Genetic and Developmental Disorders
Pediatric diseases and disorders comprise a large number of conditions. Entire volumes have been devoted just to pediatric pathologies. Given the format of this book and space limitations, in this chapter we have included as many of the more commonly encountered genetic and developmental disorders as possible. Cerebral palsy is discussed separately as a neurologic condition in Chapter 35.
A brief discussion of several other rare but important diagnoses also is included. Because physical and occupational therapy intervention is not the focus here, the reader is referred to other, more appropriate resources for specific and thorough intervention guidelines for these conditions.26,94,142,163,171
DOWN SYNDROME
Definition and Incidence
Down syndrome was the first genetic disorder attributed to a chromosomal aberration and is referred to as trisomy 21 (also Down’s syndrome). Down syndrome is characterized by muscle hypotonia, cognitive delay, abnormal facial features, and other distinctive physical abnormalities.
Down syndrome is the most common inherited chromosomal disorder, occurring once in every 700 live births. The incidence of Down syndrome rises with maternal age. Before maternal age 30 the incidence is 1 in 2000 births; it is 1 in 50 for mothers aged 35 to 39, and 1 in 20 for mothers over 40 years. There is a 2% risk of recurrence for a couple who have had a child with Down syndrome.
Etiologic Factors and Pathogenesis
The actual cause of Down syndrome is not yet known; however, some hints have begun to emerge as researchers continue to explore gene mapping and develop genetic models for Down syndrome.9 Evidence from cytogenetic and epidemiologic studies supports multiple causality.
Trisomy 21 produces three copies of chromosome 21 instead of the normal two because of faulty meiosis (cell division by which reproductive cells are formed) of the ovum or, sometimes, the sperm. This results in a karyotype (chromosomal constitution of the cell nucleus) of 47 chromosomes instead of the normal 46.
The faulty cell division can also occur after fertilization, leading to only a portion of cells being affected, with a milder clinical picture. This situation is referred to as mosaicism. Because of the positive correlation between increasing age and Down syndrome, it is hypothesized that deterioration of the oocyte (immature ovum) or environmental factors such as radiation and viruses may cause a predisposition to mistakes in meiosis and the resulting chromosomal abnormality.
In a small number of cases Down syndrome occurs as a result of a translocation of chromosome 15, 21, or 22 (i.e., the long arm of the chromosome breaks off and attaches to another chromosome). Chromosomal translocation can be hereditary or associated with advanced parental age. Although Down syndrome usually is attributed to the aging woman, evidence suggests that in 5% to 10% of cases Down syndrome is correlated with paternal age.157,195
The third copy of chromosome 21 is the cause of the phenotypic characteristics that are observed in people with Down syndrome. There are presumably many genes that are present in triplicate in individuals with Down syndrome. Only a few of these have been identified as causative of specific pathology in Down syndrome; most likely there are many more that will be identified in the future.
Alzheimer’s disease is also more common in people with Down syndrome, occurring at an earlier age than that of the Alzheimer population in general. By the age of 40 symptoms of Alzheimer can be seen in almost everyone with Down syndrome. The increased rate of Alzheimer’s in individuals with Down syndrome is a result of abnormally high production of β-amyloid. This results from the increased presence of a precursor to β-amyloid in Down syndrome and of increased amounts of β-secretase, an enzyme that divides the larger protein into β-amyloid. This is a direct result of their location on the twenty-first chromosome found in triplicate in children with Down syndrome.75
The gene for superoxide dismutase is also found on the long arm of chromosome 21 and is thought to have a role in the neuropathology of Down syndrome. It acts to break down superoxide radicals in the brain. Although it shows normal expression in the fetus of individuals with Down syndrome, it is overexpressed in the brains of adults with Down syndrome and declines as symptoms of dementia appear. This suggests that it may play a role in the developmental brain abnormalities found in people with Down syndrome.129
Astrocyte-derived neurotrophic factor is also coded for on the twenty-first chromosome. In Down syndrome this growth factor is overexpressed prior to birth in the brains of those with Down syndrome. This overexpression increases as the person ages, and in the second decade can be correlated with the degree of β-amyloid found in the brain. In addition astrocyte-derived neurotrophic factor may also play a role in neuronal injury via calcium toxicity.129
The brains of people with Down syndrome demonstrate increased levels of interleukin-1. Interleukin-1 is a cell signaling cytokine found in both the immune system and the brain. Interleukin-1 is overexpressed in Down syndrome from fetal stages of development through adulthood.
This overexpression is found in microglia and correlates with the distribution of amyloid plaques. Interleukin-1 is not coded for on the twenty-first chromosome but is modulated by both superoxide dismutase and astrocyte-derived neurotrophic factor discussed earlier. Interleukin-1 may also play a role in modulating the expression of these two factors. In addition to these indirect effects, interleukin-1 is neurotoxic in high concentrations and also plays a role in the neurofibrillary pathology described as tangles.129
The resulting gross pathology in people with Down syndrome is an overall reduction in brain weight. This especially affects the size of the cerebral and cerebellar hemispheres, the hippocampus, the pons, and the mammillary bodies. Additional abnormal findings may include smaller convolutions within the brain, structural abnormalities in the dendritic spines of the pyramidal neurons of the motor cortex, and abnormalities of the pyramidal system as a whole, including decreased pyramidal neurons in the hippocampus.
This last finding and the decreased size of the amygdala in people with Down syndrome who develop dementia have particular significance as it relates to the increased incidence of Alzheimer’s symptoms in older adults with Down syndrome.4
Clinical Manifestations
Children with Down syndrome are readily identified by their flattened nasal bridges, eye shape, short limbs, and mild to moderate hypotonia. The most frequently observed clinical characteristics are listed in Table 23-1.
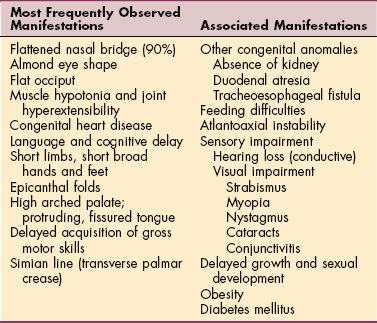
Many other associated clinical manifestations may also be present. For example, people with Down syndrome tend to exhibit increased susceptibility to infections such as otitis media (ear infection). They are frequently hepatitis B carriers and have an increased incidence of ophthalmologic disorders (35%), develop thyroid dysfunction with age (8%), experience constipation associated with gastrointestinal tract anomalies (13%), develop acute leukemia (1%), and present with congenital cardiac anomalies (50%) such as atrioventricular and ventricular septal defects. All of these conditions are present at rates that are greater than those found in the population as a whole.140
Children with Down syndrome frequently present with a variety of musculoskeletal or orthopedic problems believed to be acquired secondary to soft tissue laxity and muscle hypotonia. Some of the more common findings include recurrent patellar dislocation, excessive foot pronation, scoliosis, slipped capital femoral epiphyses (secondary to persistent hip abduction associated with hypotonia), and late hip dislocation (after 2 years).
Atlantoaxial instability (AAI) of the cervical spine (subluxation between C1 and C2) is a characteristic in some children with Down syndrome. This instability is thought to be secondary to ligamentous laxity, odontoid maldevelopment, or possibly abnormal syringomyelia in the area of the odontoid process. Syringomyelia is a slowly progressive syndrome in which cavitation (the formation of a cavity from destruction of tissue) occurs in the central segments of the spinal cord.
The majority of cases are asymptomatic; however, clinical changes indicative of symptomatic AAI include hyperreflexia, clonus, positive Babinski’s sign, torticollis, increased loss of strength, changes in sensation, loss of established bladder and bowel control, and a decrease in motor skills.
Children with Down syndrome predictably present with feeding difficulties and delayed acquisition of motor skills. These skills, however, improve with age. Because of the hypotonia and decreased strength, midline upper extremity movement is difficult, and gait usually is characterized by smaller step lengths, increased knee flexion at contact and hyperextension in stance, decreased single limb support, and an increased hip flexion posture. These children present with slower reaction times and slower postural reactions.
Secondary disorders often develop after age 30 or 35, including obesity, diabetes mellitus, and cardiovascular disease. Other significant problems can include osteoar thritic degeneration of the spine and osteoporosis with vertebral or long bone fractures.
MEDICAL MANAGEMENT
Measurement of α-fetoprotein (AFP), human chorionic gonadotropin, and unconjugated estrogen in maternal serum (triple screen) allows detection of an estimated 60% to 70% of fetuses with Down syndrome. Using this screening test prenatal diagnosis may be made during the second trimester (between 15½ and 20 weeks’ gestation).
Ultrasound identification based on nuchal translucency provides a good way to identify the fetus with Down syndrome at between 10 and 14 weeks’ gestation. Ultrasound carries a 6% false-positive rate and will identify only 77% of affected fetuses.109
Postnatal diagnosis usually begins with suspected physical findings at birth. Genetic studies showing the chromosomal abnormality can confirm the diagnosis. Specific diagnostic testing for the secondary problems discussed earlier varies depending on the involved organ systems suspected of dysfunction.
TREATMENT.
Because no known cure exists for Down syndrome, treatment is directed toward specific medical problems (e.g., antibiotics for infection, cardiac surgery, monitoring of thyroid function, and monitoring for development of Alzheimer’s disease). Larger medical centers are pursuing plastic surgery to eliminate the hypoplastic facial features based on the premise that this type of surgery has a positive influence on rehabilitation. The overall goal of treatment intervention is to help affected children develop to their full potential. This involves a team of experts, including therapists.
PROGNOSIS.
The improved life expectancy of people with Down syndrome as a result of the greater availability of surgery and advances in medical care has been documented, but life expectancy still remains lower than that for the general population.101
The presence of congenital malformations, especially of the heart and gastrointestinal tract, can result in high mortality rates in the affected population57; lack of mobility and poor eating skills are also predictors of early death.49 Respiratory tract infections are very common secondary to hypotonicity of chest and abdominal muscles and contribute significantly to morbidity and mortality. Immune system dysfunction also may be present associated with a higher incidence of acute myeloid leukemia than in the general pediatric population.72
Significant health problems contributing to mortality have been reported in the adult population with Down syndrome, including untreated congenital heart anomalies, acquired cardiac disease, pulmonary hypertension, recurrent respiratory infections and aspiration leading to chronic pulmonary interstitial changes, and complications from presenile dementia and Alzheimer’s disease.
Over the last 40 years the lifespan of those with Down syndrome has increased significantly. In 1968 the average age of death was 2 years of age and by 1997 it had increased to 50 years of age. Unfortunately this degree of improvement has not occurred for everyone with Down syndrome. There is a significant disparity that exists based on race, with Caucasians with Down syndrome enjoying a significantly longer lifespan than nonwhites.27
SCOLIOSIS
Definition
Scoliosis is an abnormal lateral curvature of the spine. The curvature may be toward the right (more common in thoracic curves) or the left (more common in lumbar curves). Rotation of the vertebral column around its axis occurs and causes the associated rib cage deformity. Scoliosis is often associated with kyphosis and lordosis.
Overview and Incidence
Scoliosis is classified as idiopathic (unknown cause; 80% of all cases), osteopathic (as a result of spinal disease or bony abnormality), myopathic (as a result of muscle weakness), or neuropathic (as a result of a central nervous system [CNS] disorder).
Age of onset can vary from birth onward and is referred to as infantile (0 to 3 years), juvenile (ages 3 to 10), adolescent (age 10 until bone maturity at between 18 and 20 years of age), or adult (after skeletal maturation). Between 0.4% and 5.5% of children may present with some type of scoliosis166 with one in four of those requiring some type of treatment intervention. The incidence is increased with associated neuromuscular impairments such as cerebral palsy, spina bifida, neurofibromatosis, and muscular dystrophy.
As a whole the overwhelming majority of cases of progressive idiopathic scoliosis are found in the adolescent age group when the growth velocity of the spine again increases after relatively slow growth period between the ages of 5 and 11 for girls (13 for boys).29
Infantile idiopathic scoliosis (rare in the United States) is characterized by curvatures that are most often thoracic and toward the left and most commonly affects males. Juvenile idiopathic scoliosis is characterized most often by a right thoracic curvature and can be rapidly progressive. Adolescent idiopathic scoliosis of greater than 30 degrees is seen most often in females without any neurologic impairments in a 10: 1 ratio.188 In its milder forms (10-degree curve or less), scoliosis affects boys and girls equally, but girls are more likely to develop more severe curvatures requiring intervention.
Adult scoliosis (curves greater than 30 degrees) affects about 500,000 people in the United States, whereas the prevalence of scoliosis above the age of 50 is reportedly between 6% and 10% (based on routine chest radiographs).16
Etiologic Factors
Scoliosis may be functional or structural. Functional (postural) scoliosis may be caused by factors other than vertebral involvement, such as pain, poor posture, leg length discrepancy, or muscle spasm induced by a herniated disk or spondylolisthesis. These curves disappear when the cause is remedied. Functional scoliosis can become structural if untreated.
Structural scoliosis is a fixed curvature of the spine associated with vertebral rotation and asymmetry of the ligamentous supporting structures. It can be caused by deformity of the vertebral bodies and may be congenital (e.g., wedge vertebrae, fused ribs or vertebrae, hemivertebrae), musculoskeletal (e.g., osteoporosis, spinal tuberculosis, rheumatoid arthritis), neuromuscular (e.g., cerebral palsy, polio, myelomeningocele, muscular dystrophy), or, most commonly, idiopathic.
At the present time, despite extensive study, the cause of idiopathic scoliosis remains unknown. Researchers hypothesize that this type of scoliosis relates to the maturation disturbances of the CNS, including neurohormonal transmitters or neuromodulators secondary to genetic defect.108
Multiple areas of research including abnormalities of connective tissue, neuromotor mechanisms, neurohormonal imbalances (e.g., melatonin, calmodulin), and biomechanics (e.g., the importance of the erect posture) have been explored for a potential relationship to the cause of idiopathic scoliosis. However, no clear evidence supports any one area as an etiologic factor of this disorder; rather, it appears to be multifactorial.105
Linkage studies have identified a genetic predisposition to scoliosis192; the probability of a person’s having idiopathic scoliosis is estimated to be 20% if other family members have scoliosis.40
Pathogenesis
The pathogenesis of scoliosis remains unclear but may be better understood in relation to the underlying cause. Abnormal embryonic formation and segmentation of the spinal column are possible pathologic pathways in congenital scoliosis. Neuromuscular scoliosis is often the result of an imbalance or asymmetry of muscle activity through the trunk and spine.
The earliest pathologic changes associated with idiopathic scoliosis occur in the soft tissues as the muscles, ligaments, and other tissues become shortened on the concave side of the curve. Some hypothesize166 that scoliosis sets up abnormal forces across the spine due to the differences in length-tension relationships, with the muscles on the convexity being in a lengthened position and those on the concavity positioned in a relatively shortened state, and as a result a muscle imbalance is present.
Evidence establishes the existence of hypertrophy of the muscles on the side of the convexity99; however, the muscles on the concavity still are at a mechanical advantage and facilitate the progression of a curve once it is established. In time, bone deformities occur as compression forces on one side of the vertebral bodies apply asymmetric forces to the epiphyseal ossification center, resulting in increased bone density on that side. The compressive force is greatest on the vertebrae in the apex of the concavity, so the apical vertebrae become most deformed (Fig. 23-1).
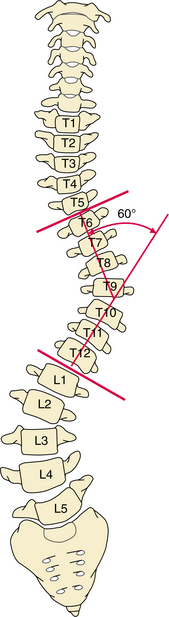
Figure 23-1 Cobb’s method of measuring scoliosis. This is the method most commonly used because it is readily reproduced. The top vertebra used in the measurement is identified as the uppermost vertebra whose upper surface tilts toward the curvature’s concave side. (The superior surface of the vertebra above it usually tilts in the opposite direction or may be parallel to it.) The bottom vertebra is the lowest vertebra whose inferior surface tilts toward the curvature’s concave side. (Likewise, the inferior surface of the vertebra below it usually tilts in the opposite direction or may be parallel to it.) A line is drawn parallel to each of these vertebrae. The angle formed by perpendicular lines drawn to each of the parallel lines is the angle of the curvature.
Clinical Manifestations
Curvatures of less than 20 degrees (mild scoliosis) rarely cause significant problems. Severe untreated scoliosis (curvatures greater than 60 degrees) may produce pulmonary insufficiency and reduced lung capacity, back pain, degenerative spinal arthritis, disk disease, vertebral subluxation, or sciatica.
Back pain is not typical in children or adolescents with mild scoliosis and should be evaluated by a physician who can rule out spondylolisthesis, tumor, infection, or occult trauma. Back pain may be associated with curve progression after institution of brace treatment for idiopathic scoliosis.146
The adult with scoliosis often presents with back pain that is considered multifactorial, arising from muscle fatigue, trunk imbalance, facet arthropathy, spinal stenosis, degenerative disk disease, and radiculopathy. Although the incidence of back pain in adults with scoliosis is similar to that in the general population, the pain in the group with scoliosis is greater and more persistent.16
Common characteristics of scoliosis are asymmetric shoulder and pelvic position, often identified when clothes do not hang evenly. Curves are designated as right or left depending on the convexity (e.g., right thoracic scoliosis describes a curve in the thoracic spine with convexity to the right). Usually one primary curvature exists with a secondary or compensatory curvature that develops to balance the body. Two primary curvatures may exist (usually right thoracic and left lumbar). If the curvatures of the spine are balanced (compensated), the head is centered over the center of the pelvis; if the spinal alignment is uncompensated, the head is displaced to one side.
Paraspinal muscles become asymmetric as the muscles on the convex side of the curve become rounded, appearing prominent or bulging, while the muscles on the concave side flatten. Rotational deformity on the convex side is observed as a rib hump (gibbus) sometimes seen in the upright position but always apparent in the forward bend position.
MEDICAL MANAGEMENT
Diagnosis by clinical examination requires the client to bend forward 90 degrees with the hands joined in the midline as if taking a dive into a swimming pool. A scoliometer also can be used to measure the angle of trunk rotation (ATR) (Fig. 23-2).
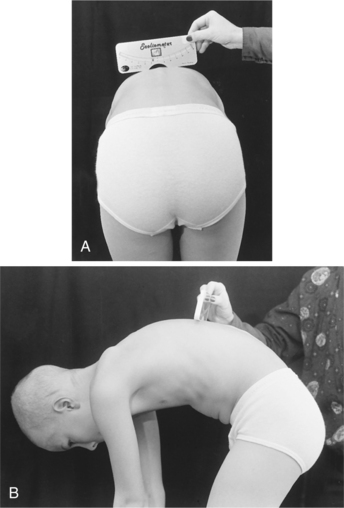
Figure 23-2 The scoliometer. This device can be used by any health care worker trained to screen for scoliosis. Some medical personnel also use this device to monitor curvatures over time, thereby avoiding unnecessary radiographs. Ask the client to bend forward slowly (Adam’s position), stopping when the shoulders are level with the hips. View the client from both the front and back, keeping your eyes at the same level as the back. Before measuring with the scoliometer, adjust the height of the person’s bending position to the level where the deformity of the spine is most pronounced. This position varies from one person to another depending upon the location of the curvature (e.g., a low lumber curvature requires further bending than an upper thoracic curvature). Lay the scoliometer across the deformity with the 0 mark over the top of the spinous process. A measurement of 5 degrees or more in the screening test is considered positive and requires medical follow-up. Visually observe for asymmetry of the ribs or paravertebral muscles. In this child, hamstring tightness (greater on the left) accounts for the positional shift to the left. The scoliometer reading was zero. (Courtesy Todd Goodrich, University of Montana, Missoula.)
An abnormal finding includes asymmetry of the height of the ribs or paravertebral muscles on one side. The examiner also checks for leg length discrepancy and other asymmetries and for the presence of hair patches, nevi, pits, or areas of abnormal skin pigmentation in the midline indicating possible underlying spinal abnormality.
Differential diagnosis is important in determining whether the scoliosis is structural or functional. Structural curvatures maintain their position irrespective of whether the spine is in an upright or forward bending position. Functional curvatures straighten when placed in a forward bend position. This is especially easy to see when the client is sitting, thereby eliminating weight bearing through the feet. The physician also performs a neurologic examination to rule out an underlying neurologic disorder, especially in the presence of left thoracic curvature.
Full-length radiographs of the spine using techniques to minimize breast radiation dosage are evaluated using the Cobb method (see Fig. 23-1) to measure the degree of curvatures. A curve must be larger than 10 degrees to be considered scoliotic.
The Risser sign is also determined from the film as an indication of maturation and prognostic predictor of progression.135 Typically, the iliac crest ossifies from anterior to posterior. Risser divided the crest into four quarters according to ossification, grading the ossification from 1 to 4, with a grade 5 indicating that the whole apophysis has ossified and is fused to the iliac crest.
Neuroimaging beyond plain films may be necessary. For example, bone scan may be used to rule out neoplasms, infections, spondylolysis, or compression fractures as the underlying cause. Magnetic resonance imaging (MRI) is used to differentiate cord lesions, disk herniations, neoplasms, infections, spondylolysis, spinal stenosis, and compression fractures.
TREATMENT.
Prevention of postural or idiopathic structural scoliosis is the key to management of the majority of scoliosis cases. Early detection allows for early treatment without surgical intervention and with good long-term results. Overall goals of management are to prevent severe and progressive deformities that might lead to decreased cardiorespiratory function.
Conservative care in the past has included exercise and electrical stimulation; however, this has not been proven efficacious.131 Observation and monitoring every 4 to 6 months for curvatures less than 25 degrees, spinal orthoses for curvatures 25 to 40 or 45 degrees (Table 23-2), and surgery for curvatures greater than 45 degrees have been recommended.123,149 The goal of the use of spinal orthoses is to serve as a passive restraint system to maintain curvatures within 5 degrees of the curvature measurement at the time of initial application. This is accomplished successfully in 85% to 88% of cases.189 Curvatures with an apex between T8 and L2 and compensated thoracolumbar curves respond the most favorably to bracing,135 whereas curvatures with an apex at T6 or above have the poorest outcome.
Table 23-2
| Brace | Use |
| Milwaukee (CTLSO) | Best with curvature at T8 or above |
| Boston (TLSO) | Best with curvature apex lower than T9 or T10 |
| Lyon | For idiopathic scoliosis with thoracic hypokyphosis |
| Charleston | For idiopathic curves, fabricated in maximum side-bend correction |
CTLSO, Cervicothoracolumbosacral orthosis; TLSO, thoracolumbosacral orthosis.
Researchers continue to explore improved dynamic orthotics and holistic treatment approaches. Exercise has not traditionally been viewed as efficacious for scoliosis; however, there is some renewed interest in its potential effect on the flexibility of an existing scoliotic curve.74 For many years, exercise has been dismissed as an effective treatment for adolescent scoliosis. More recent findings in support of exercise have been published.124a
Looking back at previous studies of exercise for scoliosis, many of the children did not do the exercises. And those who did them only did so occasionally. Unless the exercise program was designed to prepare for a sports activity, compliance was very low. Improved technology and the ability to assess muscle function have changed the picture. We now know that there is asymmetry in muscle function for everyone with idiopathic adolescent scoliosis. More specifically, there is an uneven strength in trunk rotation.124a
The former exercise programs of stretching and general strengthening may have been the wrong approach. Progressive resistive exercises specifically aimed at the trunk rotators and extensors are effective for curves less than 45 degrees.124a
Orthotic regimens have varied for late-onset idiopathic scoliosis, and the existing research on bracing is promising but does not yet establish conclusively its merit or identify the optimal regimen to be used.100
Interventions in the adult with scoliosis should follow a conservative nonoperative course of physical therapy to improve aerobic capacity, strengthen muscles, and improve flexibility and joint motion; nonnarcotic analgesics; nutritional counseling; smoking (or tobacco-use) cessation; and nerve root blocks, facet injections, and epidural steroid injections before surgery is considered. Bracing has never been shown to have an effect on the natural history of adult scoliosis but may be used for certain people who are not operative candidates.16
Surgical intervention (e.g., fusion with posterior segmental spinal instrumentation) may be necessary for curvatures greater than 45 degrees, in the presence of chronic pain, or when the curvature appears to be causing neurologic changes. Surgical goals are to halt progression of the curvature, improve alignment, decrease deformity, prevent pulmonary problems, and eliminate pain. The surgical options include a variety of segmental instrumentation systems including Luque, Cotrel-Dubousset, and unit rod instrumentation and Harrington rods. These are combined with a posterior fusion and, in more severe cases, anterior fusion. The use of growth factors to enhance spinal fusion is under investigation.
Minimally invasive surgery can be used in the population to decrease the morbidity associated with open thoracotomy in those people who need an anterior release along with a spinal fusion. This procedure is designed to maximize the stability of thoracic curves with a minimal incision. This technique uses an endoscope to enter the chest anteriorly and remove the disk material to destabilize the spine and obtain correction of the curve to the greatest degree possible.44,138 This endoscopic approach may result in better spinal alignment with faster recovery, fewer complications, and less pain.7
PROGNOSIS.
Postural curvatures resolve as the primary problem is treated. Structural curvatures are not eliminated but rather increase during periods of rapid skeletal growth. If the curvature is less than 40 degrees at skeletal maturity, the risk of progression is small. In curvatures greater than 50 degrees, the spine is biomechanically unstable, and the curvature will likely continue to progress at a rate of 1 degree/yr throughout life.16
Poor seating can contribute to this progression.93 In severe kyphoscoliosis, pain and comfortable positioning can complicate care, and pulmonary compromise can lead to death.
KYPHOSCOLIOSIS
Overview and Etiologic Factors
Scheuermann’s disease (juvenile kyphosis, vertebral epiphysitis) is a structural deformity classically characterized by anterior wedging of 5 degrees or more of three adjacent thoracic bodies affecting adolescents between the ages of 12 and 16. Scheuermann’s disease is the most common cause of structural kyphosis in adolescence. The mode of inheritance is likely autosomal dominant, but the etiologic factors and pathogenesis of this excessive kyphosis remain unknown.
Scheuermann originally proposed that vascular disturbance in the vertebral epiphyses (usually at the thoracic level) during periods of rapid growth was the underlying cause; however, this has not been subsequently supported. Scheuermann’s disease also has been associated with increased levels of growth hormone, and individuals with this disease are frequently taller than average.
In the aging population, kyphoscoliosis (adult round back) is more likely to develop as a result of poor posture, aging, degeneration of the intervertebral disks, vertebral compression fractures or osteoporotic collapse of the vertebrae, endocrine disorders (e.g., hyperparathyroidism, Cushing’s disease), arthritis, Paget’s disease, metastatic tumor, or tuberculosis.
Clinical Manifestations
Adolescent kyphosis is usually asymptomatic, although some adolescents experience mild pain at the apex of the curvature, fatigue, prominent vertebral spinous processes, and tenderness or stiffness in the involved area or along the entire spine. The pectoral, hamstring, and hip flexor muscles are often tight, producing a crouched posture with anterior pelvic tilt and lumbar lordosis. Signs and symptoms associated with adult kyphosis are similar to those of the adolescent form but rarely produce local tenderness unless caused by vertebral compression fractures.
In both adolescent kyphoscoliosis (Scheuermann’s disease) and adult kyphosis, the vertebrae are wedged anteriorly and disk lesions called Schmorl’s nodes develop. Schmorl’s nodes are localized extrusions of the nuclear material through the cartilage plates and into the spongy bone of the vertebral bodies. The cancellous bone reacts by encapsulating the herniated tissue within a wall of fibrous tissue and bone, producing the Schmorl’s node.
MEDICAL MANAGEMENT
Adolescents may be referred for medical evaluation as a result of school screening, or they may present because of concerns over posture and appearance. Adults more commonly present because of increased pain. Diagnosis is based on clinical examination and confirmed by radiographic findings, including Schmorl’s nodes, endplate narrowing, and irregular endplates.
TREATMENT.
Indications for treatment remain controversial, because the true natural history of this disease has not been clearly defined. Presently, the choice of treatment in Scheuermann’s kyphosis is based on the severity and progression of the curve, the age of the individual, and the symptomatology present.
Bracing appears to be very effective if the diagnosis is made early and in adolescents who have not reached skeletal maturity and have curves less than 50 degrees.104 Surgical management is warranted in those with more severe curves and in adults who continue to show progression of the curve or who have progressive neurologic symptoms or unmanageable pain.
SPINA BIFIDA OCCULTA, MENINGOCELE, MYELOMENINGOCELE
Definition
Congenital neural tube defects (NTDs) encompass a variety of abnormalities. The term spina bifida is the one most often used to describe the more common congenital defects of neural tube closure. Normally, the spinal cord and cauda equina are encased in a protective sheath of bone and meninges (Fig. 23-3). Failure of neural tube closure produces defects that may involve the entire length of the neural tube or may be restricted to a small area.
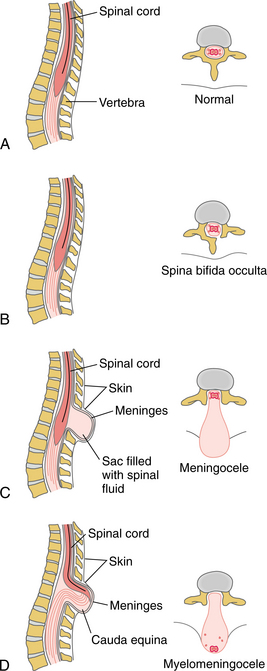
Figure 23-3 Various degrees of spina bifida. A, Normal anatomic structure. B, Spina bifida occulta results in only a bony defect, with the spinal cord, meninges, and spinal fluid intact. C, Meningocele involves the bifid vertebra, with only a cerebrospinal fluid (CSF)–filled sac protruding; the spinal cord or cauda equina (depending on the level of the lesion) remains intact. D, Myelomeningocele is the most severe form because the spine is open and the protruding sac contains CSF, the meninges, and the spinal cord or cauda equina.
The three most common NTDs presented here are spina bifida occulta (incomplete fusion of the posterior vertebral arch), meningocele (external protrusion of the meninges), and myelomeningocele (protrusion of the meninges and spinal cord). Generally these defects occur in the lumbosacral area but also may be found in the sacral, thoracic, and cervical areas (Fig. 23-4).
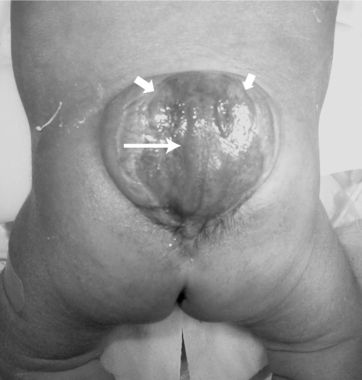
Figure 23-4 Myelomeningocele in a newborn. The neural placode is visible at the surface (long arrow) in this lumbosacral myelomeningocele. A placode is an area of thickening in the embryonic epithelial layer where the spinal cord develops later. Abnormal epithelium lines the edges of the cerebrospinal fluid (CSF)–filled cyst (short arrows). (From Burg FD, Ingelfinger JR, Polin RA, et al: Current pediatric therapy, ed 18, Philadelphia, Saunders, 2006.)
Incidence and Etiologic Factors
The incidence of NTDs varies by ethnic background, geographic area, and socioeconomic status. Data collected by the Centers for Disease Control and Prevention place the incidence in Atlanta at 5 per 10,000 live births.132 Regional variations are significant, however, and in Scandinavia the rate can be as low as 2 per 10,000, whereas in China it can be as high as 100 per 10,000.159
The incidence of spina bifida appears to be declining.194 Termination of pregnancies as a result of the wider availability of maternal serum screening and better nutrition and prenatal vitamins containing folic acid have contributed to this decline.
Evidence supports the hypothesis that the etiology of NTDs is multifactorial and related to the interaction of a genetic predisposition, teratogenic exposure, and an essential folic acid deficiency or folic acid metabolic disorder. Folic acid is a B vitamin found chiefly in yeast, orange juice, and green leafy vegetables and bread products, which are now fortified with folic acid.
Multivitamins containing folic acid taken when planning a pregnancy and during the first 6 weeks of pregnancy prevent between 50% and 70% of NTDs.122,124 Women must be cautioned that half of all pregnancies are not planned and that folic acid must be taken before conception to be effective. Taking supplements containing folic acid is the safest and most effective way of preventing NTDs.120
Genetic factors are considered important in the pathogenesis of spina bifida, and several genes have been identified in the folate-homocysteine metabolism pathway. Studies have identified a number of these individually or in combination as being associated with an increased risk of spina bifida. Couples who have had one child with spina bifida have a recurrence rate of between 3% and 8%.121
Many genetic disorders are associated with NTDs, either with recessive, dominant, or X-linked inheritance patterns. Increased rates of spina bifida are found in individuals with trisomy 13 and 18,38 and in chromosome 13q deletion syndrome.103
Teratogenic exposure also has been associated with an increased incidence of NTDs. Exposure to vitamin A, valproic acid, solvents, lead, herbicides, glycol ether, clomi phene, carbamazepine, aminopterin, and alcohol has been linked to increased rates of NTDs. A number of occupations have also been linked to NTDs, presumably because of teratogenic exposure. Finally, insulin-dependent diabetes has been associated with increased risk of NTDs as well.71
Pathogenesis
Normally, about 20 days after conception, the embryo develops a neural groove in the dorsal ectoderm. The neural groove deepens as the two edges fuse to form the neural tube. By about day 23 this tube is completely closed except for an opening at each end. The upper end closes on day 25 and continues to fold and develop, forming the brain, whereas the bottom end closes on day 27 and forms the spinal cord.
The neural groove is formed by both cell proliferation and the production of a hyaluronic acid extracellular matrix. The first opportunity for failure of vertebral architecture to develop and close normally is through abnormalities in the hyaluronic acid matrix or the actin microfilaments that support elevation of the neural crest.
A second opportunity for failed closure is slightly later in development when an abnormal overgrowth at the caudal end may develop, causing closure to fail. Just before closure of the neural tube surface glycoproteins are produced by the ectoderm and act as the glue that holds the cells together.
A third opportunity for failed closure is abnormal production of these glycoproteins, leading to failure of the neural tube to close. A final possible genesis of myelomeningocele is the rupture of the neural tube after its closure as a result of cerebral spinal fluid (CSF) pressure. In this case development of Chiari II malformation occurs, in which the cerebellar tonsils develop below the foramen magnum or are forced through the foramen magnum because of pressure leading to increased CSF pressure and forcing the neural tube open. The defect in myelomeningocele can be identified by the eighth week of gestation and is complete by the twelfth week.6
Some animal models support the presence of a defect in homocysteine metabolism in the pathogenesis of NTD, which correlates with an increased risk of NTD in some populations.112 Plasma homocysteine levels and folic acid levels show an inverse relationship, and current research is focusing on the metabolism of folic acid and its genetic determinants124 in addition to the importance of these genetic defects in spina bifida. It appears that the genetics of NTDs are multifactorial.
Clinical Manifestations
NTDs are typically divided into two groups, occulta (hidden) and aperta (visible). Approximately 75% of vertebral defects are located in the lumbosacral region, most commonly at the L5 to S1 level. Motor dysfunction depends on the level of involvement and sparing of sensory and motor innervation (Fig. 23-5 and Table 23-3).
Table 23-3
Myelomeningocele: Functional Mobility
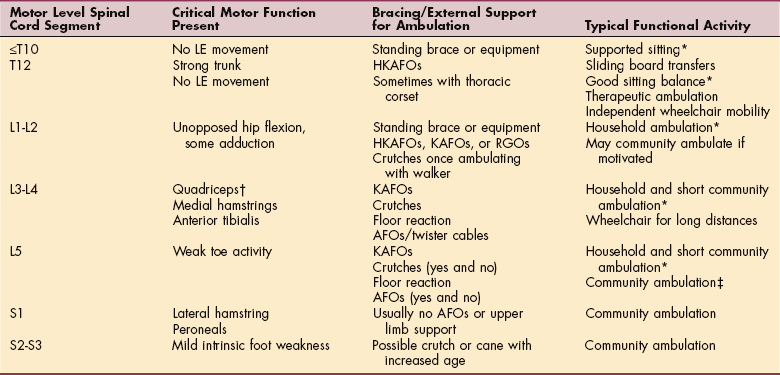
LE, Lower extremity; HKAFO, hip-knee-ankle-foot orthosis; KAFO, knee-ankle-foot orthosis; RGO, reciprocating gait orthosis; AFO, ankle-foot orthosis.
*Do not usually walk as adults.
†Approximately 50% probability of long-distance ambulation with muscle grade 4/5.
‡Able to use ambulation as the primary means of locomotion outside the home.
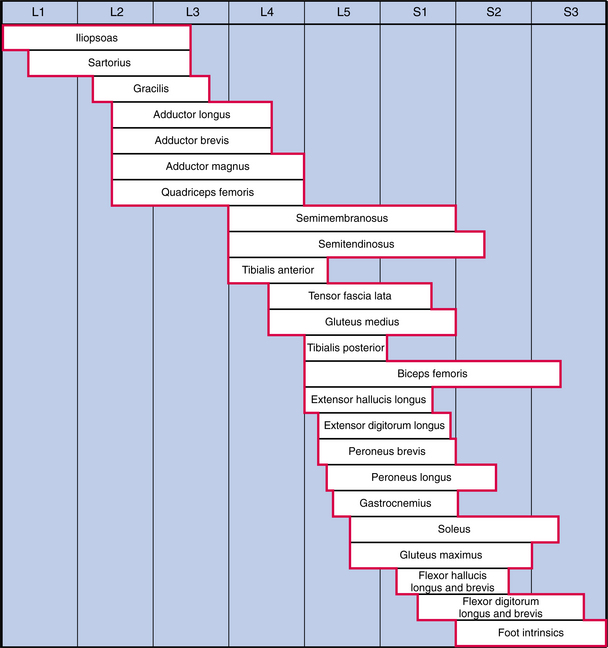
Figure 23-5 Normal lumbar and sacral segmental innervation. For the child with myelomeningocele, once the level of the lesion has been identified, the therapist can begin to assess muscle involvement above and below that level. (From Sharrard WJ: The segmental innervation of the lower limb muscles in man, Ann R Coll Surg Engl 35:106-122, 1964.)
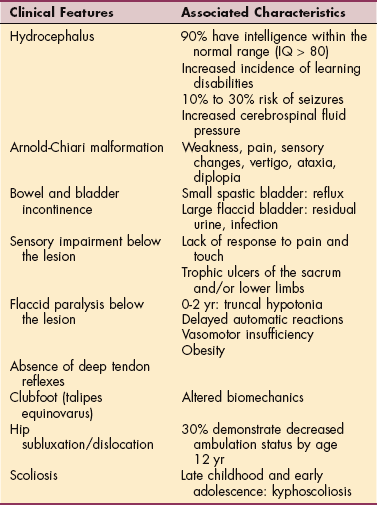
The loss of motor function is not evenly distributed over the limbs and spine, resulting in muscle imbalance contributing to the development of scoliosis and various musculoskeletal deformities that are related to the specific muscles not innervated. Clinical features and other associated characteristics are listed in Table 23-4.
Spina bifida occulta does not protrude visibly but is often accompanied by a depression or dimple in the skin, a tuft of dark hair, soft fatty deposits (subcutaneous lipomas or dermoid cyst), port-wine nevi, or a combination of these abnormalities on the skin at the level of the underlying lesion. Spina bifida occulta usually does not cause neurologic dysfunction, but occasionally bowel and bladder disturbances or foot weakness occurs.
In spina bifida aperta (meningocele and myelomeningocele), a saclike cyst protrudes outside the spine. Like spina bifida occulta, meningocele rarely causes neurologic deficits, whereas myelomeningocele causes permanent neurologic impairment depending on the level of involvement.
Myelomeningocele may be accompanied by flaccid or spastic paralysis, various combinations of bowel and bladder incontinence, musculoskeletal deformities (e.g., scoliosis, hip dysplasia, hip dislocation, clubfoot [talipes equinovarus], hip and knee contractures), hydrocephalus, and sometimes mental retardation. During the first 2 years of life, children with myelomeningocele often present with various degrees of truncal hypotonia and delayed automatic postural reactions, even those children with sacrum-level lesions.
Approximately 90% of children born with this condition have an associated hydrocephalus (Box 23-1). Hydrocephalus accompanying spina bifida usually occurs in the presence of a type I or type II Arnold-Chiari malformation; that is, the cerebellar tonsils are displaced through the foramen magnum (Fig. 23-6), resulting in obstruction of CSF flow and increased CSF pressure and hydrocephalus.
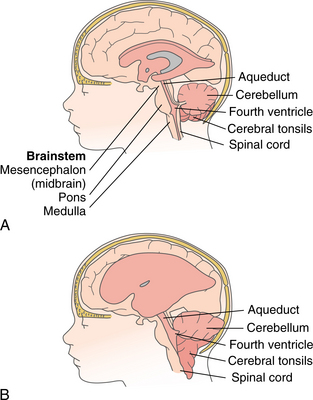
Figure 23-6 Arnold-Chiari malformation. A, Normal brain with patent cerebrospinal fluid (CSF) circulation. B, Arnold-Chiari type II malformation with enlarged ventricles, which predisposes a child with myelomeningocele to hydrocephalus. The brainstem, fourth ventricle, part of the cerebellum, and the cerebral tonsils are displaced downward through the foramen magnum, leading to blockage of CSF flow. Additionally, pressure on the brainstem housing the cranial nerves may result in nerve palsies.
Generally speaking, most children with myelomeningocele have some degree of type II Arnold-Chiari malformation, regardless of the presence of hydrocephalus. This picture has been altered by the advent of fetal repair, and the lower rates of Chiari malformation and hydrocephalus are noted after fetal closure.20,121,179 Although a Chiari malformation may be present radiographically, it may not be causing any symptoms. Syringomyelia, a cavity or syrinx present within the spinal cord or medulla, also can be present and progress, with pressure impinging on the surrounding tissue.
Severe Arnold-Chiari malformations and syrinxes are rare but can lead to potentially fatal consequences. Because of the location of the respiratory centers of the brainstem, central apnea can be serious and necessitate the use of mechanical ventilation and can potentially result in death. Sleep problems including hypersomnolence, sleep fragmentation, choking, snoring, and morning headaches are all potential clinical findings.15
Cranial nerve involvement with feeding difficulties, choking, pooling of secretions, aspiration, and stridor is also a common finding. Vertigo, ataxia, or spasticity as well as pain, progressive weakness, or diplopia can also be presenting findings in the older child. For a variety of helpful Internet websites related to spina bifida, syringomyelia, and hydrocephalus, see http://neurosurgery.mgh.harvard.edu/pedi/.
Tethered cord syndrome is also a common comorbidity following surgical closure of the primary lesion and can occur at any time during growth. As the child grows, the spinal cord can become tethered or bound down, resulting in progressive neurologic compromise. The presenting features are consistent with neurologic compromise and include incontinence, progressive weakness, and back pain. Tethered cord syndrome occurs in between 3% and 5% of children with spina bifida.85
Sensory disturbances usually parallel motor dysfunction. Pressure ulcers at the sacrum, ischial tuberosities, knees, and the dorsum of the feet can be a significant comorbidity. Factors contributing to pressure ulcers in this population are listed in Box 23-2. Many of these same risk factors are present in other conditions prone to pressure ulcers (e.g., diabetic neuropathy) (see also Box 10-16 and Table 10-10).
Bowel and bladder problems are present in virtually all children with myelomeningocele because these functions are controlled at the S2 to S4 levels. Even children with sacral lesions and normal leg movement often have bowel and bladder problems.
Problems with urinary incontinence and infection can occur if the bladder is small and spastic (bladder holds little urine) or large and hypotonic (incomplete emptying of the bladder and ureteral reflux). Bladder dyssynergy occurs with either a flaccid or spastic sphincter. Normally, when the bladder contracts, the sphincter relaxes, allowing urine to flow. In a dyssynergistic state, the bladder and sphincter contract together, predisposing the child to urethral reflux.
MEDICAL MANAGEMENT
Frequently, NTDs are detected prenatally with ultrasonic scanning and serum AFP testing. Elevated AFP usually occurs by 14 weeks’ gestation in the presence of NTDs. This type of screening will not detect skin-covered (closed) neural defects such as spina bifida occulta. The potential for false-positive results with this test may result in unnecessary intervention.
Additionally, as the incidence of this condition continues to decrease, the less reliable the test becomes, because the positive predictive value of the AFP test is dependent on the prevalence of the disease in a population. The less prevalent the disease, the less accurate laboratory results may be. See Chapter 40 for further explanation of the limitations of laboratory tests.
Amniocentesis can detect only open NTDs and is recommended for pregnant women who have previously had children with NTDs or in the case of a large lesion noted with ultrasonic scanning. The need for more accurate, noninvasive imaging of the CNS has been recognized, and fetal MRI has become an effective, noninvasive means of assessing fetal CNS anatomy with superior ability to resolve posterior fossa anatomy over ultrasonography. However, to date fetal MRI has not surpassed ultrasonography in evaluating hydrocephalus and the level and nature of the spinal lesion.111
Postnatally, meningocele and myelomeningocele are obvious on examination. Transillumination of the protruding sac usually can distinguish between these two conditions. In meningocele, the sac with its CSF contents is transilluminated (light shines through the sac); in myelomeningocele, the light does not shine through the neural bundle that is present. Spinal films can be used to detect defects, and the computerized tomographic scan demonstrates the presence of hydrocephalus. Other laboratory tests may include urinalysis, urine cultures, and tests for renal function.
TREATMENT.
Timing of the closure is important. Prenatal diagnosis has made planned cesarean sections, fetal repair, and therapeutic abortion possible. A cesarean section is the preferred method of birth to avoid trauma to the neural sac that occurs during vaginal delivery.
Prenatal closure is now available by fetal surgery and has been found to decrease the incidence of shunt-dependent hydrocephalus and reverse Chiari malformation from above 90% in each case to 59% and 38%, respectively.20,179 By interrupting the flow of CSF during gestation, intrauterine repair enables the cerebellum and brainstem to resume a normal (or nearly normal) configuration.
Prospective parents should be cautioned not to expect improvement in leg function as a result of this surgery. The potential benefits of surgery must be weighed carefully against the potential risks of preterm labor and delivery, potential infection, and blood loss.178
If postnatal closure is chosen, infection and drying of the nerve roots can lead to further loss of function and necessitates surgical closure within 48 hours of birth. Ventriculoperitoneal shunting (Figs. 23-7 and 23-8) is recommended in the presence of hydrocephalus; shunt revision is often required as the child grows or if the shunt becomes obstructed, infected, or separated.
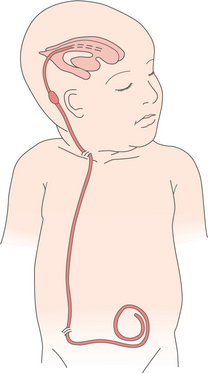
Figure 23-7 Ventriculoperitoneal (VP) shunt. This provides primary drainage of cerebrospinal fluid from the ventricles to an extracranial compartment (usually either the heart [ventriculoatrial] or the abdominal or peritoneal [ventriculoperitoneal] cavity, as shown here). Extra tubing is left in the extracranial site to uncoil as the child grows. A unidirectional valve designed to open at a predetermined intraventricular pressure and close when the pressure falls below that level prevents backflow of fluid.

Figure 23-8 Placement of the shunt. The shunt is placed very superficially, necessitating caution when handling the infant. The therapist must be careful to avoid placing pressure over the shunt, stretching the neck, or placing the child in the head-down position. Parents may be distressed initially by the cosmetic appearance of the shunt, but as the child grows, and with hair growth, the shunt is no longer visible. See Fig. 23-10 of this same child with no obvious signs of a shunt. (Courtesy Todd Goodrich, University of Montana, Missoula.)
A variety of orthopedic surgical interventions may be required throughout the child’s growing years. Surgical correction for hip dislocation rarely is indicated, except in the case of ambulatory clients with unilateral dislocation.55 Investigation has shown that a level pelvis and good ROM of the hips are more important for ambulation than is reduction of bilateral hip dislocation.76
Spinal fusion for kyphotic deformity of the spine has had mixed results and frequent complications. Hip flexion and knee flexion contractures often are addressed with muscle releases, and foot deformity correction often is achieved with soft tissue procedures and in more severe cases with bony procedures (Figs. 23-9 through 23-11).
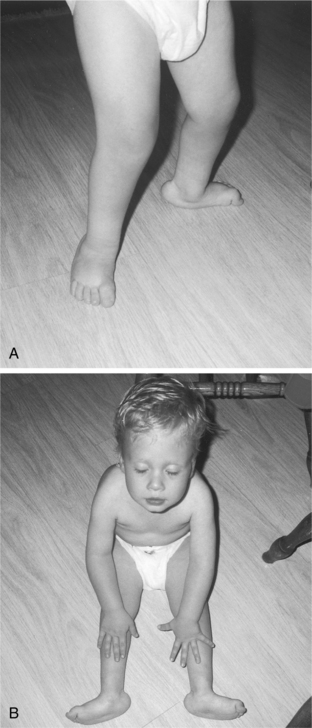
Figure 23-9 Orthopedic involvement. A, Three-year-old boy with bilateral congenital vertical talus resulting in rocker-bottom foot deformities caused by an L4 to L5 myelomeningocele. Note the compensatory knee flexion and genu valgus along with developing toe flexion contractures (the latter from loss of motor control). B, Rocker-bottom foot deformity seen more clearly in the non–weight-bearing position. (Courtesy Zane and Dianna Kuhnhenn, Missoula, Montana.)
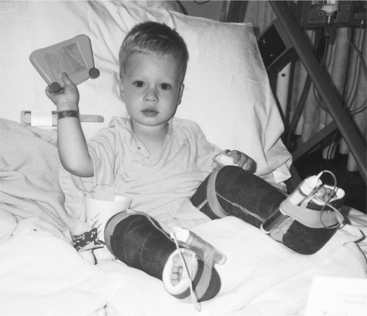
Figure 23-10 Postoperative inpatient after orthopedic reconstructive surgery for congenital vertical talus deformity. Drainage tubes directly from the incision sites were used for 12 hours. (Courtesy Zane and Dianna Kuhnhenn, Missoula, Montana.)

Figure 23-11 Postoperative result. Risk for skin breakdown is reduced around the great toe (no longer contracted into flexion), and base of support is improved for ambulation and allows the child to stand on one leg with support (note the more neutral alignment of lower extremity, especially the knee). The child wears ankle-foot orthoses (AFOs) to maintain proper alignment; there may be some regression of alignment in time because of the continued lack of motor control. (Courtesy Zane and Dianna Kuhnhenn, Missoula, Montana.)
Medical management of the bowel and bladder dysfunction is of critical importance from both a medical and social standpoint. The muscles of the bladder can show either spasticity or flaccidity, leading to either a condition where the bladder is small and under high pressure from urine or large and stretched out and under low pressure.
In children with spastic bladders under high pressure, vesicoureteral reflux and decreased bladder volume and compliance are critical factors that contribute to damage to the upper urinary tract and kidneys. Children with hypotonic bladders often have more residual urine and are more prone to infection.
Infection is treated prophylactically in most children with spina bifida, with antibiotics and high fluid intake as a critical part of an overall management program. Kidney damage is unusual in children with hypotonic bladders because bladder urine is under low pressure and reflux is less of a problem.
Complete bladder emptying using clean intermittent catheterization provides a means to manage urine flow. Manual pressure on the bladder (Credé’s method) is used less often due to its tendency to cause reflux. Implantation of an artificial urinary sphincter has been used in the older child or adolescent,137 and bladder augmentation or urinary diversion are options for the child with high pressures and insufficient volume.
In most of these devices, the opening and closing of the bladder outlet are accomplished by a cuff placed around the outlet. The cuff can be constricted to close the outlet or relaxed to open the outlet and allow urine to flow. Intravesical electrical stimulation remains controversial; however, it has been employed at some centers, with the benefits of increased bladder compliance and increased bladder volume being the most common positive outcomes.87
Stool incontinence is managed most commonly by a program to regulate bowel movements using diet, timed enemas, or suppositories. In some centers the Malone antegrade continence enema procedure is being used to aid in bowel control. This procedure places a cecostomy, bringing the cecum to the abdominal wall in a procedure similar to the placement of a percutaneous endoscopic gastrostomy (PEG) tube. Antegrade enemas are then used to control bowel function.37
PROGNOSIS.
Early, aggressive care of NTDs has now improved the overall prognosis associated with this condition. Prognosis varies with the degree of accompanying neurologic deficit, but researchers are evaluating quality-of-life issues as a possible predictor of prognosis.91
At present, prognosis is poorest for those children who have total paralysis below the lesion, kyphoscoliosis, hydrocephalus, and progressive loss of renal function secondary to chronic infection and reflux. At present, survival to adulthood is approximately 85%; most deaths occur before age 4.
Approximately two thirds of children with myelomeningocele and shunted hydrocephalus have intelligence that falls in the normal range. The remaining one third fall into the range for mental retardation, usually mild. Irrespective of IQ, children with spina bifida still have difficulties in perceptual organizational abilities, attention, speed of motor response, memory, and hand function in addition to mental flexibility, efficiency of processing, conceptualization, and problem solving. Overall cognitive delays occur less often as a result of improved medical treatment for these children.
Adult outcome data are incomplete at this time. Most adults over 40 years of age survived the preshunt era of the 1950s and are without hydrocephalus, whereas adults now in their thirties include people with more severe disabilities who benefited from the advances in medical and surgical management.
Adults with myelomeningocele continue to need therapy and medical management secondary to joint and spinal deformities, joint pain, pressure ulcers, neurologic deterioration, depression, and poor social interaction and adjustment.
Prognosis for Motor Function.
The child’s motor abilities vary according to the level of the lesion, but delay in achieving ambulation can be expected in all children with spina bifida, including those with low neurosegment-level lesions.
A child’s ability to walk outdoors and use a wheelchair by age 7 usually suggests a good ambulation prognosis.41 If functional ambulation is not present by 7 to 9 years of age, it is unlikely to occur subsequently.3,52 A third of all people with myelomeningocele demonstrate a decline in ambulatory status with increasing age, usually around age 12. These losses in ambulatory status often correlate with a variety of adolescent changes, including increasing body size and composition, loss of upper and lower extremity strength, or immobilization for varied periods of time secondary to musculoskeletal surgery or fracture healing.
Adult ambulatory status in spina bifida is highly determined by two variables, including motor level and sitting balance. Overall ambulation status declines over time.
DEVELOPMENTAL DYSPLASIA OF THE HIP
Overview
Developmental dysplasia of the hip (DDH), previously known as congenital hip dysplasia or dislocation, is a common hip disorder affecting infants and children. The change in name reflects the fact that DDH is a developmental process occurring either in utero or during the first year of life; this condition is not necessarily present at birth as the word congenital implies.
DDH can be unilateral or bilateral and occurs in three forms of varying severity: (1) unstable hip dysplasia, in which the hip is positioned normally but can be dislocated by manipulation; (2) subluxation or incomplete dislocation, in which the femoral head remains in contact with the acetabulum but the head of the femur is partially displaced or uncovered; and (3) complete dislocation, in which the femoral head is totally outside the acetabulum.
Incidence and Risk Factors
The incidence of DDH is between 8.6 and 11.5 per 1000 live births.98 About 85% of affected infants are females. The risk of hip dysplasia increases dramatically in the presence of certain obstetric conditions (e.g., breech delivery, large neonate, twin or multiple births) and other conditions such as idiopathic scoliosis, myelomeningocele (spina bifida), arthrogryposis, and cerebral palsy.
The presence of other musculoskeletal deformities such as torticollis,185 metatarsus adductus, and calcaneal valgus deformity should alert the medical practitioner to the need for further evaluation.
Other risk factors include family history, first pregnancies, multiple fetuses, and oligohydramnios (deficient volume of amniotic fluid limiting fetal movement). Certain ethnic groups (Eastern Europeans, Lapps, and Native Americans) also have an increased risk of DDH. One fourth of all cases involve both hips; when only one hip is involved, the left hip is affected three times more often than the right.
Etiologic Factors
The cause varies depending on the associated condition but is usually the result of mechanical, physiologic, or environmental factors. Hormonally derived (maternal hormone relaxin may affect the child in utero and during the neonatal period) or hereditary laxity of the ligaments about the joint and positioning are possible etiologic factors.
Infant positioning, both prenatally and postnatally, may affect the formation of the acetabular cup and hip stability because the acetabulum is formed as a result of contact with the femoral head, and this is thought to be one possible cause of DDH.39
Cultural customs of how babies are carried affect rates of DDH; those cultures that swaddle infants with the hips in extension and adduction are at greater risk of DDH. Conversely, carrying the infant or young child with hips and lower extremities abducted, flexed, and externally rotated may increase stability of the femoral head in relation to the acetabulum.
Pathogenesis
DDH can affect the acetabulum, the femoral head, and the relationship of the femoral head to the acetabulum. The femur, acetabulum, and hip joint capsule usually are well developed by approximately 10 weeks’ gestation but continue to enlarge throughout gestation and develop through contact between the femoral head and acetabulum. Most dislocations result in a progressive deformation of the femoral head and acetabulum during gestation.39
The subluxated hip maintains contact with the acetabulum but is not well seated within the hip joint. Often this occurs because the acetabulum is shallow, with the roof of the acetabulum sloping at an increased angle in people with DDH rather than showing a normal cup shape. The dislocated hip has no contact between the femoral head and the acetabulum, the femoral head sits on the iliac wing and the ligamentum teres is elongated and taut (Fig. 23-12).
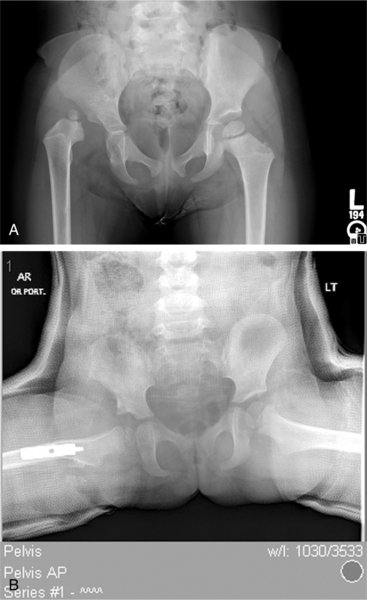
Figure 23-12 Developmental dysplasia of the hip (DDH). Three-year-old child with unilateral developmentally dysplastic hip. A, Note the head of the femur sitting lateral to the acetabulum. The roof of the acetabulum appears dysplastic and the proximal femur somewhat valgus. B, Postoperative: the femur has been relocated in the acetabulum and a varus derotation osteotomy performed. A wedge is cut from the femoral shaft, then internally rotated and positioned in varus to correct the femoral anteversion and valgus. It is also common when there is acetabular insufficiency for portion of the iliac crest to be removed and used as a wedge above the acetabulum to deepen the acetabulum. (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
If the dislocation is not diagnosed and treated early, secondary changes in both soft tissues and bony structures occur. The longer the dislocation has been present, the greater the secondary changes that occur. These changes include stretching of the hip capsule, contracture and shortening of the structures of the hip joint, changes in the blood supply to the hip, flattening of the femoral head, and acetabular dysplasia, sometimes with development of a false acetabulum.
Clinical Manifestations
Clinical manifestations of DDH vary with age. In the newborn and nonambulatory period up to 12 months of age, one or more positive signs may be present (Fig. 23-13). Any observed physical asymmetries in ROM (even as little as 10 degrees is considered significant, especially limitation of hip abduction), asymmetry in the buttock or gluteal fold (higher on the affected side), extra thigh skin folds, or leg length discrepancy requires medical evaluation.
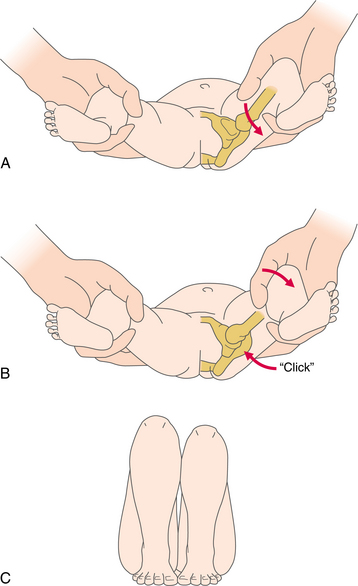
Figure 23-13 Signs of hip dislocation. A, Ortolani maneuver No. 1 (also the second part of the Barlow test): hip flexion and adduction with downward pressure dislocates the hip. B, Ortolani maneuver No. 2: gentle hip flexion, abduction, and slight traction reduce the hip with a discernible click or clunk, and increased hip abduction is possible in a positive test. This test is valid only for the first few weeks after birth. C, Galeazzi test (Allis’ sign): in the supine position, with hips and knees flexed and feet flat on the floor, the knee is lower on the dislocated side, indicating that the head of the femur is positioned posterior or superior to the rim of the acetabulum. This test is used to assess unilateral hip dislocation and can be used in older children (from 3 months on).
In the ambulating child, uncorrected bilateral dysplasia may cause a characteristic gait pattern known as a compensated Trendelenburg gait. As the child sways the torso from side to side to compensate for an ineffective gluteus medius, the child assumes a waddling gait pattern.
Unilateral dysplasia usually is characterized by a limp with a positive Trendelenburg’s sign during the stance phase of gait on the involved side (Fig. 23-14). A flexion contracture on the involved side(s) develops as a result of posterior displacement of the hips, which then contributes to marked lumbar lordosis.
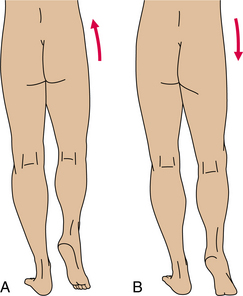
Figure 23-14 Trendelenburg’s sign. A, Negative: with the weight on one leg, the pelvis on the opposite side is slightly elevated (observed from behind the client). B, Positive: with weight on one leg, the pelvis drops on the opposite side because of muscle weakness or pain in the hip joint on the stance side. Trendelenburg’s sign measures weakness of the hip abductor muscles, especially the gluteus medius.
MEDICAL MANAGEMENT
In the newborn period, clinical examination is the most important diagnostic tool and continues to be the standard screening tool. A positive Ortolani or Barlow click confirms DDH in the first month of life (see Fig. 23-13). These tests are considered significantly less diagnostic past 2 to 4 weeks.65 As ligamentous structures become stronger or if the joints become stretched and worn, it is more difficult to elicit the characteristic popping in and out described.
In some cases, dislocation is not diagnosed by these standard tests, and the disorder may not be apparent at birth. Because a normal neonatal examination cannot guarantee that a hip will not become dysplastic, serial examination throughout infancy is essential. Well-baby checkups should include hip examination until the child begins to walk with a normal gait pattern.
The Galeazzi sign becomes positive in the older infant once shortening of the thigh becomes apparent. Radiographic examination is unreliable in the infant and is used more commonly in older infants and children. Plain radiographs are not able to image the hip adequately until the head is ossified and may not confirm the diagnosis if the unstable hip is in the reduced position at the time the film is taken.
For this reason, ultrasonography is suggested in cases of suspected but unconfirmed DDH. Ultrasonography allows visualization of the cartilaginous structures of the hip and is especially accurate during the first 6 months of life; however, its use as a screening tool remains controversial65,160
TREATMENT.
The goal of treatment for DDH is to ensure stability of the femoral head in the acetabulum, thereby encouraging the development of a normally shaped socket and femoral head. This is accomplished by replacing the head of the femur into the acetabulum with no intervening soft tissue.
The proper position then must be maintained for a period of time sufficient for the bony and cartilaginous structures to develop sufficient stability so that the hip does not subluxate or dislocate with normal movement. Treatment depends on the age of the child and the severity and duration of the dysplasia.
The most common treatment in the infant is placement of the hip in a position of 100 degrees flexion and 90 degrees abduction until the joint capsule tightens and the acetabulum is molded to assume a cup shape. This can be accomplished through the use of a hip harness such as the Pavlik harness (Fig. 23-15). The former standard of treatment with triple diapering is no longer recommended, because proper positioning cannot be ensured and the treatment results in an unacceptable incidence of aseptic necrosis of the femoral head.

Figure 23-15 Pavlik Harness. A 7-month old with developmental hip dysplasia wearing a Pavlik harness that holds her legs in flexion and abduction. The harness is worn 23 hours a day, removed only for bathing and diaper changes. The goal of treatment is to keep the femoral head in good contact with the acetabulum. A stable hip encourages the development of a normally shaped socket and rounded head of the femur. The proper hip position must be maintained for enough time to stabilize the joint. The hip should be flexed to 95 degrees and abducted (apart) at least 90 degrees. This position keeps the femoral head in the best position and allows the ligaments and joint capsule to tighten up. (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
The infant must wear the apparatus continuously for 3 to 9 months, eventually weaning its use to nighttime only before its discharge to stabilize the hip in the correct alignment. Criteria for discontinuation of the harness are not standard. Some physicians advocate complete removal of the harness 6 weeks after the hip can no longer be moved in and out of the acetabulum. Others recommend discontinuation when radiographic findings confirm hip stabilization.
Lack of contact of the femoral head in the acetabulum allows the persistence of acetabular dysplasia. This phenomenon is the basis for standing programs for nonambulatory children (i.e., standing provides mechanical forces to assist in the development of the acetabular cup, adding to the stability of the hip).
Treatment in older children who have been walking is usually surgical (e.g. traction, closed reduction [hip spica cast], open reduction, tenotomy of contracted muscles, or osteotomy of either the femur or acetabulum) (see Fig. 23-12, B) depending on the clinical presentation. Traction is used before closed reduction by some in an attempt to aid in reducing the dislocation by applying a distractive force to the joint to loosen the surrounding tissue before the closed reduction.
In the child treated between 6 months and 2 years, a closed reduction is used with an arthrogram to confirm the reduction followed by 3 to 5 months in a hip spica cast. Treatment after the age of 2 years requires surgical reduction, often with both femoral and pelvic osteotomies.
PROGNOSIS.
Outcome is directly related to the child’s age at initiation of treatment. If the dislocation is corrected in the first few weeks of life, the dysplasia is completely reversible and a normal hip can develop, with rates of success as high as 95%.110 If surgical reduction is required, 86% have a satisfactory outcome.
The two main complications include avascular necrosis (7%) and premature physeal arrest (18%), which presents during the adolescent growth spurt.197 As the child becomes older and the primary subluxation or dislocation persists, the deformity becomes more difficult to correct conservatively, and increased rates of avascular necrosis and redislocation are seen.
When the condition is untreated, long-term problems can include degenerative joint disease, hip pain, antalgic gait, scoliosis, back pain, or the need for total hip replacement.
NEUROMUSCULAR DISORDERS
Neuromuscular disorders including the muscular dystrophies, congenital myopathies, and spinal muscular atrophy are presented in this chapter. Other neuromuscular disorders such as Charcot-Marie-Tooth disease, amyotrophic lateral sclerosis (ALS), Guillain-Barré polyneuritis, and chronic inflammatory demyelinating polyneuropathy are discussed in other chapters in this text.
The Muscular Dystrophies
The muscular dystrophies (MDs) comprise the largest and most common group of inherited progressive neuromuscular disorders of childhood. They affect all population types, even animals. Signs of MD can occur at any point in the lifespan.
These disorders, in general, have a genetic origin and are characterized by ongoing, typically symmetric, muscle wasting with increasing deformity and disability. Paradoxically, in some forms (e.g., Duchenne’s, Becker’s) wasted muscles tend to hypertrophy because of connective tissue and fat deposits, giving the visual appearance of muscle strength.
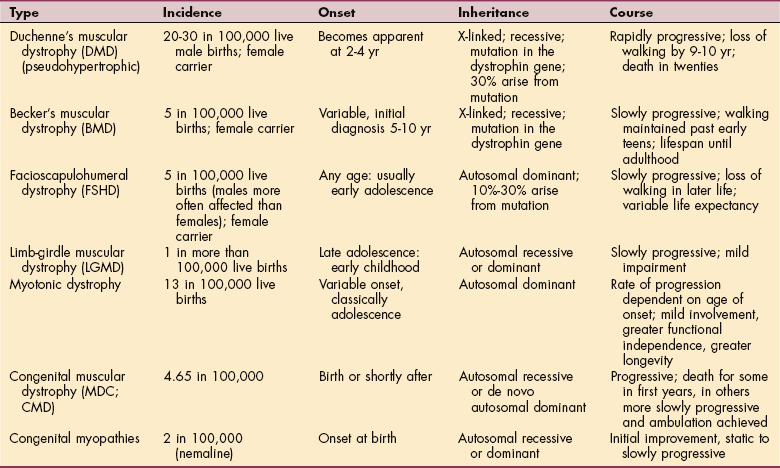
Six major types of MD are included in this text discussion: (1) Duchenne’s muscular dystrophy (DMD), (2) Becker’s muscular dystrophy (BMD), (3) facioscapulohumeral (Landouzy-Dejerine) dystrophy (FSHD), (4) limb-girdle dystrophy (LGMD), (5) myotonic dystrophy, and (6) muscular dystrophy congenita (MDC), also known as congenital muscular dystrophy (CMD). These forms of MD involve a primary degeneration of muscle with a gradual loss of strength, but each type differs as to which muscle groups are affected (Fig. 23-16).
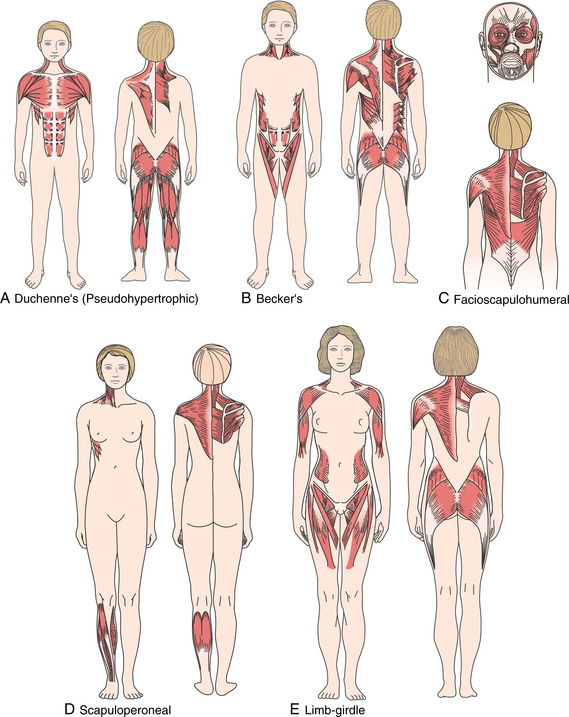
Figure 23-16 Muscle groups involved in muscular dystrophies. These are presented in relative terms; that is, unlike spinal cord injury with definitive muscle involvement, in muscular dystrophy, proximal or distal muscle groups are affected in varying ways with individual differences noted. For example, in the facioscapulohumeral form, the lower erector spinae is featured here but may be spared, and in limb-girdle dystrophy, the lower abdominal muscles may be involved but are not shown in this illustration. A, Duchenne’s: shoulder girdle (trapezius, levator scapulae, rhomboids, serratus anterior), pectoral muscles, deltoid, rectus abdominis, gluteals, hamstrings, calf muscles. B, Becker’s: neck, trunk, pelvic and shoulder girdle. C, Facioscapulohumeral: muscles of the face and shoulder girdle. D, Scapuloperoneal: muscles of the legs below the knees (first), shoulder girdle (later). E, Limb-girdle: upper arm (biceps and deltoid) and pelvic girdle.
Incidence and Etiologic Factors
The incidence of DMD is approximately 1 in 3500 live births. Rates of occurrence for each type are listed in Table 23-5. All dystrophies are genetically based disorders.
DMD and BMD are X-linked recessive disorders caused by mutations in the dystrophin gene Xp21 that codes for the muscle membrane protein dystrophin. The affected gene on the short arm of the X chromosome (Xp21) is one of the largest genes in the human genome. In these two forms of MD, males are affected clinically and females are usually only carriers.
FSHD is an autosomal dominant disorder with onset in early adolescence. The son or daughter of a person affected with FSHD is at 50% risk of inheriting the defective gene. FSHD occurs with an incidence of 1 in 20,000.
The gene for FSHD has been localized to 4q35 in most people with FSHD; however, the specific gene has not been identified, and there are some people with the FSHD phenotype in whom the defect does not localize to the fourth chromosome.
LGMD may be inherited in several ways depending on the type. LGMD type 2 (A through H) disorders are autosomal recessive disorders of late childhood or adolescence and type 1 (A through G) disorders are autosomal dominant disorders. Dominant disorders have a 50% risk of inheritance if one parent is affected; recessive disorders carry a 25% risk of disease when both parents are carriers and a 50% chance of carrier status.
Myotonic dystrophy has an incidence that varies between 1 in 5000 to 1 in 50,000, with rates in some populations that approach 1 in 550 because of local founder effects.196 The founder effect occurs when there is a loss of genetic variation such as occurs when a new colony is formed by a very small number of individuals from a larger population. For example, when a small part of a population moves to a new locale, or when the population is reduced to a small size because of some environmental change, the genes of the “founders” of the new society are disproportionately frequent in the resulting population.
Myotonic dystrophy demonstrates an autosomal dominant inheritance pattern, with each generation being somewhat more severely affected than the last. This increase in the phenotypic severity of the disease state with succeeding generations is referred to as anticipation and can be correlated with an expansion in the size of the triple-repeat genetic enlargement that is the causative factor in this disease.96
MDC represents a group of recessively inherited disorders that can be divided into two groups based on the presence of brain involvement. Only the most common forms are included here. The overall incidence has been placed at 4.65 per 100,000 in the Italian population.127 In Japan, however, Fukuyama MD, one form of MDC, is as common as DMD.
There are a number of classification schemes that have been proposed for the MDCs.130 For example, MDCs can be divided into those that are the result of extracellular protein deficits, endoplasmic reticulum protein deficits, defects of glycosylation, and defects of integrin.
Pathogenesis
Knowledge of the MDs and understanding of their increasing complexities escalated dramatically in the late 1980s when the protein dystrophin was identified as the causative factor in DMD and BMD. Subsequently, other members of the dystrophin glycoprotein transmembrane complex were identified as causative proteins in many other forms of MD.
In addition to transmembrane proteins of the dystrophin glycoprotein complex, there have been proteins in the extracellular matrix, sarcomere, and nucleus identified as causative in MD (Fig. 23-17). Recently it has become apparent that not only can structural proteins create MD but enzymatic defects and defects in glycosylation (the modification of proteins by the addition of sugars) can also create MD.
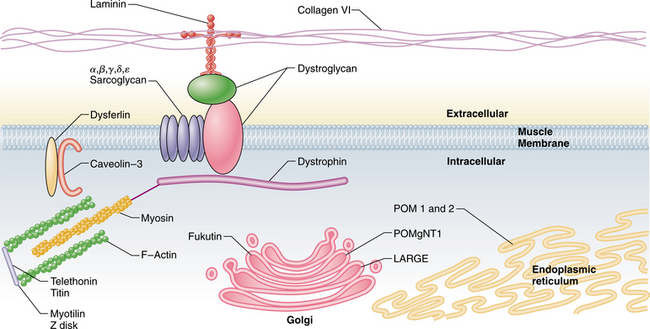
Figure 23-17 Diagram showing the most common muscle protein defects that lead to muscle disease. The dystroglycan complex spans the muscle membrane and connects dystrophin and the myosin/F-actin contractile mechanism to the extracellular matrix. The Golgi complex and endoplasmic reticulum are also displayed with the causative factors related to glycosylation defects. (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
These discoveries, along with advances in research and technology, have brought new information on the molecular pathogenesis of these disorders, including the genetic and molecular characterization of many forms of MD.
Duchenne’s and Becker’s Muscular Dystrophy.: The affected gene in DMD/BMD encodes messenger RNA (mRNA) for the adhesive protein dystrophin that is located in the muscle membrane, the sarcolemma. Muscle membrane lesions play an early role in the pathogenesis of MD, involving skeletal, cardiac, and smooth muscle membranes.
Dystrophin is the protein that links the muscle surface membrane (sarcolemma) with the contractile muscle protein (actin). Lack of normal dystrophin makes the sarcolemma susceptible to damage during contraction-relaxation cycles. Disruption of the muscle membrane and muscle fiber necrosis are initiated by muscle contraction, especially eccentric contraction.136
The underlying biochemical defect in all types of MD does not necessarily disrupt the integration of dystrophin in the membrane, and while the absence of some sarcoglycan molecules in LGMD (see later) can lead to susceptibility to mechanical trauma, the absence of others does not.69
Males with undetectable levels of dystrophin have Duchenne-type MD, whereas those with nearly normal levels but dystrophin of an abnormal size or low levels have the Becker-type MD. The absence or altered state of dystrophin destabilizes the membrane and allows the influx of Ca2+, which triggers the destruction of the cell from the inside through the activation of calpain, a calcium-activated proteinase.133
Muscle cells are replaced by fatty and connective tissues, and contractures develop. Fat cells then continue to accumulate between damaged muscle fibers as a response to the ongoing muscle atrophy. Disorganization of tendinous insertions also is associated with fat accumulation.
Limb-Girdle Muscular Dystrophy.: LGMD represents a collection of genetically heterogeneous disorders that can be broadly divided on a genetic basis into two groups. LGMD type 1 (LGMD1A through 1G) are all inherited dominantly, and LGMD type 2 (LGMD2A through 2K) are inherited recessively.
LGMD type 1A is the result of the absence of myotilin and is very rare. Myotilin is a thin filament protein associated with the Z disk and involved in assembly of the contractile mechanism of the cell.153 LGMD1B results from the absence of lamin A and lamin C. Lamins A and C are part of a large class of proteins. Lamins A and C are nuclear membrane proteins that are involved in stability of the nuclear membrane and cell differentiation.67
LGMD1C results from the absence of caveolin 3. Caveolin 3 is found as part of the muscle membrane and acts in cell signaling. The remaining dominant forms of LGMD (2D through 2F) have genetic characteristics but as yet have no protein identified.67
The second group of LGMDs (2A through 2K) are more common than the dominant forms and are inherited recessively. LGMD2A is one of the most common, with an estimated carrier frequency of 1 in 103.139 The underlying defect in LGMD2A is that of a cellular regulatory enzyme P94, calpain 3, in the calpain family of molecules (calcium-activated protein enzymes). The function of calpain 3 is not well understood, but it appears to play a role in the organization of the muscle cell as it forms.133
LGMD2B is caused by an absence of dysferlin (chromosome 2p13). This is also the protein defect in Miyoshi myopathy, which presents with a different dystrophic distal phenotype.187 Dysferlin acts as a membrane repair molecule and interacts with other molecules at the muscle membrane.25
The absent gene in LGMD2C, 2D, 2E, and 2F all code for a specific protein in the sarcoglycan-glycoprotein complex. This glycoprotein complex is found in close association with dystrophin in the muscle membrane. These proteins act to stabilize the membrane. As a group these are known as sarcoglycanopathies, because each is named by the missing sarcoglycan protein (γ, α, β, and δ).
Sarcoglycan-deficient muscle in some cases is sensitive to eccentric contraction–induced disruption of the plasma membrane.69 In fact, researchers have further identified the specific sarcoglycan deficiency to be that of δ-sarcoglycan. Reduced levels of other forms of sarcoglycan (e.g., γ, α, β) appear to be involved with the instability at the muscle membrane but do not account for contraction-induced muscle injury.69
The pathogenesis of fiber demise in some types of LGMD is presumably similar to that of DMD and BMD. The absence of some of the sarcoglycans affects the integration of other sarcoglycans and the dystroglycan complex in the muscle membrane.69,176
LGMD2G is the result of a genetic defect on the seventeenth chromosome (17q11-12), which codes for the protein telethonin. Telethonin is a protein that acts closely together with titin (discussed later) in the formation of the muscle cell. In the adult muscle it is found at the Z disk in the sarcomere.125
LGMD2H results from the absence of TRIM 32, which belongs to a family of similar proteins. TRIM 32 is not well understood but acts in conjunction with other enzymes in the muscle and is found to be up-regulated in situations where muscle remodeling is occurring, as is the case when changes in weight bearing occur. In addition, it is found in increased levels when muscle cells are differentiating. TRIM 32 is found associated with the thick myosin filaments of the sarcomere.95
LGMD2I is also a common form of LGMD caused by a mutation in the fukutin-related protein (FKRP) gene (19q13.3). FKRP encodes for an enzyme that is involved in glycosylation of α-dystroglycan.174 α-Dystroglycan is a component of the dystrophin glycoprotein complex.
The clinical picture of individuals with abnormalities of this protein is widely distributed, ranging from mild, late-onset disease in the fourth or fifth decade of life to a severe form of MDC with brain abnormalities in addition to severe weakness at birth.14
LGMD2J results from a mutation at 2q24.3 in the titin gene. Titin is a large intracellular protein that is responsible for elasticity and stability of the sarcomere. In addition it plays a role in the assembly of the sarcomere in the developing muscle cell. Titin extends from the M line and connects the thick-filament myosin to the Z line, where it forms an elastic connection.107 Clinically titinopathies can present as the LGMD2J homozygous form, resulting from mutations on both alleles, and as a milder tibial MD, a heterozygous form with mutations on only one allele.180
LGMD2K has been designated to identify the milder phenotypic variants of the POMT1 mutation. This is the same mutation that is found in some of the more severe MDC cases with Walker-Warburg syndrome (WWS). This form of LGMD is one of the only forms that has mental retardation as a component. There are some intermediate forms that fall between LGMD and WWS. Like the other glycosylation defects found in FKRP, there is a spectrum of severity found in people with POMT1 mutations. This is likely the result of still unknown modifying factors.33
Congenital Muscular Dystrophy.: The pathogenesis of the MDCs with brain involvement is related to the glycosylation of α-dystroglycan. Glycosylation is the addition of sugars or chains of sugars (glycans) to proteins and lipids. This process takes place in the endoplasmic reticulum and Golgi complex and is regulated by a series of enzymes that control the addition of glycans.
O-mannose glycans develop into very complicated structures and are carried by α-dystroglycan, which is closely associated with β-dystroglycan and is an integral part of the dystrophin-associated protein complex. The β-dystroglycan spans the muscle membrane and binds to both actin and laminin 2.
Five of the MDCs have been linked to mutations in the process of glycosylation. Different mutations in the same gene can produce widely varied phenotypes, which in some cases result from the varied amounts of residual enzyme activity. Glycosylation has been identified as necessary for the binding of laminin 2 to dystroglycan in the extracellular matrix.
WWS is the most severe MDC and is the result of defects in the first step in the glycosylation process where O-mannose is added to α-dystroglycan caused by the absence of the glycosyltransferase that facilitates this reaction. The gene that is responsible is the POMT1 gene. Because WWS is a phenotypic diagnosis, POMT1 is only responsible for about 20% of the cases. Mutations in POMT2 are also seen in WWS, and the combination of these two genes in the endoplasmic reticulum modulates transferase activity.
Additionally defects in Fukutin and Fukutin-related proteins (FKRPs) can also be seen in WWS and presumably occur in the glycosylation pathway, although FKRP defects can produce a wide range of clinical phenotypes. Muscle-eye-brain disease can be caused by mutations in the POMgNT1 gene that encodes for the transferase in the next step in the pathway.
Clinically muscle-eye-brain disease represents a spectrum of phenotypic expression from mild (those who will live into adulthood) to severe (WWS). Because of this expression variability, other factors are presumed to affect the severity of the presenting phenotype. One such factor might be overexpression of like-glycosyltransferase (LARGE).
Fukuyana muscular dystrophy is caused by a mutation in the fukutin gene; the products are expressed in muscle in the same location as dystroglycan. Fukutin is found in the cis-Golgi, but beyond that its function has not been established.
MDC type 1C has a milder phenotype and is the result of a mutation in LARGE. LARGE is also found in the Golgi complex and rate-limiting step in the glycosylation of α-dystroglycan. When it is overexpressed, α-dystroglycan is hyperglycosylated. Despite this knowledge, its exact function still remains unclear. The potent effect of LARGE makes it an ideal target for treatment, since its overexpression can rescue both in vitro and in vivo models of a variety of glycosylation-based MDs.56
Two forms of MDCs without brain involvement include Ullrich-negative and merosin-negative MDC. The Ullrich-negative form is the result of a defect in the extracellular matrix protein collagen VI. Collagen VI is made up of three strands, COL6A1, A2, and A3, which are coded for on 21q22.3 or 2q37.
Dominant mutations traditionally have been thought to result in a more Bethlehem phenotype. Recessive mutations result in the congenital Ullrich presentation. However, there have been recent cases reported where dominant mutations have presented with an Ullrich picture. The underlying defect might relate to the protein interaction with the membrane, but the exact function still remains unclear. Merosin-negative MDC type 1A results in merosin deficiency and is caused by a genetic defect on chromosome band 6q22-23.92
Facioscapulohumeral Dystrophy.: FSHD is less well understood. The genetic defect has been found at 4q35 in 90% to 95% of individuals with FSHD.24,183 However, the abnormal protein has not been identified, because it is coded for at some distance from the genetic defect and has yet to be cloned.
Myotonic Dystrophy.: There are three forms of myotonic dystrophy. The major form of myotonic dystrophy, also known as Steinert type or MD1, has been linked to chromosome band 19q13.3 and represents 98% of the cases. Two other types of myotonic dystrophy have been identified. Myotonic dystrophy type 2 (MD2) has been linked to chromosome band 3q21.3 (MD2)118 and a third type has been linked to chromosome band 15q21-24.
In MD1 the underlying defect of the gene is a trinucleotide repeat in which cytosine, thymine, and guanine (CTG) are repeated an abnormally large number of times (see Fig. 1-4) With succeeding generations this defect expands, a condition known as anticipation, also seen in other triple-repeat disorders, where each succeeding generation is more affected than the last.96
This results in the absence of myotonin protein kinase through a very complex and poorly understood pathway involving the creation of abnormal RNA transcript processing and alterations in splicing of other genes, some of which code for the chloride channel.12 In myotonic dystrophy muscle fibers demonstrate altered resting muscle membrane potentials, possibly resulting from a dysregulation of ion channel function.10
Clinical Manifestations
People with myotonic dystrophy have muscular weakness, wasting, and hypotonia. The degree of severity and age of onset vary with the type of myotonic dystrophy present (see Table 23-5).
Duchenne’s Muscular Dystrophy.: DMD is usually identified when the child has difficulty getting up off the floor (Gowers’ sign; Fig. 23-18), falls frequently, has difficulty climbing stairs, and starts to walk with a waddling gait (proximal muscle weakness) and an increased lumbar lordosis (compensation for abdominal and hip extensor weakness). At the same time, the child begins to walk on the toes because of contracture of the posterior calf musculature and weakness of the anterior tibial, peroneal, and proximal muscles.

Figure 23-18 Gowers’ sign. This boy adopts the typical movement seen with proximal weakness, such as myopathies, when arising from the floor, a chair, or even when climbing stairs. During Gowers’ maneuver the client places the hands on the thighs and walks up the legs with the hands until the weight of the trunk can be placed posterior to the hip joint. This sign is characteristic of weakness of the lumbar and gluteal muscles. (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
Hip abductor weakness produces a positive Trendelenburg’s sign (see Fig. 23-14), which eventually changes to a compensated gluteus medius gait as hip abductor weakness progresses. Classically ambulation continues to deteriorate up to the age of 10 to 12 years, at which time the majority of people with DMD are no longer able to walk.17,164 However, with more recent medical treatment with prednisone or deflazacort children may walk beyond their twelfth birthday, which previously was not seen.
The shoulder girdle becomes involved, with excessive scapular winging and muscle hypertrophy (especially the upper arms but also the calves and thighs) (Fig. 23-19). One of the major problems in gait occurs when the affected person requires upper extremity support. Shoulder girdle weakness and the need to maintain the weight line posterior to the hips and anterior to the knees often prevents the use of crutches to support the body weight.
Figure 23-19 Duchenne’s muscular dystrophy with pseudohypertrophy of calves and lordotic posture that places the weight of the trunk behind the hip joint. Even though weakness occurs symmetrically, habitual standing postures may create asymmetries in flexibility in some cases. (Courtesy Allan Glanzman, Children’s Seashore House of the Children’s Hospital of Philadelphia, PA.)
Weakness of the shoulder girdle also causes difficulty in performing overhead activities related to hygiene and work. Bicipital tendinitis or other impingement disorders at the shoulder occur as the children get older as the result of weakness and the repeated manual lifts that become necessary for transfers. Muscle imbalances create biomechanical dysfunction, and weakness impairs the ability to stabilize the shoulder girdle, contributing to shoulder problems.
With the progression of weakness, scoliosis occurs at a rate of 80% to 90% and usually progresses more rapidly after the person is wheelchair bound. Spinal fusion is usually considered when the spinal curve approaches 40 degrees. Early fusion, when a good correction and level pelvis can be obtained, and when the individual’s respiratory status is relatively more intact, produces the best result.
Common comorbidities associated with DMD include cognitive, respiratory, cardiac, and gastrointestinal dysfunction. The average IQ of individuals with DMD is 1 standard deviation below the mean, with specific reading disorders noted irrespective of IQ. Even so, many children with DMD have normal or above-normal intelligence.
Children with DMD develop a progressive restrictive respiratory impairment secondary to weakness and contracture of the respiratory muscles. Respiratory problems become more of a problem after the children become wheelchair bound. Nocturnal hypoventilation is one of the earlier manifestations of respiratory involvement and is usually accompanied by headaches, sleep disturbance, or nightmares. Chest muscle deterioration combined with joint contractures and involvement of the spine results in diminished ventilation and ability to produce pressure to cough up secretions, leading to pneumonia, other respiratory infections, and even death.
Disruption of sarcoglycan complexes in vascular smooth muscle also can result in vascular irregularities of the heart, diaphragm, kidneys, and gastrointestinal tract. Dilated cardiac myopathy and, less frequently, conduction abnormalities can be life-threatening. Gastrointestinal problems are common, and constipation and pseudo-obstruction can result from the smooth muscle deterioration and gastric dilatation that can occur.
Becker’s Muscular Dystrophy.: Signs and symptoms of BMD resemble those of DMD but with a slower progression and longer life expectancy. Ambulation is preserved into midadolescence or later but often is marked by the toe walking with bilateral calf muscle hypertrophy.
Proximal muscles tend to be affected to a greater degree and before the involvement of the distal musculature, with primary effects observed in the neck (relatively preserved in BMD versus DMD), trunk, pelvis, and shoulder girdle. Muscle cramps are a common complaint in late childhood and early adolescence.
Scoliosis and contractures (elbow flexors, forearm pronators, and wrist flexors in the upper extremity and plantar flexors, knee flexors, and hip abductors in the lower extremity) and other comorbidities also found in DMD are common; however, these occur less frequently and with less severity in the BMD type.
Limb-Girdle Muscular Dystrophy.: LGMD affects both proximal and distal muscles and follows a slow course, often with only mild impairment, although the course can vary widely even in a given family. Early symptoms develop as a result of muscle weakness in the upper arm (biceps and deltoid muscles) and pelvic muscles, usually noticed in late adolescence or early adulthood but as late as the person’s forties.
Winging of the scapulae, lumbar lordosis, abdominal protrusion, waddling gait, poor balance, and inability to raise the arms may also develop. The lack of consistent clinical features makes this type of MD more difficult to diagnose. Identification of the specific type of LGMD should be pursued to provide the best anticipatory guidance for the client and to allow the therapist and physician to treat the person proactively with full knowledge of the natural history of the specific disorder rather than approaching the case reactively.
Congenital Muscular Dystrophy.: MDC represents a spectrum of disease states that most commonly present at the more severe end of the spectrum in infancy with rapidly progressive muscle strength loss and progressive respiratory symptoms.
Clients demonstrate a mixed central and peripheral picture with involvement of both the brain and muscle in addition to involvement of the visual system. WWS is the most severe of the MDCs and presents at birth with a rapidly progressive course, with death most commonly prior to a year of age. Ocular impairments include retinal abnormalities, microphthalmia glaucoma, cataracts, and anterior chamber abnormalities.
The CNS complications include a cobblestone lissencephaly with agyria as well as areas of macrogyria and polymicrogyria. Cerebellar abnormalities are also present and include hypoplasia in addition to fourth ventricular dilatation. Muscle-eye-brain disease has similar clinical findings as WWS but a wider range of phenotypic presentations. Fukuyama MD can also present as an MDC but is more often found in its milder LGMD form.
Common phenotypic presentation includes onset at or shortly after birth with weakness and delayed gross motor skills with progressive contractures and weakness, with very few achieving ambulation and those typically requiring a wheelchair by the age of 10.
Cognitive impairment is common and often severe, with MRI findings somewhat similar to those found in WWS. Ullrich MDC can present along a spectrum of severity, with its milder form, Bethlem myopathy, representing the mildest presentation. In its more severe forms children have significant weakness that either prevents ambulation or children only walk for a short time prior to adolescence.119
The most prominent feature of the phenotype is the prominent distal laxity mixed with proximal contractures, particularly of the knee. People with merison-negative MDC typically present with weakness at birth, with respiratory insufficiency developing typically in the first decade; however, a spectrum including an LGMD phenotype has been reported.92
Facioscapulohumeral Dystrophy.: FSHD is a mild form of MD beginning with weakness and atrophy of the facial muscles and shoulder girdle, usually presenting in the second decade of life. Phenotypic expression is more common in males than in females (95% versus 69%), with more females being carriers. Inability to close the eyes may be the earliest sign; the face is expressionless even when laughing or crying, forward shoulders and scapular winging develop, and the person has difficulty raising the arms overhead (Fig. 23-20).
Figure 23-20 Facioscapulohumeral dystrophy. Weakness of subscapular musculature makes it difficult to perform overhead activities; wasting also causes the clavicles to jut forward and the shoulders to have a drooping appearance. During humeral movement, the scapulae wing and ride up over the thorax. (From Morgan-Hughes JA: Diseases of striated muscle. In Asbury AK, McKhann GM, McDonald WI, eds: Diseases of the nervous system: clinical neurobiology, ed 2, Philadelphia, 1992, Saunders, p 170.)
Other changes in the face include diffuse facial flattening, a pouting lower lip, and inability to pucker the mouth to whistle or, for the infant, an inability to suckle. Progression is descending, with subsequent involvement of either the distal anterior leg or hip girdle muscles.170 Weakness of the lower extremities may be delayed for many years. Contractures, skeletal deformities, and hypertrophy of the muscles are uncommon.
There is wide variability in age at onset, disease severity, and side-to-side symmetry, even within affected members of the same family. Associated non–skeletal muscle manifestations include high-frequency hearing loss and retinal telangiectasias, both of which are usually asymptomatic. The natural history and progression are well documented.170,183
A variation of FSHD is scapuloperoneal MD, involving the proximal muscles of the shoulder girdle with sparing of the face. The process slowly spreads to the distal part of the lower extremities in several years or decades. Early symptoms may include shoulder weakness characterized by scapular winging; foot drop develops later.