Inflammation and Healing
Injury or death of cells caused by infectious microbes, mechanical trauma, heat, cold, radiation, or cancerous cells can initiate a well-organized cascade of fluidic and cellular changes within living vascularized tissue called acute inflammation (Fig. 3-1). These changes result in the accumulation of fluid, electrolytes, and plasma proteins, as well as leukocytes, in extravascular tissue and are recognized clinically by redness, heat, swelling, pain, and loss of function of the affected tissue. Inflammation is often a protective mechanism whose biologic purpose is to dilute, isolate, and eliminate the cause of injury and to repair tissue damage resulting from the injury. Without inflammation, animals would not survive their daily interactions with environmental microbes, foreign materials, and trauma and with degenerate, senescent, and neoplastic cells.
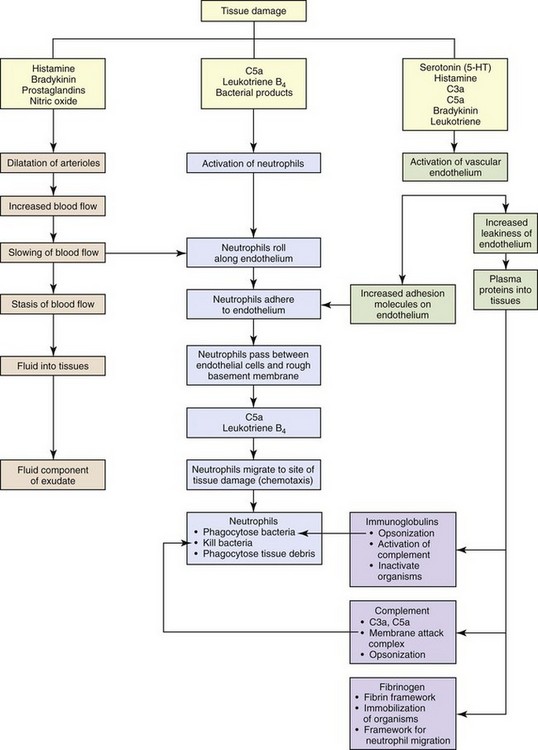
Fig. 3-1 The major steps of the acute inflammatory process. (Modified from Young B, O’Dowd G, Stewart W: Wheater’s basic pathology: a text, atlas and review of histopathology, ed 5, New York, 2010, Churchill Livingstone.)
Acute inflammation, a provoked response, is the progressive reaction of vascularized living tissue to injury over time. This process is usually a well-ordered cascade mediated by chemoattractants, vasoactive molecules, proinflammatory and antiinflammatory cytokines and their receptors, and antimicrobial or cytotoxic molecules. Acute inflammation has a short duration, ranging from a few hours to a few days, and its main characteristics are exudation of electrolytes, fluid, and plasma proteins and leukocytic emigration, principally neutrophils from the microvasculature, followed by rapid repair and healing. For convenience, acute inflammation is divided into three sequential phases: fluidic, cellular, and reparative.
Chronic inflammation is considered to be inflammation of prolonged duration, usually weeks to months and even years, in which the response is characterized predominately by lymphocytes and macrophages, tissue necrosis, and accompanied by tissue repair, such as healing, fibrosis, and granulation tissue formation, all of which may occur simultaneously. Chronic inflammation can be a sequela to acute inflammation if there is failure to eliminate the agent or substance that incites the process. With such persistent substances, the inflammatory reaction and exudates gradually transition from seroproteinaceous fluids and neutrophils to macrophages, lymphocytes, and fibroblasts with the potential for formation of granulomas. Alternatively, some inciting substances can invoke chronic inflammation directly and almost immediately. Examples include infections by Mycobacterium spp.; exposure to foreign materials, such as silicates and grass awns; and immune-mediated diseases, such as arthritis.
WEB TABLE 3-1
Highlights of the Historical Contributions to the Understanding and Characterization of Inflammation
| Contributor | Time | Contribution |
| Egyptians | 1600 bc | Descriptions of inflammation |
| Celsus (Italy) | 25 bc-50 ad | Four cardinal signs of inflammation: rubor (redness), tumor (swelling), calor (heat), dolor (pain) |
| John Hunter (Scotland) | 1793 | Inflammation is a salutary (favorable to health) effect, not a disease per se |
| Julius Cohnheim (Germany) | 1839-1884 | Observed inflamed vessels microscopically |
| Elie Metchnikoff (Russia) | 1882 | Observed and described phagocytosis |
| Rudolf Virchow (Germany) | 1821-1902 | Fifth cardinal sign of inflammation: functio laesa (loss of function); cellular injury |
| Sir Thomas Lewis (England) | 1927 | Determined that chemicals (histamine) induce vascular changes |
| Jules Bordet (Belgium) | 1898 | Antibacterial effects of serum |
| Paul Ehrlich (Germany) | 1908 | Complement mediates antibacterial effects of serum |
Evolution of the Current Understanding of Inflammation
The current understanding of inflammation has evolved over the last 3600 years of recorded history (Web Table 3-1). Clinical signs attributable to inflammation were first described in Egypt in 1600 bc. Aulus Cornelius Celsus, 25 bc to 50 ad, was a Roman writer (de Medicina) and the first individual to describe the four cardinal signs of inflammation (redness, heat, swelling, and pain) that are commonly used today to diagnose inflammation in medicine. In the mid-1800s, Rudolf Virchow, the founder of modern pathology, added the fifth cardinal sign of inflammation: loss of function. In addition to his numerous contributions to cellular pathology, Dr. Virchow, by the age of 25, had discovered fibrinogen, described the processes of leukocytosis, and later characterized pus and necrosis. In 1859, his book, Cell Pathology, became the basis for all microscopic study of disease. Phagocytosis by macrophages, an important component of inflammation and immunologic responses, was first described by Illya Mechnikov (Elie Metchnikoff) in 1883, and he was awarded the Nobel Prize in Medicine in 1908. Jules Bordet, a Belgian scientist, demonstrated antibacterial activity of serum, which Paul Ehrlich, of Germany, later defined as complement. The first experiment to demonstrate the role of a chemical mediator (histamine) in inducing vascular changes (flare and wheal reactions) was conducted by Sir Thomas Lewis in 1927. The work of these pioneers, as well as additional experimental studies conducted during the past century, has provided (1) an in-depth and clearer understanding of inflammation and (2) the foundation for development of therapeutic compounds to treat undesirable effects of inflammatory responses. In fact, such treatments are so widely used and commonplace in veterinary medicine today that the contributions and discoveries of these scientists are often taken for granted.
Beneficial and Harmful Aspects of Inflammation
As a general rule, inflammatory responses are beneficial in the following ways:
• Diluting and/or inactivating biologic and chemical toxins
• Killing or sequestering microbes, foreign material, necrotic tissue (e.g., bone sequestrum), and neoplastic cells
• Providing wound healing factors to ulcerated surfaces and traumatized tissue
• Restricting movement of appendages and joints to allow time for healing and repair
• Increasing temperature in the body or locally to induce vasodilation and inhibiting replication of some microbial agents
However, in some instances, an excessive and/or prolonged inflammatory response can be detrimental and even more harmful than that of the inciting agent/substance. In several disorders of humans, such as myocardial infarction, cerebral thrombosis and infarction, and atherosclerosis, excessive and prolonged inflammatory responses can exacerbate the severity of the disease process. In veterinary medicine, exuberant or uncontrolled inflammatory responses occurring in the diseases listed in Box 3-1 can also result in increased severity of disease.
Acute Inflammation
The acute inflammatory response (Fig. 3-2) can be initiated by a variety of exogenous and endogenous substances that injure vascularized tissue. The response to injury begins as active hyperemia, characterized by an increased flow of blood to injured tissue secondary to dilation of arterioles and capillaries (vasodilation), and it is this response that is responsible for redness and heat. It is facilitated by chemical mediators such as prostaglandins, endothelin, and nitric oxide (Box 3-2). With vasodilation, vascular flow is slowed (vascular congestion), allowing time for fluid leakage that occurs as a result of changes in junctional complexes of endothelial cells induced by vasoactive amines, complement components C3a and C5a, bradykinin, leukotrienes, prostaglandins, and platelet-activating factor (PAF), resulting in leakage of plasma and plasma proteins into the extracellular space (swelling and pain [stretching of pain receptors]) mainly from interendothelial cell gaps in the postcapillary venules.
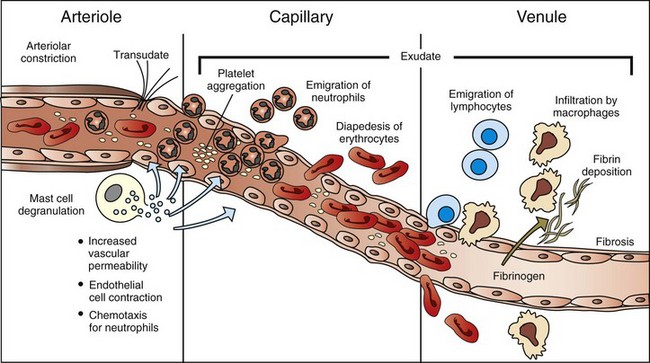
Fig. 3-2 The principal cellular and vascular responses during the inflammatory response.
The majority of leukocyte transmigration and hemorrhage occurs in the capillaries and postcapillary venules. (Modified from McCance KL, Huether SE: Pathophysiology: the biologic basis for diseases in adults and children, ed 3, St Louis, 1998, Mosby.)
The volume and protein concentration of leaked fluid is a function of the size of gaps between endothelial cells and the molecular weight, size, and charge of electrolytes and plasma proteins, such as albumin and fibrinogen. With more severe injury resulting in destruction of individual endothelial cells, hemorrhage, as well as plasma and plasma proteins, can leak directly through a breach in the wall of the capillary or venule. Once activated, endothelial and perivascular cells, such as mast cells, dendritic cells, fibroblasts, and pericytes, can produce cytokines and chemokines that regulate the expression of receptors for inflammatory mediators and adhesion molecules within the lesions.
The plasma proteins and fluid that initially accumulate in the extracellular space in response to injury is classified as a transudate (Fig. 3-3). A transudate is a fluid with minimal protein (specific gravity <1.012 [<3 g of protein/dL]) and cellular elements (<1500 leukocytes/mL) and is essentially an electrolyte solution similar to that of plasma. Most commonly, the formation of a transudate occurs with hypertension, hypoproteinemia, and/or early in the acute inflammatory response. With these conditions, there is increased permeability because of small physiologic gaps between endothelial cells. Hypertension in veins and capillaries can be secondary to arterial hypertension or venous/lymphatic obstruction. Hypoproteinemia is often due to loss of albumin, the major intravascular colloidal protein, and an inability of the liver to rapidly synthesize replacement albumin. The loss of albumin allows intravascular fluid to move toward the extravascular colloids (extravascular proteins). Loss of albumin and other intracellular proteins can occur secondary to renal disease (urinary loss), severe burns, and severe hepatic disease (decreased albumin production). During the early stages of the acute inflammatory response, intercellular gaps form between endothelial cells caused by endothelial cell contraction. The gaps are very small and only allow water and electrolytes to pass through them. With persistent and widening endothelial gap formation or with endothelial cell injury, neutrophils and additional protein can enter injured areas resulting in the formation of an exudate (see Fig. 3-3). An exudate is an opaque and often viscous fluid (specific gravity >1.020) that contains more than 3 g of protein/dL and more than 1500 leukocytes/mL. As discussed in a later section, the morphologic classification of inflammatory responses into categories, such as serous, fibrinous, and/or suppurative, is based on the character of the fluid that leaks from the vessel and of the leukocytes that migrate from the vascular lumen into the extracellular space.
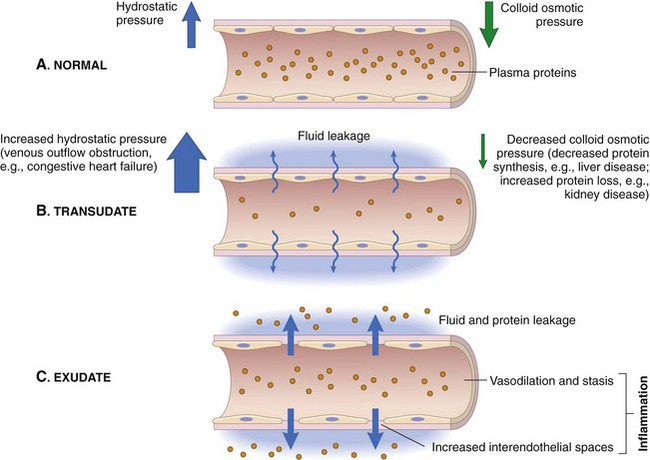
Fig. 3-3 Formation of transudates and exudates.
A, Normal hydrostatic pressure (blue arrows) is about 32 mm Hg at the arterial end of a capillary bed and 12 mm Hg at the venous end; the mean colloid osmotic pressure of tissues is approximately 25 mm Hg (green arrows), which is equal to the mean capillary pressure. Therefore the net flow of fluid across the vascular bed is minimal. B, A transudate forms when fluid leaks out because of increased hydrostatic pressure or decreased osmotic pressure. C, An exudate forms in inflammation because vascular permeability increases as a result of increased interendothelial spaces. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Fibrinogen is an important plasma protein in exudates that polymerizes in extravascular tissues to form fibrin (Fig. 3-4). Plasma dilutes the effects of the inciting stimulus, whereas polymerized fibrin confines the stimulus to an isolated area, thus preventing its movement into adjacent tissue. This confinement provides leukocytes with a well-defined target for migration during the cellular phase of the acute inflammatory response. Neutrophils are the first leukocytes to enter the exudate, and their accumulation in the exudate after they liquefy is termed pus. Neutrophils have a variety of cytoplasmic granules, such as lysosomes, that contain antimicrobial peptides and proteins, as well as matrix metalloproteinases, elastases, and myeloperoxidases. They kill pathogens and degrade foreign material by two mechanisms: (1) phagocytosis and fusion with primary and secondary lysosomes and (2) secretion of the contents of granules into the exudate. Because of the enzymes released, these cells can contribute to tissue injury. Fibrin and its products have additional activities, including chemotactic properties and blood clot formation. Fibrin also forms a framework/scaffold for fibroblast and endothelial cell migration during the initial stages of wound healing.
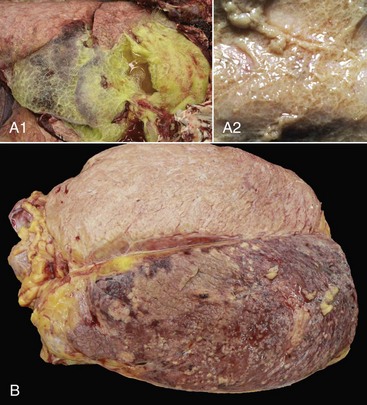
Fig. 3-4 Examples of the appearance of fibrin in acute inflammation in the lung and mammary gland.
A1, Lungs (in situ) from an ox with acute pleuritis. The lungs have a thick mat of fibrin covering the cranioventral region. A small area of fibrin is torn away revealing the subjacent clear, yellow fluid subjacent. The fibrin coating the lung surface was released as fibrinogen from inflamed pleural vessels and polymerized on the serosal surface. The remaining lung surface is less affected. A2, Higher magnification of fibrin on the surface of the lung. B, Acute fibrinonecrotic mastitis, mammary gland, horizontal section, cow. The left quarters of the mammary gland (lower half of the image) are swollen as a result of edema and fibrin exudation and are reddened because of hyperemia, vascular congestion, and hemorrhage. The right quarters (upper half of the image) are unaffected. (A1 and B courtesy Dr. J.S. Haynes, College of Veterinary Medicine, Iowa State University; A2 courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Neutrophils and other leukocytes leave capillaries and venules and migrate into tissue exudates in response to chemoattractant molecules released from host cells, microbes, foreign substances, and some neoplastic cells. As would be expected, the greatest concentration of chemoattractant exists nearest the microbes or foreign substance, and the concentration decreases in a gradient-like manner with increasing distances from the source. This forms a “chemotactic gradient” that essentially creates a pathway for leukocytes to follow to reach the site of tissue injury. Chemoattractants activate receptors and molecules on neutrophils that result in neutrophil (1) movement and attachment to the luminal surface of capillaries and venules, (2) migration through the intercellular junctions formed by gaps between endothelial cells, and (3) migration within the exudate up the concentration gradient to the source of injury. This transmigration process, called the leukocyte adhesion cascade, has a well-characterized sequence of events occurring on the luminal surface of endothelial cells. These events, discussed in detail in a later section, lead to the transmigration of leukocytes into the exudate.
The reparative phase of the acute inflammatory response begins early and is only completed after the process or substance causing injury is removed. In the reparative phase, necrotic cells and tissue are replaced by differentiation and regeneration of parenchymal and mesenchymal stem cells, coupled by filling the defect with connective tissue and covering denuded surfaces with a basement membrane and reepithelialization. When the acute inflammatory response has been completed in the proper sequence and the stimulus of injury removed, the inflammatory process is terminated. Failure to remove the stimulus can result in a persistent, unresolved lesion that becomes chronic and can form into granulation tissue or fibrosis.
Substances Inducing the Acute Inflammatory Response
There are two classes of substances, endogenous and exogenous, capable of injuring cells and tissue and inducing the acute inflammatory response. Endogenous substances include those that primarily cause autoreactive inflammatory responses, such as those induced by newly developed antigens and intracellular molecules released from degenerate, dysplastic, or neoplastic cells, and hypersensitivity reactions. Exogenous substances include microbes, such as viruses, bacteria, protozoa, and metazoan parasites; foreign bodies, such as plant fibers and suture material; mechanical actions, such as traumatic injury; physical actions, such as thermal or freezing injury, ionizing radiation, and microwaves; chemical substances, such as caustic agents, poisons, and venoms; and nutritive actions, such as ischemia and vitamin deficiencies. These substances or actions trigger cells to release mediators that lead to an acute inflammatory reaction and include preformed (in cytoplasmic granules) and synthesized (released from cell immediately after synthesis) chemical mediators from effector cells such as mast cells (histamine and tumor necrosis factor-α [TNF-α), leukocytes (cytokines, degradative enzymes), macrophages (cytokines), endothelial and epithelial cells (chemokines, interferons). These cells also produce inflammatory mediators such as prostaglandins, leukotrienes, and PAF from products released from plasma membranes.
The acute inflammatory response to either endogenous or exogenous substances occurs simultaneously with activation of the innate immune system (see Chapter 5). Innate immunity is a nonspecific defense against potentially harmful environmental substances and consists of the following:
• Physical barriers and microenvironments provided by epithelia of the skin (low pH, lactic and fatty acids) and mucosae such as in the respiratory mucociliary escalator, reproductive tracts (secretions), and alimentary system (gastric and duodenal secretions, peristalsis, saliva)
• Molecular products released by mucosae including lactoferrin, antimicrobial peptides (α- and β-defensins, cathelicidins), and collectins. These products have immune activity but also contribute to proinflammatory and antiinflammatory reactions, leukocyte activation, and wound healing.
• Effector molecules in the blood, such as plasma proteases (complement, kinin, and clotting systems), and inflammatory mediators released from nerve fibers (sensory fibers, C-reactive fibers), such as substance P.
The physical and biologic processes that activate the acute inflammatory and innate immune responses can exert their actions directly on the following:
• Effector cells in mucosae and vascularized connective tissue
The location, severity, and clinical signs of the acute inflammatory response depend on the route of exposure, such as dermal, alimentary, respiratory, urinary, or hematogenous, and the physical or biologic characteristics of the stimulus. More specifically, causes of the acute inflammatory response include but are not limited to the following:
• Visible and ultraviolet light spectra (sunburn and photosensitization)
• Radiation, blunt force trauma (abrasion, bruising, incision, and laceration)
• Thermal injury (hot and cold)
• Microbial molecules (lipids and proteins)
• Venom (insect, snake, and reptile)
• Responses of the adaptive immune system (type I to IV hypersensitivities) to microbial and environmental antigens
These must either penetrate (light spectra) or break/penetrate (microbes and foreign bodies) epithelial barriers of the skin and alimentary, urinary, and respiratory systems to irritate the tissue and incite an acute inflammatory response. Microbes have a few highly conserved ligands called pathogen-associated molecular patterns (PAMPs), and when in contact with mucosae, they immediately encounter cells that express membrane pattern-recognition receptors (PRRs), which include Toll-like receptors (TLRs), expressed on the cell surface or in its cytoplasm. These patterns include the lipopolysaccharide (LPS; Gram-negative bacterial cell wall), lipoteichoic acids (Gram-positive bacterial cell wall), mannose, peptidoglycan, bacterial DNA, N-formylmethionine (in bacterial proteins), double-stranded RNA (dsRNA; viruses), and glucans (fungal cell walls). When these molecular patterns are recognized by receptors, such as TLRs, on macrophages, leukocytes, and mucosal epithelia, they trigger the release of chemokines and cytokines and cellular activation, all of which initiate and/or participate in the acute inflammatory response and the innate immune response.
In contrast to the innate immune system, adaptive immunity results in an antigen-specific immune response characterized by the production of protective antibodies and effector leukocytes that attempt to eliminate the inciting cause of injury and the generation of memory cells that make subsequent adaptive immune responses against a specific microbial antigen more efficient and effective (see Chapter 5). Because acute inflammation is a vasocentric response, it would be reasonable to assume that just about any exogenous or endogenous cause could induce inflammation. This is true, but because the inflammatory response has numerous redundant checks and balances that regulate the occurrence and severity of expression of the response, harmful effects of inflammation are minimized.
The effects of inflammation are mediated through chemical mediators of inflammation, which include the following:
• Vasoactive amines such as histamine and serotonin
• Plasma proteases such as complement, kinin, and clotting system proteins
• Lipid mediators such as arachidonic acid metabolites and platelet-activating factor (PAF)
Mast cells are rich in histamine and many of the chemical mediators listed previously and are widely distributed in connective tissue adjacent to blood vessels. Changes in the permeability of these vessels in the fluidic phase of the acute inflammatory response often occur as the result of mast cell activation. Histamine, preformed in mast cell granules, is released through a process called degranulation. Bradykinin, another vasoactive amine, is produced where there is vascular and/or endothelial cell injury. Both histamine and bradykinin cause changes in the caliber of arterioles, capillaries, and postcapillary venules and permeability in capillaries and postcapillary venules. These occur early in the fluidic phase of the acute inflammatory response and are quickly followed by the cellular phase.
Fluidic (Exudative) Phase of the Acute Inflammatory Response
The principal function of the fluidic phase of the acute inflammatory response is to dilute and localize the inciting agent/substance. In this phase, there is an immediate vasocentric reaction (arterioles, capillaries, and postcapillary venules) to the inciting agent/substance. The sequence of vascular events in the acute inflammatory response includes the following:
• Increased blood flow (active hyperemia) to the site of injury
• Increased permeability of capillaries and postcapillary venules to plasma proteins and leukocytes through release of inflammatory mediators
• Emigration of leukocytes (via the leukocyte adhesion cascade) into the perivascular area
Initially, arterioles dilate and capillary beds in the affected area expand in volume to accommodate an increased blood flow (heat and redness) in response to the stimulus. Second, as a result of permeability changes induced by inflammatory mediators, blood flow through the capillary beds is slowed as a result of increased viscosity and hemoconcentration after leakage of water from capillary beds into extracellular space. Because of the reduced blood flow and pressure, microscopically, capillaries are often packed with erythrocytes and the microenvironment facilitates leukocytic margination along the luminal surface of endothelial cells. This stage precedes leukocyte emigration through intercellular junctions of endothelial cells into the extravascular space. Inflammatory mediators induce endothelial cell contraction, resulting in the formation of interendothelial cell gaps, which further allow fluid leakage and leukocyte migration. In general, the endothelium of the normal vascular capillaries limits exchange of molecules to those less than 69,000 MW, the size of albumin. The exchange of small molecules and water between the vessel lumen and the interstitial space is extremely rapid. For example, the water of plasma is exchanged with the water of the interstitial space 80 times before the plasma can move the entire length of the capillary. Physiologically, increased amounts of fluid can pass across the vascular wall when there is (1) excessive hydrostatic pressure caused by hypertension and/or sodium retention, (2) decreased plasma proteins (colloid), or (3) lymphatic and/or venous obstruction. If fluid leakage is excessive, edema (transudate) develops (see Fig. 3-3). If the leakage is not excessive and postcapillary venules and lymphatic vessels are functioning normally, all of the fluid released from arterioles and small capillaries is returned to the circulation via paracellular gaps of postcapillary venular and lymphatic vessels. During acute inflammatory responses, there is a net outflow of fluid from arterioles, capillaries, and venules into extracellular tissue, which overwhelms the capacity for resorption by postcapillary venules and lymphatic vessels).
Endothelial Cell Dynamics During the Acute Inflammatory Response
Endothelial cells are the interface between plasma in the lumen and the perivascular connective tissue. They are polarized cells that have specific luminal versus abluminal surfaces, which serve the physiologic needs of the vascular bed of each organ. Transport across the endothelial cell layer occurs by (1) transcytosis (transcellular passage) via small vesicles and caveolae or (2) paracellular passage. Transcytosis, the process of transporting substances across the endothelium by uptake into and release from coated vesicles, facilitates the transport of albumin, low-density lipoproteins (LDLs), metalloproteinases, and insulin. Paracellular passage allows transport of water and ions between cells (cell junctions). Paracellular passage is especially active in postcapillary venules. Roughly 30% of endothelial cell junctions in postcapillary venules can open to a width of 6 mm, roughly the width of a red blood cell. Leakage of fluid from the vasculature can occur within seconds after the acute inflammatory response is induced.
The mechanisms of leakage (Fig. 3-5) depend on the biologic and physical characteristics of the inciting agent or substance and include the following:
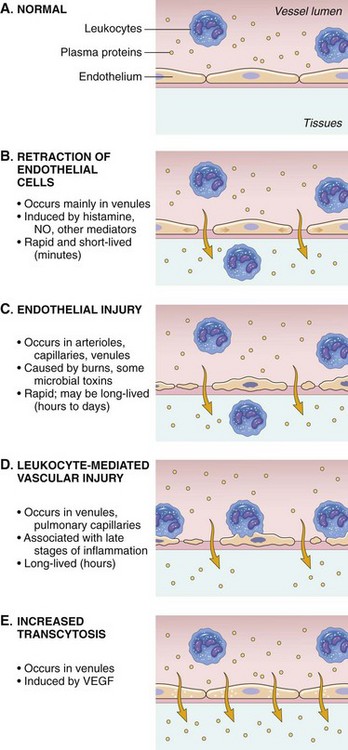
Fig. 3-5 Principal mechanisms of increased vascular permeability in inflammation and their features and underlying causes.
NO, Nitric oxide; VEGF, vascular endothelial growth factor. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
• Opening of junctional complexes (endothelial gaps) between endothelial cells responding to inflammatory mediators
• Direct injury that results in necrosis and detachment of endothelial cells, as occurs with certain viral, protozoal infections, toxins, and radiation
• Leukocyte-dependent injury that results in necrosis and detachment of endothelial cells and is induced by enzymes and mediators released from leukocytes during the transmigration phase of the acute inflammatory response
• Increased endothelial cell transcytosis mediated by vascular endothelial growth factor (VEGF)
Formation of Endothelial Cell Gaps
Endothelial gaps, resulting in vascular leakage, can occur by (1) contraction (actin/myosin) of adjacent endothelial cells and through (2) the reorganization of the cytoskeletal microtubule and microfilament proteins within the endothelial cells. With both of these changes, gaps result from the opening of junctional complexes between endothelial cells. Gaps by cell contraction in postcapillary venules where there is a high density of receptors for histamine, serotonin, bradykinin, and angiotensin II. Gaps formed by cytoskeletal reorganization occur most commonly in postcapillary venules and to a lesser extent, in capillaries in response to cytokines, such as interleukin-1 (IL-1) and TNF and hypoxia. Gap formation is transient and lasts 15 to 30 minutes after the stimulus occurs.
Vascular leakage resulting from direct injury to endothelial cells can cause detachment of the cell from the underlying basement membrane. Such damage establishes conditions favorable for the activation and attachment of platelets, clotting, and complement cascades. This type of extravasation usually occurs immediately after necrotizing injury induced by, for example, thermal injury, chemotherapeutic drugs, radiation, bacterial cytotoxins, and inhaled gases such as hydrogen sulfide. It affects arterioles, capillaries, and postcapillary venules. Vascular leakage resulting from leukocyte-induced damage occurs secondary to neutrophils and other leukocytes interacting with endothelial cells during the leukocyte adhesion cascade. Activated leukocytes release reactive oxygen species, such as singlet oxygen and oxygen free radicals, and proteolytic enzymes, such as matrix metalloproteinases and elastase from lysosomes during degranulation of the cells, which then result in endothelial cell necrosis and detachment and thus an increase in vascular permeability. This type of extravasation usually affects capillaries and postcapillary venules.
Cellular Phase of the Acute Inflammatory Response
The principal function of the cellular phase of the acute inflammatory response is to deliver leukocytes into the exudate at the site of injury so they can internalize agents/substances through phagocytosis and as required, for killing and/or degradation. Neutrophils, eosinophils, basophils, monocytes, mast cells, lymphocytes, natural killer T (NK-T) cells, and dendritic cells play an integral role in protecting mucosa, skin, and other surfaces of the body, as well as the pleura, pericardium, and peritoneum, from infection by microbes through phagocytosis or release of proteolytic degradative enzymes, chemical mediators, and reactive oxygen species. Neutrophils also have an important role in responding to foreign materials and toxins and in responding to neoplastic cells.
Leukocyte Adhesion Cascade
The movement of leukocytes from the lumina of capillaries and postcapillary venules into the interstitial connective tissue occurs through a process called the leukocyte adhesion cascade (Fig. 3-6). Chemokines, cytokines, and other inflammatory mediators influence this process by modulating the surface expression and/or avidity of adhesion molecules on both endothelial cells and leukocytes. It has a well-characterized sequence of events, including margination, rolling, activation and stable adherence (adhesion), and transmigration of leukocytes toward a chemotactic stimulus.
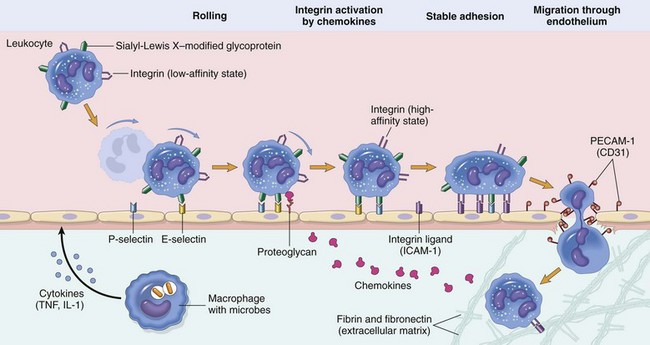
Fig. 3-6 Leukocyte adhesion cascade.
Steps involved with neutrophil migration across the vascular wall include rolling, activation, stable adhesion, and migration across the endothelial layer and vascular wall. Neutrophils express receptors for E- and P-selectin, which bind to ligands expressed on endothelial cells. L-selectin is also expressed by bovine neutrophils and to a varying degree in other species. The rolling process slows neutrophil movement within the vessel and brings the neutrophil closer to the vascular endothelial cell surface. Activation is induced by inflammatory mediators, including chemokines, such as interleukin-8 (IL-8), and cytokines, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), which are released by nearby leukocytes and endothelial cells. These activate both neutrophils and endothelial cells which then changes the conformation of integrins allowing for enhanced binding avidity to integrin receptors and increased expression of additional adhesion molecules. Binding of β2 integrins (Mac-1; CD11a/CD18) expressed by leukocytes to intercellular adhesion molecule-1 (ICAM-1) expressed by endothelial cells leads to stable adherence of the cell to the endothelial cell surface. Once leukocytes are attached to the vascular endothelium, they adhere to platelet-endothelial cell adhesion (PECAM-1) and other junctional adhesion molecules present at the endothelial cell junction and transmigrate through the junction into the perivascular tissue, where they express β1 integrins that adhere to extracellular matrix proteins such as laminin, fibronectin, vitronectin, and collagen. The process is mediated by chemokines (CXCL8; IL-8), complement fragments, vasoactive amines, cytokines, and membrane-derived mediators such as platelet-activating factor (PAF) and leukotrienes. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
• Margination. As vessels vasodilate and have reduced hydrostatic pressure and blood flow, leukocytes exit the central region of the vascular lumen and move to the periphery of the vascular lumen near the endothelial cell surface (marginization).
• Rolling. The initial contact between leukocytes and endothelial cells occurs by transient, weak binding interactions between the selectin family of adhesion molecules and their receptors. During rolling, leukocytes temporarily bind to endothelium and then release, which brings the leukocyte close to endothelial cell surface and reduced velocity of the traveling leukocyte. This process is mediated by selectins, including L-selectin expressed by neutrophils and P-selectin, a carbohydrate binding molecule stored in Weibel-Palade bodies of endothelial cells and α-granules of platelets, as well as E-selectin expressed by endothelial cells. L-selectin is expressed on all leukocytes, but at low concentrations in normal human neutrophils and binds sialyl Lewis X receptor (and other receptors) on endothelial cells. P-selectin molecules expressed on endothelial cell surfaces bind to P selectin glycoprotein ligand 1 (sialyl Lewis X–modified proteins) present on neutrophils, eosinophils, monocytes, and lymphocytes. E-selectin also mediates leukocyte-endothelial cell adherence and is expressed on endothelial cell surfaces for binding glycoprotein receptors expressed on leukocytes. Selectin-mediated attachments are formed at the leading edge of the rolling leukocyte and broken at the trailing edge. Even slight disturbances, such as surgical manipulation, heat, temporary ischemia, and mast cell products, induce rolling of neutrophils along the surface of endothelial cells. By slowing leukocyte transit time through capillaries and postcapillary venules, combined with the continual close proximity of slow rolling leukocytes to the endothelium, and the continued release of chemokines and proinflammatory cytokines, the proper microenvironment for progression to the “stable adhesion” stage occurs.
• Stable adhesion. For stable adhesion to occur, neutrophils and endothelial cells become activated by a variety of cytokines (such as IL-1, interleukin-6 [IL-6], TNF), complement factors (C5a), PAF, platelet-derived growth factor (PDGF), chemokines, and other inflammatory mediators. Once neutrophils are activated, L-selectin molecules are proteolytically cleaved from the neutrophil surface by ADAM17, and the neutrophils express a new set of membrane proteins (integrins) by rapid exocytosis of cytoplasmic vesicles. Firm adhesion of neutrophils to endothelial cells is mediated by binding of β2 integrin molecules, such as Mac-1 (CD11a/CD18), that are expressed on stimulated neutrophils in an active conformation, to intercellular adhesion molecule-1 (ICAM-1) and other ICAM molecules on endothelial cells. P- and E-selectin adherence also contributes to the process of firm adhesion. There are four β2 integrins (lymphocyte function antigen-1 [LFA-1], Mac-1, p150,95, and αdβ2), each of which are heterodimers that differ only in their subunit CD11 a, b, c, and d for LFA-1, Mac-1, p150,95, and αdβ2, respectively. CD18 (the β-subunit) is identical in all four β2 integrins. Three β2 integrins (LFA-1, Mac-1, and p150,95) are involved with leukocyte adherence; however, the β2 integrin αdβ2, which was first identified in dogs and subsequently in humans, is apparently not meaningfully involved with the adherence of neutrophils or other leukocytes to endothelium. Once stable adherence is achieved, neutrophils move to endothelial cell junctions for transendothelial cell migration.
• Transendothelial cell migration. In postcapillary venules, neutrophil movement decreases from 10 µm/sec to a complete stop after margination. Firmly adhered leukocytes emigrate (transmigrate) across the endothelial layer by passing between endothelial cells. A number of leukocyte adhesion molecules are involved in this process (Table 3-1). Adhesion molecule activity and expression differ slightly for different tissues and cell types. In noninflamed skin, for example, there is a higher level of E- and P-selectin expression on endothelial cells, which facilitates rolling of leukocytes. In inflamed liver, CD44 of neutrophils binds serum-derived hyaluronan-associated protein (SHAP) that is bound to hyaluronic acid present on the luminal surface of endothelial cells. Nonactivated lymphocytes and monocytes utilize L-selectin to mediate adherence to high endothelial venules (HEVs) in lymph nodes. These cells also utilize α4β1 (very-late antigen-4 [VLA-4]) to mediate stable adherence to the endothelial ligand, vascular cell-adhesion molecule-1 (VCAM-1). Neutrophils and other leukocytes transmigrate between endothelial cells at the intercellular junctions. Platelet-endothelial cell adhesion molecule-1 (PECAM-1), a molecule that is present on endothelial cell membranes, and junctional adhesion molecules (JAM) A, B, and C mediate adherence activities and the adherence process. Contributions to this process also include β2 integrin binding of ICAM-1 and E-selectin binding. Pseudopodia of neutrophils and other leukocytes extend between endothelial cells and come into contact with and bind to the basement membrane (composed of laminin and collagens) and subjacent extracellular matrix (ECM) proteins (proteoglycans, fibronectin, and vitronectin). This binding interaction is mediated, at least in part, by the β1 integrins. Neutrophils that pass across the vascular wall accumulate in the perivascular connective tissue stroma within the inflammatory exudate. Once within the perivascular stroma, neutrophils migrate along a pathway established by the chemotactic gradients and inflammatory mediators.
TABLE 3-1
Endothelial Cell/Neutrophil Adhesion Molecules
| Endothelial Molecule | Leukocyte Receptor | Major Role |
| P-selectin | Sialyl-Lewis X PSGL-1 |
Rolling (neutrophils, monocytes, lymphocytes) |
| E-selectin | Sialyl-Lewis X ESL-1, PSGL-1 |
Rolling, adhesion to activated endothelium (neutrophils, monocytes, T-lymphocytes) |
| ICAM-1 | CD11/CD18 (integrins) (LFA-1, Mac-1) |
Adhesion, arrest, transmigration (all leukocytes) |
| PECAM-1 | PECAM-1 | Transendothelial cell migration |
| JAM A | JAM A, LFA-1 | Transendothelial cell migration |
| JAM C | JAM B, Mac-1 | Transendothelial cell migration |
ESL-1, E-selectin ligand-1; ICAM-1, intercellular adhesion molecule-1; JAM, junctional adhesion molecule; LFA-1, lymphocyte function antigen-1; Mac-1, macrophage antigen-1; PECAM-1, platelet endothelial cell adhesion molecule-1; PSGL-1, P-selectin glycoprotein ligand-1; VCAM-1, vascular cell adhesion molecule-1; VLA, very late antigen.
From Cotran RS, Kumar V, Collins T, et al: Robbins pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.
This process is actually initiated during the fluidic phase of the acute inflammatory response and is driven by chemokines, cytokines, and chemoattractant substances such as complement. Temporally, margination, rolling, activation and firm adhesion, and transmigration all occur concurrently, involving different leukocytes in the same capillaries and postcapillary venules. This process is largely mediated by the interaction of ligands expressed on the surface of neutrophils, lymphocytes, and macrophages and their receptors expressed on luminal surfaces of activated endothelial cells (see Table 3-1). Adhesion molecules are divided into (1) selectins (E-, L-, and P-selectin), (2) integrins (VLA family of β1 integrins; β2 integrins [Mac-1, LFA-1, p150,95, αdβ2]), (3) cytoadhesin family (vitronectin, β3 integrins, and β7 integrins used predominately by lymphocytes), (4) the immunoglobulin superfamily (ICAM-1 to ICAM-3, VCAM-1, PECAM-1) and mucosal addressin adhesion molecule-1 (MAdCAM-1), and (5) other molecules such as CD44 (Web Table 3-2).
WEB TABLE 3-2
Summary of Various Leukocyte Adhesion Molecules
This table is not comprehensive for cellular expression and binding activity.
DC, Dendritic cells; EC, endothelial cells; ECM, extracellular matrix; ESL-1, E-selectin ligand; GlyCAM-1, glycoprotein cell adhesion molecule-1; HEV, high endothelial venules; L, leukocytes; lympho, lymphocytes; M, monocytes; Mac, macrophages; MAdCAM-1, mucosal addressin cell adhesion molecule-1; PMN, polymorphonuclear cells (neutrophils); PSGL-1, P-selectin glycoprotein ligand-1; SC, Schwann cells; sLex, Sialyl-Lewis X. See text for definition of adhesion molecule abbreviations.
WEB TABLE 3-3
Leukocyte Defects Identified in Animals and Humans
NADPH, Nicotinamide adenine dinucleotide phosphate.
From Cotran RS, Kumar V, Collins T, et al: Robbins pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.
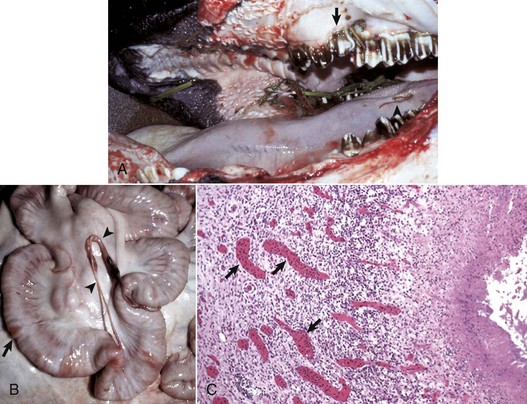
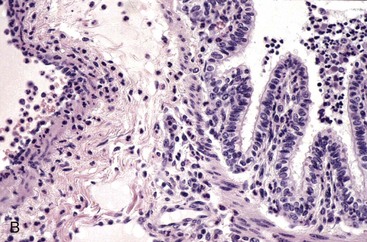
Web Fig. 3-1 Bovine leukocyte adhesion deficiency (BLAD), gross and microscopic characteristics.
A, The oral cavity has irregularly arranged molar teeth (arrow), oral ulcers (arrowhead), and grass material within the oral cavity secondary to impaired mastication. B, The serosa of the small intestine is thickened (arrow), and there are fibrous tags between serosal surfaces (arrowheads). C, Microscopically the mucosa underlying the areas of thickened serosa is ulcerated and covered by cell debris. Vascular lumina contain elevated numbers of neutrophils, which do not adhere to the vascular endothelium (arrows) despite the ulcerated and “inflamed” mucosa. Lymphocytes, macrophages, and plasma cells are present in perivascular areas of Peyer’s patches, but the surrounding lymphoid tissue virtually lacks neutrophils. The lack of neutrophil adherence to the vascular endothelium and of infiltration into the perivascular areas is caused by the lack of expression of β2 integrins in BLAD. The lumen of the intestine is to the right. H&E stain. (Courtesy Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
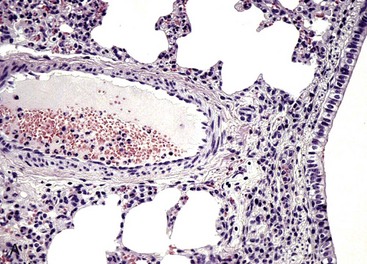
Web Fig. 3-2 Reduced neutrophil infiltration in lung of a calf with bovine leukocyte adhesion deficiency (BLAD).
Both lungs were inoculated with Mannheimia haemolytica and in the animal with normal CD18 expression, neutrophils pass across the vascular wall into the extravascular space and the nearby airway lumen. In contrast, neutrophils in the calf with BLAD are confined to the vascular lumen and do not pass across the vascular wall. Thus the lack of CD18 expression impaired neutrophil stable adherence and migration across the vascular wall, despite the bacterial stimulus. (Courtesy Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
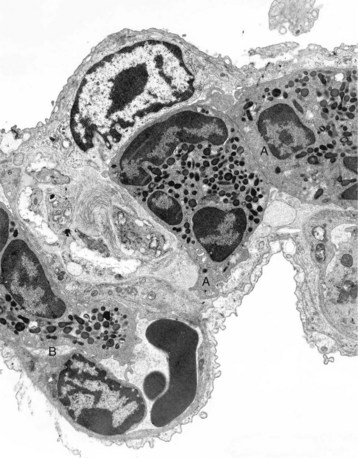
Web Fig. 3-3 Bovine leukocyte adhesion deficiency (BLAD), intravascular neutrophils partially adhered (rolling) on endothelial cells, capillary, alveolar septum, lung.
The septum is thickened because of two neutrophils (A) within the septal wall and a neutrophil (B) within the lumen of a vascular capillary (adherence). Transmission electron microscopy (TEM). Uranyl acetate and lead citrate stain. (Courtesy Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
Leukocyte Adhesion Deficiencies
The three types (I, II, and III) of leukocyte adhesion deficiencies (LADs) are the result of one or more defects in the sequence of steps leading to migration of leukocytes into the site of inflammation. These deficiencies are rare in humans, cattle, and dogs (Web Table 3-3), although debilitation often occurs in humans and animals with severe forms of LADs. These clinical conditions underscore the importance of leukocytes in host defense and the vital role that these adhesion molecules serve in the transmigration process. Cattle, dogs (Irish setters), and humans with LAD type I lack functional expression of the β2 integrins. Affected individuals often have high numbers of neutrophils, exceeding 125,000/µL in the blood; however, in cattle, these neutrophils have impaired passage across vascular walls (Web Fig. 3-1) and numerous other functional abnormalities related to inadequate membrane adherence activity. As a result, lesion sites lack significant neutrophils. Cattle with bovine LAD (BLAD) develop severe oral ulcers, gingivitis, tooth loss, enteric ulcers, cutaneous ulcers, abscesses that lack pus formation, and pneumonia (see Web Fig. 3-1). Most affected cattle die within few days or weeks after birth as the result of diarrhea and/or pneumonia. The hallmark lesion histologically is a sparse infiltration of neutrophils into ulcerated mucosal surfaces or pulmonary alveoli, despite high numbers of neutrophils within lumen of submucosal and pulmonary septal blood vessels (Web Figs. 3-2 and 3-3). There are no effective treatments for calves and Irish setters with LAD, although cattle with BLAD can survive into adulthood with intensive medical care.
The deficiency in β2-integrin expression in Holstein calves is due to a single-point mutation (adenine → guanine) at position 128 resulting in a single amino acid change (aspartic acid → glycine) in the β-subunit (CD18) of the β2 integrins. This defect occurs in a highly conserved extracellular region (amino acids 96 through 389) of the protein. A second, silent mutation has also been detected in cattle with BLAD and does not alter the amino acid sequence. In Irish setters with canine LAD (CLAD), there is a single missense mutation resulting in a G to C transversion at nucleotide 107 of the cDNA sequence, resulting in a serine that replaces a highly conserved cysteine. In 1992, a second form of LAD was discovered in humans and called LAD type II (see Table 3-3). In LAD type II, patients lack fucosylated sialyl Lewis X used for selectin-mediated adherence caused by a mutation in a fucose transporter gene. Their CD18 binding remains intact; however, these patients suffer from the same types of lesions and infections, as do patients with CD18 deficiency. Therefore these observations demonstrate the importance of CD18-independent adherence. More recently, a third type of LAD (LAD type III) has been identified. LAD type III is caused by a defect in kindlin-3, which is a cytoplasmic protein essential for integrin activity in the cytosol. LAD type III patients have defects in leukocyte function and platelet aggregation. Both LAD types II and III have not been identified in animals.
Therapeutic Strategies to Modulate Leukocyte Infiltration
Inhibition of leukocyte infiltration may be useful treating diseases such as stroke, myocardial infarction, asthma, and autoimmune diseases in humans and laminitis, reperfusion injury of intestine after colic, gastric dilation/volvulus, mastitis, enteritis, allergic lung disease, pneumonia, and autoimmune diseases in domestic animals. In animal models, antibodies and small molecule antagonists have diminished the severity of rheumatoid arthritis, type 1 diabetes, asthma, and other conditions. In humans, antibody (efalizumab) to αLβ2 integrin (LFA-1) is effective against psoriasis, whereas antibody (natalizumab) to α4β1 integrin reduces the severity of multiple sclerosis and Crohn’s disease. However, complete inhibition of integrin activity can have side effects. Additional strategies to modulate integrin function therapeutically include inhibition of integrin-associated proteins, such as paxillin, to regulate α4β1 integrin, talin effects on β2 integrins, and inhibition of membrane protein CD98 to regulate β1 and β3 integrins.
Additional Regulation of Inflammation
There are many regulatory mechanisms that closely control the onset, intensity, duration, and resolution of inflammation. In addition to those discussed, additional factors influence the type of inflammatory response by specific individuals. These include mechanisms that affect the expression level and activity of individual inflammatory genes and gene products. First, single nucleotide polymorphisms (SNPs) within the pyrin domain of the inflammasome receptor allow enhanced IL-1β production in patients with familial Mediterranean fever. SNPs in TLR are associated with altered responses to ligands and thus altered inflammatory responses. Similarly, SNP alterations in expression or regulation cytokines, chemokines, and their receptors can change the type and magnitude of inflammatory reactions. Second, premature truncation codons (PTC) are genes with stop codons within a critical location resulting in truncation of the mRNA and protein product. Thus the protein product, whether a cytokine or other inflammatory molecule, has an altered function or may even lack function. Third, gene copy number (the number of copies of a gene in a chromosome) directly affects the amount of protein produced. Some antimicrobial peptides have low gene copy numbers, therefore there is a very limited amount of peptide produced even in the presence of inflammatory stimuli. Fourth, there are latent forms of some molecules such as transforming growth factor-β (TGF-β). The latent forms are present within the tissue stroma and become functional on activation by matrix metalloproteinases. Fifth, there are isoforms of some genes that inhibit activity. VEGF-A, for example, has an isoform that when bound to the VEGF receptor-2 (VEGFR-2) reduces rather than enhances signaling. Sixth, there are soluble receptors such as soluble ICAM-1, which bind ligands but do not transmit signals because the receptor is detached from a cell.
Effector Cells of the Acute Inflammatory Response
Central to the integrity of the vasculature and any type of acute inflammation is the endothelial cell. Once considered a cell that, in the most simplistic view, forms separation between the blood and the surrounding tissue, endothelial cells are now known to have an extremely sophisticated role in regulating (1) hemostasis/coagulation, (2) vascular pressure, (3) angiogenesis during wound healing, (4) carcinogenesis, (5) leukocyte homing, and (6) inflammation. Under physiologic conditions, transcytosis (transcellular passage) of albumin, LDLs, metalloproteinases, and insulin occur via small vesicles and caveolae in the cytoplasm of the endothelial cell. Paracellular passage (between cell junctions) of water and ions occurs with endothelial cell contraction secondary to physiologic stimuli and/or inflammatory mediators. Secondary to inflammatory mediators, endothelial cells are activated and contract, allowing fluid to lead into the extravascular tissue. Vascular tone is held in check in part by endothelin, a vasoconstrictive molecule and angiotensin II, both of which are produced by endothelial cells, along with vasodilatory substances such as NO and prostacyclin (PGI2). Activated endothelial cells release these chemical mediators and express adhesion molecules and receptors, including E-selectin, P-selectin, L-selectin ligand, PECAM-1, JAM A, JAM B, and JAM C, and the immunoglobulin superfamily such as ICAM-1. These adhesion molecules serve as ligands for leukocyte adherence. With the onset of inflammation, endothelial cells tend to increase procoagulative properties through release of tissue factor and other procoagulative substances.
Mast Cells and Basophils
The origin and relationship between mast cells and basophils has been a traditional point of debate and confusion. Current research clearly indicates that mast cells and basophils represent distinct cell types even though they share several morphologic and functional characteristics. Mast cells and basophils, along with other granulocytes and monocytes, originate and differentiate in bone marrow from a common CD34+ precursor cell. Differentiation of CD34+ precursor cells into mast cells or basophils depends on stem cell factor, a glycoprotein that acts with other cytokines and is produced in the bone marrow by fibroblasts and vascular endothelial cells. There is no evidence to suggest that basophils differentiate into tissue mast cells.
Mammalian mast cells are normally distributed throughout connective tissue adjacent to small blood and lymphatic vessels of skin and mucous membranes. In this location, they respond rapidly to foreign proteins, microbes, and other substances and contribute significantly to the initiation of acute inflammation. This location also allows mast cells to interact with resident dendritic cells and release inflammatory mediators that activate endothelial cells. Experimental studies suggest that cutaneous mast cells in tissue have a lifespan of 4 to 12 weeks, depending on their location. Mast cells represent an extremely heterogeneous population of cells. In the 1960s, Enerback identified two separate types of mast cells: mucosal and connective tissue. The mucosal mast cells are typically located in the respiratory and intestinal mucosa and can increase in numbers during some types of T helper 2 (TH2) lymphocyte–dependent immune responses. In contrast, the connective tissue mast cells show little or no T lymphocyte dependence. Mast cells express high affinity receptors for immunoglobulin E (IgE; Fc ε-RI) on their surface, and the release of mast cell granules is stimulated by the cross-linking of IgE receptors by antigens such as pollens, allergens, and parasites. Substance P released from sensory (C-reactive) nerve fibers and macrophages also causes degranulation of mast cells. Degranulation results in the release of preformed TNF-α histamine, neutral proteases, proteoglycans (chondroitin sulfates and heparin), serotonin (in rodent species but not humans), tryptase, chymase, and stem cell factor into tissue. Histamine and substance P activity appears interrelated because histamine released by mast cells can downregulate the release of substance P by nerve fibers, thereby reducing excessive amounts of the two proinflammatory molecules. The mast cell–substance P fiber interrelationship is an often-cited example of the neuroinflammatory-neuroimmune pathway.
Mast cells also synthesize leukotriene (LT) C4 (LTC4), PAF, prostaglandin (PG) D2 (PGD2), numerous cytokines, serotonin (in some species), heparin, and C-C chemokines (macrophage inflammatory protein [MIP]-1-α and macrophage chemotactic protein [MCP]-1). The release of these mediators contributes significantly to the initiation of the acute inflammatory response. In addition, at physiologic concentrations these products likely counteract the effects of dense populations of mast cells in tissue and thus assist in regulating vascular permeability. Mast cells also release proteolytic enzymes, such as tryptase and chymase, which are involved with remodeling of the ECM. Tryptase is mitogenic to epithelial cells and likely contributes to proliferation of epithelial cells during wound repair.
Basophils are similar to neutrophils and eosinophils in that they mature in the bone marrow, circulate in the peripheral blood, are recruited into the tissue, and have a lifespan of several days in tissue. Basophils express high affinity IgE receptors, similar to mast cells, and release granules and inflammatory mediators. Basophils appear to lack heparin, have a more limited cytokine repertoire than mast cells, and release mainly IL-4 and IL-13. Basophils express CD40L and CCR3 (eotaxin receptor). The presence of these suggests that they have a role as a cell that can enter sites of inflammation, release regulatory cytokines where they upregulate VCAM-1 expression by endothelial cells, and switch B lymphocytes to produce IgE, further contributing to the IgE type of response. Basophils can be prominent in IgE-mediated leukocyte infiltration into the mucosa of the nose, sinuses, respiratory tract, and skin, and all of these sites are particularly predisposed to allergic conditions.
The role of mast cells and basophils in IgE-mediated hypersensitivity reactions has been known for decades. These cells are critical effector cells in disorders of IgE-dependent immediate type I hypersensitivities (see Chapter 5). The release of their granules and mediators at inflammatory concentrations in the lung, for example, results in mucus secretion, accumulation of seroproteinaceous fluid in airways, bronchoconstriction, and vasodilation. The excessive release of tryptase and chymase by mast cells may enhance degradation of the ECM, which contributes to fibrosis and tissue remodeling. Chemokines and cytokines from mast cells and basophils contribute to innate immune defenses through chemotaxis and release of antimicrobial peptides. The mediators also enhance adhesion molecule expression on endothelial cells of nearby blood vessels and leukocytes that enter the area.
Neutrophils
Neutrophils are often the first type of leukocyte recruited into the inflammatory exudate. Their purpose is to (1) kill microbes, such as bacteria, fungi, protozoa, and viruses; (2) kill tumor cells; or (3) eliminate foreign materials. The biologic activities of neutrophils are primarily designed to kill microbes through lysosomal degradation, but if killing does not occur, neutrophils can limit the growth of microbes, allowing time for adaptive immunologic responses to develop.
Neutrophils perform two important functions to accomplish their effects: (1) phagocytosis of microbes or foreign material and then fusion of the phagosome with primary lysosomes to form a phagolysosome in which the microbes or foreign material are killed or degraded, respectively (Web Fig. 3-4), and (2) secretion and/or release of the contents of their granules into the inflammatory exudate to enhance the acute inflammatory response. They also infiltrate areas of acute tissue necrosis, such as those that occur in infarcts and necrotic areas of tumors.
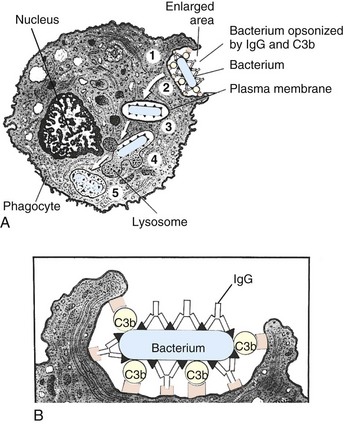
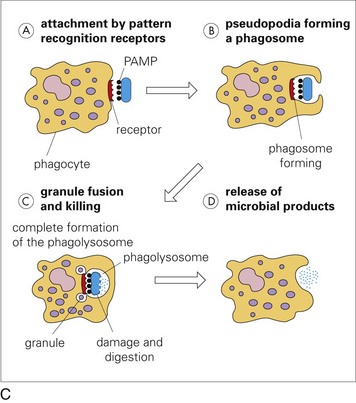
Web Fig. 3-4 Phases of phagocytosis with and without opsonization.
A, Opsonized microbes (1) bind to the surface of a phagocyte and (2) are ingested into a phagocytic vacuole or phagosome (3). Lysosomes fuse with the phagosome (4), releasing their digestive enzymes into the vacuole. This process results in the formation of a phagolysosome (5), within which the microbe is killed and digested. B, Enlargement showing microbe opsonization. IgG, Immunoglobulin G; C3b, complement component. C, Microbial pathogens express pathogen-associated molecular patterns (PAMPs), which bind receptors on neutrophil membranes that can trigger the steps of phagocytosis: attachment, pseudopodia formation, granule fusion and killing, and release of microbial products. (A and B from McCance K, Huether S: Understanding pathophysiology, St Louis, 1996, Mosby. C from Goering R, Dockrell H, Roitt I, et al: Mims’ medical microbiology, ed 4, St Louis, 2008, Mosby.)
Neutrophils are produced in the bone marrow, circulate in the bloodstream, and if not recruited into tissue by an acute inflammatory response, can enter tissue where they eventually are destroyed by macrophages via apoptosis and phagocytosis or are lost from the body by migration across mucosae such as the alimentary and respiratory tracts. The average transit time in the blood is 10 hours, and the half-life in the blood varies between species but ranges from 5 to 10 hours; neutrophils within tissue survive from 1 to 4 days. Cytokines, such as IL-1 and TNF, and growth factors, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), and IL-3 can increase release of neutrophils from the bone marrow and induce granulopoiesis in 2 to 4 days. GM-CSF and G-CSF are also present in tissue during acute inflammatory response and prevent apoptosis of tissue neutrophils. Within areas of intense inflammation, neutrophils and other leukocytes must function under hypoxic conditions and do so through the stabilization of hypoxia-inducible factor-1α (HIF-1α). HIF-1α induces transcription of genes that promote phagocytosis, inhibition of apoptosis, release of antimicrobial peptides, granule proteases, VEGF, cytokine release, and inducible NO synthetase (iNOS). Growth factor withdrawal, which occurs during resolution of acute inflammation, induces apoptosis, and this outcome can be accelerated by TNF. During apoptosis, neutrophils lose the capacity to degranulate and become activated, which prevents release of their lysosomal enzymes and thus excessive tissue damage and allows for their phagocytosis by macrophages.
Neutrophils entering tissue become activated, which enhances migration, phagocytosis, and microbial killing, through stimulation by inflammatory mediators and by adherence to ligands by surface adhesion molecules. Inflammatory mediators bind receptors on neutrophils, such as receptors for PAF, C5a, IL-8 and substance P (the neurokinin-1 receptor), leukotrienes, kallikrein, GM-CSF, and cytokines such as TNF. Many of these mediators induce chemotaxis; when leukocyte adhesion molecules, such as the selectins and integrins, bind to their respective ligands, they induce mitogen-activated protein kinase (MAPK) and G proteins, resulting in migration of neutrophils, usually toward a chemotactic gradient and activation.
Neutrophils can internalize large particles up to 0.5 µm in diameter by phagocytosis, including microbes, foreign bodies, senescent cells, and debris. Although neutrophils can internalize nonopsonized particles, opsonization greatly facilitates phagocytosis. The principal opsonin receptors present on neutrophil membranes are complement (CR1 and CR3) and Fc receptors (Fc γ-receptor I, IIA, IIIB), which bind complement fragments (C3b and C3bi) and the Fc portion of immunoglobulins such as IgG1 and IgG3. Such binding initiates activity of the guanosine triphosphatase (GTPase) Rac1 and the β2 integrin Mac-1 (CD11b/CD18), which also binds complement fragment C3bi and initiates GTPase-ρ. Such binding-inducing proteins and lipid kinases (e.g., protein kinase C and phosphatidylinositol 3-kinase) mediate actin assembly for formation of filopodia or lamellipodia, which surround and then internalize particles via phagocytosis by activated neutrophils. The activation process also leads to the release of calcium stored in the endoplasmic reticulum, which induces a respiratory (oxidative) burst (Table 3-2). Oxidative burst is the process by which nicotinamide adenine dinucleotide phosphate (NADPH) oxidase composed of five phox protein subunits in the membrane of phagosomes is formed. It catalyzes the formation of superoxide free radical that is used to kill microbes or degrade internalized material. Superoxide can react to form hydrogen peroxide, and additional free radicals such as hydroxyl radical, and hypochlorous acid. Neutrophils also express iNOS, which generates NO, and myeloperoxidase, which also produces hypochlorous acid. Superoxide anion and NO can form peroxynitrite, which is highly reactive.
TABLE 3-2
Antimicrobial Mechanisms in Phagocytic Vacuoles

Microbicidal species in bold letters. Fe/RSH, a complex of iron with a general sulfhydryl molecule; Fe(RS)2, oxidized Fe/RSH; O2−, superoxide anion; 1O2, singlet (activated) oxygen; ·OH, hydroxyl free radical; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NADP+, oxidized NADPH; H2O2, hydrogen peroxide; OCl−, hypochlorite anion; NO; nitric oxide; ·ONOO−, peroxynitrite radical.
From Goering R, Dockrell H, Roitt I, et al: Mims’ medical microbiology, ed 4, St Louis, 2008, Mosby.
Once a particle is internalized, phagosomes can “mature” by fusing with lysosomes and endosomes or remove parts of internalized particles. The fusion process is likely mediated by calmodulin, a calcium-binding protein, and soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptors (SNARE; a fusion protein) that bind ligands on another vesicle to bring the membranes together for fusion. The maturation process results in lowering of the pH within the phagosome and the activation of microbicidal enzymes, including NADPH oxidase and myeloperoxidase complexes. Smaller particles are internalized by receptor-mediator endocytosis.
The ability of neutrophils to kill microbes or to degrade foreign material depends largely on the contents of the neutrophil granules, which store degradative enzymes, peroxidative enzymes, adhesion molecules, and antimicrobial peptides and/or proteins (Web Box 3-1). Myeloperoxidase is an enzyme used to convert hydrogen peroxide to hypochlorous acid. Hypochlorous acid, hydrogen peroxide, and a halide cofactor (chloride) form the myeloperoxidase system, which is an effective microbicidal mechanism used by neutrophils to kill internalized microbes and degrade internalized substances. Defensins, cathelicidins, and antimicrobial proteins contribute to the degradation of microbes by forming pores in microbial membranes. They also affect chemotaxis and activation of the adaptive immune response. Lactoferrin inhibits the growth of phagocytosed bacteria by sequestering free iron, and elastase hydrolyzes bacterial cell wall proteins and tissue elastin. The enzymatic contents of granules, such as gelatinase (matrix metalloproteinase-9 [MMP-9]) and myeloperoxidase, and nonenzymatic substances, such as antimicrobial peptides and lactoferrin, are also commonly released by the cell into the extracellular space and contribute to killing of extracellular microbial pathogens and with degradation of the ECM. The effects of extracellular neutrophil proteases, if not inactivated, can cause serious injury to tissue; therefore protease inhibitors are present in plasma and are present in inflammatory lesions after vascular leakage.
Neutrophil granule formation begins during myeloid cell differentiation in the bone marrow (see Web Box 3-1). Granules are observed initially in myeloblasts and promyelocytes when immature transport vesicles bud from Golgi and fuse to form primary granules. Primary granules are also called azurophilic granules because of their affinity for the dye azure A. These granules contain myeloperoxidase, elastase, defensins, and small amounts of lysozyme. Myelocytes and metamyelocytes form secondary (specific) granules that contain defensins, lactoferrin, lysozyme, and lesser amounts of myeloperoxidase, CD11b/CD18, and elastase. Band cells, the penultimate stage of neutrophil development, form tertiary (gelatinase) granules that contain lysozyme, gelatinase (MMP-9), cysteine-rich secretory protein-3 (CRISP-3), and adhesion molecules CD11b/CD18 (Mac-1) but have smaller amounts of myeloperoxidase, lactoferrin, proteinase 3, elastase, and defensins. Band and mature neutrophils also have secretory vesicles that contain plasma proteins, alkaline phosphatase, and numerous CD antigens, including CD11b/CD18 adhesion molecules. The secretory vesicles are mobilized quickly after neutrophil activation resulting in rapid expression of adhesion molecules, which mediate leukocyte infiltration.
Neutrophil granules have evolved phylogenetically and are specially adapted for each species. In most mammals, enzymes released into an exudate from neutrophil granules cause liquefaction of the exudate and the process results in the formation of pus. Reptiles and birds either lack or have reduced concentrations of these enzymes, particularly myeloperoxidase, and cannot liquefy the exudate. Thus a caseous material forms to be degraded by the next available line of inflammatory cells, macrophages. Granules in chicken heterophils (the avian, rabbit, and guinea pig neutrophil equivalent is termed heterophils) have little myeloperoxidase, but concentrations are also reduced in neutrophils of cattle and pigs. Cattle and sheep neutrophils have limited lysozyme levels. α-Defensins are present in rabbits, guinea pigs, hamsters, rats, and cattle neutrophils but have not been identified in those of dogs, cats, mice, pigs, and horses. The effect of these granule differences in various animal species on host defense and neutrophil function is not fully understood.
On cell death, neutrophils can release neutrophil extracellular traps (NETs) composed of a DNA backbone embedded with antimicrobial peptides and proteins and peptides that include histones, primary granule contents, lactoferrin, gelatinase, cathelicidins, and α-defensins. NETs entrap bacteria and can be microbicidal. Actin is also released from dead neutrophils, and in the lung, it can increase the viscosity of respiratory mucus. This outcome may occlude airways in dehydrated animals.
Eosinophils
Eosinophils are recruited from the bloodstream into vascularized connective tissue of most organs in response to eosinophil chemoattractants present in allergic and parasitic diseases. Eosinophils frequently enter lesions during the transition from acute to chronic inflammation. Eosinophils have prominent granules that release basic proteins and when activated produce cytokines, chemokines, proteases, and oxidative radicals. This array of mediators is often released in response to helminthic infections, and eosinophilic infiltration has been more recently implicated in resistance to the development of some cancers. On the other hand, eosinophil products contribute to tissue damage in several organs, including the lungs (asthma), heart, skin, and gastrointestinal tract.
Eosinophils were first recognized as blood cells (leukocytes) having numerous cytoplasmic granules with affinity for acidic dyes such as eosin. Therefore the name eosinophil (“eosin-loving”) was proposed by Ehrlich in the late 1800s for these unique cells. By 1939, eosinophils were postulated to have a role in the immune response to helminths, and by the 1970s, eosinophils were well known to increase in the blood (eosinophilia) in parasitic and allergic diseases. Eosinophils are slightly larger than neutrophils. The nucleus is lobulated (bilobed) and composed primarily of heterochromatin (condensed). Eosinophil granules are known for their large size, especially in horses, and are rich in arginine with reddish brown tinctorial properties.
Eosinophils have several types of granules listed in Web Table 3-4, including large specific granules, small granules, primary granules, and secondary granules. Large specific granules contain four distinct basic proteins: (1) major basic protein, (2) eosinophil cationic protein, (3) eosinophil-derived neurotoxin, and (4) eosinophil peroxidase. These proteins exert biologic effects on microbes and on the tissue in which the microbes replicate by damaging lipid membranes. In addition, histaminase and a variety of hydrolytic lysosomal enzymes, such as collagenase and gelatinase, are also present in the large specific granules. Small granules contain enzymes such as arylsulphatases, acid phosphatases, MMPs, and gelatinases. Eosinophils also elaborate cytokines such as IL-1 to IL-6, IL-8, IL-10, IL-12, IL-16, GM-CSF, TGF-α and TGF-β, and chemokines. The content of eosinophil granules are released in response to inflammatory stimuli in a manner similar to those used to activate neutrophils. However, products of eosinophil granules can result in extensive tissue degradation, including the degradation of collagen, which is commonly seen in eosinophilic granulomas of cats, horses, and dogs. Nearly all mast cell tumors in dogs and some mast cell tumors in cats contain eosinophils.
Major chemoattractants for eosinophils include histamine and eosinophilic chemotactic factor A (from mast cells), C5a, cytokines (IL-4, IL-5, and IL-13), and chemokines (CCL-5, known as regulated on activation, normal T lymphocytes expressed and secreted [RANTES], and CCL-11 [known as eotaxin]) released from epithelial cells, eosinophils, mast cells, and helminths. 5-Oxo-6, 8, 11, 14-eicosatetraenoic acid (5-oxo-ETE) is a strong activator of human eosinophils with a chemotactic potency comparable with those of eotaxin and RANTES, both of which enhance 5-oxo-ETE–induced chemotaxis. 5-Oxo-ETE and these chemokines contribute to the accumulation of eosinophils in the respiratory system in diseases such as asthma.
Natural Killer Cells and Natural Killer T lymphocytes
Natural killer (NK) cells are sentinels of the immune system named for lysis of tumor cells and virus-infected cells without previous encounter. These cells enter regions of acute inflammation hours and even days after initiation of the lesion. NK cells kill target cells through release of perforin from cytoplasmic granules. NK cells express CD161, a C-type lectin, but do not express CD3, the T lymphocyte antigen. Roughly 95% of NK cells expression CD56 and produce interferon-γ (IFN-γ); these are type I NK cells. Type II NK cells lack CD56 expression and produce IL-4, IL-5, and IL-13, thus supporting a TH2 response.
IL-21 regulates differentiation and apoptotic death induced by NK cells. Inactive NK cells can be stimulated by Flt-3 ligand, a hematopoietic cytokine that stimulates proliferation of dendritic cells and antitumor immune responses, and also by IL-4, IL-12, IL-15, and IL-21. Once activated, IL-21 induces NK cell differentiation and upregulation of CD16, the low affinity IgG receptor necessary for antibody-dependent cellular cytotoxicity (ADCC), and also NK release of IFN-γ required for macrophage and dendritic cell activation. Finally, IL-21 initiates a delayed apoptotic program for death of the differentiated NK cell and prevents recruitment of uninvolved NK cells. NK-T lymphocytes are T lymphocytes (express CD3 antigen) that have T and NK cell properties. NK-T lymphocytes recognize CD1d molecule, which is an antigen-presenting molecule that binds self and foreign lipids and glycolipids, and on activation of the NK-T lymphocyte, induce release of IFN-γ, IL-4, and GM-CSF. Because of this close discretion of self and nonself, NK-T lymphocytes can have important roles in the development of autoimmune disease.
Monocytes and Macrophages
Macrophages arise from bone marrow–derived monocytes, which circulate hematogenously with some monocytes localizing in tissues physiologically. They enter acute inflammatory lesions roughly 12 to 48 hours after the initiation of a lesion, depending on the inciting agent/substance. Differentiation of monocyte stem cells into blood monocytes proceeds rapidly in the bone marrow (i.e., 1.5 to 3 days) and is regulated by growth and differentiating factors, cytokines, and adhesion molecules such as IL-3, CSFs, and TNF. Under physiologic conditions, monocytes in the blood localize throughout the body and differentiate into tissue macrophages. Recently, nonclassical monocytes have been identified; they migrate slowly along the luminal side of endothelium and monitor healthy tissue. When these monocytes sense damage or infection, they migrate rapidly into the tissue.
There are two types of tissue macrophages: macrophages that reside within specific organs/tissue (free macrophages and fixed macrophages) and macrophages derived from monocytes in response to inflammatory stimuli. Macrophages residing in organs/connective tissue first enter these sites as blood monocytes under physiologic (rather than inflammatory) conditions. These macrophages form the monocyte-macrophage system and include macrophages in connective tissue (histiocytes [free macrophages]), liver (Kupffer cells [fixed macrophages]), lung (alveolar macrophages [free macrophages], and intravascular macrophages [fixed macrophages]), lymph nodes (free and fixed macrophages), spleen (free and fixed macrophages), bone marrow (fixed macrophages), serous fluids (pleural and peritoneal macrophages [free macrophages]), brain (microglial cells), and skin (histiocytes [fixed macrophages]). The number of macrophages in tissue is maintained by (1) influx of monocytes from the blood, (2) proliferation of recruited monocytes locally in tissue, and (3) biologic turnover of macrophages via apoptotic cell death (lifespan in tissue of less than 3 weeks).
During inflammatory responses, monocytes express receptors (IgG Fc-domains, C3b) for chemical mediators of inflammation that exert migratory, chemotactic, pinocytic, and phagocytic activities in response to inflammatory stimuli. Once within inflammatory lesions, monocyte receptors are bound by cytokines, antigens, and other stimuli, which rapidly activate the maturation of monocytes into macrophages. This process can occur virtually anywhere in the body and often sets the stage for the development of chronic inflammation. In chronic inflammatory lesions, macrophages are the cell of last resort and accumulate in sites of persistent antigen, persistent microbes, foreign material, or repeated injury. Functionally, macrophages are a component of the innate immune system in terms of their role in phagocytosis and cytokine release during the acute inflammatory response. However, macrophages are one of the main triggers of the adaptive immune response because of their ability to process and present antigen and regulate T lymphocyte activity.
Chemical Mediators of the Acute Inflammatory Response
Chemical mediators of the acute inflammatory response include molecules such as histamine, serotonin, bradykinin, and tachykinins (Web Fig. 3-5; Web Appendix 3-2). Many are produced as preformed or synthesized molecules in the liver and in neutrophils, basophils, macrophages/monocytes, platelets, mast cells, endothelial cells, smooth muscle cells, fibroblasts, and most epithelial cells. Preformed molecules, such as histamine, are transcribed, translated, processed, and stored, often in granules or vacuoles within inflammatory cells. They can be released immediately on cellular activation and are therefore active in seconds. Other molecules, such as most cytokines, adhesion molecules, and prostaglandins, are largely synthesized after an inflammatory cell becomes activated or injured. Endothelial cells, for example, often express low, basal levels of the adhesion molecule ICAM-1, but after the cells become activated (by cytokines such as IL-1), they rapidly transcribe the ICAM-1 gene to generate ICAM-1 messenger RNA (mRNA), which is translated into ICAM-1 protein, that is, processed, transported, and expressed on the cell surface. This is a quick process, resulting in ICAM-1 expression within hours; however, it is not nearly as rapid as the release of histamine, which occurs in seconds. Inflammatory mediators originating from plasma proteins, such as kinin, and the coagulation and complement system proteins are constantly secreted by the liver in precursor forms that must be activated via proteolytic cleavage in the circulatory system to their active forms; however, once the proteolytic cleavage is initiated, kinin and complement activity is immediate, similar to histamine.
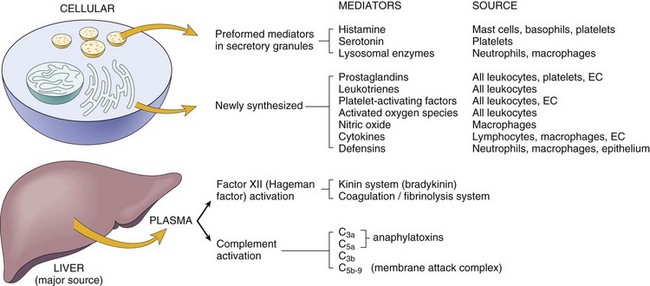
Web Fig. 3-5 Chemical mediators of inflammation.
EC, Endothelial cells. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Inflammatory mediators, whether preformed, synthesized, or derived from plasma, generally bind to receptors on target cells and often activate target cells or cause the target cell to secrete additional inflammatory mediators. In the latter case, the mediators may amplify or suppress secretion by target cells of additional mediators. Once activated and released or secreted, most inflammatory mediators:
• Have short half-lives and decay rapidly
• Are scavenged by protective mechanisms such as antioxidants
• Are blocked by endogenous inhibitors such as complement inhibitors and decoy receptors
This arrangement provides a check and balance system on the severity of the acute inflammatory response and also can be exploited in the development of drugs to inhibit excessive inflammatory responses. Inflammatory mediators, if excessively unregulated, have the potential to cause severe injury to tissue in and surrounding the acute inflammatory response.
In addition to histamine, other preformed inflammatory proteins include serotonin, bradykinin, and tachykinins (substance P and neurokinins). Mast cells and basophils are the principal sources of histamine and serotonin. Bradykinin is released by leukocytes and vascular endothelial cells, and substance P is released by mast cells, basophils, and C-reactive (sensory) nerve fibers. As indicated, the mediators are rapidly active (in seconds to minutes) and contribute to increased vascular permeability that lasts from minutes to hours.
Histamine rapidly enhances vascular permeability and is one of the earliest recognized mediators of inflammation. Experiments by Sir Thomas Lewis in 1927 and Dale and Laidlaw in 1911 indicated the potential role of histamine and other local mediators in acute inflammation. Histamine is derived from the amino acid histidine through the action of histidine decarboxylase. This enzyme catalyzes the decarboxylation of histidine to histamine and carbon dioxide. Histamine is stored in granules of mast cells, basophils, and platelets.
As a mediator of inflammation, major effects of histamine are (1) vasodilation (active hyperemia), (2) increased microvascular, (3) neural reflexes, vagal reflexes, bronchial constriction, (4) release of PGF2α, (5) pain and itching, (6) tachycardia, and (7) eosinophil chemotaxis (Web Table 3-5). The acute vascular effects of histamine are immediate (within minutes) and transient (last about 30 to 90 minutes). Whether histamine has a role in chronic inflammation is speculative, but it may act to modulate the inflammatory response and the reactivity of various leukocytes, including lymphocytes. There are four G protein–coupled receptors (GPCR) for histamine. Two of these, H1 and H4, are present on leukocytes, whereas H2 and H3 are present on gastric mucosa and nerve terminals, respectively.
WEB TABLE 3-5
Distribution and Function of Histamine Receptors
| Histamine Receptor Type | Cellular Distribution | Major Function |
| H1 | Leukocytes, endothelial cells, smooth muscle cells | Acute inflammatory/allergic reactions |
| H2 | Gastric mucosa, endothelial cells, neurons | Gastric acid secretion |
| H3 | Histamine-containing nerve terminals | Neurotransmitter modulation |
| H4 | Mast cells, dendritic cells, eosinophils, monocytes, basophils, T lymphocytes | Immunomodulator |
The release of histamine from mast cells is in response to a variety of stimuli including IgE, C3a, C5a, heat, cold, substance P, adenosine triphosphate (ATP), and products from leukocytes, endothelial cells, and platelets. Free histamine reacts within minutes with H1 receptors on venular endothelium to cause contraction and gap formation (cytoskeletal reorganization [actinomycin filaments]), resulting in increased vascular permeability. Histamine H1-receptor activation may lead to the production of cytokines and antibodies by T lymphocytes and B lymphocytes, respectively. In addition, H1 receptors are also found on a variety of blood leukocytes, such as T lymphocytes, B lymphocytes, and monocytes.
There is substantial overlap of activities by these receptors on many types of cells. Many of the actions of histamine can be mimicked by H1- and H2-receptor agonists (a molecule or drug that binds to a receptor and triggers a response by the cell), whereas these actions can be blocked by H1 and H2 antagonists, molecules or drugs that block a receptor and prevent a response by the cell. This latter effect is the basis for therapies used in veterinary medicine today. Histamine receptors, for example, are involved in the pathogenesis of allergies, and H1-receptor antagonists (antihistamines) reduce symptoms associated with allergic rhinitis such as sneezing, pruritus, and rhinorrhea (runny nose). In allergic bronchiolitis of cats and horses, H1-receptor activation results in increased vascular permeability leading to serous inflammation in the bronchi and bronchioles. If this response can be blocked, the effects of allergens can be minimized in affected patients. Eosinophils produce histaminases that degrade histamine.
Serotonin (5-hydroxytryptamine) is an important preformed vasoactive amine with actions similar to those described previously for histamine. Serotonin is also an important neurotransmitter. Serotonin is found in mast cell granules of rodents and platelets of mammals. Serotonin and histamine are released from platelets after they are activated by the following:
• Aggregation and after contact with collagen in an exposed basement membrane from areas of endothelial necrosis and detached cells
• Thrombin from activation of the coagulation cascade
• Adenosine diphosphate (ADP) released from injured endothelial cells
• Immune complex activation of the complement cascade (C3a, C5a)
Kinins, such as the tachykinins and bradykinin, are chemical mediators of the acute inflammatory response and also act by modulating the responses of the clotting and complement cascades. Kinin system activation leads ultimately to the formation of bradykinin. Bradykinin, a prototype kinin and a vasoactive peptide, has proinflammatory (makes the disease worse) properties that result in the following:
• Increased vascular permeability
• Increased sensitivity to pain
• Increased arachidonic acid metabolism (stimulation of phospholipase A2)
Kinins are formed by two distinct pathways: the plasma kinin pathway and the tissue kinin pathway. The plasma kinin pathway is activated by contact of a protein complex formed by high molecular weight kininogen (HMWK), factor XI, and prekallikrein with negatively charged surfaces, such as exposed basement membrane. When factor XII (Hageman factor [HF]) binds to this surface and interacts with the bound protein complex, there is reciprocal activation/generation of activated HF (HFa) and kallikrein (contact activation system). Kallikrein then acts on HMWK to yield bradykinin, an oligopeptide containing nine amino acid residues.
The tissue kinin pathway is generated by the action of tissue kallikrein on a low molecular weight kininogen (LMWK) to produce lysyl bradykinin and finally bradykinin. Tissue kallikrein is chemically and antigenically distinct from plasma kallikrein, although it is capable of acting on either HMWK or LMWK to generate bradykinin. Bradykinin binds to two GPCRs, B1R in inflamed tissue and B2R in normal tissues. Control of the proinflammatory effects of kinins is through rapid inactivation of bradykinin and kallikrein. Bradykinin is broken down by aminopeptidase M, neutral endopeptidase, carboxypeptidase (kininase I) and angiotensin-converting enzyme (kininase II). Plasma kallikrein is inhibited by C1-INH esterase (serum α2-macroglobulin), a member of the serpin family of proteases. This family of proteases forms approximately 20% of the proteins in blood plasma and includes α1-antichymotrypsin, α1-antitrypsin, and antithrombin III. They act to block proteolytic activity in the clotting and complement systems and thus serve as a regulatory check on these systems.
Tachykinins are a family of vasoactive neuropeptides that includes substance P, neurokinin A and B, neuropeptide Y, and hemokinin-1. Substance P and NK A and B synthesized by sensory afferent nerve fibers of the lungs and alimentary system. These substances are involved in allergic reactions and asthma. Substance P can induce vasoconstriction, vasodilation, increased permeability changes leading to edema, leukocyte activation, and chemotaxis. Substance P also induces activation and degranulation of mast cells, basophils, and eosinophils and their release of histamine and other inflammatory mediators. The released histamine, in a feedback mechanism, binds the H3 receptors of nerve fibers and partially inhibits the production of substance P, thus regulating the level of activity. One of the main receptors for substance P, neurokinin-1 receptor (NK-1R), is expressed on a variety of cells including mast cells, epithelial cells, endothelial cells, and macrophages. NK-1R is regulated by substance P expression. Frequently, increased levels of substance P result in decreased NK-1R expression. Neurokinin 2 and 3 receptors have a lower affinity for substance P than NK-1R.
The release of substance P from afferent sensory nerve fibers in skin and mucous membranes can also be induced by capsaicinoids such as capsaicin and dihydrocapsaicin. Capsaicinoids are natural compounds present in chile peppers of the genus Capsicum, which cause the burning sensation in commercial pepper sprays (less-than-lethal, self-defense weapons). Capsaicin binds the vanilloid receptor-1 of afferent sensory fibers, leading to release of substance P from these fibers. Thus the tachykinins induce inflammatory responses when released by activated and degranulated mast cells, basophils, and eosinophils and also from stimulated nerve fibers.
Complement Cascade
The complement cascade is a unique sequence of molecular events occurring within the vascular system in which inactive complement plasma proteins synthesized by the liver are activated after tissue injury (Fig. 3-7; Web Fig. 3-6), inflammation, clotting, or immune responses. This cascade results in the generation of numerous biologically active molecules, which have effects that are proinflammatory, chemotactic, opsonizing, antigen solubilizing, antibody inducing, permeability enhancing, and microbicidal (cell lysis) and usually beneficial to the animal (Table 3-3). A large number of plasma proteins make up the complement system, and nearly 10% of serum proteins are complement factors. Divided by the “classical,” “alternative,” and lectin pathways, activation or “fixation” of complement proteins eventually results in formation of a membrane attack complex (MAC) that perforates the cell membranes of foreign invaders and naïve host cells alike. In the generation of MAC, a variety of complement components are elaborated that have important inflammatory and immune effects.
TABLE 3-3
The Main Functions of Complement
Adapted from Mackay IR, Rosen FS: N Engl J Med 344:1058, 2001.
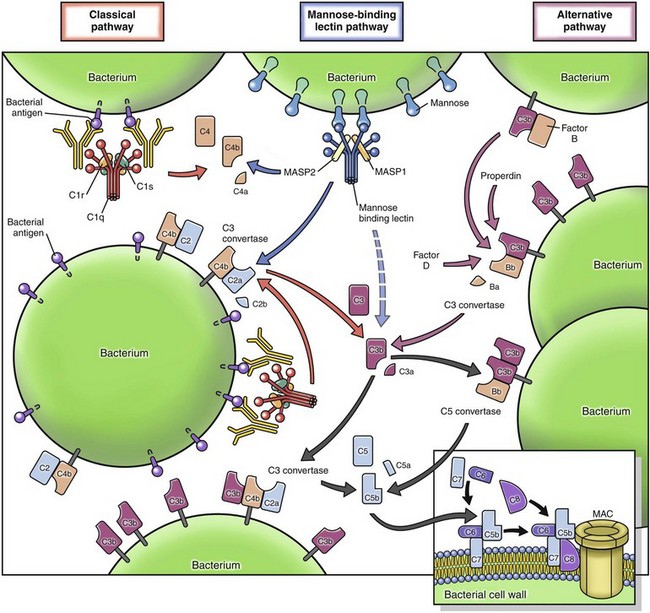
Fig. 3-7 Activation of complement cascade.
Complement is activated by classic, mannose-binding lectin and alternative pathways. The classical pathway is triggered by antibody opsonization, the mannose-binding pathway is induced by binding of the mannose-binding lectin to mannose residue on the surface of microbes, while the alternative pathway is first initiated by binding of C3b to hydroxyl group residues of carbohydrates and proteins and subsequent Factor D cleavage of C3b in plasma. After initiation by these three pathways, the complement cascade continues with formation of C5a and C3a which induce inflammation through attraction of leukocytes, C3b which opsonizes pathogens and induces phagocytosis, and formation of the membrane attack complex (MAC) that creates a pore in the microbial surface. (Redrawn from MJ Walport: Complement, N Engl J Med 344:1058-1066, 2001.)
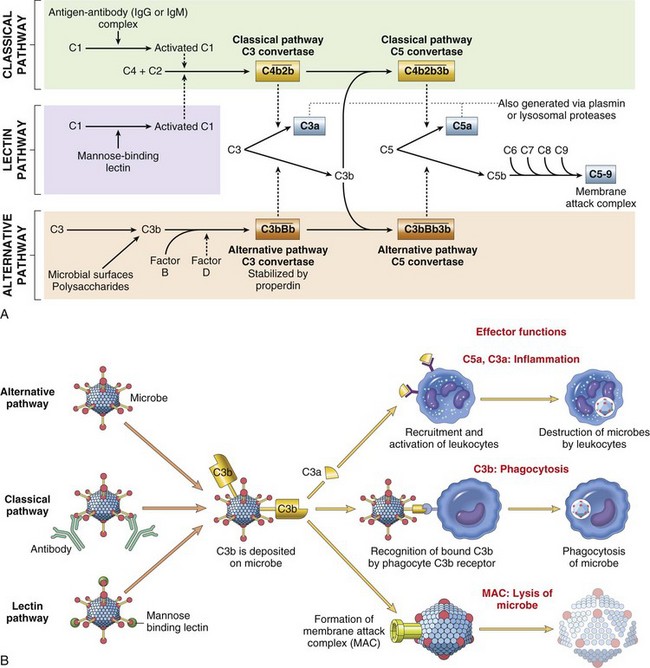
Web Fig. 3-6 Complement cascade.
A, Components and pathways of the complement cascade. B, Effector functions of the complement system. IgG, Immunoglobulin G; IgM, immunoglobulin M. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Complement proteins C1 through C9 are inactive components of plasma that are activated by substances, including microbial molecules such as endotoxins, aggregated immunoglobulin, complex polysaccharides, and venoms. The critical step in unleashing the biologic functions of the complement cascade in the classical pathway is activation of C3 through either the classical or alternative complement activation pathways. The classical pathway of the complement cascade can be activated by antibody complexes. Activation occurs when IgG and/or IgM are cross-linked with C1. C1 has three components: C1q, r, and s. C1q binds the Fc regions of IgG and/or IgM and brings C1r, which is proteolytic, into proximity to C1s, which is cleaved, through interactions with C4 and C2. This leads to the formation of classical pathway C3 convertase (C4b2a) and the eventual formation of classical pathway C5 convertase (C4b2a3b). Classical pathway C3 convertase converts C3 to C3a, and classical pathway C5 convertase converts C5 to C5a.
The alternative pathway is initiated by products from microorganisms, including LPS from Gram-negative bacteria and polysaccharides from fungal cell walls (see Web Fig. 3-6). In addition, other activated plasma proteins, including kallikrein, plasmin, and activated factor XII, can cleave C3, resulting in its activation to C3b. C3b combines with factor B and coupled with factor D activity, forms the alternative pathway C3 convertase (C3bBb). Alternative pathway C3 convertase converts C3 to C3b. C3b combines with alternative pathway C3 convertase to form alternative pathway C5 convertase (C3bBb3b), which converts C5 to C5a. Because the alternative pathway can be activated by clotting and kinin factors, once the clotting or the kinin systems are activated, complement activity follows and vice versa. Therefore the clotting, kinin, and complement systems are closely interactive, and often activation of one system leads to activation of others (Fig. 3-8). When bound to a microbial product, mannose-binding lectins (MBLs) and ficolins can also activate complement through MBL-associated serine proteases (MASPs). MASP-2 cleaves C4 and C2 to activate steps of the classical pathway.
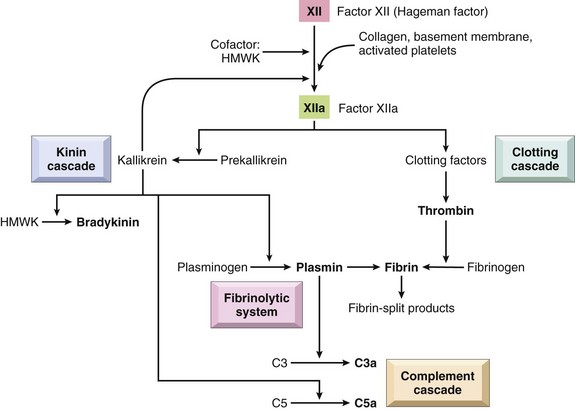
Fig. 3-8 Interrelationships between the four plasma mediator systems triggered by activation of factor XII (Hageman factor).
Note that thrombin induces inflammation by binding to protease-activated receptors (PARs) (principally PAR-1) on platelets, endothelium, smooth muscle cells, and other cells. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
C3a increases vascular permeability by inducing histamine release from mast cells. C5a, once formed, is released into the inflammatory exudate and behaves as an anaphylatoxin (a molecule that causes the release of histamine and other chemical mediators from mast cells or basophils), a chemoattractant for leukocytes, and an inducer of adhesion molecule expression by endothelial cells. C3b and C3bi are important opsonins and enhance neutrophil phagocytosis through CR1 and CR3 receptors. The plasma enzyme carboxypeptidase can degrade both C3a and C5b.
The MAC results from the cleavage of C5 by C5 convertase, leading to the formation of C5a and C5b. C5b serves as the anchor for the assembly of a single molecule composed of C6, C7, and C8. This MAC (C5b with C6, C7, C8) facilitates polymerization of C9 (up to 18 molecules of C9) into a tube that is inserted into the lipid bilayer of the plasma membrane of, for example, a bacterium. A channel is formed through the cell membrane allowing the passage of ions, small molecules, and water into the bacterium by osmosis. Bacterial lysis ensues. This process can also injure naïve host cells such as occurs in hemolytic anemia.
Arachidonic Acid Metabolites
When inflammation or inflammatory mediators injure cells, the cell membrane lipids are rapidly rearranged to create a variety of biologically active lipid mediators derived from arachidonic acid. Arachidonic acid metabolites are lipid-derived autocoid (acting as a local hormone) mediators of inflammation that serve as intracellular and extracellular signals influencing the coagulation cascade and mediating nearly every step of the acute inflammatory response (Fig. 3-9; Web Fig. 3-7). Effects are short-lived because these lipid metabolites decay rapidly or are destroyed by enzymes. Arachidonic acid metabolites include prostaglandins, leukotrienes, and lipoxins that are produced by cyclooxygenase (COX) and lipoxygenase enzyme pathways.
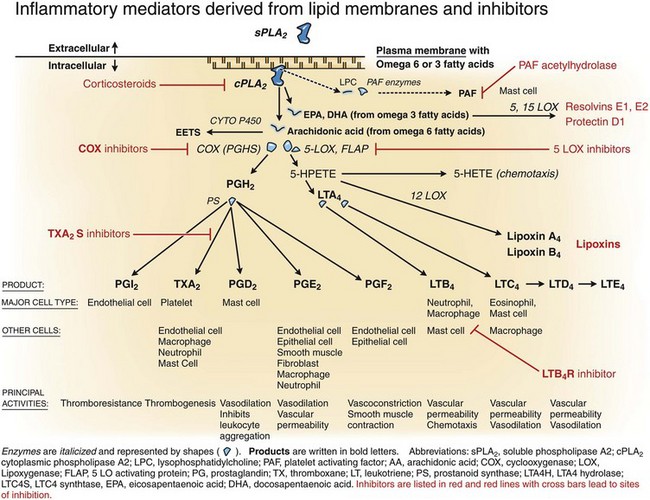
Fig. 3-9 Key inflammatory mediators derived from the plasma membrane.
Cytoplasmic phospholipase A2 (cPLA2) activity on the plasma membrane leads to free arachidonic acid (AA) and lysophosphatidylcholine (LPC). Arachidonic acid metabolites include prostaglandins and leukotrienes, and LPC is a substrate for platelet-activating factor (PAF). The type of prostaglandin formed by a cell is dependent on the cell type. Platelets, for example, form thromboxane, whereas endothelial cells form prostacyclin. Leukotrienes are formed by leukocytes. Inhibitors specific to the various enzymes or products are indicated in red text and lines. COX, Cyclooxygenase; EETS, epoxyeicosatrienoics; FLAP, 5-lipoxygenase-activating protein; HETE, hydroxyeicosatetraenoic acid; LOX, lipoxygenase; LTA4, leukotriene A4; LTB4, leukotriene B4; LTC4, leukotriene C4; LTD4, leukotriene D4; LTE4, leukotriene E4; LTA4H, leukotriene A4 hydrolase; LTC4S, leukotriene C4 synthase; PGD2, prostaglandins D2; PGE2, prostaglandins E2; PGF2, prostaglandins F2; PGH2, prostaglandins H2; PGI2, prostaglandins I2; PGHS, prostaglandin H synthase; PS, polysaccharides; sPLA2, soluble phospholipase A2; TXA2, thromboxane A2. (Modified from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
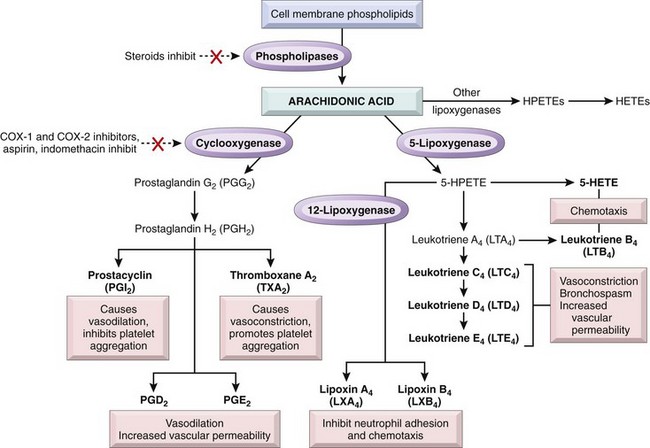
Web Fig. 3-7 Generation of arachidonic acid metabolites and their roles in inflammation.
The molecular targets of action of some anti-inflammatory drugs are indicated by a red X. Not shown are agents that inhibit leukotriene production by inhibition of 5-lipoxygenase (e.g., Zileuton) or block leukotriene receptors (e.g., Montelukast). COX, Cyclooxygenase; HETE, hydroxyeicosatetraenoic acid; HPETE, hydroperoxyeicosatetraenoic acid. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Arachidonic acid (eicosapentaenoic acid) is an essential 20-carbon, polyunsaturated fatty acid derived from linoleic acid, which is present in plasma membranes of dietary red meats. Arachidonic acid is an integral component of esterified membrane phospholipids that, when cleaved from the plasma membrane by phospholipase, serves as the major precursor for eicosanoids. Eicosanoids are synthesized by two main classes of enzymes: (1) COXs and (2) lipoxygenases and also (3) cytochrome P450 enzymes. Their respective products (eicosanoids) are (1) prostaglandins and thromboxanes and (2) leukotrienes and lipoxins. These molecules are synthesized in endothelial cells, leukocytes, and platelets and principally exert their biologic effects on vascular and airway smooth muscle cells, endothelial cells, and platelets during the acute inflammatory response.
Arachidonic acid is released from membrane phospholipids of many cell types, but particularly endothelial cells and leukocytes, through the action of cytoplasmic phospholipase A2 (cPLA2) and to a lesser degree, soluble (extracellular) phospholipase A2 (sPLA2). This occurs in response to physical and chemical stimuli, including C5a. cPLA2 is translocated from the endoplasmic reticulum to plasma membrane when intracellular calcium concentrations increase. The activity of sPLA2 also requires the participation of calcium; however, its contribution to the formation of intracellular arachidonic acid varies among the different cell types when compared with that of cPLA2. Membrane phospholipids contain a glycerol backbone attached to which is often a saturated fatty acid at the sn-1 position, an unsaturated fatty acid at the sn-2 position, and a base at the sn-3 position. Arachidonic acid is often in the sn-2 position and released by cPLA2 or sPLA2 to become free arachidonic acid.
Free arachidonic acid is metabolized in one of three pathways: (1) the COX pathway for the formation of prostaglandins and thromboxanes, (2) the lipoxygenase pathway for the formation of leukotrienes and lipoxins, and (3) the cytochrome p450 pathway for the formation of epoxyeicosatrienoic acids (hydroperoxyeicosatetraenoic acid [HPETE] and hydroxyeicosatetraenoic acid [HETE]). There are three COX isoenzymes—COX-1, COX-2, and COX-3—that are actually components of prostaglandin H synthase and work in concert with a peroxidase heme group. The COX-1 isoenzyme is constitutively expressed, is present in almost all tissues, and is considered a housekeeping enzyme with physiologic roles in hemostasis and gastric mucosa protection. COX-2 isoenzyme expression is induced by exogenous and endogenous stimuli and occurs locally in sites of inflammation. It is present in leukocytes, endothelial cells of blood vessels, and synovial fibroblasts. COX-3 isoenzyme is a splice variant of COX-1 (and also termed COX-1b or COX-1v). It is present in greatest abundance in the cerebral cortex of dogs and humans and is also detected in human aortas and rodent cerebral endothelia, heart, kidney, and neuronal tissues.
Prostaglandin Formation and Inhibition
Arachidonic acid metabolites from COX isoenzymes induce an intermediate prostaglandin, PGH2, that is converted into at least five metabolites (PGD2, PGF2, PGE2, PGI2, and thromboxane A2 [TXA2]) by prostanoid synthase enzymes unique for each of these five metabolites (see Web Fig. 3-7). The relative concentration of each of these five types of prostaglandins synthesized after stimulation depends on the cell type stimulated. For example, PGI2—a thromboresistant prostaglandin termed prostacyclin—is produced by endothelial cells via PGI2 (prostacyclin) synthase, whereas TXA2—a thrombogenic prostaglandin termed thromboxane—is produced by platelets via TXA2 (thromboxane) synthase. PGD2 is the major prostanoid produced by mast cells; PGE2 is the major prostanoid produced by epithelial cells, fibroblasts, and smooth muscle cells. Specific prostaglandins inhibit (PGI2) or induce (TXA2) coagulation/thrombosis, whereas others affect vascular permeability (PGD2 and PGE2). Prostaglandins bind GPCRs that are specific for each prostaglandin. Activated prostaglandin receptors trigger cyclic adenosine monophosphate (cAMP) or increased cytoplasmic calcium. PGE2 is long known for immunomodulatory activity; mechanistically, recent work suggests that PGE2 receptor EP4 promotes differentiation of both T helper 1 (TH1) and T helper 17 (TH17) CD4+ T lymphocytes.
Prostaglandins contribute to the following:
• Adrenocorticotropic hormone (ACTH) response (neurons of the paraventricular nucleus of the brain release ACTH)
• Behavior stress syndrome (reduced movement and loss of social contact)
Aspirin, indomethacin, ibuprofen, and naproxen are COX-1 inhibitors. Aspirin, naproxen, and ibuprofen also inhibit COX-2, as do the highly selective COX-2 inhibitor drugs celecoxib, rofecoxib, valdecoxib, lumiracoxib, and etoricoxib. Acetaminophen was thought to inhibit COX-3; however, acetaminophen’s activity in humans and rodents is not fully defined. Because COX-3 is present in greatest abundance in the cerebral cortex and acetaminophen crosses the blood-brain barrier (unlike other nonsteroidal antiinflammatory drugs [NSAIDs]), these observations were initially thought to explain why acetaminophen was sometimes more effective for treating headaches and pain relief and less effective in inhibiting inflammation in the body. Although this may be the case in dogs, the mechanistic basis of acetaminophen’s activity in humans is not fully understood at this time. Acetaminophen may inhibit COX-2 to a minor degree and also slightly inhibit COX-3 (COX-1b), and yet have other activity/activities. Much of the antiinflammatory effect of corticosteroids is due to their inhibition of phospholipase A2, the enzyme that releases arachidonic acid from membrane phospholipids. Corticosteroids signal the cell to synthesize a polypeptide known as lipocortin (lipomodulin), which then acts to inhibit phospholipase A2. Therefore the antiinflammatory effect of corticosteroids is delayed. A variety of other natural and synthetic compounds can inhibit phospholipase A2.
Leukotriene Formation and Inhibition
In leukocytes, arachidonic acid can be metabolized through the 5-lipoxygenase pathway to form leukotrienes and lipoxins (see Web Fig. 3-7), which have profound effects on inflammation. 5-Lipoxygenase works in concert with 5-lipoxygenase-activating protein as an arachidonic acid binding protein, which optimizes the proximity of 5-lipoxygenase to interact with arachidonic acid. The complex formed by 5-lipoxygenase and 5-lipoxygenase-activating protein (HPETE) is converted to an intermediate complex, LTA4, which is subsequently converted to either LTB4 or LTC4, via LTA4 hydrolase or LTC4 synthetase, respectively. LTC4 synthetase is a glutathione transferase, and once LTC4 is secreted from a cell, it is metabolized to LTD4 and LTE4 by sequential removal of the glutathione, glutamic acid, and glycine moieties.
The major effects of the leukotrienes include (1) increased vascular permeability, (2) chemotaxis for leukocytes, and (3) vasoconstriction, all of which exacerbate the acute inflammatory response. Neutrophils produce primarily LTB4, which is one of the most potent chemotactic factors for neutrophils and macrophages. LTB4 also stimulates leukocytes to release their lysosomal contents further contributing to inflammation. Mast cells and eosinophils produce primarily leukotriene C4, and the group of leukotrienes—LTC4, LTD4, and LTE4, formerly known as “slow-reacting substance”—causes intense vascular leakage from the venules. Macrophages produce both LTB4 and LTC4. In addition to the leukotrienes, 5-HPETE and its end-product 5-HETE are highly chemotactic for neutrophils and macrophages. Leukotrienes also bind GPCR. LTC4, LTD4, and LTE4 bind the leukotriene receptors CysLT1 and CysLT2, which mediate bronchoconstriction and vasoconstriction, and LTB4 binds two receptors, BLT1 and BLT2, which mediate neutrophil chemotaxis.
Lipoxins, secreted primarily by platelets, are also produced from arachidonic acid via the lipoxygenase pathway. Platelets cannot form lipoxins directly, but through cell-to-cell interactions with leukocytes and the transfer of arachidonic acid metabolites to platelets, cooperation between different cells types can result in the formation of eicosanoids via transcellular biosynthesis. Neutrophils can transfer their LTA4 to platelets. Platelets contain 12-lipooxygenase, which converts the neutrophil derived and transferred LTA4 to lipoxin A4 and lipoxin B4. Lipoxins have proinflammatory and antiinflammatory effects and appear to counteract the effects of leukotrienes. They stimulate macrophage adhesion to endothelial cells in blood vessels, but inhibit LTB4 activation of neutrophils, thus decreasing neutrophil adhesion and chemotaxis, and also inhibit angiogenesis. Lipoxin A4 causes vasodilation and dampens the actions of LTC4–induced vasoconstriction. Resolvin is a product of eicosapentaenoic acid which also has antiinflammatory effects, including inhibition of BLT1 (LTB4 receptor) and dendritic cell migration.
The 5-lipoxygenase pathway can be inhibited by a variety of new chemical agents (5-lipoxygenase inhibitors and leukotriene receptor antagonists) (see Fig. 3-9). In addition, eosinophils release arylsulfatase from granules, which inactivates leukotrienes.
Omega-3 Fatty Acids (Fish Oils) and Inhibition of Eicosanoid Activity
Many studies have demonstrated that omega-3 fatty polyunsaturated fatty acids, which are in high concentrations in certain fish, particularly salmon, can reduce inflammatory responses mediated by prostaglandin and leukotriene activity (see Web Fig. 3-7). This effect occurs because omega 3 fatty acids, when acted on by phospholipase A and subsequent enzymes, form the “3” series of prostaglandins (TXA3) and the “5” series of leukotrienes (LTB5) instead of the “2” series of prostaglandins (TXA2) and the “4” series of leukotrienes (LTB4), which occur with omega-6 fatty acids. The “3” and “5” series of prostaglandins and leukotrienes derived from omega-3 fatty acids are less biologically active than their omega-6 series counterparts. Roughly two-thirds of the polyunsaturated fatty acids in the body have an omega-6 structure; the remaining have an omega-3 structure. The two-thirds to one-third ratio can be altered by consumption of diets high in the proper fish oils and thereby result in more omega-3 in the tissue and less active prostaglandin and leukotriene products.
Platelet-Activating Factor
PAF is another potent molecule of phospholipid origin derived from cell membranes of platelets, basophils, mast cells, neutrophils, macrophages, and endothelial cells. PAF has potent pathophysiologic effects and contributes to inflammation, endotoxic shock, and allergic reactions (asthma) through vasoconstriction, bronchoconstriction, platelet aggregation, and leukocyte adhesion, chemotaxis, and degranulation. However, at low concentrations (experimentally induced), PAF can cause vasodilation and increased vascular permeability. PAF also contributes to the inflammatory response by increasing the oxidative burst in neutrophils following phagocytosis of bacteria and enhances the synthesis of eicosanoids by leukocytes. During IgE-mediated hypersensitivity reaction in the lung, PAF induces mast cell release of serotonin and histamine as well as platelet aggregation. Thus platelet aggregation can increase vascular permeability in acquired immunologic responses and result in inflammation.
Two enzymes, lysoPAF acetyltransferase (lysoPAF-AT) and PAF-synthesizing phosphocholinetransferase (PAF-PCT), control the final synthesis of PAF from lipid membranes and thus promote PAF production. PAF mediates its effects on target cells through a single GPCR. These receptors have been identified on endothelium, neutrophils, eosinophils, macrophages, smooth muscle, and glial cells in the brain. PAF activity is reduced/regulated by PAF acetylhydrolase, which is expressed by cells expressing PAF receptor. PAF acetylhydrolase degrades PAF through hydrolysis of its acetate moiety in the sn-2 position of glycerol, which inhibits the proinflammatory activity of PAF. PAF acetylhydrolase is thus potentially an enzyme that could be used for the development of drugs to reduce inflammatory responses.
Cytokine Family
Overview in Inflammation and Induction of CD4 TH Subsets
Cytokines are a group of proteins produced by many cell types including lymphocytes, macrophages, endothelial cells, neutrophils, basophils, mast cells, eosinophils, epithelial cells, and connective tissue cells (see Web Appendix 3-2). The primary purpose of cytokines is to modulate, via enhancement or suppression, the functional expression of other cell types during the inflammatory response. Chemokines are produced from almost all cell types and are cytokines that promote leukocyte chemotaxis and migration across capillaries and postcapillary venules. Cytokines also play major roles in (1) hematopoiesis, including granulopoiesis by cytokines such as IL-3, G-CSF, and GM-CSF; and (2) adaptive immunity, such as the proliferation of lymphocytes by cytokines, including IL-2, and activation of Th1 or Th2 responses. The CD4 TH cell response consists of subsets of T-lymphocytes characterized by the secretion of different cytokines, including IFN-γ–producing TH1 cells and IL-4–producing TH2 cells, which are involved in cellular or humoral immunity, respectively. The cytokines have been organized into the following categories according to their principal functional activities:
• Hematopoietic growth factors, including IL-3, G-CSF, GM-CSF, possibly IL-9, IL-11, and stem cell factor
• Inflammatory mediators, which induce acute phase reactants and natural immunity (IL-1, IL-6, TNF-α, and TNF-β)
• Chemotactic cytokines (IL-8)
• T lymphocyte proliferation, activation, and differentiation cytokines (IL-2, IL-4, IL-5, IL-7, IL-9, IL-10, IL-12, and IL-17 on up to IL-29)
Stimuli that invoke expression of these families of cytokines are varied and secondary to activation of a wide variety of receptors, including soluble receptors, receptors on the cell surface, endosomal receptors, and cytoplasmic receptors, which activate nuclear factor (NF) kappa B, extracellular signal-related kinase (ERK), Jun, p38, and other signaling pathways.
Concerning T-lymphocyte activity, there are cytokines and proteins that exert T-lymphocyte regulatory activity (IFN-γ, TGF-β). Of particular importance is the presentation of antigen by dendritic cells to T-lymphocyte cells that release cytokines, which mediate formation of lineages of T-lymphocyte helper cells (Th1, Th2, TH17, and T regulatory [T reg] cells). Dendritic cells are present in tissues that are in contact with the environment such as Langerhans’ cells of the skin and dendritic cells of the mucosal surfaces of the respiratory and alimentary systems. These dendritic cells are sentinels constantly monitoring for and phagocytosing microbes and foreign materials. In cooperation with lymphocytes and macrophages in lymphoid tissue, they act as antigen-presenting cells to activate T helper lymphocytes and cytotoxic T lymphocytes and B lymphocytes. The Th1 lineage of CD4 T lymphocytes, normally driven by IL-12, expresses the transcription factor T-bet, releases IFN-γ, and induces cell-mediated immune responses within specific diseases such as the granulomatous inflammatory response to mycobacterial infections. Whereas Th2 CD4 cells form in response to IL-4, IL-6, and thymic stromal lymphopoietin (TSLP); express transcription factor GATA-3; produce IL-4, IL-5, IL-10, and IL-13; and induce humoral responses and humoral-related diseases, such as asthma and atopy, TH17 CD4 T lymphocytes form in response to TGF-β, IL-6, and IL-23, express transcription factor Rorγt, and release IL-17. These cells bridge innate and adaptive immune responses through the production of IL-17 cytokines, which promote inflammatory responses such as neutrophil recruitment and development of autoimmune responses. T reg cells develop in the thymus and also in response to antigenic stimulation in the presence of TGF-β, which induces transient or stabile expression of transcription factor forkhead box P3 (FOXP3). T reg cells modulate adaptive responses through the release of IL-10. Follicular CD4 T lymphocytes (TFH) develop in the presence of IL-6 and IL-21, express transcription factor Bcl6 and cMaf (BLIMP-1 is inhibitory), and release IL-21. Through secretion of IL-21, TFH cells support B lymphocyte development and antibody production. Follicular dendritic cells are present in the follicles of lymph nodes and present antigen to B lymphocytes.
Biochemically, cytokines can be divided into type I and type II cytokines. Type I cytokines have four α-helix units. Type II cytokines have six α-helix units and are likely derived from a single ancestral gene. Type I and II cytokines bind receptors specific for the I and II ligand structures. Despite the similarities within type I and type II cytokines and their receptors, there is still much diversity in structure and function of cytokines within each type.
Cytokine Receptors and Signaling
Cytokine activity can be altered in several ways, usually via binding to and activating cytokine receptors. Briefly, there are immunoglobulin superfamily receptors (such as IL-1), hematopoietic growth factor family (type 1) receptors (receptors for IL-2 through IL-9, IL-11 through IL-13, IL-15, IL-21, IL-23, IL-27, erythropoietin [EPO], G-CSF, GM-CSF, leptin, and others), interferon family (type II) receptors (receptors for IFN-α, IFN-β, IFN-γ, and IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29), and TNF (type 3) receptors. Agonists and inhibitors to some of these receptors have been developed. For example, EPO agonists are commonly used for patients with anemia and are drugs abused by endurance athletes such as marathoners, cross-country skiers, and cyclists. Other G-CSF agonists, such as Neupogen (Amgen Inc., Thousand Oaks, CA), increase granulopoiesis and are commonly used for cancer patients with decreased blood neutrophil levels (neutropenia) secondary to chemotherapy.
Many cytokine receptors, including those for the interferons IL-2 through IL-7, IL-11, IL-13, IFN-γ, EPO, and GM-CSF, activate the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway (Web Fig. 3-8). Members of the JAK family are cytoplasmic protein tyrosine kinases physically associated with ligand-bound receptors. JAK phosphorylates amino acids in a protein on STAT, and STAT dimers (a molecule consisting of two identical simpler molecules) enter the nucleus to bind DNA and induce transcription and the expression of specific gene products and cellular responses to activation. There are three isoforms of JAK (JAK1 to JAK3) and a fourth JAK family member, TYK2. All of these have slightly different activities, depending on the stimulus. JAK1, for example, is integral to lymphocyte development, whereas JAK2 mediates erythropoiesis. There are several isoforms in the STAT family, including STAT1 to STAT6, which also have subtle signaling differences. STAT1 and STAT2 are especially involved with interferon signaling, including IFN-γ signaling (STAT1) for Th1 responses; STAT3 mediates responses to IL-6, IL-10, IL-23 to induce Th17 responses; STAT4 is involved with Th1/IL-12 activity; STAT5A and STAT5B mediate cell growth and proliferation, including NK growth, and inhibit apoptosis; and STAT6 is activated by IL-4 and IL-13 to mediate Th2 responses. Suppressors of cytokine signaling (SOCS) are signaling proteins that inhibit activity of some cytokines whose receptors induce the JAK/STAT pathway of signaling through the inhibition of JAK. Protein tyrosine phosphatases (PTPs) such as SH2-domain–containing PTP (SHP2) can inhibit JAK and STAT. Phosphorylated STAT proteins in the nucleus can be inhibited by protein inhibitor of activated STAT (PIAS) proteins as well. These proteins regulate the activity of cytokine activity on cells and may be potentially useful as a mechanism to target and inhibit specific components of the acute inflammatory response with existing or newly developed antiinflammatory drugs. In contrast to the Th1 and Th2 signaling that occurs through JAK/STAT signaling, Th17 cytokines signal through the adaptor protein ACT1 (also known as CIKS), TNF-receptor–associated factor 6 (TRAF 6), and NF kappa B. Thus the Th17 signaling pathway resembles receptors involved with inflammation and innate immunity (e.g., IL-1 and TLRs) and is consistent with the Th17 induction of inflammatory events and bridging of innate/adaptive responses.
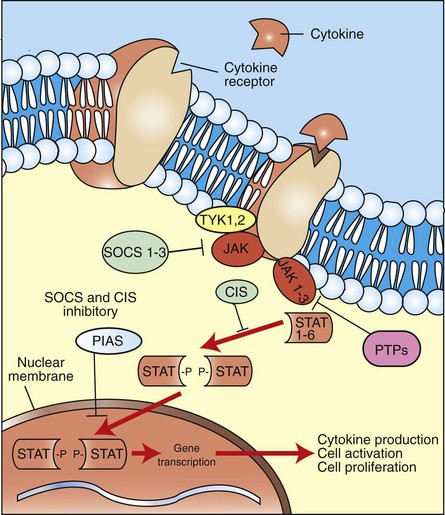
Web Fig. 3-8 Several families of cytokines activate the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway.
The STAT class of proteins function both as a signal transducer and transcription activator. Activation of STAT involves dimerization and phosphorylation by JAK, a tyrosine kinase. Signaling by JAK/STAT proteins is regulated by proteins such as the SOCS (suppressor of cytokine signaling). JAK/STAT signaling leads to transcription of genes that drive inflammatory responses, including cell activation, immune regulation, and cell proliferation. CIS, Cytokine-inducible SH2 domain-containing protein; PIAS, protein inhibitor of activated signal; PTPs, protein tyrosine phosphatases; TYK, tyrosine kinase (of the JAK family). (Modified from Leonard W: Nat Rev Immunol 1:200-208, 2001.)
TNF-α receptors are trimers of three identical cell-surface transmembrane proteins. Binding of the receptor activates the receptors intracytoplasmic domain (TRADD) to trigger signaling cascades involving IRAK and NEMO, which invoke phosphorylation of I kappa B (IKB) and which allows release of IKB from NF kappa B in the cytosol. IKB becomes ubiquinated and degraded, and NF kappa B moves to the nucleus, where it binds promoter and enhancer regions of DNA of more than 60 genes, including genes for IL-1 and other cytokines and genes for cell proliferation, cell adhesion, and angiogenesis. TNF can also bind receptor death domains that that lead to apoptosis.
Cytokine activity can also be inhibited. Nonspecific inhibitors include corticosteroids, aspirin, and ibuprofen. More precisely, “molecular” inhibitors can inhibit cytokine activity through anticytokine antibodies, such as the IL-1 receptor antagonist anakinra (Kineret, Amgen/Biovitrum, Stockholm), the IL-1 trap rilonacept (Arcalyst, Regeneron Pharmaceuticals, Tarrytown, NY), anti-TNF monoclonal antibodies (infliximab [Remicade, Johnson & Johnson, New Brunswick, NJ]; etanercept [Enbrel, Immunex Corporation, Thousand Oaks, CA]; certolizumab pegol [Cimzia, UCB Pharmaceuticals, Brussels]), and anti-IL-23 (ustekinumab, Stelara, Centocor Ortho Biotech, Horsham, PA). Specific inhibitors are also under development for cytokine receptors, including SOCS and PTP. Anti-IL-1 drugs are more effective against autoinflammatory diseases, whereas anti-TNF drugs are more effective against autoimmune diseases (e.g., rheumatoid arthritis, Crohn’s disease, psoriasis).
Inflammatory Proteins
Interferons: Interferons are glycoprotein cytokines produced by lymphocytes and many other cell types in response to viruses and virus-infected cells, parasites, and neoplastic cells. They inhibit viral replication within host cells, activate NK cells and macrophages, increase antigen presentation to T lymphocytes, and increase the resistance of host cells to viral infection. Type 1 interferons (IFN-α and IFN-β) bind IFN-α receptors (IFNAR) 1 and 2 to signal through JAK1 and STAT1 and STAT2 and mediate antiviral responses; type II interferons (IFN-γ) bind IFN-γ receptors (IFNGR) 1 and 2, signal through JAK1, JAK2, and STAT1, to induce TH1 and TH17 responses. Type III interferons bind IL-10 receptors and have been described, but their function is not fully clarified. The antiviral activity of type I interferons involves expression activation of the ISGlyation pathway, MxA protein, 2′,5′-oligoadenylate synthetase 1 (OAS1) activation of ribonuclease L (RNaseL), and protein kinase R (PKR). ISGlyation is a process in which interferon-stimulated gene 15 (ISG15) protein binds key interferon antiviral proteins (e.g., JAK1, STAT1, MxA, PKR, and RNaseL) and prevents their degradation, thus enhancing the antiviral response. MxA protein binds and traps viruses. The OAS1/RNaseL process cleaves viral RNA and PKR protein dimerizes on itself and in the presence of viral RNA phosphorylates, thus inhibiting host cell translation initiation factor-2α (EIF-2α), which blocks viral replication.
High Mobility Group Box Protein-1: High mobility group box protein 1 (HMGB-1) is a proinflammatory cytokine released by monocytes and macrophages. It is a nonhistone nuclear protein that binds DNA to regulate gene expression and the chromosomal architecture. HMGB-1 is released by nearly all cells during necrosis, and once outside the cell, it is an alarmin, which is an endogenous molecule that triggers host inflammatory responses. HMGB-1 binds macrophage receptor for advanced glycosylation end-products (RAGE) and TLRs 2 and 4, activating the innate immune system and pathologic responses that include release of IL-1, TNF-α, and IFN-γ. HMGB-1 and cytokines, such as IL-1, TNF, and IL-6, and prostaglandins, such as PGE2, are involved with hypothalamic function in food aversion, hypophagia, anorexia, weight loss, and sickness behavior (see section on Febrile Response) and serve to amplify the inflammatory response.
Chemokines
Chemokines, released in response to inflammatory stimuli, are secreted proteins that induce leukocyte chemotaxis into inflammatory exudates (Fig. 3-10). They also activate inflammatory cells, induce antiviral activity, regulate immune responses, and induce hematopoiesis, angiogenesis, and cell growth. Chemokines are produced by all nucleated cells in the body, including epithelial cells, fibroblasts, macrophages, mast cells, keratinocytes, dendritic cells, and endothelial cells. Leukocytes secrete all types of chemokines, except for fractalkine (used for monocyte chemotaxis), which is produced by nonhematopoietic cells such as vascular endothelial cells.
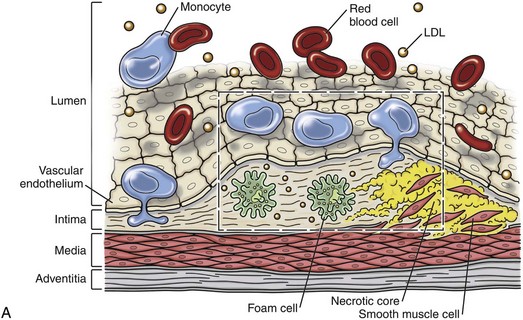
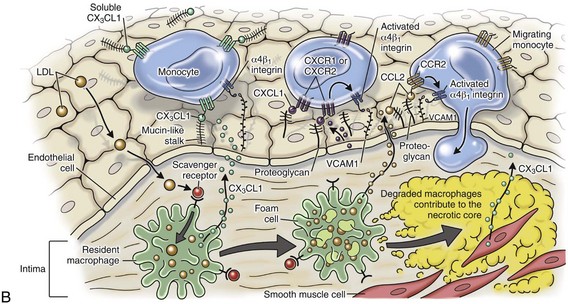
Fig. 3-10 Chemokine responses to vascular injury.
A, The atherosclerotic plaque illustrates the interactions of chemokine ligands with their receptors during an inflammatory reaction. While neutrophils respond to IL-8 (a C-C chemokine), monocytes respond to CX3CL1, CXCL1, CCL2 and their respective receptors to attract cells along the vascular wall for adhesion molecule adherence and leukocyte infiltration. B, Higher magnification of the area defined by the box in A. (Redrawn from Charo IF, Ransohoff RM: The many roles of chemokines and chemokine receptors in inflammation, N Engl J Med 354(6):610-621, 2006.)
Classification of Chemokines: Chemokines can be classified based on biochemical structure (position of their cysteine residues) or by the functional behavior they induce (inflammatory or homing). Structural classification of chemokines is based on their primary amino acid structure, depending on their cysteine residues that induce secondary folding: (1) the CC family, which bind the CCR (receptor); (2) the CXC family, which bind the CXCR; (3) the C chemokine lymphotoxin, which binds CR; and (4) the CX3C chemokine fractalkine, which binds CX3CR.
The functional classification of chemokines includes inflammatory chemokines or homing chemokines (Web Table 3-6). Inflammatory chemokines (IL-8, GCP-2, ENA-78, Gro, NAP-2, IP-10, Mig, I-Tac, RANTES, MIP-a and MIP-b, MCP-2,3, HHC-1, MCP 1 to 5, and eotaxins 1 to 3) are expressed by resident cells and infiltrating leukocytes in response to cytokines, bacterial toxins, and other agents during the acute inflammatory response. Receptors for these chemokines include CXCR1, 2, 3 and CCR1, 2, 3, and 5; these receptors often bind several different chemokines (Web Tables 3-7 and 3-8). Neutrophils express chemokine receptors CXCR1 and CXCR2, which bind CXCL8 (IL-8) and follows this ligand along a gradient. Monocytes are drawn to areas of inflammation by chemokine ligands CCL2, CCL7, CCL8, and CCL13. CCL2 and CCL13 bind CCR2; CCL8 binds CCR2 and CCR5; and CCL7 binds CCR1, CCR2, CCR3. Monocyte chemokine also attracts basophils and eosinophils. Chemokines for leukocyte homing are generally produced constitutively in lymphoid tissue. Receptors for these chemokines are usually present on lymphocytes and dendritic cells, and they typically bind only one or maybe two chemokines. The expression of receptors by these cells varies with the state of cellular differentiation, thereby preventing immature cells from entering sites lacking lesions.
WEB TABLE 3-6
Name, Abbreviation, and Common Name of Chemokines
| Name | Abbreviation | Common Name |
| CCL1 | TCA | T-lymphocyte activation (I-309) |
| CCL2 | MCP-1 | Macrophage chemotactic protein-1 |
| CCL3 | MIP-1-α | Macrophage inflammatory protein-1-α |
| CCL4 | MIP-1-β | Macrophage inflammatory protein-1-β |
| CCL5 | RANTES | Regulated on activation, normal T-lymphocytes expressed and secreted |
| CCL7 | MCP-3 | Macrophage chemotactic protein-3 |
| CCL8 | MCP-2 | Macrophage chemotactic protein-2 |
| CCL11 | Eotaxin | |
| CCL12 | MCP-5 | Macrophage chemotactic protein-5 |
| CCL13 | MCP-4 | Macrophage chemotactic protein-4 |
| CCL17 | TARC | Thymus and activated regulated cytokine |
| CCL19 | MIP-3-β | Macrophage inflammatory protein-3-β |
| CCL20 | LARC/MIP-3-α | Liver and activation-regulated chemokine/Macrophage inflammatory protein-3-α |
| CCL21 | SLC | Secondary lymphoid organ chemokine |
| CCL22 | MDC | Macrophage-derived chemoattractant |
| CCL24 | Eotaxin-2 | |
| CCL26 | Eotaxin-3 | |
| CXCL1 | Gro-α | Growth-related oncogene-α |
| CXCL2 | Gro-β | Growth-related oncogene-β |
| CXCL3 | Gro-γ | Growth-related oncogene-γ |
| CXCL5 | ENA-78 | Epithelial neutrophil-activating peptide-78 |
| CXCL6 | GCP-2 | Granulocyte chemotactic protein-2 |
| CXCL8 | IL-8 (NAP-1) | Interleukin-8 |
| CXCL9 | MIG | Monokine induced by interferon-γ |
| CXCL10 | IP-10 | Interferon-inducible protein |
| CXCL11 | ITAC | Interferon-inducible T-lymphocyte α chemoattractant |
| CXCL12 | SDF-1 | Stromal derived factor-1 |
| CXCL13 | BCA-1/BLC | B lymphocyte attracting chemokine |
| CXCL14 | BRAK | Breast and kidney expressed chemokine |
| CXCL16 |
WEB TABLE 3-8
CC, CXC, CX3C, and XC Families of Chemokine Receptors, Ligands, Cell Localization And Associated Disease Conditions In Humans
BCA-1, B lymphocyte chemoattractant 1; COPD, chronic obstructive pulmonary disease; CTACK, cutaneous T cell-attracting chemokine; ELC, Epstein-Barr I1-ligand chemokine; ENA, epithelial cell–derived neutrophil activating peptide; GCP, granulocyte chemotactic protein; GRO, growth-regulated oncogene; HHC, hemofiltrate chemokine; HIV, human immunodeficiency virus; IP-10, interferon-induced protein 10; I-TAC, interferon-inducible T lymphocyte α chemoattractant; LARC, liver and activation-regulated chemokine; MCP, monocyte chemoattractant protein; MDC, macrophage-derived chemokine; MEC, mammary-enriched chemokine; MIG, monokine induced by interferon-γ; MIP, macrophage inflammatory protein; PF, platelet factor; SDF, stromal cell-derived factor; SLC, secondary lymphoid-tissue chemokine; SR-PSOX, scavenger receptor for phosphatidylserine containing oxidized lipids; TARC, thymus and activation-regulated chemokine; TECK, thymus-expressed chemokine; TH2, type 2 helper T lymphocytes.
From Charo IF, Ransohoff RM: N Engl J Med 354: 2006.
Naïve T lymphocytes express CCD7, which binds to CCL21 expressed on HEVs of lymph nodes to localize into the parafollicular (T lymphocyte region) of the node. B lymphocytes express CXCL13, which binds to CXCR5 for entrance into the follicular area of lymph nodes. Dendritic cells and macrophages from peripheral sites of inflammation enter the lymph node through the afferent lymph vessel and express CCR7 and CCR2, respectively, which bind their ligands, CCL19 and CCL21.
Chemerin is a recently described molecule that is a chemoattractant for dendritic cells and macrophages. Chemerin is activated by proteolytic cleavage and binds chemokine-like receptor-1 (CMKLR1), which is a GPCR.
Chemokine Receptors and Signaling
Chemokines are the only cytokines that bind G protein–coupled seven transmembrane (7TM) spanning receptors, and although chemokines are relatively small (8 to 10 kDa), they are somewhat large for the 7TM receptor family, which serves as receptors for larger proteins such as histamine, dopamine, and serotonin. Once bound, the 7TM receptor activates a G protein, which initiates production of second messengers, most commonly cAMP and inositol 1,4,5-trisphosphate (IP3). cAMP and IP3, in turn, phosphorylate enzymes and stimulate the release of calcium. Enzymatic activity induced by cAMP activates the transcription factor cAMP responsive element-binding protein (CREB). Chemokine receptors bound by ligand are endocytosed and either recycled or degraded by the cell.
Experimentally, chemokines can be inhibited at several stages: (1) inhibition of interaction between the ligand and the glycosylated molecules on the cell surface, (2) inhibition of ligand/receptor binding, (3) inhibition of receptor activity, and (4) inhibition of cell signaling. The development of new drugs to treat specific aspects of inflammatory diseases will likely target one or all of these pathways in the chemokine cascade.
Oxygen-Derived Free Radicals and Nitric Oxide
Free radicals, such as superoxide anion, hydroxy radical, and NO derivatives, can (1) injure vascular endothelial cells, leading to increased vascular permeability; (2) inactivate antiproteases, such as α1-antitrypsin, resulting in damage to ECM proteins; (3) enhance cytokine and chemokine expression secondary to signaling changes and cell damage; (4) activate endothelial cells and increase adhesion molecule expression; and (5) increase the formation of chemotactic factors (LTB4). They can also inactivate neurotransmitters (adrenaline and noradrenaline), leading to hypotension.
Oxygen-derived free radicals are released from neutrophils and macrophages after exposure to chemokines and immune complexes and after phagocytosis by the leukocyte (see Table 3-2). They damage cells through peroxidation of cell membrane lipids, cross-linking of proteins and oxidizing thiol groups on methionine and cysteine, cleaving glycoconjugates, directly damaging DNA phosphate backbone and bases, and inducing the formation of DNA adducts. Because of damage to proteins, the activity of key enzymes and transcription factors needed by the cell can be impaired. Fortunately, the body has antioxidants that are (1) enzymatic, such as superoxide dismutase (SOD) isoforms 1, 2, and 3, catalase, thioredoxin, peroxiredoxins, and glutathione reductase; (2) nonenzymatic endogenous substances, such as ceruloplasmin, transferrin, metallothionein, uric acid, and melatonin; and (3) nonenzymatic dietary substances, such as vitamins A, C, and E and lycopenes, flavonoids, resveratrol genistein, anthocyanins, naringenin, and reserpines. All of these minimize the damage to tissue caused by free radicals.
NO is a chemical mediator of inflammation that causes vasodilation by relaxing vascular smooth muscle cells. In response to injury and inflammatory stimuli, NO derivatives are synthesized by endothelial cells, macrophages, and specific populations of neurons in the brain from l-arginine, molecular oxygen, NADPH, other cofactors, and the enzyme NO synthetase (NOS). There are three forms of NOS that mediate NO formation: neuronal (nNOS), iNOS, and endothelial (eNOS). In addition to its vasodilatory activities, NO inhibits platelet aggregation and adhesion, inhibits mast cell-induced inflammation, oxidizes lipids and other molecules, and regulates leukocyte chemotaxis.
Receptors for Exogenous and Endogenous Inflammatory Stimuli and Toll-Like Receptors
Inflammatory responses occur in response to endogenous and exogenous substances. The body’s inflammatory reaction to exogenous pathogens has been understood and studied for many years. Increasingly, it is clear that the body also produces inflammatory reactions to host endogenous molecules released under sterile conditions. Thus both endogenous and exogenous stimuli produce “danger” signals, termed danger-associated molecular patterns (DAMPs).
Exogenous microbial products, often with redundant molecular structure, are PAMPs and include substances such as LPS, peptidoglycan, and lipoteichoic acid. PAMPs can bind several types of PRRs (Fig. 3-11; Table 3-4; Web Fig. 3-9). These receptors include secreted receptors that circulate in the blood (LPS binding protein), surface receptors (macrophage mannose receptors; and TLRs), nucleotide-binding oligomerization domain (NOD) -like receptors (NLRs), and endosomal receptors (TLR and endosomal viral receptors). These receptors have different mechanisms by which they activate the cell and some work together. LPS, for example, is bound by LPS-binding protein (LBP), which in turn binds CD14 and TLR4. The formation of a PRR-PAMP complex initiates transmembrane signaling that often involves a MyD88 protein event leading to NF kappa B activation and MAPK (p38) signaling. The NF kappa B family of transcription factors initiates gene transcription and translation, resulting in the expression of proteins involved in many cellular processes such as cell proliferation, differentiation, apoptosis, and cell responses to injury, stress, and external pathogens. NF kappa B and p38 then can induce phagocytosis by leukocytes, dendritic cell activation, release of inflammatory cytokines and chemokines, and the activation of the innate (defensin and antimicrobial peptide release) and adaptive (Th1, Th2, Th17, TFH activity) immune systems. Alternatively, TLR4 signaling does not engage MyD88 (MyD88-independent signaling) and results in the formation of IFN-β and IFN-inducible gene products.
TABLE 3-4
Types of Pathogen-Associated Molecular Pattern (PAMP) Exogenous Ligands, Their Pattern Recognition Receptors (PRRs), and the Subsequent Action Related to Acute Inflammation
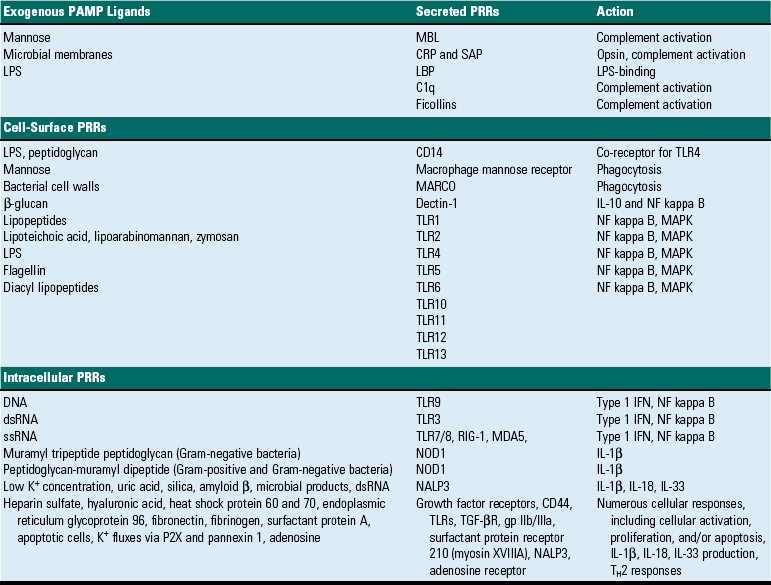
Endogenous inflammatory stimuli are those produced by host cells.
CRP, C-reactive protein; dsRNA, double-stranded RNA; gp, Glycoprotein; IFN, interferon; IL, interleukin; LBP, LPS-binding protein; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MARCO, class A scavenger receptor macrophage receptor with collagenous structure; MDA5, melanoma differentiation-associated gene 5; MBL, Mannan-binding lectin; NALP3, ; NF, nuclear factor; NOD1, nucleotide-binding and oligomerization domain; RIG-1, retinoic acid-inducible gene-I; SAP, serum amyloid protein; ssRNA, single-stranded RNA; TGF-βR, transforming growth factor-β receptor; TLR4, Toll-like receptor 4.
Data from Medzhitov R: Nat Rev Immunol 1:135-142, 2001.
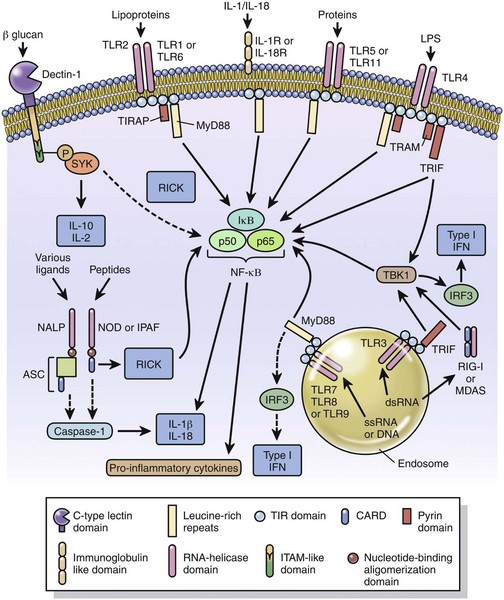
Fig. 3-11 Pathogen-associated molecular patterns (PAMPs), pattern recognition receptors (PRR), and cellular signaling.
PAMPs such as lipopolysaccharide (LPS), teichoic acid, beta-glucans, as well as microbial peptides, proteins, RNA, DNA, and CpG, bind specific receptors and trigger activation of the subjacent signaling cascade leading to transcription of pro-inflammatory genes (cytokines, interferons, chemokines). Receptors are present on the membrane surface (Dectin-1, TLR1, 2, 4, 5, 6, 11, IL-1) in endosomes (TLR3, 7, 8, 9), and in the cytoplasm (NOD, NALP, RIG-1). TLR utilize MyD88 protein for signaling, except for TLR 3 and 4 which can be MyD88-independent. All pictured trigger NF kappa B (NFκB) and some also induce interferon responses. (Redrawn from Dale DC, Boxer L, Liles WC: The phagocytes: neutrophils and monocytes, Blood 112(4):935-945, 2008.)
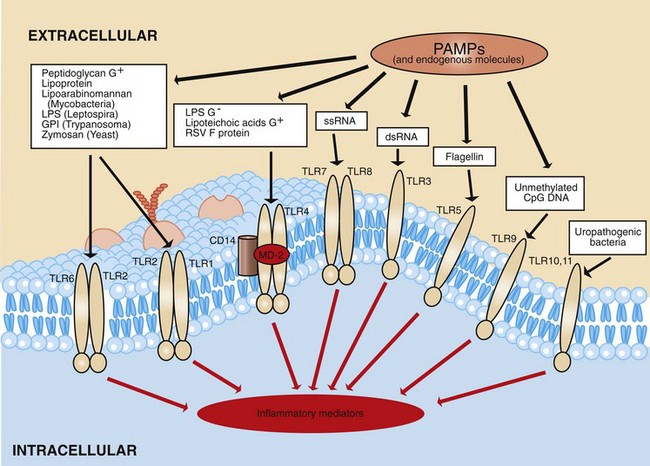
Web Fig. 3-9 Pathogen associated molecular patterns (PAMPs).
PAMPs, such as lipopolysaccharide (LPS), teichoic acid, and CpG, as well as other endogenous substances, have affinities to certain Toll-like receptors (TLRs) and result in activation of TLR signaling, as well as inflammatory and other responses. Endogenous molecules can also activate TLRs and include heparan sulfate, hyaluronan, heat shock protein 60 (mitochondria), heat shock protein 70 (cytoplasm), fibronectin, fibrinogen, and surfactant protein A. Ligands for TLR10 are not known; TLR11 ligands include uropathogenic bacteria. CD14, LPS-binding protein receptor; CpG, bacterial cytosine-phosphate-guanine motifs; GPI, glycosylphosphatidylinositol; MD-2, accessory extracellular adaptor protein that binds TLR4; RSV, respiratory syncytial virus. (Modified from Medzhitov R: Nat Rev Immunol 1:135-142, 2001.)
In addition to LBP, other secreted PRRs that can trigger inflammatory reactions through complement activation or receptor binding include collagenous lectins such as mannan-binding lectins A and C, ficolins, surfactant proteins A (SP-A) and D (SP-D), and conglutinin (cattle), which bind glycans of chitin, bacterial capsules, viruses, C-reactive protein, and serum amyloid–binding protein. SP-A, for example, can bind, aggregate, and inhibit respiratory syncytial virus. SP-A can also activate macrophages.
Leukocytes, epithelia, mucosal-lining cells, and other cells can also express PRR on the plasma membrane, including TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11. TLRs are a family of PRRs in mammals that can distinguish between chemically diverse classes of genetically conserved microbial products (see Table 3-4). For example, bacterial lipoprotein binds TLR1 and TLR2/6, flagellin binds TLR5, and LPS binds TLR4. These receptors have a central role in the release of inflammatory cytokines from the innate immune system in response to microbial structures, such as exogenous microbial substances and endogenous products (Web Fig. 3-10). Also, TLRs likely play a role in the adaptive immune response, which develops during the acute inflammatory response.
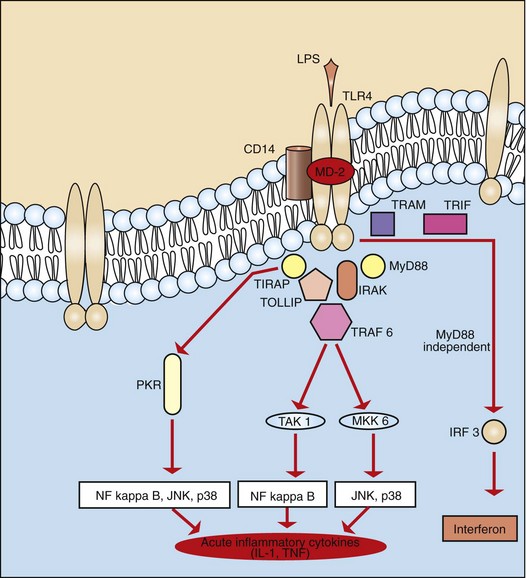
Web Fig. 3-10 Toll-like receptor 4 (TLR4).
TLR4, when bound by certain microbial or endogenous products, initiates signaling that leads to release of inflammatory cytokines, such as interleukin (IL-1) and tumor necrosis factor (TNF), antimicrobial peptides, and molecules that initiate adaptive immunity. Ligand: Lipopolysaccharide (LPS). Selected signaling molecules: Toll/IL-1 receptor (TIR) cytoplasmic domain, cytoplasmic protein adaptors to TIR (TIRAP, MyD88, TRAF), IL-1 receptor–associated kinases (IRAK), dsRNA-responsive protein kinase (PKR), TNF receptor–associated factor 6 (TRAF 6), protein kinase (TAK 1), map-kinase kinase (MKK), interferon regulatory factor 3 (IRF 3). Transcription factors: Nuclear factor kappa B (NF kappa B), c-Jun N-terminal kinase (JNK), p38. MD-2, accessory protein; TRAM, TRIF-related adaptor molecule; TRIF, TIR-containing adaptor inducing interferon-β. (Modified from Medzhitov R: Nat Rev Immunol 1:135, 2001.)
C-type lectin receptors are also present on the surface of monocytes, macrophages, neutrophils, and dendritic cells and include dectin 1 that binds beta 1,3 glucan, as well as dectin 2, MBL, and dendritic cell-specific intercellular adhesion molecule-grabbing nonintegrins (DC-SIGNs) that bind high mannose and fructose; both glucans and mannose/fructose residues are produced and released by mycobacterial and fungal organisms. Once bound, both TLR and C-type lectins regulate a wide range of innate and adaptive immune responses through NF kappa B, transcription factor activators protein-1 (TFAP-1), and interferon regulatory factors (IRFs).
Intracellular receptors, such as the NLRs, have a leucine-rich repeat domain that binds PAMPs and xenocompounds to induce and/or regulate inflammation. NOD1 binds to peptidoglycan-derived tripeptide structures, whereas NOD2 binds peptidoglycan-derived muramyl dipeptides. Bound NOD1 and NOD2 activate the receptor-interacting protein (RIP) family of serine/threonine protein kinase (RIPK2), which then induces NF kappa B activity and eventual secretion of antimicrobial peptides and inflammatory cytokines such as IL-1β and IL-18 and IL-33, invoking inflammation. Muramyl dipeptides and a variety of xenocompounds and endogenous stimuli can activate another family of NLRs that contain a pyrin domain NLRP3 (also termed NALP3), which triggers caspase 1 activation and activation of IL-1β, IL-18, and IL-33.
PRRs for certain viral infections include retinoic acid-inducible gene I (RIG-I) (e.g., paramyxoviridae, influenza viruses) and melanoma differentiation-associated gene 5 (MDA-5) (picornaviridae). These molecules sense viral RNA, alter mitochondrial activity through IPS-1, and invoke IRF3 for IFN-α and -β release and NF kappa B for inflammatory cytokine production along with antimicrobial peptides.
Endosomes express PRRs that also have important roles in sensing viral infection and triggering a cellular response that often results in inflammatory and immune activity. TLR3, TLR7, TLR8, and TLR9 are present along the inner endosomal membrane and bind double-stranded RNA (dsRNA) (TLR3) or single-stranded RNA/DNA (ssRNA/DNA) (TLR7, TLR8, TLR9) and activate IRF7 and NF kappa B.
Endogenous molecules (not produced by microbes) are released in response to noninfectious injury (trauma, toxins, neoplasia, necrosis, or irritation). Because endogenous molecules can trigger an inflammatory reaction, they have been termed alarmins. Alarmins include sulfate, hyaluronan, HSP 60 (mitochondria), HSP 70 (cytoplasm), Gp 96 (endoplasmic reticulum), fibronectin, fibrinogen, and SP-A, all of which can bind PRRs or other receptors and initiate cell signaling, inflammation, and activation of the innate immune system (see Table 3-4). These molecules are often overlooked when considering acute inflammation in the context of microbial infections because microbial products, such as teichoic acid and LPS, are very potent activators of acute inflammation. However, endogenous molecules likely have a significant role in inflammation generated against neoplastic cells, toxins, and mechanical injury. For example, a wide variety of xenocompounds, including apoptotic cells, toxins (maitotoxin, valinomycin, nigericin, aerolysin), stress molecules (ultraviolet [UV] radiation, monosodium urate, calcium pyrophosphate, beta amyloid fibrils), xenogeneic compounds (silicates, asbestos, aluminum hydroxide), as well as microbial products (lipoteichoic acid, LPS), can affect lysosomal function which in turn induces NLRP3 (NALP3) activation (Fig. 3-12). Furthermore, adenosine, a by-product of cell injury CD39 hydroxylation of ATP, can bind adenosine receptors present on the surface membrane of leukocytes and modulate inflammatory and immune responses. Endogenous molecules mitigate the intensity of the inflammatory response by regulating signaling intensity.
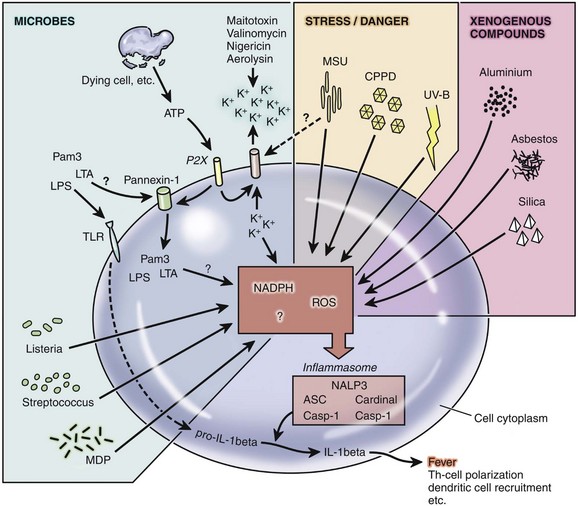
Fig. 3-12 Inflammasome activation by exogenous and endogenous stimuli.
Numerous stimuli can result in activation of the NALP3 inflammasome. Exogenous stimuli include lipopolysaccharide (LPS), lipoteichoic acid (LTA), toxins, monosodium urate crystals (MSU), calcium pyrophosphate dehydrate (CPPD) crystals, ultraviolet (UV)-B waves, aluminum, asbestos, and silica. Endogenous stimuli include ATP, potassium, NADPH and reactive oxygen species (ROS). Once engaged, NALP3 activates caspase 1 which cleaves pro-IL-beta into IL-1 beta and also activates IL-18 (not shown). (Redrawn from Benko S, Philpott DJ, Girardin SE: The microbial and danger signals that activate Nod-like receptors, Cytokine 43(3):368-373, 2008. Epub 2008 Aug 19.)
Antimicrobial Peptides and Collectins
Antimicrobial peptides, such as the α- and β-defensins, cathelicidins, and others—such as anionic peptides, histatin, and dermacidins—are small peptides with microbicidal activity against Gram-negative and -positive bacteria, fungi, mycobacteria, and some enveloped viruses such as human immunodeficiency virus (HIV) (Web Table 3-9). They are encoded by genes of the cells involved in the inflammatory response, especially neutrophils, and by epithelial cells forming the skin and the mucosal barriers of the respiratory and alimentary systems. The microbicidal activity likely occurs through the formation of pores within bacterial membranes and viral envelopes. In addition to the microbicidal activity, antimicrobial peptides are increasingly appreciated for their role in other nonmicrobial activities related to inflammation and wound repair. These activities include chemotaxis of leukocytes and dendritic cells, cell proliferation, wound repair, cytokine release, and the protease-antiprotease balance. There is also much evidence that antimicrobial peptides link the innate and adaptive immune responses.
WEB TABLE 3-9
Inflammatory and Other Activities of Antimicrobial Peptides and Surfactant Proteins
| Antimicrobial Peptide/Protein | Major Cell of Production | Activity |
| α-Defensins | Enterocytes, leukocytes | Microbicidal activity, induce interleukin-8 (IL-8) production (cryptdins), inhibit angiogenesis (neutrophil α-defensins) |
| β-Defensins | Epithelia, leukocytes | Microbicidal activity, chemotactic for neutrophils, dendritic cells, other leukocytes, induce cell proliferation |
| Cathelicidins | Leukocytes, epithelia | Microbicidal activity, induce cytokine and histamine release, regulate cell proliferation, angiogenesis, wound healing, prevent apoptosis (PR-39) |
| Surfactant proteins A and D | Epithelia | Opsonization of pathogens, macrophage activation |
Cryptdins are a type of α-defensin produced by Paneth cells; neutrophil α-defensins are α-defensins produced by human neutrophils; PR-39 is a cathelicidin molecule produced by porcine neutrophils.
There are a wide variety of antimicrobial peptides; however, the defensins have received the most attention for activities other than microbial killing. There are three types of defensins: the α-, β- and θ-defensins. α-Defensins are produced by neutrophils and Paneth cells. β-Defensins are produced by neutrophils and epithelial cells. θ-Defensins are produced in neutrophils of primates. The defensins are cationic proteins with three pairs of intramolecular disulfide bonds. α-Defensins and β-defensins can stimulate mast cell degranulation, induce IL-8 synthesis, induce T-lymphocyte chemotaxis, and activate T lymphocytes, macrophages, and dendritic cells, thus interconnecting innate and adaptive immunity. The importance of defensins in immunity is underscored by the fact that HIV-1 infected individuals that are healthy and in “remission” have high concentrations of α-defensins, which are thought to enhance T-lymphocyte activity and may also have direct anti–HIV-1 activity.
Acute Phase Proteins
Acute phase proteins are plasma proteins synthesized in the liver whose concentrations increase (or decrease) by 25% or more during inflammation. These proteins serve as inhibitors or mediators of the inflammatory processes and include C-reactive protein, α1-acid glycoprotein, haptoglobin, mannose-binding protein, fibrinogen, α1-antitrypsin, and complement components C3 and C4. The concentration of these acute phase proteins usually increases during inflammation, whereas the concentration of prealbumin and albumin (also acute phase proteins) decreases in inflammation. Acute inflammatory conditions that are severe enough to raise blood plasma concentrations of cytokines, such as IL-1 and TNF, increase the blood concentrations of acute phase proteins and elevation of fibrinogen in the blood of cattle is used clinically as an indicator of systemic inflammation. C-reactive protein has recently received attention as a marker of inflammatory conditions, especially atherosclerosis in humans. In addition to its role diagnostically, C-reactive protein binds to bacteria and fungi and also activates complement. Once acute phase proteins and systemic levels of inflammatory cytokines are elevated, they affect the heart rate, blood pressure, and the hypothalamic regulation of temperature by directly or indirectly stimulating neurons within specific hypothalamic nuclei. The previously described changes also affect respiratory rates and gaseous exchange.
Summary of the Chemical Mediators of Acute Inflammation
Various exogenous and endogenous stimuli can activate soluble, surface, or cytoplasmic and endosomal receptors or simply cause mechanical or other damage to invoke an acute inflammatory response. This response can occur very rapidly as the result of the release of preformed or rapidly activated inflammatory mediators such as histamine, kinins, complement factors like C3a and C5a, and tachykinins (substance P). These molecules generally affect vascular caliber and permeability and activate leukocytes and endothelial cells. Concurrently, lipid-based products, such as prostaglandins, leukotrienes, and PAF, contribute to chemotaxis, vascular tone, and leukocyte activity, all of which work in concert with chemokines and cytokines to activate endothelial cells and leukocytes and enhance neutrophil infiltration. NO released by macrophages and endothelia induce vasodilation and also can contribute to tissue damage caused by other reactive oxygen species. Inflammatory mediators bind receptors that subsequently induce cytoplasmic signaling and cellular activation, resulting in the production of additional cytokines, chemokines, adhesion molecules, antimicrobial peptides, and other inflammatory mediators that may either exacerbate or inhibit the inflammatory process. Activated neutrophils expressing leukocyte adhesion molecules, such as the integrins, enter inflamed tissue and can release hydrolytic enzymes and other granule contents that further damage tissue. Systemically, increases in acute phase proteins, cytokines, chemokines, complement, and inflammatory proteins can affect body temperature, cardiovascular function, locomotion, sleep, appetite, and other activities (Table 3-5).
TABLE 3-5
Examples of Differing Types of Inflammatory and Clinical Responses to Substances
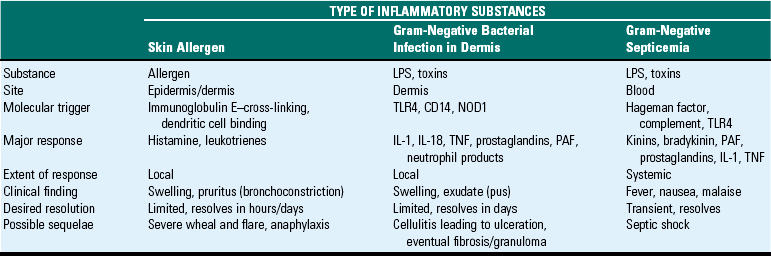
CD14 = receptor for LPS-binding protein.
Hageman factor = clotting factor.
IL-1, Interleukin-1; IL-18, interleukin 18; LPS, lipopolysaccharide (endotoxin); PAF, platelet-activating factor; NOD1, nucleotide-binding and oligomerization domain; TLR4, Toll-like receptor 4; TNF, tumor necrosis factor.
Reparative Phase of the Acute Inflammatory Response
Outcomes of the Acute Inflammatory Response
The four main outcomes of acute inflammation are as follows:
The severity of tissue damage, the ability of cells to regenerate, and the physical or biologic characteristics of the cause of the injury determine these outcomes. In the acute inflammatory response (Fig. 3-13), the desired outcome is resolution, that is, the complete return of normal structure and function. Resolution occurs if the following occur:
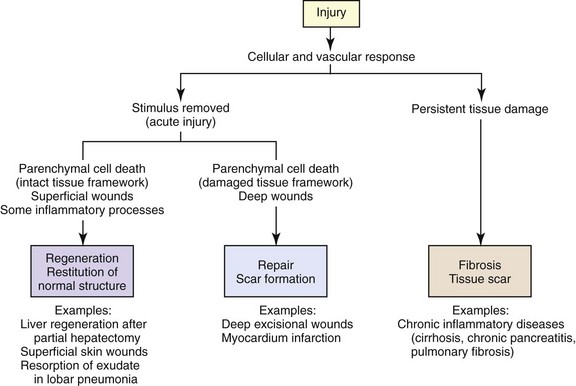
Fig. 3-13 Repair, regeneration, and fibrosis after injury and inflammation. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
• The acute inflammatory response is completed in the correct sequence.
• Macrophages and lymphatic vessels remove the exudate.
• The inciting agent or substance is eliminated.
• The stroma (connective tissue) of the affected tissue is intact and can provide support for regeneration of epithelial cells.
• Ulcerated or necrotic epithelial cells are replaced by regeneration of adjacent epithelial cells on an intact basement membrane.
Mechanistically, the critical first stage of resolution involves the killing and/or removal of the inciting cause, removal of chemical mediators by neutralization or decay, return to normal vascular flow and capillary permeability, cessation of leukocyte emigration, apoptotic cell death of remaining neutrophils in the exudate, the removal of the exudate by monocyte-macrophage phagocytosis, and drainage to regional lymph nodes (Fig. 3-14). Inflammatory responses of neutrophils, monocytes, and macrophages are further inhibited by chemokine inactivation and release of products such as lipoxin A4, resolvins, annexin A, lactoferrin, and lysophosphatidylcholine.
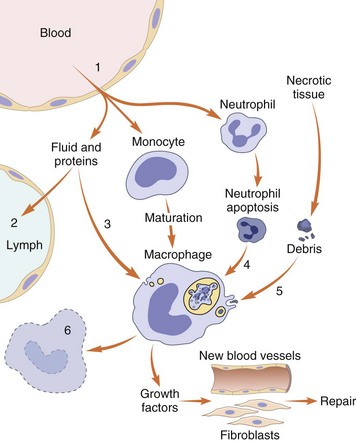
Fig. 3-14 Events in the resolution of inflammation.
1, Return to normal vascular permeability; 2, drainage of edema fluid and proteins into lymphatics or 3, by pinocytosis into macrophages; 4, phagocytosis of apoptotic neutrophils and 5, phagocytosis of necrotic debris; and 6, disposal of macrophages. Macrophages also produce growth factors that initiate the subsequent process of repair. Note the central role of macrophages in resolution. (Modified from Haslett C, Henson PM: Resolution of inflammation. In Clark R, Henson PM, editors: The molecular and cellular biology of wound repair, New York, 1996, Plenum Press.)
Regeneration is the second stage of resolution and depends on the availability of progenitor epithelial cells and the presence of a supportive stroma and intact basement membrane for orderly cellular migration. As an example, acute renal tubular necrosis can be caused by aminoglycoside antibiotics and results in detachment and necrosis of tubular epithelial cells from the tubular basement membrane. If the supporting stroma and basement membrane remain intact, progenitor epithelial cells can divide and migrate to replace lost cells and thus return the tubule to normal function. If the basement membrane is not intact to guide the proliferating cells, functional tubules will not form. Instead, regenerative tubular epithelial cells will atrophy or form into small aggregates with syncytial giant cells. Simultaneously, the microvascular supply to the region must be restored, which occurs mechanistically by the proliferation of endothelial cells in response to molecules such as VEGF.
Nomenclature of The Inflammatory Response (Morphologic Diagnoses)
Nomenclature, a system of names assigned to structures and processes in a scientific discipline, used in veterinary pathology provides clinicians with a morphologic diagnosis, a precise description of the process, type of inflammation, and disease. A morphologic diagnosis has six components listed in the following sequence: degree of severity, duration, distribution, exudate, modifier, and tissue (Table 3-6). Based on the results of a postmortem examination and/or histologic evaluation of tissue specimens, a pathologist will construct a morphologic diagnosis by including in sequential order the components of the nomenclature that best describe the specimens. For example, using the kidney as the injured tissue, the central component of a morphologic diagnosis is the name of the tissue derived from its Latin term, “nephro-.” If the kidney is inflamed, the prefix “nephro-” is combined with the suffix “itis” (inflammation or disease of) to form the word “nephritis,” meaning inflammation of the kidney. The other components of the morphologic diagnosis, such as degree, duration, distribution, exudate, and modifier, precede nephritis and are used to describe the characteristics of the inflammatory process. The pattern of distribution of the inflammatory lesion not only provides location but also in many instances implies a mechanism of injury. These patterns of distribution as shown in Fig. 3-15 include focal, multifocal, locally extensive, and diffuse. They are covered in greater detail in each organ system. The subtleties of this process are learned through advanced training in the discipline of pathology, and terms describing the degree and duration evolve from years of professional experience and are not likely to be mastered by veterinary students during their professional training.
TABLE 3-6
The Nomenclature of a Morphologic Diagnosis
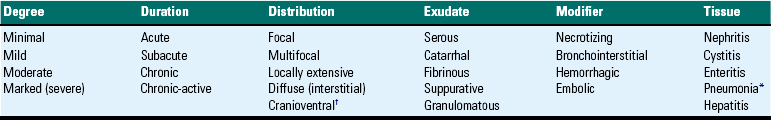
This table provides an example of how nomenclature can be used to construct a morphologic diagnosis. It is not intended to be all-inclusive and may vary from schemes used in other veterinary colleges.
*In the lung, it is customary to use the term pneumonia to indicate inflammation of the lung.
†Used only for diseases of the lungs.
Fig. 3-15 Patterns of lesion distribution used to construct morphologic diagnoses, as an example, in the kidney. (From Slauson DO, Cooper BJ: Mechanisms of disease: a textbook of comparative general pathology, ed 3, St Louis, 2002, Mosby.)
Morphologic diagnoses can also use the suffixes “osis” (diseased or abnormal condition) or “opathy” (disease). In this context, such diseases or conditions refer to those caused by degenerative or aging processes without inflammation. Therefore the same nomenclature can be used to diagnose a degenerative condition in the kidney by using the term nephrosis or the term nephropathy. The other components are then added to nephrosis or nephropathy to describe the characteristics of the degenerative process. Finally, some metabolic and neoplastic diseases do not fit into this nomenclature, but equally valid morphologic diagnoses can be constructed. An enlarged, soft and friable, yellow, and greasy liver can be morphologically diagnosed as “fatty liver” (hepatic lipidosis), whereas a solid, firm, white expansile mass in the liver could be morphologically diagnosed as “hepatic malignant lymphoma.”
Morphologic Classification of Exudates in Acute Inflammatory Lesions
The gross and microscopic appearance of different types of acute inflammatory reactions in tissue can often be classified according to the vascular and cellular components of the response, thus providing a mechanistic basis for understanding the pathogenesis. Histopathologic lesions of acute inflammation are most commonly grouped into five categories: serous, catarrhal, fibrinous, suppurative or purulent, and hemorrhagic, or a combination of these lesions such as fibrinosuppurative. Similar histopathologic patterns of lesions also occur in chronic inflammation (lymphohistiocytic or granulomatous [macrophages, multinucleate giant cells, lymphocytes, plasma cells, fibrosis]).
It should be recognized that the histopathologic lesions of acute inflammation often represent (1) a continuum of progressive changes of the same type of inflammation occurring over time or (2) different types of inflammatory responses occurring concurrently in the same or different areas of an affected tissue. Therefore rhinitis, for example, could progress in a sequence from serous to catarrhal to mucopurulent to purulent. If the inciting stimulus is severe, changes could progress rapidly from serous to fibrinous to hemorrhagic.
Serous Inflammation
Serous inflammation is the term used to describe a pattern of acute inflammation in which the tissue response consists of the leakage or accumulation of fluid with a low concentration of plasma protein and no to low numbers of leukocytes. This watery material is released from small gaps between endothelial cells and from hypersecretion of inflamed serous glands. This response is essentially a transudate (specific gravity <1.012) and is seen with (1) thermal injury to skin, such as burns and photosensitization, in which the lesion can appear as a fluid-filled blisters, or (2) acute allergic responses characterized by watery eyes and a runny nose with a clear, colorless transudate.
Grossly, lesions, for example, with serous inflammation consist of tissue that (1) contains excessive clear to slightly yellow, watery fluid that leaks from the tissue on cut section or (2) forms raised fluid-filled vesicles protruding above the surface of the mucous membrane of the nasal cavity (serous rhinitis) or of the skin (Fig. 3-16). Microscopically, connective tissue fibers are separated, often widely, and capillaries and postcapillary venules are dilated with erythrocytes (active hyperemia). Endothelial cells lining these vessels can vary from flattened to hypertrophied.
Fig. 3-16 Serous exudate/subcutaneous edema, photosensitization, skin of the nose and ears, ewe.
A, The nonhaired skin of the nose is covered by a crust resulting from dehydration of the serous exudate released from injured blood vessels following a short exposure to the sun. The ears are edematous and droopy. B, Microscopically, there is moderate expansion of the superficial dermis with edema secondary to vascular leakage (serous inflammation). The postcapillary venules are dilated (active hyperemia), and leukocytes have marginated on endothelial cells. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Catarrhal Inflammation
Catarrhal inflammation, or mucoid inflammation, is the term used to describe a pattern of acute inflammation in which the tissue response consists of the secretion or accumulation of a thick gelatinous fluid containing abundant mucus and mucins from a mucous membrane. This response occurs most commonly in tissue with abundant goblet cells and mucus glands, such as in certain types of chronic allergic and autoimmune gastrointestinal diseases and with chronic inflammation of the airways of the respiratory system (chronic asthma). Grossly, the surface or cut surface of affected tissue may be covered with or contain a clear to slightly opaque, thick fluid (Fig. 3-17). Microscopically the lesion can include hyperplastic epithelial cells of mucus glands and goblet cells, as well as connective tissue fibers separated by mucins.
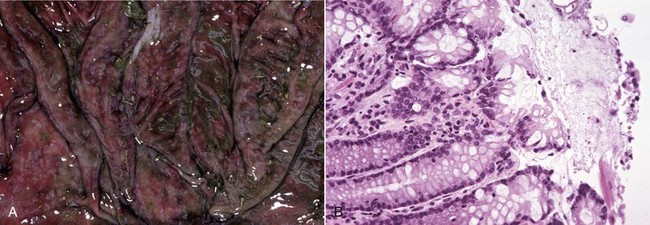
Fig. 3-17 Catarrhal inflammation.
A, Abomasum, cow. The mucosal epithelium is moderately thickened, covered by a glistening layer of clear mucus, and has a subtle nodular appearance caused by accumulation of mucinous secretory products (catarrhal exudate) in the gastric pits. B, Colon, cow. Microscopically, there is a catarrhal colitis with hyperplasia of mucosal epithelial cells and increased accumulation of mucus on the mucosal surface. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Fibrinous Inflammation
Fibrinous inflammation is the term used to describe a pattern of acute inflammation in which the tissue response consists of the accumulation of fluid with a high concentration of plasma protein (specific gravity >1.02) and no to low numbers of leukocytes. This response is an exudate. Fibrinous inflammation occurs with more severe endothelial cell injury that allows leakage of large molecular weight proteins such as fibrinogen. Fibrinogen leaks from capillaries and postcapillary venules during the fluidic phase of the acute inflammatory response and polymerizes outside of the vessels to fibrin, a homogeneous and vividly pink (eosinophilic) protein when stained with hematoxylin and eosin (H&E). This lesion is most commonly caused by infectious microbes and is seen in the serous membranes of the body cavities, such as those lined by pleura (fibrinous pleuritis), pericardium (fibrinous pericarditis), peritoneum (fibrinous peritonitis), and in the synovial membranes of joints (fibrinous synovitis) and the meninges (fibrinous leptomeningitis). Common examples include the lesions in pulmonary alveoli in fibrinous pneumonia (Mannheimia haemolytica), atypical interstitial pneumonia (3-methyl indol), and respiratory viral infections (bovine herpesvirus-1 in cattle). When fibrin forms a distinct layer covering an ulcer, it is referred to as a fibrinous pseudomembrane, whereas when it lines the pneumonocyte surface of lung alveoli in a curvilinear fashion and is mixed with necrotic cell debris, such as occurs with bovine respiratory syncytial virus (BRSV) infection, it is called a hyaline membrane.
Grossly, the surfaces of affected tissue are red (active hyperemia) and covered with a thick, stringy, elastic, white-gray to yellow exudate that can be removed from the surface of the tissue (in contrast to a fibrous response) (Fig. 3-18; see Fig. 3-4). A classic example of fibrinous inflammation occurs in fibrinous pneumonia caused by acute Mannheimia haemolytica infection, in which both alveoli and stromal connective tissue (interlobular septa and pleura) contain notable fibrinous exudate that rapidly becomes infiltrated by neutrophils, resulting in a fibrinosuppurative exudate. Another example of fibrinous inflammation occurs in bovine herpesvirus-1 infection. This virus injures epithelial cells of the respiratory tract, resulting in an acute fibrinous inflammatory response. Noninfectious causes, such as heat and smoke inhalation, can cause fibrinous exudate in the trachea. Microscopically, capillaries and postcapillary venules are dilated with erythrocytes (active hyperemia), and endothelial cells are reactive (hypertrophied). The stromal connective tissue or the mesothelial surfaces of the affected organ contains or is covered by red to vividly red layers of coagulated and/or polymerized fibrin, albumin, and other plasma proteins. The fibrinous exudate is often rapidly infiltrated by neutrophils, resulting in fibrinosuppurative inflammation.
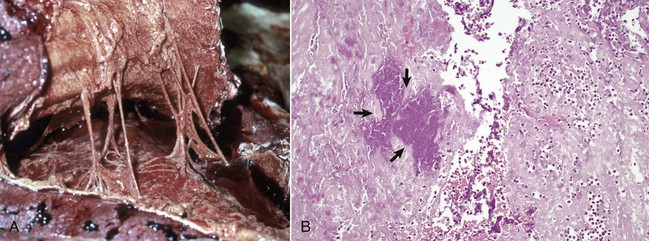
Fig. 3-18 Fibrinous inflammation, pleural cavity, visceral and parietal pleurae, horse.
A, The pleural surfaces are covered by a yellow-gray, thick, friable exudate consisting of fibrin mixed with other plasma proteins. This exudate can be easily pulled apart and should not be confused with fibrous adhesions. The latter response occurs over time and consists of a similar appearing material that contains collagen fibers, which provide tensile strength to the material and form adhesions between opposing surfaces that can only be broken with some difficulty. B, Microscopically, there are layers of homogeneous red material (fibrin-fibrinous exudate) that contain occasional neutrophils and a focus of bacteria (arrows). H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Suppurative Inflammation
Suppurative inflammation is the term used to describe a pattern of acute inflammation in which the tissue response consists of the accumulation of fluid with a high concentration of plasma protein (specific gravity >1.02) and high numbers of leukocytes, predominately neutrophils. This material is an exudate commonly known as pus. Pus can be a creamy liquid, but if dehydrated, it can be more caseous and firm in consistency and occasionally laminated in diseases such as ovine caseous lymphadenitis. A collection of pus circumscribed by a fibrous capsule that is visible grossly is called an abscess, whereas, if visible only microscopically, it is called a microabscess.
Dense neutrophil accumulation (pus) can also be distributed in tissue layers such as fascial planes and subcutaneous connective tissue and is referred to as cellulitis or phlegmonous inflammation. Instead of producing a focal abscess, the neutrophils evoke a watery suppurative exudate distributed along fascial planes and tissue spaces, as in some cases of blackleg or extensive Gram-positive (staphylococcal) infection. Suppurative inflammation, microabscesses, abscesses, and exudates are most commonly caused by bacteria, including Staphylococcus spp., Streptococcus spp., and Escherichia coli, and can occur in many organs. These genera can also cause suppurative bacterial meningitis in the central nervous system (CNS). Abscesses in the brains of horses caused by Streptococcus equi and microabscesses in the brains of cows caused by Listeria monocytogenes are good examples of suppurative inflammation. Bacteria-induced suppurative inflammation also occurs commonly in (1) the pelvis and tubules (pyelonephritis) of the kidney, (2) the bronchi of the lungs (bronchopneumonia), (3) the nasal and sinus cavities (rhinitis and sinusitis), (4) the glandular epithelium of the prostate (prostatitis), (5) the lumina of the gall bladder (cholecystis) and urinary bladder (urocystitis), and (6) the acini and ducts of the mammary gland (mastitis). Unresolved suppurative inflammation can progress to chronic inflammation.
Grossly the surfaces and/or connective tissues of affected organs are hyperemic and covered by or contain, respectively, a thick white-gray to yellow pus (Fig. 3-19). In some cases, pus will be mixed with fibrin, forming a fibrinosuppurative exudate. Microscopically, affected tissues have large numbers of neutrophils; many degenerate and often are mixed with necrotic cellular debris, bacteria, plasma proteins, and fibrin.
Fig. 3-19 Suppurative (purulent) inflammation, secondary bacterial bronchopneumonia, infectious canine distemper, puppy.
A, The cranioventral areas of the lung are firm and beige to brown. This lesion is caused by neutrophils transmigrating into alveoli in an acute inflammatory response secondary to bacterial infection of the lung. B, Microscopically, alveoli contain numerous neutrophils (suppurative exudate) and sloughed pneumocytes. H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Chronic Inflammation
Chronic inflammation is inflammation of prolonged duration (weeks to months to years) that occurs (1) when the acute inflammatory response fails to eliminate the inciting stimulus, (2) after repeated episodes of acute inflammation, or (3) in response to unique biochemical characteristics and/or virulence factors in the inciting stimulus or microbe. Table 3-7 lists some of the most common causes of chronic inflammation in domestic animals. The underlying biologic mechanisms that result in chronic inflammation include persistence/resistance, isolation in tissue, unresponsiveness, autoimmunity, and unidentified mechanisms.
TABLE 3-7
Selected Examples of Conditions That Can Cause or Lead to Chronic Inflammation in Domestic Animals
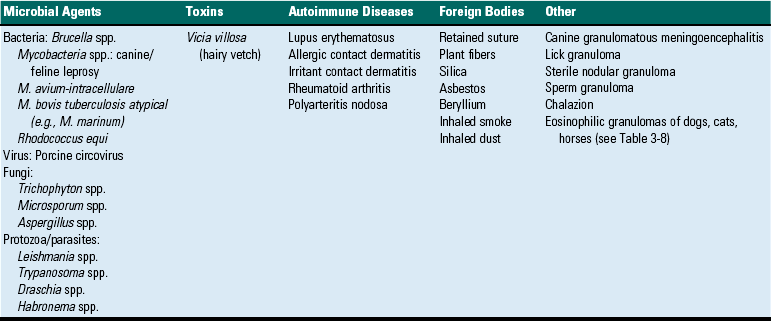
• Persistence/resistance: Persistent infections, such as those caused by Mycobacterium spp.; Nocardia spp.; deep-seated mycoses, such as Blastomyces dermatitidis (Web Fig. 3-11) and Histoplasma capsulatum; and parasites, such as Toxocara canis larvae, can avoid and/or resist phagocytosis by neutrophils and macrophages—or once internalized by these cells, can prevent fusion of primary and secondary lysosomes or killing by lysosomes. Such microbes also usually do not produce biologic molecules that cause severe tissue injury, but their presence continually incites chronic inflammatory and immune responses. Some microbial agents can induce macrophages to undergo apoptosis and subsequent internalization by adjacent macrophages. Tissue destruction, granulomatous inflammation, and fibrosis are common sequelae of persistent/resistant infectious agents.
• Isolation: Some microbes, such as Streptococcus and Staphylococcus spp., are not naturally resistant to phagocytosis and/or destruction but are able to isolate themselves from effective innate and adaptive immune responses and from antimicrobial drugs by “hiding” themselves in pus.
• Unresponsiveness: Certain foreign materials are virtually indestructible and therefore are unresponsive to phagocytosis and/or enzymic breakdown. These include plant material, grass awns, silica dust, asbestos fibers, some suture materials, and surgical prostheses.
• Autoimmunity and leukocyte defects: Alterations in the regulation of adaptive immune responses to self-antigens result in autoimmune diseases, such as polyarteritis nodosa, with a chronic inflammatory response. Defects in leukocyte function can also result in chronic inflammation. For example, loss of NADPH oxidative function in human patients with chronic granulomatous disease, impair leukocyte free radical formation and oxidative killing, thus allowing persistence of microbial agents or internalized material.
• Unidentified mechanisms: In some diseases, such as canine granulomatous meningoencephalitis, the cause of chronic inflammation remains unknown.
WEB TABLE 3-11
Composition and Tissue Specificity of Selected Glycosaminoglycans (GAGs)
| GAG | Carbohydrate Components | Localization |
| Hyaluronate | Hyaluronic acid | Synovial fluid, vitreous humor, extracellular matrix of connective tissue |
| Chondroitin sulfate | d-glucuronate + N-acetylgalactosamine | Cartilage, bone, heart valves |
| Heparan sulfate | Glucuronate + glucosamine | Basement membranes, cell surfaces, intracellular granules of mast cells |
| Heparin | Glucuronate + glucosamine | Lines arteries of lungs, liver, skin |
| Dermatan sulfate | l-idurante + N-acetylgalactosamine | Skin, blood vessels, heart valves |
| Keratan sulfate | Galactose + N-acetylglucosamine | Cartilage, aggregated with chondroitin sulfate |
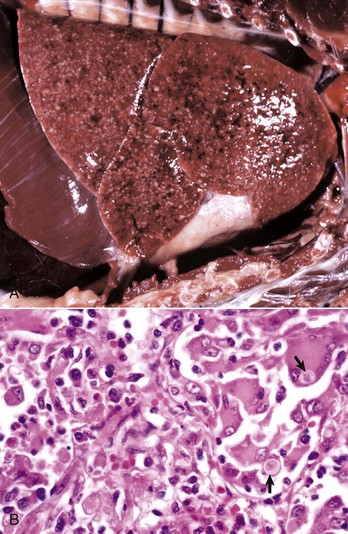
Web Fig. 3-11 Granulomas and pyogranulomas, blastomycosis, lung, dog.
A, Numerous multifocal discrete and coalescing variably sized gray-white nodules are scattered at random (embolic pattern) in the lung parenchyma. They are composed of granulomatous inflammatory cells. Note that the lung lobes fail to collapse. B, The granulomatous inflammatory exudate contains activated and multinucleate giant cells with intracellular Blastomyces dermatitidis organisms (arrows). Lymphocytes and plasma cells are also present in the exudate. H&E stain. (A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The chronic inflammatory response is maintained by cytokines, chemokines, and other inflammatory mediators that are released and incite (1) ongoing inflammation mediated by infiltration and activation of lymphocytes, macrophages, plasma cells, and multinucleated giant cells (MGCs); (2) tissue destruction (necrosis); (3) proliferation of fibroblasts and deposition of collagen (desmoplasia and/or fibroplasia); (4) angiogenesis and neovascularization (granulation tissue formation); and (5) initiation of wound healing (reepithelialization and tissue repair).
Beneficial and Harmful Aspects of Chronic Inflammation
The body initially responds to injury through acute inflammation. Once the acute inflammatory response fails to overcome the inciting agent or persistent substance, chronic inflammation ensues as the body attempts to overcome the inciting agent/substance via a variety of cells such as NK cells, lymphocytes, macrophages and the adaptive immune response. If these responses fail, the inciting agent/substance is then “walled-off” with collagen produced by fibroblasts, encapsulating the agent/substance and functionally placing it “outside” of the body. Some types of responses, such as lepromatous (diffuse) granulomatous reactions, do not form defined fibrous capsules or walls and instead separate the agent/substance by dense accumulations of macrophages and fibroblasts that are arranged irregularly. Often, this response can be beneficial and in time can lead to a return to normal activity. Small granulomas or abscesses in the lung, liver, or even in some areas of the skin, with time, eventually go unnoticed by the innate and adaptive immune systems and do not stimulate pain or mechanical interference to movement or function.
On the other hand, chronic inflammation can be detrimental. The mononuclear leukocyte infiltrates (macrophages, lymphocytes, and NK cells) within areas of chronic inflammation take up space and often displace, replace, and sometimes obliterate the original tissue. At the same time, new blood vessels form, fibroblasts proliferate and deposit collagen, and if the lesion expands, the inflammatory response can affect function of adjacent tissues and/or cells and ultimately the function of the entire organ. For example, chronic inflammatory lesions in the intestine of dogs and cats with inflammatory bowel disease (IBD) can induce progressive weight loss and debilitation. Also, chronic inflammation in the brains of dogs with granulomatous meningoencephalitis, can destroy neurons and glia, impinge and obstruct flow of cerebrospinal fluid (CSF) in the ventricular system, and elevate the intracranial pressure, all of which can impair cognition and movement.
The extent of debilitation in animals with chronic inflammatory lesions depends on the location of the lesion and extent of tissue involvement. Even very small chronic lesions in the brain can rapidly incite clinical signs through either the destruction of neuroparenchyma or, perhaps, by impairing flow or resorption of CSF. In contrast, some widespread chronic inflammatory lesions, such as those in inflammatory bowel disease of dogs and cats and Johne’s disease of cattle, can involve extensive areas of the intestine and often precede clinical signs (diarrhea) by months or even years. Yet other diseases, such as embolic hepatic or lung abscesses or disseminated tuberculoid granulomas, can be debilitating over time through the loss of parenchymal function and the continual release of inflammatory mediators, such as TNF and IL-1, both of which affect temperature and appetite.
In chronic inflammation, the first clinical intervention is to remove the inciting factor, if possible. Thus antibiotics and antifungal drugs are used against bacterial and mycotic infections. Some foreign bodies can be removed surgically, and it may be possible to identify, opsonize, chelate, or sequester immunologic allergens, antigens, and nondegradable substances. Unfortunately, few medical therapies completely resolve certain types of chronic inflammation, especially if granulomas and/or extensive scar tissue have developed. In the future, perhaps surgical debulking of large lesions may be followed by gene or stem cell therapy to effectively eliminate specific types of granulomas, such as those caused by mycobacterial infection or inducing apoptosis of fibroblasts, myofibroblasts, and macrophages.