Cerebral Palsy
CEREBRAL PALSY
Overview
Cerebral palsy (CP) is a nonprogressive lesion of the brain occurring prior to 2 years of age resulting in a disorder of posture and voluntary movement. CP may be accompanied by impairment of speech, vision, hearing, and perceptual function. Common comorbidities include visual and hearing deficits, seizure disorders, hydrocephalus, microcephaly, scoliosis, hip dislocation, and mental retardation.
Classification
CP is often classified by the type of muscle tone, distribution of limb involvement, or functional skills. The types of muscle tone include hypotonia (low tone); hypertonia (high tone, spasticity); ataxia; and choreoathetosis or dystonia.
Choreoathetosis is characterized by involuntary distal writhing movements (athetosis) and poorly graded proximal voluntary movement (chorea). Spasticity is graded most commonly by using the modified Ashworth scale and is characterized by a velocity-dependent resistance to passive stretch.10 Dystonia is characterized by sustained muscle contraction resulting in sustained end-range posture. Ataxia is characterized by diametric movement patterns. The patterns of motor involvement and distribution are described in Table 35-1.
Table 35-1
Classification of Cerebral Palsy
| Type | Distribution or Description |
| Spastic | |
| Monoplegia | Only one limb affected |
| Diplegia | Involves trunk and lower extremities; upper extremities to a lesser degree |
| Hemiplegia | Primarily one total side involved; upper extremity usually more than lower extremity |
| Quadriplegia (tetraplegia) | Involvement of all four limbs, head, and trunk |
| Ataxia | Irregularity of muscular action manifested by dysmetria; may be pure or combined with other forms |
| Dyskinesia (choreoathetosis) | Impairment of the power of voluntary movement; poor control of proximal movement (chorea) alternating with repetitive, involuntary, slow, writhing movements (athetosis); movements increase with emotional stress and around adolescence; often associated with rigidity or spastic quadriplegia or diplegia |
| Hypotonia | Abnormally reduced tension or muscle tone; accompanied by variable degrees of weakness |
Spastic CP, particularly quadriplegia and spastic diplegia, accounts for the majority of cases. Hemiplegia, ataxia, dystonia, and choreoathetoid CP affect a relatively smaller number of children. New cases of choreoathetoid CP have become rare in the United States and Canada as a result of improved prenatal care in the prevention of Rh incompatibility and hyperbilirubinemia. The disorder remains a problem in developing countries.
Functional skills can also be used to classify individuals with CP. The Gross Motor Function Classification System (GMFCS) provides a five-level system to classify motor involvement of children with CP on the basis of their functional status and their need for assistive technology and wheeled mobility (Box 35-1). The GMFCS provides a means of grading age-related developmental skill.31,32
Level I includes children with neuromotor impairments whose functional limitations are less than what is typically associated with CP. It also includes children who have traditionally been diagnosed as having “minimal brain dysfunction” or “cerebral palsy of minimal severity.” The distinctions between levels I and II therefore are not as pronounced as the distinctions between the other levels, particularly for infants less than 2 years of age.31,32
The descriptions of the five levels are broad and not intended to describe all aspects of the function of individual children. The focus is on determining which level best represents the child’s present abilities and limitations in motor function. Emphasis is on the child’s usual performance in home, school, and community settings (not best performance).32
The levels are described on a time line including before second birthday, between second and fourth birthdays, between fourth and sixth birthdays, and between sixth and twelfth birthdays. Distinctions between adjacent levels are outlined in Box 35-1.
Incidence and Etiologic and Risk Factors
The reported incidence of CP ranges from 1.5 to 4.7 cases per 1000 births in the United States.28 Advances in perinatal care have improved the chances for survival of infants of extremely low birth weight and immature gestational age.22
Despite the increased use of fertility drugs, survival of infants in multiple births, and survival of extremelylow-birth-weight infants, the incidence (number of cases occurring over a certain period) of neurodisabilities, including CP, has remained constant among surviving premature infants.26,33 In fact, incidence may have begun to show some recent decline,21 although the prevalence (overall number of cases present at a specified time) has increased because of the improved survival rates.
The cause of CP may be unknown and is often multifactorial. In children of normal birth weight who have disabilities associated with CP, 80% of the disabilities are a result of factors occurring before birth and 20% are attributed to factors occurring around the birth or in the immediate postbirth period (first 4 weeks of life).
In children of low birth weight who develop disabilities associated with CP (approximately 0.7 per 1000 live births) uncertainty remains as to when the brain damage occurred (i.e., during embryonic development or during or after birth). Any prenatal, perinatal, or postnatal condition that results in cerebral anoxia, hemorrhage, or damage to the brain can cause CP (Table 35-2). CP is the second most common neurologic impairment in childhood (mental retardation is the first).49
Pathogenesis
No consistent or uniform pathology is associated with CP. Several types of neuropathic lesions have been identified on the basis of autopsy: (1) hemorrhage below the lining of the ventricles (subependymal or interventricular), (2) hypoxia causing encephalopathy, and (3) malformations of the central nervous system (CNS).54
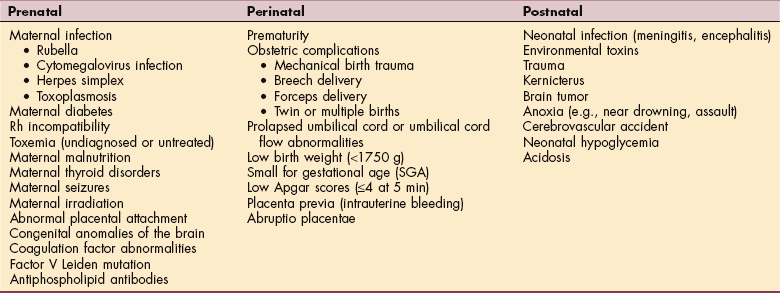
Until recently, interventricular hemorrhage (IVH) was the most common form of brain injury in the premature infant (Fig. 35-1). In recent years the incidence of IVH has declined from an incidence of 49% in very-low-birth-weight infants to 20% in the same population.35a As a result, periventricular white matter injury has become the more common cause of long-lasting brain injury in this population.
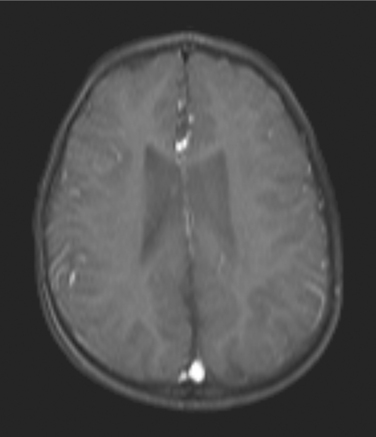
Figure 35-1 Magnetic resonance image of an interventricular hemorrhage with expansion of the lateral ventricles. This child was born at 26 weeks’ gestation and diagnosed with diplegic cerebral palsy. (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
Periventricular lesions can be either cystic, as in periventricular leukomalacia (PVL), or more diffuse and result in abnormal myelination (Fig. 35-2). Diffuse periventricular myelination abnormalities can be found in up to 65% of premature infants when they reach full-term (9 months from conception). The incidence of PVL has declined somewhat, and PVL is seen in only 5% of this population. During the preterm period infants are at heightened risk for ischemia in the periventricular areas. The heightened risk is the result of passive-pressure circulation in the premature infant. The autoregulation of CNS blood flow normally present in full-term infants is absent, and the CNS blood pressure is more dependent on peripheral pressure. The premature infant between 23 and 32 weeks’ gestation is at the highest risk of periventricular injury. As the periventricular white matter begins to myelinate, the risk of hypoxic injury declines.
Figure 35-2 Magnetic resonance image of a periventricular leukomalacia with cystic formation extending into the parenchyma in a child with quadriplegic cerebral palsy. Top and bottom are serial sections in the same brain. In this child, the ventricles are a normal size. The abnormal finding is in the bottom slice where the cystic changes (black) extend into the brain tissue. (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
Hypoxic injury can also occur in the full-term infant; however, this represents only a small portion of infants with CP. It often occurs in the presence of bradycardia, intrauterine growth retardation, and preeclampsia and may also be facilitated by the presence of infection.4a
Hypoxic-ischemic injury can be the result of three possible underlying causes: (1) decreased perfusion resulting from systemic hypotension and poor autoregulation of cerebral blood flow; (2) emboli, which block distal perfusion, and thrombosis; or (3) clot formation from polycythemia or a hypercoagulable state.35 Hypoxic-ischemic injury is known to disrupt the normal metabolic processes, starving the cells of oxygen because of poor perfusion and poor oxygen delivery to the cells, resulting in a reliance on the cells’ limited ability to maintain homeostasis through anaerobic energy metabolism.
Eventually, insufficient energy is all that is available for powering the sodium-potassium pump in the cell membrane, and the ionic gradients across the cell membranes break down. The resulting influx of calcium begins a cascade that, along with the osmotic pressure gradient which has developed, ends in cell death. The second phase of cell damage occurs with reperfusion when vasodilatation allows increased blood flow and oxygen free radicals (see Fig. 6-2) are released that trigger programmed cell death or apoptosis. The severity and topography of the damage depends on the gestational age at the time of the injury and the degree of injury sustained.7
The primary hypoxic-ischemic lesion found in the premature infant is PVL (bilateral necrosis of the white matter of the brain adjacent to the lateral ventricles), present in 42% of term and 87% of preterm infants with CP.25,52 A portion of premature infants demonstrate impaired autoregulation of cerebral circulation and also demonstrate a passive-pressure cerebral circulation (i.e., blood pressure in the CNS is not able to remain constant with fluctuations in the peripheral circulation, placing them at risk of cerebral injury when fluctuations in peripheral pressure occur). The periventricular arterial border zones are at particular risk for hypoxic-ischemic injury in these children.52
Focal injury to the brain can also result from hemorrhage and ischemia, with the resulting collection of blood creating injury from direct mechanical pressure on the tissue and secondary ischemia. In the premature infant, hemorrhage of the germinal matrix (the cells from which the nervous system arises; in the adult, these cells, called ependymal cells, lie adjacent to the ventricular system) into the lateral ventricle (see Fig. 35-1) is a common cause of CP and can result in venous infarction of the periventricular area with a resulting cystic lesion in that portion of the brain.52
Hypoxic-ischemic injury in the mature neonate most commonly results from either selective neuronal cell damage or parasagittal brain damage. These hypoxicischemic insults affect the border zones of the major cerebral arteries, either in the cerebral cortex, cerebellum, or the parietal or occipital regions. Focal or multifocal brain damage can result from either arterial embolism or venous thrombosis and is more common in the more mature neonate. The incidence of this mechanism of injury increases with gestational age greater than 28 weeks and typically presents as a unilateral injury.7
Children who develop CP fail to demonstrate normal CNS maturation after a CNS injury. Persistence of immature layers of the primary motor cortex is often present, and many of the other layers demonstrate abnormalities, particularly those with projections to the pyramidal tract.2
Clinical Manifestations
Although the neurologic manifestations of CP are nonprogressive, the motor impairments change with growth and maturation and may become more apparent as the affected child grows. Clinical manifestations of motor impairments associated with CP may include alterations of muscle tone, delayed postural reactions, persistence of primitive reflexes (Fig. 35-3), delayed motor development, and abnormal motor performance (e.g., delay in movement onset, poor timing of force generation, poor force production, inability to maintain antigravity postural control, decreased speed of movement, and increased co-contraction).14

Figure 35-3 Asymmetric tonic neck reflex. Four-year-old with quadriplegic cerebral palsy demonstrating the asymmetric tonic neck reflex. This primitive reflex contributes to an obligatory change in body posture resulting from a change in head position. With head turning to one side, the arm and leg on the same side extend while the arm and leg on the opposite side flex. This posture resembles a fencing position. (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
Persistence of primitive reflexes and impaired motor function can affect the head, neck, trunk, and extremities and impair sucking and swallowing, resulting in feeding difficulties, a major focus of occupational and speech therapists (Fig. 35-4).
Figure 35-4 Symmetric tonic neck reflex. The same 4-year old with quadriplegic cerebral palsy as in Fig. 35-3 demonstrates another primitive reflex known as the symmetric tonic neck reflex (STNR). When the head and neck are extended, the arms extend; flexion usually predominates in the lower extremities. Flexion of the head and neck causes flexion in the upper extremities and extension in the lower extremities (not shown). In the normal infant the asymmetric tonic neck reflex and STNR are typically integrated by 6 to 8 months. Integration of the STNR allows voluntary flexion of both arms and legs needed to sit comfortably. Prior to 6 to 8 months, these reflexes can be observed in developing infants but when present are not obligatory (i.e., the person can voluntarily move out of the position). (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
Associated disabilities may include cognitive impairments (e.g., mental retardation, learning disabilities, seizure disorders); sensory impairments (in vision, hearing); and constipation or bowel and bladder incontinence with their associated problems (e.g., poor hygiene, skin problems).29,34
Microcephalus and hydrocephalus are also common findings, with the latter being the result of increased intracranial pressure. Behavioral signs of increased intracranial pressure accompanying hydrocephalus may include extreme irritability, vomiting, and eventually delay in reaching developmental milestones, resulting from pressure-induced damage as discussed earlier.
Musculoskeletal problems of altered muscle tone, muscle weakness, and joint restrictions are common and can result in functional and orthopedic impairments. For example, the abnormal pull of the spastic iliopsoas and adductor muscles are the initiating deforming force in hip dislocations (Fig. 35-5).
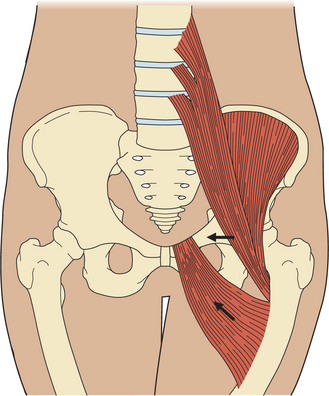
Figure 35-5 Spastic iliopsoas and adductor muscles are the initiating deforming force in acquired spastic hip dislocation.
When spasticity and contracture of the iliopsoas occur, the medial joint capsule is compressed and the femoral head is pushed laterally. As lateral drift of the femoral head occurs, the iliopsoas insertion on the lesser trochan- ter becomes the center of rotation. Acetabular development ceases when the femoral head is completely displaced laterally, and further hip flexion pushes the head posteriorly to complete the dislocation (Fig. 35-6).9
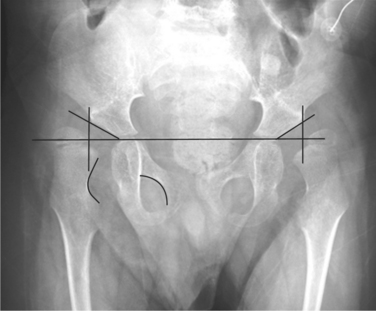
Figure 35-6 Anteroposterior radiograph of a young child with spastic quadriplegia and subsequent hip dysplasia with subluxation on the left. Note that a line drawn vertically down from the outermost edge of the acetabulum would bisect the head of the femur. Failure of the acetabulum to deepen with weight bearing resulting in hip dysplasia and subluxation occur as a result of the inability to weight bear and abnormal muscular forces pulling on the bone. The standard measurement for hip dislocation is a migration percentage. This is done by drawing Hilgenreiner’s line, which provides a horizontal reference to the pelvis and then drawing Perkin’s line perpendicular to Hilgenreiner’s line from the outermost edge of the acetabulum. (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
Joint restrictions associated with CP are a result of a decrease in the number of sarcomeres44 per muscle fiber. Muscles also demonstrate an increased variation in fiber size and type37 with both hypertrophy and atrophy present, possibly representing an ongoing dynamic process. Increases in fat and fibrous tissue and a decrease in blood flow have been identified.36 In this process, bone grows faster than muscle, resulting in a disadvantageous length-tension relationship of the muscle and an increased risk of subsequent contracture.43 A characteristic decrease in muscle mass also results in decreased muscle power and endurance.
Changes in muscle tone affect a person’s ability to control movement, resulting in poor selective control of muscles, poor regulation of activity and muscle groups, decreased ability to learn unique movements, inappropriate sequencing of movements, and delayed anticipatory postural response (Table 35-3). Most often, the timing and sequence of muscle activity are also affected.

A significant number of children with a diagnosis of spastic CP present with low muscle tone early in the first year of life and later develop spasticity. They often have insufficient flexor skills to position themselves against gravity for activities such as lifting the head, reaching, and kicking. The child will attempt to develop alternative strategies to complete the tasks. If control is not available, these strategies result in postures that allow completion of a particular sequence but do not allow for subsequent movement and transitions.
Examples of such situations are a wide-based sitting posture that allows the child to maintain sitting but decreases the ability to turn and rotate in and out of the position. Pulling to stand with the arms only (without using the lower extremities) is another example of an alternative strategy used by children with CP. If practiced and repeated over time, these abnormal movements become habitual and are difficult to change.
Each type of CP is characterized by its own clinical picture based on the presence and extent of these clinical manifestations. The progression of motor development associated with each type of CP is beyond the scope of this book. The reader is referred to other texts for a more detailed discussion.16,45
MEDICAL MANAGEMENT
Observation, a good history, and a neurologic examination will provide the physician with the information necessary to make an accurate and early diagnosis. The diagnostic studies performed depend on clinical findings. For example, electroencephalography (EEG) is indicated when seizures are present or suspected; hip radiographic films are indicated to rule out hip dislocations and should be followed over time, particularly in the presence of spasticity.
Blood or urine screening tests may be used to rule out certain metabolic diseases, and a thorough workup should be undertaken if a history reveals a progressive course of positive family history. A computed tomo- graphic (CT) or magnetic resonance imaging (MRI) scan can provide information on the location of the insult.
TREATMENT.
Comprehensive and cooperative planning with an interdisciplinary team including physicians, therapists, nurses, special educators, psychologists, social workers, nutritionists, and family members is essential.
Some of the most common medical management strategies include pharmacologic intervention, neurosurgical intervention, and orthopedic surgery. Skeletal muscle relaxants (e.g., baclofen, diazepam, dantrolene, botulinum toxin) can be used to assist in controlling increased spasticity and can be administered orally (baclofen, dantrolene, and diazepam), intrathecally (baclofen), or directly to muscles through injection at the motor point (botulinum toxin).
Intrathecal administration of baclofen (through the sheath of the spinal cord into the subarachnoid space) uses an implantable intrathecal infusion pump to deliver medication to the spinal cord without the associated CNS sedation found with oral administration. After the pump is implanted, the dosage can be titrated to the optimal level for each person. Any attempts to control excess muscle tone (pharmaceutically or otherwise) should always be paired with functional goals to take advantage of the modulated tone.1,3
Motor point blocks can also be used to control spasticity and can be paired with serial casting to increase muscle length. Muscles such as the gastrocnemius, hip adductors, or hamstrings are injected with a botulinum toxin (or phenol) to create a temporary denervation and to decrease tone and increase movement.11,47
The type A botulinum toxin (Botox) is injected directly into the muscle at the motor point and is used to blockade the neuromuscular junction by acting presynaptically to reduce the release of acetylcholine. Muscle weakness and decrease in muscle spasm occur in 3 to 7 days and gradually reappear in 4 to 6 months.
Successful use of botulinum toxin type A in the upper extremity and at lower doses has been reported.19,20,42 The effects of these injections will wear off anywhere from several weeks to several months later.
Dorsal rhizotomy (surgically identifying the posterior roots of the spinal cord and selectively resecting some of them) to reduce spasticity has been used over the past decade. This is usually performed at the L2 to L5 spinal levels for clients with spastic diplegia or mildly increased tone who are independent ambulators but who have abnormalities of posture and gait.51 A rhizotomy may also be used effectively for clients with severe positioning difficulties such as severe quadriplegia. This procedure may reduce muscle tone enough to facilitate personal hygiene and provide improved sitting and comfort.
Orthopedic surgery may include muscle lengthening or releases (e.g., adductors, iliopsoas, hamstrings) to address contracture, muscle transfers (e.g., rectus femoris or tibialis posterior) to increase control or decrease excessive muscle pull, or bone procedures (e.g., femoral derotational osteotomy [see Fig. 23-12, B]; acetabular augmentation; triple arthrodesis; spinal fusions) to correct bony deformity, hip dislocation, or scoliosis.
Orthotic intervention may be used to maintain flexibility, support or stabilize a joint, or improve alignment. The ultimate goal is to delay the development of fixed contractures and improve function.
PROGNOSIS.
Little information is available about the causes of death of people with disabilities resulting from CP. No national registry of names of people with CP is available, and cause of death is not usually listed on a death certificate even if the CP was a meaningful contributing factor to the cause of death. Currently available data indicate that common causes of death in this population are related to infection, aspiration, respiratory compromise, and heart disease.48
Most children with mild to moderate CP have normal lifespans, but there is some increased mortality in the early years (before age 4) and then again with advancing age (50 and older). In those individuals with quadriplegic CP, the excess death rate declines during childhood and adulthood only to climb again after the age of 50, as is noted in the more mildly involved population.41
Ambulation potential may be predicted based on achievement of motor milestones (Table 35-4). Independent sitting before age 2 years is a positive indicator of future ambulation.53 If it is going to occur, ambulation usually takes place by 8 years of age.
Table 35-4
Predictors of Ambulation for Cerebral Palsy
| Predictors | Ambulation Potential |
| By Diagnosis | |
| Monoplegia | 100% |
| Hemiplegia | 100%* |
| Ataxia | 100% |
| Diplegia | 60%*-90% |
| Spastic quadriplegia | 0%-70% |
| By Motor Function | |
| Sits independently by age 2 | Good† |
| Sits independently by age 3 to 4 | 50% community ambulation |
| Presence of primitive reflexes beyond age 2 | Poor |
| Absence of postural reactions | Poor |
| Independently crawled symmetrically or reciprocally by age 2.5 | Good† |
*From Pallas Alonso CR et al: Cerebral palsy and age of sitting and walking in very low birth weight infants, An Esp Pediatr 53:48-52, 2000.
†da Paz Júnior, Burnett SM, Braga LW: Walking prognosis in cerebral palsy: a 22-year retrospective analysis, Dev Med Child Neurol 36:130-134, 1994.
35-1 SPECIAL IMPLICATIONS FOR THE THERAPIST
Impaired Joint Mobility, Motor Function, Muscle Performance, and Range of Motion Associated with Bony or Soft Tissue Surgical Procedures
Impaired Motor Function and Sensory Integrity Associated with Nonprogressive Disorders of the Central Nervous System—Congenital Origin or Acquired in Infancy or Childhood
Impaired Ventilation and Respiration/Gas Exchange Associated with Ventilatory Pump Dysfunction or Failure
Primary Prevention/Risk Reduction for Integumentary Disorders
In addition to the treatment options discussed in the previous section, physical therapists are exploring a more focused and proactive approach of activity-based intervention through intense activity training protocols, lifestyle modifications, and mobility-enhancing devices. Increased motor activity has been shown to lead to better physical and mental health and improve various aspects of cognitive performance.16a
Activity-based programs for individuals with CP focus on maximizing physical function while preventing secondary musculoskeletal impairments; foster cognitive, social, and emotional development; and potentially promote or enhance neural recovery.16a
With new research information about the role of neural recovery in damaged nervous systems, therapists can expect to see continued changes in philosophy and intervention approaches with this unique population. Focus will continue with early intervention but include other phases through the lifespan. As attention is directed toward establishing, enhancing, and maintaining neural pathways, we may see changes in how CP is approached. For a more detailed discussion of traditional and emerging physical therapist practices for CP, the reader is referred to reference 16a.
When designing a therapy program for a child with CP, the therapist should take a broad view of the child’s needs and consider the interactive effects that the child’s family environment creates on the goals that have been developed. To provide family-centered care the therapist must do the following49a:
• Spend enough time with the family
• Listen carefully to the parents
• Make the parents feel like partners in the child’s care
The strengths and weaknesses of each family need to be assessed and considered when designing a given child’s program, and the therapist needs to consider what the impact of carrying out the program will be on the family and, as a result, on the child.
If the cost (emotional, social, financial) is too great, the family may choose to abandon the intervention. As a result, the child may lose ground in terms of altered musculoskeletal alignment and decreased function, and the family must bear the emotional impact.
If the therapist is able to match the program with the family’s cultural expectations, ability to participate, and emotional and financial resources, then a partnership with the family can develop that will most benefit the child in the long run. Expecting from family members only what they can succeed at and providing support and education where it is needed to help the family grow and care for the child with special needs will create the best therapeutic environment to allow the child to thrive.
There is often a fine line between balancing the natural history of the condition with the family’s commitment and understanding in maximizing the child’s quality of life. In addition, families make choices in terms of providing for the child with CP. Often these choices must take into consideration other family members, expectations of themselves, expectations of the child, and, as mentioned, cultural and ethnic beliefs that may not match up with the health care professional’s defined goals and plans for intervention.
A general review of intervention studies shows that children benefit from early intervention compared with those children not involved in specific programmed activities. Programming focused on cognitive outcomes has relatively stronger support5 than programming aimed at solely motor outcomes.23 The potential for improvement is better for children less than 9 months old but no greater than 2 years of age at a minimum frequency of intervention of two times per week.8
Early and accurate identification of CP provides the most likely opportunity for facilitation of optimal motor development.40 Many motor milestone checklists are available from which a comparison with the normal can be made.24 In fact, the gross motor function of children with CP and outcomes of intervention have often been evaluated using measures on children without motor impairment.
A more meaningful approach would be to make management decisions and evaluate intervention outcomes based on expectations for children with CP of the same age and gross motor function.31 This type of evaluation can be made by using assessment tools specifically designed to evaluate the child with CP (e.g., Gross Motor Function Assessment38; Gross Motor Function Classification System31,32). An assessment of management practices with guidelines for the management of clients with CP is available,15 as is a model for the acquisition of basic motor abilities and intervention implications.6
A significant number of children with a diagnosis of spastic CP present with low muscle tone early in the first year of life and later develop spasticity. They often have insufficient flexor skills to position themselves against gravity for activities such as lifting the head, reaching, or kicking. The child will attempt to develop alternative strategies to complete the tasks. If control is not available, these strategies result in postures that allow completion of a particular sequence but do not allow for subsequent movement and transitions.
Examples of such situations are a wide-based sitting posture that allows the child to maintain sitting but decreases the ability to turn and rotate in and out of the position. Pulling to stand with the arms only (without using the lower extremities) is another example of an alternative strategy used by children with CP. If practiced and repeated over time, these abnormal movements become habitual and are difficult to change.
After orthopedic surgery, the therapist can assist in reducing muscle spasms that increase postoperative pain by moving and turning the child carefully and slowly; however, adequate postoperative pain management should include medication (e.g., codeine and diazepam [Valium]) prescribed by the surgeon.
In the case of postoperative casting, the therapist can instruct the family to wash and dry the skin at the edge of the cast frequently, inspecting often for signs of skin breakdown. Repositioning and ventilation under the cast with a cool-air blow dryer can also assist in preventing skin breakdown. A flashlight can be used daily to inspect beneath the cast.
Surgical procedures (orthopedic or neurosurgical) may expose areas of underlying muscle weakness and instability. It is critical that an intensive therapy intervention program begin after surgery to assist with strengthening and improving functional performance.
Properly prescribed assistive technology is vital in allowing the child with CP the least restrictive access to both the physical and social environment and is a critical part of the overall management of the child with CP. Assistive technology includes any device used to increase, maintain, or improve the functional ability of a person with a disability (Fig. 35-7).
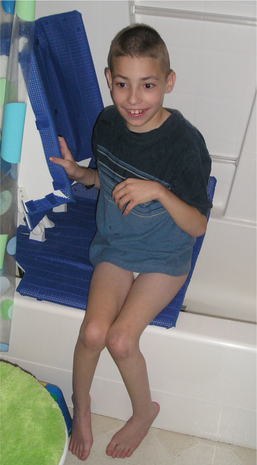
Figure 35-7 Tub lift. A battery-powered tub lift has been very successful with this child, who has spastic quadriplegic cerebral palsy. With help, he transfers from the toilet next to the tub to the tub seat. With assistance, he swings his legs into the tub, and then can independently operate the unit to lower (and later elevate) himself. (Courtesy Tamara Kittelson-Aldred, Access Therapy Services, Missoula, MT. Used with permission.)
This equipment can be either low tech (standers, positioning equipment, communication boards, or wheelchairs) or high tech (switch toys, power wheelchairs, or computer-based communication systems), as long as it is provided with a functional goal (Fig. 35-8).
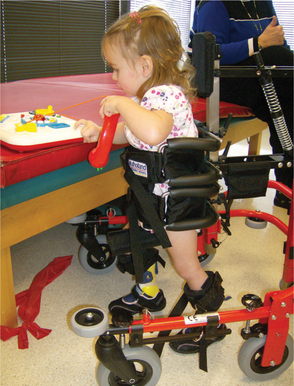
Figure 35-8 Mulholland Walkabout. This 3-year-old girl with spastic quadriplegia can propel this wheeled upright walker through space to explore her environment and play where she wants to. Although she may not become a functional ambulator, the use of this equipment is developmentally appropriate. She could be a candidate for independent wheelchair mobility, but her parents have deferred this decision for now. (Courtesy Tamara Kittelson-Aldred, Access Therapy Services, Missoula, MT. Used with permission.)
Quality of life has become a new focus in the management of all clients seeking health care services. Mobility impairment limits can negatively effect overall development, including social, cognitive, emotional, and physical development. An increased emphasis on powered mobility to increase voluntary activity, function, and independence has contributed to improved quality of life for many individuals with CP (Fig. 35-9).
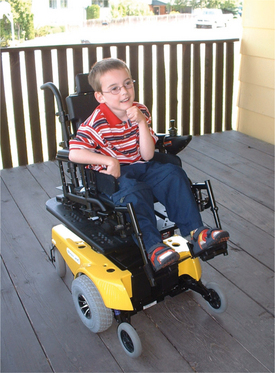
Figure 35-9 New power wheelchair. This 4-year-old child with cerebral palsy receives a new power wheelchair with a seat elevator to enable him to reach age-appropriate items on countertops and tabletops. Trunk supports and footplates help with alignment. The joystick on the left allows him to navigate independently. (Courtesy Tamara Kittelson-Aldred, Access Therapy Services, Missoula, MT. Used with permission.)
For children who are dependent for mobility, power mobility can be an option and can be successful in children as young as 2 years of age with corresponding cognitive skills.12,13,46 These systems can be controlled with a standard joystick or adapted for control with a variety of switch systems.
These systems allow control with the head (either by a switch array or by proportional control), control through the use of individual switches, or control by a single switch through a scanning program (Fig. 35-10). When computer access is educationally appropriate, the same wheelchair-based control system can be adapted through an infrared link and mouse emulator to operate the computer. The mouse emulator is an electronic link that allows use of the joystick to control the mouse, usually by infrared beam.

Figure 35-10 This young lady with spastic quadriplegic cerebral palsy uses a DynaVox speech-generating device with a Tash Microlite switch on the left. By moving her head, she is able to hit the switch with her cheek to stop the electronic scan where she wants it to create a message. Stealth neck rest provides suboccipital head support. (Courtesy Tamara Kittelson-Aldred, Access Therapy Services, Missoula, MT. Used with permission.)
Use of mobility and speech-generating (communication) devices is encouraged earlier and with children at all levels of motor disability, including those with severe involvement. Differences in clinical practice and debate continue over providing an external means of mobility in favor of promoting more voluntary activity. More definitive research guidance is needed in these areas.
Proper positioning is critical to the child with CP, both from a functional perspective and to help prevent the soft tissue limitations that can develop over time. Appropriate positioning has been shown to encourage smoother and faster reach, decrease extensor tone, increase vital capacity, and improve performance on cognitive testing.30
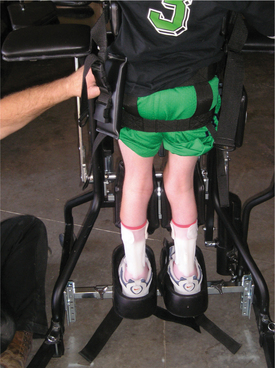
In addition to proper wheelchair position, time out of the chair to counteract the flexed posture of the body is necessary. A standing program can be initiated between 12 and 18 months of age in the child who is not pulling to stand independently to maintain flexibility and provide the normal weight-bearing forces across the hip joint.
Standing helps orient children to the upright position, assists with visual perception, and can aid in digestion and elimination. Therapists hear anecdotal reports of decreased constipation and urinary tract infections in response to standing (Fig. 35-11). Standing also provides relief from pressure and can be used for prolonged muscle stretching, especially in the older or larger child.

Figure 35-11 Standing frame. Many different types and styles of standers are available with a variety of adaptive features. A, Young girl with spastic hemiplegia from a birth injury/infection drives her power chair up to the stander. With assistance, she is able to get a seat sling under her buttocks to lift her up to standing. The sling is significant because it allows the parent to avoid lifting her into the stander. Shoe holders guide the placement of her feet. B, This standing frame offers an additional fun feature: the ability to operate a PlayStation. (Courtesy Tamara Kittelson-Aldred, Access Therapy Services, Missoula, MT. Used with permission.)
Positioning for feeding for the child with CP is often critical for his or her ultimate success and safety at this task. The child’s head and neck posture, pelvic posture, and inclination with respect to gravity are important considerations. A team approach using the skills of the physical and occupational therapist in conjunction with those of the speech therapist is essential in designing a program to optimize the child’s oral motor skills (Fig. 35-12).
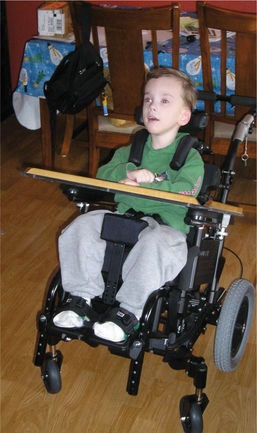
Figure 35-12 Young boy with schizencephaly and cerebral palsy with severe involvement has a planar seating system with postural components. A tilt-in-space feature and deep ischial ledge formed in the seat keep his pelvis aligned and hips back. Hip flexion is combined with the medial thigh support between his legs to keep his knees apart and allow him to relax. Shoulder pad retractors are a feature added when it was discovered that downward pressure and anterior support at the shoulders improved head control for this child. (Courtesy Tamara Kittelson-Aldred, Access Therapy Services, Missoula, MT. Used with permission.)
For children with expressive communication deficits, sign language, communication boards, and a variety of high-tech communication systems with voice synthesizers are available to augment spoken communication and can also be linked with wheelchair-based control systems in the power wheelchair user.
When evaluating a person for assistive technology, consideration should be given both to the individual’s unique abilities and challenges and to the environment in which the equipment will be used. The products should provide the person the greatest degree of functional independence in all the environmental situations encountered. The barriers in each environment may vary, and thus the solutions by necessity may be different in different environments.
Manual Passive Range-of-Motion Exercise
It is generally accepted that manual straight-plane passive range-of-motion (ROM) exercise for children with a chronic neurologic disorder such as CP is not, by itself, the best way to meet the physiologic requirements necessary to stretch a muscle. However, passive trunk rotation has been found to be useful in assisting with general flexibility and modulation of increased tone for persons with spastic quadriplegia.
Instead, splinting or positioning that offers a low-load prolonged stretch for longer than 30 minutes or that is used throughout the day is recommended. For example, splints such as lower extremity ankle-foot orthoses (AFOs) to maintain ankle ROM or supported standing to control lower extremity flexion contractures and assist with hip development and stability may be implemented. However, manual passive ROM exercise is not without its applications and is best combined with a well-thought-out positioning and splinting program.
Other interventions used by therapists to improve ROM and facilitate motor development or improve function include relaxation techniques such as neutral warmth or acupressure points; serial or tone casts (often in conjunction with botulinum toxin A injections); therapy ball activities; aquatic programs; and manual therapy techniques.
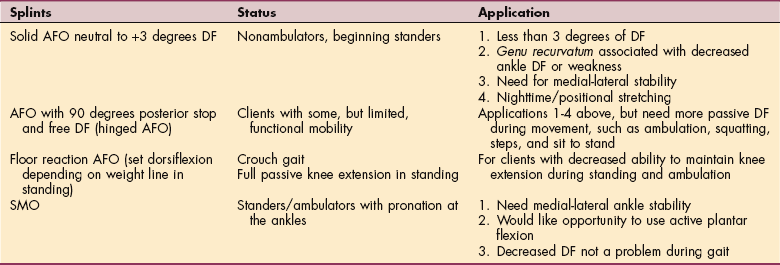
AFOs are probably the most commonly used orthoses for children with CP. A rigid polypropylene AFO is used to provide medial-lateral stability to the foot and ankle while at the same time assisting with foot clearance during gait. The AFO can be set at +3 degrees of dorsiflexion to facilitate the increased ankle dorsiflexion necessary for the swing phase of gait or to decrease genu recurvatum (hyperextension at the knee) through ground reaction forces.
Hinged AFOs may be recommended once a child is moving in the upright position, especially when the child is beginning to walk, squat, or move up and down stairs, both to allow active ankle motion and to allow normal tibial progression during the stance phase of gait. A more flexible plastic such as copolymer or a thinner polypropylene may be used in the lighter child to provide more dynamic use of the foot musculature. In this case the term dynamic ankle-foot orthotic (DAFO) is used.
In some cases dorsiflexion assist hinges may be used, either with a plantar flexion stop or in the more mild cases with free plantar flexion. Care must be taken to choose the correct degree of hinge strength (or an adjustable hinge) so as not to create a crouched posture.
A supramalleolar orthosis (SMO) provides medial-lateral stability for the foot and ankle while allowing free plantar flexion and dorsiflexion. It is always helpful to have whatever plantar flexion is available, since this motion helps decelerate the limb during middle and late stances and facilitates the initial progression of the limb during late stance and early swing.
The SMO can be used when decreased active ankle dorsiflexion and excessive genu recurvatum are not problems. Extending the SMO proximally to the malleoli provides important support, whereas support distal to the malleoli usually shifts the deformity in a proximal direction. General guidelines and recommendations for foot and ankle splinting can be found in Table 35-5 (Fig. 35-13).
Adolescents with Cerebral Palsy31a
Therapists are encouraged to include older children and teens in problem solving to help them become more self-sufficient, assuming more responsibility during this developmental phase despite their limitations in physical capability. Providing adolescents the opportunity to participate in planning and decision making is important for transition planning.
This may include decisions about assistive technology, environmental modifications, health and fitness, and prevention of secondary musculoskeletal impairments. Likewise, the therapist can work closely with those individuals interested in participating in recreation and sports activities.
Client-centered assessment of strengths and needs identifying self-care, productivity, and leisure activities is possible and has been reported with this population.31a
Therapists must also recognize and address the ongoing and unique needs of adults with CP (Fig. 35-14). With improved understanding of CP and its associated long-term complications and with improved health care, increased longevity has brought a new area of concern for children with CP living into adulthood: effects of the aging process. Decline of already impaired muscle strength and elasticity and bone density can lead to loss of ambulatory status.10a
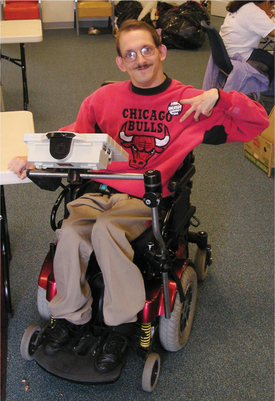
Figure 35-14 Adult with cerebral palsy. This 33-year-old man with athetoid cerebral palsy uses a speech-generating (communication) device and power chair; the joystick to operate the chair is under the client’s right hand. The communication device can be folded and moved out to the side to allow for transfers. The chair has power tilt for independent position changes and pressure relief. Ankle huggers wrapped around the ankles keep his legs from flailing. A neoprene chest harness was added later to help provide external stability and increase control of his movements (not shown). This individual has clearly communicated how much he likes having the chest support, saying he feels much more in control of his body with it on. Straps and supports are fashioned with buckles, because Velcro is not strong enough to hold this client. The additional supports help reduce athetoid movements and improve function. (Courtesy Tamara Kittelson-Aldred, Access Therapy Services, Missoula, MT. Used with permission.)
Group homes, independent living centers, and sheltered workshops are now making it possible for many nonambulatory adults with disabilities to function independently or semiindependently. Regular daily living assistance is required by adults with spastic quadriplegia, especially in the area of lifts and transfers.
Degenerative arthritis, severe joint contractures, and other orthopedic deformities present the most common and challenging problems in this population (Fig. 35-15). Moderate to intense pain is a significant problem for the majority of adults with CP, accompanied by depressive symptoms interfering with activities. Further research is needed to determine the functional impact of this problem and appropriate interventions.18,39

Figure 35-15 Adult with cerebral palsy. Adults with moderate to severe effects of cerebral palsy can face some difficult physical challenges. This 21-year-old woman has a power chair with seat elevator, power tilt in space, and power elevating leg rests she can operate herself. Each leg rest can be raised or lowered separately to her comfort. This client changed her lower extremity position frequently to manage pain related to spasticity and immobility. A spring upper extremity assist on the left helps keep her hand on a modified joystick to allow her to independently control her chair. (Courtesy Tamara Kittelson-Aldred, Access Therapy Services, Missoula, MT. Used with permission.)
Management strategies for older children and adults are different by virtue of their ability to participate and understand the aims of therapy. Therapy to maintain functional skills through the adolescent growth spurt, when weight gain, weakness, and atrophy often result in a decline in function, is essential. Aerobic training may prevent deterioration in body composition and muscle strength.50
Strengthening has become an integral part of therapy programs for individuals with CP and is especially helpful in this population. Measuring isokinetic strength is considered reliable in this population and should be used in rehabilitation protocols.4 Isokinetic strengthening three times per week for 8 weeks can improve muscle strength and gross motor skills27,50 and increase cadence17 for those people who remain ambulatory into adulthood. A traditional upper extremity strengthening program of 6 to 10 repetitions three times per week for 8 weeks has been useful in improving speed and endurance in independent wheelchair propulsion.
References
1. Albright, AL. Baclofen in the treatment of CP. J Child Neurol. 1996;11:77–83.
2. Amunts, K, Schleicher, A, Zilles, K. Persistence of layer IV in the primary motor cortex (area 4) of children with cerebral palsy. J fur Hirnkorschung. 1997;38:247–260.
3. Armstrong, RW, Steinbok, P. Intrathecally administered baclofen for treatment of children with spasticity of cerebral origin. J Neurosurg. 1997;87:409–414.
4. Ayalon, M, et al. Reliability of isokinetic strength measurements of the knee in children with cerebral palsy. Dev Med Child Neurol. 2000;42(6):398–402.
4a. Back, SA, Rivkees, SA. Emerging concepts in periventricular white matter injury. Semin Perinatol. 2004;28:405–414.
5. Barnett, WS. Long-term cognitive and academic effects of early childhood education on children in poverty. Prev Med. 1998;27:204–207.
6. Bartlett, DJ, Palisano, RJ. A multivariate model of determinants of motor change for children with cerebral palsy. Phys Ther. 2000;80(6):598–614.
7. Berger, R, Garnier, Y. Pathophysiology of perinatal brain damage. Perinat Brain Damage. 1999;30:107–134.
8. Binder, H. Rehabilitation management of children with spastic diplegic cerebral palsy. Arch Phys Med Rehabil. 1989;70:482–489.
9. Bleck, EE. Orthopedic management of cerebral palsy. Philadelphia: Saunders, 1979.
10. Bohannon, RW, Smith, MB. Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther. 1987;67:206–207.
10a. Bottos, M. Functional status of adults with cerebral palsy and implications for treatment of children. Dev Med Child Neurol. 2001;43:516–528.
11. Boyd, RN, Pliatsios, V, Starr, R, et al. Biomechanical transformation of the gastroc-soleus muscle with botulinum toxin A in children with CP. Dev Med Child Neurol. 2000;42:32–41.
12. Butler, C, Okamoto, GA, McKay, TM. Motorized wheelchair driving by disabled children. Arch Phys Med Rehabil. 1984;65:95–97.
13. Butler, C, Okamoto, GA, McKay, TM. Powered mobility for very young disabled children. Dev Med Child Neurol. 1983;25:472–474.
14. Campbell, SK. Decision making in pediatric neurologic physical therapy. New York: Churchill Livingstone, 1999.
15. Campbell, SK. Pediatric consensus statement. Phys Ther. 1990;2:121–122.
16. Campbell, SK, eds. ed 2. Physical therapy for children. Philadelphia: Saunders; 2000.
16a. Damiano, DL. Activity, activity, activity: rethinking our physical therapy approach to cerebral palsy. Phys Ther. 2006;86(11):1534–1540.
17. Damiano, DL, Abel, MF. Functional outcomes of strength training in spastic cerebral palsy. Arch Phys Med Rehabil. 1998;79:119–125.
18. Engel, JM, et al. Pain in cerebral palsy: the relation of coping strategies to adjustment. Pain. 2000;88(3):225–230.
19. Fehlings, D, et al. An evaluation of botulinum-A toxin injections to improve upper extremity function in children with hemiplegic cerebral palsy. J Pediatr. 2000;137(3):331–337.
20. Friedman, A, et al. Effects of botulinum toxin A on upper limb spasticity in children with cerebral palsy. Am J Phys Med Rehabil. 2000;79:53–59.
21. Grether, JK, Nelson, KB. Possible decrease in prevalence of cerebral palsy in premature infants. J Pediatr. 2000;136(1):133.
22. Hack, M, Fanaroff, AA. Outcomes of children of extremely low birthweight and gestational age in the 1990s. Early Hum Dev. 1999;53(3):193–218.
23. Hurr, JJ. Review of research on therapeutic interventions for children with cerebral palsy. Acta Neurol Scand. 1995;91:423–432.
24. Ketelaar, M, Vermeer, A, Helders, PJ. Functional motor abilities of children with cerebral palsy: a systematic literature review of assessment measures. Clin Rehabil. 1998;12:369–380.
25. Krageloh-Mann, I, et al. Bilateral spastic cerebral palsy-MRI pathology and origin: analysis from a representative series of 56 cases. Dev Med Child Neurol. 1995;37:379–397.
26. Lorenz, JM, et al. A quantitative review of mortality and developmental disability in extremely premature newborns. Arch Pediatr Adolesc Med. 1998;152(5):425–435.
27. Macphail, H. The effect of isokinetic strength training on functional mobility and walking efficiency in adolescents with cerebral palsy. Dev Med Child Neurol. 1995;37:763–776.
28. Manning, FA, et al. Fetal assessment based on fetal biophysical profile scoring: VIII. The incidence of cerebral palsy in tested and untested perinates. Am J Obstet Gynecol. 1998;178:696–703.
29. Mayo, ME. Lower urinary tract dysfunction in cerebral palsy. Urology. 1992;147:419–420.
30. Myhr, U. A five-year follow-up of functional sitting position in children with cerebral palsy. Dev Med Child Neurol. 1995;37:587–596.
31. Palisano, RJ, et al. Validation of a model of gross motor function for children with cerebral palsy. Phys Ther. 2000;80(10):974–985.
31a. Palisano, RJ, Copeland, WP, Galuppi, BE. Performance of physical activities by adolescents with cerebral palsy. Phys Ther. 2007;87(1):77–87.
32. Palisano, R, Rosenbaum, P, Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol 1997;39:214–223. The GMFCS is available on-line at http://www.canchild.ca/Portals/0/outcomes/pdf/GMFCS.pdf. Accessed January 13, 2007.
33. Petterson, B, et al. Adverse outcome after multiple pregnancy. Baillieres Clin Obstet Gynaecol. 1998;12(1):1–17.
34. Reid, CJ, Borzyskowski, M. Lower urinary tract dysfunction in cerebral palsy. Arch Dis Child. 1993;68:739–742.
35. Rivkin, MJ. Hypoxic-ischemic brain injury in the term newborn. Clin Perinatol. 1997;24:607–625.
35a. Roland, EH, Hill, A. Germinal matrix-intraventricular hemorrhage in the premature newborn: management and outcome. Neurol Clin North Am. 2003;21:833–851.
36. Romanini, L, et al. Histological and morphological aspects of muscle in infantile cerebral palsy. Ital J Orthop Traumatol. 1989;15:87–93.
37. Rose, J, et al. Muscle pathology and clinical measures of disability in children with cerebral palsy. J Orthop Res. 1994;12:758–768.
38. Russell, DJ, et al. Improving scaling of the gross motor function measure for children with cerebral palsy: evidence of reliability and validity. Phys Ther. 2000;80:873–885.
39. Schwartz, L, Engel, JM, Jensen, MP. Pain in persons with cerebral palsy. Arch Phys Med Rehabil. 1999;80:1243–1246.
40. Sharkey, M. The effects of early referral and intervention on the developmentally disabled infant: an evaluation at eighteen months of age. J Am Board Fam Pract. 1990;3:163–170.
41. Singer, RB, Strauss, D, Shavelle, R. Comparative mortality in cerebral palsy patients in California, 1980-1996. J Insur Med. 1998;30:240–246.
42. Suputtitada, A. Managing spasticity in pediatric cerebral palsy using a very low dose of botulinum toxin type A: preliminary report. Am J Phys Med Rehabil. 2000;79(4):320–326.
43. Tardieu, C. For how long must the soleus muscle be stretched each day to prevent contracture? Dev Med Child Neurol. 1988;30:3–10.
44. Tardieu, C, et al. Muscle hypoextensibility in children with cerebral palsy: clinical and experimental observations. Arch Phys Med Rehabil. 1982;63:97–102.
45. Tecklin, JS. Pediatric physical therapy, ed 3. Philadelphia: Lippincott Williams & Wilkins, 1999.
46. Tefft, D, Guerette, P, Furumasu, J. Cognitive predictors of young children’s readiness for powered mobility. Dev Med Child Neurol. 1999;41:665–670.
47. Ubhi, T, Bhakta, BB, Ives, HL, et al. Randomised double blind placebo controlled trial of the effect of botulinum toxin on walking in cerebral palsy. Arch Dis Child. 2000;83(6):481–487.
48. United Cerebral Palsy Research Foundation Causes of death of persons with disabilities due to cerebral palsy, 2001. Available on-line at http://www.ucpa.org Accessed June 09, 2008.
49. United Cerebral Palsy Research Foundation Research fact sheets: statistics, 2001. Available on-line at http://www.ucpa.org Accessed June 09, 2008.
49a. U.S. Department of Health and Human Services, Health Resources and Services Administration, Maternal and Child Health Bureau. The national survey of children with special health care needs chartbook 2001. Rockville, MD: U.S. Department of Health and Human Services, 2004.
50. Van den Berg-Emons, RJ, et al. Physical training of school children with spastic cerebral palsy: effects on daily activity, fat mass and fitness. Int J Rehabil Res. 1998;21(2):179–194.
51. Vaughan, CL, Subramanian, N, Busse, ME. Selective dorsal rhizotomy as a treatment option for children with spastic CP. Gait Posture. 1998;8:43–59.
52. Volpe, JJ. Brain injury in the premature infant. Clin Perinatol. 1997;24:567–587.
53. Watt, JM, Robertson, CM, Grace, MG. Early prognosis for ambulation of neonatal intensive care survivors with cerebral palsy. Dev Med Child Neurol. 1989;31:766–773.
54. Weindling, AM. Intervention after brain injury to reduce disability. Semin Neonatol. 2000;5:53–60.