Chapter 53 Immunologic Disorders
EQUINE IMMUNODEFICIENCY DISEASES
The environment in whichdomestic animals live with regard to exposure to pathogens is challenging and often life threatening. Complex defense mechanisms have developed to protect the host from outside challenges, particularly the pathogenic effects of microorganisms. It is important to understand that diseases, particularly infectious diseases, can result from failure of the host’s normal defense mechanisms, as well as from overwhelming challenge from the outside. When animals are plagued by repeated or chronic infections, the clinician should always determine whether host factors are involved.
The analysis of immunodeficiency diseases depends on an understanding of the normal immune response. The development of protective immunity is a result of the orchestration of numerous cell types and soluble serum factors (Fig. 53-1). Both innate (nonspecific) and adaptive (specific) mechanisms play a role.
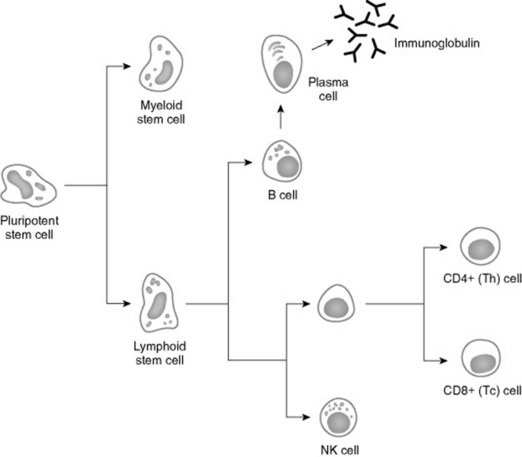
Fig. 53-1 Ontogeny of the immune response. Failures at any site in the maturation process can result in manifestation of immune deficiencies.
Two major populations of lymphocytes are involved in immune responses, T cells and B cells. Classically, T cells are associated with cell-mediated immune responses that protect against fungal, protozoal, intracellular bacterial, and many viral infections. B cells are associated with humoral immunity. T cells originate from stem cells, which probably develop in the fetal liver. These cells must undergo a maturation process in the thymus before becoming fully functional. T cells comprise about 70% to 80% of peripheral blood lymphocytes and populate the periarteriolar regions of the spleen and the paracortical regions of lymph nodes. As with T cells, B cells originate from stem cells in the fetal liver. The site of B-cell maturation varies with species and includes several different organs, such as the bursa of Fabricius in birds and the bone marrow and certain Peyer’s patches in mammals. Of peripheral blood lymphocytes, 15% to 30% are B cells. B cells populate germinal centers of spleen and lymph nodes.1,2
T lymphocytes are important in regulating the immune response, and both humoral and cellular immune responses depend on input from T cells. Initially characterized as either helper T (Th) cells or cytotoxic/suppressor T (Tc) cells based on their primary function, T cells have subsequently been differentiated on the basis of cell surface antigens, with Th cells expressing the CD4 antigen and Tc cells expressing CD8.3,4 Further work has shown that the function of these cells is complex, and that the pattern of cytokine expression is important in regulation of the immune response. Based on their cytokine expression, CD4+ cells have been further subdivided into distinct subsets, Th1 and Th2 cells (Fig. 53-2). Although there is some species variation, Th1 cells generally produce interferon-γ and interleukin-2 (IL-2) and are involved primarily in the generation of cell-mediated immune responses, whereas Th2 cells produce IL-4, IL-5, and IL-13 and are involved in humoral responses. In mice, either protection from disease or development of lesions can be associated with the particular type of Th cell response, and the clinical relevance of this is currently being investigated in a number of equine diseases.
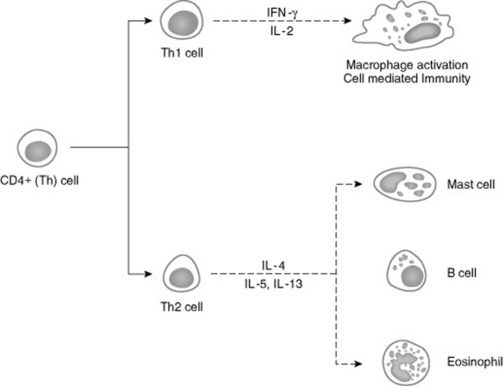
Fig. 53-2 CD4+ T cells can be subdivided into Th1 and Th2 on the basis of their cytokine profiles. Preliminary data suggest that the pathogenesis of certain diseases is associated with the particular type of Th response.
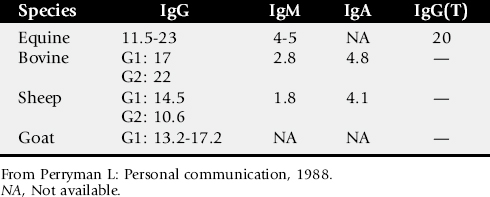
The second major class of lymphocytes is B lymphocytes, which produce immunoglobulins and are the precursors of plasma cells. Several classes of immunoglobulins are produced by B cells. There is some variation among species, but the major classes are IgG (IgG1 and IgG2), IgM, IgA, IgE, and in horses, IgG(T). Immunoglobulins provide a defense against extracellular bacterial and certain viral infections.
The innate immune system, which is nonspecific in nature, includes natural killer cells, phagocytic cells, neutrophils, eosinophils, basophils, and nonimmunoglobulin serum and cellular factors such as complement and interferon. These components play a distinct role in host defenses and work with T and B cells to produce an effective protective response.
A deficiency of functional T cells, B cells, nonspecific components, or any combination predisposes animals to infections that may result in death. Immune deficiencies can be classified according to (1) the site of defect in the host defense system and (2) whether the mechanism is primary or secondary.1,5-7 In a primary disorder there is an inherent abnormality in the immune system that has a proven or suspected genetic basis, whereas in a secondary disorder the host’s initially normal ability to respond immunologically is altered. Some factors that can produce secondary immunodeficiencies include irradiation, neoplasia, toxicities, malnutrition, and certain microbial infections.1,6,7 Physiologic stress, such as that caused by pregnancy, lactation, and exercise, can also induce transient immunosuppression.6,8 Both primary and secondary immunodeficiencies can affect various components of the immune system, and by careful dissection of the immune response, the site of the defect can often be identified.
General clinical features associated with immunodeficiencies include the following1,5-7:

Laboratory or special in vivo testing is necessary to confirm the presence of an immunodeficiency. Such testing is also important in differentiating the various immunodeficiency syndromes because clinically the presenting signs are nonspecific. In general, tests to evaluate the immune system either quantify the component or measure the functional capacity.
The enumeration of lymphocytes, and in some cases, specific lymphocyte types, can be useful in the diagnosis of immunodeficiencies. Currently, specific subsets of lymphocytes are most often identified using antibodies to cell surface markers and flow cytometry.3,4,9In normal horses, about 20% of circulating lymphocytes are B cells, and about 62% are CD4+ T cells and 18% CD8+ T cells.9,10 B cells can also be enumerated in blood and lymphoid tissue using fluorescent-labeled antibody to detect surface immunoglobulin and erythrocyte-antigen-complement rosetting techniques to detect complement receptors. Erythrocyte rosetting assays and fluoresceinated peanut agglutinin surface labeling tests have been used to enumerate T cells in peripheral blood and lymphoid tissue.
The primary clinical tests of B-cell function are quantitation of immunoglobulins and measurement of specific antibody responses. Numerous methods are available to quantitate or semiquantitate immunoglobulin levels. Semiquantitative tests are useful for some conditions, such as failure of passive transfer (FPT) after colostrum ingestion; however, they do not provide information on specific immunoglobulin classes. Some tests are species specific, whereas others can be used to detect immunoglobulins of several species. Precipitation of immunoglobulins with specific salt concentrations tends not to be species specific, although the tests may work better in some species than others. These tests include zinc sulfate and sodium sulfite precipitation and glutaraldehyde coagulation (see p. 1679).11,12 Commercial tests based on these principles are available.* Serum electrophoresis can also be used in all species to quantitate γ-globulins, which are primarily immunoglobulins. Several species-specific tests based on antigen-antibody reactions that use latex bead agglutination† or enzyme immunoassay† as the marker systems are also commercially available. These tests are semiquantitative and are primarily marketed for detection of FPT after colostrum ingestion.
Radial immunodiffusion (RID) quantitates immunoglobulins of specific classes using a precipitation reaction between antigen and antibody directed against one species-specific, class-specific immunoglobulin to be quantitated. This is an accurate method for quantitating specific classes of immunoglobulin such as IgG and IgM. RID kits are commercially available for some immunoglobulin classes for domestic animals.§ These tests require incubation for 18 to 24 hours, which is their greatest drawback for clinical use. Test reagents are prepared for use in a single species; however, some cross-reactivity does exist among species. Test reagents designed for use in another species (e.g., human) have been shown to be useful13; however, they must be standardized and calibrated for the species in which they are to be used.
Production of antibody in response to immunization with specific antigens is another way B-cell function can be evaluated, although functioning T cells are also required for this response. The only requirement to assess antibody production is an in vitro test to detect specific antibody. Serologic measurement of antibody titers before and after vaccination with commercially available vaccines is one approach. Killed infectious bovine rhinotracheitis (IBR) and bovine viral diarrhea (BVD) vaccines in cattle and influenza or rhinopneumonitis vaccines in horses are readily available, and responses are easily tested. Another approach is to look for the presence of naturally occurring antibodies that are produced without immunization (e.g., antibodies that cross-react with sheep red blood cells in horses), although the assays for these antibodies may not be readily available. Other foreign “nonvaccine” antigens can also be used if an assay is available for the detection of antibody.
One clinical in vivo test of T-cell function is intradermal skin testing with the plant lectin, phytohemagglutinin (PHA), which identifies delayed-type hypersensitivity (DTH) responses. PHA is capable of eliciting a DTH response without requirement of prior sensitization, which is required with some other antigens, such as dinitrochlorobenzene. To perform this test, the thickness of a skin fold is measured before injection. A 50-μg dose of PHA* in 0.5 ml of phosphate-buffered saline (PBS) is injected intradermally, and the same volume of PBS is injected at a control site at least 10 cm (4 inches) away from the site. Twenty-four hours later the skin thickness is measured. A 1-mm to 3-mm increase in skin thickness at the test site should normally occur. An increase of 0.6 mm or less indicates a defect in cell-mediated immunity.14
Other tests for B- and T-cell function are available primarily on a research basis. In vitro lymphocyte blastogenesis with pokeweed mitogen requires both B- and T-cell function for normal responses, whereas lipopolysaccharide requires predominantly a B-cell response. Blastogenesis with PHA and concanavalin A assess primarily T-cell function.
A variety of assays for phagocytosis and killing by neutrophils and macrophages have been developed.15-17 Recently, flow cytometric analyses of phagocytic function have been described, as well as methods for the quantitation of complement, interferon, and various lymphokines. However, these procedures are currently available only in selected research facilities.
From a practical standpoint, only a limited number of tests are available, and most are crude indicators of immune response and therefore detect only severe deviations from normal. Nevertheless, a number of immunodeficiency syndromes have been characterized in domestic animals. As methods improve, so will veterinarians’ ability to define immune disorders more precisely.
FAILURE OF PASSIVE TRANSFER
Definition and Etiology
Normal foals are immunocompetent at birth (i.e., they are capable of mounting an immune response). However, they are immunologically naive in that they have had no exposure to foreign antigens and have therefore not yet mounted any type of protective immune response or accumulated significant levels of immunoglobulins. Although foals are capable of producing antibody, they are essentially devoid of immunoglobulin at birth, with the exception of small amounts of IgM normally produced in utero. Because they are “starting from scratch,” foals are indeed more susceptible to infectious agents during the early neonatal period. Foals begin producing immunoglobulins immediately on exposure to antigens after birth, and immunoglobulins produced by the foal are detectable within 1 to 2 weeks of life and reach significant levels by 2 months.
Under normal circumstances, temporary protection against infection for the first 1 to 2 months is provided to the foal in the form of passively transferred antibody (Fig. 53-3). Because of the diffuse epitheliochorial nature of the equine placenta, no transplacental transfer of immunoglobulins occurs in horses. Instead, ingestion and absorption of immunoglobulin-rich colostrum are the sole means of passive transfer in foals. In a properly functioning system, maternal antibodies wane as levels of autologous antibodies increase, and thus the neonate is never left totally unprotected (Fig. 53-4).
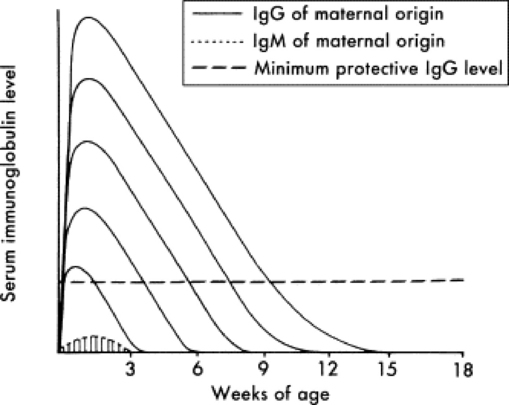
Fig. 53-3 The duration of protection provided by maternal immunoglobulins varies with the class of immunoglobulin and the quantity ingested during the first 24 hours of life.
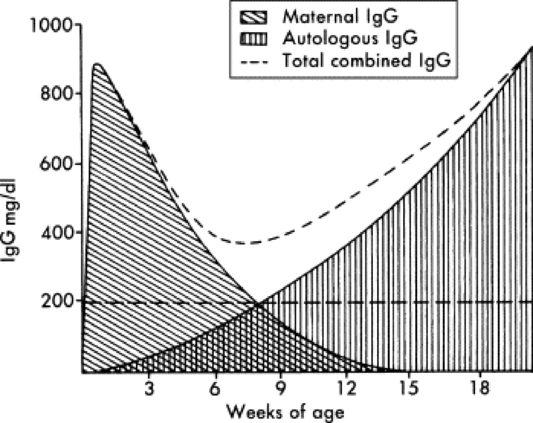
Fig. 53-4 Maternal immunoglobulin wanes, whereas the production of autologous immunoglobulin increases during the first several months of life. The combined total amount of immunoglobulin ideally remains above the level considered minimum for maintenance of good health.
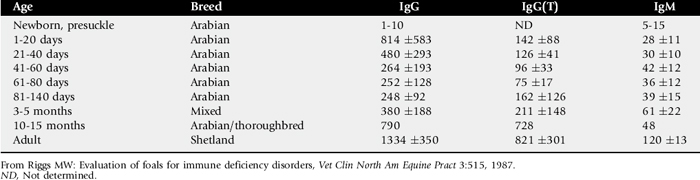
Failure of the foal to ingest or absorb sufficient quantities of colostrum, primarily as defined by absorption of IgG, is termed failure of passive transfer (FPT). Complete FPT is defined as a foal with a serum IgG concentration of less than 400 mg/dL at 24 hours of age. Partial FPT is defined as a foal with a serum IgG concentration of 400 to 800 mg/dL at 24 hours of age. The reported incidence of complete or partial FPT in foals varies from 3% to 37.8%.18-25
Immunoglobulins are not produced locally in the mammary gland, but rather are selectively concentrated from the mare’s sera into colostrum in response to hormonal changes that occur in the last 2 weeks of pregnancy. Most immunoglobulin in equine colostrum is IgG or IgG(T), with smaller quantities of IgM and IgA. At birth the neonate has specialized enterocytes in the gastrointestinal (GI) tract that are able to absorb large molecules such as immunoglobulins intact by pinocytosis. Absorbed proteins pass through the intercellular spaces and lacteals into the systemic circulation via the lymph. The window of gut absorptive capacity for immunoglobulins is narrow, lasting from birth until about 18 to 24 hours. Maximal absorptive efficiency occurs immediately after birth, declining to only 22% efficiency at 3 hours after birth and less than 1% by 20 hours.26,27 The decline in immunoglobulin absorption is accompanied by transient proteinuria that peaks at 6 to 12 hours of age and declines by 24 to 36 hours of age. This proteinuria most likely reflects absorption and excretion of low—molecular-weight milk proteins.28
Diminished immunoglobulin absorption over the first 12-24 hours of life is the result of shedding of specialized enterocytes capable of pinocytosis and replacement by more mature cells that are incapable of absorbing immunoglobulins.26,27,29 It has been hypothesized that delayed ingestion of macromolecules may prolong the duration of intestinal permeability to immunoglobulins. In one study, however, the type of fluid administered to foals before the ingestion of colostrum did not influence subsequent IgG absorption, suggesting that the process of gut closure is not mediated by a finite capacity for the uptake of macromolecules.30
The half-life for maternal antibodies in the foal’s circulation varies between 20 and 30 days.31-33 Concentrations decline as a result of normal protein catabolism, gradual dilution in an increasing plasma volume as the foal grows, and transfer of functional antibody into the GI tract. Most maternal antibodies are present in only negligible concentrations by 6 months of age, although antibodies to some infectious agents have been detected for up to 12 months after birth. As passive antibody concentrations decline, autogenous antibody production begins. There is a nadir in serum immunoglobulin concentrations in colostrum-fed foals at approximately 1 to 2 months of age, followed by gradually increasing concentrations until adult levels are reached at 5 to 10 months.2,34 Serum immunoglobulin concentrations are similar in colostrum-fed and colostrum-deprived foals by 3 to 4 months of age.
In addition to antibody, other colostral factors may be important for optimal immune protection of foals. For example, colostrum influences cell-mediated immunity and activates granulocytes. Colostrum contains many constituents of innate immunity and immunomodulating agents, such as complement, cytokines, and trace elements, that have a local protective effect in the neonatal digestive tract.35,36
In foals, FPT may occur because of ingestion of poor-quality colostrum with a low immunoglobulin content, failure to ingest a sufficient quantity of colostrum, or failure to absorb colostral immunoglobulins from the GI tract.29,37 Colostrum may have an insufficient quantity of immunoglobulin because of prelactation (lactation before parturition), premature foaling, a defect in the mare’s ability to concentrate immunoglobulin in the colostrum, ingestion of endophyte-contaminated fescue grass or hay, or other factors. Mares most likely to produce colostrum with low immunoglobulin content are those older than 15 years of age, those that foal early in the year, and standardbred mares.
Foals that are orphaned or rejected at birth, too weak to stand, or unable or lack the desire to suckle are unlikely to ingest sufficient colostrum to prevent FPT. Malabsorption is occasionally incriminated as a cause of FPT in foals that are observed to suckle adequate quantities of good-quality colostrum. This most often happens in premature or dysmature foals, possibly as a result of immature GI function, but may also occur in otherwise healthy and vigorous full-term foals.38 Glucocorticoids enhance the maturation of small-intestinal epithelial cells and thus their loss of absorptive capacity, leading to speculation that endogenous corticosteroids released secondary to stress at parturition may impair immunoglobulin absorption in foals.39,40 However, administration of adrenocorticotropic hormone (ACTH) failed to affect absorption in experimental foals, and stress has not been a consistent historic finding in foals with FPT caused by presumptive impaired immunoglobulin absorption.22,41
Clinical Signs and Differential Diagnoses
The association between FPT and infection has been investigated in numerous studies.18-20,24,25 Although the results have varied somewhat, FPT is generally considered a risk factor for infectious disease. By itself, FPT produces no clinical signs of disease and cannot be detected by physical examination. Clinical presentations that strongly suggest an underlying problem with FPT include onset of bacterial infections within the first 2 weeks of life, particularly septicemia, septic arthritis, pneumonia, and enteritis. Other immunodeficiencies or simply exposure to potent pathogens cannot be ruled out solely on the basis of the time of onset; however, even with other forms of immunodeficiency, clinical signs of infection usually do not show up for several weeks if passive transfer is adequate.
Clinical Pathology
FPT is diagnosed by the demonstration of low serum concentrations of IgG in the foal as early as 6 to 12 hours after birth and probably for as long as several weeks after birth (Table 53-1). The level of IgG considered adequate for protection against infectious disease is poorly defined and probably varies considerably with the environment. A serum IgG concentration greater than 800 mg/dL is considered adequate for most foals. However, levels of 400 mg/dL may be sufficient in healthy foals housed in clean environments. In contrast, an IgG concentration of 400 to 800 mg/dL in a foal at high risk for sepsis because of its environment or other factors indicates the need for treatment. Importantly, these figures only address total immunoglobulin content and not specific antibody titers, which also play a critical role in determining resistance to particular pathogens.
In healthy foals that nurse within 2 hours of birth, serum IgG concentrations become detectable at approximately 6 hours of age and peak at approximately 18 hours. Routine determination of serum IgG concentrations in apparently healthy foals is usually recommended at 18 to 24 hours of age. Foals considered to be at high risk for FPT and sepsis may be assessed as early as 6 to 12 hours of age.35,42
Methods available for IgG quantitation include single radial immunodiffusion, zinc sulfate turbidity, latex agglutination, glutaraldehyde coagulation, turbidometric immunoassay, and enzyme immunoassay.11,35,43-47 Single radial immunodiffusion is considered the most quantitatively accurate diagnostic test of those widely available to practitioners. However, this test is more expensive than some other screening tests, and results are not available for at least 24 hours, making it impractical when a critically ill foal needs rapid diagnosis and treatment. Total serum protein is not a reliable indicator of FPT in foals (unlike calves) because of the wide variation in total serum protein in cases of adequate transfer.
No consistent changes in the hemogram and biochemical panel are seen in foals with FPT; however, a range of abnormalities related to secondary infection (e.g., neutrophilia, neutropenia), hyperfibrinogenemia, and hypoglycemia may be present. The presence and severity of these changes depend on the organisms and systems involved.
Necropsy Findings
No specific necropsy findings are indicative of FPT. Necropsy findings reflect the site and severity of secondary infectious problems that have developed. Lymphoid tissue is normally developed, unless secondary infections have caused lymphoid necrosis or atrophy.
Treatment and Prognosis
Treatment of FPT depends on the degree of FPT, the environment in which the foal is exposed, the foal’s age at diagnosis, and the presence of secondary infectious problems. Treatment is aimed at minimizing exposure to pathogens, supplying immunoglobulins, and managing secondary infections, if present.
If FPT can be anticipated within hours of birth because of premature lactation, low—specific gravity colostrum, or a weak or orphaned foal, treatment can include the provision of an alternative source of colostrum or antibody orally. Foals with complete colostrum deprivation require approximately 1.5 g IgG/kg body weight to achieve a peak serum IgG concentration of more than 800 mg/dL. In a 45-kg foal, administration of 1 to 3 L of colostrum with a specific gravity greater than 1.060, divided into multiple hourly feedings over the first 6 to 8 hours of life, is desirable. Mares that donate colostrum for feeding should be healthy, checked for blood type, negative for anti—red blood cell (RBC) alloantibodies (especially anti-A and anti-Q) and appropriately vaccinated during the last 4 to 6 weeks of gestation.
If equine donor colostrum is not available, bovine colostrum, a commercial colostrum substitute, or equine plasma may be administered orally to the foal.48-55 Because bovine colostrum is often more readily available than equine colostrum, it may be substituted in emergency situations when equine colostrum is not available. Bovine colostrum is relatively well absorbed in the foal, but bovine immunoglobulins have a much shorter half-life in foals and do not contain antibodies specifically directed against equine pathogens. It is certainly better than no colostrum and, on the basis of a small experimental study, may be used without creating adverse reactions.48-50 Approximately 2 to 4 L should be administered orally; many foals develop transient mild diarrhea.
Lyophilized equine IgG* is available as an equine colostral substitute. A minimum of 50 to 70 g of IgG is recommended for treatment of the average 45-kg foal that receives no colostrum, but in one study this dose failed to increase serum IgG concentration to over 450 mg/dL in colostrum-deprived foals.55 A concentrated equine serum product† is also available for use in foals with FPT.56 Again, however, in one study it failed to increase serum IgG concentrations in colostrum-deprived foals to adequate levels, probably because of the relatively low total IgG dose administered.51 If the product contains 25 to 30 g of IgG per 300-mL bottle, approximately three bottles may be required to increase the serum IgG concentration of a 45-kg colostrum-deprived foal to greater than 400 mg/dL.51
If no other sources of immunoglobulin are available for a foal, oral administration of equine plasma or serum may be considered. This is an expensive source of oral immunoglobulin, however, and approximately 2 to 4 L are required to treat a colostrum-deprived 45-kg foal.
If the foal is over 6 hours old, the absorption of colostral antibody is significantly decreased, although a locally protective effect of the colostrum may still be present in the intestinal tract. If the foal is over 12 hours old, it is unlikely that sufficient colostrum will be absorbed; therefore, immunoglobulin levels should be rechecked at 24 hours and intravenous (IV) plasma transfusion given, if indicated by persistently low serum immunoglobulin levels.
Some animals with FPT, particularly partial FPT, do well without treatment if they are systemically healthy, are not heavily exposed to pathogens, and have no preexisting infections. FPT itself is not necessarily fatal. If plasma transfusions are not administered, owners should be made aware of the risks, and these foals should be maintained in an environment with minimal exposure to potential pathogens. Foals with other risk factors for septicemia (e.g., prematurity, dysmaturity, placentitis) should receive IV plasma transfusions if they have blood IgG concentrations of less than 800 mg/dL at 12 to 24 hours of age.
If the decision is made to supplement plasma parenterally, equine plasma for transfusions is commercially available† or can be collected and processed locally. Commercial sources are convenient, save time, have been screened for anti-RBC antibody, are free of diseases such as equine infectious anemia, and originate from animals with known immunoglobulin levels. The major disadvantage is that plasma may not contain antibody specific for the pathogens from the particular environment to which the foal is exposed.
Use of a local donor is desirable in that it presumably has antibody specific to the environmental pathogens to which the foal has been exposed. If a local donor is to be selected, several criteria should be met. First, the horse should be healthy, and results of agar gel immunodiffusion for equine infectious anemia should be negative. Second, no anti-RBC antibody should be detectable in the horse’s serum. The donor’s plasma should be screened for lysins and agglutinins by a blood-typing laboratory against a panel of cells representing all known blood groups. If plasma evaluated in this way is not available, the presence of anti-RBC antibody in donor plasma can be crudely evaluated with a minor crossmatch for agglutination using donor plasma and recipient blood cells. Lytic antibodies require an external source of complement for activity in vitro and may not be detected using this test.
Ideally, a third criterion is selection of a horse that is negative for blood group factors Aa and Qa. Even though there are dozens of blood group factors, Aa and Qa have been associated with the great majority of cases of neonatal isoerythrolysis (NI).57-59 If plasma is collected and separated by sedimentation, it is inevitable that some RBC contamination be present. If Aa-positive (Aa+) RBCs are given to an animal that is Aa negative (Aa−), or if Qa+ RBCs are given to an animal that is Qa−, the recipients could become sensitized to these antigens. This sensitization would probably not have any immediate consequences for the recipient foal, because the foal’s cells would not be affected by the antibodies, but it has potentially sensitized any Aa− and Qa− females for production of an NI foal later (see p. 1685). To avoid these potential complications, the ideal donor should be Aa− and Qa− and possess no anti-RBC antibody in its serum. It is desirable to have identified this type of donor to avoid the need for immediate crossmatching in every case of plasma transfusion.
The volume of plasma needed to correct the measured IgG deficit in a foal can theoretically be calculated on the basis of the blood volume of the foal and the concentration of IgG in the foal’s serum and in the donor plasma; however, these calculations do not reliably predict the actual levels of IgG achieved after transfusion.35,60 A 20-mL volume of plasma per kilogram of body weight administered intravenously routinely only raises serum IgG levels 200 to 300 mg/dL, and often two to three times this amount is needed to bring serum IgG levels into the range considered minimum for protection (e.g., 400 to 800 mg/dL).35,61 If the foal is already clinically ill, additional plasma will often be required to raise IgG levels an equivalent amount.35,44 In a newborn foal (estimated 50 kg) with complete FPT, between 2 and 4 L of plasma is frequently needed.
Plasma should be administered through an IV catheter placed aseptically in one jugular vein. Frozen plasma is thawed and warmed slowly to room temperature in a warm water bath. Microwave thawing or thawing with very high temperatures is not recommended because this may denature important plasma proteins. An appropriate in-line blood filter should be used for IV administration of any blood product to remove fibrin clumps and other debris. Initial infusion rates should be slow (0.5 mL/kg over 10 to 20 minutes) to monitor for adverse reactions. Muscle fasciculations, piloerection, increased heart or respiratory rate, fever, respiratory distress, abdominal pain, blanching of mucous membranes, and collapse are indicative of transfusion reactions. In the absence of these or other adverse effects, the remainder of the transfusion may be administered at rates up to 40 mL/kg/hr. Slower infusion rates are recommended for foals that are systemically ill. If other IV fluid therapy is being administered concurrently, slower infusion rates are also indicated to diminish the likelihood of inadvertent fluid overload.
Serum IgG concentrations in the foal should be rechecked 12 to 24 hours after plasma transfusion to confirm that the desired increase has been achieved. The delay from transfusion to IgG assessment is necessary to allow for distribution of immunoglobulin into extravascular spaces. Healthy foals transfused with plasma at 1 day of age experienced a 30% decrease in serum IgG concentrations by 7 days of age.62 This decline might be even more dramatic in septic foals with increased vascular permeability, increased catabolism, and increased demand for utilization in immune responses.
Although several equine serum-derived products are marketed for IV administration in the treatment of foals with FPT, these products have been associated with significant adverse reactions in some foals. Administration of high-quality equine plasma is preferred for treatment of foals with FPT.
Prevention and Control
Evaluation of colostral immunoglobulin content has proved to be valuable in predicting the occurrence of FPT and assessing the neonate’s risk for FPT.35,63 Colostrum with high immunoglobulin concentration tends to be sticky, yellow, and thick, but these subjective criteria are unreliable in assessing colostral quality. The quantity of immunoglobulin in colostrum may be more accurately estimated by single radial immunodiffusion (RID), refractometry, glutaraldehyde coagulation, or specific gravity.62 Because 18 hours are required to read the results of RID, it is more practical in a field situation to assess specific gravity with a refractometer, glutaraldehyde coagulation test or colostrometer. Sugar refractometry using a hand-held Brix 0-50% sugar refractometer is a simple and cost-efficient stall-side screening test for assessing colostral quality.64,65 A Brix reading of 20% to 30% correlates with adequate colostral quality; a reading greater than 30% indicates good-quality colostrum.66 A commercial kit based on glutaraldehyde coagulation of immunoglobulins* is available for screening of colostral quality. Using a colostrometer,† colostral specific gravity should be a minimum of 1.060, corresponding to an IgG concentration of greater than 3000 mg/dL; levels of 6000 mg/dL or higher are desirable. Approximately 75% of foals that ingest colostrum with a specific gravity less than 1.060 will have serum IgG concentrations under 400 mg/dL; when colostral specific gravity is greater than 1.060, foals usually attain serum IgG concentrations above 500 mg/dL.63
Some of the causes of FPT can be alleviated or recognized for early intervention by careful management. These include identification of mares that drip colostrum before parturition, attendance at foaling to ensure that foals suckle within several hours of birth or are supplemented artificially with colostrum, and screening of high-risk foals with doubtful nursing histories. Routine screening of foals at 18 to 24 hours of age allows early identification of FPT and potentially allows for therapy before the onset of infections. Although signs of septicemia secondary to FPT are often first observed on day 3 to 4 of life, a bacteremia may already be present at 24 hours of age or earlier.66,67
A colostrum bank can be established by collecting small amounts of colostrum from lactating mares (e.g., 200 to 250 mL) within the first 3 to 6 hours after foaling. This is only about 10% of the total colostrum produced by the average mare in the first 20 hours after parturition and therefore does not adversely affect the foal suckling the donor mare. Although the volumes are quite variable, mares produce about 300 mL of colostrum per hour and about 5 L during the first 18 hours. Colostrum can be stored frozen for at least 1 year at standard freezer temperatures, approximately −20° C (−4° F). Although frozen immunoglobulins are stable for much longer, the overall quality of the colostrum may deteriorate. Ideally, banked colostrum should be screened for the presence of anti-RBC antibodies as advised for plasma. Colostrum typically has low titers of agglutinins, which are probably not of significance unless present a dilutions of 1/8 or greater.
SEVERE COMBINED IMMUNODEFICIENCY
Definition and Etiology
Severe combined immunodeficiency (SCID) is a lethal, inherited condition in which both T-cell and B-cell function is absent.68 Affected foals have a stem cell defect that prevents maturation of T and B cells, resulting in a complete inability to produce antigen-specific immune responses. The condition primarily affects Arabians and part Arabians, although sporadic cases have been described in other breeds.68,69 In horses of Arabian breeding, the condition is transmitted as an autosomal recessive trait.70 Carriers of the gene are asymptomatic but can now be detected by genetic testing.71
Clinical Signs and Differential Diagnoses
Foals that are homozygous for the defective SCID gene are clinically affected. These foals generally appear physically normal at birth, but the absence of both specific humoral and cellular immune responses renders them susceptible to infections once colostral protection wanes.68 Affected foals typically develop infectious diseases between birth and 2 months of age and die before 5 months of age. The age of onset of infectious disease depends to some degree on the adequacy of passive transfer and the environmental challenge by organisms. The infections in affected animals are nonspecific and are caused by a variety of bacterial, viral, parasitic, and fungal agents, some of which rarely affect animals that are not immunocompromised.68,72,73 Many body systems may be involved, but pneumonia is a particularly common feature. Pneumocystis carinii pneumonia and adenoviral pneumonia are found often in SCID foals and rarely in other foals.
Clinical Pathology
A consistent finding on the hemogram is an absolute lymphopenia, which is consistently less than 1000 lymphocytes/μL and often much lower. The total white blood cell (WBC) count may be low, normal, or elevated, depending on the neutrophilic response; thus it is imperative that the absolute number of lymphocytes be determined. Infected and other compromised foals, as well as some normal foals, have low lymphocyte counts during the first few days of life; therefore, the clinician should establish persistent lymphopenia before considering a diagnosis of SCID.68,74
Foals affected with SCID are unable to produce immunoglobulins. Therefore, levels of autologous serum immunoglobulin for age are abnormally low. However, quantitative immunoglobulin tests do not distinguish between autologously produced and maternal-origin immunoglobulin. Thus the degree of colostral transfer and the age of the foal must be considered when interpreting the serum immunoglobulin values (see Table 53-1). Some IgM is normally produced in utero by the foal, and some IgM should be present in presuckle serum.75 Although not pathognomonic, the absence of IgM in presuckle serum is a feature of SCID. The presence of colostral antibody of maternal origin may mask low levels of autologous IgG and IgM, particularly early in life. The levels of maternal IgM in colostrum are lower than IgG, and the half-life of IgM is shorter (Table 53-2). Most maternal IgM received from colostrum is metabolized by about 3 weeks of age, whereas the age at which maternal IgG is gone is much more variable and may actually be months, depending on the original amount absorbed. The absence of serum IgM after 3 weeks of age is not pathognomonic but is consistent with SCID.
Foals affected with SCID do not respond to intradermal phytohemagglutinin (see p. 1667) by increasing skin thickness as do normal foals, nor do they respond to DTH stimulators, such as dinitrochlorobenzene.76 In vitro tests such as blastogenesis are depressed with all mitogens.77,78
The definitive diagnosis of SCID in foals of Arabian breeding is based on demonstrating that the foal is homozygous for the defective SCID gene.71,79 Blood or cheek swabs may be submitted to VetGen for DNA testing to determine if a horse is clear, heterozygous, or homozygous for the gene defect. Before the advent of genetic testing, the criteria required to confirm a diagnosis of SCID included (1) persistent lymphopenia (<1000/μL), (2) absence of serum IgM in presuckle samples or samples collected after 3 weeks of age, and (3) thymic hypoplasia and characteristic histopathologic changes in lymphoid tissue.80,81
Pathophysiology
Foals with SCID lack activity of the enzyme DNA-dependent protein kinase (DNA-PK) resulting from a mutation in the gene encoding the catalytic subunit.79,82,83 The mutation results in a five—base-pair deletion in the gene on equine chromosome 9. Without functional DNA-PK, lymphocyte precursors are unable to complete gene rearrangement events that lead to the expression of antigen-specific receptors on lymphocyte surfaces. As a result, there is an absence of mature, functional T and B lymphocytes. Interferon-γ (EqIFN-γ), which is produced by lymphocytes, is deficient.84
Neutrophils, monocytes, and natural killer (NK) cells appear to be fully functional.85,86 Complement levels are normal.78 Although these nonspecific protective mechanisms appear to be intact, the absence of both cell-mediated and antibody-mediated immunity leaves the foal vulnerable to even innocuous infectious agents.
Epidemiology
SCID has been reported in Arabians in the United States, Canada, Australia, and Great Britain.68,87,88 It is believed that the trait originated in a horse in Great Britain and was subsequently imported into the United States. The distribution of this disease provides a significant example of the founder effect in population genetics.
The prevalence of SCID at one time was estimated to be 2% to 3% of Arabian foals born in the United States. From this, it was predicted that the numbers of SCID carriers at that time could be as high as 25% of the U.S. Arabian horse population. A 1998 study of 250 randomly selected Arabian horses in the United States found the frequency of SCID gene carriers to be 8.4%.88 Based on this finding, it was predicted that 0.18% of Arabian foals would be affected with SCID, assuming a random breeding population.
Necropsy Findings
It is imperative to evaluate lymphoid tissues grossly and histologically in cases of suspected immunodeficiencies. In SCID the thymus is small and has a fatty appearance on gross examination.80 It is frequently difficult to locate, and mediastinal tissue often must be collected “blindly” for subsequent histologic evaluation and identification of thymic remnants. Histologically, the thymus is largely replaced with adipose tissue, with only islands of lymphoid cells and partially formed Hassall’s corpuscles present. The gross appearance of spleen and lymph nodes may not be dramatically abnormal; however, they have very abnormal microscopic appearances, including absence of germinal centers and periarteriolar lymphocytic sheaths in the spleen, as well as absence of germinal centers with scarcity of lymphocytes in other areas of lymph nodes.
Bronchopneumonia is a common finding in SCID. Infectious lesions in other systems are also frequently found, including colitis and hepatitis.
Treatment and Prognosis
Affected foals invariably die by about 5 months of age, despite intensive conventional therapy (e.g., antimicrobials, plasma, isolation).
There is no practical method of curing affected foals at this time. Successful bone marrow transplants have been performed between histocompatible full siblings in a research setting, but this option remains impractical.89
Prevention and Control
Production of an affected foal identifies both sire and dam as carriers of the SCID gene.70 Mating of two carriers of an autosomal recessive trait such as SCID is expected to result in one in four foals affected with SCID, one in four completely normal and not a carrier, and two in four asymptomatic carriers of the SCID trait. Mating of a carrier and a normal (noncarrier) does not produce any affected foals, but half the offspring would be expected to be carriers.
The disease can be controlled in horses of Arabian breeding by avoiding the production of affected foals, which has been simplified now that carriers can be identified by genetic testing. Mares and stallions intended for breeding should be tested to determine whether they are free or heterozygous for the defective SCID gene. Under no circumstances should two heterozygotes be mated to each other. If an owner decides to continue breeding a heterozygote, the breeding partner should be confirmed homozygous normal. All foals from such matings should be tested to determine if they are clear of or heterozygous for the defective gene (50/50 probability). Homozygous normal foals may be selected for future breeding purposes, whereas heterozygous foals should be managed for nonreproductive pursuits.
SELECTIVE IgM DEFICIENCY
Definition and Etiology
Selective IgM deficiency is an immunologic disorder characterized by absent or decreased serum IgM levels with normal or elevated levels of other immunoglobulin classes. Three presentations of the disorder have been described in horses.90-93 The most common involves foals that have severe infectious pneumonia, arthritis, or enteritis, resulting in death before 10 months of age. The second involves foals with a history of repeated episodes of infections that respond to antimicrobial therapy but that recur when treatment is stopped. These foals tend to do poorly and are stunted, although they may survive for 1 to 2 years. The third involves older horses that are usually 2 to 5 years of age at initial diagnosis. These horses do not necessarily have problems with recurrent infections, but about half ultimately have lymphosarcoma.94
It is not known whether the IgM deficiencies reported in foals are primary or secondary. The levels of IgM present before the onset of infections have not been measured. The occurrence of multiple cases within groups of related horses suggests a genetic basis. However, breeding trials have been inconclusive to date with regard to the heritability of this condition. In humans, both primary and secondary selective IgM deficiencies are known. Secondary cases are often associated with neoplasia, immunologic diseases such as Wiskott-Aldrich syndrome, and gluten-sensitive enteropathies.
Clinical Signs and Differential Diagnoses
Foals with selective IgM deficiency tend to have frequent Klebsiella infections of the respiratory tract, although enteritis and septic arthritis may also be complicating infections. The age of onset of clinical signs may be slightly later than in foals with SCID and certainly later than in foals with FPT.
Older horses with IgM deficiency should be carefully evaluated for lymphoid neoplasia, including palpation of peripheral and internal lymph nodes. Weight loss, depression, and other nonspecific signs frequently accompany lymphosarcoma (see Chapter 37).
Clinical Pathology
The only significant immunologic abnormality is a low or absent serum IgM. IgM levels are more than 2 standard deviations below the age-specific mean IgM level (see Table 53-1). Other classes of immunoglobulin are generally within normal limits or elevated for age. It is advisable to document that IgM levels are persistently depressed rather than to base a diagnosis on a single sample, because IgM levels may sporadically decrease in seriously ill foals but return to normal with recovery. Lymphocyte counts and other in vitro immunologic tests are generally normal, and these features serve to differentiate selective IgM deficiency from other immunodeficiency conditions. In one horse with selective IgM deficiency, however, lymphocytes failed to respond to the B-cell mitogen lipopolysaccharide (LPS), whereas response to the T-cell mitogens concanavalin A (ConA) and phytohemagglutinin (PHA) was normal.93 The hemogram and biochemical profile may reflect infection or inflammation, depending on the underlying infectious process (e.g., neutrophilia, hyperfibrinogenemia, anemia) and the organ system involved, but no diagnostically specific changes occur.
Pathophysiology
Whether IgM is low because of decreased production, hypercatabolism, or loss is not known. In one case with lymphosarcoma, suppressor activity was identified in the neoplastic cells, suggesting that the low IgM may be a result of suppression of B-cell function by neoplastic cells. Selective IgM deficiency has been most frequently reported in Arabians and quarter horses, although it affects other breeds as well.
Necropsy Findings
Lymphoid tissue in affected foals is grossly and histologically normal. Pneumonia is usually present and is frequently caused by Klebsiella infection. Other findings reflect secondary infections.
Treatment and Prognosis
Selective IgM deficiency has an unfavorable prognosis. Most horses eventually succumb to infection despite appropriate antimicrobial therapy. Plasma therapy may provide short-term benefit, but only small amounts of IgM are contained in transfused whole plasma, and the half-life of transfused IgM is probably quite short. No concentrated IgM preparations are commercially available for parenteral use. Relief, at best, can be considered only temporary. However, the outcome of these cases seems less certain than with SCID foals, and a rare case recovers. If persistently low IgM levels can be documented, a poor to grave prognosis can be expected.
TRANSIENT HYPOGAMMAGLOBULINEMIA
Definition and Etiology
Transient hypogammaglobulinemia is a rare disorder characterized by the delayed onset of immunoglobulin synthesis by the neonate.5,95 Foals normally begin to produce significant amounts of immunoglobulins at birth, when they are first exposed to environmental antigens (see Fig. 53-4). Assuming adequate passive transfer, maternal antibody fills the void from birth until the foal has produced sufficient autologous antibody for protection (e.g., first 1 to 2 months). In transient hypogammaglobulinemia the onset of autologous production is delayed for unknown reasons and may not begin for as long as 3 months. As maternal immunoglobulins wane, for a time the production of autologous antibody is insufficient to be protective, making the foal highly susceptible to infections.
The disorder has been described in an Arabian and a thoroughbred foal, but the low number of reported cases may not accurately reflect the prevalence. To make a diagnosis, serial samples are required to document decreasing maternal immunoglobulin and subsequent increasing autologous immunoglobulin. Many cases probably occur that are not followed this closely.
Clinical Signs and Differential Diagnoses
As with other immunodeficiency disorders, recurrent episodes of bacterial and viral infections are characteristic of transient hypogammaglobulinemia.
Clinical Pathology
The cardinal feature is low immunoglobulin levels.5,23,95 At approximately 2 to 4 months of age, the concentrations of all classes of immunoglobulin, particularly IgG and IgG(T), are substantially below the means when age-matched. At this stage, maternal antibody will have waned, but the foal should have produced significant quantities of autologous antibody, regardless of whether passive transfer occurred. Affected foals have normal cell-mediated responses in vitro and in vivo and appear to be able to respond to immunization with some antigens. Normal numbers of B cells are present in blood and lymph nodes.
Differentiation between FPT and transient hypogammaglobulinemia is based on the age of the patient at evaluation. Transient hypogammaglobulinemia must be differentiated from agammaglobulinemia. Both disorders have normal lymphocyte counts and responses to PHA skin testing and show waning levels of maternal IgG and IgG(T) if followed serially. However, transient hypogammaglobulinemia cases usually have low but detectable levels of IgM and IgA, whereas agammaglobulinemia cases usually have no detectable levels. Differentiation between the two may require serial sampling to show that immunoglobulin production does ultimately increase in the patient with transient hypogammaglobulinemia.
Necropsy Findings
No specific gross or microscopic lymphoid changes have been associated with transient hypogammaglobulinemia. Necropsy findings reflect the secondary infectious processes that have affected the foal.
Treatment and Prognosis
The goal of therapy is to minimize infections until the foal’s immune system begins to function properly. Treatment should include antimicrobial therapy and plasma transfusions. Bacterial infections may be manageable with antibiotics alone, but the effects of viral infections may be more difficult to manage and may be helped by plasma transfusions.
AGAMMAGLOBULINEMIA
Definition and Etiology
Agammaglobulinemia is a rare primary immunodeficiency disorder of horses. It is characterized by complete B-cell dysfunction with an intact cell-mediated response. This condition has been observed only in males, suggesting the possibility of an X-linked disorder. It has been described in thoroughbreds, quarter horses, and standardbreds.23,96-98 Agammaglobulinemia in a young boy was the first immunodeficiency disorder described in any species, and in human patients it is now known to be inherited as an X-linked trait.99
Horses with agammaglobulinemia illustrate the significant contribution of T and B lymphocytes to the maintenance of good health. Horses with defects in both B-cell and T-cell functions (e.g., SCID) seldom survive to 5 months of age, whereas the cases with a pure B-cell defect such as agammaglobulinemia have survived between 1 and 2 years. Recently, agammaglobulinemia with a lack of circulating B cells was diagnosed in a Pinto gelding that did not exhibit recurrent pyogenic infections until 3 years of age. Although it could not be established whether the immunodeficiency was primary or secondary, an underlying disease process was not identified.*
Clinical Signs and Differential Diagnoses
No outward physical signs suggest agammaglobulinemia other than the opportunistic infections that develop. Frequently, recurrent infections of the respiratory tract or joints with extracellular bacteria have been reported. Dermatitis, enteritis, and laminitis have also been associated with this disease.
Clinical Pathology
Total peripheral blood lymphocyte counts are within the normal range. However, there is a lack of circulating B cells with normal numbers of T cells. Neutrophil counts may be normal, low, or high, depending on the response to infection. No specific changes are noted in the biochemical profile. Serum levels of IgM, IgA, IgG(T), and IgG are persistently low or absent. Depending on the age at evaluation, maternal antibody may be present, but its decline is evident if sampled serially. The continued presence of low levels of immunoglobulin in these foals may be explained by the prolonged catabolism of antibody or by some residual B-cell activity, as in human patients with sex-linked agammaglobulinemia.23,96-99
Specific antibody responses are depressed, both for antigens to which horses naturally tend to produce antibodies, such as sheep RBCs, and for antigens administered by planned immunization, such as vaccines. Cell-mediated tests are essentially normal, including blastogenesis and DTH skin testing. Total hemolytic complement activity is also normal.
Pathophysiology
The molecular basis of agammaglobulinemia in horses is unknown, but a defect at the stem cell level that blocks early B-cell differentiation is suspected because all classes of immunoglobulin are affected. In human patients a mutation in BTK, a gene encoding tyrosine kinase, accounts for the disease. Assessment of the BTK gene in horses may help in understanding this disorder.
Necropsy Findings
Gross lymphoid changes in lymph nodes, spleen, and thymus have been observed. Lymph nodes are small. The thymus may be small and difficult to locate. Lymphoid tissue taken from the mediastinal region where the thymus should be found lacks the defined lobular structure of normal thymus. The spleen grossly may be small and contracted. Microscopically, lymph nodes are devoid of germinal centers and follicles. The spleen has no germinal centers, periarteriolar lymphocytic sheaths, or plasma cells.
The thymus does not show recognizable epithelial structure and lacks defined nodules.23,98 Why cell-mediated functions are normal while the architecture of the thymus is so abnormal remains unexplained.
FELL PONY SYNDROME: ANEMIA, IMMUNODEFICIENCY, AND PERIPHERAL GANGLIONOPATHY
Definition and Etiology
A congenital fatal syndrome characterized primarily by severe anemia and immunodeficiency has been identified in Fell Pony foals.100 In addition, peripheral ganglionopathy has been reported in some cases. Both genders are affected. The exact nature of the defect is as yet unknown, but an intrinsic genetic disorder transmitted by a single autosomal recessive gene is suspected.101 The Fell Pony is considered an endangered breed by the Rare Breeds Survival Trust, with an estimated 5000 animals worldwide. Originally described in Fell Ponies in the United Kingdom, Fell Pony syndrome has also been found in the Netherlands and North America.100-102
Clinical Signs
Affected Fell Pony foals typically develop signs of decreased suckling, diarrhea, cough, and chewing motions beginning at 2 to 3 weeks of age.100-102 The foals progressively lose condition and develop pale mucous membranes. The condition is generally fatal by 4 to 12 weeks of age. Opportunistic infections such as cryptosporidial enteritis, adenoviral pancreatitis, and adenoviral bronchopneumonia are frequently observed in affected foals and suggest an underlying immunodeficiency.
Clinical Pathology
Foals develop severe normocytic to macrocytic anemia associated with small numbers of erythroid precursor cells in the bone marrow.100-102 Myeloid/erythroid ratios in the bone marrow have ranged from 21:1 to 62:1. Although total circulating lymphocyte counts are variable, lymphopenia is described in some cases. Numbers of both CD4+ and CD8+ T lymphocytes are normal, but numbers of B cells are decreased.103,104 Consistent with this B-cell lymphopenia, serum immunoglobulin concentrations are often decreased once concentrations of maternal antibodies have declined.102-104 Because maternally derived concentrations of IgM and IgA generally do not persist as long as IgG, concentrations of these antibodies are more likely to reflect production by the foal. The responses of lymphocytes to in vitro stimulation with mitogens have been variable.105
At this time, no definitive test is available for the diagnosis of Fell Pony syndrome, and diagnosis is based on the signalment, history, clinical signs, and laboratory findings, including anemia, B-cell lymphopenia, and low concentrations of IgM after 4 weeks of age.
Pathophysiology
The precise nature of the immune defect is unknown, and thus far the characteristics identified do not conform to any known immunodeficiencies in other species. In addition to the decrease in circulating B cells, immunohistochemical staining reveals decreased B cells in the bone marrow, lymph nodes, and primary follicles of the spleen. Although analyses of cellular immunity and phagocytic activity have not revealed any consistent abnormalities, the severity of disease has led to speculation that multiple arms of the immune system are affected. The anemia is associated with severe erythroid hypoplasia of the bone marrow.
Necropsy Findings
The absence of secondary lymphoid follicles and lack of plasma cells on histologic examination is characteristic of the condition. There is also marked erythroid hypoplasia in the bone marrow. In some cases, peripheral ganglionopathy is seen, characterized by neuronal chromatolysis and nuclear pyknosis of the trigeminal, cranial mesenteric, or dorsal root ganglia. Other common necropsy findings include abnormalities associated with infections characteristic of immunodeficiency, particularly infections with Cryptosporidium parvum and adenoviral infections of the pancreas and bronchial tree.
COMMON VARIABLE IMMUNODEFICIENCY
Definition and Etiology
Common variable immunodeficiency (CVID), a heterologous syndrome of immunodeficiency, has been described in four horses.106,107 It has also been suspected in additional horses with varying immunologic deficits.108-110 CVID was initially defined in human patients, and although the syndrome is highly variable, recurrent bacterial infections and hypogammaglobulinemia are common characteristics.111,112 CVID has also been reported in miniature dachshunds with Pneumocystis carinii pneumonia.113
Clinical Findings
Information on CVID in horses is limited because of the small number of cases. The syndrome has been identified in various breeds and both genders and is characterized by a late onset, with horses ranging in age from 6 to 14 years.106,107 In human patients the disorder affects both men and women and can develop at any age, although the onset is most frequently seen during the second or third decade of life.111,112
Recurrent bacterial infection is common in CVID. Three horses with CVID diagnosed with presumptive bacterial meningitis were successfully managed medically with antibiotic therapy without immunoglobulin replacement therapy.107 Another horse with CVID presented with infection of the guttural pouch and cholangiohepatitis and was ultimately euthanized because of deterioration in the horse’s condition.106 The clinical spectrum of CVID in human patients is broad, and as expected, clinical disease is more severe in patients with more marked immunologic abnormalities.111,112
Immunologic Findings
Common features of CVID in affected people include recurrent bacterial infections and agammaglobulinemia or hypogammaglobulinemia, particularly involving IgM and IgG.111,112 The humoral response to vaccination is generally impaired. B-cell maturation may be arrested at various stages, and B-cell numbers may be normal or decreased. T-cell abnormalities may also be present. As in human patients, immunologic abnormalities identified in affected horses include hypogammaglobulinemia and failure to respond to immunization.106,107 The type and severity of the hypogammaglobulinemia vary; IgM deficiency is common. Affected horses have an abnormal lymphocyte distribution characterized primarily by B-cell lymphopenia. The lymphocyte response to mitogens varies but generally is decreased. Phagocytosis, oxidative burst activity, and serum opsonization capacity were normal in three horses tested.
The variable immunologic abnormalities associated with CVID make it difficult to define cases, and it has been suggested that additional cases of recurrent bacterial infections and immune abnormalities may represent variations of CVID. An adult Paso Fino mare with proliferative interstitial pneumonia and Pneumocystis carinii infection was found to have a complete lack of IgM, low concentrations of IgG, and decreased numbers of immune cells expressing major histocompatibility complex (MHC) class II cell surface antigens.108 In another case, a 3-year-old quarter horse with chronic diarrhea and bacterial pneumonia was diagnosed with an acquired B-lymphocyte deficiency associated with deficiencies in serum IgG, IgA, and IgM and a concurrent decrease in T-cell function.109 Additional cases with hypogammaglobulinemia and other immunologic abnormalities may fit the description of CVID.93,110,114
UNCLASSIFIED AND SECONDARY IMMUNODEFICIENCIES
Many equine patients with evidence of immunologic defects cannot be placed in a currently recognized immunodeficiency category. These animals form a diverse group that may seem to have little in common except the propensity to develop infections. Attempts to further classify these cases have been hampered by the lack of specific, practical tests that precisely define the immunologic defect. As newer testing methods are developed, this group will eventually be defined. Numerous immunodeficiency syndromes are described in humans, the counterparts of which can be reasonably assumed to exist in horses. As previously discussed, some of the previously unclassified immunodeficiencies characterized primarily by hypogammaglobulinemia are now suspected to be a form of CVID.93,107-110,114 Also, a CD4+ and CD8+ T-lymphocytopenia has been described in a filly with Pneumocystis carinii pneumonia.115
Numerous endogenous and exogenous factors can cause secondary immunodeficiency or suppression.7 These include malnutrition, specific nutrient deficiencies or excesses, microbial and parasitic agents, corticosteroids and other hormones or drugs, and neoplasia. In addition, factors such as age and pregnancy can influence the immune system. Stress, such as that associated with exercise or transport, may also have immunomodulatory effects. To complicate the issue, many of these factors can induce either suppression or stimulation under appropriate circumstances; thus their presence alone is not adequate to confirm immune compromise. In general, the effects of these factors on the immune system can be assessed by the same tests used to classify primary immunodeficiencies. Distinguishing the role of these factors from more subtle types of primary immunodeficiency is difficult.
Adult Acquired Immunodeficiency
A 7-year-old Appaloosa gelding with no history of prior illness became lethargic, anorexic, and dyspneic.114Rhodococcus equi septicemia was diagnosed on the basis of blood cultures. Immunologic evaluation of the patient revealed lymphopenia, low IgM and IgA concentrations, marginally low IgG concentrations, low R. equi antibody titer, negligible response to lymphocyte stimulation, histologic depletion of the lymphoid tissue, and failure to respond to antigenic stimulation. Marked thrombocytopenia was also present. These abnormalities suggested suppression of both humoral and cell-mediated arms of the immune system. The age of the horse, absence of previous history of illness, and histopathologic findings suggestive of atrophy of lymphoid tissue indicated the immunodeficiency was acquired. No underlying cause was identified for the immunodeficiency; specifically, no neoplasms were identified at necropsy, and there was no history of exposure to immunosuppressive toxins. This may represent CVID.
Infectious Disease
Secondary immune suppression can result from a variety of infectious or inflammatory diseases. Many viral, fungal, and bacterial infections transiently suppress specific and nonspecific immune responses, predisposing to secondary bacterial infections.116 Severe endotoxemia or septicemia may suppress neutrophil numbers and bactericidal function.
Perinatal Equine Herpesvirus Type 1 Infection
Infection of the fetus with equine herpesvirus type 1 (EHV-1, rhinopneumonitis) late in gestation has been associated with postnatal development of interstitial pneumonia, lymphopenia, marked necrosis and atrophy of the thymus and splenic lymphoid tissue, and increased susceptibility to bacterial infections.117 Affected foals may be either weak or normal at birth. Despite apparently adequate passive transfer of maternal antibody, affected foals contract a variety of infectious bacterial diseases, including colibacillosis, streptococcal septicemia, salmonellosis, and Tyzzer’s disease. EHV-1 is isolated from nasal passages of about 30% of the cases. The spleen and thymus are grossly small at necropsy. Bilateral adrenocortical hyperplasia is noted in most foals. Histologically, splenic periarteriolar lymphocytic sheaths are depleted of lymphocytes, and no lymphoid follicles are detectable. Thymic alterations vary from extreme diminutions of thymocyte numbers to complete necrosis of thymic lymphocytes with disappearance of Hassall’s corpuscles and disruption of the epithelial matrix. Lymph nodes also show lymphoid necrosis.
An immunodeficiency secondary to the marked lymphoid damage induced by the virus is credited with allowing secondary bacterial infections to become established. Immunization of broodmares against EHV-1 would seem to be the most appropriate approach to prevention.
Immunodeficiency Associated with Oral Candidiasis and Bacterial Septicemia
Definition and Etiology
A group of foals with laboratory or histologic evidence of immunodeficiency that did not fit into any recognized category of primary immunodeficiency have been described with oral candidiasis and bacterial septicemia.118 The foals shared no consistent pattern of in vitro immunologic abnormalities, but all had the clinical features of oral candidiasis and bacterial septicemia. This syndrome has some similarities to mucocutaneous candidiasis in people, which has been associated with several different subtle T-cell defects. Not all human cases are thought to result from a single immunodeficiency disorder. Candidiasis does not occur in the presence of B-cell defects alone, or in the absence of T-cell defects.
Clinical Signs
Most affected foals with oral candidiasis were about 4 months of age at onset, although several were less than 2 weeks old.118 Oral lesions ranged from discrete, focal, white, plaquelike lesions on the margins of the tongue to a generalized, thick, white, pseudomembranous coating covering the tongue and gingival mucosa. Most foals exhibited bruxism, ptyalism, fever, and depression. Other significant clinical problems included pneumonia, septic arthritis, and diarrhea. This syndrome should be distinguished from the glossitis caused by Candida infection seen in inappetent or debilitated neonatal foals, which is fairly common and generally resolves with amelioration of the primary disease.119
Clinical Pathology
Serum IgG and IgM levels were variable, but most of the older foals also had low or marginal IgG or IgM for their age.118 The younger foals all had FPT and low IgG levels. Some horses showed a transient lymphopenia, but lymphocyte counts were generally within normal limits. Almost all cases had other abnormalities, including depressed blastogenesis, suggesting a cellular defect distinct from previously described immunodeficiency cases that involved only immunoglobulin production.
Necropsy Findings
Thymic tissue was difficult to locate grossly at necropsy.118 Mediastinal tissue collected in the thymic region was confirmed to be thymus microscopically in only one of six cases. Histologically splenic lymphoid depletion was present. Evidence of disseminated bacterial infections included pulmonary abscesses, enteritis, septic arthritis, and focal hepatitis. Candida species were identified in tissues histologically or with culture in all cases. A variety of bacterial organisms were associated with the septicemias and secondary infections. Organisms usually considered minor pathogens, such as Acinetobacter and adenovirus, were identified in some cases.
Treatment and Prognosis
Affected foals did not respond to extensive parenteral antibiotic, topical antimycotic, or plasma transfusion therapy. Because antibiotic administration can predispose animals to secondary candidiasis, the role that antibiotic therapy may have played in the development of the oral lesions was considered. In light of the high frequency of antibiotic use in foals in the general population and the uncommon occurrence of oral candidiasis, additional factors such as an underlying immune defect are deemed likely. Whatever the cause, the prognosis for foals with oral candidiasis and associated bacterial septicemia is guarded to poor.
Neoplasia
Neoplastic disease can impair cell-mediated and humoral immune responses as a result of an abnormal bone marrow environment, altered patterns of cytokine production or release, or impaired proliferative responses (anergy). In horses, immunodeficiency has most often been described in association with lymphosarcoma or plasma cell myeloma.
LYMPHOSARCOMA
Immunodeficiency has been identified in some cases of equine lymphosarcoma. The immunologic abnormality most often described is a decrease in the concentrations of serum immunoglobulins, especially IgM.94,120-123 In addition, decreased lymphocyte blastogenesis in response to mitogens has been reported. Some horses with lymphosarcoma have been diagnosed with concurrent bacterial infections, which may be related to immunosuppression.122,123 A horse with myelomonocytic leukemia was found to have pulmonary aspergillosis.124
PLASMA CELL MYELOMA
Plasma cell myelomas have been diagnosed in horses of several breeds.125 Although reports of the condition are limited, there appears to be no gender predilection. Horses have ranged in age from 3 months to 22 years (median, 11 years) at diagnosis. Common clinical signs include weight loss, anorexia, fever, pneumonia, and limb edema. As a malignancy of plasma cells or lymphocytoid plasma cells, plasma cell myelomas typically produce large quantities of a homogenous immunoglobulin or immunoglobulin fragment, resulting in a monoclonal gammopathy. In both equine and human cases of myeloma, the predominant serum globulins are generally subclasses of IgG.125,126 Hyperglobulinemia is a characteristic but not invariable finding. Monoclonal gammopathies in the horse have also been reported with lymphoma and benign disorders.127,128 Both equine and human patients with plasma cell myelomas have an increased susceptibility to bacterial infections, probably as a result of a secondary immunodeficiency.125,126,129 The concentrations of normal polyclonal immunoglobulins are generally decreased in myeloma patients as a result of several mechanisms, including decreased synthesis and accelerated catabolism of antibody and suppressed clonal expansion of B cells.125,126,129 Decreased numbers of neutrophils, which may also be dysfunctional, and defective complement activation may also contribute to the impaired immune function.
Corticosteroid-Induced Immunosuppression
Increased concentrations of corticosteroids can result in varying degrees of immunosuppression. The increase in corticosteroids may be associated with stress or disease, most often pituitary pars intermedia dysfunction. In addition, corticosteroid treatment is the most common iatrogenic cause of immunosuppression. Increased concentrations of corticosteroids may exacerbate preexisting infectious diseases or decrease resistance to environmental pathogens.130,131
Corticosteroids have a number of effects on the immune system, many of which are dose dependent. They may suppress macrophage phagocytic function by impairing the killing of ingested microorganisms, decreasing secretion of monokines, and inhibiting antigen processing and presentation. Corticosteroids suppress cell-mediated immunity through induction of a T-cell lymphopenia, suppression of proliferation in response to mitogen stimulation, altered cytokine production, and decreased antigen presentation. Humoral immunity may be affected through impaired T-cell responses, enhanced catabolism of immunoglobulins, and decreased antigen presentation.132,133
Exercise
Exercise can clearly act as a stressor that may significantly alter the immune response.134,135 However, defining the precise effects of exercise on the equine immune system and susceptibility to disease has been difficult because of the complexity of the immune system, host factors (e.g., age, level of fitness), and the variable nature of exercise.134-136 In general, it appears that exercise may have both positive and negative effects on the immune response.134-140 Suppressive effects, such as a decline in the ratio of CD4+ to CD8+ T cells, decreased lymphoproliferative responses, and suppression of the innate immune system, have been associated with strenuous high-intensity exercise, prolonged exhaustive exercise, or overtraining. In contrast, moderate exercise tends to have beneficial effects on the host defense mechanisms.
Data in horses directly linking exercise-induced immunosuppression and increased susceptibility to infectious disease are limited. However, the potential immunosuppressive effects of exercise need to be recognized. In one study, unconditioned ponies vaccinated with a killed influenza vaccine and subjected to 5 days of strenuous exercise had an increased susceptibility to clinical influenza after challenge exposure compared with rested ponies.141 However, ponies with exercise-induced immunosuppression responded to the administration of an intranasal modified-live equine influenza vaccine and were protected from challenge.142 In a study of influenza infection in trained horses, moderate exercise led to increased signs of clinical disease, but the duration of disease was unaffected.143
Age
Age has been shown to affect immune function in multiple species. The increased vulnerability of foals to respiratory tract infections, especially with specific pathogens such as Rhodococcus equi, is thought to reflect an innate immunodeficiency.144 Although foals are immunocompetent at birth, it is likely that their immune responses differ from those of adults.144-146 Recently it has been shown that newborn foals have a diminished ability to express the interferon-gamma (IFN-γ) gene and produce IFN-γ protein.146 This ability increases steadily, reaching adult levels with the first year of life. These findings suggest that foals have an inherent inability to mount a Th1 immune response, which may contribute to their susceptibility to intracellular pathogens.
Aging may also be associated with a relative immunodeficiency. Data in horses are limited, but older horses have been shown to have lower proliferative responses to mitogens and antibody titers than younger horses.136-147 However, older horses were more resistant to exercise-induced immune suppression than younger horses. The high prevalence of pituitary pars intermedia dysfunction in older horses is thought to be a risk factor for infectious disease, possibly because of elevated steroid concentrations.
FAILURE OF PASSIVE TRANSFER
Definition and Etiology
At birth, ruminants leave the sterile uterus and are exposed to an environment laden with pathogens. Although capable of mounting a measurable immune response at birth or even earlier, neonates are best characterized as being “immunonaive.” A neonate’s ability to mount a protective immune response is hindered by the immaturity of the immune system and the delay between initiation of response and effective protection. Unless adequate maternal immunologic assistance is provided, neonates have an increased likelihood of succumbing to infectious diseases such as septicemia, diarrhea, enteritis, omphalitis, arthritis, and respiratory conditions.
Neonatal ruminants are protected against disease from infectious agents through passive transfer of maternal immunity by consumption of colostrum. The concept of failure of passive transfer (FPT) has largely been used to describe a neonate that does not absorb adequate levels of immunoglobulin. Immunoglobulins are a significant component of colostrum and have been the most studied constituent of colostrum. However, colostrum is a complex fluid that, in addition to immunoglobulins, contains high numbers of immune cells, immunoactive substances such as cytokines, and nutritional elements.148
Successful passive transfer of immunity can be defined as the timely ingestion and absorption of an adequate mass of colostral immunoglobulins (primarily IgG1 in calves), and possibly other colostral components as well. Successful passive transfer has been quantitatively defined as calves that have attained greater than 10 mg/mL serum IgG1 by 48 hours of age.149 The levels of serum immunoglobulins defining successful passive transfer are simply guidelines derived from statistical analysis of large populations. Numerous factors interact with the level of passively acquired immunoglobulin to determine the occurrence of disease, including management, environment, hygiene, infection pressure, virulence of infectious organisms, and antibody specificity. Although neonates with FPT are at increased risk for disease, having low serum immunoglobulin does not necessarily guarantee disease if the neonate resides in a clean environment or does not interact with highly virulent organisms. Alternatively, neonates with adequate passive transfer can readily develop disease if exposed to an environment with a high pathogen load or highly virulent organisms.
Mechanism of Passive Transfer
Two distinct processes must occur for adequate passive transfer of maternal immunoglobulins through colostrum. The first process, colostrogenesis, is the transport of immunoglobulin from maternal serum into colostrum during the last 4 to 6 weeks of gestation and is largely controlled by lactogenic hormones.150-152 Studies in cattle have demonstrated that the mechanism of immunoglobulin transfer involves an active, IgG1-specific, receptor-mediated process.153
The second process involves the transport of colostral IgG1 from the gut lumen into the neonate’s system. This process is not selective for specific immunoglobulin isotypes and does not appear to differentiate between most macromolecules. Specific receptors for immunoglobulins are absent in the calf’s gut; rather, transport occurs through nonselective pinocytosis. Absorption is initiated by the presence of macromolecules but declines over the first 24 hours of life154; absorption is finite and decreases regardless of macromolecules being present.155,156 The absorptive process also appears to be saturable; efficiency of immunoglobulin absorption decreases with increasing immunoglobulin concentration in the colostrum being fed.157 Presumably, because all macromolecules compete for the same absorptive process, the mass of immunoglobulin absorbed by the calf would be inversely proportional to the concentration of nonimmunoglobulin macromolecules present in the gut lumen.
Epidemiology
The percentage in calves with FPT at 24 hours of age varies from 15% to 68%.158 Similar findings have been recorded in other neonatal ruminants. In one report comparing FPT in beef calves and diary calves, rates of 5% and 39% were reported, respectively.159 Several interrelated factors account for the varying incidence of FPT, including the formation of colostrum with an adequate immunoglobulin concentration, ingestion of an adequate mass of immunoglobulin, and absorption of immunoglobulin in a timely manner.
Formation of Colostrum with Adequate IgG1 Concentration
The mass of immunoglobulin presented to the neonate for absorption depends on the colostral immunoglobulin concentration and volume of colostrum available. The volume of colostrum produced by the cow is usually not a limiting factor.160 However, colostrums have varying immunoglobulin concentration; IgG1 concentration in beef cow colostrum is two to three times greater than that of dairy cow colostrum.161-164 Significant differences exist not only among different breeds but also among cattle within a specific breed.165 Lactation number also influences IgG1 concentration in colostrum, although this variation is less than between cows or breeds.
Colostrum with low immunoglobulin concentration, resulting in ingestion of an inadequate mass of immunoglobulin, is a primary cause of FPT in dairy calves.161 In contrast, FPT in beef calves is unlikely to result from low colostral IgG1 concentration.
Ingestion of Adequate Mass of IgG1
Depending on the environment and pathogen load to which they are exposed, calves must ingest an adequate mass of IgG1 to obtain protective immunity. Although the actual mass of immunoglobulin needed for protection depends on many factors, studies suggest that for a 45-kg calf to attain a serum IgG1 concentration greater than 10 mg/mL, it must consume approximately 100 g of IgG1 in the first hours of life.166 Consumption of 100 g colostral IgG1 ultimately depends on colostral volume and IgG1 concentration. A calf provided colostrum with 35 mg/mL IgG1 would need to consume approximately 3 L in the first 12 to 24 hours, whereas a calf provided colostrum with 100 mg/mL IgG1 must consume only 1 L. An accepted recommendation for attaining adequate passive transfer in diary calves is to administer 4 L of colostrum in the first 12 hours of life.166
Concentration of colostral immunoglobulin is rarely a significant factor leading to FPT in beef calves. Instead, factors such as mismothering, poor udder conformation, and environmental stresses are more significant in preventing beef calves from ingesting the colostrum. Other factors that may affect passive transfer in beef calves have been reviewed.167 There is little evidence to suggest a direct effect on passive transfer when beef cows are fed protein- or energy-restricted diets. Dystocia does not appear to affect passive transfer of immunoglobulins as long as the calf receives an appropriate mass of immunoglobulin in a timely manner.
Young age of the dam is a risk factor for FPT, likely because of lower colostral immunoglobulin concentration in heifer colostrum (beef and dairy) and poorer mothering ability.
Timely Absorption of IgG1 by Gut
Cessation of absorption of colostral immunoglobulins (closure) occurs at approximately 24 hours of age.154,168 If colostrum is completely withheld, closure will still occur by 30 hours of age.155,156 Closure is minimally affected by stresses such as dystocia or cold environmental temperatures, although extremely high environmental temperature has been associated with reduced immunoglobulin absorption in calves.154,169 In a study comparing three different feeding methods of colostrum to dairy calves, FPT was more prevalent with sucking than artificial feeding methods, presumably because of inadequate colostrum volumes ingested by calves.166 That is, dairy calves consuming relatively low IgG1 concentration colostrum did not consume adequate volumes of colostrum to obtain 100 g of IgG1 in a timely manner. When artificial feeding methods are used, inadequate immunoglobulin concentration in the colostrum is the most important factor resulting in FPT, even when calves are fed in a timely manner.
Mothering
Mothering has been associated with increased efficiency of absorption of colostral immunoglobulins. This may simply be a result of the dam’s presence or may be related to the effect of the neonate physically suckling the dam.154
Clinical Signs and Laboratory Findings
Although not diagnosed by physical examination, FPT is typically suspected based on physical findings. Young ruminants devoid of passive immunity are highly susceptible to bacterial septicemia.158,170 Clinical signs of bacteremia include central nervous system (CNS) depression, weakness, injected scleral vessels, rapid or labored respiration, diarrhea, and anorexia. Fever may or may not be present. Septic arthritis, meningitis, and panophthalmitis frequently develop in many neonates with FPT that survive the initial challenge of bacteremia. Diarrhea can also result from a number of dietary and infectious agents in neonatal ruminants with normal serum immunoglobulin levels.171 Historical information of clinical disease may lead to a high degree of suspicion that FPT is present.
There are no consistent changes in the hemogram or biochemical panel of a neonatal ruminant with FPT. Abnormal laboratory findings usually reflect secondary processes involving sepsis, stress, or starvation. Laboratory evaluation is necessary to assess the degree of passive transfer.
Measurement of Passive Transfer
Several methods are available to measure passive transfer of immunity either directly or indirectly. The greatest degree of accuracy is obtained if the tests are performed during the first week of life, and most tests are optimized when serum samples are collected between 24 and 48 hours of age. After this period, neonates begin to synthesize significant amounts of immunoglobulins, although a tentative decision can still be made as to the degree of passive transfer that occurred after birth. Any question about the status of passive transfer in a newborn should trigger laboratory evaluation of the animal so that corrective measures can be undertaken.
Single radial immunodiffusion (RID) quantitatively measures serum IgG1 and is considered the standard in cattle. The semiquantitative methods for estimating passive transfer (zinc sulfate turbidity, sodium sulfite precipitation, glutaraldehyde coagulation, total serum solids) are based on the fact that IgG1 normally accounts for the majority of passively transferred proteins in colostrum. Measurement of serum γ-glutamyltransferase (GGT) is based on the findings that mammary secretory alveolar epithelial cells can secrete large amounts of GGT during colostrogenesis and that GGT is readily absorbed by the neonate before gut closure.
Single Radial Immunodiffusion
Quantification of each immunoglobulin class that is transferred can be determined using specific, commercially available antisera. Forty-eight-hour serum concentrations of less than 1000, 80, and 22 mg/dL for IgG, IgM, and IgA, respectively, are consistent with FPT.160
Zinc Sulfate Turbidity
Specific concentrations of zinc sulfate precipitate immunoglobulins.149,172 Historically, a defined volume of serum (0.1 mL) is added to zinc sulfate solution (6 mL) containing 208 mg of ZnSO4 \cdot 7H2O per liter, and the solution is allowed to incubate at room temperature for 1 hour. The turbidity of the solution is then evaluated visually or with spectrophotometric methods.149 A decrease in or an absence of turbidity (precipitation) indicates failure of immunoglobulin absorption.
A study by Tyler et al.173 suggests that the zinc sulfate test using a 208-mg/L solution has an inappropriately high endpoint, with a specificity of only 0.52. The poor predictive value of this method results in a significant misclassification of calves with adequate passive transfer as having FPT. A follow-up study suggests that higher zinc sulfate concentrations in the test solution provide a more accurate assessment of passive transfer. As zinc sulfate concentrations are increased from 200 to 400 mg/L, sensitivity decreases from 100 to 82.6, and specificity increases from 25.5 to 90.6, respectively. The concentration of zinc sulfate test solution chosen therefore depends on the goal of the test. Choosing a test solution with higher sensitivity (e.g., 250 mg/L zinc sulfate) is likely warranted when testing individual calves with high economic value. In herd-monitoring programs, however, the choice of a test solution with higher specificity (e.g., 350 to 400 mg/L zinc sulfate) will maximize the proportion of calves correctly classified.
Sodium Sulfite Precipitation
This test is similar to the zinc sulfate turbidity test and is also a precipitation test for immunoglobulins, primarily IgG1, with the results recorded visually.174 The reagents are commercially available.* Historically, three concentrations of Na2SO3− anhydrous (14%, 16%, and 18%) are used in the test. Serum (0.1 mL) is added to each concentration of sodium sulfite (1.9 mL), and these are allowed to incubate for 1 hour. Turbidity in only the 18% sample indicates transfer of less than 500 mg/dL immunoglobulin, turbidity in both the 16% and the 18% sample indicates 500 to 1500 mg/dL of transfer, and turbidity in all samples indicates greater than 1500 mg/dL transfer.
Similar to the zinc sulfate turbidity assay, the endpoint for the sodium sulfite precipitation assay is likely set too high.173 The preferred test endpoint appears to be a 1+ test result. A 1+ result equates to observing turbidity in the 18% test solution, with no turbidity in either the 16% or the 14% test solution. Tyler et al.173 demonstrated the lowest serum IgG1 concentration (measured by RID) observed in calves with a 1+ test result was 645 mg/dL (range, 645 to 2450 mg/dL), indicating at least partial successful passive transfer of immunoglobulin. Therefore, it is recommended that the sodium sulfite test be used as a single-dilution assay procedure whereby calves with 1+ test results (precipitation in only the 18% test solution) are classified as having “adequate” passive transfer.
Glutaraldehyde Coagulation Test
This test is based on 10% glutaraldehyde reagent coagulating serum immunoglobulins in concentrations greater than 600 mg/dL. When performed on serum, the glutaraldehyde coagulation test is an accurate predictor of passive transfer status.175-178 However, if whole blood is used in a similar assay, the sensitivity and specificity of the assay are deemed to be inadequate for routine diagnostic use.179
Refractometer (Total Serum Solids)
Quantification of total serum solids as an indicator of passive transfer of immunity assumes that the increase in serum proteins over that of presuckle newborn ruminants is caused by the absorption of immunoglobulin.180 In the absence of dehydration, serum protein concentration greater than 5.0 g/dL has historically been associated with successful passive transfer, and values less than 4.5 g/dL are consistent with FPT. Values between 4.5 and 5 g/dL are deemed questionable.
In a study of 242 calves, use of a 5.0-g/dL threshold resulted in a highly specific (0.96) but insensitive (0.59) test, whereas a 5.5-g/dL threshold resulted in a highly sensitive (0.94) but relatively nonspecific (0.76) test.173 The choice of threshold (5.0 or 5.5 g/dL) depends on the prevalence of passive transfer in the herd being tested and the costs associated with false-positive and false-negative results. Using regression analysis to predict serum IgG1 concentration as a function of serum protein concentrations (with a goal of 10 mg/mL IgG1), the optimal threshold value for serum concentration is calculated to be 5.2 g/dL.173
γ-Glutamyltransferase
GGT is present in colostrum at levels about 300 times higher than that normally found in serum. GGT is absorbed by the calf during the period of immunoglobulin absorption, and calf serum levels can be used as an indication of immunoglobulin absorption. Levels of GGT are maximal in the calf on the first or second day after birth.181,182 Parish et al.183 used a regression formula to set age-adjusted thresholds for serum GGT activity in order to define passive transfer status in dairy calves.183 Using serum IgG1 concentration of 10 mg/mL as an acceptable goal for passive transfer of immunoglobulins, calves at 1 day, 4 days, and 7 days of age should have serum GGT activities greater than 200, 100, and 75 IU/L, respectively. Calves in the first 2 weeks of life with serum GGT levels less than 50 IU/L can be classified as having FPT.
Prevention
The intake and absorption of adequate amounts of quality colostrum are key to the survivability of young ruminants. Documented natural sucking or force-feeding of colostrum in the first 6 hours of life helps to ensure adequate transfer. It is generally thought that a neonate should consume between 6% and 10% of its body weight as colostrum in the first 24 hours of life. Most calves consume approximately 2 L (40 to 50 mL/kg) of colostrum at the first feeding. In force-feeding practices, 2 L typically has been advocated as the proper amount for initial intake. However, the variance in colostrum quality suggests that a greater volume should be fed at the initial feeding.154 As stated earlier, to attain adequate passive transfer in dairy calves, the current recommendation is to administer approximately 4 L of colostrum in the first 12 hours of life.166 In herds or flocks where neonatal disease is a problem, levels of passive transfer should be determined, and force-feeding of all newborn animals should be instituted to ensure adequate transfer.
Whole-colostrum specific gravity can be reasonably measured with a colostrometer.184 A specific gravity less than 1.050 is associated with colostrum that has low immunoglobulin concentration. This device can eliminate 50% of the low-immunoglobulin colostrums and predict high immunoglobulin concentrations.154 At colostrum temperatures other than 20° C (68° F), less accuracy is achieved.185 Colostrum should contain greater than 6000 mg/dL total immunoglobulin. To ensure that all neonates receive adequate amounts of colostrum, producers should be encouraged to maintain a bank of frozen colostrum. Colostrum from first milkings is preferred because immunoglobulin concentrations decrease with successive milkings.
Immunoglobulins are generally unaltered by freezing, and only small amounts are inactivated by thawing. Thawing frozen colostrum in warm water is quickly accomplished for individual calf feeding. Microwave ovens have been used to thaw frozen colostrum without adverse effects.186
Pooling or mixing colostrum from several cows is a common practice to ensure adequate immunoglobulin concentrations. This practice is advocated to circumvent the problem of low-concentration colostrum from being fed. However, calves that receive pooled colostrum acquire substantially lower serum immunoglobulin levels than calves fed equivalent volumes of homologous colostrum from their own dam.160 This appears to relate to the fact that colostrums used in pooling frequently come from cows with high volumes of colostrum that tend to have lower immunoglobulin levels.187
Many commercially available colostrum substitutes can be used either to supplement available colostrum or to provide a source of immunoglobulins for calves when colostrum is not available. It is important to note the amount of immunoglobulin present in these products and administer enough product to ensure passive transfer. With some products, it may not be possible or practical to administer enough colostrum substitute to obtain 100 g IgG1. Furthermore, the antibody specificity of the immunoglobulins present in these products may be unknown.
Bovine colostrum is frequently used to supplement or replace homologous sources of colostrum in nonbovine ruminants. Neonatal lambs and kids are frequently supplemented in this way.188,189 The immunoglobulins are absorbed at rates similar to homologous colostrum. The half-life of bovine IgG1 in lambs is approximately 14 days,189 which is similar to the 13.7 days reported for ovine IgG1 in lambs190 (see Table 53-2). Occasionally, anemia has been reported in lambs and kids fed bovine colostrums as a result of immune complex attachment to erythrocytes, causing their removal from circulation.191-193 The effectiveness of bovine or caprine colostrum in other species is unknown, but the practice appears to be widespread and has been used in a wild animal park successfully hand-raising neonatal exotic ruminants.
Treatment
FPT in an otherwise normal neonate can be treated with plasma administered at 20 to 40 mL/kg intravenously (or intraperitoneally if IV administration is not possible or practical). Whole blood can also be used, although the dose should be increased to account for the presence of RBCs. Similar approaches are indicated in animals with clinical disease, but the results are often less rewarding. Continued oral colostrum supplementation of neonates with FPT beyond the period of closure may also provide some local immune protection in the gut.160 Some clinicians have advised oral supplementation with plasma after gut closure, but this practice is questionable in light of the cost of obtaining plasma, and plasma contains only about 15 mg/mL IgG1, significantly less than average colostrum.
LETHAL TRAIT A46
Lethal trait A46 is a primary immunodeficiency that is found in black, pied Danish cattle of Friesian descent.194 It is an autosomal recessive trait in which the calves appear normal at birth but develop skin disease at 2 to 8 weeks of age. Skin lesions are characterized by exanthema, alopecia, and hyperparakeratosis, with initial lesions occurring around the head, neck, and flexor surfaces of the legs and later involving the entire animal. Death within 4 months is often associated with bronchopneumonia and diarrhea. These animals do respond normally to humoral antigens but show deficits in cellular immunity. At necropsy the calves show marked lymphoid regression involving the thymus, spleen, lymph nodes, and gut-associated lymphoid tissues in the thymic-dependent regions. The condition is responsive to oral zinc oxide therapy, which suggests an underlying problem in zinc metabolism. Daily oral dosages of up to 1 g zinc oxide have been used to establish normal function.194 The primary immunologic defect is found in T lymphocytes. Because of a defect in a recessive gene, these animals apparently have an unusually high metabolic requirement for zinc ions to sustain normal T-lymphocyte development and function.
SELECTIVE IgG2 DEFICIENCY
A primary IgG2 deficiency has been described in the red Danish milk breed of cattle involving a primary, partial to complete deficit in IgG2.195,196 These animals appear to be more susceptible to gangrenous mastitis and pyogenic infections such as bronchopneumonia, peritonitis, and abomasoenteritis.197 A transient IgG2 deficiency has also been reported in neonatal lambs.196 Lambs that ingested colostrum had delayed onset of IgG2 synthesis until 5 to 6 weeks of age, with no adverse consequences.
CHéDIAK-HIGASHI SYNDROME
Chédiak-Higashi syndrome is an inherited disorder that affects neutrophil and monocyte function in Hereford cattle, mink, cats, mice, killer whales, and humans.198,199 Clinically, these patients are recognized by partial albinism that affects the skin and eyes, coagulation difficulties, and an increased susceptibility to bacterial infections. Large cytoplasmic granules are seen within neutrophils. The increased susceptibility to pyogenic infections is related to decreased neutrophil killing of ingested organisms caused by a defect in hexose monophosphate shunt activity and defective degranulation.
BOVINE LEUKOCYTE ADHESION DEFICIENCY
Bovine leukocyte adhesion deficiency (BLAD) is an autosomal recessive disorder of Holstein calves characterized clinically by chronic bacterial infections and premature death.200-202 Leukocytes of affected calves lack surface glycoproteins, termed β-2 integrins. A mutation of the gene encoding bovine CD18 causes this disorder. The gene defect is termed the D128G allele mutation.
Heterozygous carriers are clinically normal; however bulls and cows with this mutation may produce homozygous calves that manifest the deficiency. All calves with BLAD can be traced to a single male ancestor that must be present in both the sire and dam pedigree. In the United States, approximately 14% of sires and almost 6% of Holstein calves are affected with BLAD.
Homozygous calves have chronic, recurrent bacterial infections. Consistent clinical findings include fever, bronchopneumonia, stomatitis, gingivitis, recurrent or chronic diarrhea, peripheral lymphadenopathy, vasculitis, and dermatitis.203,204 Calves appear normal at birth, but clinical signs are often present within the first few weeks of life. Affected calves usually die within the first year of life. Those surviving past 1 year of age have persistent ill-thrift. Hematologic abnormalities include mature neutrophilia (>40,000/μL) without significant left shifts, lymphocytosis, and monocytosis. Abnormal serum biochemical findings include hypoalbuminemia; hyperglobulinemia; and low serum creatinine, urea nitrogen, and glucose concentrations.
Diagnostic tests are available to detect heterozygous carriers and identify homozygous affected calves. Information regarding testing is available from the Holstein Association of America, Brattleboro, VT.
VIRAL- AND BACTERIAL-INDUCED IMMUNODEFICIENCY
Decreased function of B and T lymphocytes has been associated with bovine viral diarrhea (BVD) infection in calves developing chronic BVD syndrome.205 Depression of cellular, but not humoral, immunity is seen in cattle infected with Johne’s disease (Mycobacterium avium subsp. paratuberculosis), with failure to develop or delayed-type hypersensitivity (DTH) reactions in vivo and in vitro.206 It is associated with a humoral-suppressive factor.
COMBINED IMMUNODEFICIENCY
Genetic failure of development of both T and B cell lines results in a severely compromised, immunodeficient animal. As passively acquired maternal antibody wanes, infections tend to develop. One possible case of combined immunodeficiency was reported in a 6-week-old Angus calf that had an absence of serum IgM, low serum IgG, absolute lymphopenia, and an absence of lymphoid tissue at necropsy. The calf died of bronchopneumonia and disseminated fungal infection.207
PREGNANCY-ASSOCIATED IMMUNODEFICIENCY
The areas of immunology pertaining to pregnancy, fetal survival, and postpartum periods have continued to expand over the past decade.208,209Additional input from the fields of epidemiology and evidence-based medicine has increased our understanding of the delicate balance that occurs at the maternal-fetal interface.210 In its broadest context, the immune system of the pregnant animal becomes compromised as the fetus matures.211 Although transitory, this immunodeficient state also apparently extends into the postpartum period for up to 4 weeks.212
Mechanisms
The cellular mechanisms responsible for the immunosuppression during pregnancy and postpartum involve (1) a shift of T-helper (Th) cells from a Th1 to a Th2 response and (2) a decrease in neutrophil functions.213,214 The T-cell populations are affected by progesterone, prostaglandin F2α, and α-fetoprotein.211 The Th1 cells are important effectors of the cell-mediated immune (CMI) response and interact with T-cytotoxic (Tc) cells. Tc cells are the main defense against foreign antigens, which include viral, intracellular bacterial, and protozoal pathogens. With the onset of pregnancy, the hormonal factors cause macrophages to release predominantly Th2-stimulating cytokines, which contribute to the overall dominance of humoral immunity during pregnancy and immediately postpartum. This phenomenon of the Th-cell populations is referred to as the “Th1-Th2 shift of pregnancy” and is generally regarded as a contributing factor to maternal tolerance of the fetus by suppressing the antifetal CMI response.209
The second key cell population affected during pregnancy and postpartum is the neutrophil.212 The point of maximum immunosuppression occurs when glucocorticoid levels are acutely elevated in the periparturient cow.215 Neutrophil dysfunction, as well as the effects on the Th-cell population, is considered temporary during this period. Nonetheless, with impaired neutrophil response, the animal is then vulnerable to increased bacterial infections because of compromised bactericidal functions.212
Outcome
This temporary immunodeficiency allows for fetal survival but may also result in an increased susceptibility to exterior environmental infection with viruses, bacteria, and fungi. Intracellular infections, such as with viruses and protozoans, which may have been acquired early during postnatal development, may become exacerbated during pregnancy because of the suppressive effects on the Th1 cells.211 This results in a decreased CMI response. The CMI effector cells (Tc cells) function normally to control virtually all the viral infections, such as infectious bovine rhinotracheitis virus and BVD virus.211,216 Intracellular bacterial infections, such as Brucella abortus, become more pathogenic during pregnancy. In addition to the effects on T-cell functions, macrophage function is also compromised, allowing for opportunistic bacteria and Chlamydiophila species (usually confined to external mucosal surfaces) to become systemic. Concurrent with the immunosuppression that accompanies pregnancy, there is an increased shedding of infectious microorganisms211 (Figure 53-5). This is considered to be an extension of impaired Tc-cell function. Although the pregnant animal may appear clinically normal, her altered immune response results in an increased shedding of gastrointestinal (GI) viruses such as coronavirus and rotavirus211,217 and bacteria (Mycobacterium avium subsp. paratuberculosis, Escherichia coli), usually during the periparturient period.218,219 This increased shedding of infectious microorganisms is an important factor in the management of animals through this period. The suppression of neutrophil functions during later stages of pregnancy and immediately postpartum are the subject of active investigation.215,220
Fig. 53-5 Proposed relationships among shedding of infectious microorganisms during periods of nonpregnant, pregnant, postpartum, and relative immune competence.
Modified from Evermann JF: Immunology of bovine pregnancy: vulnerability to infectious disease, Bovine Proc 26:101, 1994.
The reduced neutrophil function and its association with periparturient immune suppression are significant problems, especially for the transition dairy cow.215 This immunosuppression is directly associated with increased postpartum metritis and increased susceptibility to mastitis. As noted with the immunosuppression during pregnancy, this periparturient stage can be aggravated by both preexisting infections (e.g., bovine herpesvirus type 4)221 and environmental bacterial pathogens (e.g., Staphylococcus aureus, Klebsiella species).222
Management
Although the immunosuppressive periods during pregnancy and up to 4 weeks postpartum are well recognized, current understanding of the cellular processes is still somewhat rudimentary. However, clinicians and owners can proceed with control measures that accomplish two primary goals: maximize reproductive performance and ensure successful neonatal survival.210,223,224 Over the years, we have emphasized the importance of effective vaccination programs before breeding, clean birthing areas, and good hygiene for the lactating animal.210,222 These measures, in conjunction with good colostral management, allow owners to compensate for the temporary immunosuppressive states encountered during and immediately after pregnancy.
DISEASES CAUSED BY ALLOGENEIC INCOMPATIBILITIES
The surfaces of cells are covered with molecular structures, the presence or absence of which is determined by genes. Some of these molecules have limited heterogeneity within a species, and all members of the species share identical forms (monomorphic). In other cases a spectrum of minor structural variations or polymorphisms of a particular molecule occur within the population, so that some members of the species will have one form of the molecule, whereas others will have another. If the structural variations are such that the immune system of one individual can recognize the differences in the molecules from another individual, these are known as alloantigens. Antibodies produced against these antigens are alloantibodies. Examples of alloantigens include red blood cell (RBC, erythrocyte) antigens and lymphocyte antigens.
In nature, because few situations arise in which tissues are exchanged, there are relatively few naturally occurring diseases that involve allogeneic incompatibilities. Neonatal isoerythrolysis (NI) and neonatal alloimmune thrombocytopenia (NAIT) are two examples of such naturally occurring diseases. Blood transfusion reactions and organ transplant rejection are examples of iatrogenic diseases associated with allogeneic incompatibilities.
The same alloantibodies that mediate disease are also useful in vitro as reagents to detect the presence of alloantigens and serve as the basis for blood typing.
BLOOD TYPING AND DNA PROFILING
The blood type of an individual is actually a composite list of genetic markers that have been detected in the individual’s blood. Several types of genetic markers have been used in blood typing, including RBC alloantigens, electrophoretic markers, and lymphocyte alloantigens. Polymorphisms of the DNA itself have emerged as useful genetic markers and have essentially replaced other genetic tests for parentage verification.
Red Blood Cell Antigens
Seven independent RBC groups or systems have been internationally defined in horses under the auspices of the International Society for Animal Genetics225-228 (Tables 53-3 and 53-4). These systems are named A, C, D, K, P, Q, and U. Additional systems are recognized by individual laboratories. Each system corresponds to a particular gene for which two or more alleles exist. Some blood groups are relatively simple and contain only two alleles, whereas other systems may have over two dozen different alleles.
Table 53-3 Domestic Animal Blood Groups
| Species | Blood Group. Systems |
|---|---|
| Equine | A, C, D, K, P, Q, U |
| Bovine | A, B, C, F, J,* L, M, R′, S, T, Z |
| Ovine | A, B, C, D, M, R,* X |
| Caprine | A, B, C, M, J* |
From Suzuki: Personal communication, 1990.
Table 53-4 Blood Group Systems, Factors, and Alleles of the Horse Recognized by the International Society for Animal Genetics
| System | Factors | Recognized Alleles |
|---|---|---|
| A | a,* b,† c, d, e, f, g | Aa, Aadf, Aadg, Aabdf, Aabdg, Ab, Abc, Abce, Abe, Ac, Ace, Ae, A− |
| C | a | Ca, C− |
| D | a, b,† c,† d, e, f, g,† h, i, k, l, m, n, o, p, q, r | Dadl, Dadlnr, Dadlr, Dbcmq, Dcefgmq, Dcegimnq, Dcfgkm, Dcfmqr, Dcgm, Dcdmp,Dcgmq, Dcgmqr, Ddeklqr, Delno, Ddeloq, Ddelq, Ddfkir, Ddghmp, Ddghmq, Ddghmqr, Ddki, Ddlnq, Ddlnqr, Ddlqr, Ddno, Dq, (D−) |
| K | a | Ka, K− |
| P | a,† b, c, d | Pa, Pac, Pacd, Pad, Pb, Pbd, Pd, P− |
| Q | a,* b, c† | Qa, Qab, Qabc, Qac, Qb, Qbc, Qc, Q− |
| U | a† | Ua, U− |
* Most common factors involved in neonatal isoerythrolysis (NI).
† Previously reported to cause NI in at least one case.
From Cothran G: Personal communication.
The blood group genes produce surface molecules that contain antigenic sites known as factors. More than 30 different factors have been identified. Each factor is associated with only one system, although the same factor may be associated with more than one allele within the system. The factors are named with an uppercase letter to denote the system and a lowercase letter to designate the factor within that system. Groups of factors that are produced by a single allele are called phenogroups.
The presence of RBC antigens is detected primarily by either agglutination or complement-mediated lysis of test cells by antibodies directed against specific erythrocyte alloantigens. Antigens in some systems are detected with antiglobulin tests.
Eleven blood groups have been identified in cattle229 (Table 53-5); groups B and J have the greatest clinical relevance. The B group is a very complex system, with more than 60 different antigens.230 This complexity has been used to advantage for individual animal identification and parentage studies in the past, but it makes it difficult to match donor and recipient blood for transfusions.
Table 53-5 Blood Group Factors of Cattle Recognized by the International Society for Animal Genetics
| System | Factors |
|---|---|
| AA* | A1, A2, H, Z, a |
| F* | F, V |
| J | J, j |
| L | L, l |
| M | M, m |
| Z | Z, z |
| R″ | R′, S′ |
| B† | B, G1, G2, G3, I1, I2, K, O1, O3, OX, Pl, P2, Q, T, Y2, A′, B′, D′, E1′, E2′, E3′, G′, I1′ (I′), I2′, Jl′, J2′, K′, O′, P1′, P2′, Q′, Y′, A″, B″, G″, I″ |
| C | C1, C2, R1, R2, W, X, X2, L′, E1, C′, C″, X′, Fl, F6, F10, Fl5 |
| S | S1, S2, U, H′, U′, S″, U″ |
| T | — |
* Cases of neonatal isoerythrolysis associated with production of anti—red blood cell antibodies against factors in response to anaplasmosis and Babesia vaccines.
† Additional factors are recognized by individual laboratories.
The J antigen is a lipid that is found in body fluid and that is adsorbed to erythrocytes; thus it is not a true erythrocyte antigen. Newborn calves do not have this antigen but usually acquire it during the first 6 months of life. After acquiring it, some groups of cattle have high amounts of J antigen both in the serum and adsorbed to erythrocytes, whereas other groups have very small amounts found only on RBCs. These latter groups, often referred to as J-negative, may actually have anti-J antibodies and develop transfusion reactions when transfused with blood from J-positive donors.225
Seven blood groups have been identified in sheep (see Table 53-3). The B system is analogous to the B system in cattle and is extremely polymorphic, with more than 52 different alleles. The R system is similar to the J system in cattle in that the antigens are soluble and are passively adsorbed to erythrocytes.229 The M-L system is involved in the genetically determined RBC potassium polymorphism found in sheep.231
Blood groups of goats appear to be very similar to those of sheep, and many of the reagents used for typing sheep blood have been used to type goats’ blood. Reagents used to detect antigens of the J system of cattle have been used to detect differences in a similar system in goats.232
The biologic function of red blood cell antigens is not known for any system in any species with the exception of the M–L blood group system in sheep, which is involved in active potassium transport across red blood cell membranes.
Electrophoretic Markers
Many plasma and intracellular proteins have polymorphic forms. These variants have subtle biochemical differences, which for the most part do not alter the major characteristics of the molecules, but which can be detected using electrophoretic methods. Technically, these are not alloantigens because the differences are not detected immunologically. The various forms of the molecules are detected on the basis of their different rates of migration in electrophoresis. The specific form(s) of any particular protein that an individual possesses are determined genetically, and those used in blood typing are usually expressed co-dominantly. Currently, no diseases are clearly associated with the presence or absence of particular alleles of polymorphic proteins; however, they are useful genetic markers for identification and parentage studies and were typically included in the blood type227,232-234 (Table 53-6). Electrophoretic markers were used extensively in horses because RBC antigens did not provide the same degree of discrimination among individuals in this species as they do, for example, in cattle. The current application is limited to legacy parentage verification when parental DNA is not available for testing.
Table 53-6 Examples of Polymorphic Proteins Used in Blood Typing
| Protein | System | Source |
|---|---|---|
| A1B glycoprotein | AlB | Serum |
| Albumin | ALB | Serum |
| Transferrin | TF | Serum |
| Carboxylesterase | ES | Serum |
| Vitamin D–binding protein | GC | Serum |
| Protease inhibitor | PI | Serum |
| Peptidase A | PEPA | Serum |
| Plasminogen | PLG | Serum |
| Glucose phosphate isomerase | GPI | Red cells |
| 6-Phosphogluconate dehydrogenase | PGD | Red cells |
| Phosphoglucomutase | PGM | Red cells |
| Catalase | CAT | Red cells |
| Carbonic anhydrase | CA | Red cells |
| Acid phosphatase | AP | Red cells |
| Hemoglobin-a | HBA | Red cells |
| NADH diaphorase | DIA | Red cells |
Lymphocyte Antigens
Three systems of equine lymphocyte alloantigens have been described: ELA, ELY 1, and ELY 2.235-239 ELA is the major histocompatibility complex (MHC) of the horse. ELA is a complex system that includes several genes, each of which is polymorphic (e.g., has many different alleles). Because the MHC regulates the interactions of many cells in the immune response, disease susceptibility or resistance could be associated with ELA. An association between a particular ELA allele and equine sarcoid has been described.240,241 ELY 1 and ELY 2 have limited polymorphism and appear to be two allele systems.
The MHC of cattle is called BoLA, for bovine lymphocyte antigen. In sheep the MHC is called OLA, for ovine lymphocyte antigen, and is also known as SH-LA. In goats the comparable system is GLA.242
DNA POLYMORPHISMS
A variety of approaches have been developed for detecting genetic variation using DNA. Certainly, DNA sequencing is the most sensitive approach; however, this remains too costly for routine testing outside of research laboratories. Other approaches have been developed to detect DNA variants. Genetic variation of DNA can be detected among individuals on the basis of differences in length for a section of DNA or even for differences in base composition at specific sites. DNA length variants are usually referred to as minisatellite or microsatellite markers. Minisatellite DNA markers are based on sequences, often 15 to 100 bases long, that may be repeated hundreds of times on a chromosome. The number of repetitions can vary dramatically between individuals and can be readily detectable in Southern blot assays. These tests were originally referred to as “DNA fingerprinting.” Although powerful, these tests are costly and time-consuming.
More recently, another type of DNA length variant has been discovered. Microsatellite DNA markers are short repetitions of nucleotides, usually ranging from one to nine bases long, which may be repeated 15 to 40 times. As with minisatellites, the number of repetitions may vary between chromosomes. The development of the polymerase chain reaction (PCR) has opened the door to easy, inexpensive detection of microsatellite and other types of markers. PCR is a method for amplifying short (<3000 bases) pieces of DNA, allowing a particular gene to be easily studied. The amplification products can be separated by electrophoresis and length variants readily detected. More than 100,000 of these markers are thought to exist in each species. These markers form an important core for development of genetic maps in diverse species. The markers currently form the basis for most parentage analysis.
Variation recognizable as a change in a single base of DNA is more subtle but can be detected through a variety of techniques. DNA sequence variation is often detectable using restriction enzymes, which are isolated from bacteria and will cleave DNA when the sequence is specific for the enzyme. For example, the method to detect the gene defect causing hyperkalemic periodic paralysis (HYPP) in horses is based on PCR amplification of the gene and then detection of sequence differences that occur between the normal and mutant gene using restriction enzyme digestion of the DNA. Other approaches to identifying DNA sequence differences include single-strand conformational polymorphism (SSCP), allele-specific PCR amplification, and ligase chain reaction.
Testing based on DNA has essentially replaced blood typing for purposes of parentage analysis. Furthermore, DNA-based techniques are also being used to develop gene maps for many species of domestic animals. These gene maps can be used to identify genes that play a role in performance, production, and disease.
BLOOD-TYPING AND DNA-GENOTYPING APPLICATIONS
To determine an animal’s blood type or DNA genotype, a battery of tests are run on the blood or tissue to check for the presence or absence of each recognized allele in each of several genetic systems. Each system is controlled by a separate autosomal gene. These genes are inherited independently of one another.228,239 Most genetic markers used in blood typing are expressed co-dominantly, which means that the alleles on both chromosomes are expressed. This often allows determination of genotype (the actual genes present) from the phenotype (the antigens actually detected). Some systems have null alleles (with no detectable products) that do not directly allow inference of genotype (e.g., C, K, and U blood group systems in horses). An individual can have only two of the possible different alleles for any given gene (one on each chromosome of a pair).
Blood typing based on RBC antigens and electrophoretic markers has been used for animal identification and pedigree analysis, but currently, RBC typing is used primarily for prediction of potential for NI and crossmatching for blood transfusion.
The use of genetic markers for parentage verification is based on exclusion (i.e., markers are tested until a discrepancy is found). If no discrepancy is observed after a statistically acceptable amount of testing, the probability of a match is determined.
Because genetic testing is a composite mosaic of a large number of different, largely independent genetic markers, the odds of two individuals sharing exactly the same pattern of markers, whether RBC antigens, electrophoretic markers, or DNA polymorphisms, is remote. Thus, once an animal’s genetic type is on record, it can be a powerful identifier of an individual animal. In this sense it is an unalterable form of identification that is present from birth to death.
Because the presence or absence of markers is genetically determined and because the markers contributed by both parents are expressed in a co-dominant fashion (unless “blank” alleles exist), these markers can be used to support or refute parentage claims. Blood typing and DNA profiling cannot prove that one horse is the parent of another; however, in certain cases it can exclude with certainty a stated parent as the true parent. Two types of parentage exclusions can occur, type I and type II243 (Fig. 53-6). With type I the foal possesses a dominant or co-dominant marker that is not present in either parent. This means that at least one of the stated parents is incorrect. If one parent (usually the dam) is known with certainty on the basis of circumstances other than genetic type, the error lies with the other stated parent. With type II exclusions, the foal does not possess either of the dominant or co-dominant markers of a stated parent. Because a foal must inherit one of the two markers expressed by each parent, exclusion can be made if neither of the two markers present on the stated parent is detected in the foal. In parentage cases, each genetic system is evaluated for type I and type II exclusions. A parent can be excluded on the basis of an inconsistency in even one system. For this reason, the more systems used, the better genetic typing becomes as a tool for solution of parentage disputes. The calculated effectiveness of blood typing for detecting incorrect paternity using 20 internationally defined systems in horses is as high as 96%.227,233,244 With DNA genotyping, effectiveness is as high as 99.998%.245-247
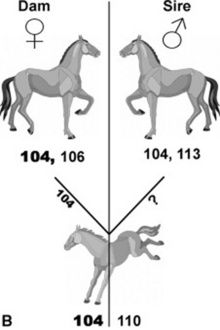
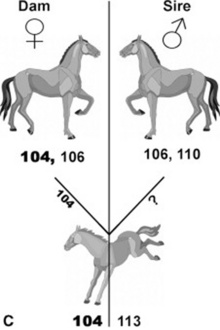
Fig. 53-6 A, The sire of this foal cannot be excluded as the sire of the foal based on the results for DNA marker VHL20. B, The sire of this foal is excluded by a type I exclusion. The foal possesses an allele for the DNA marker VHL20 not present on either parent. Assuming that the dam of the foal is known to be correct and therefore would have contributed the 104 allele to the foal, the sire must be incorrectly stated. C, The sire of this foal is excluded by both type I and type II exclusion. The foal possesses an allele not present on either parent and also does not possess either allele expressed by the putative sire.
Box 53-1 lists laboratories in North America that provide DNA genotyping.
NEONATAL ISOERYTHROLYSIS
Equine
Definition and Etiology
Neonatal isoerythrolysis (NI) is a condition characterized by the destruction of RBCs in the circulation of a foal by alloantibodies of maternal origin absorbed from colostrum.226,248,249
Production of alloantibodies can be stimulated in mares several ways, including transfusion, exposure to blood from the foal during parturition, or as a result of placental pathology during gestation. Incompatibilities of at least some blood groups between the dam and foal are the rule rather than the exception, and yet the incidence of sensitization of the dam and occurrence of NI is relatively low. In thoroughbreds the prevalence is about 1% and in standardbreds about 2%.250 The prevalence in mules (donkey sire, horse dam) has been reported to be as high as 10%.226 Most blood groups are not strongly antigenic under the conditions of exposure through previous parturition or placental leakage. Several blood group factors, however, are particularly immunogenic, and antibodies against these antigens have been reported to cause almost all cases of NI. These include factor Aa in the A system and factor Qa in the Q system. In mules a unique donkey RBC antigen named donkey factor has been associated with NI.251 Antibodies to these antigens develop after the exposure of the mare to RBCs and are not naturally occurring antibodies. In humans the absence of certain RBC antigens results in the production of natural antibodies against that antigen, presumably because of exposure to cross-reacting antigens in the diet. This does not occur in horses (with a few exceptions) and has not been associated with NI. Ca-negative (Ca−) horses frequently make anti-Ca antibody of low titer without known RBC exposure. However, anti-Ca antibody is generally not associated with NI, and mares with anti-Ca antibody actually appear less likely to develop certain antibodies responsible for NI.
For NI to occur, several conditions must be present (Figs. 53-7 and 53-8). First, the dam must be negative for the antigen in question. Because most cases of NI in horses are associated with either anti-Aa or anti-Qa antibodies, mares that are Aa− or Qa− or both are considered at risk. All horses appear to lack donkey factor; thus all mule pregnancies are considered at risk. Second, the mare must become sensitized and produce antibody to the offending antigen. Sensitization can result from exposure during a previous pregnancy, blood transfusion, or transplacental contamination with fetal RBCs earlier in the current pregnancy (rare). Third, the foal from the current pregnancy must have inherited from its sire the antigen(s) to which the mare has been sensitized. When these conditions are met, there is a significant potential that maternal antibody directed against the foal’s RBC antigens will appear in the colostrum and, if subsequently ingested and absorbed by the foal, could cause loss of RBCs. The higher the anti-RBC titer is at foaling, the higher the risk. The highest titers are likely to be produced in a previously sensitized mare that is reexposed to the same RBC antigens shortly before parturition.
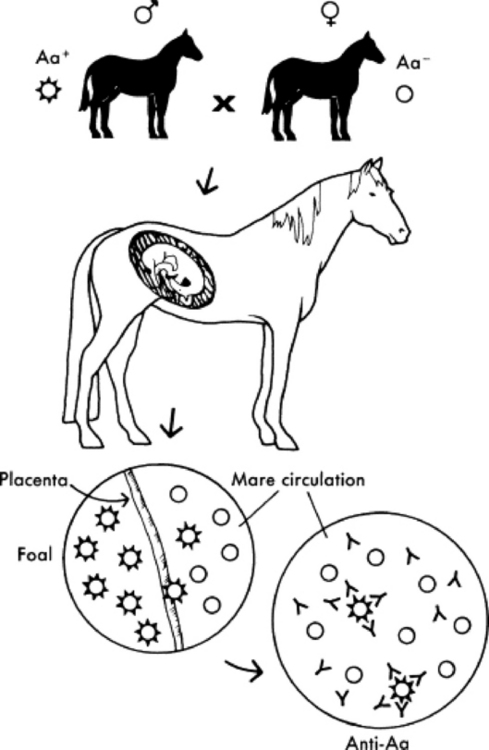
Fig. 53-7 Entrance of RBCs of paternal antigen type into maternal circulation stimulates the production of alloantibody in the mare’s serum.
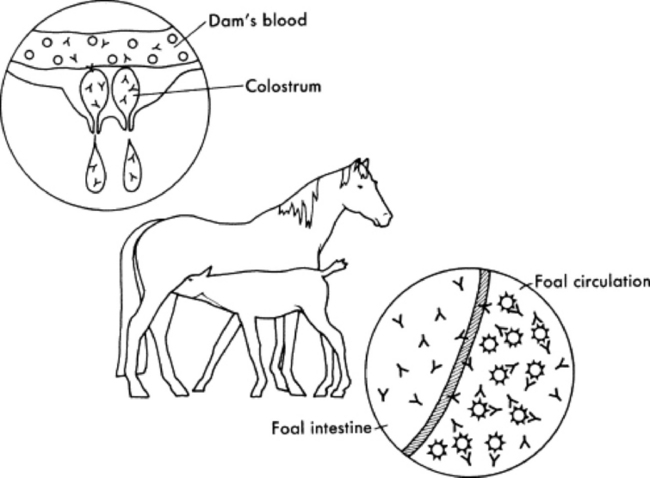
Fig. 53-8 Alloantibody in the mare’s serum is concentrated in colostrum at the end of gestation. Through passive transfer, the foal absorbs immunoglobulins, including these alloantibodies. The antibodies attach to RBCs and cause either their premature removal from circulation or intravascular lysis.
Clinical Signs and Differential Diagnoses
Foals are born healthy and usually begin to develop signs of NI at 24 to 36 hours of age, after suckling. Progressive lethargy and weakness are early signs. In acute cases, mucous membranes show initial pallor that is followed by icterus. In severe cases, hemoglobinemia and hemoglobinuria may be pronounced. In peracute cases, death may precede the development of icterus. Breathing becomes rapid and shallow, followed by labored breathing as the disease progresses. The foals may yawn repeatedly. Heart rate is elevated. Seizure-like activity may occur as the anemia becomes more severe.
Clinical Pathology
Affected foals are anemic. All indicators of RBC concentration (packed cell volume [PCV], hemoglobin, RBC count) show significant decreases. PCV values often decline to between 10% and 20%, and values as low as 5% have been observed. Hemoglobinemia and hemoglobinuria may be present. Bilirubin (mainly unconjugated) levels will be increased as a result of accelerated RBC destruction. Total bilirubin levels may be close to 20 mg/dL in severe cases. Affected foals, especially mule foals, may also be thrombocytopenic.228
Demonstration of significant amounts of antibody in the colostrum (or serum from the mare) that are directed against the RBC antigens expressed by the foal provides a definitive diagnosis of NI. These antibodies are most often demonstrated by lytic and agglutinating tests. Lytic tests are believed to be more reliable indicators of the presence of offending antibody.252 The presence of antibodies attached to the foal’s RBCs can also be demonstrated with a direct antiglobulin test (Coombs’ test). The presence of antibodies in the mare’s serum that attach to RBCs can be demonstrated with an indirect antiglobulin test.226
Pathophysiology
In mares sensitized to RBC antigens, most often Aa or Qa, alloantibodies are concentrated in the colostrum late in gestation. These antibodies are passed to the foal through passive transfer. If the foal’s RBCs carry the antigen that the antibody recognizes, the cells become antibody coated. Subsequently, they are removed prematurely by the reticuloendothelial system or lysed intravascularly by complement. A distinction has been made between antibodies that are lysins as opposed to agglutinins; however, this distinction is based on in vitro testing and may be somewhat artificial. Under appropriate laboratory conditions, offending antibodies may exhibit both abilities.250 However, with conventional agglutination tests, some alloantibodies capable of producing NI may go undetected.234
Epidemiology
The percentages of mares at risk for sensitization against the common offending antigens (Aa and Qa) vary among breeds, depending on the frequency in the population of each gene involved (Table 53-7). Increased numbers of mares at risk in a breed does not necessarily translate into higher numbers of NI cases. A corresponding number of stallions lack the factors in question and are therefore unable to sire foals with the offending antigen.249 Virtually all mule pregnancies are incompatible with regard to donkey factor.251
Table 53-7 Estimated Percentage of Mares in Selected Breeds “At Risk” for Producing Foal with Neonatal Isoerythrolysis*
| Breed | At Risk for Aa | At Risk for Qa |
|---|---|---|
| Thoroughbred | 2% | 16% |
| Standardbred, pacer | 22% | † |
| Standardbred, trotter | 3% | † |
| Saddlebred | 25% | 88% |
| Quarter horse | 25% | 68% |
| Arabian | 3% | 72% |
* Based on the lack of all alleles, including factors Aa or Qa.
† All mares lack factor Qa and are technically “at risk,” but all stallions in this breed also lack the factor.
Data from Bailey E, Conboy HS, McCarthy PF: Neonatal isoerythrolysis of foals: an update on testing, Proc Am Assoc Equine Pract 33:341, 1987.
Although more common in multiparous mares, NI can occur with the first pregnancy.
Necropsy Findings
Pale tissues with or without icterus and splenomegaly are characteristic necropsy findings in foals dying of NI. Lesions associated with RBC destruction and anemia, such as nephrosis and centrilobular hepatic necrosis, may also be present.
Treatment and Prognosis
In most cases, by the time NI is recognized clinically (e.g., when the foal is about 24 hours of age), the bulk of colostral antibody will have been depleted from the mare’s milk, and the absorptive ability of the foal’s gut will have diminished. Withholding milk at this point is of questionable benefit.
Stress should be minimized and exercise restricted. Affected foals have decreased exercise tolerance and can collapse and die if forced to follow their dams. Generalized supportive care should be administered as indicated by clinical parameters. Intravenous fluids are frequently indicated to promote diuresis to minimize the effects of the large hemoglobin load presented to the kidneys. Acid-base balance should be monitored and corrected if indicated.
If the anemia becomes severe (e.g., PCV of 10% to 15%), transfusions that provide RBCs should be considered. Unless the PCV drops below this level, transfusion may not be necessary if rest is enforced. The object of transfusion is to provide the affected foal with RBCs that will not be destroyed by the maternal alloantibodies that were absorbed from colostrum. The foal is immunologically naive and will not have autologous alloantibody directed against any RBCs that would have an immediate effect on transfused RBCs. Thus the key is to select an RBC donor whose cells will not be destroyed by the maternal antibody derived from colostrum. Crossmatching, with particular attention to the reaction of mare sera, colostrum, and foal sera (all contain the same antibody), with the donor RBCs, is important for selecting a cell that will not be destroyed by the maternal antibody.
Washed RBCs from the dam are obviously the perfect choice in terms of cells that will not react with the alloantibodies present in the foal. However, to avoid administering additional harmful antibody to the foal, the mare’s sera (containing antibody against the foal’s cells) must be removed by washing before administration. Up to 6 to 8 L of blood can be collected from the mare and anticoagulated with acid-citrate-dextrose (ACD) or sodium citrate (3.8% Na citrate solution; 1 part Na citrate/9 parts blood), although 3 to 4 L usually provides sufficient RBCs. The preparation of large volumes of washed cells is aided by a large-volume centrifuge, but the procedure can be accomplished by serial sedimentations. Anticoagulated blood from the mare is allowed to settle for 1 to 2 hours. The plasma is aseptically drawn off; a similar or greater volume of sterile isotonic saline (0.9% Na citrate) is added to the RBCs and mixed; and the RBCs are again allowed to settle. The saline is then drawn off and discarded. At this point, the RBCs can be resuspended in an equal volume of isotonic saline for administration, or the washing procedure can be repeated. The sedimentation method is less desirable than centrifugation because it is slower and does not remove as much of the offending antibody. The aim is to dilute any harmful antibody to insignificant levels.
If the dam’s RBCs cannot be used, alternative donors can be selected. The donor should lack the antigen to which the alloantibody is directed. Because it is generally not possible to blood-type donors on short notice, a previously identified horse that has been determined by blood typing to be Aa−, Qa−, and free of alloantibody is a good choice for donor, based on most cases of NI being associated with these two antigens. The odds of randomly selecting a donor that lacks the offending type (e.g., Aa−, Qa−, or both) and would therefore be a suitable donor vary significantly with the breed and would mirror the percentage of the population of mares at risk. For example, the odds of finding an Aa− thoroughbred to serve as an RBC donor would be about 1 in 50 (2%), whereas in quarter horses the odds would be about one in four (25%) (see Table 53-7). The sire of the foal is not the donor of choice. He shares the same RBC antigens as the foal, and his cells would react with the maternal alloantibody present, adding more of a load to the foal’s reticuloendothelial system as the cells are destroyed.
With mules the same considerations in the use of washed cells would be necessary if the dam’s cells were used. However, because the offending antibody is generally directed against a unique donkey antigen, RBCs from any horse apparently would be satisfactory. Horses do not appear to make naturally occurring antibodies against donkey factor; therefore, in most cases it is not necessary to wash the cells from horses that would not be likely to have been immunized by pregnancy against donkey factor.
Transfusing RBCs that will not react with maternal alloantibody means introducing an obviously incompatible cell into the foal, a cell that will probably not survive long in the circulation. Such transfusions should be considered temporary stopgap measures. Transfused cells may also sensitize the foal to future transfusions, causing reactions (perhaps not within hours or a few days, but potentially within a week). This must be considered when weighing the potential good versus potential harm of such transfusions.
From 1 to 4 L of washed RBCs or whole blood is usually adequate to produce clinical improvement, although some cases may require repeated transfusion if the anemia progresses. Exchange transfusions can be done whereby blood is administered through one jugular vein and withdrawn simultaneously from the opposite jugular vein and discarded, allowing administration of large volumes of blood without overloading the vascular system. There is no good evidence to suggest that this is more effective than simply providing a source of RBCs that are unaffected by the maternal antibody.
Limited transfusion studies in adult horses have suggested that transfused RBCs do not survive long in circulation (e.g., 2 to 4 days), whereas in foals, cells survived slightly longer (e.g., 4 to 6 days).253,254 PCVs in foals with NI usually increase after transfusion and then gradually decline. This decline is probably not a concern if it is gradual, because the PCV of the foal generally levels off as the offending maternal antibody is metabolized. However, even short-term survival of cells may be of benefit in severely affected foals and may allow them to survive until the titer of maternal anti-RBC immunoglobulin has declined in the circulation.
The prognosis varies, depending on the quantity of antibody ingested, the rapidity of onset of signs, and the degree of anemia. Foals with peracute NI may die before the problem is recognized, with no chance for administration of therapy. Foals that develop the condition more slowly may respond to supportive care or transfusion if the PCV continues to fall.
Prevention and Control
Several strategies are available for prevention of NI.255,256 First, identify broodmares at risk for development of NI by testing them for the presence of Aa and Qa. Mares negative for either antigen, which means they could potentially make antibodies against them, should be considered at risk. One subsequent strategy would be to breed at-risk mares to Aa/Qa-negative stallions, thus eliminating the possibility of the foal inheriting the offending antigens. However, in breeds in which a relatively small part of the population is negative for these antigens, identifying a stallion that is negative for these antigens and suitable on the basis of other criteria may be difficult. The percentage of at-risk females based on the presence or absence of Aa or Qa is somewhat balanced by the numbers of males able to transmit the offending antigen. For example, all standardbred mares would be considered at risk based on the absence of Qa; however, because the antigen Qa is not present in the standardbred population, no stallions have the Qa antigen to pass on to foals.
In the circumstance of an unknown or incompatible matings, sera from at-risk mares should be screened for the presence of anti-RBC antibodies within 30 days before foaling. This can be done by submitting a serum sample and an anticoagulated sample to a screening laboratory. A panel of 10 to 12 different RBCs selected to represent all major blood groups is adequate to screen for anti-RBC antibody in the absence of blood from the sire. If results of serum testing are equivocal (e.g., low but positive titer, especially if there is anti-Aa or anti-Qa activity), the test should be repeated closer to the time of parturition because the levels of offending antibody can rise very quickly late in gestation.
If anti-RBC antibody is detected in the mare before parturition, the colostrum should be checked for reactivity against the foal’s RBCs before allowing the foal to ingest colostrum. An alternative source of colostrum should be provided to the foal. Most field screening tests of colostrum have not proved to be satisfactory for practical use; however, the jaundiced foal agglutination (JFA) test described in Box 53-2 has been shown to correlate well with the standard hemolytic assay, and it may detect antibody that does not react on the standard agglutination tests.257,258
Box 53-2 Jaundiced Foal Agglutination (JFA) Test
Data from Bailey E, Conboy HS, McCarthy PF: Neonatal isoerythrolysis of foals: an update on testing, Proc Am Assoc Equine Pract 33:341, 1987; and Blackmer JM, Costa LRR, Koch C: The jaundiced foal agglutination test, Vet Tech 23:577, 2002.
METHODS
If there is a positive reaction with the foal’s cells, the test should be run with the dam’s own RBCs to ensure that the conditions of the test and the viscosity of the colostrum are not causing the agglutination.
Positive reactions at 1:16 or greater are considered significant. At levels of 1:16 or greater, this test correlates well with the standard hemolytic assay. At dilutions of less than 1:16, the correlation is not as good, and more false-positive results will be recorded. Also, other factors (e.g., viscosity of colostrum) make less diluted samples more difficult to read.
Horses negative for Ca frequently make anti-Ca antibody; however, this antibody is not known to produce adverse effects in the foal. It has clouded the issue of field screening tests for NI because it causes positive reactions in most screening tests at low dilutions. Because these tests do not differentiate between antibody to Aa, Qa, Ca, or any other blood group, anti-Ca. antibody, when present in low dilutions, is responsible for many false-positive reactions. This antibody actually appears to play a protective role in the prevention of sensitization of mares to NI through a mechanism of antibody-mediated immune suppression. Aa−/Qa− mares that are also Ca− (and thus often produce anti-Ca antibody) become sensitized to Aa and Qa at a significantly lower rate than mares that are Ca+.259 This is attributed to anti-Ca antibody being produced by Ca− mares. The anti-Ca antibody may more rapidly remove potentially sensitizing cells from the circulation before they stimulate production of antibody against Aa or Qa.
Other antigens infrequently have been associated with NI in foals. These include factors Db, Dc, Dg, Ua, Pa, Qc, and Ab.226,228,260-263 Two other factors, R and S, have been described to be associated with NI, but they are not detected by routine hemolytic or agglutinating methods and are only detected using an antiglobulin test (direct Coombs’ test).248Because of the difficulty in testing for these antigens, there has never been sufficient agreement between laboratories to allow international designation. These antigens may not be detected using the JFA screening test.249 Estimates are that 1 in 2000 pregnancies may result in sensitization against some other antigen besides Aa or Qa. Because these cases occur so infrequently, it is not practical to consider mares without these antigens to be at risk for NI. A blood type evaluation of mares with a history of production of foals with NI should be done to identify the offending antibody/antigen.
Box 53-3 lists laboratories providing NI screening tests (e.g., Aa, Qa, and Ca typing and screening of sera for alloantibody). Routine agglutinating crossmatch tests using mare serum or colostrum and foal or sire cells can be performed by most veterinary or human hematology laboratories.
Ruminants
Neonatal isoerythrolysis is not a naturally occurring disease in cattle, sheep, or goats. Its occurrence in cattle has been associated with administration of vaccines derived from blood, such as certain anaplasmosis and babesiosis vaccines.226 When used on breeding females, these vaccines may sensitize the dam to certain blood groups, most often in the A and F systems. Under chance circumstances, if the blood types of the sire and offspring reflect these systems and the dam has produced alloantibodies, an isoimmune hemolytic crisis may appear in the calf associated with successful passive transfer. Hemolytic crises are rare in sheep and are even difficult to induce experimentally.226
EQUINE NEONATAL ALLOIMMUNE THROMBOCYTOPENIA
Definition and Etiology
Neonatal alloimmune thrombocytopenia (NAIT) is a condition characterized by the destruction of platelets in the circulation of a foal by alloantibodies of maternal origin absorbed from colostrum.264 The syndrome has been observed in horse and mule foals.251,264,265 The prevalence of NAIT is not known. Some foals may be asymptomatic, and the condition may be self-limiting as alloantibody is metabolized.
Clinical Signs and Differential Diagnoses
Few clinical signs may appear unless foals are traumatized. Affected foals may have prolonged bleeding from venipuncture sites. Petechial hemorrhages may not be present. Other conditions often associated with thrombocytopenia in neonates include sepsis, disseminated intravascular coagulation, equine infectious anemia, drug-induced thrombocytopenias, and angiopathies. The challenge is to determine whether the thrombocytopenia is primarily caused by allogeneic antibodies or secondary to some other disease process. For differential diagnoses of thrombocytopenia in neonatal foals, see Chapter 19.
Clinical Pathology
Profound thrombocytopenia in the absence of other hematologic changes typifies uncomplicated NAIT. Evidence of successful passive transfer is present based on quantification of serum IgG. Affected foals are thrombocytopenic. Thrombocyte counts less than 10,000/μL have been observed. Prolonged bleeding from venipuncture sites and petechiae may be present.264 Demonstration of significant amounts of antibody in the colostrum (or serum from the mare) that are directed against platelet antigens expressed by the foal provides a definitive diagnosis of NAIT. However, assays for equine platelet-bindable and platelet-associated immunoglobulins are not routinely available.
Pathophysiology
The pathophysiology is believed to mirror that of neonatal isoerythrolysis. In mares sensitized to platelet antigens, alloantibodies are concentrated in the colostrum late in gestation. These antibodies are passed to the foal through passive transfer. If the foal’s platelets carry the antigen that the antibody recognizes, the platelets become antibody coated. Subsequently, they are removed prematurely by the reticuloendothelial system. Platelet antigens have not been characterized in horses; however, platelet-associated antibodies have been demonstrated in affected foals. Circulating antibody is removed by attachment to platelets and rapid clearance by the reticuloendothelial system.
Treatment and Prognosis
Circulating antibody is removed relatively quickly, and treatment may not be necessary. Platelet-rich plasma may be indicated in cases of serve thrombocytopenia accompanied by clinical signs of bleeding problems. As offending antibody is removed, however, the problem will tend to be self-limiting.
1 Perryman LE. Primary and secondary immune deficiencies of domestic animals. Adv Vet Sci Comp Med. 1979;23:23.
2 Perryman LE. Immunological management of young foals. Compend Cont Educ (Pract Vet). 1981;3:S223.
3 Lo JW, McClure JJ. Surface markers of lymphocytes. In: Barta O, editor. Laboratory techniques of veterinary clinical immunology. Springfield, Ill: Charles C Thomas, 1984.
4 Lunn DP, Holmes MA, Antczak DF, et al. Report of the Second Equine Leukocyte Antigen Workshop. Vet Immunol Immunopathol. 1998;42:3.
5 Perryman LE. Primary immunodeficiencies of horses. Vet Clin North Am Equine Pract. 2000;16:105.
6 Halliwell REW, Gorman NT. Diseases associated with immunodeficiency. In: Halliwell REW, Gorman NT, editors. Veterinary clinical immunology. Philadelphia: Saunders, 1989.
7 Sellon DC. Secondary immunodeficiencies of horses. Vet Clin North Am Equine Pract. 2000;16:117.
8 Hines MT, Schott HC. Exercise and immune function. Proceedings of the Twelfth Annual Veterinary Medical Forum, San Francisco. 1994.
9 Hines MT, Palmer GH, Byrne KM, et al. Quantitative characterization of lymphocyte populations in bronchoalveolar lavage fluid and peripheral blood of normal adult Arabian horses. Vet Immunol Immunopathol. 1996;51:29.
10 Smith RIII, Chaffin MK, Cohen ND, et al. Age-related changes in lymphocyte subsets of Quarter horse foals. Am J Vet Res. 2002;63:531.
11 Rumbaugh GE, Ardans AA, Ginno D, et al. Measurement of neonatal equine immunoglobulins for assessment of colostral immunoglobulin transfer: comparison of single radial immunodiffusion with zinc sulfate turbidity test, serum electrophoresis, refractometry for total serum protein, and the sodium sulfite precipitation test. J Am Vet Med Assoc. 1978;172:321.
12 Tennant B, Baldwin BH, Braun RK, et al. Use of the glutaraldehyde coagulation test for detection of hypogammaglobulinemia in neonatal calves. J Am Vet Med Assoc. 1979;174:848.
13 Buening GM, Perryman LE, McGuire TC. Practical methods of determining serum immunoglobulin M and immunoglobulin G concentrations in foals. J Am Vet Med Assoc. 1977;171:455.
14 Hodgin EC, McGuire TC, Perryman LE, et al. Evaluation of delayed hypersensitivity responses in normal horses and immunodeficient foals. Am J Vet Res. 1978;39:1161.
15 Enright FM, Jeffers GW. Function tests for mononuclear phagocytes. In: Barta O, editor. Laboratory techniques of veterinary clinical immunology. Springfield, Ill: Charles C Thomas, 1984.
16 Raidal SL, Bailey GD, Love DN. The flow cytometric evaluation of phagocytosis by equine peripheral blood neutrophils and pulmonary alveolar macrophages. Vet J. 1998;156:107.
17 Raidal SL, Bailey GD, Love DN. Flow cytometric determination of oxidative burst activity of equine peripheral blood and bronchoalveolar lavage-derived leukocytes. Vet J. 1998;156:117.
18 McGuire TC, Crawford TB, Hallowell AL, et al. Failure of colostral immunoglobulin transfer as an explanation for most infections and deaths of neonatal foals. J Am Vet Med Assoc. 1977;170:1302.
19 Baldwin JL, Cooper WL, Vanderwall DK, et al. Prevalence (treatment days) and severity of illness in hypogammaglobulinemic and normogammaglobulinemic foals. J Am Vet Med Assoc. 1991;198:423.
20 Clabough DL, Levine JF, Grant GL, et al. Factors associated with failure of passive transfer of colostral antibodies in standardbred foals. J Vet Intern Med. 1991;5:335.
21 Kohn CW, Knight D, Hueston W, et al. Colostral and serum IgG, IgA, and IgM concentrations in standardbred mares and their foals at parturition. J Am Vet Med Assoc. 1989;195:64.
22 Morris DD, Meirs DA, Merryman GS. Passive transfer failure in horses: incidence and causative factors on a breeding farm. Am J Vet Res. 1985;46:2294.
23 Perryman LE, McGuire TC. Evaluation for immune system failures in horses and ponies. J Am Vet Med Assoc. 1980;176:1374.
24 Raidal SL. The incidence and consequences of failure of passive transfer of immunity on a thoroughbred breeding farm. Aust Vet J. 1996;73:201.
25 Stoneham SJ, Wingfield Digby NJ, Ricketts SW. Failure of passive transfer of colostral immunity in the foal: incidence, and the effect of stud management and plasma transfusions. Vet Rec. 1991;128:416.
26 Jeffcott LB. Duration of permeability of the intestine to macromolecules in the newly-born foal. Vet Rec. 1971;88:340.
27 Jeffcott LB. Studies on passive immunity in the foal. II. The absorption of 125I-labelled PVP (polyvinyl pyrrolidone) by the neonatal intestine. J Comp Pathol. 1974;84:279.
28 Jeffcott LB, Jeffcott TJ. Studies on passive immunity in the foal. III. The characterization and significance of neonatal proteinuria. J Comp Pathol. 1974;84:455.
29 Jeffcott LB. Passive transfer of immunity to foals. In Robinson NE, editor: Current therapy in equine medicine, ed 2, Philadelphia: Saunders, 1987.
30 Raidal SL, McTaggart C, Penhale J. Effect of withholding macromolecules on the duration of intestinal permeability to colostral IgG in foals. Aust Vet J. 2005;83:78.
31 Higgins WP, Gillespie JH, Robson DS. Studies of maternally-acquired antibodies in the foal to equine influenza a1 and a2, and equine rhinopneumonitis. Equine Vet Sci. 1987;7:207.
32 Jeffcott LB. Studies on passive immunity in the foal. 1. Gamma-globulin and antibody variations associated with the maternal transfer of immunity and the onset of active immunity. J Comp Pathol. 1974;84:93.
33 Reilly WJ, Macdougall DF. The metabolism of IgG in the newborn foal. Res Vet Sci. 1973;14:136.
34 Rouse BT. The immunoglobulins of adult equine and foal sera: a quantitative study. Br Vet J. 1971;127:45.
35 LeBlanc MM. Immunologic considerations. In: Koterba AM, Drummond WN, Kosch PC, editors. Equine clinical neonatology. Philadelphia: Lea & Febiger, 1990.
36 Reiter B. Review of nonspecific antimicrobial factors in colostrum. Ann Rech Vet. 1978;9:205.
37 Jeffcott LB. Some practical aspects of the transfer of passive immunity to newborn foals. Equine Vet J. 1974;6:109.
38 Sandgild PT. Uptake of colostral immunoglobulins by the compromised newborn farm animal. Acta Vet Scand Suppl. 2003;98:105.
39 Halliday R. Failure of some hill lambs to absorb maternal gammaglobulin. Nature. 1965;205:614.
40 Gillette DD, Filkins M. Factors affecting antibody transfer in the newborn puppy. Am J Physiol. 1966;210:419.
41 Carrick JB, Pollitt CC, Thompson HL, et al. Failure of the administration of ACTH to affect the absorption of colostral immunoglobulin in neonatal foals. Equine Vet J. 1987;19:545.
42 LeBlanc MM. Update on passive transfer of immunoglobulins in the foal. Pferdeheilkunde. 2001;17:662.
43 Cowles RR, Copeland DD, Conboy HS, et al. A new test for the detection of immunoglobulin levels in the neonatal foal. Proc Am Assoc Equine Pract. 1983;29:415.
44 Metzger N, Hinchcliff KW, Hardy J, et al. Usefulness of a commercial equine IgG test and serum protein concentration as indicators of failure of transfer of passive immunity in hospitalized foals. J Vet Intern Med. 2006;20:382.
45 Davis R, Giguere S. Evaluation of five commercially available assays and measurement of serum total protein concentration via refractometry for the diagnosis of failure of passive transfer of immunity. J Am Vet Med Assoc. 2005;227:1640.
46 Davis DG, Schaefer DM, Hinchcliff KW, et al. Measurement of serum IgG in foals by radial immunodiffusion and automated turbidimetric immunoassay. J Vet Intern Med. 2005;19:93.
47 Erhard MH, Luft C, Remler HP, et al. Assessment of colostral transfer and systemic availability of immunoglobulin G in newborn foals using a newly developed enzyme-linked immunosorbent assay (ELISA) system. J Anim Physiol Anim Nutr (Berl). 2001;85:164.
48 Holmes MA, Lunn DP. A study of bovine and equine immunoglobulin levels in pony foals fed bovine colostrum. Equine Vet J. 1991;23:116.
49 Lavoie JP, Spensley MS, Smith BP, et al. Complement activity and selected hematologic variables in newborn foals fed bovine colostrum. Am J Vet Res. 1989;50:1532.
50 Lavoie JP, Spensley MS, Smith BP, et al. Absorption of bovine colostral immunoglobulins G and M in newborn foals. Am J Vet Res. 1989;50:1598.
51 Vivrette SL, Young K, Manning S. Efficacy of Seramune in the treatment of failure of passive transfer in foals. Proc Am Assoc Equine Pract. 1998;44:136.
52 Vivrette S. Colostrum and oral immunoglobulin therapy in newborn foals. Compend Cont Educ (Pract Vet). 2001;23:286.
53 Burton SC, Hintz HF, Kemen MJ, et al. Lyophilized hyperimmune equine serum as a source of antibodies for neonatal foals. Am J Vet Res. 1981;42:308.
54 Liu IK, Brown C, Myers RC, et al. Evaluation of intravenous administration of concentrated immunoglobulin G to colostrum-deprived foals. Am J Vet Res. 1991;52:709.
55 Franz LC, Landon JC, Lopes LA, et al. Oral and intravenous immunoglobulin therapy in neonatal foals. Equine Vet J. 1991;23:116.
56 Hammer CJ, Booth JA, Etzel L, et al. Adequacy of a concentrated equine serum product in preventing failure of immune passive transfer in neonatal foals: preliminary study. Equine Vet J. 2001;33:734.
57 Bailey E, Conboy HS, McCarthy PF. Neonatal isoerythrolysis of foals: an update on testing. Proc Am Assoc Equine Pract. 1987;33:341.
58 Scott AM, Jeffcott LB. Haemolytic disease of the newborn foal. Vet Rec. 1978;103:71.
59 Stormont C. Neonatal isoerythrolysis in domestic animals: a comparative review. Adv Vet Sci Comp Med. 1975;19:23-45.
60 Perryman LE, Crawford TB. Diagnosis and management of immune system failures of foals. Proc Am Assoc Equine Pract. 1979;25:235.
61 Koterba AM, Brewer B, Drummond WH. Prevention and control of infection. Vet Clin North Am Equine Pract. 1985;1:41.
62 White S. Exogenous IgG in the treatment of foals with failure of passive transfer and/or sepsis. Proc Am Coll Vet Intern Med. 1988;6:145.
63 LeBlanc MM, McLaurin BI, Boswell R. Relationships among serum immunoglobulin concentration in foals, colostral specific gravity, and colostral immunoglobulin concentration. J Am Vet Med Assoc. 1986;189:57.
64 Cash RSG. Colostral quality determined by refractometry. Equine Vet Educ. 1999;11:36.
65 Chavatte P, Clement F, Cash R, et al. Field determination of colostrum quality by a novel, practical method. Proc Am Assoc Equine Pract. 1998;44:206.
66 Madigan JE. Method for preventing neonatal septicemia, the leading cause of death in the neonatal foal. Proc Am Assoc Equine Pract. 1997;43:17.
67 Brewer BD. Neonatal infection. In: Koterba AM, Drummond WN, Kosch PC, editors. Equine clinical neonatology. Philadelphia: Lea & Febiger, 1990.
68 McGuire TC, Poppie MJ, Banks KL. Combined (B and T lymphocyte) immunodeficiency: a fatal genetic disease in Arabian foals. J Am Vet Med Assoc. 1974;164:70.
69 Perryman LE, Boreson CR, Conaway MW, et al. Combined immunodeficiency in an Appaloosa foal. Vet Pathol. 1984;21:547.
70 Perryman LE, Torbeck RL. Combined immunodeficiency of Arabian horses: confirmation of autosomal recessive mode of inheritance. J Am Vet Med Assoc. 1980;176:1250.
71 Shin EK, Perryman LE, Meek K: Evaluation of a test for identification of Arabian horses heterozygous for the severe combined immunodeficiency trait, J Am Vet Med Assoc 211:1268
72 Mair TS, Taylor FG, Harbour DA, et al. Concurrent Cryptosporidium and Coronavirus infections in an Arabian foal with combined immunodeficiency syndrome. Vet Rec. 1990;126:127.
73 Don-van’t Slot HP, van der Kolk JH. Severe combined immunodeficiency disease (SCID) in the Arabian horse. Tijdschr Diergeneeskd. 2000;125:577.
74 Harvey JW. Normal hematologic values. In: Koterba AM, Drummond WN, Kosch PC, editors. Equine clinical neonatology. Philadelphia: Lea & Febiger, 1990.
75 Tizard I. Immunity in the fetus and newborn. In Tizard I, editor: Veterinary immunology: an introduction, ed 3, Philadelphia: Saunders, 1987.
76 Hodgin EC, McGuire TC, Perryman LE, et al. Evaluation of delayed hypersensitivity responses in normal horses and immunodeficient foals. Am J Vet Res. 1978;39:1161.
77 Lew AM, Hosking CS, Studdert MJ. Immunologic aspects of combined immunodeficiency disease in Arabian foals. Am J Vet Res. 1980;41:1161.
78 McGuire TC, Banks KL, Poppie MJ. Combined immunodeficiency in horses: characterization of the lymphocyte defect. Clin Immunol Immunopathol. 1975;3:555.
79 Wiler R, Leber R, Moore BB, et al. Equine severe combined immunodeficiency: a defect in V(D)J recombination and DNA-dependent protein kinase activity. Proc Natl Acad Sci USA. 1995;92:11485.
80 McGuire TC, Banks KL, Davis WC. Alterations of the thymus and other lymphoid tissue in young horses with combined immunodeficiency. Am J Pathol. 1976;84:39.
81 Perryman LE. Primary and secondary immune deficiencies of domestic animals. Adv Vet Sci Comp Med. 1979;23:23.
82 Perryman LE. Molecular pathology of severe combined immunodeficiency in mice, horses, and dogs. Vet Pathol. 2004;41:95.
83 Leber R, Wiler R, Perryman LE, et al. Equine SCID: mechanistic analysis and comparison with murine SCID. Vet Immunol Immunopathol. 1998;65:1.
84 Yilma T, Perryman LE, McGuire TC. Deficiency of interferon-γ but not interferon-β in Arabian foals with severe combined immunodeficiency. J Immunol. 1982;129:931.
85 Banks KL, McGuire TC. Surface receptors on neutrophils and monocytes from immunodeficient and normal horses. Immunology. 1975;28:581.
86 Lunn DP, McClure JT, Schobert CS, et al: Abnormal patterns of equine leucocyte differentiation antigen expression in severe combined immunodeficiency foals suggests the phenotype of normal equine natural killer cells, Immunology 84:495
87 Swinburne J, Lockhart L, Scott M, et al. Estimation of the prevalence of severe combined immunodeficiency disease in UK Arab horses as determined by a DNA-based test. Vet Rec. 1999;16:79.
88 Bernoco D, Bailey E. Frequency of the SCID gene among Arabian horses in the USA. Anim Genet. 1998;29:41.
89 Bue CM, Davis WC, Magnuson NS, et al. Correction of equine severe combined immunodeficiency by bone marrow transplantation. Transplantation. 1986;42:14.
90 Perryman LE, McGuire TC, Hilbert BJ. Selective immunoglobulin M deficiency in foals. J Am Vet Med Assoc. 1977;170:212.
91 Dopson LC, Reed SM, Roth JA, et al. Immunosuppression associated with lymphosarcoma in two horses. J Am Vet Med Assoc. 1983;182:1239.
92 Perryman LE, Crisman MV. Immune deficiency syndromes. In Robinson NE, editor: Current therapy in equine medicine, ed 2, Philadelphia: Saunders, 1987.
93 Weldon AD, Zhang C, Antczak DJ, et al. Selective IgM deficiency and abnormal B cell response in a foal. J Am Vet Med Assoc. 1992;201:1396.
94 Perryman LE, Wyatt DR, Magnuson NS. Biochemical and functional characterization of lymphocytes from a horse with lymphosarcoma and IgM deficiency. Comp Immunol Microbiol Infect Dis. 1984;7:53.
95 McGuire TC, Poppie MJ, Banks KL. Hypogammaglobulinaemia predisposing to infection in foals. J Am Vet Med Assoc. 1975;166:71.
96 Banks KL, McGuire TC, Jerrells TR. Absence of B lymphocytes in a horse with primary agammaglobulinemia. Clin Immunol Immunopathol. 1976;5:282.
97 Deem DA, Traver DS, Thacker HL, et al. Agammaglobulinemia in a horse. J Am Vet Med Assoc. 1979;175:469.
98 McGuire TC, Banks KL, Evans DR, et al. Agammaglobulinemia in a horse with evidence of functional T lymphocytes. Am J Vet Res. 1976;37:41.
99 Rosen FS, Janeway CA. The γ-globulins. III. The antibody deficiency syndromes. N Engl J Med. 1966;275:709.
100 Scholes SF, Holliman A, May PD, et al. A syndrome of anaemia, immunodeficiency and peripheral ganglionopathy in Fell pony foals. Vet Rec. 1998;142:128.
101 Butler CM, Westermann CM, Koeman JP, et al. The Fell pony immunodeficiency syndrome also occurs in the Netherlands: a review and six cases. Tijdschr Diergeneeskd. 2006;131:114.
102 Gardner RB, Hart KA, Stokol T, et al. Fell Pony Syndrome in a pony in North America. J Vet Intern Med. 2006;20:198.
103 Thomas GW, Bell SC, Carter SD. Immunoglobulin and peripheral B-lymphocyte concentrations in Fell pony foal syndrome. Equine Vet J. 2005;37:48.
104 Thomas GW, Bell SC, Phythian C, et al. Aid to the antemortem diagnosis of Fell pony foal syndrome by the analysis of B lymphocytes. Vet Rec. 2003;152:618.
105 Bell SC, Savidge C, Taylor P, et al. An immunodeficiency in Fell ponies: a preliminary study into cellular responses. Equine Vet J. 2001;33:687.
106 Flaminio MJBF, LaCombe V, Kohn CW, et al. Common variable immunodeficiency in a horse. J Am Vet Med Assoc. 2002;221:1296.
107 Pellegrini-Masini A, Bentz AI, Johns IC, et al. Common variable immunodeficiency in three horses with presumptive bacterial meningitis. J Am Vet Med Assoc. 2005;227:114.
108 Franklin RP, Long MT, MacNeill A, et al. Proliferative interstitial pneumonia, Pneumocystis carinii infection, and immunodeficiency in an adult Paso Fino horse. J Vet Intern Med. 2002;16:607.
109 MacLeay JM, Ames TR, Hayden DW, et al. Acquired B lymphocyte deficiency and chronic enterocolitis in a 3-year-old quarter horse. Vet Immunol Immunopathol. 1997;57:49.
110 Boy MG, Zhang C, Antczak DF, et al. Unusual selective immunoglobulin deficiency in an Arabian foal. J Vet Intern Med. 1992;6:201.
111 Spickett GP. Current perspectives on common variable immunodeficiency (CVID). Clin Exp Allergy. 2001;31:536.
112 Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34.
113 Lobetti R. Common variable immunodeficiency in miniature dachshunds affected with Pneumocystis carinii pneumonia. J Vet Diagn Invest. 2000;12:39.
114 Freestone JF, Hietala S, Moulton J, et al. Acquired immunodeficiency in a 7-year-old horse. J Am Vet Med Assoc. 1987;190:689.
115 Flaminio MJ, Rush BR, Cox JH, et al. CD4+ and CD8+ T-lymphocytopenia in a filly with Pneumocystis carinii pneumonia. Aust Vet J. 1998;76:399.
116 Olsen RG. Immune dysfunctions associated with viral infections. Compend Cont Educ (Pract Vet). 1984;6:422.
117 Bryans JT, Swerczek TW, Darlington RW, et al. Neonatal foal disease associated with perinatal infection by equine herpesvirus 1. J Equine Med Surg. 1977;1:20.
118 McClure JJ, Addison JD, Miller RI. Immunodeficiency manifested by oral candidiasis and bacterial septicemia. J Am Vet Med Assoc. 1985;186:195.
119 Wilson JH, Cudd TA. Common gastrointestinal diseases. In: Koterba AM, Drummond WN, Kosch PC, editors. Equine clinical neonatology. Philadelphia: Lea & Febiger, 1990.
120 Ahmed SA, Furr M, Chickering WR, et al. Immunologic studies of a horse with lymphosarcoma. Vet Immunol Immunopathol. 1993;38:229.
121 Furr MO, Crisman MV, Robertson J, et al. Immunodeficiency associated with lymphosarcoma in a horse. J Am Vet Med Assoc. 1992;201:307.
122 Biberstein EL, Jang SS, Hirsh DC. Nocardia asteroides infection in horses: a review. J Am Vet Med Assoc. 1985;186:273.
123 Dopson LC, Reed SM, Roth JA, et al. Immunosuppression associated with lymphosarcoma in two horses. J Am Vet Med Assoc. 1983;182:1239.
124 Buechner-Maxwell V, Zhang C, Robertson J, et al. Intravascular leukostasis and systemic aspergillosis in a horse with subleukemic acute myelomonocytic leukemia. J Vet Intern Med. 1994;8:258.
125 Edwards DF, Parker JW, Wilkinson JE, et al. Plasma cell myeloma in the horse. J Vet Intern Med. 1993;7:169.
126 Oken MM. Multiple myeloma. Med Clin North Am. 1984;68:757.
127 Kent JE, Roberts CA. Serum protein changes in four horses with monoclonal gammopathy. Equine Vet J. 1990;22:373.
128 Traub-Dargatz J, Bertone A, Bennett D. Monoclonal aggregating immunoglobulin cryoglobulinemia in a horse with malignant lymphoma. Equine Vet J. 1985;17:470.
129 Matus RE, Leifer CE. Immunoglobulin-producing tumors. Vet Clin North Am Small Anim Pract. 1985;15:741.
130 Kono Y, Hirasawa K, Fukunaga Y, et al. Recrudescence of equine infectious anemia by treatment with immunosuppressive drugs. Natl Inst Anim Health Q Tokyo. 1976;16:8.
131 Mair TS. Bacterial pneumonia associated with corticosteroid therapy in three horses. Vet Rec. 1996;138:205.
132 Cohn LA. The influence of corticosteroids on host defense mechanisms. J Vet Intern Med. 1991;5:95.
133 Schaffner A, Schaffner T. Glucocorticoid-induced impairment of macrophage antimicrobial activity: mechanisms and dependence on the state of activation. Rev Infect Dis. 1987;9(suppl):620.
134 Wong CW, Smith SE, Thong YH, et al. Effects of exercise stress on various immune functions in horses. Am J Vet Res. 1992;53:1414.
135 Hines MT, Schott HC, Bayly WM, et al. Exercise and immunity: a review with emphasis on the horse. J Vet Intern Med. 1996;10:280.
136 Horohov DW, Dimock A, Guirnalda P, et al. Effect of exercise on the immune response of young and old horses. Am J Vet Res. 1999;60:643.
137 Robson PJ, Alston TD, Myburgh KH. Prolonged suppression of the innate immune system in the horse following an 80 km endurance race. Equine Vet J. 2003;35:133.
138 Escribano BM, Aguera EI, Vivo R, et al. Benefits of moderate training to the nonspecific immune response of colts. Equine Vet J Suppl. 2002;34:182.
139 Raidal SL, Rose RJ, Love DN. Effects of training on resting peripheral blood and BAL-derived leucocyte function in horses. Equine Vet J. 2001;33:238.
140 Raidal SL, Love DN, Bailey GD, et al. Effect of single bouts of moderate and high intensity exercise and training on equine peripheral blood neutrophils function. Res Vet Sci. 2000;68:141.
141 Folsom RW, Littlefield-Chabaud MA, French DD, et al. Exercise alters the immune response to equine influenza virus and increases susceptibility to infection. Equine Vet J. 2001;33:664.
142 Lunn DP, Hussey S, Sebing R, et al. Safety, efficacy, and immunogenicity of a modified-live equine influenza virus vaccine in ponies after induction of exercise-induced immunosuppression. J Am Vet Med Assoc. 2001;218:900.
143 Gross DK, Hinchcliff KW, French PS, et al. Effect of moderate exercise on the severity of clinical signs associated with influenza virus infection in horses. Equine Vet J. 1998;30:489.
144 Prescott JF. Immunodeficiency and serious pneumonia in foals: the plot thickens. Equine Vet J. 1993;35:88.
145 Wagner B, Flaminio JB, Hillegas J, et al. Occurrence of IgE in foals: evidence for transfer of maternal IgE by the colostrum and late onset of endogenous IgE production in the horse. Vet Immunol Immunopathol. 2006;110:269.
146 Breathnach CC, Sturgill-Wright T, Stiltner JL, et al. Foals are interferon gamma—deficient at birth. Vet Immunol Immunopathol. 2006;112:199.
147 Horohov DW, Kydd JH, Hannant D. The effect of aging on T cell responses in the horse. Dev Comp Immunol. 2002;26:121.
148 Barrington GM, Parish SM. Neonatal immunology. Vet Clin North Am Food Anim Pract. 2001;17:463.
149 McEwan AD, Fisher EW, Selman IE. A turbidity test for the estimation of immune globulin levels in neonatal calf serum. Clin Chem Acta. 1970;27:155.
150 Barrington GM, Besser TE, Gay CC, et al. Effect of prolactin on in vitro expression of the bovine mammary immunoglobulin G1 receptor. J Dairy Sci. 1997;80:94.
151 Barrington GM, Besser TE, Gay CC, et al. Regulation of the immunoglobulin G1 receptor: effect of prolactin on in vivo expression of the bovine mammary immunoglobulin G1 receptor. J Endocrinol. 1999;163:25.
152 Winger K, Gay CC, Besser TE. Immunoglobulin G1 transfer into induced mammary secretions: the effect of dexamethasone. J Dairy Sci. 1995;78:1306.
153 Butler JE. Bovine immunoglobulins: an augmented review. Vet Immunol Immunopathol. 1983;4:43.
154 Besser TE, Gay CC. Septicemic colibacillosis and failure of passive transfer of colostral immunoglobulins in calves. Vet Clin North Am Food Anim Pract. 1985;1:445.
155 Stott GH, Marx DB, Menefee BE, et al. Colostral immunoglobulin transfer in calves. II. The rate of absorption. J Dairy Sci. 1979;62:1766.
156 Stott GH, Marx DB, Menefee BE, et al. Colostral immunoglobulin transfer in calves. III. Amount of absorption. J Dairy Sci. 1979;62:1902.
157 Besser TE, Garmendia AE, McGuire TC, et al. Effect of colostral immunoglobulin G1 and immunoglobulin M concentrations on immunoglobulin absorption in calves. J Dairy Sci. 1985;68:2033.
158 Logen EF. Colostral immunity to colibacillosis in the neonatal calf. Br Vet J. 1974;130:405.
159 Gay CC. Failure of passive transfer of colostral immunoglobulins and neonatal disease in calves: a review. Proceedings of the 4th International Symposium on Neonatal Diarrhea, Saskatoon, Saskatchewan, Canada. Veterinary Infectious Disease Organization; 1983:346.
160 Gay CC. The role of colostrum in managing calf health. Bovine Pract. 1984;16:79.
161 Pritchett LC, Gay CC, Besser TE, et al. Management and production factors influencing immunoglobulin G1 concentration in colostrum from Holstein cows. J Dairy Sci. 1991;74:2336.
162 Shell TM, Early RJ, Carpenter JR, et al. Prepartum nutrition and solar radiation in beef cattle. II. Residual effects on post-partum milk yield, immunoglobulin, and calf growth. J Anim Sci. 1995;73:1303.
163 Petrie L, Acres SD, McCartney DH, et al. The yield of colostrum and colostral gammaglobulins in beef cows and the absorption of colostral gammaglobulins by beef calves. Can Vet J. 1984;25:273.
164 Field RW, Bretzlaff KN, Elmore RG, et al. Effect of induction of parturition on immunoglobulin content of colostrum and calf serum. Theriogenology. 1989;32:501.
165 Stott GH, Fleemor WA, Kleese WC. Colostral immunoglobulin concentration in fractions of first milking postpartum and five additional milkings. J Dairy Sci. 1981;64:459.
166 Besser TE, Gay CC, Pritchett L. Comparison of three methods of feeding colostrum to dairy calves. J Am Vet Med Assoc. 1991;198:419.
167 Perino LJ. A guide to colostrum management in beef cows and calves. Vet Med. 1997. January
168 Stott GH, Marx DB, Menefee BE, et al. Colostral immunoglobulin transfer in calves. I. Period of absorption. J Dairy Sci. 1979;62:1632.
169 Stott GH, Wiersma F, Menefee BE, et al. Influence of environment on passive immunity in calves. J Dairy Sci. 1976;59:1306.
170 McEwan AD, Fisher EW, Selman IE. Observations on the immune globulin levels of neonatal calves and their relationship to disease. J Comp Pathol. 1970;80:259.
171 Penhale WJ, Christie G, McEwan AD, et al. Quantitative studies on bovine immunoglobulins. II. Plasma immunoglobulin levels in market calves and their relationship to neonatal infection. Br Vet J. 1970;126:30.
172 Hudgens AR, Tyler JW, Besser TE, et al. Optimizing performance of a qualitative zinc sulfate turbidity test for passive transfer of immunoglobulin G in calves. Am J Vet Res. 1996;12:1711.
173 Tyler JW, Hancock DD, Parish SM, et al. Evaluation of 3 assays for failure of passive transfer in calves. J Vet Intern Med. 1996;10:304.
174 Stone SS, Gitler M. The validity of the sodium sulfite test for detecting immunoglobulins in calf serum. Br Vet J. 1969;125:68.
175 Tennant B, Baldwin BH, Braum RK, et al. Use of the glutaraldehyde coagulation test for detection of hypogammaglobulinemia in neonatal calves. J Am Vet Med Assoc. 1979;174:848.
176 Beetson SA, Hilbert BJ, Mills JN. The use of the glutaraldehyde coagulation test for the detection of hypogammaglobulinemia in neonatal foals. Aust Vet J. 1985;62:279.
177 Carstairs-Grant SJ, Crawshaw GJ, Mehren KG. A comparison of the glutaraldehyde coagulation test and total serum protein estimation as indicators of gamma globulin levels in neonatal ruminants. J Zoo Anim Med. 1988;19:14.
178 Clabough DL, Conboy HS, Roberts MC. Comparison of four screening techniques for detection of equine neonatal hypogammaglobulinemia. J Am Vet Med Assoc. 1989;194:1717.
179 Tyler JW, Besser TE, Wilson L, et al. Evaluation of a whole blood glutaraldehyde coagulation test for the detection of failure of passive transfer in calves. J Vet Intern Med. 1996;10:82.
180 Naylor JM, Kronfeld DS. Refractometry as a measure of the immunoglobulin status of the newborn dairy calf: comparison with the zinc sulfate turbidity test and single radial immunodiffusion. J Am Vet Med Assoc. 1977;171:1331.
181 Braun JP, Tainturier D, Laugier C, et al. Early variations of blood plasma γ-glutamyltransferase in newborn calves: a test of colostrum intake. J Dairy Sci. 1982;65:2178.
182 Paris TNN, Ouedraogo G, Rico AG, et al. Use of serum γ-glutamyltransferase and total blood proteins for monitoring the colostrum intake of newborn calves. Recueil Med Vet. 1992;168:43.
183 Parish SM, Tyler JW, Besser TE, et al. Prediction of serum IgG1 concentration in Holstein calves using serum gamma glutamyltransferase activity. J Vet Intern Med. 1997;11:344.
184 Fleenor WA, Stott GH. Hygrometer test for estimation of immunoglobulin concentration in bovine colostrum. J Dairy Sci. 1980;63:973.
185 Mechor GD, Grohn YT, McDowell LR, et al. Specific gravity of bovine colostrum immunoglobulins as affected by temperature and colostrum components. J Dairy Sci. 1992;75:3131.
186 Besser TE: Unpublished data
187 Weaver DM, Tyler JW, Van Metre DC, et al. Passive transfer of colostral immunoglobulins in calves. J Vet Intern Med. 2000;14:569.
188 Clarkson MJ, Faull WB, Kerry JB. Vaccination of cows with clostridial antigens and passive transfer of clostridial antibodies from bovine colostrum to lambs. Vet Rec. 1985;116:467.
189 Sherman DM, Arendt TD, Gay JM, et al. Comparing the effects of four colostral preparations on serum Ig levels of newborn kids. Vet Med. 1990:908. August
190 Ciupercescu DD. Dynamics of serum immunoglobulin concentrations in sheep during pregnancy and lactation. Res Vet Sci. 1977;22:23.
191 Stubbings DP, Gibbons DF, Tindall JR. Feeding cows’ colostrum to lambs. Vet Rec. 1983;112:88.
192 Perrin G. Bovine colostrum warning. Vet Rec. 1988;122:240.
193 Franken P. Feeding cows’ colostrum to lambs. Vet Rec. 1983;112:332.
194 Brummerstedt E. Immune-deficient animals in biomedical research. Proceedings of the Fifth International Workshop on Immune-Deficient Animals, Copenhagen, 1985. Basel: S Karger; 1987.
195 Mansa B. Hypo-7S-g globinemia in mature cattle. Acta Pathol Microbiol. 1965;63:153.
196 Perryman LE. Primary and secondary immune deficiencies of domestic animals. Adv Vet Sci Comp Med. 1979;23:23.
197 Kulkarni PE. IgG2 deficiency in gangrenous mastitis in cows. Acta Vet Scand. 1971;12:611.
198 Prieur DJ, Collier LL. Chédiak-Higashi syndrome. Am J Pathol. 1978;90:533.
199 Renshaw HW, Davis WC, Fudenberg HH, et al. Leukocyte dysfunction in the bovine homologue of the Chédiak-Higashi syndrome of humans. Infect Immun. 1974;10:928.
200 Hagemoser WA, Roth JA, Lofstedt J, et al. Granulocytopathy in a Holstein heifer. J Am Vet Med Assoc. 1983;183:1093.
201 Nagahata H, Noda H, Takahashi K, et al. Bovine granulocytopathy syndrome: neutrophil dysfunction in Holstein-Friesian calves. Zentralbl Vetinarmed A. 1987;34:445.
202 Kehrli MEJr, Schmalstieg FC, Anderson DC, et al. Molecular definition of the bovine granulocytopathy syndrome: identification of the deficiency of the Mac-1 (CD11b/CD18) glycoprotein. Am J Vet Res. 1990;51:1826.
203 Gilbert RO, Rebuhn WC, Kim CA, et al. Clinical manifestations of leukocyte adhesion deficiency in cattle: 14 cases (1977-1991). J Am Vet Med Assoc. 1993;202:445.
204 Ackermann MR, Kehrli ME, Morfitt DC. Ventral dermatitis and vasculitis in a calf with bovine leukocyte adhesion deficiency. J Am Vet Med Assoc. 1993;202:413.
205 Muscoplat CC, Johnson DW, Stevens JB. Abnormalities of in vitro lymphocyte responses during bovine viral diarrhea virus infection. Am J Vet Res. 1973;34:753.
206 Davies DH, Corbiel L. A humoral suppressor of in vitro lymphocyte transformation responses in cattle with Johne’s disease. Proc Soc Exp Biol Med. 1974;145:1372.
207 Bartram PA, Smith BP, Holmberg C, et al. Combined immunodeficiency in a calf. J Am Vet Med Assoc. 1989;195:347.
208 Mellor AL, Munn DH. Extinguishing maternal immune responses during pregnancy: implications for immunosuppression. Semin Immunol. 2001;13:213.
209 Jamieson DJ, Theiler RN, Rasmussen SA. Emerging infections and pregnancy. Emerg Infect Dis. 2006;12:1638.
210 Sanderson MW, Gnad DP. Biosecurity for reproductive diseases. Vet Clin North Am Food Animal Pract. 2002;18:79.
211 Evermann JF. Pregnancy-associated immunodeficiency. In Smith BP, editor: Large animal internal medicine, ed 3, St Louis: Mosby-Elsevier, 2002.
212 Burton JL, Madsen AS, Chang LC, et al. Gene expression signatures in neutrophils exposed to glucocorticoids: a new paradigm to help explain “neutrophil dysfunction” in parturient dairy cows. Vet Immunol Immunopathol. 2005;105:197.
213 Hill JA. Immunological mechanisms of pregnancy maintenance and failure: a critique of theories and therapy. Am J Reprod Immunol. 1990;22:33.
214 Ragupathy R. Pregnancy: success and failure within the Th1/Th2/Th3 paradigm. Semin Immunol. 2001;13:219.
215 Lippolis JD, Peterson-Burch BD, Reinhardt TA. Differential expression analysis of proteins from neutrophils in the periparturient period and neutrophils from dexamethasone treated dairy cows. Vet Immunol Immunopathol. 2006;111:149.
216 Evermann JF, Ridpath JF. Clinical and epidemiologic observations of bovine viral diarrhea virus in the northwestern United States. Vet Microbiol. 2002;89:129.
217 Traven M, Naslunde K, Linde N, et al. Experimental reproduction of winter dysentery in lactating cows using BCV: comparison with BCV infection in milk-fed calves. Vet Microbiol. 2001;81:127.
218 Wren G. Immunology of the calving cow I. Bovine Vet. 2003:16. October
219 Wren G. Immunology of the calving cow II. Bovine Vet. 2003:25. November-December
220 Kimura K, Goff JP, Kehrli ME, et al. Decreased neutrophil function as a cause of retained placenta in dairy cattle. J Dairy Sci. 2002;85:544.
221 Monge A, Elvira L, Gonzalez JV, et al. Bovine herpesvirus 4 associated with post-partum metritis in a Spanish dairy herd. Res Vet Sci. 2006;80:120.
222 Dinsmore RP. Biosecurity for mammary diseases in dairy cattle. Vet Clin North Am Food Animal Pract. 2002;18:115.
223 Kinsel ML. An epidemiologic approach to investigating abortion problems in dairy herds. Compend Cont Educ (Pract Vet). 2002;24:S43.
224 Evermann JF. Immunology of bovine pregnancy: vulnerability to infectious disease. Bovine Proc. 1994;26:101.
225 Bowling AT, Clark RS. Blood group and protein polymorphism gene frequencies for seven breeds of horses in the United States. Anim Blood Groups Biochem Genet. 1985;16:938.
226 Scott AM, Jeffcott LB. Haemolytic disease of the newborn foal. Equine Vet J. 1978;103:71.
227 Stormont C. Current status of equine blood groups and their applications. Proc Am Assoc Equine Pract. 1972;18:401.
228 Stormont C, Suzuki Y, Rhode EA. Serology of horse blood groups. Cornell Vet. 1964;54:439.
229 Tizard I. Erythrocyte antigens and type II hypersensitivity. In: Tizard I, editor. Veterinary immunology: an introduction. ed 3. Philadelphia: Sanders; 1987:297.
230 Grosclaude F, Guerin G, Houlier G. The genetic map of the B system of cattle blood groups as observed in French breeds. Anim Blood Groups Biochem Genet. 1979;10:199.
231 Tucker EM, Ellory JC. The M-L blood group system and active potassium transport in sheep reticulocytes. Anim Blood Groups Biochem Genet. 1971;2:77.
232 Schmid DO, Odermatt K, Kunz H. Vergleichende analyse der Blutghruppen und einiger polymorpher Proteinsysteme bei funf verschiedenen Ziegenrassen der Schweiz. Schweiz Arch Tierheikld. 1975;117:31.
233 Scott AM. Immunogenetic analysis as a means of identification in horses. Bryans JT, Gerber H, . Equine infectious diseases IV: Proceedings of the Fourth International Conference on Equine Infectious Diseases, Princeton, NJ. Veterinary Publications; 1978:259.
234 Kaminski M. Distribution of genetic variants of blood proteins and enzymes in horses of various breeds. Bryans JT, Gerber H, . Equine infectious diseases IV: Proceedings of the Fourth International Conference on Equine Infectious Diseases, Princeton, NJ. Veterinary Publications; 1978:243.
235 Antczak DF. Lymphocyte alloantigens of the horse. III. ELY-2.1: a lymphocyte alloantigen not coded for by the MHC. Anim Blood Groups Biochem Genet. 2006;15:103.
236 Bailey E, et al. Joint report of the Second International Workshop on Lymphocyte Alloantigens of the Horse, 1982. Anim Blood Groups Biochem Genet. 1984;15:123.
237 Bailey E, Henney PJ. Comparison of ELY-2.1 with blood group and ELY-1 markers in the horse. Anim Blood Groups Biochem Genet. 1984;15:117.
238 Lazary S, Gerber H, deWeck AL, et al. Arnold P: Equine leucocyte antigen system. III. Non-MHC linked alloantigenic system in horses. J Immunogenet. 1982;9:327.
239 Matthews SMS. Equine leucocyte antigen system: progress and potential. Equine Vet J. 1985;17:265.
240 Lazary S, Gerber H, Glatt PA, et al. Equine leucocyte antigens in sarcoid-affected horses. Equine Vet J. 1985;17:283.
241 Meredith D, Elser AH, Wolf B, et al. Equine leukocyte antigens: relationships with sarcoid tumors and laminitis in two pure breeds. Immunogenetics. 1986;23:221.
242 Newman MJ, Antczak DF. Histocompatibility polymorphisms of domestic animals. Adv Vet Sci Comp Med. 1983;27:1.
243 Bailey E. Usefulness of lymphocyte typing to exclude incorrectly assigned paternity in horses. Am J Vet Res. 1984;45:1976.
244 Bowling AT, Egglestone-Stott ML, Byrns G, et al. Validation of microsatellite markers for routine horse parentage testing. Anim Genet. 1997;28:247.
245 Tozaki T, Kakoi H, Mashima S, et al. Population study and validation of paternity testing for thoroughbred horses by 15 microsatellite loci. J Vet Med Sci. 2001;63:1191.
246 Lee SY, Cho GJ. Parentage testing of Thoroughbred horse in Korea using microsatellite DNA. J Vet Sci. 2006;7:63.
247 Luis C, Cothran EG, Oom MM. Microsatellites in Portuguese autochthonous horse breeds: usefulness for parentage testing. Genet Mol Biol. 2002;25:131.
248 Stormont C. Neonatal isoerythrolysis in domestic animals: a comparative review. Adv Vet Sci Comp Med. 1975;19:23.
249 Bailey E. Prevalence of antired blood cell antibodies in the serum and colostrum of mares and its relationship to neonatal isoerythrolysis. Am J Vet Res. 1982;43:1917.
250 Brumpt E. Considerations defavorables a l’hypothese de l’etiologie parasitaire de la jaunisse des muletons: jaundice in the newly born mule foals not caused by Babesia infections. Ann Parasit Hum Comp. 1947;22:5.
251 Traub-Dargatz JL, McClure JJ, Koch C, et al. Neonatal isoerythrolysis in mule foals. J Am Vet Med Assoc. 1995;206:67.
252 Becht JL, Page EH, Morter RL, et al. Experimental production of neonatal isoerythrolysis in the foal. Cornell Vet. 1983;73:380.
253 Kallfelz FA, Whitlock RH, Schultz RD. Survival of Fe-labeled erythrocytes in cross-transfused equine blood. Am J Vet Res. 1978;39:617.
254 Smith JE, Dever M, Smith J, et al. Post-transfusion survival of 50Cr-labeled erythrocytes in neonatal foals. J Vet Intern Med. 1992;6:183.
255 McClure JJ. Strategies for prevention of neonatal isoerythrolysis in horses and mules. Equine Vet Educ. 1997;9:118.
256 Blackmer JM, Paccamonti D, Eilts B, et al. Strategies for preventing neonatal isoerythrolysis. Comp Contin Educ (Pract Vet). 2002;24:562.
257 Bailey E, Conboy HS, McCarthy PF. Neonatal isoerythrolysis of foals: an update on testing. Proc Am Assoc Equine Pract. 1987;33:341.
258 Blackmer JM, Costa LRR, Koch C. The jaundiced foal agglutination test. Vet Tech. 2002;23:577.
259 Bailey E, Albright AG, Henney PJ. Equine neonatal isoerythrolysis: evidence for prevention by maternal antibodies to the Ca blood group antigen. Am J Vet Res. 1988;49:1218.
260 Boyle AG, Magdesian KG, Ruby RE. Neonatal isoerythrolysis in horse foals and a mule foal: 18 cases (1988-2003). J Am Vet Med Assoc. 2005;227:1276.
261 MacLeay JM. Neonatal isoerythrolysis involving the Qc and Db antigens in a foal. J Am Vet Med Assoc. 2001;219:79.
262 Zaruby JF, Hearn P, Colling D. Neonatal isoerythrolysis in a foal, involving anti-Pa alloantibody. Equine Vet J. 1992;24:71.
263 Noda H, Watanabe Y. Relationships between groups and hemolytic disease of newborn foal. Jpn J Zootech Sci. 1975;46:180.
264 Buechner-Maxwell V, Scott MA, Godber L, et al. Neonatal alloimmune thrombocytopenia in a quarter horse foal. J Vet Intern Med. 1997;11:304.
265 Ramirez S, Gaunt SD, McClure JJ, et al. Detection and effects of platelet function of anti-platelet antibody in mule foals with experimentally induced neonatal alloimmune thrombocytopenia. J Vet Intern Med. 1999;13:534.