 CHAPTER 171 Diabetes Mellitus
CHAPTER 171 Diabetes Mellitus
Diabetes mellitus (DM) is characterized by hyperglycemia and glycosuria and is an end point of a few disease processes (Table 171-1). The most common type occurring in childhood is type 1 DM, which is caused by autoimmune destruction of the insulin-producing beta cells (islets) of the pancreas leading to permanent insulin deficiency. Type 2 diabetes mellitus (DM2) is less common in children and often results from insulin resistance and relative insulin deficiency, usually in the context of exogenous obesity. The incidence of DM1 and DM2 in the United States is increasing. Less common subtypes of DM2 result from genetic defects of the insulin receptor or inherited abnormalities in sensing of ambient glucose concentration by pancreatic beta cells (see Table 171-1).
TABLE 171-1 Classification of Diabetes Mellitus in Children and Adolescents
| Type | Comment |
|---|---|
| TYPE 1 (INSULIN-DEPENDENT) | |
| Transient neonatal | Manifests immediately after birth; lasts 1–3 months |
| Permanent neonatal | Other pancreatic defects possible |
| Classic type 1 | Glycosuria, ketonuria, hyperglycemia, islet cell antibody to glutamic acid decarboxylase positive; genetic component |
| TYPE 2 (NON–INSULIN-DEPENDENT) | |
| Secondary | Cystic fibrosis, hemochromatosis, drugs (L-asparaginase, tacrolimus) |
| Adult type (classic) | Associated with obesity, insulin resistance; genetic component |
| Maturity-onset diabetes of youth | |
| Mitochondrial diabetes | Associated with deafness and other neurologic defects, maternal transmission—mtDNA point mutations |
| OTHER | |
| Gestational diabetes | Abnormal glucose tolerance only during pregnancy, which reverts to normal post partum; increased risk for later onset of diabetes |
mtDNA, mitochondrial DNA.
DEFINITION
A diagnosis of diabetes mellitus is made when a fasting serum glucose concentration is at or above 126 mg/dL or a 2-hour postprandial serum glucose concentration is at or above 200 mg/dL on two separate occasions. A patient is considered glucose intolerant if fasting serum glucose concentrations is 100 to 125 mg/dL and 2-hour plasma glucose following an oral glucose tolerance test (OGTT) is 140 to 199 mg/dL. Sporadic hyperglycemia can occur in children, usually in the setting of an intercurrent illness. When the hyperglycemic episode is clearly related to an illness or other physiologic stress, the probability of incipient diabetes is small (< 5%). Sporadic hyperglycemia occurring without a clear precipitating physiologic stress is of more concern because diabetes develops in at least 30%.
INSULIN-DEPENDENT (TYPE 1) DIABETES MELLITUS
Etiology
DM1 results from the autoimmune destruction of insulin-producing beta cells (islets) of the pancreas. In addition to the presence of diabetes susceptibility genes, an unknown environmental insult presumably triggers the autoimmune process. A variety of studies have produced conflicting data regarding a host of environmental factors. These include cow’s milk feeding before age 2 years, viral infectious agents (coxsackie B virus, cytomegalovirus, mumps, and rubella), and possibly vitamin D deficiency. DM1 is thought to be primarily a T cell–mediated disease; however, that knowledge is evolving.
Antibodies to islet cell antigens may be seen months to years before the onset of beta cell dysfunction (Fig. 171-1). These include islet cell antibodies, insulin autoantibodies, antibodies to tyrosine phosphatase IA-2, antibodies to glutamic acid decarboxylase, and others. The risk for diabetes increases with the number of antibodies detected in the serum. In individuals with only one detectable antibody, the risk is only 10% to 15%; in individuals with three or more antibodies, the risk is 55% to 90%. When 80% to 90% of the beta cell mass has been destroyed, the remaining beta cell mass is insufficient to maintain euglycemia, and clinical manifestations of diabetes result (see Fig. 171-1).
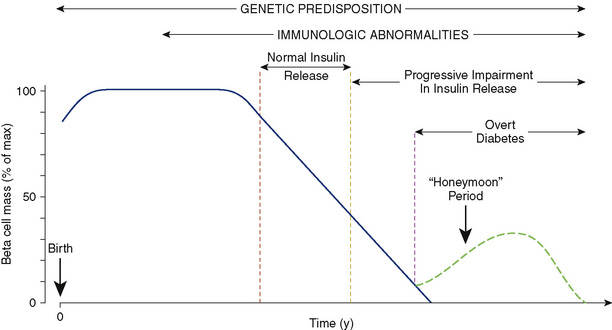
FIGURE 171-1 Schematic representation of the autoimmune evolution of diabetes in genetically predisposed individuals.
Diabetes in childhood also can result from rare inherited defects in mitochondrial genes. Other rare subtypes of DM2 are syndromes of severe insulin resistance caused by mutations in the insulin receptor gene and diabetes resulting from the secretion of abnormal forms of insulin or insulin action (e.g., lipoatrophic DM). Diseases of exocrine pancreas (e.g., cystic fibrosis related diabetes); endocrinopathies (e.g., Cushing syndrome); and drug therapies (e.g., glucocorticoids, chemotherapeutics) are other rare causes of diabetes or glucose intolerance.
Epidemiology
In the United States, the annual incidence is approximately 20 in 100,000. The annual incidence in children ranges from a high of 40 in 100,000 among Scandinavian populations to less than 1 in 100,000 in China. The annual incidence of DM1 is increasing steadily but with significant geographic differences. The prevalence of DM1 in the United States is highest in whites and is lower in African Americans and Hispanic Americans.
Genetic determinants play a role in the susceptibility to DM1, although the mode of inheritance is complex and clearly multigenic. Siblings or offspring of patients with diabetes have a risk of 3% to 6% for development of diabetes; an identical twin has a 30% to 50% risk. The HLA region on chromosome 6 provides the strongest determinant of susceptibility, accounting for approximately 40% of familial inheritance of DM1. Specific class II DR and DQ HLA alleles (HLA DR3 and HLA DR4) increase the risk of developing DM1, whereas other specific HLA alleles exert a protective effect. More than 90% of children with DM1 possess HLA DR3 alleles, HLA DR4 alleles, or both. The insulin gene region VNTR on chromosome 11 is also linked to DM1 susceptibility. There is evidence for association, beyond HLA, of more than 100 other loci with DM1. Genetic factors do not fully account for susceptibility to DM1; environmental factors also play a role.
Clinical Manifestations
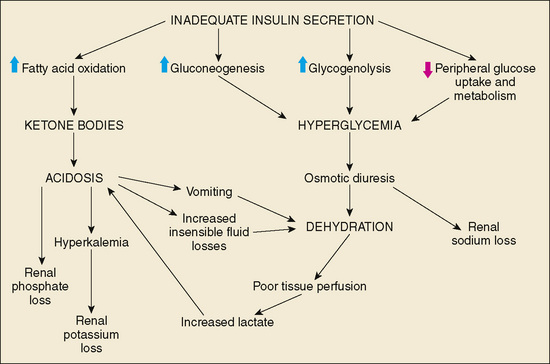
Hyperglycemia results when insulin secretory capacity becomes inadequate to enhance peripheral glucose uptake and to suppress hepatic and renal glucose production. Insulin deficiency usually first causes postprandial hyperglycemia, then fasting hyperglycemia. Ketogenesis is a sign of more complete insulin deficiency. Lack of suppression of gluconeogenesis and glycogenolysis further exacerbates hyperglycemia while fatty acid oxidation generates the ketone bodies: β-hydroxybutyrate, acetoacetate, and acetone. Protein stores in muscle and fat stores in adipose tissue are metabolized to provide substrates for gluconeogenesis and fatty acid oxidation.
Glycosuria occurs when the serum glucose concentration exceeds the renal threshold for glucose reabsorption (approximately 180 mg/dL). Glycosuria causes an osmotic diuresis (including obligate loss of sodium, potassium, and other electrolytes), leading to dehydration. Polydipsia occurs as the patient attempts to compensate for the excess fluid losses. Weight loss results from the persistent catabolic state and the loss of calories through glycosuria and ketonuria. The classic presentation of DM1 includes polyuria, polydipsia, polyphagia, and weight loss.
Diabetic Ketoacidosis
If the clinical features of new-onset DM1 are not detected, diabetic ketoacidosis (DKA) will occur. DKA also can occur in patients with known DM1 if insulin injections are omitted or during an intercurrent illness when greater insulin requirements are unmet in the presence of elevated concentrations of the counter-regulatory and stress hormones (glucagon, growth hormone [GH], cortisol, and catecholamines). DKA can be considered to be present if (1) the arterial pH is below 7.25, (2) the serum bicarbonate level is below 15 mEq/L, and (3) ketones are elevated in serum or urine.
Pathophysiology
In the absence of adequate insulin secretion, persistent partial hepatic oxidation of fatty acids to ketone bodies occurs. Two of these three ketone bodies are organic acids and lead to metabolic acidosis with an elevated anion gap. Lactic acid may contribute to the acidosis when severe dehydration results in decreased tissue perfusion. Hyperglycemia causes an osmotic diuresis that is initially compensated for by increased fluid intake. As the hyperglycemia and diuresis worsen, most patients are unable to maintain the large fluid intake, and dehydration occurs. Vomiting, as a result of increasing acidosis, and increased insensible water losses caused by tachypnea can worsen the dehydration. Electrolyte abnormalities occur through a loss of electrolytes in the urine and transmembrane alterations resulting from acidosis. As hydrogen ions accumulate as a result of ketoacidosis, intracellular potassium is exchanged for hydrogen ions. Serum concentrations of potassium increase initially with acidosis, then decrease as serum potassium is cleared by the kidney. Depending on the duration of ketoacidosis, serum potassium concentrations at diagnosis may be increased, normal, or decreased, but intracellular potassium concentrations are depleted. A decreased serum potassium concentration is an ominous sign of total body potassium depletion. Phosphate depletion also can occur as a result of the increased renal phosphate excretion required for elimination of excess hydrogen ions. Sodium depletion also is common in DKA, resulting from renal losses of sodium caused by osmotic diuresis and from gastrointestinal losses from vomiting (Fig. 171-2).
Presentation
Patients with DKA present initially with polyuria, polydipsia, and nausea and vomiting. Abdominal pain occurs frequently and can mimic an acute abdomen. The abdomen may be tender from vomiting, or distended secondary to a paralytic ileus. The presence of polyuria, despite a state of clinical dehydration, indicates osmotic diuresis and differentiates patients with DKA from patients with gastroenteritis or other gastrointestinal disorders. Respiratory compensation for acidosis results in tachypnea with deep (Küssmaul) respirations. The fruity odor of acetone frequently can be detected on the patient’s breath. An altered mental status can occur, ranging from disorientation to coma.
Laboratory studies reveal hyperglycemia (serum glucose concentrations ranging from 200 mg/dL to > 1000 mg/dL). Arterial pH is below 7.25, and the serum bicarbonate concentration is less than 15 mEq/L. Serum sodium concentrations may be elevated, normal, or low, depending on the balance of sodium and free water losses. The measured serum sodium concentration is artificially depressed, however, because of hyperglycemia (see Chapter 35). Hyperlipidemia also contributes to the decrease in measured serum sodium. The level of blood urea nitrogen (BUN) can be elevated with prerenal azotemia secondary to dehydration. The white blood cell count is usually elevated and can be left-shifted without implying the presence of infection. Fever is unusual and should prompt a search for infectious sources that may have triggered the episode of DKA.
Treatment
Therapy for patients with DKA involves careful replacement of fluid deficits, correction of acidosis and hyperglycemia via insulin administration, correction of electrolyte imbalances, and monitoring for complications of treatment. The optimal approach to management of DKA must strike a balance between adequate correction of fluid losses and avoidance of rapid shifts in osmolality and fluid balance. The most serious complication of DKA and its treatment is cerebral edema and cerebral herniation.
Dehydration
A patient with severe DKA is assumed to be approximately 10% dehydrated. If a recent weight measurement is available, the precise extent of dehydration can be calculated. An initial intravenous (IV) fluid bolus of a glucose-free isotonic solution (normal saline, lactated Ringer’s solution) at 10 to 20 mL/kg should be given to restore intravascular volume and renal perfusion. The remaining fluid deficit after the initial bolus should be added to maintenance fluid requirements, and the total should be replaced slowly over 36 to 48 hours. Ongoing losses resulting from osmotic diuresis usually do not need to be replaced unless urine output is large or signs of poor perfusion are present. Osmotic diuresis is usually minimal when the serum glucose concentration decreases to less than 300 mg/dL. To avoid rapid shifts in serum osmolality, 0.9% sodium chloride can be used as the replacement fluid for the initial 4 to 6 hours, followed by 0.45% sodium chloride.
Hyperglycemia
Fast-acting soluble insulin should be administered as a continuous IV infusion (0.1 U/kg/hour). Serum glucose concentrations should decrease at a rate no faster than 100 mg/dL/hour. When serum glucose concentrations decrease to less than 250 to 300 mg/dL, glucose should be added to the IV fluids. If serum glucose concentrations decrease to less than 200 mg/dL before correction of acidosis, the glucose concentration of the IV fluids should be increased, but the insulin infusion should not be decreased by more than half, and it should never be discontinued before resolution of acidosis.
Acidosis
Insulin therapy decreases the production of free fatty acids (FFAs) and protein catabolism, and enhances glucose usage in target tissues. These processes correct acidosis. Bicarbonate therapy should be avoided unless severe acidosis (pH < 7.0) results in hemodynamic instability, or when symptomatic hyperkalemia is present. Potential adverse effects of bicarbonate administration include paradoxical increases in central nervous system (CNS) acidosis caused by increased diffusion of carbon dioxide across the blood-brain barrier, potential tissue hypoxia caused by shifts in the oxyhemoglobin dissociation curve, abrupt osmotic changes, and increased risk of development of cerebral edema.
As acidosis is corrected, urine ketone concentrations may appear to rise. β-Hydroxybutyrate, which is not detected in urine ketone assays, is converted with treatment to what the assay most detects, acetoacetate. Hence, minute to minute urine ketone concentrations are not a required index of the adequacy of therapy.
Electrolyte Imbalances
Regardless of the serum potassium concentration at presentation, total body potassium depletion is likely. Potassium concentrations can decrease rapidly as insulin, and then glucose, therapy improves acidosis and potassium is exchanged for intracellular hydrogen ions. When adequate urine output is shown, potassium should be added to the IV fluids. Potassium replacement should be given as 50% potassium chloride and 50% potassium phosphate at a concentration of 20 to 40 mEq/L. This combination provides phosphate for replacement of deficits, but avoids excess phosphate administration, which may precipitate hypocalcemia. If the serum potassium level is greater than 6 mEq/L, potassium should not be added to IV fluids until the potassium level decreases.
Monitoring
A flow sheet should be used to record and monitor fluid balance and laboratory measurements. Initial laboratory measurements should include serum glucose, sodium, potassium, chloride, bicarbonate, BUN, creatinine, calcium, phosphate, and magnesium concentrations; arterial or venous pH; and a urinalysis. Serum glucose measurement should be repeated every hour during therapy; electrolyte concentrations should be repeated every 2 to 3 hours. Calcium, phosphate, and magnesium concentrations should be measured initially and every 4 to 6 hours during therapy. Neurologic and mental status should be assessed at frequent intervals. Any complaints of headache or deterioration of mental status should prompt rapid evaluation for possible cerebral edema. Indicative symptoms include a decreased sensorium, sudden severe headache, vomiting, change in vital signs (bradycardia, hypertension, apnea), a dilated pupil, ophthalmoplegia, or seizure.
Complications
Clinically apparent cerebral edema occurs in 1% to 5% of cases of DKA. Cerebral edema is the most serious complication of DKA, with a mortality rate of 20% to 80%. The pathogenesis of cerebral edema likely involves osmolar shift resulting in fluid accumulation in the intracellular compartment and cell swelling. Subclinical cerebral edema is common in patients with DKA, but the factors that exacerbate this process leading to symptomatic brain swelling and possible cerebral herniation are not clearly defined. Cerebral edema typically occurs 6 to 12 hours after therapy for DKA is begun, often following a period of apparent clinical improvement. Factors that correlate with increased risk for cerebral edema include higher initial BUN concentration, lower initial PCO2, failure of the serum sodium concentration to increase as glucose concentration decreases during treatment, and treatment with bicarbonate.
Signs of advanced cerebral edema include obtundation, papilledema, pupillary dilation or inequality, hypertension, bradycardia, and apnea. Treatment involves the rapid use of IV mannitol, endotracheal intubation, and hyperventilation, and may require the use of a subdural bolt. Other complications of DKA are intracranial thrombosis or infarction, acute tubular necrosis with acute renal failure caused by severe dehydration, pancreatitis, arrhythmias caused by electrolyte abnormalities, pulmonary edema, and bowel ischemia. Peripheral edema occurs commonly 24 to 48 hours after therapy is initiated and may be related to residual elevations in antidiuretic hormone and aldosterone.
Transition to Outpatient Management
When the acidosis has been corrected and the patient tolerates oral feedings, the IV insulin infusion can be discontinued and a regimen of subcutaneous (SC) insulin injections can be initiated. The first SC insulin dose should be given 30 to 45 minutes before discontinuation of the IV insulin infusion. Further adjustment of the insulin dose should be made over the following 2 to 3 days. A patient already diagnosed with DM1 may be restarted on the prior doses if they were adequate. For a patient with new-onset DM1, typical starting total daily doses are approximately 0.7 U/kg/24 hours for prepubertal patients and approximately 1 U/kg/24 hours for adolescents, using any number of the available insulin combinations. The best and most common choice for making the transition to SC insulin is to begin by giving injections of fast-acting insulin (lispro or aspart or glulisine insulin) with each meal and long- or basal-acting insulin (glargine or detemir) at bedtime. This regimen of multiple daily injections (MDIs) provides the most flexibility but requires the patient to administer many injections per day and to count carbohydrates in food. An alternative to MDIs is a fixed mixed split dosing regimen, with two daily injections. Two thirds of the total daily dose is given in the morning, at breakfast, and one third given in the evening, with dinner (Fig. 171-3). The ratio of insulins in the morning dose should be two-thirds intermediate-acting insulin (NPH), and one-third fast-acting insulin. In the evening, one half of the evening insulin dose should be given as intermediate-acting insulin (NPH), and one half should be given as fast-acting insulin.
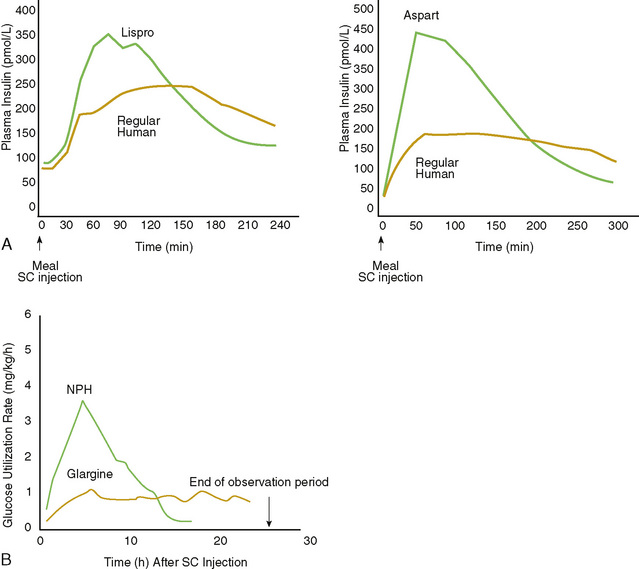
FIGURE 171-3 Representative profiles of insulin effect using combined injection regimens: intermediate-acting insulin (NPH or lente) and regular (short-acting) insulin. A, Short-acting lispro, regular, or aspart insulins. B, Long-acting NPH or glargine profiles. SC, subcutaneous.
(Adapted from Mudaliar SR, Lindberg FA, Joyce M, et al: Insulin aspart [B28 asp-insulin]: A fast-acting analog of human insulin: Absorption kinetics and action profile compared with regular human insulin in healthy nondiabetic subjects. Diabetes Care 22:1501–1506, 1999; and Lepore M, Kurzals R, Pampanelli S, et al: Pharmacokinetics and dynamics of S.C. injection of the long-acting glargine [HOE 901] in T1DM. Diabetes 48[Suppl 1]:A97, 1999.)
Externally worn pumps that provide a continuous SC infusion of fast-acting insulin are available, although not usually at the very onset of DM1. They may be used by all age groups who are highly motivated to achieve tight control.
Serum glucose concentrations should be assessed before each meal, at bedtime, and at 2 to 3 AM to provide information for adjustment of the regimen. Patients and their families should begin learning the principles of diabetes care as soon as possible. Demonstration of ability to administer insulin injections and test glucose concentrations using a glucometer is necessary before discharge, as is knowledge of hypoglycemia management. Meal planning is crucial to control of glucose in DM1, and nutrition services must be part of the care delivered to the families from the outset.
Honeymoon Period
In patients with new onset of DM1 who do not have DKA, the beta cell mass has not been completely destroyed. The remaining functional beta cells seem to recover function with insulin treatment. When this occurs, exogenous insulin requirements decrease. This is a period of stable blood glucose control, often with nearly normal glucose concentrations. This phase of the disease, known as the honeymoon period, usually starts in the first weeks of therapy and often continues for 3 to 6 months, but can last 2 years.
Outpatient Type 1 Diabetes Mellitus Management
Management of DM1 in children requires a comprehensive approach, with attention to medical, nutritional, and psychosocial issues. Therapeutic strategies should be flexible, with the individual needs of each patient and the family taken into account. Optimal care involves a team of diabetes professionals, including a physician, a diabetes nurse educator, a dietitian, and a social worker or psychologist.
Goals
The Diabetes Control and Complications Trial established that intensive insulin therapy, with the goal of maintaining blood glucose concentrations as close to normal as possible, can delay the onset and slow the progression of complications of diabetes (retinopathy, nephropathy, neuropathy). Attaining this goal using intensive insulin therapy can increase the risk of hypoglycemia. The adverse effects of hypoglycemia in young children may be significant because the immature CNS may be more susceptible to glycopenia. Although the risk for diabetic complications increases with duration of diabetes, there is controversy as to whether the rate of increase of risk may be slower in the prepubertal years than in adolescence and adulthood. It is safe to establish goals to achieve tight control of blood glucose concentrations in childhood.
The goals of therapy differ, depending on the age of the patient. For children younger than 5 years old, an appropriate goal is maintenance of blood glucose concentrations between 80 and 180 mg/dL. For school age children, 80 to 150 mg/dL is a reasonable target range. For adolescents, the goal is 70 to 150 mg/dL. Goals of therapy also should take into account other individual characteristics, such as a past history of severe hypoglycemia and the abilities of the patient and family.
Insulin Regimens
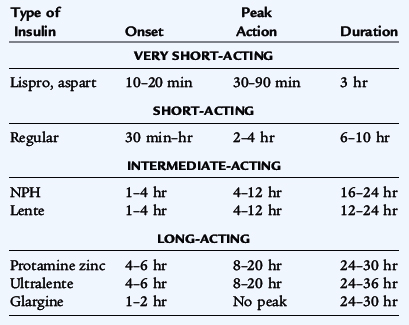
Many types of insulin differ in duration of action and time to peak effect (Table 171-2). These insulins can be used in various combinations, depending on the needs and goals of the individual patient. The most commonly used regimen is that of multiple injections of fast-acting insulin given with meals in combination with long-acting basal insulin given at bedtime. This regimen provides flexibility but requires administration of many injections per day and will require assistance for young children. After the total daily dose of insulin is determined, 50% is given usually at bed time as long-acting insulin, while the remainder is given as fast-acting insulin, divided according to the need for corrections of high glucose levels and for meals. To correct for hyperglycemia, one determines the insulin sensitivity using the 1800 rule, by dividing 1800 by the total daily dose of insulin to determine how many mg/dL of glucose will decrease with 1 unit of insulin. The insulin:carbohydrate ratio is used to calculate insulin for the carbohydrate content of food; 450 divided by the total daily dose determines the number of grams of carbohydrate that requires 1 unit of insulin.
When it is not possible for the patient to receive an insulin injection at midday (e.g., during school hours) one can use intermediate-acting insulin (NPH) in the morning along with the fast-acting insulin (see Fig. 171-3). Patients using this regimen must adhere to a meal schedule with consistent breakfast, lunch, and dinner and snacks as needed, and must receive a second evening injection of NPH; this can be difficult to coordinate with variations in daily activities. Pumps that provide a continuous SC infusion of short-acting insulin also are available and are being used by children and adolescents who are highly motivated to achieve tight control.
Lispro and aspart and glulisine insulins are synthetic human insulin analogs in which amino acid alterations result in fast absorption and onset of action (see Table 171-2). Because of the short duration of action, these are used in combination with long-acting insulin. Glargine and detemir are insulin analogs in which the amino acid alterations result in increased solubility at acidic pH and decreased solubility at physiologic pH. Glargine and detemir have been shown to have a duration of greater than 24 hours and act as a basal insulin.
Newly diagnosed patients in the honeymoon period may require 0.4 to 0.6 U/kg/24 hours. Prepubertal patients with diabetes longer than 1 to 2 years typically require 0.5 to 1 U/kg/24 hours. During middle adolescence, when elevated growth hormone (GH) concentrations produce relative insulin resistance, insulin requirements increase by 40% to 50%, and doses of 1 to 2 U/kg/24 hours are typical.
Nutrition
Balancing the daily meal plan with the dosages of insulin is crucial for maintaining serum glucose concentrations within the target range and avoiding hypoglycemia or hyperglycemia. The content and schedule of meals vary according to the type of insulin regimen used; it is recommended that carbohydrates contribute 50% to 65% of the total calories; protein, 12% to 20%; and fat, less than 30%. Saturated fat should contribute less than 10% of the total caloric intake, and cholesterol intake should be less than 300 mg/24 hours. High fiber content is recommended.
Patients using MDIs or the insulin pump can maintain a more flexible meal schedule with regard to the timing of meals and the carbohydrate content. These patients give an injection of insulin before or immediately after each meal, with the total dose calculated according to the carbohydrate content of the meal. Further adjustments in the dose can be made based on the measured serum glucose concentration and plans for exercise during the day. Children using a twice a day combination of intermediate-acting and fast-acting insulins need to maintain a relatively consistent meal schedule so that carbohydrate absorption and insulin action peaks correspond. A typical meal schedule for a patient using this type of regimen involves three meals and three snacks daily. The total carbohydrate content of the meals and snacks should be kept constant.
Blood Glucose Testing
Blood glucose should be routinely monitored (with rapid glucose meters) before each meal and at bedtime. Hypoglycemia during the night or excessive variability in the morning glucose concentrations should prompt additional testing at 2 or 3 AM to ensure that there is no consistent hypoglycemia or hyperglycemia. During periods of intercurrent illness or when blood glucose concentrations are higher than 300 mg/dL, urine ketones also should be tested.
Long-Term Glycemic Control
Measurements of glycohemoglobin or hemoglobin A1c reflect the average blood glucose concentration over the preceding 3 months and provide a means for assessing long-term glycemic control. Glycohemoglobin should be measured four times a year, and the results should be used for counseling of patients. Table 171-3 summarizes the correlation between hemoglobin A1c or glycohemoglobin and daily blood glucose measurements. Measurements of glycohemoglobin or hemoglobin A1c are inaccurate in patients with hemoglobinopathies. Glycosylated albumin or fructosamine can be used in these cases.
TABLE 171-3 Biochemical Indices of Glycemic Control
| Poor Control | Average Control | Intensive Control |
|---|---|---|
| HgbA1c > 10% | HgbA1c > 8–10% | HgbA1c 6–8% |
| Average blood glucose > 240 mg/dL | Average blood glucose 180–240 mg/dL | Average blood glucose 120–180 mg/dL |
HgbA1c, hemoglobin A1c.
Complications
Patients with DM1 for more than 3 to 5 years should receive an annual ophthalmologic examination for retinopathy. Urine should be collected annually for assessment of microalbuminuria; if present, microalbuminuria suggests early renal dysfunction and indicates a high risk of progression to nephropathy. Treatment with angiotensin-converting enzyme inhibitors may halt the progression of microalbuminuria. In children with DM1, annual cholesterol measurements and periodic assessment of blood pressure are recommended. Early detection of hypertension and hypercholesterolemia with appropriate intervention can help to limit future risk of coronary disease.
Other Disorders
Chronic lymphocytic thyroiditis is particularly common and can result in hypothyroidism. Because symptoms can be subtle, thyroid function tests should be performed annually. Other disorders that occur with increased frequency in children with DM1 include celiac disease, IgA deficiency, Addison disease, and peptic ulcer disease.
Special Problems: Hypoglycemia
Hypoglycemia occurs commonly in patients with DM1. For patients in adequate or better control, it is expected to occur on average once or twice a week. Severe episodes of hypoglycemia, resulting in seizures or coma or requiring assistance from another person, occur in 10% to 25% of these patients per year.
Hypoglycemia in patients with DM1 results from a relative excess of insulin in relation to the serum glucose concentration. This excess can be caused by alterations in the dose, timing, or absorption of insulin; alterations of carbohydrate intake; or changes in insulin sensitivity resulting from exercise. Defective counter-regulatory responses also contribute to hypoglycemia. Abnormal glucagon responses to falling serum glucose concentrations develop within the first few years of the disease, and abnormalities in epinephrine release occur after a longer duration.
Lack of awareness of hypoglycemia occurs in approximately 25% of patients with diabetes. Recent episodes of hypoglycemia may play a role in the pathophysiology of hypoglycemia unawareness; after an episode of hypoglycemia, autonomic responses to subsequent episodes are reduced. A return of symptoms of hypoglycemia can be exhibited in these patients after 2 to 3 weeks of strict avoidance of hypoglycemic episodes.
Symptoms of hypoglycemia include symptoms resulting from neuroglycopenia (headache, visual changes, confusion, irritability, or seizures) and symptoms resulting from the catecholamine response (tremors, tachycardia, diaphoresis, or anxiety) (see Chapter 172). Mild episodes can be treated with administration of rapidly absorbed oral glucose (glucose gel or tablets, fruit juices, and non-diet or non–artificially sweetened sodas). More severe episodes that result in seizures or loss of consciousness at home should be treated with glucagon injections. IV glucose should be given in hospital settings.
Prognosis
Long-term complications of DM1 include retinopathy, nephropathy, neuropathy, and macrovascular disease. Evidence of organ damage caused by hyperglycemia is rare in patients with duration of diabetes of less than 5 to 10 years, and clinically apparent disease rarely occurs before 10 to 15 years’ duration. The eventual morbidity and fatality attributable to these disorders are substantial. Diabetic retinopathy is the leading cause of blindness in the United States. Nephropathy eventually occurs in 30% to 40% and accounts for approximately 30% of all new adult cases of end-stage renal disease. Neuropathy occurs in 30% to 40% of postpubertal patients with DM1 and leads to sensory, motor, or autonomic deficits. Macrovascular disease results in an increased risk of myocardial infarction and stroke among individuals with diabetes.
Intensive control of diabetes, employing frequent blood glucose testing and multiple daily injections of insulin or an insulin pump can reduce substantially the development or progression of diabetic complications. Intensive management resulted in a 76% reduction of risk for retinopathy, a 39% reduction in microalbuminuria, and a 60% reduction in clinical neuropathy. For pubertal and adult patients, these benefits of intensive therapy likely outweigh the increase in risk for hypoglycemia. For younger patients, in whom the risks for hypoglycemia are greater, and the benefits of tight glucose control may be lower, a less intensive regimen may be appropriate.
NON–INSULIN-DEPENDENT (TYPE 2) DIABETES MELLITUS
Pathophysiology
DM2 can occur as the result of various pathophysiologic processes; however, the most common form results from peripheral insulin resistance and compensatory hyperinsulinemia followed thereafter with failure of the pancreas to maintain adequate insulin secretion (see Table 171-1). The precise defects underlying the insulin-resistant state and the eventual pancreatic beta cell failure are complex and, while still poorly understood, are beginning to be understood at the genetic level. Other subtypes of DM2 also can occur in children. Maturity-onset diabetes of youth (MODY) comprises a group of dominantly inherited forms of relatively mild diabetes. Insulin resistance does not occur in these patients; instead the primary abnormality is an insufficient insulin secretory response to glycemic stimulation.
Epidemiology
The prevalence of DM2 in children is increasing in parallel with childhood obesity and is highest in children of ethnic groups with a high prevalence of DM2 in adults, including Native Americans, Hispanic Americans, and African Americans. Obesity, the metabolic syndrome, ethnicity, and a family history of DM2 are thus risk factors. Among some clinically assumed to have DM2, autoantibodies to the pancreas are present, compounding the difficulty in differentiating DM1 from DM2 at the outset.
Clinical Manifestations and Differential Diagnosis
Fasting and postprandial glucose levels for diagnosis of DM2 are the same as those for DM1. The diagnosis of DM2 may be suspected on the basis of polyuria and polydipsia and in a background of the metabolic syndrome. Differentiating DM2 from DM1 in children on clinical grounds only can be challenging. The possibility of DM2 should be considered in patients who are obese, have a strong family history of DM2, have other characteristics of the metabolic syndrome and acanthosis nigricans on physical examination, or have absence of antibodies to beta cell antigens at the time of diagnosis of diabetes. Acanthosis nigricans, a dermatologic manifestation of hyperinsulinism, presents as hyperkeratotic pigmentation in the nape of the neck and in flexural areas. Although ketoacidosis occurs far more commonly in DM1, it also can occur in patients with DM2 under conditions of physiologic stress and cannot be used as an absolute differentiating factor. DM2 can be confirmed by evaluation of insulin or C-peptide responses to stimulation with oral carbohydrate and in the absence of islet cell autoreactivity.
Therapy
DM2 is the result of a combination of insulin resistance and relative insulin deficiency, and a secretory beta cell defect. Asymptomatic patients with mildly elevated glucose values (slightly > 126 mg/dL for fasting or slightly > 200 mg/dL for random glucose) may be managed initially with lifestyle modifications, including nutrition therapy (dietary adjustments) and increased exercise. Exercise has been shown to decrease insulin resistance. In most children with new-onset, uncomplicated DM2, oral hypoglycemic agents are usually the first line of therapy. These medications include insulin secretagogues and insulin sensitizers. The most common treatment is either metformin or one of the thiazolidinediones. A rare side effect of metformin is lactic acidosis, which occurs mainly in patients with compromised renal function. The most common side effect is gastrointestinal upset. If ketonuria or ketoacidosis occurs, insulin treatment is necessary at first to achieve adequate glycemic control, but may be discontinued within weeks with continuation of oral medications. Oral drugs may be used as combinations.
Because DM2 may have a long preclinical course, early diagnosis is possible, including prevention in subjects at risk who have the metabolic syndrome. Emerging data indicate that treatment with insulin sensitizers may delay or prevent development of the full-blown disease. Significant lifestyle modifications, such as improved eating habits and increased exercise, have a role in preventing or decreasing the morbidity of DM2. Finally, it is critical to monitor and manage the other components of the metabolic syndrome, such as advanced pubertal development, hypertension, hyperlipidemia, and polycystic ovary syndrome in females.