18C). Differentiation of the characteristic suprarenal cortical zones begins during the late fetal period (Fig. 13-16C to E). The zona glomerulosa and the zona fasciculata are present at birth, but the zona reticularis is not recognizable until the end of the third year (Fig. 13-16H). Relative to body weight, the fetal suprarenal glands are 10 to 20 times larger than the adult glands because of the extensive size of the fetal cortex. The suprarenal medulla remains small until after birth (Fig. 13-16F). The suprarenal glands rapidly become smaller as the cortex regresses during the first year of infancy (Fig. 13-16G).
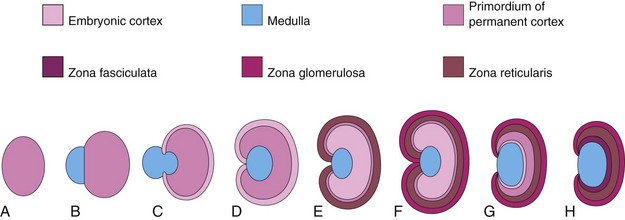
Figure 13–16 Illustrations of the development of the suprarenal glands. A, At 6 weeks, showing the mesodermal primordium of the fetal cortex. B, At 7 weeks, showing the addition of neural crest cells. C, At 8 weeks, showing the fetal cortex and the early permanent cortex beginning to encapsulate the medulla. D and E, Later stages of encapsulation of the medulla by the cortex. F, Neonatal period, showing the fetal cortex and two zones of the permanent cortex. G, At 1 year of age. Note that the fetal cortex has almost disappeared. H, At 4 years of age. Note the adult pattern of cortical zones. Observe that the fetal cortex has disappeared and that the gland is smaller than it was at birth (F).
Congenital Adrenal Hyperplasia
Adrenogenital Syndrome
Congenital adrenal hyperplasia represents a group of autosomal recessive disorders in which an abnormal increase in the cells of the suprarenal cortex results in excessive androgen production during the fetal period. In female infants, this usually causes masculinization of the external genitalia and enlargement of the clitoris (Fig. 13-17). Affected male infants have normal external genitalia and may remain undiagnosed in early infancy. Later in childhood, in both sexes, excess androgen leads to rapid growth and accelerated skeletal maturation. Congenital adrenal hyperplasia (CAH) is usually caused by a genetically determined mutation in the cytochrome-P450c 21-steroid, 21-hydroxylase gene, which results in a deficiency of suprarenal cortical enzymes. These are necessary for the biosynthesis of various steroid hormones. The reduced hormone output results in increased release of adrenocorticotrophic hormone by the anterior pituitary, which causes CAH and overproduction of androgens by the hyperplastic suprarenal glands.
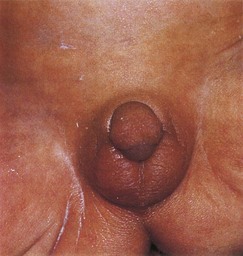
Figure 13–17 External genitalia of a newborn female infant with congenital adrenal hyperplasia (CAH). The virilization was caused by excessive androgens produced by the suprarenal glands during the fetal period. Note the enlarged clitoris and fusion of the labia majora to form a scrotum.
(Courtesy of Dr. Heather Dean, Department of Pediatrics and Child Health, University of Manitoba, Winnipeg, Manitoba, Canada.)
Development of Genital System
The early genital systems in the two sexes are similar; therefore, the initial period of genital development is referred to as the indifferent state of sexual development.
Development of Gonads
The gonads (testes and ovaries) are derived from three sources (Fig. 13-18):
Indifferent Gonads
Gonadal development begins during the fifth week, when a thickened area of mesothelium develops on the medial side of the mesonephros (Fig. 13-18A to C). Proliferation of this epithelium and the underlying mesenchyme produces a bulge on the medial side of the mesonephros—gonadal ridge (Fig. 13-18A and C). Finger-like epithelial cords—gonadal cords—soon grow into the underlying mesenchyme (Fig. 13-18D). The indifferent gonads now consist of an external cortex and an internal medulla. In embryos with an XX sex chromosome complex, the cortex of the gonad differentiates into an ovary and the medulla regresses. In embryos with an XY sex chromosome complex, the medulla differentiates into a testis and the cortex regresses, except for vestigial remnants (Table 13-1).
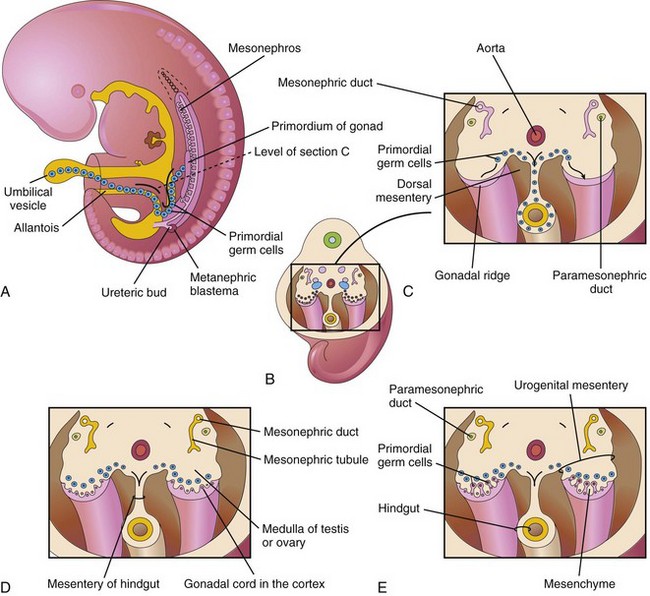
Figure 13–18 A, A 5-week embryo, showing migration of primordial germ cells from the umbilical vesicle into the embryo. B, Three-dimensional sketch of the caudal region of the 5-week embryo, showing the location and extent of the gonadal ridges. C, Transverse section, showing the gonadal ridges and the migration of primordial germ cells into the developing gonads. D, Transverse section of a 6-week embryo, showing the gonadal cords. E, Similar section at a later stage, showing the indifferent gonads and the paramesonephric ducts.
Primordial Germ Cells
The primordial germ cells originate in the wall of the umbilical vesicle (yolk sac) and migrate along the dorsal mesentery of the gut to the gonadal ridges (Fig. 13-18A). During the sixth week, the primordial germ cells enter the underlying mesenchyme and are incorporated into the gonadal cords (see Fig. 13-18D and E). They eventually differentiate into oocytes or sperms.
Sex Determination
Chromosomal and genetic sex, established at fertilization, depends on whether an X-bearing or a Y-bearing sperm fertilizes the X-bearing oocyte. The type of gonads that develop is determined by the sex chromosome complex of the embryo (XX or XY). Before the seventh week, the gonads of the two sexes are identical in appearance and are called indifferent gonads (Fig. 13-19).
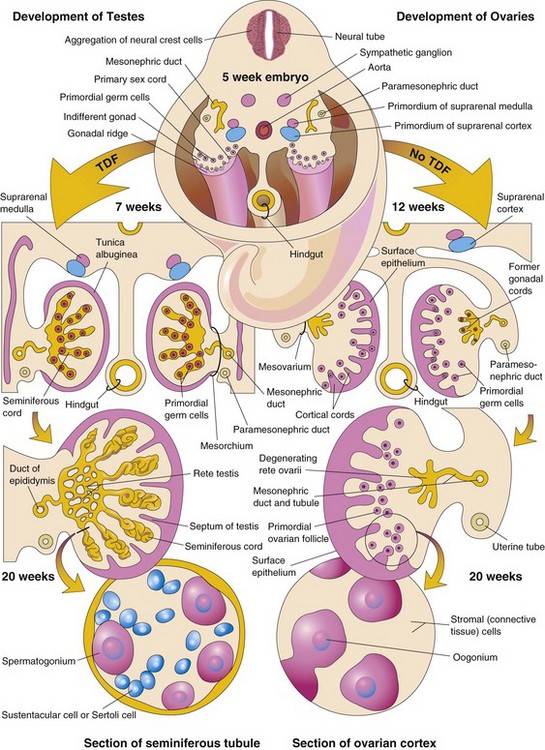
Figure 13–19 Differentiation of the indifferent gonads of a 5-week embryo (top) into ovaries or testes. The left side of the drawing shows the development of testes resulting from the effects of the testis-determining factor (TDF) located on the Y chromosome. Note that the gonadal cords become seminiferous cords, the primordia of the seminiferous tubules. The parts of the gonadal cords that enter the medulla of the testis form the rete testis. In the section of the testis at the bottom left, observe that there are two kinds of cells: spermatogonia, derived from the primordial germ cells and Sertoli cells, derived from the mesenchyme. The right side shows the development of ovaries in the absence of TDF. Cortical cords have extended from the surface epithelium of the gonad, and primordial germ cells have entered them. They are the primordia of the oogonia. Follicular cells are derived from the surface epithelium of the ovary. The arrows indicate the changes that occur as the gonads (testes and ovaries) develop.
Development of a male phenotype requires a Y chromosome. Two X chromosomes are required for the development of the female phenotype.
Abnormal Sex Chromosome Complexes
In embryos with abnormal sex chromosome complexes, such as XXX or XXY, the number of X chromosomes appears to be unimportant in sex determination. If a normal Y chromosome is present, the embryo develops as a male. If no Y chromosome is present or if the testis-determining region of the Y chromosome has been lost, female development occurs. The loss of an X chromosome does not appear to interfere with the migration of primordial germ cells to the gonadal ridges because some germ cells have been observed in the fetal gonads of 45, XO females with Turner syndrome. Two X chromosomes are needed, however, to bring about complete ovarian development.
Development of Testes
A coordinated sequence of genes induces the development of testes. The SRY gene for the testis-determining factor (TDF) on the short arm of the Y chromosome acts as the switch that directs the development of the indifferent gonad into a testis. Expression of the transcription factor SOX9 is also essential for testicular determination. TDF induces the gonadal cords to condense and extend into the medulla of the indifferent gonad, where they branch and anastomose to form the rete testis (Fig. 13-19). The connection of the prominent gonadal cords—the seminiferous cords—with the surface epithelium is lost when the tunica albuginea develops. This dense tunica, a thick, fibrous capsule, is a characteristic feature of testicular development. Gradually, the testis separates from the degenerating mesonephros and becomes suspended by its own mesentery, the mesorchium. The seminiferous cords develop into the seminiferous tubules, the straight tubules (tubuli recti), and the rete testis.
The seminiferous tubules are separated by the mesenchyme, giving rise to the interstitial cells (of Leydig). By the eighth week, these cells secrete androgenic hormones—testosterone and androstenedione—that induce masculine differentiation of the mesonephric ducts and the external genitalia. Testosterone production is stimulated by human chorionic gonadotropin, which reaches peak amounts during the 8- to 12-week period of embryonic and fetal development. The fetal testes also produce a glycoprotein known as müllerian-inhibiting substance (MIS) or antimüllerian hormone. MIS is produced by the sustentacular (Sertoli) cells, which are present until puberty, at which time the levels of MIS decrease. MIS suppresses the development of the paramesonephric ducts, which form the uterus and uterine tubes. The seminiferous tubules remain until puberty (i.e., without lumina), when lumina begin to develop. The walls of the seminiferous tubules are composed of two kinds of cells (Fig. 13-19):
• Sertoli cells, supporting cells derived from the surface epithelium of the testis
• Spermatogonia, primordial sperms derived from the primordial germ cells
Sertoli cells constitute most of the seminiferous epithelium in the fetal testis (Fig. 13-19). The rete testis becomes continuous with 15 to 20 mesonephric tubules that become efferent ductules. These ductules are connected with the mesonephric duct, which becomes the ductus epididymis (Figs. 13-19 and 13-20A).
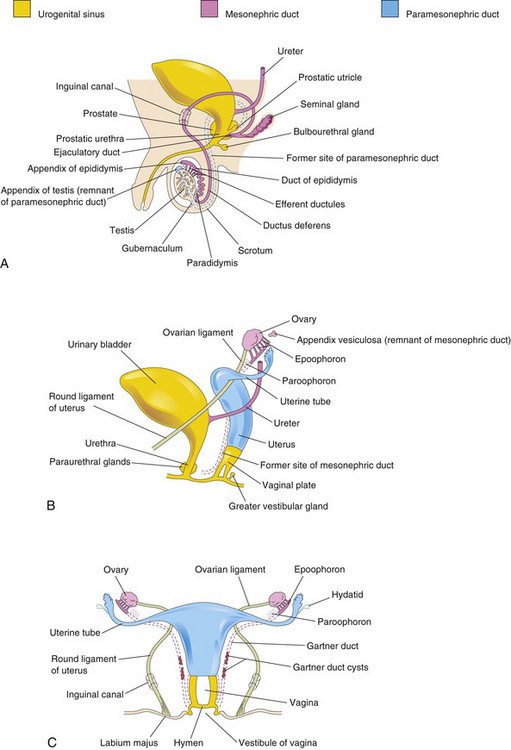
Figure 13–20 Development of male and female reproductive systems from the genital ducts and the urogenital sinus. Vestigial structures are also shown. A, Reproductive system in a newborn male infant. B, Female reproductive system in a 12-week fetus. C, Reproductive system in a newborn female infant.
Development of Ovaries
The X chromosomes have genes for ovarian development; autosomal genes also appear to play a role in ovarian organogenesis. The ovary is not identifiable by histologic examination until approximately the 10th week of development. Gonadal cords extend into the medulla of the ovary and form a rudimentary rete ovarii (Figs. 13-18D and 13-19). The rete ovarii normally degenerate. Cortical cords extend from the surface epithelium of the developing ovary into the underlying mesenchyme during the early fetal period. As the cortical cords increase in size, primordial germ cells are incorporated into them. At approximately 16 weeks, these cortical cords begin to break up into isolated cell clusters—primordial follicles—each of which consists of an oogonium (derived from a primordial germ cell), surrounded by a single layer of follicular cells derived from the surface epithelium (see Fig. 13-19). Active mitosis produces many oogonia during fetal life.
No oogonia form postnatally. Although many oogonia degenerate before birth, 2 million or so enlarge to become primary oocytes before birth. After birth, the surface epithelium of the ovary flattens to a single layer of cells that is continuous with the mesothelium of the peritoneum at the hilum of the ovary. The surface epithelium becomes separated from the follicles in the cortex by a thin, fibrous capsule, the tunica albuginea. As the ovary separates from the regressing mesonephros, it is suspended by its mesentery, the mesovarium.
Development of Genital Ducts
Both male and female embryos have two pairs of genital ducts: the mesonephric ducts (Wolffian ducts) and the paramesonephric ducts (müllerian ducts) (Fig. 13-21A).
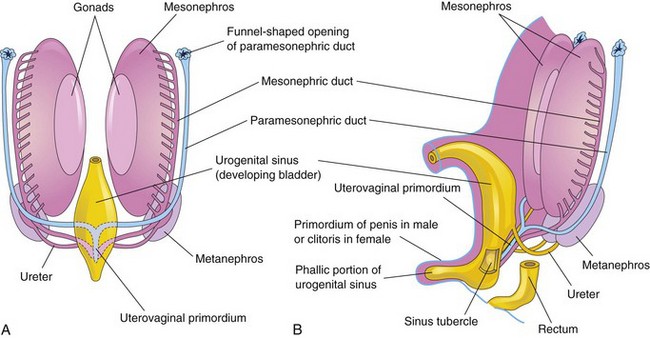
Figure 13–21 A, Ventral view of a 7-week embryo, showing the two pairs of genital ducts that are present during the indifferent state of sexual development. B, Lateral view of a 9-week fetus, showing the sinus tubercle on the posterior wall of the urogenital sinus. It becomes the hymen in females (Fig. 13-20C) and the seminal colliculus in males.
The mesonephric ducts play an essential role in the development of the male reproductive system (Fig. 13-20A) while the paramesonephric ducts play an essential role in the development of the female reproductive system (Table 13-1 and Fig. 13-20B and C). During conversion of the mesonephric and paramesonephric ducts into adult structures, some parts of the ducts remain as vestigial structures. These vestiges are rarely seen unless pathologic changes develop in them.
Development of Male Genital Ducts
The fetal testes produce testosterone and müllerian-inhibiting substance (MIS). Testosterone stimulates the mesonephric ducts to form male genital ducts; MIS causes the paramesonephric ducts to disappear by epithelial-mesenchymal transformation. As the mesonephros degenerates, some mesonephric tubules persist and are transformed into efferent ductules (Fig. 13-20A). These ductules open into the mesonephric duct, which has been transformed into the duct of the epididymis in this region. Distal to the epididymis, the mesonephric duct acquires a thick investment of smooth muscle and becomes the ductus deferens. The part of the mesonephric duct between the duct of this gland and the urethra becomes the ejaculatory duct.
Seminal Gland
A lateral outgrowth from the caudal end of each mesonephric duct gives rise to the seminal gland (vesicle). The secretions of this pair of glands nourish the sperms.
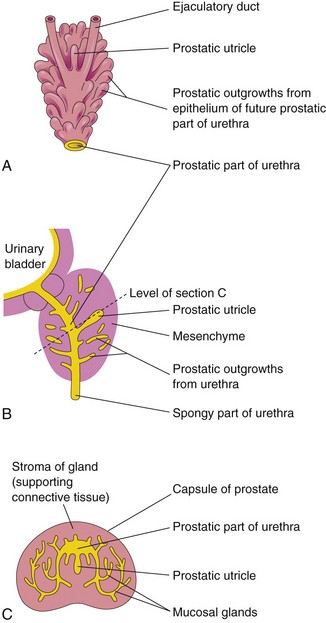
Prostate
Multiple endodermal outgrowths arise from the prostatic part of the urethra and grow into the surrounding mesenchyme (Fig. 13-22). The glandular epithelium of the prostate differentiates from these endodermal cells and the associated mesenchyme differentiates into the dense stroma and the smooth muscle of the prostate. Secretions from the prostate make up a portion of the fluid in the ejaculate.
Bulbourethral Glands
The bulbourethral glands are pea-sized structures that develop from paired outgrowths from the spongy part of the urethra (Fig. 13-20A). The smooth muscle fibers and the stroma differentiate from the adjacent mesenchyme. The secretions of these glands and the previous ones, mix with the sperms to form the semen (ejaculate).
Development of Female Genital Ducts and Glands
In female embryos, the mesonephric ducts regress because of lack of testosterone, and the paramesonephric ducts develop because of the absence of MIS. Female sexual development does not depend on the presence of ovaries or hormones. The paramesonephric ducts form most of the female genital tract. The uterine tubes develop from the unfused cranial parts of the paramesonephric ducts (Fig. 13-20B and C). The caudal, fused portions of these ducts form the uterovaginal primordium, which gives rise to the uterus and the superior portion of the vagina (Fig. 13-21). Expression of Hox genes in the paramesonephric ducts regulates the development of the female genital ducts. The endometrial stroma and myometrium are derived from splanchnic mesenchyme. Fusion of the paramesonephric ducts also brings together two peritoneal folds that form the right and left broad ligament and two peritoneal compartments, the rectouterine pouch and the vesicouterine pouch (Fig. 13-23B to D).
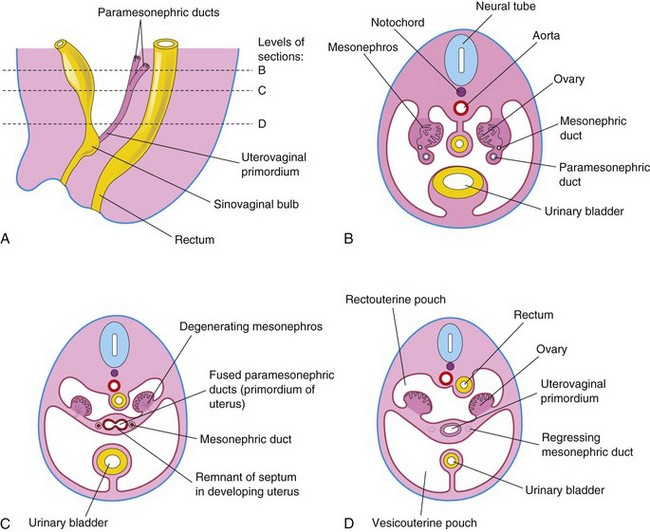
Figure 13–23 Early development of the ovaries and uterus. A, Sagittal section of the caudal region of an 8-week female embryo. B, Transverse section, showing the paramesonephric ducts approaching each other. C, Similar section at a more caudal level, showing fusion of the paramesonephric ducts. D, Similar section, showing the uterovaginal primordium, broad ligament, and pouches in the pelvic cavity.
Development of Vagina
The vaginal epithelium is derived from the endoderm of the urogenital sinus. The fibromuscular wall of the vagina develops from the surrounding mesenchyme. Contact of the uterovaginal primordium with the urogenital sinus, forming the sinus tubercle (Fig. 13-21B), induces the formation of paired endodermal outgrowths—sinovaginal bulbs (Fig. 13-23A). They extend from the urogenital sinus to the caudal end of the uterovaginal primordium. The sinovaginal bulbs fuse to form a vaginal plate (Fig. 13-20B). The central cells of this plate break down, forming the lumen of the vagina. The peripheral cells of the plate form the vaginal epithelium or lining (see Fig. 13-20C). Until late in fetal life, the lumen of the vagina is separated from the cavity of the urogenital sinus by a membrane—the hymen (Fig. 13-24H; see also Fig. 13-20C). The hymen is formed by invagination of the posterior wall of the urogenital sinus.
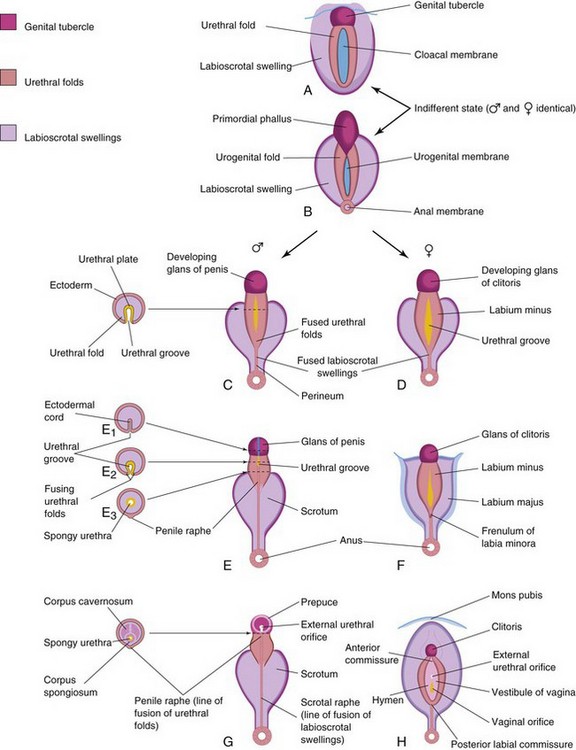
Figure 13–24 Development of the external genitalia. A and B, Appearance of the genitalia during the indifferent state (fourth to seventh weeks). C, E, and G, Stages in the development of the male external genitalia at 9, 11, and 12 weeks, respectively. On the left are schematic transverse sections of the developing penis, showing the formation of the spongy urethra and scrotum. D, F, and H, Stages in the development of the female external genitalia at 9, 11, and 12 weeks, respectively.
Female Auxiliary Genital Glands
Buds grow from the urethra into the surrounding mesenchyme, forming the mucus-secreting urethral and paraurethral glands (Fig. 13-20B). Outgrowths from the urogenital sinus form bilateral greater vestibular glands (of Bartholin) in the lower one third of the labia majora. These tubuloalveolar glands also secrete mucus (see Table 13-1).
Development of External Genitalia
From the fourth week to the early part of the seventh week, the external genitalia are sexually undifferentiated (Fig. 13-24A and B). Distinguishing sexual characteristics begin to appear during the 9th week, but the external genitalia are not fully differentiated until the 12th week. Early in the fourth week, the proliferating mesenchyme produces a genital tubercle in both sexes at the cranial end of the cloacal membrane. Fgf8 is involved in the signaling pathways in the early development of the external genitalia. Labioscrotal swellings and urogenital folds soon develop on each side of the cloacal membrane. The genital tubercle soon elongates to form a primordial phallus (Fig. 13-24B). When the urorectal septum fuses with the cloacal membrane at the end of the sixth week, it divides the cloacal membrane into a dorsal anal membrane and a ventral urogenital membrane The urethral membrane lies in the floor of a median cleft, the urethral groove, which is bound by the urogenital folds (Fig. 13-24C and D). The anal and urogenital membranes rupture approximately 1 week later, forming the anus and the urogenital orifice, respectively. In the female fetus, the urethra and vagina open into a common cavity, the vestibule of the vagina.
Determination of Fetal Sex
Assessment of fetal sex by transabdominal ultrasound is important for decision making especially in pregnancies at risk of serious X-linked abnormalities. Assessment is based on the direct visualization of the external genitalia. By the 12th week of gestation the genital tubercle has differentiated to form the penis. Several studies indicate that sex assignment is highly accurate in most cases (99%–100%) after 13 weeks of gestation providing the external genitalia are not malformed. The accuracy of diagnosis increases with gestational age and depends on the experience of the sonographer, the equipment, the position of the fetus, and the amount of amniotic fluid.
Development of Male External Genitalia
Masculinization of the indifferent external genitalia is induced by dihydrotestosterone (Fig. 13-24C, E, and G). As the primordial phallus enlarges and elongates to become the penis, the urogenital folds form the lateral walls of the urethral groove on the ventral surface of the penis. This groove is lined by a proliferation of endodermal cells, the urethral plate (Fig. 13-24C), which extends from the phallic portion of the urogenital sinus. The urethral folds fuse with each other along the ventral surface of the penis to form the spongy urethra (Fig. 13-24E1 to E3). The surface ectoderm fuses in the median plane of the penis, forming the penile raphe and enclosing the spongy urethra within the penis. At the tip of the glans penis, an ectodermal ingrowth forms a cellular ectodermal cord, which extends toward the root of the penis to meet the spongy urethra (Fig. 13-15A). This cord canalizes and joins the previously formed spongy urethra (Fig. 13-15B). This juncture completes the terminal part of the urethra and moves the external urethral orifice to the tip of the glans penis (see Fig. 13-15C). During the 12th week, a circular ingrowth of ectoderm occurs at the periphery of the glans penis (Fig. 13-15B). When this ingrowth breaks down, it forms the prepuce (foreskin) (Fig. 13-15C). The corpora cavernosa penis and the corpus spongiosum penis develop from mesenchyme in the phallus. The labioscrotal swellings grow toward each other and fuse to form the scrotum (Fig. 13-24E). The line of fusion of these folds is clearly visible as the scrotal raphe (Fig. 13-24G).
Development of Female External Genitalia
Growth of the primordial phallus in the female fetus gradually decreases as it becomes the clitoris (Fig. 13-24D, F, and H). The clitoris develops in the same way as the penis, except that the urogenital folds do not fuse, but for posteriorly, where they join to form the frenulum of the labia minora. The unfused parts of the urogenital folds form the labia minora. The labioscrotal folds fuse posteriorly to form the posterior labial commissure and anteriorly to form the anterior labial commissure and the mons pubis. Most parts of the labioscrotal folds remain unfused and form two large folds of skin, the labia majora.
Intersex Disorders
Advances in molecular genetics have led to a better understanding of abnormal sexual development and ambiguous genitalia. Because of psychosocial stigma and in order to provide better clinical management for infants born with atypical chromosomal constitution or gonads, a new nomenclature has been introduced to describe these conditions, which are now called disorders of sex development (DSD). The new classification avoids using the term “hermaphrodite.” (See Lee PA, Houk CP, Ahmed SF, Hughes IA: Consensus statement on management of intersex disorders. Pediatrics 118:e488, 2006.)
Ovotesticular DSD (True Hermaphroditism)
Persons with the extremely rare intersexual condition of ovotesticular DSD (true hermaphroditism) usually have a 46, XX sex chromosome constitution. Ovotesticular DSD results from an error in sex determination, and these individuals have both testicular and ovarian tissue. The phenotype may be male or female, but the external genitalia are always ambiguous.
46, XX DSD (Female Pseudohermaphroditism)
Females with 46, XX DSD (female pseudohermaphroditism) result from exposure of a female fetus to excessive androgens, the principal effect of which is virilization of the external genitalia (Fig. 13-25). Persons with this intersexual condition have chromatin-positive nuclei and a 46, XX chromosome constitution. The common cause of 46, XX DSD is congenital adrenal hyperplasia. There is no ovarian abnormality, but the excessive production of androgens by the fetal suprarenal glands causes masculinization of the external genitalia, varying from enlargement of the clitoris to almost masculine genitalia. Commonly, clitoral hypertrophy, partial fusion of the labia majora, and a persistent urogenital sinus are noted.
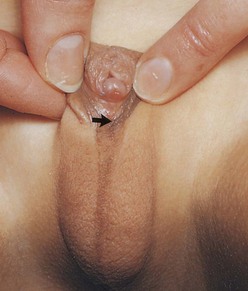
Figure 13–25 External genitalia of a 6-year-old girl, showing an enlarged clitoris and a scrotum-like structure formed by fusion of the labia majora. The arrow indicates the opening into the urogenital sinus (see Fig. 13-11C). This extreme masculinization is the result of congenital adrenal hyperplasia.
(Courtesy of Dr. Heather Dean, Department of Pediatrics and Child Health, University of Manitoba, Winnipeg, Manitoba, Canada.)
46, XY DSD (Male Pseudohermaphroditism)
Males with 46, XY DSD (male pseudohermaphroditism) have sex chromatin-negative nuclei and a 46, XY chromosome constitution. The external and internal genitalia are variable, owing to varying degrees of development. These anomalies are caused by inadequate production of testosterone and MIS by the fetal testes. Testicular development ranges from rudimentary to normal.
Androgen Insensitivity Syndrome
Androgen insensitivity syndrome (AIS)—also called testicular feminization syndrome—occurs in 1 in 20,000 live births. Individuals with this unusual condition are usually normal-appearing females, despite the presence of testes and a 46, XY chromosome constitution. The external genitalia are female but the vagina usually ends in a blind pouch and the uterus and the uterine tubes are absent or rudimentary. At puberty, there is normal development of breasts and female characteristics, but menstruation does not occur and pubic hair is scanty or absent. In some cases, the external genitalia are abnormal (e.g., enlarged clitoris and a scrotum-like structure; see Fig. 13-25). The failure of masculinization to occur in these individuals results from a resistance to the action of testosterone at the cellular level in the genital tubercle and the labioscrotal and urogenital folds.
Hypospadias
There are four types of hypospadias: glanular, penile, penoscrotal, and perineal hypospadias. Hypospadias is the most frequent anomaly involving the penis and is found in 1 in 125 male infants. In glanular hypospadias, the external urethral orifice is on the ventral surface of the glans penis. In penile hypospadias the external urethral orifice is on the ventral surface of the body of the penis. The glanular and penile types of hypospadias are the most common types (Fig. 13-26). In penoscrotal hypospadias, the urethral orifice is at the junction of the penis and the scrotum. In perineal hypospadias, the external urethral orifice is located between the unfused halves of the scrotum. Hypospadias results from inadequate production of androgens by the fetal testes. It is believed that environmental factors may disrupt testosterone-related gene expression.
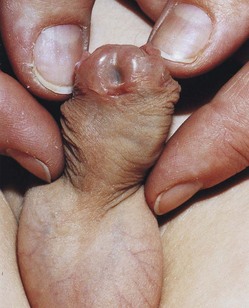
Epispadias
In a rare condition known as epispadias, the urethra opens on the dorsal surface of the penis. It is often associated with exstrophy of the bladder (see Fig. 13-13). Epispadias may result from inadequate ectodermal-mesenchymal interactions during development of the genital tubercle. As a consequence, the genital tubercle develops more dorsally than in normal embryos. Consequently, when the urogenital membrane ruptures, the urogenital sinus opens on the dorsal surface of the penis. Urine is expelled at the root of the malformed penis.
Anomalies of Female Genital Tract
Various types of uterine duplication and vaginal anomalies result from developmental arrest of the uterovaginal primordium during the eighth week of development (Fig. 13-27B to G). The main developmental anomalies are:
• Incomplete fusion of the paramesonephric ducts
• Incomplete development of one or both paramesonephric ducts
• Failure of parts of one or both paramesonephric ducts to develop
• Incomplete canalization of the vaginal plate that forms the vagina
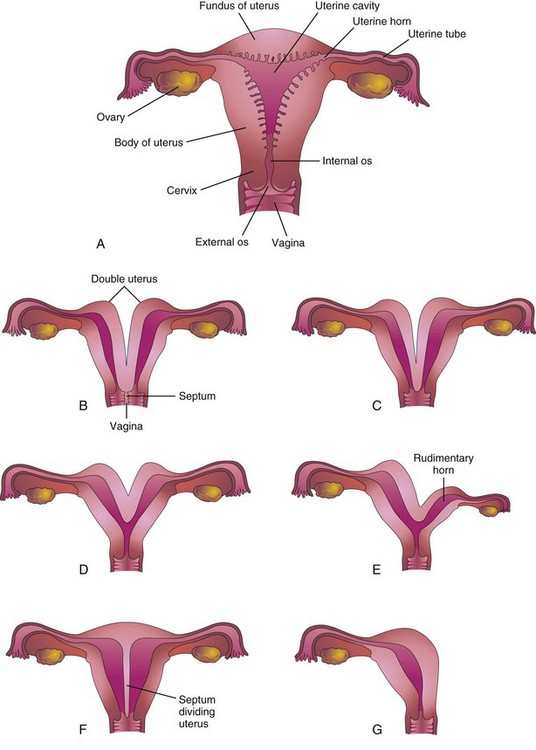
Figure 13–27 Various types of congenital uterine anomalies. A, Normal uterus and vagina. B, Double uterus (uterus didelphys) and double vagina. Note the septum dividing the vagina. C, Double uterus with a single vagina. D, Bicornuate uterus (two uterine horns). E, Bicornuate uterus with a rudimentary left horn. F, Septate uterus. Note the septum dividing the uterus. G, Unicornuate uterus. Note that only half of the uterus exists.
In some cases, the uterus is divided internally by a septum (Fig. 13-27F). If the duplication involves only the superior part of the body of the uterus, the condition is called bicornuate uterus (Fig. 13-27D and E). If growth of one paramesonephric duct is retarded and the duct does not fuse with the other one, a bicornuate uterus with a rudimentary horn develops (Fig. 13-27E). The rudimentary horn may not communicate with the cavity of the uterus. A unicornuate uterus develops when one paramesonephric duct does not develop; this results in a uterus with one uterine tube (Fig. 13-27G). In many of these cases, the individuals are fertile, but may have an increased incidence of premature delivery.
A double uterus (uterus didelphys) results from failure of fusion of the inferior parts of the paramesonephric ducts. It may be associated with a double or a single vagina (Fig. 13-27B and C).
Agenesis of the vagina results from failure of the sinovaginal bulbs to develop and form the vaginal plate (Fig. 13-20B). When the vagina is absent, the uterus is usually absent also, because the developing uterus (uterovaginal primordium) induces the formation of sinovaginal bulbs, which fuse to form the vaginal plate. Failure of canalization of the vaginal plate results in blockage of the vagina. Failure of the inferior end of the vaginal plate to perforate results in an imperforate hymen (Fig. 13-20C).
Development of Inguinal Canals
The inguinal canals form pathways for the testes to descend from their intra-abdominal position through the anterior abdominal wall and into the scrotum. Inguinal canals develop in both sexes because of the morphologically indifferent state of sexual development. As the mesonephros degenerates, a ligament called the gubernaculum develops on each side of the abdomen from the inferior pole of the gonad (Fig. 13-28A). The gubernaculum passes obliquely through the developing anterior abdominal wall at the site of the future inguinal anal. The gubernaculum attaches caudally to the internal surface of the labioscrotal swellings.
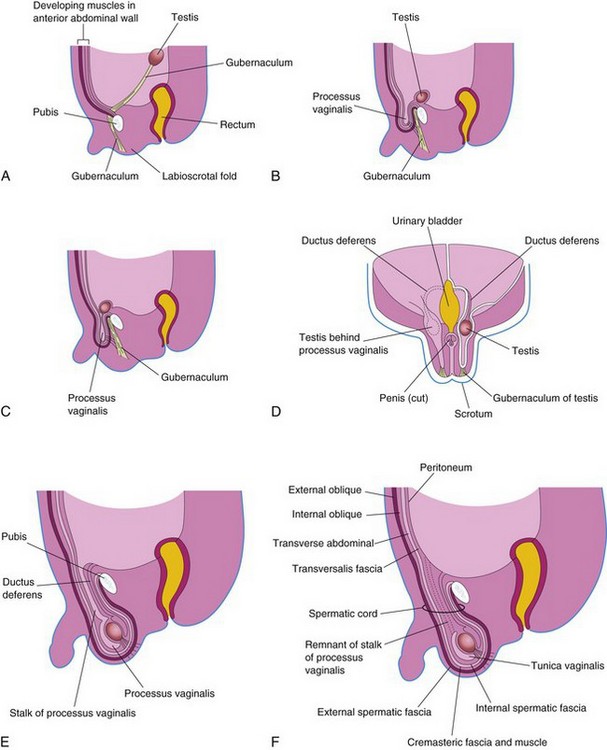
Figure 13–28 Formation of the inguinal canals and descent of the testes. A, Sagittal section of a 7-week embryo, showing the testis adjacent to the dorsal abdominal wall. B and C, Similar sections at approximately 28 weeks, showing the processus vaginalis and the testis beginning to pass through the inguinal canal. Note that the processus vaginalis carries fascial layers of the abdominal wall before it. D, Frontal section of a fetus approximately 3 days later, showing descent of the testis posterior to the processus vaginalis. The processus vaginalis has been cut away on the left side to show the testis and ductus deferens. E, Sagittal section of a newborn male infant, showing the processus vaginalis communicating with the peritoneal cavity by a narrow stalk. F, Similar section of a 1-month-old male infant after obliteration of the stalk of the processus vaginalis. Note that the extended fascial layers of the abdominal wall now form the coverings of the spermatic cord.
The processus vaginalis, an evagination of peritoneum, develops ventral to the gubernaculum and herniates through the abdominal wall along the path formed by the gubernaculum (Fig. 13-28B to E). The processus vaginalis carries extensions of the layers of the abdominal wall, which form the walls of the inguinal canal. In males, these layers also form the coverings of the spermatic cord and the testis (Fig. 13-28E and F). The opening in the transversalis fascia produced by the vaginal process becomes the deep inguinal ring, and the opening created in the external oblique aponeurosis forms the superficial inguinal ring.
Descent of Testes
By 26 weeks, the testes have descended retroperitoneally from the posterior abdominal wall to the deep inguinal rings (see Fig. 13-28B and C). This change in position occurs as the fetal pelvis enlarges and the trunk of the embryo elongates. Transabdominal movement of the testes is largely a relative movement that results from growth of the cranial part of the abdomen away from the future pelvic region.
Testicular descent through the inguinal canals and into the scrotum is controlled by androgens (e.g., testosterone) produced by the fetal testes. The gubernaculum appears to guide the testes during their descent. Descent of the testes through the inguinal canals and into the scrotum usually begins during the 26th week and takes 2 to 3 days. When the testis descends, it carries its ductus deferens and vessels with it. As the testis and the ductus deferens descend, they are ensheathed by the fascial extensions of the abdominal wall (Fig. 13-28F):
• The extension of the transversalis fascia becomes the internal spermatic fascia.
• The extensions of the internal oblique muscle and the fascia become the cremasteric muscle and fascia.
• The extension of the external oblique aponeurosis becomes the external spermatic fascia.
Within the scrotum, the testis projects into the distal end of the processus vaginalis. During the perinatal period, the connecting stalk of the process is usually obliterated, isolating the tunica vaginalis as a peritoneal sac related to the testis (Fig. 13-28F).
Cryptorchidism
Cryptorchidism (undescended testes) occurs in up to 30% of premature male infants and in approximately 3% to 4% of full-term male infants. Cryptorchidism may be unilateral or bilateral. In most cases, the testes descend into the scrotum by the end of the first year. If both testes remain within or just outside the abdominal cavity, they do not mature and sterility is common. If uncorrected, there is a significantly higher risk for the development of germ cell tumors, especially in cases of abdominal cryptorchidism. Cryptorchid testes may be in the abdominal cavity or anywhere along the usual path of descent of the testis, but they are usually in the inguinal canal (Fig. 13-29A). The cause of most cases of cryptorchidism is unknown, but a deficiency of androgen production by the fetal testes is an important factor.
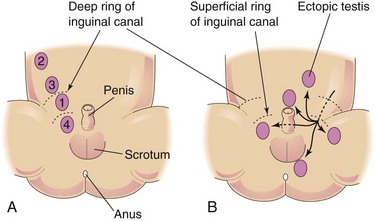
Ectopic Testes
After traversing the inguinal canal, the testis may deviate from its usual path of descent and lodge in various abnormal locations (Fig. 13-29B):
• Interstitial (external to the aponeurosis of the external oblique muscle)
All types of ectopic testis are rare, but interstitial ectopia occurs most frequently. Ectopic testis occurs when a part of the gubernaculum passes to an abnormal location and the testis follows it.
Descent of Ovaries
The ovaries also descend from the posterior abdominal wall to the pelvis, just inferior to the pelvic brim. The gubernaculum is attached to the uterus near the attachment of the uterine tube. The cranial part of the gubernaculum becomes the ovarian ligament and the caudal part forms the round ligament of the uterus (Fig. 13-20C). The round ligaments pass through the inguinal canals and terminate in the labia majora. The relatively small processus vaginalis in the female is usually obliterated and it disappears long before birth.
Congenital Inguinal Hernia
If the communication between the tunica vaginalis and the peritoneal cavity does not close, a persistent processus vaginalis occurs. A loop of intestine may herniate through it into the scrotum or labia majora (Fig. 13-30A and B). Embryonic remnants resembling the ductus deferens or the epididymis are often found in inguinal hernial sacs. Congenital inguinal hernia is much more common in males than in females and it is often associated with cryptorchidism and, in females, with androgen insensitivity syndrome.
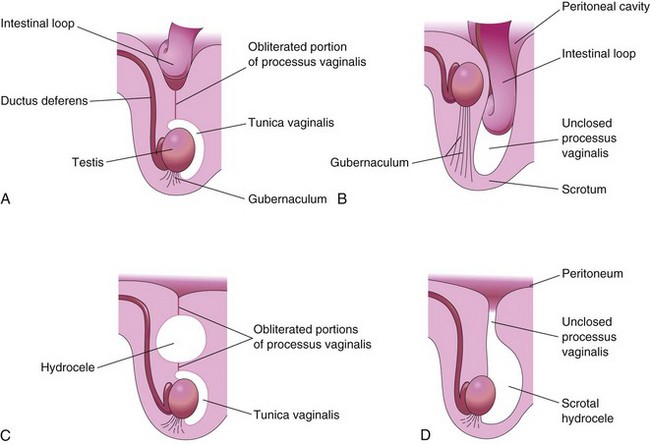
Figure 13–30 Sagittal sections showing conditions resulting from failure of closure of the processus vaginalis. A, Incomplete congenital inguinal hernia resulting from persistence of the proximal part of the processus vaginalis. B, Complete congenital inguinal hernia entering the unclosed processus in the scrotum. Cryptorchidism, a commonly associated condition is also shown. C, Large hydrocele that arose from an unobliterated portion of the processus vaginalis. D, Hydrocele of the testis and the spermatic cord resulting from peritoneal fluid passing into a patent processus vaginalis.
Hydrocele
Occasionally, the abdominal end of the processus vaginalis remains open, but is too small to permit herniation of the intestine (Fig. 13-30D). In such cases, peritoneal fluid passes into the patent processus vaginalis and forms a hydrocele of the testis. If the middle part of the processus vaginalis remains open, fluid may accumulate and give rise to a hydrocele of the spermatic cord (Fig. 13-30C).
Clinically Oriented Questions
1. Does a horseshoe kidney usually function normally? What problems may occur with this anomaly and how can they be corrected?
2. A patient was told that he has two kidneys on one side and none on the other. How did this abnormality probably happen? Are there likely to be any problems associated with this condition?
3. Are individuals with ovotesticular DSD (true hermaphrodites) ever fertile?
4. When a baby is born with ambiguous external genitalia, how long does it take to assign the appropriate sex? What does the physician tell the parents? How is the appropriate sex determined?
5. What is the most common type of disorder that produces ambiguous external genitalia? Will masculinizing, or androgenic, hormones given during the fetal period of development cause ambiguity of the external genitalia in female fetuses?