Progression of the Acute Inflammatory Response to Chronic Inflammation, Fibrosis, and Abscess Formation
Acute inflammatory responses can either fully resolve with return of the tissue to normal structure and function or repair by healing (Fig. 3-20). If conditions do not allow for complete resolution of the acute inflammatory response, four outcomes can result: (1) progression to chronic/granulomatous inflammation, (2) healing by fibrosis, (3) healing with increased cellularity (cerebral gliosis, Kupffer cell hyperplasia, or glomerular mesangial cell hyperplasia), or (4) abscess formation. These outcomes are determined by the severity of tissue damage, the ability of cells to regenerate, and the biologic characteristics of the agent/substance (e.g., mycobacterial waxes, poorly degradable plant fibers) that caused the injury.
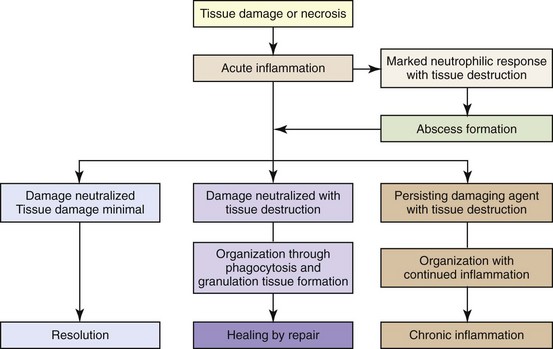
Fig. 3-20 The outcomes of tissue injury and unresolved acute inflammation. (Modified from Young B, O’Dowd G, Stewart W: Wheater’s basic pathology: a text, atlas and review of histopathology, ed 5, New York, 2010, Churchill Livingstone.)
Progression to Chronic/Granulomatous Inflammation
Progression to chronic/granulomatous inflammation occurs when the acute inflammatory response fails. Failure is characterized by the following:
• The inciting stimulus persisting for a long period of time (weeks to months)
• Extensive tissue injury and necrosis (third-degree burn)
• A shift of the cellular elements of the inflammatory response from neutrophils to lymphocytes, macrophages, and sometimes multinucleate giant cells
• Extensive connective tissue reorganization followed by fibrosis (fibroplasia)
Examples of agents and substances that often result in chronic inflammatory responses include systemic mycoses, such as Blastomyces dermatitidis and Histoplasma capsulatum; intracellular bacterial pathogens, such as Nocardia, Brucella, Mycobacterium, or Salmonella spp.; protozoa, such as Leishmania or Trypanosoma spp.; parasites, such as Toxocara larva or Habronema; autoantigens, such as those occurring in sperm granulomas or in autoimmune diseases like lupus erythematosus; and foreign bodies (plant awns, sticks, metals, asbestos, or suture material). Such agents continually induce the release of inflammatory mediators from indigenous parenchymal cells and leukocytes, leading to macrophage infiltration and activation; T-lymphocyte, NK cell, and perhaps mast cell or eosinophil infiltration; and fibroblast and endothelial cell proliferation. Some of the inflammatory cytokines, such as TGF-β, may interfere with regeneration of epithelial and parenchymal cells (see section on Wound Healing and Angiogenesis).
Healing by Fibrosis
Healing by fibrosis occurs after tissue injury in which there is necrosis of the tissue framework provided by stromal elements (connective tissue) and of the epithelial cells required to regenerate and successfully reconstitute the parenchymatous elements of the tissue. After necrosis, dead tissue and the acute inflammatory exudate are removed by macrophages (phagocytosis by cells of the monocyte-macrophage system), and the space is filled with fibrovascular tissue (granulation tissue) commonly seen in the healing process. Granulation tissue is eventually replaced by immature fibrous connective tissue that is poorly collagenized and then by mature connective tissue that is well collagenized, healing the wound and forming a scar (cicatrix). Structural integrity may be reestablished, but functional integrity depends on the extent of the loss of parenchymal cells. For example, with severe skin burns or extensive lacerations, dermal scarring eventually replaces lost dermal structures and to a limited extent restores structural integrity; however, the skin’s functional integrity is extremely limited because of the loss of adnexal glands, hair follicles, and scar tissue that reduces the range of motion in joints of the limbs and digits. The degree and extent of fibroblast and myofibroblast proliferation in such wounds largely depends on mediators such as TGF-β and IL-13 (see the section on Wound Healing and Angiogenesis).
Abscess Formation
Abscess formation (Fig. 3-21) occurs when the acute inflammatory response fails to rapidly eliminate the inciting stimulus and the enzymes and inflammatory mediators from neutrophils in the exudate liquefy the affected tissue and neutrophils to form pus. Myeloperoxidase enzyme in neutrophils contributes to neutrophil necrosis and liquefaction. The presence of myeloperoxidase is an evolutionary phenomena and reptiles and birds lack this enzyme and are therefore unable to liquefy neutrophils into pus. Abscesses can have a septic or sterile origin. Septic abscesses most commonly originate from bacterial infection, whereas sterile abscesses arise from incompletely degraded foreign bodies or from the failure of injected medications to be completely absorbed. Pyogenic bacteria, such as Staphylococcus and Streptococcus spp., commonly cause septic abscesses. They enter tissue hematogenously or by direct extension from the skin following trauma. The pus within an abscess can range in consistency from serous to purulent to caseous and in color from white, to yellow, to green, depending on the inciting agent/substance. The color of the exudate often depends on the pigment produced by the inciting bacterium and the species; for example, yellow exudates are caused by abscesses formed by Staphylococcus, Streptococcus spp., and Corynebacterium ovis; green exudate is caused by abscesses formed by Pseudomonas aeruginosa; and red exudate is caused by abscesses formed by Serratia marcescens.
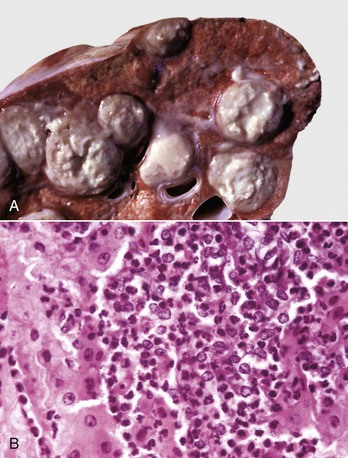
Fig. 3-21 Abscess formation.
A, Abscess, lung, cow. A cut section of lung has numerous abscesses. Note the white-to-gray exudate and how it bulges from the cut surface. B, The exudate in A consists of cell debris and a large number of neutrophils admixed with lesser numbers of degenerating macrophages and lymphocytes, and bacteria (the latter not visible with H&E stain). H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
After an acute inflammatory response has been established, the development of an abscess in this site consists of a collection of neutrophils admixed with cell debris, macrophages, and fibroblasts with variable infiltrates of lymphocytes. Experimentally, such a site can form in 2 to 3 days, depending on the agent/substance. Fibroblasts in this site begin to produce collagen and ECM proteins that can form into a “thin” vascularized connective tissue area. At this point, antibiotics can penetrate this area and enter the exudate. If the septic abscess persists, the initial “thin” connective tissue area surrounding the exudates can mature into a fibrous capsule, which is thick and largely impermeable, in an attempt to “wall-off” the exudates from normal tissue. A capsular wall takes weeks to form. Abscesses with this response can present serious problems in systemic (hematogenous) or local (topical diffusion) antibiotic treatments. In large abscesses with abundant pus, the pus itself may dilute the antibiotic and further prevent the drug from reaching the optimal concentration required to kill the bacteria. It is for these reasons that larger abscesses are often lanced to drain the pus. Sterile abscesses do not require antibiotics or other drugs to kill an inciting agent/substance, but they do require breakdown of the capsule by lancing or some other means.
Granulomatous Inflammation and Granuloma Formation
Granulomatous inflammation is a distinct type of chronic inflammation in which cells of the monocyte-macrophage system are predominant and take the form of macrophages, epithelioid macrophages (activated macrophages), and MGC. In granulomatous inflammation, the cells are dispersed as sheets of cells distributed at random (diffuse or lepromatous) within parenchymal and connective tissue planes (Fig. 3-22, A), whereas in a granuloma (tuberculoid granuloma), they are arranged in distinct masses or nodules (Fig. 3-22, B).
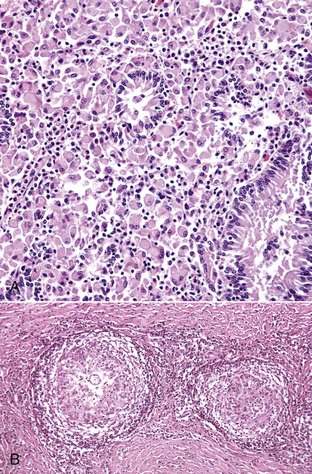
Fig. 3-22 Granulomatous inflammation and granulomas.
A, Granulomatous inflammation in Johne’s disease, ileum. The lamina propria contains a solid sheet of granulomatous inflammatory cells, which is characteristic of lepromatous (diffuse) granulomatous inflammation, H&E stain. B, Nodular (tuberculoid) granuloma in coccidioidomycosis. Granulomas are round to oval with a central core of granulomatous inflammatory cells and a peripheral zone of fibroblasts, which may produce a fibrous capsule. The granuloma on the left contains a single central fungal element. H&E stain. (A courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Granulomatous inflammation occurs secondarily in response to endogenous or exogenous antigens or idiopathically as in granulomatous meningoencephalitis of dogs. Development and regulation of granulomatous inflammation requires multiple factors: (1) the inciting agent, usually with indigestible, poorly degradable, and persistent antigens (e.g., Mycobacteria spp.); (2) the host immune response (e.g., TH and macrophage response); and (3) the interplay of various cytokines, chemokines, and other proinflammatory and antiinflammatory mediators produced by cells within the chronic inflammatory lesion.
Classification of granulomatous inflammation by pathologists has evolved over the years because of the increased understanding of disease pathogenesis and advances in molecular biology. For simplicity, this chapter discusses two morphologic forms of granulomatous inflammation: diffuse (lepromatous) granulomas, which are currently thought to be consistent with a TH2-biased immunologic response, and nodular (tuberculoid) granulomas, which are currently thought to be consistent with a TH1-biased immunologic response. Both of these terms are derived from granulomatous lesions in humans and are increasingly becoming defined and seen as distinct, both immunologically and molecularly.
Nodular (Tuberculoid) Granulomas (TH1-Biased Granulomas)
Examples of nodular (tuberculoid) granulomas are those that have been classically caused by Mycobacterium bovis or Mycobacterium tuberculosis (Fig. 3-23) and by some deep fungal infections, such as coccidioidomycosis (Web Fig. 3-12). Grossly, nodular granulomas are often gray to white, round to oval, and firm to hard, whereas diffuse granulomatous inflammation is often gray to white; expansile but poorly demarcated from adjacent tissue; and firm. Tuberculoid (nodular) granulomas develop with a TH1-type lymphocytic response and occur in many species but have been described extensively from lesions of infected humans, cattle, and rhesus monkeys. Because the portal of entry is often the respiratory tract, these lesions involve the lung with secondary involvement of other parenchymal organs and induce the formation of granuloma. Microscopically, nodular (tuberculoid) granulomas may or may not have a central core of necrotic cell debris (caseating and noncaseating granulomas) (see Fig. 3-23). Granulomas of either type are often oval to round and can be irregular as well as multinodular, vary in size from microscopic to macroscopic, and very large.
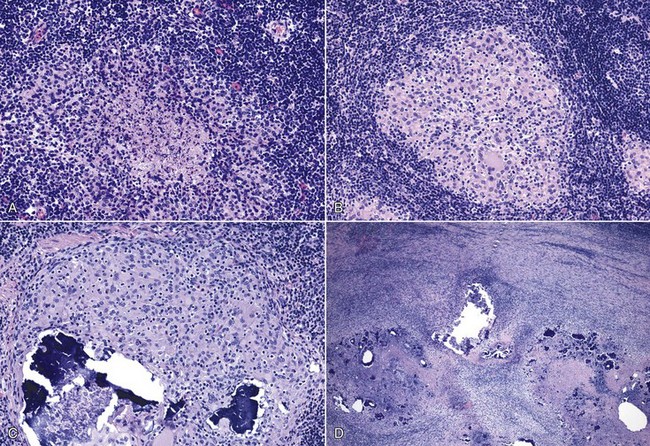
Fig. 3-23 Nodular (tuberculoid, TH1) type of granulomatous inflammation.
A series of micrographs from lymph nodes of cattle experimentally infected with Mycobacterium bovis illustrate stages of TH1 granuloma formation. A, Stage I granuloma. Initial lesions have a central region of cell debris with occasional neutrophils and macrophages that are surrounded by a zone of macrophages that are irregularly arranged and further surrounded by densely associated lymphocytes. B, Stage II granuloma. Days later, granulomas are composed of numerous macrophages aggregated in an oval region with occasional epithelioid macrophages and small multinucleate giant cells. This core is surrounded by dense infiltrates of lymphocytes. C, Stage III granuloma. The mature granuloma has a central area of mineralization along with numerous macrophages, multinucleate giant cells and epithelioid macrophages. D, Stage IV granuloma. With long-term persistence of antigen, the granulomatous reactions with areas of mineralization coalesce and overtake additional surrounding tissue and are bordered by dense infiltrates of lymphocytes. H&E stain. (Courtesy Dr. M. Palmer, USDA/ARS-National Animal Disease Center, Ames, Iowa.)
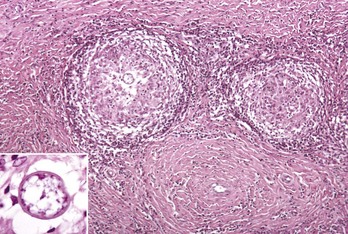
Web Fig. 3-12 Nodular (tuberculoid) type of granulomatous inflammation, coccidioidomycosis.
Granulomas are round to oval with a central core of numerous macrophages surrounded by lymphocytes, plasma cells, macrophages, and a peripheral zone of fibroblasts, which produce a fibrous capsule. The granuloma on the left contains a single central fungal element (inset). H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Noncaseating granulomas are often round to oval and microscopically composed of numerous macrophages with variable numbers of epithelioid macrophages, perhaps multinucleate giant cells, with a peripheral zone of fibroblasts, lymphocytes, and plasma cells. Caseating granulomas have the same morphologic features as noncaseating granulomas; however, the center is formed by a core of gray-white-yellow pasty (thick-dehydrated) necrotic debris resembling cheese (Latin caseus = cheese). Caseating granulomas most commonly occur in tuberculosis. Microscopically, caseating granulomas have a central core of cell debris, surrounded by a dense zone of macrophages that may contain epithelioid macrophages that are admixed increasingly in the outer layers of granuloma with lymphocytes, plasma cells, and fibroblasts.
The outermost zones of either a noncaseating or caseating granuloma are frequently similar and composed of fibroblasts that deposit collagen and ECM proteins that create a dense fibrous region that can form into a capsule. Thus a well-formed granuloma has three distinctive morphologic areas. The innermost area is often but not always a centrally located region of macrophages and multinucleate giant cells in a noncaseating granuloma and cellular necrosis in a caseating granuloma, which is surrounded by a middle area containing macrophages, epithelioid macrophages, and multinucleate giant cells. The outermost area surrounding the entire lesion consists of T and B lymphocytes, plasma cells, macrophages, and a fibrous capsule (Fig. 3-24).
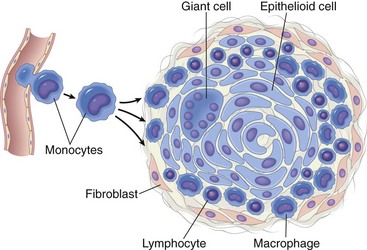
Fig. 3-24 Schematic illustration of the formation of a granuloma. (Modified from Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Mycobacterial organisms and their antigens are very sparse in these granulomas and not commonly detected with acid-fast stains and immunohistochemical stains for mycobacterial antigens. Mineralization can occur in tuberculoid granulomas, but this outcome depends on the species of animal affected. It is common in cattle, present to a lesser degree in pigs, and uncommon in sheep. “Atypical” mycobacteria, such as Mycobacterium marinum, can also cause nodular (tuberculoid) granulomas in the subcutaneous tissues of dogs, cats, and other species, and very few organisms are detected with stains. Certain persistent, poorly degradable antigens, such as those in foreign bodies and in microbes, such as Nocardia spp., can have eosinophilic proteinaceous aggregates of immunoglobulin on their outer surfaces, which can be seen histologically and are termed Splendore-Hoeppli proteins.
Steps in formation of a tuberculoid (nodular) granuloma (see Fig. 3-23) are as follows:
1. Stage I granuloma. Days after infection, the lesion site is infiltrated by neutrophils, monocytes, macrophages, γ/δ T lymphocytes, and NK cells. Epithelioid macrophages also form.
2. Stage II granuloma. From roughly 48 hours to multiple days and weeks, lesions contain macrophages, epithelioid macrophages, thin rims of fibrous connective tissue, variable numbers of NK cells, and γ/δ T lymphocytes, as well as α/β T lymphocytes and B lymphocytes. MGCs can also form.
3. Stage III granuloma. From weeks to 1 month, the central area can caseate or become dense with macrophages and mineralize. Lymphocytes, plasma cells, a zone of fibroblasts, and a fibrous connective tissue capsule surround this core.
4. Stage IV granuloma. From several weeks to months, the lesion can be walled off by a dense capsule and regions within the lesion can become mineralized and overtake the surrounding tissue. The capsule wall can sometimes become degraded when microorganisms are released from the inner regions of the lesion.
Diffuse (Lepromatous) Granulomas (TH2-Biased Granulomas)
Mycobacterium leprae, the cause of human leprosy, produces noncaseating aggregates of macrophages and chronic inflammatory cells often around nerve fibres in the distal extremities and upper respiratory tract mucosa (sites in the body with temperatures lower than core body temperature) of infected humans. This type of granulomatous inflammation appears to form with a predominantly TH2-type of adaptive immune response that is seen in veterinary medicine during the clinical stages of Johne’s disease in cattle and sheep.
These lesions can be poorly delineated (e.g., poorly defined borders) and have a widespread distribution, a heavy intracellular bacterial burden, relatively few lymphocytes, numerous macrophages that extend into surrounding tissue often without a distinct capsule, variable degrees of fibrosis, and lack caseation. Similar granulomatous lesions are seen in animals. Feline leprosy and canine lepromatous-like granulomas are somewhat similar to human leprosy in lesion formation. Mycobacterium avium-intracellulare paratuberculosis—the cause of Johne’s disease in cattle, sheep, and goats—also induces a diffuse (lepromatous) type of granulomatous inflammation consisting of diffuse sheets of macrophages with few lymphocytes and plasma cells. This lesion most commonly occurs in the lamina propria of the ileum and colon (Fig. 3-25) and in mesenteric lymph nodes. Special stains, such as acid-fast and immunohistochemical stains specific to bacterial antigens, can be used to identify these bacteria within the cytoplasm of macrophages and those that are extracellular (Web Fig. 3-13). Because bacteria are present in large numbers in these diseases, they are commonly identified by these techniques. Nodular (tuberculoid) granulomas, defined later, are not seen in Johne’s disease lesions. Finally, Mycobacterium avium ssp. paratuberculosis infection also occurs in the lung and other organs of birds often induces lesions with similar sheets of macrophages containing abundant bacteria detectable with special stains.
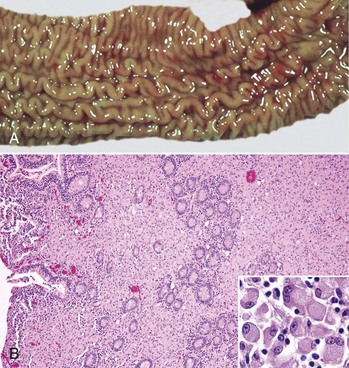
Fig. 3-25 Diffuse (lepromatous) type of granulomatous inflammation, Johne’s disease (Mycobacterium avium ssp. paratuberculosis), ileum, cow.
A, The mucosa is thickened because of a dense infiltrate of granulomatous inflammatory cells in the lamina propria. The lumen of the intestine is to the left. B, The lamina propria contains numerous macrophages arranged in sheets. The ileal lumen is to the left; scattered crypts still remain in the central area of the specimen. H&E stain. Inset, Higher magnification of macrophages present in the granulomatous inflammatory exudate. H&E stain. (A courtesy Dr. M.D. McCracken, College of Veterinary Medicine, University of Tennessee; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. J. Hostetter, College of Veterinary Medicine, Iowa State University. Inset courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
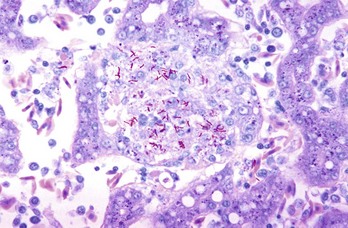
Web Fig. 3-13 Mycobacterium avium ssp. paratuberculosis bacilli.
Diffuse (lepromatous) type of granulomatous inflammation with numerous macrophages and multinucleate giant cells that contain abundant bacilli stained red. Acid-fast stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Sarcoids of Horses
Sarcoids of human patients are granulomatous-like lesions. In contrast, sarcoids of horses are not a correlate to sarcoids in humans. Sarcoids that occur in the skin of horses are not granulomas as occur in humans but are locally aggressive skin tumors, and the most common dermatologic neoplasm reported in horses. They are composed of proliferating fibroblasts and do not contain the numerous macrophages, lymphocytes, and plasma cells seen in human sarcoids. Bovine papillomavirus (BPV) types 1 and 2 and the major transforming protein, E5, are associated with equine sarcoids but appear not to produce infectious virions. E5 may contribute to the persistence of the virus and development of the lesion by downregulating major histocompatibility complex (MHC) class I expression and thereby reducing immunosurveillance. The mode of transmission of BPV infection has not been determined.
Eosinophilic Granulomas
Certain types of chronic inflammation have dense infiltrates of eosinophils with macrophages, and varying numbers of lymphocytes and plasma cells (Web Fig. 3-14). Because of the presence of eosinophils, they are termed eosinophilic granulomas (Table 3-8). Some granulomas with numerous eosinophils develop in response to migrating parasites, such as Toxocara canis (larval migrans). In eosinophilic granuloma of cats, eosinophilic stomatitis of dogs, and equine eosinophilic dermatitis of horses, it is suspected these conditions form in response to antigen in a TH2-directed manner; however, no specific antigen has been identified.
TABLE 3-8
Eosinophilic Granulomas of Domestic Animals
| Species | Type of Eosinophilic Granuloma |
| Feline | Eosinophilic plaque, granuloma, and dermatitis |
| Canine | Eosinophilic granuloma of the oral cavity of huskies and other dogs |
| Equine | Equine collagenolytic granuloma, axillary nodular necrosis, and unilateral papular dermatosis |
| All species | Eosinophilic (TH2) granulomas secondary to parasitic infections |
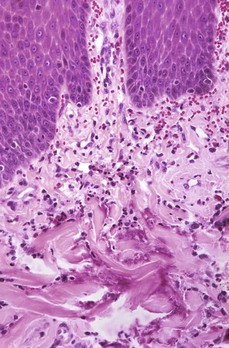
Web Fig. 3-14 Eosinophilic granuloma, oral mucosa, cat.
Note the mixture of eosinophils, macrophages, and lymphocytes in the superficial dermis accompanied by collagenolysis (bottom half of illustration). H&E stain. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Grossly, eosinophilic granulomas in cats appear as papules, nodules, plaques (sometimes linear), and ulcers in the skin. They also occur as nodular or ulcerated lesions in the oral mucosae and footpads. Microscopically, the inflammatory response consists of eosinophils, macrophages, and areas of dense eosinophilia around collagen (Fig. 3-26). For many years, the densely eosinophilic, collagen-rich areas were considered regions of collagen degradation; however, the eosinophilic material is composed largely of major basic protein (MBP), a protein present in large amounts in the granules of eosinophils. Apparently eosinophils degranulate in these regions, releasing MBP, which accumulates over time. Some eosinophilic granulomas do not form distinct nodular granulomas and the cellular content of the lesions (e.g., the numbers of macrophages and eosinophils) can vary widely. But the lesions are chronic in nature and contain enough macrophages and other chronic inflammatory cells to be classified as granulomas by most pathologists.
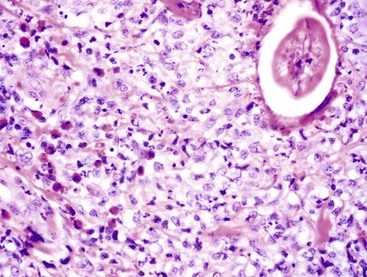
Fig. 3-26 Granuloma with eosinophils.
A region of skin from a horse in which there is a parasitic organism (Habronema sp) that has invoked a marked granulomatous reaction of closely associated macrophages admixed with eosinophils. H&E stain. (Courtesy Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
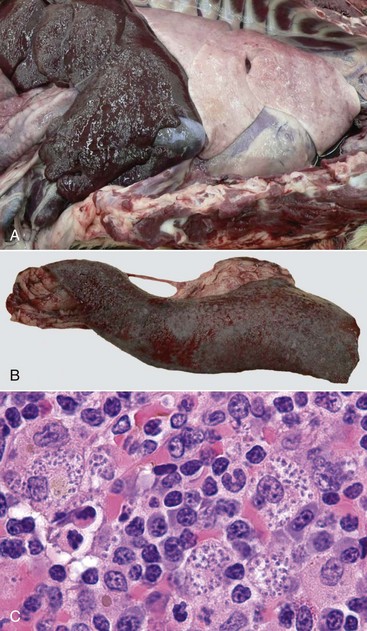
Web Fig. 3-15 Granulomatous inflammation causing organ enlargement.
A dog with leishmaniasis has hepatomegaly (A) and splenomegaly (B). C, Microscopically, these organs have dense infiltrates of macrophages, lymphocytes, and plasma cells. Macrophages contain numerous amastigotes within the cytoplasm. (Courtesy Dr. K. Gibson-Corley and Dr. C. Petersen, College of Veterinary Medicine, Iowa State University.)
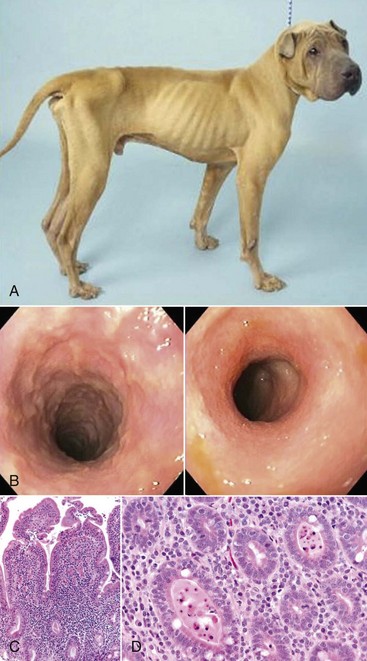
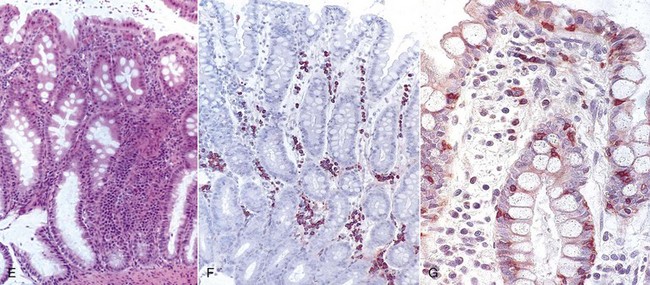
Web Fig. 3-16 Chronic inflammation resulting in severe weight loss and clinical disease, dog.
The dog is severely thin due to inflammatory bowel disease, a condition characterized by chronic inflammation in the intestinal mucosa. A, The dog is thin with accentuation of the ribs and musculature due to loss of subcutaneous adipose tissue. The ventral abdominal wall spans directly upward toward the ventral pelvis and is not pendulous due to loss of omental and enteric adipose tissue. B, Endoscopy of the intestinal mucosa reveals granulation. Endoscopic image of a normal dog lacks such alterations. C, Duodenal mucosa also contains lymphocytes, plasma cells, and macrophages, which widen the intestinal villi. Enterocytes lining the villi are cuboidal and have few intervening goblet cells. D, Some regions of the small intestine have dilated crypts that contain neutrophils and cell debris. E, The colon contains diffuse infiltrates of lymphocytes and plasma cells in the lamina propria that slightly separate colonic glands. The enteric lumen contains mucin and cell debris. F, Immunohistochemical stains detect T lymphocytes (CD3). G, IgA-secreting cells. (Courtesy Dr. A. Jergens, College of Veterinary Medicine, Iowa State University.)
Other Chronic Inflammatory/Granulomatous Conditions
With chronic inflammation, organs can become enlarged by the granulomatous inflammation which, once formed, persists. For example, intense macrophage infiltration in the liver and spleen of Foxhounds with visceral leishmaniasis result in hepatomegaly and splenomegaly that can be detected clinically and at postmortem (Web Fig. 3-15). In other conditions, chronic inflammation results in chronic weight loss and debilitation. Dogs with inflammatory bowel disease often lack granulomatous inflammation within the intestinal mucosa, but instead the mucosa is characterized by chronic inflammation composed of lymphocytes, plasma cells, and macrophages within the lamina propria of the intestinal mucosa (Web Fig. 3-16). This lesion alters intestinal function that can result in severe weight loss and vomiting. Several conditions of humans and animals are characterized by chronic inflammatory lesions, but the cause of these lesions is poorly understood. One such condition, polyarteritis nodosa, is seen in humans and rats and only rarely in other species. It is characterized by perivascular infiltrates of lymphocytes, plasma cells, and macrophages. Dogs can develop idiopathic canine polyarteritis, a condition in which arterial lesions not only involve coronary arteries but also medium-to-small arteries of other organs. As indicated, the cause of eosinophilic granulomas has not been determined but is thought to occur secondary to parasitic larval migration, microbial infections, or foreign material. Finally, a variety of other human diseases are characterized by varying degrees of perivascular granulomatous inflammation but to date have no or very rarely seen veterinary correlates. These diseases occur around blood vessels, may be secondary to immunologic mechanisms, and include large-vessel vasculitis (giant cell [temporal] arteritis, Takayasu arteritis), medium-sized vessel arteritis (Kawasaki disease, [and polyarteritis nodosa]), and small-vessel vasculitis (e.g., Wegener’s granulomatosis, Churg-Strauss syndrome).
Gross and Microscopic Lesions and Nomenclature of the Chronic Inflammatory Response
The term chronic inflammation implies two underlying and often concurrently occurring processes: fibroplasia and cellular infiltration. Often, one of these types of responses predominates. Fibroplasia, the formation of fibrous connective tissue, includes any stage of the process from the formation of fibrous connective tissue that includes newly formed “immature” connective tissue with newly formed blood vessels to “mature” connective tissue that contains well-collagenized and remodeled granulation tissue. Cellular infiltrates are composed of predominantly macrophages, lymphocytes, and plasma cells, depending on the inciting agent/substance and the duration of the inflammatory process. It is important to understand the mechanism of these processes to apply gross and histopathologic diagnoses. To clinicians, these terms imply duration of illness, whereas to pathologists, they imply the characteristics of the tissue’s response to injury.
Grossly, chronic inflammatory lesions are often gray-to-white and firm and have either a nodular surface in the case of granulomas or an indented or pitted surface in the case of fibrosis. The gray-to-white color is largely a result of the infiltrates of macrophages and lymphocytes, proliferation of fibroblasts, and deposition of fibrous connective tissue. The firm texture is attributable to fibrous connective tissue (fibroblasts and endothelial cells) and the consolidation (i.e., solidification) of the leukocytes in the exudate. The irregular shape occurs because of the haphazard accumulation of leukocytes and the fibrosis/scarring and contraction of the lesion by myofibroblasts within the fibrous connective tissue (see section on Wound Healing and Angiogenesis).
The lungs of dogs with Blastomyces dermatitidis infection often have a nodular appearance because of the formation of numerous granulomas and/or pyogranulomas. The use of and distinction between terms such as granuloma and pyogranuloma depend on the number of neutrophils in the overall inflammatory exudate or in the center of the granuloma and often reflect the interpretation of the examining pathologist.
The pitted surface of kidneys of dogs, cats, or other species can be seen, for example, with chronic interstitial nephritis or chronic pyelonephritis. Frequently, the pitted surfaces correspond to areas where fibrous tissue formed within renal parenchyma during the chronic inflammatory response that pulls the renal capsule into the parenchyma as part of the healing process. In chronic pyelonephritis, the inflammatory bands often radiate from the renal medulla into the cortex and to the renal capsule and obliterate or surround and separate cortical tubules and glomeruli. Fibrous adhesions between the renal cortex and the capsule can occur. This fibrous connective tissue may also contain lymphocytes, plasma cells, and macrophages.
Grossly, abscesses, granulomas, and areas of fibrosis that occur with the persistence of chronic inflammation are often easily seen. Severe fibrosis results in an area that is generally gray to white with extensive contraction, whereas abscesses are often round with a fibrous capsule and a central area of pus. Grossly, the three main differential diagnoses for a white, firm, oval-to-irregular nodular mass are abscess, granuloma, and neoplasm. Most commonly, histopathology is required to differentiate the three because they can appear very similar grossly.
Microscopically, chronic inflammatory responses are classified into categories based on the types and distribution of the inflammatory cells in the exudate. These categories include (1) chronic lymphocytic/lymphohistiocytic inflammation, (2) fibrosing chronic inflammation, (3) chronic-active (purulent) inflammation, (4) granulomatous (noncaseating) inflammation, (5) pyogranulomatous inflammation, (6) granulomas, and (7) pyogranulomas.
• Chronic inflammation composed of lymphocytes and plasma cells admixed with macrophages occurs commonly in the body (Fig. 3-27). Sometimes, lymphocytes and macrophages predominate over plasma cells, and such lesions can be called lymphohistiocytic. Histiocytic is a term used for macrophage infiltration; however, some pathologists use the term macrophagic. This type of inflammatory response is characteristically seen in the early stages of the chronic inflammatory response and in response to specific microbes, such as viruses, and in mucosal surfaces in response to antigenic stimulation.
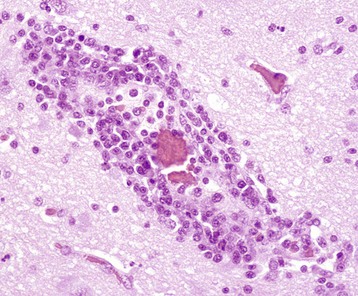
Fig. 3-27 Chronic inflammation, distemper, brain, raccoon.
In its simplest form, chronic inflammation, as seen with some viral infections, consists of an exudate of lymphocytes with occasional macrophages and plasma cells. In many tissues, especially in the central nervous system, these cells can have a perivascular pattern of distribution. In certain animal species (exotic wildlife species, horses) and specific disease categories (parasitic, protozoal, viral), perivascular chronic inflammatory exudates may also contain variable numbers of eosinophils. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
• Fibrosing chronic inflammation is a region of chronic inflammation that is predominantly composed of fibrous connective tissue. This can occur in chronic traumatic pericarditis (hardware disease) of cattle where there are areas of pericardial fibrosis covered by fibrin and in the chronic fibrosis that surround necrotic regions of lung and form along the pleura of cattle with contagious bovine pleuropneumonia and chronic Mannheimia haemolytica pneumonia.
• Chronic-active inflammation has the same cellular components as chronic inflammation but also contains neutrophils, fibrin, and plasma proteins that are constituents of the acute inflammatory response. Chronic-active inflammation occurs when the inciting stimulus has not been removed from the exudate in the chronic inflammatory response, and it continues to elicit an acute inflammatory response. One should be careful not to confuse the morphologic diagnosis of chronic-active inflammation with the hepatic disease entity of dogs termed chronic-active hepatitis.
• Granulomatous inflammation has a basic cellular exudate consisting predominantly of activated macrophages and in some cases also epithelioid macrophages, multinucleate giant cells, and lesser numbers of lymphocytes and plasma cells. Granulomatous inflammation can be arranged in a diffuse or haphazard manner as seen in the thickened intestinal mucosa (i.e., lamina propria) of cattle with Johne’s disease (see Fig. 3-25). While the presence of a few macrophages in a lesion is indicative of chronic inflammation, these are considered to be granulomatous inflammation when the macrophages aggregate and begin to replace portions of the normal stroma.
• Pyogranulomatous inflammation has the same cellular exudate as granulomatous inflammation but also contains multifocal/random infiltrates of neutrophils, fibrin, and plasma proteins, which are constituents of the acute inflammatory response. Pyogranulomatous inflammation occurs when the inciting stimulus has not been removed from the exudate in the granulomatous inflammatory response, and it continues to elicit an acute inflammatory response. A nodular-like granulomatous area with neutrophils is termed a pyogranuloma (see later). Pyogranulomatous inflammation is often seen with infections caused by Blastomyces dermatitidis and is frequently multinodular. Pyogranulomatous is sometimes used loosely, with little discretion. For example, it can be used in lesions that are in transition from acute inflammation to chronic inflammation or during the “clean-up” phase of healing.
• Granulomas are a distinct type of granulomatous inflammatory response that occurs when macrophage infiltration is present in a well-defined area and thus form a distinct mass on gross observation. Granulomas can occur as noncaseating and caseating types, as described.
• A pyogranuloma is a nodular granuloma with a central area of neutrophils.
Cellular Mechanisms of Chronic Inflammatory Responses
Lymphocytes play a key role in most chronic inflammatory lesions, especially in autoimmune diseases and in diseases with persistent antigen. As with macrophages, lymphocytes enter unresolved areas of acute inflammation within 24 to 48 hours, being attracted by chemokines, cytokines, and other stimuli. Histologically, they are often aggregated around blood vessels and surround granulomas or are distributed haphazardly within injured tissue (see Fig. 3-27). In viral encephalitides, lymphocytes are commonly distributed in a perivascular pattern, primarily in the gray matter. In other types of conditions, such as lymphoplasmacytic stomatitis and pododermatitis of cats, lymphocytes and plasma cells are the predominant cell types in the lesions.
γ/δ–T Lymphocytes
γ/δ–T lymphocytes are often the first type of T lymphocyte to arrive in the lesion of chronic inflammation and can contribute to the development of granulomas. This mechanism is supported by the fact that mice lacking γ/δ–T lymphocytes have defects in granuloma formation. However, the role of γ/δ–T lymphocytes in the establishment and persistence of granulomas has not been fully determined. Cattle have high numbers of circulating γ/δ–T lymphocytes, relative to other T lymphocytes, and are a useful model for assessing the role of γ/δ–T lymphocytes, in the formation of classic granulomas in Mycobacterium tuberculosis and Mycobacterium bovis infections and in the formation of diffuse granulomatous lesions in Mycobacterium avium-paratuberculosis infections.
α/β–T Lymphocytes (CD4/CD8)
α/β–T lymphocytes (CD4 and CD8 lymphocytes) enter areas of chronic inflammation and are key to the regulation of the type of adaptive immune response that ensues, albeit a TH1, TH2, or TH0 response of chronic inflammation and/or granuloma formation. Under the influence of cytokines, these lymphocytes can further differentiate into (1) effector memory lymphocytes, which enter extralymphoid sites of inflammation; and (2) central memory lymphocytes, which settle into the blood and lymphoid organs. Memory lymphocytes contribute to the persistence of the chronic inflammatory response and granuloma formation.
TH1, TH2, TH17, and T Reg Immunologic Responses
The mechanistic basis for chronic inflammation to develop and persist is slightly to profoundly different, depending on the inciting agent or condition. While the cell types (e.g., monocytes, macrophages, T and B lymphocytes, and NK cells) are consistent features of chronic inflammation, their relative numbers and function can vary widely. T lymphocytes expressing CD4+ (T helper lymphocytes) and CD8+ T lymphocytes (cytotoxic cells) play important roles in the adaptive immune response that shapes chronic inflammation. When CD4+ T lymphocytes bind foreign antigens presented by macrophages, dendritic cells, or B lymphocytes, they release lymphokines that attract leukocytes into the lesion site; some of these cells are directed toward TH1 (cell mediated), TH2 (humoral), TH17, TFH, and T reg responses (Web Fig. 3-17). Dendritic cells generally present antigen to naïve CD4 T lymphocytes, whereas macrophages present antigen to committed T lymphocytes. Thus antigen presentation by macrophages is especially active in long-standing regions of chronic inflammation. In the presence of poorly degradable exogenous or endogenous antigens, the biologic characteristics of antigens, including the amount and structure of protein, polysaccharide, and lipid, contribute to the extent to which the T lymphocyte response will be between a TH1, TH2, TH17, TFH, or T reg. However, in most chronic inflammatory conditions, there is a mixed TH1, TH2, and TH17 type cellular response. T reg lymphocytes, regulatory dendritic cells, and other cells (NK type 1 cells, mast cells, eosinophils, or basophils) can affect the strength and the balance of TH1, TH2, and TH17 responses through inhibition of the responses. TH1 immunologic responses often occur in response to (1) foreign bodies; (2) endogenous antigens, such as those in MBP (which can occur in murine experimental allergic encephalomyelitis [EAE] and is an experimental model of multiple sclerosis); and (3) endogenous antigens, such as those in intracellular microbes like Mycobacteria spp., Listeria monocytogenes, Histoplasma capsulatum, and Leishmania spp. TH1 chronic inflammatory responses are composed, histologically, of macrophages with variable numbers of T and B lymphocytes, dendritic cells, and occasionally fibroblasts. The TH1 response is induced by IL-12, IFN-γ, IL-18, IL-23, and IL-27. Briefly, antigen-presenting cells release IL-12 (also IL-18, IL-23, and IL-27) that induces pre-TH/CD4+ cells to commit to the TH1 pathway. The TH1-committed CD4+ cells then release IL-2 for proliferation of T lymphocytes that produce IFN-γ and TNF-β, which activates macrophages. In addition, IFN-γ can inhibit the commitment of pre-ThH/CD4+ cells to the TH2 pathway.
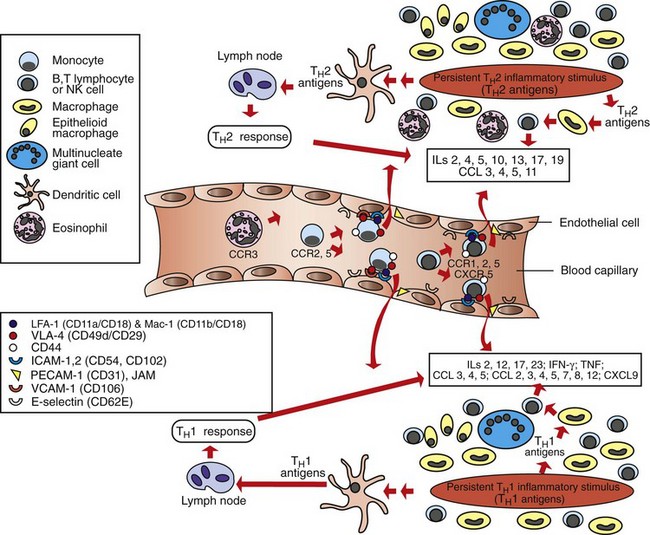
Web Fig. 3-17 Schematic illustration of the TH1- and TH2-mediated chronic inflammation.
TH1-mediated (below the blood vessel) and TH2-mediated (above the blood vessel) types of chronic inflammation and some of the key inflammatory mediators, chemokines, and adhesion molecules that mediate the process. CCL, CXCL, Chemokine ligand; ICAM-1, intercellular adhesion molecule-1; ICAM-2, intercellular adhesion molecule-2; IL, interleukin; INF-γ, interferon-γ; JAM, junctional adhesion molecule; LFA-1, lymphocyte function antigen-1; Mac-1, macrophage-1 antigen; NK, natural killer; PECAM-1, platelet endothelial cell adhesion mlecule-1; TNF, tumor necrosis factor; VCAM-1, vascular cell adhesion molecule-1; VLA-4, very late antigen-4. (Redrawn with permission from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
In contrast, theTH2-predominant responses are often seen in chronic inflammatory conditions with an allergic basis, such as asthma, chronic allergic inhalant, or food allergic dermatitis, or inflammatory bowel disease. The mucosa and skin dermis of these conditions contain T and B lymphocytes, macrophages, dendritic cells, and fibroblasts in the TH2 inflammatory milieu that leads to increased numbers of mast cells, basophils, and eosinophils associated with IgE and other humoral responses. TH2 responses are induced by high concentrations of IL-4 and IL-10, as well as IL-5, IL-9, IL-13, IL-17, and to some degree, perhaps IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. Briefly, TH2 dendritic cells present antigen to pre-T helper/CD4+ lymphocytes committing them to the TH2 pathway. TH2-committed CD4+ lymphocytes release IL-4 and IL-5, IL-10, IL-13, IL-17, and IL-19 (also IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29) to act on B lymphocytes, thus resulting in antibody production and in some cases, increased activity of eosinophils, mast cells, and basophils that release mediators of acute inflammation and TH2 cytokines such as IL-4. In addition, IL-4, IL-5, IL-10, and IL-13 can also inhibit pre-T helper/CD4+ lymphocytes from committing to the TH1 pathway.
TH17 immunologic responses form in the presence of IL-23 and induce secretion of IL-17A, IL-17F, IL-21, IL-22, IL-26, and CCL20, which are associated with development of autoimmune responses in conditions such as allergic airway inflammation, psoriasis, inflammatory bowel disease, arthritis, and multiple sclerosis (humans). TH17 lymphocytes function predominantly at mucosal surfaces and trigger neutrophil activation and release of antimicrobial factors to bridge innate and adaptive immune responses. Other cell types, such as γδ T lymphocytes, NK cells, NK-T lymphocytes, and lymphoid tissue inducer (LTi) cells, produce a range of cytokines similar to TH17 lymphocytes.
Monocytes/Macrophages
Monocytes/macrophages are the signature cell types of chronic inflammation. They produce a wide variety of inflammatory mediators, including chemokines, cytokines, and NO, and are often situated at strategic locations within tissues of the body to (1) quickly sense the initial activity of acute inflammation, (2) migrate in response to chemotaxins, (3) remove and kill microbial agents by phagocytosis, (4) remove and degrade particulate matter by phagocytosis, (5) process antigens for presentation to effector cells of the adaptive immune response, and (6) facilitate angiogenesis and remodel the ECM (Fig. 3-28).
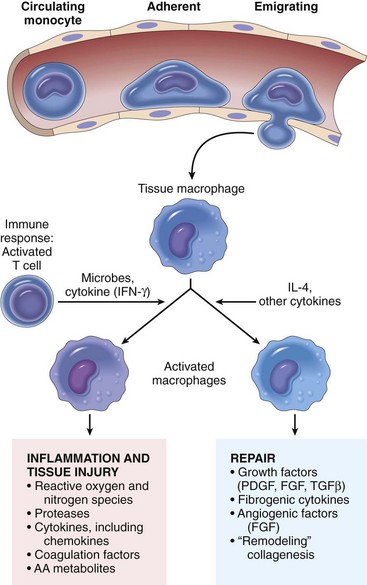
Fig. 3-28 The roles of activated macrophages in chronic inflammation.
Macrophages are activated by nonimmunologic stimuli such as endotoxin or by cytokines from immune-activated T lymphocytes (particularly interferon-γ [IFN-γ]). The products made by activated macrophages that cause tissue injury and fibrosis are indicated. AA, Arachidonic acid; PDGF, platelet-derived growth factor; FGF, fibroblast growth factor; TGF-β, transforming growth factor-β. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Epithelium
Epithelial cells can contribute to chronic inflammatory responses in a variety of manners. For example, experimental models of granuloma formation have shown that Mycobacteria release an early secretory antigen-6 (ESAT-6), which induces release of MMP-9 from epithelia that is vital for recruitment of macrophage infiltration. Epithelia can also release type I interferons, cytokines, antimicrobial peptides, and chemokines and express adhesion molecules.
Mononuclear Cell Maturation and Trafficking in the Chronic Inflammatory Response
The macrophage is key to the development and persistence of chronic inflammation. Monocytes, derived from the bone marrow, form the monocyte/macrophage system by entering tissues and differentiating into macrophages (e.g., Kupffer cells, alveolar macrophages, and microglial cells) and are central to innate and adaptive immune systems. Recent work also shows that the spleen is a significant reservoir of monocytes, which can exit en masse on tissue injury. Monocytes are then recruited from the bloodstream to enter tissue and differentiate into macrophages that can also respond to tissue injury. Under noninflammatory conditions, the replenishment of tissue macrophages occurs through local proliferation and not via monocyte influx. However, with inflammatory stimuli, monocytes are recruited from the blood into tissue in response to inciting agents/substances.
In noninflamed tissue, monocytes expressing CX3CR1 and CCR5 chemokine receptors are attracted to tissues expressing their respective ligands (i.e., fractalkine (CX3CL) and MIP-1-a [CCL3]). In areas of inflammation, monocytes expressing CCR2 chemokine receptors are attracted by MCP-1 (CCL2). Attracted monocytes enter these areas in a manner similar to that described for the leukocyte adhesion cascade outlined for neutrophils. Slow rolling is mediated by E and P selectins and firm adherence by monocytes to endothelial cells is largely mediated by LFA-1 (CD11a/CD18), VLA-4 (α4β1-integrin), and also Mac-1 (CD11b/CD18) molecules that adhere to the respective endothelial cell ligands: ICAM-1/2, VCAM-1, and ICAM-1/2. Transmigration of monocytes between endothelial cells is mediated by leukocyte adhesion molecules expressed on the monocyte such as LFA-1, VLA-4, Mac-1, PECAM-1, and JAM A, JAM B, and JAM C. These molecules bind to adhesion molecules, such as PECAM-1, and the JAM molecules expressed by endothelial cells at the intracellular junction. As monocytes and other leukocytes pass between endothelial cells, they separate the tight junctions and vascular-endothelial (VE)-cadherins to allow the passage of the leukocyte.
TH1, TH2, TH17, and T reg lymphocytes, along with inflammatory mediators released by somatic cells and macrophages during injury and/or infection, affect the differentiation of monocytes and noncommitted macrophages to macrophages with specific function (see Web Fig. 3-21). In conditions favorable to TH1 influence, macrophages respond to IFN-γ released by TH1, NK lymphocytes, and TNF from antigen-presenting cells to form classically-activated macrophages (Table 3-9). Under TH2 conditions, macrophages respond to IL-4 released by TH2 lymphocytes and granulocytes (e.g., mast cells, basophils) by forming wound-healing (tissue repair) type macrophages. Under T-reg secretion of IL-10 and other substances, such as immune complexes, prostaglandins, glucocorticoids, and apoptotic cells, macrophages differentiate into regulatory macrophages (antiinflammatory cells), which secrete IL-10 to suppress inflammation. Macrophages committed to the classical-activation, wound-healing/tissue repair, or regulatory pathways greatly influence the anatomic (histopathologic) structure and function of the chronic inflammatory response and granuloma formation (Fig. 3-29; Web Fig. 3-18).
TABLE 3-9
Stimuli That Affect the Activation Status of Macrophages
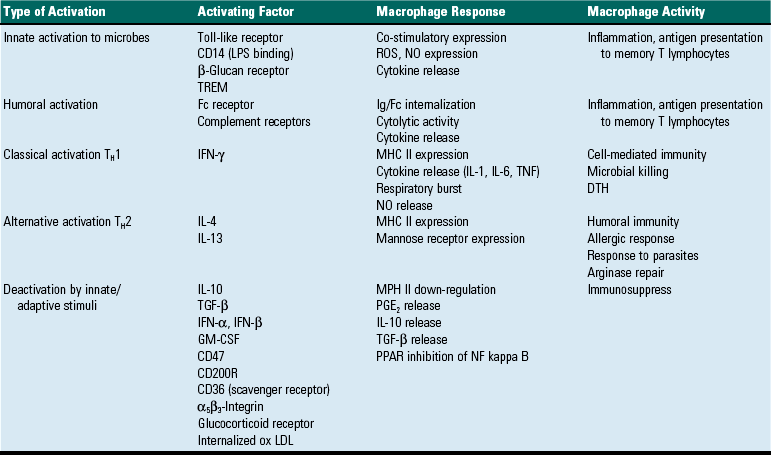
GM-CSF, Granulocyte-macrophage colony-stimulating factor; DTH, delayed type hypersensitivity; IFN, interferon; Ig, immunoglobulin; IL, interleukin; MHC, major histocompatibility complex; NF, nuclear factor, NO, nitric oxide; ox LDL, oxidized low-density lipoprotein; PGE2, prostaglandin type E2; PPAR, peroxisome proliferator-activated receptor; ROS, reactive oxygen species; TGF, transforming growth factor; TH1, T helper cell type 1 lymphocyte response; TH2, T helper cell type 2 lymphocyte response; TREM, triggering receptor expressed on myeloid cells.
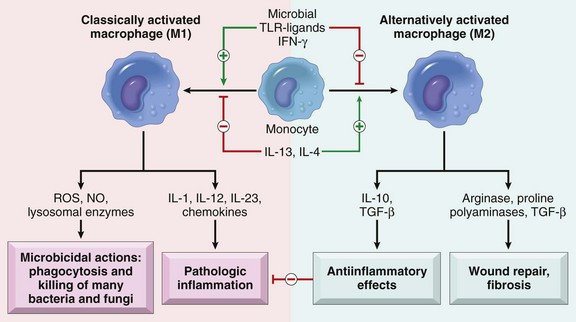
Fig. 3-29 Subsets of activated macrophages.
Different stimuli activate monocytes/macrophages to develop into functionally distinct populations. Classically activated macrophages are induced by microbial products and cytokines, particularly interferon-γ [IFN-γ], and are microbicidal and involved in potentially harmful inflammation. Alternatively activated macrophages are induced by other cytokines and in response to helminths (not shown), and are important in tissue repair and the resolution of inflammation (and may play a role in defense against helminthic parasites, also not shown). (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
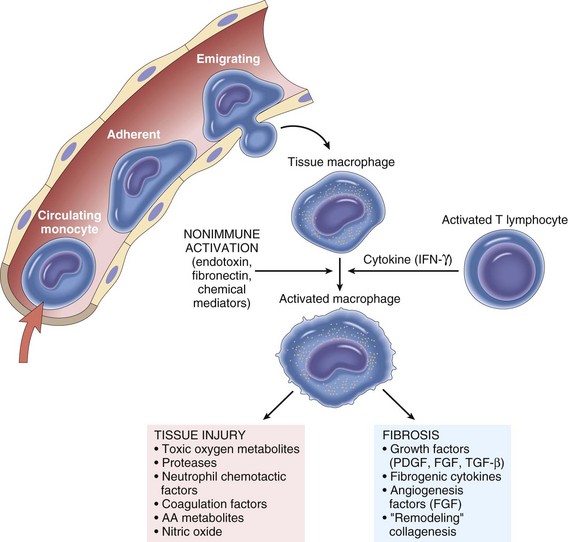
Web Fig. 3-18 The roles of activated macrophages in chronic inflammation.
Macrophages are activated by cytokines from immune-activated T lymphocytes (particularly interferon-γ [IFN-γ]) or by nonimmunologic stimuli such as endotoxin. The products made by activated macrophages that cause tissue injury and fibrosis are indicated. AA, Arachidonic acid; FGF, fibroblast growth factor; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-β. Also see Table 3-8. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Macrophages activated within lesions enter lymphatic vessels that drain into nearby lymph nodes via the afferent lymphatic vessels. In addition, some macrophages from lesions within the caudal body can enter the thoracic duct, which drains into the cranial vena cava or one of its branches. Macrophages within the head and neck can enter right or left tracheal lymphatic ducts. The right tracheal lymphatic duct empties into the cranial vena cava, whereas the left tracheal duct empties into the thoracic duct. Once in the blood, the macrophages are disseminated throughout the body.
The outcome of these different types of macrophage activation leads to the following specific responses (see Fig. 3-29):
• Innate activation can lead to release of reactive oxygen species (ROS), NO, and IFN-α and IFN-β.
• Classic activation with IFN-γ leads to expression of MHC II antigen, respiratory burst, release of IL-1 and TNF for microbial killing, cellular immunity, and delayed type hypersensitivity.
• Alternative activation enhances MHC class II expression and mannose receptor expression for humoral immunity and allergic responses.
• Innate deactivation and reduced inflammatory responses can occur with the uptake of apoptotic cells or storage of oxidized LDLs in lysosomes and release of IL-10.
In addition to T reg lymphocytes and regulatory macrophages, recent work has shown that sialylated IgG molecules can regulate macrophage function. This stems from the observation that certain types of chronic inflammation, particularly autoimmune conditions of humans, have responded favorably to polyclonal IgG given intravenously. The mechanism by which this therapy works is poorly understood. Recently, evidence suggests that the polyclonal IgG activity may be due to a subset of IgG molecules that are sialylated on the Fc chain of the IgG. The sialylated IgGs are thought to interact with sialic acid–specific receptors on regulatory macrophages and upregulate expression of Fc receptor IIB, which is inhibitory, on effector macrophages.
Formation of Epithelioid Macrophages and Multinucleate Giant Cells
Activated macrophages within tissues are relatively large cells histologically (20 to 25 mm in diameter) with abundant, often clear cytoplasm and a single, oval-to-polygonal, often slightly eccentric, reniform nucleus (Fig. 3-30). With time, activated macrophages can sometimes further differentiate into epithelioid macrophages and MGC (Fig. 3-31).
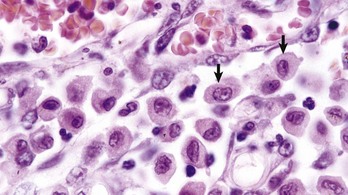
Fig. 3-30 Macrophages, lung, dog.
Macrophages have abundant cytoplasm and slightly eccentric, often reniform nuclei (arrows). Note the small vacuoles in the cytoplasm, probably phagocytosed material. H&E stain. (Courtesy Dr. N. Cheville, College of Veterinary Medicine, Iowa State University.)
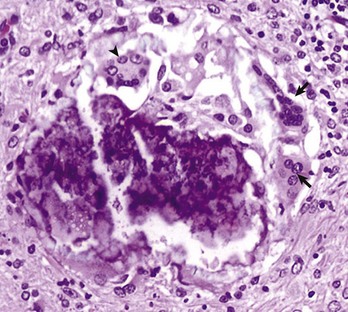
Fig. 3-31 Multinucleate giant cells, chronic granulomatous inflammation, central nervous system, rabbit.
This focus contains both foreign body-type (arrows) and Langhans’-type (arrowhead) multinucleate giant cells. H&E stain. (Courtesy Dr. A. Loretti, Ontario Veterinary College, University of Guelph.)
Both epithelioid macrophages and multinucleate giant cells often form in response to foreign bodies or persistent intracellular pathogens. The molecular mechanisms by which epithelioid macrophages and multinucleate giant cells form are poorly understood. It is a fascinating biologic phenomenon that requires membrane fusion and integration of the cytoplasm and nuclei of multiple cells. Studies in human patients with sarcoidosis, a special type of granulomatous inflammation in humans (see section on Sarcoidosis) have elucidated some of the important factors and conditions that contribute to MGC formation. Epithelioid macrophages are larger than activated macrophages. They have abundant cytoplasm, and the cell membrane occasionally assumes a polygonal to elongated shape, forming sheets, and thus can resemble to a limited degree squamous epithelium. These cells have diminished phagocytic capacity, but they contain large amounts of rough endoplasmic reticulum (RER), Golgi, vesicles, and vacuoles. These latter structures suggest that the main function of epithelioid macrophages involves extracellular secretion; however, the physiologic activity of epithelioid macrophages is poorly understood and requires additional investigation.
MGC are seen frequently in granulomatous inflammation. MGC are syncytial cells formed by the fusion of two or more activated macrophages into one large cell with two or more nuclei (see Fig. 3-31). These nuclei can be distributed in the cell in a haphazard manner or aggregated in the center of the cytoplasm. This form is called a foreign body type of MGC. The nuclei can also be arranged in a horseshoe-like semicircle at the periphery of the cell. This form is called a Langhans’-type cell. The Langhans’-type giant cell should not be confused with Langerhans’ cells, which are dendritic cells (see next discussion) of the skin. Although epithelioid macrophages, foreign body MGC, and Langhans’ MGC have distinct cellular morphology, their physiologic activity is poorly understood.
The process of MGC formation (macrophage fusion) is not fully understood. It requires that macrophages be present within a chronic inflammatory milieu and thus likely bathed in cytokines, such as IFN-γ, IL-3, IL-4, IL-13, and GM-CSF; pathogen factors, such as muramyl dipeptide, a peptidoglycan portion of bacterial cells walls; and other inflammatory mediators. In this close proximity, the membranes of adjacent macrophages express fusionogenic molecules such as DC-STAMP (a seven transmembrane receptor), β1 and β2 integrins, CD44 (hyaluronic acid receptor), CD47 (integrin-associated protein), macrophage fusion receptor (MFR), fusion regulatory protein (FRP-1; CD98), and P2X7 (a ligand-gated ion channel activated by ATP that forms a pore). The fusion process has similarities to osteoclast formation.
Dendritic Cells
Dendritic cells are central to antigen processing, presentation, and the stimulation of adaptive immunity (Fig. 3-32). Functionally, they serve as sentinel cells of the adaptive immune response. Nearly all tissues and organs contain dendritic cells; however, they are most plentiful in tissues that cover the body, such as the skin and the mucous membranes that line the respiratory and alimentary tracts. Although dendritic cells have some resemblance to macrophages, they have numerous distinct filopodia, which extend from their surface (Web Fig. 3-19). Increasingly, several subtypes of dendritic cells are being identified (Table 3-10). In general, immature dendritic cells (CD34+) migrate to sites of antigen exposure, take up antigen, and migrate to and mature in a lymphoid organ in which they present antigen to T and B lymphocytes (Web Fig. 3-20). This migration process is mediated by chemokines and adhesion molecules, and most dendritic cells enter lymph nodes via the afferent lymphatic vessels in lymph fluid under the influence of chemokines, particularly CCL21. Once in the lymph node, dendritic cells often localize in the parafollicular (T lymphocytes) area in the vicinity of HEVs, a site in which naïve T lymphocytes enter the node. In this location, dendritic cells activate naïve T lymphocytes. Follicular dendritic cells, localized in lymph node follicles, take up antigen from the lymphatic fluid for presentation to follicular B lymphocytes. In addition to their activity as antigen-presenting cells, dendritic cells also contribute to adaptive immune responses through release of chemokines and cytokines. Outside of lymph nodes, dendritic cells, cytokines, and chemokines also contribute to inflammatory and immune responses; however, their contribution to these responses are dwarfed by macrophages, since there are many more macrophages within inflammatory lesions than dendritic cells.
TABLE 3-10
Subpopulations of Dendritic Cells (DCs) and Activities
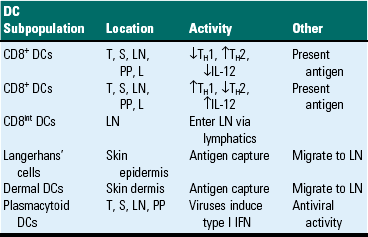
IFN, Interferon; IL-12, interleukin-12; L, liver; LN, lymph node; PP, Peyer’s patch; S, spleen; T, thymus; TH1, T helper cell type 1 lymphocyte response; TH2, T helper cell type 2 lymphocyte response.
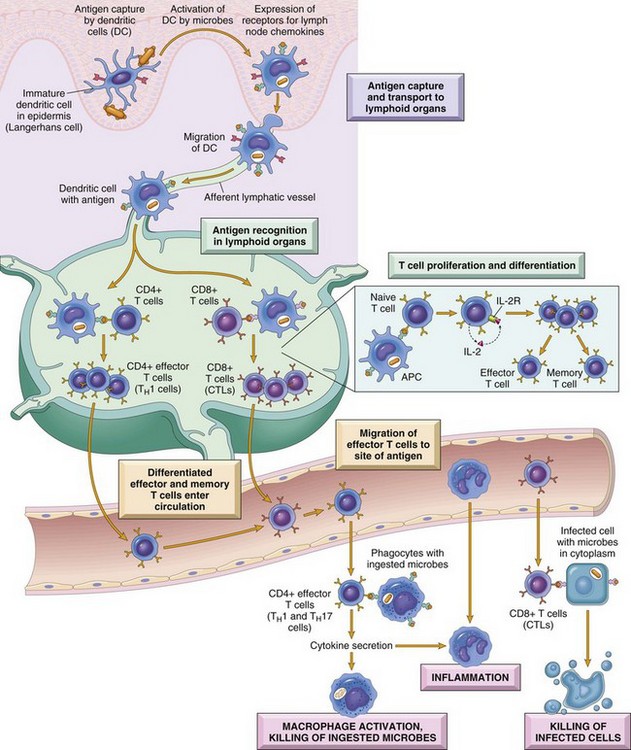
Fig. 3-32 Dendritic cells (DCs).
They capture microbial antigens from epithelia and tissues and transport the antigens to lymph nodes. During this process, the DCs mature, and express high levels of MHC molecules and costimulators. Naïve T lymphocytes recognize MHC-associated peptide antigens displayed on DCs. The T lymphocytes are activated to proliferate and to differentiate into effector and memory cells, which migrate to sites of infection and serve various functions in cell-mediated immunity. CD4+ effector T lymphocytes of the TH1 subset recognize the antigens of microbes ingested by phagocytes, and activate the phagocytes to kill the microbes. CD4+ T lymphocytes also induce inflammation. CD8+ cytotoxic T lymphocytes (CTLs) kill infected cells harboring microbes in the cytoplasm. Not shown are TH2 lymphocytes, which are especially important in defense against helminthic infections. Some activated T lymphocytes differentiate into long-lived memory cells. APC, Antigen-presenting cell. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
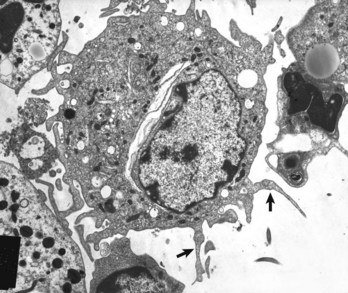
Web Fig. 3-19 Dendritic cell.
Note the numerous filopodia that extend from the cell surface (arrows). TEM. Uranyl acetate and lead citrate stain. (Courtesy Dr. S. Sacco and S. Fach, USDA/ARS-National Animal Disease Center.)
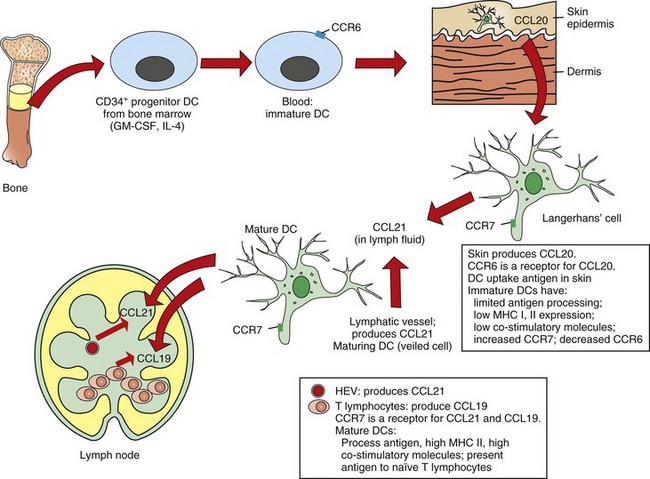
Web Fig. 3-20 Dendritic cell (DC) precursor migration.
In this example, migration to the skin is mediated by chemokine receptors located on the dendritic cell and by chemokine ligands (CCL) in the skin. Eventually, the dendritic cell takes up antigen and migrates to the lymph node under chemokine influence to present the antigen. In the lymph node, the dendritic cell localizes to the parafollicular area and presents antigen to lymphocytes. CCR, Chemokine receptor; GM-CSF, granulocyte-macrophage colony-stimulating factor; HEV, high endothelial venule; IL-4, interleukin-4; MHC, major histocompatibility complex. (Redrawn with permission from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
Although macrophages also present antigen to naïve T lymphocytes, macrophage antigen presentation is more efficient for memory T lymphocytes than for naïve T lymphocytes, the forte of dendritic cells. Through recruitment of naïve T lymphocytes within and outside of the lymph node and presentation of antigen, dendritic cells contribute to the ongoing persistence of a stimulus in chronic inflammatory lesions. In contrast, tolerogenic dendritic cells can suppress immune responses. They accomplish this activity through the sampling of small amounts of self-antigens, harmless environmental antigens, and inciting the deletion of self-reactive T lymphocytes.
Dendritic Cell Trafficking: Trafficking immature monocytic dendritic cells released from the bone marrow express chemokine receptors CCR1 and CCR5 and are recruited by chemokine ligands CCL3 and CCL4 released from lymphocytes and macrophages in tissue (see Fig. 3-32). There are several subtypes of dendritic cells, and those dendritic cells that express CD11c antigen express CCR2, which responds to CCL2, CCL7, CCL8, CCL12, and CCL13. After dendritic cells take up antigen and become exposed to an endogenous (e.g., TNF-α) or exogenous (a ligand for a TLR) mediator, the dendritic cell matures and expresses CCR7 (see Web Fig. 3-20). Mature dendritic cells expressing CCR7 migrate from the site of the inflammatory lesion into the vasculature and then spread hematogenously throughout the body until they are recruited by HEVs in the paracortical areas of lymph nodes in which lymphocytes express CCL19 and CCL20. In this location, dendritic cells present antigen and thus contribute to the amplification of the adaptive immune response. Mature dendritic cells from lesion sites that enter the lymphatic vessels travel to the subtrabecular sinus of the lymph node, draining the lesion site and presenting antigen within paracortical regions. Some dendritic cells also drain via the lymphatic vessels to the thoracic duct and enter the blood. Follicular dendritic cells reside in the lymph node follicle and take up antigen present in the lymphatic fluid for presentation to B lymphocytes.
B Lymphocytes
B lymphocytes contribute to chronic inflammation in at least two major ways. B lymphocytes can (1) take up and present antigen and (2) differentiate into immunoglobulin-producing cells (plasma cells or immunocytes), which secrete immunoglobulins that bind to and opsonize antigens facilitating phagocytosis. B lymphocytes are present within chronic inflammatory lesions and granulomas. They also populate the medullary sinus of lymph nodes in which they produce immunoglobulin locally or leave the medullary sinus through efferent lymphatic flow.
Plasma Cells
Under appropriate stimuli, such as intense antigenic stimulation and B lymphocyte presentation of antigens, B lymphocytes differentiate into plasma cells, which can secrete immunoglobulins that bind to and opsonize antigens and facilitate phagocytosis. Plasma cells form within lymph nodes, mucosal surfaces, and wound sites. The bone marrow also contains a resident population of plasma cells, which can increase in certain disease conditions. Clusters of these cells must be differentiated from neoplastic accumulations as can occur with multiple myelomas. Bone marrow plasma cells can easily migrate into venular walls in the bone marrow to enter the vasculature. Similarly, plasma cells within the medullary sinus of lymph nodes can enter efferent lymphatic vessels and eventually drain into the blood; however, peripheral blood often contains few plasma cells. In the chronic inflammatory exudate, plasma cells are usually found mixed with lymphocytes and macrophages, although in lesser numbers. Plasma cells predominate in certain chronic inflammatory conditions such as inflammatory bowel disease of dogs and cats, lymphoplasmacytic stomatitis and pododermatitis of cats, chronic dermatitis of any domestic animal species, and interstitial nephritis of dogs and cats.
Eosinophils
Different types of chronic inflammatory conditions and granulomas contain a low to high number of eosinophils. Eosinophils are recruited into and stimulated to proliferate within chronic inflammatory exudates by several mediators, most notably IL-5 and eotaxin. In some chronic inflammatory conditions that contain eosinophils, such as asthma in humans, there is a TH2 shift, resulting in increased concentrations of chemokines, such as eotaxin, in the tissue that contribute to the recruitment of additional eosinophils and exacerbates the TH2 response. The same result is likely true for other as yet poorly characterized conditions such as the eosinophilic complex of cats, eosinophilic infiltrates in the base of the tongue of Siberian huskies and other dogs, eosinophilic enteritis in boxer dogs, and eosinophilic inflammatory lesions in the skin of horses. For these conditions, it may be that some type of persistent yet unidentified TH2-inducing antigen is present locally.
Mast Cells
Mast cells have a central role in triggering acute inflammatory reactions. In chronic inflammation, mast cells tend to look similar to macrophages in H&E-stained tissue sections and therefore are often not considered to be a part of the chronic inflammatory lesion. However, special stains, such as a Giemsa stain, of chronic or granulomatous inflammation frequently reveal a surprisingly large number of mast cells identified by their characteristic metachromatic granules. For example, chronic lung lesions (e.g., fibrosis and alveolar epithelial hyperplasia) that develop following severe Mannheimia haemolytica pneumonia often contain increased numbers of mast cells and reduced levels of substance P fibers resulting in persistently altered immune responses.
The reason for the presence of mast cells in chronic inflammatory conditions likely relates to their production of proteolytic enzymes such as chymase and tryptase. Such enzymes likely help physiologically in remodeling and fine tuning components of the ECM. With persistent inflammation and fibrosis, there can be increased proliferation of mast cells. Increased mast cell numbers in such lesions occur by increased infiltration and also by increased proliferation of mast cells in situ. With severe inflammation, there can be loss of substance P fibers, and mast cells can respond to this loss by increasing their expression of c-kit, an important regulator of mast cell proliferation.
Natural Killer Cells
NK cells are present in chronic inflammatory lesions, but their role varies based on the characteristics of the inflammatory stimulus. NK cells can kill cells recognized as foreign without prior exposure to antigen and thereby lacking antigen specificity as required by T lymphocytes. NK cells are activated by type I interferons and IL-12 and can activate macrophages and dendritic cells, thus contributing to chronic inflammation. NK-T lymphocyte activation can be triggered by lipid antigens in the presence of CD1d and can contribute to autoimmune responses.
Fibroblasts
Fibroblasts are multipurpose cells whose function is often overlooked in tissue responses to injury. Fibroblasts are elongated cells that contribute to the structural integrity of tissue and have abundant RER, which is used for the synthesis of collagen and ECM proteins. In addition, they also produce cytokines, MMPs, and chemokines that regulate the composition of the extracellular microenvironment in physiologic and pathologic conditions.
With tissue injury or certain hypoxic conditions, fibroblasts undergo proliferation in response to the release of fibroblast growth factors (FGFs), TGF-β, IL-13, PDGF, VEGF, and other mediators/molecules. Continued release of these substances in response to chronic inflammatory stimuli leads to the extensive fibrosis characteristic of chronic inflammation (Fig. 3-33; Web Fig. 3-21).
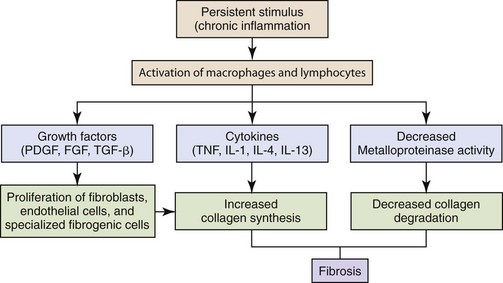
Fig. 3-33 Development of fibrosis in chronic inflammation.
The persistent stimulus of chronic inflammation activates macrophages and lymphocytes, leading to the production of growth factors and cytokines, which increase the synthesis of collagen. Deposition of collagen is enhanced by decreased activity of metalloproteinases. (Modified from Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
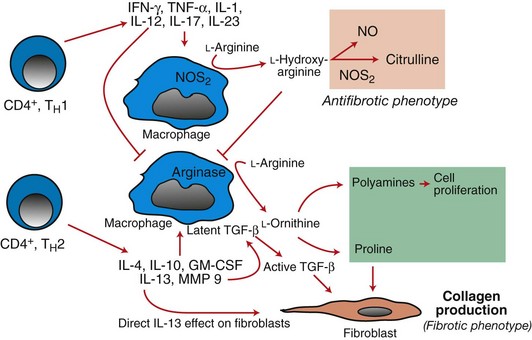
Web Fig. 3-21 Mechanistic basis for fibrosis in experimentally induced TH1- and TH2-mediated responses by macrophages.
In TH1-mediated responses, nitric oxide synthase 2 (NOS2) enzyme is activated by interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukin-1 (IL-1) produced by T lymphocytes. The NOS2 enzyme converts l-hydroxy-arginine to citrulline, which is antifibrotic. With the TH2 pathway, NOS2 is not activated in the presence of the TH2 cytokines (ILs 4, 10, and 13) and granulocyte-macrophage colony-stimulating factor (GM-CSF), and ornithine is converted to polyamines and praline, which induce cell proliferation of fibroblasts and collagen production, respectively, thereby contributing to fibrosis. TGF-β, Transforming growth factor-β. (Redrawn with permission from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
Endothelial Cells
Endothelial cells are essential for neovascularization of chronic inflammatory lesions. The process of angiogenesis (neovascularization) in chronic lesions is similar to that which occurs during wound healing (see section on Wound Healing and Angiogenesis) and is induced by hypoxia and release of endothelial cell growth factors, such as FGF, VEGF, and PDGF.
Endothelial cells are interconnected by tight junctions composed of occludins, claudin, and JAMs, as well as adherens junctions composed of VE-cadherins. As leukocytes migrate between endothelial cells, leukocyte adhesion molecules bind some of these intercellular molecules. For example, LFA-1 molecule (CD11-α/CD18) binds JAM A, VLA-4 molecule (α-4/β-1) binds VCAM-1 and JAM B, and Mac-1 (CD11b/CD18) binds JAM C to mediate leukocyte passage between endothelial cells. These molecules are especially important for the transmigration of monocytes and lymphocytes through endothelial cell junctions into sites of chronic inflammation, by providing a stable yet temporary site of attachment of leukocyte filopodia and lamellipodia.
Trafficking of Naïve and Activated T and B Lymphocytes
Homing of Naïve Lymphocytes Via High Endothelial Venules: After naïve T lymphocytes are formed in the thymus and B lymphocytes are formed in the bone marrow, they traffic (home) to various secondary lymphoid organs, including lymph nodes, lymphoid nodules in the mucosal surfaces of the colon and cecum, Peyer’s patches in the mucosal surfaces of the small intestine, and organs such as the spleen. These cells express L-selectin and migrate in the blood and often enter these areas through specialized vessels termed high endothelial venules (HEVs), which are postcapillary venules that have a thick basal lamina, and plump endothelial cells with abundant cytoplasm. HEVs produce certain chemokines constitutively (CCL19, CCL21, CXCL12, and CXCL13) to attract naïve T and B lymphocytes expressing the corresponding chemokine receptors CCR7 (receptor for CCL19, CCL21) and/or CXCR4 (receptor for CXCL12) and/or CXCR5 (receptor for CXCL12) (Web Table 3-10). HEVs are located principally in T-lymphocyte zones (paracortical areas of lymph nodes, interfollicular areas of Peyer’s patches), but some are located in the B-lymphocyte zones, especially the periphery of B-lymphocyte follicles. Both peripheral lymph nodes and Peyer’s patch HEVs express the adhesion molecules that mediate T and B lymphocyte adherence, including peripheral node addressin ligand for L-selectin, ICAM 1, ICAM-2, VE-cadherins; however, only Peyer’s patch HEVs express MAdCAM-1, a receptor for the α-4/β-7 adhesion molecule expressed by T and B lymphocytes destined for Peyer’s patches and mesenteric lymph nodes.
WEB TABLE 3-10
Key Adhesion Molecules and Chemokines Involved with Homing of Naïve Lymphocytes, Adherence/Transmigration of Monocytes, and Activated T Lymphocytes in Sites of Chronic Inflammation
Slow rolling is the process of decreased vascular transit of leukocytes within the blood by selectin-mediated adherence/tethering; Firm adhesion is the stable binding of leukocytes to endothelial cells (see Chapter 3). L-selectin–PNAD denotes the leukocyte adherence molecule (L-selectin) and the endothelial cell molecule (PNAD) as does LFA-1 (leukocyte adherence molecule) and ICAM-1 (endothelial cell molecule). E-selectin and P-selectin R are expressed on monocytes at low levels and increase with activation; E- and P-selectin are expressed at increased levels on endothelial cells in areas of chronic inflammation.
α4β7, α4 β7-Integrin; CCL, CC-type chemokine ligand; CCR, CC-type chemokine receptor; HEV, high endothelial venule; ICAM, intercellular adhesion molecule; JAM, junctional adhesion molecule; LFA, lymphocyte function antigen; MAdCAM, mucosal addressin cellular adhesion molecule; PECAM, platelet endothelial cell adhesion molecule; PNAD, peripheral lymph node addressin; R, receptor; VCAM, vascular cell adhesion molecule.
*Activated T lymphocyte in this case is a generic term for TH1, TH0, TH2 effector memory and central memory cells, and T-lymphocyte clones. Note that effector memory cells have only minimal to no CCR7 expression. Activated B lymphocytes likely migrate similarly.
In addition to chemokines, sphingosine-1-phosphate (S1P) is produced by endothelial cells and by local mast cells and platelets and accumulates in the fluid of lymphatic vessels at high levels. S1P receptors are present on T and B lymphocytes, NK-T lymphocytes, and dendritic cells, as well as eosinophils, mast cells, NK cells, and macrophages. S1P mediates homing and also egress of naïve T and B lymphocytes and dendritic cells from the lymph node into the lymphatic fluid.
Adherence and Transendothelial Migration of Activated T Lymphocytes
Lesions developing into sites of chronic inflammation release chemokines, S1P, and other chemoattractant substances that attract and activate T lymphocytes. Although naïve T lymphocytes express high concentrations of L-selectin and the antigen CD45RB, in recently activated T lymphocytes, in effector/memory T lymphocytes, and T-lymphocyte clones, L-selectin expression decreases along with expression of the CD45RB antigen. Instead of L-selectin, acutely activated TH1 and TH2 cells, effector and central memory lymphocytes, and T-lymphocyte clones begin to express high to low levels of E- and P-selectin ligands, which bind to E- and P-selectin receptors, which are expressed by endothelial cells, activated in sites of chronic inflammation. Lymphocytes adhere to these areas of endothelium by eventually binding firmly to the vascular wall via adhesions between the LFA-1 adhesion molecule expressed on lymphocytes, which binds to ICAM-1 and ICAM-2 expressed on endothelial cells. Lymphocytes transmigrate across the vascular wall by adherence between LFA-1 integrins and the ICAM-1 molecules, JAM molecules, and also contributions from PECAM-1.
Inflammation and the Sensation of Pain
Several features of inflammatory response activate sensory nerve fibers that mediate the sensations of swelling, thermal change, and pain. Mechanoreceptors can sense changes in tissue swelling secondary to the accumulation of edema fluid and/or exudates. Thermoreceptors can sense changes in localized or systemic temperatures. Pain receptors can also become activated. Concerning pain, release of PGE2 is a crucial mediator of the inflammatory pain sensation. PGE2 mediates this activity through a specific glycine receptor subtype (GlyR α-3) of the dorsal gray horn of the spinal cord. Inhibition of PGE2 expression through inhibition of COX enzymes reduces pain and future therapies may target GlyR α-3 receptors.
The Effect of Inflammation on the Febrile Response and Other Activities
Cytokines such as IL-1, TNF, and IL-6, as well as high mobility group protein box-1 (HMGB-1) molecules are often produced during acute inflammatory responses. These inflammatory mediators, along with LPS (microbial factor) and other potentially toxic microbial or chemical molecules are important regulators of body temperature (fever), malaise, headache, confusion, anorexia, locomotion, and unconsciousness. Many of these actions occur through (1) the effects of these cytokines on sensory fibers in the vagus nerve, which extends to the brainstem, and (2) their effects on cerebral endothelial cells, perivascular microglia, and meningeal macrophages and the ability of the cells to induce COX-1, COX-2, and COX-3 activity for the formation and release of prostaglandins such as PGE2. PGE2 then activates various hypothalamic nuclei, including the ventromedial preoptic nucleus, paraventricular nucleus, solitary tract nucleus, ventrolateral medulla, and parabrachial nucleus, which regulate fever and the other clinical responses listed previously.
Unique Types of Inflammation
Septicemia and Endotoxic Shock
Septicemia: Septicemia is a clinically significant form of bacteremia complicated by toxemia, fever, malaise, and often shock (see Table 3-5). Septicemia is characterized by the multiplication of microorganisms within the bloodstream and “seeding” into blood from fixed microcolonies present in one or more tissues. In septicemia, inflammation is not a localized reaction to injury, but instead mediators of inflammation are generated systemically leading to diffuse “leakage” of plasma into the interstitium and sequestration of leukocytes in the microvasculature. Generation of cytokines, kinins, vasoactive amines, and lipid mediators of inflammation, combined with widespread endothelial damage, leads to profound circulatory disturbances. Because of the systemic nature of this host-microbial interaction, quantities of phagocytic cells, antibody, complement components, coagulation proteins, and platelets may become depleted unless septicemia is controlled in the early stages. Septic shock and disseminated intravascular coagulation (DIC) are the usual sequelae of advanced bacterial septicemia.
Septicemia should be differentiated from a bacterial embolism. For example, some strains of Streptococcus spp. may break free from vegetative lesions (valvular endocarditis), as large colonies are protected by cell debris and fibrin. The bacterial emboli may then mechanically lodge in the lung, liver, kidney, or brain to produce a secondary focus of infection (abscess), but the whole process remains subclinical. In such a case, the blood sample taken for culture often lacks viable bacteria.
Septic (Endotoxic) Shock: The systemic interaction of microorganisms and their products (toxins) with a spectrum of host cells and chemical mediators results in a clinical syndrome recognized as sepsis or septic shock (see Table 3-5). The host mediators and amplification systems initiating the syndrome vary with the type of organism and the nature of the infectious process (local or systemic). Regardless of the specific cause, the major elements of septic shock form a continuum, including (1) hemodynamic derangements (reduced blood pressure and increased heart rate), (2) abnormal body temperature, (3) progressive hypoperfusion of the microvasculature, (4) hypoxic injury to susceptible cells, (5) quantitative adjustments in blood leukocytes and platelets, (6) DIC, (7) multiple organ failure, and (8) death.
Bacterial endotoxin, the LPS from the outer membrane of Gram-negative bacteria, has been studied extensively as an initiator of septic shock. The peptidoglycan layer of Gram-positive bacteria and bacterial exotoxins can initiate many of the same host responses. Other important initiators are products of the interaction of neutrophils, macrophages, and platelets with microorganisms in the tissue. Endotoxin is bound in the serum by LBP that binds CD14. Endotoxin has numerous ways to induce systemic activation of inflammatory mediators. Three direct effects of endotoxin are the activation of Hageman factor (a clotting factor), the complement cascade, and induction of the TLR4 pathway. These pathways can ultimately activate bradykinin, PAF, arachidonic acid metabolites, and cytokines (IL-1 and TNF), all of which have a role in many of the coagulation, hemodynamic, thermoregulatory, and leukocyte derangements observed in septic shock. TNF is capable of producing most clinical and pathologic features of septic shock, including hypotension, metabolic acidosis, hemoconcentration, intestinal hemorrhage, fever, neutrophil and endothelial activation, and predisposition to thrombosis. IL-1 shares many of the biologic activities of TNF in the mediation of septic shock. Secretion of TNF by activated macrophages can be partially inhibited by pretreatment with glucocorticoids, such as dexamethasone, which have been used therapeutically, often with limited success. In addition, lethal shock is prevented with anti-TNF antibody or TNF-receptor inhibitors. Chelators of endotoxin and other bacterial products are also under development for use in therapeutic intervention.
In severe septicemia, a systemic inflammatory response syndrome (SIRS) can develop in which there is extensive accumulation of cytokines, activated neutrophils, and platelets in the circulatory system. This result leads to multiple organ failure (MOF) and shock. Most patients survive initial SIRS insults, but these individuals are at increased risk of secondary or opportunistic infections termed compensatory antiinflammatory response syndrome (CARS). The initial activation of innate immunity can lead to decreased macrophage activity, T lymphocyte anergy, and apoptosis of lymphocytes contributing to CARS.
Multiple organ dysfunction syndrome (MODS) represents a late stage in septic shock and accounts for much of the irreversibility of organ failure. Systemic tissue ischemia and hypoxia, associated with progressive cardiovascular derangements, increased vascular leakage, and DIC, lead to generalized organ failure. Organs that are particularly sensitive to these effects include heart, brain, kidney, lung, liver, and intestinal mucosa. Cells injured by ischemia revert to anaerobic energy production (glycolysis), resulting in rapid depletion of substrates (glycogen, glucose), accumulation of lactate, and a deficiency of ATP. Without sufficient ATP, cell membrane ion pumps fail to maintain electrolyte balances, membrane integrity, and protein synthesis. The influx of sodium into cells with water causes cells to swell with further loss of function. Influx of calcium ions activates many intracellular enzymes, including phospholipase, which breaks down cellular membranes and generates arachidonic acid products. Loss of the proton gradient of the inner mitochondrial membrane makes oxidative phosphorylation impossible. Irreversible cell injury is believed to be closely related to generalized failure of mitochondria and loss of selective permeability of cell membranes. In dogs, sepsis of abdominal origin can induce MODS, and dysfunction of any organ system is associated with increased risk of death and mortality rates increase as the number of affected organs increase.
Animals dying of septic shock typically have evidence of fluid in the body cavities, pulmonary edema, petechial hemorrhages, congestion of the liver and intestines, and dehydration. Common microscopic lesions include acute necrosis of renal tubules, centrolobular hepatocytes, cardiac myocytes, adrenals, and tips of intestinal villi.
Wound Healing and Angiogenesis
Almost immediately after a wound develops, the process of healing begins. Injured tissue goes through four temporal phases to repair the wound: hemostasis, acute inflammation, proliferation (granulation), and remodeling (maturation, contraction) (Fig. 3-34). These phases occur in this sequence but may progress at different rates. Even in one lesion site, different areas may be in different phases of repair.
Fig. 3-34 Phases of cutaneous wound healing.
Inflammation, proliferation, and maturation (see text for details). (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Hemostasis occurs immediately after injury unless there is a clotting disorder. Initially after injury, hemostasis is controlled via vasospasm, a process in which blood vessels constrict in response to injury. But this spasm subsides rapidly, and the injured (transected) blood vessels will subsequently relax, allowing additional bleeding if platelets do not become involved. During the initial period of vasoconstriction, platelets aggregate and adhere to exposed collagen, especially collagen in the basement membrane underlying injured endothelial cells. Once adhered, platelets secrete vasoconstrictive substances to (1) maintain constriction of the transected vessels, (2) initiate the process of thrombogenesis to “plug” the leak in the vessel and prevent additional bleeding, and (3) initiate blood vessel healing (angiogenesis). This process also occurs with large blood vessels, but additional physiologic factors, such as blood shunting and decreased blood pressure. are involved.
By 24 hours after vascular injury, the inflammation phase (acute inflammation) of wound healing is fully established and can last up to 96 hours or longer if the healing process is disrupted by infection, trauma, or some other perturbation. It is in this phase that the “cardinal signs” of inflammation—redness, swelling, heart pain, and loss of function are observed. Neutrophils and macrophages, through phagocytosis and their degradative enzymes, breakdown and remove (“clean-up”) the cell debris resulting from tissue injury. Macrophages secrete a variety of chemotactic and growth factors that establish the microenvironment for the proliferation (granulation) phase. The “clean-up” activity of neutrophils and macrophages within wounds is necessary, although excessive inflammatory cell infiltration reduces healing.
Some ECM molecules, such as some proteoglycans, have a negative charge thus attracting and binding to more positive-charged growth factors, chemokines, cytokines, MMPs, and other molecules. In addition, fragments of collagen, fibrin, and other molecules in wounds can induce chemotaxis, cell proliferation, and angiogenesis. Thus, with degradation of the ECM in wounds, there is release of these molecules, which contribute to matrix degradation, chemotaxis, and cell proliferation.
In tissues lined by epithelium, migration of basal cells from the overlying epithelium begins early in the healing process and does not require an underlying collagenous matrix. These cells arise from the transected edges of epithelium that border the wound, which rapidly undergo hyperplasia. The migrating basal cells proliferate, spreading in attempt to bridge the wound, and some of these cells differentiate; however, once the cells differentiate, they cease to proliferate and migrate. The proliferation phase can last up to 3 to 4 weeks or longer depending on the size of the wound. This phase is characterized by the generation of new endothelium (angiogenesis), epithelium (epithelialization), and connective tissue stroma (fibroplasia/desmoplasia) to restore normal structure and function to the injured tissue. The healing of skin after third-degree burns or severe ulcerations is an excellent example of this process. The return to normal structure and function depends on (1) the retention of normal stromal elements of the ECM to provide the structural framework for repair, and (2) normally functioning fibroblasts, myofibroblasts (contractile fibroblasts), endothelial cells, pericytes (nonendothelial components of blood vessels), and epithelial cells. Initial deposition of collagen and other key ECM molecules begins after fibroblasts have proliferated and fortified regions of the wound. Collagen fiber deposition alone is not sufficient for complete matrix repair without proliferative and functional fibroblasts.
The role of stem cells of epithelia and mesenchymal stroma in wound healing is increasingly understood. For epithelia, stem cells reside along the basal layer and are more clustered in specific regions. In the cornea of the eye, stem cells are located in the limbus. In skin, clusters are present in the bulge region of the hair follicle (roughly half-way down the follicular wall in the dermis). In lung, stem cells are present along the bronchiole-alveolar junction. In intestine, stem cells are present within the crypts. The stem cells are often in a quiescent (senescent) stage by the influence of bone morphogenic protein (BMP), which inhibits proliferation. Beta catenin (wnt) released from active stem cells in the dermal papilla of hair follicles, for example, induces proliferation of the quiescent cells that form new structures. Mesenchymal stromal cells subjacent to epithelia also undergo proliferation and communicate between epithelial cells and the subjacent inflammatory cells and stroma. The mesenchymal stromal cells function similarly to those of embryonic blastema cells.
The remodeling (maturation, contraction) phase begins approximately 3 to 4 weeks after injury, but only after the inflammation and proliferation phases have been successfully completed. This phase includes remodeling of granulation tissue by immature connective tissue and the conversion of immature connective tissue to mature connective tissue through extracellular collagen formation. Remodeling can last for 2 or more years. It essentially provides the time some tissues and organs, such as bone, need to return to the near-normal tensile strength required for normal axial and appendicular skeletal function.
A key component of wound repair is the ECM and stromal stem cells (fibroblasts, myofibroblasts). In mild or moderate injury, partially degraded collagen, proteoglycans, and elastin are completely degraded by MMPs and other enzymes removed by macrophages and then resynthesized by surviving fibroblasts. Simultaneously, fibroblasts and endothelial cells proliferate to fill tissue defects (granulation tissue) and epithelial cells, endothelial cells, and some parenchymal cells proliferate along basement membranes to restore the normal structure of the tissue. If basement membranes are degraded in the injury and the healing process disrupted, then complete healing is delayed because of a requirement for a new basement membrane to be deposited by endothelial cells that attach to and line the contiguous remaining basement membrane. Should healing be continually delayed (infection) or prevented (large tissue defect with loss of stroma and basement membrane), then dysregulated healing can occur in the form of extensive fibrosis (scars and hypertrophic scars) with haphazard arrangement and/or metaplasia of the overlying epithelial cells.
The four phases of wound healing described are applicable to all tissues and organ systems, but each system has its unique mesenchymal and parenchymal cell types that influence the process of healing. Bone healing with callus formation and skin healing with reepithelialization are good examples of healing and the specialization of cell types involved (see Chapters 16 and 17). Overall, the success of wound healing, especially in the skin, is often determined by whether the process occurs via first or second intention healing. Healing in other tissue is similar. In bone, for example, optimal healing occurs in bone fragments that are stabilized and in direct apposition.
First and Second Intention Healing
First intention healing in skin occurs when the edges of a wound site are directly apposed and reattach and heal to each other rapidly (Fig. 3-35). Wounds lacking such close, intimate apposition are termed second intention healing. In the simplest type of wound, such as a cut or incision in the skin by a surgeon, there is initial hemorrhage from the damaged vasculature and retraction and constriction of blood vessels. In the wound area, there is deposition of fibrin, leakage of plasma proteins, clot formation, platelet aggregation, and neutrophil infiltration. The nature of repair depends on several factors, including the proximity of the cut edges to each other, the presence or absence of foreign bodies or infectious microbes, and the general health capacity of the animal to repair wounds. Under ideal conditions, such as in surgery, first intention healing is desired (see Fig. 3-35). First intention healing occurs in nonseptic wounds, whereas second intention healing occurs in septic wounds, with foreign bodies, or gaping wounds with nonopposed edges. If the wound healing process is disrupted or delayed, the process is shifted to second intention healing. First and second intention healing are also discussed in Chapter 17.
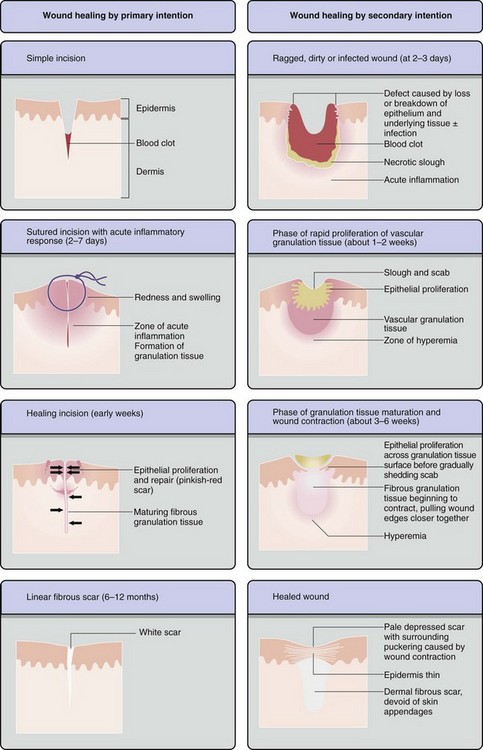
Fig. 3-35 Wound healing.
Steps in wound healing by primary intention (left) and secondary intention (right). Note large amounts of granulation tissue and wound contraction in the healing by second intention. (From Young B, O’Dowd G, Stewart W: Wheater’s basic pathology: a text, atlas and review of histopathology, ed 5, New York, 2010, Churchill Livingstone.)
First Intention Healing
First intention healing occurs in 2 to 3 days in the skin, if the cut edges of a nonseptic wound are positioned in close proximity to each other by sutures or bandages. During this time, the hemorrhage, plasma proteins, and cell debris within the wound are phagocytosed and removed by macrophages, new blood vessels sprout and grow into the lesion, and the ECM is synthesized to fill the gap between the apposed tissue edges. With time (weeks), this stable interconnection in the dermis is replaced by collagen fibers that undergo continual maturation, thus providing the skin with near-normal tensile strength after wound healing. Concurrently, basal cells of the squamous epithelium will undergo hyperplasia and cover the defect in 3 to 5 days. This type of repair leaves little trace of the wound, except for perhaps mild fibrosis in the superficial dermis and loss of adnexa (e.g., hair follicles, sebaceous glands, and sweat glands) at the site of the wound. The tensile strength is nearly the same as that of adjacent tissue. First intention healing is the goal of the surgeon for repair of incision sites made during surgery.
Second Intention Healing
Second intention healing occurs when the cut edges of the skin, for example, are not brought into appropriate apposition for healing (see Fig. 3-35). In such wounds, connective tissue is haphazardly synthesized and arranged and there is little or no organization in the healing process; however, fibrous connective tissue fills the defect in the superficial and deep dermis. This disorganization can also delay or prevent the migration of epithelial cells that attempt to cover the surface of the wound and disrupt the orderly deposition of ECM in the wound. In addition, new fibrous connective tissue lacks adnexa (hair follicles, sebaceous and sweat glands). In some cases, fibrous connective tissue can form into granulation tissue (see later section) in which histologically, proliferating fibroblasts are arranged perpendicular to new capillaries, and the long axes of the new capillaries are arranged perpendicular to the surface of the skin (Fig. 3-36). Tensile strength of granulation tissue is diminished and the lesions can tear or split. Thus with second intention healing, the site can remain ulcerated, lack hair, and in some cases, the fibrous connective tissue can undergo continuous proliferation and protrude from the skin surface as a hyperplastic scar.
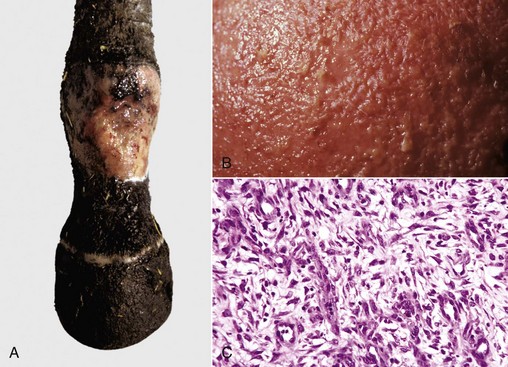
Fig. 3-36 Granulation tissue, nonhealing ulcer, skin, distal limb, horse.
A, In the bed of the ulcer, there is extensive fibrosis and granulation tissue. B, Gross photograph of the surface of the granulation tissue. Note the fine nodules or “granulations” on the surface that gave rise to the term granulation tissue. These are a mixture of newly formed blood vessels, extracellular matrix (ECM), and fibroblasts, with minimal or no collagen deposition. It provides the support for wound repair and remodeling via fibroplasia and reepithelialization. C, Photomicrograph of granulation tissue. Note how the new fibroblasts are arranged perpendicularly to the newly formed blood vessels in a rich bed of ECM (clear spaces). (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Impaired Wound Healing
In addition to spontaneously occurring impairments of wound healing, such as foreign bodies, infection, and neoplasms, certain other conditions can prevent or impair wound healing, even first intention healing. For example, altered deposition of collagen and ECM proteins can occur with osteogenesis imperfecta because of impaired production of type I collagen. Similarly, impaired synthesis, cross-linking, hydroxylation, or posttranslational processing of collagen can delay wound healing in individuals with Ehlers-Danlos syndrome. Hyperglycosylation of proteins, which can occur with prolonged diabetes mellitus, can alter the vasculature, lead to diabetic ulcers, and inhibit wound healing.
Chemotherapeutic drugs can also prevent cellular proliferation and may reduce healing. Several new chemotherapeutic drugs specifically target endothelial cell proliferation, which may greatly influence the process of neovascularization so vital to efficient wound repair. In humans, guinea pigs, and other species that require dietary vitamin C, deficiencies in intake can lead to scurvy, a disease in which there is decreased collagen hydroxyproline synthesis and poor wound healing. Extreme starvation, malnutrition, and cachexia from cancer or severe weight loss from chemotherapy can impair the synthesis and deposition of ECM proteins as a result of negative energy balance and a lack of amino acid substrates, normally synthesized in the liver. Additionally, such individuals and severe burn victims often lack adequate levels of serum proteins such as albumin, which results in lowered osmotic plasma pressure, impaired fluid resorption from the wound site, and enhanced edema fluid accumulation.
Expression of Genes Responsible for Wound Repair
Wound repair requires activation of genes of the viable cells such as macrophages, fibroblasts, and endothelial cells adjacent to the sites of tissue injury. As indicated, macrophages internalize through phagocytosis cell debris to “clean-up” an area and degrade the ECM. In concert with fibroblasts, macrophages release growth factors that enhance the proliferation of (1) endothelial cells for neovascularization, (2) fibroblasts for deposition of a new ECM, (3) myofibroblasts for wound contraction, and (4) parenchymal cells for return to normal structure and function of the affected tissue.
Gene expression by cells in a wound is regulated to a large degree by oxygen levels (Fig. 3-37; Web Fig. 3-22). In the milieu of a wound, there is generally a reduced oxygen tension caused by vascular damage. Normal tissues have oxygen levels >90% oxygen saturation, and there is increased activity of non-heme iron-containing 2-oxglutarate (2-OG)-dependent oxygenases that sense oxygen levels and use dioxygen as a cosubstrate. These include prolyl hydroxylase domain-containing protein-1 (PHD-1), PHD-2, PHD-3, and factor-inhibiting HIF (FIH). These enzymes place a hydroxyl group on proline and asparagine amino acids in HIF-1α protein. Hydroxylated HIF-1α is degraded by the ubiquitin pathway when oxygen levels are high. In hypoxic tissue, however, as occurs in wounds, within neoplastic masses, and areas of inflammation, there is reduced activity of PHDs and FIH and thereby less hydroxylation of HIF-1α. Nonhydroxylated HIF-1α aggregates with HIF-1β and induces transcription of hypoxia-responsive elements (HREs) in the genome.
Fig. 3-37 Regulation of hypoxia-inducible factor (HIF) transcriptional activity by prolyl hydroxylase protein domain-containing proteins (PHDs) and factor-inhibiting HIF (FIH).
With sufficient oxygen (left), HIF-1α protein is hydroxylated by PHDs and FIH resulting in degradation. With insufficient oxygen (right) HIF-1α protein is not hydroxylated and forms an active complex with HIF-1β resulting in transcriptions of genes that contribute to wound healing an angiogenesis, including the transcription of vascular endothelial growth factor (VEGF). (Modified from Fraisl P, Aragones J, Carmeliet P: Nat Rev Drug Discovery 8(2):139-151, 2009.)
Web Fig. 3-22 Hypoxia-inducible factor (HIF).
Initiation of wound healing and regulation of gene expression by HIF. When oxygen levels are normal, HIF is not active, and hypoxia-responsive genes are not activated. With hypoxia, however, HIF activates transcription of genes of the hypoxia-response elements (HREs). UB, Ubiq, Ubiquitin; VHL, von Hippel-Lindau disease. These genes increase blood vessel formation, iron sequestration, and hypoxia metabolism. (Redrawn with permission from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
The HREs include genes for growth factors, including VEGF, iron-binding proteins, regulators of apoptosis, erythropoiesis, angiogenesis, pH regulation, and glucose and energy metabolism. Early growth response gene-1 (EGR-1) is another transcription factor activated in wounds that leads to expression of growth factors and cytokines. Therefore both HIF-1α and EGR-1 activity in hypoxic conditions lead to increased cellular transcription that upregulates genes for energy (glucose transporters, hexokinase 1 and 2, lactate dehydrogenase, phosphofructokinase), endothelial and fibroblast proliferation (TGF-β, VEGF), and iron sequestration (ceruloplasmin, transferring receptor). These genes promote cell survival in hypoxic conditions, enhance cell proliferation, especially of cells vital to repair (endothelial cells, fibroblasts), and delay or alter differentiation of other cells (epithelia or parenchymal cells) until endothelial and fibroblast proliferation is well established.
Degradation of Cells and Tissue Components in Wounds
Wounds generally have a central core composed of (1) degenerate and/or necrotic cells, such as parenchymal cells, fibroblasts, and endothelial cells, as well as infiltrating leukocytes, such as neutrophils, platelets, lymphocytes, mast cells, and macrophages; (2) inflammatory products (cytokines, eicosanoids, chemokines, and their respective receptors); (3) serum proteins (albumin, acute phase proteins, complement); (4) clotting proteins (fibrin); and (5) ECM proteins and substances. Many of these cells and mediators need to be removed before optimal healing takes place. Phagocytic cells, such as neutrophils and macrophages, are very important in the clean-up process through phagocytosis of particulate matter and subsequent lysosomal degradation and the release of digestive enzymes into the tissue. In addition, macrophages have a major role in the uptake of apoptotic cells that form in response to TNF-α or other proapoptotic inflammatory stimuli. The ECM can be especially difficult to degrade. However, macrophages and fibroblasts are key to this process through the release of matrix metalloproteinases that degrade the ECM.
Degradation of the Extracellular Matrix In Wounds
The ECM is composed of (1) proteins and (2) the hydrated gel of proteoglycans in which they lie. It surrounds and interconnects cells in connective tissue such as fibroblasts, blood vessels, lymphatic vessels, resident mast cells, macrophages, dendritic cells, and nearby parenchymal cells and/or epithelia (Web Fig. 3-23). The ECM influences cellular development, polarity (organization), and function of epithelial cells (Web Fig. 3-24). Soluble proteoglycans and fragments of glycosaminoglycans can activate Toll-like receptors and proteoglycans and hyaluronan can facilitate leukocyte adhesion. Also, ECM binds and sequesters cytokines, chemokines, and growth factors that are released during ECM degradation.
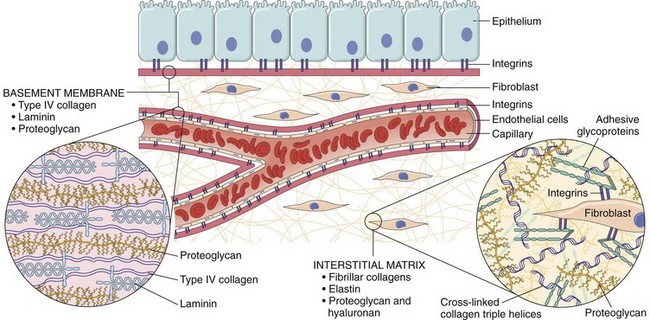
Web Fig. 3-23 Extracellular matrix (ECM).
Main components of the extracellular matrix (ECM), including collagens, proteoglycans, and adhesive glycoproteins. Both epithelial and mesenchymal cells (e.g., fibroblasts) interact with ECM via integrins. Basement membranes and interstitial ECM have different architecture and general composition, although there is some overlap in their constituents. For the sake of simplification, many ECM components (e.g., elastin, fibrillin, hyaluronan, and syndecan) are not included. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
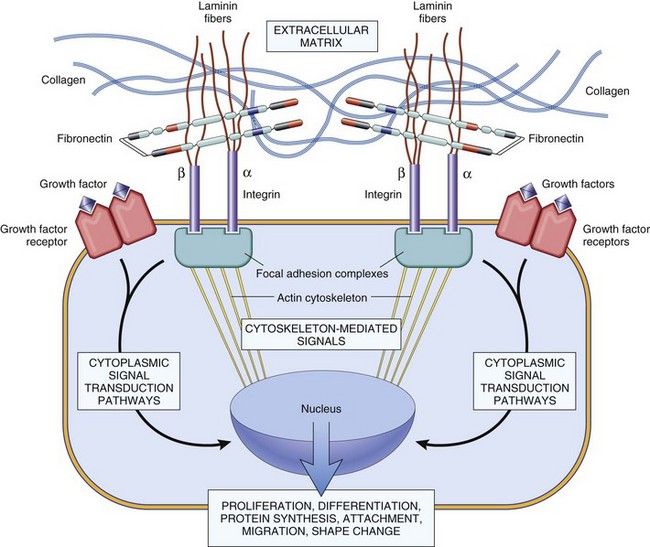
Web Fig. 3-24 Extracellular matrix (ECM) regulation of cell functions.
Mechanisms by which ECM (e.g., fibronectin and laminin) and growth factors can influence cell growth, motility, differentiation, and protein synthesis. Integrins bind ECM components and interact with the cytoskeleton at focal adhesion complexes (protein aggregates that include vinculin, α-actin, and talin). This can initiate the production of intracellular messengers or can directly mediate nuclear signals. Cell-surface receptors for growth factors may activate signal transduction pathways that overlap with those activated by integrins. Collectively, these are integrated by the cell to yield various responses, including changes in cell growth, locomotion, and differentiation. (From Kumar V, Abbas A, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
With tissue injury, there is often destruction and degradation of the ECM. This process occurs through physical separation or tearing, dilution from plasma proteins, infiltration by inflammatory cells, and degradation by enzymes, largely the MMPs (Web Fig. 3-25). Macrophages, fibroblasts, mast cells, and most leukocytes produce MMPs (Table 3-11). Many MMPs were initially named after the type of ECM protein that they were found to degrade (e.g., collagenase), but because the MMPs are now known not to be uniquely specific for a particular ECM substrate, they have been reclassified in a numeric manner, MMP-1 through MMP-20.
TABLE 3-11
Matrix Metalloproteinase (MMP) Activity, Regulation, and Cellular Production
| Type of MMP | Cell Type |
| MMP 1, 2, 3, 11, 14 | Fibroblasts |
| MMP 9, 12 | Macrophages |
| MMP 9 | Neutrophils |
| MMP 2, 3, 9 | Endothelial cells |
| MMP 9 | Pericytes |
| MMP 1, 3, 7, 9, 13 | Some cancer cells |
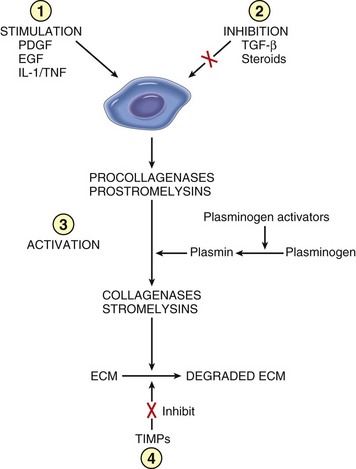
Web Fig. 3-25 Matrix metalloproteinase (MMP) activities.
Four mechanisms are shown: 1, regulation of synthesis by growth factors or cytokines; 2, inhibition of synthesis by corticosteroids or transforming growth factor-β (TGF-β); 3, regulation of the activation of the secreted but inactive precursors; and 4, blockage of the enzymes by specific tissue inhibitors of metalloproteinase (TIMPs). ECM, Extracellular matrix; EGF, epidermal growth factor; IL-1, interleukin-1; PDGF, platelet-derived growth factor. (Modified from Matrisian LM: Trends Genet 6:122, 1990.)
For example, collagenase is MMP-1, gelatinase is MMP-2, stromelysin is MMP-3, and matrilysin is MMP-7. MMPs degrade collagen, gelatin, elastin, aggrecan, versican, proteoglycan, tenascin, laminin, fibronectin, and other ECM components. The MMP enzymatic domain contains three histidine residues that form a complex with zinc. A regulatory domain is responsible for latency and allows activation in the presence of zinc. MMP activity is also regulated by tissue inhibitors of MMP (TIMP). ADAM (a disintegrin and metalloproteinase) are zinc proteinases capable of degrading matrix molecules as can cathepsin G, tissue plasminogen-activator (tPA), and urokinase plasminogen activator (uPA) (Web Box 3-2). Fragments of proteins degraded by MMP, tPA, uPA, and other degradative processes are removed from wounds by lymphatic drainage and phagocytosis by macrophages and neutrophils. Proteoglycans are largely degraded by lysosomal enzymes of macrophages and neutrophils that include hyaluronidases, heparinases, and galactosidases. As indicated, ECM degradative enzymes also (1) release latent growth factors and other latent molecules bound to ECM molecules, (2) inactivate some molecules present within the region, (3) break down basement membranes, and (4) cleave intercellular adhesion molecules between epithelial cells.
Resynthesis of the Extracellular Matrix with Wound Healing
Synthesis of Collagen and Matrix Proteins
As wounds repair, the body attempts to reestablish the ECM. The structural proteins of the ECM include several types of collagens, elastin, and adhesive type proteins including fibronectin, laminin, versican, tenascin, and vitronectin. The fibrillar collagens (types I, II, III, V, and XI) are triple-stranded helical structures aggregated into fibrils in the extracellular space and surrounded by collagens IX and XII, which interconnect the collagen fibrils with one another and the ECM. Most tissues have a predominance of one collagen type. For example, collagen type I is present in bone, skin, and tendon; collagen type II is present in cartilage and vitreous humor; collagen type III is present in skin, around vessels, and in newly formed wounds; collagens type V and VI are present in interstitial tissues; collagen type VI is present near epithelia; collagen type VIII is present near endothelial cells; and collagens type X and XI are present in cartilage.
Collagen type IV is largely present in basal lamina along with laminin, entactin, a heparin sulfate proteoglycan, and perlecan. Throughout the ECM are molecules of elastin, which stretch, recoil, and allow flexibility in the tissue. Collagen fibers, laminin, fibronectin, tenascin, and other ECM proteins bind to cells in the connective tissue via extracellular domain of integrin molecules of cells by means of a specific amino acid sequence, the RGDS sequence. For example, laminin binds α2β1-integrins of endothelial cells, some collagens bind α6β1-integrins of epithelial cells, and fibronectin and vitronectin bind α5β3-integrins. The intracellular portion of integrin molecules interact with the cellular cytoskeleton (i.e., actin assembly) and thereby link the extracellular milieu with cellular activities such as cell growth, differentiation, proliferation, and senescence.
Collagen Production by Fibroblasts
Collagen deposition within a site of wound repair provides a scaffold for reestablishment of the ECM and stroma (Web Fig. 3-26). Fibroblasts are induced by TGF-β and other cytokines to synthesize collagen. Ribosomes in fibroblasts produce approximately 30 types of collagen α-chains that are composed of repetitive glycine-x-y segments. Although within the RER, praline and lysine residues in these chains are hydroxylated, and this hydroxylation process requires vitamin C. The chains are then glycosylated, arranged in a triple helix, and eventually released into the extracellular space as procollagen. The ends of procollagen are cleaved enzymatically resulting in the formation of fibrils termed tropocollagen. Cross-linkages between collagen fibrils occur at lysine and hydroxylysine residues through the activities of the enzyme lysyl oxidase, and this cross-linking process provides the tensile strength of collagen.
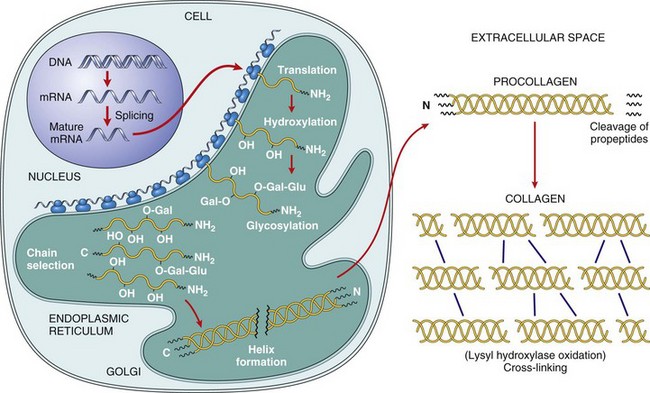
Synthesis of Proteoglycans
Proteoglycans are produced by fibroblasts. They retain water and are vital to the hydration of the ECM. Proteoglycans have a protein backbone surrounded by a network of glycosaminoglycan (GAG) chains (Web Fig. 3-27). The GAGs are negatively charged, often highly sulfated, polysaccharide chains covalently linked to the serine residues on a protein backbone. Most GAGs contain high concentrations of N-acetylglucosamine (Web Table 3-11). Hyaluronic acid lacks sulfation and is not connected to the protein backbone. GAGs are key to the water retention properties of proteoglycans and thus, hydration of the extracellular milieu. Proteoglycan hydration of the ECM allows tissues to be pliable and have elasticity.
Web Fig. 3-27 Proteoglycan molecule.
Proteoglycans have a central protein backbone that supports glycosaminoglycan (GAG) side chains. The GAGs retain water and contribute to the hydration of tissue. (Redrawn with permission from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
Heparin sulfate proteoglycans, such as syndecan, decorin, and perlecan, encircle and surround cells and basal laminae. Syndecan is an integral transmembrane protein that can bind chemokines. With inflammation, syndecan can release the chemokine, which then induces leukocyte infiltration.
Fibroblasts and the Mechanistic Basis of Fibrosis
Fibroblasts align along planes of tissue stress during development (Langer’s lines or tension lines). In quadrupeds, these lines are generally dorsoventral over the thorax and abdomen (axial body plane) and parallel to the long axis of the limbs (appendicular body plane). Surgical incisions along Langer’s lines extend between, rather than transect, bands of fibrous connective tissue and tend to pull the margins of surgical skin incisions together. Such incisions reduce the degree of postsurgical scar formation.
Fibroblasts of cats appear to be especially responsive to injury and inflammation. In fact, injury of fibroblasts has been associated with their neoplastic transformation in cats. For example, traumatic lens rupture can lead to intraocular inflammation and fibroblast proliferation, and in some cases, fibrosarcomas. In addition, fibroblast proliferation and fibrosarcomas are common in cats at vaccination sites.
Initially during the hemostasis and inflammation phases of wound repair, fibrin and serum proteins form a loose gel-like framework for the migration of fibroblasts and endothelial cells into the wound to form granulation tissue. Simultaneously, leukocytes and other cells, such as fibroblasts and endothelial cells, are stimulated by HIF-α and epidermal growth factor (EGF) to synthesize and release a variety of growth factors that result in fibroblast proliferation and migration. These factors include FGF-1 and FGF-2, PDGF, EGF, and TGF-β1, 2, and 3. FGF, PDGF, IL-13, and TGF-β induce fibroblasts to produce collagen, whereas FGF, VEGF, TGF-β, angiopoietin, and mast cell tryptase induce endothelial cells to proliferate and migrate and produce basement membrane for formation of new capillaries (Web Fig. 3-28).
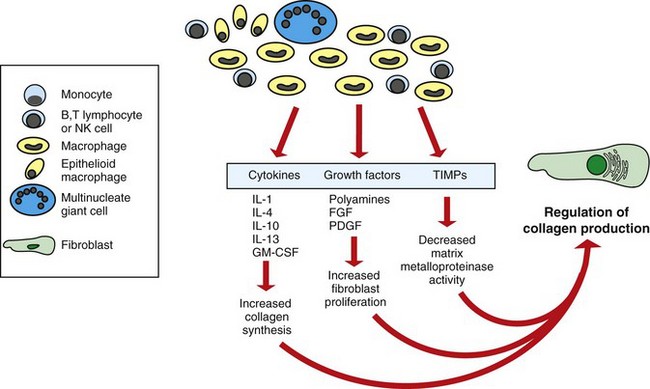
Web Fig. 3-28 Development of fibrosis in chronic inflammation.
In most chronic inflammatory lesions, activated macrophages, lymphocytes, and other cells secrete cytokines, growth factors, and tissue inhibitors of matrix metalloproteinases (TIMPs) that lead to fibroblast proliferation and collagen synthesis. FGF, Fibroblast growth factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; NK, natural killer; PDGF, platelet-derived growth factor. (Redrawn with permission from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
With time, the newly formed, provisional connective tissue is remodeled into a more mature matrix. In the entire process, TGF-β has a central role in fibroblast activity and collagen deposition, because it is produced by platelets and macrophages and induces macrophage chemotaxis, fibroblast migration and proliferation, and synthesis of collagen and ECM proteins. TGF-β binds TGF-β receptor II (TGF-βRII), which dimerizes with TGF-βRI. The TGF receptor then phosphorylates R-SMAD and Co-SMAD to overcome inhibition of SMAD 7. This signaling process induces fibroblast activity, and regulation of the signaling may be useful in therapeutic strategies to control scarring and/or fibrosis (Web Fig. 3-29).
Web Fig. 3-29 Transforming growth factor-β (TGF-β) signaling.
This process results in fibroblast and myofibroblast activation and collagen deposition. R, Receptor. (Redrawn with permission from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
In addition to producing collagen, fibroblasts can migrate to a certain degree, and this process is mediated by adhesion molecules that bind to the ECM. This binding is a complicated event in which the adherence process is essential for migration of the cell and its anchoring to extracellular proteins. During wound repair, proliferating fibroblasts often align themselves parallel with lines of tension stress.
Morphology of Granulation Tissue and Fibrous Connective Tissue
Some lesions develop a distinctive type of arrangement of connective tissue fibers, fibroblasts, and blood vessels termed granulation tissue. Granulation tissue is the exposed connective tissue that forms within a healing wound. It is often red and hemorrhagic and bleeds easily when bumped or traumatized because of the fragility of the newly formed capillaries (see Fig. 3-36). It is especially common in horses. When viewed with a magnifying glass, the surface of granulation tissue has a granular appearance and thus the term granulation tissue arose. In granulation tissue, fibroblasts and connective tissue fibers grow parallel to the wound surface and are arranged perpendicularly to the proliferating capillaries. Often, the penetrating blood vessels are evenly spaced. Excessive granulation can lead to a type of hypertrophic scar called proud flesh. In cats, fasciotomy and fascial excision induce formation of early granulation tissue in cutaneous wounds and may be effective in enhancing closure of secondary wounds.
Hypertrophic Scars: Hypertrophic scars occur as a result of exuberant proliferation of fibroblasts and collagen in wounds that fail to heal properly. The best example of this condition occurs in skin wounds of the distal limbs of horses and is known as “proud flesh,” as indicated proliferating connective tissue forms a large cauliflower-like mass that cannot be covered by epithelium (Fig. 3-38). Why this lesion most commonly occurs in horses is unclear; however, the epidermis of horses is often very “tight” with limited elasticity.
Fig. 3-38 Exuberant granulation tissue (proud flesh), chronic ulcer, skin, distal hindlimb, horse.
Note the large proliferating mass of fibrous tissue on the lower portion of the left hindlimb. It often lacks superficial epithelium. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Keloid is a special type of excessive connective tissue deposit that occurs in humans. It has an incidence of 5% to 16% after skin trauma in high-risk populations, such as blacks, Hispanics, and Asians. Clinical management of hypertrophic scars, proud flesh, and keloids can be difficult but includes intralesional corticosteroids, compression, occlusive dressings, pulsed-dye laser therapy, cryosurgery, surgical excision, radiation, fluorouracil chemotherapy, topical silicone, interferons, and drugs, such as imiquimod, that induce IFN-γ.
Fibrous Connective Tissue
Fibrous connective tissue is the dense accumulation of fibroblasts and collagen formed within a wound site. Histologic characteristics depend on wound severity and duration. Fibrous connective tissue contains variable numbers of fibroblasts and collagen along with inflammatory cells (Fig. 3-39). In recently formed wounds, the collagen can be very immature and edematous with a variety of inflammatory cells, perhaps neutrophils. With time, the fibrous connective tissue progresses into mature, densely packed collagen with few inflammatory cells. Once formed and matured, fibrous connective tissue often persists for years, perhaps life.
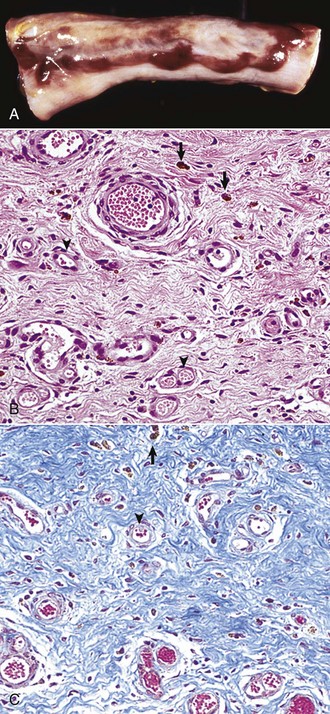
Fig. 3-39 Fibrous connective tissue.
A, Hemomelasma ilei, ileum, antimesenteric serosal surface, horse. This lesion is approximately 1 to 2 weeks old. Strongylus edentatus-induced injury to the serosal vasculature results in hemorrhage followed by wound healing. Note the raised areas of fibrosis (raised gray-white areas), hemosiderosis (yellow-brown areas), and hemorrhage (red-brown areas). B, Healing response in hemomelasma ilei. Note the abundant newly formed capillaries (arrowheads) and intervening fibrous connective tissue (bands of red fibers). This healing response is the next step following the granulation tissue phase demonstrated in Fig. 3-35. Hemosiderin (arrows) is present in the connective tissue and is indicative of hemorrhage having occurred in the injury at an earlier time (weeks). H&E stain. C, Fibrous connective tissue in the healing response. Collagen is readily demonstrated in fibrous connective tissue by a Trichrome stain (blue-stained fibers). Masson trichrome stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Wound Contraction
With severe thermal/chemical burns or extensive abrasions of a large surface area of the skin, the healing process and the formation of connective tissue becomes extensive. In time, these areas of connective tissue contract and place tension on the surrounding normal skin, resulting in a scirrhous reaction that can cause immobility of the surrounding skin and perhaps limbs along with pain and deformation. Contraction of such wounds is mediated largely by myofibroblasts.
Similarly, within areas of necrosis and/or inflammation in the liver, lung, spleen, and kidney, excessive fibrosis in parenchymal areas can result in the formation of connective tissue tracts between the healing area and capsular and interstitial connective tissue. When this new connective tissue contracts during the healing process, it grossly results in local indentation or pitting on the organ surface, such as occurs with chronic renal cortical infarcts. If there are multiple such areas, the organ surface develops an undulating and/or nodular appearance such as occurs in a cirrhotic liver. Contraction of such wounds is again mediated largely by myofibroblasts.
Myofibroblasts: Myofibroblasts are specialized fibroblasts with contractile activity. They form within wounds in response to tissue plane stress and the secretion of TGF-β by platelets and macrophages as wounds develop, and they increase in number with time and severity. Their function is to contract the wound and thus bring together injured tissue separated by edema and inflammation. Physiologically, myofibroblasts also occur in tissues with contractility such as uterine submucosa, intestinal villi, testicular stroma, the ovary, periodontal ligament, bone stroma, capillaries, and pericytes.
Myofibroblasts have stress fibers, actin and myosin fibers, gap junctions, and a fibronexus. The fibronexus is a mechanotransduction region of the plasma membrane, which is rich in integrin molecules. The fibronexus interconnects intracellular actin fibers with extracellular proteins such as fibronectin. This connection provides an anchor point during myofibroblast contraction. In contrast, fibroblasts lack contractile myofilaments and a fibronexus. Actin polymerization and contractility in myofibroblasts is stimulated by Rho GTPases. The Rho signaling that induces contractility in myofibroblasts results in continual contraction of filaments in myofibroblasts. Continual contraction by myofibroblasts differs from the periodic contractility that occurs in smooth muscle cells. Such contractions condense wound sites and are frequently beneficial to repair. But excess, as in severe burns, induces excessive contraction and sometimes loss of mobility of nearby joints requiring patients to undergo physical therapy to maintain the range of motion for limbs extending from affected joints.
Angiogenesis in Wound Repair
Angiogenesis is the formation of new blood vessels from preexisting vessels. It is a process essential for all living organisms with a cardiovascular system and involves a series of steps, as illustrated in Fig. 3-40, for the formation of new capillaries, including the following:
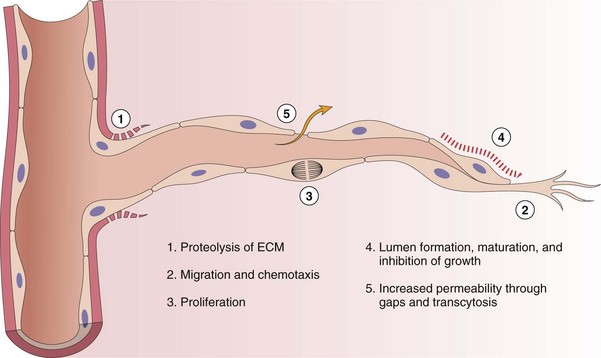
Fig. 3-40 Steps in the process of angiogenesis.
Extracellular matrix (ECM). (Modified from Motamed K, Sage EH: Kidney Int 51:1383, 1997.)
• Proteolysis of the ECM and basement membrane of parental vessels at the margins of the wound so a new capillary “bud” can form and initiate cellular migration
• Migration of immature endothelial cells into the wound
• Proliferation of endothelial cells to form solid “endothelial tubes”
• Maturation of endothelial tubes into new capillaries with the formation of lumina
• Formation of stalk cells (proliferative endothelial cells lining developing vessels) and tip cells at the end of vascular buds
• Establishment of endothelial cell adhesion to adjacent cells and basal lamina and expression of the receptors/ligands responsible for the leukocyte adhesion cascade along the luminal surface of the endothelial cells
• Recruitment of pericytes and smooth muscle cells to support the final differentiation stage of the newly formed vessel
This process occurs because as wounds heal, new vessels are necessary to supply the injured site with oxygen, remove carbon dioxide and other waste products, drain excess fluid, and provide a vascular pathway for cells and stem cells into the wound. This same beneficial process has also been adapted by primary and metastatic neoplastic cells to grow and spread throughout tissues of the body.
Initiation of Endothelial Cell Proliferation
Endothelial Cell Growth Factors: The formation of new blood vessels in wounds begins from the proliferation of endothelial cell buds from blood vessels in viable tissue adjacent to the wound or can be derived from bone marrow endothelial precursor cells (EPCs) (Fig. 3-41). These buds grow into the “healing” wound, form elongated vascular tubular structures within the wound, interconnect and revascularize the wound, and then eventually differentiate into mature vessels. Initially, endothelial cell buds form, and cells migrate into wounds under the autocrine influence of HIF-α and EGF (see section on Expression of Genes Responsible for Wound Repair), which enhance expression of genes that improve cell survival in hypoxic conditions.
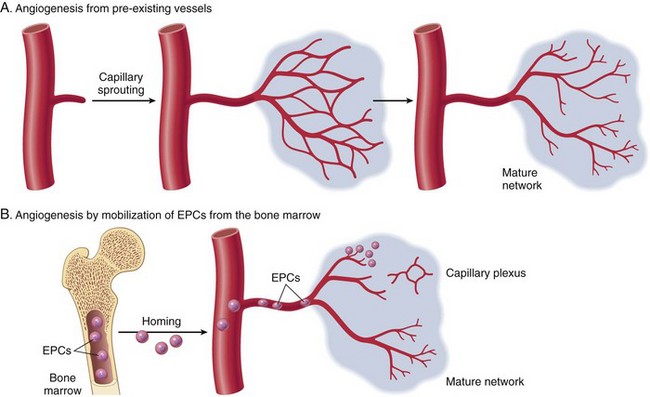
Fig. 3-41 Angiogenesis by mobilization of endothelial cell precursors (EPCs).
A, Bone marrow. EPCs are mobilized from the bone marrow and may migrate to a site of injury. The homing mechanisms have not yet been defined. At these sites, EPCs differentiate and form a mature network by linking with existing vessels. B, Preexisting vessels (capillary growth). In angiogenesis from preexisting vessels, endothelial cells from these vessels become motile and proliferate to form capillary sprouts. Regardless of the initiating mechanism, vessel maturation (stabilization) involves the recruitment of pericytes and smooth muscle cells to from the periendothelial layer. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Concurrently, growth factors such as PDGF, FGF, VEGF-A, angiogenins, and ephrins released from macrophages, endothelial cells, and fibroblasts bind receptors on endothelial cells and induce vascular formation (Fig. 3-42). VEGF A and its various isoforms stimulate the initial stages of endothelial cell proliferation through binding the VEGF-R2 receptor on endothelial cells. Proliferative effects of VEGF are regulated by Notch ligands and receptors. VEGF enhances expression of DII4, a ligand in vascular tip cells produced by tip cells that bind to Notch receptors expressed by stalk cells. DII4 binding of Notch receptor leads to expression of genes by the stalk cells that reduce VEGF-R expression and cellular proliferation. The secondary stages of endothelial cell proliferation involve angiopoietin 1 and its receptor, Tie2, both of which establish vascular stabilization through the recruitment of pericytes and smooth muscle cells and deposition of ECM proteins. Vascular stabilization is further advanced by PDGF and TGF-β. Recent work has shown that specific microRNA molecules, such as microRNA-92a (MiR-92a), control angiogenesis in mice. MiR-92a targets mRNAs with pro-angiogenic activity, binds these, and reduces their activity.
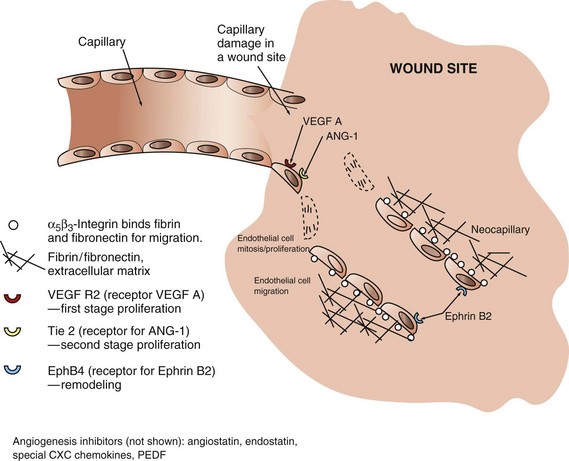
Fig. 3-42 Molecular mechanisms of angiogenesis.
Growth factors, such as vascular endothelial growth factor (VEGF-A) and angiopoietin (ANG-1), bind receptors on endothelial cells that induce proliferation and migration. The migration is mediated by α5β3-integrins expressed by endothelial cells that bind molecules such as fibrin and fibronectin. Factors such as Ephrin B2 bind endothelial cell receptors Ephrin B4 and mediate vascular remodeling. (Redrawn from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
Endothelial Cell Migration Is Mediated by Integrins: Newly formed endothelial cells and fibroblasts migrate into wound sites and bind to fibrinogen and plasma proteins, as well as newly deposited ECM substances, such as heparin sulfate, chondroitin sulfate, type III collagen, laminin, vitronectin, and fibronectin. This adherence is mediated by adhesion molecules expressed by new endothelial cells and fibroblasts. These adhesion molecules include α5- and β3-integrins, which bind fibrin and fibronectin. It is interesting that for wound repair, enhancement of angiogenesis is beneficial and vital; however, in neoplasia, inhibition of angiogenesis and thus the growth of the tumor have potential therapeutic benefits.
Vascular Remodeling: Once blood vessels are initially formed, they are loosely arranged and require remodeling to become mature. With remodeling, endothelial cells produce a mature basement membrane. In addition, smooth muscle cells and pericytes can form within the wall, and fibroblasts can form adventitial fibers, depending on whether the vessel is a capillary, artery, vein, or lymphatic vessel. Other endothelial cell growth factors and receptors involved with vascular remodeling include angiopoietin 2, which also binds Tie2 and ephrin B2 (EphB2) and its receptor, EphB4. Proliferation of lymphatic endothelial cells is mediated largely by VEGF-C and its receptor, VEGF-R3, as well as prox 1 gene expression.
Regulators/Inhibitors of Endothelial Cell Growth: Inhibitors of angiogenesis are produced by endothelial cells, macrophages, and fibroblasts. These inhibitors balance the proliferative healing responses of angiogenesis and prevent overexuberant proliferation of endothelial cells. These inhibitors include angiostatin, endostatin, thrombospondin, and specialized CXC chemokines (lacking ELR motif). In addition, certain isoforms of VEGF can bind VEGF receptors and reduce VEGF signaling and activity. Such inhibitors of angiogenesis are being studied intensely for their potential chemotherapeutic role against certain types of cancer and for exuberant vascularization that can occur in the retina, for example. Avastin (bevacizumab, Genetech, Inc., San Francisco) is an inhibitor of VEGF and can reduce vascularization in cancer and in the retina.
Epithelialization in Wound Repair
Epithelialization (reepithelialization) is the process by which the skin and mucous membranes replace superficial epithelial cells damaged or lost in a wound. Epithelial cells at the edge of a wound proliferate almost immediately after injury to cover the denuded area. Under normal conditions, this process is rapid, and first intention healing occurs in 3 to 5 days to repair the wound. During wound repair, keratinocytes and mucosal epithelial cells must move laterally across the wound surface to fill the void. Before this lateral movement can occur, epithelial cells must disassemble their connections to the underlying basement membrane and their junctional complexes with neighboring cells. They must also express surface receptors that permit movement over the ECM of the wound surface.
Intact Basement Membranes Enhance Reepithelialization
The presence or rapid deposition of basement membrane into the wound greatly facilitates proliferation of viable epithelial cells at the margins of the wound. For example, with initial loss of enterocytes that cover the surface of intestinal villi or renal tubular cells that line proximal convoluted tubules, the immediate response is for the adjacent normal epithelial cells to extend over the denuded basement membrane and to cover the area, if it is larger, by becoming thin, elongated cells. At the same time, there is proliferation (mitosis) of viable adjacent epithelial cells, and these cells migrate along the basement membrane to cover the denuded surface and replace lost cells. Without a basement membrane, proliferative cells lack a clear path of migration. The immature cells may loiter at the site of proliferation and fuse, thus forming syncytial cells, as can be seen with renal tubular injury and the failure of the tubular epithelium to migrate.
Similarly, regenerating skeletal muscle cells and transected axons will regenerate inside a tube surrounded by basal lamina and endoneurium. Components of the basement membrane, including laminin, type III collagen, and the associated proteoglycans, provide a substratum for epithelial and other cells to bind the basement membrane via integrins, proliferate, and migrate along the basement membrane surface.
Initiation of Cell Proliferation in Epithelia
Growth factors are vital for the proliferation of keratinocytes, mucosal epithelia, renal tubular cells, and other parenchymal epithelial cells. In skin and other surface epithelia, for example, keratinocyte growth factor (KGF) and EGF bind receptors on epithelial cells and induce signal transduction, which activates MAPK that induces cells in the nonproliferating Go phase of the cell cycle to enter the cycle and proliferate (see Chapter 6). Hepatocyte growth factor (HGF) induces proliferation of hepatocytes, and nerve growth factor (NGF) enhances growth of nerve fibers. Cell proliferation is regulated by (1) the amount of growth factor produced; (2) the level of expression of the growth factor receptor; (3) inhibitory signals from other growth factors; (4) the microenvironment, including the availability of oxygen and nutrients; and (5) integrin attachment to an established basement membrane. Although TGF-β induces fibroblast proliferation and collagen deposition, TGF-β inhibits proliferation of epithelial cells in many parenchymal organs.
Differentiation of Epithelia
Once epithelial cells have filled in a gap in the epithelium of a tissue or an organ, cellular differentiation is required for return of the tissue or organ to normal function. FGF-10 is a key initiator of wound repair in skin and lung epithelia. FGF-10 binds FGF-RIII, which through BMP4 and sonic hedgehog (a signaling protein for developmental patterning) enhances expression of several transcription factors, including GATA-6, thyroid transcription factor-1 (TTF-1), hepatocyte nuclear factor-β (HNF-β), and hepatocyte factor homolog-4 (HFH-4). Each of these transcription factors enhances expression of genes, which regulate a specific function for a particular cell (Web Fig. 3-30). In the lung, for example, TTF-1 induces production of surfactant proteins A, B, and C, and HFH-4 stimulates cilia formation. Activity of these transcription factors is reduced in the presence of NF kappa B, an important mediator of inflammation. Therefore concurrent inflammation can impair differentiation of epithelial and parenchymal cells and thus inhibit or delay reepithelialization.
Web Fig. 3-30 Epithelial cell differentiation, growth, and transcription factors.
Fibroblast growth factor-10 (FGF-10) binds epithelial receptor fibroblast growth factor receptor IIIB (FGF-RIIIB), which activates bone morphogenic protein and sonic hedgehog that induce activity of transcription factors for epithelial cell differentiation. This process is inhibited with inflammatory conditions that have increased levels of nuclear factor (NF) kappa B. HFH-4, Hepatocyte factor homolog-4; HNF-β, hepatocyte nuclear factor-β; TTF-1, thyroid transcription factor-1. (Redrawn with permission from Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
Metaplasia in Wound Repair
Some wounds do not heal properly and can turn into hypertrophic scars that impair epithelial and parenchymal cell growth. Such wounds may remain ulcerated or in parenchymal organs; the injured site may be replaced by fibroblasts and inflammatory cells rather than parenchymal cells. In either case, epithelial and parenchymal stem cells may continually attempt to cover or fill wound defects. With time, these cells may convert to another cell or tissue type. For example, regions of the lung constantly exposed to smoke can change from pseudostratified epithelium to stratified squamous epithelium, or regions of lower esophagus continually exposed to gastric acidity can undergo metaplasia into squamous cells. Osseous and chondroid metaplasia can occur in persistent wounds. In general, cells that undergo metaplasia have either (1) enhanced expression of an altered set of transcription factors and/or (2) decreased expression of transcription factors generally active for the affected tissue. The result is conversion of the cell’s phenotype into a new phenotype. Often, if the initiating stimulus is removed, cells can revert to the original phenotype.
Aderem, A, Ulevitch, RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787.
Anthony, RM, Nimmerjahn, F, Ashline, DJ, et al. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–376.
Benko, S, Philpott, DJ, Girardin, SE. The microbial and danger signals that activate Nod-like receptors. Cytokine. 2008;43:368–373.
Cantor, JM, Ginsberg, MH, Rose, DM. Integrin-associated proteins as potential therapeutic targets. Immun Rev. 2008;223:236–251.
Charo, IF, Ransohoff, RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621.
Etzioni, A. Leukocyte adhesion deficiencies: molecular basis, clinical findings, and therapeutic options. Adv Exp Med Biol. 2007;601:51–60.
Geddes, K, Magalhaes, JG, Girardin, SE. Unleashing the therapeutic potential of NOD-like receptors. Nat Rev Drug Disc. 2008;8:465–479.
Gibson-Corley, KN, Hostetter, JM, Hostetter, SH, et al. Disseminated Leishmania infantum infection in two sibling foxhounds due to possible vertical transmission. Can Vet J. 2008;49:1005–1008.
Gill, S, Wight, TN, Frevert, CW. Proteoglycans: key regulators of pulmonary inflammation and the innate immune response to lung infection. Anat Rec. 2010;293:968–981.
Helming, L, Gordon, G. The molecular basis of macrophage fusion. Immunobiology. 2008;212:785–793.
Kanneganti, T-D. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10:688–698.
Kawai, T, Akira, S. Toll-like receptor and RIG-1-like receptor signaling. Ann NY Acad Sci. 2008;1143:1–20.
Kenny, EM, Rozanski, EA, Rush, JE, et al. Association between outcome and organ system dysfunction in dogs with sepsis: 114 cases (2003-2007). J Am Vet Med Assoc. 2010;236:83–87.
Klune, JR, Dhupar, R, Cardinal, J, et al. HMGB1: Endogenous danger signaling. Mol Med. 2008;14:476–484.
Petersen, CA. Canine leishmaniasis in North America: emerging or newly recognized. Vet Clin North Am. 2009. (in press)
Soehnlein, O, Lindom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439.
Swirski, FK, Nahrendorf, M, Etzrodt, M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616.
Thurmond, RL, Gelfand, WE, Dunford, PJ. The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat Rev Drug Disc. 2008;7:41–53.
Yao, C, Sakata, D, Esaki, Y, et al. Prostaglandin E2-EP4 signaling promotes immune inflammation through TH1 cell differentiation and TH17 cell expansion. Nature Med. 2009;15:633–640.
Zabel, BA, Zuniga, L, Ohyama, T, et al. Chemoattractants, extracellular proteases, and the integrated host defense response. Exp Hematol. 2006;34:1021–1032.
Bonauer, A, Carmona, G, Iwasaki, M, et al. MicroRNA-92a controls angiogenesis and function recovery of ischemic tissues in mice. Science. 2009;324:1710–1714.
Braiman-Wiksman, L, Solomonik, I, Spira, R, et al. Novel insights into wound healing sequence of events. Toxicol Pathol. 2007;35:767–779.
Elmquist, JK, Scammell, TE, Saper, CB. Mechanisms of CNS response to systemic immune challenge: the febrile response. Trends Neurosci. 1997;20(12):565–570.
Faurschou, M, Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5:1317–1327.
Foster, D, Parrish-Novak, J, Fox, B, et al. Cytokine-receptor pairing: accelerating discovery of cytokine function. Nat Rev Drug Discov. 2003;3:160–170.
Fraisl, P, Aragones, J, Carmeliet, P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nature Reviews Drug Discovery. 2009;8:139–151.
Gangur, V, Birmingham, NP, Thanesvorakul, S. Chemokines in health and disease. Vet Immunol Immunopathol. 2002;86:127–136.
Giger, U, Boxer, LA, Simpson, PJ, et al. Deficiency of leukocyte surface glycoproteins Mo1, LFA-1, and Leu M5 in a dog with recurrent bacterial infections: an animal model. Blood. 1984;69:1622–1630.
Hong, HS, Lee, J, Lee, E, et al. A new role of substance P as an injury-inducible messenger for mobilization of CD29+ stromal-like cells. Nature Med. 2009;15:425–435.
Kehrli, ME, Jr., Ackermann, MR, Shuster, DE, et al. Animal model of human disease: bovine leukocyte adhesion deficiency: beta-2 integrin deficiency in young Holstein cattle. Am J Pathol. 1992;140:1489–1492.
Kilarski, WW, Samolov, B, Petersson, L, et al. Biomechanical regulation of blood vessel growth during tissue vascularization. Nature Med. 2009;15:657–662.
Li, L, Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327:542–545.
Mayadas, T, Cullere, X. Neutrophil β2 integrins: moderators of life or death decisions. Trends Immunol. 2005;26(7):388–395.
Mitsui, A, et al. Effects of fascial abrasion, fasciotomy, and fascial excision on cutaneous wound healing in cats. Am J Vet Res. 2009;70:532–538.
Palmer, MV, Waters, WR, Thacker, TC. Lesion development and immunohistochemical changes in granulomas from cattle experimentally infected with Mycobacterium bovis. Vet Pathol. 2007;44:863–874.
Ramirez-Romero, R, Brogden, KA, Gallup, JM, et al. Mast cell density and substance P-like immunoreactivity during initiation and progression of lung lesions in ovine Mannheimia haemolytica pneumonia. Microb Pathog. 2001;30:325–335.
Rottman, JB. Key role of chemokines and chemokine receptors in inflammation, immunity, neoplasia, and infectious disease. Vet Pathol. 1999;36:357–367.
Simionescu, M, Gafencu, A, Antohe, F. Transcytosis of plasma macromolecules in endothelial cells: a cell biological survey. Microsc Res Tech. 2002;57:269–288.
Stappenbeck, TS, Miyoshi, H. The role of stromal stem cells in tissue regeneration and wound repair. Science. 2009;324:1666–1669.
Ulbrich, H, Eriksson, EE, Lindobom, L. Leukocyte and endothelial cell adhesion molecules as targets for therapeutic interventions in inflammatory disease. Trends Pharm Sci. 2003;24:640–647.
Volkman, HE, Pozos, TC, Zheng, J, et al. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science. 2010;327:446–469.
Wedemeyer, J, Tsai, M, Galli, SJ. Roles of mast cells and basophils in innate and acquired immunity. Current Opin Immunol. 2000;12:624–631.
Yang, D, Biragyn, A, Hoover, DM, et al. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Ann Rev Immunol. 2004;22:181–215.
WEB APPENDIX 3-1
| Acronym | Term | Function |
| Ach | Acetylcholine | Parasympathetic and antiinflammatory responses |
| ADAM9 | A disintegrin and metalloproteinase | Cell fusion |
| Arg-1 | Argininase-1 | TH2 fibrotic response |
| BPV | Bovine papillomavirus | Papilloma in cattle, sarcoid in horses |
| CD4 | Cluster determinant 4 lymphocyte | Lymphocyte responses |
| CD8 | Cluster determinant 8 lymphocyte | Lymphocyte killing |
| CD36 | Cluster determinant 36 | Monocyte scavenger receptor |
| CCL, CX3CL | Chemokine ligand | Chemotaxis |
| CCR, CX3CR | Chemokine receptor | Chemotaxis |
| CSF | Cerebrospinal fluid | Central nervous system function |
| ECM | Extracellular matrix | Proteinaceous matrix between cells |
| EPC | Endothelial precursor cell | Endothelial precursor cell |
| FGF | Fibroblast growth factor | Promotes proliferation of many cell types |
| GAG | Glycosaminoglycan | Extracellular glycoconjugates |
| GATA-6 | Transcription factor | Transcription factor for differentiation |
| GM-CSF (see Web Table 3-6) | Granulocyte-macrophage colony-stimulating factor | Cell proliferation |
| HEV | High endothelial venule | Lymphocyte trafficking |
| HFH-4 | Hepatocyte factor homolog-4 | Growth factor |
| HGF | Hepatocyte growth factor | Transcription factor for cell differentiation |
| HIF | Hypoxia-inducing factor | Regulator of cell activity in hypoxic conditions |
| HNF | Hepatocyte nuclear factor | Transcription factor for cell differentiation |
| HRE | Hypoxia response elements | Response elements to HIF |
| ICAM-1 | Intercellular adhesion molecule-1 | Leukocyte adhesion |
| IFN (see Web Table 3-6) | Interferon | Antiviral and TH1 responses |
| IL (see Web Table 3-6) | Interleukin | Cytokine |
| JAM | Junctional adhesion molecule | Cell/leukocyte adhesion |
| LDL | Low-density lipoprotein | Vascular lipid transport |
| LFA-1 | Lymphocyte function antigen-1 | Leukocyte adhesion |
| Mac-1 | Macrophage-1 antigen | Leukocyte adhesion |
| MAPK | Mitogen-activated protein kinase | Cell signaling |
| MBP | Major basic protein | Eosinophil granule product |
| MCP (see Web Table 3-6) | Macrophage chemotactic protein | Macrophage chemotaxis |
| MFPR | Macrophage fusion protein receptor | Receptor for CD47 ligand |
| MGC | Multinucleate giant cell | Sequester persistent antigen |
| MHC | Major histocompatibility complex | Immunity |
| MIP (see Web Table 3-6) | Macrophage inflammatory protein | Macrophage chemotaxis |
| MMP | Matrix metalloproteinase | Degradation of ECM |
| NF kappa B | Nuclear factor kappa B | Inflammation transcription factor |
| NGF | Nerve growth factor | Growth factor |
| NK | Natural killer | Specialized lymphocyte-like cell |
| NOS | Nitric oxide synthase | Nitric oxide and TH1 response |
| PDGF | Platelet-derived growth factor | Growth factor |
| PECAM-1 | Platelet endothelial cell adhesion molecule-1 | Leukocyte adhesion |
| PPAR | Peroxisome proliferator activated receptor | Transcription factor that reduces inflammatory response by cells |
| STAT | STAT kinase receptor | Cytokine receptor signaling |
| T reg | Regulatory T lymphocyte | Regulated dendritic cell activity |
| TGF-α | Transforming growth factor-α | Stimulates the growth of parenchymal cells |
| TGF-β | Transforming growth factor-β | Stimulates the growth of fibroblasts |
| TH1 | T helper lymphocyte type 1 | Cell-mediated lymphocyte responses |
| TH2 | T helper lymphocyte type 2 | Humoral lymphocyte responses |
| TIMP | Tissue inhibitor of matrix metalloproteinase | Reduction of ECM degradation |
| TNF (see Web Table 3-6) | Tumor necrosis factor | Inflammation |
| TTF | Thyroid transcription factor | Transcription factor for cell differentiation |
| VCAM-1 | Vascular cell adhesion molecule-1 | Adhesion molecule for monocytes |
| VEGF | Vascular endothelial cell growth factor | Angiogenesis |
| VLA-4 | Very late antigen-4 | Leukocyte adhesion |
WEB APPENDIX 3-2
COX, Cyclooxygenase; CRF 2, cytokine receptor family 2; CSF, colony-stimulating factor; DD, death domain; FADD, Fas-associated death domain; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; GPCR, G protein–coupled receptors; HMGB-1, high-mobility group box protein-1; HMWK, high molecular weight kininogen; IFN, interferon; IgE, immunoglobulin E; IL-1 through IL-29, interleukin 1 through interleukin-29; JAK/STAT, Janus kinase/signal transducer and activator of transcription; LTB4, leukotriene B4; LTC4, leukotriene C4; LTD4, leukotriene D4; LTE4, leukotriene E4; LOX, lipoxygenase; M-CSF, macrophage colony-stimulating factor; MHC, major histocompatibility complex; MORT, mediator of receptor-induced toxicity; N/A, not applicable; NK, natural killer; NO, nitric oxide; NOD, nucleotide-binding oligomerization domain; NOS, nitric oxide synthetase; PAF, platelet-activating factor; PGE2, prostaglandin E2; PGF2, prostaglandins F2, PGI2, prostaglandins I2; PLA2, phospholipase A2; RAGE, receptor for advanced glycosylation end-products; RNA, ribonucleic acid; S1P, sphingosine-1-phosphate; TGF-β, transforming growth factor-β; T reg, T regulatory; TNF, tumor necrosis factor; TXA2, thromboxane A2.
*A = vasodilation/vasoconstriction, B = vascular permeability, C = chemotaxis.