2 The glomerulus
By the end of the chapter you should be able to:
• Describe the structure of the glomerular filter
• Explain how the molecular size and charge of particles affects filtration
• Define clearance. Explain how it is measured and what its units are
• Describe how to measure the glomerular filtration rate and renal blood flow. Discuss how this varies with age
• State the four variables commonly used to estimate the glomerular filtration rate.
• Define autoregulation and explain how it is achieved
• Discuss the main clinical syndromes associated with glomerular disease, giving examples of typical clinical manifestations
• Explain how acute nephritic syndrome differs from nephrotic syndrome
• State four systemic diseases associated with glomerulonephropathy
Glomerular structure and function
Structure of the glomerular filter
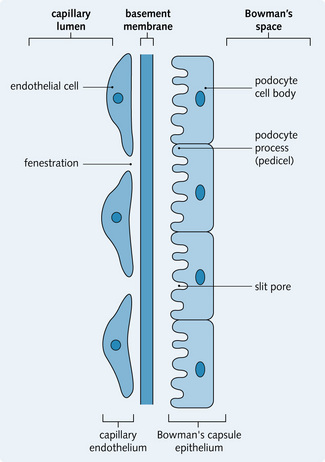
The renal corpuscle consists of a ball of capillaries, called the glomerulus, invaginated into the start of the nephron, the Bowman's capsule. This is where the first stage of urine production takes place. Plasma is filtered through the glomerular capillary wall into the Bowman's capsule. The composition of the plasma ultrafiltrate that enters the Bowman's capsule depends on the filtration barrier, which has three layers (Fig. 2.1):
Endothelial cells
The endothelial cells lining the glomerular capillaries are thin and flat with a large nucleus. The cells are perforated by numerous fenestrae (pores), which have a diameter of 60 nm. This allows plasma components to cross the vessel wall, but not blood cells or platelets.
Basement membrane
The basement membrane is a continuous layer of connective tissue and glycoproteins. It is a non-cellular structure that prevents any large molecules from being filtered.
Epithelial lining
The epithelial lining of Bowman's capsule consists of a single layer of cells (podocytes), which rest on the basement membrane. The podocytes have large extensions or trabeculae, which extend out from the cell body and are embedded in the basement membrane surrounding a capillary. Small processes called pedicels extend out from the trabeculae and interdigitate extensively with the pedicels of adjacent trabeculae. This leads to the formation of slit pores, which control the movement of substances through the final layer of the filter. The podocytes have a well-developed Golgi apparatus, used to produce and maintain the glomerular basement membrane. Podocytes can also be involved in phagocytosis of macromolecules (Figs 2.1 and 2.2).
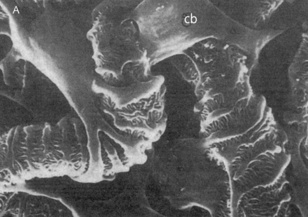
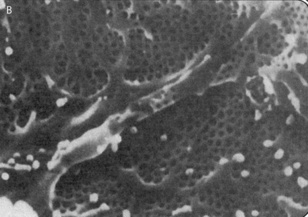
Fig. 2.2 Electron micrographs showing the arrangement of podocytes and glomerular capillaries as seen from Bowman's capsule. (A) Processes of podocytes run from the cell body (cb) towards the capillaries where they ultimately split into foot processes (pedicels). (B) Inner surface of a glomerular capillary.
(From Koeppen BM, Stanton B, 1996. Renal physiology, 2nd edn. Mosby Year Book.)
Mesangium
The mesangium is also part of the renal corpuscle and consists of two components:
The mesangial cells surround the glomerular capillaries and have a function similar to monocytes. They provide structural support for the capillaries, exhibit phagocytic activity, secrete extracellular matrix, and secrete prostaglandins. The cells are contractile, which helps regulate blood flow through the glomerular capillaries (Fig. 2.3).
Process of glomerular filtration
This is a passive process that involves the flow of a solvent through a filter. The glomerular filter allows only low-molecular-weight substances in plasma to pass through it, forming the glomerular ultrafiltrate.
Molecular size and electrical charge
Molecular weight is the main factor in determining whether a substance is filtered or remains in the capillaries. The maximum molecular weight of substances able to pass through the filter is 70 kDa. Any molecule with a molecular weight of less than 70 kDa passes freely through the filter (e.g. glucose, amino acids, Na+, urea, K+). The shape and electrochemical charge of macromolecules also affect filterability. All three layers of the filter are coated with anions and these repel negatively charged macromolecules. Smaller, positively charged molecules pass through the filtration barriers relatively easily, unless they are protein-bound.
Albumin has a molecular weight of 69 kDa and is a negatively charged protein. Only very tiny amounts pass through the glomerular filter because of the repelling effect of its negative charge, and all of this is reabsorbed in the proximal tubule. A total of 30 g of protein a day enters the renal lymph vessels. Significant amounts of protein in the urine (proteinuria) are usually indicative of damage to the glomerular filter.
Glomerular filtration rate
The glomerular filtration rate (GFR) is the amount of filtrate that is produced from the blood flowing through the glomerulus per unit time.
Forces governing tissue fluid formation
The movement of fluid between plasma and tissue fluid is determined by Starling's forces:
Changes in these forces alter the GFR. The following factors affect tissue fluid formation:
• At the arteriole end of the capillary, hydrostatic pressure is greater than colloid oncotic pressure as a result of resistance to flow due to the narrowing of the vessel, and fluid is forced out of the capillary
• As the fluid moves out of capillaries via the highly permeable wall, oncotic pressure increases and the pressure forces are reversed (most apparent at the venous end of the capillary) so there is net movement of fluid back into the capillaries.
Forces governing glomerular filtration rate
GFR is also driven by Starling's forces. However, in the renal vascular bed the surface area of the glomerular capillaries is much larger than that of normal capillary beds, so there is less resistance to flow. The hydrostatic pressure falls less along the length of the capillary because the efferent arterioles, which act as secondary resistance vessels, maintain a constant pressure along the entire length of the glomerular capillary.
Tissue fluid in a vascular bed is the equivalent of glomerular filtrate in Bowman's space, produced as a result of the glomerular capillary hydrostatic pressure (50 mmHg). This is opposed by the hydrostatic pressure in Bowman's capsule (12 mmHg) and the colloid oncotic pressure (25 mmHg) within the capillary. When these forces are equal the filtration equilibrium is reached, with very little fluid movement after this.
Fluid is reabsorbed into the peritubular capillaries as a result of high colloid oncotic pressure (35 mmHg) and low hydrostatic pressure. This reabsorption causes a fall in colloid oncotic pressure as plasma proteins become diluted.
The pressures controlling glomerular filtration into Bowman's capsule are illustrated in Fig. 2.4 and the composition of the glomerular filtrate is shown in Fig. 2.5.
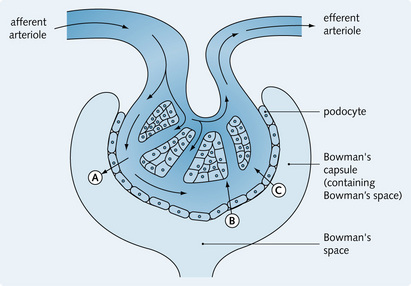
Fig. 2.4 Pressures controlling glomerular filtration into Bowman's capsule. A, Hydrostatic pressure of glomerular capillary = 50 mmHg; B, hydrostatic pressure in Bowman's space = 12 mmHg (increases fluid uptake into capillary); C, oncotic pressure of glomerular capillary = 25 mmHg (increases fluid uptake into capillary).
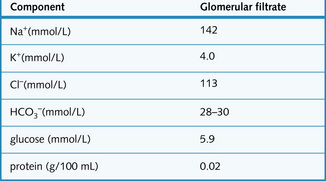
Measurement of glomerular filtration rate
Clearance
Clearance (C) is the volume of plasma that is cleared of a substance in a unit time. It is a measure of the kidney's ability to remove a substance from the plasma and excrete it. The clearance of a substance x is:
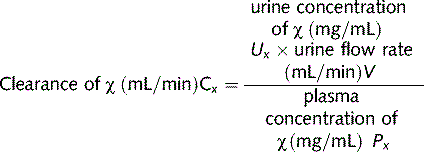
Clearance of a substance will provide an accurate estimate of the glomerular filtration rate (GFR) if that substance ‘follows’ the filtrate without being altered by the kidney (i.e. is not reabsorbed, secreted, synthesized or metabolized by the kidney). Inulin is such a substance:
• It is a polysaccharide of molecular weight 5500
• It is not normally found within the body, so is introduced into the body by injection or intravenous infusion
• It passes into the glomerular filtrate but is not reabsorbed, secreted, synthesized or metabolized by the kidney – so all inulin filtered by the glomerulus is excreted in the urine. Inulin clearance can be used to assess glomerular function in disease.
Inulin clearance is equal to the GFR, i.e. 120 mL/min/1.73 m2 body surface area (this varies with body size). GFR is widely accepted as the best measure of kidney function; however, it is difficult to measure since techniques such as inulin clearance are complicated and cannot be used to assess GFR in routine clinical practice. Instead, creatinine clearance can be used as an estimate of GFR. Creatinine is found in the body; it is produced during muscle metabolism:
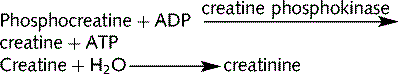
Like inulin, creatinine is filtered freely and is not affected or produced by the kidney. A small contribution to the clearance of creatinine comes from tubular secretion, however with this caveat, creatinine clearance approximates to GRF. Plasma creatinine is commonly used as a marker of renal function since plasma creatinine levels will only vary with renal function as long as muscle mass and metabolism are stable. It has a reciprocal relationship with GFR (see Fig. 2.6).
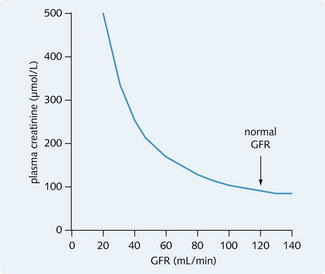
Estimated glomerular filtration rate (eGFR)
The exact relationship between plasma creatinine and GFR depends upon muscle mass and therefore age, sex and size, thus plasma creatinine levels alone cannot be used as an accurate predictor of renal function. Instead the GFR may be estimated from the plasma creatinine using a formula, the most commonly used of which is the Modification of Diet in Renal Disease (MDRD) equation.
Regulation of renal blood flow and glomerular filtration rate
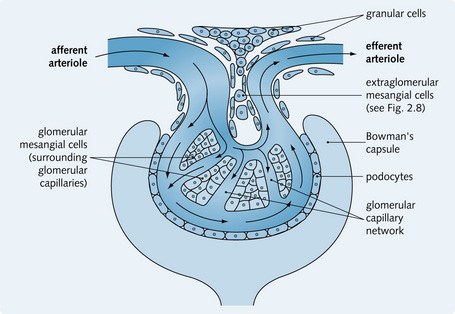
Both remain fairly constant because of autoregulation, which involves changes in tone of the afferent and efferent arterioles (Fig. 2.7). Over the autoregulatory range of perfusion pressures (90–200 mmHg), blood flow is independent of perfusion pressure so, as the perfusion pressure increases, resistance to flow increases (Fig. 2.8). Two mechanisms are involved in autoregulation:
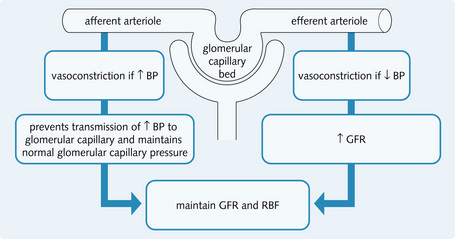
Fig. 2.7 The regulation of renal blood flow (RBF) and glomerular filtration rate (GFR) by vasoconstriction of arterioles.
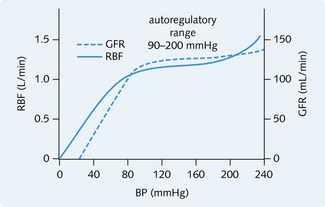
Myogenic mechanism
An increase in pressure (caused by an increase in blood flow) stimulates stretch receptors in smooth muscle fibres in the vessel wall. This causes reflex contraction of smooth muscle fibres, resulting in vessel vasoconstriction. There is an increased resistance to flow so, overall, renal blood flow remains constant.
Tubuloglomerular feedback mechanism
When GFR increases, the increased flow through the tubules leads to proportionally less NaCl being reabsorbed. Hence, when the filtrate reaches the distal convoluted tubule it has a higher NaCl concentration. This is the trigger for the tubuloglomerular feedback mechanism which has three components within the juxtaglomerular apparatus:
1. The macula densa cells of the distal convoluted tubule epithelium. These detect osmolality or the rate of Na+ or Cl– movement into the cells. The higher the flow of the filtrate the higher the Na+ concentration in the cells
2. A signal is sent via the juxtaglomerular cells, triggered by a change in the NaCl concentration of distal tubular fluid
3. Angiotensin II or prostaglandins (see Chapter 4) act to vasoconstrict the smooth muscle of the adjacent afferent arterioles and therefore decrease renal plasma flow, which in turn reduces GFR.
This mechanism maintains a constant GFR, thus preventing nephron overload because a high NaCl load decreases the filtration capacity of that nephron. A number of factors can modulate the sensitivity of the tubuloglomerular feedback mechanism including ANP and NO (decrease sensitivity) and Angiotensin II (increase sensitivity).
Renal blood flow and systemic blood pressure
Glomerular filtration and renal blood flow remain relatively constant over a wide physiological range of blood pressure due to autoregulation. For example, in acute severe haemorrhage increased sympathetic activity leads to vasoconstriction and consequently decreased blood flow. However, in response intrarenal vasodilator prostaglandins are produced to prevent excessive vasoconstriction within the kidney and renal perfusion is thus maintained.
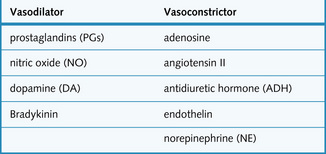
Regulation of GFR involves other vasoactive substances, which are found in the walls of blood vessels. These are summarized in Fig. 2.9.
Diseases of the glomerulus
Overview
Glomerular disease can be classified as:
• Hereditary (e.g. Alport's and Fabry's syndromes)
• Primary (most common): disease process originates from the glomerulus
• Secondary to systemic diseases: e.g. systemic lupus erythematosus (SLE), diabetes mellitus, bacterial endocarditis.
Disease of the glomerulus is frequently called a glomerulonephritis. Glomerulonephritis can result in clinical presentation with either the nephritic syndrome (haematuria with or without proteinuria salt and water retention and hypertension), or the nephrotic syndrome (heavy proteinuria sufficient to cause a low serum albumin, oedema and hypercholesterolaemia). Presentation can be acute, or chronic, rapidly or slowly progressive and may lead to chronic kidney disease. Glomerulonephritis can be asymptomatic.
The mechanisms of glomerular injury
Circulating immune complex nephritis
This is the most common mechanism of immune-mediated damage. Immune complexes form outside the kidney and become trapped in the glomerulus after travelling to the kidney via the renal circulation (Fig. 2.10B). The antigen can be:
• Exogenous: bacteria (e.g. group A streptococci), viruses (surface antigen of hepatitis B, hepatitis C virus antigen), tumour antigens
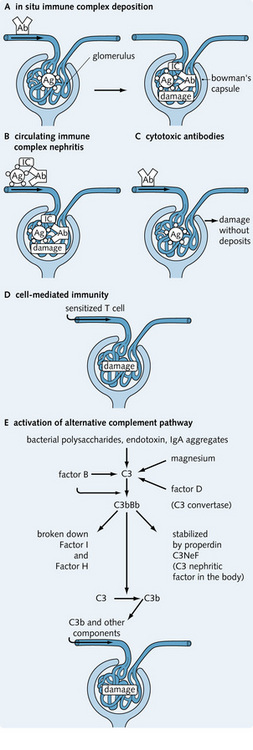
Fig. 2.10 (A–E) The five mechanisms of immune complex renal disease. Ab, antibody; Ag, antigen; IC, immune complex.
When trapped in the glomerulus, the immune complexes activate the classical complement pathway, causing acute inflammation of the glomerulus. Immunofluorescence microscopy demonstrates immunoglobulin deposits along the basement membrane and/or in the mesangium. These increase vascular permeability.
In situ immune complex deposition
Antigen–antibody immune complexes form within the kidney when antibodies react with intrinsic or planted antigens within the glomerulus (Fig. 2.10A).
Antiglomerular basement membrane (anti-GBM) disease is an example of reaction to intrinsic antigens. Antibodies are formed against an antigen in the GBM, to form a complex which stimulates the complement cascade. This damages the glomerulus and leads to rapidly progressing renal failure. The anti-GBM antibodies also attack the basement membrane of the alveoli in the lungs. The triad of anti-GBM antibodies, GN and pulmonary haemorrhage is known as Goodpasture's syndrome.
Reaction to planted antigens occurs when circulating antigens are deposited within the glomerulus. They can be:
Cell-mediated immunity
Sensitized T cells from cell-mediated immune reactions play a role in the progression of acute GN to chronic GN (Fig. 2.10D). Glomerular damage is thought to be mediated by macrophages and T lymphocytes.
Activation of alternative complement pathway
Bacterial polysaccharides, endotoxins, and IgA aggregates can stimulate the alternative complement pathway – the products of which deposit in the glomeruli, impairing glomerular function (Fig. 2.10E). This occurs in membranoproliferative GN.
Cytotoxic antibodies
Antibodies to glomerular cell antigens cause damage without the formation and deposition of immune complexes (Fig. 2.10C). This is uncommon. An example would be antibody fixing to mesangial cells, resulting in complement-mediated mesangiolysis and mesangial cell proliferation.
Clinical manifestations of glomerular disease
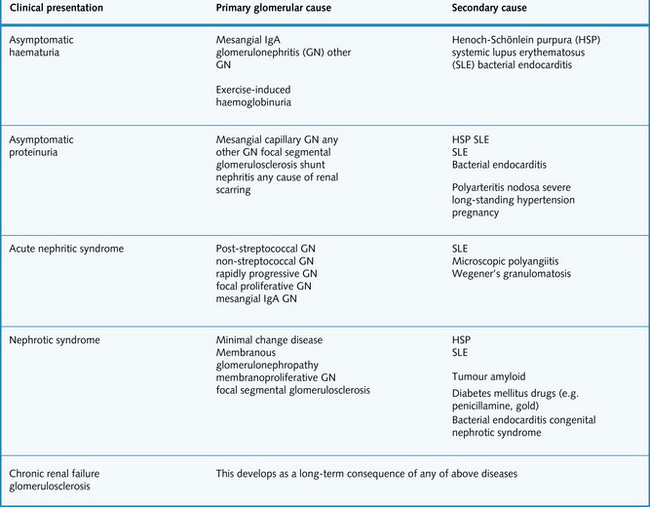
Glomerular disease usually presents in one of the following five ways (Fig. 2.11):
Asymptomatic haematuria
Haematuria due to glomerular disease is often invisible and painless and can be continuous or intermittent. Glomerular disease can also result in visible haematuria. Primary and secondary causes are summarized in Fig. 2.11. It should be noted that heavy exercise can result in haemoglobinuria (not haematuria).
Asymptomatic proteinuria
This is characterized by proteinuria (usually 0.3–3 g protein daily) with no other symptoms. Possible causes are summarized in Fig. 2.11.
Nephrotic syndrome
• Proteinuria (typically > 3 g/24 h) – sufficient to cause:
• Hypoalbuminaemia (serum albumin typically < 25 g/L) – sufficient to cause:
• Hyperlipidaemia (increased hepatic lipoprotein synthesis secondary to protein losses).
There is increased permeability of the glomerular filter to albumin as a result of glomerular basement membrane damage and increase in pore size. The capillary wall becomes permeable to proteins of higher molecular weight as the severity of injury increases. Heavy proteinuria leads to low plasma albumin and therefore tissue oedema.
In an adult, a loss of more than 3–5 g of albumin per day can cause hypoalbuminaemia, but some patients can have nephrotic-range proteinuria without being overtly nephrotic, because the rate of albumin synthesis compensates for the albumin loss. A limited amount of filtered protein can be reabsorbed by endocytosis, but if this is exceeded, protein is lost in the urine. A normal albumin concentration within the capillary maintains the colloid osmotic pressure and when this is decreased, less fluid moves back into the capillaries, causing oedema in the peripheral tissues. The decreased circulating volume stimulates the renin–angiotensin–aldosterone system, leading to further sodium and water retention, and further oedema.
Other proteins can be lost from the plasma, including immunoglobulins and proteins controlling coagulation. Complications of nephrotic syndrome include:
• Immunosuppression: increases risk of infection
• Hypercoagulable state: increases risk of deep vein thrombosis, pulmonary embolus and renal vein thrombosis
• Hyperlipidaemia: increases risk of vascular disease and ischaemic heart disease.
• Reduction of proteinuria, using angiotensin-converting enzyme (ACE) inhibitors
• Anticoagulation if hypercoagulable (risk of thrombosis increases as albumin decreases)
• Treatment of underlying causes when possible, e.g. in minimal change disease high-dose corticosteroid therapy will eliminate proteinuria in up to 90% of cases.
Primary and secondary causes are summarized in Fig. 2.11.
Acute nephritic syndrome
The symptoms and signs include:
Patients might also complain of loin pain, headache and general malaise. Primary and secondary causes of acute nephritic syndrome are summarized in Fig. 2.11.
Rapidly progressive GN
Rapidly progressive GN, or crescentic GN, occurs when there is severe glomerular injury. It presents with haematuria, oliguria and hypertension, eventually causing renal failure. This may be seen in a renal vasculitis such as Wegner's granulomatosis or microscopic polyangiitis.
Chronic kidney disease (CKD)
Chronic glomerulonephritis causes CKD as a result of progressive nephron loss. The kidney shrinks, with cortical thinning and granular scarring. Histological examination of chronic glomerulonephritis reveals:
This is often asymptomatic in the early stages. Later, symptoms of CKD develop as waste products accumulate and erythropoietin or vitamin D production is reduced. Symptoms and signs of CKD (discussed in detail in Chapter 7) include:
• Salt and water retention causing oedema
• Paraesthesiae (due to polyneuropathy)
• Mental slowing and clouding of consciousness (terminal stage).
In extreme cases, oliguria results. Dialysis or renal transplantation are effective treatments. The glomerular causes of CKD are given in Fig. 2.11, however, when advanced it is usually not possible to determine the nature of the initial insult.
Primary glomerulonephritis typically presenting with heavy proteinuria or the nephrotic syndrome
Minimal change disease
This is the most common cause of nephrotic syndrome in children under the age of 6 years, and more commonly affects males. No significant renal changes are seen under the light microscope (hence the name). Electron microscopy shows podocyte fusion, i.e. foot process effacement. The cause is unknown, but potential mechanisms include a post-allergic reaction, circulating immune complexes, or altered T-cell immunity. Treatment involves corticosteroid therapy and ciclosporin or cyclophosphamide (if resistant). The prognosis is good in children and variable in adults, but usually good with it only very rarely causing end-stage renal failure.
Membranous glomerulonephropathy
This is a chronic disease characterized by:
It accounts for 40% of adult nephrotic syndrome and is more common in males. Causes are idiopathic (85%), primary or secondary. Secondary causes include:
• Infections: syphilis, malaria, hepatitis B
• Tumours: melanoma, carcinoma of the bronchus, lymphoma
Histological examination reveals widespread glomerular basement thickening caused by immunoglobulin deposition. Over time, the abnormal excess mesangial matrix causes hyalinization of the glomerulus and death of individual nephrons. Treatment could involve immunosuppressants. Prognosis depends on the cause; 30% of idiopathic cases develop CKD and require dialysis or transplantation. In secondary membranous glomerulonephropathy, treatment of underlying disease causes disease remission.
Focal segmental glomerulosclerosis
This accounts for 10% of childhood and up to 30% of cases of adult nephrotic syndrome. It is more common in males and its causes are:
Histological examination reveals focal collapse and sclerosis, with hyaline deposits in glomerular segments. Presentation is with proteinuria or nephrotic syndrome, later developing haematuria and hypertension. Most develop CKD within 10 years. Treatment of the idiopathic form might involve steroids, cyclophosphamide, ciclosporin, dialysis and renal transplantation. Recurrence can be seen after a renal transplant.
Primary glomerulonephritis typically presenting with the nephritic syndrome
IgA nephropathy (Berger's disease)
This is the most common primary glomerular disease worldwide, causing recurrent haematuria. There is some association with geographical location (more common in France, Australia and Singapore) and human leucocyte antigen (HLA) DR4. It typically affects young men after an upper respiratory tract infection. Presentation is with microscopic haematuria and proteinuria and renal impairment. There is hypertension and plasma IgA levels are raised. Histologically IgA and C3 deposits are seen in the mesangium of all the glomeruli, with some mesangial proliferation (this is similar to the histological picture seen in HSP). Eventually, there is sclerosis of the damaged segment. There is no effective treatment. Patients with late onset, proteinuria, increased blood pressure and increased creatinine at presentation have a worse prognosis – up to 20% of patients develop advanced CKD.
Rapidly progressive (crescentic) glomerulonephritis
This results in severe glomerular injury. Histologically, glomerular injury results in leakage of fibrin, which stimulates epithelial cells and macrophages within Bowman's capsule to proliferate and form crescent-shaped masses, reducing glomerular blood supply. It can be seen as part of systemic illnesses such as SLE, Wegener's granulomatosis and microscopic polyangiitis. As the name suggests, the disease progresses rapidly and there is a loss of renal function within days to weeks. Prompt diagnosis and treatment is therefore required to prevent hypertension, kidney scarring and renal failure. Treatment consists of high-dose steroids, immunosuppressants and plasma exchange.
Focal proliferative glomerulonephritis
Focal proliferative GN results in inflammation of some parts of some glomeruli. Its presentation is less acute. It might affect only the kidney (IgA nephropathy, see below) or be secondary to systemic illnesses such as Henoch–Schönlein purpura (HSP), Goodpasture's syndrome, subacute bacterial endocarditis, vasculitis and other connective tissue diseases (e.g. SLE). Treatment with immunosuppression can be effective. The prognosis is variable. Necrotizing GN is also often seen in malignant hypertension.
Membranoproliferative glomerulonephropathy
Also known as mesangiocapillary glomerulonephritis, this is rare. It occurs most often in children and females. It is characterized histologically by diffuse global basement membrane thickening and mesangial proliferation. It is usually primary, but can be secondary to disorders such as SLE and malaria. Primary membranoproliferative GN is classified as:
• Type I (more common): immune complexes deposit in the subendothelium, causing inflammation and capillary thickening
• Type II: caused by activation of the alternative complement pathway following an infection. Histological examination reveals thickened capillaries caused by C3 deposition.
It can present with asymptomatic haematuria or combined nephrotic/nephritic syndrome.
Secondary glomerulonephritis
Systemic disorders can cause glomerular disease. They can be:
• Immune complex mediated (e.g. SLE, HSP, bacterial endocarditis)
• Vascular (e.g. microscopic polyangiitis, Wegener's granulomatosis)
• Metabolic (e.g. diabetes mellitus, amyloidosis)
• Drug treatment (e.g. penicillamine, gold, captopril, phenytoin)
• Infections (e.g. hepatitis B, leprosy, syphilis, malaria).
Post-streptococcal glomerulonephritis
This presents 1–3 weeks following a group A ß-haemolytic streptococcal infection of the tonsils, pharynx or skin. Clinical features include proteinuria, haematuria and a low glomerular filtration rate (GFR) (this causes fluid retention, oligaemia and hypertension). All the glomeruli are involved thus resulting in a diffuse proliferative GN – ‘proliferative’ because there is an increase in the cellularity in the glomerulus (Fig. 2.12). Treatment is usually conservative, with antibiotics to treat any remaining infection. The prognosis is excellent in children but only 60% of adults recover completely; the rest develop hypertension or renal impairment.
Non-streptococcal glomerulonephritis
Non-streptococcal GN follows a similar process to that for post-streptococcal GN except that the causative organism is not a Streptococcus. It can be triggered by:
Systemic lupus erythematosus
SLE is an autoimmune vasculitis characterized by antinuclear antibodies and widespread immune-complex-mediated inflammation. It is more common in females, Asians and if an individual is HLA B8-, DR2- or DR3-positive. It is a relapsing and remitting condition, usually diagnosed between 30 and 40 years of age. It affects many systems and organs in the body; for example, the joints, skin, heart, lungs and the kidneys (75% of cases). The renal lesions are the most important clinically and affect prognosis. Glomerular changes vary from minimal involvement to diffuse proliferative disease with:
• Immune complex deposition in glomerulus (frequently all classes of immunoglobulin and complement)
This results in focal or diffuse proliferative GN, or membranous glomerulopathy. Patients present with proteinuria, oedema and hypertension. There may be extrarenal systemic symptoms. Patients may develop CKD, but the prognosis is improved with immunosuppressive treatment (steroids, azathioprine or cyclophosphamide).
Henoch–Schönlein purpura
HSP is seen predominantly in children, affecting males more than females. It is an immune-mediated systemic vasculitis that affects many parts of the body including:
• Skin: a purpuric rash is seen over on the extensor surface of the legs, arms and buttocks
• Intestine: resulting in abdominal pain, vomiting, bleeding
• Kidney: resulting in GN (a third of patients develop glomerular lesions histologically identical to IgA nephropathy).
HSP can follow an upper respiratory tract infection. It has an excellent prognosis in children.
Bacterial endocarditis
Glomerular disease in bacterial endocarditis is caused by:
• Immune complex deposits in the glomerulus
• Embolism-mediated infarction – emboli break away from the heart valves.
The main histological diagnoses are focal, segmental and diffuse proliferative GN. Presentation is with microscopic haematuria, fluid retention and renal impairment. Renal lesions resolve on antibiotic therapy.
Diabetic glomerulosclerosis
Diabetes mellitus affects several organs including the kidneys, which are the most commonly and severely damaged organs in diabetes. Diabetic nephropathy is the most common cause of CKD requiring dialysis treatment in developed countries. Renal manifestations include nodular glomerulosclerosis and arteriosclerosis, including benign nephrosclerosis with hypertension (Fig. 2.13 presents a summary of the natural history of diabetes). Histological features can include:
• Thickening of the capillary basement membrane
• Increase in the matrix of the mesangium
• A diffuse or nodular pattern of glomerulosclerosis (also known as Kimmelstiel–Wilson syndrome)
• Arterial hyalinosis of both the afferent and efferent arterioles – this predisposes to vessel occlusion.
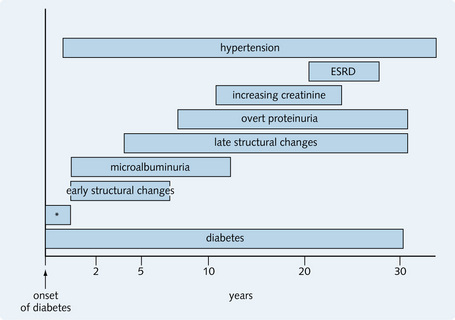
Fig. 2.13 Summary of the natural history of diabetes. ‘Asterisk’ (*) indicates functional changes in kidney size (increased) and short-term glomerular filtration rate (increased). ESRD, end-stage renal disease (or CKD stage 5).
The arteries can also be affected (especially in type II diabetes) and severe atheroma in the renal artery leads to renal ischaemia and hypertension. Papillary necrosis is a recognized complication, especially in the presence of infection.
Diabetic nephropathy presents with microalbuminuria, which increases progressively to nephrotic range proteinuria (i.e. > 3 g/24 h). Consequently, GFR gradually declines, leading to CKD, which can be seen in 30% of cases of insulin-dependent type 1 diabetes mellitus. It is also associated with diabetic complications elsewhere (e.g. retinopathy in the eyes). Chronic renal damage as a result of diabetes is associated and accelerated by hypertension.
Treatment includes ACE inhibitors to reduce proteinuria (these are discussed later), strict blood pressure and glycaemic control, and dialysis for stage 5 CKD.
Amyloidosis
This disorder involves deposition of amyloid (an extracellular fibrillar protein) in the glomeruli, usually within the mesangium and subendothelium, and sometimes in the subepithelial space. Deposits can also be found in the walls of the blood vessels and in the interstitium. Clinically, this results in heavy proteinuria or the nephrotic syndrome, eventually leading to CKD (due to ischaemia and glomerulosclerosis). Dialysis or transplant is required to prevent death from uraemia.
Goodpasture's syndrome
In Goodpasture's syndrome, autoantibodies to type IV collagen in the glomerular basement membrane develop, causing inflammation. Presentation is with a rapidly progressive crescentic GN and acute renal failure (ARF) and lung haemorrhage. Prognosis is poor without treatment, which involves:
Microscopic polyarteritis nodosa (also known as microscopic polyangiitis)
This is a necrotizing vasculitis affecting the small arteries of the body; it is more common in males. Initially, there is a focal, segmental or necrotizing GN followed by rapidly progressive GN. Histological examination reveals extensive necrosis, fibrin deposition and epithelial crescents. Microscopic polyarteritis nodosa (PAN) is associated with circulating antineutrophil cytoplasmic antibodies (ANCA), which complex with a perinuclear antigen (myeloperoxidase) in fixed neutrophils (pANCA).
Wegener's granulomatosis
This is a rare necrotizing vasculitis affecting the nose, upper respiratory tract and kidneys. It typically presents between 40 and 50 years of age. The glomerular disease is similar to that in microscopic PAN, with granuloma formation. Presentation is with asymptomatic haematuria or nephritic syndrome (focal segmental GN) or rapidly progressive GN. It is associated with ANCA, which characteristically recognizes a cytoplasmic antigen (proteinase 3) in fixed neutrophils (cANCA).
Hereditary nephritis
Alport's syndrome
This is usually X-linked, affecting mainly males (females are usually asymptomatic carriers). Autosomal dominant and autosomal recessive patterns of inheritance have also been described. An abnormality of basement membrane collagen IV is found in all patients, and they lack the Goodpasture's antigen.
Presentation is with glomerulonephritis and haematuria, ocular abnormalities and sensorineural deafness. Ocular lesions include lens dislocation, cataract and conical cornea. It is also associated with platelet dysfunction and hyperproteinaemia.
A few patients develop end-stage renal failure in childhood and adolescence. Females might have microscopic haematuria, but rarely develop end-stage renal failure. Treatment involves dialysis and/or transplantation.
Fabry's syndrome
This is a rare X-linked disorder, with a glycolipid metabolism defect due to the deficiency of galacto-sidase A. As a result, ceramide trihexoside (a glyco-sphingolipid) accumulates and is deposited in the kidneys, skin and vascular system. This disorder is associated with cardiac problems such as angina and cardiac failure – consequently, most patients die in the fifth decade of life.