Neurocutaneous disorders and other syndromes
Certain inherited skin disorders also have significant involvement of internal organs. The neurocutaneous disorders, the inherited diseases of connective tissue and the premature ageing syndromes are included.
Neurofibromatosis
von Recklinghausen’s neurofibromatosis (NF1) is relatively common, affecting about 1 in 3000 births. Café-au-lait spots, cutaneous neurofibromas and other bony or neurological abnormalities characterize NF1. The disease shows autosomal dominant inheritance, although 50% of cases are new mutations.
Aetiopathogenesis
The NF1 gene is a tumour suppressor gene, mapped to chromosome 17. This finding offers the prospect of devising a prenatal DNA screening test.
Clinical presentation
The two main cutaneous features are:
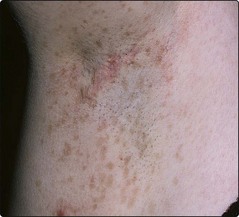
1. Café-au-lait spots: round or oval coffee-coloured macules, due to increased melanin pigment. They often appear in the first year of life. One or two café-au-lait spots are seen in 10% of normal people but, in neurofibromatosis, six or more are usually present. Freckling of the axilla is also found (Fig. 1).
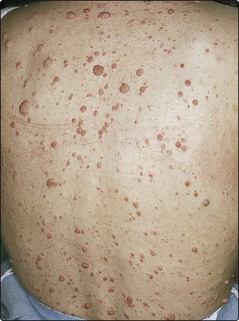
2. Dermal neurofibromas: small nodules that appear during childhood and increase in number at the time of puberty (Fig. 2). Their number varies from a few to several hundred.
A proportion of patients with NF1 have short stature and macrocephaly. Rare variants of the disease are occasionally seen. The commonest is NF2 (central neurofibromatosis) in which patients have bilateral acoustic neuromas but few, if any, café-au-lait spots or dermal nodules. NF2 also shows autosomal dominant inheritance. The NF2 gene is on chromosome 22.
Complications
Complications develop in many cases and include the following:
 Plexiform neurofibromas are larger than their dermal counterparts and measure up to several centimetres in size. They are associated with pigmentation and hypertrophy of the overlying skin or underlying bone, and present a cosmetic problem.
Plexiform neurofibromas are larger than their dermal counterparts and measure up to several centimetres in size. They are associated with pigmentation and hypertrophy of the overlying skin or underlying bone, and present a cosmetic problem.
Benign tumours of the nervous system may develop. These include optic gliomas, acoustic neuromas and spinal neurofibromas that arise from nerve roots of the spinal cord.
Sarcomatous change in a neurofibroma, typically non-cutaneous, occurs in 1.5–15% of cases.
Kyphoscoliosis (in 2%) or bowing of the tibia and fibula may occur.
Other problems include iris hamartomas (Lisch nodules), hypertension, epilepsy and learning difficulties.
Management
Once the diagnosis has been made, genetic counselling and the exclusion of any complicating factors are important. Troublesome nodules can be excised, and larger disfiguring neurofibromas removed by plastic surgery. Patients are often helped by contact with a patient support group (p. 132).
Tuberous sclerosis complex
Tuberous sclerosis complex is an autosomal dominant condition of variable expression with an incidence of 1 in 10 000. About 60–70% of patients have new mutations. Hamartomas occur in several organs. The abnormal genes have been mapped to chromosomes 9 and 16.
Clinical manifestations
The features may not appear until puberty. Patients typically show the following:
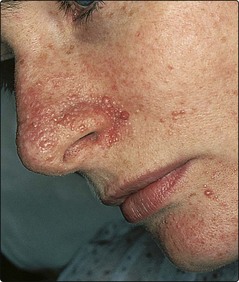
Adenoma sebaceum: red–brown angiofibromatous papules that are usually found around the nose (Fig. 3). They appear in childhood.
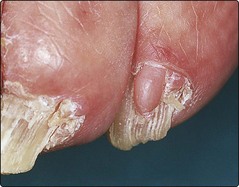
Periungual fibromas: pink fibrous projections are seen under the nail folds (Fig. 4).
Shagreen patches: connective tissue naevi, soft, yellowish with a cobblestone surface, are found on the lumbosacral region.
Ash-leaf macules: small (1–3 cm long) white oval macules, sometimes present at birth, and best seen with a Wood’s light.
Neurological involvement: learning difficulties and epilepsy affect 60–70% of cases. Intracranial calcification is seen.
Other features: retinal phacomas, cardiac rhabdomyomas and renal tumours are found.
Management
An affected individual should have a full clinical examination, often with radiographs and magnetic resonance imaging (MRI) of the head. Children are screened for ash-leaf macules using a Wood’s light. The angiofibromas may be improved by hyfrecation or laser, but tend to recur. Genetic counselling is given once the diagnosis is made. The support group is helpful (p. 132).
Incontinentia pigmenti
Incontinentia pigmenti is a rare X-linked dominant condition that is usually lethal in utero in males. In females, it presents within a few days of birth as a widespread blistering eruption (p. 13). Warty papules follow, but are replaced by hyperpigmentation, which appears in a whorled pattern. Skeletal, ocular, neurological and dental abnormalities are associated.
Xeroderma pigmentosum
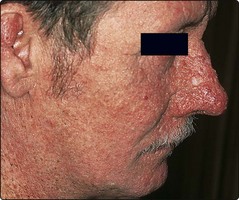
Xeroderma pigmentosum is a group of rare autosomal recessive conditions characterized by defective repair of ultraviolet (UV)-damaged DNA. Photosensitivity begins in infancy, and freckles and keratoses appear on exposed skin in childhood. Squamous cell and basal cell carcinomas, keratoacanthomas and malignant melanomas subsequently develop in the UV-damaged skin (Fig. 5). Strict sunlight avoidance is necessary but, in its severe form, the disease can be fatal in the first or second decade. The gene loci are known (p. 12). Prenatal diagnosis is possible (p. 91) and is used when parents have already had one affected child.
Ehlers–Danlos syndrome
At least 10 inherited disorders of defective collagen structure and biochemistry are included in this group of conditions (gene loci: p. 12). The diseases may be dominant, recessive or X-linked, and present in varying degrees of severity. The features include:
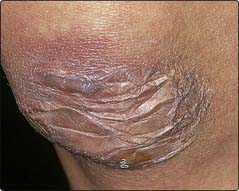
In the more severe types, aneurysms and rupture of large arteries may be found.
Pseudoxanthoma elasticum
Pseudoxanthoma elasticum describes a group of at least five disorders. The inheritance is autosomal recessive. The affected gene (chromosome 16) controls transmembrane peptide transport and results in calcified elastic fibres. The skin is loose, wrinkled and yellow and contains small papules (resembling xanthomas), giving a ‘chicken skin’ appearance. These changes are most obvious at the neck and flexures. Angioid streaks in the retina are seen in more than 50% of cases. Arterial involvement may result in gastrointestinal or cerebral haemorrhage.
The premature ageing syndromes
The features of ageing include an increased susceptibility to neoplasia, dementia, diabetes, autoimmune disease, cataracts, premature alopecia and hair greying, osteoporosis and degenerative vascular disease. Down syndrome shows several of these stigmata and is the most common condition in which premature ageing occurs. Many of the other disorders of premature ageing, such as Werner syndrome or progeria, are very rare and often show autosomal recessive inheritance.
Aged skin is dry, wrinkled, atrophic, shows loss of elasticity and uneven pigmentation and is susceptible to the development of benign and malignant tumours. Photoageing from chronic sun exposure (p. 106) can produce similar changes, although certain of the features are more prominent. Some conditions, such as pseudoxanthoma elasticum or xeroderma pigmentosum, have the signs of aged skin without necessarily showing more generalized features of ageing.
Neurocutaneous disorders and other syndromes
NF1 neurofibromatosis is a relatively common autosomal dominant condition characterized by café-au-lait spots, dermal neurofibromas and often skeletal or neurological anomalies. The abnormal gene is on chromosome 17.
Tuberous sclerosis complex is a not infrequent autosomal dominant disorder with prominent skin signs (e.g. facial angiofibromas and periungual fibromas), neurological problems (mental retardation and epilepsy) and ocular, cardiac and renal tumours.
Incontinentia pigmenti is a rare X-linked dominant disease, present at birth, which evolves through vesicular and warty stages to leave whorled patterns of pigmentation. Skeletal, ocular and neurological defects are associated.
Xeroderma pigmentosum represents a group of rare recessive conditions showing defects of DNA repair, characterized by skin tumours and premature death.
Ehlers–Danlos syndrome is a group of at least 10 inherited collagen disorders in which skin elasticity and scarring and joint hypermobility are found.
Pseudoxanthoma elasticum represents at least five disorders that result in calcification of elastin. The skin is wrinkled with yellowish papules. Retinal angioid streaks are seen.