Chapter 17 Antihistamine drugs
Introduction
Histamine (Fig. 17.1) was isolated in ca. 1908 from an extract of the ergot fungus which had been found to stimulate the uterine muscle of the cat to contract. It was already a known compound but its pharmacological action had not been previously assessed. It was found that histamine was present in the human body and was widely distributed in connective tissues and in the capsular membranes of organs. Histamine is a very polar molecule with two amine functionalities. The primary amine in the side chain has a pKa value of around 9.7 and thus is completely protonated at physiological pH, while the nitrogen in the imidazole ring is a weaker base having a pKa value of around 6, which means that at physiological pH the lone pair on the nitrogen is readily available to interact with electophilic groups. Histamine is stored in mast cells in the form of granules in association with the polysulphate molecule heparin and it seems that electrostatic interaction with the heparin is an important factor in granule formation. Histamine also occurs in basophils in association with another polysulphated polysaccharide chondroitin sulphate. Release of histamine occurs as part of the inflammatory response and is provoked by mediators such as the chemotaxins (small peptides) or by IgE. Histamine acts via four different receptor subtypes: H1–H4. The effects of its interaction with the different receptors are summarised in Table 17.1.
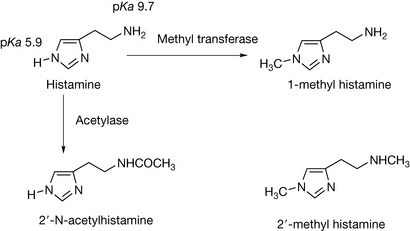
Table 17.1 Histamine receptors
| Receptor | Tissue/cell | Physiological effects |
|---|---|---|
| H1 | Smooth muscle, endothelium, neurological tissue | Dilates blood vessels and increases their permeability, contracts lung and other smooth muscle tissues |
| H2 | Parietal cells Heart tissue | Increased stomach acid secretion, stimulates heart |
| H3 | Located in neural tissue in the CNS | Reduces release of: adrenaline, serotonin and acetyl choline and histamine |
| H4 | Found in immune cells | Modulates the immune response |
The development of drugs affecting the histaminergic response has focused on interactions with H1 and H2 receptors, leading to two main categories of drugs: the antihistamines which are antagonists of the H1 receptor and reduce allergic reactions, and the H2 antagonists which reduce the secretion of stomach acid. The structural requirements for histamine are quite specific and thus its closely related metabolite 1-methylhistamine (Fig. 17.1) is inactive, while substitution with a single methyl group on the side chain producing 2′-methylhistamine reduces activity to about 20%. Other metabolic inactivation products include imidazole acetic acid and 2′-N-acetylhistamine (Fig. 17.1).
Antagonists of histamine H1 receptors
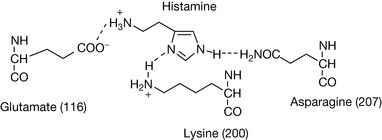
Figure 17.2 shows the amino acid sequence of the histamine H1 receptor. It is a typical transmembrane G-protein with seven membrane-spanning helices (five of the putative hydrophobic/transmembrane regions are shown in blue). Three important amino acid residues have been found to play a role in the binding of histamine: the aspartic acid residue at 116, the lysine residue at 200, and the asparagine residue at 207.1,2 These residues occur in the middle of the hydrophobic transmembrane regions which group together quite closely in space, and the force exerted by the binding of histamine changes the conformation of the G-protein. In the case of the H1 receptor, the G-protein is coupled to a Gq protein which results in the activation of phospholipase C which cleaves phosphatidyl inositol. Figure 17.3 shows the hypothetical interaction of histamine with its receptor.

Figure 17.2 Primary amino acid sequence of H1 receptor showing transmembrane regions in blue and important binding residues for histamine and its antagonists in red.
After the identification of the effects of histamine in the early twentieth century, screening for potential histamine antagonists was carried out. The first effective antihistamine compound to be found was cyclizine, which was discovered in the 1940s (Fig. 17.4). It was effective both in treating allergy and as an antiemetic. Investigations of the phenothiazine class of compounds led to the discovery of promethazine (Fig. 17.4) which is a very effective long-acting antihistamine. Diphenhydramine (Fig. 17.4) was also synthesised in the 1940s. These compounds defined a distinct pharmacophore shown in Figure 17.4 where the important interaction with the aspartate residue at 116 is present but the interactions promoting binding with the lysine and asparagine residues have been replaced by π–π/lipophilic interactions between the aromatic rings in the antagonist and aromatic rings in the fourth and sixth membrane-spanning helices (Fig. 17.5). Thus the agonist binds but does not cause the conformational change required for receptor activation.
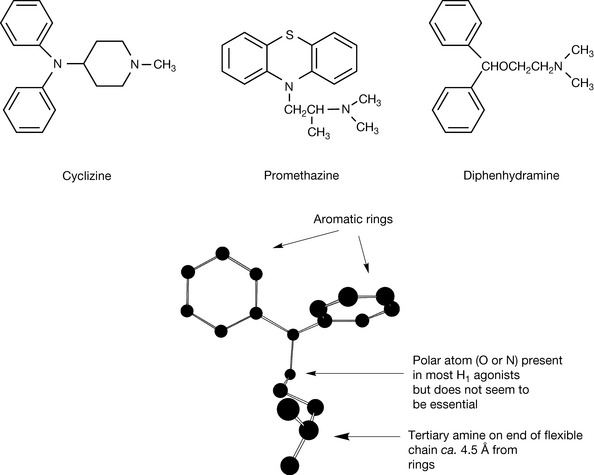
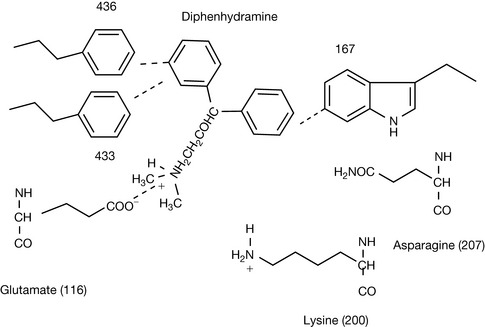
Figure 17.5 Putative interaction of diphenhydramine with glutamate 116 and tryptophan and phenyalanine residues in the fourth and sixth helices.
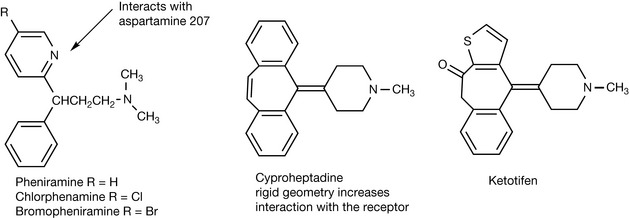
The pharmacophore present in the first generation of antihistamines was more or less carried forward into the next generation with the development of pheniramine, bromopheniramine and chlorphenamine (Fig. 17.6) except that in these compounds there is a nitrogen atom in one of the aromatic rings, which may enhance the interaction of one of the aromatic rings with asparagine 207. Chlorphenamine is the only compound still used. It is a more potent antagonist than the first generation of antihistamines and its high partition coefficient ensures that, like the first-generation antihistamines, it penetrates into the CNS and causes sedation in addition to having antihistamine activity. The sedating activity may be useful in some cases but if the patient needs to be alert, then obviously this is a disadvantage. A series of stereochemically rigid antihistamines which included cyproheptadine and ketotifen (Fig. 17.6) was produced. The rigid stereochemistry of these analogues favours stronger interaction with the phenylalanine and tryptophan residues in the fourth and sixth helices and these are more potent analogues. However, they are lipophilic and retain the sedating activity of the first-generation antihistamines.
Thus, the next generation of antihistamines were designed to have a lower partition coefficient whilst retaining their activity at the H1 receptor. The initial discovery of non-sedating antihistamines was accidental. Terfenadine (Fig. 17.7) was designed to be a sedative but was found to be a non-sedating antihistamine. Its structure looks similar to that of diphenhydramine and it might be expected to penetrate into the CNS but that it does not is in part due to the fact that it is metabolised via CyP450 activity to fexofenadine (Fig. 17.7) which has a much reduced partitioning into the CNS due to the presence of a carboxylate group in its structure.
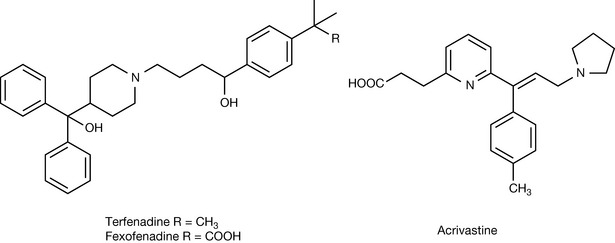
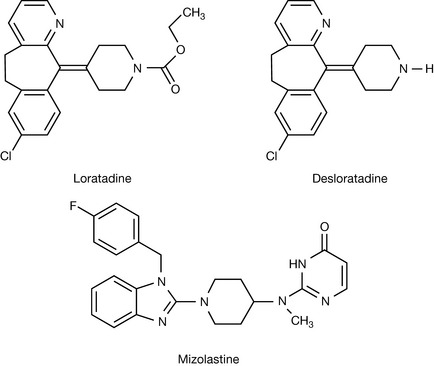
Fexofenadine has replaced terfenadine as a drug and is one of the most popular antihistamines. Acrivastine (Fig. 17.7) is another antihistamine which obviously derived from the observation that terfenadine was converted to its carboxy metabolite. It has been proposed that the presence of the carboxy group in some of the non-sedating antihistamines increases binding to the H1 receptor via interaction with lysine 200 (see Fig. 17.2). Loratadine and its metabolite desloratadine (Fig. 17.8), sold separately as a drug, are also non-sedating antihistamines but it is difficult to explain the lack of CNS penetration from their structures, which are more like those of sedating antihistamines. Mizolastine is a relatively new non-sedating antihistamine, and its lack of CNS penetration might be attributed to its rapid and extensive metabolism.
 Self Test 17.1
Self Test 17.1
Preparations of antihistamines
Histamine H2 antagonists
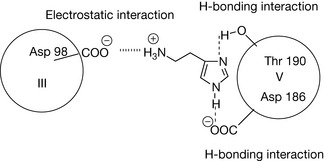
Classical antihistamines were found to have no effect on the secretion of gastric acid and this led Sir James Black to speculate that there might be a separate histamine receptor controlling gastric acid secretion which could be exploited in order to treat peptic ulcers and reflux oesophagitis. Classical antihistamine receptors were subsequently termed H1 receptors and the receptors controlling gastric acid secretion were termed H2 receptors. Like the H1 receptor, the H2 receptor has seven transmembrane-spanning helices. However, it only has 33% amino acid similarity within its primary structure to the H1 receptor. The primary amino acid sequence is shown in Figure 17.9 with the transmembrane helices and binding sites for histamine highlighted in red and the binding sites for the coupled G-protein on the third intracellular loop highlighted in blue. The proposed mode of binding3 is shown in Figure 17.10 where the primary interaction with charged side chain on histamine is with the aspartic acid residue at position 98 in the third helix. The two nitrogen atoms in the imidazole ring interact with the aspartic acid and threonine residues at 186 and 190, respectively. The interaction with the aspartic acid residue might be either hydrogen bonding or electrostatic since at physiological pH the imidazole nitrogen (pKa 5.9) is barely charged. As with all G-coupled receptors, binding of the histamine agonist causes a change in the conformation of the receptor which then activates the G-protein and the chain of events resulting from this activation. Site-directed mutagenesis studies4 indicated that the aspartic acid residue at position 98 was essential for histamine activity but that mutation of the aspartic acid and threonine residues at 186 and 190 did not cause complete loss of histamine agonist activity but did reduce the binding affinity of H2-receptor antagonists. Later studies5 have suggested that the tyrosine residue at position 182 (Fig. 17.9) might also be important in the binding of histamine to the receptor.

Figure 17.9 Primary structure of the histamine H2 receptor with the binding sites for histamine and the coupled G-protein highlighted in red and blue, respectively.
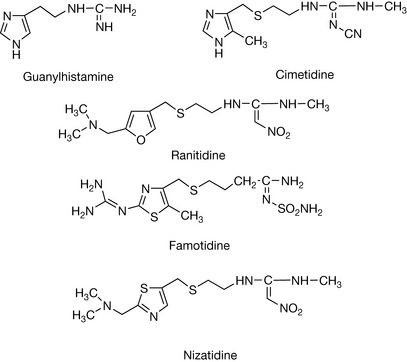
The two major H2-receptor blocking drugs cimetidine and ranitidine (Fig. 17.11) were discovered before anything was known about the structure of the binding site of the H2 receptor. The lead compound produced by Sir James Black and his team was guanylhistamine (Fig. 17.11), where the primary amine group in histamine has been replaced by the more strongly basic guanine group. This compound functions as a partial agonist, binding to the receptor and blocking histamine binding but at the same time producing a lower level of acid secretion. Presumably, the presence of the guanine group reduces the binding to the two other points of attachment in helix V since it would be likely to bind strongly to asp 98. It was assumed that by reducing the interaction of the basic side chain with the aspartic acid residue at 98 it would be possible to get the same antagonism without the partial agonist activity. The first commercially successful H2 receptor antagonist was cimetidine and in this structure imidazole ring is retained but the side chain is lengthened and the basic guanidine group is substituted with a cyano group which more or less abolishes its basicity due to the electron withdrawing effect of the CN group. Despite the substituted guanine group no longer being basic, cimetidine binds strongly to the receptor without exhibiting any agonist activity. Cimetidine exhibited side effects due to absorption into the CNS and also interferes with CyP450 oxidation of other drugs, and thus ranitidine (Fig. 17.11) was developed. The ranitidine structure is quite remote from the structure of histamine but is an effective antagonist. It retains the modified guanidine group of cimetidine which in the case of ranitidine is rendered non-basic via substitution of a strongly electron withdrawing nitro group, but the histidine ring has been replaced by a side chain containing a tertiary amine group. The tertiary amine group has a higher pKa value than the imidazole ring thus ensuring the molecule is completely ionised at physiological pH and reducing penetration into the CNS and also increasing the binding to the H2 receptor. Ranitidine is about three times more potent than cimetidine in vivo. Following on from ranitidine, two more H2-receptor antagonists were launched on to the market: famotidine (Fig. 17.11), which is about 20 times more potent than cimetidine, and nizatidine (Fig. 17.11) which has similar potency to ranitidine. There are no clinical advantages of famotidine and nizatidine over ranitidine.
Other agents used in treatment of peptic ulcers
The use of proton pump inhibitors in the treatment of ulcers is covered in Chapter 12. However, there is a disparate collection of compounds used in therapy of ulcers which do not fall into a particular class. These are described below.
Bismuth salts
Bismuth salts were used in the eighteenth and nineteenth centuries as antimicrobial compounds and also as antacids. With the discovery that Helicobacter pylori was the causative agent of ulcer formation, there was revival of interest in their use in the 1990s. The most commonly used water-soluble form of bismuth is tripotassium dicitratobismuthate ([Bi2(C6H4O7)]2−2K+) which has a complex structure.6 This salt is unstable in the stomach acid and precipitates out to form complex cationic species which form complexes with proteins in the stomach wall protecting ulcers from gastric acid and pepsin, and these complexes are also toxic to H. pylori. The basis of the toxicity of bismuth salts to the bacteria may be due to formation of complexes with the urease (which produces ammonia from urea) that the bacteria produce in large amounts in order to maintain the pH of their environment at around 6 so that they can survive under the acidic conditions in the stomach.7 Bismuth salts are usually used in combination with an H2-receptor antagonist and a tetracycline (see Ch. 22) which is also used to kill the bacteria.
Sucralfate
Sucralfate is a basic salt of aluminium with sucrose octasulphate. Its mechanism of action is not fully understood but it appears to strengthen the mucosal layer in the stomach by binding to proteins on the stomach wall. In addition, there is some evidence that it stimulates PGE2 formation which in turn strengthens the mucosal layer.8
Misoprostol
Misoprostol is a synthetic prostaglandin which is used to treat gastric and duodenal ulcers. It is the methyl ester of PGE2, which has various activities including promotion of the secretion of mucus. The ester is rapidly converted into the biologically active free acid in the body. It is used particularly to mitigate the side effects of non-steroidal anti-inflammatory drugs.
Preparations of H2-receptor antagonists and other drugs promoting ulcer healing
 Self Test 17.1
Self Test 17.11. Leurs R., Smit M.J., Meeder R., Ter Laak A.M., Timmerman H. Lysine 200 located in the fifth transmembrane domain of the histamine H1 receptor interacts with histamine but not with all H1 agonists. Biophysical Res Comm. 1995;214:110-117.
2. Wieland K., Ter Laak A.M., Smit M.J., Kuhne R., Timmerman H., Leurs R. Mutational analysis of the agonist-binding site of the histamine H1 receptor. J Biol Chem. 1999;274:29994-30000.
3. Gantz I., Del Valle J., Wang L., et al. Molecular basis for the interaction of histamine with the histamine H2 receptor. J Biol Chem. 1992;267:20840-20843.
4. Nederkoorn P.H.J., van Gelder E.M., Donné-Op den Kelder G.M., Timmerman H. The agonistic binding site at the H2 receptor II. Theoretical investigations of histamine binding to the seven α-helical transmembrane domain. J Comp Aided Mol Des. 1996;10:479-489.
5. Del Valle J., Gantz I. Novel insights into histamine H2 receptor biology. Am J Physiol Gastrointest Liver Physiol. 1997;273:987-996.
6. Sandler P.J., Guo Z. Metal complexes in medicine: Design and mechanism of action. Pure Appl Chem. 1998;70:863-871.
7. Zhang L., Mulrooney S.B., Leung A.F.K., et al. Inhibition of urease by bismuth(III): Implications for the mechanism of action of bismuth drugs. Biometals. 2006;19:503-511.
8. Slomiany B.L., Murty V.L.N., Piotrowski E., Morita M., Piotrowsky J., Slomiany A. Activation of arachidonoyl phospholipase A, in prostaglandin-mediated action of sucralfate. Gem Pharmar. 1994;25:261-266.