 CHAPTER 172 Hypoglycemia
CHAPTER 172 Hypoglycemia
Hypoglycemia in infancy and childhood can result from a large variety of hormonal and metabolic defects (Table 172-1). Hypoglycemia occurs most frequently in the early neonatal period, often as a result of inadequate energy stores to meet the disproportionately large metabolic needs of premature or small for gestational age newborns. Hypoglycemia during the first few days of life in an otherwise normal newborn is less frequent and warrants concern (see Chapter 6). After the initial 2 to 3 days of life, hypoglycemia is far less common and is more frequently the result of endocrine or metabolic disorders.
TABLE 172-1 Classification of Hypoglycemia in Infants and Children
ABNORMALITIES IN THE HORMONAL SIGNAL INDICATING HYPOGLYCEMIA
Counterregulatory Hormone Deficiency
Hyperinsulinism
INADEQUATE SUBSTRATE
DISORDERS OF METABOLIC RESPONSE PATHWAYS
Glycogenolysis
Gluconeogenesis
FATTY ACID OXIDATION
Other
DRUGS/INTOXICATIONS
ACTH, adrenocorticotropic hormone; acyl-CoA, acyl coenzyme A; IGF-2, insulin-like growth factor-2.
DEFINITION
The diagnosis of hypoglycemia should be made on the basis of a low serum glucose concentration, symptoms compatible with hypoglycemia, and resolution of the symptoms after administration of glucose.
Serum glucose concentrations less than 45 mg/dL are considered to be abnormal and necessitate treatment. Serum glucose concentrations greater than 55 mg/dL occasionally can occur in normal individuals, especially with prolonged fasting, but should be considered suspect, particularly if there are concurrent symptoms of hypoglycemia (Table 172-2).
TABLE 172-2 Symptoms and Signs of Hypoglycemia
| Features Associated with Epinephrine Release⁎ | Features Associated with Cerebral Glycopenia |
|---|---|
⁎ These features and perceptions of the features may be blunted if the patient is receiving β-blocking agents.
CLINICAL MANIFESTATIONS
The symptoms and signs of hypoglycemia result from direct depression of the central nervous system (CNS) owing to lack of energy substrate and the counter-regulatory adrenergic response to low glucose via catecholamine secretion designed to correct hypoglycemia (see Table 172-2). Compared with older children, infants do not usually show adrenergic symptoms. The neuroglycopenic symptoms and signs of hypoglycemia in infants are relatively nonspecific and include jitteriness, feeding difficulties, pallor, hypotonia, hypothermia, episodes of apnea and bradycardia, depressed levels of consciousness, and seizures. In older children, symptoms and signs include confusion, irritability, headaches, visual changes, tremors, pallor, sweating, tachycardia, weakness, seizures, and coma.
Failure to recognize and treat severe prolonged hypoglycemia can result in serious long-term morbidity, including mental retardation and nonhypoglycemic seizures. Younger infants and patients with more severe or prolonged hypoglycemia are at greatest risk for adverse outcomes.
PATHOPHYSIOLOGY
Hormonal Signal
Normal regulation of serum glucose concentrations requires appropriate interaction of a number of hormonal signals and metabolic pathways. An overview of these pathways is presented in Figure 172-1. In a normal individual, a decrease in serum glucose concentrations leads to suppression of insulin secretion and increased secretion of the counter-regulatory hormones (growth hormone [GH], cortisol, glucagon, and epinephrine) (see Fig. 172-1). This hormonal signal promotes the release of amino acids (particularly alanine) from muscle to fuel gluconeogenesis and the release of triglyceride from adipose tissue stores to provide free fatty acids (FFAs) for hepatic ketogenesis. FFAs and ketones serve as alternate fuels for muscle; the brain cannot transport FFAs across the blood-brain barrier. This hormonal signal also stimulates the breakdown of hepatic glycogen and promotes gluconeogenesis. Failure of any of the components of this hormonal signal can lead to hypoglycemia.
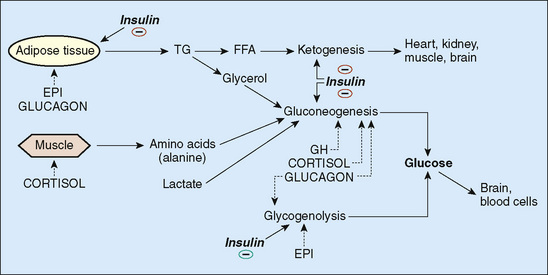
FIGURE 172-1 Regulation of serum glucose. — represents inhibitions. EPI, epinephrine; FFA, free fatty acid; GH, growth hormone; TG, triglyceride.
Hyperinsulinemia
A lack of suppression of insulin secretion in response to low serum glucose concentrations can occur in infants, but is uncommon beyond the neonatal period. This situation arises most frequently in infants of diabetic mothers who were exposed to high concentrations of maternally derived glucose in utero, resulting in fetal islet cell hyperplasia. The hyperinsulinemic state is transient, usually lasting hours to days.
Hyperinsulinism that persists beyond a few days of age can result from distinct genetic disorders affecting glucose-regulated insulin release. These were previously referred to as nesidioblastosis, or persistent hyperinsulinemic hypoglycemia of the newborn. In these infants, hyperplasia of the pancreatic islet cells develops in the absence of excess stimulation by maternal diabetes. Some patients with persistent hyperinsulinemic hypoglycemia of the newborn have genetic abnormalities of the islet cell sulfonylurea receptor or other genetic defects that alter the function of the adenosine triphosphate (ATP)-sensitive potassium channel that regulates insulin secretion. Hyperinsulinism also can occur in Beckwith-Wiedemann syndrome, a condition characterized by neonatal somatic gigantism: macrosomia, macroglossia, omphalocele, visceromegaly, and earlobe creases.
Regardless of the cause, neonates with hyperinsulinism are characteristically large for gestational age (see Chapter 60). Hypoglycemia is severe and frequently occurs within 1 to 3 hours of a feeding. Glucose requirements are increased, often two to three times the normal basal glucose requirement of 6 to 8 mg/kg/min. The diagnosis of hyperinsulinism is confirmed by the detection of serum insulin concentrations greater than 5 μU/mL during an episode of hypoglycemia. The absence of serum and urine ketones at the time of hypoglycemia is an important diagnostic feature, distinguishing hyperinsulinism from defects in counter-regulatory hormone secretion.
Treatment initially involves the infusion of intravenous (IV) glucose at high rates and of diazoxide to suppress insulin secretion. If diazoxide therapy is unsuccessful because the receptor on which it acts is dysfunctional, long-acting somatostatin analogs can be tried. Often, medical therapy for persistent hyperinsulinemic hypoglycemia of the newborn is unsuccessful, and subtotal (90%) pancreatectomy is required to prevent long-term neurologic sequelae of hypoglycemia.
In children, hyperinsulinemia is rare and usually results from an islet cell adenoma. Children with this condition characteristically have a voracious appetite, obesity, and accelerated linear growth. As in infants, diagnosis requires demonstration of insulin concentrations greater than 5 μU/mL during an episode of hypoglycemia. Computed tomography (CT), magnetic resonance imaging (MRI), or radioisotope imaging of the pancreas should be attempted, but visualization of an adenoma is usually difficult. Surgical removal of the adenoma is curative.
Factitious Hyperinsulinemia
In rare cases, insulin or a hypoglycemic medication is administered by a parent or caregiver to a child as a form of child abuse, which is a condition referred to as Munchausen syndrome by proxy. This diagnosis should be suspected if extremely high insulin concentrations are detected (>100 μU/mL). C-peptide concentrations are low or undetectable, which confirms that the insulin is from an exogenous source.
Defects in Counter-regulatory Hormones
Abnormalities in the secretion of counter-regulatory hormones that produce hypoglycemia usually involve GH, cortisol, or both. Deficiencies in glucagon and epinephrine secretion are rare. GH and cortisol deficiency occur as a result of hypopituitarism. Hypopituitarism results from congenital hypoplasia or aplasia of the pituitary or, more commonly, from deficiency of hypothalamic releasing factors (see Chapter 173). Clues to this diagnosis in infants include the presence of hypoglycemia in association with midline facial or neurologic defects (e.g., cleft lip and palate or absence of the corpus callosum), pendular nystagmus (indicating visual impairment from possible abnormalities in the development of the optic nerves, which can occur in septo-optic dysplasia), and the presence of microphallus and cryptorchidism in boys (indicating abnormalities in gonadotropin secretion). Jaundice and hepatomegaly also can occur, simulating neonatal hepatitis. Despite the presence of GH deficiency, these infants are usually of normal size at birth. Older children with hypopituitarism usually have short stature and a subnormal growth velocity.
Deficient cortisol secretion also can occur in primary adrenal insufficiency resulting from a variety of causes. In infants, it often results from congenital adrenal hyperplasia (CAH), most frequently as a result of 21-hydroxylase deficiency (see Chapters 177 and 178). In older children, primary adrenal insufficiency is seen most frequently in Addison disease, but it also can occur in adrenoleukodystrophy and other disorders (see Chapter 178). Addison disease should be suspected if there is hyperpigmentation of the skin, a history of salt craving, and confirmation of hyponatremia, and hyperkalemia.
Confirmation of GH or cortisol deficiency as the cause of hypoglycemia requires the detection of low serum GH and cortisol concentrations during an episode of hypoglycemia or after other stimulatory testing. In contrast to hyperinsulinism, serum and urine ketones are positive at the time of hypoglycemia, and FFAs are elevated. Treatment involves supplementation of the deficient hormones, GH or cortisol, in physiologic doses.
ENERGY STORES
Sufficient energy stores in the form of glycogen, adipose tissue, and muscle are necessary to respond appropriately to hypoglycemia. Deficiencies in these stores are a common cause of hypoglycemia in neonates who are small for gestational age or premature (see Chapter 60). Beyond the early neonatal period, energy stores are usually sufficient to meet the metabolic requirements except in malnourished children. Release of substrate from energy stores, for gluconeogenesis and fatty acid oxidation, is thought to be abnormal in one common form of childhood hypoglycemia, ketotic hypoglycemia.
IDIOPATHIC KETOTIC HYPOGLYCEMIA
Idiopathic ketotic hypoglycemia is usually seen in children between 18 months and 5 years of age. It is a common cause of new-onset hypoglycemia in this age group. Patients have symptoms of hypoglycemia after a period of prolonged fasting, often in the setting of an intercurrent illness with decreased feeding. Children with this disorder are often thin and small and may have a history of being small for gestational age. Defective mobilization of alanine from muscle to fuel gluconeogenesis is thought to be the cause, although the condition may derive mostly from having lower fuel reserves. Because there are no specific diagnostic tests for this disorder, ketotic hypoglycemia is a diagnosis of exclusion.
Treatment involves avoidance of fasting and frequent feedings of a high-protein, high-carbohydrate diet. Patients may require hospitalization for IV glucose infusion if they cannot maintain adequate oral intake during a period of illness. The disorder usually resolves spontaneously by 7 to 8 years of age.
METABOLIC RESPONSE PATHWAYS
Maintenance of normal serum glucose concentrations in the fasting state requires glucose production via glycogenolysis and gluconeogenesis and the production of alternative energy sources (FFAs and ketones) via lipolysis and fatty acid oxidation.
Glycogenolysis
Glycogen storage diseases occur in a variety of subtypes that differ in severity (see Chapter 52). Among the subtypes that result in hypoglycemia, the most severe form is glucose-6-phosphatase deficiency which is characterized by severe hypoglycemia, massive hepatomegaly, growth retardation, and lactic acidosis. In contrast, deficiencies in the glycogen phosphorylase enzymes may cause isolated hepatomegaly with or without hypoglycemia.
The diagnosis of glycogen storage disease is suggested by a finding of hepatomegaly without splenomegaly. Ketosis occurs with hypoglycemic episodes. Confirmation of the diagnosis requires specific biochemical studies of leukocytes or liver biopsy specimens. Treatment involves frequent high-carbohydrate feedings during the day and continuous feedings at night via nasogastric tube. Feedings of uncooked cornstarch during bedtime are sufficient to maintain serum glucose concentrations in some patients. Treatment can prevent night-time hypoglycemia.
Gluconeogenesis
Defects in gluconeogenesis are uncommon and include fructose-1,6-diphosphatase deficiency and phosphoenolpyruvate carboxykinase deficiency. Affected patients exhibit fasting hypoglycemia, hepatomegaly caused by fatty infiltration, lactic acidosis, and hyperuricemia. Ketosis occurs and FFA and alanine concentrations are high. Treatment involves frequent high-carbohydrate, low-protein feedings (see Chapter 52).
Fatty Acid Oxidation
Fatty acid oxidation disorders of ketogenesis include the fatty acid acyl-coenzyme A (CoA) dehydrogenase deficiencies; long-chain, medium-chain, and short-chain acyl-CoA dehydrogenase deficiencies; and hereditary carnitine deficiency (see Chapter 55). Of these disorders, medium-chain acyl-CoA dehydrogenase deficiency is the most common; it occurs in 1 in 9000 to 1 in 15,000 live births. Patients often are well in infancy and have the first episode of hypoglycemia at 2 years of age or older. Episodes of hypoglycemia usually occur with prolonged fasting or during episodes of intercurrent illness.
Mild hepatomegaly may be present along with mild hyperammonemia, hyperuricemia, and mild elevations in hepatic transaminases. Ketone concentrations are low or undetected. The diagnosis is confirmed by the finding of elevated concentrations of dicarboxylic acids in the urine. Treatment involves avoidance of fasting.
OTHER METABOLIC DISORDERS
Many metabolic disorders can lead to hypoglycemia, including galactosemia, hereditary fructose intolerance, and disorders of organic acid metabolism (see Table 172-1). Hypoglycemia in these disorders is usually a reflection of global hepatic dysfunction secondary to the buildup of hepatotoxic intermediates. Many of these disorders present with low concentrations of ketone bodies because ketogenesis also is affected. The finding of non–glucose-reducing substances in the urine suggests a diagnosis of galactosemia or hereditary fructose intolerance. Occurrence of symptoms after ingestion of fructose or sucrose suggests hereditary fructose intolerance. Treatment requires dietary restriction of the specific offending substances.
MEDICATIONS AND INTOXICATION
Hypoglycemia can occur as an adverse effect of numerous medications, including insulin, oral hypoglycemic agents, and propranolol, and as a result of salicylate intoxication. Valproate toxicity can cause a disorder similar to that seen in the fatty acid oxidation defects. Ethanol ingestion also can cause hypoglycemia, especially in younger children, because the metabolism of ethanol results in the depletion of cofactors necessary for gluconeogenesis.
DIAGNOSIS
While the list of causes of hypoglycemia is long and complex, establishing the etiology in a particular patient is important. Frequently, it is difficult to make an accurate diagnosis until one can obtain a critical sample of blood and urine at the time of the hypoglycemic episode. In a child with unexplained hypoglycemia, a serum sample should be obtained before treatment for the measurement of glucose and insulin, GH, cortisol, FFAs, and β-hydroxybutyrate and aceto-acetate. Measurement of serum lactate levels also should be considered. A urine specimen should be obtained for measuring ketones and reducing substances. Hypoglycemia without ketonuria suggests hyperinsulinism or a defect in fatty acid oxidation. The results of this initial testing can establish whether endocrine causes are responsible and, if not, provide initial information regarding which types of metabolic disorders are most likely. Whenever possible, additional samples of blood and urine should be frozen for further analysis if necessary.
EMERGENCY MANAGEMENT
Acute care of a patient with hypoglycemia consists of rapid administration of IV glucose (2 mL/kg of 10% dextrose in water). After the initial bolus, an infusion of IV glucose should provide approximately 1.5 times the normal hepatic glucose production rate (8–12 mg/kg/min in infants, 6–8 mg/kg/min in children). This infusion allows for suppression of the catabolic state and prevents further decompensation in patients with certain metabolic disorders. Higher infusion rates may be needed for hyperinsulinemic states. If adrenal insufficiency is suspected, stress doses of glucocorticoids should be administered.