Chapter 21 Connective tissue disorders
KEY POINTS
 Connective tissue disorders are uncommon conditions compared to other rheumatic diseases such as rheumatoid arthritis, osteoarthritis and fibromyalgia but just as debilitating. Fatigue is a universal symptom of all of the disorders and thus patients could benefit from instruction in energy conservation. Range of motion and strengthening exercises are appropriate for all patients; however, extreme care should be taken during periods of flare as in myositis and dermatomyositis. During a flare, rest and active-assisted to active exercises are indicated. As symptoms subside, gentle strengthening may be incorporated into the programme.
Connective tissue disorders are uncommon conditions compared to other rheumatic diseases such as rheumatoid arthritis, osteoarthritis and fibromyalgia but just as debilitating. Fatigue is a universal symptom of all of the disorders and thus patients could benefit from instruction in energy conservation. Range of motion and strengthening exercises are appropriate for all patients; however, extreme care should be taken during periods of flare as in myositis and dermatomyositis. During a flare, rest and active-assisted to active exercises are indicated. As symptoms subside, gentle strengthening may be incorporated into the programme.INTRODUCTION
This chapter will provide an overview of the connective tissue disorders including scleroderma, systemic lupus erythematosus, dermatomyositis, polymyositis, and mixed connective tissue disease. These diseases are characterized by the presence of spontaneous overactivity of the immune system which results in the production of extra antibodies into the circulation. Each of these diseases has a ‘classic’ presentation with typical findings and can evolve slowly or rapidly from very subtle abnormalities before demonstrating the classic features which help in the diagnosis.
SCLERODERMA
Systemic sclerosis (SSc) or scleroderma is a connective tissue disease characterized by thickening of the skin, fibrosis, and vascular and internal organ involvement. Scleroderma literally means ‘thick skin’.
PREVALENCE AND INCIDENCE
The prevalence of scleroderma is estimated to be 300,000 in the USA; 4000 to 5000 new cases are diagnosed each year. The disease is four times more common in women than men and the average age of onset is between the third and fifth decade of life (Silman 1997). Scleroderma affects all racial groups but the onset of scleroderma is more likely to occur at a younger age in African American women who are more likely to develop diffuse disease, and have a poorer age-adjusted survival rate (Greidinger et al 1998, Laing et al 1997). In the UK, the prevalence has been reported to range from 3.08 per 100,000 (West Midlands area) (Silman et al 1988) to 8.8/100,000 in Northeast England (Allock et al 2004) to 14.6 per 100,000 in south and west London (Silman et al 1990).
AETIOLOGY, PATHOLOGY, IMMUNOLOGY
The cause of scleroderma is not known. Some precipitating event causes cells to start making collagen as if some injury has occurred. However, once the production has begun, the cells do not turn off. The excess collagen interferes with functioning of the skin, lungs, heart, muscles and gastrointestinal tract as well as other organs. Factors that are related to higher rates of scleroderma include ethnicity, geography, gender, and age.
DIAGNOSIS, DIFFERENTIAL DIAGNOSIS, SPECIAL TESTS
Many of the symptoms of scleroderma are similar to other diseases and there are no definitive blood tests that confirm a diagnosis of scleroderma. However, serum anti-topoisomerase (SCL-70) is present in 30% of people with diffuse scleroderma and anticentromere antibody (ACA) is present in 70–80% of people with limited cutaneous scleroderma (Medsger 2004). Other special tests that may be indicated for those with scleroderma are pulmonary function tests, echocardiograms and visualization of nailfold capillaries by microscope.
CLINICAL PRESENTATION, CLINICAL FEATURES, CLINICAL SUBSETS
The two main forms of scleroderma are localized scleroderma and systemic scleroderma (Box 21.1). In localized scleroderma, the skins changes are confined to a specific area of the skin and the internal organ systems are not involved (Medsger 2004). In systemic scleroderma, the internal organ systems are involved and the skin involvement is less localized. There are two subsets of systemic scleroderma: diffuse scleroderma and limited cutaneous scleroderma.
BOX 21.1 Clinical subsets of scleroderma
Systemic scleroderma
Limited cutaneous scleroderma
 Other early symptoms include Raynaud’s phenomenon, puffiness in the fingers and heartburn (Medsger 2004).
Other early symptoms include Raynaud’s phenomenon, puffiness in the fingers and heartburn (Medsger 2004).
SYMPTOMS AND SIGNS
Common symptoms of scleroderma include Raynaud’s phenomenon (Fig. 21.1), skin thickening, and involvement of the musculoskeletal, pulmonary, gastrointestinal, cardiac and renal systems. Musculoskeletal involvement, such as non inflammatory arthralgias and myalgias, can be an early symptom in people with scleroderma (Blocka 2004). Skin tightening and fibrosis can lead to contractures in the hand (Entin & Wilkinson 1973). The most common contractures are a loss of flexion of the metacarpal phalangeal joints, loss of extension of the proximal interphalangeal joints and a loss of thumb abduction. Mandibular resorption is also a common symptom as well as thickening of the periodontal membrane. These oral changes may lead to microstomia (Fig. 21.2) and dental problems.
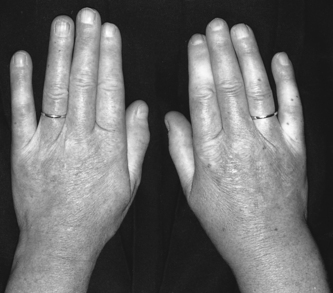
Figure 21.1 Raynaud’s phenomenon in a patient with scleroderma (Note: white discoloration and telangectasia).
With permission from Al-Alluf AW, Belch JF 2003 Ch. 136 Raynaud’s Phenomenon. In: Hochberg MC et al (eds) Rheumatology (3rd edn), Elsevier, London, Fig. 136.1 p1509.
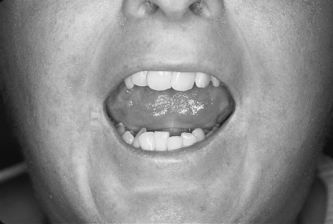
Figure 21.2 Microstomia in sclerdoderma (Note the taut smooth skin and reduced oral aperture).
With permission from: Wigley FM, Hummers LK 2003 Ch. 133 Clinical features of systemic sclerosis. In: Hochberg MC et al (eds.) Rheumatology (3rd edn), Elsevier, London, Fig. 133.12 p1469.
Symptoms of lung involvement in scleroderma are shortness of breath and coughing. Some persons develop pulmonary fibrosis of the lungs, that in the later stages, may cause death (White 2004). Initial gastrointestinal symptoms include gastroesophegeal reflux disease (GERD) which consists of difficulty swallowing, nausea, vomiting and bloating after meals (Weinstein & Kadell 2004). Symptoms progress from the upper to the lower GI tract with symptoms of diarrhoea and/or constipation. Cardiac problems are more subtle and symptoms may not occur until later in the disease (Follansbee & Marroquin 2004). Later cardiac involvement may consist of arrythmias, heart failure and pericarditis. Many people with scleroderma also have kidney involvement. However, the most important clinical manifestation is an accelerated hypertension with resultant decrease or lack of urine output precipitating a scleroderma renal crisis (Steen 2004, Wollheim 2004).
DISEASE ACTIVITY, PROGRESSION, PROGNOSIS
The course of scleroderma is variable and prognosis dependent on the subtype of scleroderma and timing, site and degree of internal organ involvement (Medsger 2004). With early diagnosis and the introduction of new medications such as ACE inhibitors, some of the complications from scleroderma, such as renal crisis, have been decreased.
CLINICAL EVALUATION/ASSESSMENT/EXAMINATION – SUBJECTIVE AND OBJECTIVE
A clinical evaluation must consist of range of motion, hand function, pain, fatigue, and ability to perform daily activities (ADL), including basic ADL, work and leisure activities. In addition, the patient’s hand should be inspected for digital ulcers, calcium deposits, extent of skin thickness, and Raynaud’s phenomenon. Table 21.1 describes assessments that have been shown to be reliable and valid with persons with scleroderma.
Table 21.1 Assessments specific to scleroderma
| ASSESSMENT | WHAT IS MEASURED? | TYPE OF ASSESSMENT |
|---|---|---|
| Health Assessment Questionnaire (Fries et al 1980, Poole & Steen 1991) | Ability to perform daily tasks | Self-report |
| United Kingdom Scleroderma Questionnaire (Poole & Brower 2004, Silman et al 1998) | Ability to perform daily tasks | Self-report |
| The Hand Mobility in Scleroderma Test (Sandqvist & Eklund 2000a, 2000b) | Functional joint motion test for the fingers and wrists and forearm | Performance test |
| The Arthritis Hand Function Test (Backman & Mackie 1997, Poole et al 2000) | Assesses hand strength (grip and pinch), dexterity, applied dexterity and applied strength. Normative data provided. Some training and equipment are required | Performance test |
AIMS AND PRINCIPLES OF MANAGEMENT
Exercise and splinting
Range of motion exercises should be started before there is any observed loss of motion. The exercises should be performed frequently and aggressively and patients are encouraged to maintain a position of stretch. Specific exercises for the hand and face can be found in Poole (2004). In general the exercises should emphasize flexion of the metacarpophalangeal joint, extension of the proximal interphalangeal joints, and flexion and abduction of the thumb. Stretching exercises for the hand were shown to increase motion and subsequent function in daily tasks (Mugii et al 2006) and several studies have found stretching exercises for the mouth increase oral aperture and ease of oral hygiene (Naylor & Douglass 1984, Poole, Cante & Brewer, et al in press). Hand splints to increase joint motion must be used very carefully as dynamic splints were shown to exacerbate Raynaud’s (Seeger & Furst 1987). Static splints may be useful if clients have inflammation but should only be worn at night until the inflammation decreases.
Those patients with weakness can benefit from strengthening and conditioning programmes monitoring blood pressure and other vital signs. Warm water swimming programmes can be especially helpful. The only concern has been that chlorine in the water may act as a drying agent for skin. To minimize this, it is recommended to shower after being in the water, rinse off, and rub the skin with moisturizing skin creams. There is scant information regarding the effectiveness of conditioning exercises in people with scleroderma but because of limitations in physical capacity and decreased lung functioning, general conditioning exercise programs are recommended in moderation with periodic rests.
Energy conservation
People with scleroderma should be instructed in energy conservation due to the fatigue associated with pulmonary and cardiac involvement. The general principles of pacing, planning, prioritizing and positioning are appropriate for persons with scleroderma (see Ch. 10).
Modalities
Heat modalities, such as paraffin, in conjunction with exercise programmes, have been shown to be effective in increasing or maintaining joint motion and hand function (Mancuso & Poole 2009, Pils et al 1991, Sandqvist et al 2004) (see Ch. 8).
Assistive/adapted devices
Persons with scleroderma have been shown to have difficulty performing daily tasks and may benefit from devices to compensate for decreased manipulation (built up handles, button hooks, electric can openers), reach (long handled equipment, reachers), and weakness (raised toilet seats, grab bars) (Poole 2004). Patients and families/caregivers also need education about devices, preventative techniques for Raynaud’s, fatigue and wound care. Splints may help protect digital ulcers.
Some people diagnosed with scleroderma have psychosocial impairments due to uncertainties regarding disease progression and ability to work, disfigurement, and impact on their families. Malcarne (2004) provides some suggestions ranging from support groups, patient self-management programmes, individual/family therapy and/or pharmacotherapy. Although there are few studies examining the effectiveness of self-management programmes for people with scleroderma, two studies found that these provided practical information to manage activities, stress, fatigue and improved sense of control (Brown et al 2004, Samuelson & Ahlmen 2000).
SYSTEMIC LUPUS ERYTHEMATOSUS
Systemic lupus erythematosus (SLE) is a multi-system, highly inflammatory autoimmune disease. The disease can be mild or life threatening. The course is unpredictable in that different systems can be affected at different times.
PREVALENCE AND INCIDENCE
The incidence of SLE varies between and within countries. In the United States is estimated to be 9.4 per 100,000) in women and 1.54 per 100,000 in men. In the UK, incidence is 7.89 per 100,000 in women and 1.53 per 100,000 in men. SLE is more common in women with a ratio of 9:1 in the US and 5.2:1 in the UK (Petri, 2006, Somers et al 2007). Disease onset usually occurs between the ages of 20–30 years of age but may occur at any other point across the lifespan as well. In the USA, SLE is more common in African Americans and Hispanics than in Caucasians.
AETIOLOGY, PATHOLOGY, IMMUNOLOGY
The cause of SLE is not known. However, genetic factors and environmental factors such as ultraviolet light, medications, smoking, infections and toxin exposure, have been implicated (Petri 2006).
DIAGNOSIS, DIFFERENTIAL DIAGNOSIS, SPECIAL TESTS
Although the majority of persons with SLE have a positive antinuclear antibody (ANA) test, a positive test is not sufficient for diagnosis (Petri 2006) as this is seen in other diseases and can be caused by medications. Therefore, other criteria such as organ involvement and other immunological tests help establish a diagnosis of SLE. To be classified as having SLE, four or more of the 11 symptoms in Box 21.2 must be present (Tan et al 1982). These are intended as guidelines not diagnostic criteria.
BOX 21.2 Guidelines for classification of systemic lupus erythematosus
Adapted from Tan et al (1982)
To be classified as systemic lupus erythematosus, four or more of the following 11 symptoms must be present:
CLINICAL PRESENTATION, CLINICAL FEATURES, SIGNS AND SYMPTOMS
The clinical presentation and features of SLE are listed above under Classification and in Box 21.2. The major symptoms are the facial rash over the cheeks and bridge of the nose (i.e. the malar or ‘butterfly rash’; Fig. 21.3), photosensitivity and ulcers in the nose and mouth. The major organ system manifestations are musculoskeletal, renal, neuropsychiatric, serous, gastrointestinal, pulmonary and cardiac (Buyon 2008). Musculoskeletal manifestions may consist of arthralgias, arthritis and muscle weakness. Some patients develop ulnar deviation and swan neck deformities of the fingers, called ‘Jaccoud’s arthopathy’ (Fig. 21.4) (Petri 2006). Clinical features of renal involvement may not be noticed until there is advanced kidney disease. A common neuropsychiatric manifestation is headache. Less common are seizures, strokes, cranial and peripheral neuropathy, organic brain syndrome and psychosis. Studies of neurocognitive function report that more than 80% have active or inactive neuropsychological involvement (Denburg et al 1993). Serositis in SLE may present as pleurisy, pericarditis or peritonitis. Gastrointestinal manifestations are common and include abdominal pain, anorexia, and nausea. Pulmonary manifestions include pneumonitis, pulmonary haemorrhage, pulmonary embolism, and pulmonary hypertension. Cardiac manifestations include pericarditis, myocarditis, endocarditis or coronary artery disease.
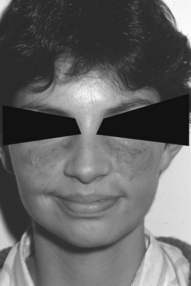
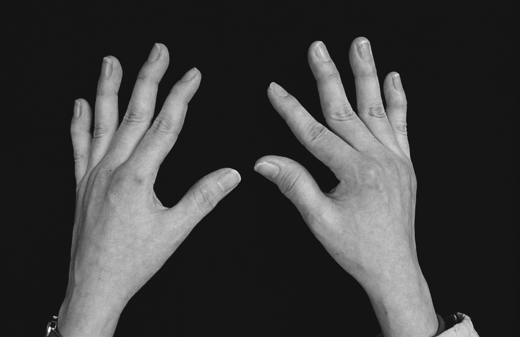
DISEASE ACTIVITY, PROGRESSION, PROGNOSIS
The progression of SLE is highly variable and depends on the organs involved. Early detection and aggressive intervention before major organ damage occurs increases life expectancy (Hahn 2001).
CLINICAL EVALUATION, ASSESSMENT AND EXAMINATION – SUBJECTIVE AND OBJECTIVE
A clinical evaluation must consist of range of motion, pain, fatigue, stress, cognitive function, and ability to perform daily activities, including basic ADL, work and leisure activities. There are some specific assessments for people with SLE but the majority of these are measures of organ damage (Ramsey-Goldman & Isenberg 2003). The Fatigue Severity Scale (Krupp et al 1989) was developed especially to measure fatigue in persons with SLE and the Health Assessment Questionnaire (Fries et al 1980, Milligan et al 1993) has been widely used to measure disability in persons with SLE.
AIMS AND PRINCIPLES OF MANAGEMENT
The management of SLE is challenging due to the involvement of multiple systems and unpredictable nature of the disease. Patient education is crucial as well as empowering the person with SLE to ensure open communication between the patient, family and health professionals.
Exercise
Before starting an exercise programme, people with SLE should have their physician’s approval. In clinically stable patients with mild to moderate disease activity, conditioning exercises of high to moderate intensity have been shown to be effective in increasing aerobic capacity without exacerbating symptoms (Carvalho et al 2005, Clarke-Jenssen et al 2005, Daltroy et al 1995, Ramsey-Goldman et al 2000, Robb-Nicholson et al 1989, Strombeck & Jacobsson 2007, Tench et al 2003). Heart rate and blood pressure should be monitored. Petri (2006) provided the following considerations for rehabilitation. For patients with positive antiphospholipid antibody or cardiac history, patients should be monitored for deep vein thrombosis. For patients with avascular necrosis, loading weight bearing joints should be avoided; aquatic exercises would be indicated. In persons with muscle involvement, use eccentric exercise with caution (Petri 2006).
Energy conservation
Fatigue is a common debilitating complaint in persons with SLE. A healthy diet, exercise, rest and managing stress may lessen the fatigue. In addition, incorporating energy conservation techniques into daily life can also help manage fatigue. Patients can identify activities that cause fatigue and create a plan to complete activities and pace more fatiguing ones throughout a day or week. Energy conservation is also important when patients have cognitive impairments. When cognitive impairment is present, patients should plan their days so that they perform more cognitively challenging tasks earlier in the day when they are more fresh and alert. Using memory devices such as schedule books or palm pilots can also help to compensate for memory impairments. Several studies have shown that aerobic conditioning exercise programs reduce fatigue (Robb-Nicholson et al 1989, Tench et al 2003). Self-management programmes that teach coping and techniques to manage fatigue have resulted in decrease fatigue (Sohng 2003).
Modalities
For patients who have arthralgias, heat modalities such as hot packs and paraffin wax treatments may be helpful. Those with ulnar deviation and swan neck deformities of the fingers (‘Jaccoud’s arthopathy’) should be taught joint protection. Entrapment of the median nerve (carpel tunnel) occurs in some patients. In this case, wrist splints may help alleviate symptoms.
Assistive/adapted devices
Assistive devices to compensate for reach (long handled equipment, reachers), and weakness (raised toilet seats, grab bars) may be indicated. Electrical appliances such as dishwashers, food choppers and processors, microwaves and crockpots (slow cookers) may help save energy.
DERMATOMYOSITIS
Dermatomyositis is classified as an idiopathic inflammatory myopathy. It is clearly related to polymyositis (see later). It is characterized by chronic muscle inflammation and muscle weakness. The accompanying rash looks patchy. There are bluish-purple or red discolorations, classically on the eyelids) and over muscles which extend joints.
PREVALENCE AND INCIDENCE
Because it is rare and does not have a universally accepted diagnostic criteria, it is difficult to estimate its prevalence. A rough estimate would be between 5.5 cases per million individuals. The incidence seems to be increasing, but this may be reflective of increased awareness of its presence (Callen 2009).
AETIOLOGY, PATHOLOGY, IMMUNOLOGY
The pathology is similar to polymyositis. It shows a perivascular infiltration of inflammatory cells composed of higher percentages of B lymphocytes and CD4+ T-helper lymphocytes. Biopsies reveal perifascicular atrophy. This may in fact be a diagnostic criteria. About 10% of all cases have no evidence of muscle disease. Fatigue may be a dominant complaint and testing by magnetic resonance spectroscopy shows abnormal muscle energy metabolism and altered exercise capacities when energy containing compounds (ATP) are examined. There may be an increased prevalence of neoplasia associated with this presentation of the disease.
DIAGNOSIS, DIFFERENTIAL DIAGNOSIS, SPECIAL TESTS
Three of four of the following criteria plus the rash must be present to be definitely diagnosed as dermatomyositis.
CLINICAL PRESENTATION, CLINICAL FEATURES, CLINICAL SUBSETS
In adult dermatomyositis, rashes may precede the muscle weakness by a year or so. Skin involvement varies widely from person to person. This tends to be symmetrical and classically includes the heliotrope (lilac) discoloration on the eyelids (Fig. 21.5) with oedema and macular erythema of the back of the shoulders and neck (shawl sign). Other dermatologic features include: scaly skin patches over the knees, elbows, and medial malleoli; face, neck and upper torso erythematosus; and dermatitis over the dorsum of the hands, especially the MCP and PIP joints (Fig. 21.6). Symptoms may include telangectasias periungually and nail-fold capillary changes similar to those in scleroderma or systemic lupus erythematosus. Raynaud’s phenomenon may also be seen. Hand deformities may also occur (Fig. 21.7A,B).
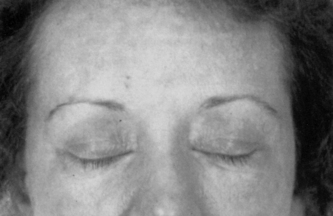
Figure 21.5 Heliotrope rash of dermatomyositis (Note: the rash over the eyelids is a characteristic feature).
With permission from: Oddis CV, Medsger TA (2003) Ch.139 Inflammatory muscle disease: clinical features. In: Hochberg MC et al (eds.) Rheumatology (3rd edn), Elsevier, London. Fig. 139.3 p1540.
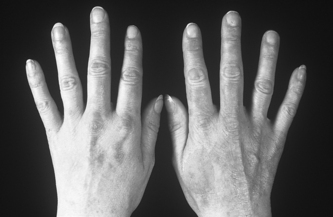
Figure 21.6 Scaling rash over the knuckles and dorsum of the hand in dermatomyositis.
With permission from: Oddis CV, Medsger TA (2003) Ch.139 Inflammatory muscle disease: clinical features. In: Hochberg MC et al (eds.) Rheumatology (3rd edn), Elsevier, London, Fig. 139.1 p1540.
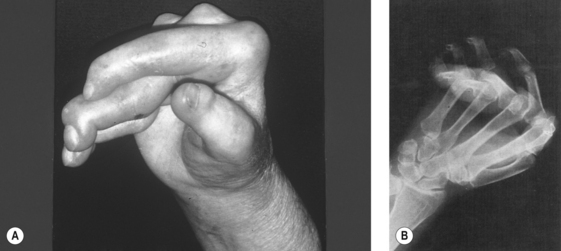
Figure 21.7 (A), (B) Deforming arthropathy of polymyositis. (Note: the rheumatoid-like deformities. The radiograph shows numerous subluxations but minimal erosive changes).
With permission from Oddis CV, Medsger TA (2003) Ch.139 Inflammatory muscle disease: clinical features. In: Hochberg MC et al (eds.) Rheumatology (3rd edn), Elsevier, London, Fig. 139.7 p1542.
Many adults also experience low-grade fevers, have inflamed lungs, and may be sensitive to light (National Institute of Neurological Disorders and Stroke 2009). Juvenile cases generally show muscle inflammatory processes first. These include vasculitis, ectopic calcification, lipodystrophy, and muscle weakness. There is great variety from patient to patient.
SYMPTOMS AND SIGNS
As stated above, the rash and muscle weakness are generally indicative that a problem exists. Calcinosis often occurs 1–3 years after the beginning of the disease. The deposits are more often seen in children than adults. They appear as hard bumps under the skin or in the muscle.
DISEASE ACTIVITY, PROGRESSION, PROGNOSIS
Disease activity is varied in both adults and juveniles. In some cases remission is complete with little or no therapy. Most cases do respond to therapies. When accompanied with vasculitis the disease may be devastating despite therapies. It is also generally more severe and therapy resistant for individuals with cardiac or respiratory problems.
CLINICAL EVALUATION, ASSESSMENT AND EXAMINATION – SUBJECTIVE AND OBJECTIVE
Physical evaluation must include range of motion, strength as well as assessment of functional activities. Patient complaints and information are important to guide development of therapeutic goals. Knowledge of muscle enzyme activity is imperative to drive any therapeutic intervention. During periods of severe inflammation bed rest may be required with passive range of motion to preserve joint integrity.
AIMS AND PRINCIPLES OF MANAGEMENT
Exercise
This must be targeted at information gained during evaluation and to the patient’s disease activity. When appropriate, patients can progress to active and gentle resisted exercises targeted at maintaining or improving function. Assistive devices for gait may be helpful and require proper fitting and instruction. Patient and family education must include instructions for all levels of the disease process. They need to understand how to modify activity levels appropriately accounting for disease activity at the time. For instance, resisted activities should not be included during periods of active muscle inflammation.
Energy conservation
Instruction is imperative for these patients. It is important to decrease activity in times of flare. Use of assistive devices for ADL will allow best use of muscle strength and energy.
Modalities
Gentle use of mild heat is helpful for painful muscles with consideration to the skin in affected areas.
Medical management
Corticosteroids are the standard first-line medication. Initially prednisolone is generally given daily, but may be switched to IV methylprednisolone if needed. Improvements may be noted in the early weeks and gradually over 3 to 6 months. As many as 90% of patients improve at least partially and 50–75% achieve complete remission of symptoms. If the patient does not respond, immunosuppressant drugs such as methotrexate or azathioprine may be added by the physician if symptoms do not subside. Topical ointments may be helpful for the skin symptoms.
POLYMYOSITIS
Polymyositis is another inflammatory muscle disease and is characterized by symmetrical proximal muscle weakness. It can be accompanied by systemic symptoms like fatigue, morning stiffness, and anorexia.
PREVALENCE AND INCIDENCE
Polymyositis is uncommon, being seen in approximately eight individuals per 100,000.
AETIOLOGY, PATHOLOGY, IMMUNOLOGY
At this time, there is no known cause of polymyositis, but consideration is given to environmental factors triggering the disease in genetically susceptible individuals. The autoimmunity factor is supported by its association with other autoimmune diseases, including Hashimoto’s thyroiditis, Grave’s disease, myasthenia gravis, type I diabetes mellitus and connective tissue diseases. Also the high prevalence of circulating autoantibodies associated with polymyositis and dermatomyositis include the myositis-specific autoantibodies (MSAs) found commonly in myositis. These are nonspecific and are also found in overlap syndromes.
Genetic factors are evident in mouse models. Individuals with HLS-DR3 are at increased risk for developing inflammatory muscle diseases. Also suspicious are those carrying anti-Jo-1 antibodies, as well as HLS-138, HLA-DR3 and DR6, HLA-DR1, DR6, AND DQ1. Pathologic changes in muscle provide the strongest evidence these diseases are immune-mediated. Research findings suggest the pathology of polymyositis involves recognition of an antigen on the surface of muscle fibres by antigen-specific T cells.
DIAGNOSIS, DIFFERENTIAL DIAGNOSIS, SPECIAL TESTS
Diagnosis is initiated by the patient complaining of muscle weakness and fatigue. Laboratory investigation reveals elevated serum enzymes derived from skeletal muscle, especially creatine kinase (CK), CPK, aldolase, SGOT, SGPT, and LDH. Electromyography (EMG) demonstrates myopathic changes consistent with inflammation which is also shown in muscle histology. Muscle biopsy is used to confirm muscle inflammation. Muscles commonly used for biopsy include quadriceps, biceps, and deltoid. A pathologist examines the tissue under a microscope. No single feature is specific or diagnostic, and a variety of patterns can occur.
CLINICAL PRESENTATION, CLINICAL FEATURES AND CLINICAL SUBSETS
Weakness of proximal muscles is most commonly seen in this disease. Patients report difficulty with sit-to-stand, navigating stairs, and lifting above shoulder height. It could also include difficulty swallowing or lifting the head from the pillow. Heart and lung involvement can lead to irregular heart rhythm, heart failure and shortness of breath. It may sometimes be associated with cancers, including lymphoma, breast, lung, ovarian and colon cancer. Patients must be monitored for possible cancer occurrence.
SYMPTOMS AND SIGNS
Muscle weakness is the most common symptom, generally in muscles close to the trunk. About 25% of patients complain of fatigue, a general feeling of discomfort and weight loss and low grade fever.
DISEASE ACTIVITY, PROGRESSION AND PROGNOSIS
Onset can be gradual or rapid, with varying degrees of loss, muscle power and atrophy.
MEDICAL MANAGEMENT
Patients are treated with high doses of corticosteroids either by mouth or intravenously, in order to decrease the inflammatory process in muscles. Patients must be monitored for the many potential side effects of steroids. If management is not effective on corticosteroids alone, immunosuppressive medications may be added. These could include methotrexate, azathioprine, cyclophosphamide, chlorambucil or cyclosporine. These also have side effects that require monitoring.
CLINICAL EVALUATION, ASSESSMENT AND EXAMINATION – SUBJECTIVE AND OBJECTIVE
Evaluations by therapists include examination of medical information and physical examination including range of motion, muscle strength and physical function. Subjective information from the patient is helpful to guide treatment and appropriate goal setting. Exercise management will need to be lifelong, and patient education is paramount to successful management. Patients need to understand appropriate lifestyle adjustment depending on symptoms present at any given time.
AIMS AND PRINCIPLES OF MANAGEMENT
Exercise
Therapeutic exercises are dependent on patient’s symptoms and muscle enzymes at the time. When in flare, rest and active-assisted to active exercises are indicated. As symptoms subside, gentle strengthening may be incorporated into the programme.
MIXED CONNECTIVE TISSUE DISEASE
Mixed Connective Tissue Disease (MCTD) was first described in 1972 and is considered to be an ‘overlap’ or ‘mix’ of three specific connective tissue diseases. These are systemic lupus erythematosis (SLE), scleroderma and polymyositis. Patients with MCTD display features of the three conditions. Some patients even demonstrate features of rheumatoid arthritis.
PRESENTATION OF MCTD SYMPTOMS
Symptoms include high quantities of antinuclear antibodies (ANAs), and antibodies to ribonucleoprotein (anti-RNP) detectible by blood testing. In most cases the symptoms become dominated by features of one of the above three diseases, most commonly scleroderma.
MEDICAL MANAGEMENT
Management is similar to that for the most common diseases which are presenting. MCTD is often considered as a subset of SLE. Children with MCTD tend to have an increased incidence of Raynaud’s phenomenon and hypergammaglobulinemia, but a lower incidence of hypocomplementemia. They are less likely to develop severe nephritis or require immunosuppressive therapies. A significant number of children with MCTD develop scleroderma. As above, treatment of children with MCTD is identical to that for SLE.
THERAPEUTIC EVALUATION AND MANAGEMENT
The therapist’s evaluation of clients with MCTD must include evaluation of medical information, patient’s subjective information, and objective measurements of range of motion, strength, endurance and physical function. Treatment is designed to improve the deficits noted and includes patient education about the disease process and careful monitoring of symptoms to determine the appropriate activity level. Management will follow the advice given earlier for management of SLE, scleroderma and polymyositis.
CONCLUSION
This chapter has provided an overview of the connective tissue disorders including scleroderma, systemic lupus erythematosus, dermatomyositis, polymyositis, and mixed connective tissue disease. Although these disorders are very different, persons with these disorders could benefit from occupational and physical therapy to improve the performance of daily tasks, manage fatigue and maintain or improve joint motion and muscle strength.
Alyson is a 38-year-old white female referred to physical therapy and occupational therapy. She was diagnosed with systemic sclerosis (scleroderma). She was an insurance underwriter, married and lived out of town. Three months later, she stopped working, went on disability and returned to Pittsburgh with her husband to be closer to her family and medical care. At that time, she was disheartened about her disease and needed assistance with many activities of daily living (ADLs).
Alyson’s disease was progressing rapidly from diagnosis to evaluation in physical therapy. She also had involvement in kidneys, hypertension and problems with swallowing in addition to the tight skin over hands, face and upper extremities to the shoulder. She complained of joint stiffness which limited her functional activities mostly in the upper extremities, but also in her right hip. She has problems with sit to stand and with gait due to her hip. Her complaint as she stated was “I cannot do anything, dressing or any household chores and the itching is driving me crazy”. Management of her disease to this time has been medical. She has stopped all physical activities and has help with all ADLs.
Therapy intervention - Range of motion measurements were taken of all joints while instructing her in a programme of range of motion/stretching exercises. She had limitations of 45 degrees in all shoulder planes, elbows were −40 degrees of extension and −30 degrees of full flexion bilaterally. Both wrists flexed 70 degrees and lacked hyperextension. Supination and pronation were about half of her range and she was 1” from touching fingers to palm. She had similar degrees of lower extremity limitations, but was not as aware of these until the evaluation. She had hip flexion contractures of 30 degrees and knee contractures of 20 degrees, therefore walking forward flexed. Her posture was poor with rounded shoulders and forward head. Her general strength fell in the 3+ to 4− range in her available ranges. Her endurance was not tested, but was reported to be poor. Alyson reported her pain in the 7-9/10 range most of the time. She was emotionally frustrated and hopeless. Her family was supportive, perhaps too much so. Alyson was seen for five therapy sessions focusing on patient education to teach her a programme of heat (electric heating pad) to larger joints and paraffin wax for the hands prior to exercise to decrease pain, stiffness and skin tightness. She was then instructed in range of motion with hold at maximum end range to all joints. We suggested 15–20 second hold beginning with two to three repetitions increasing to 10. We also instructed her in a walking programme, beginning with 5 minutes two to three times daily.
Alyson was also observed performing her ADLs and it was recommended that she use a long handled sponge for bathing and to put a bathroom caddy over her showerhead to hold shampoo, conditioner, and soap. Alyson had difficulty with dressing and was encouraged to wear pull over tops and pants. She was issued a button hook to help with buttoning some of her favorite shirts. Alyson was also provided with education in energy conservation to pace herself throughout the day, delegate tasks to family members and consider using smaller appliances such as the microwave and table top (or toaster) oven in the kitchen. Although Alyson was not working, her computer set up at home was evaluated and recommendations made to lower her keyboard and to try a smaller size keyboard.
Alyson returned for a ‘tune-up’ a year later. At that time, she was still stiff, but more positive emotionally, she had increased her physical activity and reported decreased itching and improved kidney function. Her shoulder, elbow and hip mobility had increased at that time. She was also independent in dressing and bathing and doing most of her home management tasks. She was also enjoying using the computer and thinking of trying to work part time. Stretches to hip, hamstring, cervical and chest areas and fingers were increased at that time.
CASE STUDY 21.2 DERMATOMYOSITIS
Emma presented to physical therapy and occupational therapy with a prescription from her Rheumatologist for ‘strengthening exercises and ADL training’ Dx. Dermatomyositis.
Emma is 58 years old and has had complaints of fatigue, generalised muscle aches and weakness off and on for several years. Recently, she had developed a red/purple rash on her eyelids and muscles. Three months ago, her GP suggested she see a rheumatologist to better diagnose her problems.
After a series of tests, including blood tests, finally a magnetic resonance spectroscopy showed abnormal muscle metabolism and Emma was diagnosed with Dermatomyositis. Her past medical history is significant for weight gain, mild hypertension and the above rash and fatigue.
On evaluation, Emma’s proximal muscles of both upper and lower extremities scored 3/5 in shoulders and hips, but the distal muscles were in the 4/5 range. She needed to use hand assist to rise from a chair and used the rails to go up and down stairs at home. She required assistance with chores for overhead activities and had difficulty putting her dishes in the cabinets. She felt she needed a rest by mid-day and was exhausted following shopping trips. Emma demonstrated difficulty trying to stand on one leg and she was nervous walking.
Following a call to Emma’s rheumatologist to check she could perform strengthening exercises, she was instructed in range of motion exercises to all joints and isometric exercises for gluteal and quadriceps muscles. Her programme was to be done daily beginning with five repetitions and increasing slowly to ten. She was shown use of a cane for longer distances, but had already learned to use a shopping trolley (or buggy) on shopping trips. It was also recommended that Emma get a raised toilet seat and grab bars for her bathroom and a long sponge for bathing. To conserve energy and compensate for her shoulder weakness, it was recommended that her kitchen be arranged so that the more frequently used items were put on lower shelves that could be reached easily. Emma was also issued and shown how to use a dressing stick to get clothes over her shoulders, and to get her trousers over her feet and a long handed shoe horn to put on her shoes.
Emma was seen once a week for 3 weeks and once a month for two more sessions to gently increase her programme. Gradually, she was able to add resistance bands three times per week and begin a walking programme. She was cautioned many times to go slow, pace herself, use energy saving appliances, and pay attention to her fatigue level. Emma was quite pleased to transfer sit to stand without use of hands.
References and further reading
Allock R.J., Forrest I., Corris P.A., et al. A study of the prevalence of systemic sclerosis in northeast England. Rheumatology. 2004;43(5):596-602.
Backman C., Mackie H. Arthritis hand function test: test manual. Vancouver, BC: University of British Columbia; 1997.
Blocka K.L.N. Musculoskeletal involvement in systemic sclerosis. In: Clements P.J., Furst D.E., editors. Systemic sclerosis. Philadelphia: Lippincott Williams & Wilkins; 2004:249-260.
Brown S.J., Somerset M.E., McCabe C.S., et al. The impact of group education on participants’ management of their disease in lupus and scleroderma. Musculoskelal Care. 2004;2(4):207-217.
Buyon J.P. Systemic lupus erythematosus: clinical and laboratory features. In: Klippel J.H., editor. Primer on the Rheumatic Diseases. New York: Springer; 2008:303-318.
Callen, J.P., 2009. Dermatomysositis. http://emedicine.medscape.com/article/332783-overview/ (accessed 9.2.09).
Carvalho M.R., Sato E.I., Tebexreni A.S., et al. Effects of supervised cardiovascular training program on exercise tolerance, aerobic capacity, and quality of life in patients with systemic lupus erythematosus. Arthritis Rheum.. 2005;53:838-844.
Clarke-Jenssen A., Fredriksen P.M., Lilleby V., et al. Effects of supervised aerobic exercise in patients with systemic lupus erythematosus: a pilot study. Arthritis Care Res.. 2005;53(2):308-312.
Daltroy L.H., Robb-Nicholson C., Iversen M.D., et al. Effectiveness of minimally supervised home aerobic training in patients with systemic rheumatic disease. Br. J. Rheumatol.. 1995;34:1064-1069.
Denburg S.D., Denburg J.A., Carbotte R.M., et al. Cognitive deficits in systemic lupus erythematosus. Rheum. Dis. Clin. North Am.. 1993;19:815-831.
Entin M.A., Wilkinson R.D. Scleroderma hand: a reappraisal. Orthop. Clin. North Am.. 1973;4:1031-1038.
Follansbee W.P., Marroquin O.C. Cardiac involvement in systemic sclerosis. In: Clements P.J., Furst D.E., editors. Systemic sclerosis. Philadelphia: Lippincott Williams & Wilkins; 2004:195-220.
Fries J.F., Spitz P.V., Kraines R.G., et al. Measurement of patient outcome in arthritis. Arthritis Rheum.. 1980;23:137-145.
Greidinger E.L., Flaherty K.T., White B., et al. African-American race and antibodies to topoisomerase I are associated with increased severity of scleroderma lung disease. Chest. 1998;114(3):801-807.
Hahn B.H. Management of systemic lupus erythematosus. In: Ruddy S., Harris E.D., Sledge C.B., editors. Kelly’s textbook of rheumatology. 6th ed. Philadelphia: WB Saunders; 2001:1125-1143.
Krupp L.B., LaRocca N.G., Muir-Nash J., et al. The fatigue severity scale: application to patients with multiple sclerosis and systemic lupus erythematosus. Arch. Neurol.. 1989;46:1121-1123.
Laing T.J., Gillespie B.W., Toth M.B., et al. Racial differences in scleroderma among women in Michigan. Arthritis Rheum.. 1997;40:734-742.
Malcarne V.L. Psychosocial adjustment in systemic sclerosis. In: Clements P.J., Furst D.E., editors. Systemic sclerosis. Philadelphia: Lippincott Williams & Wilkins; 2004:331-350.
Mancuso T., Poole J.L. The effect of paraffin and hand exercises on hand function in persons with scleroderma. J. Hand Ther.. 2009;22(1):71-78.
Medsger T.A.Jr. Classification, prognosis. In: Clements P.J., Furst D.E., editors. Systemic sclerosis. Philadelphia: Lippincott Williams & Wilkins; 2004:17-28.
Milligan E.D., Horn D.L., Ballou S.P., et al. An assessment of the Health Assessment Questionnaire functional Ability Index among women with systemic lupus erythematosus. J. Rheumatol.. 1993;20:972-976.
Mugii N., Hasegawa M., Matsushita T., et al. The efficacy of self-administered stretching for finger joint motion in Japanese patients with systemic sclerosis. J. Rheumatol.. 2006;33(8):1586-1592.
Naylor W.P., Douglass S.W. The non-surgical treatment of microstomia in scleroderma: a pilot study. Oral Surg. Oral Med.Oral Pathol.. 1984;57(5):508-511.
National Institute of Neurological Disorders and Stroke, 2009. Dermatomyositis information page. http://www.ninds.nih.gov/disorders/dermatomyositis/dermatomyositis.htm/ (accessed 9.2.09).
Petri M.A. Systemic lupus erythematosus. In: Bartlett S.J., editor. Clinical care in the rheumatic diseases. Atlanta, GA: ACR; 2006:187-191.
Pils K., Graninger W., Sadil F. Paraffin hand bath for scleroderma. Phys. Med. Rehabil.. 1991;1:19-21.
Poole J.L. Occupational and physical therapy in systemic sclerosis: an experiential approach. In: Clements P.J., Furst D.E., editors. Systemic sclerosis. Philadelphia: Lippincott Williams & Wilkins; 2004:261-268.
Poole J.L., Steen V.D. The use of the Health Assessment Questionnaire (HAQ) to determine physical disability in systemic sclerosis. Arthritis Care Res.. 1991;4:27-31.
Poole J.L., Brower L.M. Validity of the scleroderma functional assessment questionnaire. J. Rheumatol.. 2004;31(2):402-403.
Poole J.L., Gallegos M., O’Linc S. Reliability and validity of the Arthritis Hand Function Test in adults with systemic sclerosis (scleroderma). Arthritis Care Res.. 2000;13:69-73.
Poole, J.L., Conte, C., Brewer, C., et al., In press. Oral hygiene in scleroderma: The effectiveness of a multi-disciplinary intervention program. Disabil. Rehabil.
Ramsey-Goldman R., Isenberg D.A. Systemic lupus erythematosus measures. Arthritis Care Res.. 2003;49:S225-S233.
Ramsey-Goldman R., Schilling E.M., Dunlop D., et al. A pilot study on the effects of exercise in patients with systemic lupus erythematosus. Arthritis Care Res.. 2000;13:262-269.
Robb-Nicholson C., Daltroy L., Eaton H., et al. Effects of aerobic conditioning in lupus fatigue: a pilot study. Br. J. Rheumatol.. 1989;28:500-505.
Samuelson U.K., Ahlmen M.E. Development and evaluation of a patient education programme for persons with systemic sclerosis. Arthritis Care Res.. 2000;13(3):141-148.
Sandqvist G., Akesson A., Eklund M. Evaluation of paraffin bath treatment in patients with systemic sclerosis. Disabil. Rehabil.. 2004;26:981-987.
Sandqvist G., Eklund M. Hand mobility in scleroderma (HAMIS) test: the reliability of a novel hand function test. Arthritis Care Res.. 2000;13:382-387.
Sandqvist G., Eklund M. Validity of HAMIS: a test of hand mobility in scleroderma. Arthritis Care Res.. 2000;13:382-387.
Seeger M.W., Furst D.E. Effects of splinting in the treatment of hand contractures in progressive systemic sclerosis. Am. J. Occup. Ther.. 1987;41:118-121.
Silman A.J. Scleroderma-demographics and survival. J. Rheumatol.. 1997;48:58-61.
Silman A.J., Howard Y., Hicklin A.J., et al. Geographical clustering of scleroderma in south and west London. Br. J. Rheumatol.. 1990;29(2):93-96.
Silman A., Akesson A., Newman J., et al. Assessment of functional ability in patients with scleroderma: a proposed new disability instrument. J. Rheumatol.. 1998;25:79-83.
Silman A., Jannini S., Symmons D., et al. An epidemiological study of scleroderma in the West Midlands. Br. J. Rheumatol.. 1988;27(4):286-290.
Sohng K.Y. Effects of a self-management course for patients with systemic lupus erythe-matosus. J. Adv. Nurs.. 2003;42(5):479-486.
Somers E.C., Thomas S.L., Smeeth L., et al. Incidence of systemic lupus erythematosus in the United Kingdom 1990–1999. Arthritis Rheum.. 2007;57(4):612-618.
Strombeck B., Jacobsson L.T.H. The role of exercise in the rehabilitation of patients with systemic lupus erythematosus and patients with primary sjogren’s syndrome. Curr. Opin. Rheumatol.. 2007;19:197-203.
Tan E.M., Cohen A.S., Fries J.F., et al. The 1982 revised criteria for the classification of systemic lupus erythematosis. Arthritis Rheum.. 1982;25:1271-1277.
Tench C.M., McCarthy J., McCurdie I., et al. Fatigue in systemic lupus erythematosus: A randomized controlled trial of exercise. Rheumatology. 2003;42:1050-1054.
Weinstein W.M., Kadell B.M. The gastrointestinal tract in systemic sclerosis. In: Clements P.J., Furst D.E., editors. Systemic sclerosis. Philadelphia: Lippincott Williams & Wilkins; 2004:293-308.
White B. Pulmonary fibrosis in systemic sclerosis. In: Clements P.J., Furst D.E., editors. Systemic sclerosis. Philadelphia: Lippincott Williams & Wilkins; 2004:163-183.
Wollheim F.A. Scleroderma renal crisis. J. Clin. Rheumatol.. 2004;10(5):234-235.