The Urinary System*
Structure of the Kidney
Mammalian kidneys are paired organs present in the retroperitoneum, ventrolateral and adjacent to the lumbar vertebral bodies and their corresponding transverse processes. These complex organs, which function in excretion, metabolism, secretion, and regulation, are susceptible to disease insults that affect the four major anatomic structures of the kidney: the glomeruli, tubules, interstitium, and vasculature. Because of the limited ways that renal tissue can respond to injury and the limited patterns of injury, in severe and prolonged disease the endpoint will be similar—chronic renal disease and failure. Interdependence between components of the nephron also are responsible for producing a narrow range of repeatable injury patterns, which students can come to recognize on gross or histologic assessment.
Macroscopically, kidneys are organized functionally and anatomically into lobules. Each lobule represents collections of nephrons separated by the medullary rays (Fig. 11-1). Renal lobules should not be confused with renal lobes. Each lobe is represented by a renal pyramid (see Fig. 11-1). Among domestic animals, carnivores and horses have unilobar (or unipyramidal) kidneys. Porcine and bovine kidneys are multilobar (or multipyramidal), but only bovine kidneys have external lobation (Fig. 11-2). A diffuse fibrous capsule that in normal kidneys can be easily removed from the renal surface covers the kidneys. The renal parenchyma is divided into a cortex and medulla (Fig. 11-3). The corticomedullary ratio is usually approximately 1 : 2 or 1 : 3 in domestic animals. The ratio varies among species; for example, those adapted to the desert have a far larger medulla and thus a corticomedullary ratio that can approach 1 : 5. Normally the cortex is radially striated and dark red-brown except in mature cats, in which the cortex is often yellow because of the large lipid content of tubular epithelial cells. The renal medulla is pale gray and has either a single renal papilla, as in cats; a fused, crestlike papilla (renal medullary crest), as in dogs, sheep, and horses; or multiple renal papillae, as in pigs and cattle. The medulla generally can be subdivided into an outer zone, that portion of the medulla close to the cortex, and an inner zone, that portion closer to the pelvis. Papillae are surrounded by minor calyces that coalesce to form major calyces, which empty into the renal pelvis, where urine collects before entry into the ureters.
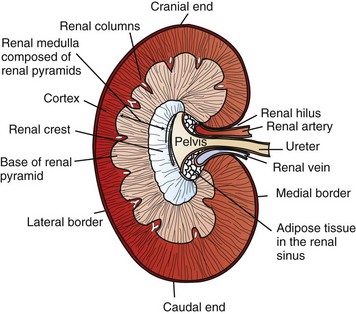
Fig. 11-1 Schematic diagram, kidney, dorsal section, dog. (Based on Schaller O, Constantinescu GM, eds: Illustrated veterinary anatomical nomenclature, Stuttgart, Germany, 2007, Enke Verlag.)
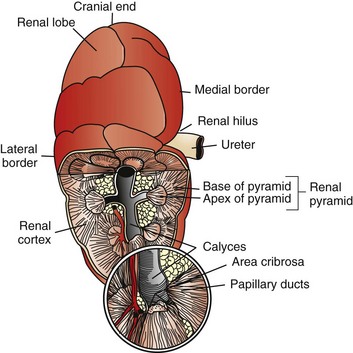
Fig. 11-2 Schematic diagram, kidney, dorsal surface and partial dorsal section, cow. (Based on Schaller O, Constantinescu GM, eds: Illustrated veterinary anatomical nomenclature, Stuttgart, Germany, 2007, Enke Verlag.)
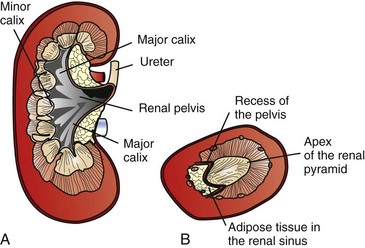
Fig. 11-3 Schematic diagram, kidney.
A, Dorsal section through hilus, pig. B, Transverse section through hilus, dog. (Based on Schaller O, Constantinescu GM, eds: Illustrated veterinary anatomical nomenclature, Stuttgart, Germany, 2007, Enke Verlag.)
Microscopically, for ease of discussion, the kidney (and nephron) can be divided into four structural units: renal corpuscle (glomerulus and Bowman’s capsule), tubules, interstitium, and vasculature. The functional unit of the kidney is the nephron, which includes the renal corpuscle and renal tubules (the tubular system includes the proximal convoluted tubules, the loop of Henle, and the distal convoluted tubule). The uriniferous tubule is composed of the nephron and the collecting ducts, which are embryologically distinct from the renal tubules (Fig. 11-4). The uriniferous tubule is embedded structurally in the renal interstitium formed by a meshwork composed of stromal cells such as fibroblasts. The interstitium also contains the renal vasculature, which supplies blood first to the glomerulus and then to the renal tubules.
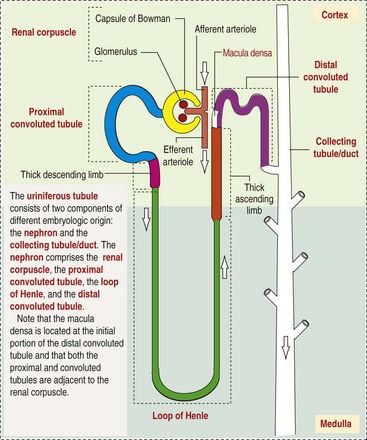
Fig. 11-4 Schematic diagram of the uriniferous tubule. (From Kierszenbaum AL: Histology and cell biology: an introduction to pathology, ed 2, St Louis, 2007, Mosby.)
Glomerulus (Glomerular Tufts)
Macroscopically, glomeruli are difficult to detect in the normal kidney but can be accentuated by lesions that allow them to be identified on cut section as randomly distributed granular foci or as red dots throughout the cortex. Microscopically, the glomerulus is a complex, convoluted tuft of fenestrated endothelial-lined capillaries held together by a supporting structure of cells in a glycoprotein matrix, the mesangium (Fig. 11-5). The entire glomerulus is supported by mesangial matrix that is secreted by the mesangial cells, a type of modified pericyte (Fig. 11-6). Mesangial cells are pluripotential mesenchymal cells, which are contractile and phagocytic and capable of synthesizing collagen and mesangial matrix, as well as secreting inflammatory mediators.
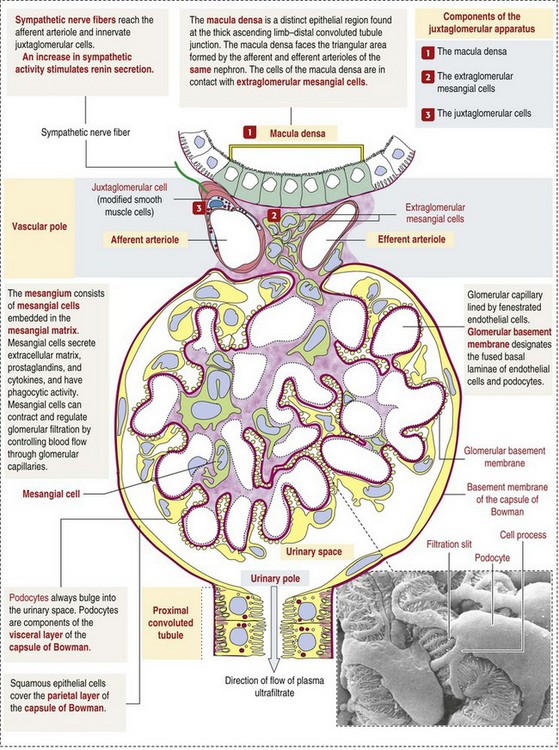
Fig. 11-5 Schematic diagram of the renal corpuscle. (From Kierszenbaum AL: Histology and cell biology: an introduction to pathology, ed 2, St Louis, 2007, Mosby. Scanning electron micrograph from Kessel RG, Kardon RH: Tissues and organs, New York, 1979, WH Freeman.)
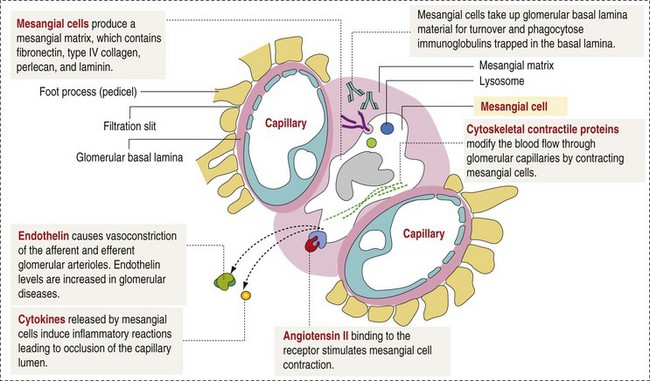
Fig. 11-6 Schematic diagram of the functions and organization of the mesangium. (From Kierszenbaum AL: Histology and cell biology: an introduction to pathology, ed 2, St Louis, 2007, Mosby.)
Glomerular Filtration Barrier
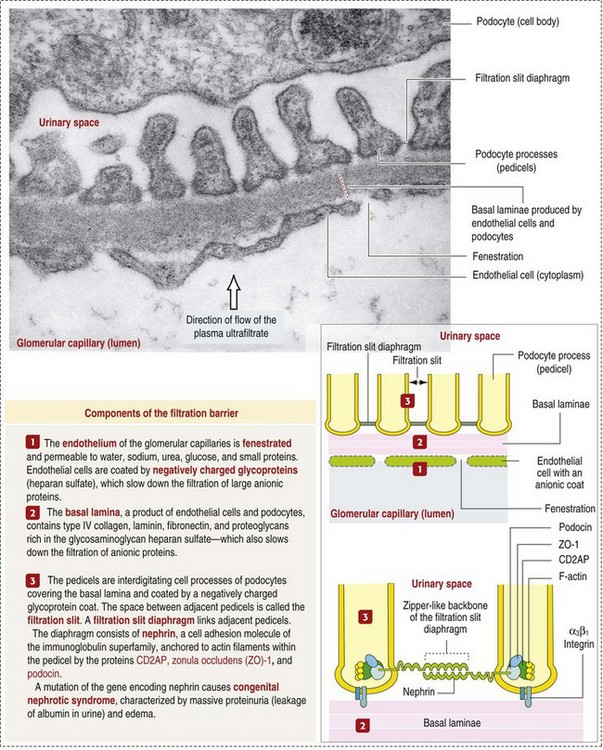
The glomerular filtration barrier is composed of (1) pedicles of podocytes (visceral epithelium of Bowman’s capsule), (2) basal lamina (produced by both endothelial and epithelial cells), and (3) the fenestrated endothelium of glomerular capillaries (Fig. 11-7).
Visceral Epithelium (Podocytes)
Visceral epithelial cells (podocytes), aligned on the external surface of the basement membrane, are responsible for synthesis of basement membrane components and have special cytoplasmic processes (foot processes) that are embedded in the lamina rara externa. Negatively charged glycoproteins overlying the endothelial cells and the podocytes contribute to the charge differential of the glomerular basement membrane (GBM). Foot processes from adjacent visceral epithelium interdigitate to form filtration slits between them. Filtration slit diaphragms are composed of nephrin, a cell adhesion molecule of the immunoglobulin superfamily, which controls slit size by its connection to podocyte actin (see Fig. 11-7). The glomerular filtration barrier selectively filters molecules based on size (70,000 Da), electrical charge (the more cationic, the more permeable), and capillary pressure. In summary, both size-dependent and charge-dependent filtration is possible because of the porous structure of capillary walls, which is a function of endothelial fenestrations, a basement membrane formed of type IV collagen, basement membrane anionic glycoproteins, and filtration slits of the visceral epithelium.
Glomerular Basement Membrane (Basal Lamina)
The glomerular basement membrane has a thick, dense central layer, the lamina densa, which is covered by thinner, more electron-lucent inner and outer layers, the lamina rara interna and lamina rara externa, respectively. The basement membrane has a network of type IV collagen, which forms a tetrameric porous infrastructure. Numerous glycoproteins, such as acidic proteoglycans and laminin, together with the collagen fibers form the complete structure of the membrane.
Bowman’s Capsule
Bowman’s capsule is a cup-shaped sac at the beginning of the nephron that encloses the glomerulus (see Fig. 11-4).
Parietal Epithelium
The capillary tuft (glomerulus) is covered by visceral epithelial cells (podocytes) and is contained within the membranous “cap” (Bowman’s capsule) that is lined by parietal epithelial cells resembling squamous epithelium (see Fig. 11-5).
Tubules
The renal tubular system (in the order of flow of urine) consists of a proximal tubule, loop of Henle, and distal tubule (see Fig. 11-4). The tubules connect to the renal pelvis at the distal end of the collecting ducts, and the whole structure, including the renal corpuscle, renal tubules, and collecting ducts is referred to as the uriniferous tubule (see Fig. 11-4). Macroscopically, the proximal and distal convoluted tubules are linked by the loop of Henle, which is divided into a descending and an ascending limb. The wall of the descending limb and initial portion of the ascending limb is thin (permeable), whereas the cortical portion of the ascending limb is thick (impermeable). Microscopically, the proximal tubule is lined by columnar epithelial cells that have a microvillous (brush) border. This arrangement greatly increases their absorptive surface, and their numerous intracellular mitochondria supply energy for the various secretory and absorptive functions. Distal tubules, collecting tubules, and the loop of Henle are lined by cuboidal epithelial cells that contribute to the concentration of urine by absorptive and secretory activities.
Interstitium
Macroscopically, the interstitium consists of a relatively scant connective tissue stroma, primarily present as a reticular meshwork found around and between uriniferous tubules. Microscopically, fibroblasts and connective tissue provide most of the interstitial tissue support. Glycosaminoglycans secreted as part of the extracellular matrix (ECM) increase with age and ischemic damage. Cells in the interstitium, particularly in the medulla, are responsible for local production of prostaglandins. Blood vessels, nerves, and lymphatic vessels are present in the renal interstitium.
Vasculature
Macroscopically, knowledge of the normal renal blood supply is important in understanding the pathogenesis and distribution of various renal lesions, especially renal infarcts. The kidneys receive blood primarily through the renal artery. An interlobar artery extends along the boundary of each renal lobe (renal column) and then branches at right angles to form an arcuate artery that runs along the corticomedullary junction (Fig. 11-8). Interlobular arteries branch from the arcuate artery and extend into the cortex. They have no anastomoses, making them susceptible to focal ischemic necrosis (infarct) as in any organ with end arteries.
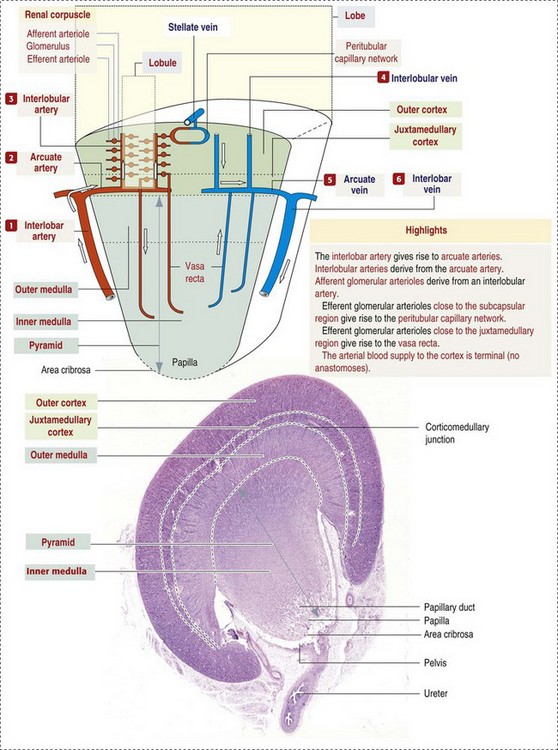
Fig. 11-8 Schematic diagram of the vasculature of the kidney. (From Kierszenbaum AL: Histology and cell biology: an introduction to pathology, ed 2, St Louis, 2007, Mosby.)
Microscopically, interlobular arteries have small branches that become afferent glomerular arterioles, which enter the renal corpuscle and subsequently exit at the vascular pole as efferent glomerular arterioles (see Fig. 11-8). Efferent arterioles supply the blood for the extensive network of capillaries that surround the cortical and medullary tubular system of the kidneys, known as the peritubular capillary network. The latter surrounds cortical segments of the tubules and then drains into the interlobular vein, arcuate vein, interlobar vein, and ultimately the renal vein. Additionally, the vasa recta are formed from the deeper portions of the peritubular network and descend into the medulla and around the lower portions of the loop of Henle before ascending to the cortex and emptying into venous vessels that connect to the interlobular and arcuate veins. The vasa recta parallel the descending and ascending limbs of the loop of Henle and the collecting ducts (see Fig. 11-4). Hence the blood supply to the tubules depends on passage through the glomerular vessels.
Function of the Kidney
The kidney has the following five basic functions:
• Formation of urine for the purpose of elimination of metabolic wastes.
• Acid-base regulation, predominantly through reclamation of bicarbonate from the glomerular filtrate.
• Conservation of water through reabsorption by the proximal convoluted tubules, the countercurrent mechanism of the loop of Henle, antidiuretic hormone (ADH) activity in the distal tubules, and the urea gradient in the medulla. The tubular system is capable of absorbing up to 99% of the water in the glomerular filtrate.
• Maintenance of normal extracellular potassium ion concentration through passive reabsorption in the proximal tubules and tubular secretion in the distal tubules under the influence of aldosterone.
• Control of endocrine function through three hormonal axes: renin-angiotensin (see Fig. 11-8), most importantly, but also erythropoietin and vitamin D. Erythropoietin, produced in the kidneys in response to reduced oxygen tension, is released into the blood and stimulates bone marrow to produce erythrocytes. Vitamin D is converted in the kidneys to its most active form (1,25-dihydroxycholecalciferol [calcitriol]), which facilitates calcium absorption by the intestine.
Function of the Glomerular Basement Membrane
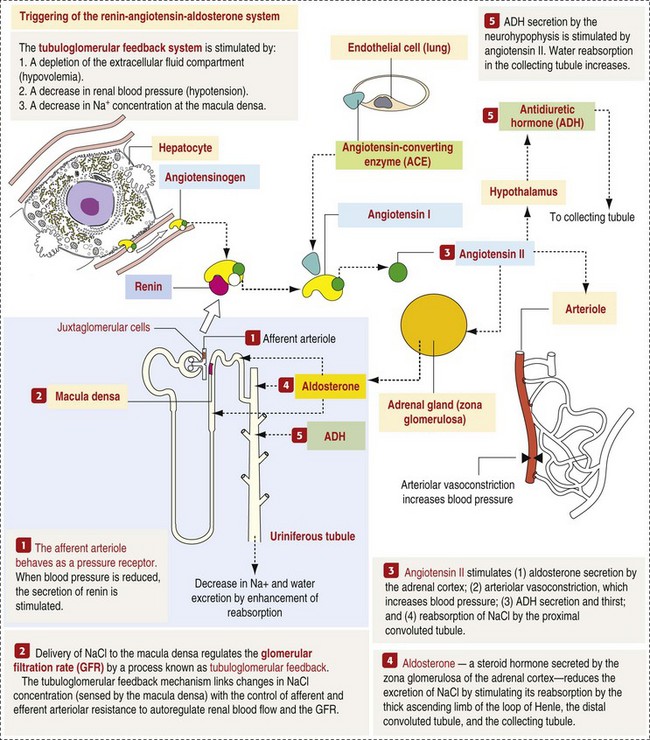
The GBM is structurally adept at separating substances based on size and charge. Additionally, the glomerulus is equipped with its own specialized mesangial cells, a component of the monocyte-macrophage system (see Figs. 11-6 and 11-7). Both size-dependent and charge-dependent filtration is possible because of the porous structure of capillary walls, which is a function of endothelial fenestrations, a basement membrane formed of type IV collagen, basement membrane anionic glycoproteins, and filtration slits of visceral epithelium. In addition to the principal glomerular function of plasma filtration, glomerular functions also include regulation of blood pressure by means of secreting vasopressor agents and/or hormones, regulation of peritubular blood flow, regulation of tubular metabolism, and removal of macromolecules from circulation by the glomerular mesangium. Integral to these functions is the juxtaglomerular apparatus, which functions in tubuloglomerular feedback by autoregulating renal blood flow and glomerular filtration rate. The juxtaglomerular apparatus is composed of four components: (1) an afferent arteriole whose smooth muscle is modified to form myoepithelial cells, which are the juxtaglomerular cells that secrete renin; (2) an efferent arteriole; (3) the macula densa; and (4) the extraglomerular mesangium. Renin, produced by cells of the juxtaglomerular apparatus, stimulates the production of angiotensin I from circulating angiotensinogen. The angiotensin-converting enzyme in the macula densa converts angiotensin I to angiotensin II, which then functions to constrict afferent renal arterioles; maintain renal blood pressure; stimulate aldosterone secretion from the adrenal gland, thus increasing sodium (Na+) reabsorption; and stimulate ADH release (Fig. 11-9). ADH principally increases the permeability of collecting tubules to water and increases the permeability of the medullary region to urea.
Function of the Proximal Tubules
A key function of the proximal tubules is to reabsorb Na+, chloride (Cl−), potassium (K+), albumin, glucose, water, and bicarbonate. This is facilitated by luminal brush border, basolateral infoldings, magnesium-dependent Na+ and K+ pumps, and transport proteins. The proximal tubule is continuous with the loop of Henle that is in close physiologic and anatomic association with the peritubular capillary network (within the cortex) and the vasa recta (within the medulla). The loop of Henle, via a countercurrent mechanism and Na+/K+-adenosine phosphatase (ATPase) pumps, absorbs Na+ and Cl− ions, producing a hypotonic filtrate that flows into the next portion of the nephron—the distal convoluted tubule. Here, water is reabsorbed from the tubule into the interstitium because of a solute concentration gradient and by the effects of ADH. The filtrate is further concentrated in the collecting ducts by water and sodium reabsorption by a Na+/K+-ATPase pump and additional water reabsorption into the medullary interstitium by a urea gradient. Intercalated cells of the collecting tubule regulate acid-base balance and reabsorb potassium. Thus the final excretory product, urine, is formed (Fig. 11-10).
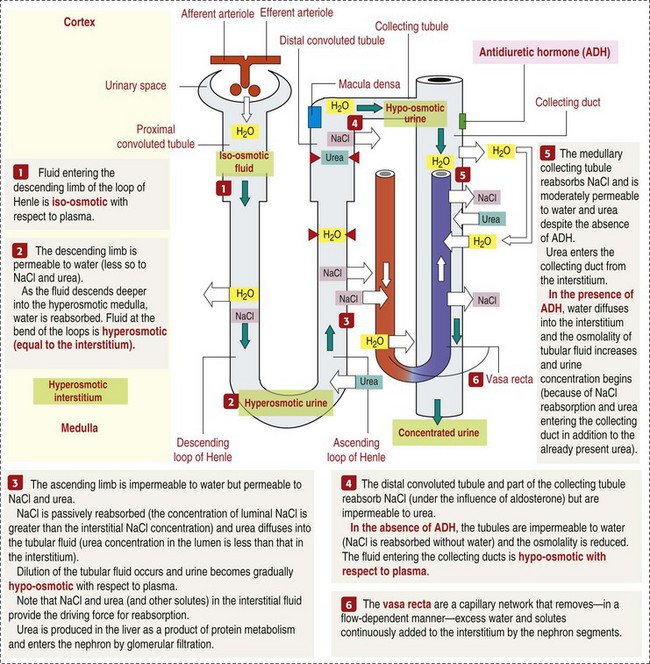
Fig. 11-10 Schematic diagram of the counter-current multiplier and exchanger. (From Kierszenbaum AL: Histology and cell biology: an introduction to pathology, ed 2, St Louis, 2007, Mosby.)
Renal Failure (Loss of Function)
Renal failure occurs when one or more of the functions previously listed are altered. When renal functional capacity is abruptly impaired (loss of 75%), such that the kidneys fail to carry out their normal metabolic and endocrine functions, acute renal failure can ensue. It is important to remember that the glomerulus, tubules, collecting ducts, and capillary blood supply in each nephron are closely interrelated, both anatomically and functionally. The fluid that filters through the glomerulus into Bowman’s space is called glomerular filtrate, and it arises after passage through the glomerular filtration barrier. This ultrafiltrate of plasma (primary urine), which contains water, salts, ions, glucose, and albumin, passes into the capsular space (Bowman’s space) and then empties into the proximal convoluted tubule at the urinary pole to traverse through and be acted on by the tubular system. Alterations in tubular structure or function influence glomerular structure and function and vice versa. For example, necrosis or atrophy of renal tubules results in loss of function of the affected nephrons and secondary atrophy of the glomerulus. In addition, because most of the capillary blood supply to tubules is through postglomerular capillaries, a reduction in glomerular blood flow consequently reduces the blood supply to the tubules.
Acute Renal Failure: Acute renal failure can be caused by (1) tubular necrosis from infectious agents, such as bacteria (Leptospira spp., Escherichia coli, Streptococcus spp., Staphylococcus spp., and Proteus spp.) or viruses (infectious canine hepatitis virus, canine distemper virus, and canine herpesvirus); (2) obstructive nephropathy from urolithiasis, transitional cell neoplasms of the lower urinary system, or trauma; (3) renal ischemia with tubular necrosis from occlusive vasculitis/vasculopathy caused by bacteria, bacterial toxins, or tumor emboli; (4) tubular necrosis from nephrotoxic drugs, such as aminoglycoside-based antimicrobial drugs or antineoplastic drugs; and/or (5) tubular necrosis from chemicals, such as ethylene glycol and heavy metals.
Functionally, acute renal failure can be caused by prerenal (compromised renal perfusion), intrarenal (compromised kidney function), or postrenal (obstruction of the urinary tract) factors. Prerenal factors include reduced renal blood flow, whether secondary to circulatory collapse (shock, severe hypovolemia) or local obstruction of vascular supply (thrombus or lodgement of embolus). Acute tubular necrosis, a form of intrarenal acute renal failure, induces clinical oliguria (decrease in urine production) or anuria (absence of urine production) by one or several mechanisms. These mechanisms include the following:
• Leakage of tubular ultrafiltrate from damaged tubules across disrupted basement membranes into the renal interstitium.
• Intratubular obstruction resulting from sloughed necrotic epithelium.
The latter mechanism is less well accepted, but both mechanisms result in decreased glomerular filtration rate.
Prerenal and intrarenal factors are most responsible for episodes of acute renal failure, with prerenal azotemia and ischemic tubular damage actually being a continuum. Postrenal obstructive diseases are discussed in the lower urinary tract section. Intrarenal disease can target tubules by the following three main mechanisms:
• Ascending disease, such as pyelonephritis
• Intraluminal toxic metabolites derived from glomerular filtrate
• Ischemia (Fig. 11-11)
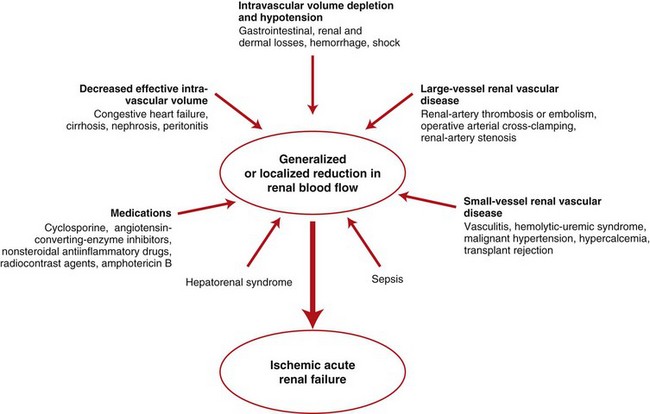
Fig. 11-11 Schematic diagram of ischemic renal failure.
A wide spectrum of clinical conditions can result in a generalized or localized reduction in renal blood flow, thus increasing the likelihood of ischemic acute renal failure. The most common condition leading to ischemic acute renal failure is severe and sustained prerenal azotemia. Kidney ischemia and acute renal failure are often the result of a combination of factors. (Redrawn from Thadhani R, Pascual M, Bonventre JV: N Engl J Med 334(22):1448-1460, 1996.)
Acute renal failure occurs when the kidney fails to excrete waste products and to maintain fluid and electrolyte homeostasis. The four main pathologic alterations in acute renal failure are as follows:
These alterations can occur after many insults, including the following:
Animals that die of acute renal failure often do so because of the cardiotoxicity of elevated serum potassium, metabolic acidosis, and/or pulmonary edema. Hyperkalemia results from decreased filtration, decreased tubular secretion, and decreased tubular sodium transport. Cell lysis and the extracellular shift of fluid in acidic environments also contribute to the increased serum potassium concentrations. These alterations are reflected clinically by signs, such as polyuria and polydipsia, vomiting and diarrhea, and ammoniacal-smelling breath, and an array of nonrenal lesions described later and can be detected and monitored with biochemical tests of serum, plasma, and urine for azotemia and uremia.
Azotemia and Uremia: Assays for plasma or serum concentrations of urea, creatinine, and the nitrogenous waste products of protein catabolism, are routinely used as indices of diminished renal function. The intravascular increase of these nitrogenous waste products is referred to as azotemia. Renal failure can result in the following:
• Intravascular accumulation of other metabolic wastes such as guanidines, phenolic acids, and large molecular weight alcohols (example: myoinositol)
• Reduced blood pH (metabolic acidosis)
• Alterations in plasma ion concentrations, particularly potassium, calcium, and phosphate
The result of renal failure is a toxicosis called uremia. Uremia can therefore be defined as a syndrome associated with multisystemic lesions and clinical signs because of renal failure. These multisystemic lesions are discussed in greater detail in the section on Disorders of Domestic Animals.
Chronic Renal Failure: Similar to acute renal failure, chronic renal failure occurs over a longer time duration and results in several additional hematologic and biochemical alterations. In the diseased kidney, production of erythropoietin, a stimulant of erythropoietic maturation, is reduced and contributes to nonregenerative anemia, as does uremia-associated increased erythrocytic fragility. Most animals in renal failure have hyperphosphatemia and low to normal calcium levels, although variations exist, depending on species and stage of the disease. Alterations in calcium-phosphorus metabolism in the uremic animal are a hallmark of chronic renal failure and result from a complex set of events as outlined in the following:
• When the glomerular filtration rate is chronically reduced to less than 25% of normal, phosphorus is no longer adequately secreted by the kidneys and hyperphosphatemia results.
• Because of the mass law interactions between serum calcium and phosphorus, ionized calcium concentration in serum is reduced as a result of precipitation of calcium and phosphorus.
• Reduced ionized serum calcium stimulates parathyroid hormone (PTH) secretion, causing calcium release from the readily mobilizable calcium stores in the bone and from osteoclastic bone resorption.
These changes in calcium-phosphorus metabolism are made more severe by the reduced ability of the diseased kidneys to hydroxylate 25-hydroxycholecalciferol to the more active 1,25-dihydroxycholecalciferol (calcitriol), resulting in decreased intestinal absorption of calcium. Calcitriol production is further inhibited by hyperphosphatemia. In addition, calcitriol normally suppresses PTH secretion; therefore reduced calcitriol production further increases PTH secretion. With time, these events lead to parathyroid chief cell hyperplasia (renal secondary hyperparathyroidism), fibrous osteodystrophy (renal osteodystrophy), and soft tissue calcification.
Renal secondary hyperparathyroidism is further thought to perpetuate and enhance renal disease by stimulating nephrocalcinosis (see Fig. 11-31; also see Chapter 12), the process by which renal tubular epithelium is damaged by an increase in intracellular calcium. Calcium is precipitated in mitochondria and in tubular basement membranes. Soft tissue calcification associated with uremia occurs in numerous sites and represents both dystrophic and metastatic calcification. These lesions are discussed in greater detail in the section on Disorders of Domestic Animals.
Portals of Entry
The urinary system and especially the kidney can be exposed to injurious stimuli and agents via a number of routes (Box 11-1), including the following:
Tubules
Substances secreted into the glomerular filtrate can produce localized trauma to tubular lining cells such as crystalline salt oversaturation (e.g., oxalate crystals). Filtered preformed toxins or metabolized substances processed by the tubular lining epithelium exert their effect principally on the proximal tubular epithelium.
Ascending Injury
Ascension from the exterior via the urethra and urinary bladder is relatively unique in that few body systems have an opening to the exterior, and these are rarely blind-ended as in the urinary system. Thus ascending infection from the exterior can localize and be perpetuated in the kidneys. Common etiologic agents in this process, such as bacteria, may originate from the exterior skin surface and the adjacent orifices of the intestinal tract or the genital tract.
Interstitium
The interstitium can be entered most successfully by the associated interstitial blood supply, allowing for interstitial localization of blood-borne pathogens, similar to those seen with glomerular blood-borne infections.
Lymphoid Nodules
Although often present as aggregates or less commonly as nodules, lymphoid infiltrates within the interstitium are not normal but the result of previous insults typically from infectious etiologies, such as Leptospirosis, or as a result of ascension secondary to long-standing pyelonephritis.
Vasculature
As in all visceral organs, the sustaining blood supply can provide a portal of hematogenous entry for infectious organisms, which in the case of the kidney principally leads to arterial localization at one of several gradated sites as follows:
• Localization within large renal vasculature. Massive infarction of the kidney is the result of large-bore renal vessel disease.
• Localization within corticomedullary vessels. In the case of associated bacterial spread, septic embolic nephritis can occur. In those examples in which the embolus is nonseptic, the result is infarctive necrosis.
• Localization within glomerular tufts. In this example, the lesions are localized to the vasculature of the small vessels within the glomerular tuft.
• Localization within interstitial vessels. In this example, the lesions are localized to necrosis of interstitial tissues and tubules.
Defense Mechanisms
Defense mechanisms unique to the renal system have evolved to counteract the routes of typical exposure to injurious agents and include those localized to the renal corpuscle, tubules, interstitium, and vasculature (Box 11-2).
Renal Corpuscle
The most important of these barrier systems is the glomerular filtration membrane (see Fig. 11-7). The glomerular membrane is structurally adept at separating substances based on size and charge. Both size-dependent and charge-dependent filtration is possible because of the porous structure of capillary walls, which is a function of endothelial fenestrations, a basement membrane formed of type IV collagen, basement membrane anionic glycoproteins, and filtration slits of visceral epithelium. This inherent function of the glomerulus can also protect other regions of the nephron from damage by circulating inflammatory cells and their cytokines, as well as infectious agents that are present in the systemic circulation (i.e., bacteria in bacteremia).
Glomerular Mesangium
Additionally, the glomerulus is equipped with its own specialized mesangial cells, a component of the monocyte-macrophage system (see Fig. 11-6), which can remove macromolecules from circulation.
Tubules
The most effective barrier system associated with the tubules is the tubular basement membrane. Intact basement membranes prevent intraluminal organisms, such as ascending bacteria, from gaining easy access to the interstitium. They also provide the scaffolding for reepithelialization of the tubule, which follows tubular necrosis in association with many toxic principles but not in instances of renal ischemia.
Interstitium
Innate humoral and cellular responses by the immune system contribute to protection of the kidney. Humoral antibodies may protect mucosal surfaces such as those of the renal pelvis and less commonly the tubular epithelial cell lining, especially against insults such as ascending bacterial infections. Cell infiltrates are typically localized to the interstitial tissues. Lymphocytes and plasma cells within the interstitium provide either cell-mediated surveillance against invasive pathogens (i.e., Leptospira) and in the case of plasma cells, can produce local antibodies.
Vasculature
Intact endothelial lining of the renal vasculature functions as a localized defense mechanism (barrier system) to prevent access by intravascular pathogens, many of which produce toxic by-products that can damage endothelium and allow for localized vasculitis and colonization. The result is often embolic septic nephritis. Intact endothelium also prevents activation of the clotting cascade and thus reduction in the likelihood of thrombus formation.
Responses to Injury
The response of the urinary system to injury is the response of each of its components—kidney, ureter, bladder, and urethra—to injury. Additionally, components within the kidney, such as the renal corpuscle, tubules, interstitium, and vasculature, have their own unique responses to injury. Responses to injury are described sequentially in this section and are summarized in Box 11-3.
Responses of the Kidney to Injury
The functional unit of the kidney is the nephron, and damage to any component of the nephron (renal corpuscle and tubules) results in diminished function and progressive damage to the kidney. Renal disease can be best summarized by dividing it into general tissue responses that affect the primary anatomic components: glomeruli, tubules, interstitium, and vasculature. In the early stages of disease, specific anatomic components may be targeted by specific insults: glomeruli in immune-mediated disease and tubules in toxin-induced necrosis. But in the more chronic stages of disease, the kidney undergoes changes related to nephron loss that are not specific to the original cause but are considered common end-stage responses to many injurious stimuli.
Renal Corpuscle
Primary glomerular damage often occurs as a result of deposition of immune complexes, entrapment of thromboemboli and bacterial emboli, or direct viral or bacterial infection of glomerular components. Such insults are reflected morphologically by necrosis, thickening of membranes, or infiltration of leukocytes, and functionally by reduced vascular perfusion. Continued or severe injury can result in chronic changes characterized at first by atrophy and fibrosis of the glomerular tuft (sclerosis) and secondarily by atrophy of renal tubules, as the nephron dies. Similar chronic glomerular changes can result from reduced blood flow or chronic loss of tubular function.
Damage to the glomerular filtration barrier can result from several causes and produce a variety of clinical signs. The major clinical finding of glomerular disease is the leakage of various low molecular weight (small molecule size) proteins, such as albumin, into the glomerular filtrate. As a result, large quantities of albumin overload the protein reabsorption capabilities of the proximal convoluted tubular epithelium to such an extent that protein-rich glomerular filtrate accumulates in the variably dilated tubular lumina and protein subsequently appears in the urine. Renal diseases that result in proteinuria are called protein-losing nephropathies. Protein-losing nephropathy is one of several causes of severe hypoproteinemia in animals. Prolonged, severe renal protein loss results in hypoproteinemia, reduced plasma colloid osmotic (oncotic) pressure, and loss of antithrombin III. The nephrotic syndrome is further characterized by generalized edema, ascites, pleural effusion, and hypercholesterolemia.
The functions of the glomerulus listed in the following are affected by processes that injure it in disease:
The pathophysiologic mechanisms of glomerular injury from infectious or chemical insults have been summarized by the following three theories:
The intact nephron hypothesis proposes that damage to any portion of the nephron affects the entire nephron function. This is seen when glomerular damage interferes with peritubular blood flow and results in decreased tubular resorption or secretion. Not all nephron damage is irreversible, for example, renal tubular epithelium can regenerate but whole nephrons are not capable of regeneration. Thus the outcomes for the nephrons vary from hypertrophy to repair.
Unlike the intact nephron hypothesis, the hyperfiltration hypothesis helps explain the progressive nature of glomerular disease. Glomerular hyperfiltration is a result of increased hydrostatic pressure that damages delicate glomerular capillaries and in cases of prolonged hypertension produces a sustained repeating deleterious effect on the glomerulus, ultimately resulting in glomerulosclerosis. Increased dietary protein can produce a transient increase in glomerular hyperfiltration and if persistent can result in glomerulosclerosis. There may be a species effect, as dogs that undergo experimental hyperfiltration are much less prone to development of progressive glomerular disease than are rats.
The theory of complex deposition is derived from the fact that glomeruli are the primary site for removal of macromolecules (principally immune complex) from the circulation. Complexes may be deposited in subepithelial, subendothelial, or mesangial locations. These immune complexes are capable of triggering a sequence of inflammatory responses including the following:
• Recruitment and localization of inflammatory cells at the site.
• Release of inflammatory mediators and enzymes.
• Destruction of glomerular structures such as the basement membrane.
• Further compromise of nephron function.
• Continuing damage by altered transglomerular hyperfiltration and perfusion shifts between nephron populations, so the less affected become overworked and succumb to the same fate.
Kidney lesions differ slightly, depending on the duration of the glomerular disease. Acute disease may be identified by pallor of the parenchyma and accentuation of the glomerular tufts as fine red dots. Accompanying petechial hemorrhages may be noted. In the more chronic stage, the kidney can be shrunken and show a fine granularity to the cortical cut section. The capsule may be adherent.
Responses of Tubules to Injury
Renal tubular epithelial cells can respond to injury by undergoing degeneration, necrosis, apoptosis, and/or atrophy. The basement membrane can respond by rupturing or thickening. Tubular disease occurs as a result of tubular epithelial damage from the following:
• Blood-borne infections (innocent bystander effect)
• Ascending infections (intratubular pathogens)
When nephrons are lost because of injury, remaining tubules can undergo compensatory hypertrophy in an attempt to maintain overall renal function, but there is no regeneration of nephrons. In many instances of tubular epithelial cell necrosis, particularly as a response to toxins, tubular epithelium has an incredible capacity to regenerate and contribute to restoration of function, providing the tubular basement membrane scaffolding remains intact. Severe damage to or loss of tubular basement membranes, as occurs after ischemic damage, results in necrosis and loss of tubular segments, failure of functional repair, and permanent loss of function of the entire nephron, despite the potential for tubular epithelial hyperplasia.
Atrophy
Tubular atrophy can occur secondary to the following:
• External compression of tubule by a space-occupying mass, neoplasm, or abscess
• Interstitial fibrosis as the end result of ischemia
• Intratubular obstruction and backpressure
If the insult to the renal tubules is not lethal and is removed, some forms of acute tubular degeneration are reversible. The success of reparative regeneration is affected by several variables, including severity of degeneration.
Apoptosis
Single cell loss (apoptosis) from the tubular epithelial lining is handled efficiently by adjacent viable tubular epithelial cells, which by mitotic division fill the epithelial gap. The cells that are lost slough into the lumen to form cellular casts within the lumens of renal tubules.
Acute Tubular Degeneration
More severe generalized loss of tubular epithelial lining cells is repaired by proliferation of the remaining viable epithelial cells over an intact tubular basement membrane to form a low cuboidal rather than a mature columnar epithelial lining. This appears as an ectatic proximal tubule. Restitution of renal function eventually results, despite the presence of replacement low cuboidal epithelium, which is not identical to the tubular lining cell (with microvilli) present before the injury. The exact mechanism of this return to function is not entirely known. The main determinant of this regenerative ability is the viability of tubular basement membranes, which are retained more consistently after toxic rather than ischemic insults.
Tubular Regeneration
Because the regenerative process is reliant on many factors for its success, things often go awry. Examples of adverse outcomes include the following:
• Focal loss of basement membrane scaffolding allows for a bulge defect to occur where the regenerative population of proliferating tubular epithelial cells coalesce to form well-differentiated syncytial cells (giant cells) at certain levels of the tubule.
• Regenerative epithelial cells fail to regain all cytoplasmic structural aspects of the original columnar epithelial cells (e.g., microvilli and luminal enzymes) because of a failure to fully differentiate, and thus function may be affected.
• If there is excessive tubular epithelial loss, the potential for regeneration is lost and repair proceeds by replacement fibrosis and scarring.
• Reperfusion is necessary for cell viability following ischemia, but reperfusion injury occurs when activated endothelial cells produce proinflammatory mediators, such as reactive oxygen species, proteolytic enzymes, and cytokines, which result in further renal injury. Recent evidence indicates that epidermal growth factor secreted by distal convoluted tubules mediates the tubular repair process. The sequence of events in tubular regeneration after necrosis has been well documented in experimental model systems using mercuric chloride in mice, rats, and rabbits. In that system, morphologic evidence of regeneration of proximal convoluted tubules is seen within 3 days after a toxic dose. At this time, basement membranes are partially covered with low cuboidal to flattened and elongated epithelial cells that are more basophilic than normal because of the increased concentrations of cytoplasmic ribosomes and rough endoplasmic reticulum producing protein for repair. Nuclei are hyperchromatic and mitotic figures are present. Regenerating tubules do not function normally because they lack both a brush border and normal tubular membrane function, and this is evident clinically as polyuria. Normal-appearing tubular epithelium subsequently reappears between 7 and 14 days after toxin exposure. Normal renal structure without residual evidence of tubular damage is restored between 21 and 56 days after exposure to the nephrotoxin. Similar time frames for tubular regeneration have been described through sequential renal biopsies from human patients naturally exposed to inorganic mercury and in experimental systems using other nephrotoxins.
Acute Tubular Necrosis
Acute tubular necrosis is the single most important cause of acute renal failure. Acute tubular degeneration and necrosis, often referred to as nephrosis, lower nephron nephrosis, tubular nephrosis, tubular dysfunction, or acute cortical necrosis, is principally the result of nephrotoxic injury to the renal tubular epithelial cells or ischemia (Box 11-4). This was borne out by a recent study of cats with renal failure in which 18 of 32 cases were the result of nephrotoxin exposure and four of 32 cases were the result of ischemia.
Acute tubular necrosis induces clinical oliguria (decrease in urine production) or anuria (absence of urine production) by one or several mechanisms. These mechanisms include the following:
• Leakage of tubular ultrafiltrate from damaged tubules across disrupted basement membranes into the renal interstitium
• Intratubular obstruction resulting from sloughed necrotic epithelium
The latter mechanism is less well accepted, but both mechanisms result in decreased glomerular filtration rate.
Nephrotoxic Injury: Nephrotoxic injury can be caused by a large group of substances that are called nephrotoxins. They preferentially damage kidneys because (1) 20% to 25% of cardiac output goes to the kidney, (2) the substance is filtered into the urine by the glomerulus, and (3) the toxin or its metabolites within the renal tubular lumens are concentrated. Nephrotoxins can do the following:
• Directly damage renal epithelial cells, particularly those of the proximal convoluted tubules, after their intracellular conversion via enzyme pathways to reactive metabolites.
• Produce reactive metabolites in the tubular filtrate, which can cause renal tubular epithelial necrosis after reabsorption.
• Cause renal tubular epithelial necrosis after diffusion through the intertubular capillary walls and basement membranes into the tubule epithelial cell.
• Indirectly stimulate vasoconstriction of the intertubular capillaries, thus causing ischemia, which further compromises renal function.
One of the first events in renal tubular cell damage is altered ion transport at the luminal surface (uptake). This process results in decreased sodium absorption and increased sodium ions in the lumens of the distal tubules, which stimulate the renin-angiotensin mechanism, causing vasoconstriction and reduced blood flow that result in ischemia and tubular cell damage. Nephrotoxins usually do not damage the tubular basement membranes, and thus regeneration (repair) of tubules can occur in an orderly and expeditious manner. The intact basement membrane acts as a scaffold over which regenerating epithelial cells may slide. Exposure to a variety of nephrotoxins, either from the vasculature (including certain chemicals [glycolaldehyde, glycolic acid, and glyoxylic acid]) or from the tubular lumen (including certain antibiotics [aminoglycosides], pigments [hemoglobin], metals [lead], or chemicals [ethylene glycol–induced calcium oxalate crystals]), or their metabolites cause cells to undergo degeneration followed by necrosis and desquamation into the tubular lumen.
Cell death results from decreased adenosine triphosphate (ATP) production, which is central to many of the secondary metabolic derangements, including calcium ion influx, purine depletion, metabolic acidosis, and generation of oxygen radicals. Increased intracellular calcium is associated with degenerative changes in renal tubular cells, smooth muscle cells, and mesangial cells. Oxygen radicals activate phospholipase, which subsequently increases membrane permeability. Because mitochondrial respiration is disrupted, further cell membrane damage occurs.
Notably reduced renal perfusion from any cause can result in tubular necrosis. Severe hypotension associated with shock results in preglomerular vasoconstriction and reduced glomerular filtration. The resulting renal ischemia can produce sublethal tubular cell injury and dysfunction or cause cell death by necrosis or apoptosis. Following less severe insults and within different portions of the renal tubule, apoptosis may occur in lieu of necrosis. The apoptotic pathway can be triggered by the following:
• Binding of ligands to the tumor necrosis factor (TNF) superfamily
• Deficiency of cellular growth factors
• Imbalance between proapoptotic and antiapoptotic oncogenes
• Alteration of other mediators of apoptotic signaling pathways such as reactive oxygen metabolites, caspases, and ceramide
Proximal tubular epithelium has a microvillous border, which amplifies absorptive surface area and cellular junctional complexes that structurally polarize the cell so that membrane phospholipids and specialized proteins remain in the appropriate domains. The integrity of these cellular structures is critical to absorption and secretion. Early structural changes after ischemic insult include formation of apical blebs, loss of brush border, loss of cellular polarity, disruption of tight junctions, and sloughing of cells, which result in intratubular cast formation (Fig. 11-12).
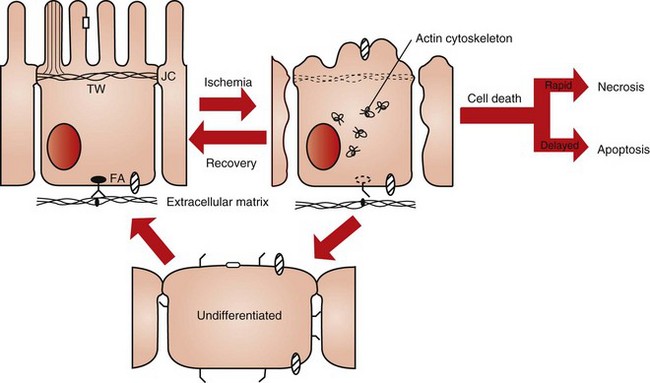
Fig. 11-12 Schematic diagram of the effect of ischemia on cell structure and function.
A polarized renal proximal tubule cell with a well-developed actin cortical cytoskeleton is shown on the left. Also shown is attachment to the extracellular matrix (ECM) via integrins. Following ischemic injury there is extensive disruption, redistribution, and aggregation of the actin cytoskeleton resulting in loss of microvilli structure, blebbing of microvilli into the lumen, detachment of cells from the ECM, and opening of junctional complexes (JC). Injured proximal tubule cells can undergo primary repair and recover directly into a polarized epithelial cell. Cells can also go through an undifferentiated phase followed by redifferentiation, or cells can die either rapidly via necrosis or in a much slower programmed fashion known as apoptosis. The primary route of cellular repair involves direct recovery. The percentage of cells reverting to an undifferentiated state or dying depends on the severity of the injury and the location within the kidney. FA, Focal adhesions; TW, terminal web. (Redrawn from Molitoris BA, Marrs J: Am J Med 106:583-592, 1999.)
Damage to the cellular cytoskeleton modifies cell polarity, cell-to-cell interactions, and cell-matrix interactions. Initially, ischemic damage modifies cell polarity by disruption of the terminal web and disassembly of the microvillar actin cores. This is followed by conversion of G actin to F actin and its redistribution from the apical cell component to form diffuse aggregates throughout the cytoplasm (Figs. 11-13 and 11-14). Cells are attached to each other by junctional complexes, tight junctions, and adherens junctions and to the ECM by integrins. Several mechanisms contribute to tight junction disruption, which is manifested as alteration in cellular permeability and cell polarity. The contributing mechanisms include redistribution of membrane lipids and proteins, such as Na+/K+-ATPase, to the apical membrane after alteration of the actin cytoskeleton and redistribution of integrins to the apical cell surface so that cell desquamation occurs. The former results in deranged sodium handling by the proximal tubular cell.
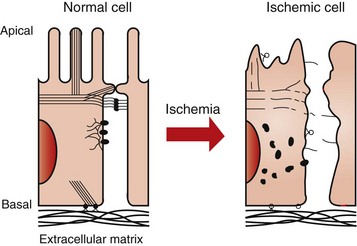
Fig. 11-13 Schematic diagram of the effect of ischemia on the actin cytoskeleton and the cytoskeletal-surface membrane interactions in proximal tubule cells.
During ischemia, alterations in the actin cytoskeleton involve disruption of the actin cytoskeleton with redistribution and aggregation of actin throughout the cytoplasm. Consequently, notable alterations occur in cytoskeletal-surface membrane interactions. Loss of cell-cell adhesion, cell-matrix adhesion, and polarity of surface membrane proteins during ischemia play a role in the diminished glomerular filtration rate that is the hallmark of ischemic acute renal failure. See Fig. 11-14 for more detail about the actin cytoskeleton and the cytoskeletal-surface membrane. (Redrawn from Sutton TA, Molitoris BA: Sem Nephr 18(5):490-497, 1998.)
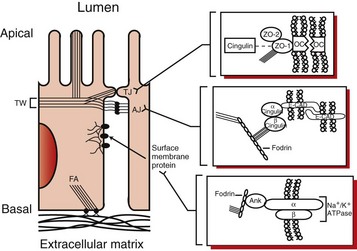
Fig. 11-14 Schematic diagram of the actin cytoskeleton and the cytoskeletal-surface membrane interactions in proximal tubule cells.
Microvillar F-actin filaments extend into the apical actin network termed the terminal web (TW) and are bound together by villin and attached to the surface membrane by myosin and ezrin to form the structural core of the apical microvilli. The actin cytoskeleton associates with junctional complexes involved in cell-cell interactions, including the tight junction and the adherens junction. More detailed schematics of the tight junction (TJ) and the adherens junction (AJ) appear to the right and demonstrate the interaction of F-actin filaments with TJ and AJ protein complexes. OC represents occludin in the schematic of the TJ, and E-CAD represents E-cadherin in the schematic of the AJ. The cortical actin network associates with surface membrane proteins, such as the sodium-potassium adenosinetriphosphatase, which is demonstrated by the detailed schematic to the lower right. Ank represents ankyrin in this schematic. Finally the actin cytoskeleton associates with structures involved in cell-matrix interactions, including focal adhesions (FA). F-actin filaments (stress fibers) possibly bundled together by myosin II associate with a protein complex at sites where integrins bind to the extracellular matrix (ECM). (Redrawn from Sutton TA, Molitoris BA: Sem Nephr 18(5):490-497, 1998.)
Animals with severe tubular necrosis have accompanying functional derangements of vascular, tubular, and/or glomerular origin. Vascular derangements include the following:
Prolonged ischemia can produce a paradoxic response of the autoregulatory system, where increased glomerular capillary resistance from tubular fluid stasis results in activation of afferent arteriolar vasoconstriction. Decreased production of or response to vasodilative factors, such as prostaglandin and atrial natriuretic peptide, also contribute. Afferent arteriole vasoconstriction, back leak of fluid, and tubular obstruction account for decreased glomerular filtration rate (GFR) (Fig. 11-15).
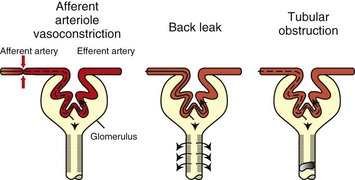
Fig. 11-15 Schematic diagram of the mechanisms of decreased glomerular filtration rate (GFR) during ischemic acute renal failure.
Proposed mechanisms for the decrease in GFR that occurs during ischemic acute renal failure include afferent arteriole vasoconstriction, back leak of glomerular filtrate, and tubular obstruction. All three of these mechanisms relate to ischemia-induced alterations in proximal tubule cells. Deranged proximal tubule handling of sodium leads to a high delivery of sodium to the macula densa, which in turn causes afferent arteriole vasoconstriction via tubuloglomerular feedback. Afferent arteriole vasoconstriction reduces glomerular capillary pressure and therefore GFR. Altered cell-cell adhesion results in an open tight junction that leads to increased paracellular permeability and subsequent back leak of glomerular filtrate from the tubular lumen into the extracellular space and ultimately into the bloodstream. Disrupted cell-matrix adhesion and abnormal cell-cell adhesion leads to cellular cast formation, which obstructs the tubular lumen and causes increased tubular pressure resulting in diminished or no GFR. (Redrawn from Sutton TA, Molitoris BA: Sem Nephr 18(5):490-497, 1998.)
Tubuloglomerular feedback is the mechanism by which GFR is matched to the solute load and the solute handling characteristics of the tubules. Because of altered sodium handling, increased levels reach the macula densa, and activation of the renin-angiotensin system occurs. This is followed by intrarenal vasoconstriction, particularly affecting outer cortical nephrons, and results in decreased glomerular blood flow, decreased filtration, and reduced formation of urine.
Gross Lesions of Acute Tubular Necrosis: On gross necropsy, the recognition of acute tubular necrosis is often difficult. Nevertheless, initially the cortex is swollen, pale mahogany to beige, and with a slightly translucent smooth, thinned, capsular surface. The cut surface of the renal cortex bulges and is excessively moist; striations are muted or accentuated by radially oriented opaque, white streaks, presumably related to the stage of the necrosis, with coagulation necrosis being responsible for the white streaks. The medulla is either pale or diffusely congested.
Microscopic Lesions of Acute Tubular Necrosis: The microscopic appearance of kidneys with acute tubular necrosis varies, depending on the following:
• The extent of the tubular necrosis
• The duration of exposure to the damaging agent
• The length of time between the injury and death, in other words the stage of necrosis or dissolution of necrotic epithelium
Initially, tubular necrosis is randomly distributed in nephrons, but the proximal convoluted tubules are most severely affected because of their high metabolic demands and first line of exposure (Fig. 11-16). Prolonged ischemia can produce necrosis of epithelium of the proximal and distal convoluted tubules, the loops of Henle, and the collecting ducts throughout the cortex and to a lesser extent, the medulla. Glomeruli are resistant to ischemia and often remain morphologically normal, even when ischemia is prolonged. Initially, proximal tubular epithelium is swollen, and the cytoplasm is vacuolated or granular and intensely eosinophilic, all features indicative of necrosis (Fig. 11-17). In such cells, the nuclear changes are pyknosis, karyorrhexis, or karyolysis. Necrotic tubular epithelium is subsequently sloughed into tubular lumens resulting in dilated, notably hypocellular tubules that contain necrotic cellular debris and hyalinized or granular casts.
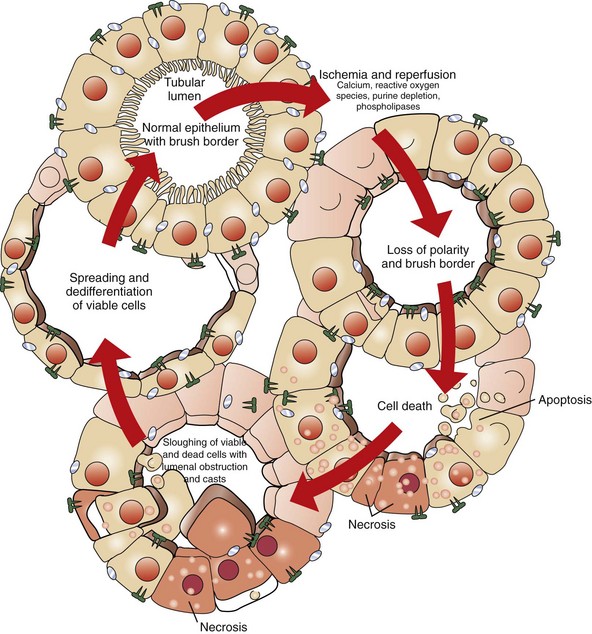
Fig. 11-16 Schematic diagram of the effects of ischemia and reperfusion.
After ischemia and reperfusion, morphologic changes occur in the proximal tubules, including loss of the brush border, loss of polarity, and redistribution of integrins and sodium-potassium adenosinetriphosphatase to the apical surface. Calcium, reactive oxygen species, purine depletion, and phospholipases probably have a role in these changes in morphology and polarity and in the subsequent cell death that occurs as a result of necrosis and apoptosis. There is a sloughing of viable and nonviable cells into the tubular lumen, resulting in the formation of casts and luminal obstruction and contributing to the reduction in the glomerular filtration rate. The severely damaged kidney can completely restore its structure and function. Spreading and dedifferentiation of viable cells occur during recovery from ischemic acute renal failure, which duplicates aspects of normal renal development. A variety of growth factors probably contribute to the restoration of a normal tubular epithelium. (Redrawn from Thadhani R, Pascual M, Bonventre JV: N Engl J Med 334(22):1448-1460, 1996. Color from Molitoris BA, Finn WF, eds: Acute renal failure: a companion to Brenner and Rector’s the kidney, Philadelphia, 2001, Saunders.)
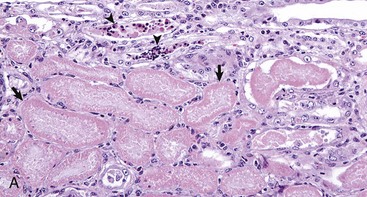
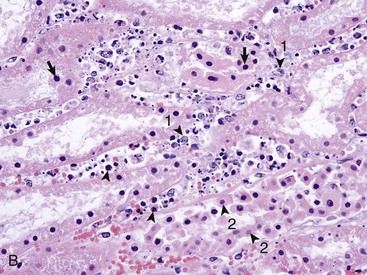
Fig. 11-17 Acute tubular necrosis, kidney, proximal tubules, cat.
A, This lesion is characterized primarily by coagulation necrosis of tubular epithelial cells (arrows) and nuclear pyknosis and intratubular nuclear and proteinaceous debris (arrowheads). H&E stain. B, This lesion is characterized primarily by nuclear pyknosis (arrows), karyorrhexis (arrowheads), and karyolysis (arrowheads 1) with intratubular nuclear and proteinaceous debris and coagulation necrosis with detachment of the epithelium from the tubular basement membrane (arrowheads 2). H&E stain. (Courtesy Dr. J. F. Zachary, College of Veterinary Medicine, University of Illinois.)
A characteristic histologic lesion of ischemic tubular necrosis is possible disruption of the tubular basement membranes, referred to as tubulorrhexis (Fig. 11-18). Tubular repair in these kidneys is imperfect because regenerating epithelial cells do not have their normal scaffolding. Tubules that remain in an affected site are less functional in resorption, can be dilated and lined by flattened epithelium, or are notably atrophic, appearing shrunken with a collapsed lumen lined by flattened epithelium and fail to heal completely by regeneration, resulting in tubular atrophy.
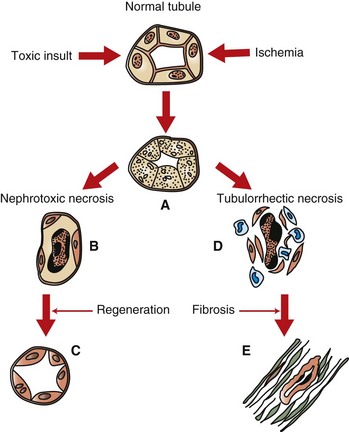
Fig. 11-18 Schematic diagram of acute tubular necrosis, kidney, proximal tubules.
Acute tubular necrosis results from a nephrotoxin or ischemia. A, Both insults cause acute necrosis characterized by cellular swelling, pyknosis, karyorrhexis, and karyolysis. B, Subsequent to nephrotoxic necrosis, there is sloughing of necrotic epithelium into the tubular lumina. The basement membranes remain intact and act as a scaffold for (C) tubular epithelial regeneration to occur. D, Ischemia may result in tubulorrhexis. Necrotic epithelial cells slough into the tubular lumen, the basement membrane is disrupted, macrophages infiltrate, and fibroblasts proliferate. E, Fibrosis with tubular atrophy results.
Nephrosis
Nephrosis is a form of acute tubular injury that is not caused by inflammation. Most commonly, it is caused by tubular ischemia or nephrotoxins as described in the sections on acute tubular necrosis and is often exacerbated by hemoglobin (hemoglobinuric nephrosis) or myoglobin pigments (myoglobinuric nephrosis).
Miscellaneous Responses to Injury of Renal Tubules
Lipofuscinosis: Fine golden granules of brown iron free pigment with the staining characteristics of lipofuscin (“wear and tear pigment”) can accumulate in renal epithelial cells of old cattle and in striated muscle, resulting in lipofuscinosis. Grossly, the renal cortex can have streaks of brown discoloration, but renal function is not affected. Microscopically, the accumulations are noted most prominently within the proximal convoluted epithelial cells. The direct cause is not known, but it is suspected that accumulations result from previous cell membrane breakdown and subsequent storage of end-product in the tubular epithelial cells.
Hydropic Degeneration: Hydropic degeneration is defined as cloudy swelling and because it is a degenerative change can sometimes be reversed. While rare in the kidney, a severe hydropic degeneration of the proximal convoluted tubules and the ascending loop of Henle, has been observed after intravenous administration of hypertonic solutions such as dextrose.
Glycogenic Degeneration: Abundant cytoplasmic vacuolation of tubular epithelium of the outer medulla and inner cortex is seen in dogs and cats with diabetes mellitus. Glycogen can be demonstrated as the accumulating compound within the cells of the ascending limb. Treatment with insulin relieves the deposition. The change is not thought to affect renal function.
Fat: Proximal tubular epithelial cell cytoplasm is often expanded by accumulations of lipid especially in cats. This is a normal storage location and is recognized as large clearings within the renal tubular epithelial cells, most prominently the proximal tubules. There is not thought to be any significant alteration to renal function.
Hemosiderin and Ferritin: Pigment can be present in the renal tubules. The origin of hemosiderin pigment is most likely from degradation of hemoglobin resorbed from the glomerular filtrate by proximal tubular epithelium. However, a history or concurrent lesions of a prior hemolytic crisis are often lacking. In dogs, microscopic granules of hemosiderin are frequent incidental findings in the cytoplasm of proximal convoluted tubular epithelial cells in kidneys that are otherwise normal.
Cloisonné kidneys, which occur in goats, are the result of proximal tubular basement membrane thickening as a result of deposits of ferritin and hemosiderin. Grossly, these kidneys have diffuse, intense, black or brown discoloration of the cortex (Fig. 11-19). The medulla is spared. Although this lesion is striking, renal function is normal.
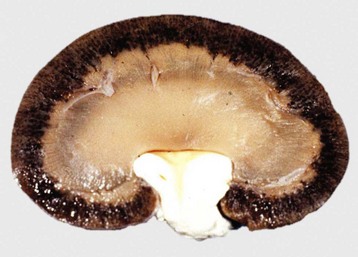
Fig. 11-19 Cloisonné kidney, dorsal section, goat.
The cortex is diffusely black; the medulla is unaffected. (Courtesy Dr. J. King, College of Veterinary Medicine, Cornell University.)
Other causes of incidental tubular changes include the following:
• Vacuolation of renal tubular epithelium in the lysosomal storage diseases, such as feline sphingomyelinosis and ovine GM1 gangliosidosis
• Intranuclear eosinophilic crystalline pseudoinclusions (so-called crystalloids), which occur in renal tubular epithelium of old dogs. These can be round or rectangular and often greatly distort the nuclei.
Responses of the Interstitium to Injury
In instances of acute interstitial nephritis, especially those caused by septicemia, there can be an accompanying hyperemia of renal vasculature within the interstitium.
Edema
Similarly, in instances of acute inflammation targeting the interstitium, vascular leakage of high protein fluid (edema) can be the end result. Additionally, tubular damage, especially with basement membrane breach, allows for more extensive interstitial edema accumulation.
Inflammatory Infiltrates
Interstitial inflammation is a consistent component of injury to the interstitium, whether acute or chronic, focal or generalized, or suppurative or nonsuppurative. Suppurative interstitial inflammation is typically seen in instances of hematogenous bacteremia. A variety of renal insults result in release of a wide array of cytokines and growth factors that stimulate interstitial inflammation, particularly monocyte infiltration. Fibroblasts often become activated and fibrosis ensues.
Fibrosis
Once interstitial fibroblasts become activated, fibrosis can ensue. Often, at this stage, the underlying or inciting pathogen is not present and thus its role is not determined. Recurrent bouts of fibrosis continue and set up a vicious cycle of loss and scarring, so that the result is a common endpoint, known as end-stage kidney.
Lymphofollicular Inflammation
Lymphofollicular inflammation is the most common response to Leptospira infection of the kidney. Severe multinodular lymphocytic inflammatory reaction is confined to the cortex. The reaction subsides slowly and inflammatory cells decrease in numbers and the degree of fibrosis increases. Similarly, with chronic recurrent bouts of pyelonephritis, lymphocyte-rich inflammatory infiltrates are noted within the interstitium.
Interstitial Nephritis
When interstitial inflammation, mounted against the veins, arteries, lymphatic vessels, or connective tissues of kidneys appears to be a primary lesion, it has traditionally been called interstitial nephritis and may have an infectious or noninfectious cause and is acute, subacute, or chronic in its duration. Interstitial nephritis is traditionally associated with a lymphoplasmacytic infiltrate; however, other types of leukocytes can also be present. In many of these diseases, the inflammatory cell infiltrates are visible only microscopically, are not associated with renal failure, and are generally inconsequential (examples are canine ehrlichiosis and equine infectious anemia). When the renal interstitium is the site of moderate-to-severe, grossly visible, interstitial inflammatory cell infiltrates and fibrosis, renal failure can occur.
The renal interstitium is the fibrovascular stroma that surrounds the nephron and is significantly involved in renal diseases, whether this is of primary interstitial origin as in interstitial nephritis or subsequent to tubular damage, when it is referred to as tubulointerstitial disease. Interstitial inflammation occurs as the result of ascending urinary tract infections (pyelonephritis), systemically derived infections of tubules and interstitium, toxins, or secondary to injury of tubules or glomeruli. Common acute lesions of the interstitium in response to toxins and tubular necrosis include edema, hemorrhage, and inflammation characterized by infiltration of neutrophils. As lesions become subacute to chronic, neutrophils become less prominent, and in several diseases, infiltrates of macrophages, lymphocytes, and plasma cells predominate. With chronic injury to or after atrophy of nephrons, fibrosis of the interstitium can be severe, resulting in notable reduction in nephron function and accentuation of renal disease.
Tubulointerstitial Nephritis
More recently, the term tubulointerstitial nephritis has been used to characterize a group of inflammatory diseases that involve the interstitium and tubules. Acute tubulointerstitial disease includes a group of processes, namely, inflammation secondary to acute tubular necrosis, whereas chronic tubulointerstitial processes include the progression with time or instances in which the interstitium is the primary target.
Tubulointerstitial nephritis can result from bacterial or viral septicemias, in which these infectious agents first infect the kidney tubules and damage them, which then incites an inflammatory response in the interstitium (Box 11-5). Acute tubulointerstitial nephritis is characterized by the presence of inflammatory cells (principally neutrophils) within the interstitium and may result from toxicoses or from acute infection with agents such as Leptospira (see Fig. 11-73), adenoviruses, lentiviruses, or herpesviruses. Chronic tubulointerstitial nephritis (Fig. 11-20, A) is a less well-characterized entity in dogs, but atrophy of tubular segments is a significant feature of this syndrome along with sparse mononuclear cell infiltration, cortical and medullary fibrosis (Fig. 11-20, B and C), variable degrees of tubular and glomerular atrophy and/or sclerosis, and compromised nephron function.
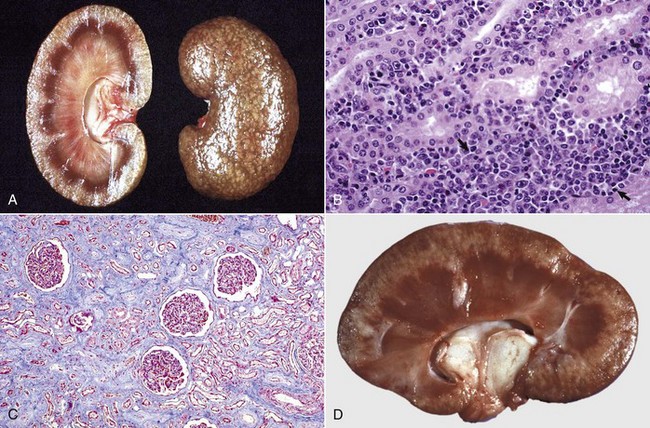
Fig. 11-20 Chronic tubulointerstitial nephritis.
A, Kidney, dorsal surface and dorsal section, dog. Note the nodularity of the capsular surface (right) from cortical interstitial fibrosis and the reduced width of the cortex (atrophy) (left). B, Kidney, dorsal section, dog. There is an intense lymphoplasmacytic interstitial infiltrate (arrows). H&E stain. C, Exotic zoo animal. This lesion is characterized by cortical and medullary fibrosis, variable degrees of tubular atrophy, and mononuclear cell interstitial infiltrate. Masson trichrome stain. D, Leptospirosis, dog. The pale streaks and foci in the cortex are chiefly interstitial lymphoplasmacytic infiltrates. (A and C courtesy Dr. A. Confer, College of Veterinary Medicine, Oklahoma State University. B courtesy Dr. Abdy, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. D courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
The pathogenesis of leptospirosis is discussed as an example of acute bacterial tubulointerstitial nephritis, the most well-understood causes of which include the following serovars of Leptospira interrogans (Fig. 11-20, D):
• Serovars canicola and icterohaemorrhagiae are the most common causes of canine leptospirosis (see the section on Disorders of Dogs).
• Serovar pomona is the most common cause of the lesion in pigs and less consistently in cattle (see the section on Disorders of Ruminants).
• Other serovars, such as grippotyphosa and bratislava, have also been associated with renal leptospirosis in several animal species.
Mechanism of Injury of Tubulointerstitial Nephritis: Three theories on the cause of chronic tubulointerstitial nephritis are (1) focal acute interstitial nephritis evolves into the chronic form, (2) it is produced as a secondary manifestation of chronic glomerulonephritis (GN) or chronic pyelonephritis, or (3) it is produced after immune-mediated damage to the renal tubules and interstitium. There are few acute tubulointerstitial diseases to account for the large number of cases of chronic tubulointerstitial nephritis seen in dogs, and thus the first theory cannot account for a high percentage of the cases. As diagnostic techniques improve, some cases of chronic tubulointerstitial nephritis may be reclassified more specifically as either chronic pyelonephritis or secondary to chronic GN.
The mechanism of injury is known in a few instances, most specifically with Leptospira infections. After exposure to the organism, leptospiremia occurs and then organisms do the following:
• Localize in the renal interstitial capillaries
• Migrate through vascular endothelium
• Persist in the interstitial spaces
• Migrate via the lateral intercellular junctions of tubular epithelial cells to reach renal tubular lumina
• Associate with epithelial microvilli
• Persist within phagosomes of the epithelial cells of the proximal and distal convoluted tubules
• Induce tubular epithelial cells to undergo degeneration and necrosis as a result of either direct toxic effects of the leptospires or the accompanying interstitial inflammatory reaction
Although neutrophils can be present in tubular lumina, the predominant chronic lesion is an infiltrate of monocytes, macrophages, lymphocytes, and plasma cells in the interstitium (chiefly cortical) (see Fig. 11-20, D). In affected dogs, interstitial plasma cells secrete Leptospira-specific antibodies. However, the role of those antibodies in pathogenesis or resolution of the lesion is not known.
Another well-documented mechanism for the production of tubulointerstitial nephritis is the immune response that develops secondary to canine adenoviral infection. The sequence of events includes the following:
• Localization of virus in the glomeruli (viral glomerulitis) during the viremic phase of the disease
• Production of a transient immune-complex GN
• Recovery from the acute phase of the disease
• Onset of the systemic immune response
• Disappearance of virus from the glomeruli only to reappear in tubular epithelial cells in various portions of the nephron in basophilic intranuclear viral inclusions
• Persistence of virus in tubular epithelium for weeks to months
• Production of tubular epithelial necrosis as a result of viral-induced cytolysis
• Production of chronic lymphocytic, plasmacytic, and less commonly histiocytic interstitial nephritis
Infection with equine arteritis virus or porcine reproductive and respiratory syndrome (PRRS) virus often results in multifocal lymphohistiocytic chronic tubulointerstitial nephritis with interstitial edema. Lesions can involve any area of the cortex but are especially intense in the medulla and at the corticomedullary junction. A severe vasculitis, characterized by fibrinoid necrosis and lymphohistiocytic infiltrates that involve the adventitial and medial layers of cortical and medullary arteries and veins, is present. Virus can be found in endothelium and in macrophages.
Deposition of immune complexes in or interactions between antibasement membrane antibodies and tubular basement membranes can initiate immune-mediated tubulointerstitial disease in humans and laboratory animals. Depositions of immunoglobulin (Ig) and complement have rarely been identified in renal tubular basement membranes in domestic animals, but administration of preformed complexes (bovine serum albumin and antibody) to dogs demonstrated that these complexes interacted with proximal renal tubules not glomeruli. Damaged tubules respond with epithelial cell proliferation, basement membrane thickening, and peritubular fibrosis. At present, the role of immune-mediated mechanisms in tubulointerstitial nephritis in domestic animals is unclear.
Gross Lesions of Tubulointerstitial Nephritis: Gross lesions can be classified as acute, subacute, or chronic; chronic tubulointerstitial nephritis is discussed in more detail later (see the section on Renal Fibrosis). The distribution of lesions can be diffuse as in canine leptospirosis (see Fig. 11-73) or multifocal as in “white spotted kidneys” of calves as the result of Escherichia coli septicemia (see Fig. 11-70), infectious canine hepatitis (see Fig. 11-43), canine herpesvirus infection (see Fig. 11-74), malignant catarrhal fever, or porcine and bovine leptospirosis (see Fig. 11-73). In diffuse tubulointerstitial nephritis, kidneys can be swollen and pale tan with a random gray mottling of the capsular surface. The cut surface bulges; gray infiltrates of varying sizes and intensities obscure the normal radially striated cortical architecture. These renal lesions usually are manifested as coalescing gray foci that are particularly intense in the inner cortex. Focal lesions of tubulointerstitial nephritis are less extensive and composed of more discrete gray areas in the cortex and outer medulla.
Microscopic Lesions of Tubulointerstitial Nephritis: Microscopically, aggregates of lymphocytes, plasma cells, monocytes, and fewer neutrophils are randomly scattered or intensely localized throughout the edematous interstitium. Tubular epithelial cells within severely inflamed areas can be degenerate, necrotic, or both, and profound tubular loss is usually accompanied by eventual replacement fibrosis.
Renal Fibrosis (Scarring)
The alternative to regeneration is irreparable damage that results in functional tubular loss when cuboidal epithelium is replaced by nonabsorptive cuboidal or squamous cells or actual physical loss of tubules so that the nephron is lost. This can occur after either an ischemic insult or exposure to a limited number of nephrotoxins. The ultimate result is replacement fibrosis/scarring. This is seen most commonly if the following occur:
• The basement membrane does not remain intact.
• Adequate tubular epithelium does not survive the toxic dose to allow for complete repair.
Fibrosis with a finely granular pattern can occur subsequent to widespread necrosis of renal tubular epithelium (acute tubular necrosis). An example is oak poisoning of cattle (see Fig. 11-69), in which severe tubular necrosis extends to the level of the renal tubular basement membrane, resulting in leakage of tubular contents. The loss of the continuity of the basement membrane prevents orderly tubular epithelial cell regeneration, which can be followed by interstitial fibrosis. Recent experimental studies have demonstrated that after severe nephrotoxin-induced tubular epithelial cell injury, the remaining cells undergo accelerated apoptosis resulting in tubular atrophy, interstitial fibroblast proliferation, and eventual fibrosis.
Renal fibrosis is the replacement of renal parenchyma, including tubules, glomeruli, and interstitium with mature fibrous connective tissue. It can occur as a primary event, but more frequently is a manifestation of the healing phase of a preexisting tubular or glomerular lesion. It is the common endpoint of all reparative stages and results when conditions are not conducive for healing of the tubular epithelium by regeneration. Regeneration of nephrons as a whole is not possible. Renal fibrosis follows many renal lesions, including primary inflammation of glomeruli (GN), tubules, or interstitial tissue (tubulointerstitial nephritis) (Fig. 11-21) and necrosis of renal tubules. Its severity usually parallels the intensity of the primary renal disease. The mechanisms by which fibrosis is induced are related to destruction and loss of nephron components by inflammatory or less commonly noninflammatory processes. T lymphocytes and interleukin-6 (IL-6) play important roles in renal fibrosis. Renal fibrosis is seen commonly following any number of renal insults and includes the following:
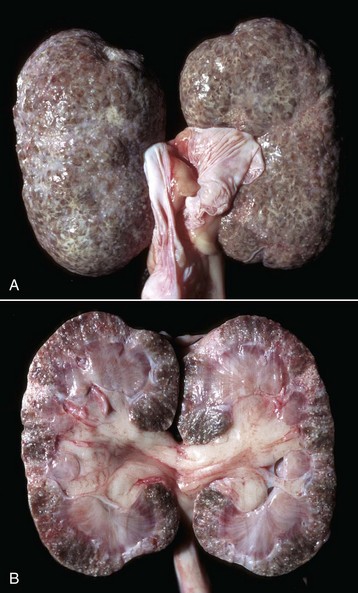
Fig. 11-21 Chronic interstitial nephritis, kidney, dog.
A, Diffuse interstitial fibrosis is responsible for the fine pitting of the capsular cortical surface, which is stippled red, the result of bands of fibrous tissue (gray) surrounding islands of renal cortex. B, Dorsal section. The cortex is pitted and granular because of multiple linear and focal scars, and it is also thinner than normal (atrophic). (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Renal fibrosis may manifest in a multitude of grossly recognizable forms as described previously. Generally, fibrotic kidneys are recognized grossly by the pale, tan-to-white, shrunken, pitted, and firm consistency, along with excessive adhesions of the capsule to the underlying cortex. Fibrosis can be diffuse and finely stippled with pinpoint dimpling and granularity on the capsular surface, or it can be coarser as seen by deep and irregularly shaped depressions of the capsular surface in either a diffuse, multifocal, or patchy distribution. In addition to these changes of the capsular surface, the cut surface of the cortex is thinned beneath the capsular surface depressions, and these fibrotic areas are pale tan when compared with more normal parenchyma.
Microscopically, renal fibrosis is characterized by an increase in interstitial connective tissue and absence of renal tubules (Fig. 11-22, A). Remaining tubules are usually atrophic and have a reduced luminal diameter or can appear ectatic because they are lined by flattened epithelium, producing an enlarged luminal diameter. A thickened hyalinized basement membrane and a lining of flattened epithelium (squamous or low cuboidal) are also characteristic. Multiple acquired cysts can be present throughout the cortex and medulla and can be a result of either dilated Bowman’s capsules and associated atrophic glomerular tufts or nephrons whose tubules have segments compressed by connective tissue (Fig. 11-22, B). Even in fibrotic lesions that are not the result of an infectious disease or inflammation, foci of lymphocytes and plasma cells can be seen randomly scattered throughout the interstitium (Fig. 11-22, C). In areas of severe interstitial fibrosis, glomerulosclerosis (as an end-stage of isolated glomeruli) is common. Calcification of vessels, tubular basement membranes, Bowman’s capsules, and degenerate tubular epithelium are common in fibrotic kidneys because of alterations in calcium-phosphorus metabolism associated with chronic renal failure.
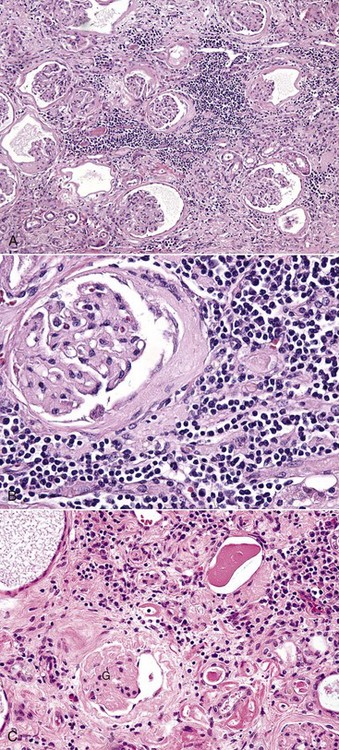
Fig. 11-22 Chronic interstitial nephritis, kidney, dog.
A, Dorsal section, cortex. This lesion is characterized by interstitial fibrosis, tubular atrophy, and interstitial inflammatory cell (lymphocytes and plasma cell) infiltration. The renal corpuscles have contracted glomeruli with increased volume of mesangial matrix and thickened Bowman’s capsules. H&E stain. B, Higher magnification of A. H&E stain. C, Cortex. Higher magnification showing interstitial fibrosis, lymphocytic inflammatory infiltrates, sclerotic glomerular tufts (G), and ectatic tubules and Bowman’s spaces. H&E stain. (A and B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois. C courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Renal fibrosis and chronic renal disease are the most frequently recognized renal pathologic processes in mature or aging domestic animals, particularly dogs and cats. When renal fibrosis and loss of nephrons are severe, these lesions can be manifested clinically as chronic renal failure and uremia. One of the most common expressions of this chronic disease is the inability of an animal to concentrate urine, resulting in frequent urination (polyuria) of dilute urine (isosthenuria). Polyuria is accompanied by dehydration and excessive water drinking (polydipsia). Hypoplastic anemia occurs as a result of the kidneys’ failure to synthesize and secrete erythropoietin. Fibrous osteodystrophy can develop because of abnormal calcium-phosphorus metabolism and renal secondary hyperparathyroidism.
End-Stage Kidneys: Without careful attention to the pattern of resultant fibrosis, such kidneys can be indiscriminately termed as end-stage kidneys; however, fibrosis generally follows a pattern characteristic of the antecedent injury and is described here for tubulointerstitial disease.
A coarser pattern of diffuse renal fibrosis occurs in chronic interstitial nephritis and certain progressive juvenile nephropathies of dogs. Both cortex and medulla can be fibrotic, cortical striations are severely distorted or effaced, and the formation of multiple cortical cysts is common. End-stage kidneys are those referred to as a result of fibrosis, mineralization, sclerotic glomeruli, and foci of hyperplastic and hypertrophic tubules. Progressive interstitial fibrosis is thought to be the final common pathway to chronic renal failure.
Responses of the Vasculature to Injury
Hyperemia refers to an increase in arterial blood flow, and congestion is an increase in venous blood pooling within the vasculature of the kidney. Renal hyperemia is an active process usually secondary to acute renal inflammation.
Renal congestion can be the following:
Hyperemic kidneys are darker red than normal, can be perceptibly swollen, and ooze blood from the cut surface. Congested kidneys are dark purple and ooze blood from the cut surface as the result of the accumulation of unoxygenated blood in the renal venous system. At necropsy, unilateral renal hypostatic congestion is present in animals that die in lateral recumbency, after which the force of gravity pulls the unclotted blood downward. Microscopically, the arterial and venous vessels are distended with blood, and if there has been sufficient time for the blood to clot, serum and blood cells may be present.
Hemorrhage and Thrombosis
Hemorrhage occurs when red blood cells extend beyond the vessel walls. Large intrarenal hemorrhages can result from direct trauma, renal needle biopsy, and systemic bleeding disorders, such as factor VIII deficiency. Subcapsular and renal cortical hemorrhages occur in association with septicemic diseases, vasculitis, vascular necrosis, thromboembolism, and disseminated intravascular coagulation (DIC). Perirenal hemorrhage has been noted with ovine herpesvirus arteritis (malignant catarrhal fever [MCF]) and of course, abdominal trauma.
Petechial hemorrhages are commonly seen on the surface and throughout the cortex of kidneys from pigs that die of viremia or septicemia caused by diseases such as hog cholera (swine fever), African swine fever, erysipelas (see Fig. 11-71), streptococcal infections, salmonellosis, and other embolic bacterial diseases (e.g., Actinobacillus spp.). Renal cortical ecchymotic hemorrhages associated with multifocal tubular and vascular necrosis are salient and diagnostically important lesions of viremia in neonatal puppies infected with herpesvirus. Portions of thrombus that break free from affected heart valves in valvular endocarditis can lodge in the glomeruli or interstitial capillaries in any species.
When DIC causes widespread thrombosis in the glomerular capillaries (Fig. 11-23), the afferent arterioles and most importantly the interlobular arteries, widespread cortical infarction results and is designated renal cortical necrosis. This lesion is not to be confused with ischemic acute tubular necrosis discussed in this chapter (see the section on Acute Tubular Necrosis). Partial or complete renal cortical necrosis is usually a bilateral lesion that occurs in all animal species, especially as a result of Gram-negative septicemias or endotoxemias, and is related to the following:
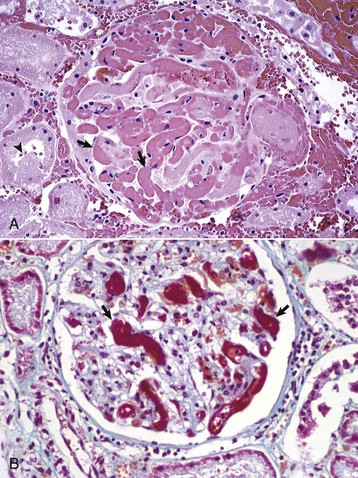
Fig. 11-23 Glomerular capillary thrombosis, kidney, glomerulus, dog.
A, Microthrombi. Capillary lumens are occluded by microthrombi (arrows) caused by disseminated intravascular coagulation. Adjacent cortical tubular epithelial cells are undergoing coagulation necrosis with nuclei undergoing pyknosis and karyolysis (arrowhead), the result of ischemia from reduced blood flow to the peritubular capillaries, which are downstream from the glomerulus. H&E stain. B, Fibrinous microthrombi. A glomerulus similar to the one shown in Fig. 11-23, A, stained to demonstrate fibrinous thrombi (arrows). Fibrin is red. Lendrum-Fraser stain for fibrin. (A courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy College of Veterinary Medicine, University of Illinois.)
The lesion can be induced experimentally in animals by two endotoxin injections 24 hours apart and is a manifestation of the generalized Shwartzman reaction. The resulting microthrombosis of vessels throughout the renal cortex results in widespread ischemia and small and large infarcts of coagulation necrosis and hemorrhage. The renal cortex can be diffusely pale with a zone of hyperemia separating the necrotic cortex from the viable medulla, or more often the cortex is a mosaic of large, irregular hemorrhagic areas, resembling hemorrhagic infarcts interspersed with large yellow-gray areas resembling pale infarcts. The necrotic tissue can involve the full width of the cortex or only the outer portion. Thrombi are demonstrable mainly in the interlobular arteries.
Infarction
Renal infarcts are areas of coagulative necrosis that result from the local ischemia of vascular occlusion and usually are due to thromboembolism.
Renal emboli are derived from the following:
• Mural thrombi on heart valves in valvular endocarditis
• Endarteritis in parasitic diseases such as canine dirofilariasis and equine strongylosis
Because of the high circulating blood volume (20% to 25% of cardiac output) through the kidney, renal infarcts from thromboemboli derived from lesions of mitral or aortic valvular vegetative endocarditis or mural endocarditis are common in many species, especially in cattle with Arcanobacterium colonization, in pigs with Erysipelas or in cats with left atrial thrombosis associated with cardiomyopathy. Rarely, emboli may occlude the renal artery, causing infarction of the entire kidney. Sometimes, emboli occlude the interlobar/arcuate arteries, causing infarction of triangular (in cross-section of the kidney) segments of the cortex and medulla. Most commonly, emboli obstruct many smaller vessels (e.g., interlobular arteries), causing multiple smaller infarcts involving only the renal cortex. In general, renal infarction can occur because of thrombosis resulting from endothelial damage of glomerular capillaries associated with a vascular disease (as in Alabama rot in greyhounds; see Fig. 11-44). Renal infarcts in horses can result from emboli lodging in the renal vasculature after mural thrombosis of the aorta, from aortic wall damage, caused by migrating larvae of Strongylus vulgaris. Thrombosis of pulmonary, coronary, splenic, or renal arteries and the resultant infarction are common in dogs with glomerular amyloidosis, which results in loss of plasma anticoagulants, such as antithrombin III, through damaged glomeruli. Endotoxin-mediated arterial or capillary thromboemboli are a common cause of infarction in association with Gram-negative sepsis or endotoxic shock. Septic emboli, particularly those from bacterial valvular endocarditis, caused by Arcanobacterium pyogenes in cattle, Erysipelothrix rhusiopathiae in pigs, and Staphylococcus aureus in small animals, can cause renal infarcts and can progress to microabscesses or granulomas, depending on the microorganism involved.
Grossly, renal infarcts appear red or pale white depending on several factors, including the interval after vascular occlusion (i.e., age of infarct) (Figs. 11-24 and 11-25). Occlusion of small interlobular arteries results in infarcts that are initially slightly swollen and red because of hemorrhage (Fig. 11-25, A) and later become pale yellow-gray within 2 to 3 days because of lysis of erythrocytes and loss of hemoglobin (Fig. 11-25, B). Initially, large infarcts develop when an embolus lodges in the interlobular artery closer to its origin from the arcuate artery. These larger infarcts have a central area of pallor (coagulative necrosis) and are usually surrounded by a peripheral red zone of congestion and hemorrhage along with a pale margin because of a surrounding zone of leukocytes (Fig. 11-25, C). Because of the loss of parenchyma, during healing infarcts are depressed below the cortical surface and later become pale and shrunken as a result of fibrosis (Fig. 11-25, D).
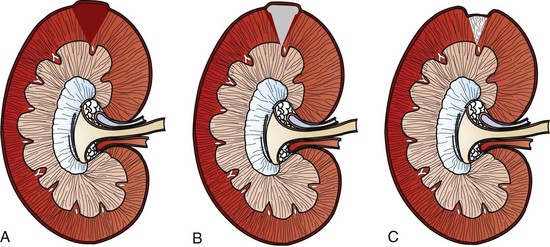
Fig. 11-24 Schematic diagram of renal infarction.
The normal progression of renal infarcts is outlined. A and B, Acute renal infarcts. Initially, renal infarcts are swollen and hemorrhagic (A). In 2 to 3 days, infarcts become pale (B), surrounded by a zone of hyperemia and hemorrhage. C, Chronic infarcts are pale, shrunken, and fibrotic, resulting in distortion and depression of the renal contour.
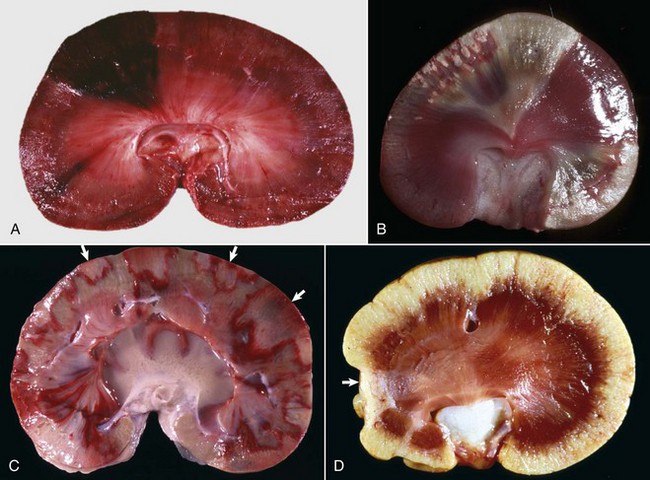
Fig. 11-25 Gross appearance of renal cortical infarcts with age, kidney, dorsal sections.
A, Acute (early) hemorrhagic infarct, dog. Focal wedge-shaped area of cortical necrosis. Note how the infarct bulges above the capsular surface due to cell swelling and hemorrhage. B, Acute pale infarcts, rabbit. There are two pale white to tan wedge-shaped infarcts (top, lower right). Note how the infarct (top) bulges above the capsular surface, indicating cell swelling. C, Subacute infarcts, dog. Multiple renal cortical infarcts are pale and surrounded by a red rim of active hyperemia (arrows). The cortical surface of many, but not all, of the infarcts is even with that of the adjacent unaffected cortex, indicating that cell swelling has subsided. D, Chronic infarct, cat. A focal pale truncated wedge-shaped scar of fibrous connective tissue has replaced the pole (arrow) of the renal cortex. Note that the surface of the infarct is below that of the adjacent normal kidney because of the loss of tissue, fibrosis, and contraction of the fibrous scar. (A courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. C courtesy Dr. K. Read, College of Veterinary Medicine, Texas A&M University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. D courtesy Dr. J. Sagartz, College of Veterinary Medicine, The Ohio State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
The scarring that follows infarction is related to several variables, including the size of the ischemic area caused by vascular compromise. Healing is by fibrosis and results in large, deeply depressed wedge-shaped (on cross-section of the kidney) scars that involve primarily the cortex but can extend into the medulla. Obstruction of smaller arterioles results in smaller areas of more superficial coagulative necrosis that heal as small-diameter pits in the renal surface, which correspond to pale white, linear scars on the cut surface.
Infarcts are often wedge-shaped in a cross-section of kidney, with the base against the cortical surface and the apex pointing toward the medulla, conforming to a zone of cortical parenchyma supplied by the site of the obstruction. Usually, infarcts are the result of blockage of an interlobular artery and exactly how large that infarct depends on where the artery is blocked. Blockage of the interlobular artery at its point of origin causes the largest infarcts, blockage toward its end in the outer cortex causes the smallest. Infarcts can involve the cortex only or the cortex and medulla depending on the size of the occluded vessel and the site of obstruction. For example, thrombosis of an arcuate artery, which supplies both cortex and medulla, would result in an infarct involving the cortex and extending partway into the medulla (see Fig. 11-8). Thrombosis of a cortical interlobular artery, which supplies mainly the cortex, would result in an infarct of the cortex only (see Fig. 11-8). Although less common, hypoxic renal necrosis as the result of venous occlusion and/or infarction is seen occasionally, and they retain their hemorrhagic appearance longer than infarcts resulting from arterial occlusion because of the continued arterial blood flow into the area.
Microscopically, in an acute infarct, nephrons (including tubules, glomeruli, and interstitium) in the central zone of the infarct become necrotic (Fig. 11-26). At the periphery of the infarct, only the proximal tubules, because of their high metabolic rate, are necrotic; the glomeruli tend to be spared. After a couple of days, the margin of the necrotic zone contains an inflammatory infiltrate consisting largely of neutrophils and fewer macrophages and lymphocytes. Capillaries adjacent to the necrotic area are notably engorged with blood (hyperemia). Healing of the infarcted area occurs by lysis and phagocytosis of the necrotic tissue and replacement by fibrous connective tissue, which matures to a discrete scar. Scars range from linear to broad depending on the size of the acute infarct. Septic infarcts are initially hemorrhagic, but because of the presence of pyogenic bacteria, the necrotic tissue undergoes liquefactive necrosis and the infarcts can eventually develop into abscesses and ultimately into substantial scars. Septic infarcts often fail to respect a solely cortical or medullary pattern of distribution caused by the extensive local inflammation that they generate.
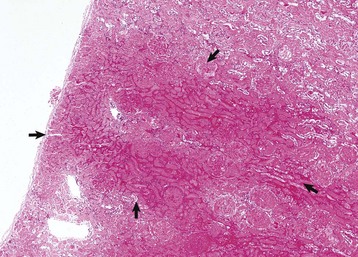
Papillary (Medullary Crest) Necrosis
See the section on Disorders of Domestic Animals for a discussion of papillary necrosis.
Structure of the Lower Urinary Tract
The lower urinary tract is the conduit for the transport of urinary waste from the kidney to the exterior through paired ureters, the urinary bladder, and the urethra.
Ureters
Macroscopically, the ureters enter the bladder wall obliquely and are covered by a mucosal flap, the vesicoureteral valve, which is an important structure because it normally prevents reflux of urine from the bladder into the ureter and renal pelvis. The normal mucosa of the ureter should be smooth and glistening. Microscopically, the ureters are lined by transitional epithelium. Histologically, the ureteral mucosa is folded longitudinally; there are poorly defined internal and external longitudinal muscle layers, a prominent middle circular muscle layer and externally, either adventitia or peritoneal serosa.
Urinary Bladder
Macroscopically at death, the urinary bladder can contract to such a degree that the normal bladder wall appears thick on necropsy. The normal mucosa of the urinary bladder should be smooth and glistening. Urine should be clear except in horses, where it is cloudy because of the normal presence of mucus and crystalline material produced by the branched tubuloalveolar mucous glands in the submucosa of the renal pelvis and proximal ureter. Microscopically, as in other mucous membranes, the lamina propria has small lymphoid follicles, which, after inflammation or antigenic stimulation, can be large enough to be seen grossly as discrete, circular, white foci (1 to 2 mm) in the mucosa. Histologically, the bladder is an expanded ureter, lined by pseudostratified transitional epithelium ranging from 3 to 14 cells thick, depending on the species and degree of distention. Histologically, the urinary bladder wall is composed of poorly defined internal and external longitudinal muscle layers, a prominent middle circular muscle layer, and externally either adventitia or peritoneal serosa.
Function of the Lower Urinary Tract
Portals of Entry
Common portals of entry for the lower urinary tract are shown in Box 11-6.
Ascending Infection
Extension from the exterior secondary to contamination from the gastrointestinal tract, the genital tract, or severe bacterial dermatitis can result in damage secondary to ascension of bacteria. This represents a distinctly unique mechanism because the lower urinary tract represents a tubular system that is blind-ended and has only one exit to the exterior (i.e., through the urethra), unlike the intestinal system, which is a continuous tubular system. This predisposes to bacterial ascension and colonization by adherent bacteria, especially in females. In males, end-stage obstruction is due partially to the narrow diameter and length of the urethra. Etiologic agents, such as bacteria, may originate from the exterior skin surface or the closely located openings of the intestinal tract and the female genitourinary tract. Bacteria with adhesion capabilities may overcome peristalsis and periodic urine flushing. Regular complete emptying will minimize risks of pathologic changes, in contrast to stasis, retention of urine, and infrequent urinations, which predispose to ascending disease.
Descending Infection
Extension from disease processes that occur within the kidney and renal pelvis, such as pyelonephritis, may account for extension of inflammation into the lower urinary tract. The presence of renal pelvic calculi may also be seen concurrently.
Direct Penetration From the Lumen
When toxic principles are excreted in the urine, they may accumulate to toxic levels because urine is stored in the urinary bladder for extended periods of time. These agents may produce lesions to the lower urinary tract as a result. Additionally, the presence of a urolith anywhere within the lower urinary tract can result in trauma to the mucosa, with accompanying edema, hemorrhage, ulceration, and in the most severe cases, rupture caused by pressure necrosis.
Defense Mechanisms
Defense mechanisms unique to the lower urinary system have evolved to counteract the typical forms of injury (Box 11-7). The most notable of these defense mechanisms of the lower urinary tract, which includes the ureters, urinary bladder, and urethra are as follows:
Responses to Injury
Ureter, Urinary Bladder, and Urethra
The predominant responses to injury include dilation and pressure necrosis caused by obstruction to the lower urinary tract system, inflammation after exposure to infectious etiologies, and/or neoplastic transformation. Additionally, concentration of excreted urinary substances, such as drug metabolites, pesticides, and other toxins, can damage the surface of the lower urinary system and predispose it to infection, secondary hyperplasia, or neoplastic responses.
Disorders of Domestic Animals
Nonrenal lesions of uremia identified clinically or at necropsy are useful indicators of renal failure (Table 11-1). The severity of nonrenal lesions of uremia depends on the length of time that the animal has survived in the uremic state. Therefore, in acute renal failure, nonrenal lesions are few, whereas many lesions can be present in chronic renal failure. Typically, nonrenal lesions can be attributed to either one of the following mechanisms:
TABLE 11-1
| Lesion | Mechanism |
| Pulmonary edema | Increased vascular permeability |
| Fibrinous pericarditis | Increased vascular permeability |
| Ulcerative and hemorrhagic gastritis | Ammonia secretion and vascular necrosis |
| Ulcerative and necrotic stomatitis | Ammonia secretion in saliva and vascular necrosis |
| Atrial and aortic thrombosis | Endothelial and subendothelial damage |
| Hypoplastic anemia | Increased erythrocyte fragility and lack of erythropoietin production in the kidney |
| Soft-tissue mineralization | Altered calcium-phosphorus metabolism (stomach, lungs, pleura, kidneys) |
| Fibrous osteodystrophy | Altered calcium-phosphorus metabolism |
| Parathyroid hyperplasia | Altered calcium-phosphorus metabolism |
• Endothelial degeneration and necrosis, resulting in vasculitis with secondary thrombosis and infarction in a variety of tissues (i.e., intestinal tract).
• Caustic injury to epithelium of the oral cavity and stomach, which results in ulcer formation, is secondary to the production of large concentrations of ammonia after splitting of salivary or gastric urea by bacteria.
Systemic nonrenal lesions of uremia include one or more of the following:
• Ulcerative and necrotic glossitis/stomatitis characterized by a brown, foul-smelling, mucoid material adherent to the eroded and ulcerated lingual and oral mucosa; ulcers are most commonly bilateral (symmetric) and present on the underside of the tongue (Fig. 11-27).
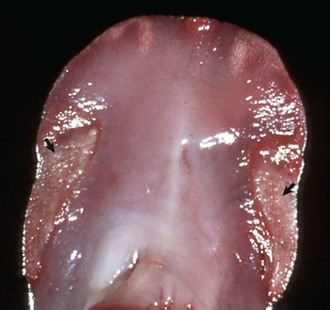
Fig. 11-27 Ulcerative glossitis, uremia, tongue, ventral surface, cat.
Bilaterally symmetrical ulcers (arrows) are present on the rostrolateral borders of the ventral surface of the tongue. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
• Ulcerative and hemorrhagic gastritis in dogs and cats (Fig. 11-28), often with secondary midzonal mineralization (Fig. 11-29). The gastric wall can be gritty when cut because of calcification of the inner and middle layers of the mucosa and the submucosal arterioles. This lesion is less commonly seen in horses and cattle, in which intestinal lesions predominate.
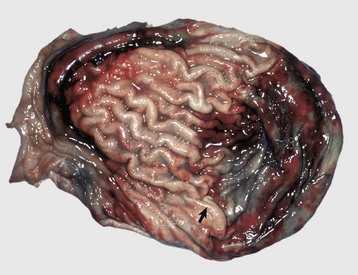
Fig. 11-28 Uremic gastritis, stomach, dog.
Because of uremia, the stomach wall is hemorrhagic (right) and the stomach contents may contain blood and mucus (not shown here). Note the edematous mucosal thickening (arrow). (Courtesy Dr. A. Confer, College of Veterinary Medicine, Oklahoma State University.)
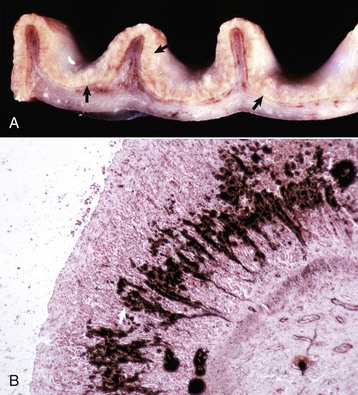
Fig. 11-29 Uremic gastritis, stomach, dog.
A, There is accentuation of the gastric rugae and calcification in the deep mucosa (arrows). B, The mucosa has laminar mineralization (black color) of gastric glands (arrow), von Kossa stain. (A courtesy Dr. J. King, College of Veterinary Medicine, Cornell University. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
• Ulcerative and hemorrhagic colitis in horses and cattle, in which large areas of colonic mucosa are often edematous and dark red because of hemorrhage. The gastrointestinal contents can be bloody and smell of ammonia. Microscopically, coagulative necrosis, hemorrhage, and a neutrophilic infiltrate occur in the intestinal mucosa. Degeneration, necrosis, and mineralization of the arteriolar intima and media are often present in the gastric mucosa and submucosa (Fig. 11-29, B).
• Intercostal mineralization/uremic mineralization is characterized, particularly in dogs, by calcification of the subpleural connective tissue of the cranial intercostal spaces (Fig. 11-30). These lesions are white-gray granular pleural thickenings with a horizontal “ladderlike” arrangement. The intercostal muscles are only superficially calcified. Patchy or diffuse pulmonary calcification of the lungs results in their failure to collapse, areas of paleness, and mild-to-moderate firmness and crunchiness and can occur occasionally in conjunction with the lesions of uremic pneumonitis and localized emphysema.

Fig. 11-30 Thoracic cavity, parietal pleura, cat.
Horizontally oriented streaks (arrows) of mineral (intercostal mineralization) are present in the subpleural intercostal connective tissue as a result of chronic uremia. (Courtesy Dr. J. King, College of Veterinary Medicine, Cornell University.)
• Fibrinous pericarditis is characterized by fine granular deposits of calcium on the epicardium (visceral pericardium).
• Diffuse pulmonary edema is characterized by alveoli that contain fibrin-rich fluid and often a mild infiltrate of macrophages and neutrophils. This lesion is also called uremic pneumonitis. The underlying lesion is a vasculitis affecting the alveolar capillaries, which results in increased vascular permeability and high protein effusion.
• Arteritis is characterized macroscopically by finely granular roughened plaques within the left atrial endocardium and less frequently in the proximal aorta and pulmonary trunk. Arteritis in conjunction with loss of the anticoagulant antithrombin III by glomerular leakage is conducive to the formation of large mural thrombi at these sites.
• Nephrocalcinosis (calcification), although usually not visible at necropsy and not a true nonrenal lesion of uremia, occurs in the kidneys (Fig. 11-31). The kidneys can be gritty when cut because of calcification of tubular basement membranes, Bowman’s capsules, and necrotic tubular epithelium, especially in the medulla and inner cortex.
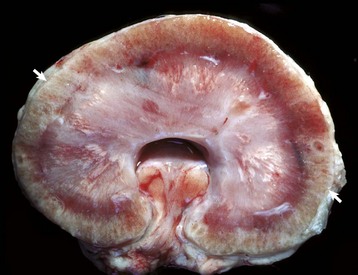
Fig. 11-31 Nephrocalcinosis, kidney, dorsal section, dog.
Note the white streaks (arrows) in the cortex and medulla attributable to mineralization of the interstitium, basement membranes, and tubules. This lesion results from diseases that increase plasma calcium concentrations (e.g., hyperparathyroidism). Renal tubular epithelium is damaged by an increase in intracellular calcium, which is initially precipitated in mitochondria and tubular basement membranes. (Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)