Degenerative Diseases of the Central Nervous System
Section 1: amyotrophic lateral sclerosis
Section 2: Alzheimer’s disease
Section 3: Huntington’s disease
After studying this chapter, the student or practitioner will be able to do the following:
1 Describe the course of amyotrophic lateral sclerosis.
2 Describe the differences between familial and sporadic amyotrophic lateral sclerosis.
3 Describe the role of the occupational therapist for a client with amyotrophic lateral sclerosis.
4 Describe the three subtypes of amyotrophic lateral sclerosis.
5 Identify the symptoms and incidence of Alzheimer’s disease.
6 Describe the pathophysiology of Alzheimer’s disease.
7 Describe the overall model of medical management of Alzheimer’s disease used by primary care providers and other health professionals.
8 Describe an approach to evaluation used by occupational therapists.
9 Identify stages of disease progression and general methods of treatment interventions associated with stages of dementia.
10 Describe the course and stages of Huntington’s disease.
11 Identify current research regarding the etiology of the disease.
12 Describe the medical management of Huntington’s disease.
13 Describe the purpose of occupational therapy for a client with Huntington’s disease.
14 Describe the three typical forms of multiple sclerosis.
15 Describe current research regarding the etiology of the disease.
16 Describe the symptoms of multiple sclerosis.
17 Describe complications that may occur as a result of the disease.
18 Describe the role of the occupational therapist for a person with multiple sclerosis.
19 Describe the course and stages of Parkinson’s disease.
20 Identify current research on the etiology of the disease.
21 Describe the medical management of Parkinson’s disease.
22 Describe the role of the occupational therapist for a client with Parkinson’s disease.
 Case Study
Case Study
Marguerite
Marguerite is a 35-year-old woman who was diagnosed with multiple sclerosis (MS) when she was 26. Although Marguerite was initially identified as having the relapsing-remitting form, the secondary progressive form of multiple sclerosis was recently diagnosed by her neurologist, who referred her for occupational therapy. Marguerite now uses an ankle-foot orthosis on her right lower extremity when walking and has diminished sensation and dexterity in her nondominant left hand.
An occupational profile revealed the following background information about Marguerite. She is married and has two boys aged 8 and 6. Both children are involved in soccer and swimming on a weekly basis. Her husband travels every month in his job as a sales manager for an insurance company. Marguerite is a special education teacher in an elementary school and works full-time, although she was unable to work during relapses. She cares for her 69-year-old mother, who has been diagnosed with Alzheimer’s disease. Her mother lives alone in her own apartment in the same city. Although Marguerite has two sisters, neither lives within driving distance; Marguerite has the primary responsibility of caring for her mother and her children.
Marguerite was asked to identify what areas of occupational performance were problematic or successful. She quickly replied that being a chauffeur for her children, a manager of her mother’s medical care, and a housekeeper for her family was problematic. She felt as though she were constantly juggling schedules; recently, her children’s swimming class changed to a different day, which caused a big change in her schedule. Even though she wants her children to have the opportunity to pursue sports, Marguerite finds it difficult to supply snacks every month for these extracurricular activities, an obligation that each participant’s mother is expected to fulfill. Her husband does try to help with household chores when he is home, but he works long hours and does not have time to shop for the family’s groceries. Marguerite is also responsible for arranging all her mother’s medical appointments; shopping for her mother, who no longer drives; and visiting her mother daily. Marguerite reports that she often feels so tired after teaching all day and running all her errands that she has trouble making dinner when she returns home.
Marguerite was most comfortable with her work situation and reported that many of her colleagues had offered to help with various tasks such as playground duty or monitoring the students during lunchtime. This allowed Marguerite to have a brief break during the day to rest.
1. What evaluations would you perform to collect additional data for developing an intervention plan?
2. Where would you start your intervention plan in light of Marguerite’s report of her occupational profile?
3. How would the change in Marguerite’s diagnosis from the relapsing-remitting form to the secondary progressive form of multiple sclerosis affect her current occupational roles?
4. How would the strategies that you select for Marguerite be applied to other clients who have degenerative neurologic disorders?
Introduction
This chapter addresses the impact of degenerative neurologic disorders on a person’s occupational performance and outlines the role of occupational therapy (OT) in providing services to clients with these disorders. The specific disorders discussed in this chapter are amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Huntington’s disease (HD), multiple sclerosis (MS), and Parkinson’s disease (PD).
In degenerative neurologic disorders, the disease progresses and an individual’s occupational performance is often increasingly compromised. OT aims to help the client compensate and adapt as function declines secondary to the disease process. Environmental adaptations and modifications are frequently necessary to maintain functional skills for as long as possible.
Degenerative neurologic diseases may occur because of structural or neurochemical changes within the central nervous system (CNS).72 In the disorders discussed in this section, the client’s CNS usually functions normally during childhood and adolescence. After these years, the client experiences signs and symptoms indicating that CNS functions are deteriorating. The progressive nature of the disorder varies from person to person. Some clients have a rapid decline in function, whereas others maintain functional skills for many years.
The decline in function may compromise the individual’s sense of self-efficacy in performing various tasks.195 No longer is the individual able to perform personal or instrumental activities of daily life at the same level of independence. Dependence on others can alter the client’s concept of self-worth and self-control. The OT practitioner serves an important role in reframing the client’s sense of self even though functional independence may be deteriorating. A man with PD who is unable to dress independently may now direct a personal care attendant or home health aide to perform these tasks. A woman with MS who was previously responsible for household finances may need to instruct a member of the family to complete these activities.
The disorders discussed in this chapter are most often diagnosed during adult or later adult life, after habits, routines, and patterns of independent behavior are well established. A client may encounter a significant change in social relationships and interactions secondary to a decline in functional abilities. The OT practitioner must consider the ways in which progressive loss of function affects the client’s social and occupational roles, whether those roles are as husband, wife, parent, adult child, worker, sibling, or friend. OT must address the needs of the client within the context of his or her social, physical, and cultural environment.
OT intervention aims to support the client’s ability to function within his or her environment. The rate at which the client’s symptoms progress influences the intervention plan. A client who displays progressive loss of fine motor skills over a 20-year period has a much different profile than one who loses all upper extremity function within 2 years. Use of adaptive equipment must be weighed carefully against the rate of deteriorating skills.
The OT practitioner must be knowledgeable about the support services and respite care available to clients with a degenerative neurologic disorder. A PD support group may provide the necessary social support for both a man with this disorder and his family. MS support groups may offer clients information on new intervention methods available, along with the opportunity to share life experiences.
An OT intervention plan should address not only the physical limitations associated with various disorders but also their cognitive, social, and emotional implications. Many individuals with neurodegenerative disorders have concomitant depression. Depression can be a reaction to the loss of function associated with some disorders or the primary symptom of other disorders. Occupational therapists should regularly screen for depressive features. An instrument such as the Beck Depression Inventory can effectively evaluate this component.25,26 In addition to evaluation of depression in clients with neurodegenerative disorders, cognitive abilities should be assessed. Clients may have concomitant cognitive problems because of the destruction of neurologic structures, and these deficits can have a dramatic effect on intervention. Brief assessments such as the Mini-Mental State Examination (MMSE)65 or the Cognistat154 can be used to determine cognitive abilities and establish a baseline of performance.
In most cases, the occupational therapist is a member of a team providing services to the individual with a degenerative neurologic disorder.98 As a team member, the occupational therapist must consider the roles that other professionals and family members play in the client’s life and incorporate this knowledge into the intervention plan. OT practitioners provide a unique and needed service to individuals with degenerative neurologic disorders. A client who is able to engage in meaningful occupations despite deteriorating skills reflects the significant contribution of OT.
Two case studies are presented to illustrate the similarities and differences among clients faced with degenerative neurologic disorders. The first case concerns a woman who has MS. That case is presented at the beginning of the chapter. The second case concerns a man with PD and is presented at the end of the chapter to serve as a review. The cases should serve to prompt clinical reasoning and decision making as you read this chapter.
Section 1 Amyotrophic Lateral Sclerosis
This section of the chapter focuses on both areas of occupation and performance skills. Diagnosis and progression of disease are described in terms of the loss of performance skills, whereas Box 35-1 describes the OT interventions in relation to both occupation and performance skills as identified in the Occupational Therapy Practice Framework.
The term amyotrophic lateral sclerosis is used to identify a group of progressive, degenerative neuromuscular diseases. The underlying neurologic process involves destruction of motor neurons within the spinal cord, brainstem, and motor cortex.28,35 Affected individuals exhibit a combination of both upper motor neuron (UMN) and lower motor neuron (LMN) deficits at some point in the progression of the disease.
In the United States, ALS is also known as Lou Gehrig’s disease; in France, it is referred to as Charcot’s disease.27 The term motor neuron disease refers to a group of diseases that includes ALS, progressive bulbar palsy, progressive spinal muscular atrophy, and primary lateral sclerosis.106,151 Table 35-1 provides a description of each of these distinct subtypes of ALS. The classic forms of ALS are presented in this section.
TABLE 35-1
Clinical Subtypes of Amyotrophic Lateral Sclerosis
| Name | Area of Destruction | Symptoms |
| Progressive bulbar palsy (bulbar form) | Corticobulbar tracts and brainstem motor nuclei involved | Dysarthria, dysphagia, facial and tongue weakness, and wasting |
| Progressive spinal muscular atrophy (lower motor neuron form) | Lower motor neurons in the spinal cord and sometimes the brainstem | Marked muscle wasting of the limbs, trunk, and sometimes the bulbar muscles |
| Primary lateral sclerosis (upper motor neuron form)* | Destruction of cortical motor neurons; may involve both the corticospinal and corticobulbar regions | Progressive spastic paraparesis |
*The World Federation of Neurology classification of spinal muscular atrophy and other disorders of the motor neurons does not identify primary lateral sclerosis as a subtype of amyotrophic lateral sclerosis.27 This author is including primary lateral sclerosis in the list in recognition of the many other articles and books that recognize it as a subtype of amyotrophic lateral sclerosis.
From Belsh JM, Schiffman PL, editors: ALS diagnosis and management for the clinician, Armonk, NY, 1996, Futura; and Guberman A: An introduction to clinical neurology, pathophysiology, diagnosis, and treatment, Boston, Mass, 1994, Little, Brown.
The incidence of ALS is about 2.0 per 100,000 people, and it is diagnosed in approximately 5000 people in the United States each year.146 Two forms of ALS are recognized: sporadic and familial. Sporadic ALS makes up about 90% to 95% of the ALS population. Between 5% and 10% of individuals with ALS are found to have a family history of the disease. There is no difference in the symptoms of clients with the familial and sporadic types.
The diagnosis is primarily determined by clinical symptoms and ruling out other neurologic disorders.27 Multiple tests and a thorough neurologic examination provide a differential diagnosis. Tests include electromyography and nerve conduction velocity; blood and urine studies, including high-resolution serum protein electrophoresis, thyroid and parathyroid hormone levels, and 24-hour urine collection to assess for heavy metals; spinal tap; radiographs; magnetic resonance imaging; myelography of the cervical spine; and muscle and/or nerve biopsy.1,27,28,35
Pathophysiology
The etiology of ALS has not been established. Multiple theories have been proposed as the cause of the motor neuron destruction, including metabolic disorders of glutamate insufficiency, metal toxicity, autoimmune factors, genetic factors, and viral infection.187
Clinical Picture
The symptoms of ALS are variable, depending on the initial area of motor neuron destruction. An individual with ALS typically has a focal weakness beginning in the arm, leg, or bulbar muscles. The individual may trip or drop things and may have slurred speech, abnormal fatigue, and uncontrollable periods of laughing or crying (i.e., emotional lability). As the disease progresses, marked muscle atrophy, weight loss, spasticity, muscle cramping, and fasciculations (i.e., twitching of the muscle fascicles at rest) ensue. The individual may have greater difficulty with walking, dressing, fine motor activities, swallowing, and breathing. In the end stages the individual may require tube feedings and the support of a ventilator for respiration. ALS is a rapidly progressive disease with the majority dying of respiratory failure in 3 to 5 years after the onset of symptoms if no tracheostomy is performed. Approximately 10% of the population will live 10 years or longer.146 As ALS progresses, the disorder does not affect a person’s eye function, bowel and bladder function, or sensory function. Recent research has found that cognitive changes occur in 20% to 25% of the population with ALS. Specific cognitive deficits have been identified such as frontal temporal lobe dysfunction. As many as half may have mild to moderate cognitive or behavioral abnormalities. “Risk factors for developing cognitive problems, behavior changes or a full-fledged dementia syndrome may include: older age, bulbar onset ALS, a reduction in FVC (functional vital capacity) and a family history of dementia.”1,125,177
The prognosis is difficult to predict. Generally, individuals with early bulbar involvement have a poorer prognosis. A more positive prognosis is usually associated with the following factors: younger age at onset; onset involving LMNs located in the spinal cord; deficits in either UMNs or LMNs, not a combination of both areas; absent or slow changes in respiratory function; fewer fasciculations; and a longer time from the onset of symptoms to diagnosis. In some cases, the client’s condition stabilizes, with little progression of the disease.86
A client with ALS differs from a client, such as Marguerite, with MS; with ALS the loss of function is more rapid, without episodes of remission. A person with ALS must cope with a fatal disease, whereas a person with MS must cope with a chronically disabling condition.
Medical Management
The American Academy of Neurology has established practice parameters and standards to address major management issues for persons with ALS. An OT practitioner working with this population should be familiar with the standards to better understand the approaches and rationale for intervention. The parameters cover the following topics: how to inform the client of the diagnosis, when to consider noninvasive and invasive ventilator support, evaluation of dysphagia and intervention with a feeding tube, management of saliva and pain, and use of hospice services.133
Symptoms such as muscle cramping, excessive saliva, depression, and pain are managed with medication. The client’s respiratory status should be re-evaluated frequently to determine when noninvasive and invasive ventilator support is necessary. Swallowing function should also be evaluated frequently to prevent aspiration and to determine when and whether a feeding tube should be placed.
The Food and Drug Administration approved the drug riluzole (Rilutek) in 1995. Riluzole, an antiglutamate, was the first drug used specifically to alter the course of the disease by prolonging survival. In addition to inhibiting the release of glutamate from nerve endings, it blocks amino acid receptors on the cell bodies.187 Researchers believe that the success of riluzole indicates that an excess of glutamate leads to the death of motor neurons. Research has shown that riluzole prolongs the life of clients with ALS by at least a few months.142
Other medications and stem cell research204 for the treatment of ALS are currently in clinical trial.28,217 The clinical trials are focused on extending survival and slowing the decline associated with the disease.
Occupational Therapy Evaluation and Intervention
It is essential to work with the client and family throughout progression of the disease as needs change. Cultural, social, and spiritual values must be understood because these factors will influence ongoing decisions about personal care and life support. The client and family members must regularly update decisions about care. Decisions range from when or whether a wheelchair or adaptive eating device should be used to whether the client should undergo a tracheostomy, choose tube feeding, or use a ventilator. Psychosocial support regarding decisions about the extent of life support and medical intervention should be provided by the entire health care team, with the physician and client having primary responsibility. Several studies have shown that caregivers and patients have different needs and perceptions of quality of life. It may be beneficial for patients and caregivers to have time for education and support separately to meet their individual needs.32,88,162
Initial and ongoing OT assessment is essential to educate the client about ways to adapt functional activities as the disease progresses. Education is needed for nursing staff and physicians to understand the role of the occupational therapist in the treatment of clients with ALS.
Steady disintegration in the ability to speak, swallow, move, and perform activities of daily living (ADLs) makes it easy to overlook the presence of some common signs of cognitive or behavioral dysfunction, such as poor insight and deficits in planning.1 Multiple cognitive domains may be affected in persons with ALS, including psychomotor speed, fluency, language, visual memory, immediate verbal memory, and executive function.177
With impaired executive function, a person with ALS may have difficulty handling the visual, auditory, and other sensory data required for complex decision making.125 These cognitive and behavioral issues are pertinent when considering teaching strategies and the need for caregiver counseling and support.
Role of the Occupational Therapist
ALS progresses rapidly, with ongoing deterioration in physical status and possibly cognitive deficits. The intervention plan should focus on the client’s participation in occupational performance because the client’s functional status changes frequently and intervention focused on physical performance is limited. As the client’s function declines, there is a greater need for environmental support through providing durable medical equipment, modifying the home, and providing adaptive equipment. Depending on the client’s level of understanding, life support choices, and acceptance of the disease, the OT intervention may focus on structuring the client’s environment to support independence.45 Some clients with ALS may choose to have the maximum environmental and life support available to extend life. In this case, the occupational therapist may provide periodic re-evaluations to determine the client’s need for adapting self-care, work, and leisure activities. Other clients may request that no extraordinary life support be used, in which case the occupational therapist would assume a supportive role, perhaps helping these clients create a memory book to give to their loved ones. Table 35-2 provides a list of the functional deficits at various stages of the disease and interventions that may be required. When referring to this table, the OT practitioner must remember that each client’s clinical picture is unique and that symptoms may appear in a different sequence than the table indicates. For example, a client with early-onset bulbar signs may require earlier intervention regarding swallowing assessment and communication devices; another client may not need a wheelchair until the very late stages. Olney’s resource manual, Daily Activities Made Easier for People with Amyotrophic Lateral Sclerosis, provides practical solutions for ADLs and a variety of resources that address many of the problems that this population faces and is an invaluable resource for practitioners, patients, and families.32,131
TABLE 35-2
Interventions for Amyotrophic Lateral Sclerosis
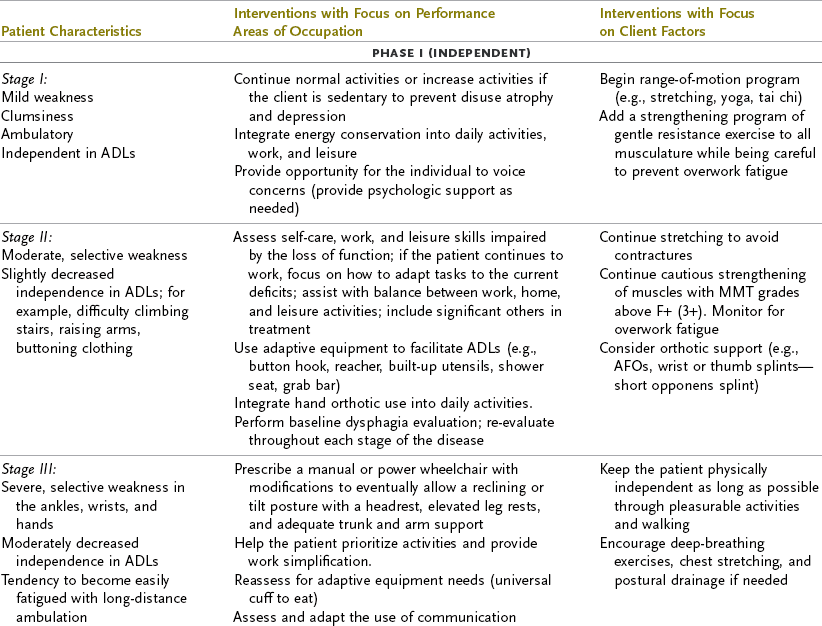
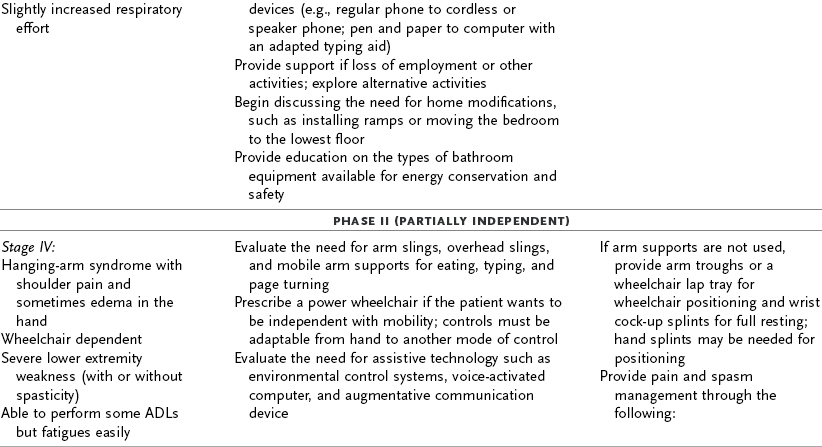
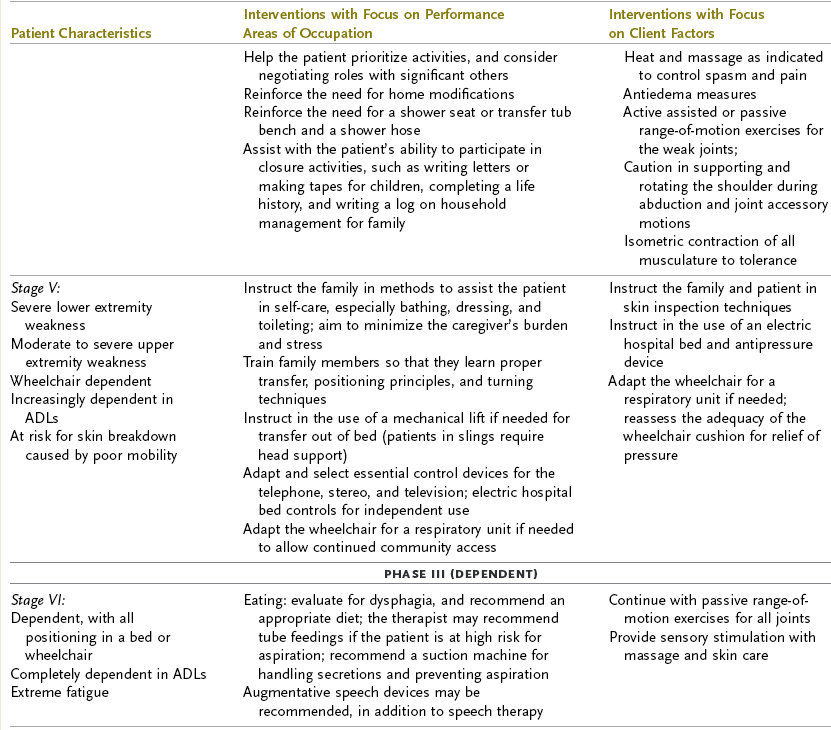
ADL, activities of daily living; AFO, ankle-foot orthosis; MMT, manual muscle test.
Modified from Yase Y, Tsubaki T, editors: Amyotrophic lateral sclerosis: recent advances in research and treatment, Amsterdam, 1988, Elsevier; and Umphred DA, editor: Neurological rehabilitation, ed 3, St. Louis, Mo, 1995, Mosby.
Individuals and their families require an interdisciplinary approach to the rapid changes in function, complex psychosocial factors, and quality-of-life issues associated with ALS. The impact of the disease on the client’s quality of life has been examined frequently. One study indicated that those who were less distressed and less depressed129,131,133 and had a more positive attitude lived longer.91 Fatigue and depression have been associated with a poor quality of life for an individual with ALS.121 The same study found that progression of the disease was not necessarily associated with a poor quality of life. Hecht and colleagues91 found that social withdrawal correlated with levels of disability; based on the results of this investigation, the authors recommended that mobility be improved with power wheelchairs and public transportation to prevent social withdrawal. Studies have also examined the impact of hope, spirituality, and religion as a means of coping with the disease.
Summary
ALS is a rapidly progressing, fatal condition of unknown etiology. OT aims to maximize the client’s function by providing interventions and periodic reassessment to compensate for declining motor function through modification of the environment, roles, and tasks and by helping the client and family achieve self-identified goals.
Section 2 Alzheimer’s Disease
Dementia is the general word used for a group of symptoms caused by serious disorders of the brain. Memory loss and other related problems in language, perception, thinking, and judgment interfere with learning, communicating, relating, and even caring for self. Dementia is not just one disease but includes a number of diseases (e.g., vascular dementia, Lewy body dementia, frontotemporal dementia). The most common dementia is Alzheimer’s disease, especially in persons older than 65 years.
AD is considered a primary dementia because it does not result from other diseases. Secondary dementia may be used to refer to symptoms of dementia that are associated with other physical diseases (PD, ALS, MS, HD) and are discussed elsewhere in this chapter. AD, unlike the other neurologic disorders discussed in this chapter, is formally classified as a mental disorder by the American Psychiatric Association.11 The exact cause of AD is still unknown. Because of the damage to brain cells and irreversible cognitive decline, the disease results in impaired higher mental processes, altered behavior, and disturbances in mood. The disorder progresses gradually and produces multiple cognitive deficits, a significant decline from previous levels of functioning, and noticeable impairment in social and occupational functioning. Effects on the motor and sensory systems are not apparent until later in the disease process. Dementia is a significant health care problem because of the increasing number of individuals who are living longer, the higher incidence of dementia in older persons, the very high cost of supervised care, and the extensive use of medical resources.93 Almost three times as much money is spent by Medicare for persons with AD and other dementias as for beneficiaries without AD.4,6 Early recognition of cognitive decline by physicians, occupational therapists, and all other health care professionals is critical. Slower progression of the disease, greater understanding of functional decline, improved quality of life, and time to prepare for the future for older adults and their families could be an outcome of early recognition. AD is often overlooked or mistaken for other disorders, especially in the early stages.
 OT Practice Notes
OT Practice Notes
Occupational therapists have an essential role in helping a client with Alzheimer’s disease enjoy life and remain as self-sufficient as possible and in supporting families and caregivers over the course of this difficult disease.
Incidence
AD accounts for more than two thirds of cases of dementia, and the incidence increases dramatically as people age.4,6,41,61,184,225 It is estimated that close to 13% of adults 65 years or older have AD. The disease affects more than 5 million people in the United States, and 4.9 million are older than 65 years. Age is the strongest primary risk factor. The incidence of AD more than doubles for every 5 years above the age of 65. It is estimated that 40% to 50% of the population of old-old (85+ years) adults have AD. Many more Americans are surviving into their 80s and 90s because of advances in medical technology. In a recent study of 97-year-olds, 61% of the population had some form of dementia.104 As the population of old-old adults continues to grow, the number of persons with AD is also expected to grow. Family history and genetics are other risk factors for AD.70 Early-onset, familial forms of AD are linked to genetic mutations on chromosomes 1, 14, and 21.81,117,150,199 Late-onset AD has been linked to the apolipoprotein E-4 (APOE-4) allele on chromosome 19, but it should be noted that this allele has also been found in older persons who do not have AD.150,151,172 It is generally agreed that although genetic factors pose some risk for late-onset AD, it is the interactive effects of diet, lifestyle, and environment affecting each person differently. This field of research is known as epigenetics. Three new genes thought to have a role in late-onset AD have recently been discovered by researchers.69,150 Two of these genes are responsible for protein encoding (CLU—clearing β-amyloid; PICALM—recycling cell membrane proteins at synapses), and a third gene, CR1 (complement receptor 1), is responsible for the inflammatory response and clearing of β-amyloid. Scientists conducting AD research are currently exploring the idea that changes in the regulation of genes—turning them on and off through the interactive effects of nature and nurture—and not just gene mutations can make someone more or less susceptible to AD.150 Previous head trauma is a well-established risk factor for AD; other factors that result in increased risk include diabetes mellitus, APOE gene variation, current smoking, and depression. Limited evidence shows increased risk associated with use of estrogens and nonsteroidal anti-inflammatory drugs, and the evidence is inconsistent for risk factors associated with heart disease, high blood pressure, and obesity. Researchers are also exploring the effects of certain inhaled anesthetics, lead exposure, and timing of the use of hormone replacement therapy after menopause.148–150
Although the incidence of dementia is growing rapidly, it does not occur in all older adults. Many older adults experience a normal slowing of information processing, called age-related cognitive decline. Clinically significant cognitive deficits do not develop in this population.11 Mild cognitive impairment (MCI), a condition that involves problems with memory, language, or other essential cognitive functions that is serious enough to be noticeable to others and to show up on tests, develops in a proportion of the older adult population, but this impairment is not severe enough to interfere with daily life.148 In some, but not all persons with MCI, AD eventually develops. A study by Driscoll and colleagues found that persons with MCI showed brain atrophy, but the group in whom AD eventually developed exhibited a pattern of atrophy in the region of the temporal lobes.58 A term sometimes used by laypersons to refer to older adults with memory loss or cognitive impairments is senility. Senility is not a medical term. Use of the word senility perpetuates stereotypic impressions that progressive cognitive decline occurs in normal aging. Such ideas prevent early recognition and accurate diagnosis of dementia.
Pathophysiology
AD is the result of degenerative changes in the CNS. Neuroanatomic (structural) and neurochemical changes occur in genetically and/or environmentally susceptible brains.134 Many neurons die, stop functioning, or lose their connections with other neurons. Disruption of communication, metabolism, and neuron repair occurs. The result of these changes is progressive and diffuse neuronal loss in the cerebral cortex and hippocampus.150,194 Three noticeable pathologic changes have been found through microscopic examination of brain tissue after death: accumulation of amyloid in the space between neurons, increased neuritic plaques and neurofibrillary tangles, and loss of neurons and synapses. Early AD is associated with decreased cholinergic markers in areas of the brain with an increased distribution of plaques and tangles. Many of the changes in the brains of persons with AD can be seen only at autopsy, although neuroimaging techniques (e.g., computed tomography [CT], magnetic resonance imaging [MRI], and positron emission tomography [PET]) provide further diagnostic information such as enlarged ventricles. The degenerative changes in the brain involve several processes that affect neurotransmission and result in neuronal death.194 Studies suggest that the sequence of disease events in individuals with AD involves amyloid deposits in the earliest stages that are not yet associated with actual cognitive impairment.136,150,209 Later, abnormal tau protein accumulates and causes loss of synapses, neurons, and brain volume An inflammatory process causes the tau proteins in cortical and limbic neurons to undergo microtubular dysfunction, which prevents neurons from sending nutrients and hormones along the axons. The paired filaments of these intracellular proteins actually become cross-linked in an abnormal metabolic process. These filaments form neurofibrillary tangles that eventually lead to neuron death as the neuron transport system collapses. Neurofibrillary tangles are also seen in the temporal areas and to a lesser degree in the parietal association areas.
Neuritic plaques are large, extraneuronal bodies consisting of accumulated β-amyloid and neuronal debris—small axons and dendrites. Neuronal plaques predominate in the temporal and parietal areas in early AD. This material degenerates and takes up cellular space. Extracellular accumulation of too much insoluble β-amyloid in neuritic plaques contributes to the neuron degeneration. When neurons lose their connections, they cannot function and eventually die. The neuron degeneration and death spread through the brain, connections between other neurons break down, and affected regions begin to shrink in a process called brain atrophy. By the final stage of AD, damage is widespread, and brain tissue has shrunk significantly. The production of high levels of insoluble β-amyloid is associated with early-onset familial AD and has been linked to genetic markers on chromosomes 1, 14, and 21.81,117,199 In late-onset AD, the accumulation of amyloid deposits may be affected by APOE-4 on chromosome 19 and can also affect the development of neurofibrillary tangles.197 Scientists anticipate that an understanding of the mutation in chromosomes associated with early-onset AD will help them better understand late-onset AD.150 Late-onset AD is more likely related to a combination of genetic, environmental, and lifestyle factors that affect the regulation of gene expression, which can differ from person to person.51
Cholinergic dysfunction also occurs in individuals with AD, and this depletion in neurotransmitter is the process responsible for the expression of clinical symptoms, such as deficits in memory and word-finding problems, in early AD. Specifically, cholinergic deficits, thought to be linked to APOE-4, include less choline acetyltransferase activity in the frontal cortex, hippocampus, and temporal cortex.169,171,194 These areas of the brain are associated with such symptoms of AD as recent memory impairment and problems in executive functions.
Clinical Picture
Initially, the signs and symptoms of AD may seem puzzling because the disease can affect different people in different ways.4 The symptom that is most prominent is the progressive inability to remember new information. Recently, the Alzheimer’s Association compiled a checklist, “Know the 10 Signs,”5 an Early Detection Matters education campaign that assists older adults and their families in early recognition and contact with their primary care physician. The symptoms and patterns of behavior in persons with AD are often described in terms of stages, but it is important to recognize that it can be difficult to place a person with AD in a specific stage because stages may overlap.196 The simplest description of staging, useful for caregivers and consumers, defines the progression of AD in terms of a three-point stage scale: early, middle, and late stages.80 A more clinically and diagnostically complex scale, such as the seven-point Global Deterioration Scale (GDS),182,183 is used in research or modified for diagnostic purposes and is often used as part of an assessment battery. It is important to realize that not all persons with AD will progress at the same rate or have the same pattern of symptoms despite the GDS staging framework.
The primary symptom of AD is impairment in recent memory that worsens with time, with at least one other cognitive deficit being present, such as apraxia, aphasia, agnosia, or impaired executive function, according to the American Psychiatric Association.11 Memory impairment involves increased difficulty learning new information and recalling information after more than a few minutes.150,205 Over time, the ability to learn deteriorates further and the ability to recall old memories also declines. Symptoms such as speech and language problems, impaired recognition of previously familiar objects, and impaired ability to perform planned motor movement are more variable and may not be seen in all persons with AD. Expression of symptoms depends on the areas of the brain most affected by the disease. Executive function (the ability to initiate, plan, organize, implement safely, and judge and monitor performance) inevitably deteriorates as AD progresses. Visuospatial dysfunction is common. Changes in mood and behavior consisting of personality shifts and the development of depression, anxiety, and increased irritability are often observed in the early stages of AD.130 Later in the course of the disease, troubling behavioral problems such as agitation, psychosis (i.e., delusions and hallucinations), aggression, and wandering can emerge.4,6,148,150 Motor performance areas such as gait and balance may become impaired, and sensory changes usually arise in the middle to later stages of the course of AD (see Table 35-3). Frequently, delirium and depression complicate the clinical picture. The life expectancy following the diagnosis of AD is typically 8 to 10 years but can range from 3 to 20 years, with a variable rate of progression.137
TABLE 35-3
Progression of Alzheimer’s Disease and Intervention Considerations
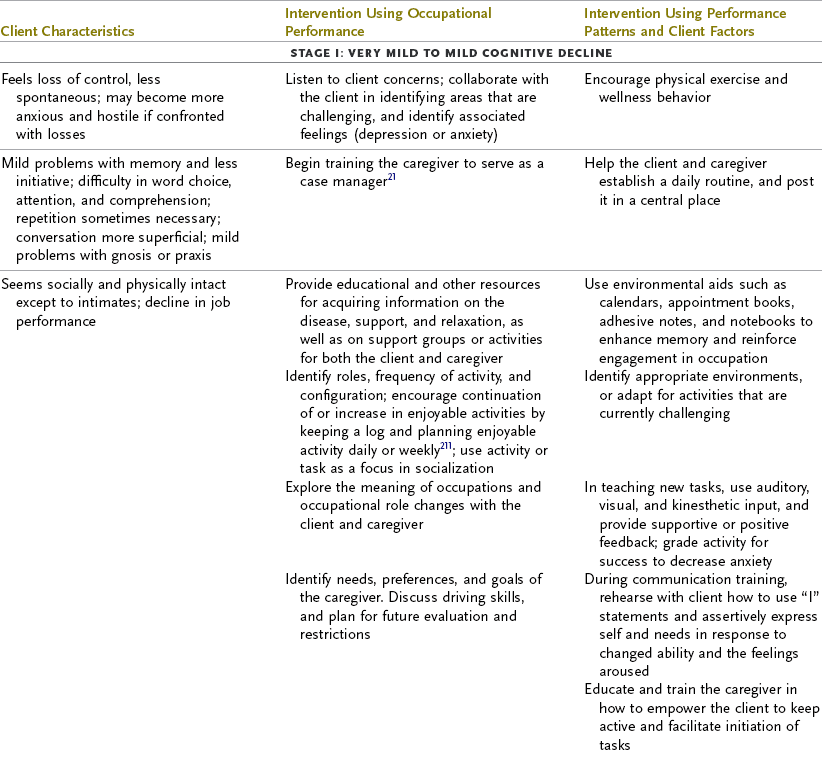


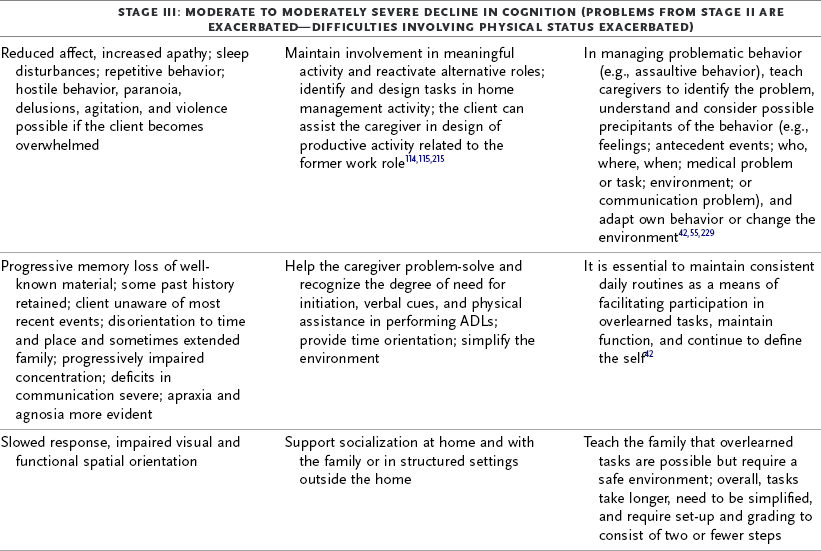
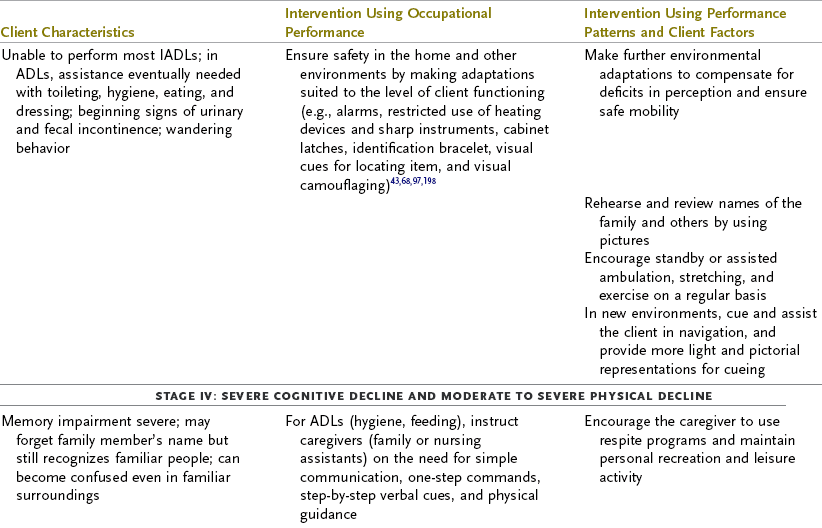
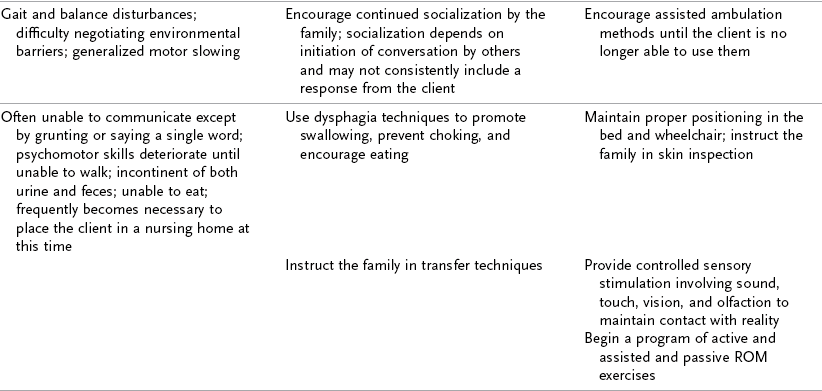
ADL, activities of daily living; AFO, ankle-foot orthosis; IADLs, instrumental activities of daily living; MMT, manual muscle test; ROM, range of motion.
Adapted from Baum C: Addressing the needs of the cognitively impaired elderly from a family policy perspective, Am J Occup Ther 45:594, 1991; Morscheck P: An overview of Alzheimer’s disease and long term care, Pride J Long-Term Health Care 3:4, 1984; Glickstein J: Therapeutic interventions in Alzheimer’s disease, Gaithersburg, Md, 1997, Aspen; Gwyther L, Matteson M: Care for the caregivers, J Gerontol Nurs 9, 1983.
Deterioration in the individual’s functional performance usually occurs in a hierarchic pattern. This pattern of decline initially consists of a gradual progression from mild impairments in work and leisure performance to more moderate difficulty in performing instrumental activities of daily living (IADLs), especially finances and driving. Eventually, there is progressive loss of the ability to perform even basic self-care ADL tasks. The pattern of disability is usually characterized by lower extremity ADL problems (walking) before decline in upper extremity ADL functions.87,208 The trend in AD is for cognitive deficits to increase and executive function to become more impaired (see Table 35-3). Motivation and perception can influence functional performance but may not be routinely considered in individuals with AD (Levy, 2005).68
Medical Management
According to Larson,109 medical management of individuals with AD in primary care settings generally includes several areas. Many aspects of what is termed medical management may also be performed by certain other members of an interdisciplinary health care team, including the nurse, social worker, physical therapist, or occupational therapist. First, early recognition and diagnosis of AD are needed.109,148,193 Second is the issue of how to intervene on behalf of a person with AD who is living in the community before institutionalization or more restrictive care becomes necessary. The third area concerns intervention issues as the disease progresses. Last is the role of health care providers in recognizing and addressing the treatment of other conditions that lead to excess disability in a person with AD.
The brain damage caused by AD begins much earlier than the appearance of cognitive impairment. Although dementia is relatively common in persons older than 80 years, it is often not diagnosed in such individuals until approximately 2 to 4 years after the onset of dementia symptoms.108,109,111,128 Recent evidence suggests that AD eventually develops in as many as 23% of persons with MCI.38,148,220 Wilson and colleagues identified olfactory impairment in persons in whom AD later developed.226 Identification of motor, visuospatial, and other sensory deficits, even before impaired memory is evident, is a current area of research that may aid in earlier detection of AD.148,150 The Alzheimer’s Association has made a concerted effort to help in the earlier diagnosis of older adults through their Early Detection Matters program.6 A comprehensive physical examination, laboratory evaluation, mental status examination, brief neurologic examination, and informant and family interview are essential in diagnosing AD. It is important to identify and treat medical conditions (e.g., metabolic disturbances, infections, alcohol use, vitamin deficiencies, chronic obstructive pulmonary disease, heart disease, and drug toxicity) that can contribute to comorbidity. Findings on MRI, PET, and CT can be useful, but overreliance on these techniques should be avoided because their value lies in identifying relatively uncommon, treatable causes of cognitive impairment. A comprehensive and skillful interview with a reliable informant is essential to the evaluation and diagnostic process to recognize decline by comparing current changes with past performance. Informant questionnaires, interviews, and screening measures may be performed by many health care professionals other than physicians and are important in the diagnostic process. National Institute on Aging145–funded Alzheimer’s Disease Centers can diagnose AD with up to 90% accuracy,150 although autopsy is the only diagnostic tool with 100% accuracy. Cognitive testing and testing of cerebrospinal fluid are two developing areas for earlier detection and diagnosis.
The goal of health care providers in the successful management of an individual with dementia, whether in the community or in a semi-institutional or institutional setting, is to “minimize behavior disturbances, maximize function and independence, and foster a safe and secure environment”205 (p. 1367). Dementia is associated with increased mortality.34 Regular health maintenance visits in primary care settings to identify treatable illnesses such as depression, PD, low folate levels, arthritic conditions, urinary tract infections, and other conditions that may complicate dementia are important for all older adults, especially those with AD.113,194 New evidence suggests that the cellular abnormalities of vascular disease (high blood pressure, cerebrovascular disease, cardiovascular disease, diabetes) exacerbate the cellular abnormalities found in AD, increase the risk, and may speed the progression of AD.150 Depression and dementia may easily be mistaken for each other, or they may coexist.205 Careful attention to whether the onset of symptoms has been gradual (dementia) or more recent (depression) is an important diagnostic issue because affective and cognitive symptoms frequently occur together.181 The cognitive impairments and especially functional performance may improve in individuals with both dementia and depression after they are treated for depression. Delirium (i.e., impairment in attention, alertness, and perception) and dementia frequently coexist as well, especially in hospital settings.205 Both conditions involve global cognitive impairment, but delirium is usually acute in onset, shows fluctuating symptoms, disrupts consciousness and attention, and interferes with sleep. Adverse drug reactions are more common in AD because of the vulnerable, impaired brain.110 Drug toxicity, often a cause of delirium, is treatable.
Hearing, vision, and other sensory impairments are known to make dementia worse and cause greater strain on the caregiver.218,219 Falls with hip fractures are 5 to 10 times more common in persons with AD than in normal persons of the same age and frequently result in earlier institutionalization for the individual and the need for higher levels of care.36 Unsafe mobility quickly becomes an overwhelming burden for caregivers, especially those who are aged. Occupational therapists can assess the tasks, context, and environmental features of the living situation in which the person with AD resides and find ways to analyze activities and make compensations so that living is more meaningful and safe.
According to Small and colleagues205 and the most recent progress report on AD from the National Institutes of Health,150 pharmacotherapy for the treatment of individuals with AD should be assessed carefully and justified at regular intervals.192 Although OT practitioners do not prescribe medications, knowledge of pharmacotherapy is useful. Cholinesterase inhibitors such as galantamine and donepezil may improve cognition and functional performance, at least in the short term. The drug memantine blocks receptors for glutamate and has been used in individuals with moderate to severe AD.150 Promising research is under way in this area. Cognitive health is highly related to the health of blood vessels in the brain. Controlling blood pressure, using cholesterol-lowering drugs, eating a Mediterranean diet, and maintaining physical activity are strategies for vascular health and are also thought to reduce the risk for AD. Antidepressant medications, especially selective serotonin reuptake inhibitors, are often prescribed to manage the behavioral manifestations of AD—but it should be noted that these medications are not approved by the Food and Drug Administration for the treatment of AD. It is important to note that some of the tricyclic antidepressants (e.g., amitriptyline, imipramine, and clomipramine) and monoamine oxidase inhibitors can have troublesome side effects in older adults. Atypical antipsychotics such as clozapine, risperidone, and olanzapine may be used to reduce agitation and psychosis.123,205 Benzodiazepines are prescribed for treating anxiety and infrequent agitation but have been found to be less effective than antipsychotics when the symptoms are severe.205
Role of the Occupational Therapist
Most individuals with AD live in the community, alone or with family or friends, rather than in institutions. A predominant feature of AD is significant and progressive deterioration in function from previous levels of performance because of advancing brain atrophy and pathologic tissue changes. Families and significant others associated with persons with AD become progressively involved as the disease itself progresses and provide increasing oversight and personal assistance.77 Changes in the brain caused by AD result in deficits in client factors, which in turn lead to deterioration in occupational performance skills and occupational performance areas and major changes in occupational roles. Over time, more structured, supervised living environments are usually needed. Increased difficulty in performing everyday functions creates challenges for the individual with AD and has an impact on quality of life for the client, family, and caregivers as the disease progresses. Effective OT interventions must be directed at supporting occupational performance for the individual and creating as much quality of life as possible. Intervention should focus on supporting and maintaining capabilities and adapting tasks and environments or otherwise compensating for declining function in individuals with AD while trying to help them retain as much control as possible over their lives in the least restrictive environment.10 Adult day care centers can often provide an alternative to assisted living or skilled nursing facilities; they offer a safe, structured day program for persons with cognitive impairments who still reside at home and allow them to be in the least restrictive setting and able to return to their caregivers in the evenings. The Program of All-Inclusive Care is one model of day care available in some communities and offers innovative, high-quality care for older adults at high risk for needing institutional care (Trice, 2006).79
 Ethical Considerations
Ethical Considerations
Support for the caregiver is a must. Collaboration with and training of the caregiver is essential in the management of clients with dementia. Family members should encounter an open and encouraging environment in which to discuss safety, security, and dependence issues. Legal, financial, and health concerns that require advance directives (e.g., medical and legal), trusts, restrictions in activity (e.g., driving, financial matters, and medication management), and contingency and transitional care plans (e.g., day care, residential care, and long-term care) are important in preparation for the inevitable progression of the disease.109,204
Behavioral problems can be expected in clients with AD until the terminal or bed-bound stage. Encouragement to use respite care, in-home support services, and caregiver support groups is important. Caregivers also need effective strategies for dealing with behavioral disturbances and disruptions in mood. The use of environmental adaptations, therapeutic interpersonal approaches, referral to other disciplines, and sharing of resources helps in collaborating with the client’s family and handling these problems. Health professionals use education, training, counseling, and support to help caregivers deal with their feelings, manage behavior, and maintain quality of life for themselves and for the client with AD. Awareness of the multidimensional effects of this illness on the individual, the family, and the society at large is important to promote more effective and efficient care.178 Specialized homes and units known as Alzheimer’s Care Centers are available.79 The model of care is one of wellness support instead of an illness model of care. The idea is to maximize and support independence, maintain safety, and focus on the strengths and remaining skills of the individual. The Alzheimer’s Association5 has developed “Dementia Care Practice Recommendations” for dementia care centers, and these guidelines are supported by the American Occupational Therapy Association. The fundamentals are provision of food and fluid, management of pain, resident wandering, falls, physical free restraint, and social engagement.5,79
Evaluation
An OT screening is often performed before the evaluation. OT services are indicated for individuals who have demonstrated a recent decline in function; whose behavior poses a safety hazard to family, staff, other residents, or self; or who may experience improved quality of life.30 Much of the therapist’s time in community settings and in long-term care is spent helping families and caregivers develop strategies and environmental adaptations to cope with the overwhelming stress of safely managing a cognitively impaired individual.21 The other major focus is promoting the best quality of life that is possible while ensuring that no matter the setting, a person with AD has choice, dignity, and self-determination.
The type of assessment and the depth of the evaluation process used are influenced by the setting, the stage of progression of AD, the reimbursement process, the presence of other medical and mental health disorders, and the cooperation and interest of the caregiver or care staff. The consequences of caregiving and the needs of the caregiver can vary greatly, depending on gender, family relationships, culture, and ethnicity. The caregiver’s understanding of dementia, reaction to dementia-related behavior, use of problem-solving skills, use of the environment, use of formal and informal support systems, and decision-making style greatly affect the caregiver’s ability to participate in the care plan and treatment of persons with dementia.19,49,60,118,227 In some cases the occupational therapist must engage in the dual role of serving as an advocate for the person with AD.
Evaluation should be comprehensive despite changing reimbursement. Much information can be gathered before an interview and intervention session by asking caregivers, family members, and staff informants to complete questionnaires and rating scales. These scales assess occupational performance, functional abilities, and skills with the use of measures such as the Functional Behavior Profile,23 the Activity Profile,21 the Caregiver’s Strain Questionnaire,188 the Katz Activities of Daily Living Scale,103 and the Instrumental Activities of Daily Living Scale.112 Informant rating measures should routinely be followed by an interview either before or during the first visit. The use of a few brief screening instruments for mental status (e.g., the MMSE),65 depression,26,228 and anxiety113 provides baseline data and a wealth of information about factors that are likely to influence performance. It is always useful, with any evaluation, to triangulate information to achieve reliability. This means talking with other family members, roommates, and care staff and, of course, the occupational therapist’s own observations.40
The functional evaluation of an individual with AD depends on the stage of cognitive decline.10 The American Occupational Therapy Association’s statement on services for persons with AD suggests that tasks involving work, home management, driving skills, and safety should be targeted in the early stages of the disease. In the later stages, the focus shifts to self-care, mobility, communication, and leisure skills.
The concerns and observations of the caregiver are important, but observation of task performance by the therapist is also necessary. Unfortunately, many of the functional ADL scales developed for use in older adults have targeted physical performance and are not appropriate for persons experiencing cognitive decline.73 Fortunately, several excellent, standardized measures that determine whether individuals are able to use their cognitive skills to perform ADL and IADL tasks have been developed over the last 15 years. The Kitchen Task Assessment determines the level of cognitive support that a person with AD needs to complete a cooking task successfully.22 More recently, Baum and colleagues compiled the Executive Function Performance Test,24 a performance-based standardized assessment of cognitive function that has been standardized in administration and scoring.2 The Allen Cognitive Level (ACL) test determines the quality of problem solving that an individual uses while engaged in perceptual motor tasks.2 Levy has written at great length about use of the ACL test for clients with cognitive impairments.115,116 Consistent with the Allen theoretic approach, the Cognitive Performance Test was developed to identify cognitive deficits that are predictive of functional capacity by using several ADL and IADL tasks.3,37 Another measure, the Assessment of Motor and Process Skills (AMPS), has been used in individuals who have dementia.63,158 The AMPS measures motor (e.g., posture, mobility, and strength) and process (e.g., attentional, organizational, and adaptive) skills by using task performance in IADLs. The Disability Assessment for Dementia (DAD) uses informant ratings to determine the ability of an individual with AD to complete tasks in both ADLs and IADLs.73 The DAD also provides information relevant to executive functioning, such as the person’s ability to initiate, plan, and execute the activity. The Independent Living Scales was originally developed to assess older adults and assist in making a determination of an individual’s problem solving and performance as applied to memory and orientation, money management, home maintenance and transportation, health and safety, and social development.120 Further information regarding the evaluation of cognitive function and ADL performance is provided in Chapters 10 and 26. After obtaining a thorough evaluation and a good understanding of the disease process and the functional level of the person with AD, the therapist can begin to look at the all-important question of what aspects of the occupational performance context, especially the environment and care provider interactions, must be modified to optimize safety, function, and quality of life of the person with AD.158
Intervention Methods
The goals of OT are to provide services to persons with dementia and their families and caregivers that emphasize remaining strengths, maintain physical and mental activity for as long as possible, decrease caregiver stress, and keep the person in the least restrictive setting possible.12,14,89,101 Although AD is the primary problem, the occupational therapist has to consider the complexities of making interventions in an older adult who may be experiencing additional sensory loss and numerous medical problems such as arthritis, orthopedic issues, chronic obstructive pulmonary disease, diabetes, and heart disease in addition to AD. Intervention planning takes into account the physical issues associated with aging, co-occurring disorders such as depression and anxiety, the progressive nature of the disorder, the expected decline in function, and the care setting itself. OT interventions for clients with dementia are directed toward maintaining, restoring, or improving functional capacity; promoting participation in occupations that are satisfying and that optimize health and well-being; and easing the burdens of caregiving.21 Gitlin and colleagues recently instituted a program to improve the overall pleasure of persons living at home who have dementia.78 This program was designed to decrease the distress of caregivers when caring for a person with AD who exhibits erratic and disturbing behavior. A program called the Tailored Activity Program (TAP) is an OT service that evaluates the interests and capabilities of a person with dementia and provides customized activities for each individual. TAP then trains families to use these activities as part of the daily care routines. Caregivers reported “being less upset with the behavioral symptoms” when using the tailored activities (p. 428). Methods that therapists use in the intervention process include activity analysis, caregiver training, behavior management techniques, environmental modification, use of purposeful activity, and provision of resources and referrals. Services are provided in many different settings, such as home care, adult day care, and semi-institutional or institutional long-term care. The intervention setting and the stage of the illness help frame the focus of intervention, determine the recipients of service, and prescribe the methods used (see Table 35-3). A useful role for an occupational therapist working with an individual with dementia and his or her family is to assist the individual with AD in making the many transitions that will probably take place—from home to hospital to assisted living, residential care, or skilled nursing home—as the disease progresses, as well as medical problems that bring about crises. With each move to a new residential setting the therapist can determine the interrelationship of the client’s physical, cognitive, sensory, and social world. OT can be instrumental in determining how this newest transition might influence occupational performance, health, and well-being. At each transition, interventions can target arriving at the best person-environment fit, recommending environmental adaptations, providing assistive devices, and promoting collaborative relationships between care providers to support occupational engagement and desired roles while enabling independence and safety at each transition79
Summary
AD is a neurologic condition characterized by the gradual development of multiple cognitive impairments. The effect of these impairments is a significant and progressive decline from previous levels of functioning. The course of the disorder is variable, but loss of function generally occurs in a hierarchic pattern, beginning with work and progressing to difficulties with home management, driving, and safety, until even basic self-care skills such as dressing, functional mobility, toileting, communication, and feeding are affected.
OT interventions should be directed at enhancing the abilities of a client with AD by continually adapting tasks of daily living and modifying the physical and social environment as the client experiences progressive loss of function. Given many of the current limitations in treatment time imposed by third party payment, therapists may find it helpful to use some of the self-report and informant report measures identified in this chapter as a means of gathering information more efficiently during the evaluation process. Several standardized measures have also been identified to assist in the assessment of functional performance and establishment of a baseline of performance. Recommendations for OT treatment of AD have been identified. The focus of intervention must be flexible and depends on an understanding of the particular expression of the disease process in the client, the specific treatment setting, and the needs of the caregiver. Generally, the goals of OT services for persons with dementia are to maintain or enhance function, promote continued participation in meaningful occupation, optimize health and quality of life, and work collaboratively with the caregiver to ease the burden of caregiving.
Section 3 Huntington’s Disease
Incidence
HD is a fatal, degenerative neurologic disorder that affects 5 to 10 of every 100,000 individuals.76,167 The disorder is transmitted in an autosomal dominant pattern. Each offspring of an affected parent has a 50% chance of having HD. Genetic studies have identified a mutation on chromosome 4 as the cause of this disease.144,167,176,180 Presymptomatic diagnosis of HD is possible with genetic testing when the family history shows this disease.165,167 Diagnosis is also made through clinical examination when the family history is unavailable or unknown.
Pathophysiology
The neurologic structure associated with HD is the corpus striatum. Deterioration of the caudate nucleus is more severe and occurs earlier than atrophy of the putamen.44,163 The corpus striatum plays an important role in motor control, and deterioration in this area contributes to the chorea associated with HD. The caudate nucleus is also linked to cognitive and emotional function through connections with the cerebral cortex. Progressive loss of tissue occurs in the frontal cortex, globus pallidus, and thalamus as the disease advances.167 Degeneration of the corpus striatum results in a decrease in the neurotransmitter γ-aminobutyric acid. Additional deficiencies in acetylcholine and substance P, both neurotransmitters, are noted in clients with HD. The triggering mechanism for the neuronal degeneration has not been clearly identified, but it is linked to genetic coding on chromosome 4.180
Clinical Picture
HD is characterized by progressive disorders in both voluntary and involuntary movement, in addition to a significant deterioration in cognitive and behavioral abilities.167,200 A client usually experiences an insidious onset of symptoms in the third to fourth decade of life, but cases have been reported in teenage and younger clients.224 Clients who are positively identified by genetic testing should be carefully monitored for the first indications of HD symptoms. Not all individuals who have potential for the development of HD because of a family history have elected to undergo genetic testing, a fact that may affect determination of the time at which the first symptoms emerged. The course of progression of HD is often divided into three stages—early, middle, and late—but with the advent of genetic testing, an additional presymptomatic stage has been added.221 During the presymptomatic stage, monitoring of symptoms is critical, and a decrease in the speed of finger tapping (an item on the Unified Huntington’s Disease Rating Scale [UHDRS]) may mark the beginning of the early stage of HD. The symptoms progress over a 15- to 20-year period, with long-term care or hospitalization of the client ultimately being necessary.167,203 Death is often the result of secondary causes, such as pneumonia.165
The initial symptoms vary but are most often reported as alterations in behavior, changes in cognitive function, and choreiform movements of the hands.221,224 The early symptoms of cognitive disturbances are most likely related to degeneration of the caudate nucleus. The client may appear forgetful or display difficulty concentrating. During the early stages of HD a client may have difficulty maintaining adequate work performance. Family members frequently identify the initial behavioral changes seen in a person with HD as increased irritability or depression. Irritability and depression may be attributed inappropriately to the decline in work performance rather than to the disease process. Emotional and behavioral changes are often the earliest symptoms of HD.17,66 Chorea, seen in clients with HD, consists of rapid, involuntary, irregular movements.166 During the early stages of HD, chorea is often limited to the hands. A client may mask the initial chorea by engaging in behavior such as manipulating small objects with the hands. These irregular movements are exacerbated during stressful conditions and decrease during voluntary motor activities in the early stages of HD. Chorea is absent when the client is sleeping. Onset of HD in the teenage years is associated more frequently with early symptoms of rigidity rather than chorea.221,224
Cognitive and emotional abilities progressively deteriorate over the course of the disease.224 Disturbances in memory and decision-making skills become more apparent during the middle stages of HD. Establishing and maintaining meaningful habits and routines for an individual who has HD are important ways to support continued participation in occupational pursuits. A client may be able to complete familiar tasks at work or in the home, but if the environment is changed or if additional demands are placed on the individual, task performance is significantly compromised. Further deterioration in cognitive abilities in a person with HD may result in dismissal from employment. The cognitive deficits most frequently associated with HD are problems involving mental calculations, performance of sequential tasks, and memory.66 Verbal comprehension is often spared until the middle or later stages of the disease, and even then, dysarthria is a more significant issue than difficulty in comprehension until the late stage of HD.
As HD progresses, depression often worsens and suicide is not uncommon.221,224 Clients with HD are frequently hospitalized because of various psychiatric problems, including depression, emotional lability, and behavioral outbursts. Although the loss of function may contribute to the client’s level of depression, depression is clearly identified as a specific characteristic of HD.66,143 This affective disorder is frequently treated with various antidepressants. Periods of mania have also been reported in approximately 10% of individuals with HD.
As the disease progresses, the chorea becomes more severe and may involve the entire body, including the face.167 Disturbances in gait are often observed during the middle stages of the disease, and balance is frequently compromised.66 An individual with HD may display a wide-based gait pattern and have difficulty walking on uneven terrain. This staggering gait is at times misinterpreted by others in the client’s life as evidence of alcoholism.166 The client also has progressive difficulty in performing voluntary movements.167 Performance of voluntary motor tasks is slowed (bradykinesia), and initiation of movement is compromised (akinesia). Although handwriting ability may be spared initially, the client displays increasing difficulty with this task as the disease progresses. Letter size is enlarged, and letter formation, such as slant and shape, is distorted. Saccadic eye movements and ocular pursuits may be slowed at this stage of HD.165 Slight dysarthria compromising communication may be noted.66 Dysphagia occurs, and the client may choke on various foods. Difficulty may be noted in coordination of both chewing and breathing while eating.
In the later stages of HD, choreiform movements may be reduced because of further deterioration of the corpus striatum and globus pallidus.167 Hypertonicity and rigidity often replace the chorea as the disease progresses, and the client experiences a severe reduction in voluntary movements. Severe difficulties in eye movement are common during the final stage of the disease.165 At this stage the client often needs significant support from others or resides in a long-term care facility. The client is usually unable to talk, walk, or perform basic ADLs without significant assistance.144
Medical Management
Medical management of clients with HD can address the symptoms, but no effective course of treatment has been identified to arrest progression of this disease.179 Intervention based on replacing the deficient neurotransmitters has not been effective in changing the course or rate of progression of HD. Tricyclic antidepressants are often used to treat the depression seen in clients with HD, but monoamine oxidase inhibitors are contraindicated because of possible exacerbation of chorea.224 Haloperidol may be used to decrease the negative effects of chorea on the performance of functional activities.167 Haloperidol is prescribed cautiously and only when the chorea significantly compromises a person’s daily activities. Medical management is focused on three areas: managing symptoms and reducing the burden of the symptoms, maximizing function, and providing education to the client and significant others regarding the course of progression of the disease.143 A team of professionals, including an occupational therapist, is advocated when working with a client who has HD.
A team of various medical professionals is needed to support an individual with HD, as well as family members and significant others in the client’s life.92,102 Perception of the illness experience and the coping strategies used by a client with HD are an important part of the overall medical services provided.92 Likewise, the significant others in the client’s life need support and guidance in developing coping strategies as their loved one who has HD experiences progressive deterioration.102
Systematic evaluation of a client with HD must be performed at regular intervals to identify the rate of symptom progression and modify intervention strategies. Standardized instruments are available for determining the presence and severity of various symptoms.96,200 One evaluation tool, the UHDRS, combines aspects from several instruments into a scale that can be administered within 30 minutes. The UHDRS is often administered by a team. This tool provides an accurate means of determining a change in the areas of “motor function, cognitive function, behavioral abnormalities, and functional capacity.”96 The UHDRS has been used to assess the rate of decline and demonstrates good reliability (Klempir et al, 2006). The occupational therapist should perform additional assessments before an intervention plan is developed. An evaluation would address functional daily living skills; cognitive abilities such as problem solving, motor performance, and strength; and personal interests and values. The occupational therapist must consider the client’s role within the family and community and incorporate these data into the intervention plan. An evaluation at both the home and work site would provide needed information that could be modified if necessary.
Role of the Occupational Therapist
The role of the OT practitioner varies depending on the stage of the disease.98 During the early stages of HD, an occupational therapist should address the cognitive components of memory and concentration. A client may still be employed at this stage. Strategies such as establishment of a daily routine, use of checklists, and task analysis to break tasks down into manageable steps can be very helpful. These strategies provide the external structure and support needed to help a person with HD maintain functional abilities at both the workplace and home. A work site evaluation can identify changes that would allow a person with HD to continue working. Modifications may include the use of tools such as organizers, electronic planners, and reminders to prompt an individual to complete regularly occurring tasks in the workplace. Family members should also be instructed in the use of these techniques. Environmental modifications such as providing a quiet workplace and reducing extraneous stimuli will decrease the impact of compromised memory and concentration on performing functional tasks in the workplace. Even during the early stages, work performance may deteriorate and the client may be dismissed because of an inability to meet the job essentials. This increases the stress experienced by the client from a financial perspective and from loss of a role as a worker.
Psychologic issues during this stage of the disease frequently include anxiety, depression, and irritability.66,90 A client may express guilt that any of his or her children have a 50% chance of HD developing.166 The diagnosis of HD is often not confirmed until a person is 30 to 40 years old unless genetic testing is performed and confirms HD. The client may already be married and have children by that time. Decisions whether children should undergo complete predictive genetic testing may be a significant stressor for a client with HD and for his or her family members. As mentioned previously, not all individuals elect to be genetically tested, and sporadic cases of HD do arise when no family member has HD. During this early stage of HD, clients may use denial as a coping strategy even though choreiform movements are present in the hands.92
Maintaining social contacts and engaging in purposeful activities are important in designing interventions for clients with HD.166 Changes in cognitive abilities and unpredictable or exaggerated emotional responses may result in the loss of a job and decreased income for the family, even during this early stage of the disease.66 This additional stress should also be considered when developing an intervention plan. The OT intervention plan must include community support services for clients with HD. OT services should include providing clients with information regarding support groups, opportunities to engage in community activities, and connecting with virtual resources through the Internet.
The motor disturbances during the early stages of HD are usually limited to fine motor coordination problems.66 The characteristic chorea may be noticed only as a twitching of the hands when the client is anxious. OT should provide modifications to diminish the effect of chorea and fine motor incoordination on performance of functional activities.98 This would include modifications in clothing and selection of clothing that does not require small fasteners such as small buttons, snaps, or hooks. Shoes with Velcro or elastic closure are recommended to compensate for diminished fine motor skills. Home modifications should be instituted at this stage to allow a client with HD to become familiar with the changes. Developing the skills for using adaptive equipment or modifications and then converting these skills into habits are critical during the early stage of HD. Typical modifications are the use of cooking and eating utensils with built-up handles, unbreakable dishes, a shower bench or seat with tub safety bars, and sturdy chairs with high backs and armrests. Throw or scatter rugs should be eliminated wherever possible in the home, and walkways should be kept free of clutter. The occupational therapist should establish a home exercise program with the client to address flexibility and endurance of the entire body. These exercises will be incorporated into the client’s daily routine. As the movement disorder progresses with increased chorea and difficulty in oculomotor control, the client will no longer be able to safely drive a car, and further loss of community mobility may be experienced. This further loss of independent function and control must be considered within the OT intervention plan. Alternative methods of community mobility must be explored.
As HD progresses, the role of OT changes to meet the client’s needs.98 During the middle stages of HD, further deterioration in cognitive abilities often requires the person to resign from a job. Engagement in purposeful activities is greatly needed at this stage and should be a focus of the OT intervention plan.90,166 Decision-making and arithmetic skills show further deterioration, and family members may need to arrange for others to handle the client’s financial matters.66,147 Generally, comprehension of verbal information is better preserved than the ability to perform sequential tasks during this stage. The occupational therapist should encourage the family to use simple written cues or words to help a family member with HD complete self-care and simple household activities. For example, selecting clothing items for a person with HD and placing the clothes in a highly visible area can provide the prompt to change from pajamas to clothes in the morning. Arranging the bathroom with visual cues, such as putting the toothpaste and toothbrush by the sink, can remind the client to brush his or her teeth in the morning and evening.
During the middle stage of HD, the client may display increasing levels of irritability and depression.224 Clients with HD may attempt suicide. The OT intervention plan should focus on the client’s engagement in purposeful activities, particularly leisure activities. When selecting craft activities, the therapist should always consider the client’s interests but should also strive to ensure that no sharp instruments are required.90,147 Modification of craft activities allows a client with HD to successfully complete a task with minimal support.31 Materials often require additional stabilization to compensate for the client’s movement disorders, and any tools used in the leisure activity, such as a wood sander and paintbrushes, should have enlarged or built-up handles.
Motor problems become more apparent during the middle stage of HD and necessitate further modifications in daily living tasks.98,200 The client’s compromised balance may require that tasks such as dressing, brushing teeth, shaving, and combing hair be performed while the client is seated. The client may require the use of a walker or wheelchair at this stage. A Rollator Walker is preferred over a standard walker without wheels. The walker may need to be fitted with forearm supports to provide additional support when the client is ambulating. Once a wheelchair becomes necessary, it should have a firm back and seat; however, additional padding is often required on the armrests because of the client’s chorea. Many clients with HD are better able to move the wheelchair with their feet than with their hands. The height of the wheelchair seat should be adjusted to allow clients to use their feet to move the chair if possible.
Fatigue is a common issue during the middle stage of HD and can be addressed by taking frequent breaks during the day. Breaks must be scheduled because a person with HD may not readily recognize fatigue. Clothing should have few or no fasteners, and shoes should be sturdy with low heels.147 Additional adapted equipment that may prove helpful for clients with HD includes shower mitts, electric razor or chemical hair removal method, covered mugs, and nonslip placemats.98 The choreic movements may become so severe that use of a bed with railings is necessary. Padding should be used on the railings, and additional cushions should be used on the bed.
Because of the excessive movements associated with chorea, clients with HD often need to consume 3000 to 5000 calories per day to maintain weight.147 Clients with HD display higher energy expenditure than do individuals without HD and consequently have issues with weight loss and difficulty maintaining appropriate weight.216 Smaller, high-calorie meals should be provided five times a day. This schedule may require additional support from family members or a personal care attendant. Dysphagia, poor postural control, and deficient fine motor coordination compromise the client’s ability to eat.98 Positioning during feeding is crucial, and the trunk should be well supported during mealtime. Clients with HD should be able to support their arms on the table while the feet are stabilized. The feet may be supported on the floor or wrapped around the legs of the chair for additional support. Problems with dysphagia can be addressed with positioning, oral motor exercises, and changes in diet consistency. Soft foods and thickened fluids are preferable to chewy foods, foods with mixed texture, and thin liquids. Nutritional support has been used successfully for clients with HD to maintain appropriate weight.216 Appropriate body weight is important to maintain overall health of clients with HD.
During the final stages of HD, the client often depends on others for all self-care tasks because of the lack of voluntary motor control.98,144 In some clients, the chorea may diminish and be replaced by rigidity. The occupational therapist provides important input on positioning and the use of splints to prevent contractures at this stage. Because of the risk for aspiration, oral feedings are provided by trained personnel; alternatively, the client may receive nutrition through a feeding tube.147 A combination of oral feedings and tube feedings may be used during this stage.
Although cognitive abilities continue to deteriorate, the level of functional decline is difficult to assess because of dysarthria and loss of motor control.66 Dementia is part of the HD profile and must be considered during development of the intervention plan. For example, a person in the late stage of HD may still recognize family members and enjoy watching television. The occupational therapist should explore the use of various environmental controls to allow the client control of and access to the immediate environment.16 Providing a touch pad or switch for selecting television channels may prove beneficial for the client.
Behavioral outbursts have been reported in approximately a third of clients with HD living in long-term care facilities.144 OT can decrease the frequency of these outbursts by organizing consistent daily schedules and routines for clients with HD.
Summary
Although HD is a progressive, degenerative process, OT has much to offer clients with this disease.90,98,147,166 The diminishing ability to control the environment has been identified as one of the variables contributing to the deterioration in function in clients with HD. Throughout the course of the disease, OT addresses the client’s ability to exercise a degree of control over the environment and engage in purposeful activity.
Section 4 Multiple Sclerosis
Incidence
MS is a progressive, inflammatory neurologic disease that damages the myelin sheath in the CNS. Onset usually occurs between the ages of 20 and 45 years. The prevalence in the United States is approximately 350,000.119 It is more prevalent in women than in men.39 The highest prevalence of the disease is in Caucasians of northern European ancestry. Retrospective studies show that MS also occurs in people younger than 20, with 2% to 10% having a history of MS symptoms before the age of 18.48,213
The myelin is typically damaged in discrete regions of the white matter, with the axon remaining preserved. Disruption of the myelin sheath has differing effects on axonal conduction, depending on the degree of breakdown and the length of the damaged segment.39 When axons are conducting impulses in a slower manner because of inflammation of the myelin sheath, a person with MS may have intermittent symptoms of sensory distortion, incoordination, visual loss, or weakness. This inflammatory process accounts for the unpredictability of the disease.
In advanced cases of MS, acute and chronic plaques develop throughout the white matter, especially in the spinal cord, optic nerve, and periventricular white matter, including the corpus callosum. Axons may be damaged and severed in advanced cases and result in extensive loss of function. MRI may show lesions and changes in brain volume.29,100
Etiology
The specific cause of MS is unknown, although it is suspected to be the result of a combination of environmental and genetic factors. The current theory is that MS is an immune system reaction that acts on the nervous system.126 Studies have shown that 30% to 60% of new clinical attacks of the disease occur after a cold, flu, or common viral illness. Some researchers theorize that the immune system mistakes a portion of the myelin protein for a virus and destroys it. Other researchers believe that the viral infection damages the myelin and releases small amounts into the body, which results in an autoimmune reaction.202
Clinical Picture
The symptoms that occur in individuals with MS are related to the area of the CNS affected.9 Early symptoms may consist of paresthesias, diplopia, visual loss in one eye, fatigability, emotional lability, and sensory loss in the extremities. Other initial symptoms are trigeminal neuralgia and worsening of symptoms when body temperature is elevated. Symptoms that occur with an elevated body temperature are generally temporary and resolve in time. Cognitive deficits are reported to occur in 30% to 70% of persons with MS but do not necessarily correlate with a physical decline.7,85,156 Cognitive deficits have been documented in individuals with a disease duration of less than 2 years and with few neurologic signs.8
In advanced stages of the disease process, the individual may have varying degrees of paralysis from total lower extremity paralysis to involvement of the upper extremities, dysarthria, dysphagia, severe visual impairment, ataxia, spasticity, nystagmus, neurogenic bladder, and impaired cognition.
The course of MS is unpredictable. It is marked by episodes of exacerbation and remission.156 An exacerbation may be an episode as minor as fatigue and sensory loss or as extensive as total paralysis of all extremities and loss of bladder control. Remission may involve total resolution of the symptoms, slight return of some function with the symptoms remaining, or a short plateau in which no new symptoms occur but the current symptoms remain.
The three typical patterns seen in MS are (1) relapsing and remitting, (2) secondary progressive, and (3) primary progressive.9 The relapsing and remitting form of MS involves 85% of the MS population and leads to episodes of exacerbation and remission that result in a slow, stepwise progression as the deficits accumulate. The secondary progressive course begins with a pattern of relapses and remissions but evolves into the progressive form of the disease. Before the introduction of disease-modifying drugs, approximately 50% of clients with relapsing and remitting MS progressed to the secondary progressive form of the disease. The relapsing remitting form of MS was first diagnosed in Marguerite and, recently, the secondary progressive course of the disease. She is now experiencing sustained diminished fine motor abilities and sensation in her left hand. She also has weakness in one lower extremity requiring the use of an ankle-foot orthosis.
Though not as common as adult-onset MS, children account for 3% to 10% of the MS population.74,168 Ninety-five percent of children with MS initially experience a relapsing and remitting course of the disease. Over time, secondary progressive MS will develop in approximately 60% of children.213
The primary progressive form of MS (10% of the MS population) is distinguished by a downward course and little recovery after exacerbations. Individuals with this form eventually become nonambulatory and incontinent of urine and may have dysphagia and dysarthria, severely compromised lower extremity function, and varying limitations in upper extremity function.
Studies vary on precisely how life expectancy is affected, but for most people with MS, life expectancy is not greatly reduced. Approximately 50% live for at least 30 years after onset of the disease, and 50% die of complications of MS. Most people with MS will live into the years when they are experiencing other changes related to normal aging, which can complicate the clinical picture.7,9,120,126 There are two atypical patterns of MS, a benign course and the progressive relapsing form. Clinical signs of the benign course of MS are a younger age at onset, female sex, and onset with sensory symptoms. The symptoms usually resolve within 6 weeks, and there may be only a few residual deficits. If the disability is minimal after 5 years, the course is considered benign.65a The progressive relapsing form of MS is rare (5% of the MS population). It is identified as steadily progressive but has specific relapses.9 Typically, the course of the disease can generally be determined after 5 years of the initial symptoms (Box 35-2).
Medical Management
Medical management is dependent on the type of MS. Disease-modifying drugs are used in those with relapsing and remitting MS, and for all forms of the disease anti-inflammatories are used to treat exacerbations.156 For the relapsing and remitting form of MS, disease-modifying medications are thought to have an effect in slowing progression of the disease. Four different medications for this group have recently been introduced: interferon beta-1b (Betaseron), interferon beta-1a (Avonex or Rebif), glatiramer (Copaxone), and Rebif.95,156 Patients treated with these medications, administered by self-injection, showed a one-third reduction in frequency of exacerbations. Studies regarding the effectiveness of these medications in individuals with the progressive form of the disease are ongoing. The anti-inflammatory medications used to treat exacerbations, such as prednisone and methylprednisolone, help reduce symptoms and shorten the duration of the exacerbation.39
Mitoxantrone (Novantrone) is the first drug approved in the United States for secondary progressive MS. It is a form of chemotherapy that can also be offered as a treatment option for those with secondary progressive, progressive relapsing, or worsening relapsing remitting MS. Novantrone is reported to help in reducing neurologic disability and the frequency of clinical relapses.
Medical management is focused primarily on treating the symptoms of the disease. Symptom management includes treatment of spasticity, bladder management, prevention of bladder infection, and management of pain and fatigue. Spasticity is often managed with medications; unfortunately, this may also worsen the muscle weakness. Bladder management may involve the use of incontinence pads or catheters, along with prevention of bladder infections. Fatigue should be managed with good nutrition, prevention of overfatigue with energy conservation methods, regular exercise, establishment of routines for rest and sleep, and control of stress.140 Bowel incontinence is rarely a problem related to a neurologic deficit but is usually a functional impairment resulting from immobility.
Changes in cognition and mood often occur in persons with MS. Cognitive deficits are reported to occur in 30% to 70% of the MS population but do not necessarily correlate with a physical decline.7,8,85,157 The volume of lesions seen on MRI correlates with cognitive decline in complex attention, processing speed, and verbal memory.206 One investigation evaluated overall brain atrophy and size of the ventricles in relation to cognitive abilities. The results suggested a relationship between an increase in the size of the third ventricle and a decrease in cognition.29 Emotional changes such as depression may also be present. Persons with MS and other chronic conditions experience a higher rate of depression than the general population does.152 Depression may have different causes and should be assessed by the team. Depression may be caused by a physiologic response to the disease process, may be a psychologic response to the diagnosis, or might be a side effect of one of the disease-modifying medications. Depression may occur at any stage of the disease process. It should be addressed promptly because it may contribute to fatigue and an inability to cope with challenges and make adaptations as needed.152 Dementia may develop in individuals who exhibit euphoria and indifference. Dementia occurs in less than 5% of the population with MS.86
The most widely accepted tool to measure clinical impairment in a person with MS is the Expanded Disability Status Scale (EDSS).164 The scale should be completed by a physician because it includes a detailed neurologic examination. The EDSS combines an assessment of neurologic function and a scale to measure a client’s ambulatory and functional mobility status. There are limitations with this tool; it does not allow specific assessment of all ADLs and is not sensitive to potential cognitive and sexual deficits in MS.164 The OT practitioner should be familiar with the EDSS because it is often mentioned in the literature as a baseline for evaluating disability and has been adopted by the International Federation of MS Societies.99,222 The MS Functional Composite was developed to measure leg function and ambulation, arm and hand function, and cognition.46,141 It can be used for periodic baseline function and may be more sensitive for use as baseline cognitive evaluation than the EDSS.
Role of the Occupational Therapist
OT practitioners provide services for persons with MS in a number of settings. The type and degree of intervention provided will be determined by the setting, the type of reimbursement, and the client’s and caregiver’s responses to intervention.
Evaluation includes an occupational profile to guide the evaluation process. The occupational therapist then selects the necessary instruments to assess occupational performance of ADLs, IADL education, work, play, leisure, and social participation. During the performance of various occupations, the following skills should be evaluated: motor and praxis, sensory-perceptual, emotional regulation, cognitive, and communication skills. This is generally accomplished with a combination of standardized and nonstandardized assessments, through the use of interviews with the family and client, and by observation. The MS Society recommends a number of standardized evaluations appropriate for the OT evaluation.153 Optimally, a home evaluation should be completed. Because not all settings allow a home evaluation, the occupational therapist should interview the client and caregiver regarding the home environment and potential barriers. Since MS has an unpredictable course, the client may need referral for other resources and periodic re-evaluation by an occupational therapist. If the client has a cognitive deficit, a family member or significant other should be included in the evaluation process to provide accurate information. It is essential that actual performance evaluations be used for persons with MS because studies have shown that self-report is frequently inaccurate.84 Having an understanding of the client’s cultural, social, and spiritual perspective will provide insight into available support systems and their impact on the client’s adaptation to the disability.
Assessment of sensorimotor skills is discussed thoroughly in previous chapters. Because endurance and fatigue are such significant factors, it is important to not rely solely on the results of assessment of a specific client factor. For example, a manual muscle test is not likely to accurately reflect the degree of weakness experienced throughout the day. Observing a client performing a functional activity over a certain period or gathering information about the client’s daily activity patterns will provide the clinician with a more accurate evaluation of fatigue.95 The National MS Society recommends use of the Modified Fatigue Scale (MFS) or the Fatigue Questionnaire to understand specific problems resulting from fatigue.140 In the case of Marguerite, the MFS assessment may help determine the impact of fatigue on her daily activities.
When evaluating a client’s activity patterns, the OT practitioner should also ask about sleep patterns. Disrupted sleep patterns sometimes contribute to fatigue in the MS population. The information can be shared with the client’s physician so that appropriate intervention can be undertaken either medically or with counseling that addresses the habits and routines that may contribute to the client’s disrupted sleep.15
MS may also affect visual abilities, and compromises may be noted in visual tracking, scanning, and acuity. An objective evaluation will help determine the type of deficits, when the deficits occur, and more specifically how they affect ADLs and IADLs.
Perceptual processing and cognitive status should be reassessed periodically. The data gathered will help identify specific deficits and their potential impact on functional activities so that the OT practitioner can incorporate this information into family training.155 Cognitive deficits vary from a slight decrease in short-term memory and attention span to poor orientation and severely impaired short-term memory. The literature is mixed on the relationship between cognitive deficits and the degree of physical deficits.53 Assumptions should not be made regarding the presence or absence of cognitive deficits based on physical deficits or abilities or on an individual’s basic social skills. A person with significant physical deficits may have fewer cognitive deficits than an ambulatory person with MS. The client’s perceptual and cognitive deficits are factors that must be considered when deciding whether the client needs close, constant supervision or can stay home alone. Various standardized cognitive and perceptual assessments are included in previous chapters of this text. Basso developed a screening tool for cognitive dysfunction in individuals with MS. Basso’s tool was found to be both sensitive to functional impairment and cost-effective.20 This tool can be used by an occupational therapist or practitioners in other disciplines when evaluating a person with MS for cognitive deficits. ADLs may be evaluated with a check-off list, a standardized evaluation such as the Assessment of Motor and Process Skills,56 or other standardized assessments for ADLs.13
Evaluation of the cultural, social, and physical environment is important to consider with each client. MS is usually identified during the phase of life in which a person is raising a family and developing a career, as in the case study example of Marguerite. She is in the “sandwich” generation, simultaneously caring for her children and her mother. Because the disease is unpredictable and fluctuating, it leads to disruptions in normal daily activities and in family life. These disruptions create stress for the spouse or partner, children, and other family members. The occupational therapist must determine the type of support that the client can expect from family members.
The occupational performance assessment should include listening to the client and family describe performance patterns to identify typical daily habits, routines, and roles. Evaluating and treating the client at different times during the day will probably reflect different levels of fatigue. Understanding the performance patterns of rest periods, quality and amount of sleep, exercise patterns, intensity of activity (activity demands) during various times of the day, week, or month, and the time of the day when activities are most challenging is critical in developing treatment planning strategies. Understanding the performance patterns may help the OT practitioner understand how the side effects of the disease-modifying medications affect activity and also how to encourage integrating use of the medication into daily routines while managing side effects.
Ethical Considerations
A client with multiple sclerosis is able to ambulate to the car but has cognitive deficits that are exacerbated by fatigue in the afternoon. Because the client has discontinued work, the family members do not want to take away driving privileges as well. However, by continuing to drive, the client places himself and others at risk for injury. The occupational therapist may need to educate the family about the deficits by providing examples of the ways in which cognition is evaluated and by discussing the ways in which cognitive deficits affect driving and other daily activities. The occupational therapist is responsible for reporting the driving risk to the client’s physician.
Emotional and behavioral issues vary depending on the premorbid personality, progression of the disease, coping skills, and social environment of the person with MS. Cognitive deficits and denial of the progressive nature of the disease may lead to behavior that places the client at risk and makes management difficult. If family members do not understand or recognize the client’s behavioral problems, further complications may arise when the behavior is not restricted or modified by the family. Other emotional and cognitive issues include a client who is depressed or labile, has poor memory, refuses assistance from outside caregivers, or uses poor judgment regarding safety with medications and transfers. Each client demonstrates a unique set of behavioral issues and requires individual evaluation and an intervention approach that encompasses the family, client, and caregivers.
Goal Setting
For a client with a progressive disease such as MS, goal setting focuses on the client’s need to adapt as the disability progresses. Families often need to negotiate role changes to accommodate the person with MS, who may not be able to participate consistently in a previously established family role. The client’s ability to adapt depends on the family and client’s acknowledgment of deficits and willingness to consider alternatives. OT intervention may include (1) problem-solving compensatory strategies; (2) time and energy management; (3) role delegation; and (4) the use of adaptive equipment to compensate for motor, sensory, endurance, cognitive, and visual deficits. The National Multiple Sclerosis Society has developed guidelines and recommendations for care at home that address many ADL issues requiring OT treatment. These guidelines are by no means all-encompassing regarding interventions for ADLs and IADLs but provide a solid starting point to problem-solve with the patient and family.155 For specific techniques related to the individual’s deficits, see Chapter 10.
Marguerite’s OT intervention may involve analyzing her daily and weekly routine to identify activities that can be eliminated, modified, or delegated. She may consider grocery shopping online or delegating this task to her husband. Marguerite will need to evaluate the number of activities in which her children participate and the possibility of carpooling. She may need adaptive equipment for activities that require bilateral strength and dexterity because of the loss of sensation and dexterity in her left hand. Marguerite may be referred to an OT group designed for persons who have MS to learn energy management techniques.127
The benefits of an exercise program should be discussed with clients who have MS. An individual may already have an exercise program recommended by a physical therapist, and coordination would be important so that the client is able to engage in a manageable exercise program. The OT role may involve helping clients with energy management issues fit the program into their usual routine. It has long been thought that exercise will result in exacerbation and worsening of symptoms. Recent studies report benefits such as improved quality of life, reduced fatigue, and improved ambulation from a regular exercise program.138,159,223 Energy management recommendations should include assessment of the client’s typical exercise routine.
Summary
Because MS affects each person in a unique way, individual evaluation is essential to determine the client’s deficits and strengths. Individuals with MS may range from being ambulatory with primarily fatigue and energy management issues to partially limited in mobility and hand function to an individual who is a full-time wheelchair user requiring assistance with ADLs. Working with clients who have MS requires the practitioner to use expertise in evaluation and intervention from all areas of OT. With a pattern of relapses and remissions over the course of the disease process, development of an OT intervention plan is particularly challenging. Because the client may expect return of function, he or she may deny deficits and refuse to adapt to a change in status; this attitude can create safety problems. The OT practitioner focuses on assessing the current level of functioning and the best methods for the client to adapt to current changes in status.132 The occupational therapist may also assist the family in making long-range, realistic plans. For example, if the family is planning to remodel the bathroom, the therapist may encourage consideration of a roll-in shower and not just a standard shower stall with a shower seat.
Working with clients with MS requires a multidisciplinary approach. In addition to the occupational therapist, a physician, physical therapist, registered nurse, social worker, and psychologist may be involved as team members. Because the social environment may create complex and difficult problems, good communication among all team members is needed to ensure that the team goals are congruous. The occupational therapist has a unique perspective to offer the team regarding the client’s strengths and weaknesses since cognitive, perceptual, psychosocial, and motor abilities are assessed and treated in a functional context.
Section 5 Parkinson’s Disease
Incidence
PD is one of the most common adult-onset, degenerative neurologic disorders,57 second only to AD.201 Three classic symptoms are associated with PD: tremor, rigidity, and bradykinesia. The incidence rate for PD varies greatly, from 10 to greater than 400 per 100,000.230 Prevalence increases with age, and the disease affects 1.4% of the population older than 55 years,186 but approximately 3% of all PD cases are initially recognized in individuals younger than 50 years.201 Approximately 10% to 30% of clients with PD report first-degree relatives who also have PD. Gender differences have been noted; the prevalence of PD in men between the ages of 55 and 74 is slightly higher than that in women of the same age. After the age of 74, however, women show a slightly greater prevalence of PD than do men. The diagnosis is most often made after the age of 60.
The etiology of PD has not been definitively established.18,83 Previous literature referred to familial and sporadic forms of PD, but those distinctions are being revised in light of current genetic research. Although a positive family history has been established as a risk factor for PD, a single predictive genetic marker has not been absolutely identified. To date, mutations have been found in several genes that are associated with PD, including α-synuclein, parkin, ubiquitin carboxyl terminal hydrolase, SCA2, and DJ-1.201 These mutations have been found to have a role in abnormal protein processing in cells.18 Current genetic work has identified a gene mutation in familial PD, and this genetic mutation has also been identified in clients with sporadic PD, thus providing further evidence of a genetic determination for this disease process.201
Previously, environmental factors were considered a possible cause of PD.83 The possibility of an exogenous agent producing PD gained considerable recognition when narcotic addicts began using 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). After using MPTP, many addicts quickly exhibited parkinsonism that “strictly mimics the clinical and anatomical features of Parkinson’s disease”83 (p. 143). Although environmental factors have not been completely eliminated as contributing factors in the etiology of PD, they appear to play less of a role than do various genetic mutations.
Pathophysiology
The neurologic structure associated with PD is the substantia nigra, specifically the pars compacta portion.161 The pars compacta receives input from other basal ganglia nuclei and appears to serve as a modulator of striatal activity.167 The substantia nigra nuclei undergo significant deterioration as the disease progresses. The significant reduction in dopaminergic neurons in the substantia nigra pars compacta produces a decrease in activity within the basal ganglia and an overall “reduction in spontaneous movement”167 (p. 426). The substantia nigra serves as one of the major output nuclei for the basal ganglia to other structures.163 In addition to the loss of dopaminergic neurons, intracytoplasmic inclusions are found on postmortem examination within the substantia nigra.161 These intracytoplasmic inclusions are also known as Lewy bodies.59 Although the greatest amount of neurodegeneration is found in the pars compacta substantia nigra, destruction of other neurologic structures has been reported.161 Deterioration is also seen in the remainder of the substantia nigra, locus caeruleus, nucleus basilis, and hypothalamus.
Clinical Picture
PD is a slowly progressive, degenerative movement disorder.167 The diagnosis of PD is usually made after the age of 55. Although PD is not considered fatal, the degeneration of various neurologic structures severely compromises performance of functional tasks. A person in whom PD is diagnosed may live an additional 20 to 30 years, with a progressive loss of motor function that eventually requires specialized care.59 A person with PD has increased risk for pneumonia, which may be fatal.
PD is characterized by dysfunction in both voluntary and involuntary movements.167 The classic triad of symptoms includes tremor, rigidity, and a voluntary movement disorder. By the time that these “motor symptoms are clinically recognized, 60% of the dopaminergic” cells of the substantia nigra have deteriorated.201 The disturbances in voluntary movement are identified as difficulty initiating movement (akinesia) and slowness in maintaining movement (bradykinesia). The bradykinesia and akinesia are often the most disabling motor symptoms for a client with PD.82,83 The delay in initiating movement patterns and the slowness in executing the motion compromise functional tasks such as driving, dressing, and eating.
In addition to the slowness of movement, rigidity is a characteristic of PD. Rigidity is the stiffness within a muscle that impedes smooth movement. This stiffness occurs in both directions for each plane of motion at a specific joint.175 The characteristic resting tremor with a rate between 4 and 5 Hz is a disturbance in involuntary movement.135 This tremor often diminishes with activity, but in some clients the tremor persists during performance of functional activities.
Additional symptoms of PD are disturbances in gait and postural reactions, a masked face with decreased facial expressions, and emotional disturbances, including depression and psychosis.82,167 Deterioration in gait is seen throughout the course of the disease.190 Initially, gait may be fairly normal, but as the disease progresses, changes in stride length and speed of gait are apparent. The characteristic festinating gait is often seen; as the client walks, stride length decreases in length and speed slightly increases, thereby creating a shuffling effect. Reduced arm swing during ambulation is evident, and trunk rotation is markedly decreased during walking. Another motor disturbance associated with gait is the phenomenon of “freezing.”75 Freezing occurs when the person ceases to move, often after attempting to initiate, maintain, or alter a movement pattern. During gait, freezing may be seen as the client attempts to change directions or approach a narrow hallway or stairs. As the client attempts to alter the trajectory of walking to turn and enter another room, he or she may cease moving. Freezing can also be seen during other motor tasks, such as writing, brushing teeth, and speaking.
Postural abnormalities associated with PD include a flexed, stooped posture with the head positioned forward.139 The client tends to stand with flexion at the knees and hips. In addition to the stooped posture, balance reactions are compromised.174 Righting and equilibrium reactions are markedly reduced in effectiveness, and a person with PD may experience frequent falls.
Approximately 50% of individuals with diagnosed PD exhibit depression,174 which is not merely reactive to the severity of the symptoms or the chronic nature of the disease.59 The depression seen in individuals with PD appears to be related to a serotonergic deficit, similar to that in clients without PD who have depression. Complicating the feature of depression is a decrease in facial expressiveness caused by akinesia.174 This decrease in spontaneous facial expression, or the so-called masked face, is characteristic of clients with PD. Initially, decreased facial expressions are seen unilaterally, but as the disease progresses, spontaneous expression decreases on both sides of the face.59 Individuals with PD may also self-limit social interaction because they are embarrassed by their decreased facial expressions and movement disorders. Psychosis is also a common complication for clients with PD, and they may experience hallucinations and delusions.82 These disturbances compromise cognitive abilities and often limit functional skills.
Mental status is fairly normal throughout the early stages of PD, but visual-spatial perception is frequently compromised.174 Higher-order cognitive disorders are common in clients with PD. Clients with PD often have difficulty shifting attention among various stimuli. Processing simultaneous information is frequently difficult for an individual with PD, and tasks that require a sequential process are somewhat easier to perform. Driving a car presents a particular challenge because it necessitates processing of multiple forms of information in a simultaneous manner. Some self-care tasks, such as brushing teeth, have a clear sequence that can be followed; these activities can be performed with less demand on cognitive functioning. Although dementia is seldom seen in clients with an earlier-onset form of PD, approximately a third of people older than 70 years who have PD display dementia.
Additional symptoms associated with PD include autonomic dysfunction, dysphagia, and dysarthria.174 A client with PD may have bowel and bladder problems, with reduced intestinal motility producing constipation. Clients often report an increase in the frequency and urgency of urination. Clients also frequently complain of orthostatic hypotension, but syncope is rare.59 Clients with PD occasionally report periods of sweating and abnormal tolerance of heat and cold.174 Because speech volume is often decreased, persons with PD frequently seem to whisper. Articulation is imprecise, and speech is monotone. Dysphagia tends to occur in the later stages of PD, and clients may be at risk for choking and aspiration pneumonia as a result of the dysphagia.
The course of the disease varies from person to person, but the first clinical symptom identified is typically a unilateral resting tremor in the hand.135 Hoehn and Yahr established a scale identifying the progression of symptoms in PD.94 A client in stage I exhibits unilateral involvement, typically a hand tremor, but no impairment in functional abilities is reported. Such a client is able to complete personal ADLs and IADLs, but performance often requires additional effort and energy. Depending on job demands, a client with stage I PD is frequently employed but may require modifications of the work site. During this stage the client’s handwriting may become very small, with letters that are cramped together.59 This change in handwriting is referred to as micrographia. The client may also complain of muscle cramping when required to write for extended periods. Slight rigidity may be seen when the client is asked to rapidly open and close the involved hand.
Stage II denotes a progression of symptoms and the development of bilateral motor disturbances.94 Although the course of PD is variable, this stage is usually seen 1 to 2 years after initial diagnosis. Even though tremors or rigidity may be noted bilaterally, the client can still perform ADL skills. Performance of IADL skills may require modification because of motor difficulties. Work, depending on the job requirements, often requires additional modifications, and the client may need several rest breaks during the day. The client should make decisions at this point regarding the benefits of remaining employed relative to the energy expenditure. Posture becomes slightly stooped, with flexion at the knees and hips. A person with stage II PD is still able to ambulate independently. As PD progresses to stage III, the client experiences delayed righting and equilibrium reactions. Balance is impaired; the client has difficulty performing daily tasks that require standing, such as showering and meal preparation. Employment may be difficult because of its energy demands. Safety while walking is a concern because of the client’s reduced balance, and home modifications are necessary at this stage. A person with stage IV PD has significant deficits in completing daily living tasks. The client is still able to ambulate at this stage, but motor control is severely compromised and negatively affects dressing, feeding, and hygiene skills. Stage V is the final stage of PD. The client is typically confined to a wheelchair or bed and depends on others for most self-care activities. The rate of progression through these stages varies from person to person, but PD is a slowly progressive disorder.
The extent of PD symptoms in individual clients has been measured with the Unified Parkinson’s Disease Rating Scale.62 This scale evaluates a client’s motor skills, functional status, and extent of disability. Motor skills are evaluated by a trained observer.185 Functional status and extent of disability are measured through a client interview that includes items addressing ADL skills and cognitive and emotional factors.122 This instrument has been used for research and clinical practice to measure the effectiveness of various interventions in reducing PD symptoms (Table 35-4).
TABLE 35-4
Progression of Symptoms in Parkinson’s Disease
| Stage | Symptoms | Occupational Therapy Management |
| I | Unilateral tremor, micrographia, poor endurance for previous occupations, fatigue | Work evaluation if the client is employed; work simplification for work and home settings; develop the habit of taking frequent rest breaks; use of utensils with enlarged handles |
| II | Bilateral motor disturbances, mild rigidity reported, difficulties with simultaneous tasks, difficulties with executive function | Energy conservation techniques related to ADLs; develop daily flexibility exercises focused on trunk rotation; driving assessment and alternatives for community mobility; use of task analysis to structure sequential tasks |
| III | Balance problems with delayed reactions, difficulties in skilled sequential tasks | Environmental modifications in the home, including raised toilet seats, chairs with armrests, removal of throw rugs; use of visual cues and supports for sequential tasks |
| IV | Fine motor control severely compromised, oral motor deficits | Modifications to support participation in self-care tasks, changes in food textures |
| V | Client severely compromised motorically, dependent with ADLs | Use of environmental controls to allow access to the environment |
Medical Management
The most frequently used medical management strategy for PD is prescription of a dopamine agonist to make up for the depletion of dopamine caused by destruction of the substantia nigra.57,191 Levodopa is the medication most commonly used for the treatment of PD.170 This oral medication is actually a precursor to dopamine because dopamine is too large to cross the blood-brain barrier. Levodopa provides substantial relief from tremors and rigidity during the initial stages of PD. After approximately 5 to 10 years of chronic use of levodopa, motor side effects are reported.174 Those most often reported are dyskinesias and motor fluctuations. This so-called on-off phenomenon is related to the levodopa dosage. A decrease in tremors and rigidity occurs during the “on” period after the administration of levodopa, but the client may also experience various dyskinesias, such as abnormal movements of the limbs. As the dosage of levodopa wears off, the motor symptoms, specifically tremors and rigidity, associated with PD return. Timing of the medication and the periods of “on-off” are important considerations in planning the client’s daily activities. Even though abnormal movements are observed during the “on” period, the client has greater freedom of movement to complete functional activities.
As PD progresses, control of various motor symptoms through the use of levodopa becomes less effective.174 Surgical intervention, known as stereotactic surgery, has been used. In this surgery, specific lesions are made in neurologic structures to decrease the severity of PD symptoms. Stereotactic surgery on the globus pallidus internus has been used to decrease the severity of motor symptoms associated with PD and thus reduce the dosage of levodopa needed.105,160 This surgical procedure is known as pallidotomy. Pallidotomies have also been shown to reduce the dyskinesias associated with long-term use of levodopa.174 Stereotactic surgery has likewise been used to create lesions in portions of the thalamus to reduce the tremor and rigidity associated with PD.107
Neural transplantation has been used selectively for clients with PD.33 This process involves harvesting fetal mesencephalic neural tissue and then transplanting this tissue into the basal ganglia of clients with PD.47 The results of fetal brain transplants have been varied. In some clients the tremors and rigidity were substantially reduced, but other clients experienced dyskinesias following transplantation of such tissue.173 The best success with this procedure has been reported when bilateral implants are placed in the putamen from multiple fetuses. The transplanted fetal tissue produces dopamine and thereby reduces the debilitating symptoms of progressive PD. Clients must continue to use levodopa, but at a reduced dosage.
Role of the Occupational Therapist
OT services vary depending on the client’s stage of PD. Typically, an OT program would provide compensatory strategies, client and family education, environmental and task modifications, and community involvement.
During the initial stages of the disease, the occupational therapist should develop an occupational profile with the client and significant others to establish intervention priorities.189 Clients have expressed the desire to retain a sense of self and normalcy within their family, even in the face of deteriorating abilities. The focus of intervention is on developing habits and routines to foster participation in desired occupations as the disease process progresses. Educating the client and significant others regarding the course of the disease is an important step in this process, one that aids in selection of occupations. For example, the case study at the end of this chapter introduces Carl. He identified traveling and painting as important occupations, and interventions were designed to help him continue participating in these occupations as the disease progressed.
During the early stages of the disease, the client and family should be informed of community resources and support groups. In one study, clients with PD were found to be far more dependent on others for personal care and household activities than were same-age peers without PD.214 This dependence can place additional stress on the family. Involvement in a community-based group may provide the support needed to accommodate the changes in family roles and interaction.189
Even during the early stages of the disease process, clients with PD have difficulty in executive functioning.67 An investigation by Foster and Hershey compared clients who had PD, but no dementia, with age-matched controls on several assessments of executive functioning. Clients with PD were in stage I or II. Clients with PD performed significantly worse on working memory and executive functioning. Furthermore, lower executive functioning “was associated with reduced activity participation after controlling for motor dysfunction and depressive symptoms.” The authors urge occupational therapists to assess executive functioning even during the early stages of PD and develop strategies to support client participation in desired occupations.
Modification of household items may decrease the impact of tremors during the initial stage of the disease process. The use of built-up handles for eating and writing utensils should be introduced during the initial stages of PD. Handwriting often becomes small and difficult to read during the initial stage of PD. Time management techniques should be introduced at this stage. Paying bills, signing forms, or doing other written work should be completed soon after taking levodopa with the use of utensils that have built-up handles. Even though the tremors are not severe during the early stages of PD, clothing fasteners should be modified. The use of slip-on shoes or Velcro closures for clothing should be considered at this time. Although a client may be able to complete the fastening of clothing during this stage of PD, the occupational therapist must consider the amount of energy and time needed to perform such a task. In addition to the modification of specific tasks, household changes should be made at this time. Loose rugs should be removed from floors and furniture placed close to the wall to decrease obstacles. Chairs should have armrests to allow the client to push up from the chair to stand. Although balance is not significantly compromised during the early stages of PD, the family and client should become familiar with the new arrangement of furniture before it becomes a necessity. Bath and toilet railings and a raised toilet seat should be provided within the home. Because fatigue is a common complaint, clients should develop the habit of taking frequent breaks during the day. Modifying the household setting early in the course of PD allows the client and family members to adjust to changes and incorporate these changes into daily routines before they become a necessity.
A work evaluation should be performed during the early stage of the disease process to assess safety risks, potential hazards, and work simplification techniques that could be used. An ergonomic assessment of the work site and modifications in tools would be appropriate. In the case of Carl, computer modifications were considered. A client may have the option of reducing the number of work hours, but that decision may result in reduced medical benefits. These decisions and available options are part of the OT intervention process during the early stage of the disease.
During the initial stage of PD, the occupational therapist should establish a daily exercise program that addresses full range of motion.207 It is preferable to have a client with PD perform a short exercise program for 5 to 10 minutes daily rather than a longer program three times a week. Exercises should include alternating movements from various planes since many clients with PD display difficulty in smooth shifting of movements.124 Postural flexibility exercises should be included in the program, with specific attention directed to trunk extension. The most common postural change noted with progression of PD is a stooped stance. In addition to flexibility exercises, occupational therapists should instruct clients in the use of relaxation techniques and controlled breathing. Inhaling slowly through the nose and exhaling through pursed lips two or three times in succession, combined with improved postural alignment, can promote relaxation.
As the disease progresses, additional exercises can improve gait.212 Rhythmic auditory stimulation in the form of music with an accentuated initial beat has been found to significantly improve stride length and speed in clients with PD. Dancing can also enhance gait patterns, in addition to providing a social environment for clients with PD. As akinesia becomes more apparent, a client with PD should be instructed to use a rocking motion to begin movement activities. Rocking forward and backward a few times while seated can produce the momentum needed to rise from a chair.
As a person with PD progresses to the middle stages of the disease, the client experiences further deterioration in motor skills, particularly the execution of skilled sequential movements.52 These types of movements are needed to complete personal care and household tasks. Curra and associates found that external cues improve the speed and sequential performance of novel motor tasks.52 The occupational therapist should suggest modifying activities to include visual cues, verbal prompts, and rehearsal of movements. These strategies increase a client’s ability to perform personal care and household activities.
During the middle stages of PD, clients may have decreased oral motor control.174 Dysphagia and drooling may embarrass them and further restrict social engagements. The occupational therapist should encourage oral motor exercises and provide education regarding food selection. Food consistencies can be altered to improve the client’s ability to eat.
The ability to complete personal care tasks has been identified as a critical variable in a client’s perception of quality of life.64 Although progressive movement problems are characteristic of PD, the occupational therapist can minimize the impact that the movement disorder has on functional activities. Tremors have more negative effects on postural control compared to self-care tasks since compensatory strategies can reduce the impact of tremors on completion of ADLs.71 The use of group OT sessions has been demonstrated to be effective in reducing the impact of postural instability in clients with PD. An additional benefit of group sessions is the reported improvement in perception of quality of life in clients attending the sessions.
Access to community mobility and support programs should be included in the OT intervention plan during the middle stages of PD. A client with PD is often dependent on others for transportation. The use of community mobility services can decrease the client’s dependence on family members for shopping and errands.
During the last stages of PD, movement disorders and rigidity may eliminate the client’s ability to perform personal care tasks such as dressing and grooming.94 Depression caused by decreased ability to perform these tasks can significantly compromise a person’s quality of life.64 OT services should be provided to further modify the home environment for access and control. The use of environmental control units, such as a switch-operated television or radio, can be helpful. The switch plate should be activated with only light touch. Voice- or sound-activated environmental control units may not be as useful because of decreased vocal volume and poor control of articulation during speech production. The client’s ability to control the immediate environment can compensate for the losses experienced during the final stages of PD. The client may no longer be able to dress himself or herself, but through the use of various switches the client can select preferred television or radio programs, control room lighting, and operate a computer with minimal motor action.
Summary
Although PD is a progressive, neurodegenerative disorder, OT has much to offer clients with this disease.64,71 The diminishing ability to perform personal care activities and engage in self-selected tasks has been identified as one of the variables contributing to depression and the decreased quality of life in clients with PD. Throughout the progressive course of PD, OT addresses the ability of the client to engage in meaningful activities. The client’s wishes and family circumstances are incorporated into the OT intervention plan at every stage of the disease process.
Case Study
Carl
Carl is a 62-year-old college professor in whom PD was diagnosed at the age of 57. He is married and lives in a small one-story home with his wife. He has two adult children who live in another state. Carl reports that he enjoys traveling, reading, painting, and attending concerts.
Carl recently considered taking early retirement because of the increase in tremors in both his hands, which made correcting papers difficult. He also reports some problems with endurance as a result of stiffness. Carl indicates that he is no longer able to paint because of the tremors in his hands. He also reports that he is unsure whether he should continue driving because of these tremors.
The results of OT evaluation indicate that Carl is cooperative and motivated for therapy. Although he does not state that he is depressed, his wife reports features of depression, such as decreased interest in going to concerts and planning summer vacations to see their adult children and grandchildren. His wife also reports that Carl seems depressed about his possible early retirement and loss of status as a college professor.
Carl is able to complete most personal ADLs independently but has difficulty stepping into and out of the tub and shower. His wife reports that she is afraid that he will fall and that she often assists him in getting into and out of the shower. Carl also has difficulty tying his tie and buttoning his shirt. Tremors are noted bilaterally in his hands, and slight rigidity is present during passive range of motion. Dynamic balance is slightly compromised on uneven surfaces and stairs.
Carl has been taking Sinemet (levodopa and carbidopa medication) for the past 3 years to decrease the rigidity and tremors. He does not report any dyskinesias.
When asked about his personal goals, Carl replies, “I guess I’ll have more time to read now.”
OT was initiated to accomplish the following:
• Instruct in the use of a buttonhook.
• Make suggestions regarding clothing modifications such as clip-on ties and slip-on shoes.
• Instruct in the use of momentum to initiate movement, such as rocking back and forth to rise from a chair.
2. Modify the home environment:
• Remove throw rugs and obstacles in walkways.
• Provide a tub seat and shower extension hose.
3. Assess the work setting for modifications:
• Instruct in energy conservation and urge him to take frequent breaks and schedule activities during the “on” phase of medications.
4. Investigate leisure pursuits:
• Provide modifications in his easel such as using forearm supports to allow him to continue to paint.
• Provide information regarding community-based PD support groups.
5. Instruct in a daily active range-of-motion exercise program:
Carl responded well to OT intervention. Although he was able to complete the academic school year, he decided to retire afterward. He stopped driving, but his wife began to drive them to concerts and art exhibits. He was able to complete personal ADLs safely with the use of adapted equipment and home modifications. He resumed painting during the “on” periods of his medication schedule by using forearm supports attached to an angled table. He attended a Parkinson’s support group two times a week and began to socialize with members of that group. Carl reported that the daily exercises seemed to decrease his stiffness, and he and his wife took frequent strolls in the park when weather permitted. He and his wife also joined a book club.
1. What are the symptoms of ALS at onset?
2. What is the underlying neurologic process in ALS?
3. What body functions remain intact throughout the ALS disease process?
4. What is the prognosis for ALS? Given this prognosis, what is the goal of the occupational therapist?
5. What are the symptoms of ALS at each stage of the disease?
6. What interventions are appropriate at each stage of ALS?
7. What are the initial symptoms of AD?
8. What is the underlying degenerative neurologic process associated with AD?
9. What changes in symptoms occur over the course of AD?
10. How do the changes in symptoms affect occupational performance in clients with AD?
11. What is the prognosis for a client with AD?
12. What OT interventions are appropriate for a client at each stage of AD?
13. What environmental modifications should be made to accommodate a client with AD?
14. What are the symptoms of MS at onset?
15. What is the underlying neurologic process in MS?
16. What are the three typical patterns of MS? How do they differ?
17. What symptoms of MS are managed with medication? What are the side effects of medication management?
18. How is medication management of the relapsing and remitting form of MS different from that in the other forms of MS?
19. What does the OT evaluation include for a person with MS?
20. Why is it important to include the family in the evaluation and treatment process for a client with MS?
21. What are the initial symptoms of HD?
22. What is the underlying degenerative neurologic process associated with HD?
23. What changes in symptoms occur over the course of HD?
24. How do the changes in HD symptoms affect occupational performance?
25. What is the prognosis for a client with HD?
26. What OT interventions are appropriate for a client with HD at the various stages of the disease?
27. What environmental modifications should be made to accommodate a client with HD?
28. What are the initial symptoms of PD?
29. What is the underlying degenerative neurologic process associated with PD?
30. What changes in symptoms occur over the course of PD?
31. How do the changes in PD symptoms affect occupational performance?
32. What is the prognosis for a client with PD?
33. What OT interventions are appropriate for a client with PD?
34. How does the medication schedule of levodopa affect the daily routine of a client with PD?
35. What environmental modifications should be made to accommodate a client with PD?
References
1. ALS Association. ALS, cognitive impairment (CI) and frontotemporal lobar dementia (FTLD): a professional’s guide. alsinfo@alsa-national.org/www.alsa.org, 2005. [27001 Agoura Road, Suite 250, Calabasas Hills, CA 91301-5104, Phone: (800) 782-4747. Website].
2. Allen, C. Allen Cognitive Level (ACL) test. Rockville, Md: American Occupational Therapy Foundation; 1991.
3. Allen, CK, Earhart, CA, Blue, T. Occupational therapy treatment goals for the physically and cognitively disabled. Rockville, Md: American Occupational Therapy Association; 1992.
4. Alzheimer’s Association. Alzheimer’s disease facts and figures. Washington DC: Alzheimer’s Association; 2007.
5. Alzheimer’s Association. Campaign for quality residential care; dementia care practice recommendations for assisted living residences and nursing homes, 2009. Cited January 29, 2011. Available at www.alz.org/professionals_and_researchers_dementia_care_practice_recommendations.asp.
6. Alzheimer’s Association. Early onset dementia: a national challenge, a future crisis. Washington DC: Alzheimer’s Association; 2006.
7. Amato, MP, Ponziani, G, Guiseppina, MD, et al. Cognitive impairment in early-onset multiple sclerosis: a reappraisal after 10 years. Arch Neurol. 2001;58(10):1602.
8. Amato, MP, Ponziani, G, Pracucci, G, et al. Cognitive impairment in early-onset multiple sclerosis. Arch Neurol. 1995;52(2):168–172.
9. American Occupational Therapy Association. Occupational therapy practice guidelines for adults with neurodegenerative diseases: multiple sclerosis, transverse myelitis, and amyotrophic lateral sclerosis. Bethesda, Md: American Occupational Therapy Association; 1999.
10. American Occupational Therapy Association. Statement: occupational therapy services for persons with Alzheimer’s disease and other dementias. Am J Occup Ther. 1994;48(11):1029.
11. American Psychiatric Association. Diagnostic and statistical manual of mental disorders: fourth edition text revised. Arlington, Va: American Psychiatric Association; 2004.
12. American Psychiatric Association. DSM IV: Diagnostic and statistical manual of mental disorders, ed 4. Washington, DC: American Psychiatric Association; 1994.
13. Asher, IE. Occupational therapy assessment tools: an annotated index, ed 2. Bethesda, Md: American Occupational Therapy Association; 1996.
14. Atchison, P. Helping people with Alzheimer’s and their families preserve independence. OT Week. 1994;8:16.
15. Attarian, HP, Brown, KM, Duntley, SP, et al. The relationship of sleep disturbances and fatigue in multiple sclerosis. Arch Neurol. 2004;61(4):525–528.
16. Bain, BK. Switches, control interfaces, and access methods. In: Bain BK, Leger D, eds. Assistive technology. New York, NY: Churchill Livingstone, 1997.
17. Baker, M, Blumlein, D. Huntington’s disease part 1: what is it? Br J Healthcare Assistants. 2009;35:223–227.
18. Bandmann, O, Marsden, CD, Wood, NW. Genetic aspects of Parkinson’s disease. Mov Disord. 1998;13(2):203.
19. Barusch, A, Spaid, W. Gender differences in caregiving: why do wives report greater burden? Gerontologist. 1989;29(5):667.
20. Basso, MR, Beason-Hazen, S, Lynn, J, et al. Screening for cognitive dysfunction in multiple sclerosis. Arch Neurol. 1996;53(10):980–984.
21. Baum, C. Addressing the needs of the cognitively impaired elderly from a family policy perspective. Am J Occup Ther. 1991;45(7):594.
22. Baum, C, Edwards, D. Cognitive performance in senile dementia of the Alzheimer’s type: the kitchen task assessment. Am J Occup Ther. 1993;47(5):431.
23. Baum, C, Edwards, D. Identification and measurement of productive behaviors in senile dementia of the Alzheimer’s type. Gerontologist. 1993;33(3):403.
24. Baum, C, Morrison, T, Hahn, M, Edwards, D. Executive function performance test. Program in occupational therapy. St. Louis: Washington University School of Medicine, Test Protocol Booklet; 2008.
25. Beck, AT, Steer, RA, Beck depression inventory. rev ed, San Antonio, Tex, Psychological Corporation, 1987.
26. Beck, AT, Ward, CH, Mendelson, M, et al. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571.
27. Belsh, JM. Definitions of terms, classifications, and diagnostic criteria of ALS. In: Belsh JM, Schiffman PL, eds. ALS diagnosis and management for the clinician. Armonk, NY: Futura Publishing, 1996.
28. Belsh JM, Schiffman PL, eds. ALS diagnosis and management for the clinician. Armonk, NY: Futura Publishing, 1996.
29. Berg, D, Mäurer, M, Warmuth-Metz, M, et al. The correlation between ventricular diameter measured by transcranial sonography and clinical disability and cognitive dysfunction in patients with multiple sclerosis. Arch Neurol. 2000;57(9):1289–1292.
30. Birnesser, L. Treating dementia: practical strategies for long-term care. OT Pract. 1997;2(6):16.
31. Blacker, D, Broadhurst, L, Teixeira, L. The role of occupational therapy in leisure adaptation with complex neurological disability: a discussion using two case study examples. NeuroRehabiltiaiton. 2008;23:313–319.
32. Bolmsjö, I, Hermerén, G. Interviews with patients, family, and caregivers in amyotrophic lateral sclerosis: comparing needs. J Palliat Care. 2001;17(4):236–240.
33. Borlongan, CV, Sanberg, PR, Freman, TB. Neural transplantation for neurodegenerative disorders. Lancet. 1999;353(suppl 1):29.
34. Bowen, JD, Malter, AD, Sheppard, L, et al. Predictors of mortality in patients diagnosed with probable Alzheimer’s disease. Neurology. 1996;47(2):433–439.
35. Brooks, BR, Miller, RG, Swash, M, Munsat, TL, for the World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299.
36. Buchner, D, Larsen, E. Falls and fractures in patients with Alzheimer’s-type dementia. JAMA. 1987;257(11):1492.
37. Burns, T, Mortimer, JA, Merchak, P. Cognitive performance test: a new approach to functional assessment in Alzheimer’s disease. J Geriatr Psychiatry Neurol. 1994;7(1):46.
38. Busse, A, Bischkopf, J, Riedel-Heller, SG, Angermeyer, MC. Subclassifications for mild cognitive impairment: prevalence and predictive validity. Psychol Med. 2003;33(6):1029–1038.
39. Calabresi, P. Diagnosis and management of multiple sclerosis. Am Fam Physician. 2004;15(20):1935.
40. Carswell, A, Eastwood, R. Activities of daily living, cognitive impairment and social function in community residents with Alzheimer disease. Can J Occup Ther. 1993;60:130.
41. Centers for Disease Control and Prevention, Alzheimer’s disease, Healthy Brain Initiative (HBI) Program. Atlanta, Ga: Centers for Disease Control and Prevention, 2010.
42. Cherry, D. Teaching others how to manage the challenging behaviors of dementia. In: Summer series on aging. San Francisco, Calif: American Society on Aging; 1997.
43. Christenson, M. Environmental design, modification and adaptation. In: Larson O, et al, eds. Role of occupational therapy and the elderly. Rockville, Md: American Occupational Therapy Association, 1996.
44. Cicchetti, F, Parent, A. Striatal interneurons in Huntington’s disease: selective increase in the density of calretinin-immunoreactive medium-sized neurons. Mov Disord. 1996;11(6):619.
45. Cobb, AK, Hamera, E, Festoff, BW. The decision-making process in amyotrophic lateral sclerosis. In: Charash L, et al, eds. Coping with progressive neuromuscular diseases. Philadelphia, Pa: Charles Press, 1987.
46. Cohen, JA, Fischer, JS, Bolibrush, DM, et al. Intrarater and interrater reliability of the MS functional composite outcome measure. Neurology. 2000;54(4):802–806.
47. Collier, TJ, Kordower, JH. Neural transplantation for the treatment of Parkinson’s disease: present-day optimism and future challenges. In Jankovic J, Tolosa E, eds.: Parkinson’s disease and movement disorders, ed 3, Baltimore, Md: Williams & Wilkins, 1998.
48. Comptson A, ed. McAlpine’s multiple sclerosis, ed 3, New York, NY: Churchill Livingstone, 1998.
49. Corcoran, M, Gitlin, L. Dementia management: an occupational therapy home based intervention for caregivers. Am J Occup Ther. 1992;46(9):801.
50. Corcoran, M, Gitlin, L. Family acceptance and use of environmental strategies provided in an occupational therapy intervention. Occup Phys Ther Geriatr. 2001;19(1):1–20.
51. Corder, EH, Saunders, AM, Strittmatter, WJ, et al. Gene dose of apolipoprotein E type allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923.
52. Curra, A, Berardelli, A, Agostino, R. Performance of sequential arm movements with and without advanced knowledge of motor pathways in Parkinson’s disease. Mov Disord. 1997;12(5):646.
54. Deloire, M, Ruet, A, Hamel, D, et al. Early cognitive impairment in multiple sclerosis predicts disability outcome several years later. Mult Scler. 2010;16(5):581–587.
55. Dixon, C. Preventing striking out behavior by a geriatric resident. OT Pract. 1996;1(2):39.
56. Doble, SE, Fisk, JD, Fisher, AG, et al. Functional competence of community-dwelling persons with multiple sclerosis using the Assessment of Motor Process Skills. Arch Phys Med Rehabil. 1994;75(8):843–851.
57. Dodel, RC, Eggert, KM, Singer, MS, et al. Cost of drug treatment in Parkinson’s disease. Mov Disord. 1998;13(2):249–254.
58. Driscoll, I, Davatzikos, C, An, Y, et al. Longitudinal pattern of regional brain volume change differentiates normal aging from MCI. Neurology. 2009;72(22):1906–1913.
59. Duvoisin, RC, Sage, JI. The spectrum of parkinsonism. In: Chokroverty S, ed. Movement disorders. New Brunswick, NJ: PMA Publishing, 1990.
60. Edwards, D, Baum, C. Caregiver burden across stages of dementia. OT Pract. 1990;2:17.
61. Evans, D. Estimated prevalence of Alzheimer’s disease in the U.S. Milbank Q. 1990;68:267.
62. Fahn, S, Elton, RL. The Unified Parkinson’s Disease Rating Scale. In: Fahn, S, et al, eds. Recent developments in Parkinson’s disease, vol 2. Florham Park, NJ: Macmillan Healthcare Information; 1987.
63. Fisher, A. The assessment of motor and process skill (AMPS) in assessing adults: functional measures and successful outcomes. Rockville, Md: American Occupational Therapy Foundation; 1991.
64. Fitzpatrick, R, Peto, V, Jenkinson, C. Health-related quality of life in Parkinson’s disease: a study of outpatient clinic attenders. Mov Disord. 1997;12(6):916.
65. Folstein, MF, Folstein, SE, McHugh, PR. Mini-Mental State: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189.
66. Folstein, SE. Huntington’s disease: a disorder of families. Baltimore, Md: Johns Hopkins University Press; 1989.
67. Foster, ER, Hershey, T. Everyday executive function is associated with activity participation in Parkinson disease without dementia. Occup Ther J Res. 2011;31(1S):S16–S22.
68. Foti, D. Gerontic occupational therapy: specialized intervention for the older adult. In: Larson O, et al, eds. Role of occupational therapy and the elderly. Rockville, Md: American Occupational Therapy Association, 1996.
69. Galasko, D, Edland, SD, Morris, JC, et al. The consortium to establish a registry for Alzheimer’s disease (CERAD). XI: Clinical milestones in your patients with Alzheimer’s disease followed over 3 years. Neurology. 1995;45(8):1451–1455.
70. Galvin, JE, Fagan, AM, Holtzman, DM, et al. (2010) Relationship of dementia screening tests with biomarkers of Alzheimer’s disease. Brain. 2010;133(11):3290–3300.
71. Gauthier, L, Dalziel, S, Gauthier, S. The benefits of group occupational therapy for patients with Parkinson’s disease. Am J Occup Ther. 1987;41(6):360.
72. Gelb, DJ. Introduction to clinical neurology, ed 3. St. Louis, Mo: Mosby; 2005.
73. Gélinas, I, Gauthier, L, McIntyre, M, Gauthier, S. Development of a functional measure for persons with Alzheimer’s disease: the disability assessment for dementia. Am J Occup Ther. 1999;53(5):471–481.
74. Ghezzi, A. Multiple sclerosis and the aging process, Formulary, Proquest Nursing & Allied Health Source, Nov 17, 2005. Therapeutic strategies in childhood multiple sclerosis. Ther Adv Neurol Dis. 2010;3(4):217–228.
75. Giladi, N, Kao, R, Fahn, S. Freezing phenomenon in patients with parkinsonian syndromes. Mov Disord. 1997;12(3):302.
76. Gilliam, TC, Kandel, ER, Jessell, TM. Genes and behavior. In Kandel ER, Schwartz JH, Jessell TM, eds.: Principles of neural science, ed 4, New York, NY: McGraw-Hill, 2000.
77. Gitlin, L, Corcoran, M. Occupational therapy & dementia care. Bethesda, Md: AOTA Press; 2005.
78. Gitlin LN Winter, L, Vause Earland, T, et al, The Tailored Activity Program to reduce behavioral symptoms in individuals with dementia: feasibility, acceptability and replication potential. Gerontologist, 2009;49(3):428–439. Available at, www.ncbi.nlm.nih.gov/pubmed/19420314.
79. Glantz, C, Richman, N. Promoting occupational therapy for older adults where they live and throughout the continuum of care. In: Coppola S, Elliot S, Toto P, eds. Strategies to advance gerontology excellence. Bethesda, Md: AOTA Press; 2008:475–496.
80. Glickstein, J. Therapeutic interventions in Alzheimer’s disease. Gaithersburg, Md: Aspen; 1997.
81. Goate, A, Chartier-Harlin, MC, Mullan, M. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):973.
82. Goldman, JG. New thoughts on thought disorders in Parkinson’s disease: review of current research strategies and challenges. Parkinsons Dis. 2011;2011:675630.
83. Goldman, SM, Tanner, C. Etiology of Parkinson’s disease. In Jankovic J, Tolosa E, eds.: Parkinson’s disease and movement disorders, ed 3, Baltimore, Md: Williams & Wilkins, 1998.
84. Goverover, Y, O’Brien, AR, Moore, NB, DeLuca, J. Actual reality: a new approach to functional assessment in persons with multiple sclerosis. Arch Phys Med Rehabil. 2010;91(2):252–260.
85. Grigsby, J, Kravcisin, N, Ayarbe, SD, Busenbark, D. Prediction of deficits in behavioral self-regulation among persons with multiple sclerosis. Arch Phys Med Rehabil. 1993;74(12):1350–1353.
86. Guberman, A. An introduction to clinical neurology, pathophysiology, diagnosis, and treatment. Boston, Mass: Little, Brown; 1994.
87. Gure, TR, Kabeto, MU, Plassman, BL, et al. Differences in functional impairments in subtypes of dementia. J Gerontol A Biol Sci Med Sci. 2010;65(4):431–441.
88. Gwyther, L, Matteson, M. Care for the caregivers. J Gerontol Nurs. 1983;9(2):93.
89. Hasselkus, B. Occupation and well being in dementia: the experience of day-care staff. Am J Occup Ther. 1998;52(6):423.
90. Hayden, MR. Huntington’s chorea. New York, NY: Springer-Verlag; 1996.
91. Hecht, M, Hillemacher, T, Gräsel, E, et al. Subjective experience and coping in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:225–231.
92. Helder, DI, Kaptein, AA, van Kempen, GMJ, et al. Living with Huntington’s disease: illness perceptions, coping mechanisms, and patients’ well-being. Br J Health Psychol. 2002;7:449–462.
93. Hendrie, H. Epidemiology of dementia and Alzheimer’s disease. Am J Geriatr Psychiatry. 1998;6(2):S3.
94. Hoehn, MM, Yahr, MD. Parkinsonism: onset, progression and mortality. Neurology. 1967;17(5):427.
95. Hugos, C, Copperman, L. Workshop: the new multiple sclerosis guidelines, delivering effective comprehensive therapy services. Monterey: Calif; 1999.
96. Huntington Study Group. Unified Huntington’s Disease Rating Scale: reliability and consistency. Mov Disord. 1996;11(2):136.
97. Hussian, R. Modification of behaviors in dementia via stimulus manipulation. Clin Gerontol. 1988;8:37.
98. Imbriglio, S. Physical and occupational therapy for Huntington’s disease. New York, NY: Huntington’s Disease Society of America; 1997.
99. International Federation of Multiple Sclerosis Societies. Symposium on a minimal record of disability for multiple sclerosis. Acta Neurol Scand. 1984;70:169.
100. Janardhan, V, Bakshi, R. Quality of life and its relationship to brain lesions and atrophy on magnetic resonance images in 60 patients with multiple sclerosis. Arch Neurol. 2000;57(10):1485.
101. Joiner, C, Hansel, M. Empowering the geriatric client. OT Pract. 1996;1:34–39.
102. Kaptein, AA, Scharloo, M, Helder, DI, et al. Quality of life in couples living with Huntington’s disease: the role of patients’ and partners’ illness perceptions. Qual Life Res. 2007;16:793–801.
103. Katz, S, Ford, AB, Moskowitz, RW, et al. Studies of illness in the aged. The index of ADL: a standardized measure of biological and psychological function. JAMA. 1963;185:914.
104. Kaye, J, Michael, Y, Calvert, J, et al. Exceptional brain aging in a rural population–based cohort. J Rural Health. 2009;25(3):320–325.
105. Kelly, PJ. Pallidotomy in Parkinson’s disease. Neurosurgery. 1995;36(6):1154.
106. Kim, WK, Liu, X, Sandner, J, et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology. 2009;73(20):1686–1692.
107. Krauss, JK, Grossman, RG. Surgery for hyperkinetic movement disorders. In Jankovic J, Tolosa E, eds.: Parkinson’s disease and movement disorders, ed 3, Baltimore, Md: Williams & Wilkins, 1998.
108. Kukull, WA, Larson, EB, Teri, L, et al. The Mini-Mental Status Examination and the diagnosis of dementia. J Clin Epidemiol. 1994;47(9):1061–1067.
109. Larson, E. Management of Alzheimer’s disease in primary care settings. Am J Geriatr Psychiatry. 1998;6(2):S34.
110. Larson, EB, Kukull, WA, Buchner, D, Reifler, BV. Adverse drug reactions associated with global cognitive impairment in elderly persons. Ann Intern Med. 1987;107(2):169–173.
111. Larson, EB, Reifler, BV, Featherstone, HJ, English, DR. Dementia in elderly outpatients: a prospective study. Ann Intern Med. 1984;100(3):417–423.
112. Lawton, M, Brody, E. Assessment of older people: self-maintaining and IADL. Gerontologist. 1969;9(3):179.
113. LeBarge, E. A preliminary scale to measure degree of worry among mildly demented Alzheimer disease patients. Phys Occup Ther Geriatr. 1993;11:43.
114. Levy, LL. Activity, social role retention and the multiply disabled aged: strategies for intervention. Occup Ther Mental Health. 1990;10:1.
115. Levy, LL. Cognitive integration and cognitive components. In: Larson KO, et al, eds. The role of occupational therapy with the elderly. Bethesda, Md: American Occupational Therapy Association, 1996.
116. Levy, LL. Cognitive treatment. In: Davis LJ, Kirkland M, eds. The role of occupational therapy with the elderly. Rockville, Md: American Occupational Therapy Association, 1988.
117. Levy-Lahad, E, Wasco, W, Poorkaj, P, et al. Candidate gene for chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977.
118. Lewis I, Kirchen S, eds. Dealing with ethnic diversity in nursing homes. Washington, DC: Taylor & Francis, 1996.
119. Burks, J, Bigley, GK, Hill, H. Multiple sclerosis. In: Lin V, Cardinas DD, Cutter NC, et al, eds. Spinal cord medicine principles and practice. New York, NY: Demos Medical, 2003.
120. Loeb, P. Independent living scales. Austin, Tex: Psychological Corporation; 2003.
121. Lou, JS, Reeves, A, Benice, T, Sexton, G. Fatigue and depression are associated with poor quality of life in ALS. Neurology. 2003;60:122–123.
122. Louis, ED, Lynch, T, Marder, K, Fahn, S. Reliability of patient completion of the historical section of the Unified Parkinson’s Disease Rating Scale. Mov Disord. 1996;11(2):185–192.
123. Madhusoodanan, S, Brenner, R, Araujo, L, Abaza, A. Efficacy of risperidone treatment for psychoses associated with schizophrenia, bipolar disorder or senile dementia in 11 geriatric patients: a case series. J Clin Psychiatry. 1995;56(11):514–518.
124. Maitra, KK, Dasgupta, AK. Incoordination of a sequential motor task in Parkinson’s disease. Occup Ther Int. 2005;12:218–233.
125. Gordon, PH, Goetz, RR, Rabkin, JG, et al. A prospective cohort study of neuropsychological test performance in ALS. Amyotroph Lateral Scler. 2010;11(3):312–320.
126. Matthews WB, Acheson ED, Batchelor JR, Weller RO, eds. McAlpine’s multiple sclerosis. Edinburgh, Scotland: Churchill Livingstone, 1985.
127. Matuska, K, Mathiowetz, V, Finlayson, M. Use and perceived effectiveness of energy conservation strategies for managing multiple sclerosis fatigue. Am J Occup Ther. 2007;61:62–69.
128. McCormick, WC, Kukull, WA, van Belle, G, et al. Symptom patterns and co-morbidity in the early stages of Alzheimer’s disease. J Am Geriatr Soc. 1994;42:517–521.
129. McDonald, ER, Wiedenfeld, SA, Hillel, A, et al. Survival in amyotrophic lateral sclerosis. Arch Neurol. 1994;51:17–23.
130. Mega, MS, Cummings, JL, Fiorello, T, Gornbein, J. The spectrum of behavioral changes in Alzheimer’s disease. Neurology. 1996;46(1):130–135.
131. Miller, RG, Rosenberg, JA, Gelinas, DF, et al. Practice parameter: the care of the patient with amyotrophic lateral sclerosis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1999;52:1311–1323.
132. Miller, DM, Rudick, RA, Cutter, G, et al. Clinical significance of the Multiple Sclerosis Functional Composite: relationship to patient-reported quality of life. Arch Neurol. 2000;57(9):1319–1324.
133. Miller, RG, Jackson, CE, Kasarskis, EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutrition and respiratory therapies (an evidence based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1218–1226.
134. Mirra, SS, Heyman, A, McKeel, D, et al. The consortium to establish a registry for Alzheimer’s disease (CERAD). II: standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41(4):479–486.
135. Misulis, KE. Neurologic localization and diagnosis. Boston, Mass: Butterworth-Heinemann; 1996.
136. Morris, J, Roe, CM, Grant, EA, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66(12):1469–1475.
137. Morscheck, P. An overview of Alzheimer’s disease and long term care. Pride J Long-Term Health Care. 1984;3:4.
138. Motl, RW, Gosney, JL. Effect of exercise training on quality of life in multiple sclerosis: a meta-analysis. Mult Scler. 2008;14(1):129–135.
139. Müller, V, Mohr, B, Rosin, R, et al. Short-term effects of behavioral treatment on movement initiation and postural control in Parkinson’s disease: a controlled clinical study. Mov Disord. 1997;12(3):306–314.
140. Multiple Sclerosis Council. Fatigue and multiple sclerosis. Washington, DC: Paralyzed Veterans of America; 1998.
141. Multiple Sclerosis Council. MS Functional Composite, Oct 2001, National MS Society. Available at www.nationalmssociety.org.
142. Munsat, TL. Slowing down ALS—is this good or bad? Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(suppl 1):S19.
143. Nance, MA. Comprehensive care in Huntington’s disease: a physician’s perspective. Brain Res Bull. 2007;72:175–178.
144. Nance, MA, Sander, G. Characteristics of individuals with Huntington’s disease in long-term care. Mov Disord. 1996;11(5):542.
145. National Institute of Aging. 2009 progress Report on Alzheimer’s Disease. Bethesda, Md: National Institute of Aging Health, Alzheimer’s Disease Education & Referral Center; 2009.
146. National Institutes of Health. Amyotrophic lateral sclerosis. Available at http://www.ninds.nih.gov, 2010.
147. National Institutes of Health, Huntington’s disease: hope through research. NIH Publication No. 98-19, Bethesda, Md, National Institutes of Health, 1998.
148. National Institutes of Health. NIH Consensus Development Conference Statement on Preventing Alzheimer’s Disease and Cognitive Decline. Bethesda, Md: National Institutes of Health; 2010.
149. National Institutes of Health. NIH Senior Health Alzheimer’s Disease. 2002 March 30, 2010. Cited November 5, 2010. Available at http://nihseniorhealth.gov/alzheimersdisease/causesandriskfactors/05.html.
150. National Institutes of Health. 2009 Progress Report on Alzheimer’s Disease: translating New Knowledge. Bethesda, Md: National Institute of Aging; 2010.
151. National Institute of Neurological Disorders and Stroke. Available at http://www.ninds.nih.gov/disorders/motor_neuron_diseases, 2010.
152. National Multiple Sclerosis Society. Depression and multiple sclerosis. http://www.nationalmssociety.or/brochures, 2004. [Available at].
153. National Multiple Sclerosis Society. Rehabilitation Assessment Measures in MS, 2010. Available at http://www.nationalmssociety.org.
154. Northern California Neurobehavioral Group. Cognistat: the neurobehavioral cognitive status examination. Fairfax, Calif: Northern California Neurobehavioral Group; 1995.
155. Northrup, D, Frankel, D, Guidelines & recommendations for home care providers and personal care assistants, National Multiple Sclerosis Society Clinical Programs Department, 2008. www.nationalmssociety.org.
156. Noseworthy, JH, Lucchinetti, C, Rodriguez, M, Weinshenker, BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952.
157. Nuyens, G, Van Asch, P, Kerckhofs, E, et al. Predictive value of SF-36 for MS-specific scales of the MS Quality of Life Inventory. Int J MS Care. 2003;5(1):8.
158. Nygård, L, Bernspång, B, Fisher, AG, Winblad, B. Comparing motor and process ability of persons with suspected dementia in home and clinic settings. Am J Occup Ther. 1994;48(8):689–696.
159. Oken, BS, Kishiyama, S, Zajdel, D, et al. Randomized controlled trial of yoga and exercise in multiple sclerosis. Neurology. 2004;62(11):2058–2064.
160. Olanow, CW. Gpi pallidotomy—have we made a dent in Parkinson’s disease? Ann Neurol. 1996;40(3):341.
161. Olanow, CW, Jenner, P, Tatton, N, Tatton, W. Neurodegeneration and Parkinson’s disease. In Jankovic J, Tolosa E, eds.: Parkinson’s disease and movement disorders, ed 3, Baltimore, Md: Williams & Wilkins, 1998.
162. Olsson, AG, Markhede, I, Strang, S, Persson, LI. Differences in quality of life modalities give rise to needs of individual support in patients with ALS and their next of kin. Palliat Support Care. 2010;8(1):75–82.
163. Parent, A, Cicchetti, F. The current model of basal ganglia organization under scrutiny. Mov Disord. 1998;13(2):199.
164. Paty, D, Willoughby, E, Whitaker, J. Assessing the outcome of experimental therapies in multiple sclerosis patients. In: Rudick RA, Goodkin DE, eds. Treatment of multiple sclerosis: trial design, results, and future perspectives. London, UK: Springer-Verlag, 1992.
165. Penney, JB, Young, AB. Huntington’s disease. In Jankovic J, Tolosa E, eds.: Parkinson’s disease and movement disorders, ed 3, Baltimore, Md: Williams & Wilkins, 1998.
166. Phillips, DH. Living with Huntington’s disease. Madison, Wisc: University of Wisconsin Press; 1982.
167. Phillips, JG, Stelmach, GE. Parkinson’s disease and other involuntary movement disorders of the basal ganglia. In: Fredericks CM, Saladin LK, eds. Pathophysiology of the motor systems. Philadelphia, Pa: Davis, 1996.
168. Pinhas-Hamiel, O, Sarova-Pinhas, I, Achiron, A. Multiple sclerosis in childhood and adolescence: clinical features and management. Paediatr Drugs. 2001;3(5):329.
169. Pittock, SJ, Mayr, WT, McClelland, RL, et al. Quality of life is favorable for most patients with multiple sclerosis: a population-based cohort study. Arch Neurol. 2004;61(5):679–686.
170. Poewe, W, Wenning, G. Levodopa in Parkinson’s disease: mechanisms of action and pathophysiology of late failure. In Jankovic J, Tolosa E, eds.: Parkinson’s disease and movement disorders, ed 3, Baltimore, Md: Williams & Wilkins, 1998.
171. Poirier, J. Apolipoprotein E in animal models of CNS injury and in Alzheimer’s disease. Trends Neurosci. 1994;17(12):525.
172. Poirier, J, Delisle, MC, Quirion, R, et al. Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer’s disease. Proc Natl Acad Sci U S A. 1995;92(26):12260–12264.
173. Politis, M. Dyskinesias after neural transplantation in Parkinson’s disease: what do we know and what is next? BMC Med. 2010;8:80.
174. Pollak, P. Parkinson’s disease and related movement disorders. In: Bogousslasky J, Fisher M, eds. Textbook of neurology. Boston, Mass: Butterworth Heinemann, 1998.
175. Prochazka, A, Bennett, DJ, Stephens, MJ, et al. Measurement of rigidity in Parkinson’s disease. Mov Disord. 1997;12(1):24–32.
176. Quinn, N, Brown, R, Craufurd, D, et al. Core assessment program for intracerebral transplantation in Huntington’s disease (CAPIT-HD). Mov Disord. 1996;11(2):143–150.
177. Raaphorst, J, de Visser, M, Linssen, WH, et al. The cognitive profile of amyotrophic lateral sclerosis: a meta-analysis. Amyotroph Lateral Scler. 2010;11(1-2):27–37.
178. Rabins, P, Cummings, J. Introduction. Am J Geriatr Psychiatry. 1998;6(2):S1.
179. Ranen, NG, Peyser, CE, Coyle, JT, et al. A controlled trial of idebenone in Huntington’s disease. Mov Disord. 1996;11(5):549–554.
180. Reddy, PH, Williams, M, Tagle, DA. Recent advances in understanding the pathogenesis of Huntington’s disease. Trends Neurosci. 1999;22(6):248.
181. Reifler, B. Detection and treatment of mixed cognitive and affective symptoms in the elderly: is it dementia, depression or both? Clin Geriatr. 1998;6:17.
182. Reisberg, B, Ferris, S, Anand, R. Functional staging of dementia of the Alzheimer’s type. Ann N Y Acad Sci. 1984;435:481.
183. Reisberg, B, Ferris, SH, de Leon, MJ, Crook, T. The Global Deterioration Scale for assessment of primary degenerative dementia. Am J Psychiatry. 1982;139(9):1136–1139.
184. Reitz, C, Brayne, C, Mayeux, R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137–152.
185. Richards, M, Marder, K, Cote, L, Mayeux, R. Interrater reliability of the Unified Parkinson’s Disease Rating Scale Motor Examination. Mov Disord. 1994;9(1):89–91.
186. de Rijk, MC, Breteler, MM, Graveland, GA, et al. Prevalence of Parkinson’s disease in the elderly: the Rotterdam study. Neurology. 1995;45(12):2143–2146.
187. Robberecht, W, Brown, RH. Etiology and pathogenesis of ALS: biochemical, genetic, and other theories. In: Belsh JM, Schiffman PL, eds. ALS diagnosis and management for the clinician. Armonk, NY: Futura, 1996.
188. Robinson, B. Validation of caregiver strain index. J Gerontol. 1983;38(3):99.
189. Roger, KS, Medved, MI. Living with Parkinson’s disease—managing identity together. Int J Qualitative Stud Health Well-being. 2010;5:5129.
190. Rosin, R, Topka, H, Dichgans, J. Gait initiation in Parkinson’s disease. Mov Disord. 1997;12(5):682.
191. Sage, JI, Duvoisin, RC. The modern management of Parkinson’s disease. In: Chokroverty S, ed. Movement disorders. New Brunswick, NJ: PMA Publishing, 1990.
192. Sano, M, Ernesto, C, Thomas, RG, et al. A controlled trial of selegiline, alpha-tocopherol or both as treatment for Alzheimer’s disease. N Engl J Med. 1997;336(17):1216–1222.
193. Santacruz, KS, Swagerty, D. Early diagnosis of dementia. Am Fam Physician. 2001;63(4):703–714.
194. Schneider, LS. Cholinergic deficiency in Alzheimer’s disease: pathogenic model. Am J Geriatr Psychiatry. 1998;6(2):S49.
195. Schwartz, CE, Coulthard-Morris, L, Zeng, Q, Retzlaff, P. Measuring self-efficacy in people with multiple sclerosis: a validation study. Arch Phys Med Rehabil. 1996;77(4):394–398.
196. Sclan, S, Reisberg, B. Functional assessment staging (FAST) in Alzheimer’s disease: reliability, validity, and ordinality. Int Psychogeriatr. 1992;4(Suppl 1):55.
197. Seshadri, S, Drachman, D, Lippa, C. Apolipoprotein E epsilon 4 allele and lifetime risk of Alzheimer’s disease: what physicians know and what they should know. Arch Neurol. 1995;52(11):1074.
198. Shamberg, S, Shamberg, A. Blueprints for independence. OT Week. 1996:24. [June].
199. Sherrington, R, Rogaev, EI, Liang, Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760.
200. Shoulson, I, Fahn, S. Huntington’s disease: clinical care and evaluation. Neurology. 1979;29(1):1.
201. Shulman, JM, DeJager, PL, Feany, MB. Parkinson’s disease: genetics and pathogenesis. Annu Rev Pathol. 2011;6:193–222.
202. Sibley, W. Therapeutic claims in multiple sclerosis: a guide to treatments, ed 4. New York, NY: Demos Medical; 1996.
203. Siesling, S, Zwinderman, AH, van Vugt, JP, et al. A shortened version of the motor section of the Unified Huntington’s Disease Rating Scale. Mov Disord. 1997;12(2):229.
204. Silani, V, Fogh, I, Ratti, A, et al. Stem cells in the treatment of amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:173–181.
205. Small, GW, Rabins, PV, Barry, PP, et al. Diagnosis and treatment of Alzheimer’s disease and related disorders. Consensus statement of the American Association of Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA. 1997;278(16):1363–1371.
206. Sperling, RA, Guttmann, CR, Hohol, MJ, et al. Regional magnetic resonance imaging lesion burden and cognitive function in multiple sclerosis: a longitudinal study. Arch Neurol. 2001;58(1):115–121.
207. Stern, G, Lees, A. Parkinson’s disease. Oxford, UK: Oxford University Press; 1990.
208. Stern, Y, Hesdorffer, D, Sano, M, Mayeux, R. Measurement and prediction of functional capacity in Alzheimer’s disease. Neurology. 1990;40(1):8–14.
209. Storandt, M, Mintun, MA, Head, D, Morris, JC. Cognitive decline and brain volume loss as signatures of cerebral amyloid-beta peptide deposition identified with Pittsburgh compound B: cognitive decline associated with Abeta deposition. Arch Neurol. 2009;66(12):1476–1481.
210. National Institute of Neurological Disorders and Stroke. Dementia: hope through research. Available at http://www.ninds.nih.gov/disorders/dementias/detail_dementia.htm.
211. Teri, L, Logsdon, R. Identifying pleasant activities for Alzheimer’s disease patients: the pleasant events schedule. Gerontologist. 1990;31:124.
212. Thaut, MH, McIntosh, GC, Rice, RR, et al. Rhythmic auditory stimulation in gait training for Parkinson’s disease patients. Mov Disord. 1996;11(2):193–200.
213. Thomas, T, Banwell, B. Multiple sclerosis in children. Semin Neurol. 2008;28(1):70.
214. Tison, F, Barberger-Gateau, P, Dubroca, B, et al. Dependency in Parkinson’s disease: a population-based survey in nondemented elderly subjects. Mov Disord. 1997;12(6):910–915.
215. Trace, S, Howell, T. Occupational therapy in geriatric mental health. Am J Occup Ther. 1991;45(9):833.
216. Trejo, A, Boll, MC, Alonso, ME, et al. Use of oral nutritional supplements in patients with Huntington’s disease. Nutrition. 2005;21:889–894.
217. Turner, MR, Parton, MJ, Leigh, PN. Clinical trials in ALS: an overview. Semin Neurol. 2001;21(2):167.
218. Uhlmann, R, Larson, E, Koepsell, T. Visual impairment and cognitive dysfunction in Alzheimer’s disease. J Gen Intern Med. 1991;6(2):126.
219. Uhlmann, R, Larson, E, Rees, T. Relationship of hearing impairment to dementia and cognitive dysfunction in older adults. JAMA. 1989;261(13):1916.
220. Unverzagt, FW, Gao, S, Baiyewu, O, et al. Prevalence of cognitive impairment: data from the Indianapolis Study of Health and Aging. Neurology. 2001;57(9):1655–1662.
221. Walker, FO. Huntington’s disease. Lancet. 2007;369:218–228.
222. Weinshenker, BG, Issa, M, Baskerville, J. Long-term and short-term outcome of multiple sclerosis. Arch Neurol. 1996;53(4):353.
223. White, LJ, McCoy, SC, Castellano, V, et al. Resistance training improves strength and functional capacity in persons with multiple sclerosis. Mult Scler. 2004;10(6):668–674.
224. Wiederholt, W, Parkinson’s disease and other movement disorders. Neurology for non-neurologists, ed 4, Philadelphia, Pa, WB Saunders, 2000.
225. Wilson, RS, Aggarwal, NT, Barnes, LL, et al. Cognitive decline in incident Alzheimer’s disease in a community population. Neurology. 2010;74(12):951–955.
226. Wilson, RS, Arnold, SE, Schneider, JA, et al. Olfactory impairment in presymptomatic Alzheimers disease. Ann N Y Acad Sci. 2009;1170:730–735.
227. Yeo G, ed. Background. Washington, DC: Taylor & Francis, 1996.
228. Yesavage, JA, Brink, TL, Rose, TL, et al. Development and validation of a geriatric depression scale: a preliminary report. J Psychiatr Res. 1982-1983;17(1):37.
229. Zarit, S, Orr, N, Zarit, J. The hidden victims of Alzheimer’s disease. New York, NY: New York University Press; 1985.
230. Zhang, Z, Roman, GC. Worldwide occurrence of Parkinson’s disease: an updated review. Neuroepidemiology. 1993;12(4):195–208.
Bello-Haas, VD, Kloos, AD, Mitsumoto, H. Physical therapy for a patient through six stages of amyotrophic lateral sclerosis. Phys Ther. 1998;78(12):1312.
Borasio, GD, Votz, R, Miller, RG. Palliative care in amyotrophic lateral sclerosis. Neurol Clin. 2001;19(4):829–847.
Frankel, D. Multiple sclerosis. In Umphred DA, ed.: Neurological rehabilitation, ed 3, St. Louis, Mo: Mosby, 1995.
Kraft, GH, Freal, JE, Coryell, JK. Disability, disease duration, and rehabilitation service needs in multiple sclerosis: patient perspectives. Arch Phys Med Rehabil. 1986;67(3):353.
LaBan, MM, Martin, T, Pechur, J, et al. Physical and occupational therapy in the treatment of patients with multiple sclerosis. Phys Med Rehabil Clin North Am. 1998;9(3):603.
Lechtzin, N, Rothstein, J, Clawson, L, et al. Amyotrophic lateral sclerosis: evaluation and treatment of respiratory impairment. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:5.
Newman, EM, Echevarria, ME, Digman, G. Degenerative diseases. In Trombly CA, ed.: Occupational therapy for physical dysfunction, ed 4, Boston, Mass: Williams & Wilkins, 1995.
Pulaski, KH. Adult neurological dysfunction. In Neistadt ME, Crepeau EB, eds.: Willard & Spackman’s occupational therapy, ed 9, Philadelphia, Pa: Lippincott, 1998.
Struifbergen, AK, Rogers, S. Health promotion: an essential component of rehabilitation for persons with chronic disabling conditions. Adv Nurs Sci. 1997;19(4):1.
Verma, A, Bradley, WG. Atypical motor neuron disease and related motor syndromes. Semin Neurol. 2001;21(2):177.
Walling, AD. Amyotrophic lateral sclerosis: Lou Gehrig’s disease. Am Fam Physician. 1999;59(6):1489.
Amyotrophic Lateral Sclerosis Association
Brunel University, Centre for the Study of Health and Illness
www.brunel.ac.uk/about/acad/sssl/ssslresearch/centres/cshi/.
International Journal of MS Care