Chapter 7 DNA structure and its importance to drug action
Introduction
The elucidation of the structure of deoxyribonucleic acid (DNA) in 1953 by James Watson and Frances Crick was one of the major scientific events of the last century. The recent unravelling of the human genome would not have been possible today without Watson and Crick’s fundamental descriptions of the role of complementary base pairing and the organisation of the component deoxynucleotides into a double helical structure. Since Rosalind Franklin’s groundbreaking research using X-ray crystallography to define two general forms of helical DNA, a whole variety of experimental techniques have shown that the structure of DNA is far more complex than originally proposed. Not only are there different morphological states (e.g. A, B, Z), the structure is also sequence dependent, where the order of the nucleotides can influence the three-dimensional shape in different regions of the helix. Such variations in structure according to sequence are fundamental to the function of DNA and its interactions with the many different proteins that seek to influence its role in cellular biochemistry. It is not the purpose of this chapter to discuss in depth the minutiae of DNA structural variations; there is already a wealth of literature available which describes these phenomena. Here, we provide the basics of DNA structure and function in order to lay foundations for later chapters where DNA plays a part in the pharmacological action of specific drugs. These work through a variety of chemical mechanisms including DNA cleavage and cross-linking, or by reversible association, usually by intercalation or binding in one of the DNA grooves. It is fair to say that a number of drugs whose cellular target is DNA were in use before the structure of DNA had been solved, or even before it was recognised as the repository of the genetic code, e.g. the nitrogen mustards in the treatment of cancer. However, this does not detract from the fact that in the design of new drugs which target DNA, and to grasp the mechanisms of action of current DNA-targeting drugs already in the clinic, an understanding of the target’s structure, function and chemistry is necessary.
DNA is a polymeric molecule composed of subunits called deoxynucleotides in whose sequence is stored our genetic code. The information necessary for a cell to function and replicate in a programmed manner is ultimately determined by this code, and which parts of it are turned on or off. Essentially, the specific order of the deoxynucleotides within the DNA sequence, codes for specific proteins, which when synthesised, perform explicit functions of a cell’s biochemistry. Sometimes the code is continually turned on (expressed) to produce proteins which are in constant demand, whereas for proteins which are required in response to a particular signal, their code will be turned off until such a time as that signal in question is initiated, e.g. release of a hormone such as oestrogen in puberty. Considering that it is the DNA sequence that holds the code for proteins to be synthesised, and that it is the interactions of specific proteins with particular code sequences that turn on and off these sequences, we can already appreciate the highly complex role of DNA within cellular function and replication. In fact, with DNA and proteins it is the proverbial chicken and egg problem; without DNA, proteins cannot be synthesised, yet without proteins, our genetic code cannot be expressed. Ultimately, drugs that exert their pharmacological effect at the DNA level act by altering the interactions between these two classes of biological macromolecule. So how is this code stored and how is it translated?
The structural components of DNA – DNA primary structure
The structure of DNA is like a long piece of string, composed of two strands wound around each other like strands in a rope. Each strand consists of subunits or monomers linked together like beads, and each subunit is called a deoxynucleotide. DNA is therefore a biological polymer, and it is the order of the deoxynucleotide monomers and their chemical nature and linkage that is responsible for the genetic code, and is referred to as the primary structure of DNA.
The deoxynucleotide monomers in DNA are made up of the same chemical moieties, namely a phosphate group, a deoxyribose sugar and a heteroaromatic base. The latter moiety can be one of four different bases and it is this chemical diversity that enables a code to be constructed. The sugar and the phosphate groups of the deoxynucleotides form the backbone of each polymeric strand, being linked together through phosphodiester bonds.
DNA bases
The DNA bases, as their name suggests, are mainly basic molecules, and fall into two categories of aromatic heterocycle: the pyrimidines and purines. Although some of the DNA bases are bases, none of them is a strong enough base or acid to carry a charge at physiological pH (7.4) and their pKa values are at least 2 units above or below physiological pH.1
Pyrimidine bases
The bases thymine and cytosine are derivatives of the heterocycle pyrimidine. Thymine and cytosine are examples of tautomers, and the structures shown in Figure 7.1 are the major, stable tautomeric forms of these bases. Those stable forms have an amino substituent in the amino form as opposed to the imino form, and the oxygen atoms prefer to exist as the keto rather than the enol form at physiological pH. However, since the nitrogens in the molecule are weakly acidic at very high pH the oxygen atoms can bear a negative charge (see Ch. 4). The fact that the major tautomeric form is found in DNA is crucial to the existence of DNA as a double-stranded, self-replicating structure.
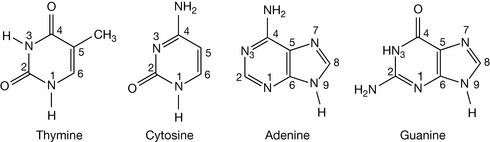
Thymine is a very weak base pKa ca. 4.5 and cytosine is not a base at all, being a very weak acid.
Purine bases
The bases adenine and guanine are heterocyclic purine derivatives. Again, they are the major tautomers that are present in DNA (Fig. 7.1), and are weak bases, having their pKa values around 5.
Deoxynucleosides
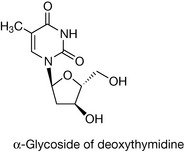
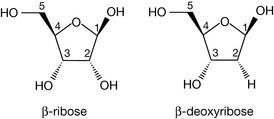
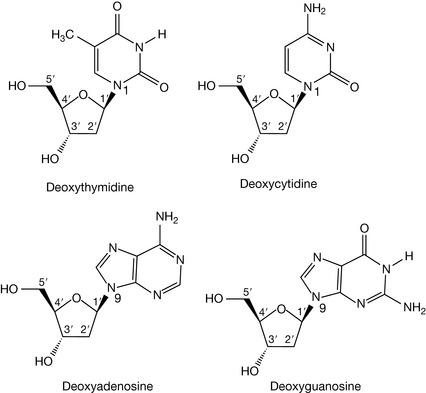
The sugar unit found in the backbone linkages of DNA is the furanose, deoxyribose (Fig. 7.2), which differs from its parent sugar ribose at the 2 position; the hydroxyl group has been replaced by a hydrogen and, in other words, has lost an oxygen, hence the prefix deoxyribose. Attachment of the pyrimidine or purine bases via the 1 or 9 position, respectively, to the 1 position of deoxyribose (through the loss of water) yields the four deoxynucleosides. The bond linking the base to the sugar is called a β-glycosidic link, β because the base is above the plane of the sugar, in common with the carbon at the 5′ position (if the base were below the sugar plane, it would be an α-glycoside). Note how the numbering system changes for the sugar in the deoxynucleoside, with each number having a ‘prime’ associated with it to distinguish it from the positions in the covalently attached base. In nomenclature terms, because the deoxyribose sugars are associated with these molecules, they are given the deoxy prefix in their names. The suffix to the base name also changes, to indicate that it is part of a base-sugar conjugate, hence we have deoxythymidine, deoxycytidine, deoxyadenosine and deoxyguanosine (Fig. 7.3).
Deoxynucleotides
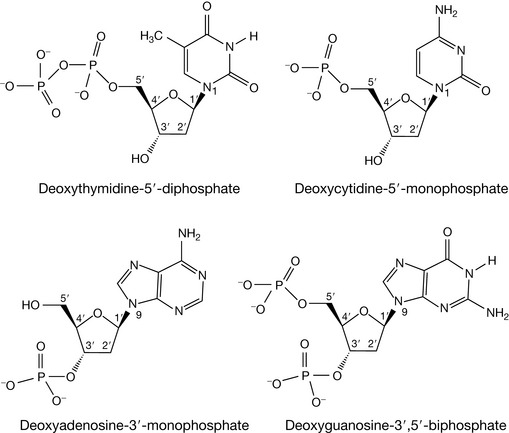
Deoxynucleotides are the phosphate esters of the deoxynucleosides. Just as a carboxylic ester can be considered as the product of a carboxylic acid and alcohol, then a phosphate ester is the product of phosphoric acid and an alcohol, where the alcohol, in this instance, is the sugar moiety of the deoxynucleoside. As there are two alcoholic functionalities on the deoxynucleoside sugar at the 5′ and 3′ positions, the corresponding deoxynucleotides can be 5′-monophosphates, 3′-monophosphates, or even 3′,5′-biphosphates (Fig. 7.4). If two or three phosphate groups were attached via the 5′ hydroxyl, the deoxynucleotide would be given the 5′-di- or triphosphate respectively, e.g. deoxythymidine-5′-diphosphate.
 Self Test 7.2
Self Test 7.2
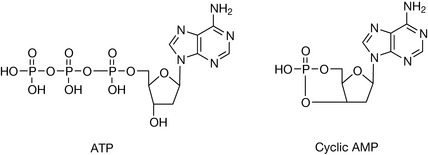
The universal energy cofactor ATP is an abbreviation of adenosine-5′-triphosphate – draw the structure.
Cyclic AMP is a secondary messenger in cellular biochemistry and is an abbreviation for cyclic adenosine-3′,5′-monophosphate – draw the structure (hint: one phosphate group is bonded to two positions).
If the deoxynucleotides are the monomeric units that compose DNA and their order along its backbone is the foundation of the genetic code, how are they linked together? Phosphoric acid is a tribasic acid and therefore has the capacity to form more than one ester linkage with alcohols. There are two alcoholic groups on each deoxyribose, so a phosphodiester linkage between two separate deoxynucleosides is possible. Within DNA, there is an order to such linkages, with a phosphodiester bond only forming between the 5′ and 3′ hydroxyl groups of different monomers in a contiguous fashion. Formation of phosphodiester groups from phosphoric acid leaves one free acidic oxygen on the phosphate which is fully ionised at physiological pH. Consequently, DNA strand backbones will have an overall net negative charge. We can now see the route of the name for unabbreviated deoxyribonucleic acid; deoxyribose is the sugar present in each of the linked deoxyribonucleotides, and the acid arises from the acidity of the phosphodiester backbone.
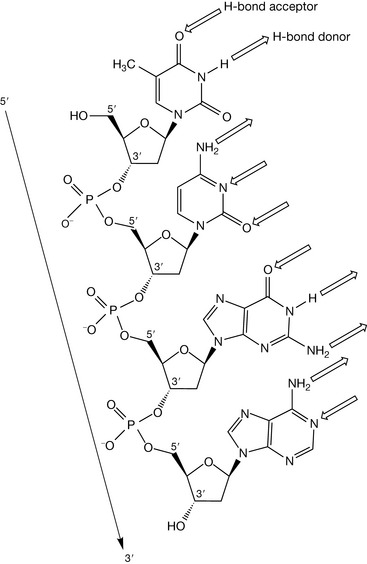
Examination of the four deoxynucleotides linked together by phosphodiester bonds shown in Figure 7.5 reveals that DNA strands have directionality, i.e. we can move in a 5′ to 3′ direction along the strand, or in a 3′ to 5′ direction. Such directionality features in the secondary structure of DNA, and is vital to the replicatory and decoding process of the genetic code.
Complementary hydrogen bonding between DNA strands
Within the cell nucleus, DNA is present not as a single polymeric strand, but as a double strand, where two chains of deoxynucleotides are bound together. The binding interaction between the strands is not covalent in nature, but involves weaker forces that can be overcome by thermal heating. Raising the temperature of a solution of double-stranded DNA results in strand separation at about 100°C. If such a solution is then cooled, the strands anneal to the same original, double-stranded structure, which indicates that the forces that bind the strands together are highly specific and discriminatory. Inspection of the single-stranded structure shown in Figure 7.5 reveals the presence of hydrogen-bonding functional groups on the bases which project away from the sugar–phosphate backbone. It is these hydrogen bond donors and acceptors that are instrumental in holding the two strands together in a highly specific manner.
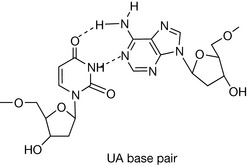
The hydrogen-bonding compatibilities between deoxyadenosine (dA) and deoxythymidine (dT) ensures that these two bases pair with each other in a complementary fashion via two hydrogen bonds. For deoxycytidine (dC) and deoxyguanosine (dG), three hydrogen bonds between compatible hydrogen bond donors and acceptors give rise to the second type of base pair. The arrangement of the respective hydrogen bond donor and acceptor groups on each base means that under normal conditions, dA will only bind with dT, and dC with dG. The nature and positions of the hydrogen bonds ensures that each base pair is planar. In Figure 7.6, position 1 of the pyrimidine deoxynucleotides (dC and dT) and position 9 of the purines (dA and dG) are the points of attachment of each base to the deoxyribose sugar in the strand backbone. Significantly, the distance between these attachment points for each base pair is practically the same because each base pair consists of a purine and pyrimidine base. The consequent effect of such an arrangement ensures that the backbone of each strand remains more or less equidistant along the whole double-stranded structure, thus imparting a degree of regularity which is irrespective of the sequence of the bases within that structure.
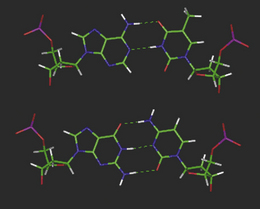
Figure 7.6 Molecular models of the DNA base pairs illustrating ‘Watson–Crick’ hydrogen bonding complementarity.
Self Test 7.3
You have two separate solutions of double-stranded DNA: one solution contains DNA composed only of dG and dC as self-complementary strands, and the other only of dA and dT (shown below). Heating the solutions will result in strand separation. Which strands will separate at the lower temperature, and why?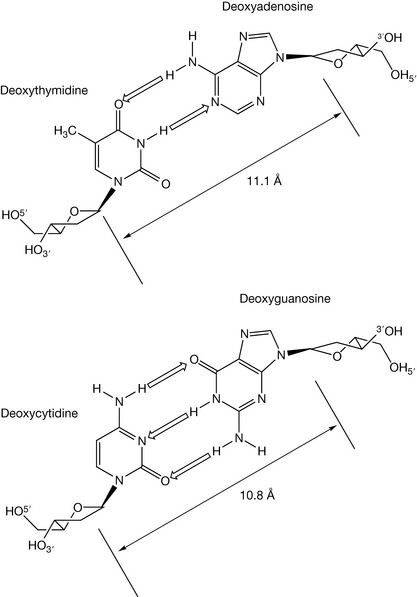
For the base pairs to hydrogen bond in the manner shown in Figure 7.6, one base from each pair must be rotated by 180° to ensure that the hydrogen bonding groups are directed towards each other. Because the opposing complementary bases are not isolated, but linked together in a strand, in order to rotate the opposite base to ensure complementarity, the whole strand must have a directionality that is opposite to the strand with which it is bound (Fig. 7.6). Two strands having opposing directionality are said to be antiparallel, where direction is defined by the phosphodiester links in the sugar–phosphate backbone i.e. 5′ to 3′ versus 3′ to 5′ (Fig 7.7).
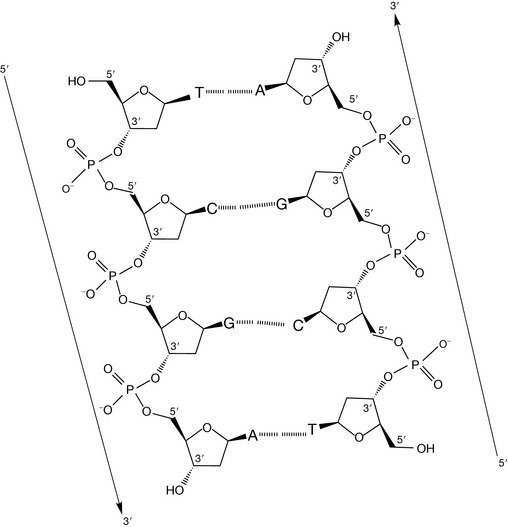
Figure 7.7 A schematic section of a DNA double strand illustrating antiparallel directionality and interstrand hydrogen bonding.
Before discussing the three-dimensional secondary structure of double-stranded DNA, it is worth making a few observations of the two-dimensional representation of the primary structure illustrated in Figure 7.7.
DNA secondary structure – the double helix
The three-dimensional secondary structure of DNA is determined by the conformational preferences and restrictions of the relatively rigid individual deoxynucleotides which are linked together in each of the strands, which are, in turn, influenced by the properties of the DNA primary structure listed above. Thus, the inspection of some of the factors that influence deoxynucleotide conformation can help in the extrapolation to the more complex three-dimensional nature of the double-stranded polymer itself.
Conformation of the deoxyribose sugar
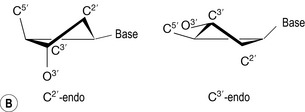
The pentagonal representations of the deoxyribose sugar moieties have so far been as a ‘plan’ view. However, they are saturated rings (as illustrated by the stereochemical centres at the 1′, 3′ and 4′ positions) and will be twisted out of plane to reduce the non-bonded repulsions between the ring substituents in order to adopt low energy conformations. Two low energy structures for free deoxynucleotides in solution are the C2′ endo and C3′ endo conformations, endo referring to the position of the carbon atom most distorted from planarity in the ring structure and on the same side of the ring as the attached base (Fig. 7.8). The two different conformations will influence the relative spatial positions of phosphate groups attached at the 5′ and 3′ hydroxyls (Fig. 7.8). C2′ endo sugar pucker produces an interphosphorus distance of 7.0 Å, whilst the C3′ endo conformation results in an equivalent distance of 5.9 Å. If these conformations were adopted within the deoxynucleotide strand, then an exclusively C2′ endo backbone would be more elongated than a corresponding C3′ endo chain.
Conformation of the base with respect to the sugar
Low energy structures of the base with respect to the deoxyribose sugar are achieved when the base plane is perpendicular to the sugar plane and bisects the C2′-O-O4′ angle to produce a syn or anti conformation. Syn or anti refers to the base with respect to this angle at the β-glycosidic bond (Fig. 7.9). Of the two, the anti conformation is more stable because of lower steric repulsion between the base substituents and the sugar ring. The pyrimidine bases have an oxygen atom in position 2 which, in an anti conformation, is projected away from the sugar. In the corresponding syn conformation, this oxygen is in direct conflict with the sugar ring and 5′-phosphate atoms (Fig 7.9). The effect is more pronounced with the purine bases, where the syn conformation places an aromatic ring directly over the sugar. The net effect of the deoxynucleotides preferring an anti conformation is to direct the hydrogen bond donor and acceptor groups of the bases away from the backbone and into a position where complementary hydrogen bonding with an antiparallel strand is favoured.
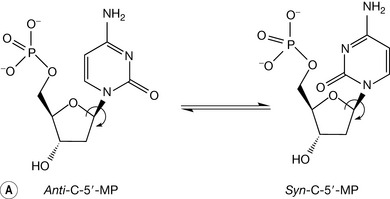
Figure 7.9 The possible anti and syn conformations of cytosine with respect to the deoxyribose sugar unit in a cytidine deoxynucleotide.
The anti conformations of the bases are stabilised further by a weak electrostatic interaction between the electron-deficient acidic hydrogen in position 6 (pyrimidines)/position 8 (purines) and the O5′ oxygen of the deoxyribose sugar. These hydrogens are electron deficient (acidic) because of their proximity to the electronegative nitrogen and oxygen atoms within the heteroaromatic bases. Such an interaction helps ensure that the hydrogen bonding functions of the bases are directed away from the backbone in the DNA structure.
The phosphodiester bonds
The P–O bonds in the phosphodiester backbone are the positions of greatest flexibility within the DNA strands, and are the main pivots affecting polydeoxynucleotide structure and allow for some degree of flexibility. If a degree of strain is introduced into the double stranded structure, it will more than likely be relieved at these bonds initially (Fig. 7.10). The phosphodiester P–O bonds are also orientated so that the negatively charged oxygens are removed from the hydrophobic core interior where the bases lie, and project out into the water which surrounds the exterior, where they can interact with hydrophilic cationic counter-ions and water molecules.
Base pair stacking
The complementary base pairs in the centre of the double-stranded DNA structure are planar yet perpendicular to the plane of the sugars in the backbone. The optimum orientation relative to each other is therefore for them to stack on top of each other, subject to the restrictions of the sugar–phosphate backbone to which they are joined on opposite strands (Fig. 7.11). In fact, single deoxynucleotides will aggregate in aqueous solution by stacking their planar, heteroaromatic bases on top of each other. Van der Waals forces and charge transfer interactions between π-electron systems of the bases drive the hydrophobic stacking process, and such self-stacking of the rings into aggregates removes them from the hydrophilic aqueous environment. The optimum balance between the attractive and repulsive forces between the base pairs in this stacking environment is achieved at a distance of 3.4 Å between the respective planes.
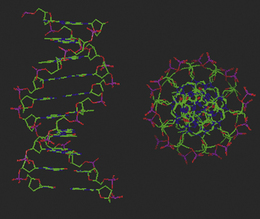
Figure 7.11 Molecular model of double-stranded DNA illustrating base pair stacking from the side and above the helix.
Taking into account the conformational preferences and restrictions of the individual deoxynucleotides in both strands, hydrogen bonding between the bases of opposite strands, the equidistance between those strands, the stacking of the base pairs within the hydrophobic interior, and the flexible P–O bonds directing the negatively charged oxygens towards the structural exterior, the most stable conformation for DNA is a right-handed double helix (Fig. 7.11). The 3.4 Å optimum distance between the base pair planes is determined by the sugar pucker of the deoxyribose in the backbone to which the bases are attached, which in turn is dependent on the degree of hydration by water molecules and the salt content of the solution in which the DNA is present. The backbone of the DNA helix is negatively charged, and the mutual repulsion between the anionic oxygens to a distance of 7 Å, promotes the C2′-endo sugar conformation within the deoxynucleotides, which subsequently enables an optimum base pair separation of 3.4 Å. Water molecules and cations such as sodium ions, which are attracted towards the sugar–phosphate backbone, mask the interanionic repulsive forces and stabilise the helical structure by forming a hydration sheath.
Why a helix, not a ladder?
It is apparent that planar base pairs stacked on top of each other and attached at each side to the sugar–phosphate backbones could adopt a structure akin to a ladder, as opposed to the spiral staircase dimensions of a double helix. Here the analogy is appropriate, considering that DNA contains millions of base pairs, so in terms of simple packing, more base pairs can be stacked on top of each other in a helical form. To adopt a ladder, the sugar–phosphate backbones would have to be extended, pulling the base pairs further apart to a distance approaching 7.0 Å, which would also reduce the attractive stacking interactions between the bases. Because the sugar–phosphate backbones twist around each other, the long base pair axes are rotated with respect to each other’s long axis by an angle of 36° for each step up the staircase (Fig 7.12). This is referred to as the helix twist or winding angle, and is an average value for the whole helix. There will be small variations of this angle, depending on the base pair sequence, within specific regions of the helix.
Helical grooves – major and minor
One of the most significant structural properties of the double-stranded DNA is the presence of two differently sized grooves that plough furrows along and around the double helix. It is these grooves that provide access to the genetic code, and form the basis of recognition with the proteins that bind and process the DNA during the various cellular events in which it is involved. The presence of two grooves of different sizes can be explained by inspecting the positions of attachment of the base pairs to the sugar–phosphate backbone. In Figure 7.13, the sugars from opposite backbones are not attached to the bases directly opposite each other, but displaced from the central helical axis. If the backbones were attached at the 6 position of the pyrimidine bases whilst still at the 9 position of the purines, i.e. opposite each other, then the two grooves between the backbones would be the same size, width and depth. The fact that they are attached off-centre means that there are two different-sized grooves, one wide, known as the major groove, and one narrow, referred to as the minor groove. When viewed from the side of the helix (Fig. 7.14), the edges of the base pairs can be seen on the groove floors. It is here that the genetic code can be accessed, because the functional groups exposed on the groove floor are totally dependent upon the base sequence/code (Fig. 7.15). Both grooves also have water molecules associated with them, hydrogen bonded to the base pairs and sugar–phosphate backbone, which form spines of hydration along the groove floors.
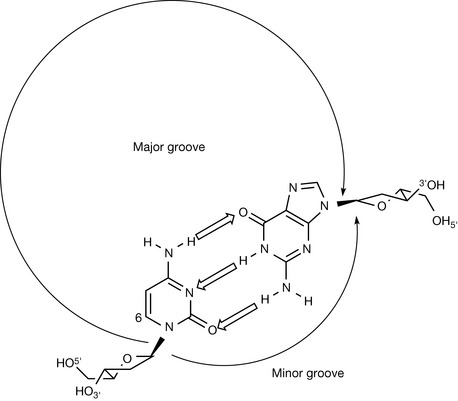
Figure 7.13 Schematic representation of a dc:dG base pair from above the helix illustrating that off-centre points of attachment between the backbones and the bases produce different-sized grooves.


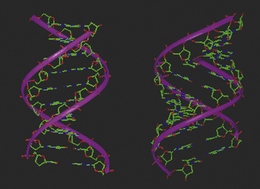
Helical repeat/pitch
The dimensions and parameters described above for double-helix DNA are associated with one particular polymorph or conformation of DNA known as B-DNA. In this form, one turn, or repeat of the helix contains ten base pairs, and the length of that turn is 34 Å (Fig. 7.16). To define a helical repeat, we can use the spiral staircase analogy again: if you stand on the bottom stair (base pair) and climb upwards until you reach the stair that is directly above the starting position, you have moved through one helical repeat, having climbed ten base pairs. B-DNA is the most common form of DNA, the structure of which was elucidated by Watson and Crick, and is generally associated with the physiological conditions of the cell. It must be stressed, however, that during cellular processing, DNA is a dynamic structure, and can morph between different structural motifs, depending upon the environment to which it is being subjected. Additionally, the dimensions and parameters so far discussed are considered as average values, because structural variations in the helical parameters are sequence dependent. New polymorphs of DNA are continually being discovered, and it is not in the remit of this chapter to discuss all of these forms. However, to illustrate the flexibility of the structure, a brief description of A-DNA is appropriate.
A-DNA
Low levels of hydration and higher salt concentrations will convert B-DNA to its A form by increasing the screening of the repulsive forces between the anionic oxygens in the phosphate backbone. Consequently, the deoxyribose sugars tend to adopt a C3′-endo pucker which reduces the interatomic distance between the phosphorus atoms of the phosphate groups in the backbone to 5.9 Å, thus compressing the structure compared with B-DNA. Drawing the phosphate groups closer together along the backbone forces the base pairs into closer proximity from 3.4 Å to 2.6 Å, which increases their ring stacking net repulsive forces. Whilst the helical twist angle remains around 36°, a parameter, known as base pair tilt, becomes predominant within the structure. In order to reduce the repulsion between the base pairs, they become tilted through 20° with respect to each other’s planarity: in other words, the stairs in the spiral staircase are no longer even, but are tilted as you climb them (Fig. 7.17). The overall effect on the structure is to compress the helix, giving it a smaller helical pitch of 2.8 Å in comparison with B-DNA, with eleven base pairs in each turn (Fig. 7.16). Within A-DNA, the groove sizes are different from those of B-DNA: the major groove is deeper while the minor groove is more shallow.
RNA
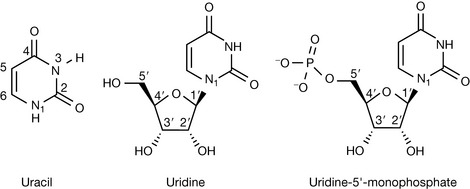
Another class of nucleic acid which is important in gene expression is RNA. Chemically, RNA and DNA are very closely related, but with two significant differences, one of which can be deduced from the expansion of the abbreviated name of RNA: ribonucleic acid. As the name suggests, RNA contains a different sugar within its subunits, namely ribose as opposed to deoxyribose. The 2′ position is no longer ‘deoxy’, but has a hydroxyl group oriented in the same way as the 3′ hydroxyl, below the plane of the sugar ring. Consequently, all subunits found in RNA drop the ‘deoxy’ prefix, and are referred to as nucleosides/nucleotides, or alternatively as ribonucleosides/ribonucleotides. The other essential chemical difference with DNA is the replacement of the thymine base within the structure by the equivalent pyrimidine base, uracil. Uracil has the same properties as thymine, forming two hydrogen bonds in a complementary base pair with adenine, but does not have a methyl group in the 5 position of the pyrimidine ring (Fig. 7.18).
Whilst the chemical distinctions between the nucleic acids may be small, their structural and functional differences are more significant. Whilst DNA may be responsible for storing the genetic code, in order to turn the genetic information into a protein two steps are required: transcription and translation. During transcription the template of the DNA is used to produce messenger RNA (mRNA) and the mRNA template is used to assemble a protein using transfer RNA (tRNA) which literally fetches the amino acids coded for by the mRNA. These are then used to assemble the peptide chain (see Ch. 6). Table 7.1 shows the genetic code triplets in RNA corresponding to the different amino acids used to assemble peptides.
Table 7.1 Three base codons used to code for the 20 amino acids found in proteins
| Amino acid | Codes |
|---|---|
| Glycine | GGU, GGC, GGA, GGG |
| Alanine | GCU, GCC, GCA, GCG |
| Serine | UCU, UCC, UCA, UCG, AGU, AGC |
| Proline | CCU, CCC, CCA, CCG |
| Valine | GUU, GUC, GUA, GUG |
| Threonine | ACU, ACC, ACA, ACG |
| Cysteine | UGU, UGC |
| Leucine | CUU, CUC, CUA, CUG, UUA, UUG |
| Isoleucine | AUU, AUC, AUA |
| Aspargine | AAU, AAC |
| Aspartic acid | GAU, GAC |
| Glutamine | CAA, CAG |
| Lysine | AAA, AAG |
| Glutamic acid | GAA, GAG |
| Methionine | AUG |
| Histidine | CAU, CAC |
| Phenylalanine | UUU, UUC |
| Tyrosine | UAU, UAC |
| Arginine | GCU, CGC, CGA, CGG, AGA, AGG |
| Tryptophan | UGG |
Table 7.1 shows 61 codons used to code for 20 amino acids. As can be seen, some amino acids have several different codes. In addition to the codons shown in the table there are three more codes from the permuations of four bases: UAA, UGA and UAG. These codons are stop signals indicating where the ribosome should stop translating the RNA since the C-terminus of the protein has been reached. Methionine is one of two amino acids which have only one codon. In addition, it shares this codon with a start signal indicating where the N-terminus of a protein is; thus all proteins in eucaryotes (organisms with complex cell structures) have methionine at their N-terminus. The methionine is usually removed post-translationally.
RNA generally does not form a double-stranded structure with a complementary strand of RNA, although its polymeric nature does allow for complementary base pairing within its single-stranded structure, i.e. it can fold back on itself. Messenger RNA (mRNA) can form double-stranded helices with the region of DNA being transcribed, where the two strands of the DNA have become separated for transcription, and the dimensions of the DNA–RNA helix in these regions are reminiscent of A-DNA. The presence of the 2′-hydroxyl group of the ribose in RNA hinders the formation of a B-type helix. Once released from the DNA, mRNA is a particularly flexible polymer, but can form transient single-stranded helices and complementary hairpins and loops in particular regions (Fig. 7.19), in order to aid recognition with the various biological macromolecules involved in the translation of the code into the amino acid sequence of the new protein.
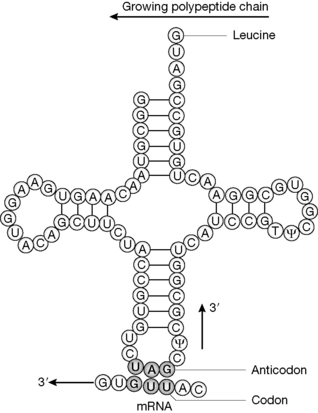
Figure 7.19 tRNA, showing the characteristic cloverleaf structure formed through base pairing within complementary regions within the one strand. N.B. uridine can pair with guanidine as well as adenine in tRNA.
tRNA has a characteristic three-dimensional ‘cloverleaf’ motif associated (Fig. 7.19). Such structures are held in place by base pairing (C with G and U with A) formed by hydrogen bonding between regions of the polymer that have complementary sequences when folded. There are certain nucleotide sequences which are highly conserved between different tRNAs and form the characteristic ‘double-stranded’ regions which stabilise the three-dimensional structures of the tRNA molecules. Figure 7.20 illustrates the role of tRNA in translating the mRNA originally transcribed from the DNA genetic template. The tRNA has an anticodon which recognises the codon in mRNA and then adds the corresponding amino acid to the growing polypeptide chain. The recognition process by the anticodon during translation is more flexible than the recognition during transcription, and this means that it is not necessary to have 61 different tRNA molecules to recognise the 61 codons for 20 amino acids. In the example shown, the anticodon shown will also recognise UUA in the mRNA which codes for leucine. The bases in tRNA may be modified post-translationally, e.g. T= thymidine and ψ = pseudouridine.
Nucleic acid processing
The biochemical mechanisms for converting DNA into RNA into protein, and the processes of DNA replication, are covered in more detail in undergraduate biochemistry textbooks, and do not require detailed examination here. We make the assumption that the reader is aware of the role of enzymes such as DNA polymerase and RNA polymerase in the overall schemes of these processes. There are, however, specific chemical mechanisms performed by certain enzymes in DNA replication, repair, transcription and translation, which are fundamental to the chemical modes of action of specific classes of drugs, which necessitate a closer inspection. Such mechanisms will now be discussed within the context of these processes, without a detailed examination of the overall biochemical processes themselves.
Nucleic acid processing enzyme targets for drug action
Nucleic acid polymerase enzymes
When DNA is replicated during cell division, or complementary mRNA is transcribed from a DNA sequence for protein synthesis, or viral RNA or DNA is replicated in order to promote cellular invasion and viral regeneration, a new nucleic acid is synthesised. A polymerase enzyme such as DNA polymerase, RNA polymerase, or RNA directed DNA polymerase (viral reverse transcriptase) performs the actual elongation process, all are basically involving the same chemical mechanism. In order to synthesise a new nucleic acid polymer, a complementary DNA template within the genome has to be exposed on which the new polymer is to be based. This template may be a gene that is to be expressed, a region of a chromosome to be replicated, or viral or bacterial RNA or DNA. The processes which initiate the process of template preparation are extremely complex, but generally involve the association of proteins such as recognition factors which bind to specific DNA sequences, e.g. a promoter sequence upstream from the sequence to be transcribed. These sequences are recognised within the major groove of the DNA and the complex formed acts as a signal for the polymerase to bind to the sequence to be transcribed (Fig. 7.20) and begin either transcription (RNA production) or DNA replication (as part of the cell division process). Polymerisation is a unidirectional procedure, meaning that the new nucleic acid can only be synthesised in one direction, and that is 5′ to 3′.
The polymerase enzymes require two substrates for chain elongation: the template and the new nucleotides (RNA) or deoxynucleotides (DNA) from which they synthesise the new polymer in a complementary and antiparallel manner. For example, if the template sequence were as follows:
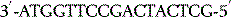
then a new DNA sequence would be synthesised as follows:
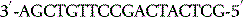
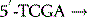
The enzyme catalyses nucleophilic attack by the terminal free 3′ hydroxyl of the growing strand at the electrophilic phosphorus of the triphosphate group directly bonded to the 5′ hydroxyl (Fig. 7.21). The leaving group in the reaction is a diphosphate group, because the substrates the enzymes use for chain elongation are the 5′-triphosphate nucleotides. The triphosphate group is very reactive if Mg2+ which is used to stabilise it is removed and this is often a component in the mechanism of phosphorylating enzymes (see Ch. 26). The active sites of these polymerase enzymes can only bind 5′-triphosphate (deoxy) nucleotides and the 3′-terminal hydroxyl group of the growing chain and hence will only synthesise the new polymer in a 5′→3′ direction.
DNA topoisomerase enzymes
DNA topoisomerases are essential enzymes that play a role in virtually every cellular DNA process; they solve the topological problems in DNA that are generated by nuclear processes such as DNA replication, transcription, recombination, repair, chromosome segregation and chromatin assembly by introducing transient breaks in the sugar–phosphate backbone of the helix. Strand cleavage by all topoisomerase enzymes involves nucleophilic attack by a catalytic tyrosine residue in the active site of the enzyme on a phosphodiester bond in the helix backbone, resulting in a covalent linkage between the enzyme and one end of the broken strand. The essential difference between the two main types of topoisomerase is that topoisomerase I breaks one strand of duplex DNA, whilst topoisomerase II breaks both strands of the backbone to generate a gate through which another region of DNA can be passed. The latter enzyme requires the energy cofactor ATP to perform its function, whilst the former does not. During transcription and replication, DNA topoisomerase I acts to reduce the torsional stress which arises within the DNA helix during these processes. DNA topoisomerase II, on the other hand, by virtue of its double-stranded DNA passage reaction, is able to regulate DNA over- and underwinding, and can resolve knots and tangles in the genetic material.
The origins of torsional strain in DNA
In the preceding sections that describe the structure of DNA, the associated figures show short sections of DNA. DNA within cells is, in fact, millions of base pairs long and is bound up within chromosomes to a variety of storage proteins such as histones. Such proteins are rich in positively charged lysine and arginine amino acid residues which, by electrostatic interactions with the negatively charged backbones of the DNA, fold it into an ordered compact form known as chromatin. In human somatic cells, a single DNA duplex of about 4 cm in length is found in each of the 46 chromosomes. During replication and transcription, only short sequences of DNA are exposed for the processing enzymes to act upon, so essentially, the ends of the DNA of the exposed section are fixed. Replication and transcription require strand separation in order to expose the template for the polymerases to work with. One way of aiding strand separation is to increase the strain within the helix so that the strands will overcome the hydrogen-bond attractive forces between the complementary base pairs, and naturally move apart. Such strain must be significantly greater than can be relieved by rotation around the flexible P–O bonds in the sugar–phosphate backbone. Topoisomerase II, by breaking both strands in the exposed regions, generates a gate, and passes another section of the exposed DNA through it. The more times the DNA is passed through the gate, the more ‘wound up’ the exposed DNA becomes because the tension cannot be relieved at the fixed ends. The resultant effect of this increase in strain into the helix forces it to adopt a new tertiary structure, known as supercoiled DNA, which is a higher-energy structure than relaxed, double-helical DNA. The hydrolysis of ATP by the topoisomerase II enzyme provides the energy input for this process. Because supercoiled DNA is a high-energy structure, the strands will separate more easily for the polymerase enzymes to replicate or transcribe the exposed DNA. Topoisomerase II performs other DNA processing functions, but the above description serves to illustrate its essential role in transcription and replication.
Supercoiling by topoisomerase II and the action of the various replication and transcription proteins and enzymes on the exposed DNA can result in strand knotting which will hinder process progression. Topoisomerase I, by breaking one of the sugar–phosphate backbones in the knotted region, can relieve the excess strain by allowing the second strand to unravel, driven by its own high, inherent energy. The conversion of the superhelical DNA back to its relaxed form after processing has taken place is also catalysed by topoisomerase I by the same mechanism. DNA replication and transcription is therefore controlled, in part, by a balance of the actions of topoisomerases I and II. Interference with these enzyme mechanisms would have implications for nucleic acid processing and this is the basis for the action of a number of drugs including the anticancer drugs camptothecin and etoposide (Ch. 21) and the isoquinolone antibiotics (Ch. 22).
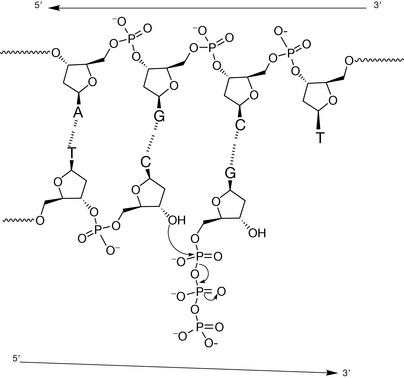
The mechanism of action of both the topoisomerase enzymes involves a tyrosine residue in the active site of the enzyme which, by nucleophilic attack, becomes covalently linked to the sugar–phosphate backbone via a phosphotyrosyl bond, releasing the sugar hydroxyl to generate a nick. Topoisomerase II attacks both strands, from opposite sides with two tyrosine residues, to generate two 5′-phosphotyrosyl links with each strand, and two free 3′ ends in the nicked strands. After another region of the DNA has been passed through the gate, the strands are resealed by the reverse action of nucleophilic attack by the free 3′ hydroxyls on the phosphotyrosyl groups to release the enzyme from the complex (Fig. 7.22). Topoisomerase I produces a single-stranded break by the action of one tyrosine residue, but generates a single 3′-phosphotyrosyl link between the enzyme and the DNA, and produces a free 5′ end in the cleaved strand. After release of torsional strain by passing the intact single strand through the gap, resealing follows by reversing the nucleophilic attack (Fig. 7.23). For the topoisomerase enzymes to perform their function, they need to bind double-stranded DNA around the phosphate backbone to perform the strand-cleavage reactions. Figure 7.24 shows schematically how topoisomerase I approaches and binds to a segment of DNA.
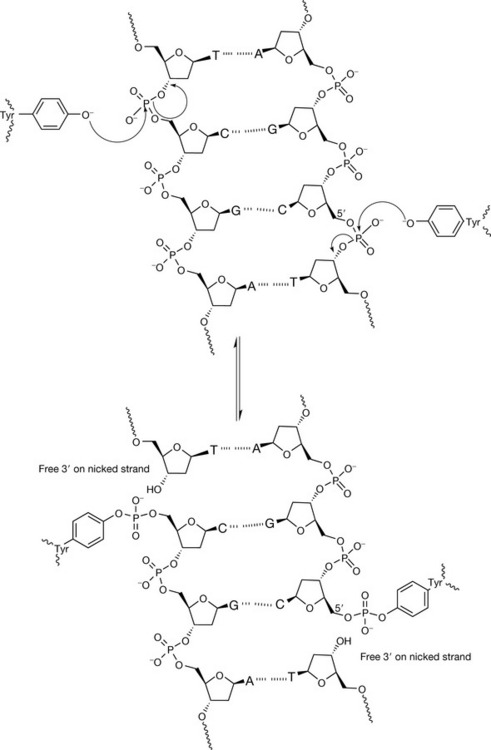
Figure 7.22 The mechanism of action of topoisomerase II, an enzyme which catalyses the cleavage of both backbones of DNA four bases apart to generate a staggered gate within the helix for double strand passage. The DNA strands are covalently linked via a 5′-phosphotyrosyl to the enzyme, generating two free 3′-termini on the scissile strands.
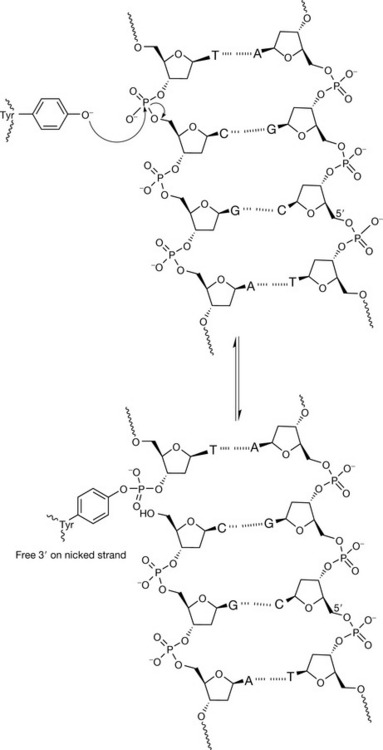
Figure 7.23 The mechanism of action of topoisomerase I. Cleavage of the phosphate backbone occurs when one DNA strand is covalently linked via a 3′-phosphotyrosyl to the enzyme, generating a free 5′-termini on the scissile strand.
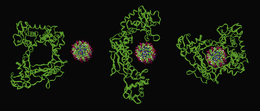
Figure 7.24 Molecular models illustrating how topoisomerase I, which contains a central pore of the same dimensions as the DNA helix, unfolds in order to bind prior to strand cleavage.