Bones, Joints, Tendons, and Ligaments
The skeleton consists of bones and joints and their supporting structures and is responsible for supporting and protecting the body and enabling movement initiated by the nervous system and facilitated by muscles. The skeleton can be divided into axial skeleton (head, vertebrae, ribs, and sternum) and appendicular skeleton (thoracic and pelvic limbs). Information on the methods for postmortem examination and evaluation of the skeleton is available in Web Appendix 16-1, which can be found at evolve.elsevier.com/Zachary/McGavin/.
Structure and Function of Bone
Structure and function can be discussed at the organ, tissue, and cellular levels. In this section, normal structure and function are briefly reviewed, beginning at the cellular level and including bone matrix and mineral. Cells directly involved with the structural integrity of bone include osteoblasts, osteocytes, and osteoclasts (Box 16-1).
Osteoblasts are cells on any bone surface (periosteal, endosteal, trabecular, intracortical) that produce bone matrix (osteoid), initiate the mineralization of this matrix (deposition of hydroxyapatite), and seemingly paradoxically initiate the resorption of this matrix by osteoclasts. Osteoblasts are derived from mesenchymal stem cells. Active osteoblasts are plump (Fig. 16-1), with abundant basophilic cytoplasm that is rich in rough endoplasmic reticulum and contains prominent Golgi apparatus and numerous mitochondria (Web Fig. 16-1). Inactive osteoblasts are disk-shaped with little cytoplasm because fewer organelles are needed for matrix synthesis and secretion (Fig. 16-2 and Web Fig. 16-2). Osteoblasts likely interact with osteocytes to assist in fine control of calcium homeostasis and detection of mechanical use and microscopic damage to bone, as discussed later. Indirect measurements of osteoblast activity are reflected in blood concentrations of the bone-specific isoform of alkaline phosphatase, an enzyme on the cell membrane of osteoblasts, and the noncollagenous protein, osteocalcin, that is secreted by osteoblasts and is present in bone matrix. Both are thought to have roles in mineralization and calcium ion homeostasis.
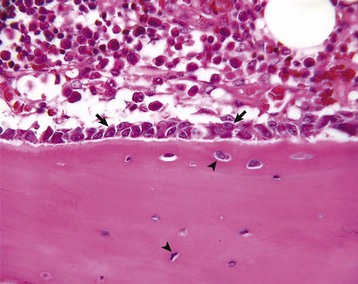
Fig. 16-1 Osteoblasts and osteocytes, long bone, young dog.
Prominent cuboidal osteoblasts (arrows) line the endosteal surface. Hematopoietic marrow is present (above the endosteal surface). More mature (older) osteocytes are present deeper in the cortex (below the endosteal surface) and are recognizable as small elliptical cells (arrowheads) residing in lacunae. The more recently embedded (younger) osteocytes are in larger lacunae closer to the endosteal surface. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
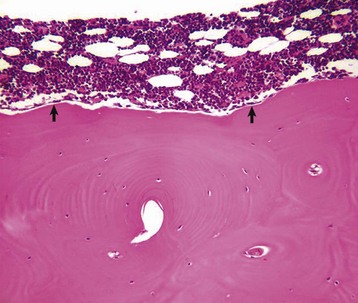
Fig. 16-2 Osteoblasts, vertebra, adult dog.
Quiescent osteoblasts (arrows) on the endosteal surface of adult bone are fusiform with little cytoplasm and form an inconspicuous layer between the cortical bone (below the endosteal surface) and active hematopoietic marrow (above the endosteal surface) of the adult axial skeleton. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
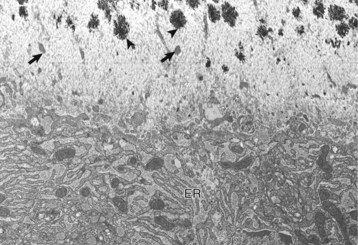
Web Fig. 16-1 Osteoblast, bone, tibia, rodent.
Osteoblast (below) with abundant endoplasmic reticulum (ER) on an actively mineralizing surface. Only a portion of the cell, not including the nucleus, is shown. Cell processes (arrows) of the osteoblasts extend out into the osteoid (above). Mineralization (black spicules) is initiated within matrix vesicles (arrowheads), then extends onto the adjacent collagen. TEM. Uranyl acetate and lead citrate stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
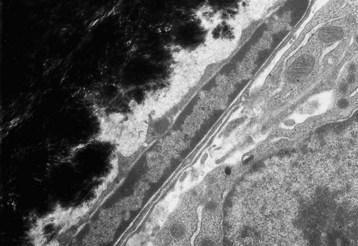
Web Fig. 16-2 Inactive osteoblast, bone, tibia, rodent.
Quiescent osteoblast has reduced cytoplasmic volume due to the less-developed or less-active organelles, which are necessary for collagen synthesis. The narrow rim of unmineralized (light gray) bone matrix between the osteoblast and the mineralized bone (black) is termed the lamina limitans. TEM. Uranyl acetate and lead citrate stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Osteocytes are osteoblasts that have been surrounded by mineralized bone matrix (Web Fig. 16-3) and are the most numerous and longest living of the bone cell types. They occupy small spaces in the bone called lacunae (singular: lacuna) and make contact with osteoblasts and other osteocytes by means of long cytoplasmic processes that pass through thin tunnels in the bone called canaliculi (singular: canaliculus). Under conditions of extreme stress to calcium homeostasis, osteocytes might have the ability to resorb perilacunar mineral and matrix, thus enlarging the lacuna (osteocytic osteolysis). This process apparently is rare and likely does not contribute significantly to development of osseous lesions. Osteocytes also retain a limited capacity to form bone and are exquisitely sensitive to mechanical strain in the form of shear stress. Other functions of osteocytes are somewhat speculative and are presented later in osteoblast-osteocyte interactions.
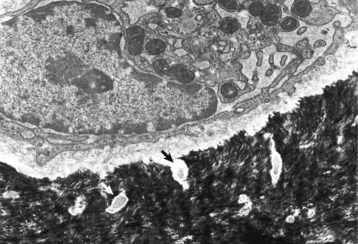
Web Fig. 16-3 Osteocyte, bone, tibia, rodent.
A recently embedded osteocyte still has residual rough endoplasmic reticulum and Golgi apparatus that were used during its osteoblastic period. Cell processes extend into the mineralized matrix (black) through tunnels called canaliculi (arrows). TEM. Uranyl acetate and lead citrate stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Osteoclasts are multinucleated cells that are derived from hematopoietic stem cells of the granulocyte-monocyte series and are responsible for bone resorption (Fig. 16-3). They have abundant eosinophilic cytoplasm and a specialized brush border along the margin of the cell that is adjacent to the bone surface that is being resorbed (Web Fig. 16-4). For osteoclasts to resorb bone, they must gain access to the bone surface that is usually covered by osteoblasts, and they must attach to the mineralized surface by transmembrane receptors in their sealing zones. These receptors bind to specific ligands in the matrix, likely residing in noncollagenous proteins. Osteoclasts are not able to bind to unmineralized bone matrix even though it contains these same ligands. Once bound to the matrix, the osteoclast resorbs bone in two stages. First, the mineral is dissolved by secretion of hydrogen ions through a proton pump located in the brush border. These hydrogen ions are derived from carbonic acid produced within the osteoclast from water and carbon dioxide by the enzyme carbonic anhydrase. Second, the collagen of the matrix is cleaved into polypeptide fragments by cysteine and metalloproteinases and cathepsins, particularly cathepsin K, released from the numerous lysosomes in the osteoclast and secreted through the brush border. The concavity in the bone created by the resorbed bone matrix is called a Howship’s lacuna, or a resorption lacuna. Physiologically, osteoclast activation is controlled by osteoblasts and bone marrow stromal cells (see later discussion of interactions). Calcitonin is a systemic inhibitor of osteoclasts. Osteoclasts have receptors for calcitonin and respond to this hormone by involuting their brush border and detaching from the bone surface. The activity of osteoclasts can be indirectly measured by determining serum concentrations of collagen degradation products (pyridinoline and deoxypyridinoline cross-links) or tartrate-resistant acid phosphatase (TRAP) activity. TRAP is a glycosylated monomeric metalloenzyme that is highly expressed by osteoclasts. TRAP staining also may be used as a cytochemical marker of osteoclasts in histologic sections (Web Fig. 16-5).
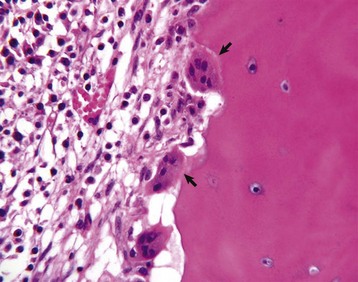
Fig. 16-3 Osteoclasts, long bone, young dog.
Several multinucleated cells (osteoclasts) (arrows) are resorbing endocortical bone and creating a scalloped endosteal surface. These scalloped cavities of resorbed bone are called Howship’s (or erosion) lacunae. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
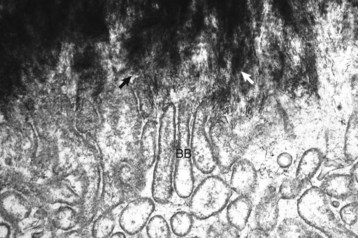
Web Fig. 16-4 Osteoclasts, brush border, bone, tibia, rodent.
Spicules of hydroxyapatite (arrows) are apparent between the projections of the osteoclast brush border (BB). The crystals have been separated from the matrix and are in the process of being dissolved by acid and enzyme secretions across the brush border. TEM. Uranyl acetate and lead citrate stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
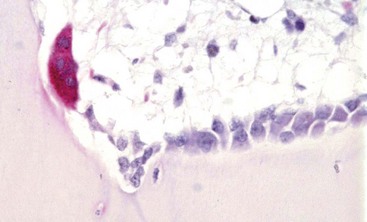
Web Fig. 16-5 Osteoclast, bone, pig.
Tartrate-resistant acid phosphatase-positive osteoclast (multinucleated cell with red cytoplasm) on the surface of mineralized bone in an area of active remodeling. Plump, cuboidal osteoblasts line the remainder of the bone surface. Marrow space is above, bone is below. TRAP stain. (Courtesy Dr. C.S. Carlson, College of Veterinary Medicine, University of Minnesota.)
Osteoblast-osteocyte interactions are apparent from their connections to each other by thin, tortuous, cytoplasmic processes. This network of osteoblasts and osteocytes forms a functional membrane that separates the extracellular fluid bathing bone surfaces from the general extracellular fluid and can regulate the flow of calcium and phosphate ions to and from the bone fluid compartment (Fig. 16-4). Because of the large surface area of perilacunar and canalicular bone available for rapid ion exchange, significant amounts of calcium can be shifted from the bone fluid compartment to the extracellular fluid compartment without structural changes within the bone. In addition, this network allows osteocytes to detect alternations in the fluid flow within the bone extracellular fluid compartment. It is thought that such flow contributes to electric currents called streaming potentials. Changes in these streaming potentials caused by altered stress and strain on the bone or disruption of these potentials by microcracks (minuscule fractures within the bone visible only microscopically) might be detected by osteocytes with subsequent signaling to the overlying osteoblasts to initiate bone formation or resorption.
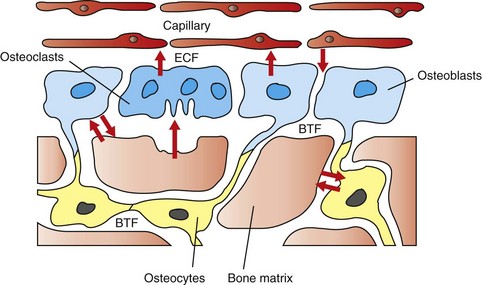
Fig. 16-4 Schematic diagram of the movement of calcium.
The hypothesized intracellular and extracellular movement of calcium (arrows) in bone and the relationships of osteoblasts (blue, single nuclei), osteocytes (yellow), and osteoclasts (blue, multiple nuclei) to blood vessels, extracellular fluid (ECF), and bone tissue fluid (BTF) compartment is shown. (Redrawn from Matthews JL, Vander Wiel C, Talmage RV: Adv Exp Med Biol 103:456, 1978.)
Osteoclast-osteoblast/stromal cell interactions are required for physiologic resorption of bone. The bone surface is protected from osteoclastic resorption by a continuous layer of osteoblasts and also by a very thin layer of unmineralized bone matrix normally present beneath resting osteoblasts (lamina limitans) (see Web Fig. 16-2). Osteoclasts are not able to bind to unmineralized bone matrix. For parathyroid hormone (PTH) to initiate bone resorption, receptors on the osteoblast bind PTH (osteoclasts do not usually express receptors for PTH). The binding of PTH to osteoblasts signals them to retract and secrete collagenases, which erode the unmineralized layer of matrix and allow osteoclasts access to a mineralized bone surface. In addition, osteoblasts and stromal cells that are activated by binding PTH and in response to a variety of other bone-resorbing stimuli (1, 25 dihydroxyvitamin D3; interleukin [IL]-1, IL-6, and IL-11; tumor necrosis factor-α [TNF-α]; prostaglandin E2 [PGE2]; and glucocorticoids) express or secrete receptor activator for nuclear factor κ B ligand (RANKL), also called osteoclast differentiation factor (ODF). RANKL binds to the RANK receptor on osteoclasts and activates the resorption process. Osteoblasts and bone marrow stromal cells also can secrete osteoprotegerin (OPG), a RANK homolog that works by binding to RANKL, thus blocking the RANK-RANKL interaction and inhibiting the differentiation of osteoclast precursors into mature osteoclasts. OPG expression can be stimulated by transforming growth factor-β (TGF-β). Therefore osteoblasts and stromal cells have the ability to both up- and down-regulate osteoclastic bone resorption (Fig. 16-5). In conditions of inflammation and necrosis, inflammatory mediators, such as IL-1 and TNF-α, can stimulate osteoclasts directly, causing bone resorption independent of the presence of viable osteoblasts.
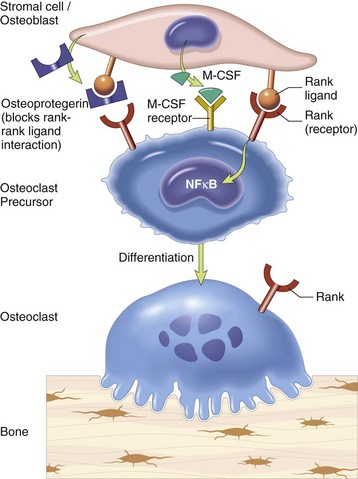
Fig. 16-5 Schematic diagram showing interaction between stromal cells/osteoblasts and osteoclast/osteoclast precursors.
Stromal cells and osteoblasts produce “rank ligand,” which binds to the “rank” receptor on the osteoclast and its precursors; this binding stimulates osteoclasis. Conversely, osteoblasts and stromal cells can inhibit the activation of osteoclasts by secreting osteoprotegerin, which can bind to “rank ligand” and block its binding to “rank receptor.” M-CSF, Macrophage colony-stimulating factor; NFκB, nuclear factor κ B. (From Kumar V, Abbas AK, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Bone at the Organic Matrix and Mineral Level
The mineralized matrix of bone provides the organ’s strength. Bone organic matrix consists of type I collagen and “ground substance” (the noncollagenous extracellular matrix: water, proteoglycans, glycosaminoglycans, noncollagenous proteins, and lipids, which are discussed later). Type I collagen polymers are secreted by osteoblasts and assembled into fibrils that are embedded in the ground substance and then mineralized. The type I collagen molecule is composed of three intertwined amino acid chains, unique to which is the hydroxylated form of the amino acid proline (hydroxyproline). Type I collagen molecules have extensive cross-linkages among the amino acid chains within the molecule and between adjacent molecules. Collagen molecules are deposited in rows with a gap between each molecule and with the rows staggered so that the molecules overlap by one-quarter of their length. This specific packing of the collagen molecules and the cross-linkages contribute to the strength and insolubility of the fibrous component of the bone matrix. Other than in rapidly deposited bone (woven bone found in primary trabeculae, bone of early fetal development, and pathologic conditions such as fracture repair), collagen fibers are arranged in parallel lamellae (singular: lamella) and the tissue is called lamellar bone. In cortical (compact) bone, lamellae are arranged concentrically (Fig. 16-6). In trabecular bone, the lamellae usually are arranged parallel with the bone surface. The collagen content of bone and its lamellar arrangement give bone its strength and flexibility. The ground substance of bone, which also is synthesized by osteoblasts, consists of noncollagenous proteins, proteoglycans, and lipids. Many of the noncollagenous proteins are cytokines that are capable of influencing bone cell activity. These cytokines, such as TGF-β, may play pivotal roles in controlling the extent of bone formation and resorption in normal remodeling and in disease (Fig. 16-7). Also, among the noncollagenous proteins are enzymes that can function in the degradation of collagen (e.g., matrix metalloproteinases) and can destroy inhibitors of mineralization (e.g., pyrophosphatases). Other noncollagenous proteins in the matrix can function as adhesion molecules and help bind cells to cells, cells to matrix, and mineral to matrix. Examples of these are osteonectin and osteocalcin. The role of proteoglycans in bone matrix is uncertain; however, there is some evidence that they influence bone cell differentiation and proliferative activity. Lipids may assist in binding calcium to cell membranes and in promoting calcification.

Fig. 16-6 Osteonal remodeling, cortical bone. Endocortical surface of bone has undergone extensive osteonal remodeling.
Collagen fibers are birefringent when viewed in appropriate plane with polarized light; alternate lamellae polarize when viewed in the same plane. This alternating pattern of birefringence demonstrates the parallel arrangement of collagen layers in lamellar bone. Much of the cortex has been remodeled into osteonal bone (concentric layers with central channel for vessels and nerves), but there are a few small areas of cortex that remain unosteonized (unremodeled). Unstained and fully mineralized. Polarized light micrograph. (Courtesy Dr. L. P. Krook, College of Veterinary Medicine, Cornell University.)
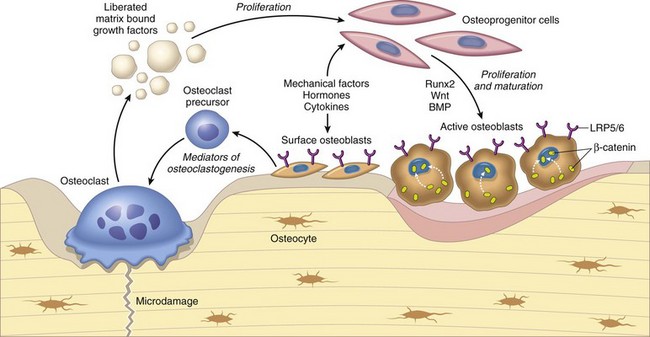
Fig. 16-7 Schematic diagram of the relationship between osteoclasts, osteoblasts, and growth factors.
Osteoclasts are able to liberate and activate growth factors from the matrix that are stimulatory to osteoblast progenitor cells and “couple” the process of osteoclastic bone lysis with subsequent bone formation. (From Kumar V, Abbas AK, Fausto N et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Bone mineral is in the form of a crystal called hydroxyapatite. Fully mineralized bone composes approximately 65% of the bone by weight and consists in part of calcium, phosphorus, carbonate, magnesium, sodium, manganese, zinc, copper, and fluoride. The mineral content gives bone its hardness. The production of osteoid (unmineralized organic matrix) by osteoblasts is followed by a period of maturation, after which mineral is deposited in exchange for water. Mineralization in woven bone is initiated within cytoplasmic blebs (matrix vesicles) of osteoblasts in the osteoid (see Web Fig. 16-1). Initiation of mineralization involves concentrating calcium, phosphorus, and other elements in these matrix vesicles to a level that causes precipitation of the mineral in the form of amorphous (not yet crystalline) hydroxyapatite. Matrix vesicles contain phospholipids and enzymes such as alkaline phosphatase and adenosine triphosphatase in their membranes. It is speculated that the membrane phospholipids attract calcium and phosphorus to the surface of the vesicle and that the alkaline phosphatase and adenosine triphosphatase enzymes might function in the pumping of these ions into the cell against a concentration gradient.
On reaching a critical mass, the amorphous mineral becomes crystalline. The crystalline hydroxyapatite pierces the lipid membrane of the matrix vesicle and extends to the gaps (holes) between collagen molecules. It is within these holes that the mineral crystals are first deposited in collagen. For the mineralization to spread beyond these gaps between the tropocollagen molecules, it is necessary for naturally occurring inhibitors of mineralization, such as inorganic pyrophosphate, in the matrix to be destroyed. Inorganic pyrophosphates are normal by-products of cellular metabolism and are deposited in unmineralized matrix by osteoblasts. The phosphatase enzymes described previously on the matrix vesicles have the ability to cleave these inorganic pyrophosphates and in doing so, destroy these inhibitors. Once the gaps are filled with mineral and the inhibitors of mineralization are destroyed, the process continues so that eventually the surfaces of collagen fibers, as well as spaces between collagen fibers, are mineralized. Initiation of mineralization in lamellar bone might not require matrix vesicles because glycoproteins, such as sialoprotein and osteonectin, can act as the nidus for the mineralization process.
Bone as a Tissue
In the cortex and subjacent to articular cartilage (subchondral bone), bone is organized into osteons (also called Haversian systems), which are cylinders of concentric layers of lamellae that are oriented parallel to the longitudinal axis of the bone and contain centrally located vessels and nerves (Fig. 16-8; see also Fig. 16-6). The bone between the osteons is called interstitial lamellae. Layers of bone oriented parallel to the internal and external circumference of the bone (beneath the endosteal and periosteal surfaces) are called circumferential lamellae. The osteonal system provides channels for the vascular supply to the cortex and also acts as tightly bound cables, giving the cortical bone strength and limited flexibility. This osteonal system also might be important in limiting propagation of microcracks in bone by diverting cracks along cement lines, which are collagen-poor, proteoglycan-rich seams between adjacent remodeling or modeling units (see later discussion). In H&E-stained histologic sections of decalcified bone, cement lines appear as basophilic lines.
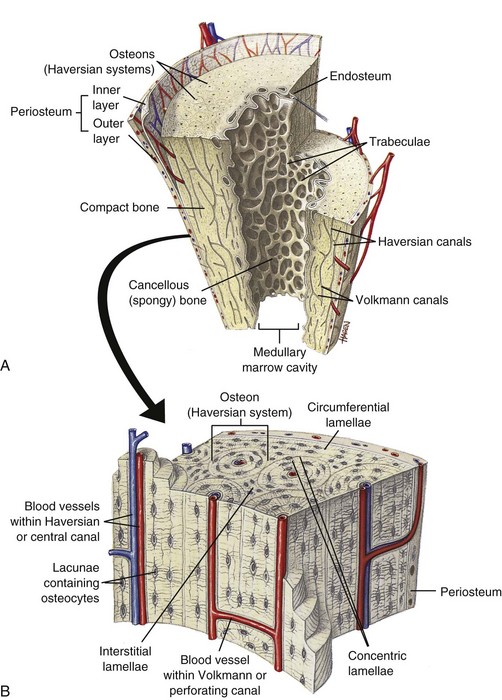
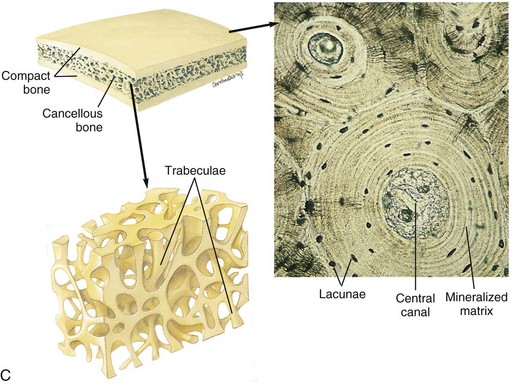
Fig. 16-8 Schematic diagram of the structure of compact and cancellous bone.
A, Longitudinal section of a long bone showing both cancellous and compact bone. B, Magnified view of compact bone. C, Section of a flat bone. Outer layers of compact bone surround centrally located cancellous bone. Fine structure of compact and cancellous bone is shown at a higher magnification. (From Thibodeau GA, Patton KT: Anatomy and physiology, ed 6, St Louis, 2007, Mosby.)
In contrast to the dense compact bone of the cortex and the subchondral bone plates, the bone in the medullary cavity is in the form of anastomosing plates or rods and is called cancellous, trabecular, or spongy bone (see Figs. 16-8 and 16-10). The orientation of the trabeculae usually reflects adaptation (modeling) to mechanical stresses applied to the bone. This is readily apparent when examining the trabeculae in the femoral neck, which are oriented in lines and arcs that are perpendicular to the stress applied and are thicker and more numerous on the ventral aspect (side of compression) compared with the dorsal aspect (side of tension). The lamellae within a trabecula usually are arranged parallel to the surface of the trabecula and are not arranged into tubes or osteons, as they are in cortical bone.
In most species, bone undergoes a low but constant replacement called remodeling: old bone is resorbed and replaced by new bone. The basal level of this remodeling activity (number of sites in the skeleton being remodeled at any one time) is likely “programmed” for each species. The number of active remodeling sites, however, can be markedly increased or decreased in response to altered mechanical use (see later discussion). This turnover of old bone to new bone allows for the repair of accumulated microscopic injury in the bone (microfractures). In normal bone remodeling, slightly less bone is replaced than removed, leaving a small negative net change in bone mass with each remodeling cycle and explaining in part the reduced bone mass in aged animals. Furthermore, in disease states, such as hyperparathyroidism, resorption is often increased and formation decreased, leaving a significant net negative bone balance. Interestingly, in small, short-lived animals, such as the mouse and rat, cortical bone is not remodeled. In addition, not all areas of bone in larger species undergo remodeling.
The remodeling unit of cortical bone is called the osteon, whereas the remodeling unit of trabecular bone is called the basic structural unit. The shape of the osteon is cylindric; however, the basic structural unit has the contour of a shallow trench filled with parallel lamellae. The time required from initiation to completion of these remodeling units (from initiation of bone removal by osteoclasts to complete filling of the defect by osteoblasts), regardless of the type of bone, is estimated to be 3 to 4 months in humans. Basophilic cement lines that mark the limit of previous resorptive activity are usually somewhat scalloped, following the contours of the Howship’s lacunae created by osteoclasts, and are called reversal lines (where resorption stopped and the process was reversed by formation) (Fig. 16-9). Cement lines also can occur when osteoblast formation ceases and subsequently resumes, and these are called “resting lines” and are usually smooth, following the contour of the overlying surface.
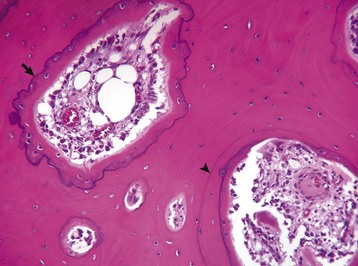
Fig. 16-9 Remodeling in compact subchondral bone adjacent to a joint with bacterial infection, bone, horse.
Resting cement lines appear as basophilic smooth lines indicating where formation has temporarily stopped (arrowhead). Reversal lines are scalloped basophilic lines (arrow) indicating where bone resorption stopped and was followed by formation. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
The term modeling is used to describe change of the shape or contour of a bone in response to normal growth, altered mechanical use, or disease. In modeling, bone surfaces (periosteal, endosteal, intracortical, and trabecular) can go directly from resting to either formation or resorption, depending on the stimulus. This process allows the shape or size of bone to change, enables the medullary cavity to enlarge, and allows the overall shape of the bone to be maintained while it is growing. Modeling is in contrast to remodeling, in which resorption must precede formation to keep bone mass and shape constant.
Bone as an Organ
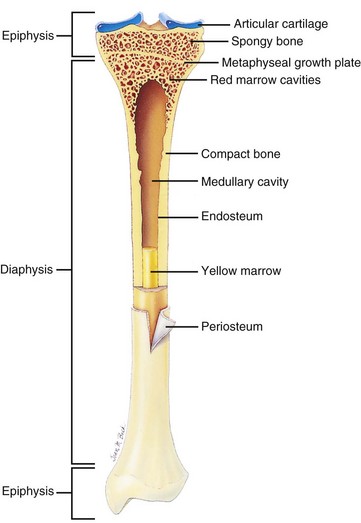
Individual bones of the skeleton vary in their manner of formation, growth, structure, and function. Flat bones of the skull develop by the process of intramembranous ossification, in which mesenchymal cells differentiate into osteoblasts and produce bone directly, in the absence of a preexisting cartilage model. In contrast, most bones develop from cartilaginous models by the process of endochondral ossification, in which cartilage is invaded by vessels, undergoes mineralization, and forms primary (diaphyseal) and secondary (epiphyseal) centers of ossification. Bones forming by endochondral ossification, such as the long bones of the appendicular skeleton and the vertebral bodies, are divided anatomically into epiphyses, metaphyseal growth plates (physes), metaphyses, and diaphyses (Fig. 16-10).
Blood Supply to Bone
Arterial blood from the systemic circulation enters bones through nutrient, metaphyseal, periosteal, and epiphyseal arteries (Fig. 16-11). Nutrient arteries penetrate the diaphyseal cortex through a nutrient foramen covered by strong, protective fascial attachments; once within the medulla, these arteries divide into proximal and distal intramedullary branches. Other arteries penetrating the cortex are the proximal and distal metaphyseal arteries, which are smaller and more numerous than the nutrient arteries. They penetrate the cortex and anastomose with the terminal branches of the nutrient arteries in the medullary cavity, protecting against infarction in case of obstruction of a nutrient artery.
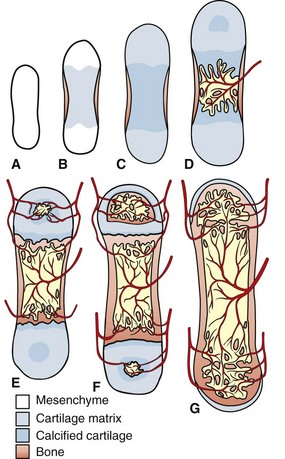
Fig. 16-11 Correlation of long bone development and vascularization.
The primitive mesenchyme that makes up the skeletal primordia contains no blood vessels (A). This mesenchyme condenses and undergoes central mineralization; a bony collar forms in the periosteum of the diaphysis (B and C). The nutrient artery enters the mineralized cartilaginous tissue in the diaphysis, bringing osteogenic and osteoclast precursors and enabling endochondral ossification to occur (primary center of ossification) (D). Similarly, the epiphyseal arteries bring these cells to the secondary centers of ossification in the epiphyses (E). Extensive anastomoses develop as the bone continues to develop and as the growth plates close (F and G). (Redrawn from Banks WJ: Applied veterinary histology, ed 3, St Louis, 1993, Mosby.)
Small periosteal arteries also pass through the diaphyseal cortex at sites of fascial attachment and can supply one quarter to one third of the outer cortex. The remainder of the cortex is supplied by the nutrient artery and its anastomotic branches. This blood flow is centrifugal (from medulla to periosteum) because of greater pressures in the intramedullary vessels. The chondrocytes of the physis nearest the epiphysis are supplied by epiphyseal arteries, whereas the chondrocytes of the physis nearest the metaphysis are supplied by branches of metaphyseal and nutrient arteries. As capillaries from these vessels approach the metaphyseal side of the physis, they make abrupt turns (loops), which are sites of predisposition to bacterial embolization in neonatal sepsis, sometimes resulting in osteomyelitis.
Bone Growth
Bone grows in length by interstitial growth within the metaphyseal growth plates (physes) (Fig. 16-12). The mineralized longitudinal septa of the growth plates serve as struts on which bone is deposited, a process called endochondral ossification. The metaphyseal growth plate is primarily responsible for lengthening the bone and is divided into a reserve or resting zone, a proliferative zone, and a hypertrophic zone (Fig. 16-13). The hypertrophic zone is sometimes further subdivided into zones of maturation, degeneration, and calcification. The resting or reserve zone serves as a source of cells for the proliferating zone in which cells multiply, accumulate glycogen, produce matrix, and become arranged in longitudinal columns. In the hypertrophic zone, chondrocytes secrete macromolecules that modify the matrix to allow capillary invasion and initiate matrix mineralization. The hypertrophic zone chondrocytes eventually die. The overall lengthening of the bone is due both to chondrocyte proliferation and hypertrophy. Calcification begins in the longitudinal septa of cartilaginous matrix between columns of chondrocytes. Matrix vesicles derived from chondrocytes (analogous to those described previously for mineralization of bone) form in the hypertrophic zone and initiate the mineralization process. The processes of mineralization and vascular invasion of the growth plate are codependent events. To supply salts for the mineralization, a nearby blood supply is necessary, and vascular invasion, a critical step in endochondral ossification, does not take place in mammalian growth plates unless there is mineralization of the longitudinal septum. Blood vessels from the metaphysis invade into the advancing growth plate, providing an entryway for osteoblasts that form bone on the cartilage spicules (see Fig. 16-13). The chondro-osseous junction in the metaphysis is a fragile lattice of bone-covered spicules of calcified cartilage (primary trabeculae). As the growth plate advances and elongates the metaphysis, the more mature trabeculae deeper in the metaphysis become fewer and thicker (secondary and tertiary trabeculae) and often contain residual fragments of cartilage.
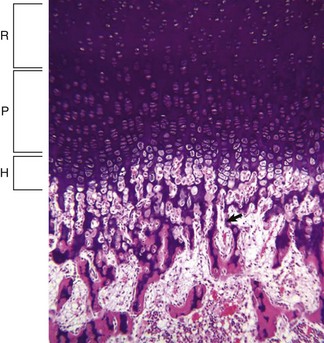
Fig. 16-12 Growth plate, long bone, dog.
Resting (R), proliferating (P), and hypertrophic (H) zones of the growth plate are visible. Apoptotic chondrocytes are released from their lacunae by invading vessels and chondroclasts leaving only the longitudinal septa (arrow) as a template on which bone will be deposited to form a primary trabeculae. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
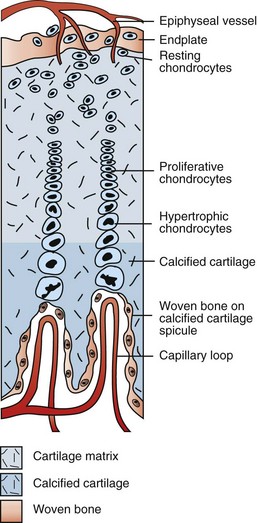
Fig. 16-13 Schematic diagram of the major blood supply to the physis.
Branches of the epiphyseal artery supply the resting zones of the growth plate. Branches of the metaphyseal artery form capillary loops at the metaphyseal side of the physis where endochondral ossification is occurring. (Redrawn from Banks WJ: Applied veterinary histology, ed 3, St Louis, 1993, Mosby.)
Growth plates are thickest when growth is most rapid; as growth slows, the plate becomes thin and “closes” (becomes replaced by bone) at skeletal maturity. The age at which a growth plate closes varies, depending on site, species, and gender. For example, the physes of vertebrae usually remain open longer than the physes of the long bones. Androgens and estrogens play a major role in determining the time of growth plate closure, and early castration results in delayed growth plate closure with subsequent increased length of bones compared with intact individuals.
Growth of the epiphysis contributes both to the overall length of the bone and to the shape of the ends of the bone. This is accomplished by endochondral ossification at the articular-epiphyseal complex (AEC). The AEC is composed of permanent articular cartilage, as well as a subjacent temporary growth/epiphyseal cartilage that is vascularized and contains the same zones as the growth plate (Fig. 16-14). In the mature individual, endochondral ossification no longer occurs at the AEC. Although the articular cartilage remains throughout life, the epiphyseal cartilage is completely replaced by bone once growth has ceased (Fig. 16-15). Bone grows in width by intramembranous bone formation. Except for articular surfaces (including the ends of the vertebral bodies), the surfaces of bones are covered by periosteum. This covering is a thin membrane that is loosely attached to underlying bone except at heavy fascial attachments on bony prominences and at tendon insertions, where its attachments are strong and are associated with large vessels penetrating the underlying bone. Microscopically, the periosteum is composed of an outer fibrous layer that provides structural support and an inner osteogenic or cambium layer, capable of forming normal lamellar appositional bone on the cortex of growing bones and of forming abnormal woven bone formation in response to injury (Fig. 16-16). The periosteum is well supplied with lymph vessels and with fine myelinated and nonmyelinated nerve fibers that account for the intense pain that occurs with periosteal injury.
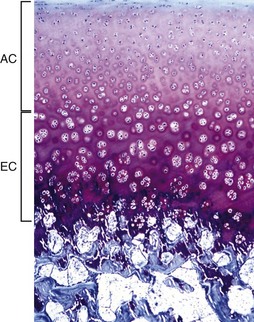
Fig. 16-14 Articular-epiphyseal complex (AEC), bone, immature pig.
The AEC is composed of a layer of articular cartilage (AC) and a subjacent layer of growth (epiphyseal) cartilage (EC). The growth cartilage is present only in immature individuals, and its structure and function is similar to that of the physis. Compare with Fig. 16-15. Toluidine blue stain. (Courtesy Dr. C.S. Carlson, College of Veterinary Medicine, University of Minnesota).
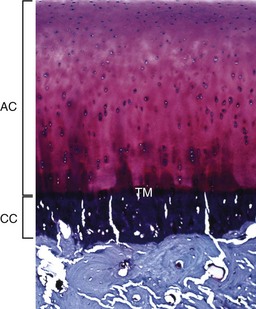
Fig. 16-15 Articular cartilage, bone, adult dog.
The articular cartilage is present throughout life; however, the epiphyseal cartilage is absent in adults. In adults, endochondral ossification has ceased and the tidemark (TM) delimits the boundary between the uncalcified articular cartilage (AC) and the calcified cartilage (CC). Compare with Fig. 16-14. Toluidine blue stain. (Courtesy Dr. C.S. Carlson, College of Veterinary Medicine, University of Minnesota.)
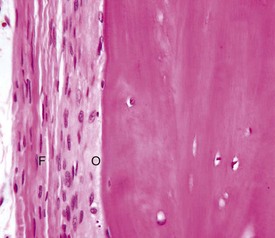
Fig. 16-16 Periosteum, bone, dog.
The outer fibrous layer (F) and inner osteogenic layer (O) line the periosteal surface. The osteogenic layer is able to rapidly deposit woven bone as a nonspecific response to injury. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
The periosteum covering the physis is called the perichondrial ring. The perichondrial ring adds new cartilage to the periphery of the physis, enabling it to expand in width as the animal grows. The metaphyseal cortex immediately adjacent to the perichondrial ring is normally very thin in the growing bone because its surfaces are the sites of very active osteoclastic bone resorption. Structurally, this area is the weakest part of the bone.
Structure and Function of Joints
Joints (articulations) join skeletal structures, provide for movement, and in some cases have shock-absorbing functions. This section of the chapter is primarily devoted to synovial joints, also called movable or diarthrodial joints.
Synovial joints occur in both the axial and appendicular skeleton. These joints allow for a variable degree of movement, and anatomically, they are composed of two bone ends joined together by a fibrous capsule and ligaments. The inner surface of the articular capsule is lined by a synovial membrane, and the bone ends are covered by articular cartilage. The joint space contains synovial fluid, and fibrocartilaginous menisci or disks are present at some sites (e.g., femorotibial and temporomandibular joints). Synovial joints operate with very low coefficients of friction and are self-lubricating. Articular cartilage serves as the bearing substance and subchondral bone as the supporting material. Articular cartilage functions to minimize friction created by movement to transmit mechanical forces to underlying bone and to maximize the contact area of the joint under load. Joints receive and absorb energy of impact. Both articular cartilage and subchondral bone deform under pressure, but it is the subchondral bone that has the most significant force-attenuating properties.
Articular Cartilage
Articular (hyaline) cartilage is normally a white to blue-white material with a smooth, moist surface. Cartilage thickness is greatest in the young and at sites of maximal weight bearing. Thinning and yellow discoloration occur in old age. At its margins, articular cartilage merges with a periosteal surface that is lined by fibrous tissue contiguous with the synovial membrane. Synovial fossae are normal depressions on non–weight-bearing articular cartilage surfaces (Fig. 16-17) that develop bilaterally in the larger appendicular joints of the horse, pig, and ruminant. These fossae are not present at birth but are fully formed by skeletal maturity. The function of synovial fossae is not known; however, they are significant because they are often mistaken for lesions. Adult articular cartilage contains no nerves or blood or lymph vessels, and its nutrients are obtained by diffusion from synovial fluid and to a lesser extent from subchondral vessels. In the immature skeleton, articular cartilage overlies the temporary growth cartilage of the epiphysis (epiphyseal cartilage). The epiphyseal cartilage is highly dependent on a network of blood vessels that originate from the perichondrium and the subchondral bone. Similar to the physis, it undergoes endochondral ossification and thereby contributes to the growth/development of the epiphysis. At skeletal maturity, the epiphyseal cartilage has been entirely replaced by bone, and the only joint cartilage remaining is the adult articular cartilage, which includes a thin subjacent zone of mineralized cartilage. In skeletally mature individuals, the junction between the unmineralized articular cartilage and the deeper mineralized cartilage forms a thin basophilic line in H&E stained sections called the tidemark (see Fig. 16-15). As animals age, multiple tidemarks can be formed, indicating an advance (thickening) of the mineralized layer of articular cartilage and thinning of the overlying nonmineralized articular cartilage. The mineralized cartilage serves to anchor articular cartilage to subchondral bone and limits the diffusion of substances between bone and cartilage.
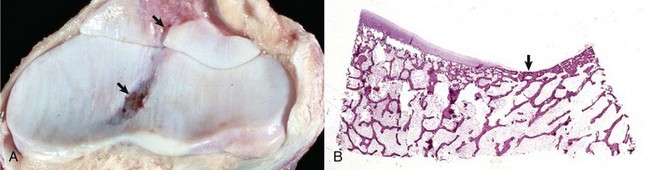
Fig. 16-17 Synovial fossa, joint, radius and ulna, bone, proximal end, adult horse.
A, Synovial fossae are depressions in the cartilage on the non–weight-bearing surfaces of the sagittal ridge of the radius and in the semilunar notch of the ulna (arrows). The parallel linear grooves apparent on the weight-bearing surface (articular cartilage) of the radius are the result of degenerative joint disease. B, Histologically, the surface of the synovial fossae is covered by a thin fibrous membrane (arrow) rather than articular cartilage. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Articular cartilage is 70% to 80% water by weight. It is a viscoelastic, hydrated fiber-reinforced gel that contains chondrocytes, collagen fibers (mostly type II), noncollagenous proteins, and proteoglycan aggregates. The largest proteoglycan in articular cartilage is aggrecan, which is composed of the immensely long, nonpolysulfated glycosaminoglycan, hyaluronan, to which core proteins are attached in a perpendicular arrangement like bristles on a brush. Similarly, smaller, negatively charged polysulfated glycosaminoglycans (chondroitin sulfate and keratan sulfate) are attached to the core proteins. The large content of negatively charged polysaccharide chains in the aggrecan aggregate is responsible for the extremely high osmotic swelling pressure of cartilage. The swelling pressure provided by aggrecan is counteracted by the resistance of the intact type II collagen fibers, giving cartilage its characteristic properties of being able to resist compressive forces and having a high tensile strength. Collagen fibers are arranged in arcades so that the tops of the arcades are parallel to the articular surface, and the sides are perpendicular to the surface and parallel with the radial or intermediate zone of chondrocytes. Functionally, the superficial zone of articular cartilage resists shearing forces, the middle zone functions in shock absorption, and the mineralized deep zone of cartilage serves to attach articular cartilage to the subchondral bone by its irregular (and therefore interlocking) interfaces. By scanning electron microscopy, the surface of articular cartilage is not smooth, but rather contains numerous depressions that can serve as reservoirs for synovial fluid.
Articular cartilage contains a single population of cells called chondrocytes that are responsible for the production, maintenance, and turnover of intercellular substances. In routine histologic sections, chondrocytes appear to be present within lacunae. However, although the lacunae within which osteocytes reside contain fluid spaces, the apparent lacunae of chondrocytes have been clearly demonstrated to be the result of shrinkage artifact. In fact, the chondrocyte cell membrane is in direct contact with the pericellular matrix and contains surface receptors for matrix components (e.g., hyaluronan).
Normal matrix turnover is enzymatic and this process is balanced by enzyme inhibitors. Proteases capable of degrading aggrecans are called aggrecanases and are members of the ADAM (A Disintegrin And Metalloprotease) protein family, whereas proteases capable of breaking the peptide bonds in collagen are called collagenases. Disease occurs if there is increased destruction or decreased synthesis of matrix components. It is important to remember that compared with bone that normally renews itself by remodeling, articular cartilage has extremely poor regenerative capabilities. Although there is evidence for proteoglycan synthesis in normal articular cartilage, the turnover of cells and type II collagen occurs at an extremely low rate. Correspondingly, the cellularity of articular cartilage declines with age.
Articular Capsule/Synovium/Synovial Fluid
The articular capsule consists of outer fibrous and inner synovial tissue layers. The outer layer is a heavy sheath that contributes to joint stability and at its insertion, attaches to bone at the margins of the joint, thereby enclosing a segment of bone of variable length within the joint cavity. It is well supplied with blood vessels and nerve endings. The inner synovial tissue layer is called the synovial membrane and covers all the inner surfaces of the joints except for the surface of the articular cartilage. The synovial membrane is normally very thin, lacks a basement membrane, and is barely visible grossly. In fact, collection of normal synovial membrane for histologic evaluation is best done immediately after opening the joint because otherwise, it will retract and become difficult to locate. The inner surface of the synovial membrane may be flat or may contain tiny projections (villi). Synovial intimal or lining cells, one to four cells thick, form a discontinuous surface layer and include two cell types: (1) A cells, which are macrophages that are responsible for the removal of microbes and the debris resulting from normal wear and tear in the joint (Fig. 16-18) and (2) B cells, which are fibroblast-like cells that produce synovial fluid. Normal synovial fluid contains hyaluronic acid, lubricin (a water-soluble glycoprotein), proteinases, and collagenase and is clear, colorless to pale yellow, and viscous. In addition to lubricating the joint surfaces, it supplies oxygen and nutrients to and removes carbon dioxide and metabolic wastes from the chondrocytes in articular cartilage. The synovial subintima can be classified according to the type of tissue that predominates (areolar, adipose, or fibrous), and contains blood and lymph vessels that supply and drain the intraarticular structures. Adipose tissue sometimes accumulates in the deep layers of the synovium, forming fat pads that serve as soft cushions in joint cavities (e.g., the infrapatellar fat body of the stifle joint).
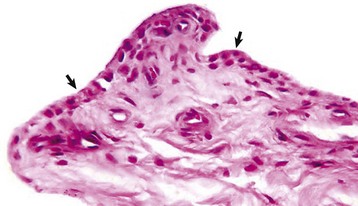
Fig. 16-18 Synovial membrane, joint, dog.
The normal synovial membrane (arrows) consists of an incomplete layer of histiocytes (phagocytic cells) and fibrocytes with subjacent loose fibrous and/or fibrofatty tissue. The joint lumen is at the top of the figure. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Joint lubrication depends on the microscopic roughness, elasticity, and hydration of articular cartilage and on the presence of hyaluronic acid and lubricin in synovial fluid. The lubricating properties of lubricin depend on its ability to bind to articular cartilage, where it retains a protective layer of water molecules. In contrast, hyaluronic acid, the molecule that makes synovial fluid viscous, has largely been excluded as a lubricant of the cartilage-on-cartilage bearing and instead, lubricates the site of surface contact between synovium and cartilage. When pressure is applied to the joint, such as during weight bearing, the articular surface is supplied with pressurized fluid that carries most of the load. This fluid is synovial fluid admixed with water that is released from the underlying cartilage, subsequent to pressure of weight bearing. When the load is removed, the water returns to the cartilage because of the hydrophilic properties of proteoglycans. This flushing of fluid in and out of the articular cartilage enables nutrients to enter cartilage from the synovial fluid and waste products to be removed.
Subchondral Bone
The subchondral bone acts to support the overlying cartilage and dissipate concussive forces to the peripheral cortical bone (Fig. 16-19). The thickness of the subchondral bone plate varies in proportion to the degree of weight bearing. Increased subchondral bone thickness is a common pathological finding in osteoarthritis. In larger animals (nonrodent species), subchondral bone often is composed of compact osteonal bone rather than trabecular bone.
Structure and Function of Tendons and Ligaments
A tendon is a unit of musculoskeletal tissue that transmits force from muscle to bone. Morphologically, tendons are similar to ligaments and fascia; however, ligaments join bone to bone and fascia connects muscle to muscle. The musculotendinous unit is composed predominately of parallel arrays of closely packed collagen fibers and rod- or spindle-shaped fibroblast-like cells (tenocytes) within a well-ordered extracellular matrix (Fig. 16-20). Collagen fibrils are bundled into large fibers that are evident throughout the tendon and are visible under light microscopy as a crimped or a sinusoidal pattern that facilitates a 1% to 3% elongation of the tendon. This elongation of the individual fibers serves to buffer the tendon from sudden mechanical loading. Water represents approximately 55% of the weight of tendon, is present mainly in the extracellular matrix, and is believed to reduce friction, facilitating the gliding of fibrils in response to mechanical loading. The major fibrillar component of tendon is type I collagen, which constitutes approximately 80% of the dry weight, whereas type III collagen is present in the endotenon and epitenon. The remainder of the tendon components include elastin, proteoglycans, and inorganic components. Collagen is synthesized by the tenocytes and constitutes the basic structural unit of tendon. The collagen polypeptides form a triple helix, which self-assembles into collagen fibrils with intermolecular cross-links that form between adjacent helixes. The collagen polypeptides and the ensuing triple helix are synthesized inside the cell, secreted into the extracellular matrix, and assembled into the microfibrillar units that constitute the collagen fibers. This step is promoted by a specialized enzyme called lysyl oxidase, which promotes cross-link formation—a process involving placement of stable cross-links within and between the molecules. Crosslink formation is the critical step that gives collagen fibers their strength. Tendon is stronger per unit area than muscle, and its tensile strength equals that of bone, although it is flexible and slightly extensible. The parallel arrangement of tendon collagen fibers resists tension so that contractile energy is not lost during transmission from muscle to bone. The mechanical properties of tendons depend on the collagen fiber diameter and orientation; however, the proteoglycan components of tendons also are important to the mechanical properties. While the collagen fibrils allow tendons to resist tensile stress, the proteoglycans allow them to resist compressive stress.
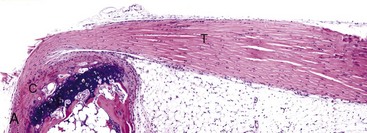
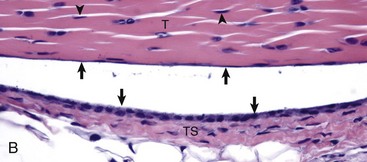
Fig. 16-20 Tendon, joint, mouse.
A, Gastrocnemius tendon (T). The tendon is composed of parallel arrays of closely packed collagen fibers and low numbers of fibroblast-like cells called tenocytes (see higher magnification in B; arrowheads). The tendon is covered by flattened synoviocytes (see higher magnification in B), so it may glide smoothly over the synoviocytes of the tendon sheath. C, Calcaneus insertion. H&E stain. B, Higher magnification of tendon sheath. The enveloping structure enclosing the tendon is the tendon sheath (TS), and it is lined by synoviocytes (arrows). T, Gastrocnemius tendon. H&E stain. (Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Skeletal ligaments are defined as dense bands of collagenous tissue that span a joint and then become anchored to the bone at either end. They vary in size, shape, orientation, and location. Their bony attachments are called insertions. Biochemically, ligaments are approximately two-thirds water and one-third solid. Type I collagen accounts for 85% of the collagen (other collagen types include III, VI, V, XI, and XIV) and the collagens account for approximately 75% of the dry weight, with the balance being made up by proteoglycans, elastin, and other proteins. While the ligament appears as a single structure, during joint movement some fibers appear to tighten or loosen depending on the bone positions and the forces that are applied, confirming the complexity of these structures. Histologically, ligaments are composed of fibroblasts that are surrounded by matrix. The cells are responsible for matrix synthesis and are relatively few in number, representing a small percentage of the total ligament volume. Recent studies have indicated that normal ligament cells may communicate by means of prominent cytoplasmic extensions that extend for long distances and connect to cytoplasmic extensions from adjacent cells, thus forming an elaborate three-dimensional architecture. Gap junctions have also been detected in association with these cell connections, raising the possibility of cell-to-cell communication and the potential to coordinate cellular and metabolic responses throughout the tissue. Ligament microstructure reveals collagen bundles aligned along the long axis of the ligament and displaying an underlying “waviness” or crimp along the length, similar to that present in tendon. Crimp is thought to play a biomechanical role, possibly relating to the ligament’s loading state with increased loading likely resulting in some areas of the ligament uncrimping, allowing the ligament to elongate without sustaining damage.
One of the main functions of ligaments is mechanical as they passively stabilize joints and help guide those joints through their normal range of motion when a tensile load is applied. Another function of ligaments relates to their viscoelastic behavior in helping provide joint homeostasis. Ligaments “load relax,” which means that loads/stresses decrease within the ligament if they are pulled to constant deformations. A third function of ligaments is their role in joint proprioception. In joints such as the knee, proprioception is provided principally by joint, muscle, and cutaneous receptors. When ligaments are strained, they invoke neurologic feedback signals that then activate muscular contraction, which appears to play a role in joint position sense.
Tendons and ligaments attach to bone in a similar manner, through osteotendinous or osteoligamentous junctions, respectively. These junctions are known as entheses and are distinguished as fibrous and fibrocartilaginous according to the type of tissue present at the attachment site. At fibrous entheses, the tendon or ligament attaches either directly to the bone or indirectly to it via the periosteum. Fibrocartilaginous entheses are sites in which chondrogenesis has occurred and commonly contain four tissue types: dense fibrous connective tissue, uncalcified fibrocartilage, calcified fibrocartilage, and bone.
Responses of Bone to Injury
Mechanical forces that can affect bone are both internal and external. Internal forces associated with extremes in work or exercise can influence modeling or remodeling (see previous discussion) and occasionally cause fracture (failure of the bone). External forces (trauma) are more commonly associated with damage to the periosteal surface or fracture of the cortex and/or trabecular bone.
Hormonal agents, particularly calcitriol and PTH, enter the bone by the bloodstream as described previously. Infectious agents are discussed later in the section on Inflammation of Bone.
Defense against mechanical forces includes the structure of the bone and the bone’s ability to model and remodel to adapt to chronic changes in forces applied to it. The dense bone of the cortex enables the bone to resist most external forces. The formation of osteons and cement lines in the cortex (primary remodeling) facilitates dissipation of microcracks and bending of the bone in response to stress.
Defense against hormonal agents that are capable of resorbing bone include the protective covering on mineralized surfaces by the lamina limitans and osteoblasts. In addition, hormonal signaling to resorb bone is done through the osteoblast, which has the ability to modulate the resorption signal. Defense against infectious agents is discussed later in the section on Inflammation of Bone.
The hard tissue of bone can be likened to the rings in a tree that leave clues to the history embedded in its hard structure. Interpreting these clues requires understanding the ways in which bone uniquely responds to injury (Box 16-2).
Disruption of endochondral ossification can alter the appearance of the primary trabeculae. Examples of this are growth retardation lattice and growth arrest lines. Growth arrest lines can be seen in cases in which multiple nutrient deficiencies are present such as occur in debilitating disease or general malnutrition. The growth plate becomes narrow (growth is impaired), and the metaphyseal face of the plate can be sealed by a layer of bone as a result of transverse trabeculation (trabecular bone forming parallel versus perpendicular, immediately subjacent to the growth plate). If endochondral ossification resumes, this layer of bone becomes separated from the physis and becomes located more distally in the metaphysis (Fig. 16-21, A and B). The resulting bony trabeculae parallel to the growth plate that also can be seen grossly and radiographically are called growth arrest lines.
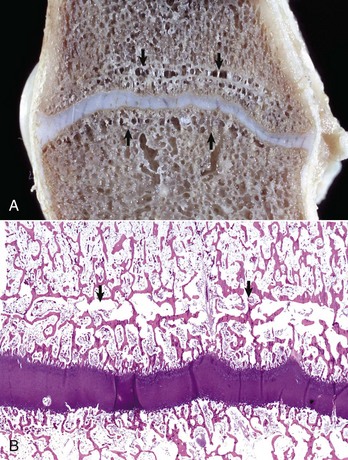
Fig. 16-21 Growth arrest lines, long bone, metaphysis, young zebra.
Gross (A) and microscopic (B) appearance of growth arrest lines. The zebra was ill from a bacterial renal infection and became anorexic. The animal made a brief clinical recovery before euthanasia. The period of inappetence is responsible for the parallel horizontal lines of bone in the metaphysis (growth arrest lines; arrows). These lines are the result of transverse (horizontal) orientation of the trabeculae during the period of slowed growth. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
A growth retardation lattice results from acquired impairment of osteoclastic resorption of bone within the primary trabeculae, resulting in retention of primary trabeculae. These trabeculae elongate because of continued endochondral ossification. The retention of the primary trabeculae results in a dense band of vertically oriented trabecular bone subjacent to the growth plate called a growth retardation lattice (Fig. 16-22). The band is apparent because the normal modeling process that resorbs many primary trabeculae completely and molds the remaining ones into progressively fewer but thicker secondary and tertiary trabeculae is impaired. Diseases that cause growth retardation lattices include canine distemper and bovine viral diarrhea, in which viral infection of osteoclasts results in defects in cell function. Similarly, toxic damage to osteoclasts, such as in lead poisoning, can cause a growth retardation lattice (“lead line”). Abnormal retention of primary trabeculae also can be seen with congenital defects in the function of osteoclasts (see later discussion of osteopetrosis). The phrase growth retardation lattice is perpetuated here because it is in common use; however, it is important to understand that the lesion is caused by a failure in modeling of the trabeculae rather than by a reduction in longitudinal growth. Weakening or destruction of the matrix of the physeal cartilage, such as occurs in animals with hypervitaminosis A and with manganese deficiency, can lead to premature closure of growth plates. If the entire plate is affected, no further longitudinal growth is possible. If closure is focal, such as can be seen subsequent to localized inflammation or traumatic damage to vessels, the remainder of the growth plate continues to undergo endochondral ossification, resulting in an angular limb deformity.
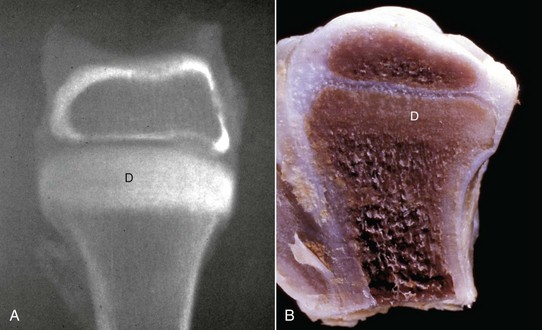
Fig. 16-22 Growth retardation lattice, bone, radius, distal end, dog.
Radiograph (A) and longitudinal section (B) of a growth retardation lattice. The increased bone density (D) of the metaphysis represents failure of osteoclasts to resorb unnecessary primary trabeculae. In this case, the failure of osteoclastic resorption was caused by canine distemper virus infection of osteoclasts. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Osteochondrosis is an important example of a disease caused by disruption of endochondral ossification and is discussed later under disorders of endochondral ossification.
The ability of bone to change its shape and size (modeling) to accommodate altered mechanical use is called Wolff’s Law (see example in Fig. 16-23). Tension and compression are important mechanical factors that affect bone modeling with formation favored at sites of compression and resorption favored at sites of tension. In addition, trabecular bone aligns along lines of stress. The ways in which bone cells detect altered mechanical use are not precisely known but likely include input from a variety of signals, including stretch receptors on bone cells, streaming potentials, and piezoelectrical activity. Streaming potentials are electrical currents in bone that are detected by the osteocyte-osteoblast network and are caused by fluid fluxes through canalicular spaces. Piezoelectrical activity refers to the production of electrical currents in bone as a result of deformation of collagen fibers and mineral crystals. Electrical currents can affect cell function (bone formation and resorption) and thus influence bone modeling. In fact, several types of portable devices that are designed to produce an electrical current are available for use in humans to promote fracture healing.
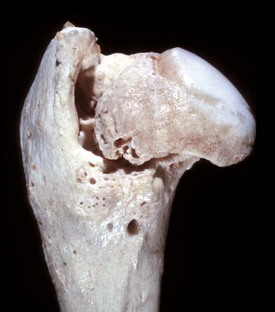
Fig. 16-23 Hip dysplasia, bone, femoral head and neck, dog.
The femoral head is severely flattened and periosteal new bone and coalescing osteophytes have formed a massively thickened femoral neck. Macerated and bleached specimen. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Bone mass can be altered to accommodate mechanical use. Normal mechanical use suppresses programmed bone resorptive activity and is required for maintenance of bone mass. Decreased mechanical use reduces this inhibition (allowing resorption to proceed) and also suppresses bone formation. The net effect of decreased mechanical use therefore is a reduced amount of bone as a result of increased resorption and decreased formation (Fig. 16-24). In contrast, increased mechanical use suppresses bone resorption and allows bone mass to increase (Fig. 16-25). Chronic suppression of remodeling, however, could result in retention of aged bone and lead to accumulated microcracks, which in turn could override the suppression and stimulate remodeling in these regions.
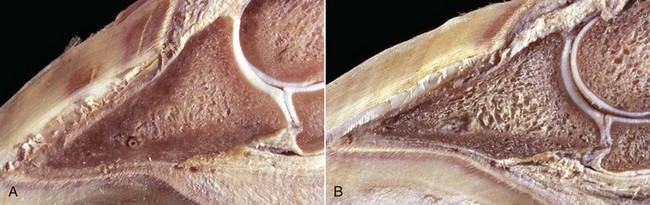
Fig. 16-24 Bone, third phalanx, left rear leg, foal.
The leg was in a cast for 2 months to repair an avulsion of gluteal muscles from their insertions. A, Normal right third phalanx. B, Left third phalanx. There is pronounced disuse osteopenia (atrophy) compared with the right third phalanx shown in A. The increase in resorption and decrease in formation associated with disuse has resulted in marked porosity of the cortical and subchondral bone. The cortex now has the appearance of trabecular bone (trabeculation of the cortex). (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
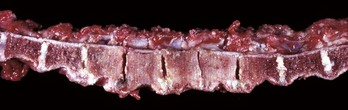
Fig. 16-25 Osteosclerosis, bone, vertebrae, dog.
Osteosclerosis (increased bone per unit of area) is evident in the four vertebrae in the center of the vertebral column. The cancellous bone of the medullary cavities has been obliterated by newly formed compact bone. The osteosclerosis is in response to increased mechanical stress in the bodies of the vertebrae as a result of degeneration and loss of the intervertebral disks between these vertebrae. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Woven bone is newly-formed, hypercellular bone that is deposited in reaction to injury. Although woven bone is normally present in immature individuals, the presence of woven bone in the adult skeleton is considered to be pathologic. The collagen fibers in woven bone are irregularly/randomly arranged rather than being oriented in a lamellar pattern, a change that is best appreciated using polarized light microscopy (Fig. 16-26). In addition, osteocytes are larger and more numerous per unit area than in lamellar bone, and there is no preferred orientation to their lacunae versus in lamellar bone in which the alignment of the elliptical lacunae is parallel to the lamellae. Over time, woven bone remodels into lamellar bone.
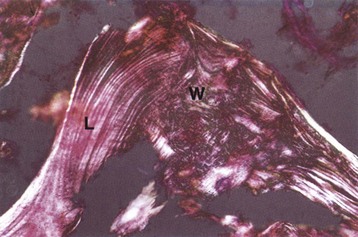
Fig. 16-26 Cancellous and woven bone, polarized microscopy.
A spicule of bone composed of areas of lamellar (L) and woven (W) bone. The woven bone, which may represent the site of a healing fracture, exhibits a disorganized orientation of collagen fibers compared with the adjacent lamellar bone, in which the collagen fibers are arranged in parallel layers. Unstained and fully mineralized. Polarized light micrograph. (From Burkitt HG, Young B, Heath JW: Wheater’s functional histology: a text and color atlas, ed 3, New York, 1993, Churchill Livingstone.)
The periosteum is programmed to respond to injury by producing woven bone, which usually is oriented perpendicularly to the long axis of the cortex. Although nodular periosteal new bone is sometimes referred to as osteophytes, the use of this term should be restricted to periarticular new bone that occurs in response to joint injury/instability. Reactive periosteal woven bone may be admixed with hyaline cartilage and sometimes is composed predominantly of cartilage. The extent to which cartilage is produced by the periosteum is thought to be a result of the available oxygen. When oxygen tension is low, cartilage proliferation may predominate; however, when oxygen tension is normal, there may be no cartilage present. Cartilage produced in such circumstances can eventually undergo endochondral ossification. In addition, woven bone produced by the periosteum may be remodeled to lamellar bone or may be removed by osteoclastic resorption. In addition to producing woven bone in response to injury, the periosteum also is capable of producing lamellar bone during slower stages of appositional growth of the diaphysis, and this bone also is subject to osteoclastic resorption. During growth, the regions of the cut-back zone of the metaphysis, in which the bone diameter is reduced from a larger diameter at the physis to the narrower diameter of the diaphysis, exhibit marked osteoclastic bone resorption at the periosteal bone surface. Infectious inflammation of the periosteum also can lead to marked osteoclastic bone resorption at the periosteal bone surface.
Responses of Joints to Injury
Although articular cartilage contains metabolically active cells, it has a limited response to injury and minimal capacity for repair that is a result in large part to its lack of a blood supply. Superficial cartilage defects (cartilage erosions) that do not penetrate into subchondral bone persist for long periods with few or no histologic changes. Clusters or clones of chondrocytes (evidence of local chondrocyte replication in response to injury) may be present, particularly along the margins of the defect, but are ineffective in filling it. Progression of the lesion, with matrix degeneration and eventual loss of the remaining articular cartilage, may occur over time, particularly if thickened (sclerotic) subchondral bone is present subjacent to the defect. In contrast, if a cartilaginous defect extends into subchondral bone (cartilage ulceration), allowing mesenchymal cells access to the defect, it is quickly filled with vascular fibrous tissue that often undergoes metaplasia to fibrocartilage but rarely, if ever, to hyaline cartilage. Formation of fibrocartilage can be hastened in full-thickness cartilaginous defects by exercise or prolonged passive motion. Injury to articular cartilage is not painful unless the synovium or subchondral bone is involved. Having no blood supply of its own, articular cartilage does not participate directly in the inflammatory response but is very much affected by inflammation in the synovium, subchondral bone, or subarticular growth (vascularized) cartilage of the epiphysis in young animals. Given that alternating compression and release of normal weight bearing facilitates the diffusion of fluid with nutrients into the articular cartilage and fluid with metabolic waste products out of articular cartilage, it follows that constant compression or lack of weight bearing leads to atrophy (thinning) of articular cartilage.
Sterile injury to cartilage can be a consequence of trauma, joint instability, or lubrication failure because of changes in synovial fluid, synovial membrane, or incongruity in joint surfaces. Destruction of articular cartilage in response to sterile injury and infectious inflammation is mediated by a combination of enzymatic digestion of matrix and failure of matrix production when chondrocytes become degenerate or necrotic. These changes can be initiated by damage to the cartilage directly or may occur indirectly, secondary to lesions in the synovium.
Matrix metalloproteinases are enzymes capable of matrix digestion; they are normal constituents of the matrix, but are present in an inactive form. Matrix metalloproteinases can be broadly categorized as gelatinases, collagenases, and stromelysins. Collagenases are most capable of digestion of collagen fibers; gelatinases digest type I collagen and basement membrane collagens but are less effective against type II collagen of cartilage. Stromelysins destroy noncollagenous proteins. Matrix metalloproteinases can be activated by products of degenerating or reactive chondrocytes and inflammatory cells. In addition, tissue inhibitors of metalloproteinases (TIMPs) are present in the matrix, acting as a control on the destructive effects of activated metalloproteinases. As mentioned earlier, proteases capable of degrading aggrecans are called aggrecanases and are members of the ADAM protein family. The loss of proteoglycans from cartilage alters the hydraulic permeability of the cartilage, thereby interfering with joint lubrication and leading to further mechanically induced injury to the cartilage. The loss of proteoglycans, with subsequent inadequate lubrication of the articular surface, leads to disruption of collagen fibers on the surface of articular cartilage. Grossly, the surfaces of affected areas of cartilage are yellow-brown and have a dull, slightly roughened appearance. As more proteoglycans are lost, the collagen fibers condense and fray (fibrillation) with multiple clefts and/or fissures forming along the vertical axis of the arcades of collagen fibers (Figs. 16-27 and 16-28). The vertical axis of the collagen fibers in these arcades is perpendicular to the plane of movement of the joint. Fibrillation is accompanied by loss of surface cartilage (erosion) and eventual thinning of the articular cartilage. Subsequent to fibrillation and erosion, necrosis of individual chondrocytes occurs in the radial zone, resulting in hypocellularity of the remaining cartilage. In response to the fibrillation, erosion, and necrosis of chondrocytes, remaining chondrocytes can undergo regenerative hyperplasia (cluster or clone formation), but the ability of chondrocytes in the adult to divide is limited and the regenerative attempt is almost always ineffective. Loss of articular cartilage can become complete, resulting in exposure of subchondral bone (ulceration), which typically is eburnated (thickened/sclerotic) (Fig. 16-29).
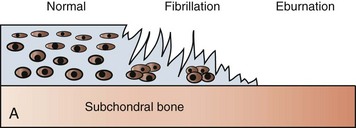
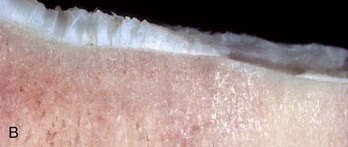
Fig. 16-27 Fibrillation.
A, Schematic diagram showing the structural changes that characterize fibrillation and loss of articular cartilage and adjacent eburnated subchondral bone. In the area of eburnation, the cartilage is missing, and the exposed subchondral bone has increased density. B, Fibrillation and ulceration, degenerative joint disease, tibia, proximal articular cartilage, sagittal section, bull. To the left, the cartilage is frayed (fibrillation) and its surface has the appearance of a shag rug. To the right of the fibrillated region is an ulcer (full-thickness loss of articular cartilage). The cartilage to the right of the ulcer is very thin, indicating erosion. (B courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
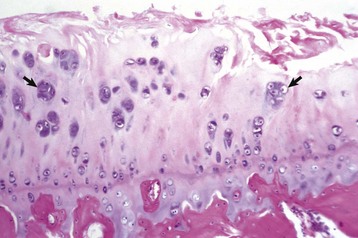
Fig. 16-28 Degenerative joint disease, hip dysplasia, femoral head, articular cartilage, dog.
The superficial cartilage is fibrillated and hypocellular (necrosis) with clusters of chondrocytes (arrows) representing ineffectual attempts at repair. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
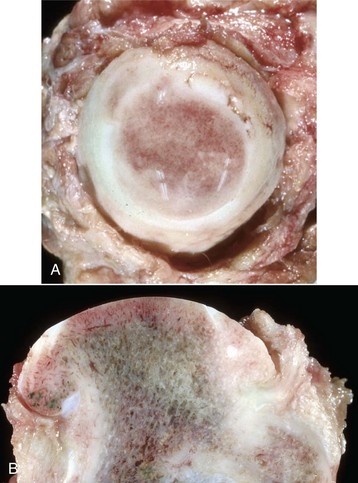
Fig. 16-29 Degenerative joint disease, hip dysplasia, bone, femoral head with eburnation, dog.
The femoral head viewed from the medial aspect (A) and in a sagittal plane (B). The head is flattened and ulcerated. The ulcerated region appears darker because of congestion of blood vessels in the marrow spaces of the subchondral bone. The zone of attachment of the round ligament to the head of the femur has been destroyed. Note in A how the subchondral bone has become smooth and shiny (eburnation). (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University).
Articular Capsule/Synovium/Synovial Fluid
Intraarticular prostaglandins, nitric oxide, TNF-α, IL-1, and neurotransmitters—such as substance P, among other cytokines and chemokines—are increased in degenerative and inflammatory joint disease. Prostaglandins and nitric oxide inhibit proteoglycan synthesis in synovium and chondrocytes; this reduction in proteoglycan content can lead to degeneration and loss of the cartilage (see previous discussion). IL-1 and TNF-α are cytokines secreted by activated macrophages (synovial type A cells or subintimal macrophages); they promote secretion of prostaglandins, nitric oxide, and neutral proteases from synovial fibroblasts and chondrocytes. Increasing concentrations of these agents decreases matrix synthesis and increases matrix destruction. Cytokines and growth factors that can be anabolic to cartilage include IL-6, TGF-β, and insulin-like growth factor (IGF).
Lysosomal enzymes (collagenase, cathepsins, elastase, and arylsulfatase) and neutral proteases, which are capable of degrading proteoglycans or collagen, can be derived from inflammatory cells, synovial lining cells, and chondrocytes.
The synovial membrane commonly responds to injury by villous hypertrophy and hyperplasia (Fig. 16-30), hypertrophy and hyperplasia of synoviocytes, and pannus formation (see later discussion). Villous hypertrophy occurs with or without synovitis. The proportions of A (macrophages) and B (fibroblast-like) cells in the synovium also can change in various disease processes. Fragments of articular cartilage can adhere to the synovium, where they are surrounded by macrophages and giant cells. Larger pieces of detached cartilage (as in osteochondrosis) can float free and survive as chondral or osteochondral fragments (“joint mice”) that continue to remain viable through nutrient supply from synovial fluid.
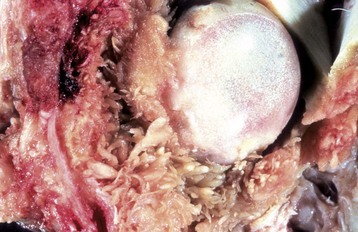
Fig. 16-30 Synovial villous hyperplasia, hip dysplasia, coxofemoral joint, articular capsule, and femoral head, dog.
There is marked villous synovial hyperplasia. The extent of the proliferation is unusually severe for hip dysplasia. Microscopically, villous synovial hyperplasia is routinely accompanied by variable lymphoplasmacytic inflammation that is independent of the cause of the articular damage. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Inflammatory cell infiltrates (see the discussion on infectious and noninfectious arthritis in the section on Inflammatory Lesions) in the synovial membrane can impair fluid drainage from the joint and can cause joint fluid to lose some of its lubricating properties as the result of degradation of hyaluronic acid by the superoxide-generating systems of neutrophils.
Pannus can develop in association with chronic infectious fibrinous synovitis and with some immune-mediated diseases; the classic example is rheumatoid arthritis. Pannus is a fibrovascular and histiocytic tissue (also called an inflammatory granulation tissue) that arises from the synovial membrane and spreads as a membrane over articular cartilage (Figs. 16-31 and 16-32). In the pannus, tissue histiocytes and monocytes of bone marrow origin transform into macrophages and they, along with the collagenases from fibroblasts, cause lysis and destruction of the underlying cartilage (Fig. 16-33). In time, if both opposing cartilaginous surfaces are involved, the fibrous tissue can unite the surfaces, causing fibrous ankylosis (fusion of the joint). In some cases of immune-mediated arthritis, pannus is present in the subchondral bone marrow (bone marrow pannus), as well as in the synovium, and may penetrate into the overlying articular cartilage.
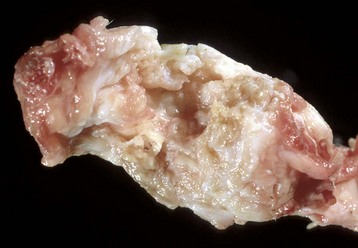
Fig. 16-31 Pannus, rheumatoid-like arthritis, proximal radius and ulna, dog.
Fibrovascular tissue (pannus) covers all articular surfaces. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
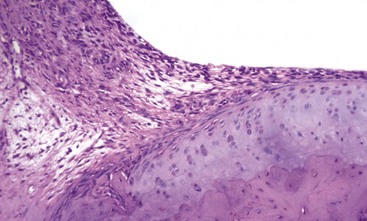
Fig. 16-32 Pannus, rheumatoid-like arthritis (experimentally induced), distal articular cartilage, tibia, rat.
The experimentally induced rheumatoid-like arthritis was produced by injecting Freund’s adjuvant and Mycobacterium butyricum into the subcutis at the base of the tail. The pathogenesis of “adjuvant arthritis” is uncertain, other than it appears to be T lymphocyte mediated. Here, fibrovascular repair tissue (pannus) is seen arising from the synovium (left) and growing onto the surface of the articular cartilage, which is relatively undamaged at this stage of the disease. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
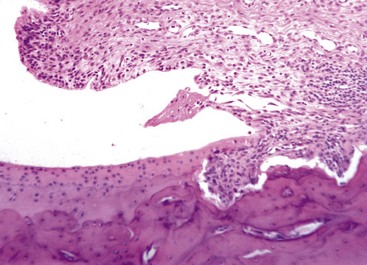
Fig. 16-33 Pannus, rheumatoid-like arthritis (experimentally induced), articular cartilage, distal tibia, rat.
Pannus originating from the synovium (right) is invading and destroying the articular cartilage and subchondral bone. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Sterile degenerative changes in articular cartilage are often accompanied by the formation of periarticular osteophytes (Fig. 16-34) and by some degree of secondary synovial inflammation and hyperplasia. The synovitis is characterized by the presence of variable numbers of plasma cells, lymphocytes, and macrophages in the synovial subintima (beneath the layers of synoviocytes) and by hyperplasia and hypertrophy of synovial lining cells. The pathogenesis of this synovitis is not known but is suspected to be the result in part of the presence of degenerate cartilage debris within the joint. The pathogenesis of osteophytes also is unclear. They can arise from mesenchymal cells with chondro-osseous potential within the synovial membrane at the junction of the synovial membrane with the perichondrium/periosteum, just peripheral to the articular cartilage, or on the surface of the bone where the articular capsule and the periosteum merge. Osteophytes do not grow continuously, but once formed, they persist as multiple periarticular spurs of bone. These spurs can be confined within the joint cavity if they arise from the perichondrium or can be protrusions from the periosteal surface of the bone if they arise from the insertion site of the joint capsule with the periosteum. Osteophytes can result from mechanical instability within the joint, causing stretching or tearing of the insertions of the articular capsule or ligaments, or they can form from stimulation by cytokines such as TGF-β that are released from reactive or degenerating mesenchymal cells within the joint.
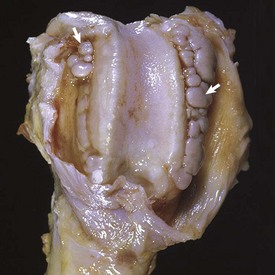
Fig. 16-34 Osteophytes, degenerative joint disease, distal femur, dog.
Note the large number of osteophytes (arrows) along the lateral and medial margins of the trochlear ridges. (Courtesy Dr. H. Leipold, College of Veterinary Medicine, Kansas State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)
Because of their antiinflammatory effects, glucocorticoids often are therapeutically injected into joints. Although the usual result is decreased pain and inflammation, glucocorticoid injection sometimes is followed by a rapid progression of degenerative changes within the joint that is designated “steroid arthropathy.” These degenerative changes relate to the antianabolic effects of glucocorticoids on chondrocytes, in which synthesis of cartilaginous matrix is reduced, proteoglycans are depleted, repair is retarded, and the mechanical strength of cartilage is reduced.
Subchondral Bone
With the loss of matrix proteoglycans in degenerate articular cartilage, a greater component of concussive forces is transmitted to the subchondral bone. The bone responds to the increased mechanical use by decreasing resorption and increasing formation, resulting in a net increase in amount of bone per unit area (increased subchondral bone density or subchondral sclerosis). If the cartilage ulcerates to the level of the bone and the joint is still being used, the surface of the dense subchondral bone can become smooth and shiny (eburnation) (see Fig. 16-29, A). There is interest in the possible role subchondral bone might play in initiating cartilage damage in degenerative joint disease, as some studies have reported an increase in subchondral bone thickness/density before the development of articular cartilage lesions. Some believe that this dense bone is less effective in dissipating normal concussive forces and causes some of the impact to be deflected back into the articular cartilage, causing injury to the chondrocytes.
Responses of Tendons and Ligaments to Injury
The site where ligament or tendon attaches to bone is known as an enthesis but is also called an insertion site or an osteotendinous or osteoligamentous junction. Entheses are vulnerable to acute or overuse injuries in sports as the result of stress concentration at the hard-soft tissue interface. Osteophytes located in these regions are termed enthesophytes.
The initial stage of repair of tendons and ligaments involves formation of scar tissue to provide continuity at the injury site. For tendons in particular, mobility must be maintained during healing to prevent or at least decrease the formation of adhesions and to increase strength. The sequence of repair includes three phases: (1) tissue inflammation, (2) cell and matrix proliferation, and (3) remodeling and maturation. The inflammatory stage usually involves the formation of a hematoma, which activates the release of chemotactic factors, including TGF-β, IGF-1, platelet-derived growth factor (PDGF), and basic fibroblast growth factor (bFGF). Inflammatory cells are attracted from the surrounding tissues to engulf and resorb the clot, cellular debris, and foreign material. Fibroblasts are recruited to the site to begin to synthesize components of the extracellular matrix and angiogenic factors that are released during this phase initiate the formation of a vascular network. During the cell proliferation phase, recruitment and proliferation of fibroblasts continues because these cells are responsible for the synthesis of collagens, proteoglycans, and other extracellular matrix components, which initially are arranged randomly. At this point in the repair process, the extracellular matrix is composed largely of type III collagen. At the end of the proliferative stage, the repair tissue is highly cellular and contains relatively large amounts of water and an abundance of extracellular matrix components. The remodeling stage begins 6 to 8 weeks after injury and is characterized by a decrease in cellularity, reduced matrix synthesis, a decrease in type III collagen, and an increase in type I collagen synthesis. The type I collagen fibers are organized longitudinally along the tendon axis and are responsible for the mechanical strength of the regenerating tissue. Despite multiple ongoing phases of remodeling, the repair tissue never achieves the characteristics of normal tendon.
Portals of Entry into Bone
Infectious agents can enter bone directly through the periosteum and cortex or through the vasculature. Agents can gain access through the periosteum by means of trauma that may or may not break the bone or by extension from adjacent inflammation, as in periodontal tissue (e.g., periodontitis progressing to mandibular or maxillary osteomyelitis) or the middle ear (otitis media extending into the bone, resulting in osteomyelitis of the tympanic bulla). Blood vessels gain access to the marrow cavity of the diaphysis and metaphysis through the nutrient foramen. The primary blood supply of the epiphysis in young animals is the epiphyseal artery, multiple branches of which arborize into the growth plate, providing vascularization of the proliferative zone. Blood-borne bacterial infection of bone in the perinatal animal may originate from the umbilicus or more commonly, by the oral-pharyngeal route. In theory, hematogenous osteomyelitis can begin in any capillary bed in bone in which viable bacteria localize. In practice, it occurs most commonly in young animals and is localized typically at the zone of vascular invasion on the metaphyseal side of the growth plate (Fig. 16-35) or immediately subjacent to the AEC (Fig. 16-36) where capillaries in both sites make sharp bends to join medullary veins (Fig. 16-37). In these locations, bacterial localization is apparently facilitated by slow flow and turbulence of blood, a lower phagocytic capacity, and a discontinuous endothelial lining. In addition, no vascular anastomoses are located in this region, therefore thrombosis of these capillaries results in bone infarction that is a predisposing factor for bacterial localization. From this nidus, the inflammation can extend into other structures, including the overlying joint cavity for AEC lesions (see Fig. 16-36) and the epiphysis, periosteum, or joint cavity for physeal lesions (Fig. 16-38; see also Fig. 16-37).
Fig. 16-35 Embolic (suppurative) osteomyelitis and physitis, bone, distal radius, foal.
The pale region in the metaphysis extending upward to the top middle border of the illustration represents suppurative inflammation and necrosis. It is bordered by a red rim of active hyperemia. A fissure (the linear space along the metaphyseal margin of the growth plate) and the porosity (darker regions within the growth plate, right) are the result of bone lysis (primary trabeculae) caused by the infection. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Fig. 16-36 Embolic (suppurative) osteomyelitis, bone, distal femur, medial trochlear ridge, transverse section, horse.
A, Oblique view of articular cartilage and transverse section. B, The suppurative osteomyelitis has extended from its site of origin in the cancellous bone into the articular-epiphyseal complex, with resulting collapse and fragmentation of the articular cartilage. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
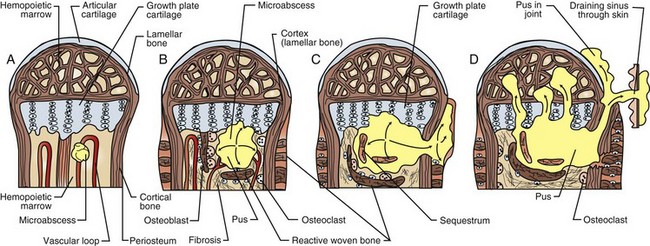
Fig. 16-37 Schematic diagram showing patterns of spread of embolic osteomyelitis from the physis.
The septic embolus lodges in capillary loops at the metaphyseal side of the physis (A). The inflammation causes lysis of metaphyseal bone and growth plate cartilage which can cause mechanical instability, to which the periosteum responds by producing reactive bone (B). The exudates can lyse the cortex at its thinnest point (the metaphyseal cut back zone) and extend into the periosteum (periostitis) or into the joint (arthritis) (C and D). (Redrawn from Rubin E: Essential pathology, ed 3, 2001, Lippincott Williams & Wilkins).
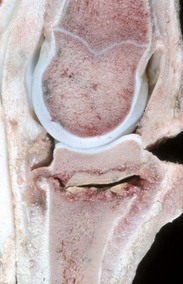
Fig. 16-38 Embolic bacterial physitis, osteomyelitis, periostitis, proximal first phalanx and arthritis, metacarpal-phalangeal joint, horse.
Bacterial inflammation has destroyed the physis and has extended into the periosteum and joint cavity (left). (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Portals of Entry into Joints
Microbial and other agents enter the joints via hematogenous spread (Fig. 16-39). For example, neonatal bacteremia secondary to omphalitis or oral-intestinal entry commonly leads to polyarthritis. Bacteria can also reach the joint by direct inoculation, as in a puncture wound, by extension from adjacent periarticular soft tissues, or by extension from adjacent bone.
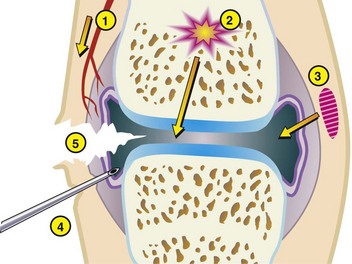
Fig. 16-39 Schematic diagram of the routes of infection for a joint in an adult.
1, The hematogenous route. 2, Extension from osteomyelitis. 3, Spread from an adjacent soft tissue infection. 4, Diagnostic or therapeutic procedures. 5, Penetrating damage from puncture or cutting. (From Huether SE, McCance KL: Understanding pathophysiology, ed 4, St Louis, 2008, Mosby.)
Portals of Entry into Tendons/Ligaments
Infection of tendons and ligaments usually requires a laceration or puncture wound because of the dense connective tissue covering of the tendon (epitenon) or ligament (epiligamentum). Less commonly, there may be extension of infection from infected skin or from an adjacent joint. The sequelae of bacterial infection of tendons and ligaments most often include adhesions and accompanying loss of function.
Defense Mechanisms of Bone
The defense against infectious agents is no different in bone than in other tissues. However, the consequences of an inflammatory response can greatly affect the structure and function of bone. Many soluble inflammatory mediators can increase both bone formation and resorption, resulting in varying degrees of reactive bone formation and bone lysis. The exudate associated with some acute infections in the medullary cavity can increase the pressure in this region and cause compression of the nutrient artery, resulting in ischemic necrosis. If resorbed cortical bone is replaced by fibrous repair tissue, the bone can become unstable.
Defense Mechanisms of Joints
The cellular and humoral defenses against infectious agents are no different in the joint from those in other tissues. However, because articular cartilage has such limited ability to regenerate, the consequence of inflammation that destroys the ability of synovium to provide nutrients and synovial fluid to the cartilage or that destroys areas of the cartilage could be the subsequent progression to degenerative joint disease.
Defense Mechanisms of Tendons/Ligaments
The cellular and humoral defenses against infectious agents are no different in tendons and ligaments from those in other tissues.
Disorders of Bone in Domestic Animals (Horses, Ruminants [Cattle, Sheep, and Goats], Pigs, Dogs, And Cats)
Abnormalities of Growth and Development
Disorders of Bone Resorption
Osteopetrosis: Osteopetrosis is a heterogenous group of heritable conditions that result in diffuse osteosclerosis (increased bone matter per unit area) caused by a defect in bone resorption by osteoclasts. Although bone mineral density is increased, the bones can be more fragile. Pathologic fractures are reported in animals that survive birth. The cause of the fragility is not certain but is likely related to failure of normal bone modeling in both cortex and medulla. The disease occurs in humans and in multiple animal species, including dogs, sheep, horses (Peruvian Paso breed), cattle (Angus and Herefords), rats, and several strains of mice. Specific immune regulators and growth factors that influence osteoclast ontogeny and/or activation have been implicated in the pathogenesis of some of the naturally occurring mutations associated with osteopetrosis in animals. In severe forms of osteopetrosis, the insufficient bone marrow cavity is unable to support hematopoiesis, resulting in thrombocytopenia, anemia, and infectious complications. Because osteoclasts arise from the hematopoietic precursors, bone marrow transplantation (in which normal donor marrow components are injected) is curative for some types of osteopetrosis in humans.
Growth plate: There are no primary lesions in the growth plate in osteopetrosis.
Trabecular bone: Osteoclasts are unable to resorb and shape (model) the primary trabeculae (Fig. 16-40). As a result, spicules of bone with central cores of calcified cartilage fill the medullary cavity. This process affects all bones that develop in a cartilaginous model (elongate by endochondral ossification from a growth plate). Affected bones are dense and have no medullary cavity. The defect in Angus cattle is inherited as an autosomal recessive trait. Affected calves are typically stillborn a few weeks premature and also have brachygnathia inferior, impacted molar teeth, and deformed cranial vaults that compress the brain.
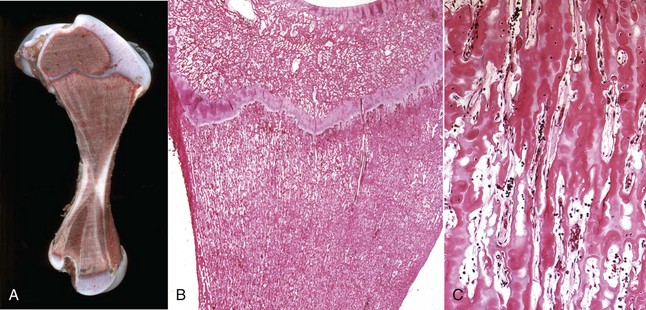
Fig. 16-40 Osteopetrosis, bone, femur, calf (Aberdeen Angus).
A, The primary trabeculae are retained and fill the entire medullary cavity. B and C, Retained straight unremodeled trabeculae containing cartilaginous cores fill the medullary cavity. H&E stain. (A courtesy Dr. H. Leipold, College of Veterinary Medicine, Kansas State University. B and C courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Cortical bone: Cortical bone lesions in osteopetrosis are not reported consistently but can range from the cortex being too thin to it being inadequately compacted in animals that have plexiform (laminar) cortical bone growth.
Disorders of Bone Formation
Osteogenesis Imperfecta: Osteogenesis imperfecta is a hereditary osteopenic (reduced bone mass) disease that is characterized clinically by multiple fractures, joint laxity, and defective dentin. It is the most common heritable connective disorder in humans and has been described in the veterinary literature in calves, lambs, kittens, and puppies. The tissues involved include bone, dentin, tendons, and sclera, and clinical signs include bone fractures, joint laxity, dental abnormalities, and blue sclerae. In humans, the disease is most commonly caused by mutations in one or both of the genes that code for type I collagen, and this also has been reported in animals. Genes coding for enzymes responsible for posttranslational modification of collagen are mutated less commonly. Mild forms of the disease, caused by inactivation of an allele of one the genes, result in reduced amounts of normal collagen. Conversely, mutations causing substitutions in critical amino acids necessary for collagen helix formation and cross-linking can result in production of normal amounts of structurally inferior collagen. Interestingly, the clinical manifestations of these mutations are usually limited to the bone, teeth, and eyes, even though type I collagen is also the major structural collagen in skin. A mutation in the SERPINH1 gene has been reported in osteogenesis imperfecta in dachshund dogs. SERPINH1 is present in high concentration in the endoplasmic reticulum and specifically binds to and stabilizes the triple helices of nascent collagens.
Growth plate: Growth plates are not affected in osteogenesis imperfecta (primary collagen of hyaline cartilage is type II collagen).
Trabecular bone: In severe cases, there is greatly reduced trabecular bone without evidence of marked osteoclasis or proliferation of fibrous tissue, as would be expected in severe cases of fibrous osteodystrophy (see later discussion). Osteoblasts may appear normal or small. In some cases, the amount of bone and its microscopic appearance are normal, but there is evidence of fractures. These fractures may occur in utero and are recognized at birth by callus formation. Bone fragility in cases in which bone morphology and mass are normal is likely to be caused by errors in helix formation or cross-linking of tropocollagen molecules. These individuals may become osteopenic with age, possibly the result of decreased mechanical use because of bone pain from fractures.
Cortical bone: There is a delay in the compaction of cortical bone. During normal development, species-dependent spaces of varying size and contour remain between laminae of woven bone in the developing cortex. The filling of these spaces by bone to solidify the cortex is called compaction. The cortices in osteogenesis imperfecta may be composed of woven bone containing large vascular spaces that remain empty. If the animal survives, these vascular spaces may eventually fill in with bone to form compact cortical bone.
Teeth: In some cases, bones may be structurally normal at birth other than the presence of fractures; therefore examination of teeth can be important in reaching a diagnosis of osteogenesis imperfecta. Grossly, teeth in cases of osteogenesis imperfecta may be pink because of visibility of the dental pulp through the thin crown. Dental fractures also may be present. Histologically, the dental tubules are short, tortuous, and sometimes absent. The disorganization of the dentin is a qualitative change that allows confirmation of the diagnosis without requiring an age-matched control animal.
Disorders of Bone Modeling
Congenital Cortical Hyperostosis: Congenital cortical hyperostosis is an autosomal recessive hereditary disease of newborn pigs that is characterized by abnormal periosteal bone formation involving the major long bones. Lesions may be a consequence of disorganization of the perichondral ossification groove (the chondrogenic membrane surrounding the growth plate that enables the growth plate to expand in diameter), and one or several limbs may be affected. Piglets are stillborn or die shortly after birth because of other defects; the pathophysiology of the bone lesions is not understood.
Growth plate: Growth plates are not involved.
Trabecular bone: Trabecular bone is not involved.
Cortical bone: The underlying cortex is normal; however, trabeculae of woven bone, oriented perpendicularly to the long axis of the cortex, radiate outward from its periphery. These trabeculae arise from the cambium layer of the periosteum and other than extent of the change, are typical of the nonspecific reaction of the periosteum to injury described previously.
Craniomandibular Osteopathy: Craniomandibular osteopathy, also known as lion jaw, typically occurs as an autosomal recessive condition in West Highland white terriers; however, similar lesions have been reported as isolated occurrences in other breeds and also have been associated with a leukocyte adhesion molecule deficiency in a colony of Irish setter pups that had concurrent metaphyseal osteopathy (see the discussion of noninfectious inflammation in the section on Inflammation of Bone). Lesions are bilaterally symmetric, resulting in diffuse, irregular thickening of the mandibles, occipital and temporal bones, and occasionally, other bones of the skull (Fig. 16-41). The tympanic bullae are often severely affected. Less commonly the disease can affect the appendicular skeleton. The disease often becomes apparent at 4 to 7 months of age and can regress. For affected dogs, mastication is painful and difficult, and the muscles of the skull become atrophic from disuse. The etiopathogenesis of this disease is unknown. A similar self-limiting disease has recently been reported in the calvaria of young Bullmastiff dogs.
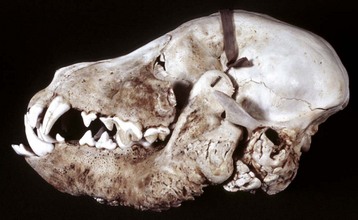
Fig. 16-41 Craniomandibular osteopathy, bone, skull, dog (West Highland white terrier).
Extensive periosteal new bone formation on the lateral surfaces of the body and ramus of the mandible, caudolateral maxilla, bulla tympanica, and occipital condyle. Macerated and bleached specimen. (Courtesy Dr. H. Leipold, College of Veterinary Medicine, Kansas State University.)
Growth plate: Growth plates are not involved.
Trabecular bone: In the medullary cavities of the bones of the skull and jaw, trabeculae become osteosclerotic (increased bone density per unit area) because of proliferation of woven bone by the osteoblasts of the endosteum with subsequent modeling and remodeling (see next discussion on cortical bone).
Cortical bone: In the skull and jaw, the cortices are thickened because of proliferation of woven bone by the periosteum. Characteristic of the disease is rapid disorganized modeling and remodeling, causing a mosaic of reversal cement lines with regions of lamellar bone adjacent to regions of woven bone.
Disorders of Endochondral Ossification
Chondrodysplasias: Chondrodysplasias (also called chondrodystrophies) are hereditary disorders of bone growth that occur as a result of primary lesions in growth cartilage. Growth cartilage is present in the physis (responsible for longitudinal bone growth) and the epiphyseal cartilage of the AEC (growth cartilage at the ends of growing long bones; responsible for development of epiphysis), both of which are replaced by bone in the adult individual. Chondrodysplasias can result in disproportionate dwarfism. Affected animals usually are short-legged with normal-sized heads because the bones of the calvarium (but not the maxilla and mandible) arise from intramembranous, rather than by endochondral, ossification. Primordial dwarfism, in which the limbs are proportional to body length, may occur secondary to underlying endocrine disease (pituitary dwarfism) or malnutrition but also may be genetically determined. Both types of dwarfism are represented by distinct breeds. For example, dachshund, Pekingese, and Bassett hounds are examples of chondrodysplastic dwarfs and miniature poodles, miniature schnauzers, and miniature pinschers are examples of primordial dwarfs.
The mechanisms of chondrodysplasias in animals are becoming better understood through genetic studies and often involve inherited errors in genes that control chondrogenesis. In humans, the most common dwarfism is called achondroplasia (a misnomer because growth cartilage is present). The condition is inherited as an autosomal dominant trait caused by a single point mutation in the fibroblast growth factor receptor 3 (FGFR3) gene, which is a negative regulator of bone growth. This mutation results in constant activation of this receptor causing downregulation of chondrocyte proliferation. Conversely, spider lamb chondrodysplasia, which occurs in Suffolk and Hampshire sheep, is the result of a single-base change in the tyrosine kinase II domain of FGFR3, which removes the FGFR3-induced inhibition of chondrocyte proliferation and results in elongation of the limbs and the presence of multiple secondary centers of ossification in the epiphyses (Fig. 16-42). Recently, the genetic mutation for the chondrodysplasia typifying the commonly recognized chondrodysplastic canine breeds, including dachshunds, Pekingese, and Bassett hounds has been identified as a conserved fibroblast growth factor 4 (fgf4) retrogene on canine chromosome 18 that likely arose before the division of early dogs into modern breeds Atypical expression of the FGF4 transcript in the chondrocytes may cause inappropriate activation of one or more of the FGF receptors, such as FGFR3.
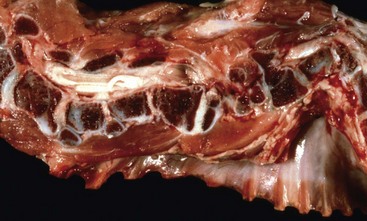
Fig. 16-42 Spider lamb chondrodysplasia, spine, longitudinal section, Suffolk lamb.
The vertebral column has multiple, disorganized ossification centers resulting in variation of the size, shape, and orientation of the vertebrae. (Courtesy Dr. Cathy Carlson, College of Veterinary Medicine, University of Minnesota.)
Skeletal dysplasias have also been reported in animals with inherited lysosomal storage diseases such as mucopolysaccharidosis and gangliosidosis. In these individuals, chondrocytes may be vacuolated due to cytoplasmic retention of glycosaminoglycans and lipid.
Growth plate: Growth plate widths can be normal or reduced depending on the disease. Likewise, chondrocytes can be arranged in normal columns or markedly disorganized. The cartilage matrix also may have a normal appearance or may be rarefied (less compact or dense, characterized by reduced staining intensity).
Trabecular bone: Trabeculae in the primary and secondary spongiosa may be thickened and irregular and may exhibit lateral bridging between trabeculae. Cartilage cores may be larger than normal and may be retained in the secondary spongiosa. These changes may vary greatly, depending on the type of chondrodysplasia.
Cortical bone: Lesions in cortical bone are greatly variable and are reflected primarily by grossly apparent abnormalities in shape or size. Histologically, this tissue usually has a normal appearance.
Osteochondrosis Latens and Manifesta: The osteochondroses consist of a heterogeneous group of lesions in growth cartilage of young animals and are characterized by focal or multifocal failure (or delay) of endochondral ossification. The sites where these lesions occur are the metaphyseal growth plate and the epiphyseal cartilage of the AEC. The lesions are common, often occur in bilaterally symmetric sites and represent an important orthopedic entity that has a number of different clinical manifestations in pigs, dogs, horses, cattle, and poultry. The hallmark of the uncomplicated gross lesions of osteochondrosis is focal retention of growth cartilage because of its failure to become mineralized and replaced by bone (a failure of endochondral ossification). In the growth plate, this is the result of an accumulation of viable hypertrophic chondrocytes, whereas in the AEC, it results from an area of necrosis of epiphyseal cartilage.
The etiology of osteochondrosis appears to be multifactorial, with genetics, rapid growth rate, vascular factors, and trauma all being implicated. Trauma is the most widely proposed etiology in both humans and animals; however, its most likely role is as a final insult to compromised epiphyseal cartilage rather than as an initiating factor in the development of early lesions. Rapid growth and nutritional factors also are cited. Although osteochondrosis occurs during the period of rapid growth and is most common in species that grow rapidly, most experimental work has failed to document rapid growth as a cause of the disease. In addition, little evidence is available to indicate that the lesions of osteochondrosis result from a specific nutritional deficiency. Copper deficiency, perhaps induced by excess dietary zinc, produces lysis of the AEC and formation of thin flaps of cartilage in thoroughbred suckling foals; however, the distribution of these lesions and their extent and severity distinguish them from the multifocal failure of endochondral ossification that is the hallmark of osteochondrosis.
Recent experimental work in pigs and horses has demonstrated that a defect (unknown cause) in vascular supply to growth cartilage, resulting in localized areas of ischemic necrosis, is important in the initiation of preclinical lesions. Although articular cartilage is avascular throughout life, subarticular epiphyseal cartilage of the AEC and growth plate cartilage both are highly vascularized tissues and depend on a vascular supply for viability (Fig. 16-43). The blood vessels supplying growth cartilage run in channels that are termed cartilage canals. Both growth plate and epiphyseal (growth) cartilage are temporary, by definition. During growth, the epiphyseal cartilage gradually becomes reduced in volume (until it disappears at maturity) and requires the nutritional support of fewer vascular channels, until it becomes entirely avascular and eventually is completely replaced by bone. It is during the time frame in which the epiphyseal cartilage is supplied by blood vessels that subclinical lesions of osteochondrosis occur. This time frame varies, based on site, age, and species, which likely explains, along with biomechanical factors, why different species have different lesion predilection sites for the development of osteochondrosis. Detailed studies in pigs and horses have revealed that these subclinical lesions occur at a much younger age in horses than in pigs. The finding of extensive areas of necrosis of epiphyseal cartilage in 2-week-old foals suggests that the underlying lesion may, in some cases, even occur in utero or shortly after birth. Although the nature of the insult to the vessel(s) is unclear, the result is a well-demarcated area of necrosis of epiphyseal cartilage that is centered on necrotic blood vessels, is visible only microscopically, and is termed osteochondrosis latens (Fig. 16-44). When the ossification front of the secondary center of ossification reaches the area of necrosis, failure of endochondral ossification occurs, resulting in an area of retained necrotic epiphyseal cartilage that is visible grossly (Fig. 16-45). The lesion at this stage is termed osteochondrosis manifesta. This lesion is highly vulnerable to traumatic clefting through the area of necrosis, termed osteochondrosis dissecans, even with physiologic pressure. Once clefting occurs and subchondral bone is exposed, the lesion is highly painful and results in clinical signs of lameness. Cleft formation most commonly does not occur, and in most cases, the necrotic epiphyseal cartilage becomes surrounded by subchondral bone, gradually diminishes in size, and eventually becomes completely replaced by bone. In horses, particularly in the medial femoral condyle (weight-bearing location), some of these early lesions of osteochondrosis have been hypothesized to develop into subchondral bone cysts.
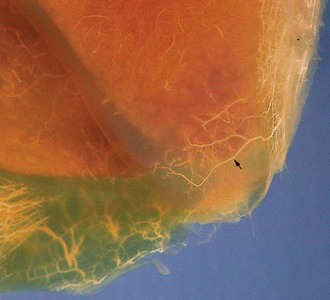
Fig. 16-43 Vascular supply to cranial aspect of distal intermediate ridge, tibia, 14-day-old Standardbred foal.
The arteries have been perfused with micronized barium and the tissues cleared by the modified Spälteholz technique. The cartilage has become translucent, and the subchondral bone is reddish-brown. The image shows a perfused vessel coursing distally on the cranial aspect of the tibia before turning, branching, and continuing within the cartilage on the articular aspect of the distal tibia (arrow). (Courtesy Dr. Kristin Olstad, Norwegian College of Veterinary Medicine, Oslo, Norway.)
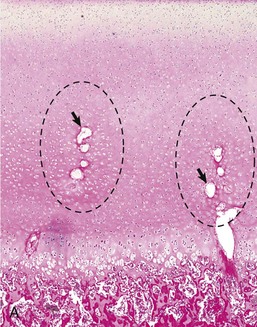
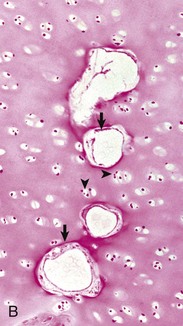
Fig. 16-44 Osteochondrosis latens, lateral femoral condyle, 12-week-old pig.
A, Locally extensive area of chondronecrosis (within ellipses) within the epiphyseal cartilage of the articular-epiphyseal complex (AEC), centered on necrotic cartilage canal blood vessels (arrows). Neither the overlying articular cartilage or the subjacent subchondral bone are involved at this stage of the disease. H&E stain. B, Higher magnification of A showing necrotic cartilage canal blood vessels (arrows) and necrotic chondrocytes (arrowheads). H&E stain. (Courtesy Dr. Cathy Carlson, College of Veterinary Medicine, University of Minnesota.)

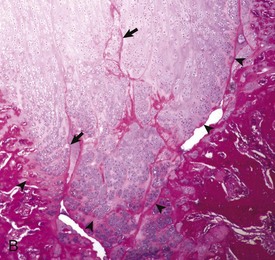
Fig. 16-45 Osteochondrosis manifesta, bone, distal femur, lateral trochlear ridge, horse.
A, The cartilage (C) extending into the epiphysis is composed of necrotic epiphyseal cartilage that has failed to undergo endochondral ossification and been retained, whereas the remainder of the epiphysial cartilage has been converted to bone. The retained cartilage has a yellowish appearance compared with the overlying articular cartilage due to degeneration/necrosis of the retained cartilage. B, Histologically, there is evidence of a marked cartilage repair process that is characterized by chondrocyte proliferation. The chondro-osseous junction (arrowheads) contains a thin margin of acellular, eosinophilic matrix that is associated with several small clefts and is interpreted as necrotic cartilage; eosinophilic streaks (arrows) within the retained cartilage may represent collapsed, necrotic vascular channels. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University).
Physeal lesions of osteochondrosis also are likely have a vascular etiology but are characterized by multifocal areas of retained, viable, hypertrophic cartilage. If these involve an extensive area of the growth plate, the growth plate may fracture, either partially or completely, resulting in a variety of clinical sequelae.
Growth plate and AEC: Lesions of osteochondrosis are not recognized grossly until they result in a focal failure of endochondral ossification, at which time they are composed of a well-demarcated wedge of retained cartilage involving the epiphyseal cartilage at the AEC (osteochondrosis manifesta) or physis (physeal osteochondrosis) (Fig. 16-46). Hemorrhage and mineralized debris may be present at the junction of the retained cartilage and the underlying bone. Small lesions that have not undergone clefting produce no clinical signs and are much more common than those causing clinical disease. Predilection sites for both clinical and subclinical lesions include the AEC of the distal femur (especially the medial femoral condyle) and humerus (condyles and head) of pigs; the distal femur (medial condyle and both trochlear ridges), distal tibia (cranial intermediate ridge and medial malleolus), talus (trochlear ridges), and articular processes of the cervical vertebrae of horses; the humerus (humeral head and medial humeral condyle), distal femur (both condyles) and tibiotarsal bone (medial trochlear ridge) of dogs; the distal talus (medial and lateral trochlear ridges), distal femur (medial and lateral trochlear ridges) of cattle; and the proximal tibia of rapidly growing birds.
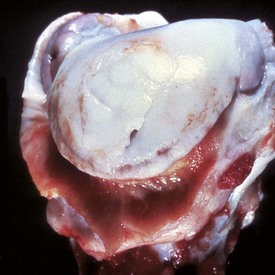
Fig. 16-46 Osteochondrosis dissecans, bone, articular cartilage, humerus, proximal end, dog.
A flap of thickened cartilage has detached from the underlying bone. The synovium is hyperemic and hyperplastic. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Osteochondrosis latens lesions are only recognized histologically in the AEC and occur as well-demarcated areas of necrosis of epiphyseal cartilage, usually adjacent to or surrounding one or more necrotic cartilage canal blood vessels (see Fig. 16-44). The overlying articular cartilage and subjacent subchondral bone are not affected at this stage of the disease. Osteochondrosis manifesta lesions also are composed of necrotic epiphyseal cartilage, but at this point of the disease, the ossification front has reached and partially surrounded the area of necrosis so that it is grossly visible (see Fig. 16-45). Lesions of physeal osteochondrosis, in contrast to those occurring in the AEC, are composed of columns of retained, viable, hypertrophic chondrocytes without evidence of mineralization or vascular invasion.
Trabecular bone: In lesions of osteochondrosis latens, trabecular bone is normal. Osteochondrosis manifesta lesions, however, include direct contact of the subchondral bone with an area of necrotic cartilage. Marked myelofibrosis and bony remodeling, with formation of locally extensive areas of woven bone, are nearly always present; however, inflammatory cells are rarely a feature of the lesion. A closely similar bony reaction is seen in lesions of physeal osteochondrosis. In both sites, cleft formation may occur at the junction of the retained cartilage and the underlying bone (see Figs. 16-46 and 16-47, A and B).
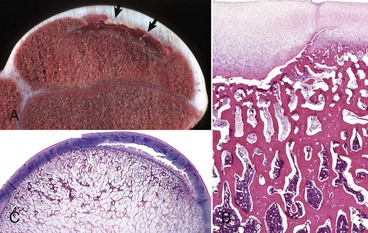
Fig. 16-47 Osteochondrosis of the articular-epiphyseal complex (AEC), bone.
A, Humerus, pig. The retained cartilage has separated from the subchondral bone (arrows) but a fissure extending to the articular surface (osteochondrosis dissecans) has not formed. B, AEC, distal femur, horse. A linear fissure between the thickened epiphyseal cartilage and subjacent underlying (subchondral) bone is extending into adjacent normal (right) articular cartilage. H&E stain. C, Humerus, horse. Note the linear fissure within the epiphyseal cartilage of the AE complex, which likely occurred secondary to ischemic necrosis of cartilage before the occurrence of a failure of endochondral ossification. H&E stain. (A and B courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University. C courtesy Dr. C. Bridges, College of Veterinary Medicine, Texas A&M University; and Dr. J. King, College of Veterinary Medicine, Cornell University.)
Cortical bone: Lesions in cortical bone are not expected at any stage of osteochondrosis of the AE complex or physis.
Osteochondrosis Dissecans: Osteochondrosis dissecans (OCD) is the name given to osteochondrosis at the AEC that forms clefts in the necrotic cartilage with subsequent fracture of the overlying articular cartilage (Fig. 16-47). The result is a cartilaginous or osteochondral flap, depending on whether bone is present with the cartilage. This may change with the duration of the lesion because cartilaginous flaps that have a blood supply may ossify over time. Should this flap fracture off and become free in the joint space, it is sometimes referred to as a joint mouse. OCD can be accompanied by pain, joint effusion, and nonspecific secondary lymphoplasmacytic synovitis. Free-floating chondral or osteochondral fragments (“joint mice”) occasionally interfere with mechanical movement of the joint. The lesions of OCD develop in various synovial joints, including those of the facets of the vertebral column. Common sites of OCD are the same as those of osteochondrosis manifesta (see previous section). The disease is extremely common in young breeding pigs and is a significant cause of lameness in this species. Although less common in horses and dogs, it remains an important cause of lameness in these species. The articular cartilage defect in OCD has poor healing capabilities, and such joints commonly develop some degree of degenerative joint disease. Surgical removal of the flap reduces the long-term clinical consequences; however, the defect repairs by the formation of fibrocartilage, which provides suboptimal biomechanical properties to the joint surface compared with articular cartilage.
For the development of OCD, clefts must develop in the epiphyseal cartilage of the AEC, usually at the stage of osteochondrosis manifesta (see Fig. 16-45). Cleft formation can cause mechanical instability and lead to separations within cartilage or between cartilage and the underlying bone. “Trauma” (most commonly normal physiologic activity) can cause the cleft to extend through the overlying articular cartilage, resulting in the formation of a flap. At this stage of the disease, the affected animal has acute pain.
Cartilage flap formation involving extensive areas of multiple joints can develop in horses without predisposing lesions of osteochondrosis latens or manifesta. In these cases, articular cartilage of normal thickness can be peeled from the subjacent bone because of fissuring in the deeper layers of the AEC. Such lesions have been seen in foals that have licked fences with zinc-based white paint. The pathogenesis of the lesion is thought to involve copper deficiency induced by the zinc excess. Copper is a required cofactor for enzymes that facilitate cross-linkages between tropocollagen molecules; however, these foals do not appear to have a generalized collagen dysplasia. The extensive distribution of the articular lesions distinguishes them from OCD, which occurs multifocally in species-dependent predilection sites.
Growth plate and AEC: The growth plate is not involved in OCD. The lesion in the AEC includes a locally extensive area of fibroplasia, neovascularization, and bony remodeling at the chondro-osseous junction. Even in the most chronic lesions, variably sized areas of necrosis of epiphyseal cartilage often can be identified at the chondro-osseous junction or along the deep surface of the articular cartilage. The superficial zones of the articular cartilage may be normal, other than the presence of a fissure that extends from the necrotic zone of epiphyseal cartilage to the articular surface. In cases in which the cartilage fragment has been displaced, all or part of the AEC is replaced by an area of cartilage ulceration overlying reactive and fibrotic subchondral bone.
Trabecular bone: The severity of changes in trabecular bone of the epiphysis reflects the extent of the cleft formation and depends on whether the flap has detached. Marked fibrosis and formation of reactive woven bone is expected at the base of the flap.
Cortical bone: Lesions in cortical bone are not expected with OCD.
Epiphysiolysis: Epiphysiolysis is the separation of the epiphysis from the metaphysis as the result of the formation of a horizontal fissure through an abnormal physis. The condition is most common in pigs and dogs. In market-weight pigs and in young gilts, the femoral head may be involved, whereas, in sows, separation of the ischial tuberosity (site of insertion of semimembranosus, semitendinosus, and biceps femoris muscles) at its growth plate (often bilaterally) is a common cause of caudal weakness and inability to stand and often occurs at parturition. In dogs, the process of epiphysiolysis can affect the anconeal process of the ulna, resulting in a condition known as ununited anconeal process. This may occur as one aspect of a condition known as the elbow dysplasia syndrome, which includes one or more of the following: OCD of the medial humeral condyle, fragmentation of the coronoid process, ununited anconeal process, and elbow incongruity (shortening of the ulna relative to the radius secondary to lesions in the distal ulnar growth plate). Because these lesions have primarily been studied in their chronic stages, the nature of the initial lesion is unclear; however, it is strongly suspected that they occur secondary to an extensive area, or to multifocal coalescing areas, of physeal osteochondrosis. A somewhat similar disease, known as diffuse physeal dysplasia with slipped capital femoral epiphysis, is described in heavy young adult castrated male cats. The growth plates in these cats appear to remain open longer than expected, are widened, and histologically, have a disorganized appearance in which chondrocytes are present in clusters, rather than columns, and are separated by variable amounts of matrix. It has been hypothesized that this dysplastic lesion may be present in all growth plates in affected animals, but fractures occur only in sites (e.g., femoral head) where shear forces occur during weight bearing. Osteochondrosis is rarely reported in cats, and the histologic changes in affected and contralateral growth plates are more characteristic of a physeal dysplasia (diffuse process) than of osteochondrosis (focal to multifocal process) in this species.
Several types of underlying conditions may result in partial closure of the physis, causing an angular limb deformity in the affected limb. This condition is most common in horses, where it usually occurs secondary to lesions of physeal osteochondrosis. Other causes include trauma and inflammation of the growth plate and/or primary trabeculae.
Growth plate: Grossly there is a horizontal fissure/fracture through the physis with complete or partial separation of the epiphysis from the metaphysis. In cats, the remaining physeal cartilage is dysplastic, as described above (disorganized arrangement of chondrocytes with separation by variable amounts of matrix). In pigs and dogs, the remaining physeal cartilage contains areas of delayed endochondral ossification. The chronicity of the lesions (with accompanying secondary changes) complicates their interpretation.
Trabecular bone: The response to trabecular bone in epiphysiolysis is characteristic of reaction to trauma and fracture and includes myelofibrosis, proliferation of reactive woven bone, and variable hemorrhage.
Cortical bone: There are no primary changes in cortical bone with this condition; any lesions that develop would be secondary to altered mechanical use.
Cervical Vertebral Myelopathy: Cervical vertebral stenotic myelopathy (CVSM) is a neurologic disease of horses and dogs (usually giant breeds) that occurs secondary to static or dynamic compression of the spinal cord by abnormally developed cervical vertebrae. This compression can be constant as the result of an anatomic stenosis of the canal termed cervical vertebral static stenosis (CVS), or may occur only during movement (usually flexion), which is termed cervical vertebral instability (CVI). CVS involves the less mobile caudal cervical vertebrae, mainly C5-C6 and C6-C7, and most commonly affects older horses (usually 1 to 4 years old). In contrast, the spinal cord lesions of CVI are centered on the more mobile cranial cervical vertebrae, primarily at C3-C4 and C4-C5, in younger horses (6 to 15 months of age). In both syndromes, anatomic localization of the lesion by neurologic examination, myelography, or computed tomography is very helpful to the pathologist. In thoroughbred and standardbred horses and in dogs, the static compression is usually the result of a grossly appreciable narrowing of the spinal canal from caudal to cranial within a single vertebral body that likely occurs during development (Fig. 16-48). In adult quarter horses, localized hyperplasia and fibrocartilaginous metaplasia of the ligamentum flavum (the ligament between the dorsal lamina), likely secondary to chronic mechanical irritation and/or trauma, can protrude into the spinal canal causing static compression of the spinal cord and closely similar clinical signs.
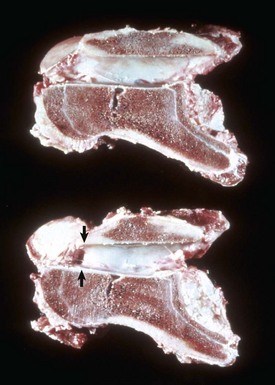
Fig. 16-48 Static stenosis, bone, cervical vertebra, sagittal section, horse.
Static stenosis (narrowing) of the vertebral canal caused by malformation of the vertebra (bottom vertebra, arrows). The top vertebra is normal. (Courtesy College of Veterinary Medicine University of Illinois.)
The lesions associated with dynamic compression of the cord are more variable and less definitive because identical lesions often occur in asymptomatic animals. In horses, the most common lesions are those of osteochondrosis (see previous discussion) of the cervical facets. Osteochondrosis can cause abnormal and asymmetric development of the facets or abnormally large cranial vertebral epiphyses. With either lesion, the dorsal aspect of the vertebral body may compress the ventral aspect of the cord during flexion, causing transient ischemia. Such compression may initially cause functional deficits without lesions in the spinal cord but also may progress to produce notable Wallerian degeneration. The frequency of osteochondrosis of the cervical vertebrae in clinically normal horses is the same as in horses with clinical signs of dynamic cervical vertebral myelopathy; however, the severity is worse in the latter cases. In addition, the frequency and severity of osteochondrosis lesions in the appendicular skeleton are greater in horses with clinical signs, further supporting the hypothesis that osteochondrosis may be an important underlying cause for this condition. The diameter of the vertebral canal is smaller in horses in which there is dynamic compression of the cord, suggesting that this change contributes to the development of clinical signs.
Metabolic Bone Diseases
Metabolic bone diseases are systemic skeletal diseases that are generally of nutritional, endocrine, or toxic origin (Table 16-1 and Web Table 16-1) and occur in both growing and adult skeletons. Metabolic bone diseases are often called osteodystrophies, which is a general term that implies defective bone formation. The classic metabolic osteodystrophies are osteoporosis, fibrous osteodystrophy, rickets, and osteomalacia. These terms imply specific pathologic changes but do not necessarily imply specific causes. For example, osteoporosis can be the result of a calcium deficiency, glucocorticoid therapy, or physical inactivity. In addition, different osteodystrophies can coexist in the same skeleton. For example, the skeleton of an animal with a severe calcium deficiency accompanied by excess dietary phosphorus might have features of both osteoporosis and fibrous osteodystrophy. In practice, most nutritional deficiencies in domestic animals do not involve a single element. More often, deficiencies are multiple, relatively mild, and do not produce the “classic” lesions that occur under experimental conditions.
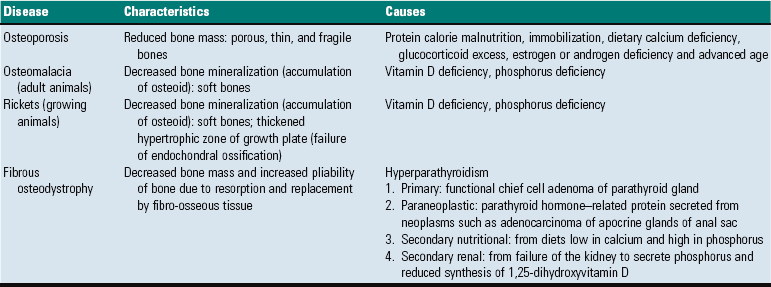
Osteoporosis
Osteoporosis is a disease in which bone fractures occur secondary to a reduction in bone density or mass (Fig. 16-49). The bone present, although reduced in amount, is normally mineralized. When there is reduced bone mass but no clinical disease (fractures), the term osteopenia is more appropriate; however, once fractures occur, the disease is called osteoporosis. In senile osteoporosis in humans, in addition to decreased bone density, the turnover rate is reduced, allowing microcracks (small cracks in the bone visible only microscopically) to accumulate. These microcracks, superimposed on the reduced bone mass, make bones more brittle than would be predicted from the reduced mass alone. In growing animals, osteoporosis is potentially reversible. In adults, however, loss of trabecular bone is generally considered to be permanent. Although osteoporosis affects both cortical and trabecular bone, the loss of trabecular bone occurs earlier because of its higher surface area. Although the thickness and density of cortical bone generally determines bone strength, trabecular bone contributes significantly to bone strength in some locations, including the femoral neck, vertebral bodies, and distal radius. This explains why osteoporotic fractures in humans occur most commonly in these locations. Some causes of osteopenia include calcium deficiency, starvation, physical inactivity, hypogonadism, and chronic glucocorticoid administration. A calcium deficiency can result in hypocalcemia, which is compensated for by increased PTH output and increased bone resorption. Starvation and malnutrition can result in arrested growth and osteoporosis, largely because of reduced bone formation resulting from deficiencies of protein and mineral. Reduced physical activity (disuse or immobilization osteoporosis) causes increased bone resorption and decreased bone formation. This loss of bone from disuse might be mediated through changes in piezoelectrical activity, streaming potentials, and stretch receptors that are able to detect decreased mechanical use of the skeleton. Loss of bone mass associated with long-term paralysis or immobilization is not necessarily progressive; rather, the skeleton stabilizes at a new (reduced) level. Postmenopausal osteoporosis is a common and important disease in women, often resulting in vertebral deformity or collapse and pathologic fractures of the femoral neck and distal radius (wrist). Declining concentrations of estrogens, physical inactivity, reduced muscle tone, and inadequate calcium intake are factors that can be responsible for causing osteopenia to progress to osteoporosis; however, in postmenopausal women, estrogen deficiency is the most important. Estrogen generally potentiates bone formation and inhibits bone resorption, likely by altering cytokine production from osteoblasts and monocytes. Estrogens promote secretion of TGF-β (an anabolic cytokine) and decrease production of IL-1, IL-6, and TNF-α (cytokines that promote osteoclasis). The loss of estrogen after menopause has the opposite effect. For example, IL-6 knockout mice do not develop osteopenia after ovariohysterectomy, confirming the critical role of cytokines in this process. Interestingly, ovariohysterectomy in the bitch is associated with only a transient osteopenia that does not develop into clinical osteoporosis. Thus dogs are not particularly useful as an animal model of this disease. Osteopenia associated with reduced estrogens from ovarian atrophy or ovariectomy appears to be greatest in animals with estrous cycles that extend throughout the year (e.g., rat, pig, and primate), rather than being seasonal.
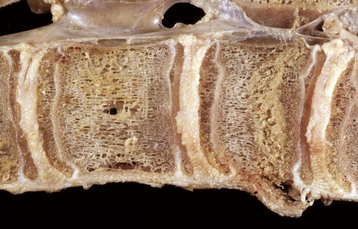
Fig. 16-49 Osteoporosis, bone, cervical vertebrae, sagittal section, horse.
Note the markedly thin cortices, particularly dorsally. The thickness of trabeculae also has been reduced, but this is difficult to appreciate grossly. The vertebra to the right has a compression fracture, causing shortening of the length of the vertebral body between the growth plates and fracture of the ventral cortex. The marrow has been flushed from the specimen in order to illustrate the bone changes. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Growth plate: Lesions in the growth plate are not expected in osteoporosis unless the disease is related to pituitary dysfunction or protein calorie malnutrition, in which case the plate would be thin.
Trabecular bone: Trabeculae become thinner, decrease in number, and develop perforations (loss of continuity) within the plates. With time the normal structure of trabecular bone (anastomosing plates) is replaced by strut or rodlike bone. The loss of these anastomoses results in reduced ability of the trabeculae to withstand stress. In most long bones of adults, trabeculae extend from the epiphysis to the metaphyseal-diaphyseal junction. The degree to which the trabeculae fail to reach this distance is an indication of the severity of the osteopenia (Fig. 16-50, A).
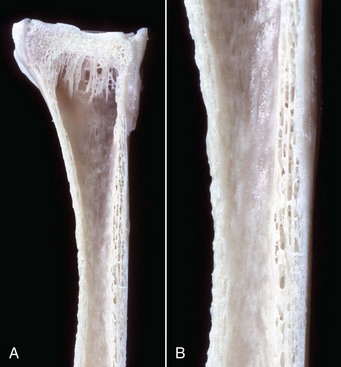
Fig. 16-50 Osteopenia, bone, metatarsal, sheep.
A, There is marked reduction in the number and length of metaphyseal trabeculae. B, The cranial cortex (right) is markedly porous (trabeculated), and the caudal cortex (left) is thin. The marrow has been flushed from the specimen. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Cortical bone: Cortical bone becomes thin because of osteoclastic resorption on the endosteal surface, with corresponding enlargement of the medullary cavity. The porosity of the cortex also increases because of increased osteoclastic resorption within the cortical vascular spaces and Haversian systems and/or decreased osteoblastic activity and may more closely resemble trabecular bone than cortical bone (Fig. 16-50, B). In severe cases, loss of cortical bone results in an increased susceptibility to fracture.
Rickets and Osteomalacia
Failure of mineralization with subsequent bone deformities and fractures is called rickets in the growing skeleton and osteomalacia (soft bone) in the adult. Rickets is a disease of bone and epiphyseal (growth) cartilage in immature animals, whereas osteomalacia is a disease where the lesions are confined to bone and only occurs in adults. Affected animals have bone pain, pathologic fractures, and deformities such as kyphosis and scoliosis.
The most common causes of rickets and osteomalacia are deficiencies of vitamin D or phosphorus. However, both diseases can occur in chronic renal disease and in chronic fluorosis. Dietary phosphorus deficiency is not common but can occur in herbivores grazing on phosphorus-deficient pastures. Phosphorus-deficient animals often have reduced feed intake, are unthrifty, and have impaired reproductive performance. Vitamin D deficiency is rare because of the common addition of this vitamin to commercial feed. In addition, animals exposed to adequate sunlight should be able to synthesize vitamin D if their kidneys are normal. Although calcium deficiency is not commonly considered to be a cause of rickets in animals, studies in humans provide evidence that some cases of rickets may be attributable to extremely low dietary calcium intakes in the presence of adequate vitamin D intake, particularly in infants and younger children (versus adolescents). In addition, low dietary calcium is known to exacerbate the development of vitamin D deficiency rickets in children. The mechanisms of this phenomenon are not known; however, it has been shown that knockout mice with no receptors for vitamin D develop rickets that can be corrected by feeding a high-calcium diet.
Growth plate: The growth plates in rickets are diffusely but irregularly thickened because of failure of cartilage matrix mineralization and endochondral ossification (Fig. 16-51). This lesion may be evident in the costal-chondral junctions as prominent, nodular thickenings that historically have been called rachitic rosary due to its resemblance to a string of prayer beads. This thickening is apparent grossly as retained epiphyseal cartilage. Microscopically, the columns of chondrocytes in the plate are somewhat disorganized, and in mammals, there is a marked increase in the number of chondrocytes in the zone of hypertrophy compared with normal. It is uncertain if the disorganization of chondrocytes in vitamin D-deficiency rickets is caused by a primary effect of a lack of vitamin D metabolites (specifically, 24,25-dihydroxyvitamin D) or to a mechanical consequence of the failure of endochondral ossification. Because of the inadequate absorption of calcium, the ricketic growth plate does not undergo mineralization. In mammals, when mineralization of the cartilage matrix does not occur, blood vessels with accompanying chondroclasts do not invade the physis and the process of endochondral ossification does not proceed.
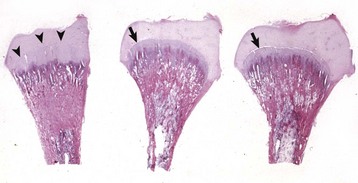
Fig. 16-51 Vitamin D deficiency (experimental), bone, tibia, chicken.
Left specimen: rachitic tibia; middle and right specimens: normal chicken and a chicken fed a vitamin D–deficient diet supplemented with calcitriol. The latter two specimens appear normal and are indistinguishable from each other. The growth plate in the rachitic bird is thickened. The arrowheads indicate the junction between the growth plate and the epiphyseal cartilage. The metaphysis has not undergone modeling (“cutback”). In the normal-appearing bones, notice the tapering of the metaphysis (“cutback” zone) and the thickness of the growth plate. In these normal chickens, the cleft (arrows) separating the growth plate from the epiphyseal cartilage is an artifact. There is no ossification center present in the epiphysis, which is normal for young broilers. H&E stain. (Courtesy Dr. L. Nagode.)
Trabecular bone: Grossly and radiographically, the metaphyses are “flared” in rickets because of failure of bone and cartilage removal in the cutback zones (see Fig. 16-51). Poorly mineralized matrices cannot be resorbed because osteoclasts cannot bind to an unmineralized matrix (see previous discussion). Microscopically, the surfaces of trabecular bone in rickets and osteomalacia contain excessive amounts of osteoid (unmineralized matrix) (Fig. 16-52). Because osteoclasts are not able to adhere to or resorb osteoid, bone modeling and remodeling is impaired. Hypocalcemia can develop concurrently with vitamin D deficiency, and lesions of secondary hyperparathyroidism (fibrous osteodystrophy [see later discussion]) also can develop.
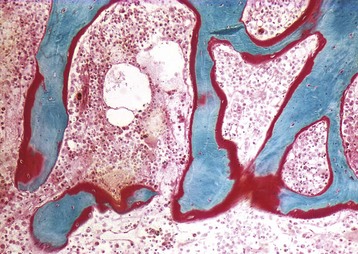
Fig. 16-52 Osteomalacia, bone, cross-section, metaphysis, trabeculae, human.
This person had osteomalacia secondary to malabsorption of fat soluble vitamins (including vitamin D) associated with gastrectomy. This section was not demineralized and was stained to show the difference between osteoid (red) and fully mineralized bone (blue-green). Normally, no more than 10% of trabecular surfaces should be covered by osteoid seams; however, in this case almost all surfaces are covered by osteoid. Goldner’s trichrome stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Cortical bone: Grossly, cortical bone can appear normal or the softened bone can be deformed by weight bearing. In severe cases, the bones are so soft that they can be cut with a knife. Microscopically, endocortical and trabecular surfaces in the cortex can have numerous wide seams of unmineralized osteoid. Osteomalacic bone is susceptible to accumulation of microcracks because of the impaired remodeling and modeling secondary to the failure of osteoclasts to bind to osteoid surfaces.
Fibrous Osteodystrophy
Fibrous osteodystrophy is the name given to the skeletal lesions that result from primary hyperparathyroidism, secondary hyperparathyroidism, and pseudohyperparathyroidism. These disorders are characterized by increased, widespread osteoclastic resorption of bone and replacement by primitive fibro-osseous tissue, which results in a weakened bone structure. Sequelae include lameness, pathologic fractures, and deformities. As described previously, osteoblasts—but not osteoclasts—have receptors for PTH and respond by upregulating production of RANKL and downregulating secretion of OPG. Bone marrow stromal cells also have receptors for PTH, and when the hormone is expressed constantly at high levels, these cells differentiate into fibroblasts. Paradoxically, intermittent dosing of PTH has the opposite effect on bone and is used clinically in humans for the treatment of osteoporosis.
In domestic animals, primary hyperparathyroidism, which includes functional parathyroid adenoma, parathyroid carcinoma, and idiopathic bilateral parathyroid hyperplasia, is rare. Secondary hyperparathyroidism is more common and can be either nutritional or renal in origin. Nutritional hyperparathyroidism is caused by dietary factors that tend to decrease the concentration of serum ionized calcium, to which the parathyroid glands respond by increasing output of PTH. Nutritional hyperparathyroidism is most common in young, growing animals that are fed rations that are deficient in calcium and have a relative excess of phosphorus. Unsupplemented cereal grain rations fed to pigs, all-meat diets fed to dogs and cats, and bran fed to horses are examples of low-calcium, high-phosphorus diets that can cause secondary nutritional hyperparathyroidism and eventually, fibrous osteodystrophy. Increased concentrations of dietary phosphorus are important in the evolution of fibrous osteodystrophy, perhaps by interfering with the intestinal absorption of calcium. Pseudohyperparathyroidism is also known as humeral hypercalcemia of malignancy and is discussed in Chapter 12.
Growth plate: No lesions are expected in the growth plate or AEC.
Trabecular bone: Trabeculae are replaced by irregular spicules of woven bone that are separated by variable amounts of fibrous connective tissue and are accompanied by numerous osteoclasts. Osteoblastic proliferation can be marked but ineffectual in that little osteoid is produced. Proliferation of fibrous tissue in the marrow space is usually marked but may be subtle in early stages, consisting of only two or three layers of fibroblasts between the bone lining cells and the marrow space (Fig. 16-53). In severe cases, the marrow cavity is filled with fibrous tissue.
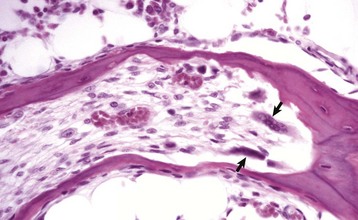
Fig. 16-53 Tunneling resorption (within a trabecula), fibrous osteodystrophy, bone, longitudinal section, dog.
Osteoclasts (arrows) have resorbed the central portion of the trabecula, and this tissue is being replaced by fibrous tissue rather than bone. H&E stain. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Cortical bone: Bone resorption and replacement by fibrous tissue can be so extensive that the bone becomes pliable (Fig. 16-54). Clinically and at postmortem examination, severely affected bones can be bent at a right angle without fracture. Osteoclastic resorption begins on the endocortical bone surface, but any vascular spaces within the wall of the bone can undergo marked enlargement by osteoclastic resorption and replacement by fibrous tissue. In advanced disease, the entire width of the cortex may be replaced by reactive woven bone and fibrous tissue. Sometimes the proliferation of fibrous tissue is so exuberant that the external dimension of the bone is increased (Fig. 16-55). This process is most common in the maxilla and mandible and may reflect the response of the weakened bone to the intense mechanical stress of mastication.
Fig. 16-54 Fibrous osteodystrophy, bone, maxillae and mandible, transverse section, dogs.
A, The pliable nature of the bone can be seen by the collapsed and folded (not fractured) appearance of the nasal septum and maxilla. B, Radiograph. There is marked loss of bone density around the teeth in both maxillae and mandibles. (Courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University.)
Fig. 16-55 Fibrous osteodystrophy, bone, maxillae and mandibles.
A, Nutritional fibrous osteodystrophy, horse. Note the swelling of the facial crest. B, Maxillae, cross-section, horse. Proliferating and poorly organized fibroosseous tissue in the maxillae has distorted and extended their contours laterally and compressed the nasal cavity medially. Note the absence of normal bone. C, Renal fibrous osteodystrophy, dog. Exuberant poorly mineralized woven bone and fibrous tissue has caused the maxillary bones to expand and distort the positioning of the teeth. Similar changes affect the mandible, and the teeth at this site are also malpositioned. D, Radiograph, renal fibrous osteodystrophy, dog. Note the reduced bone density (radiodensity) of the proliferative woven bone and fibrous tissue that has replaced the preexisting maxillary bones and the absence of normal radiodense bone. Postmortem transverse slab section. (A and D courtesy Dr. S.E. Weisbrode, College of Veterinary Medicine, The Ohio State University. B courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia. C courtesy Department of Veterinary Biosciences, The Ohio State University; and Noah’s Arkive, College of Veterinary Medicine, The University of Georgia.)