Chapter 23 Antiviral drugs
Introduction
In patients who are not immunocompromised, most virus infections are overcome by the host’s immune system, and are resolved without treatment. Thus other than alleviation of some of the more uncomfortable symptoms such as aching muscles, headaches and congestion direct treatment of the virus is not necessary. Specific treatments for viral infections in the immunocompromised are available, such as HIV or herpes virus infections, hepatitis and influenza. The role of vaccines in such treatments is not covered in this chapter. Here we are concentrating on the role of drugs in the alleviation of viral infections. Before discussing particular diseases and their treatments, it would be appropriate to remind ourselves of what we are combating, namely the causative organisms of the infections in question. The following section is a synopsis of viral biology.
Viruses
Viruses are obligate intracellular parasites that can infect all life forms. They contain either an RNA or DNA genome surrounded by a protective, virus-coded protein coat. Viruses have both an extracellular and an intracellular existence. In the extracellular form, the virus particle (virion) is the structure by which the virus genome is carried from the cell from which it has been reproduced to an uninfected cell. The genome contains the code for the proteins and enzymes required to construct a new virion from the infected cell. The basic structure of two common types of virus particles is illustrated in Figure 23.1.
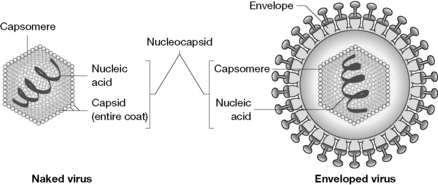
The capsid is composed of one or more individual proteins, known as structural subunits, which are arranged in a precise and highly repetitive manner around the nucleic acid. The small genome size of most viruses limits the number of proteins manufactured. A few viruses contain only one protein in their capsid, but most viruses have several chemically distinct kinds of subunits. The subunits form higher assemblies called capsomeres. The capsomere structure is formed and maintained by non-covalent intermolecular forces. It is the capsomere structure that is observed with the electron microscope. The combined assembly of the nucleic acid and the capsid is referred to as the nucleocapsid.
Certain viruses have more complex structures in which the nucleocapsid is enclosed in an envelope (Fig. 23.1). During envelope biosynthesis, the virus uses lipids derived from the host membrane to create the lipid bilayer prior to incorporating virus-specific proteins, usually glycoproteins, into the membrane. The envelope controls viral specificity and certain aspects of cellular penetration.
Viral multiplication in humans
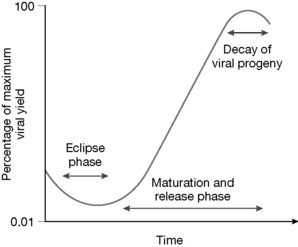
The multiplication cycle of all viruses exhibits several common features (Fig. 23.2). A short period after infection has occurred only small amounts of parenteral infectious material can be detected in the host. This interval is known as the eclipse phase. At this point replication has been initiated but progeny viruses have not been released from infected cells. This is followed by the maturation phase, an interval in which the viral progeny accumulate in the host cell at an exponential rate. Host cells infected with lytic viruses terminate their metabolic activity and viral production stops. The viral numbers start to slowly fall. Host cells infected with non-lytic viruses may, in contrast, continue to synthesise viruses indefinitely. Mature viruses that are released from the host cell are able to infect further host cells. The reproductive cycle of viruses ranges from around 6–8 hours (e.g. picornaviruses), to greater than 72 hours (e.g. certain herpes viruses). The viral yield from an infected cell ranges from several thousand copies (e.g. poxvirus) to greater than 100 000 copies (e.g. poliovirus).
Consequences of a viral infection
Epidemiological studies have indicated that viral infections are the most common cause of acute disease that does not require hospitalisation in developed countries, the most common being upper respiratory tract infections. In contrast, viral infections such as measles, mumps, rubella and rotavirus infections, which cause diarrhoea, are important causes of infant mortality, whilst the viral infection HIV remains a major cause of adult mortality and morbidity in developing countries. Host cells that allow viral replication are termed permissive. The production of infective progeny usually results in host cell death. For this reason, the infections are termed cytocidal (or cytolytic).
The viral infection process
In order to infect a cell, a virus must attach itself to the cell surface and penetrate into the cell. Once internalised, the virus must become sufficiently uncoated to make its genome accessible to host biosynthetic machinery so that transcription and translation can proceed to produce new viruses (Fig. 23.3).
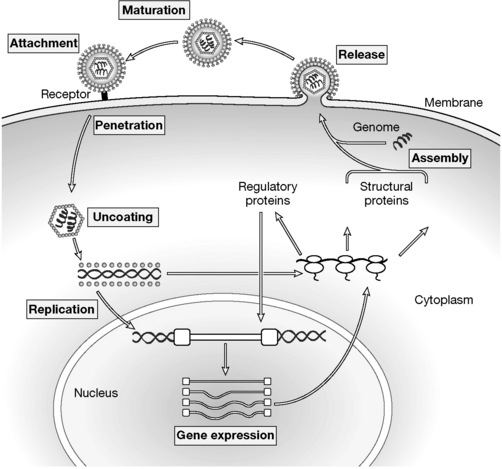
Figure 23.3 The infection cycle by which a virus enters a host cell and utilises host cellular machinery to reproduce itself.
1 Attachment
Attachment of the virus to the host cell membrane is through the specific binding of a virus protein (termed the anti-receptor) to a receptor on the host cell membrane surface. The anti-receptors are distributed throughout the viral surface. Complex viruses, such as the herpes simplex virus or poxvirus, may possess more than one type of anti-receptor. The majority of host receptors involved in the attachment process are glycoproteins, but other cell membrane constituents such as sialic acid and heparin sulphate have been identified. Attachment appears primarily to be dependent on electrostatic interactions that are temperature and energy independent. The susceptibility of a host cell to a particular virus is therefore dependent on whether it expresses a receptor that would enable the virus to attach via its specific anti-receptor. This is why viral infectivity is often limited to a particular cell type and cells lacking specific receptors are resistant. For example, a glycoprotein called gp120 on the surface of the human immunodeficiency virus (HIV), the cause of AIDS, specifically binds to the CD4 molecule found on certain human T lymphocytes. Cells that do not have surface CD4 molecules generally cannot be infected by HIV. In immunoresistant individuals, attachment is blocked by antibodies that bind to the viral or cellular receptor sites involved, which have been raised by the host’s immune system as a response to the infection or prior immunisation.
2 Penetration
Penetration occurs almost instantaneously after attachment between the receptors. It involves energy-dependent mechanisms, the most common of which is receptor-mediated endocytosis, the process by which many hormones and toxins enter cells. The virion is endocytosed and contained within a cellular cytoplasmic vacuole called an endosome. In the case of viruses that penetrate as a consequence of fusion of their envelopes with the plasma membrane, for example, herpesviruses, the envelope remains in the cell membrane and the nucleocapsid enters the cytoplasm. Fusion of viral envelopes with host cell membrane requires the interaction of specific viral proteins in the envelope with proteins in the host cell membrane.
3 Uncoating
Uncoating is a general term applied to the events occurring after penetration which enables the viral genome to be expressed. In most cases the virus disassembles either spontaneously or with the aid of host cell enzymes until only the viral nucleic acid remains. A key step in uncoating is the acidification of the content of the endosome to a pH of about 5, owing to the activity of a proton pump present in the surrounding membrane. The low pH causes rearrangement of coat components, in order to expose normally hidden hydrophobic sites, which then bind to the lipid bilayer of the membrane, causing the extrusion of the viral core into the cytosol.
4 Replication
Pathogenic viruses, either DNA or RNA, shut off cellular protein synthesis and disaggregate cellular polyribosomes so that the host cell processes are shifted exclusively to viral synthesis. A general sequence of events can be observed during viral replication:
The viral proteins synthesised once the viral genome has become integrated into the host genome and expressed have three main functions:
Viruses can be either DNA or RNA containing, and consequently employ different mechanisms of replication.
DNA viruses
The viral genome is coded for by the same nucleic acids as the host (DNA). After integration of the viral DNA into host DNA by the viral integrase enzyme, transcription occurs in the nucleus and translation in the cytoplasm. Generally, transcription of the viral DNA into mRNA is by the host RNA polymerase systems. The translation of the viral mRNA of early proteins is the key initial step in viral DNA replication to establish the replicatory process. After DNA synthesis, the remainder of the genome is transcribed into late messengers. Regulation is carried out by proteins present in the virions, or specified by viral genes transcribed and translated by the cellular host.
RNA viruses
In order to replicate the RNA viral genome, the viral RNA can act directly as the mRNA and be translated to form viral proteins. Alternatively, in the case of retroviruses, which are unique in that their genomes are transcribed into DNA from RNA by an RNA-directed DNA polymerase or reverse transcriptase, so called because information is going from RNA into DNA, which is the opposite to the conventional process of transcription (DNA into mRNA). Once transcribed into DNA, it is integrated into the cellular DNA by the viral integrase enzyme. Transcription of the viral genome by the cellular RNA polymerases yields the viral molecules that end up in virions.
5 Viral assembly
The viral proteins and genome are assembled to form the nucleopcapsid structure of a mature virion. The viral assembly can occur at different intracellular sites: in the cytoplasm, in the nucleus or on the inner surface of the cell membrane.
6 Release
The release mechanism for lytic viruses (most naked viruses) is simple: the cell breaks open (lyses) and the viruses are released. Enveloped viruses acquire their lipid membrane coat as the virus crosses the host cell membrane by a process known as budding. Budding is controlled by the virus and involves the physical interaction of the capsid proteins with the inner surface of the host cell membrane. Budding may or may not result in host cell death.
7 Maturation
The virus has to mature before it becomes infectious. Maturation usually involves a structural change to the virus resulting from the cleavage of a capsid protein. The maturation process may occur during viral assembly or later once the virus has left the host cell.
Antiviral drugs
Introduction
Antiviral drugs are among the most recently developed drugs and perhaps more than any other group of drugs are rationally designed. The strategies for halting viral replication are limited and the most common strategy is to inhibit replication of viral DNA using analogues of DNA bases which become incorporated into viral DNA and prevent thus inhibit its production. Thus the window for therapeutic intervention is relatively narrow since the drugs are also likely to interfere with the production of human DNA because the enzymes used by the virus and host to activate nucleosides and incorporate them into DNA are very similar. The other major class of antiviral drugs are protease inhibitors which are specifically concerned with inhibition of protein processing by the HIV virus.
HIV infections
Since the early 1980s, acquired immunodeficiency syndrome (AIDS) has evolved into a worldwide epidemic. Death is not caused directly by the virus, the human immunodeficiency virus (HIV), but the severe impairment of the immune system that is a consequence of the viral infection results in infection by opportunistic pathogens which ultimately kill the host. Glycoprotein spikes on the protein coat of the HIV virus have a high affinity for CD4 receptors mainly found on the surface of helper T lymphocytes, but also present in macrophages, dendritic cells, and neuroglial cells. All of these are involved in the immune response. The HIV-CD4 complex penetrates the infected host cell by endocytosis and then the virus replicates and destroys the host cell. Virus is then released into the circulation. The diagnostic ‘CD4 cell count’ drops as the infection spreads, ultimately destroying the host cellular immune system. In 1983, the causative strain responsible for this process, HIV-1, was isolated independently by two groups, and, in 1986, a genetically distinct virus, HIV-2, which occurs in different geographic locations (West Africa) was isolated. When reverse transcriptase activity was detected in a sample of lymph tissue from a patient at risk of AIDS, HIV was confirmed as a retrovirus. This means it contains RNA rather than DNA, and hence it must form viral DNA if it is to take over control and replicate inside the host cell. Once inside the host cell, the protein coating is removed from the HIV, releasing its RNA. The viral enzyme known as reverse transcriptase then transcribes this RNA into a DNA copy, forming a RNA-DNA hybrid. The RNA component of this hybrid is destroyed in the presence of ribonuclease H, leaving the DNA strand to replicate and form double-stranded DNA. This double-stranded DNA enters the nucleus of the host cell and is inserted into host DNA by the viral integrase enzyme. The host DNA now acts as the vector for the regeneration of new viral DNA, which is packaged by structural proteins coded for by the viral DNA, and released from the cell when it lyses.
For viral DNA synthesis and integration following retroviral infection, the genes of the retroviral genome are expressed together as a fusion protein. This multi-domain precursor is then processed by a viral protease enzyme at specific sites to the mature proteins essential for HIV multiplication. These enzymes are reverse transcriptase, integrase, (more) HIV protease and structural internal proteins of the new virion. All these catalytic proteins are potential targets for drugs to inhibit the viral replication process in this devastating disease. For an animated description of the HIV replication cycle, see http://www.hopkins-aids.edu/hiv_lifecycle/hivcycle_txt.html.
Reverse transcriptase (RT) inhibitors
Nucleoside reverse transcriptase inhibitors (NRTIs)
Screening of the NCI drugs database in the 1980s identified several compounds capable of preventing HIV replication in vitro, and one of these, 3′-azido-3′-deoxythymidine (AZT or zidovudine, Fig. 23.4) was the first drug to be used for the treatment of AIDS. In January 1986, a double-blind clinical trial began on 282 patients. After 16 weeks, the monitoring board stopped the trial when it had become apparent that amongst those receiving the drug, 1 out of 145 had died, compared with 16 deaths amongst those receiving a placebo. Supplies of zidovudine were released for treatment on a named-patient basis, with the FDA granting registration the following spring. Several trials confirmed that zidovudine used on its own delayed death by 12 months. If therapy was initiated before AIDS developed, it typically delayed onset of AIDS by 12 months.
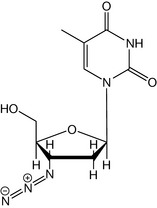
Zidovudine (AZT)
Zidovudine inhibited RT, but treatment was of limited efficacy, mainly because of dose-limiting side effects caused by affects against host metabolism, and the development of resistance by the virus. However, the principle that RT could be a target against the disease was established, and indicated the need for novel antiretroviral agents that could be developed with two primary objectives in mind:
Before considering the mode of action of these NRTIs, we may need to remind ourselves of the mechanism by which nucleic acid polymerase enzymes work to produce new DNA or RNA strands. This is described in Chapter 7.
Mechanism of inhibition of RT by NRTIs
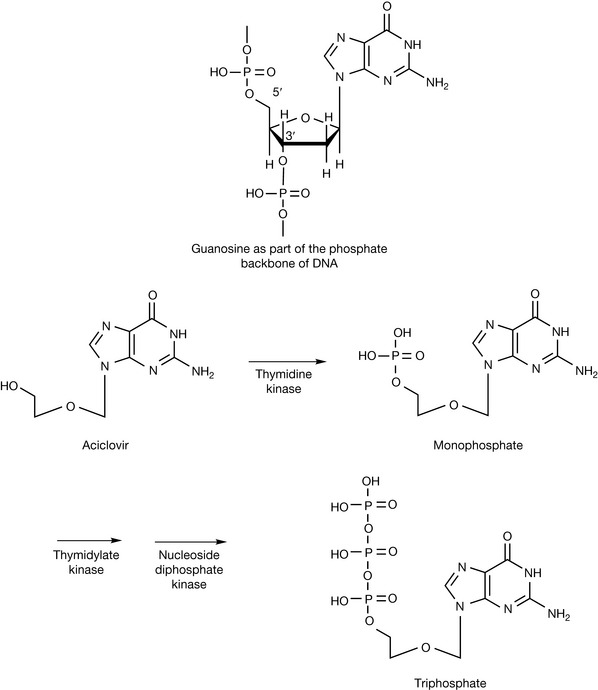
For zidovudine to act as an inhibitor of RT, it must first be converted to the monophosphate by thymidine kinase, which in turn is more slowly converted to the triphosphate by thymidylate kinase (Fig. 23.5). The bulk of the azido group is believed to interfere with the activity of thymidylate kinase, thereby slowing the rate of phosphorylation. By occupying the active site of these kinase enzymes, zidovudine also slows the generation of the endogenous thymidine triphosphate, therefore lowering the reservoir of thymine base substrates for chain elongation. This is not the main mechanism of action, however. The zidovudine triphosphate is incorporated in the viral DNA chain in place of thymidine (the base it is an analogue of) and no more nucleotides can be added since the azido group is non-nucleophilic, and blocks the creation of further phosphodiester linkages which are needed to continue with the elongation of the nucleic acid chains (Fig. 23.6). Thus, reverse transcriptase is inhibited because it cannot conjugate further nucleotides onto the 3′-end of the new chain. Zidovudine triphosphate also affects mammalian DNA polymerase, for which it has one hundredth the affinity of that for viral reverse transcriptase. This causes considerable toxicity, notably dose-dependent suppression of bone marrow, resulting in anaemia and leucopenia which causes treatment to be abandoned by many patients.
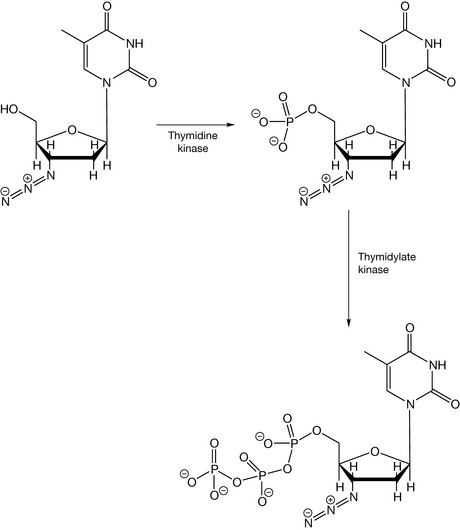
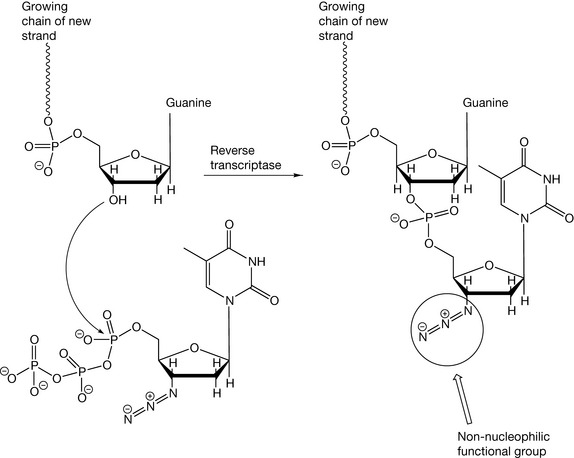
Figure 23.6 Zidovudine cannot be conjugated with further nucleotides because the 3′-azido group is non-nucleophilic, and will not displace biphosphates from incoming triphosphate substrates.
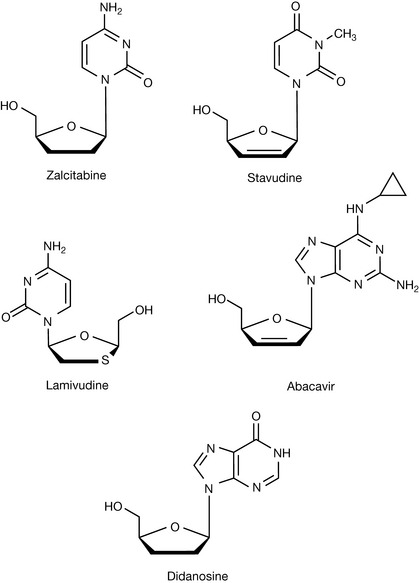
Following on from zidovudine, a number of other NRTIs have been produced (Fig. 23.7). Zalcitabine is a NRTI analogue of cytidine, and its triphosphate prevents nucleic acid chain extension because absence of the 3′-hydroxy group on the sugar ring stops formation of the phosphodiester linkages which are needed for the completion of nucleic acid chains. Stavudine acts in a similar manner. Zalcitabine is used for treatment of HIV infection when therapy with zidovudine has failed or has produced unacceptable toxicity. Lamivudine, another cytidine analogue (formerly known as 3TC), slows the reproduction of HIV by inhibiting reverse transcriptase in a similar way; it is further distinguished by the fact that it is an L-nucleoside (based on a modified L ribose molecule). Currently, there is research going on into the use of L-nucleosides as antiviral agents since they have been found to retain antiviral activity but are less toxic with respect to incorporation into the host DNA. Abacavir has been found to be a very effective reverse transcriptase inhibitor. It is an analogue of guanosine but participates in the same type of chain termination reactions as the thymidine and cytosine analogues. Didanosine is an analogue of dexoyadenosine, which, again, causes chain termination in DNA synthesis by the virus.
Non-nucleoside reverse transcriptase inhibitors (NNRTIs)
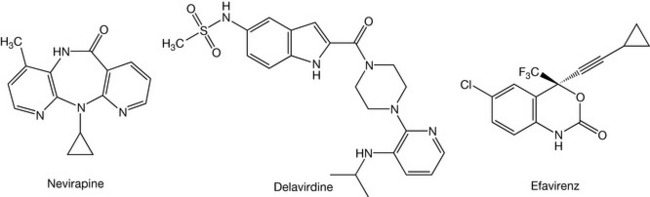
The idea behind NNRTIs started at the beginning of the 1990s with the discovery of 1-(2-hydroxyethoxymethyl)-6-(phenylthio)thymine (HEPT) and tetrahydroimidazo [4,5,1-jkj][1,4]benzodiazepine-2(1H)-one and -thione (TIBO) as specific HIV-1 inhibitors, targeted against HIV-1 RT. These three compounds had a unique specificity against HIV-1, but were inactive against HIV-2 or any other retrovirus, and they acted through the inhibition of RT. However, although the interaction was specific to RT with the consequent prevention of viral replication, they did not act at the substrate binding site associated with the NRTIs. Whereas the latter, following their intracellular phosphorylation, act as competitive inhibitors by interacting at the site where the natural nucleotide substrates bind and subsequently prevent chain elongation by incorporation into the nucleic acid, NNRTIs block the HIV-1 RT mechanism through binding to an allosterically located, non-substrate binding site. The NNRTI binding site is functionally and spatially associated with the substrate binding site, but is distinct (0.1 nm away), and through a cooperative mechanism, binding with the NNRTI site produces a conformational change in the enzyme that locks it into an inactive conformation that is unable to perform the catalytic event of elongating the new viral nucleic acid. Since HEPT and TIBO were discovered, a variety of NNRTIs have been licensed for treating HIV infection. Whilst they and the new classes currently undergoing clinical trials can be considered structurally and chemically diverse, they occupy the same binding site that causes a repositioning of the catalytic residues that locks the enzyme in its inactive conformation. Structural modelling studies have demonstrated that, despite their diverse structures (Fig. 23.8), the positions and conformations adopted by the NNRTIs, when bound to HIV-1 RT seem to be quite similar. For example, TIBO and nevirapine maintain a ‘butterfly’ shape that roughly overlay each other and appear to stack with π-electron aromatic side-chain residues that line the pocket (Fig. 23.9), and whilst delavirdine occupies the same pocket, the complex is stabilised quite differently by hydrogen bonding to the Lys 103 and hydrophobic contacts with Pro 236.
HIV protease inhibitors
A key step in the establishment of infection is the processing of the fusion proteins that produce the enzymes required for the reverse transcription of the viral RNA into DNA. If the fusion protein is not hydrolysed into its active constituents, the enzymes required to initiate reproduction of the viral genome are halted. Workers at Roche in 1987 discovered that HIV retroviral proteases contain a highly conserved Asp-Thr-Gly motif, which had a similar catalytic mechanism to cellular aspartic proteases. Most aspartic proteinases belong to the pepsin family, which includes digestive enzymes such as pepsin and chymosin as well as lysosomal cathepsins D and processing enzymes such as renin, and certain fungal proteases. Crystallographic studies have shown that these enzymes are bilobed molecules with the active site located between two homologous lobes. Each lobe contributes one aspartate residue of the catalytically active diad of aspartates. These two aspartyl residues are in close geometric proximity in the active molecule; one aspartate has a pKa of 4.5, whereas the second one has an unusually low pKa of 1.2 (influenced by the protein environment), making it a very strong acid. These differences in pKa values means that one residue can act as a proton donor, whilst the other is a proton acceptor, and through the involvement of a water molecule, can achieve acid- and base-catalysed hydrolysis at the same time (Fig. 23.10). In contrast to serine and cysteine proteases, catalysis by aspartic proteinases does not involve a covalent intermediate, although a tetrahedral intermediate exists. The nucleophilic attack is achieved by two simultaneous proton transfers: one from a water molecule to the diad of the two carboxyl groups, and a second one from the diad to the carbonyl oxygen of the substrate with the concurrent CO–NH bond cleavage. This general acid–base catalysis, which may be called a ‘push-pull’ mechanism, leads to the formation of a non-covalent neutral tetrahedral intermediate.
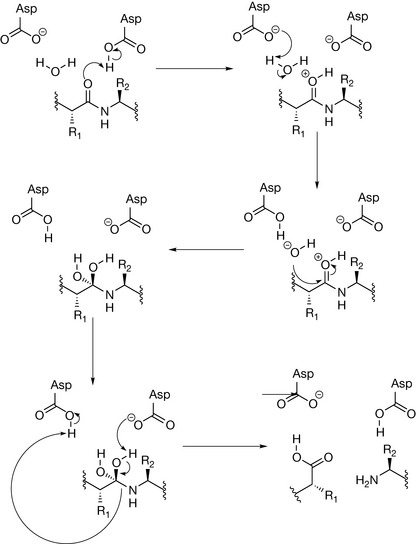
Figure 23.10 Mechanism of action of aspartate proteases, showing the generation of a tetrahedral unstable transition state through the addition of a water molecule to the amide bond where cleavage takes place.
When the first inhibitors for this protease were developed, very little was known about the enzyme other than its aspartate-catalysed mechanism of action: there was no crystal structure to propose inhibitors based on modelling, and differences with the host proteases were unclear. All that was known was that the HIV protease was a much smaller enzyme than the equivalent host aspartate proteases (around 100 amino acids as opposed to 200), and was able to cleave substrates N-terminal to proline residues, and since mammalian proteases were unable to carry out such cleavages, the possibility of selectivity for the virus over the host arose. Additionally, even before the protease had been isolated, a study of peptides obtained from infected cells suggested that the Met-Met and Tyr-Pro sites were the likely positions of cleavage in the fusion protein.
The approach taken was not unlike that used in designing the ACE-inhibitors and on previous attempts to design inhibitors of renin, another protease enzyme. Given that Tyr-Pro had been identified as a cleavage site for the HIV protease, the rationale for inhibitor design was based around the Phe-Pro or Tyr-Pro motif, given the specificity of the enzyme in hydrolysing between these residues. To establish the activity of inhibitors, rather than using the HIV fusion protein itself in the assay (expensive and difficult to isolate at the time), a series of peptides that contained the Tyr-Pro cleavage consensus sequence were prepared, with the N- and C-termini protected to prevent cleavage by exopeptidases that were present in the partially purified enzyme preparation from an Escherichia coli vector. The hexapeptide, succinyl-Ser-Leu-Asn-Tyr-Pro-Ile-isobutylamide was used as the substrate for all routine screening assays in the Roche inhibitor design project (Fig. 23.11).
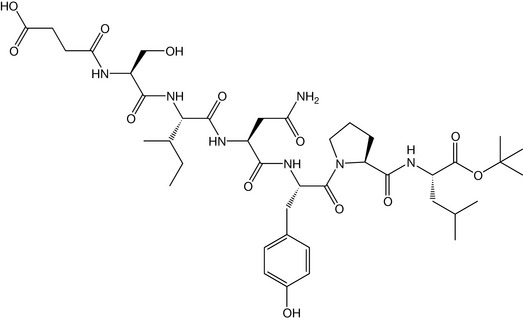
Figure 23.11 The peptide substrate Succinyl-Ser-Leu-Asn-Tyr-Pro-Ile-isobutylamide used to assay the effectiveness of inhibitors against HIV protease.
As mentioned above, aspartic proteases hydrolyse peptides and proteins through the addition of water to the target amide bond, and the reaction proceeds via an unstable tetrahedral transition state. In order to progress a reaction, enzymes, being catalysts, are known to undergo conformational changes to facilitate the stability of the transition state to lower the activation energy of the reaction from reactants to products. The design of ‘transition-state’ inhibitors is a commonly used strategy, because compounds that resemble transition states, but are within themselves stable, bind very tightly to the active site of the enzyme, lock it in its intermediate conformational state, dissociate very slowly and act as efficient inhibitors. Within the HIV protease project, of the possible transition-state mimetics that were chemically stable, the reduced amide ketomethylene derivatives and hydroxyethylamines were deemed the most appropriate, and were incorporated into an Asn-Phe-Pro motif that resembled the substrates. The most potent inhibitors from this series (Compounds 1–4) were found to be the hydroxyethylamines, with an IC50 of 140 nM (Fig. 23.12). Although this was impressive, greater potency was required for clinical evaluation, so a move away from peptide motifs through a systematic structure–activity relationship modification program was initiated.
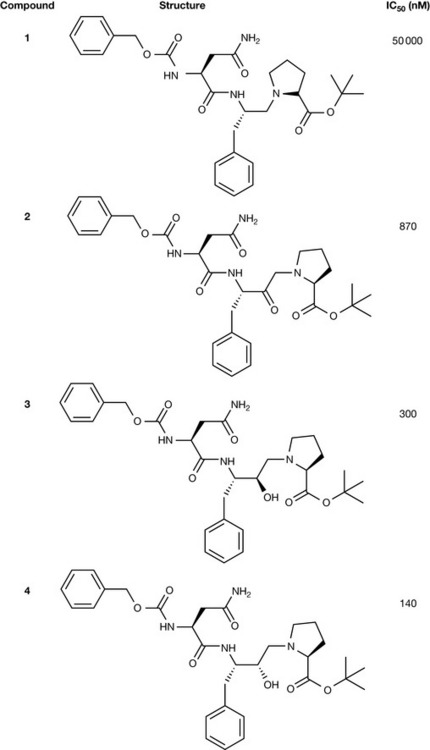
Figure 23.12 Comparison of the first transition-state mimetics based on the Asn-Phe-Pro substrate motif.
In order to examine the effect of size, protected smaller dipeptides (see Figs 23.5, 23.6) showed much reduced activity compared with the tripeptide equivalents (see Figs 23.3, 23.4), while addition of residues to the N-terminus (see Figs 23.7, 23.8), the C-terminus (see Figs 23.9, 23.10), or both ends of the molecule (see Figs 23.11, 23.12) gave no improvement in activity or potency (Fig. 23.13).
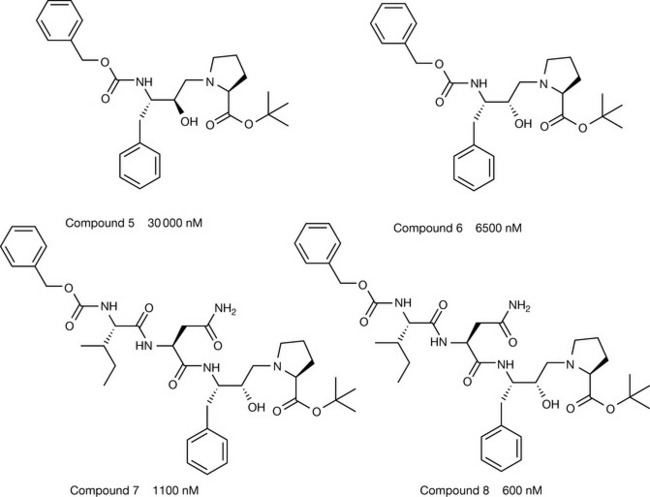
Figure 23.13 The effect of inhibitor size on potency, comparing dipeptides, tetrapeptides and pentapeptides to the optimum tripeptides.
At the C-terminus, medium-sized lipophilic residues appeared to be preferred, with little difference between esters and amides. The t-butyl amide group was chosen as the C-terminal residue for subsequent compounds on the basis of metabolic stability (see Fig. 23.13), amides being more stable than the corresponding esters (see Figs 23.3, 23.4 and Fig. 23.14). Replacement of the N-terminal benzyloxycarbonyl group by smaller non-aromatic groups, such as acetyl or t-butoxycarbonyl reduced activity, but introduction of bicyclic aromatic groups such as β-napththoyl or quinoline-2-carbonyl (see Fig. 23.14) led to compounds with significantly improved activity.
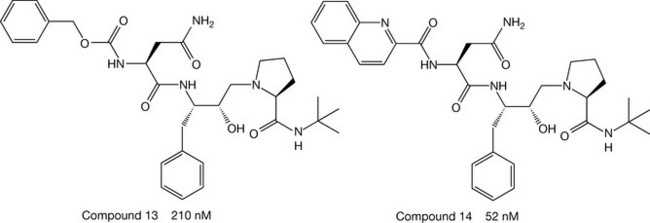
The most dramatic changes in activity were seen with modifications to the proline residue – ring size was found to be very important for activity – replacing a 5-membered ring with a 4-membered ring (see (Fig. 23.15) abolished activity, whilst incorporation of a 6-membered ring improved potency 12-fold (see Figs 23.15, 23.16). Replacement of proline by two fused 6-membered rings led to the greatest enhancement activity, and the S,S,S-decahydroisoquinoline carboxylic acid was the best replacement of proline of all (see Fig. 23.17). Within the series of compounds prepared, the order of potencies against the virus also paralleled the enzyme-inhibitory potency, which indicated good cellular penetration across lipid membranes, an essential characteristic for any effective drug. The first compound to be marketed in the USA based on this research programme, in December 1995, was Roche’s saquinavir (Fig. 23.15). A dose of 600 mg is administered three times a day by mouth in combination with a reverse transcriptase inhibitor such as lamivudine, or with both an NRTI and an NNRTI.
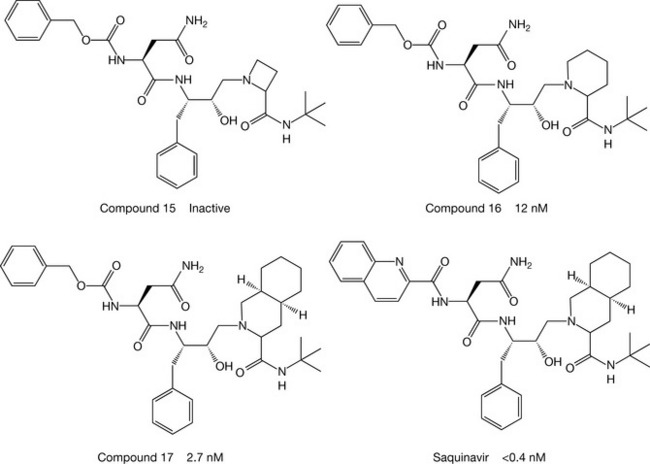
Figure 23.15 The effect of substituents on potency, with saquinavir, the most potent, being marketed.
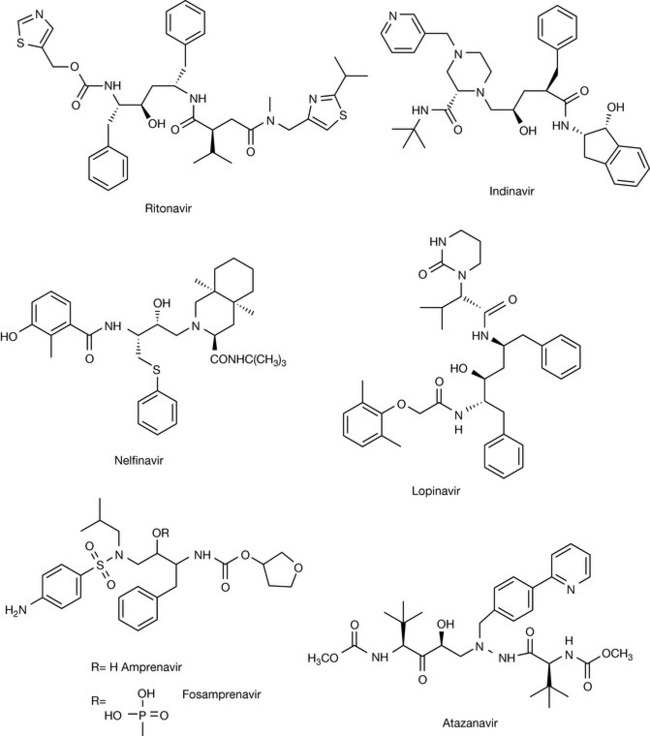
The establishment of the structure of HIV protease was determined by X-ray crystallography by a Merck team in the United States. As expected, the active site lay at the junction of two lobes joined like butterfly wings, which enabled researchers to design inhibitors of HIV protease based on molecular modelling studies. Saquinavir is rapidly metabolised, disappearing from the body in about 90 minutes. Abbott’s ritonavir (Fig. 23.16) is longer lasting and need only be administered twice daily. It was the first protease inhibitor to be licensed in the UK. Merck’s indinavir is less protein bound and so penetrates tissues more effectively than the first two drugs. It is administered three times a day by mouth. All compounds contain the essential hydroxyethylamine transition-state motif essential for effective inhibition of HIV protease.
A number of other protease inhibitors (Fig. 23.16) have been developed in the last few years with the aim of improving pharmacokinetic profile. One of the first of these was amprenavir, which does not contain peptide bonds which slows down its clearance from the body. Its pharmokinetics are further improved when it is administered as its phosphate prodrug fosamprenavir. Lopinavir was also developed to give improved pharmacokinetics. Its effectiveness is improved by co-administration with ritonavir, which is a potent inhibitor of cytochrome P450. A serious side effect of protease inhibitors is elevation of cholesterol and lipid levels in the body. This may be due to the proteases inhibiting enzymes which are involved in lipid metabolism. Atazanavir has both improved pharmacokinetics enabling it be administered once a day and it has less effect on patient’s lipid profiles. Clinical trials have shown that these protease inhibitors, when used in combination with an NRTI and an NNRTI, dramatically reduce the HIV viral load. In such a combination, indinavir, for example, was so effective that after treatment it proved impossible to detect virus in 85% of patients. Since an estimated 1012 viruses are estimated to be formed daily before treatment, this is remarkable. As a consequence, AIDS can now be held at bay for many years if chemotherapy is continued. The cost of treatment is very high and in many parts of the world it is quite unaffordable, and so patients continue to die of AIDS. The recent supply of generic forms of the anti-HIV medicines to sub-Saharan Africa, where AIDS is an epidemic, bodes well for people suffering from this disease, but strict compliance with the dose regimens must be adhered to if drug resistance by the virus is to be prevented.
Antiretroviral therapy and drug resistance
Hitting different targets in the HIV replication cycle by using combination therapy can result in a greater than 10 000-fold reduction in the viral load being achieved. Under such conditions, sustained suppression of the virus in the plasma does not necessarily eliminate it from the protected sanctuaries such as the CSF and the lymph nodes, where drugs penetrate less readily. Consequently, drug resistance is an inevitable development of the incomplete suppression of the replication of the causative organism, and under such conditions, mutations in the viral genome that produce enzymes resistant to inhibition will arise. Specific mutations that allow for viral survival and drug resistance are generally due to amino acid residues in either RT or HIV-protease which will enable catalysis to continue, whilst hindering the binding of the drug to the target.
Herpes, varicella zoster, cytomegalovirus, hepatitis
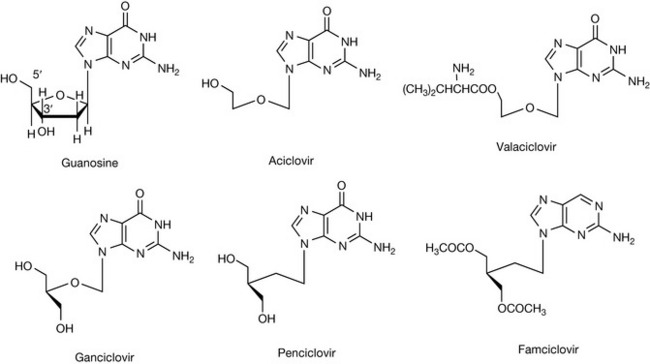
Figure 23.17 shows some antiviral drugs related to the purine base guanosine. These drugs work by blocking the synthesis of viral DNA or by becoming incorporated into viral DNA, thus preventing its transcription. In living systems, the triphosphate of guanosine becomes incorporated in a DNA strand linked to the strand via its 5′ and 3′ positions (Fig. 23.18). Two such DNA strands give the DNA double helix.
Aciclovir
In 1975, at the Wellcome Laboratories in the USA, researchers in the course of screening analogues of guanine arabinoside found that an acyclic derivative, aciclovir, had outstanding activity against the herpes virus. It is administered to treat superficial herpes simplex infections such as cold sores (by topical application) and genital herpes (by oral administration), or for treating life-threatening herpes varicella-zoster (chickenpox) infections in immunocompromised patients. Its safety has resulted in it being available over the counter for the treatment of cold sores.
Aciclovir was one of the first antiviral drugs. As can be seen from its structure in Figure 23.17, it lacks a hydroxyl attached to the 3′ carbon and thus, when it becomes incorporated into a DNA strand, it prevents it from extending further. In addition, it remains bound to the viral DNA polymerase enzyme, preventing it from synthesising further DNA. Its therapeutic selectivity is based upon two factors: (1) herpes simplex virus (HSV) and varicella-zoster virus (VZV) express high levels of thymidine kinase (see Fig. 23.2), and (2) aciclovir has a greater affinity for the viral DNA polymerase than the DNA polymerase present in human cells. Aciclovir is used to treat herpes simplex virus. It has low toxicity but a narrow spectrum of activity and is only used in the treatment of genital and labial herpes and varicella-zoster (shingles) infections. Valaciclovir is prodrug of aciclovir, containing a readily cleavable ester group, which has an improved oral availability of 54% compared with 20% for aciclovir. Like many of the antiviral nucleosides, infusions of the drug are formulated as the strongly alkaline sodium salt (Fig. 23.19). Aciclovir and its analogues do not cure viral infection, but do effectively suppress the replication of viruses. Thus, patients should be warned that cold sores will reappear in the future and further medication will be necessary. It should be noted that aciclovir is only effective when administered at the onset of infection. The selectivity of aciclovir is enhanced by the very fact that once it is phosphorylated in the virally infected cell, it becomes too polar to diffuse out of the cell.
Ganciclovir
The structure of ganciclovir (Fig. 23.17) is closely related to that of aciclovir. Ganciclovir has the equivalent of 3′ and 5′ hydroxyl groups within its structure and thus, unlike aciclovir, it can become incorporated into DNA and thus has a broader spectrum of activity. The absence of a 5-membered ribose or deoxyribose sugar to maintain the structural rigidity of the sugar-phosphate backbone of the newly synthesized nucleic acid means that its structure becomes non-functional as a template for further replication or transcription. Ganciclovir is effective against HSV and VZV but it is also effective against cytomegalovirus (CMV). Ganciclovir is more readily phosphorylated in CMV-infected cells by a kinase encoded by the viral DNA, and the drug is then converted to its triphosphate by enzymes within the infected host cell. The triphosphate is a selective inhibitor of the formation of viral DNA by competitive inhibition of the incorporation of guanosine triphosphate into DNA or through itself becoming incorporated into DNA, thus preventing viral replication. Since the drug can become incorporated into human DNA, it is very toxic and can cause myelosuppression. The drug is only recommended for use for the treatment of CMV in immunocompromised patients. Ganciclovir has poor oral bioavailability and is administered by intravenous infusion as the strongly alkaline sodium salt; thus infusion has to be carried out slowly. Ganciclovir is also available in the form of its ester valine ester, valganciclovir (cf. aciclovir), which has better oral bioavailability.
Penciclovir and famciclovir
Penciclovir (Fig. 23.17) is closely related to ganciclovir in structure and has the same mechanism of action. Penciclovir has similar activity to aciclovir against herpes simplex infection. Its safety is similar to that of aciclovir due to its lack of effect on DNA synthesis in uninfected cells. A closer structural similarity to deoxyguanosine makes penciclovir a superior substrate in comparison to aciclovir for viral thymidine kinase, resulting in an enhancement of phosphorylation. This is offset by reduced activity of penciclovir triphosphate as an inhibitor of DNA synthesis. It has poor oral bioavailability and is used in the form of a cream (1% w/w) to treat labial herpes. Famciclovir (Fig. 23.17) is a prodrug of penciclovir which has good oral bioavailability (73%) and is deacetylated and oxidised (aldehyde oxidase) in the liver to penciclovir. The toxicity of famciclovir is low. It is administered three times a day by mouth, whereas treatment of herpes with aciclovir by the oral route requires five daily doses.
Cidofovir
Cidofovir (Fig. 23.20) is a phosphonic acid derivative and is not susceptible to hydrolysis by cellular enzymes. It does not require initial phosphorylation by thymidylate kinase for activation and is converted by cellular kinases to its diphosphate derivative which acts as a competitive inhibitor for the incorporation of deoxcytosine triphosphate into DNA. It may become incorporated into DNA itself, causing chain termination. It is also used in the treatment of CMV in AIDS patients.
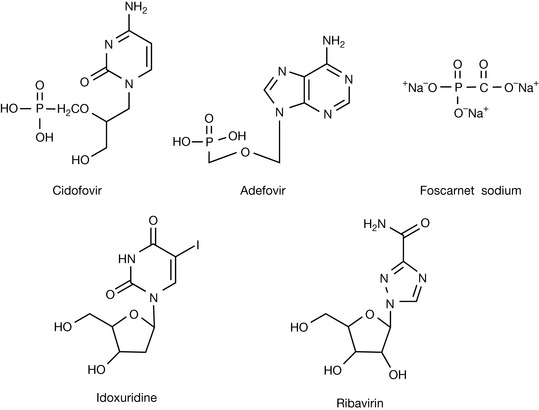
Adefovir
Like cidofovir, adefovir (Fig. 23.20) is a derivative of phosphonic acid and acts via a similar mechanism. It is used to treat chronic hepatitis B which has become resistant to the antiretroviral drug lamivudine.
Foscarnet sodium
Foscarnet sodium (Fig. 23.20) mimics pyrophosphate and inhibits the binding of nucleoside triphosphates to DNA polymerase. Thus it has a relatively broad spectrum of activity, but is used to treat CMV and herpes in AIDS patients. Since it has a different mode of action from drugs such as aciclovir, it can be used in combination with them to increase the effectiveness of treatment. Since it forms insoluble salts with bases it is incompatible with basic drugs such as pentamidine and vancomycin, which may be used to treat secondary infections in AIDS.
Idoxuridine
Idoxuridine (Fig. 23.20) was a very early viral antimetabolite which is incorporated into DNA in place of thymidine. It was believed to be of value in treating ocular herpes simplex but is currently regarded as being ineffective both for this and for treatment of the skin.
Ribavirin
Respiratory syncytial virus (RSV) is a major cause of mortality in young infants and the elderly. The mechanism of action of ribavirin is not entirely clear although it is probable that it acts as an inhibitor of inosine dehydrogenase which is involved in the introduction of a carbonyl group into the inosine structure en route to the synthesis of guanosine triphosphate (GTP). GTP is required for viral RNA transcription. Ribavirin may also interfere with the action of the viral RNA polymerase. There is some debate over the effectiveness of ribavirin in treating RSV. It is also used in combination with interferon in the treatment of hepatitis C. RSV is also treated with monoclonal antibody therapy and this is discussed in the Chapter 26.
Influenza chemotherapy
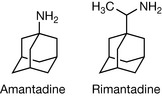
Influenza is one of the most deadly infectious diseases. For many outbreaks, it is the old or very young who are at risk of death, but lethal mutants of the virus can arise such as the outbreak in 1918 which killed more than 50 million people worldwide, more than the First World War. Even in an average year, 500 million people are affected by influenza and hundreds of thousands die. Thus health authorities are keen to avoid a recurrence of the 1918 pandemic. In fact, some aspects of the 1918 epidemic are now manageable since the complication of pneumonia which was responsible for many deaths in that pandemic is now more readily treatable. There are three types of influenza: A, B and C. Type A is the most common source of epidemics and pandemics and it is highly genetically variable. Every so often the type A virus manages to completely reorganise its genetic make-up so that the human immune system is not able to recognise it. The best way to protect against influenza is by vaccination but this only applies to established strains of the virus. Since a vaccine for a new strain takes 6 months to produce, an alternative line of defence is important. The first chemotherapeutic agent against flu was amantadine (Fig. 23.21) which was a serendipitous rather than a rational discovery. Amantadine and the related compound rimantadine (Fig. 23.21) have activity against the early stages of influenza. They are believed to interfere with the M2 transmembrane ion channel protein in the virus and inhibit uncoating of the virus which must occur before the viral DNA can be integrated into the host cell nucleus. The compounds have some use in prophylaxis but resistance develops readily and the two drugs have CNS side effects.
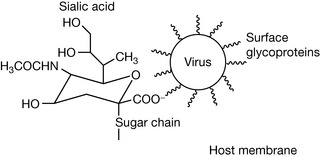
The influenza virus enters the host cell by initially binding, via its cell surface glycoproteins, to receptors bearing sialic acid residues on the surface of the host cell (Fig. 23.22). It then inserts a short fusion peptide into the host cell membrane which triggers fusion of the viral and host cell membranes (endocytosis). The virus then accomplishes the other steps in its reproduction cycle and the new viruses produced then leave the host cell by budding off the cell surface. However, the sialic acid residues on the cell surface which were useful for the initial attachment hinder the new viruses when it comes to leaving the host cell surface.
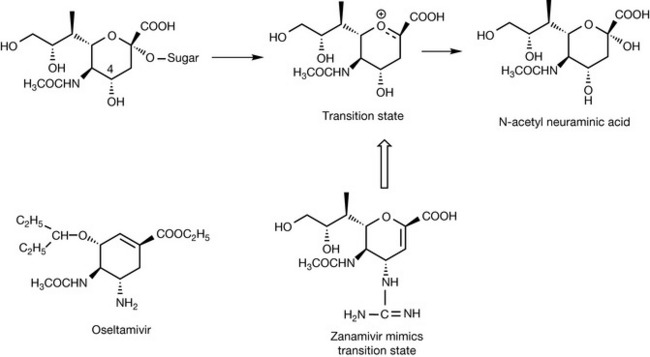
Thus the virus employs the enzyme neuraminidase (NA), which cleaves the negatively charged sialic acid residue off the host cell membrane (Fig. 23.23). The search for inhibitors of the viral NA began in the 1960s. Once the crystal structure for the viral NA was determined in the 1980s, more rapid progress in the rational design of an inhibitor was made. It was observed that within the enzyme site the OH group in the 4 position on the sialic was positioned near two negatively charged aspartic acid residues. When the 4 OH was replaced with a positively charged guanidine group, the strongly binding inhibitor zanamivir (Relenza) was produced. As might be judged from the structure, its oral bioavailability is poor, and it has to be administered via an inhaled aerosol since it also has only a short half-life in plasma. Oseltamivir was developed as an orally bioavailable NA inhibitor. The replacement of the glycol side chain with a more lipophilic hydrocarbon side chain did not reduce binding, and removal of the oxygen from the ring of the molecule improved stability in vivo. Zanamivir and oseltamivir are moderately effective in inhibiting influenza infection. Zanamivir taken prophylactically inhibits infection by 82% but this falls to 30% if it is taken after the onset of infection. The drugs are useful in preventing or treating influenza in the elderly and in infants.
There are a number of other potential targets in the life cycle of the influenza virus, such as the initial cell surface binding stage, but as yet there are no other drugs close to market.
De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30(2):115-133.
De Clercq E. New approaches toward anti-HIV chemotherapy. J Med Chem. 2005;48(5):1297-1313.
Menendez-Arias L. Targeting HIV: antiretroviral therapy and development of drug resistance. Trends Pharmacol Sci. 2002;23(8):381-388. and references therein
Tomasselli A.G., Heinrikson R. Targeting the HIV-protease in AIDS therapy: a current clinical perspective. Biochemica Biophysica Acta. 2000;1477:189-214.
Vacca J.P., Condra J.H. Clinically effective HIV-1 protease inhibitors. Drug Discov Today. 1997;7:261-272.
Herdewijn P. Structural requirements for antiviral activity in nucleosides. Drug Deliv Today. 1997;2:235-242.
Mathe C., Gosselin G. L-nucleoside enantiomers as antiviral drugs: A mini-review. Antiviral Res. 2006;71:276-281.
Luscher-Mattli M. Influenza chemotherapy: a review of the present state of art and of new drugs in development. Arch Virol. 2000;145:2233-2248.
Wade R.C. Flu’ and structure based drug design. Structure. 1997;5:1139-1145.
Wyde P.R. Respiratory syncytial virus (RSV) disease and prospects for its control. Antiviral Res. 1998;29:63-79.