Diseases of Immunity
General Features of the Immune System
The immune system is a defensive system whose primary functions are to protect against infectious organisms, such as bacteria, viruses, fungi, and parasites, and the development of cancer. The complexity by which these functions occur is evidenced not only by the cell types, recognition molecules, and soluble factors involved and interactions with other systems (e.g., endocrine, nervous) but also by the ability to recognize virtually any foreign antigen. Immunologic responses result in pathologic processes, primarily inflammatory responses, as a result of either normal immune responses to foreign antigens (e.g., microbial pathogens) or from aberrations of the immune system as in the case of hypersensitivity reactions and autoimmune diseases. Finally, the importance of a normal functional immune system cannot be more evident than in instances in which it is deficient as the result of a genetic defect or as the result of an acquired immunodeficiency disease.
Immunity is the result of nonspecific (innate) and specific (adaptive) responses that together provide effective protection. The immune system’s recognition and response functional capabilities are key components of both innate and adaptive immune responses. The recognition capabilities are highly specific and allow immune responses to develop against a diverse group of foreign (nonself) antigens and prevent the development of immune responses to self-antigens. Innate and adaptive immune responses feature effector mechanisms for eliminating or neutralizing the antigen, whereas adaptive immunity has the additional feature of memory. The emphasis of this chapter is on diseases that are the result of inadequate or inappropriate immune responses. Before one can understand the pathogenic mechanisms of these diseases, one must first have an understanding of the basic elements of the immune system. The chapter begins with an overview of our current understanding of innate and adaptive immunity, cells of the immune system, cytokines, and major histocompatibility complex (MHC) molecules. This overview facilitates the discussion of disorders of the immune system that includes hypersensitivity reactions, autoimmunity, and immunodeficiency. This chapter concludes with a discussion of amyloidosis, a diverse group of conditions characterized by the deposition of a pathologic extracellular protein. One of the conditions is associated with the deposition of immunoglobulin components. Although the focus of this text is on the pathologic basis of veterinary diseases, with an emphasis on domestic species, in this chapter we use the vast knowledge base regarding human and rodent immunology (applicable to most mammalian species studied to date) as our basis and interject major known relevant species differences as appropriate.
Innate Immunity (Nonspecific Immunity)
As stated previously, the function of the immune system is to protect against infectious pathogens and the development of cancer. There are two categories of immune responses that are based in part on their specificity for the antigen: (1) innate immunity and (2) adaptive immunity (Fig. 5-1). Innate immune responses are considered the first-line defense mechanisms, are not specific to the antigen, and lack memory. These defense mechanisms are the result of anatomic (e.g., skin, mucosal epithelia, cilia) and physiologic (e.g., stomach pH, body temperature) properties, and phagocytic and inflammatory responses. Major components of innate immunity are intact epithelial barriers, phagocytic cells, natural killer (NK) cells, and a number of plasma proteins, the most important of which are the proteins of the complement system. Phagocytic cells are recruited to sites of infection during an inflammatory response where they have a number of functions, two of which are to ingest and destroy pathogenic organisms and neutralize toxins. Neutrophils, monocytes, and tissue macrophages are the major cells involved in phagocytosis. These cells recognize components of microbial pathogens through the expression of several membrane receptors, including receptors for mannose residues and N-formyl-methionine containing peptides and a family of pattern recognition receptors (PRRs) that, when activated by microbial components, signal the activation of transcription factors that facilitate the microbicidal mechanisms of the phagocytic cell and are discussed later. NK cells are the cytotoxic cells of innate immunity and are also discussed later. The complement system, discussed in Chapter 3, is a complex cascade of proteins that has a number of biologic functions, including the formation of the membrane attack complex that efficiently lyses plasma membranes of microbial pathogens. The complement system can be activated by either the innate immune system (alternative and mannose/lectin pathways) or the adaptive immune system (classical pathway). Other important plasma proteins of the innate immune system include mannose-binding protein and C-reactive protein; two of the functions of these proteins are to facilitate phagocytosis through opsonization of pathogens and complement activation. Inflammatory responses comprise vascular, permeability, and cellular phases that act in response to damage to vascularized tissue. The features of the inflammatory response are also presented in Chapter 3.
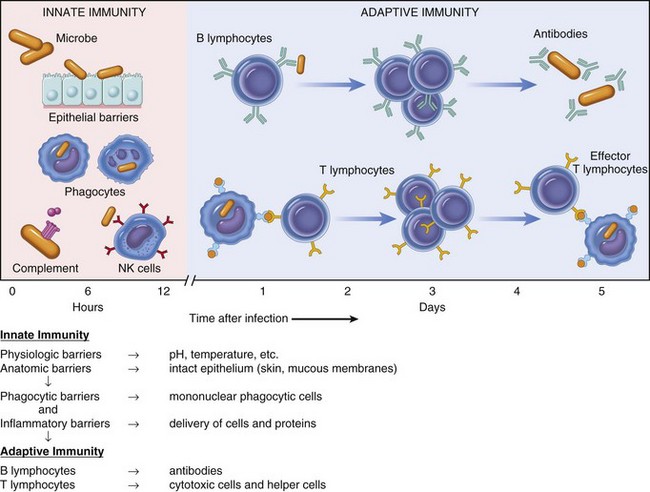
Fig. 5-1 Innate (nonspecific) and adaptive (specific) immunity are depicted in relation to the time course of an infection.
NK, Natural killer. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Recognition Molecules of Innate Immunity
The recognition molecules of innate immunity provide an opportunity to recognize pathogens by cells other than lymphocytes. These germline-encoded molecules function to sense molecular structures shared by microbes and endogenous molecules associated with inflammation. The recognition molecules associated with microbes are generally referred to as pathogen-associated molecular patterns (PAMPs). The function of the innate immune system extends beyond microbial pathogen recognition to include endogenous molecules associated with cell damage and inflammation which are generally referred to as danger or damage associated molecular patterns (DAMPs). The activation of the innate immune system through these invariant PRRs not only precedes lymphocyte activation but also is required to initiate the adaptive immune responses. PRRs molecular patterns are associated with microbial pathogens and classified as secreted, transmembrane, and cytosolic forms. Collectins and pentraxin are examples of secreted PRRs that largely function to bind to microbial surfaces and activate the complement system. Collectins, having properties of collagen and lectins, include mannose binding lectins and pulmonary surfactants A and D. Pentraxin is comprised of five identical subunits to form a pentamer and include C-reactive protein, an activator of the classical pathway of complement. Toll-like receptors (TLRs) and C-type lectins are examples of the transmembrane PRRs, and they have limited cellular distribution, including macrophages, NK cells, and dendritic cells. TLRs are expressed on the plasma membrane or in endosomal/lysosomal organelles. The cytosolic PRRs are more widely distributed, including all nucleated cells, and include retinoic acid–inducible gene (RIG)-1–like receptors (RLRs) and nucleotide-binding domain (NOD) and leucine-rich repeat–containing receptors (NLRs). These recognition molecules provide the host with the ability to sense “danger” either through PAMPs in the case of microbial infections or through DAMPs in the presence of cellular damage or stress. This allows the innate immune system to detect and initiate immune responses in response to infectious and noninfectious causes. NLR molecules as immune sensors are not unique to mammals because they have been found in plants and urchins. The molecules are designated based on the structure of the molecule NLR with a suffix of P (pyrin domain) or C (caspase activation and recruitment domains [CARD]), referring to the N-terminal moiety followed by a number (e.g., NLRP1, NLRP2, or NLRP3). The function of these pattern recognition molecules is also extended beyond the initiation of adaptive immunity to also include regulation of cell death (apoptosis). As is often the case in immunology, our understanding of immune responses is greatly enhanced through the identification of genetic immunologic disorders. Deficiencies in components of the NLRs have been described in humans and are most frequently associated with inflammatory disorders (e.g., Crohn’s disease and inflammatory bowel disease).
Analogous to the adaptive immune response, which has developed the ability to defend the host against a diverse array of microbial pathogens (humoral versus cell-mediated responses), the innate immune system is now seen as also having an ability to distinguish specific types of pathogens. Intracellular (e.g., viruses) and extracellular (extracellular bacteria) pathogens require different types of immune responses to control them. The innate immune system has developed cell-intrinsic and cell-extrinsic recognition mechanisms dependent on if it is mediated by an infected cell or an uninfected cell. Cell-extrinsic recognition mechanisms are a way in which an uninfected cell can participate in the immune response and are mediated through transmembrane receptors (e.g., TLRs) on specialized cells of the innate immune system such as macrophages and dendritic cells. Cell-intrinsic recognition mechanisms are essential for recognizing intracellular pathogens and involve type I interferon (IFN) gene transcription signaling by members of the RLRs. Three members of the RLR family are RIG-1, melanoma differentiation-associated gene-5 (MDA-5), and laboratory of genetics and physiology gene 2 (LPG-2). In viral sensing, the RLRs are highly discriminating with regards to the cytoplasmic localization and sensing of specific RNAs. They specifically detect RNA molecular patterns not normally present in the cytoplasm. Abnormal RNA patterns include chemical modifications, secondary or tertiary RNA conformations, specific RNA sequences, or double-stranded RNA. While much of what is currently known about RLRs is centered around the sensing of RNA viruses, there is preliminary evidence that similar intracellular mechanisms may exist for sensing DNA viruses and some intracellular bacteria.
Members of the NLR are central regulators of immunity and inflammation largely through activation of transcription factors like nuclear factor (NF) kappa B, interferon regulatory factor (IRF), or nuclear factor of activated T lymphocytes (NFAT). Some members of the NLR family form multiprotein complexes with the cysteine protease pro-caspase-1 and the adapter molecule ASC (apoptosis-associated speck-like protein containing a CARD) referred to as inflammasomes (also see Fig. 3-12). The inflammasome is a multiprotein complex that activates caspase-1. PAMPs and DAMPs are sensed through activation of the inflammasome complex, resulting in activation of caspase-1, which elicits effector functions through proteolytic cleavage of cytosolic proinflammatory cytokines (e.g., pro-interleukin [IL]-1β and pro-IL-18), which are then secreted in their active form. The inflammasome model is analogous to the model for the activation of the apoptotic caspases by CD95/Fas death–inducing signaling complex (DISC) and the Apaf-1 apoptosome. IL-1β functions in localized and systemic responses to infections and injury, to cause fever, activation of lymphocytes, and the extravasation of leukocytes at sites of injury or infection. IL-18 functions to induce IFN-γ by activated T lymphocytes and NK cells during a TH1 response and to induce secondary inflammatory cytokines, chemokines, cell adhesion molecules, and nitric oxide (NO) synthesis. Other members of the NLR family are involved in inflammasome-independent (noninflammasome) innate immune responses and signal through different multicomponent signalosomes such as nodosomes, transcriptosomes, mito-signalosomes. Noninflammasome-mediated innate immune responses occur through NF kappa B activation, mitogen-activated protein kinase (MAPK) activation, cytokine and chemokine production, antimicrobial reactive oxygen species production, type I interferon (IFN-α and IFN-β) production, and ribonuclease L activity.
Toll-Like Receptors
TLRs are the mammalian homologue of the Toll receptor originally identified in Drosophila. It has not only an embryologic function but also an immunologic function. In mammals, TLRs are membrane molecules that function in cellular activation by a wide range of microbial pathogens. TLRs are classified as PRRs because they recognize PAMPs and signal to the host the presence of an infection. Pathogen associated molecules include lipopolysaccharide (LPS) from Gram-negative bacteria, peptidoglycan from Gram-positive bacteria, double-stranded RNA from viruses, or α-glucans from fungi (Table 5-1). In general, TLRs 1, 2, 4, and 6 recognize unique bacterial products that are found on the cell surface, and TLRs 3, 7, 8, and 9 are involved in viral detection and nucleic acid recognition within endosomes. The specificity of TLRs for microbial products depends on interactions between TLRs and non-TLR adapter molecules. All TLRs contain an extracellular domain characterized by a leucine-rich repeat motif flanked by a cysteine-rich motif (Fig. 5-2). They also contain a conserved intracellular signaling domain, Toll/IL-1 receptor (TIR), that is identical to the cytoplasmic domain of the IL-1 and IL-18 receptors. Fig. 5-2 illustrates how TLRs function in the recognition of LPS. In the blood or extracellular fluid, the binding of LPS to LPS-binding protein (LBP) facilitates the binding of LPS to CD14, a plasma protein and glycophosphatidylinositol-linked membrane protein present on most cells. The binding of LPS to CD14 results in the dissociation of LBP and the association of the LPS-CD14 complex with TLR4. An accessory protein, MD2, complexes with the LPS-CD14-TLR4 molecule and results in LPS-induced cell signaling.
TABLE 5-1
Toll-Like Receptors (TLRs) and TLR Ligands and Their Microbial Source
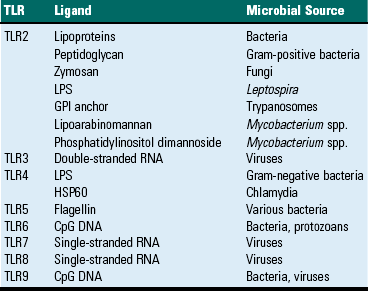
CpG, Cytosine and guanine linked oligonucleotide; GPI, glycosyl phosphatidyl inositol; HSP60, heat shock protein 60; LPS, lipopolysaccharide.
Modified from Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.
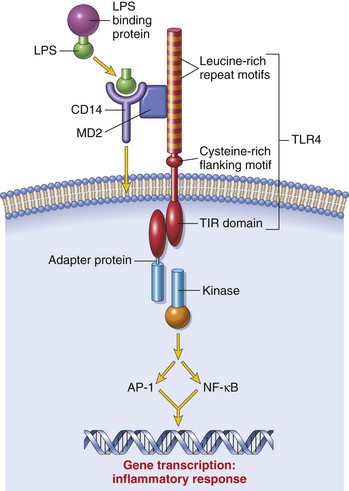
Fig. 5-2 Signaling pathway for Toll-like receptor 4 (TLR4) in response to bacterial lipopolysaccharide (LPS).
The binding of LPS to TLR4 results in activation of a signal transduction pathway, leading to gene transcription and the elicitation of an inflammatory response. AP-1, Activating protein 1; NF-κB, nuclear factor-kappa B; TIR, Toll/interleukin-1 receptor. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Briefly, TLR signaling through the binding of PAMP to a TLR leads to the activation of TIR, which forms a complex with the cytoplasmic adapter protein MyD88, an IL-1 receptor-associated kinase (IRAK), and tumor necrosis factor (TNF) receptor–associated factor 6 (TRAF6). Activated TRAF then activates the MAPK cascade leading to the activation of NF kappa B, a transcription factor. MyD88 is a universal signaling molecule for NF kappa B activation, and MyD88-deficient mice are incapable of activation by TLR, IL-1, and IL-18. Recent information suggests that there are also signaling mechanisms unique to individual TLRs.
TLRs and their pathogen-associated ligands are important recognition molecules for the innate immune system and trigger a number of antimicrobial and inflammatory responses. Up to 15 different TLR genes have been identified. The importance of these receptors in immunity is further supported by the observation of polymorphisms in the genes encoding them.
Although the individual TLRs exhibit ligand specificity, they differ in their cellular expression patterns and the signal pathways they activate, similar to that described for cytokines, which exhibit pleiotropy, redundancy, synergy, and antagonism. There are constitutively and inducibly expressed TLRs in different tissues. TLRs regulate cell recruitment to sites of infection through the upregulation of the expression of adhesion molecules, chemokines, and chemokine receptors during an inflammatory response. TLRs activate leukocytes (primarily neutrophils and NK cells of the innate immune system) and epithelial, endothelial, and hematopoietic cells. TLRs are also hypothesized to be essential for linking the innate immune response to the adaptive immune responses. Central to this hypothesis is the TLR-dependent dendritic cell-mediated control of T lymphocyte activation. Dendritic cells are important antigen-presenting cells for T lymphocyte activation. Dendritic cells uptake microbial antigens in the peripheral tissues and migrate to regional lymph nodes where they present peptide fragments, in the context of MHC molecules, to naïve T lymphocytes. In addition to the expression of the peptide-MHC signal, dendritic cells are also required to provide a second, co-stimulatory signal through the expression of B7, the ligand for the CD28 molecule on naïve T lymphocytes. The activation and maturation pathway related to the co-stimulatory signal occurs through TLR recognition of PAMPs.
There are species differences in the ligand specificity of TLRs and in the cellular responses elicited. Although sequences for canine, feline, and chicken TLR4 have been identified, no functional data have been published. With regard to domestic animals, a significant body of literature exists on TLRs of cattle.
Finally, TLRs have been implicated in “innate autoimmunity,” with several reports of TLRs recognizing fibrinogen, heat-shock proteins, or DNA. There are also reports of TLR binding of DNA as a factor in directing antibody production by autoreactive B lymphocytes contributing to the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus (SLE). Further studies are necessary to more fully understand these observations and the underlying immunopathogenesis.
Adaptive Immunity (Specific Immunity)
Adaptive immunity in general consists of cell-mediated immunity, mediated by T lymphocytes against intracellular pathogens, and humoral immunity, mediated by B lymphocytes against extracellular pathogens and toxins (Fig. 5-3). The adaptive immune response is the second-line defense mechanism and is characterized by antigen specificity, diversity, memory, and self-/nonself-recognition. Antigen specificity and self-/nonself-recognition are the result of distinct membrane molecules. Mature B lymphocytes are activated by a specific antigen-binding molecule on its membrane. The antigen receptor is membrane-bound immunoglobulin. Mature T lymphocytes express a specific antigen-binding molecule, the T lymphocyte receptor (TCR), on their membrane. Unlike membrane-bound immunoglobulin on the B lymphocyte, which can recognize antigen alone, TCRs can recognize only antigens that are associated with cell membrane proteins called MHC molecules. Self-/nonself-recognition is the result of MHC molecules. There are two major classes of MHC molecules. Class I molecules are present on all nucleated cells, and class II molecules are present primarily on antigen-presenting cells. T lymphocytes and B lymphocytes are the major cells of adaptive immunity.
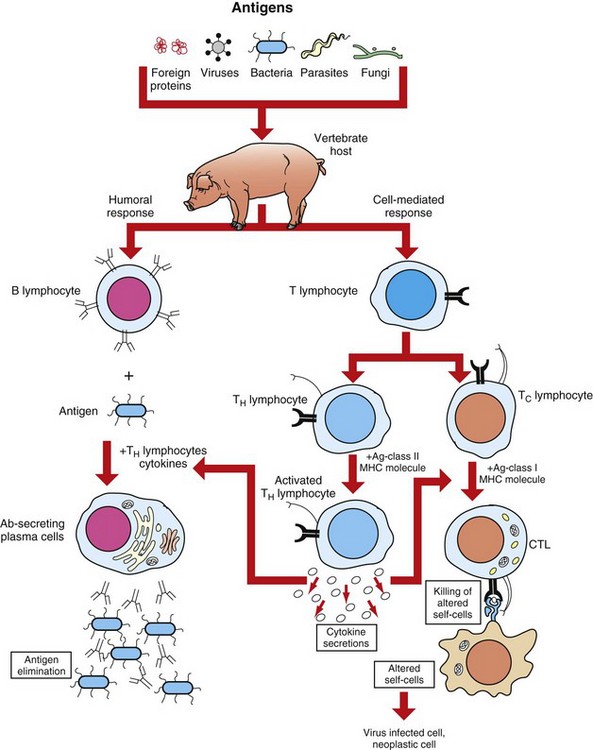
Fig. 5-3 Overview of humoral and cell-mediated arms of adaptive immunity.
CTL, Cytotoxic T lymphocyte. (Adapted from Goldsby RA, Kindt TJ, Osborne BA: Kuby immunology, ed 4, New York, 2000, WH Freeman.)
Cells and Tissues of the Immune System
T lymphocytes are small nongranular cells that constitute 50% to 70% of the peripheral blood mononuclear cells. They originate in the bone marrow and migrate to the thymus (thus the “T” designation), where they undergo differentiation, selection, and maturation processes before exiting to the periphery as effector lymphocytes. In secondary lymphoid tissues, they are located primarily in the paracortical regions of lymph nodes and the periarteriolar sheaths (PALS) of the spleen. These specific anatomic sites elaborate chemoattractant cytokines (chemokines), for which the T lymphocytes express receptors. The definitive marker for T lymphocytes is the TCR, the polymorphic antigen-binding molecule. The antigen specificity of individual lymphocytes is attributed to their respective TCR, which is genetically determined. TCRs are classified as either αβ-TCR or γδ-TCR based on the composition of their disulfide-linked heterodimers. The individual polypeptide chains of the heterodimers contain variable (antigen-binding) and constant regions. In mammals, most peripheral blood T lymphocytes express αβ-TCR; however, in ruminants and swine, these lymphocytes make up only 10% to 50%, and 10% of peripheral blood T lymphocytes, respectively. Both TCRs are associated with CD3, and together they form the TCR-CD3 complex. There are significant activation and functional differences between αβ-TCR– and γδ-TCR–expressing lymphocytes. Unlike membrane-bound immunoglobulin on B lymphocytes that can recognize soluble antigen, the αβ-TCR can only recognize antigen after it has been processed into peptide fragments and associated with MHC molecules (the MHC is discussed later). In most instances, the antigen is associated with the MHC on the surface of an antigen-presenting cell, a virally infected cell, a neoplastic cell, or a cell of a foreign tissue graft. Individual αβ-TCRs are covalently linked to a cluster of five polypeptide chains, three comprising the CD3 molecule and two comprising the β-chain. The CD3 molecule and the β-chain are invariant, and although they do not bind antigen, they do function in the signal transduction after antigen binding by the TCR. Each T lymphocyte expresses a unique TCR with regard to structure and antigen specificity. The genes that encode α-, β-, γ-, and δ-chains of the TCR can undergo somatic rearrangements during their development in the thymus, resulting in tremendous diversity for antigen recognition. Not only are these rearrangements important for diversity, but also they can be used to molecularly phenotype proliferating populations of T lymphocytes as a diagnostic tool for the identification of clonal populations (neoplastic) and polyclonal populations (nonneoplastic) (see Chapter 6).
In most species, a minority of T lymphocytes express γδ-TCR. The γδ-TCR lymphocytes develop in the thymus and migrate to the epithelium of the skin and intestine, mammary gland, and reproductive organs. Although these cells can be found within regional lymph nodes and the lamina propria, in these organs they primarily reside as intraepithelial lymphocytes (IELS). As stated, in some species, notably ruminants, γδ-TCR lymphocytes are the predominant circulating population of T lymphocytes. In contrast to the αβ-TCR lymphocytes, γδ-TCR lymphocytes can recognize native antigen in the absence of MHC binding and they do not rely exclusively on the δ-chain as a signal transducer. Most γδ-TCR lymphocytes use the γ-chain for signal transduction after activation. The diversity of antigens recognized by γδ-TCR is limited in most species except ruminants and swine, indicating their importance in these species. Some have suggested that they may provide early cell-mediated immune responses in neonates. The precise function of γδ-TCR lymphocytes remains unknown. Another small subset of T lymphocytes, called NK-T lymphocytes, expresses molecules found on NK cells in addition to a limited diversity of TCRs. NK-T lymphocytes primarily recognize glycolipids that are associated with an MHC-like molecule, CD1. The function of NK-T lymphocytes remains unknown.
Although all T lymphocytes express the TCR-CD3 complex, they are further classified according to accessory CD4 and CD8 molecules. These nonpolymorphic accessory molecules include CD4, CD8, CD2, integrins, and CD28. CD4 and CD8 functionally subdivide T lymphocytes into CD8+ cytotoxic T lymphocytes (CTL [TC]) and CD4+ helper T lymphocytes (TH). During antigen presentation, CD4+ lymphocytes only recognize antigen bound to MHC class II molecules (Fig. 5-4), whereas CD8+ lymphocytes only recognize antigen bound to MHC class I molecules. This coreceptor requirement is commonly referred to as MHC class I and MHC class II restriction, the basis for positive selection in the thymus. Although there have been reports in some species of CD4 lymphocytes that are functionally cytotoxic and CD8 lymphocytes that are functionally “helper”-like, these appear to be anomalies and for the purposes of this text are excluded. In most species, peripheral blood T lymphocytes express either CD4 or CD8. Except for ruminants and swine, lymphocytes negative for both CD4 and CD8—“double negative” lymphocytes—are rare in the peripheral blood. So-called “double positive” lymphocytes, positive for both CD4 and CD8, are rare, except in swine, where they can approach 25% of the T lymphocytes in the peripheral circulation. T lymphocytes require two signals for activation. Signal 1 is provided by the TCR and the MHC-antigen complex and the CD4 or CD8 MHC complex. Signal 2 is provided by another accessory molecule expressed by T lymphocytes, the CD28 molecule. The ligands for CD28 are B7-1 (CD80) and B7-2 (CD81) expressed on activated dendritic cells, B lymphocytes, and macrophages (see Fig. 5-4). An inability to deliver the second signal results in an unresponsive T lymphocyte that either undergoes apoptosis or remains anergic. These molecules provide an important co-stimulatory signal for T lymphocyte activation, and are discussed in more detail later in the chapter regarding anergy and the development of tolerance with regard to autoimmunity. When T lymphocytes are activated by antigen and receive the appropriate co-stimulatory signals, they clonally expand as a result of their secretion of IL-2. This clonally expanded population of T lymphocytes, of the same antigen specificity, differentiates into populations of effector lymphocytes and memory lymphocytes.
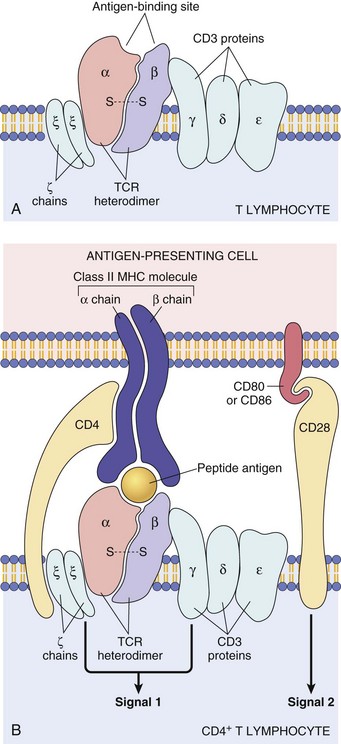
Fig. 5-4 The T lymphocyte receptor complex (T lymphocyte receptor [TCR]).
A, TCR-α and TCR-β chains complexed with CD3 γ-, δ-, and ε-chains and the invariant, ζ-chains. B, Illustrating how the TCR recognizes antigen in the context of major histocompatibility complex on the antigen-presenting cell (top) and how the ζ-chains and CD3 γ-, δ-, and ζ-chains deliver one of the two required signals for activation of the T lymphocyte. The second required signal is delivered by the co-stimulator molecules CD28 on the T lymphocyte and B7 on the antigen-presenting cell. MHC, Major histocompatibility complex. (A from Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders. B from Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
TH lymphocytes can be classified based on their functional capacity and ability to elicit primarily an antibody response or a cell-mediated immune response. After activation of TH lymphocytes, by recognition of antigen bound to MHC class II molecules on the surface of an antigen-presenting cell, there is clonal expansion of TH lymphocytes of the same antigen specificity. These clonally expanded lymphocytes are important in directing the immune response as either primarily an antibody response or a cellular response. The type of response is dictated by a restricted cytokine profile that primarily activates B lymphocytes in the case of an antibody response or activates CTL (Tc) and macrophages in a cellular response. The restricted cytokine profile for TH lymphocytes allows for their classification as either TH1 or TH2 lymphocytes (Web Table 5-1). TH1 lymphocytes synthesize and secrete IL-2 and interferon-γ (IFN-γ), stimulating CTL (Tc) and macrophages, and induce a cell-mediated immune response. TH2 lymphocytes synthesize and secrete IL-4, IL-5, IL-6, and IL-13 that stimulate B lymphocytes to develop into antibody-secreting plasma cells and inhibit macrophage functions, and induce an antibody response. The type of immune response (antibody versus cell-mediated) can have a profound influence on the outcome of a disease. In the instance of an intracellular protozoal infection, a TH2 type response results in rapid proliferation of the organism and death of the host, whereas a TH1 type response results in elimination of the organism and survival of the host. Similarly, a TH2 response to an allergen results in the elaboration of immunoglobulin (Ig) E, through IL-4 production, stimulation of eosinophils, through IL-5 production, and the development of an allergic reaction. The exact regulation of the TH1 versus the TH2 lymphocyte response is unknown, but studies suggest that IL-12 produced by activated macrophages stimulates the TH1 response, whereas IL-4 inhibits the TH1 response, allowing the TH2 response to dominate. TH lymphocytes predominately drive the immune response to microbial pathogens by activating macrophages or B lymphocytes. Another functionally distinct subpopulation of CD4+ T lymphocytes is the regulatory T lymphocyte (T reg). T reg lymphocytes function to suppress the response of self-reactive CD4 lymphocytes that have escaped the negative selection process in the thymus. They are distinguished from other CD4 T lymphocytes by the expresson of CD25 on the cell surface. Like TH1 and TH2 lymphocytes, T reg lymphocyte differentiation is driven by cytokine environments; however, where TH1 and TH2 lymphocyte activation occurs through transcripional activators T-bet and GATA-3, respectively, T reg lymphocytes are activated through the transcriptional repressor FoxP3 (Fig. 5-5). This subpopulation of T reg lymphocytes are often referred to as Fox3P lymphocytes and produce the immunosuppressive and antiinflammatory cytokines IL-4, IL-10 and TGF-β. FoxP3 lymphocytes are an intense area of investigation for their role as suppressor lymphocytes of immunity and inflammation. T reg lymphocytes have been shown to have a role in the prevention of organ-specific autoimmune diseases and in modulation of immune responses to microbial pathogens to prevent overwhelming inflammatory reactions. Finally, a subpopulation of CD4 lymphocytes characterized by the ability to produce IL-17 is designated as TH17 lymphocytes. TH17 lymphocyte differentiation is driven by TGF-β, IL-6, IL-1, and IL-23. TH17 lymphocytes, through production of chemokines IL-17 and IL-22, induce the recruitment of monocytes and neutrophils to sites of inflammation. Again, one must recognize that this is an oversimplification of a complex regulatory mechanism and that as additional knowledge is gained about TH1, TH2, T reg, and TH17 lymphocyte responses, we will be able to understand pathogenic mechanisms of diseases, which will lead to the development of more specific therapeutic targets.
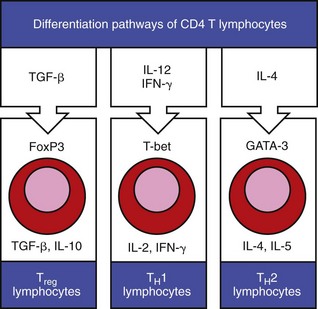
Fig. 5-5 Differentiation and expression of CD4 T lymphocytes.
Cytokines in the extracellular tissue fluids direct the differentiation of CD4 T lymphocytes to specific secretory types, such that they secrete specific cytokines that have specific functions on other lymphocytes in the environment. (Adapted from Parham P: The immune system, ed 3, 2009, Garland Science.)
WEB TABLE 5-1
Basic Cytokine and Functional Profiles of TH1 and TH2 Lymphocytes
| Cytokine | TH1 | TH2 |
| IL-2 | + | |
| IFN-γ | ++ | |
| TNF-β | ++ | |
| GM-CSF | ++ | + |
| IL-3 | ++ | ++ |
| IL-4 | ++ | |
| IL-5 | ++ | |
| IL-13 | ++ | |
| Function | ||
| Antibody* | + | ++ |
| IgE | ++ | |
| Eosinophils, mast cells | ++ | |
| Macrophages | ++ | |
| Type IV hypersensitivity | ++ | |
| Cytotoxic T lymphocytes | ++ |
GM-CSF, Granulocyte-macrophage colony-stimulating factor; IFN-γ, interferon-γ; IgE, immunoglobulin E; IL, interleukin; TNF-β, tumor necrosis factor-β.
B Lymphocytes
B lymphocytes constitute 5% to 20% of the peripheral blood mononuclear cells. B lymphocyte development occurs in two phases, an antigen-independent phase in the primary lymphoid tissues, followed by an antigen-dependent phase in secondary lymphoid tissues. B lymphocytes can be found in primary lymphoid tissues, such as the bone marrow and ileal Peyer’s patches (a primary lymphoid tissue in some species because it is the site of B lymphocyte development, rather than the bone marrow), and in secondary lymphoid tissues, such as the spleen, lymph nodes, tonsils, and Peyer’s patches. Within secondary lymphoid tissues, B lymphocytes are aggregated in the form of distinct lymphoid follicles, which on activation expand to form prominent pale regions called germinal centers (Fig. 5-6). This anatomic localization, similar to T lymphocytes in the PALS and paracortex, is the result of elaboration of chemokines for which the B lymphocyte has receptors. The antigen receptor of the B lymphocyte is the membrane bound immunoglobulin. After the antigen-independent phase of development, B lymphocytes express IgM and IgD on their surface that signifies a mature B lymphocyte. In the antigen-dependent phase, antigen-activated mature B lymphocytes differentiate into IgM-secreting plasma cells or switch to another antibody isotype. Immunoglobulins can be generated against an almost unlimited number of antigenic determinants through the rearrangement of genes encoding the light chain and heavy chain components. As in the case of the TCR, an evaluation of the rearranged genes of a B lymphocyte can be used to molecularly phenotype B lymphocyte neoplasms (see Chapter 6).
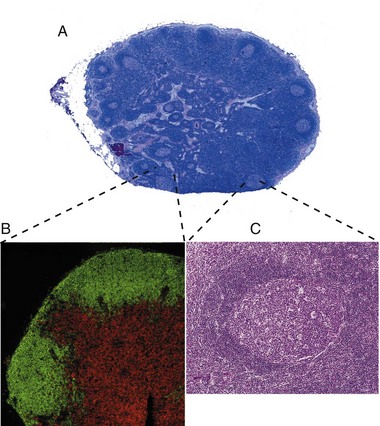
Fig. 5-6 Histology of a hyperplastic lymph node.
A, Outer cortex, containing numerous secondary lymphoid follicles with characteristic pale centers, and inner medulla are easily identifiable. B, Localization of B lymphocytes (labeled with a B lymphocyte marker conjugated with a green fluorochrome) and T lymphocytes (labeled with a T lymphocyte marker conjugated with a red fluorochrome). Immunofluorescence photomicrograph. C, Higher magnification of a secondary lymphoid follicle illustrating the central pale, germinal center, which contains primarily proliferating B lymphocytes, CD4+ lymphocytes and dendritic cells, surrounded by densely packed small B lymphocytes. H&E stain. (A, B, and C from Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Like the T lymphocyte, the B lymphocyte also has accessory molecules that function to form the antigen receptor complex (Fig. 5-7). These nonpolymorphic molecules are nonpolymorphic heterodimers composed of Ig-α (CD79a) and Ig-β (CD79b) that do not bind antigen but do interact with the transmembrane portion of surface immunoglobulin involved in cell activation. B lymphocytes, unlike T lymphocytes, can recognize soluble antigens. Additional nonpolymorphic molecules that are important to B lymphocyte functions are CD21 and CD40. The CD21 molecule is the complement receptor-2 molecule whose ligands are C3b and C3d. B lymphocyte responses to protein antigens are dependent on cytokines produced by activated T lymphocytes (CD4+). The CD40 molecule interacts with CD40 ligand on the surface of TH lymphocytes and functions to allow B lymphocyte development into antibody-secreting plasma cells. A failure to express CD40 ligand has been associated with an inability to isotype switch, resulting in a hyper-IgM syndrome. B lymphocytes activated by antigen develop into antibody secreting plasma cells and memory lymphocytes of the same antigenic specificity.
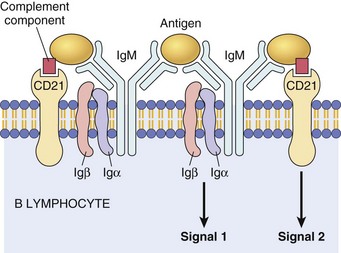
Fig. 5-7 The B lymphocyte antigen receptor complex.
Membrane IgM (or IgD, not shown) and the signaling molecules Igα and Igβ. CD21, also known as complement receptor-2, binds complement components and activates B lymphocytes. Ig, Immunoglobulin. (Courtesy Dr. Alex McPherson, University of California, Irvine.)
Mononuclear Phagocytic System (Monocyte-Macrophage System)
The preferred term today for the functionally and phenotypically diverse population of mononuclear phagocytic cells is the mononuclear phagocytic system (MPS). It is also referred to as the monocyte-macrophage system. These cells have a broad range of functions contributing to immunity, inflammation, and tissue remodeling and repair. A brief history on the recognition of the defensive mechanism of phagocytosis not only facilitates our current understanding of how these cells play a crucial role in innate and adaptive immunity but also explains some of the terminology that often leads to confusion for students. Elie Metchnikoff (1845-1916), a comparative developmental zoologist, is credited with recognizing and establishing the process of phagocytosis, an important defensive mechanism for organisms. He recognized the association between the systemic clearance of microorganisms and the presence of the microorganisms in the spleen and liver. Subsequent studies evaluating the systemic clearance of dyes suggested that Kupffer cells and endothelial cells lining the sinusoids of the liver were responsible because both cell types internalized the dyes. Because the investigators considered macrophages (Kupffer cells) and endothelial cells to have a common biologic function, phagocytosis, they proposed that they be encompassed as a system, the reticuloendothelial system. We now understand that the mechanism of uptake by endothelial cells is not by phagocytosis and as a result the term reticuloendothelial system is no longer used. The term mononuclear phagocytic system was developed to differentiate lymphoid cells (mononuclear T and B lymphocytes), granulocytes (polymorphonuclear leukocytes), and endothelial cells from the lineage-committed precursors in the bone marrow, blood monocytes and tissue macrophages, and dendritic cells that currently comprise the MPS. It is very likely that as we learn more about the precursors and differentiated cells that constitute the MPS, there will be proposals for new, more specific classification schemes.
In general, a circulating MPS cell in the blood is designated as a monocyte, whereas the tissue-based cell is designated as a macrophage. The blood monocyte-to-tissue macrophage is well recognized, although the mechanisms that allow for differentiation of the circulating monocyte pool into the tissue-based pool are still largely unknown. The monocyte is a bone marrow–derived cell of the myeloid lineage that is the precursor cell to the terminally differentiated macrophage that has limited recirculation and replication capacity. The myeloid dendritic cell represents a specific type of mononuclear cell present in nonlymphoid tissues with unique migratory properties and is discussed separately. As opposed to granulocytic myeloid cells, macrophages are long-lived (days to months) and can exist as quiescent “resident” cells widely distributed throughout the body. It is becoming increasingly clear that there is heterogeneity in the circulating monocyte pool that corresponds with the ultimate tissue localization of resident macrophages. The phenotypic characterization of the cells the MPS contains is often used in an attempt to identify specific populations of MPS (Fig. 5-8). Morphologically, monocytes are variably sized with an irregular shape, oval or kidney-shaped nucleus, prominent cytoplasmic vesicles, and a high cytoplasm to nucleus ratio. These features are not unique to monocytes, and as a result, they are difficult to distinguish from circulating dendritic cells, activated lymphocytes, and NK cells based on morphology or by light scatter (flow cytometry). This section covers basic concepts attributable to monocytes, tissue macrophages, and myeloid dendritic cells and refers to specific organ systems. See additional chapters in the section on pathology of organ systems for details on organ-specific cells of the MPS. Certain organ systems have specific names for their resident macrophages, whereas other organ systems only refer to them as macrophages (Table 5-2).
TABLE 5-2
Nomenclature and Location of Nonlymphoid Monocyte-Macrophage Cell Types
| Organ/Tissue | Name | Location |
| Lung | Alveolar macrophages Pulmonary intravascular macrophages |
Alveolar spaces Capillaries of the lung |
| Connective tissues | Histiocytes | Interstitium |
| Kidney | Mesangial cells | Glomerular tuft |
| Brain | Microglial cells | Neuroparenchyma and perivascular areas |
| Bone | Osteoclasts | Bone marrow |
| Blood | Monocytes | Circulation |
| Liver | Kupffer cells | Hepatic sinusoids |
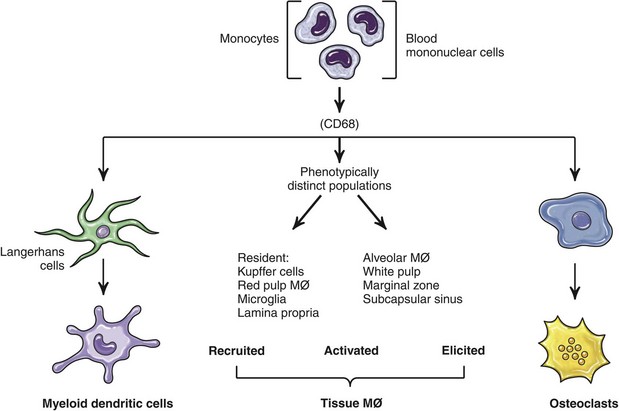
Fig. 5-8 Differentiation of mononuclear phagocytes based on antigen markers. (Adapted from Paul WE: Fundamental immunology, ed 6, Philadelphia, 2008, Lippincott Williams & Wilkins.)
Growth and differentiation of monocytes is regulated by specific growth factors, such as IL-3, colony-stimulating-factor-1 (CSF-1), granulocyte-macrophage CSF (GM-CSF), IL-4, and IL-13, and inhibitors such as type I interferons and transforming growth factor-β (TGF-β). CSF-1 is the most important because it controls the proliferation, differentiation, and survival of the monocyte. Monocytes represent approximately 4% to 10% of blood leukocytes and are identified in mammals, birds, amphibians, and fish. They are largely viewed as accessory cells that importantly link inflammation and innate immune responses to adaptive immunity. Hematopoietic stem cells (HSC) give rise to the common myeloid progenitor (CMP), the precursor cell to the granulocyte/macrophage progenitor (GMP) and macrophage/dendritic cell progenitor (MDP). The MDP is the common progenitor cell for monocytes, macrophages, and conventional dendritic cells. Monocytes express the CSF-1 receptor (CD115) and the chemokine receptor CX3CR1, are differentiated from polymorphonuclear cells (PMNs), NK cells, and lymphoid cells, and do not express CD3, CD19, or CD15. Monocyte heterogeneity based on surface marker expression and function has identified subsets that are an area of intense investigation and are best characterized in humans and rodents. Subpopulations of blood monocytes can be phenotyped based on the expression of surface markers CD14 and CD 16 (FcγR-III). Two additional subpopulations of blood monocytic cells are the myeloid blood dendritic cells, which are negative for CD14 and CD16. In the mouse, the main subset of CD115+ monocytes are characterized as large cells expressing Ly6C, the chemokine receptor CCR2 and the adhesion molecule L-selectin (CD62L), and CX3CR1 (Fig. 5-9). These are referred to as inflammatory monocytes or inflammatory-monocyte-derived macrophages that are preferentially recruited to inflamed tissues and lymph nodes where they produce high levels of TNF-α and IL-1. The emigration of Ly6C+ monocytes from the bone marrow to the periphery depends on the chemokine receptor CCR2 and its ligands CCL7 and CCL2. Ly6C negative monocytes have less of an influence on inflammatory reactions and appear to function more importantly as tissue resident cells and as cells involved in healing and regeneration associated with vascular injury. In those species characterized to date, there appear to be two major functional subpopulations of monocytes, one that is recruited and differentiated into macrophages at the site of inflammation and expresses higher levels of MHC class II and adhesion molecules and one that is responsible for repopulating resident tissue macrophages. Both populations can give rise to dendritic cells (see Fig. 5-8). Classification schemes are forever evolving, and there are definite species differences that may explain species differences in rates of infection and types of clinical disease associated with specific microbial agents. Similar phenotypic and functional heterogeneity exists with tissue-based macrophages.
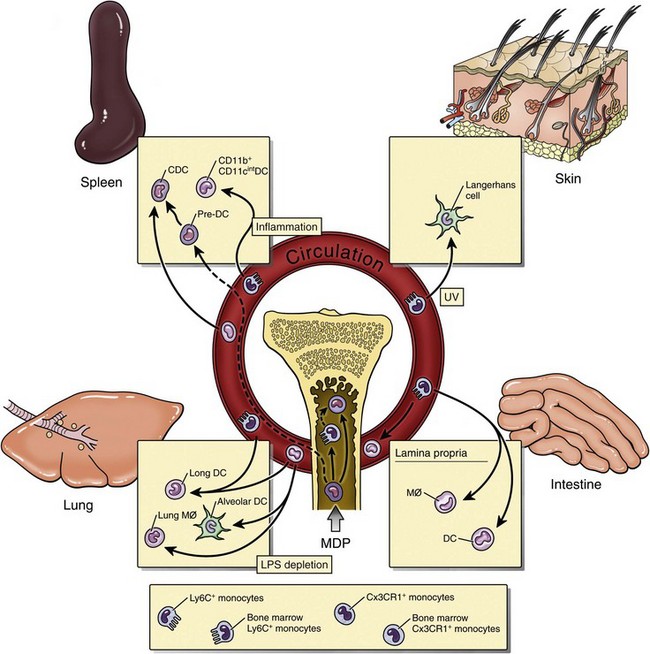
Fig. 5-9 Monocyte differentiation into dendritic cells and tissue macrophages. (Adapted from Serbina NV, Jia T, Hohl HM, et al: Ann Rev Immunol 26:421-452, 2008.)
The MPS of hematopoietic and lymphoid organs is diverse with functionally and phenotypically distinct parallel subpopulations in humans, rodents, and pigs. In primary lymphoid organs, mature macrophages are involved in the production, differentiation, and destruction of all lineages of hematopoietic cells in the bone marrow, and in positive and negative selection processes in the thymus. In secondary lymphoid organs, macrophages are uniquely positioned to enhance their phagocytic properties to endogenous and exogenous material and to influence other cell types through their products. The resident macrophages in the spleen differ in microscopic localization, phenotype, and function. The reason for this complexity is largely attributable the spleen’s role as a hematopoietic organ and as a secondary lymphoid organ. Distinct subpopulations of macrophages are present in the red pulp, white pulp, and marginal zone with some recognized species differences (see Chapter 13). The red pulp macrophage functions in the phagocytosis of hematogenous pathogens and senescent erythrocytes. Erythrophagocytosis primarily occurs in the spleen and liver and allows for the removal of senescent erythrocytes and recycling of iron, often evident as cytoplasmic accumulations of pigments. The white pulp macrophages are also actively phagocytic, and a morphologically distinct cell, the “tingible body macrophage,” can often be easily identified histologically and represents macrophages involved in the uptake and removal of apoptotic T and B lymphocytes. The marginal zone of the spleen provides a complex environment for the interface for the red and white pulp, specifically an important transit area for cells leaving circulation and entering the white pulp, and for B lymphocyte differentiation. The development of granulomatous inflammatory reactions in the spleen as a response to hematogenous microorganisms often begins in the marginal zone. The two subpopulations of macrophages present in the marginal zone are the metallophilic macrophage and the marginal zone macrophage. Marginal zone macrophages express high levels of PRRs and scavenger receptors that facilitate the clearance of pathogens from the circulation. The function of metallophilic macrophages is unknown. Tissue macrophages are derived from a combination of precursors in the blood (monocytes) and from local proliferation of precursors that varies for individual organ systems.
Much has been written about the relationship between monocytes, macrophages, and dendritic cells. The inclusion of dendritic cells as a component of the MPS is in part attributable to the fact that they are derived from a common myeloid precursor, influenced by similar growth factors (e.g., CSF-1), express common surface markers, and have no unique properties as antigen-presenting cells that allow them to be differentiated from macrophages. Like macrophages, dendritic cells in specific organ systems may have a specific name or designated only as dendritic cells preceded by the organ or in some instances a specific anatomic location within an organ. Dendritic cell subsets are also an intense area of investigation with regards to phenotypic and functional heterogeneity. The so-called conventional dendritic cells are present as immature cells in the interstitial tissues of all organs except the brain.
Macrophages
Mononuclear phagocytic cells include circulating monocytes and tissue-based macrophages. In the spleen, macrophages are located in the marginal zone, white pulp, and red pulp, where they function primarily as phagocytic cells. In the lymph node, macrophages are located in the subcapsular sinus, which is analogous to the marginal zone of the spleen, and the medulla. These physical locations, the subcapsular sinus of lymph nodes and marginal zone of the spleen, facilitate their exposure to potential antigens. Nonlymphoid tissue-based macrophages have different functions and are named according to the tissue in which they reside (see Table 5-2). One primary function of these cells is phagocytosis, as discussed in Chapter 3. Macrophages express Fc receptors (FcR) for antibody and can phagocytose antigens opsonized by antibody or complement components. Another primary function is their involvement in the immune response as antigen-presenting cells. In this instance, they phagocytose antigen and process it into peptide fragments, which are then presented to T lymphocytes, and the induction of cell-mediated immune responses. Although all nucleated cells express MHC class I molecules and could be considered antigen-presenting cells, only three cell types normally express MHC class II molecules and are regarded as the major antigen-presenting cells. They are the macrophage, dendritic cell, and B lymphocyte. Whereas B lymphocytes and dendritic cells constitutively express MHC class II molecules, macrophages express MHC class molecules on activation.
Macrophages also have an important role in the generation of a cell-mediated immune response and are essential to type IV hypersensitivity reactions. Activated TH1 lymphocytes synthesize IFN-γ, a potent activator of macrophages. Under the influence of IFN-γ, macrophages have increased phagocytic activity and are more efficient at killing.
Dendritic Cells
Dendritic cells comprise a distinct population of cells that are characterized by elongate cell processes. Most dendritic cells are antigen-presenting cells, which process antigens and present fragments to T lymphocytes. They are more efficient than macrophages and B lymphocytes at antigen presentation. Antigen-presenting dendritic cells are nonphagocytic, bone marrow–derived cells. They are the most important antigen-presenting cell for initiating primary immune responses to protein antigens (Fig. 5-10). Antigen-presenting dendritic cells express a number of molecules, such as TLRs and mannose receptors, that make them efficient at capturing and responding to antigens. They also express high concentrations of MHC class II molecules and B7 co-stimulatory molecules. By expressing chemokine receptors similar to T lymphocytes, they have the ability to localize in T lymphocyte regions of lymphoid tissue. By colocalizing to these areas, they are uniquely positioned to present antigens to recirculating T lymphocytes. Antigen-presenting dendritic cells function to capture antigen and then migrate to T lymphocyte areas of secondary lymphoid organs, where they present fragments of the antigen on their surface and increase their expression of co-stimulatory molecules that activate T lymphocytes. Specifically, migrating dendritic cells, derived from Langerhans’ cells that have captured antigen, enter the lymph node through efferent lymphatics and localize in lymphoid organs, where they present antigenic peptides to T lymphocytes that facilitate B lymphocyte activation and the production of antibody-secreting plasma cells. In addition to their function as antigen-presenting cells, they are also important in the process of negative selection in the thymus and in the maintenance of peripheral tolerance. The four types of antigen-presenting dendritic cells and their locations are listed in Table 5-3. Circulating dendritic cells, also known as veiled cells, make up less than 1% of peripheral blood mononuclear cells. The second type of dendritic cell, the follicular dendritic cell, is primarily located in lymphoid follicles. These cells are not derived from the bone marrow, do not express MHC class II molecules, and do not function as an antigen-presenting cell. Follicular dendritic cells have FcR and receptors for C3b. They store antigen-antibody and antigen-C3b complexes and are thought to be involved in the development and maintenance of memory B lymphocytes.
TABLE 5-3
Antigen-Presenting Dendritic Cells and Their Primary Location
| Dendritic Cells | Location |
| Langerhans’ cells | Skin, mucous membranes, iris, ciliary body |
| Interstitial dendritic cells | Most major organs |
| Interdigitating dendritic cells | T lymphocyte area of secondary lymphoid tissue and thymic medulla |
| Circulating dendritic cells | Peripheral blood |
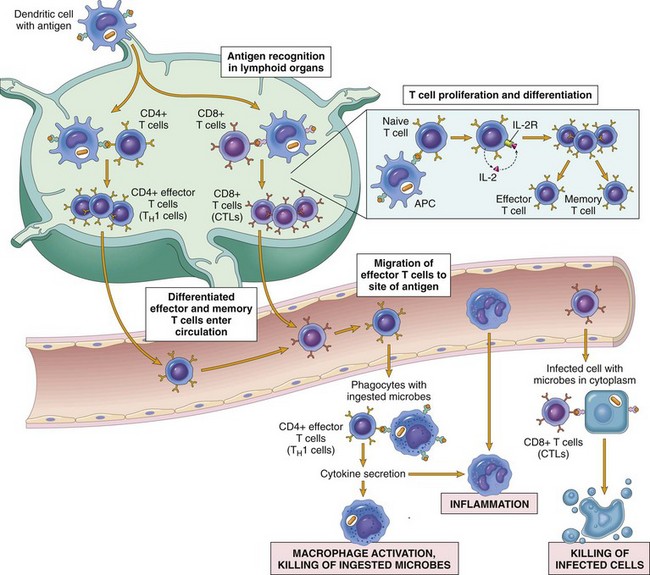
Fig. 5-10 Dendritic cell functions.
Specialized dendritic cells in the epidermis (Langerhans’ dendritic cells) capture antigen via phagocytosis or endocytosis and migrate to regional lymph nodes, where they present peptide fragments of the antigen to naïve T lymphocytes. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Natural Killer Cells
NK cells are nonspecific cytotoxic cells that are important in early responses to tumor cells and viral infections. NK cells are bone marrow–derived, large granular lymphocytes that make up 5% to 15% of the peripheral blood mononuclear cells. Their size, slightly larger than that of a small lymphocyte, and the presence of abundant granular cytoplasm distinguish them from T lymphocytes (Fig. 5-11). They are commonly referred to as large granular lymphocytes. The cytoplasm of NK cells and CTL (Tc) is characterized by cytotoxic granules that contain perforin and granzymes, two potent pathways mediating lysis of the target cell. NK cells and T lymphocytes express numerous similar surface molecules and kill virus infected cells and tumor cells by similar mechanisms. Two membrane molecules, CD16 and CD56, are commonly used to identify NK cells. NK cells express FcγR (CD16) and the β-subunit of the IL-2 receptor (CD2). They do not express antigen-specific TCR or CD3 molecules. In contrast to cytotoxic lymphocytes, NK cells are not MHC restricted, are constitutively cytolytic, and do not develop memory cells. Because NK cells are activated early in an immune response and do not require a previous sensitization phase to develop memory cells after activation, they are the cytotoxic cell of innate immunity, the counterpart to the adaptive immune response’s CTL (Tc).
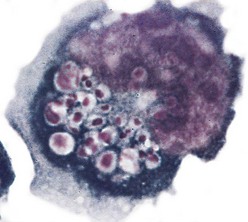
Fig. 5-11 Activated natural killer cell with numerous cytoplasmic granules that are characteristic of these large granular lymphocytes. (Courtesy Dr. Noelle Williams, Department of Pathology, University of Texas Southwestern Medical School, Dallas.)
Although NK cells do not express any antigen-specific molecules, they are very efficient at recognizing and killing altered or virally infected cells. NK cell activity is regulated through activating and inhibitory receptor molecules expressed on their cell surface (Fig. 5-12). These NK cell receptor molecules fall into two distinct categories: the immunoglobulin-like NK receptors and the C-type lectin-like NK receptors. Ligands for these receptors are cell surface molecules whose expression has been altered as a result of infection or damage. Ligands for activating receptors that stimulate NK cell activity commonly include viral and stress-induced proteins. Ligands for inhibitory receptors that block NK cell activity most commonly involve class I MHC molecules. A decreased expression of class I MHC molecules makes cells susceptible to NK cell–mediated lysis. A decrease in MHC class I expression often occurs in virus-infected cells and in neoplastic cells, making them susceptible to attack by NK cells. Normal cells are protected from NK cell killing because all nucleated cells express class I MHC molecules. This is an oversimplification of the “opposing-signals” model of NK cell regulation of how cytotoxic activity is limited to altered self-cells. Recent studies on the molecular mechanisms of NK cell regulation indicate that the absence of an inhibitory stimulus by itself is insufficient for triggering NK cell killing. NK cells also require triggering of activating receptors. Several activating receptors have been identified. One is the NKG2D receptor, a C-type lectin-like molecule that recognizes a number of stress-induced proteins. These stress-induced proteins are normally only constitutively expressed in the intestinal epithelium or as a result of cellular distress caused by infection or neoplastic transformation. There are a number of additional activating receptors, some of which recognized viral proteins, that are structurally similar to class I MHC molecules.
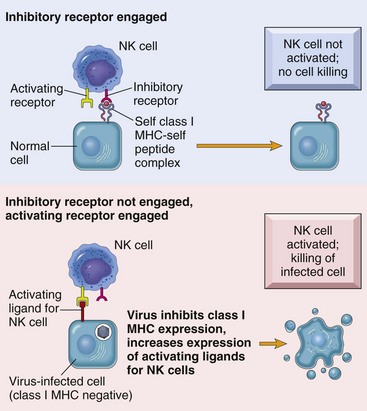
Fig. 5-12 Regulation of natural killer (NK) cell activity through activating and inhibitory receptors.
MHC, Major histocompatibility complex. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Because NK cells express FcγR (CD16), they can also function in antibody-dependent cellular cytotoxicity (ADCC). In the case of NK cells, ADCC allows for antibody-bound targets to be identified and targeted for NK cell–induced lysis.
NK cells also facilitate the early response to viral infections not only by responding to cytokines produced early in a viral infection, but also by producing cytokines that help direct the immune response. NK cells are activated by IFN-α and IFN-β, released by virus-infected cells, and by IL-12, released by macrophages. After activation, NK cells have the ability to produce IFN-γ, a major cytokine directing the development of TH1-type immune response early in the infection. IL-2 and IL-15 stimulate NK cell proliferation, and IL-12 enhances NK cell killing.
Cytokines: Messenger Molecules of the Immune System
General Properties of Cytokines
Cytokines make up a vast group of low molecular weight soluble glycoprotein proteins that are produced by immune and nonimmune cells, are largely produced locally, and act locally to direct the immune response. The expression of cytokine receptors and their respective ligands is highly regulated and contributes to the complexity of the systemic organization of the immune response. Cytokines are involved in every aspect of leukocyte biology and the immune response and are essential to leukocyte development, recirculation, differentiation, and activation and in maintaining self-tolerance. Cytokines can influence the cytokine-producing cell itself (autocrine), other cells present locally (paracrine), or distant cells (endocrine) (see Fig. 12-1). Cytokines use common signal transducing pathways, converting an extracellular signal through a cell surface receptor to activate or inhibit the target cell.
The nomenclature of cytokines has evolved from a system that originally named them according to their cellular source (e.g., lymphokine from lymphocytes and monokines from monocytes) to one that named them according to not only the cellular source but also the target cell (e.g., interleukin, a cytokine produced by a leukocyte that influences another leukocyte) or to the primary function (e.g., chemokine, a cytokine that affects chemotaxis). As cytokines were characterized, it became apparent that they have pleiotropic, redundant, synergistic, and antagonistic features that do not allow for such a simplistic classification scheme. More recently, cytokines and their receptors have been classified based on their molecular structure and common signaling pathways. Many cytokines share a similar, α-helix structure that is also shared by their respective receptors and are classified as type I cytokines and receptors. As is discussed later, the classification of cytokines and cytokine receptors according to their structural similarities allowed for the identification of the cause of the profound cytokine defects associated with X-linked severe combined immunodeficiency disease in humans and dogs. Unfortunately, type I cytokines also have now been found to include other regulatory proteins such as growth hormone, prolactin, erythropoietin, thrombopoietin, and leptin. Type II cytokines include type I interferons (IFN-α and IFN-β), type II interferon (IFN-γ), and the IL-10 family of cytokines.
The breadth and depth of knowledge regarding the function, regulation, and control of cytokines is overwhelming; however, an overview of the major cytokines and their primary functions is important for understanding the pathogenic mechanism of many disease processes. Some of the major cytokines and their primary biologic activities are presented here and in Chapter 3 as they pertain to acute and chronic inflammatory responses.
Cytokines that broadly influence innate and adaptive immune responses include IL-1, interferons (type 1), IL-6, and TNF-α. These cytokines are produced and influence a wide array of cell types. Cytokines that are involved in hematopoiesis and lymphocyte development include IL-2, IL-3, IL-4, IL-5, IL-12, IL-15, TGF-β, and GM-CSF to mention a few. Chemokines are a large group of cytokines that influence leukocyte development, trafficking, and function. They are organized into subfamilies, with distinct functions, based on the position of cysteine residues. The C-X-C subfamily of chemokines is primarily produced by activated macrophages and tissue cells (e.g., endothelium), and the C-C subfamily is largely produced by activated T lymphocytes. Chemokines are responsible for the anatomic localization (“homing”) of lymphocytes within lymphoid and nonlymphoid tissue. Chemokines and the other proinflammatory cytokines are more thoroughly covered in Chapter 3. The most important functional group of cytokines related to the pathogenesis of a number of diseases of immunity are those involved in the regulation of TH lymphocytes. As discussed previously, TH lymphocytes are classified based on their functional capacity and ability to elicit primarily an antibody response or a cell-mediated immune response rather than on their expression of specific cell markers (Fig. 5-13). TH1 lymphocytes are activated by IL-12 and IL-18 and produce primarily IL-2, IFN-γ, and TNF-β to direct a cell-mediated immune response. TH2 lymphocytes are activated by IL-4 and produce primarily IL-3, IL-4, IL-5, IL-6, IL-10, and IL-13 to direct a humoral immune response. As discussed later in the chapter, the type of response (TH1 versus TH2 type) may determine if a diseased state will occur. IL-15 regulates the growth and activity of NK cells. As indicated in Fig. 5-13, some cytokines, such as IL-10 and TGF-β, downregulate immune responses. In summary, cytokines produced by the TH1 or TH2 subset of lymphocytes not only promote the activation and functional capacities of the subset that produces them (autocrine effect) but also inhibit the development and activity of the other subset. This is known as cross-regulation and has important implications with regard to protective immune responses and adverse immune responses, as is discussed later.
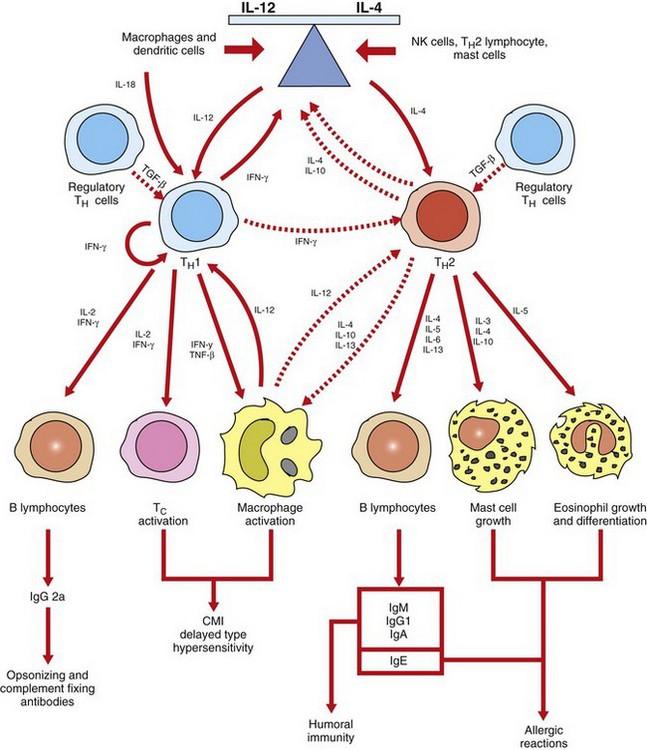
Fig. 5-13 Cross-regulation of immunity.
Cross-regulation of TH1 and TH2 lymphocytes in part determines if an immunity is primarily a cell-mediated response or a humoral response. TH1 lymphocytes, activated primarily by IL-12 and IL-18, promote cell-mediated immunity (CMI) by activating macrophages and cytotoxic T lymphocytes. TH2 lymphocytes, activated primarily by IL-4, promote humoral immunity by producing cytokines that activate B lymphocytes to develop into antibody-secreting plasma cells. TH2 lymphocytes also produce cytokines that activate mast cells and eosinophils in the pathogenesis of allergic diseases. The cross-regulation of TH1 and TH2 lymphocytes provides an inverse relationship between cell-mediated and humoral immunity. IFN-γ, Interferon-γ; Ig, immunoglobulin; IL, interleukin; NK, natural killer; TNF-β, tumor necrosis factor-β. (Adapted from Goldsby RA, Kindt TJ, Osborne BA: Kuby immunology, ed 4, New York, 2000, WH Freeman.)
Finally, a number of cytokine inhibitors were identified, and one of the more studied ones includes a factor called IL-1 receptor antagonist, which is produced by macrophages, hepatocytes, and keratinocytes. This factor inhibits the local and systemic effects of IL-1 by blocking the IL-1 receptor. Another group of inhibitors are the soluble cytokine receptors, which are the enzymatic product of cleavage of the extracellular domain of cytokine receptors that bind their respective cytokines, preventing interaction with the membrane-bound receptor form. The best characterized soluble cytokine receptor is the soluble IL-2 receptor. A number of pathogenic organisms have adapted this as an evasion strategy by producing cytokine-binding proteins or mimics to influence the development of the immune response. Although soluble receptors have been identified in a number of human diseases, their exact role remains to be determined.
Structure and Function of Histocompatibility Antigens
The MHC represents a complex of genes that encode specialized molecules involved in intercellular recognition and the distinguishing of self from nonself. These cell surface molecules have immunologic and nonimmunologic functions. The histocompatibility designation originated from the identification of these molecules in determining the compatibility of transplanted tissues. The MHC is an essential component in humoral and cell-mediated immunity. Most T lymphocytes only recognize fragments of antigen when they are bound to MHC molecules, and this requirement is the basis for MHC restriction. The repertoire of MHC molecules is genetically controlled and determines an individual’s ability or inability to respond to specific antigens. MHC molecules are present throughout vertebrates and are maintained in gene clusters, each of which encodes different MHC products, which are linked. The primary function of cell surface MHC molecules is to bind peptide fragments of foreign proteins for presentation to antigen-specific T lymphocytes. There are three major classes of genes, and these encode for MHC molecules that are grouped according to their structure, tissue distribution, and function. Class I and class II genes encode cell surface molecules. Class III genes encode components of the complement system, enzymes 21-hydroxylase A and B, cytochrome p450, TNF-α and TNF-β, and heat shock protein 70.
MHC class I molecules are present on all nucleated cells (and platelets in some species). Their major function is the presentation of peptide fragments of antigens to CTL (Tc) (CD8+). This requirement results in CD8+ lymphocytes being MHC class I restricted. MHC class I molecules are further subdivided into highly polymorphic loci, referred to as Ia, and relatively nonpolymorphic loci, referred to as Ib, Ic, and Id. Each class I molecule is composed of a heterodimer consisting of a polymorphic α-chain that is linked to a nonpolymorphic β2-microglobulin. The extracellular region of the α-chain consists of three domains (α1, α2, and α3). The α1 and α2 domains form a groove where the peptide fragments bind the MHC molecule (Web Fig. 5-1). Although MHC class I molecules differ in their ability to bind peptide fragments, they are not as restrictive as antibodies and TCRs. The intracellular processing of antigen into peptide fragments, the association of those fragments with MHC class I molecules, and their transport to the cell surface is a complex process.
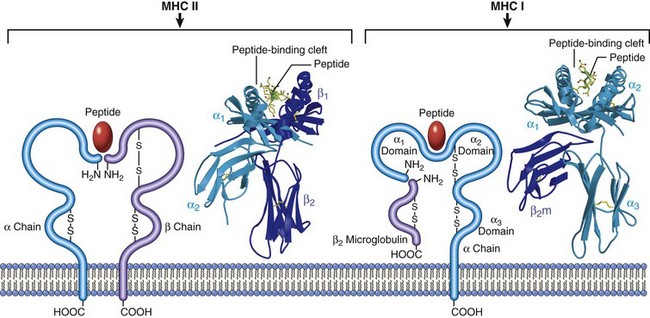
Web Fig. 5-1 Schematic diagram and crystal structures of class I and II major histocompatibility complex (MHC) molecules. (Courtesy Dr. P. Bjorkman, California Institute of Technology, Pasadena.)
Antigen uptake by antigen-presenting cells is by phagocytosis or endocytosis. Antigen processing is the degradation of an antigen into peptide fragments, which are complexed with MHC molecules. Antigen presentation is the transport of the peptide-MHC complex to the membrane, where they are displayed. Antigens can arise intracellularly (endogenous) and extracellularly (exogenous), and the immune system most effectively eliminates these antigens through the elaboration of CTL (Tc) or secretion of antibody. The immune system uses two different processing pathways for antigen processing and antigen presentation. Intracellular antigens are processed in a cytosolic pathway and presented in association with MHC class I molecules (Fig. 5-14). Extracellular antigens are processed in an endocytic pathway and presented in association with MHC class II molecules. Endogenous antigens are degraded into small peptide fragments within the cytoplasm by the proteasome complex. Peptide fragments are transported to the endoplasmic reticulum (ER) by an adenosine triphosphate–binding peptide transporter (TAP). Within the ER, the newly synthesized MHC class I α-chain and associated β2-microglobulin bind the antigenic peptide and form a complex that is transported from the ER to the Golgi and then to the plasma membrane for presentation to CD8+ CTL (Tc). The antigen recognition molecule of the cytotoxic T lymphocyte (TC) recognizes the MHC-peptide complex by means of its CD8 molecule, which functions as a co-receptor, binding to the nonpolymorphic α3 domain of the MHC class I heavy chain. Because CTL (Tc) only recognize peptides when they are presented as a complex with MHC class I molecules, they are referred to as being MHC class I restricted. MHC restriction is the result of positive selection of lymphocytes during T lymphocyte development in the thymus. Portions of some antigens, those portions not processed for presentation, and in some cases, entire antigens are completely degraded by exopeptidases of the antigen-presenting cell into amino acids and do not initiate an immune response. Endogenous antigens are most frequently encountered during viral infections and thus CTL (Tc) are an important defense mechanism for eliminating virally infected cells.
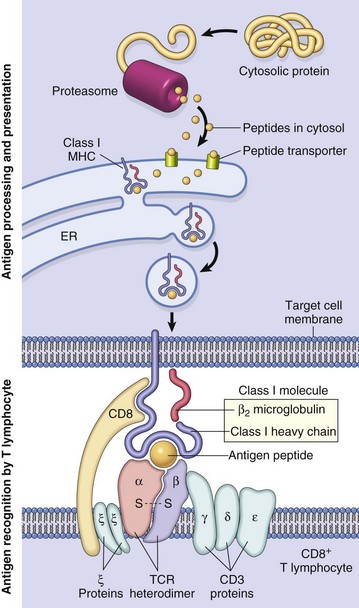
Fig. 5-14 Antigen processing and presentation by an antigen-presenting cell, and antigen recognition by T lymphocytes.
A class I major histocompatibility complex (MHC)–restricted CD8+ lymphocyte is depicted. ER, Endoplasmic reticulum; TCR, T lymphocyte receptor. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Class II molecules have a variable tissue distribution depending on the species of domestic animal, but in general they are present on antigen-presenting cells (B lymphocytes, dendritic cells, and macrophages) and can be induced on T lymphocytes, keratinocytes, and endothelial cells by IFN-γ. There are significant species differences with regard to the constitutive expression of MHC molecules. The major function of MHC class II molecules is the induction of TH lymphocytes. Class II MHC molecules are heterodimers consisting of an α-chain and a β-chain. The antigen-binding site of the class II molecule, unlike that of the class I molecule, is formed by portions of both the α1 and β1 domain. Additionally, as with the class I MHC molecules, polymorphism of the class II MHC molecules is associated with determining an individual’s response to antigens of infectious organisms (see next section). Peptides that bind to class II MHC molecules are in general derived from exogenous antigens, which have been internalized and processed within endosomes and lysosomes of antigen-presenting cells. Antigen-presenting cells can internalize antigen by phagocytosis or endocytosis (receptor mediated or pinocytosis). Macrophages are the only cell type capable of both because other antigen-presenting cells are poorly phagocytic. Extracellular antigens are processed into peptide fragments in an endocytic pathway and presented in association with MHC class II molecules on the cell membrane. During the synthesis of a class II molecule within the ER, it associates with another protein called the invariant chain, which prevents the molecule from binding endogenously derived peptides. The complex is then transported from the ER to the Golgi complex and into an endocytic compartment (vesicles) that contains the antigenic peptide fragments derived from exogenous antigens. Proteolytic cleavage of the invariant chain allows for the association of the peptide fragment with the class II MHC molecule. The peptide-class II MHC complex is then transported to the cell surface for presentation to CD4+ TH lymphocytes. The CD4 molecule acts as a coreceptor for induction of TH lymphocyte activation. Because TH lymphocytes only recognize peptides when they are presented as a complex with class II MHC molecules, they are referred to as class II MHC restricted. It is worth mentioning that of the two scientists, Peter Doherty and Rolf Zinkernagel, who received the Nobel Prize in Medicine in 1996 for discovering MHC restriction, Dr. Doherty is an Australian veterinarian.
MHC molecules are important in regulating T lymphocyte development in the thymus and in the peripheral lymphoid tissues determining specific responses to different forms of antigens. During the development and maturation process in the thymus, only T lymphocytes capable of recognizing self-MHC molecules are selected (positive selection) for export to the peripheral lymphoid tissues. These developmental processes influence an individual’s T lymphocyte repertoire, which is the functional population that influences immunity. An individual’s ability to mount an effective immune response is determined in part by his or her ability to recognize endogenous and exogenous antigens and their MHC haplotype. Thus, in one case, the association of an antigenic peptide fragment with a specific MHC molecule may result in a protective antibody response with elimination of an infectious agent, whereas in another case, the association of an antigenic peptide fragment with a specific MHC molecule may result in an inappropriate immune response to an innocuous antigen, producing an allergic reaction.
The organization (chromosomal location) and characterization (e.g., number of loci) of the MHC of each species of domestic animals appears to be fairly conserved and present in higher vertebrates and mammals. Most mammalian species studied have class I, II, and III genes, with differences between species being the arrangement and number of genetic loci comprising the MHC. In general, class I genes are more closely related within a species than between species, and the avian MHC is smaller and less complex, with many genes found in the mammalian MHC being absent. Although it is beyond the scope of this chapter to discuss the details regarding the differences between domestic species, the preceding overview is relevant to most domestic species.
Major Histocompatibility Complex and Disease Association
The MHC influences transplant acceptance or rejection, immune responsiveness, and the pathogenesis of a number of diseases. The MHC represents a complex of genes that encode specialized molecules involved in antigen presentation and thus regulate immune responses. The ability of the immune system to respond to an antigen is determined in part by the binding of peptide fragments to MHC molecules, which are then presented on the surface of antigen-presenting cells. There is a growing body of information associating certain MHC alleles with increased or decreased susceptibility to certain diseases (Web Table 5-2). Because the MHC genes of most domestic species are not well characterized, it is difficult to distinguish if the observed effects are due to the MHC itself or to other tightly linked genes. These conclusions are generally the result of an observation that some MHC alleles occur at a higher frequency among individuals affected with the disease, as compared with the general population. The association of an increased risk with certain MHC alleles is never the sole basis for determining if an individual will develop the disease, as often other hereditary and environmental factors also play an important role. The diseases most often associated with certain MHC alleles have a pathogenesis that incorporates a significant immunologic component. The types of diseases identified are diverse; however, they frequently include autoimmune, infectious, and allergic diseases. Additionally, MHC diversity may increase or decrease the susceptibility to infectious diseases. There is evidence that MHC polymorphisms may significantly impact disease resistance and is best illustrated in species in which there is a loss of MHC diversity attributed to a limited breeding pool of animals. In the case of the cheetah and Florida panther, the current breeding stocks of both species are derived from a limited genetic pool, thus resulting in reduced MHC diversity. In both species, there is an increased susceptibility to infectious agents that is not seen in other species of big cats.
WEB TABLE 5-2
Major Histocompatibility Complex (MHC) Polymorphisms Related to Resistance or Susceptibility to Diseases
Although there are a number of hypotheses to account for the role of MHC molecules in disease susceptibility, the actual mechanisms remain elusive. The most often hypothesized mechanism is attributed to the role of MHC alleles in determining responsiveness or nonresponsiveness to a particular pathogen. On the one hand, through antigen presentation and activation of cytotoxic or T lymphocytes, it determines whether a protective immune response is generated to a particular pathogen. On the other hand, certain MHC alleles may encode molecules that are used by infectious agents, as in the case of receptors for viruses or bacterial toxins, and facilitate their infectivity or pathogenicity.
Disorders of the Immune System
As has been discussed, immunity is a complex defensive system of recognition and effector mechanisms for protecting the host from infectious pathogens and cancer. During the normal immune response, there are mechanisms for eliminating the inciting foreign antigen, and associated with this is some degree of tissue damage that elicits an inflammatory response of appropriate duration and severity for the antigen. However, there are a number of instances in which the immune response elicits an inflammatory response that is not appropriate to the inciting antigen and these fall into three general categories. The largest category is the hypersensitivity reactions, which are associated with a large number of diseases covered throughout this text. The second category is the autoimmune diseases, in which the immune response is inappropriately directed at a self-antigen, resulting in damage to normal organs or tissue. The third category is the immunodeficiency diseases, in which a genetic or acquired defect results in an inability to mount an immune response and thus control infections, resulting in severe systemic inflammation. The chapter now focuses on general features of immunologic tissue injury, with discussion of some specific immunologic diseases that are attributable to disorders of the immune system. Finally, we conclude with a discussion of amyloidosis, a condition that is the result of a number of mechanisms, some of which have an immunologic basis.
Mechanisms of Immunologic Tissue Injury: Hypersensitivity Reactions
A hypersensitivity reaction is defined as the altered reactivity to a specific antigen that results in pathologic reactions upon the exposure of a sensitized host to that specific antigen. The designation of these immune responses as “hyper” is somewhat of a misnomer because the reactions elicited are better characterized as inappropriate or misdirected responses. An immune response can be either beneficial or harmful. By characterizing hypersensitivity responses as inappropriate or misdirected, we are not implying that these responses are any different from those that occur as a normal “beneficial” defense mechanism. To state it more clearly: If the immune response is beneficial it is immunity and if it is harmful, it is hypersensitivity. All hypersensitivity reactions are characterized by sensitization and effector phases. The sensitization phase requires that the host must have had either a previous exposure or a prolonged exposure to the antigen so that he or she can develop an immune response to the inciting antigen. The pathology associated with hypersensitivity reactions occurs in the effector phase and is most commonly manifested as an inflammatory reaction or as cell lysis.
Hypersensitivity reactions have historically been classified on the basis of the immunologic mechanism that mediates the disease as type I, type II, type III, or type IV. Types I, II, and III are mediated by antibody, and type IV is mediated by macrophages and T lymphocytes. Type I is also known as immediate type hypersensitivity and most often is the result of an IgE response that is directed against an environmental or exogenous antigen (also known as an allergen). The result is the release of vasoactive mediators from IgE-sensitized mast cells, and these mediators produce an acute inflammatory response. Type II is also known as cytotoxic hypersensitivity and most often occurs when IgG or IgM is directed against either an altered self-protein or a foreign antigen bound to a tissue or cell. The result can lead to either destruction of the tissue or cell by ADCC, complement-mediated lysis, or altered cellular function without evidence of tissue or cell damage. Type III is also known as immune complex hypersensitivity and is due to the formation of insoluble antibody-antigen complexes (also known as immune complexes). The result is activation of the complement system and the development of an inflammatory reaction at the sites of immune complex deposition. Type IV is also known as delayed-type hypersensitivity (DTH) and is the result of activation of sensitized T lymphocytes to a specific antigen. The resulting immune response is either mediated by direct cytotoxicity or by the release of cytokines that act primarily through macrophages. This original classification, as proposed by Gell and Coombs, was based largely on the primary initiating event involved in the individual reactions and not on the actual pathogenesis as it relates to what is seen clinically or pathologically. Although the original classification of hypersensitivity reactions is still valid, “newer” versions that are based on the pathogenesis better illustrate the complexity of these reactions and the specific pathology (lesions) associated with them. For the purposes of our discussion, we will use the original version of the Gell and Coombs classification presented in Table 5-4, understanding that many of the diseases associated with hypersensitivity reactions are actually complex and may involve more than one type. In humans, genetic mapping studies of most diseases characterized by a hypersensitivity reaction suggest that there are disease-associated susceptibility genes, further supporting the complex pathogenesis of these diseases. Finally, the pathogenesis of many diseases rarely involves a single hypersensitivity reaction, and in fact, some diseases may begin as an immediate hypersensitivity but progress to be predominantly DTH. For clarity, the hypersensitivity diseases are discussed in the context of their predominant mechanism except when it is appropriate to elaborate on the progression of a disease.
TABLE 5-4
Mechanisms of Hypersensitivity Diseases
IgE, Immunoglobulin E; IgG, immunoglobulin G; IgM, Immunoglobulin M.
Type I Hypersensitivity (Immediate Hypersensitivity)
Type I hypersensitivity reactions are most commonly the result of an IgE-mediated immune response directed against environmental antigens (i.e., allergens) and parasite antigens. Harmful IgE-mediated responses to innocuous environmental antigens resulting in allergic reactions are termed hypersensitivity, whereas similar IgE-mediated protective responses to parasite antigens are considered immunity. This distinction emphasizes the fact that these are not unique immunologic reactions but rather misdirected or inappropriate “normal” immune responses. Type I hypersensitivity occurs in a previously sensitized host and is initially manifested as acute inflammatory process that occurs within minutes (“immediate hypersensitivity”) of exposure to the specific antigen. In many instances, the reaction progresses from an early acute inflammatory response to a late-phase response and/or chronic inflammatory lesion that persists (Figs. 5-15 and 5-16). The basic pathogenesis involves a sensitization phase and an effector phase. The sensitization phase occurs during the initial exposure to an antigen when the host develops an antigen-specific IgE response, which results in sensitization of the host by the binding of the antigen-specific IgE to Fcε receptors on the surface of mast cells (Fig. 5-17). The host is now sensitized, and either through a second exposure or prolonged initial exposure to the IgE-specific antigen, there is cross-linking of two or more IgE molecules on the surface of the mast cell. This results in its activation and release of preformed and newly synthesized mediators, resulting in the effector phase. The effector phase can be limited to an acute inflammatory reaction (occurring within minutes), resulting primarily from the release of mast cell mediators, or can progress to a late-phase reaction (over a period of hours), or to a chronic reaction (persisting for days to years). The acute reaction is characterized by responses associated with release of preformed vasoactive amines from the mast cell and includes increased vascular permeability, smooth muscle contraction, and influx of inflammatory cells. The late-phase and chronic reactions, often associated with repeated or prolonged antigen exposures, are largely the result of a more intense inflammatory cell infiltration (primarily eosinophils, neutrophils, macrophages, and T lymphocytes) and tissue damage. Because the mast cell is central to the pathogenesis of a type I hypersensitivity reaction, their biologic features and primary functions are reviewed.
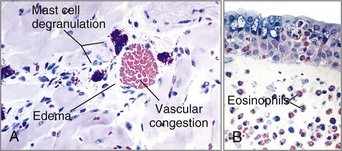
Fig. 5-15 Immediate hypersensitivity reaction.
A, Early reaction (minutes) is characterized by mast cell degranulation and release of preformed vasoactive substances that cause vasodilation and increased vascular permeability, resulting in edema of interstitial tissue. B, As the lesion progresses to the late phase (hours), the inflammatory infiltrate is primarily composed of eosinophils and fewer lymphocytes and neutrophils. (A and B courtesy Dr. Daniel Friend, Department of Pathology, Brigham and Women’s Hospital, Boston.)
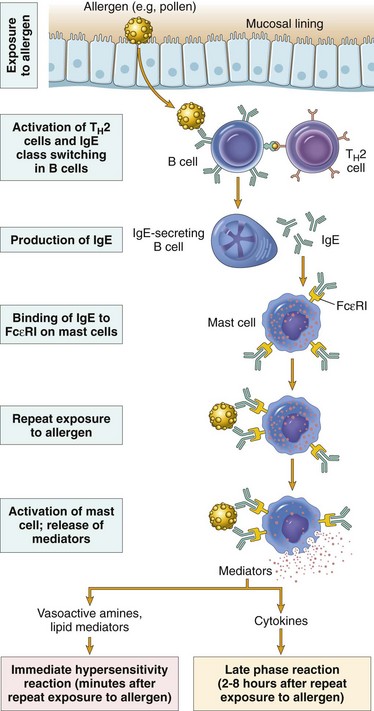
Fig. 5-16 Type I hypersensitivity reaction.
Pathogenesis of early and late responses of the type I hypersensitivity reaction. IgE, Immunoglobulin E. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
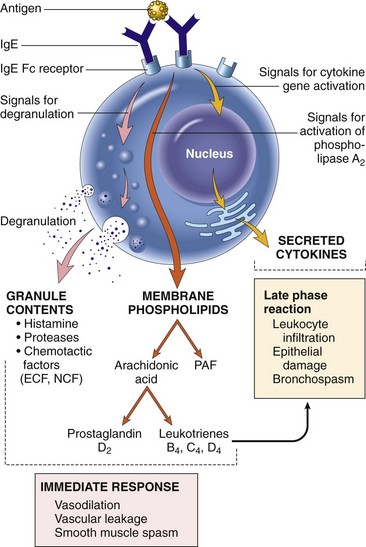
Fig. 5-17 Mast cell degranulation and activation.
Cross-linking of a sensitized (immunoglobulin E [IgE] bound to membrane Fcε receptors) mast cell by antigen results in mast cell activation and degranulation. Degranulation results in release of preformed mediators (histamine, proteases, and chemotactic substances). Activation results in synthesis of arachidonic acid from the plasma membrane and the production of prostaglandins and leukotrienes. ECF, Eosinophil chemotactic factor; NCF, neutrophil chemotactic factor; PAF, platelet-activating factor. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Mast cells are a heterogeneous population of bone marrow–derived cells that reside in vascularized tissue. Mast cells are easily identified by their abundant metachromatic cytoplasmic granules. Metachromasia is defined as the staining of a tissue component so that the color (absorption spectrum) of the tissue-dye complex differs from the color of the original dye and of the other stained tissue. In other words, the metachromatic substance is a different color from those of the dye and the other stained tissue. For example, toluidine blue is a metachromatic dye, and it stains most tissues blue, but mast cell granules are purple. Other commonly used metachromatic dyes include methylene blue and thionine. Wright’s and Giemsa stains are dye mixtures that include a metachromatic dye. Mast cells can be divided into mucosal and connective tissue subpopulations, not only based on their location but also on their phenotypic, morphologic, histochemical, and functional characteristics. This suggests that individual subpopulations of mast cells may have specific functions in normal and pathologic responses that are a result of their activation. The tyrosine kinase receptor, c-kit, expressed on mast cells, their precursors, and its ligand—stem cell factor (SCF)—is essential to mast cell development and function. Alterations in c-kit have been used to molecularly identify poorly differentiated mast cell tumors.
Mast cell activation can occur through a number of immunologic and nonimmunologic mechanisms. In addition to the activation of mast cells through cross-linking of membrane-bound IgE by antigen, other substances and stimuli can also activate mast cells. Mast cells can be activated by Fcε receptor–independent mechanisms, including cytokines (IL-8), complement products (the anaphylatoxins C3a and C5a), drugs (nonsteroidal antiinflammatory drugs, codeine, and morphine), and physical stimuli (heat, cold, and trauma). Non–IgE-mediated activation of mast cells is referred to as an anaphylactoid reaction, whereas the IgE-mediated activation is referred to as type I hypersensitivity. There are species and tissue differences in how type I reactions are manifested, and these are attributable to the types and proportions of mediators produced by the mast cell. Mast cells are a heterogeneous population of cells with regard to their structure and function. Although they are generally divided into mucosal-based and connective tissue–based populations, in either case they are primarily found adjacent to blood vessels and nerves where their mediators have their greatest influence. Mediators released by mast cells are broadly classified as preformed (primary) or newly synthesized (secondary), and as presented in Web Table 5-3 and Fig. 5-17, they influence local tissues and other cell types. Primary mediators are stored in cytoplasmic granules and include the vasoactive amines histamine, serotonin, and adenosine; chemotactic factors for eosinophils and neutrophils; enzymes, including neutral proteases and acid hydrolases; and proteoglycans, such as heparin and the chondroitin sulfates. Newly synthesized mediators consist largely of the lipid mediator products of cyclooxygenase and lipoxygenase metabolism of arachidonic acid (see Chapter 3), a number of cytokines and platelet-activating factor (PAF). The major products of arachidonic metabolism are the prostaglandins and leukotrienes, of which prostaglandin D2 and leukotrienes C4, D4, and E4 are most important. The major cytokines released from mast cells during a type I reaction include IL-4, IL-5, IL-6, and TNF-α. IL-4 and IL-5 contribute to B lymphocyte activation and IgE synthesis. IL-5 is chemotactic for eosinophils. IL-6 and TNF-α are involved in the pathogenesis of shock during a systemic type I (anaphylactic) reaction. The biochemical events involved in IgE-mediated activation and mediator release by mast cells are similar to those described for leukocyte activation in Chapter 3. The primary actions of preformed and newly synthesized mediators are attributable to cellular infiltration, vasoactive responses, and smooth muscle contraction. PAF, which was first identified as an initiator of platelet aggregation and degranulation, functions not only in the acute phase by increasing vasodilation and vascular permeability but is also important early in the late phase by recruiting and activating inflammatory cells. Finally, it is of note that recent studies have identified TLR pathways that mediate interactions between dendritic cells, T lymphocytes, and mast cells, thus modulating type I responses.
WEB TABLE 5-3
Summary of Mast Cell Mediators and Their Actions
PAF, Platelet-activating factor; TNF, tumor necrosis factor.
A type I reaction begins as an acute inflammatory reaction mediated largely by the vasoactive amines released by degranulation of mast cells. It is during this early stage that mast cells also release large quantities of chemotactic factors and cytokines. These mediators recruit and activate the inflammatory cells that will not only sustain the inflammatory response in the absence of antigen but also cause tissue damage. The immediate response is characterized by increased blood flow, increased vascular permeability (edema), and smooth muscle spasm. As the reaction progresses, additional leukocytes are recruited, and they release biologically active substances that cause cell damage. Of these leukocytes, eosinophils are particularly important.
Eosinophils are recruited to the sites of type I hypersensitivity reactions by chemokines, such as eotaxin, and their survival is influenced by IL-3, IL-5, and GM-CSF, which are largely derived from TH2 lymphocytes. Eosinophils recruited during the early response play an active role in the late-phase response by releasing components of their granules, synthesizing lipid mediators, and producing cytokines. The basic proteins released by eosinophils are toxic to parasites and host tissue. In particular, eosinophil major basic protein is toxic not only to parasites but also to tumor cells and normal cells. These proteins contribute to the epithelial cell damage associated with chronic type I reactions. Lipid mediators synthesized by activated eosinophils include PAF, leukotrienes, and lipoxins. Cytokines produced and released by eosinophils include growth factors, chemokines, cytokines involved in inflammation and repair, and regulatory cytokines. Macrophages and lymphocytes also participate in the late-phase response to varying degrees.
Epithelial cells further contribute to the inflammation by becoming activated and producing factors that recruit and activate additional inflammatory cells. It is this complex series of cell activation, recruitment, and mediator release that amplifies the immune response and sustains the inflammatory reaction long after the antigen has gone.
The factors that determine whether a host will develop a type I hypersensitivity reaction are complex. The genetic makeup of the host and the dose and route of antigen exposure are most important. These factors influence whether the individual will have a TH1 or TH2 response. The development of an IgE-secreting B lymphocyte from an immature (naïve) B lymphocyte depends on activated CD4+ lymphocytes of the TH2 type. The cytokines that define a TH2-lymphocyte response have important roles in regulating the cells involved in a type I hypersensitivity reaction. IL-3, IL-4, and IL-10 influence mast cell production; IL-4 is involved in isotype switching to IgE; and IL-3 and IL-5 influence eosinophil maturation and activation. IL-13 promotes the production of IgE. The major cytokine that defines a TH1 response, IFN-γ, inhibits the TH2 response. Thus an animal that develops predominantly a TH2 response to a particular antigen would be more likely to develop a type I hypersensitivity reaction as compared with one that develops predominantly a TH1 response. The CD4+ T lymphocyte plays a central role in the pathogenesis of a type I hypersensitivity. In humans, additional genetic influences can be linked to the human leukocyte antigen (HLA)–linked immune response genes. These genes appear to control allergen-specific IgE responses. As mentioned previously, the association of specific class I MHC molecules with an increased susceptibility to atopy in the dog has been proposed. As with the mast cell and the eosinophil, a role for the CD4+ T lymphocyte in the late-phase response has also been described. Studies suggest that the continued production of TH2 cytokines contributes to the chronic inflammation associated with some chronic type I hypersensitivity reactions.
In summary, type I hypersensitivity is a complex disease process that occurs in sensitized hosts, which can result in three types of responses: (1) an acute inflammatory response, (2) a late-phase response, and (3) a chronic inflammatory response. In sensitized hosts, the cross-linking of IgE on the surface of mast cells results in the immediate release of mediators that influence local tissue and recruit additional inflammatory cells. The acute response is dependent on resident mast cells, whereas the late-phase and chronic responses are dependent on recruited cells, especially the eosinophil. Central to the pathogenesis of a type I hypersensitivity reaction are the TH2 lymphocytes and the cytokines they produce, which influence IgE production and the recruitment and activation of leukocytes.
Systemic and localized type I hypersensitivity reactions occur in animals. The pathogenesis of many infectious and noninfectious diseases involves the production of IgE and the development of a type I hypersensitivity reaction. Type I hypersensitivity is an allergic reaction that occurs within minutes of exposure to an antigen to which the host has been previously sensitized. Allergy has become synonymous with type I hypersensitivity. By definition, type I hypersensitivity reactions are mediated by IgE. Systemic type I hypersensitivity reactions are called anaphylaxis. Atopy is the genetic predisposition to develop localized type I hypersensitivity reactions to innocuous antigens. Atopy is often limited to an organ or tissue such as in allergic dermatitis and rhinitis, food allergies, and asthma. Non–IgE-mediated allergic-like reactions are referred to as anaphylactoid reactions.
Systemic Type I Hypersensitivity (Anaphylaxis): Anaphylaxis refers to an acute systemic hypersensitivity reaction to an antigen that is mediated by IgE and involves mast cell activation, resulting in a shocklike state often involving multiple organ systems. The clinical signs and pathology attributable to a systemic anaphylactic reaction vary by species and often correlate to the primary shock organ in its most severe manifestation—death. This variation reflects differences in the distribution of the mast cells, the mediator content of their granules that are unique to individual species, and the primary target tissue. The primary target tissues are blood vessels and smooth muscle. Blood vessel beds and smooth muscles vary in their histamine receptor content, and as such some are more susceptible than others to the influences of histamine. Because of the aforementioned, the early signs of anaphylaxis can be varied. Cutaneous signs include pruritus, hyperemia, and angioedema. Cardiovascular signs include hypotension and an accompanying sinus tachycardia (characteristic of a vasovagal response). Respiratory signs include bronchospasms, laryngeal edema, and dyspnea. As the anaphylactic reaction progresses, hypotension or hypoxia may lead to unconsciousness. Fatal anaphylaxis may occur as the result of asphyxiation secondary to edema of the upper airway, circulatory failure as a result of dilation of the splanchnic vascular bed, or hypoxemia as a result of severe bronchospasms. In humans, a body of evidence also implicates human heart mast cells (HHMCs) in myocardial anaphylaxis as a primary mechanism. Other than in cases with upper airway edema or pulmonary hyperinflation (emphysema), there are no pathognomonic lesions of anaphylaxis. The species most sensitive to the development of anaphylaxis is the guinea pig. The most common pathologic findings in most species are pulmonary edema and emphysema, except for dogs, for which the major shock organ is the liver, and severe hepatic congestion and visceral hemorrhage are the most common findings.
The types of antigens that can elicit a systemic anaphylactic reaction are diverse, but most commonly include drugs (especially penicillin-based antibiotics), vaccines, venom of stinging insects, and heterologous sera. Although the greatest risk for the development of an anaphylactic reaction occurs during parenteral administration, it must be noted that in some cases even a small quantity of antigen in a highly sensitized host can elicit a systemic response.
Localized Type I Hypersensitivity: In a localized type I hypersensitivity reaction, the clinical signs and pathologic findings are restricted to a specific tissue or organ. Localized reactions most commonly occur at epithelial surfaces such as the surfaces of the skin and mucosa of the respiratory and gastrointestinal tract. As discussed previously, species differences on the location of mast cells, the mediators contained within them, and the histamine receptor distribution on target tissue may explain the different spectra of diseases seen among individual species.
Allergic dermatitis is a cutaneous manifestation of a type I hypersensitivity reaction that results in inflammation of the skin. The route of exposure to the antigen may be by inhalation, ingestion, or percutaneous absorption. If the allergic dermatitis is thought to have a genetic predisposition, then the disease is referred to as atopic dermatitis. Dietary type I hypersensitivity reactions in the dog and cat more commonly present as a cutaneous disease rather than a gastrointestinal disease. Other common cutaneous manifestations of type I hypersensitivity are flea and other arthropod bites and urticaria and angioedema (hives). All of these diseases are characterized by an acute inflammatory reaction, often perivascular, caused by mediators released from sensitized mast cells. In some instances, as in atopic dermatitis, the lesion may progress to a late-phase response or chronic inflammation characterized by more intense inflammatory infiltrates (e.g., atopic dermatitis) or to a type IV hypersensitivity reaction (arthropod bites). Other secondary changes, such as acanthosis, hyperpigmentation, sebaceous gland metaplasia, and pyoderma, occur in long-standing cases or in animals that have significant trauma related to pruritus.
Allergic rhinitis is a respiratory manifestation of a type I hypersensitivity reaction that most commonly develops in ruminants. The most common antigens are grass and weed pollens and mold spores (Micropolyspora faeni). This disease also frequently progresses from an acute inflammatory disease to a late-phase response and chronic inflammation. In cattle, long-standing allergic rhinitis may progress to a type IV hypersensitivity reaction with the formation of nasal granulomas. Mold spores (Micropolyspora faeni) are more frequently associated with a type III hypersensitivity reaction, resulting in an allergic pneumonitis (extrinsic allergic alveolitis).
Although an inherited predisposition has been implicated in some species, the exact mode of inheritance remains to be determined. In humans, a link to genes encoding IL-4 and certain MHC antigens, important components of allergic diseases, has been made.
Type II Hypersensitivity (Cytotoxic Hypersensitivity)
In the original Gell and Coombs classification, the type II hypersensitivity reaction was designated as antibody-mediated cytotoxic hypersensitivity. This type of hypersensitivity most often occurs as the result of the development of antibodies directed against antigens on the surface of a cell or in a tissue, with the result that the cell or tissue is destroyed. Antigens may be either endogenous (normal cellular or tissue protein) or exogenous (e.g., a drug or microbial protein adsorbed to the cell). In some instances, the antigen may be a cell surface receptor and the antibody may activate or block the activation of the cell rather than cause cytotoxicity. The pathogenesis of many immune-mediated and autoimmune diseases is centered on the development of antireceptor or antisurface antigen antibodies and a type II hypersensitivity reaction. The largest group of “cytotoxic” hypersensitivity reactions involves the hematologic diseases, with antibodies directed against antigens present on the surface of red blood cells and platelets. Type II hypersensitivity reactions are mediated by antibodies directed against antigens on the surface of tissue or cells so that the tissue or cell is destroyed or the function of the cell is altered. Type II hypersensitivity reactions most frequently involve IgM and IgG and occur within hours after exposure in a sensitized host.
There are three basic antibody-mediated mechanisms that result in type II hypersensitivity (Fig. 5-18). Complement-dependent reactions occur as a result of the complement activating capability of IgG and IgM. Complement activation can mediate cytotoxicity by either the formation of the membrane attack complex, resulting in cell lysis, or the fixation of C3b fragments (opsonization) to the surface facilitating phagocytosis (see Chapter 3). Antibody-dependent reactions can similarly opsonize cells, facilitating phagocytosis, or result in cell lysis by antibody-dependent cellular cytotoxicity. Opsonization of cells by antibody makes them susceptible to destruction by macrophages, neutrophils, NK cells, and eosinophils, all of which bear FcR. This is commonly referred to as antibody-dependent cellular cytotoxicity (ADCC). Finally, antibodies directed against surface receptors may result in altered cell or tissue function. The antireceptor antibodies can function as agonists, stimulating cell function, or as antagonists, blocking receptor function.
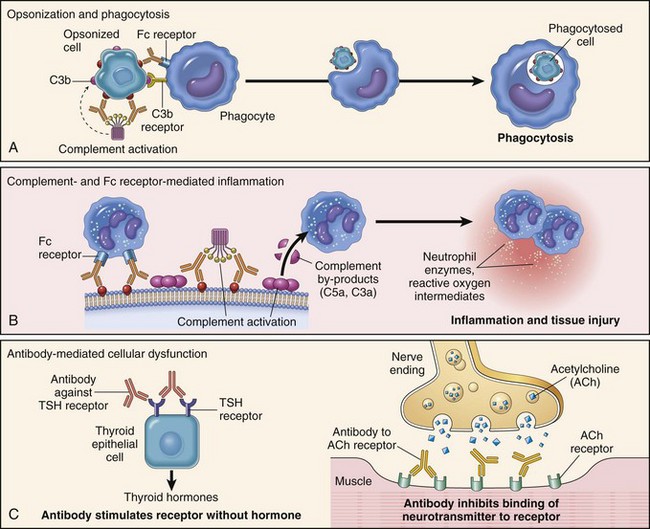
Fig. 5-18 Schematic depiction of the three major mechanisms of an antibody mediated injury.
A, Opsonization by antibodies (or complement) enhances phagocytosis of antigen by monocyte-macrophage cells. B, Antibody can activate the complement system, via the classical pathway, resulting in the elaboration of inflammatory mediators, such as C5a and C3a. C, Antibodies against cell receptors can activate (depicted) or inhibit (not depicted) cell functions. TSH, Thyroid-stimulating hormone. (A, B, and C from Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Diseases with a type II hypersensitivity pathogenesis are presented in Table 5-5. The physical and biochemical properties of red blood cells, platelets, and leukocytes make them susceptible to cytotoxic reactions. Two properties of red blood cells make them uniquely susceptible to being involved in type II reactions. First, their surface contains a complex array of blood group antigens that can become targets of antibody responses as is commonly the case in transfusion reactions or immune-mediated hemolytic disease of the newborn. Second, the biochemical properties of red blood cells make them prone to adsorb substances such as drugs or antigenic components of infectious agents or tumors. In these instances, the red blood cell may be either directly targeted because the substance alters a surface protein to an extent that it is now recognized as foreign, or indirectly targeted if there is an antibody response to the substance itself. Finally, in autoimmune forms of hemolytic anemia, agranulocytosis, and thrombocytopenia, there is a breakdown of tolerance and the subsequent development of antibodies to normal cells, and as a result they are destroyed.
TABLE 5-5
Diseases with a Primary Cytotoxic Hypersensitivity (Type II Hypersensitivity) Pathogenesis
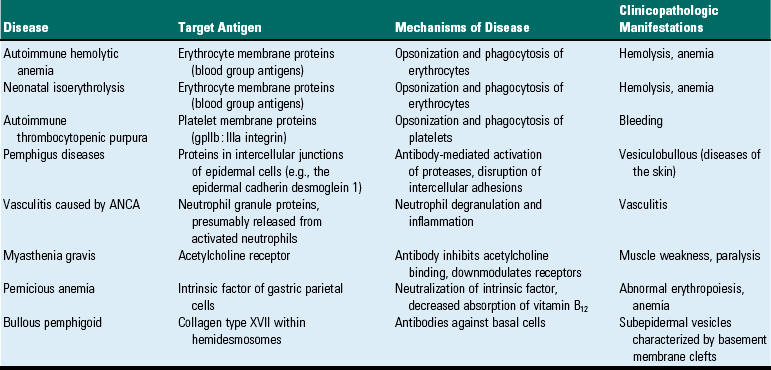
The majority of cytotoxic type II diseases result in a decrease or loss of a population of cells (e.g., anemia, thrombocytopenia). Noncytotoxic type II diseases are initially characterized by activation or inhibition of cell or tissue function followed by inflammation, which may cause inflammatory damage to the targeted organ. In a type II reaction, the pathogenesis commonly begins with cell surface antigens eliciting an antibody response, whereby the antibodies bind to the cell and the cell is either lysed or complement components attract phagocytic cells that damage tissues by releasing proteolytic enzymes.
Type III Hypersensitivity (Immune Complex Hypersensitivity)
Type III hypersensitivity is designated as immune complex hypersensitivity. This reaction occurs through the formation of antigen-antibody complexes that activate complement and result in tissue damage (Fig. 5-19). The cell or tissue injury is similar to a type II hypersensitivity reaction, although the underlying pathogenesis is different. With a type III reaction, the cell or tissue is being destroyed not because the antibody is being directed against that cell or tissue, but rather because immune complexes either become “stuck” to that cell or are deposited in that tissue. Think of it as an “innocent bystander” reaction: The targeted tissue is not a direct target of the immune response. The pathogenesis begins with the formation of immune complexes that become lodged or are formed in or deposited in tissue and are capable of activating the complement system. Products of complement activation such as anaphylatoxins and chemotactic factors result in neutrophil infiltration and activation. On activation, neutrophils release their enzymes and these result in tissue damage. Like type II hypersensitivity reactions, type III hypersensitivity reactions most frequently involve IgM and IgG and occur within hours after exposure in a sensitized host.
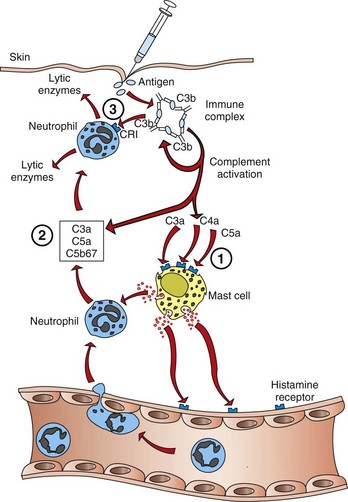
Fig. 5-19 A localized type III hypersensitivity reaction (Arthus reaction) in the dermis.
Antigen-antibody complexes, formed at the site of injection, activate the complement system to elaborate components that activate resident mast cells (1) and attract circulating neutrophils (2). Inflammation is the result of tissue damage caused by mediators and enzymes released from both cell types (3). CRI, Complement receptor 1. (Adapted from Goldsby RA, Kindt TJ, Osborne BA: Kuby immunology, ed 4, New York, 2000, WH Freeman.)
Antigen-antibody complexes form as a part of a normal immune response and usually facilitate the clearance of antigen by the phagocytic system without resulting in a type III hypersensitivity reaction. Although a number of factors determine whether a type III reaction will occur, the most important is the relationship of the antibody response to the quantity of antigen. When antibody is in great excess of antigen, the antigen-antibody complexes formed are large and insoluble, and easily removed by the phagocytic system. When antigen is in great excess of the quantity of antibody, the antigen-antibody complexes formed are too small to be capable of becoming lodged in tissues or of activating the complement system. However, when antigen is in slight excess of antibody, these small soluble complexes can become lodged in tissue and activate the complement system. When this type of small soluble antigen-antibody complex is formed in the circulation, their accumulation in tissue is essentially the result of anatomic and physiologic processes and has no immunologic basis. Finally, it has also been suggested that in some instances immune complex hypersensitivity may be the result of the normal phagocytic system being overwhelmed. Immune complex deposition can be localized to a tissue or generalized if the complexes are formed in circulation. Blood vessels, synovial membranes, glomeruli, and the choroid plexus are particularly vulnerable to deposition of immune complexes. The concentration and size of the complexes determine the sites of deposition.
Type III reactions can develop from antibody responses to endogenous or exogenous antigens and immune complexes can be deposited in a number of tissues (Table 5-6). Although a number of diseases of domestic species involve a type III hypersensitivity pathogenesis, a majority of diseases are the result of persistent infections, autoimmune disease, or inhalation of foreign antigen. Organisms that result in persistent infections are often characterized by a weak antibody response and the development of immune complex formation. A number of autoimmune and immune-mediated diseases result in the development of antibody responses to self-antigens or antigens complexed to self-proteins, and these are capable of generating complement-activating immune complexes. Immune complexes formed against commonly inhaled environmental antigens can lead to the development of an allergic alveolitis. Type III hypersensitivity reactions are mediated by the formation of antigen-antibody complexes, which results in complement activation leading to an influx of neutrophils and subsequent cell or tissue destruction. Antigen-antibody complexes may be formed in the circulation and lodge in tissue or may be formed in the tissue directly. The cell or tissue injury is largely determined by physiologic or anatomic properties rather than an immunologic basis. The pathogenesis of a number of diseases of domestic animals have a type III hypersensitivity basis.
TABLE 5-6
Diseases with a Primary Type III Hypersensitivity (Immune Complex Hypersensitivity) Pathogenesis
| Disease | Antigen Involved | Clinicopathologic Manifestations |
| Systemic lupus erythematosus | DNA, nucleoproteins, others | Glomerulonephritis, arthritis, vasculitis |
| Blue eye | Canine adenovirus 1 antigen | Anterior uveitis |
| Equine infectious anemia | Viral antigens | Anemia, thrombocytopenia |
| Poststaphylococcal hypersensitivity | Staphylococcal cell wall antigens | Dermatitis |
| Cutaneous vasculitis | Bacterial antigens, viral antigens, drugs | Vasculitis |
| Poststreptococcal (Streptococcus equi ssp. equi) hypersensitivity | M protein | Purpura hemorrhagica, glomerulonephritis |
| Acute glomerulonephritis | Bacterial antigens; parasite antigens; viral antigens; tumor antigens | Nephritis |
| Reactive arthritis | Bacterial antigens | Acute arthritis |
| Arthus reaction | Various foreign proteins | Cutaneous vasculitis |
| Serum sickness | Various proteins (e.g., foreign serum) | Arthritis, vasculitis, nephritis |
| Hypersensitivity pneumonitis | Fungal spores, dust | Alveolitis, vasculitis |
| COPD | Fungal spores, dust | Bronchiolitis |
| Aleutian mink disease | Viral antigens | Glomerulonephritis, vasculitis |
| Rheumatoid arthritis | IgG | Erosive polyarthritis |
COPD, Chronic obstructive pulmonary disease; IgG, immunoglobulin G.
Localized Type III Hypersensitivity: Localized type III hypersensitivity reactions are best exemplified by the Arthus reaction (see Fig. 5-19). The parenteral administration of an antigen to an animal that has a circulating antibody specific for that antigen results in a localized acute inflammatory response. The complexes are formed either within the tissue at the site of antigen deposition or localized within blood vessels, as the antigen and antibody diffuse into the vascular wall. Early, within hours, the reaction is characterized by margination and emigration of neutrophils to and from the blood vessels and progressively results in tissue and vascular damage. The quantity of antigen-antibody complexes formed in the wall of the vessel determines the extent of the tissue damage. Small quantities of complexes may result in only mild hyperemia and edema. Large quantities of complexes may result in tissue and vascular necrosis as a result of neutrophils releasing the contents of their granules. In some cases, the damage to the wall may be so severe as to cause thrombosis and localized ischemic injury. The Arthus reaction is still used today as an experimental model of a localized type III reaction. Recent studies, using the cutaneous Arthus reaction in complement-deficient mice, document the requirement of FcR activation for eliciting an inflammatory response and a revision of the hypothesis of the mechanism of immune complex-mediated inflammation. Complement components, such as C5a, are generated as a result of FcR activation. Conversely, the use of FcR-deficient mice and the Arthus reaction establish the requirement of this receptor because immune complexes and C3 alone are not sufficient to trigger an inflammatory response and tissue damage.
Many diseases have a progressive clinical course, and immune complex reactions often play a role, even though they may not be involved in the initial immunologic response. There are limited clinical examples of diseases characterized primarily by a localized immune complex reaction. One dramatic example is blue eye in the dog, which is an anterior uveitis that develops in a small percentage of dogs naturally infected with or vaccinated against canine adenovirus type I. Other organs commonly affected by localized immune complex disease include the lung and skin. In the lung, chronic exposure of the lower airways to inhaled antigens can lead to the development of antigen-specific antibodies that form complexes within alveolar walls. This form of allergic lung disease is commonly referred to as allergic pneumonitis (extrinsic allergic alveolitis). Common antigens include spore-forming organisms (e.g., some actinomycetes and fungi). Allergic diseases of the lower airways frequently lead to type II pneumocyte hyperplasia, emphysema, and fibrosis, which are all secondary to inflammation and tissue damage mediated by type III hypersensitivity. Chronic obstructive pulmonary disease (COPD) in horses may be caused in part by a localized type III reaction to spore-forming organisms or dust that results in bronchiolitis (see Chapter 9). In dogs, staphylococcal infections of the skin may develop a type I, III, or IV reaction. In the case of a type III reaction, a neutrophilic dermal vasculitis is often evident (see Chapter 17).
Generalized Type III Hypersensitivity: When antigen is present in the circulation at appropriate concentrations relative to circulating antibody concentrations (as discussed previously), the result is the formation of immune complexes capable of generating a type III hypersensitivity reaction. Serum sickness is the prototypical disease with a type III hypersensitivity pathogenesis. Early examples of this disease were the result of the administration of heterologous serum, which led to the formation of circulating immune complexes that became lodged primarily in blood vessels, glomeruli, and joints. The blood vessel, glomerulus, or joint was not a target of the immune response but rather an “innocent bystander” because the resulting inflammation occurred as a result of the complement activating capacity of the immune complexes that lodged there.
The pathogenesis of a systemic immune complex disease is best illustrated in three phases as depicted in Fig. 5-20. The first phase, as discussed previously, occurs when the host develops an antibody response to an antigen so that the ratio of antigen to antibody is appropriate for the formation of small, soluble circulating complexes that are not adequately cleared by the monocyte-macrophage system. Because the formation of antigen-antibody complexes can be a normal component of an immune response, the presence of immune complexes in circulation by itself is not sufficient to diagnose an immune complex disease. In the second phase, the complexes adhere to cells or lodge in tissues that are uniquely susceptible to circulating immune complexes. The biochemical properties of the antigen-antibody complexes (e.g., overall quantity and size, charge) and the physiologic and anatomic characteristics of some cells and tissues account for their unique susceptibility to immune complex deposition. Other factors may also contribute to the formation or deposition of immune complexes in certain tissues. For example, in rheumatoid arthritis, it has been proposed that lymphocytes within the joint may produce an altered IgG molecule that stimulates the production of rheumatoid factor (anti-IgG). Complexes become lodged within blood vessel walls and extravascular tissues as a result of the increased vascular permeability caused by the anaphylatoxins and vasoactive amines released from neutrophils, activated through the binding of antigen-antibody complexes to complement and FcR on their surface. The result is phase three: the activation of the complement system and the development of an acute inflammatory reaction centered on the vasculature. Neutrophils and macrophages are activated similarly through FcR and produce a number of inflammatory cytokines that attract and activate additional inflammatory cells. The inflammatory cells and mediators have been thoroughly discussed in Chapter 3. Immune complexes that lodge in blood vessels, glomeruli, or joints result in vasculitis, glomerulonephritis, and arthritis, respectively. The damage to the vessels also results in damage to the intima and exposure of collagen, which initiates the formation of microthrombi by the activation of the coagulation cascade and platelets.
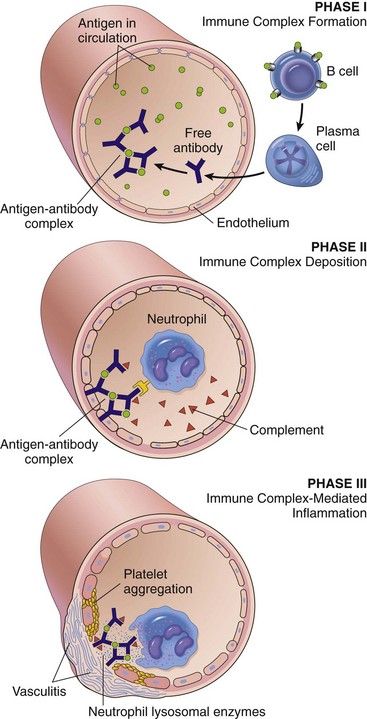
Fig. 5-20 Schematic depiction of the three phases of a systemic type III hypersensitivity reaction.
The first phase results in immune complex formation. In the second phase, the antigen-antibody complexes become lodged in the vessel wall and activate inflammatory cells. The end result, the third phase, is the elicitation of tissue damage and an inflammatory response. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
The two primary cell types involved in a type III hypersensitivity reaction are FcR–bearing neutrophils and macrophages (Fig. 5-21). Complement activation leads to the elaboration of factors (primarily C5a) that are chemotactic and attract neutrophils and macrophages to the site. These cells are activated and produce a number of proinflammatory cytokines. Early in the response, these cells release vasoactive amines that cause increased vascular permeability, allowing the immune complexes to lodge within the vessel wall. Many of these phagocytic cells are also stimulated to release their proteolytic enzymes and toxic free radicals, and these processes result in tissue and vascular damage. Platelets also contribute to the developing inflammatory reaction by releasing vasoactive amines and other proinflammatory constituents.
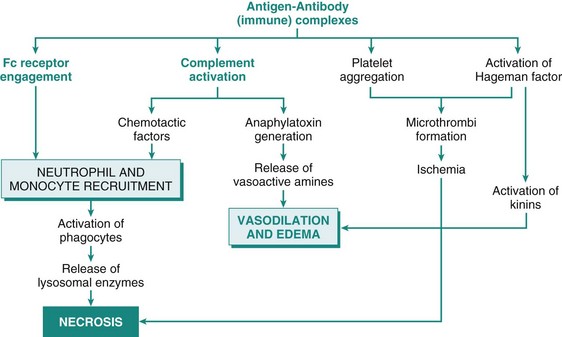
Fig. 5-21 Pathogenesis of type III hypersensitivity reactions and the morphologic consequences.
Locally or systemically deposited immune complexes result in tissue damage and inflammation by activation of the complement system and through activation of neutrophils and macrophages through their Fc receptors. Activation of a component of the coagulation system also contributes to the tissue damage. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
Diseases associated with type III hypersensitivity reactions are most commonly associated with a single exposure to a large quantity of antigen (e.g., administration of heterologous serum or from an immune response to systemic infections) or from continuous exposures to small quantities of antigen as in the case of autoimmune diseases (e.g., rheumatoid arthritis and systemic lupus erythematosus). In either of these instances, the development of type III hypersensitivity depends on antigen in excess of antibody.
Type IV Hypersensitivity (Delayed-Type Hypersensitivity)
Type IV hypersensitivity is also known as cell-mediated hypersensitivity because it is the result of the interaction of T lymphocytes and the specific antigen to which they have been sensitized. The resulting immune response is mediated either by direct cytotoxicity by CD8+ T lymphocytes or by the release of soluble cytokines from CD4+ lymphocytes, which act through mediator cells (primarily macrophages) to produce chronic inflammatory reactions (Fig. 5-22). Because these responses are dependent on sensitized T lymphocytes and require 24 to 48 hours to develop, they are also referred to as delayed-type hypersensitivity (DTH). Unlike type I, II, and III hypersensitivity reactions, type IV hypersensitivity is not dependent on an antibody. We first discuss the response mediated primarily by activated CD4+ lymphocytes. The prototypical DTH reaction is the localized tuberculin response. After an intradermal exposure of tuberculin, a purified protein derivative (PPD) of the tubercle bacillus, a previously sensitized host will develop a localized type IV reaction at the site of inoculation at 24 to 72 hours. The intradermal antigens are taken up and processed by dendritic Langerhans’ cells, which present antigenic peptides to antigen-specific CD4+ lymphocytes that are activated to produce and secrete cytokines that attract and activate other inflammatory cells. Grossly the site appears as a swollen, firm nodule. Microscopically the nodule is composed of interstitial edema and a mononuclear infiltrate that is primarily centered around blood vessels. Early (<12 hours), the infiltrate is primarily neutrophilic, which is replaced largely by macrophages and lymphocytes (>12 hours). The DTH response is generally minimal and short lived because the concentration of PPD injected is small and rapidly degraded. A similar DTH reaction can be used to test for previous exposures to a number of intracellular organisms.
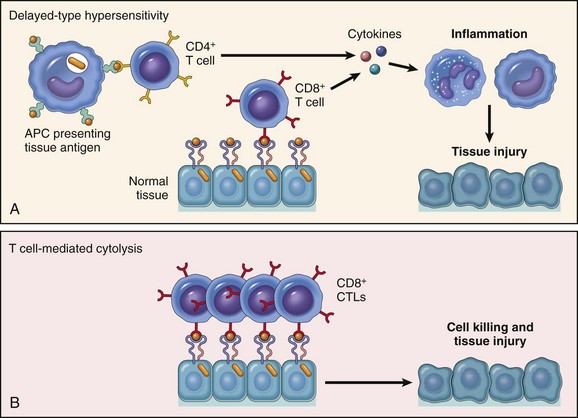
Fig. 5-22 The two primary mechanisms of T lymphocytes that cause type IV hypersensitivity reaction.
A, CD4+ T lymphocytes (and occasionally CD8+ lymphocytes) are activated by antigen and produce cytokines that attract other cell types and promote an inflammatory response. B, CD8+ T lymphocytes (cytotoxic T lymphocytes [CTLs]) are also activated by antigen and can cause inflammation by killing targeted cells and tissue. APC, Antigen-presenting cell. (A and B from Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
In addition to the tuberculin response, type IV hypersensitivity is the underlying pathogenesis for allergic contact hypersensitivity and granulomatous inflammatory responses. As mentioned with the other hypersensitivity reactions, the components of a type IV hypersensitivity reaction can be considered beneficial (protective immunity) when they occur as an appropriate response to intracellular organisms, or they can be considered harmful (hypersensitivity), for example, when they occur as an inappropriate response to exogenous chemicals or substances that are complexed with proteins, as in the case of allergic contact hypersensitivity.
In the tuberculin reaction, the quantity of antigen limits the extent of the inflammatory response, and resolution of the inflammation generally occurs in 5 to 7 days. This is in contrast to chronic infections with persistent intracellular organisms or poorly degradable intracellular antigens (Table 5-7) that develop into a specific type of chronic inflammatory response called granulomatous inflammation. DTH reactions frequently occur in response to intracellular organisms and cause extensive tissue damage. These diseases are characterized by granulomatous inflammation. In this type of response, the host is unable to destroy or eliminate the organism, resulting in antigen persistence. Compared with the tuberculin reaction, the type of inflammatory infiltrate is different. As discussed in Chapter 3, granulomatous inflammation designates that the inflammatory infiltrate has specific attributes, notably the presence of morphologically transformed macrophages into epithelial-like cells commonly called epithelioid macrophages (Figs. 5-23 and 5-24). Concurrently, there may be many multinucleated giant cells that represent fused macrophages. A number of fusion-related monocyte-macrophage surface proteins have been identified and include receptors for mannose and β1 integrin, Src homology 2 domain-containing protein tyrosine phosphatase substrate 1 (SHPS-1), and the chemoattractant chemokine ligand 2. Lymphocytes can also represent a significant component of the inflammatory infiltrate. Generally, CD4+ lymphocytes are interspersed with the macrophages, and CD8+ lymphocytes are localized to the periphery. As these lesions progress, they may become organized into nodules commonly called granulomas (see Fig. 5-23, A). Depending on the inciting antigen, there may also be varying proportions of necrosis (often as a necrotic center), calcification of the necrotic tissue, and peripheral fibrous encapsulation. These features are largely the result of lytic enzymes released from activated macrophages. Nonimmunologic granulomas can occur in cases of foreign-body type granulomas, which typically have fewer lymphocytes. In either case, the body is trying to limit the spread or wall-off the inciting antigen.
TABLE 5-7
Diseases with a Primary Type IV Hypersensitivity (Delayed-type Hypersensitivity) Pathogenesis
| Disease | Specificity of Pathogenic T Lymphocytes | Clinicopathologic Manifestations |
| Tuberculosis | Mycobacteria spp. antigens | Granuloma formation |
| Allergic contact dermatitis | Haptens | Perivascular dermatitis |
| Rheumatoid arthritis | Unknown antigen in joint synovium (type II collagen?); role of antibodies and type III hypersensitivity? | Chronic arthritis with inflammation, destruction of articular cartilage and bone |
| Johne’s disease | Mycobacterium paratuberculosis antigens | Granulomatous enteritis |
| Allograft rejection | MHC molecules | Inflammation of graft tissue |
| Equine recurrent uveitis | Unknown | Uveitis |
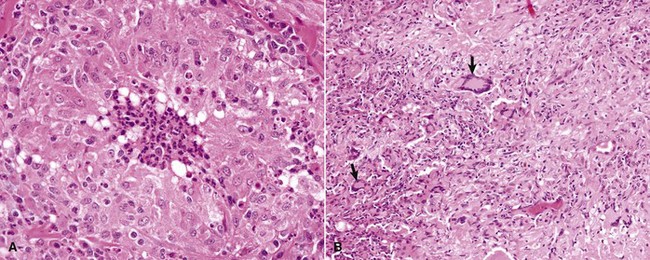
Fig. 5-23 Granulomatous inflammation associated with chronic infections.
A, Blastomycosis, skin, dog. Note the granuloma composed of sheets of epithelioid macrophages and the central focus of neutrophils. H&E stain. B, Mycobacteriosis, lung, gazelle. Numerous epithelioid macrophages and multinucleated Langhans’-type giant cells (arrows) constitute the granulomatous tissue that has replaced normal lung parenchyma. H&E stain. (A and B courtesy Dr. P.W. Snyder, School of Veterinary Medicine, Purdue University.)
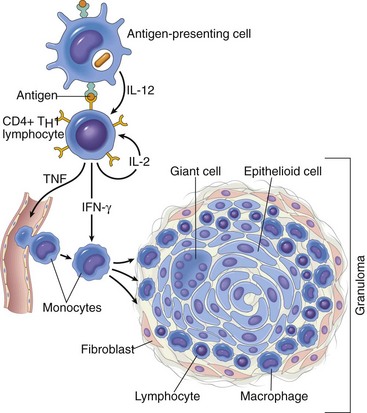
Fig. 5-24 Schematic depiction of granuloma formation in a type IV hypersensitivity reaction.
A TH1 lymphocyte synthesizes cytokines in response to interleukin-12 production and antigen presentation by an antigen-presenting cell. The cytokines activate additional TH1 lymphocytes (IL-2) and monocyte-macrophage cells (IFN-γ), and promote localized and systemic inflammatory responses (TNF-α). As the response develops, macrophages fuse to form multinucleated giant cells, and fibroblasts are stimulated to synthesize collagen, resulting in the formation of a granuloma. IL, Interleukin; IFN-γ, interferon; TNF-α, tumor necrosis factor-α. (From Kumar V, Abbas AK, Fausto N: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders.)
The type IV hypersensitivity reaction is immunologically specific and like all the hypersensitivity reactions involves a sensitization phase and an effector phase. The sensitization phase occurs with the initial exposure to the antigen and results in the development of antigen-specific memory T lymphocytes. These CD4+ lymphocytes recognize peptides presented in the context of class II molecules on the surface of antigen-presenting cells. In this context, the naïve CD4+ T lymphocytes develop into functional TH1 lymphocytes. These activated TH1 lymphocytes are sometimes designated as TDTH lymphocytes. Once the host is sensitized, a prolonged exposure or repeat exposure to the antigen results in the development of an effector phase. The effector phase can occur as a cytotoxic response mediated by CD8+ lymphocytes or more commonly as a TH1 response through the elaboration of cytokines by CD4+ lymphocytes (see Fig. 5-24). TH1 cytokines (most importantly, IL-2, IL-3, IFN-γ, and TNF-β) and chemokines (IL-8, macrophage chemotactic and activating factor, and macrophage-inhibition factor) enhance the function of cytokine-producing T lymphocytes (autocrine and paracrine fashion) and attract and activate macrophages. IL-2 induces the proliferation and long-term survival of T lymphocytes. IL-3 supports the growth and differentiation of TH1 lymphocytes and NK cells. IFN-γ, the key mediator of type IV hypersensitivity, activates macrophages not only to enhance their phagocytic and killing mechanisms but also to enhance their ability to present antigen by inducing increased expression of class II MHC molecules. Activated macrophages and dendritic cells produce IL-12, which also facilitates the development of TH1 lymphocytes. Activated macrophages also produce IL-1 and TNF-α, both of which act locally to increase the expression of adhesion molecules on endothelial cells, which further facilitates the extravasation of additional inflammatory cells. The production of cytokines and chemokines by the CD4+ TH1 lymphocytes influences macrophage function and mediates the production of cytokines that influence CD4+ lymphocytes, resulting in a response that potentially goes from a beneficial protective response (immunity) to a harmful response that results in tissue damage (hypersensitivity).
The beneficial protective response of T lymphocyte–mediated hypersensitivity is not limited to intracellular organisms. It also can be a primary component of transplant rejection and immunity to cancer. There are other harmful T lymphocyte–mediated responses that result in disease. One example is allergic contact hypersensitivity. In allergic contact hypersensitivity, the antigen is often too small to elicit an immune response by itself. These antigens must be complexed with other, larger proteins to become antigenic and are specifically referred to as haptens or generally called contact antigens (Box 5-1). Allergic contact hypersensitivity also depends on processing and presentation of the antigen by dendritic Langerhans’ cells to CD4+ lymphocytes in regional lymph nodes. In the case of allergic contact dermatitis, the keratinocyte may also participate by producing a number of cytokines that activate Langerhans’ cells, mast cells, and other inflammatory cells. In the sensitization phase, the protein-hapten complex is taken up and processed by Langerhans’ cells that migrate to regional lymph nodes. In the paracortex region of the lymph node (T lymphocyte area), they present antigenic components to CD4+ lymphocytes. The host develops a population of memory lymphocytes and is now sensitized to the antigen. In a sensitized host, continuous exposure to the antigen, or more commonly, repeat exposure to the antigen, results in an effector phase response seen as epidermal vesicle formation with dermal and epidermal infiltrates of mononuclear inflammatory cells. The result is tissue damage that is disproportionate to any beneficial effects of the immune response.
Finally, as mentioned earlier, another form of DTH can occur that is mediated by direct cytotoxicity by CD8+ T lymphocytes. This response is most commonly associated with viral infections. CD8+ T lymphocytes, bearing viral antigen-specific TCRs, kill antigen-expressing target cells. These cells are commonly referred to as CTL (Tc). The expression of viral proteins on the surface of an infected cell in association of class I MHC molecules serves as the recognition signal for the TCR-CD3 membrane complex. Following recognition of antigen by the CTL (Tc), there is upregulation of adhesion molecules on the CTL (Tc) and the target cell, resulting in a CTL (Tc)–target cell conjugate. This stimulates an activating signal pathway that results in death of the target cell by apoptosis. The two principal mechanisms of CTL (Tc)-mediated apoptosis are (1) the directional delivery of cytotoxic proteins and (2) the interaction of membrane-bound Fas ligand on the CTL (Tc), with the Fas receptor on the target cell. Both depend on the activation of caspases. Perforins and granzymes are preformed cytotoxic proteins contained in the cytoplasmic granules of CTL (Tc). Perforin, released between the conjugated CTL (Tc) and the target cell, is polymerized in the presence of Ca2+ and forms pores in the plasma membrane of the target cell, not only causing lysis but also permitting the delivery of granzymes. Granzymes activate caspases, normally present in an inactive proenzyme form, that ultimately result in apoptotic death of the cell. The cross-linking of Fas by its ligand, membrane-bound Fas ligand, results in the activation of the extrinsic (death-receptor–initiated) pathway of apoptosis, which is covered in greater detail in Chapter 1.
Cytokine-Related Diseases
A number of diseases are characterized by severe disruptions, either overproduction or underproduction, of cytokines or cytokine receptors. One of the most profound examples is the excessive elaboration of cytokines during bacterial septicemia and shock. The basic pathogenesis involves an infection with a Gram-negative, endotoxin-producing bacterium that stimulates macrophages to overproduce IL-1 and TNF-α. High concentrations of these two cytokines in circulation cause septic shock (see Chapters 2 and 3). A number of microbial pathogens also produce toxins or other antigenic molecules that are referred to as superantigens. Superantigens bind to class II MHC molecules and Vβ domains of the TCR. This binding is outside the normal antigen binding site and activates all T lymphocytes expressing the same Vβ domains, irrespective of their antigen specificity (Fig. 5-25). The result is the activation of numerous T lymphocytes (between 5% and 20% versus a normal response of <0.01%) and the elaboration of cytokines. Superantigens are subclassified as exogenous or endogenous. Exogenous superantigens are produced by bacteria and include some enterotoxins, exfoliating toxin, and toxic shock syndrome toxin 1 (TSST-1). Endogenous superantigens are specific cell-membrane molecules produced during viral infections of cells. The best characterized is minor lymphocyte-stimulating (MLs) determinants associated with mouse mammary tumor virus infections. Similar to bacterial septic shock, increased serum concentrations of IL-1 and TNF-α cause systemic responses such as fever, disseminated intravascular coagulation (DIC), and shock.
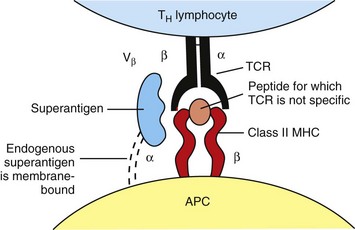
Fig. 5-25 Superantigens.
Schematic depiction of superantigens that bind to the Vβ domain of the T lymphocyte receptor (TCR) and the α-chain of a class II major histocompatibility complex (MHC) molecule and activate large numbers of T lymphocytes irrespective of their antigen specificity. APC, Antigen-presenting cell. (Adapted from Goldsby RA, Kindt TJ, Osborne BA: Kuby immunology, ed 4, New York, 2000, WH Freeman.)
Transplant Rejection
Immunologic responses are responsible for transplant rejection, and some of the basic features of those responses are discussed here. Although the frequency and success of transplantation of tissues in humans is increasing, the frequency and success in domestic species remains very low. Most of the literature on domestic species is related to the development and characterization of animal models rather than as a form of medical treatment. The most common tissue transplants in veterinary medicine are kidney grafts in dogs and cats with end-stage renal disease. The most common graft is an allograft, which is defined as a graft between two individuals of the same species. Graft rejection largely occurs as a result of the recipient recognizing the grafted tissue as foreign. The antigens responsible are the MHC molecules and blood group antigens of the graft. As discussed previously, MHC molecules are widely distributed and function as specialized molecules involved in intercellular recognition and the differentiation of self and nonself. Blood group antigens in domestic species are far more complex than those in humans. Transplant rejection is a complex process that involves both cell-mediated immunity and humoral immunity.
As we have discussed throughout this chapter, the cell-mediated immune response is primarily composed of two mechanisms that are mediated by T lymphocytes. Both mechanisms are involved in graft rejection (Web Fig. 5-2). The first mechanism, referred to as the direct pathway of graft rejection, is mediated by CD8+ CTL (Tc). In this instance, the recipient’s CTL (Tc) recognize allogenic MHC molecules expressed by antigen-presenting cells within the graft (donor origin). The molecular basis for the recognition signals are complex and poorly understood. The second mechanism, referred to as the indirect pathway of graft rejection, is mediated by the recipient’s T lymphocytes that recognize antigens of the graft presented by the antigen-presenting cells of the recipient. In this mechanism, the uptake of antigen (recipient MHC molecules), processing, and presentation to CD4+ lymphocytes is identical as for any other foreign peptide, as discussed previously. However, in this instance, the CTL (Tc) generated cannot directly kill graft cells because they are only recognizing graft antigens presented by the recipient’s antigen-presenting cells. The indirect pathway depends on the activation of CD4+ lymphocytes and the elaboration of cytokines and development of a DTH reaction. These activated CD4+ lymphocytes also facilitate the development of an antibody (humoral) response against alloantigens in the graft. Generally, graft rejection reactions are classified based on the pathologic findings within the rejected tissue as hyperacute, acute, and chronic. The human classification scheme for grading rejected renal transplants has been applied to feline renal transplant rejection reactions and was found to be less reliable in accurately characterizing the severity of the rejection reaction.
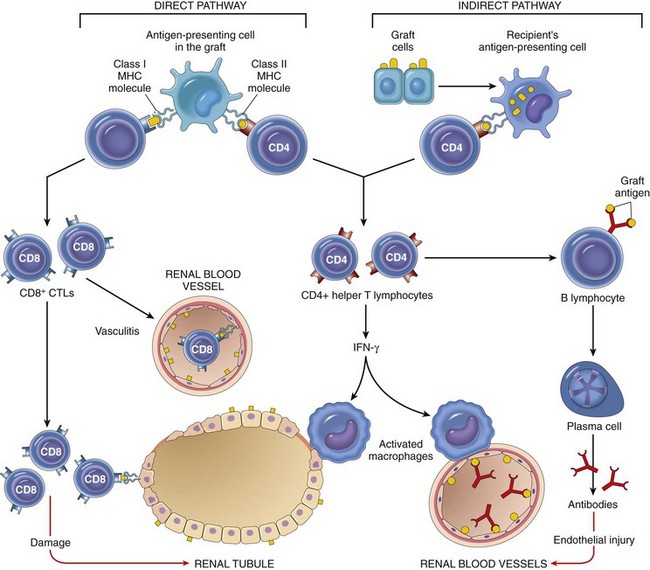
Web Fig. 5-2 Schematic depiction of the two primary pathways of graft rejection.
The direct pathway is mediated by antigen-presenting cells within the graft recognizing major histocompatibility complex (MHC) molecules of the recipient and are recognized by CD4+ and CD8+ T lymphocytes. The indirect pathway is mediated by graft antigens presented by antigen-presenting cells of the recipient that activate CD4+ and CD8+ T lymphocytes, resulting in a type IV hypersensitivity reaction. CTLs, Cytotoxic T lymphocytes; IFN-γ, interferon-γ. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Autoimmune Disease
Autoimmunity is by definition a specific immune response to self-antigens. Autoimmunity reflects a loss of immunologic tolerance to self-tissue or cellular antigens and is characterized by abnormal or excessive activity of self-reactive immune effector cells. Autoimmunity can be organ specific, localized, or systemic. Autoimmunity can be mediated by both autoantibodies and by self-reactive T lymphocytes. The etiology of most autoimmune diseases remains elusive because they are often multifactorial and have a genetic and an environmental component. Criteria for diagnosing an autoimmune disease may include (1) direct proof, such as the fact that the disease can be transferred through cells or autoantibodies; (2) indirect proof, such as in identifying the antigen, then isolating the homologous antigen in an animal model, and reproducing the disease through administration of the antigen; (3) isolating self-reactive antibodies or T lymphocytes; and (4) circumstantial evidence, such as familial occurrence, lymphocyte infiltrate, MHC associations, and clinical improvement with immunosuppressive therapy. The complexity of autoimmune diseases is also supported by the fact that nonpathologic autoreactive T lymphocytes and antibodies can be found in normal individuals. Most autoimmune diseases have a tendency to be characterized by cyclic periods of alternating clinical disease and convalescence, an increased susceptibility of the female gender, and a predisposition to multiple autoimmune phenomena as in the case of the mixed connective tissue disorders.
How does a loss of self-tolerance occur? To understand the mechanisms related to a loss of self-tolerance, one must first understand the basic concepts of maintaining immunologic tolerance to self-antigens.
Immunologic Tolerance
When exposed to an antigen, the immune system can be responsive and develop an immune response or it can be nonresponsive and develop a state of tolerance. In either case, responsive or nonresponsive, the reaction is immunologically specific and has to be carefully regulated, as a response to a self-antigen or a nonresponse to a pathogen could be equally detrimental. Immunologic tolerance is an active physiologic process and is not simply the lack of an immune response. Immunologic tolerance is defined as a failure of the immune system to respond to a specific antigen after previous exposure to that antigen. It is an absence of a functional response rather than a lack of any response at all. The development of autoimmunity can be simply described as an escape from the mechanism by which self-tolerance is maintained.
Tolerance is maintained by a number of mechanisms including deletion, anergy, and suppression. Deletion, or self-tolerance, is the process of clonally eliminating self-reactive lymphocytes. For T lymphocytes, self-tolerance occurs as a developmental process of immature lymphocytes in the thymus, termed central tolerance, and as a component of mature effector cells on exposure to antigens in the peripheral tissues, known as peripheral tolerance (Fig. 5-26). Although immature B lymphocytes undergo selection processes in the bone marrow, independent of antigen, these are not referred to as central tolerance. Not all self-reactive B and T lymphocytes are deleted during their development. However, there are mechanisms of anergy and suppression that prevent the activation of self-reactive T or B lymphocytes outside the thymus and bone marrow.
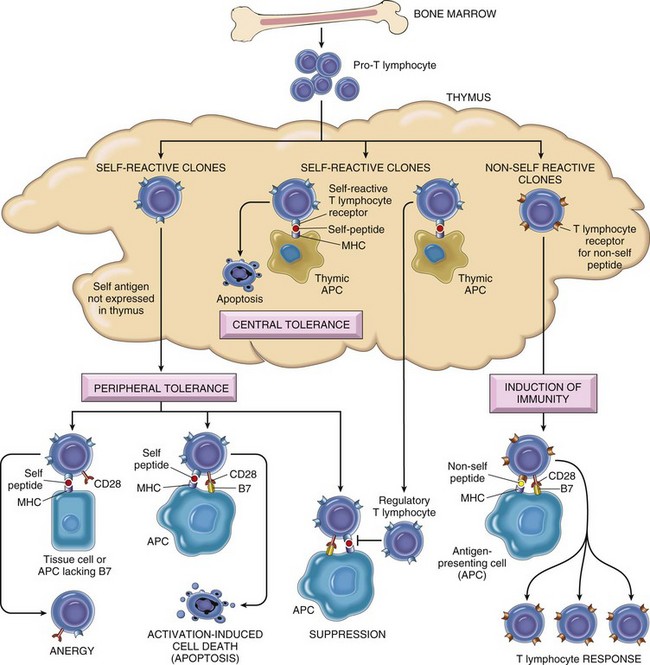
Fig. 5-26 Central and peripheral tolerance.
Schematic illustration of the mechanisms of central and peripheral tolerance of T lymphocytes. APC, Antigen-presenting cell; FasL, Fas ligand; MHC, major histocompatibility complex. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Central Tolerance: Central tolerance occurs during T lymphocyte development in the thymus, where self-reactive T lymphocytes are clonally eliminated. Central tolerance has been most extensively studied in the thymus (see Fig. 5-26), where developing T lymphocytes undergo two selection processes that are essential for their development into mature effector cells and are based on the ability of developing lymphocytes to recognize self-peptides in association with MHC molecules. Positive selection is the clonal expansion of those cells capable of self-MHC restriction. Negative selection is the clonal deletion of those cells expressing TCRs capable of recognizing self-antigens in association with MHC molecules. Developing T and B lymphocytes expressing high avidity receptors for self-antigens are deleted from further development, resulting in a peripheral effector cell population lacking self-reactive cells. Self-reactive lymphocytes are eliminated by an apoptotic mechanism. For T lymphocytes, it is the interaction of an immature lymphocyte and an antigen-presenting cell that triggers the process of clonal deletion of self-reactive T lymphocytes. This process of clonal deletion involves an apoptotic pathway mediated by the Fas-Fas ligand. The exact molecular signals that trigger this apoptotic pathway remain elusive. The expression of peripheral antigens in the thymus is thought to be partially mediated by a protein called autoimmune regulator (AIRE), which is thought to be essential for the deletion of immature self-reactive T lymphocytes. Negative selection for developing B lymphocytes also occurs through a clonal deletion process involving an apoptotic pathway for those cells that have “excessive” stimulation of their antigen receptor molecules during development. Although the mechanisms regulating tolerance during lymphocyte development are very effective at identifying and eliminating self-reactive T and B lymphocytes, they are not perfect because self-reactive lymphocytes can be identified in normal individuals. Finally, the development of central tolerance requires exposure to the antigen during lymphocyte development, and many self-antigens are not present in the thymus or bone marrow. These self-antigens are commonly referred to as sequestered antigens because they are not seen by the developing lymphocytes. Some of the tissue antigens that fall into this class of antigens include myelin basic protein, lens proteins, and sperm protein, to mention a few. These antigens can be released as a result of infection or trauma and result in an immunologic response by self-reactive lymphocytes against myelin, lens, and sperm, respectively.
Because the development of self-reactive lymphocytes may escape the mechanisms of central tolerance, the immune system has developed peripheral tolerance mechanisms to prevent these cells from becoming activated and developing into effector cells capable of causing autoimmunity.
Peripheral Tolerance: In peripheral tolerance, self-reactive T lymphocytes that are not eliminated as a result of negative selection processes in the thymus have the potential to cause tissue injury when they exit the thymus and enter the peripheral tissues (see Fig. 5-26). Within the peripheral tissues, there are mechanisms to prevent the activation of these self-reactive lymphocytes, and these occur as a consequence of the normal immune response to antigen and involve the same signals required for activation of lymphocytes during an immune response. Regulation of cellular activation occurs primarily by three mechanisms, which are briefly discussed next.
Anergy: Anergy is the functional inactivation of lymphocytes that encounter antigen. As previously discussed, two signals are required for the activation of naïve T lymphocytes by antigen-presenting cells. The first is generated by interaction of peptide antigen in association with MHC molecules on the surface of antigen-presenting cells within the TCR-CD3 complex, and the second is generated by the presence of co-stimulatory molecules. Co-stimulatory molecules are essential for the activation of naïve T lymphocytes and involve the interaction between T lymphocyte molecules (CD28) and their ligands (B7-1 and B7-2) on antigen-presenting cells. The interaction of CD28 with B7 results in T lymphocyte activation and its survival. However, if an antigen-presenting cell does not provide the co-stimulatory signal, the T lymphocyte receives a negative signal, and the cell becomes anergic (see Fig. 5-26). Another mechanism for inducing anergy involves the delivery of a specific inhibitory signal by CTLA-4 molecules on T lymphocytes that also bind to B7 molecules. The interaction of CTLA-4 with B7 results in inhibition of activation by blocking IL-2 production. The process of anergy is irreversible. The limited expression of co-stimulatory molecules by normal tissue facilitates the maintenance of peripheral tolerance to self-reactive lymphocytes. In general, CD28 is expressed on resting and activated T lymphocytes, whereas CTLA-4 is only expressed on activated T lymphocytes. What drives a T lymphocyte, expressing CD28 molecules, to recognize B7 molecules that lead to activation or to express CTLA-4 molecules that recognize the same B7 molecules that leads to anergy is unknown. Anergy of B lymphocytes occurs largely through the absence of specific TH lymphocyte activation, although negative selection of mature self-reactive B lymphocytes is known to occur. An inability of B lymphocytes to receive appropriate signals from TH lymphocytes, subsequent to antigen exposure, results in their deletion from lymphoid tissues.
Suppression by Regulatory T Lymphocytes: This mechanism of peripheral tolerance occurs through the activation of regulatory cells that prevent immune reactions to self-antigens. Suppression can occur as a result of cross-regulation of CD4+ TH1 lymphocytes by a specific population of CD4+ T reg lymphocytes that were discussed previously. Specifically, CD25+ and CD4+ T reg lymphocytes producing IL-4, IL-10, and TGF-β downregulate TH1 responses, effectively inhibiting lymphocyte activation and its effector function.
Clonal Deletion by Activation-Induced Cell Death: As discussed previously, one of the possible outcomes after lymphocyte activation as a result of antigen recognition during an immune response is lymphocyte proliferation. A second possible outcome after antigen exposure is cell death. For CD4+ T lymphocytes, both outcomes—proliferation and death—are largely regulated by the expression of accessory co-stimulatory molecules. Activation-induced cell death (AICD) of T lymphocytes occurs by Fas-Fas ligand signaling following persistent stimulation by antigen-presenting cells expressing antigen. During the normal immune response, AICD functions to down-regulate immune responses and results in the return to immune homeostasis. Lymphocytes can be induced to express Fas (CD95), a member of the TNF-receptor family. The ligand for Fas, Fas ligand (FasL), is expressed primarily on activated T lymphocytes. The binding of Fas to FasL results in apoptosis of activated T lymphocytes. Antigens that are expressed to a high level in normal tissue would result in persistent stimulation of self-reactive T lymphocytes, thus resulting in deletion through Fas-FasL–mediated apoptosis. In the case of an autoreactive B lymphocyte exposed to soluble antigen in the periphery, the cell becomes anergic. If the anergic autoreactive B lymphocyte is recognized by a T lymphocyte specific for the autoantigen, the interaction of the Fas ligand on the T lymphocyte binding to the Fas molecule on the B lymphocyte results in activation-induced cell death of the B lymphocyte. Two strains of mice have been identified with a mutation in either the Fas molecule (lpr mice) or the FasL (gld mice). The lpr and gld strains have severe autoimmune disease develop with a phenotype similar to that of humans with SLE.
Antigen Sequestration: Antigens that are not expressed in the thymus or are “cryptic” in nature have the potential to induce a self-reactive immune response. Certain physiologic characteristics of some tissue (e.g., testis, eye, and brain) are considered to render them as “immunologically privileged sites” because of the difficulty in eliciting an immune response to antigens in these tissues. Antigens in these sites cannot be seen by the immune system because they are sequestered. The sequestering of antigens may occur through the blood-brain barrier, an absence of lymphatic drainage, or the limited ability to express MHC molecules. A mechanism for the eye is referred to as the anterior chamber–associated immune deviation (ACAID), thought in part to be the result of inhibitory cytokines, such as TGF-β, produced by the cells of the iris and ciliary body. However, if the antigens in these tissues are released as the result of trauma or infection, they have the potential to cause a severe immune response as a consequence of activating self-reactive lymphocytes. Posttraumatic uveitis and orchitis are thought to be the result of the release of sequestered antigens.
Mechanisms of Autoimmunity
Although central tolerance is important in lymphocyte development, it is the mechanisms of peripheral tolerance that have a greater influence on the development of autoimmunity. We have described the complexities of central and peripheral tolerance, and thus it is understandable that the mechanisms responsible for allowing autoreactive lymphocytes to become activated and develop into self-reactive T lymphocytes or autoantibody-producing plasma cells are equally diverse and complex (Fig. 5-27). Although autoantigens have been described for a number of autoimmune diseases, it is the identification of the initiating antigen that remains elusive. The cause of most autoimmune diseases remains unknown, as they often are multifactorial and have genetic and environmental components (Fig. 5-28).
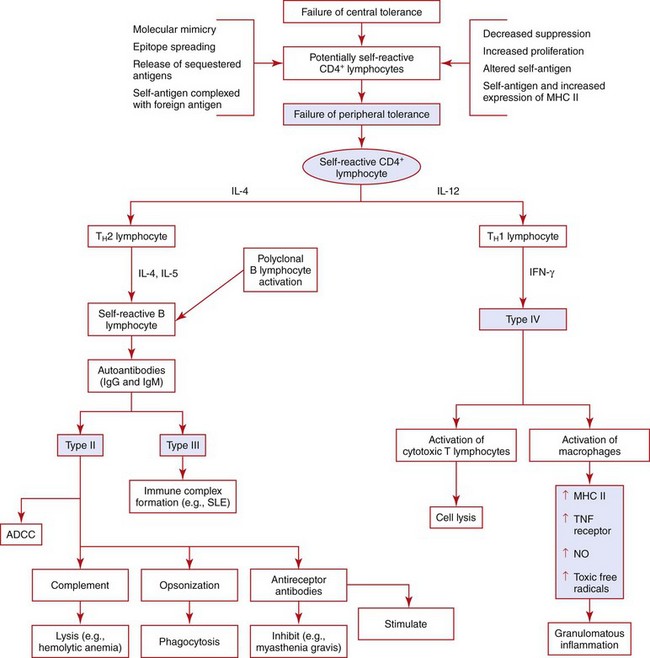
Fig. 5-27 Autoimmunity.
Pathogenetic mechanisms of autoimmunity mediated by activation of T lymphocytes (CD4+). ADCC, Antibody-dependent cellular cytotoxicity; IFN-γ, interferon-γ; Ig, immunoglobulin; IL, interleukin; MHC, major histocompatibility complex; NO, nitric oxide; SLE, systemic lupus erythematosus; TNF, tumor necrosis factor.
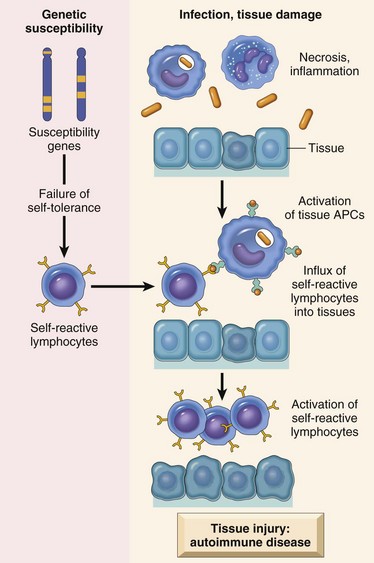
Fig. 5-28 Pathogenesis of autoimmunity.
APCs, Antigen-presenting cells. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Failure of Peripheral Tolerance
The mechanisms of self-tolerance, as previously discussed, serve as a basis for presenting how a failure to maintain those mechanisms can contribute to the pathogenesis of autoimmunity.
Genetic Factors in Autoimmunity
The majority of autoimmune diseases in humans have a strong genetic predisposition. The most well-studied genetic component centers around the MHC molecules. As previously discussed, MHC molecules are important in the development of lymphocytes and in the regulation of peripheral effector lymphocytes. Just as autoreactive lymphocytes have been identified in normal individuals without autoimmune disease, the presence of certain MHC molecules themselves is not sufficient to result in autoimmune disease. These observations would suggest that the expression of an autoimmune phenotype is not likely to be the result of a single-gene defect. Other genes that regulate proteins involved in other aspects of the immune response, or that are involved in the inflammatory or healing response, may also be involved. Additionally, experimental variations in the expression and activity of transcription factors can influence the expression of certain autoimmune diseases.
Several strains of mice with specific genetic mutations of factors involved in the maintenance of central and peripheral tolerance that results in autoimmune disease have been identified. Mice with defects of Fas or FasL have disruption of the activation-induced cell death signal in lymphocytes, resulting in autoimmune disease. Mice lacking the transcriptional factor AIRE, which is responsible for thymic expression of self-antigens, and mice defective in the expression of CTLA-4, the inhibitory receptor involved in T lymphocyte anergy, also develop autoimmunity. An important regulatory cytokine—IL-2, the major growth factor for lymphocytes—is also required for the development and function of regulatory T lymphocytes. Mice lacking either IL-2 or the IL-2 receptor develop autoimmune disease characterized by inflammatory bowel disease, anti-DNA antibodies, and autoimmune hemolytic anemia. The proposed mechanism of autoimmunity in these mice is thought to involve T lymphocytes and be a result of a failure of suppression by regulatory T lymphocytes and a failure of activation-induced cell death, two mechanisms of peripheral tolerance. These mouse models of autoimmune disease have facilitated the identification of pathogenic mechanisms of autoimmunity. Although all of these mechanisms have now been identified in human autoimmune diseases, it is likely only a matter of time before they are identified in other species.
Of the domestic species, there have been a number of autoimmune diseases in dogs documented to have a familial tendency, and the mechanism is in part attributed to certain MHC alleles. These recognized associations of specific autoimmune diseases and MHC molecules have been limited to a few specific breeds. It should also be noted that other diseases of immunity, for example, immunodeficiency and atopy, also have a higher incidence in some breeds.
Microbial Agents in Autoimmunity
The recognition that certain infections may result in the development of an autoimmune disease is the result of two observations. First, experimentally, one can induce autoimmunity in specific strains of mice by infection with certain strains of virus. Second, many spontaneous autoimmune diseases occur after viral infections, although attempts to isolate and identify viral agents in patients with autoimmune diseases have been equivocal. Again, these observations would suggest that these diseases have a complex pathogenesis with a genetic and an environmental component.
The role of infections as environmental factors in the pathogenesis of autoimmune diseases may be explained by understanding how infectious agents may cause a breakdown of anergy to self-molecules (loss of self-tolerance). As discussed earlier, not all self-reacting B and T lymphocytes are eliminated during the differentiation and development process. These potentially self-reacting lymphocytes are regulated in the periphery by clonal anergy. Therefore a loss of this regulation could explain how autoimmune diseases develop. Plausible mechanisms of why infectious agents cause aberrations of peripheral clonal anergy are twofold (Fig. 5-29). One mechanism may be the result of nonspecific disruption of the regulatory cells, which results in the induction of co-stimulatory molecules on antigen-presenting cells that are expressing self-molecules. This mechanism is not specific to the antigens of the infectious agent and is likely the result of the overall inflammatory response to the pathogen. Additionally, during an inflammatory response, some cells are induced by the inflammatory cytokine IFN-γ to increase their expression of MHC molecules. This can result in the expression of MHC molecules by cells that normally do not express them. Although the expression of MHC molecules in the absence of co-stimulatory molecules will not activate T lymphocytes, it does increase the potential to do so if co-stimulatory molecules are inappropriately expressed. The second mechanism is specific to the antigens of the infectious agent and is the result of cross-reactivity of T lymphocytes with an infectious agent’s antigen and a self-antigen. Many infectious agents express antigens that have similar peptide sequences to those of normal peptides as a part of their immune evasion mechanism. Therefore the potential exists for any immune response to an infectious agent to cross-react with a normal peptide, resulting in an immune response directed against self-cells or self-tissues. This mechanism is called molecular mimicry. The result is the activation of T lymphocytes that recognize the infectious agent peptide–MHC complex. These T lymphocytes can potentially attack self-peptide–MHC complexes that are cross-reactive.
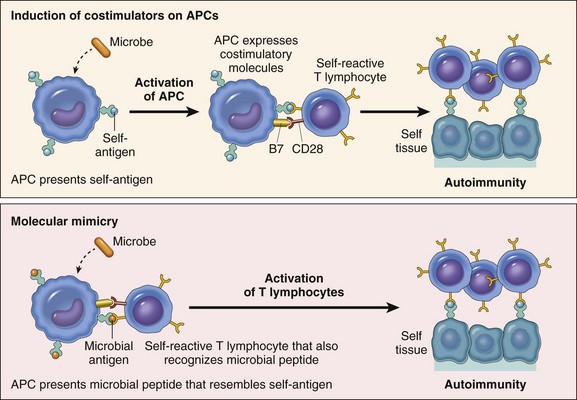
Fig. 5-29 Autoimmunity and the role of infections in its pathogenesis.
APC, Antigen-presenting cell. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia, 2004, Saunders.)
Once an autoimmune disease is initiated, the clinical course is generally progressive and characterized by cyclical periods of exacerbation and remission. As with any immune-mediated disease, antigen persistence is required to maintain the functional immune response. In autoimmune diseases, antigen persistence is thought in part to occur through epitope spreading. Epitope spreading is the process by which the immune response spreads from one epitope of an antigenic molecule to another, non–cross-reacting, epitope of the same antigenic molecule, or from one epitope of different peptides that are a part of a large complex. The epitopes involved are frequently ones that the immune response has not developed tolerance against because they are not normally presented by MHC molecules in sufficient concentrations. These so-called cryptic epitopes are normally not expressed at sufficient concentrations or are “hidden” during differentiation and development of lymphocytes. However, during an infection or inflammatory response, there may be tissue or cell damage that results in the release or expression of cryptic or hidden epitopes of self-antigens, and these become targets for the immune response. Because these epitopes were hidden, the immune system has not developed tolerance to them. Epitope spreading is thought to maintain the initiated immune response by means of the continued recruitment of autoreactive T lymphocytes specific for normal “cryptic” self-peptides.
With this basic understanding of self-tolerance and the possible molecular mechanisms involved in the pathogenesis of autoimmunity, we can present some of the more common autoimmune diseases of domestic species. Autoimmune diseases can be organ specific or systemic. With many of the organ-specific diseases, an immunologically mediated pathogenesis is suspected because of the finding of a lymphocytic inflammatory reaction within the affected tissue. Rarely, autoantibodies may be identified in the circulation. The organ-specific autoimmune diseases are discussed in the appropriate chapters covering individual organ systems. This chapter focuses on some of the systemic autoimmune diseases of domestic species, beginning with the multisystemic disease SLE.
Systemic Lupus Erythematosus
SLE is one of the most well-studied systemic autoimmune diseases in humans and is characterized by the production of autoantibodies directed against a wide array of normal tissue and cellular components. The predominant autoantibody, commonly known as antinuclear antibody (ANA), is directed against nuclear antigens. The disease has been described in humans, nonhuman primates, mice, horses, dogs, cats, snakes, and iguanas. As with many autoimmune diseases, SLE has a highly variable and often progressive clinical course characterized by a variety of clinical and immunologic abnormalities. Unlike humans, in whom the disease predominantly affects women, there is no clear sex predilection in domestic species. There are certain breeds of dogs that have a higher incidence. The average age of diagnosis is approximately 5 years. Epidemiologic data on other species is limited.
Etiology and Pathogenesis
The cause of SLE remains undetermined, although the presence of autoantibodies directed against a number of tissues and cellular components suggests that the underlying immunologic abnormality is a failure to maintain self-tolerance. Antibodies against nuclear and cytoplasmic components, which are neither organ specific nor species specific, and those directed against cell surface antigens, particularly red blood cell antigens, are central to the pathogenesis of the disease. Detection of autoantibodies also facilitates the diagnosis and monitoring of human patients with SLE. The autoantibodies and self-antigens form immune complexes, which can be deposited in glomeruli (glomerulonephritis), blood vessels (vasculitis), skin (dermatitis), and joints (arthritis), resulting in the major clinical signs associated with the disease.
ANAs are found in a high percentage of patients with SLE. In humans, antinuclear antibodies are grouped into four categories: (1) antibodies against DNA, (2) antibodies against histones, (3) antibodies to nonhistone proteins bound to RNA, and (4) antibodies against nucleolar antigens. The most common method of measuring antinuclear antibodies is indirect immunofluorescence. The pattern of immunofluorescence is used to help identify the type of autoantibody present. Other methods can be used to more specifically identify the target of ANA. In humans, the majority of ANAs are directed against nucleic acids in native double-stranded DNA, in contrast to the dog, in which the majority of ANAs are directed against nuclear proteins such as histones and extractable nuclear antigens (ENAs). ANAs are also found in normal patients and in patients with other diseases; however, their frequency is much lower. In the dog, the incidence of ANA in normal dogs and dogs with other canine diseases is 16% and 20%, respectively, compared with 97% to 100% in dogs with SLE. The indirect immunofluorescence test for ANA is sensitive but not specific because of the relatively high incidence in normal dogs and those with non-SLE diseases. Two anti-ENA antibodies appear to be specific for canine SLE. These are the anti-Sm and the anti-T1 antibodies.
Patients with SLE frequently have autoantibodies in an array of tissue and cells. Many patients have rheumatoid factors and thus have a positive Coombs’ test for anti-IgG antibodies. Antibodies directed against cellular antigens on red blood cells, platelets, and lymphocytes are frequently noted. In these instances, they may lead to clinical signs of hemolytic anemia (anti-RBC antibodies), thrombocytopenia (antiplatelet antibodies), and immune system abnormalities (antilymphocyte antibodies). Other autoantibodies to components of muscle (myositis) and skin (dermatitis) are also frequently detected. While there can be an extensive number of autoantibodies identified in patients with SLE, the major clinical signs are attributed to the deposition of immune complexes in the joint, skin, and kidney and the elaboration of a type III hypersensitivity reaction. The tissues most frequently involved are in the joint, skin, and kidney.
Canine lupus primarily affects middle-aged dogs and in some studies has been reported to occur more frequently in males than females. Breeds overrepresented are the Shetland sheepdog, German shepherd, Old English sheepdog, Afghan hound, beagle, Irish setter, and poodle. Affected dogs usually present with a spectrum of clinical signs and the disease has a progressive clinical course. Common clinical findings include fever, nonerosive polyarthritis, glomerulonephritis, mucocutaneous lesions, lymph node and splenic enlargement, and hematologic abnormalities (e.g., anemia, thrombocytopenia, and leukopenia). ANAs are the most common immunologic finding. In some reports, up to 100% of affected animals have been positive for ANAs. Dogs can also have antierythrocyte antibodies (Coombs’ positive), anti-IgG antibodies (rheumatoid factor positive), circulating immune complexes and deposits of these in the skin. Only the direct Coombs’ test is valid in the dog. In the dog, as in other species, the immunologic abnormalities involve both humoral and cellular immunity. The abnormalities in humoral immunity are largely attributed to the already discussed presence of autoantibodies and are centered on the activation of self-reactive B lymphocytes. The abnormalities in cellular immunity include a lymphopenia that is characterized by a decrease in the percentage and absolute number of CD8+ lymphocytes and a concurrent increase in the percentage and a decrease in the absolute number of CD4+ lymphocytes. This translates into a high CD4+:CD8+ ratio (as high as 6 in dogs with SLE versus <2 in normal dogs).
Lesions of Systemic Lupus Erythematosus
A wide spectrum of morphologic lesions is associated with canine SLE. The most common finding, polyarthritis, is characterized as a nonerosive lesion that commonly affects the intervertebral, carpal, tarsal, and temporomandibular joints. The acute arthritis is characterized by exudation of neutrophils and fibrin into the synovial membrane and concurrent perivascular cuffing by mononuclear cells. The primary differential diagnosis for the arthritis is rheumatoid arthritis, which is an erosive lesion. The renal lesion, the result of immune complex deposition, involves the glomerulus, blood vessels, and the basement membranes of renal tubules (Web Fig. 5-3). The resulting glomerulonephritis is variable in appearance and ranges from slight mesangial alterations to diffuse proliferative lesions. The renal lesions have a common pathogenic mechanism that is a result of the deposition of immune complexes and the activation of complement (type III hypersensitivity). Glomerular lesions are often indicated by a persistent proteinuria (>0.5 g/dL).
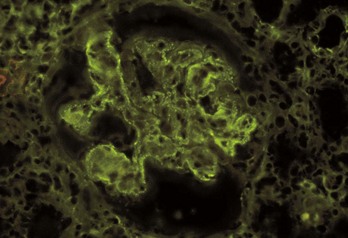
Web Fig. 5-3 Immune-complex glomerulonephritis, Aleutian mink disease, kidney, glomerulus, mink.
Intraglomerular immunoglobulin deposits demonstrated by immunofluorescence. Note the granular (“lumpy-bumpy”) pattern of fluorescence in this case of Aleutian mink disease. Immunofluorescence microscopy. (Courtesy Dr. S.J. Newman, College of Veterinary Medicine, University of Tennessee.)
Skin lesions are highly variable and nonspecific in canine SLE. The face, ears, and digital extremities are frequently involved and characterized by erythema, ulceration, and exfoliative dermatitis. The distribution of the lesions suggests that photosensitization may play a role. Histologically, the epidermis is characterized by basal cell vacuolation and necrosis, and the dermis is variably edematous with a superficial infiltration of mononuclear inflammatory cells at the dermal-epidermal junction (interface dermatitis). Additionally, there may be a panniculitis, composed primarily of lymphocytes and plasma cells, and vasculitis, with fibrinoid necrosis of the vessel wall. By indirect immunofluorescence, there are deposits of immunoglobulin and complement components at the dermal-epidermal junction. Other dermatologic variants of lupus are covered in Chapter 17. The definitive diagnosis of SLE is based on accepted criteria rather than pathognomonic findings. Using 11 established criteria for the dog, modified from the American Rheumatism Association criteria for humans, a definitive diagnosis requires the presence of four or more criteria. A “probable” diagnosis is based on the presence of three criteria or the presence of polyarthritis with the identification of ANAs. Other diagnostic schemes, using “major” and “minor” signs along with a positive ANA or SLE prep test, are also used to make definitive and probable diagnoses.
In the cat, SLE is less well recognized and manifests with fever, glomerulonephritis, dermatitis, and hemolytic anemia. The ANA test in cats is less reliable because many normal cats can have positive tests results. Horses with SLE similarly present with generalized skin disease and may also have glomerulonephritis, arthritis, and hemolytic anemia.
Genetic Factors
SLE in humans is characterized as a disease with a complex genetic component with MHC and multiple non-MHC genes involved. Extensive genetic studies in domestic animals are lacking, yet it is reasonable to suggest that the disease in other species is also characterized by involvement of multiple genes. The association of SLE with certain MHC alleles in human beings indicates that the MHC genes that regulate the production of specific antibodies are involved—specifically, MHC alleles that are linked to the production of anti–double-stranded DNA, anti-Sm, and antiphospholipid antibodies. Other than the observation of breed predilections for the dog and cat and the report of an association with the canine MHC allele DLA-A7, there are no definitive genetic studies similar to those reported in humans. Interestingly a low percentage of humans with SLE also have inherited deficiencies of complement components such as C2, C4, or C1q. Because complement components are important in the removal of circulating immune complexes by the monocyte-macrophage system, such a deficiency may contribute to the deposition of circulating complexes into tissue rather than to their removal. There is an increase in lupus-like autoimmunity in mice lacking certain complement components. Finally a well-described animal model of SLE is the NZB ∞ NZW mouse strain, in which a number of genetic loci have been identified as being associated with the development of the disease.
Environmental Factors
In addition to the genetic factors, SLE in humans has also been associated with a number of environmental factors. Specifically, drugs such as hydralazine, procainamide, and d-penicillamine can induce an SLE-like disease. In domestic animals, exposure to specific drugs and viral infections are suspected. Exposure to ultraviolet (UV) light is known to exacerbate the disease in the dog. These patients present with dermatologic manifestations localized to areas exposed to sunlight (e.g., face and dorsal regions) or areas lacking adequate hair coat (e.g., axillary region). A similar association has been noted in humans with SLE. The influence of UV radiation may be attributed to tissue damage and inflammation that result in activation of keratinocytes and the elaboration of IL-1, or the modification of DNA through the induction of apoptosis that renders the DNA immunogenic. The influence of sex hormones on the occurrence and manifestations of SLE in humans has not been documented in domestic species.
Immunologic Factors
As discussed previously, SLE is characterized by a number of immunologic abnormalities and is clinically noted to have manifestations attributable to specific immune components. It is therefore reasonable to suggest that the pathogenesis of SLE involves aberrations of humoral and/or cell-mediated immunity. Previously, as a result of the documentation of autoantibodies in patients with SLE, it was hypothesized that the pathogenesis centered on an intrinsic B lymphocyte defect. Additionally, polyclonal B lymphocyte activation is a common immunologic abnormality seen in patients with SLE and in animals that are models for the disease. Recent studies, however, indicate that the autoantibodies associated with the development of clinical SLE are not the result of polyclonal B lymphocyte activation but rather the result of antigen-specific TH lymphocyte–dependent B lymphocyte responses. This observation is consistent with our overall understanding of autoimmunity, and the current hypothesis is that these diseases are more likely to be the result of an immunologic dysregulation centered on TH lymphocytes. The currently proposed model for the pathogenesis of SLE is presented in Fig. 5-30. This model is an oversimplification of a complex disease with genetic and nongenetic factors that contribute to the development of a complex multisystemic disease with numerous clinical presentations, a progressive disease course, and an absence of specific cause.
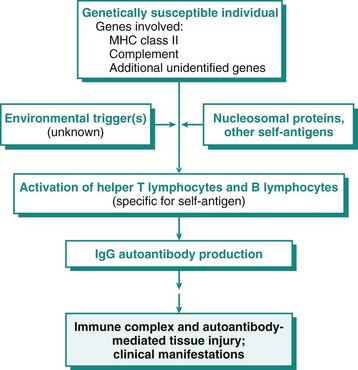
Fig. 5-30 Systemic lupus erythematosus.
Proposed model for the pathogenesis of systemic lupus erythematosus. IgG, Immunoglobulin G; MHC, major histocompatibility complex. (Modified from Kotzin BL: Cell 65:303-306, 1996.)
Although the underlying cause of autoantibody production in SLE remains unknown, the elaboration of antibody-peptide complexes is central to the mechanism of tissue damage. The majority of lesions in SLE are the result of immune complex disease (type III hypersensitivity). Antibodies directed against cell surface antigens also lead to the destruction of leukocytes, red blood cells, and platelets through direct cell lysis and enhanced removal by opsonization and phagocytosis. ANAs bind to cell-free nuclei to produce characteristic hematoxylin bodies or lupus erythematosus (LE) bodies. These bodies are frequently found in the skin, kidney, lung, lymph node, spleen, and heart of patients with SLE. These ANAs can also lead to the formation of LE cells, which are typically present in the bone marrow and are a phagocytic cell (macrophage or neutrophil) that has engulfed an opsonized nucleus. This phenomenon is also used as an in vitro diagnostic test to demonstrate the presence of ANA (LE test or LE prep).
In summary, SLE represents the prototypical multiorgan autoimmune disease with a highly variable clinical presentation, a complex cause, and pathogenesis involving multiple genetic and environmental factors and a multitude of immunologic abnormalities. The current pathogenesis suggests that these factors contribute to the activation of T and B lymphocytes, resulting in the production of autoantibodies directed against a number of self-constituents, namely molecules within the nucleus, in the cytoplasm, or on the cell surface.
Rheumatoid Arthritis
Rheumatoid arthritis is an autoimmune disease characterized by the presence of rheumatoid factors (anti-IgG antibodies) and is recognized in most species. The disease is discussed in Chapter 16.
Sjögren-Like Syndrome
Sjögren-like syndrome is a systemic autoimmune disease characterized by keratoconjunctivitis sicca, xerostomia, and lymphoplasmacytic adenitis. In humans, Sjögren syndrome can manifest itself alone or in association with other autoimmune or immune-mediated diseases, such as rheumatoid arthritis, pemphigus, SLE, polymyositis, and immune-mediated thyroiditis. A Sjögren-like syndrome has been described in the dog and cat.
Etiology and Pathogenesis
The keratoconjunctivitis sicca (dry eyes) and xerostomia (dry mouth) result from the lymphocytic infiltration and fibrosis of lacrimal and salivary glands (lymphoplasmacytic sialoadenitis). In humans the infiltrate is primarily composed of activated CD4+ lymphocytes and fewer B lymphocytes and plasma cells. Affected dogs are frequently hypergammaglobulinemic and less frequently have identifiable ANAs and rheumatoid factors. Many human patients have ANAs, rheumatoid factors, and nonorgan specific autoantibodies. Two autoantibodies specific to Sjögren syndrome in humans are directed against ribonucleoproteins, SS-A (Ro) and SS-B (La), which are considered serologic markers of the disease. Although autoantibodies can be identified, there is no direct evidence that they are the primary cause of tissue injury in any species evaluated to date. With the identification of autoantibodies and the presence of T lymphocytes within affected tissues, it is likely that the disease is the result of immunologic dysregulation centered on helper T lymphocytes. Sjögren syndrome in humans also is weakly correlated with certain MHC alleles, suggesting that as in SLE, the presence of certain MHC alleles may predispose the development of the disease.
As with many autoimmune diseases, viruses are suspected as potential causal agents. In most species in which viruses have been implicated, the evidence is largely circumstantial and Koch’s postulates have rarely been satisfied. The mechanisms by which infectious agents can induce autoimmunity were discussed earlier.
Clinical Signs and Lesions
Dogs with Sjögren-like syndrome have an adult onset of conjunctivitis and keratitis. Other findings include gingivitis and stomatitis. A case reported in the cat was characterized by dry eyes and enlarged salivary glands. The keratoconjunctivitis frequently leads to blepharospasm and conjunctival hyperemia. The xerostomia leads to dysphagia. Involvement of tissues other than salivary and lacrimal glands, as is reported in about one-third of human cases, has not been documented in the dog and cat. Human patients have lymph node involvement that is characterized as a pleomorphic infiltrate with increased mitoses and are at a fortyfold increased risk for developing lymphoid malignancies. Microscopically the salivary and lacrimal glands are infiltrated predominately by lymphocytes (Fig. 5-31). In the cat, immunohistochemical analysis of lesions in these glands indicated that the predominant cell type was positive for CD79 (B lymphocyte marker) with fewer, scattered CD3+ lymphocytes (T lymphocyte marker) and plasma cells. A mild interstitial fibrosis was also noted.
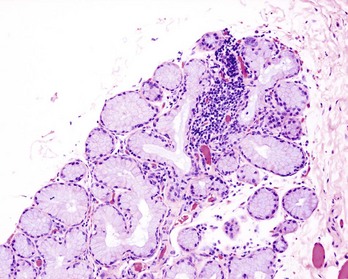
Inflammatory Myopathies
Inflammatory myopathies in domestic species comprise an uncommon, heterogeneous group of disorders that are characterized by skeletal muscle damage and inflammation. An immune-mediated pathogen is suspected. Four distinct disorders—masticatory muscle myositis, generalized inflammatory myositis, dermatomyositis, and extraocular myositis—are included in this category. They may occur alone or in association with other immune-mediated or autoimmune diseases. Of these diseases, only dermatomyositis is covered here; the remaining diseases are covered in Chapter 15.
Dermatomyositis
Dermatomyositis is an inflammatory disease of the skin, muscles, and vasculature affecting primarily young dogs. The cause and pathogenesis are unknown. The disease has a higher incidence in the collie and Shetland sheepdog breeds, and in these breeds, it is often referred to as canine familial dermatomyositis, an inheritable inflammatory disease of the skin and muscle. The disease has also been diagnosed in a number of other breeds. A sex predilection has not been reported. The clinical and pathologic findings suggest that an immune-mediated or an autoimmune mechanism is involved in the pathogenesis.
Lesions: The dermatologic manifestations of the disease are variable but generally begin at an early age (between 2 and 6 months) and are characterized by alopecia and erythematous dermatitis that involve the face, ears, and bony prominences of the distal extremities. Erosions and ulcers are common early in the disease and scarring and pigmentary changes are seen in chronic cases. Histopathologically, degeneration and necrosis of basal cells of the epidermis and follicular epithelium are characteristic. Frequently, vacuolation of the basal epithelium leads to subepidermal cleft formation. Infiltration of the superficial dermis is often composed of lymphocytes, plasma cells, and macrophages with fewer mast cells and neutrophils. Follicular atrophy and secondary ulceration and fibrosis can also be noted. The skeletal muscle manifestations of the disease are a myositis composed of a variable infiltrate of primarily mononuclear inflammatory cells and occasional neutrophils. There are varying degrees of myofiber degeneration characterized by myofiber fragmentation, vacuolation, and hyalinization. In chronic cases, there may be fibrosis and evidence of myofiber regeneration. The temporal and masseter muscles are commonly involved, although in severe cases there may be generalized muscle involvement. Involvement of the esophageal muscles can lead to the development of megaesophagus.
Vasculitis
Vasculitis is inflammation of the walls of blood vessels. It is most often seen as a component of an underlying systemic disease process (e.g., infectious or neoplastic) or as an adverse reaction to drug or vaccine administration. In these instances, the pathogenesis involves a type III hypersensitivity reaction with the formation of immune complexes that are either formed in the vessel wall or formed in the circulation and lodge in the vessel wall. The inflammation of the blood vessel is not the result of an immunologic response to components of the blood vessel but rather an “innocent bystander” phenomenon with the formation or deposition of complexes in the wall and then activation of the complement system. The pathogenesis of a type III reaction was covered earlier in the chapter. An idiopathic febrile disease characterized by a systemic necrotizing vasculitis that occurs primarily in young (4 to 10 months of age) beagle dogs is suspected to be immune-mediated, based on clinical signs, immunologic abnormalities, and pathologic findings. The syndrome has been designated as juvenile polyarteritis or beagle pain syndrome, and there appears to be a familial predisposition in some colonies. A similar syndrome has been reported in other breeds. Males and females are equally affected.
Clinical Signs and Immunologic Abnormalities
The classic presentation is a febrile (104° F to 107° F) young dog with anorexia, hunched stance, cervical pain, and an unwillingness to move the head and neck. The disease has a cyclic course, with two to seven periods of signs that resolve. They have a moderate to notable leukocytosis, with neutrophilia, nonregenerative anemia, and hypoalbuminemia. Cerebral spinal fluid analysis indicates a neutrophilic pleocytosis with mild to moderate increases in microprotein. On serum protein electrophoresis, they have a high α2-globulin fraction. ANA tests, LE preparations, and rheumatoid factor tests are generally negative. Immunologic abnormalities include an increase in serum IgA concentration, an increase in the percentage of peripheral B lymphocytes, and a decrease in the percentage of total peripheral T lymphocytes, a marked suppression of the blastogenic response to mitogenic stimulation, an inability to generate immunoglobulin-secreting plasma cells after polyclonal activation, and evidence of monocyte-macrophage activation. There are increased concentrations of IL-6 in the serum of acutely affected dogs, and these return to base-line concentrations during periods of remission or after corticosteroid therapy.
Lesions
Severe necrotizing vasculitis and perivasculitis with thrombosis of small- to medium-sized blood vessels in the leptomeninges of the cervical spinal cord, cranial mediastinum, and heart are commonly seen (Fig. 5-32). In severe cases, a more widespread distribution of vascular lesions occurs and commonly involves the thyroid gland, small intestine, testes, diaphragm, esophagus, and urinary bladder. Most patients experience multiple episodes, but some may have only one to two episodes before becoming normal, and clinical signs appear to resolve in all but the most severely affected patients by 12 to 18 months of age. Some dogs that experience repeated acute episodes develop splenic, hepatic, and renal amyloidosis. The pathogenesis of amyloidosis is attributed to serum amyloidal A production and the development of reactive systemic amyloidosis secondary to the vascular inflammation.
Immunodeficiency Syndromes
Immunodeficiency diseases occur when there is a failure of the immune system to protect the host from infectious organisms or the development of cancer. An immunodeficiency syndrome that is the result of a congenital or genetic defect in a component of the immune system is called a primary immunodeficiency. Although the defect may be present at birth, the disease may not be manifested until later in life. An immunodeficiency syndrome that is an acquired loss of immune function as a complication of infections, malnutrition, or aging or a side effect of immunosuppression, irradiation, or chemotherapy for cancer or autoimmune disease is called a secondary immunodeficiency. It is important to differentiate between a primary and secondary state of immunodeficiency with respect to treatment and prognosis. In some instances, it is useful to subclassify immunodeficiency diseases into those that affect adaptive (specific) or noninnate (nonspecific) immune responses. The study of immunodeficiency diseases has provided valuable insights and has enhanced our understanding of the complexities of the immune system. The ability to specifically identify the defective component provides great potential for the development of screening tests and of effective therapies. Naturally occurring and experimentally induced defects have made significant contributions to the field of immunology. There are a greater number of well-characterized immunodeficiency diseases in humans that likely have a domestic animal counterpart that have not yet been recognized. With the advent of new reagents and methodologies for characterizing cells and components of the immune system of domestic species, there should be a better chance in identifying immunodeficiency syndromes. Most primary immunodeficiency diseases are inherited, and the gene defect has been identified. There are additional forms of immunodeficiency that are the result of developmental defects that impair the function of an organ of the immune system. Finally, secondary immunodeficiency is a component of a large number of diseases of domestic species (ranging from malnutrition to viral infections that target lymphoid cells) and is beyond the scope of this chapter. This portion of the chapter covers some of the better characterized primary immunodeficiency diseases of domestic animals.
Primary Immunodeficiencies
Most primary immunodeficiency diseases are the result of a genetic defect (inherited or congenital) and affect specific (i.e., humoral and cell-mediated arms of the adaptive immune response) or nonspecific immunity (i.e., components of innate immune responses, such as complement, phagocytosis, NK cells, and so forth). Specific defects in adaptive immune responses can be divided into those affecting T lymphocytes or B lymphocytes or both (Fig. 5-33). As already discussed, interactions between B and T lymphocytes are necessary for the development of many immune responses. In some instances, the clinical distinction between a primary B lymphocyte defect and a T lymphocyte defect may not be obvious with respect to humoral immunity. For example, T lymphocyte defects almost always result in impaired antibody synthesis and as such are indistinguishable from B lymphocyte defects or combined B lymphocyte and T lymphocyte defects. In general, most primary deficiencies manifest themselves early in life and affected patients clinically have a failure to thrive and a susceptibility to recurrent infections. In many instances, the type of infection suggests to some extent the likely component of the immune system that is defective (Table 5-8). It is beyond the scope of this chapter to present all of the forms of human and rodent immunodeficiency, and thus only a few are referenced for clarity to facilitate understanding diseases that have been characterized in domestic species.

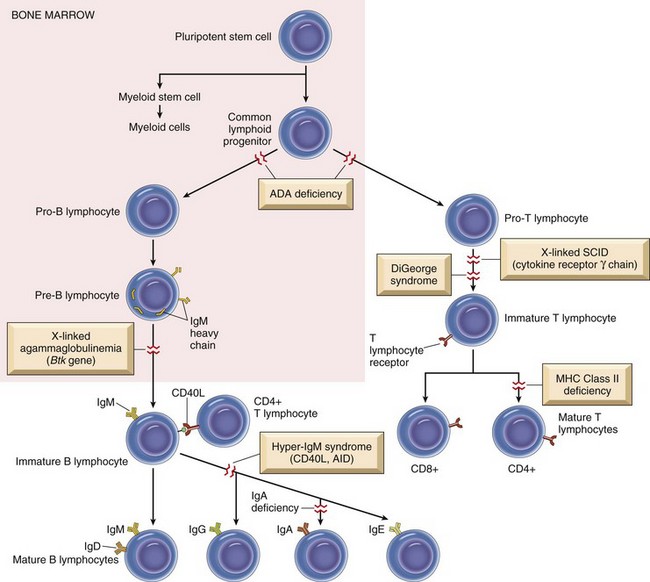
Fig. 5-33 Primary immunodeficiency diseases.
Schematic illustration of lymphocyte development and differentiation with known sites of disruption that cause primary immunodeficiency diseases. ADA, Adenosine deaminase; Ig, immunoglobulin; MHC, major histocompatibility complex; SCID, severe combined immunodeficiency disease. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Primary Immunodeficiencies of Specific Immunity
Severe Combined Immunodeficiency Disease: Severe combined immunodeficiency disease (SCID) is a family of genetic defects that have in common deficiencies in both humoral and cell-mediated immunity. In its extreme form, SCID results from a defect in the common lymphoid stem cell that results in defective cell-mediated and humoral immune responses. More commonly, SCID defects affect either T lymphocytes or B and T lymphocytes and are best characterized in humans, mice, dogs, and horses. T lymphocyte defects often clinically have a combined immunodeficiency because there is a secondary impairment of humoral immunity that is the result of an inability of the T lymphocyte to provide the necessary signals for B lymphocyte activation. These defects result in an inability to generate a specific immune response and can have an autosomal recessive, X-linked, or sporadic inheritance pattern. The types of underlying defects are diverse and may involve enzyme systems or proteins necessary for lymphocyte development and differentiation, or signal transduction pathways involved in lymphocyte activation. Often the most common infectious manifestation of SCID is a viral or fungal infection. Immunity to viral and fungal infections is largely dependent on cell-mediated immunity. Immunity to most bacterial infections (especially extracellular bacteria) largely depends on humoral immunity, and neonates have adequate humoral immunity from the passive transfer of maternal antibodies that protect them.
SCID in horses is an autosomal recessive disorder described in the Arabian or Arabian-cross breed. The defect results in a severe lymphopenia (less than 1000/mm3) attributed to an inability to produce functional T and B lymphocytes. At birth, before they acquire maternal antibodies via colostrums, affected animals are deficient in serum IgM, and after catabolism of passively transferred antibodies, they have agammaglobinemia develop. Recurrent infections are typical and death is commonly the result of infection with equine adenovirus, Pneumocystis carinii, Cryptosporidium parvum, or a variety of common equine bacterial pathogens. Grossly the thymus is small and maybe undetectable. Microscopically, there is profound lymphoid hypoplasia of primary and secondary lymphoid tissue. Thymuses contain small lobules with no corticomedullary differentiation, few lymphocytes, Hassall’s corpuscles, and occasional cysts. Spleens are characterized as having no lymphoid follicles, periarteriolar lymphoid sheaths, or plasma cells. Additionally, it has been noted that the lymphoid follicle sites in the spleen lack connective tissue stroma, and this characteristic can be used to differentiate hypoplasia from atrophy. Lymph nodes lack lymphoid follicles, plasma cells, and corticomedullary differentiation. The molecular basis for the defect has been identified as a spontaneous mutation in the gene encoding, the catalytic subunit of a DNA-dependent protein kinase (DNA-PKcs) that is located on chromosome 9. Affected foals have a complete absence of functional DNA-PKcs. DNA-PK is required for the recombination of immunoglobulin heavy chain and TCR genes during development, which when defective results in an inability to form functional V regions. Interestingly, as a result of the DNA-PKcs mutation, affected horses also are defective in their DNA repair mechanisms. The importance of DNA repair mechanisms in preventing the development of cancer (see Chapters 1 and 6) would suggest that affected foals and heterozygous carriers would be at an increased risk for cancer. Although affected foals rarely live beyond 5 months of age, heterozygous carriers do, and they have been observed to be at a greater risk for developing sarcoids, a locally aggressive fibroblastic cutaneous neoplasm.
SCID in dogs was first described in Basset hounds as an X-linked defect (XSCID) characterized by lymphopenia, with increased numbers of B lymphocytes and few to no T lymphocytes. The lymphopenia is not as profound as it is in foals with SCID. At approximately 6 to 8 weeks of age, as maternally-derived antibody concentrations decline, they develop recurrent infections of the skin, respiratory, or gastrointestinal system. Phenotypically, there is a decrease in the number of circulating CD8+ lymphocytes resulting in a CD4+:CD8+ ratio of approximately 15 : 1 (compared with 2 : 1 in normal dogs) and normal percentages of B lymphocytes. Dogs with XSCID are hypogammaglobulinemic with normal serum IgM concentrations and decreased concentrations of IgG and IgA. Affected dogs rarely live past 4 months of age, and death is frequently attributed to septicemia or systemic viral infections. At necropsy, lymph nodes, tonsils, Peyer’s patches, and the thymus are extremely small and may be undetectable. Microscopically, there is severe lymphoid hypoplasia with similar characteristic features as described previously for lymphoid tissues of the foal with SCID. Thymuses of affected puppies are small (approximately 10% the weight of age-matched controls) and are characterized by a lack of corticomedullary demarcation and small lobules with few to no lymphocytes (Fig. 5-34). Thymuses have markedly increased percentages of CD8/CD4 lymphocytes (46% versus 16% in age-matched controls) compatible with a block in T lymphocyte differentiation. Canine XSCID is due to a mutation in the common gamma (γc) subunit of the IL-2, IL-4, IL-7, IL-9, and IL-15 receptors. These receptors, for five immunologically different cytokines, belong to the type I cytokine receptor family. A four-base pair deletion results in a stop codon that prevents full translation of the γc messenger RNA (mRNA). T lymphocytes are nonfunctional because of an inability to express a functional IL-2 receptor. B lymphocytes are only activated by T lymphocyte–independent antigens, and although they are capable of synthesizing IgM, they are incapable of class-switching to IgG. A similar molecular form of XSCID has also been described in the Cardigan Welsh Corgi breed. An insertional mutation, caused by the insertion of a cytosine in exon 4, also results in a stop codon preventing complete translation of γc mRNA. In human SCID, mutations of the γc are the most common molecular mechanism with numerous distinct point, insertion, and deletion mutations identified. An autosomal recessive form of SCID has been described in Jack Russell terriers that is the result of a mutation in DNA-PKcs similar to that described previously in the Arabian horse and the CB-17 mouse.
Fig. 5-34 X-linked severe combined immunodeficiency disease (XSCID) thymuses, normal puppy and littermate.
A, The gland from the normal puppy (left) weighed 7.4 g, and the gland from the XSCID puppy (right) weighed 0.2 g. B, Thymus from a normal neonatal dog (left) and from a neonatal dog with XSCID (right). The normal thymic lobule has a pale medullary region and a dark cortical region that is densely packed with small lymphocytes. The XSCID thymus has small lobules with no corticomedullary distinction and a paucity of small lymphocytes. H&E stain. (A and B courtesy Dr. P.W. Snyder, School of Veterinary Medicine, Purdue University.)
SCID in mice occurs in the CB-17 strain and is an autosomal recessive trait characterized by an absence of mature B and T lymphocytes. The molecular basis for the SCID phenotype is attributed to a defect resulting in decreased DNA-PK enzyme activity. Although mice are highly susceptible to opportunistic infections, they can be maintained in environments that minimize their exposure to pathogenic agents and kept alive for more than a year of age. An interesting, although poorly understood, immunologic finding in older (greater than 6 months of age) mice with SCID is the ability to produce small quantities of immunoglobulin and low numbers of mature T lymphocytes. This phenotype is referred to as leaky SCID. This leaky phenotype has not been described in other forms of SCID. As in other species with defective DNA repair mechanisms, there is an increased sensitivity to ionizing radiation damage.
Common Variable Immunodeficiency: Common variable immunodeficiency is a primary immunodeficiency disease characterized by an adult onset of hypogammaglobulinemia attributed to an intrinsic B lymphocyte defect, which results in an inability to produce much of the antibody. The disease has been described in a litter of miniature Dachshund dogs. The dogs were characterized as having an absence of B lymphocytes in lymphoid tissues and little to no serum immunoglobulins. At necropsy, the affected animals had lesions characterized as atrophy of lymphoid tissue and pneumonia. Lymph nodes were further characterized as lacking lymphoid follicles. The pneumonia was caused by Pneumocystis carinii, a common opportunistic pathogen in immunocompromised animals and humans. In humans, there have been a number of identified defects intrinsic to B lymphocytes that prevent their terminal differentiation and affect their ability to produce immunoglobulin, thus the designation as variable immunodeficiency. A similar disease has also been described in a 12-year-old Quarter Horse, which was characterized by hypogammaglobulinemia and an absence of B lymphocytes in circulation, bone marrow, and spleen.
Agammaglobulinemia: Agammaglobulinemia is a primary immunodeficiency characterized by an inability to produce immunoglobulins and an absence of mature B lymphocytes and plasma cells. The disease has been described in Thoroughbred, Quarter Horse, and Standardbred breeds of horses. To date, all cases have been males, suggesting that, as in the human disease, it is an X-linked trait. Microscopically, there was an absence of plasma cells, primary follicles, and germinal centers in lymph nodes. Affected horses commonly have extracellular bacterial infections of the joints and respiratory system. In humans, there is a mutation of the BTK gene located on the X chromosome that encodes a tyrosine kinase that results in an arrest of B lymphocyte development at the pre-B stage. The disease in humans is referred to as X-linked agammaglobulinemia (XLA).
Selective Immunoglobulin Deficiencies: Selective deficiencies are represented by a number of diseases characterized by a deficiency of an individual class of immunoglobulin. Forms of these diseases have been identified and described in horses and dogs. The most common forms are selective IgM deficiency and selective IgA deficiency, characterized by a serum level of IgM or IgA, respectively, that is at least two standard deviations below normal. Serum concentrations of other classes of immunoglobulin are normal, and B lymphocyte numbers are normal. These deficiencies may not result in clinical signs until there is degradation of passively transferred maternal antibody. In horses, there are distinct forms of selective IgM deficiency with most affected animals succumbing to septicemia or pneumonia by 10 months of age. A few affected foals live beyond 10 months and commonly die of recurrent respiratory infections before reaching adulthood. Some horses reach adulthood before they show clinical signs of selective IgM deficiency. Selective IgA deficiency has been described in a number of breeds of dogs, including the German shepherd, Shar-Pei, Irish setter, and beagle. Because of the importance of IgA in mucosal immunity, many affected animals have respiratory, gastrointestinal, and skin infections. Some affected dogs, like human patients with IgA deficiency, have a predisposition to developing atopic disease. In most instances there are normal numbers of IgA-producing plasma cells, suggesting an inability to synthesize or secrete IgA. Although the molecular basis for selective immunoglobulin deficiencies is unknown, they are thought to relate to the differentiation of naïve B lymphocytes into immunoglobulin-secreting plasma cells.
Thymic Hypoplasia: Thymic hypoplasia represents a number of immunodeficiency diseases characterized by a failure to develop a functional thymus, resulting in a T lymphocyte deficiency. Mice homozygous for the genetic trait nu (nu/nu) are hairless, and this athymic strain is commonly referred to as the nude mouse. Affected mice have a developmental arrest of the thymus, which occurs around day 12 of gestation and results in an absence of a functional thymus. Nude mice have defective cell-mediated immune responses and are unable to develop antibody responses. The immunologic abnormalities are attributed to a deficiency of T lymphocyte responses. Heterozygous (nu/+) animals are normal. The few circulating T lymphocytes in affected mice have TCRs of the γ/δ-type rather than the α/β-type. Under conventional housing conditions, mortality is high during the first 2 weeks of life; however, when maintained in germ-free environments, they can survive longer. This strain of mice is an important animal model, as they can tolerate allografts and xenografts. Similar, although less well-characterized, hairless and athymic conditions have been described in other species. A condition in humans characterized by thymic hypoplasia is referred to as DiGeorge syndrome. DiGeorge syndrome is a T lymphocyte deficiency resulting from an embryologic defect affecting the development of the third and fourth pharyngeal pouches. Affected humans have thymic (fourth pharyngeal pouch) and parathyroid (third pharyngeal pouch) defects. They have decreased circulating T lymphocytes, and T lymphocyte areas of lymphoid tissues (paracortical areas of lymph nodes and periarteriolar sheaths of the spleen) are depleted of lymphocytes. There is an absence of cell-mediated immune responses. DiGeorge syndrome is not familial but rather the result of the deletion of a specific gene that is a member of the T-box family of transcription factors. Specifically, how or why this transcription factor influences the development of the thymus and parathyroid is unknown.
Primary Immunodeficiencies of Nonspecific Immunity
Deficiencies of the Complement System: The complement system contains more than 30 soluble and cell-bound proteins that influence immune and inflammatory responses. The pathways of complement activation, regulation of the complement system, and biologic consequences of complement activation have been previously covered in Chapter 3. In humans, inherited deficiencies have been described for nearly all components and two of the inhibitors. Although a deficiency of C2 is the most common, humans with deficiencies in components of the classical pathway have little to no increased risk for infections, suggesting that the alternative and lectin pathways of activation are sufficient for controlling infections. Deficiencies of the classical pathway components are associated with an increased incidence of SLE-like autoimmune disease, which has been attributed to impaired clearance of immune complexes by the monocyte-macrophage system. Although deficiencies of components of the alternative pathway (properdin and factors D and H) are rare, when they do occur they are associated with recurrent pyogenic infections. A deficiency of C3, which is required for all three pathways of complement, is the most serious deficiency and results in serious recurrent infections. An autosomal recessive trait resulting in a genetically determined deficiency of C3 has been described in the Brittany spaniel dog. Homozygous dogs have serum concentrations of C3 and also opsonic and chemotactic activities that are severely decreased compared with normal dogs. Affected dogs are predisposed to recurrent infections and type 1 membranoproliferative glomerulonephritis. Bacterial infections with Clostridium spp., Escherichia coli, and Klebsiella spp. resulting in pneumonia, septicemia, and pyometra, respectively, are the most common clinical manifestations.
The molecular basis for the deficiency has been identified as a deletion mutation that results in a premature stop codon, preventing adequate translation and resulting in decreased concentrations of mRNA. Heterozygous dogs have serum concentrations of C3 that are approximately 50% of normal, but these dogs are clinically normal. C3 deficiency has also been described in guinea pigs and rabbits.
An autosomal recessive trait resulting in a deficiency of factor H, a component of the alternative pathway of complement, has been described in the Norwegian Yorkshire breed of pig. Factor H is a regulator of complement activation that blocks the formation of C3 convertase and is a cofactor for cleavage of C3b by factor I. Deficiencies of factor H result in unregulated elaboration of C3b on activation of the alternative pathway. The most common clinical manifestation is renal disease. Affected pigs develop a type II membranoproliferative glomerulonephritis characterized by glomerular changes consisting of thickened capillary walls, proliferation of mesangial cells, dense intramembranous deposits, and glomerular deposits of C3 components. The disease is commonly referred to as porcine dense deposit disease.
The molecular basis for the decreased serum concentrations of factor H has been reported to be the result of nucleotide sequence alterations in the factor H gene that cause a block in protein secretion. Hepatocytes of affected animals have increased intracellular concentrations of factor H. Hereditary factor H deficiency in humans can be characterized by a variety of clinical manifestations ranging from recurrent bacterial infections to glomerular disease to hemolytic uremic syndrome. Deficiencies of the terminal components (C5, C6, C7, C8, and C9), which are required for the formation of the membrane attack complex and the lysis of cell membranes, do occur and generally result in increased bacterial infections.
Deficiencies of C1 inhibitor and other complement regulatory proteins, although described in humans, have yet to be described in domestic animals. C1 inhibitor deficiency is an autosomal dominant trait that causes hereditary angioedema. C1 inhibitor is a protease inhibitor that targets C1r and C1s enzymes of the classical pathway of complement, Hageman factor (factor XII) of the coagulation pathway, and the kallikrein system. As indicated in Chapter 3, these three pathways are closely linked and result in the elaboration of vasoactive amines, notably bradykinin. Affected human patients develop episodes of edema involving the skin and mucosal membranes such as those of the larynx and gastrointestinal system. Deficiencies of membrane-bound regulator proteins, such as decay-accelerating factor and homologous restriction factor, result in paroxysmal nocturnal hemoglobinuria. In the absence of these regulatory factors, red blood cells can be more easily lysed by concentrations of complement that are much lower than normally required. The increased lysis of red blood cells causes chronic hemolytic anemia and hemoglobinuria.
Chédiak-Higashi Syndrome: Chédiak-Higashi syndrome is an inherited disease caused by defective lysosomes, melanosomes, platelet-dense granules, and cytolytic granules. The disease has been described in cats, cattle, killer whales, beige mice, rats, Aleutian mink, and humans. Common clinical manifestations of the disease may include hypopigmentation, a bleeding tendency, ocular abnormalities, and recurrent infections. Some species are more susceptible to recurrent infections than others. The hallmark of the disease is the presence of enlarged granules within melanocytes, neutrophils, eosinophils, and monocytes. The enlarged granules are melanosomes (melanocytes), lysosomes (many cell types), or cytoplasmic granules (e.g., fused primary and secondary granules of neutrophils). Neutrophils containing giant granules have impaired functions such as defective chemotaxis and intracellular killing. NK cells are also defective and may also contribute to the increased susceptibility to infections reported in some species. Mink with Chédiak-Higashi syndrome have an increased susceptibility to Aleutian mink disease virus. The hypopigmentation is the result of an inability of melanocytes, containing abnormally large melanosomes, to migrate and release their contents, resulting in a deficiency of pigment most commonly evident in the skin, hair, and eye. The bleeding tendency is a coagulopathy resulting from defective platelets. Platelet counts are generally normal. In most species studied, most recently cattle, there is insufficient platelet aggregation because of a decreased response to collagen. Research suggests that the GPIa/Iia (α2β1-integrin) glycoprotein or rhodocytin pathway of platelet activation may be defective. Ocular abnormalities identified in cattle, cats, mink, mice, and humans are similar and are characterized by abnormal ocular pigmentation and an associated photophobia. Cats with Chédiak-Higashi syndrome frequently develop cataracts. The molecular basis for Chédiak-Higashi syndrome has been identified in some human forms and in beige mice and cattle to be a result of a mutation of the Lyst gene. The Lyst gene encodes a membrane-associated protein, which is thought to regulate intracellular protein trafficking. Exactly how the protein regulates intracellular trafficking remains unknown.
Leukocyte Adhesion Deficiency: Leukocyte adhesion deficiency (LAD) is a primary immunodeficiency disease characterized by the inability of leukocytes to migrate from circulation into sites of inflammation, resulting in recurrent bacterial infections (see Chapter 3). The disease is best characterized in humans and in bovine (bovine LAD [BLAD]) and canine (canine LAD [CLAD]) species, in which the defect is inherited as an autosomal recessive trait. The Irish setter breed of dogs and Holstein breed of cattle are commonly affected domestic species. The hallmark of the disease is a profound leukocytosis characterized by a marked neutrophilia, and the disease was initially referred to in these species as canine granulocytopathy syndrome and bovine granulocytopathy syndrome, respectively. In Irish setters, CLAD-affected puppies often initially have omphalophlebitis just after birth and by 4 months of age commonly have episodes of gingivitis, osteomyelitis, or lymphadenopathy. An inability to form pus and delayed wound healing may also be observed. Dogs rarely survive beyond 6 months of age as a result of the recurrent infections. In affected calves, BLAD causes a similar spectrum of recurrent infections and leads to death by 7 months of age. The molecular basis for the disease is a defective expression of the β2-integrin subunit of a large group of heterodimeric (α- and β-subunits) leukocyte adhesion molecules. The β2-integrins are a family of adhesion molecules that all share the same β-subunit (CD18) but have unique α-subunits (CD11a, b, c). Members of the β2-integrin family include leukocyte function–associated antigen, LFA-1 (CD11a/CD18); Mac-1 or complement receptor 3 (CD11b/CD18); and p150,95 or CR4 (CD11c/CD18). As discussed in Chapter 3, these surface molecules are in part required for neutrophil adhesion to the endothelium and extravasation into the tissue, migration to sites of inflammation or infection (chemotaxis), and phagocytosis of C3b-opsonized bacteria. The binding of neutrophils to cytokine-activated endothelial cells at sites of inflammation is also mediated in part by selectins, which are not affected in LAD. Therefore in LAD, neutrophils marginate but fail to migrate from the endothelium. Human LAD, BLAD, and CLAD are caused by missense mutations in the β2-integrin subunit gene, resulting in a functionally defective β2-integrin protein.
Amyloidosis
Although there is no evidence to suggest that amyloidosis is always the result of a primary immunologic abnormality, the pathogenesis of amyloidosis may involve components of the immune system and as such is discussed here. Amyloidosis represents a broad spectrum of clinical and pathologic conditions that all have in common the deposition of amyloid material. Amyloid is a pathologic proteinaceous substance of different chemical entities with an identical conformational property of forming β-pleated sheets of nonbranching fibrils as identified by x-ray crystallography and infrared spectroscopy (Fig. 5-35). By light microscopy and standard hematoxylin and eosin (H&E) staining, amyloid appears as an amorphous, eosinophilic, hyaline, extracellular substance that with progressive accumulation results in pressure atrophy of adjacent cells and tissue (Fig. 5-36). Although amyloid can be derived from different chemical entities, all deposits have the same appearance and tinctorial characteristics. In humans, there are three major and several minor biochemical forms. Amyloidosis is a feature of several different pathologic mechanisms and as such should not be considered a single disease but rather a group of diseases having in common the deposition of similar-appearing proteins. It is often necessary to differentiate amyloid from other similar-appearing extracellular deposits such as collagen and fibrin. Differentiating these proteins is readily accomplished using histochemical techniques. The most common technique for identifying amyloid is the Congo red stain, which does not have chemical specificity for amyloid but depends on the conformational property of being arranged in β-pleated sheets. Congo red stains amyloid an orange-to-red color, and when viewed by polarized light, reveals a characteristic green birefringent material (see Chapters 1, 8, and 11). A loss of Congo red staining by pretreating sections with potassium permanganate may suggest that the amyloidogenic protein is of amyloid-associated origin (see next section).
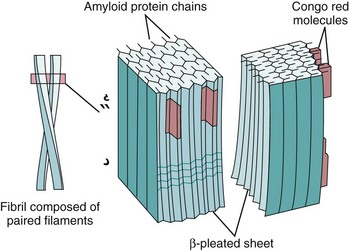
Fig. 5-35 Schematic depiction of amyloid fibrils, β-pleated sheets, and binding sites for Congo red dye. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
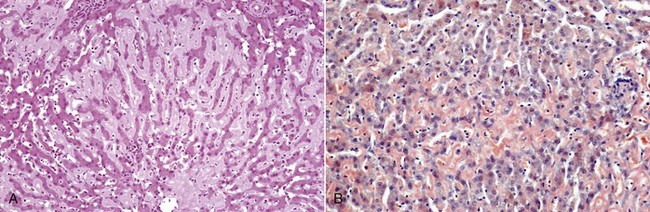
Fig. 5-36 Amyloidosis, liver, dog.
This dog had repeated episodes of vasculitis. The amyloid was attributed to systemic reactive amyloidosis (AA amyloid) caused by increased concentrations of serum amyloid A (SAA). A, Liver. The spaces of Disse are expanded by eosinophilic proteinaceous material (amyloid) that had caused atrophy of the hepatic cords. H&E stain. B, The deposits of amyloid are stained orange by the Congo red stain. (A and B courtesy Dr. P.W. Snyder, School of Veterinary Medicine, Purdue University.)
Chemical Nature of Amyloid
Amyloid contains approximately 95% fibrillar proteins and 5% P component and other glycoproteins. Of the 15 biochemically distinct forms of amyloid recognized in humans, 3 are most common: (1) amyloid light chain (AL), derived from immunoglobulin light chains of plasma cells; (2) amyloid associated (AA), derived from the acute phase protein serum amyloid A (SAA); and (3) Aβ amyloid, derived from amyloid precursor protein (APP). These forms of amyloid are also recognized in domestic species.
The AL form of amyloid can contain complete immunoglobulin light chains, the NH2-terminus portion of immunoglobulin light chains, or both. Of the two immunoglobulin light chains, λ and κ, most AL forms consist of λ-light chains or their fragments. Immunoglobulin-secreting cells, B lymphocytes, and plasma cells are associated with deposition of AL amyloid.
The AA form of amyloid consists of a proteolytic fragment of a larger acute phase protein SAA, which is found in the plasma and synthesized in the liver and released during systemic inflammatory reactions.
The Aβ form of amyloid contains a proteolytic fragment of the APP and is associated with the cerebral amyloid angiopathy of Alzheimer’s disease in humans and with some forms of neurodegeneration in the canine brain.
Of the large number of other biochemically distinct proteins that have been associated with the formation of amyloid in a number of human clinical diseases, only a few are relevant to domestic species. One of these is islet amyloidosis, affecting the feline pancreas. Islet amyloid is derived from islet amyloid polypeptide (IAPP), which is a hormone synthesized by the β cells of the pancreatic islets. All forms of amyloid contain other minor components, including serum amyloid P protein, proteoglycans, and glycosaminoglycans. Serum amyloid P may function to stabilize the fibrils and decrease their susceptibility to proteolysis.
Classification of Amyloidosis
In addition to the classification based on the primary biochemical constituent as discussed previously, it is also useful from a pathologist’s perspective to categorize these conditions according to their clinical disease and pathogenesis (Table 5-9). Amyloid can be systemic (generalized), with deposits in many organ systems, or it can be localized, with deposits limited to a single organ. The systemic or generalized category can be further subclassified into primary amyloidosis (AL), when associated with immunocyte dyscrasia, or secondary amyloidosis (SAA), when associated with a chronic inflammatory or destructive tissue process. Hereditary or familial amyloidosis is in a separate heterogeneous category with several distinctive patterns of organ involvement that are primarily recognized in humans.
TABLE 5-9
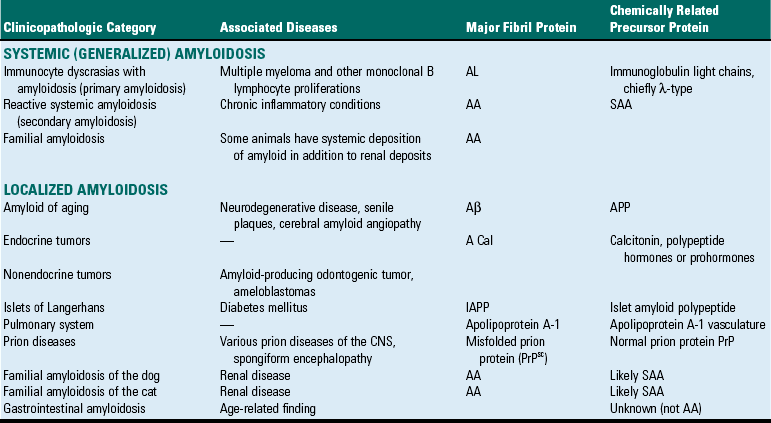
A Cal, Amyloid of hormone origin; AA, amyloid associated; AL, amyloid light chain; APP, amyloid precursor protein; CNS, central nervous system; IAPP, islet amyloid polypeptide; SAA, serum amyloid A.
Primary Amyloidosis: Primary amyloidosis is the most common systemic form of amyloidosis in humans and is of the AL type. Many cases of AL amyloidosis are attributable to the presence of some type of immunocyte dyscrasias. The most common immunocyte dyscrasias associated with AL amyloidosis in domestic species is a neoplasm of plasma cells. Plasma cell–derived neoplasms include the extramedullary plasmacytoma and the myeloma or more common multiple myeloma that are most often limited to the bone marrow. Although neoplastic plasma cells often synthesize abnormal amounts of complete immunoglobulin or components of immunoglobulins (light or heavy chain only), only a small percentage of patients with these neoplasms develop amyloidosis. On serum protein electrophoresis, there may be a monoclonal gammopathy that is compatible with the presence of a single specific immunoglobulin. Additionally, in some cases, the serum may contain only the light chain component (referred to as Bence-Jones protein). Because an individual plasma cell can only synthesize either the λ- or κ-type of light chain and because plasma cell–derived neoplasms are of clonal origin, only one light chain type will be present. The λ-light chain is most often identified in AL-type amyloidosis. Because light chains are small proteins (approximately 50 kD), they are frequently detected in the urine of affected patients. The mere presence of Bence-Jones protein itself is not sufficient to produce AL amyloidosis because many cases of plasma cell neoplasms with identifiable Bence-Jones proteins occur in most domestic species without the development of amyloidosis. In contrast to amyloidosis in humans, in which the majority of patients with AL amyloid do not have any overt B lymphocyte or plasma cell neoplasm but do have monoclonal antibodies or light chains in their serum or urine, domestic species rarely have AL-type amyloid without evidence of an immunocyte dyscrasias. A more complete discussion of these neoplasms may be found in Chapter 13.
Reactive Systemic Amyloidosis: Reactive systemic amyloidosis is the most common form in animals. This form of amyloidosis has a systemic distribution and is also referred to as secondary amyloidosis because it is often secondary to a chronic inflammation of the destructive tissue process. Reactive systemic amyloidosis in domestic animals is a form of AA amyloid, which is most often either secondary to chronic inflammatory or neoplastic (nonimmunocyte dyscrasias) conditions or idiopathic, where no underlying disease is found. A prolonged increased serum concentration of SAA protein is necessary but by itself insufficient for the development of AA amyloid. This form of amyloidosis is recognized in most species. A form of the disease is also recognized in captive cheetahs and closely related Siberian tigers. In the cheetahs, in which renal deposits were primarily in the medullary interstitium, inflammatory conditions, most often gastritis, were found in all affected cats.
Familial Amyloidosis: Familial amyloidosis is a systemic form of AA amyloidosis that is hereditary in some breeds of cats and dogs. Familial AA amyloid has been described in the Abyssinian and Siamese breeds of cat and the Shar-Pei breed of dog. The organ systems affected are varied, but deposits most commonly occur in the kidney (primarily a glomerular deposition in the Abyssinian cat and medullary interstitium deposition in the Shar-Pei dog) and liver (Siamese cat). The amyloid fibrils consist of AA proteins, suggesting that this form may be related to chronic inflammatory conditions. Preliminary analysis of the different isoforms of SAA in the cat suggests that certain SAA genes may contribute to the development of this form of amyloid. Most domestic species have at least three isoforms of SAA.
Localized Amyloidosis: Occasionally, amyloid deposits are limited to a single organ or tissue. In many instances, the localized deposits are grossly visible as masses. Localized amyloidosis is present in calcifying epithelial odontogenic tumors (amyloid-producing odontogenic tumor) of the cat and dog. Of the prominent features of these rare neoplasms, one is the presence of amyloid, which often calcifies. Some forms of naturally occurring transmissible spongiform encephalopathies, such as chronic wasting disease (CWD), are characterized by amyloid plaques in the brain in addition to the intraneuronal vacuolation. The lesions of CWD and other prion diseases are discussed in Chapter 14.
Endocrine Amyloidosis: Deposition of amyloid in the pancreas of cats, nonhuman primates (macaques and baboons), and humans can lead to the development of type 2 diabetes mellitus. The amyloid is deposited in the pancreatic islets and is derived from islet amyloid polypeptide (IAPP), a normal protein secreted by the β cell of the pancreas. How a normal protein product becomes amyloidogenic in some instances remains unknown. It is not known if the deposition of amyloid and the development of clinical diabetes mellitus is the result of progressive loss of β cells from the amyloid or if the deposition of amyloid occurs as a result of prolonged stimulation of the β cells as a consequence of insulin resistance. The disease in cats is discussed in Chapter 12. Amyloid deposition can also occur in the cortex of the adrenal gland; however, it is not associated with any functional deficiencies.
Amyloid of Aging: Amyloid deposition can occur as an age-related change in a number of organ systems. Similar to senile systemic amyloidosis in humans, old dogs can develop neurodegenerative changes that may include cerebrovascular amyloidosis or the formation of senile plaques. These deposits can consist of a number of extracellular proteins but most commonly contain Aβ type amyloid. Other organ systems rarely containing amyloid deposits in the aged dog are heart, gastrointestinal tract, and lungs. The deposition of amyloid in the pulmonary vasculature of aged dogs was reported to be derived from apolipoprotein AI (Apo AI). These forms of amyloidosis are discussed in the appropriate chapters covering these respective organ systems.
Pathogenesis of Amyloidosis
Amyloid deposition is the result of abnormal folding of pathogenic proteins, deposited extracellularly as fibrils organized into β-pleated sheets. Although there are many diverse proteins associated with the formation of amyloid, they are all characterized by misfolded proteins leading to the formation of fibrils that are unstable and self-associated. Equally diverse are the number of conditions that can be associated with the formation of amyloid, and each of these conditions is characterized by excessive production of amyloidogenic proteins that are prone to misfolding. The two general categories of amyloidogenic proteins are (1) those that are normal proteins that have an inherent tendency to misfold and form fibrils when produced in excess quantities and (2) those that are abnormal protein products as a result of genetic mutations that are structurally unstable and form fibrils that self-associate. Misfolded proteins are normally degraded intracellularly within proteasomes or extracellularly by macrophages. In amyloidosis, these degradative processes are inadequate and may be responsible for the accumulation of misfolded proteins extracellularly. A simplified pathogenic scheme for the major forms of amyloidosis is presented in Fig. 5-37. As discussed previously, the overproduction of a precursor protein, although necessary, by itself is not sufficient to result in the formation of amyloid. In secondary reactive systemic amyloidosis, a component of a systemic inflammatory reaction results in the activation of macrophages that elaborate endogenous pyrogens IL-1 and IL-6, which stimulate hepatocytes to synthesize and secrete SAA.
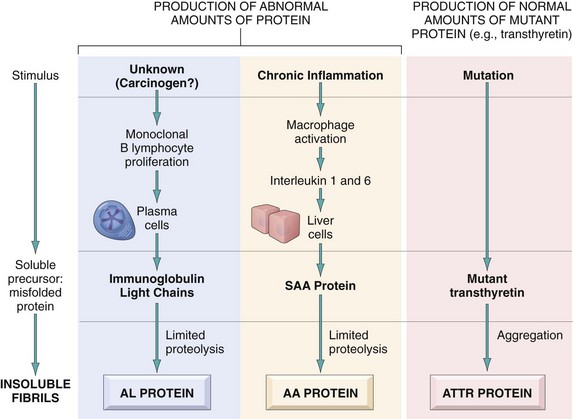
Fig. 5-37 Pathogenesis of the major forms of amyloidosis. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
During an inflammatory reaction, the quantity of SAA in the serum may increase several hundred times normal concentrations; however, not all systemic inflammatory reactions result in the formation of AA amyloid. Why is it that only some inflammatory reactions lead to amyloidosis? Two theories, one based on a defective degradation system for SAA and one based on abnormal SAA protein that is resistant to degradation, have been proposed. SAA is normally degraded to soluble protein products by enzymatic components of the monocyte-macrophage system. An enzymatic defect in this system may result in incomplete degradation and the production of insoluble AA molecules that form fibrils. Alternatively, a mutation of an SAA gene may result in the synthesis of an abnormal SAA protein that is resistant to degradation and prone to producing insoluble AA molecules that form fibrils. Finally, in the AL form of amyloidosis, plasma cells synthesize excessive quantities of immunoglobulin light chains that are resistant to complete degradation and susceptible to forming insoluble fibrils. Although there have been numerous attempts to identify specific sequence motifs unique to AA and AL forms of amyloid, to date none has been identified. Another research interest has focused on the existence of the diversity of SAA isoforms and their relationship to the formation of AA amyloid.
Morphology of Amyloidosis
Although there are no consistent or characteristic patterns for the distribution of amyloid deposition in any of the categories discussed, a few generalizations can be made. Reactive systemic amyloidosis, secondary to chronic inflammatory conditions, is often the most severe of the systemic forms. Liver, kidney, spleen, lymph nodes, and adrenal glands are most commonly affected. Animals with renal amyloidosis frequently die from renal failure.
Grossly affected organs are often enlarged, moderately firm, and abnormally discolored. In some instances, the application of an iodine solution (e.g., Lugol’s iodine) to an affected tissue may stain the deposits brown, which develops into a blue-violet color when exposed to dilute sulfuric acid solution.
Microscopically the diagnosis of amyloid is based on its extracellular location and staining characteristics. As discussed previously, Congo red is the most commonly used stain for the identification of amyloid. Congo red stains amyloid orange to red under light microscopy, which is seen under polarized light as an apple-green birefringent material. All forms of amyloid stain with Congo red, as the reaction is based on the presence of fibril organized into β-pleated sheets. Immunohistochemistry can also be used not only to identify amyloid deposits but also to identify the specific constituents composing the deposits (e.g., anti–β-light chain antibodies). By electron microscopy, amyloid fibrils are characterized as nonbranching, 7.5- to 10-nm diameter tubules. Descriptions of individual organ system involvement are covered in their respective chapters.
Abbas, AK. Diseases of immunity. In Kumar V, Abbas AK, Fausto N, eds.: Robbins & Cotran pathologic basis of disease, ed 7, Philadelphia: Saunders, 2005.
Akira, S, Takeda, K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511.
Bauer, TR, Jr., Gu, YC, Creevy, KE, et al. Leukocyte adhesion deficiency in children and Irish setter dogs. Pediatr Res. 2004;55:363–367.
Buckley, RH. Primary immunodeficiency diseases: dissectors of the immune system. Immunol Rev. 2002;185:206–219.
Chabanne, L, Fournel, C, Rigal, D, et al. Canine systemic lupus erythematosus. Part I. Clinical and biological aspects. Comp Cont Ed. 1999;21:135–141.
Goldsby, RA, Kindt, TJ, Osborne, BA, et al. Immunology, ed 5. New York: WH Freeman; 2003.
Janeway, CA, Jr., Medzhitov, R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216.
Kyewski, B, Derbinski, J. Self-representation in the thymus: an extended view. Nat Rev Immunol. 2004;4:688–698.
Leonard, WJ. Type I cytokines and interferons and their receptors. In: Paul WE, ed. Fundamental immunology. New York: Lippincott Williams & Wilkins, 2004.
Merlini, G, Bellotti, V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596.
Mok, CC, Lau, CS. Pathogenesis of systemic lupus erythematosus. J Clin Pathol. 2003;56:481–490.
O’Garra, A, Vieira, P. Regulatory T cells and mechanisms of immune system control. Nat Med. 2004;10:801–805.
Pedersen, NC. A review of immunologic diseases of the dog. Vet Immunol Immunopathol. 1999;69:251–342.
Perryman, LE. Primary immunodeficiencies of horses. Vet Clin North Am Equine Pract. 2000;16:105–116.
Perryman, LE. Molecular pathology of severe combined immunodeficiency in mice, horses, and dogs. Vet Pathol. 2004;41:95–100.
Schwartz, RH. T cell anergy. Annu Rev Immunol. 2003;21:305–334.
Sharpe, AH, Freeman, GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126.
Simonte, SJ, Cunningham-Rundles, C. Update on primary immunodeficiency: defects of lymphocytes. Clin Immunol. 2003;109:109–118.
Takeda, K, Kaisho, T, Akira, S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376.
Tizard, IR. Veterinary immunology: an introduction. Philadelphia: Saunders; 2000.
Ward, DM, Shiflett, SL, Kaplan, J. Chédiak-Higashi syndrome: a clinical and molecular view of a rare lysosomal storage disorder. Curr Mol Med. 2002;5:469–477.
Werling, D, Jungi, TW. Toll-like receptors linking innate and adaptive immune response. Vet Immunol Immunopathol. 2003;91:1–12.
Wills-Karp, M, Hershey, GKK. Immunologic mechanisms of allergic disorders. In: Paul WE, ed. Fundamental immunology. New York: Lippincott Williams & Wilkins, 2004.