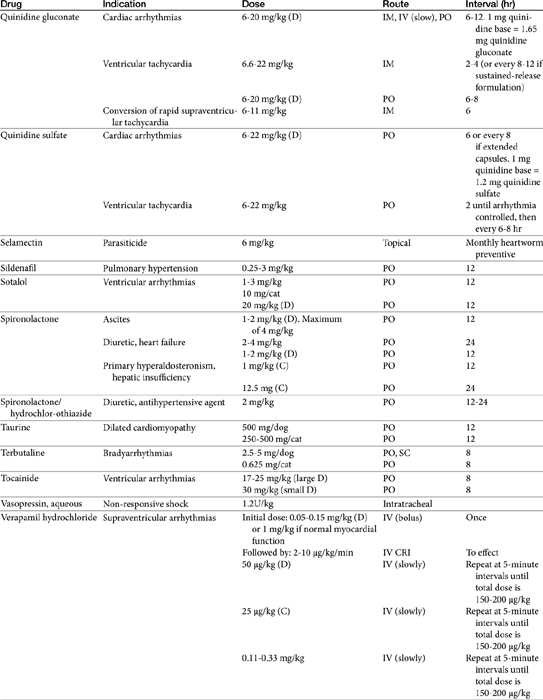
Chapter 14 Therapy of Cardiovascular Diseases
Cardiovascular Physiology as it Pertains to Cardiovascular Drugs
Membrane Ion Movements and the Action Potential
Predicting the nuances of pharmacodynamic responses to cardiac drugs depends, in part, on understanding the electrophysiology of myocardial cells. Selected mechanisms of ion movement into the cell are demonstrated in Figure 14-1. The action potential duration (APD) of myocardial cells is long compared with that of nerves, reflecting the well-orchestrated coordination of multiple ion channels and associated transport proteins.1 The magnitude and direction of the ion flow, and thus current, depends on both transmembrane voltage and ion concentration gradients. Two types of myocardial cells will be discussed: those capable of automaticity (i.e., spontaneous depolarization), exemplified by cells that normally serve as pacemakers (e.g., sinoatrial [SA] node) and those cells not normally capable of automaticity (e.g., atrial and ventricular myocardial cells). Although overlap exists, the electrophysiology of each differs from one another (Figure 14-2).
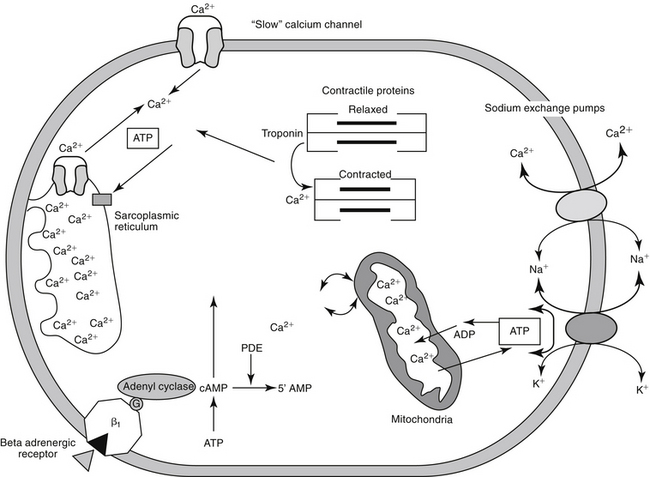
Figure 14-1 Calcium can enter the myocardial cell through several mechanisms, including the slow calcium channel, the sodium–calcium exchange ATPase pump, and beta 1 adrenergic receptor stimulation. Increased intracellular concentrations of calcium lead to the release of sarcolemmal calcium. Calcium leads to the interaction between actin and myosin, causing myocardial contractility. Sequestration of calcium in the sarcoplasmic reticulum causes myocardial relaxation. Sodium and potassium channels are not shown in this diagram. ATP, Adenosine triphosphate; cAMP, cyclic adenosine monophosphate; PDE, phosphodiesterase.
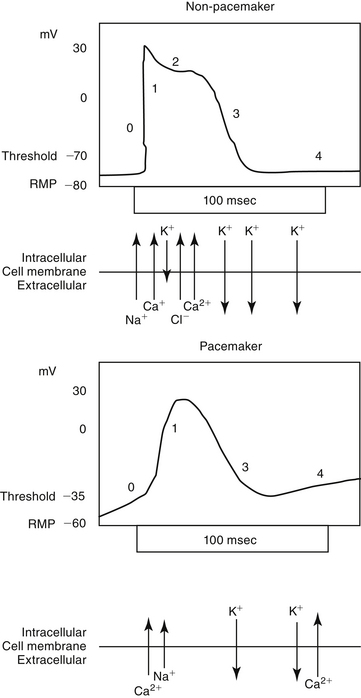
Figure 14-2 The action potential (AP) occurs in four phases. The ions responsible for and the configuration of each phase differ between cells capable of automaticity (i.e., pacemaker) and non-pacemaker (i.e., nondepoloarizing) cells. Phase 0 occurs when the resting membrane potential (RMP) reaches threshold, resulting in the generation of the AP. The rapid upswing in the membrane potential reflects sodium (non-pacemaker) and, to a lesser degree, calcium influx. In cells capable of automaticity (bottom), calcium is the primary ion moving inward during phase 0. This calcium influx stimulates release of calcium from the sarcoplasmic reticulum. The small influx of calcium in non-depolarizing cells is important to intracellular calcium fluxes. Phase 1 represents the early phase of repolarization. An influx of chloride and decreased efflux of potassium lead to reestablishment of the membrane potential. During phase 2 electrogenic movement of calcium through “slow” channels prolongs repolarization, causing a plateau phase. With phase 3, the membrane potential reaches the diastolic resting level. In cells capable of automaticity, this phase is characterized by a gradual depolarization, probably because of calcium influx, until threshold is reached. The heart rate is determined by the slope of phase 4; tissues with the steepest phase 4 slope will serve as the cardiac pacemaker.
Nondepolarizing cells
In nondepolarizing cells, the resting membrane potential (RMP) of -80 to -90 (inside compared with outside) reflects the relative distribution of sodium, potassium, and chloride across the cell membrane. The external and internal concentration of sodium approximates 145 and 15 mmol/L, respectively (ratio of 9.7), whereas that of potassium approximates 4 (range 3 to 6) and 145 mmol/L respectively (ratio of 0.027). Chloride ions contribute to the RMP only by virtue of their influence on cellular electrical responses to incoming signals. At rest the distribution of ions is not at equilibrium but is in a dynamic state, constantly subject to internal and external influences. The major driving force for the negative RMP is ion movements through channels (molecular pores) that span the myocardial cell membrane. These channels generally exist in either an open (conducting) or closed (resting) state, with a third state of “inactivation” reflecting a period in which the nonconducting channel cannot be activated. The state of channels targeted by cardiac drugs markedly influences their impact on the action potential. Drugs with greater affinity for inactivated channels generally are more influential than those that target other states.
KEY POINT 14-1
Differences in response of pacemaker versus nonpacemaker cells to cardioactive drugs reflects, in part, differences in membrane electrophysiology and ion flow.
Movement of ions through the channels follows both concentration and electrical (i.e., electrochemical) gradients. Ion flow is also influenced by energy-dependent pumps that actively and selectively direct ion movement against the electrochemical gradients. A “leak current” allows a constant, albeit smaller, ion flux. At rest the status of all channels influencing the membrane potential is static, save for a specific (“inward rectifier”) channel that is permeable only to potassium, allowing it to efflux down an electrochemical gradient. This efflux is countered by a Na+,K+-ATPase pump, which exchanges 3Na+ for 2K+, thus maintaining the intracellular concentration necessary for the gradient. Accordingly, potassium has the greatest influence on the RMP, and very slight changes in extracellular potassium can influence potassium flux, the RMP, and the cardiac cell cycle.
KEY POINT 14-2
Potassium has the greatest influence on the resting membrane potential. As such, even slight changes in extracellular potassium can influence potassium flux, the resting membrane potential, and the cardiac cell cycle.
Myocardial sodium channels are the primary gatekeepers of the action potential in nonpacemaker cells (see Figure 14-1). They are voltage gated and closed at rest. Membrane depolarization in nonpacemaker cells (see Figure 14-2) increases selective permeability in sodium channels. The concentration gradient causes an initial sodium influx, stimulating more voltage-gated sodium channels to open, thus perpetuating a self-regenerating action potential (phase 0, depolarization). Sodium channels inactivate when depolarization is complete and must go through a resting (recovery) period before they can be reactivated. Membrane permeability reestablishes itself largely in response to potassium ion efflux, and the membrane potential begins to decline back toward the RMP (phase 1, early repolarization). As the membrane potential continues to repolarize (phase 3), the ability of sodium channels to reactivate increases, until the RMP is once again reached (phase 4). Because sodium influx and sodium channel recovery is rapid, sodium channels are referred to as “fast,” in contrast to “slow” calcium channels, which take longer to recover. Drugs with greater affinity for inactivated sodium channels generally are more able to affect the refractory period and the APD compared with those that target either resting or open channels. Sodium influx during phase 0 depolarization influences other ion channels and their ion movements.
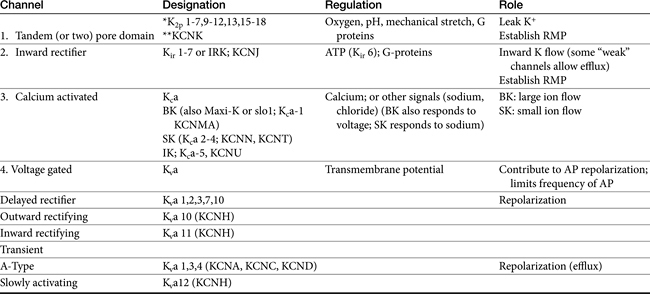
Among the ions most influenced by sodium flux in nonpacemaker cells are potassium channels, of which multiple types, exist, varying in tissue and intracellular location, and control (Table 14-1). Inward rectifier potassium channels are among the channels that determine the RMP. Depolarization causes conformational changes in transient potassium channels, which rapidly inactivate, and delayed rectifier potassium channels, which include both slow and rapid inactivators (each voltage gated). Efflux of potassium through these channels causes the membrane to begin to repolarize. Initially, the outward potassium current is balanced by an inward depolarizing current of calcium through L-type channels, causing the plateau, or phase 2, of repolarization in nonpacemaker (but not pacemaker) cells. Increased activity of delayed rectifier channels increases potassium efflux, which, when coupled with closing calcium channels, results in late repolarization (phase 3; see Figure 14-2). Slower delayed rectifier potassium channels remain open to ensure net outward potassium efflux such that the membrane potential becomes more negative, causing rapid delayed rectifier and inwardly rectifying potassium channels to open. The delayed rectifier K+ channels close when the membrane potential is restored to about -80 to -85 mV, but the inwardly rectifying potassium channels remain open throughout phase 4, thus maintaining the RMP. Because potassium channels generally remain open as long as the membrane is not at its resting potential (i.e., is in some state of depolarization), potassium channels also influence the duration of the action potential. Thus the influence of potassium channels in nondepolarizing cells includes the RMP and the rate of repolarization. In pacemaker cells (discussed in greater depth later in this chapter), potassium channels influence the RMP, the rate of repolarization, and the initial rate of phase 4. Consequently, potassium and drugs that target potassium channels potentially influence all cardiac (e.g., depolarizing and nondepolarizing) tissues.
KEY POINT 14-3
Inward flux of sodium is the primary ion responsible for phase 0 of nondepolarizing cells, whereas calcium influx is the primary ion in pacemaker cells.
Calcium also plays a major role in both nondepolarizing and depolarizing myocardial cells. The extracellular to intracellular calcium ratio of myocardial cells approximates 10, generating a calcium ion flow down both a concentration and electrical gradient. In contrast to the vasculature, in which channel movement is predominantly receptor mediated, calcium flux in the myocardium is predominantly voltage dependent. At least three types of voltage-gated calcium channels exist in the cardiovascular system, with differences reflecting conductance and sensitivity to voltage: T, N, and L types.2 The N-type (neuronal) calcium channels are located predominantly in neural tissues and markedly differ in responsiveness compared with L and T calcium channels. The best characterized of the calcium channels are of the L-type (long-lasting, large), and they are the predominant influencing channel during the states of the cardiac action in which the membrane potential is positive. Despite being voltage gated, calcium L-channels contain receptors (similar to those in the vasculature) that influence calcium channel flow and are subject to drug-induced blockade. In contrast to L-type channels, T-type (transient, tiny) channels are more active at negative potentials and are the predominant channel of influence at rest. As such, whereas L- channels are activated at high voltages, T-channels are activated at low voltages.
In nonpacemaker cells, in response to sodium influx, extracellular calcium enters the cell through L-type channels as repolarization begins. The influx is sufficient to stimulate release of intracellular calcium from the sarcoplasmic reticulum and other Ca2+ stores (e.g., mitochondria). This (initial sodium and subsequent) calcium ion flow links the action potential to excitation–contraction coupling in nondepolarizing cells.3 The duration of contraction is determined by the rate of intracellular calcium removal from actin and myosin sites in the cytosol by either resequestration into the sarcolemma (or other stores) or efflux from the cell. Efflux is accomplished by at least two pumps, both of which exchange sodium for calcium. The Na+/Ca2+ ATP-ase pump exchanges 1 Na+ for 3 Ca2+ but has only a minor influence on phase 2 of repolarization. The plateau of phase 2 repolarization in nondepolarizing cells is primarily influenced (sustained) by a balance between inward movement of calcium through L-type channels and outward movement of K+ through the slow delayed rectifier potassium channels.4
As with sodium channels, calcium channels exist in resting (closed, but responsive), open, or inactivated (unresponsive) states, and recovery must occur before the resting (responsive) state is achieved. Because both current movement and recovery of calcium channels are slower compared with those of sodium channels, the term slow channels is used to refer to ion movement through calcium channels. Hormones, neurotransmitters, and inorganic ions influence calcium channels. As with sodium channels, those drugs with a greater affinity for inactivated calcium channels are more effective.
Pacemaker Cells
Ion flow in cells capable of automaticity inherently is different from that of nondepolarizing cells to allow for spontaneity. Spontaneous depolarization occurs in response to ion fluxes across the cell membrane. The spontaneity of pacemaker cells reflects several electrophysiologic differences between pacemaker and nonpacemaker cells. In contrast to nonpacemaker cells, pacemaker cells have only a few sodium channels. They tend to be in an inactivated state, rendering the cell less responsive to sodium-induced depolarization. However, sodium can influence depolarization, albeit slightly, because more sodium channels are open in the resting state for pacemaker fibers compared with nonpacemaker cells, allowing sodium ions to continually flow down the concentration gradient into the cell. As a result, the maximum RMP of pacemaker cells is only about -60 mV. Second, calcium channels, rather than sodium channels, are responsible for phase 0 of the action potential in pacemaker cells. Third, depolarization is slow, beginning with calcium influx through T-channels until L-channels are activated to initiate phase 0 (depolarization). Fourth, phase 2 is not apparent in pacemaker cells. Fifth, as with nonpacemaker tissues, repolarization in pacemaker cells is initiated by potassium efflux. However, in contrast to nonpacemaker cells, repolarization continues beyond the RMP (-60 mV), resulting in a hyperpolarized (phase 4) membrane. As such, phase 4 in pacemaker cells is a “prepotential,” consisting of an initial reduction in potassium efflux, followed by opening of transient T calcium channels. Calcium influx decreases the membrane potential, causing it to become increasingly positive until the firing level (approximately -40 mV) is reached and L-channels are opened.
Automaticity
The rate (slope) of phase 4 in pacemaker cells is influenced by a number of factors. The slope determines the rate at which the action potential threshold of pacemaker cells is reached. Thus the slope of phase 4 determines the rate of spontaneous depolarization, and the cells that set the pace of the heart, the pacemaker cells, are those tissues with the steepest slope. Under normal circumstances, because it is characterized with the steepest slope, the SA node sets the pace. It is followed by conducting tissues, the Purkinje fibers, and, finally, myocardial tissue. As the default pacemaker, the SA node is innervated by both sympathetic and parasympathetic fibers. Vagal activation associated with acetylcholine (ACh) decreases the slope of phase 4 and thus SA nodal pacemaker rate; these actions reflect increased potassium conductance (efflux) and decreased slow inward Ca2+ and Na+ movement. Vagal activity also causes the cell to become hyperpolarized, increasing the time needed to reach threshold. However, in atrial fibers, vagal tone facilitates K+ channel-mediated repolarization, thus shortening the APD. Finally, vagal tone inhibits sympathetic activity, but the heart rate will increase only if sympathetic outflow (norepinephrine under normal conditions and epinephrine with pathologic conditions) is sufficient. Sympathetic tone, in turn, inhibits vagal tone. Changes in the serum concentration of ions, particularly potassium, also influence SA pacemaker activity. Hyperkalemia increases K+ conductance and thus efflux, resulting in bradycardia, whereas hypokalemia increases the rate of phase 4 depolarization (causing tachycardia), presumably by decreasing potassium conductance during phase 4. The slope of phase 4 can change in diseased myocardial tissue (e.g., hypoxia, acidosis, conditions that alter membrane permeability), and the generation of faster impulses may allow these tissues to take over as pacemakers.
Conduction Velocity
The interrelationship between phase 4 and phase 0 of the action potential (the rate of depolarization) determines the conduction velocity—that is, the speed of impulse propagation through pacemaker and nonpacemaker cardiac fibers. Conduction depends on the magnitude of the depolarizing current and the geometric (physical) relationship between myocardial cells. Conduction velocity is directly proportional to the rate and magnitude of phase 0 depolarization. Thus factors that slow the rate or magnitude of sodium (for nondepolarizing cells) or calcium (for pacemaker cells) flow during phase 0 also influence conduction velocity. Conduction velocity, in turn, influences the ability of the impulse to depolarize surrounding cardiac fibers. The faster and the greater the magnitude of depolarization, the more likely the impulse will depolarize surrounding cardiac fibers. For nonpacemaker cells, sodium is the major determinant of conduction velocity by virtue of its impact on the rate and extent of phase 0 upswing. However, calcium conductance during phase 0, albeit slight, also is able to influence phase 0 of nondepolarizing cells. For pacemaker cells, calcium is the major determinant of conduction velocity, with sodium an influencing factor.
Refractory Periods
Myocardial cells are neither excitable nor responsive to additional stimuli during the early and intermediate phases of the action potential cycle. Further, they are only partially responsive if stimulated before complete repolarization has occurred—that is, as the RMP returns to normal. Refractory periods (RPs) describe the period of time (or proportion of the APD) in which cells are nonresponsive. During the absolute refractory period (ARP), cells are totally nonresponsive. In contrast, cells may be partially (effective; ERP) or relatively (RRP) refractory to stimuli, with the state dependent on the number of open (i.e., recovered from inactivation) sodium channels. The ARP occurs when sodium channels are closed. Tissues will not respond to any stimuli, no matter how strong. The ERP is the shortest interval that can occur before a premature impulse can propagate a response.5 It includes the ARP plus a shorter period following the ARP that reflects the opening of some sodium channels but not enough to transmit an impulse. The RRP follows the ERP and represents a state in which a sufficient number of sodium channels are open such that a very strong stimulus might be propagated. The refractory periods are protective in that they limit the rate at which myocardial tissues can respond to impulses, thus ensuring sufficient time for cardiac filling and ejection to occur before the next contraction occurs. Proper direction of impulse propagation is also facilitated. Because sodium channels during these periods are refractory to reopening, unilateral (one direction only) conduction is ensured along a myocardial fiber, precluding the premature regeneration of an action potential and inappropriate coordination between excitation–contraction coupling. Loss of unilateral conduction is a contributing factor to re-entrant or circus rhythms.
Afterload and Preload
Both afterload and preload are variably defined depending on the source. Each is determined by the law of Laplace (T = PR/2, where T is wall tension, P is chamber or vessel pressure, and R is chamber or vessel radius). Preload occurs just before (end-diastolic) and afterload just after contraction. Preload is often referred to as the end-diastolic filling pressure, or the end-diastolic volume. Preload reflects the combined factors that influence passive (i.e., relaxed) ventricular wall stress at the end of diastole. Among the most important factors is the volume of blood that fills the ventricles. Increased filling volume enlarges the ventricular chamber, causing either tension or pressure or both to increase. A greater force is necessary for contraction to overcome the tension. Preload pressure occurs in the heart, is highest at the end of filling, or the end of diastole, and is virtually the same in the ventricle and its atria. Other measures of preload have included end-diastolic fiber length or stretch. Afterload is presented outside the heart (aorta) and is the pressure against which the heart must pump to effectively empty the ventricular chamber. At the point that ejection begins (beginning of systole), the aortic valves open and aortic pressure = peripheral resistance = arterial pressure = ventricular pressure. Afterload is also referred to as myocardial wall tension, or the force that must be generated by the heart for myocardial fiber shortening (contraction) to occur such that blood is ejected from the ventricle. Among the most important factors influencing afterload is peripheral resistance.
Myocardial Contractility
Contraction of both cardiac and smooth muscle depends on Ca2+. The inotropic state of the muscle reflects the relationship between resting fiber length and peak isometric tension. The myocardium develops force for contraction and thus the strength to pump blood by forming cross-bridges between actin and (tropo) myosin myofilaments in cardiac muscle. The amount of force that the muscle can generate depends on the number of cross-bridges that form when myosin engages actin. Energy in the form of adenosine triphosphate (ATP) causes a sliding motion between the proteins of the myofilaments and cardiac muscle to shorten and develop force. The interaction between proteins in the myofilament is regulated by troponin, which is found at regular intervals on the tropomyosin fibers. Troponin is formed from three subunit proteins: T binds to tropomysin, I inhibits the actin-binding site on tropomyosin, and C binds to calcium.4 Calcium binding of troponin C forces a conformational change in the troponin complex, causing troponin I to move away from tropomyosin, thus allowing cross-bridging between actin and myosin. The force that develops as actin and myosin interact depends on both the affinity and amount of calcium binding to troponin. The amount is regulated by the concentration of intracellular calcium. It is only as intracellular calcium is removed that troponin I moves back into position; thus contraction will continue until all intracellular calcium is removed.
Multiple mechanisms influence myocardial intracellular (cytosolic) calcium (see Figure 14-1). Extracellular calcium can enter through two sources: movement through (slow) electrogenic or voltage-gated calcium L channels embedded in T-tubules, which ensures that calcium is delivered in close proximity to the sacroplasmic reticulum, and movement though Na+-Ca2+ ATP channels, which is dependent on cell membrane ATPase.3,6 Cytosolic calcium flow is modulated primarily by β-adrenergic receptors; increased cyclic adenosine monophosphate (cAMP) activates protein kinase, which in turn increases calcium movement through the L-channels.4 Opening of slow calcium channels in response to depolarization causes intracellular calcium to rise rapidly, stimulating subsequent release from intracellular storage sites (sarcoplasmic reticulum and, to a lesser degree, mitochondria). The contracted myocardial muscle relaxes as intracellular calcium concentration falls as a result of resequestration into the sarcoplasmic reticulum and efflux from the cell, both of which are energy (ATP) dependent. Disorders of lusitropy (i.e., disorders of diastolic relaxation) occur if intracellular calcium does not decrease. The velocity and extent of cardiac muscular contraction are regulated by sarcomere length.4 Length (stretch) reflects preload, or the transmural filling pressure. An optimal stretch maximizes the relationship between actin and myosin filaments, allowing more Ca2+-activated cross-bridges and more forceful contractions. In the normal cat and dog, the upper limit of filling pressure in the left ventricle stretches the sarcomere to the length that generates peak tension during contraction. With sustained systolic overloading of the heart, however, the ideal sarcomere stretch is exceeded, cross-bridging decreases and myocardial contractility declines. Abnormalities of the excitation–contraction coupling mechanism contribute to the pathogenesis of cardiomyopathies and chronic hemodynamic overloading.
KEY POINT 14-6
Both cardiac contraction—mediated by intracellular release of calcium—and cardiac relaxation– mediated by reuptake of intracellular calcium—increase myocardial energy and thus oxygen demands.
The most important factor regulating myocardial contractility is stimulation of cardiac sympathetic nerves; cAMP serves as the secondary messenger, altering intracellular calcium flux and myocardial contractility. Myocardial cAMP is produced by adenylyl cyclase, which in turn is regulated by either stimulation or inhibition of adenine or guanine nucleotide proteins. Many cell surface receptors interact with proteins that regulate adenylyl cyclase. An increase in intracellular cAMP causes phosphorylation of proteins that increase calcium influx through the “slow” calcium channels, and the release, reaccumulation, and storage of calcium in the sarcoplasmic reticulum. Cyclic AMP is degraded by several phosphodiesterases (PDEs) isoenzymes, each of which has been associated with specific pharmacodynamic actions. At least 11 isoforms have been named. Inhibition of these enzymes causes the same effect as an increase in either adenylyl or guanylyl cyclase and thus cAMP or cGMP, respectively (see Figure 14-1). The pharmacodynamic response varies with tissue site: PDE II is located in smooth muscle of the urinary bladder detrusor muscle; PDE III in the heart, systemic vascular smooth muscle, and platelets (cAMP); PDE IV in bronchial smooth muscle and pulmonary circulation (cAMP); PDE V in the smooth muscle of the corpus cavernosum, visceral smooth muscle, skeletal muscle, platelets, kidney, lung, cerebellum, and pancreas (cGMP); and PDE VI in the retina (responsible for transduction).
Adrenergic Receptors
The adrenergic nervous system has a major physiologic role in modulating normal myocardial inotropic and chronotropic states and the time-variable tension that develops as ventricles contract. Both the myocardium and peripheral vasculature are innervated with sympathetic nerve terminals. Under normal conditions norepinephrine released from nerve endings in the heart acts as the primary regulator. Circulating catecholamines released from the adrenal gland play a less important role in normal conditions, but their influence increases as myocardial failure progresses. Molecular cloning techniques have identified nine subclasses of receptors: alpha (α) 1 (three subclasses); α 2 (three subclasses); and β 1, 2, and 3.7 Adrenergic receptors are linked to different G protein–coupled receptors, which differentially influence secondary messenger systems (sometimes the same one). Beta receptors are linked to adenylyl cyclase through Gs proteins. Beta agonists regulate cell processes by increasing cAMP, thus influencing downstream effects through cAMP-dependent protein kinases (see the discussion of smooth muscle). In contrast, α receptors are linked to Gi proteins, which oppose the actions of Gs proteins, thus decreasing cAMP.
KEY POINT 14-8
Norepinephrine from local nerve endings is the primary regulator of the heart under normal circumstances, but circulating catecholamines (e.g., epinephrine) predominate with myocardial failure.
In the normal heart, stimulation of the sympathoadrenal system is the primary method by which the heart adjusts to transient changes in workload. The myocardium possesses predominantly β receptors whereas vascular smooth muscle is rich in α receptors. Both β receptors and α receptors are subdivided into two types. Beta-1 receptors predominate in the myocardium, increasing inotropy (strength of contraction) and chronotropy (rate of impulse generation) (see Figure 14-1). Myocardial effects of adrenergic receptors are achieved through increased magnitude of the calcium current, slowed channel inactivation, and increased magnitude of K+ and Cl– repolarizing currents. Pacemaker current and thus sinus rate increase.5 Beta-2 receptors (and recently described β-3 receptors) are also located in the heart, but their function is not clear. Disease affects the state of receptors. Continued stimulation of adrenergic receptors, such as that which accompanies diseases states (e.g., congestive heart failure) or long-term adrenergic therapy, results in a dampening or desensitization of response to receptor stimulation. Desensitization reflects internalization and destruction of cell surface receptors. For example, β-1 receptors decrease up to 75% (β-2 receptors are spared) in human patients with congestive heart failure, leading to a compensatory increase in sympathetic signal outflow, which likely contributes to the pathophysiology (see the discussion of myocardial remodeling).7 The function of myocardial β-3 receptors is not clear, but they may provide feedback inhibition of contractility; an imbalance in myocardial disease may contribute to the pathophysiology of myocardial disease.8
In vascular smooth muscle, α-2 receptors mediate vasodilation (Figure 14-3). Most α activity in the cardiovascular system is mediated by way of α-1 receptors. Effects include contraction of vascular (and nonvascular) smooth muscle. A-2 receptors inhibit neurotransmitter release but also mediate vascular contraction (as do α-1 receptors). Subtypes 1 and 2 of either α or β receptors can be selectively pharmacologically stimulated (agonists) or inhibited (antagonists) to manage cardiovascular disease.
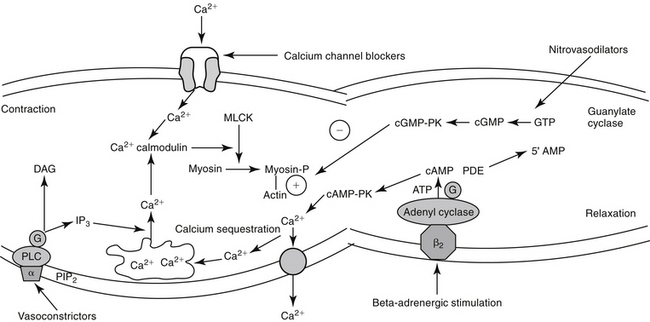
Figure 14–3 Contraction of vascular smooth muscle (vasoconstriction) reflects an influx of calcium, although mechanisms may differ from those in the myocardial cell. Calcium influx occurs through receptor-mediated channels or, less commonly, voltage-gated channels. Intracellular calcium combines with calmodulin. Myosin light-chain kinase (MLCK) is activated, and myosin light chain is phosphorylated (Myosin-P), promoting the interaction between myosin and actin. Cyclic adenosine monophosphate (cAMP) appears to stimulate sequestration and efflux of intracellular calcium and through cAMP protein kinase (cAMP-PK) decreases MLCK, causing vascular smooth muscle to relax (an action opposite to that in the myocardial muscle cell). Cyclic guanosine monophosphate (cGMP) also causes relaxation, probably through nitric oxide–mediated mechanisms. Intracellular calcium also can be released from the sarcoplasmic reticulum after hydrolysis of membrane phosphatidylinositol (PIP2) and subsequent formation of the secondary messenger inositol triphosphate (IP3). ATP, Adenosine triphosphate; DAG, diacylglycerol; GTP, guanosine triphosphate; PDE, phosphodiesterase; PLC, phospholipase C.
Smooth Muscle of the Vasculature
Myocardial oxygen demand is directly related to heart rate, myocardial wall tension, and the inotropic state of the myocardium.3,9 Myocardial wall tension is determined by the size (diameter) of the ventricle and intraventricular pressure. Thus tension is affected by preload (end-diastolic volume and stretch) and afterload (aortic blood pressure). Drugs that decrease systemic arterial pressure through dilation of arterioles decrease left ventricular afterload. Following the path of least resistance, a larger volume of blood will be ejected from the ventricular chamber into systemic circulation, thus diverting blood from the pulmonary vasculature. Left ventricular filling (preload) and thus myocardial wall size and tension will decrease, as will myocardial oxygen demand. An advantage of preload or afterload is that the decrease in cardiac work occurs without affecting myocardial contraction.
KEY POINT 14-9
A number of mediators act to cause arterial constriction and thus increase peripheral resistance.
The excitation–contraction coupling in vascular smooth muscle depends on calcium influx, which enters the cell through either voltage-sensitive (electrogenic) signals associated with depolarization or, more commonly, through receptor-operated Ca2+ channels. Intracellular calcium also is released from the sarcoplasmic reticulum in response to membrane phosphatidylinositol hydrolysis and formation of the secondary messenger inositol triphosphate.2 Intracellular calcium interacts with calmodulin, activating myosin light-chain kinase (MLCK) to phosphorylate myosin light chain. Cross-bridging between myosin and actin causes smooth muscle to contract. Cyclic AMP decreases both MLCK and intracellular calcium, causing relaxation of vascular smooth muscle. As with cAMP, the secondary intracellular messenger cGMP causes vascular smooth muscle relaxation, although the mechanism is different (see Figures 14-1 and 14-3). Endothelium-derived relaxing factor (EDRF; chemically related to nitric oxide) and endothelium-derived constricting factor (EDCF) are among the vasoactive substances released by the endothelial cell that control the hemodynamics of the cardiovascular system. Intracellular response to EDRF (or nitric oxide) is probably signaled by cGMP. Other mediators of vascular smooth muscle response include, but are not limited to, prostacyclin, histamine, and acetylcholine. Mediators released from the endothelial cell generally act locally on vascular smooth muscle (Figure 14-4); an exception might include mediators released from the pulmonary vasculature, which may be sufficient to modulate a systemic response.
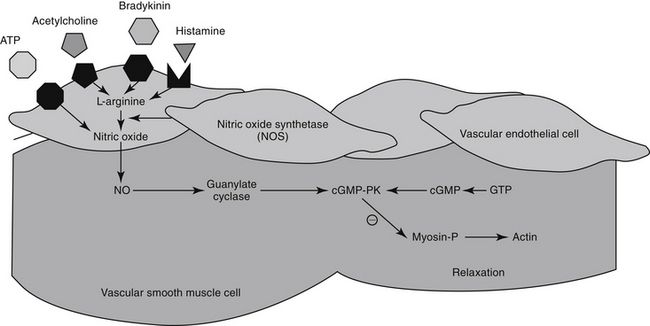
Figure 14-4 Vasoactive mediators responsible for vasodilation (e.g., prostacyclin, histamine, acetylcholine) stimulate nitric oxide synthetase to convert L-arginine to nitric oxide (NO) in the endothelial cells. Nitric oxide enters the smooth muscle cell and stimulates guanylate cyclase such that cyclic guanosine monophosphate (cGMP) is released. ATP, Adenosine triphosphate; cGMP-PK, cGMP-dependent protein kinase; GTP, guanosine triphosphate; Myosin-P, phosphorylated myosin.
Systolic and diastolic pressures are, respectively, the upper and lower limits of the oscillations around mean arterial pressure. The mean arterial pressure is the arterial pressure over time and is defined as the diastolic pressure plus one third of the pulse pressure. Arterial blood pressure is the product of cardiac output (determined by stroke volume and heart rate) and total peripheral resistance. Total peripheral resistance is the sum of resistance in all vascular beds. It is also affeccted by aortic impedance (resistance to flow) and diastolic arterial pressure, which in turn is determined by the sympathetic nervous system, the renin–angiotensin–aldosterone system and arginine vasopressin system, vascular (extracellular fluid) volume, and aldosterone or other volume active hormones.43
The Renin–Angiotensin–Aldosterone System
The renin–angiotensin–aldosterone system (RAAS) plays an important role in regulating blood volume, arterial pressure, and cardiac and vascular function (Figure 14-5).10 An additional but critically important role in cardiac repair and remodeling has recently emerged. RAAS regulation of arterial pressure is accomplished through constriction of resistance vessels, mediated by several mechanisms. Included are direct stimulation of AGII receptors and indirect stimulation through facilitation of norepinephrine. Vasopressin also is a potent mediator of peripheral vasoconstriction by way of V-1 receptors. Its increase reflects either increased release from sympathetic nerve terminal endings or decreased reuptake.11
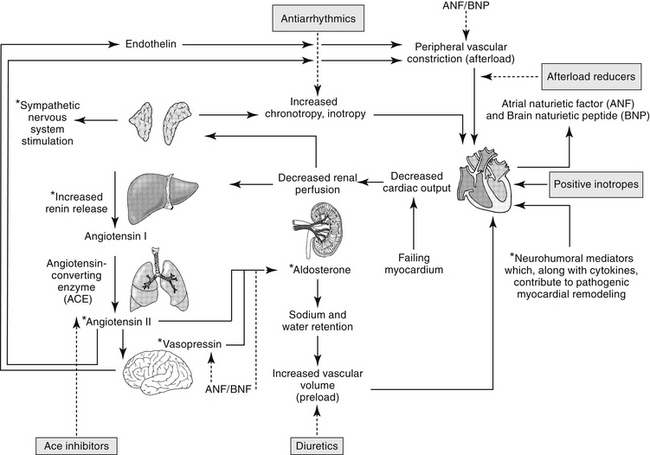
Figure 14-5 Neuroendocrine responses to decreased peripheral perfusion associated with the failing left ventricle may initially result in increased contractility, increased cardiac output, and increased tissue perfusion. Systems activated include the sympathetic adrenergic system, the renin–angiotensin–aldosterone system, and the arginine vasopressin system. Compensatory mechanisms, however, lead to increased afterload (adrenergic stimulation, angiotensin release) and increased preload (aldosterone, release of vasopressin), both of which may detrimentally increase the workload on the failing heart. Mediators signaling these responses contribute to myocardial remodeling (indicated by asterisk∗), leading to progressive myocardial failure. Therapeutic approaches that target these mediators (shaded boxes) are intended to not only alter the negative sequlae of increase preload, afterload and cardiac response, but also remodeling. ACE, Angiotensin-converting enzyme; dashed line, inhibited.
Other neurohumoral–endocrine mediators of RAAS are produced by a number of organs, resulting in both local and systemic responses. Contributing organs include the kidney, brain, heart, vasculature, adipose tissue, gonads, placenta, and pancreas. In the kidney, renin is produced in the juxtaglomerular cells, and angiotensinogen in the proximal tubular cells. The majority of the effects of RAAS reflect its most potent mediator, angiotensin II (AGII), which in turn is dependent on renin. Renal renin release is stimulated by hypotension, decreased sodium delivery to the distal tubules, or direct stimulation of β-1 adrenergic receptors. Renin catalyzes proteolytic cleavage of circulating angiotensinogen to the decapeptide, angiotensin I. Angiotensin I is then converted to AGII by angiotensin-converting enzyme (ACE), located in vascular endothelium with the majority of systemic release coming from the lungs. AGII is further degraded by angiotensinoginases located in red blood cells AG III and IV.
ACE is a kinase II metallopeptidase enzyme bound to the membrane of a variety of cells, but particularly endothelium, epithelium, neuroepithelium, and brain cells.12 Organs respond to both systemic and local renin, with local response influenced by local concentrations of ACE and angiotensin receptors. Concentrations of ACE differ among tissues, with that in the renal tubular brush border the greatest (300- and 10-fold higher than the left ventricle and lung, respectively). Thus, although the kidney is the most important site of renin release, it also is a target of the RAAS, responding to both systemic and urinary renin. Renal AGII concentrations exceed circulating AGII more than 1000-fold, causing renal vasoconstriction and sodium retention. Degradation of AGII yields angiotensin III (AgIII), which has 40% of the pressor and 100% of the aldosterone effects of AGII.
Through AGII, RAAS modulates responses to low sodium intake and provides for long-term control of renal function, body fluid volumes, and arterial pressure. In the healthy canine kidney, response to AGII results in increase in both preglomerular and postglomerular vascular resistance, although the predominant effect is on the efferent rather than afferent arteriole. Renal blood flow consequently decreases, but glomerular capillary pressure increases.12 AGII regulates body fluid content by directly stimulating thirst centers in the brain; adrenal release of aldosterone, which mediates increased renal sodium and fluid retention; and posterior pituitary release of vasopressin (antidiuretic hormone, ADH), a component of the arginine vasopressin system (AVP). Vasopressin increases renal fluid retention through V2 receptors in renal tubular cells. In addition to its vascular effects, AGII released from endothelial cells also facilitates cardiac hypertrophy and vascular hypertrophy. Notably, production of inflammatory cytokines is increased from both normal and abnormal (damaged) myocardial tissue, contributing to the negative sequelae of cardiac remodeling.13 AGII is prothrombotic and may induce cardiac muscle hypertrophy.
KEY POINT 14-10
The effect of the renin–angiotensin–aldosterone system is complex. It comprises both systemic as well as local systems, multiple receptors and mediators, and complex receptor–mediator interactions.
KEY POINT 14-11
Ultimately, the negative sequelae of compensation contribute to the progression of myocardial disease and failure
Renal vascular response to AGII is mediated by AGII receptors (ARs), a transmembrane G-coupled protein receptor consisting of several subtypes that vary in location, numbers, affinity for AGII, and secondary messenger systems. The most well known of the subtypes, AR-1, preferentially binds AGII and AGIII, mediating the classic angiotensin RAAS responses on blood pressure and water and electrolyte balance. Included are water, sodium intake, renal sodium retention, secretion of vasopressin and aldosterone, and cell growth/proliferation. In addition to AR-1, AR-2 receptors also bind AII and AIII.14-16 However, AR-2 receptor density is greatest in the brain, including areas involved with fluid and electrolyte regulation and balance, arterial pressure, cognition, behavior, and locomotion. Concentrations of AR-2 also are high in steroid-producing glands, including the adrenal glands and ovaries.16 Because of the central location of AR-2, AGII and AGIII can modulate many body functions and responses. Expression of AR-2 is particularly high during fetal development, but it persists in the adult brain, supporting a role in neuronal function. In the brain, AR-2 appears to oppose the traditional RAAS effects of AR-1 on drinking behavior and vasopressin secretion. Other effects mediated by AR-2 receptors are regulation of cell proliferation, apoptosis, and cellular differentiation.16 Again, AR-2 appears to attenuate AR-1 mediated apoptosis, pressor, and chronotropic effects. Like AR-1 receptors, several secondary systems appear to signal AR-2 effects, including the mitogen-activated protein kinase (MAPK) and nitric oxide/cGMP pathways.16
Regulation of RAAS effects through angiotensin receptors is complex, possibly involving feedback inhibition pathways, responses that vary with duration of exposure, and systems that may oppose one another. Likewise, pharmacologic management of heart disease through manipulation of AR receptors may be complex.15 Disease is likely to contribute to variability in response to AGII and its modulating drugs. For example, in the failing heart the expression of AR-1 decreases, whereas that of AR-2 does not change or increases. Both receptors are associated with effects that initially, in moderation, might benefit the patient, but with progression become detrimental. Not surprisingly, a link has been described between AR and β-adrenergic receptors. Both AR-1 and β receptors interact with G proteins, with AR-1 through activation of phospholipase C and β-receptors through activation of adenylyl cyclase. Diamerization of the two receptors has been described in vitro and occurs in vivo.17 This integration will further complicate pharmacologic manipulation.
Among the mediators stimulated by AGII are vascular endothelial production of nitric oxide and endothelin (ET-1), both of which contribute to regulation of renin release. Nitric oxide appears to oppose, whereas endothelin appears to reinforce, the vascular effects of AGII. Endothelins are peptide vasoconstrictors released from endothelial cells; three have thus far been identified: ETs 1 through 3. Endothelins exert their effects through a number of endothelin receptors (ET) including ET-A, associated with vasoconstriction and vascular smooth muscle proliferation, and ET-B, which promotes both constriction and dilation, clearance of endothelin, and production of endothelial cell prostacyclin and nitric oxide. Endothelin receptor antagonists have facilitated understanding of the role of endothelins in vascular regulation and ultimately may offer a mechanism of pharmacologic manipulation. Endothelin is the most potent vasoconstrictor known. In addition to direct vascular effects, endothelin modulates plasma concentrations of both atrial natriuretic factor (ANF), arginine vasopressin (AVP), and aldosterone. Additionally, endothelin contributes to vascular remodeling.
Mechanisms other than ACE modulate formation of AGII. Opposing effects of ACE are regulated in part by AR, but other body systems also modulate the influence of ACE. For example, ACE also inhibits breakdown of bradykinin. Bradykinin consequently increases, resulting in vasodilation and naturiesis.18 The vasodilatory effects of bradykinin are mediated in the vascular endothelium through arachidonic acid derivatives, nitric oxide, and endothelium-derived hyperpolarizing factor in the vascular endothelium. The mechanism of natriuresis is not clear.12 Bradykinin also has beneficial effects on cardiac remodeling, which helps oppose the negative sequelae of AGII.
The Role of Nitric Oxide
For decades, researchers have attempted to identify a factor released from endothelial cells, referred to as EDRF, which is responsible for mediating a number of stimuli causing vasodilation. Ultimately, nitric oxide (NO) was recognized to be the smallest and most basic mediator of vascular response.19 Released as a gas (and thus often mistaken for nitrous oxide [N2O], or “laughing gas”), it is synthesized in response to NO synthetase (NOS) enzymes from L-arginine and oxygen or by sequential reduction of inorganic nitrate (see Figure 14-4). However, as a free radical, NO is very reactive and unstable and interacts with oxygen on exposure to air to form the pollutant nitrogen dioxide (NO2). Two major classes and three isoforms of NOS have been identified.19-21 Constitutive NOS (cNOS or NOS-1) is continuously produced and includes two isoforms synthesized either by vascular endothelial cells (eNOS) or neurons (nNOS). Constitutive NOS, which is calcium dependent, tends to mediate cell responses through cellular receptors. Responses of cNOS include that mediated by vascular mediators such as acetylcholine, norepinephrine, histamine, and substance P. Not surprisingly, response is rapid. Inducible NOS (iNOS or NOS-2) is produced as needed by inflammatory cells (e.g., macrophages, neutrophils, and Kuppfer cells), generally after exposure to cytokines (e.g., tumor necrosis factor or interleukins) or bacterial lipopolysaccharides. Production of NO from iNOS requires new protein synthesis and is characterized by a delay of several (2 to 4) hours.
Regardless of origin, NO causes its effect by diffusing across cellular membranes to intracellular targets. Cytosolic cGMP is the major intracellular messenger (see Figure 14-4) causing physiologic response to NO. Responses include dilation of blood vessels, inhibition of thrombogenesis, cytotoxic responses, and neuronal signaling. However, because NO contains an unpaired electron in its outer orbit, it is a free radical. As such, it can contribute to the formation of other radicals while simultaneously scavenging oxygen radicals. The half-life of NO is so short that studies involving NO generally are based on its oxidation end products nitrates and nitrites.20 However, despite its very short half-life, NO has many important and complex actions in the body. Under basal conditions, peripheral vasoconstriction is locally relieved by intermittent cNOS-induced NO in response to sheer stress and endothelial cell receptor stimulation. Inflammation and immune signals also induce NO release by way of iNOS. NO inhibits platelet aggregation and adhesion, contributing to antithrombogenic mechanisms in the vascular endothelium. NO may ameliorate the detrimental effects of norepinephrine on the growth of cardiac myocytes and fibroblasts, suggesting that increased NO bioavailability may prevent or reverse remodeling in patients that have experienced heart failure.22 Modulation of inflammation varies, however, with cell type and the source of NO production (i.e., iNOS versus cNOS). Although targeting NO production through drug therapy may appear to be a reasonable approach to the treatment of a variety of cardiovascular disorders, the complex nature of its release and the events leading to its release currently preclude predictable and safe modulation. It is likely, however, that selective modulation of NO ultimately will provide a therapeutic approach to many disorders.
Pathophysiology of Cardiac Disease as it Relates to Drug Therapy: Congestive Heart Failure
Neural–Humoral–Endocrine Compensatory Mechanisms in the Myocardium and Vasculature
Congestive heart failure (CHF) refers to the inability of the heart to deliver blood necessary to meet the metabolic demands of body tissues. Backward failure is the most common form, reflecting increased end diastolic pressure and atrial pressures. Venous and capillary pressures increase to the point that fluid transudates into interstitial tissues, resulting in the clinical manifestations that result from heart failure, including (left sided) pulmonary and circulatory edema or (right-sided) ascites. Patients generally are hypervolemic and thus are referred to as “wet.” Less commonly, forward failure reflects decreased cardiac output and poor periperhal perfusion (“cold”) resulting in exercise intolerance or cool extremities. Backward failure ultimately may lead to forward failure.
Mechanisms that compensate for loss of contractility or abnormal loading on the heart initially maintain cardiac output in the normal range either at rest or with limited exercise. Clinical signs of disease may not be evident (preclinical stage). However, the negative sequelae of compensation ultimately contribute to the progression of myocardial disease and failure. Decompensation occurs when cardiac output is no longer sufficient to support circulation despite compensatory mechanisms
Regardless of the cause of cardiac failure, decreased blood pressure and compromised organ perfusion initiate complex interactive compensatory responses of the neural, hormonal, and endocrine systems.11,23 Neuroendocrine changes reflect “fight or flight” stimuli, affecting blood pressure and fluid volume (see Figure 14-5). Baroreceptors and the vasomotor center interact with the sympathetic and parasympathetic systems to increase heart rate, myocardial contractility, and blood pressure and to activate the RAAS. Although cardiac output may increase, the responses contribute to fluid accumulation and myocardial remodeling, which ultimately lead to irreversible myocardial failure.
The kidney directs responses designed to increase arterial blood volume in response to poor renal perfusion accompanying the failing heart. Renal compensatory mechanisms are mediated by RAAS in the juxtaglomerular apparatus. Renal glomerular arterioles are exquisitely sensitive to catecholamines; their reflex vasoconstriction exacerbates diversion of blood flow from the glomerulus. However, renal arteriolar underperfusion coupled with adrenergic stimulation causes the release of renin in pressure-volume–sensitive receptors of the afferent arterioles. In response to decreased renal plasma flow and glomerular filtration rate (GFR), the filtration fraction increases, normalizing renal excretory function. Proximal tubular function is maintained (and possibly enhanced) such that a greater percentage of sodium and water is reabsorbed from the filtrate. Decreased sodium in the filtrate causes further renin release. Fluid retention initiated by these changes increases ventricular filling. Actin and myosin interaction are optimized with myocardial cell stretching (sarcomere length–active tension relationship or Frank–Starling phenomenon), leading to improved contractility. Stroke volume and cardiac output increase, as does cardiac work. Effective restoration of blood volume and ventricular filling will improve renal perfusion but at a new equilibrium characterized by increased ventricular filling pressures and intravascular and interstitial fluid volumes.
Myocardial disease is accompanied by changes in concentrations of components of the RAAS. For example, AGII increases twofold to threefold in the left ventricle and kidneys and tenfold in plasma. Because AGII is produced by mechanisms other than ACE (e.g., chymase), the impact of ACE inhibitors on resolution of increased AGII is variable among tissues. Inhibitors of ACE may vary in their relative impact on efferent or afferent arterioles; the impact may also vary within renal zones.
Other systems influenced by RAAS also change in the diseased heart. ANF and atrial natriuretic peptide (ANP, or atriopeptin) production by atrial myocytes is stimulated by atrial stretch in the heart and a number of other signals (adrenergic stimulation, AGII, endothelin, increased sodium) associated with congestive heart failure. Brain natriuretic factor, although originally identified in brain tissue, is secreted by ventricular myocytes and interacts with ANF receptors, causing similar effects. The effects of ANF are mediated by at least three receptors (NPRA1-3 or A-C), two (NPRA 1 and 2) of which are linked to cGMP and the third to G-protein. ANF inhibits AVP activated by RAAS. Renal response to ANF includes dilation of the afferent glomerular arteriole, renal sodium wasting, and decreased renin secretion; aldosterone secretion from the adrenal gland also decreases. Vascular smooth muscle is relaxed. As such, normally, ANF induces natriuresis, diuresis, and vasodilation, inhibits renin and aldosterone secretion, and appears to attenuate vasoconstriction.. Interestingly, ANF also influences adipose tissue, causing, among other things, release of amino acids. Although plasma concentrations of ANF are increased in heart failure, response is blunted for reasons that are not clear. Diagnostically, detection of ANF has been used to differentially diagnose acute dyspnea. e.g., that associated with pulmonary edema.
Circulating ET-1 increases in both plasma and the left ventricle in response to a number of signals, including norepinephrine, AVP, and interleukin-1. Increases parallel the progression of myocardial injury and correlate with increased pulmonary arterial pressures. ET-1 may play a role in the pathophysiology of pulmonary hypertension of heart failure in humans.24
Heart failure not only is the result of dysfunction of the RAAS but also reflects several abnormalities of the second major compensatory system, the adrenergic nervous system.17,25,26 Indeed, the two systems appear to influence each other, affecting both the heart and peripheral vasculature. Loss of myocardial contractile support is partially compensated for by increases in plasma catecholamines released from the adrenal gland; autonomic imbalance occurs as the parasympathetic system fails to “check” adrenergic response. This may reflect the reduced sensitivity of baroreceptors.26 The failing heart becomes increasingly dependent on circulating, rather than local catecholamines. In the failing heart, maximum contractile and heart rate response are decreased for several reasons. In the later stages of failure, myocardial response to sympathetic nerve stimulation is blunted as a result of decreased synthesis, storage, and release of myocardial norepinephrine. Beta-1 receptors decrease and inhibitory guanine nucleotide–binding proteins (Gi) increase.27 Sustained adrenergic signaling associated with myocardial injury causes downregulation of β-1 receptors and uncoupling of both β-1 and β-2 receptors from G proteins. Because β receptors are diamerized with AR, AR-1 decreases in concert with β receptors.17
KEY POINT 14-12
Heart failure is the result of integrated altered functions of the renin–angiotensin–aldosterone system; the adrenergic nervous system; and their integrated influences on afterload, preload, heart rate, and myocardial remodeling.
As the heart fails, α-adrenergic–mediated vasoconstriction in response to circulating catecholamines causes regional peripheral vasoconstriction, ensuring preservation of arterial blood pressure. Differential vasoconstriction among the vascular beds causes blood flow to be redistributed to organs with the highest metabolic requirements (i.e., brain, heart, and active skeletal muscles). Accordingly, renal blood flow is restricted, resulting in activation of the RAAS. Autoregulation of intrarenal blood flow (e.g., efferent renal arteriolar constriction) helps maintain glomerular filtration despite systemic redistribution. Venoconstriction and fluid retention increase preload, providing some compensation for decreased cardiac output.
Although the goal of compensatory mechanisms is to increase cardiac output, eventually the secondary sequelae prove detrimental and both diastolic and systolic cardiac dysfunctions emerge. In the peripheral vasculature, vasoconstriction mediated by AGII, circulating catecholamines, AVP, and ET-1 results in persistent and significant increased systemic vascular resistance. Mechanical vascular stiffness, reflecting intramural sodium and water content, worsens resistance. The vasculature, particularly in skeletal muscle, can no longer autoregulate. Increased resistance tends to raise (maintain) blood pressure and organ perfusion, but at a cost: the marked increase in cardiac afterload causes a proportionate decrease in stroke volume. The heart must work harder, using more oxygen to affect the same cardiac output. Because stroke volume is less, the end-diastolic volume (preload) in the heart is greater, increasing myocardial wall tension. Myocardial diastolic relaxation, necessary for myocardial perfusion, is impaired. Increasing myocardial oxygen and energy needs cannot be met as myocardial perfusion decreases. Thus increased peripheral resistance represents a vicious cycle as it worsens the failing heart. Abnormal relaxation during diastole also has been associated with direct changes in the myocardial cell. These include abnormal sarcoplasmic reticulum regulation of intracellular Ca2+ and decreased density of Ca2+-ATPase.27 Drugs that increase cAMP thus may influence either contractility (inotropy) or diastolic relaxation (lusitropy).
Negative Sequelae of Myocardial Remodeling
In the last decade, as more data regarding the role of RAAS and related systems in the progression of myocardial failure have emerged, therapies have been designed to minimize the negative sequelae of compensatory mechanisms. Accordingly, the traditional goals of therapy have been to lower venous pressure (diuretics), decrease afterload on the failing heart (vasodilators), decrease heart rate (e.g., β blockers, calcium channel blockers [CCBs], or digoxin), and increase myocardial contractility (positive inotropes).28 However, the traditional view of CHF as a hemodynamic syndrome characterized by fluid retention, high venous pressure, and low cardiac output has been modified over the last decade. This change reflects a response to a number of unanticipated findings in reviews of clinical trials testing traditional therapies targeting neurohumoral responses in humans. Notably, clinical trials failed to demonstrate long-term survival with traditional therapies. Further, drugs that initially caused a favorable response often shortened, rather than lengthened, survival time. For example, vasodilators such as α-adrenergic blockers, short-acting L-type CCBs, inoxidil, prostacyclin, and phosphodiesterase inhibitors failed to prolong survival despite effective afterload reduction. Inotropic agents increased contractility by increasing cAMP but shortened long-term survival as cardiac energy needs and arrhythmias increased. Increased calcium flux associated with their use may also have contributed to diastolic dysfunction.28 In concert with these findings, selected drugs that initially worsened clinical signs (e.g., β-adrenergic drugs) were associated with improved long-term survival. Finally, several classes of drugs were associated with improved survival through mechanisms other than that expected on the basis of their known pharmacologic effects. For example, selected diuretics (e.g., spironolactone) and drugs active in the RAAS, including ACE inhibitors and angiotensin II–receptor blockers, appeared to slow myocardial deterioration and remodeling.28-30
The findings of the clinical trials reoriented investigators to the potential impact of disease and drug therapies on myocardial deterioration, progressive remodeling, and maladaptive hypertrophy. For example, worsening of disease despite effective afterload reduction appears to have reflected, in part, increased release of neurohumoral mediators (norepinephrine, AGII, and endothelin), stimulating further proliferation and remodeling. Inhibition of inappropriate mediators through drugs such as β blockers may decrease maladaptive myocardial proliferation, and progressive dilation of the heart was proposed as a mechanism.28 The impact of remodeling and its prevention on the progression of CHF is now a well-recognized target of therapy. The extent of progressive remodeling is associated with clinical outcome, and the key to improved survival for CHF that has emerged as a result of these findings is the blunting of the progressive deterioration, remodeling, and proliferation associated with disease.28
Remodeling reflects a number of cellular and biochemical activities that lead to myocardial hypertrophy, fibrosis, altered excitation–contraction coupling, apoptosis, altered cellular metabolism, and discordant electrophysiologic responses. The negative sequelae of remodeling include altered ventricular myocardial wall and chamber dimensions and altered geometry. Although myocyte hypertrophy does not appear to be associated with negative sequelae in the failing heart,31 the responses also include maladaptive proliferation and chronic dilation, leading to eccentric hypertrophy. Myocardial cell life span is shortened, initiating a vicious cycle of myocardial cell death, increased load on surviving myocytes, and compensatory proliferation. Consequently, myocardial cell death and remodeling and dilation increase. Elongation of cardiac myocytes increases cardiac chamber size but also increases individual myocyte tension, further stimulating hypertrophy.24 Several mediators recognized for their neurohumoral compensatory responses contribute to cardiac remodeling, offering a target of therapy. In particular, AG II contributes to several aspects of cardiac remodeling through AG II type 1 (AT1) receptors.32 Fibroblast gene expression increases, leading to increased density and proliferation, and myocyte hypertrophy.31 Aldosterone activates several genes responsible for synthesis of myocardial extracellular matrix.33 The myocardium appears to include a local RAAS regulated in part by AG II that supports cardiac fibrosis; the more severe the failure, the more aldosterone is activated, with local activation occurring independently of systemic effects.34 Underlying proliferation and remodeling is inflammation; both are associated with increased gene expressions of proinflammatory cytokines.35 Inflammatory cytokines, NO, and reactive oxygen species act as negative inotropes, contributing to cardiac remodeling.35 NO also impairs mechanical myocardial function by increasing intracellular cGMP, reducing calcium current and desensitizing myofilaments. NO has both negative inotropic and chronotropic effects on the heart and has been associated with myocardial necrosis.24 Free radicals also decrease calcium sensitivity and calcium accumulation in the sarcoplasmic reticulum.36 Calcium sensitization is further reduced by pathologic conditions such as acidosis or hypoxia. These changes determine the long-term prognosis in patients with heart failure.28 In the heart damaged by myocardial infarction, proinflammatory cytokines (e.g., tumor necrosis factor [TNF]-α, IL-1b, and others) stimulate cardiac fibroblasts to alter the extracellular matrix (ECM), primarily through AGII and AR-1 receptors. Plasma TNF-α has been positively correlated with the severity of CHF in humans and is increased in dogs and cats with heart failure.24 Eventually, damaged and normal tissue is replaced with scar tissue that maintains structural integrity but limits chamber size. Remodeling involves production of structural proteins, including fibronectin, collagens (Col) I and III, tissue inhibitors of matrix metalloproteinases (TMPs) and “secondary” growth factors. Apoptosis may contribute to progressive left ventricular dysfunction, as is supported by increased plasma apoptosis–signaling surface receptors that trigger programmed cell death in patients with heart failure.24 Angiotensin receptor density also increases in area macrophages.37
KEY POINT 14-13
Several mediators recognized for their neurohumoral compensatory responses contribute to cardiac remodeling, with angiotensin and its subsequent influences playing a major role.
As the understanding of the pathophysiology of CHF has advanced, the tools with which disease and response to therapy can be monitored also will advance. Preferred biomarkers have been circulating molecules associated with neurohumoral responses to the failing heart, such as endothelin, natriuretic peptides, AGII, and endothelin or markers of myocardial damage (e.g., creatine kinase isoenzymes and troponins). Increased cardiac troponin (I or T) has emerged as the preferred gold-standard marker for acute events involving the myocardium, whereas increased B-type natriuretic peptide may be preferred for identification of cardiac diseases as a cause of dyspnea. Boswood38 has addressed the status of biomarkers in feline and canine cardiac disease.
Management should attempt to correct maladaptive responses in cardiovascular function and inflammatory and proliferative responses (Tables 14-2 and 14-3). Functional changes reflect short-term hemodynamic neurohumoral responses initiated by the endocrine system that are intended to improve cardiac performance, vascular tone, and salt and water excretion. As such, traditional approaches to treatment of congestive heart failure have included drugs that decrease preload (e.g., diuretics), afterload (e.g, ACE inhibitors), and heart rate (e.g., beta-blockers). However, drugs that slow the progression of myocardial disease by virtue of their effects on myocardial inflammation, remodeling, necrosis, or apoptosis represent the newest group of drugs used to treat the failing heart. Selected drugs (including those in current use) that improve hemodynamic effects by blocking neurohumoral stimuli may prove most beneficial because of their simultaneous inhibition of proliferative stimuli. Accordingly, attempts should be made to select those drugs in each category with demonstrable muting of myocardial remodeling.
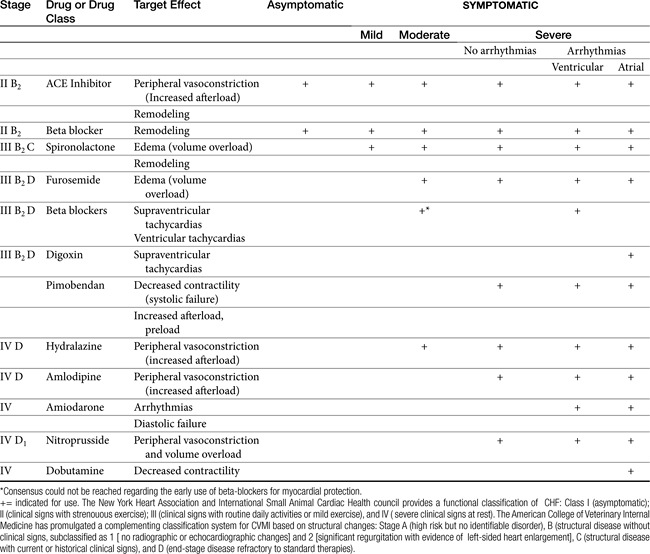
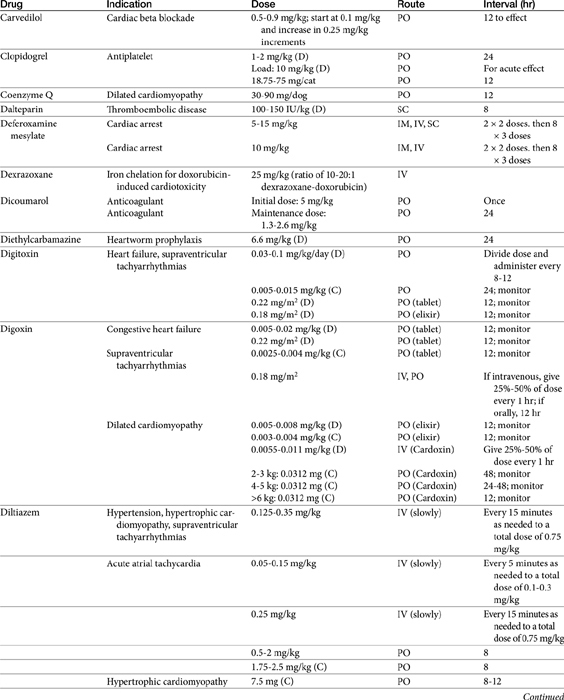
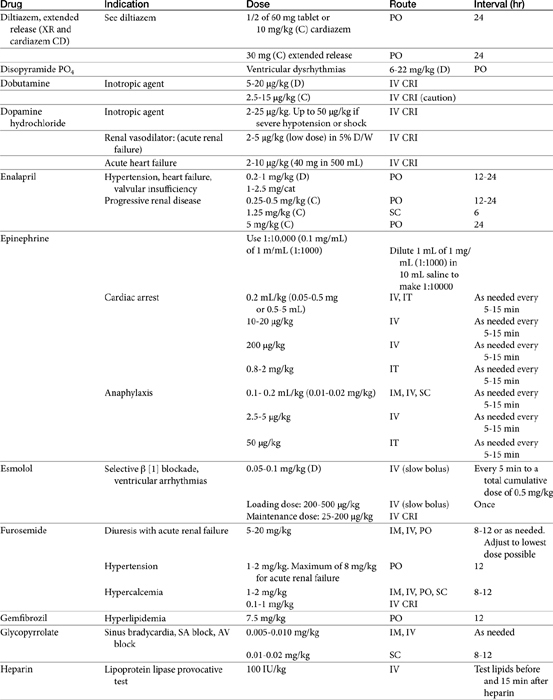
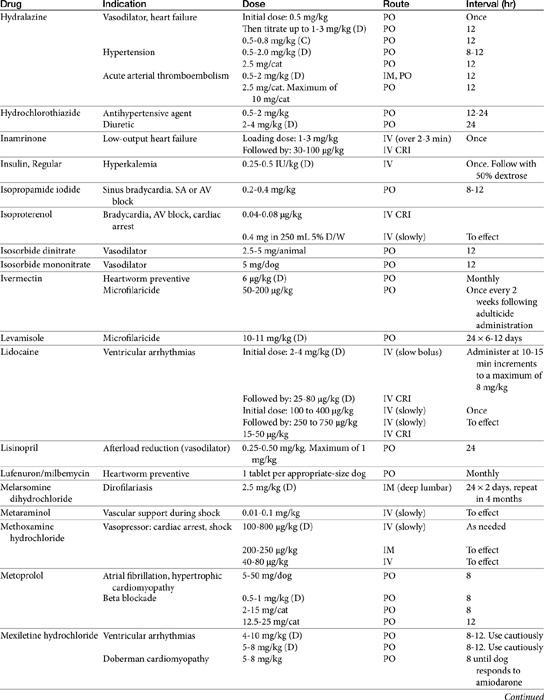
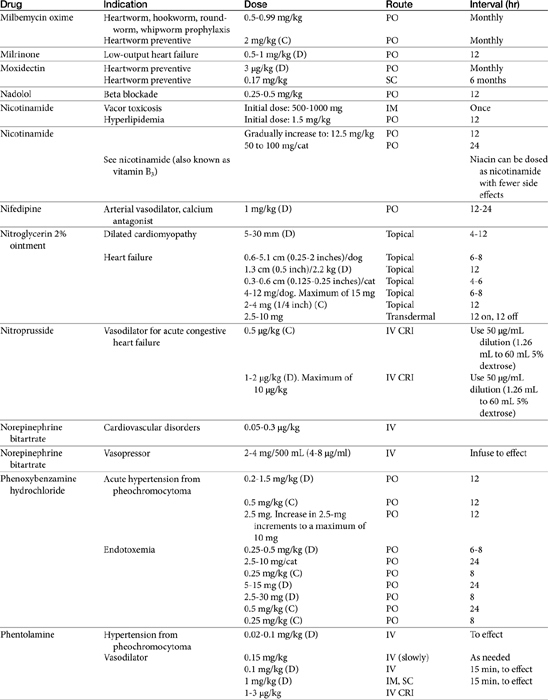
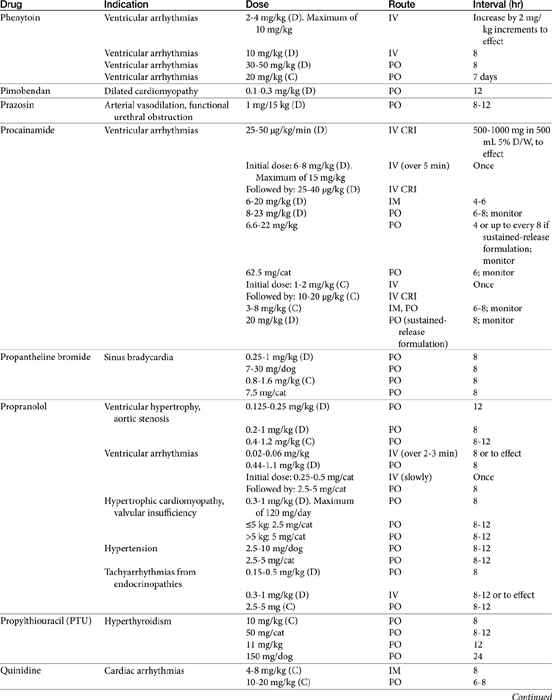
Vasodilator Therapy
Vasodilator drugs can be categorized according to the type of vessels that they dilate: arterioles (i.e., resistance vessels: arterial dilators), veins (i.e., capacitance vessels: venodilators), or both. All three types of vasodilators can be useful in the patient with CHF. Arterial vasodilators target resistance vessels, relieving vasoconstriction that accompanies CHF or primary hypertension. Normally, vasoconstriction maintains systemic pressure in the normal range of 100 to 110 mm Hg. However, the critical organs (i.e., the brain, kidneys, and heart) are effectively perfused at pressures 20 to 30 mm Hg less than normal. This “reserve” allows arterial dilating agents to decrease systemic blood pressure and cardiac afterload without compromising critical organ blood flow. As peripheral resistance decreases, in the patient with CHF, stroke volume increases. In the presence of mitral insufficiency, the regurgitant fraction that enters the pulmonary circulation and reenters the heart is reduced. Finally, with reduction in volume overload, the end-diastolic volume of the left ventricle is reduced, wall tension is reduced, and myocardial perfusion increases. Venodilators increase the volume of the capacitance vessels, also reducing preload to the right, and subsequently left, ventricle. Preload reducers may also relieve some pulmonary vascular congestion. Potential negative inotropic effects of peripherally acting drugs tend to be masked by baroreceptor-mediated increase in sympathetic tone2 in the normal animal.
KEY POINT 14-14
More than any class of drugs, cardioactive drugs are associated with adverse effects, and their use should be implemented only if they are well understood and proper monitoring tools are available.
Arterial Vasodilators
In the 1980s the role of increased resistance in cardiac failure became a focus of therapy. Drugs that decrease peripheral resistance do so by dilating arterial or resistance vessels. The inclusion of peripheral vasodilators in the armament of treatment for CHF, particularly in its early stages, has proved useful in reducing the dependency on digitalis for long-term treatment. However, their efficacy as venodilators increasingly is being challenged. Those drugs whose mechanisms also contribute to the inhibition of neurohumoral endocrine compensatory responses are more likely to address both the hemodynamic and proliferative alterations that accompany the progressively failing heart compared with those drugs whose actions decrease only resistance. For the latter group, although initially beneficial, their use may contribute to a worsening of disease, particularly if the degree of afterload reduction stimulates a hemodynamic response that counters decreased resistance. Thus care should be taken in the timing of therapy and its implementation.
Hydralazine
Hydralazine is a pure arterial vasodilator whose mechanism is not completely understood. Arteriolar smooth muscle is directly relaxed, perhaps by inhibiting calcium fluxes into the cell.9 Conversion to NO and increased cGMP also have been suggested.39 The decrease in peripheral vascular resistance caused by hydralazine is associated with an increase in cardiac performance.40 Coronary and venous vasculature is not affected. Hydralazine lowers mean arterial pressure, total systemic resistance, and left ventricular filling pressures, causing an overall increase in cardiac performance in dogs with left ventricular failure.40 In addition to its vasodilatory effects, hydralazine has been associated with a number of other effects that might benefit the patient with CHF. A positive inotropic effect has been described, perhaps reflecting stimulation of cAMP through β-receptors.41 Hydralazine acts as an antioxidant by inhibiting membrane-bound enzymes that form free radicals, including super oxide.42 More recently, hydralazine inhibition of prolyl hydroxylase domain (PHD) enzymes has been described, ultimately leading to an increase in vascular endothelial growth factor [VEGF], which has a number of positive effects. Endothelial cell growth is associated with angiogenesis, coronary vessel density, and improved myocardial perfusion;39 VEFG also is antiapoptotic and cardioprotective in animal models.
Hydralazine binds to smooth muscle, resulting in a biological half-life that is longer than its plasma half-life. The drug is well absorbed after oral administration in both dogs and humans. However, in humans it is subject to first-pass metabolism with elimination by acetylation. The extent of first-pass metabolism in the dog, which is deficient in acetylation, is not described. Peak effects occur in the dog at 3 to 5 hours.3 The incidence of adverse reactions may be significant. Hydralazine frequently causes increased heart rate; this effect may prove to be detrimental to the patient with CHF because of increased myocardial oxygen demands. β-blocker therapy (or, historically, in the case of myocardial failure, digitalis therapy) may be indicated to slow the heart rate. Hypotension may occur but is largely prevented by proper dose titration.3 If sufficient, hypotension may activate the RAAS.43 In humans, hydralazine has been associated with a variety of immune-mediated reactions, including a well-described drug-induced lupuslike syndrome.9 A previous indication for hydralazine include afterload reduction in patients with moderately early to late signs of CHF. Hydralazine should be administered in small increments until an effective dose is reached. The advent of the ACE inhibitors has largely replaced the use of hydralazine, which currently is limited to animals that cannot tolerate or respond to ACE inhibitor therapy. In a canine model of chronic left ventricular dysfunction, however, hydralazine combined with nitrate therapy can cause a more marked increase in stroke volume compared with ACE inhibitors alone.44 As the beneficial effects of hydralazine on the failing heart are realized, its use may increase.
Calcium Channel Blockers
Structure–Activity Relationship
Five types of calcium channels have been identified, with the L, N, and T subtypes the best characterized (the other two being P/Q and R subtypes). Each comprises a major subunit, α1, which is the major pore-forming unit of the channel, and associated subunits α2, β, γ and δ, which modulate α1. Calcium channels can be broadly blocked by large divalent (cadmium and manganese). Currently, 10 CCB cations have been approved for use in human medicine, all targeting the α-1 subunit.45 Three categories target L-channel blockers, each targeting different domains of the α1 subunit: phenylalkylamines, represented by verapamil; benzothiazepines, represented by diltiazem (Figure 14-6); and the dihydropyridines, represented by nifedipine (including amlodipine, felodipine, nicardipine, and others). A newer category of CCBs, represented by mibefradil, selectively block T-type channels.45 The drugs vary among classes in pharmacodynamic effects and adverse events.
Pharmacodynamic Effects
Although calcium channel (or entry) blockers are also referred to as calcium antagonists, they do not directly antagonize calcium. Rather, they inhibit the entry of calcium into the cell or inhibit its mobilization from intracellular stores. CCBs inhibit the voltage-dependent channels in vascular smooth muscle at significantly lower concentrations than that necessary to interfere with the release of intracellular calcium or receptor-operated channels.2 The pharmacodynamic effects of the CCBs reflect differences in potency at the various tissue receptors (i.e., either cardiac or vascular).
The effects of calcium entering cells by way of L-type channels is better documented than that entering T-type channels. Most clinically used CCBs block exclusively L-type channels, which are the most effective in the vasculature. Vasodilator effects of CCBs are primarily arterial, with little to no venodilator effects. Coronary vasodilation is significant but variable among drugs. The order of vasodilator potency of prototypical drugs from each class is nifedipine > verapamil > diltiazem. This may be balanced by differences in oral bioavailability; as such, the magnitude of the hemodynamic effects of the calcium channel antagonists also reflects the route of administration. Bioavailability is reduced (in humans) as a result of first-pass metabolism for nifedipine > verapamil > diltiazem. The impact of bioavailability on therapy can be complex. For example, whereas oral bioavailability of diltiazem is only 50%, chronic therapy is facilitated by decreased metabolism, which increase bioavailability. Diltiazem (discussed in greater depth as a class IV antiarrhythmic) is metabolized by acetylation, a phase II conjugation system; however, deficiencies in clearance in the dog have not been described.
All three prototypic drugs are available as oral preparations. Both verapamil and diltiazem are available as an intravenous solution for the rapid treatment of supraventricular arrhythmias. Hypotension, bradycardia, and tachycardia (generally reflex) are the predominant clinical indicators. In patients with poor myocardial reserve, exacerbation of CHF may result in peripheral or pulmonary edema. Further clinical pharmacology and side effects may be addressed for specific drugs under the appropriate category.
Amlodipine is a congener of nifedipine. Nifedipine causes vasodilation at concentrations that have little effect on the heart. Like nifedipine, amlodipine affects predominantly smooth rather than cardiac muscle and decreases total peripheral resistance. However, even at doses causing vascular effects, amlodipine has little effect on sinus node function and cardiac conduction. Thus a major advantage to amlodipine compared with other CCBs is that it may not cause reflex cardiac stimulation.
Vasodilator effects of selected CCBs may reflect modulation of NO. For example, amlodipine, but neither nifedipine nor diltiazem, experimentally causes NO release from canine coronary microvessels.46 The clinical relevance of this finding is not yet clear but may imply that such CCBs are particularly effective for treatment of heart failure. Calcium channel blockade appears to have no effect on thrombus formation.47,48 The effect of amlodipine on myocardial contractility is not clear, but most evidence to date does not support a clinically relevant positive inotropic effect.
At physiologic pH, with a pKa of 8.6, amlodipine is largely ionized, which contributes to a gradual association with the calcium channel receptor. Onset of action is thus a gradual event if therapy is begun with a loading dose. Among its peripheral vasodilatory effects, amlodipine prevents coronary vasospasm in response to a number of vasoconstrictive stimuli. Amlodipine has a protective effect against myocardial injury in an animal model of heart failure; the mechanism may be inhibition of NO induction mediated, in turn, by cytokines. Amlodipine inhibits ouabain-induced production of IL-1a, IL-1b, and IL-6, an action that appears to be calcium dependent in mononuclear cells.35
Clinical Pharmacology
In humans, amlodipine disposition is markedly variable. Peak concentrations following single oral administration do not occur for 6 to 12 hours; bioavailability ranges from 64% to 90% and is not affected by food. Protein binding is approximately 95% in human hypertensive patients. The elimination half-life is long. Approximately 90% of the drug is metabolized to inactive products. Based on limited information, the disposition of amlodipine in dogs appears similar to that in humans. Oral bioavailability in dogs is 88%; at least 50% of the drug is cleared by nonrenal mechanisms, and renal clearance includes that of both parent compound and its metabolites. The elimination half-life of amlodipine in dogs is 30 hours; little of the drug is excreted unchanged in the kidneys.49 As with many cardiovascular drugs, amlodipine contains a chiral carbon. In humans each contributes to approximately 50% of the total area under the curve (the balance leaning toward S), but only the S enantiomer is pharmacologically active. The S-isomer is characterized by a longer half-life (49.6 hours) in humans compared with the R-isomer (35 hours); the long half-life allows once-daily dosing.50 Enantiomer information is not available for dogs. Steady-state concentrations are not achieved for 5 to 10 days. Although administration of a loading dose may decrease the time to reach steady state, delayed response also reflects drug-receptor binding kinetics. Despite its use in cats, no pharmacokinetic data could be found for amlodipine in cats.
Drug Interactions and Side Effects
As a class, CCBs are involved in a number of drug interactions. Drugs that generally inhibit drug-metabolizing enzymes (e.g., cimetidine, chloramphenicol) will prolong the elimination and thus the cardiovascular effects of several CCBs. Selected CCBs can, in turn, prolong the elimination of drugs (e.g., cyclosporine, theophylline, and digoxin). The effects vary with the drug and are more likely with diltiazem and verapamil, but are largely absent for amlodipine. Side effects of CCBs vary with the primary pharmacodynamic effect. The major toxicities associated with CCBs are excessive vasodilation, which may activate the RAAS; negative inotropy; and depression of sinus nodal rate and atrioventricular conduction (negative chronotropy). These latter effects may be of therapeutic benefit (see the discussion of antiarrhythmics). Overzealous therapy associated with activation of the RAAS has been demonstrated in normal dogs receiving 0.57 mg/kg orally twice daily for 6 days.53 A number of oral adverse drug events have been attributed to cardiovascular drugs.51 Among them is gingival hyperplasia, particularly by nifidipine and its congeners, including amlodipine.52 Amlodipine has been associated with gingival hyperplasia in dogs; the risk may be worsened in animals receiving other drugs associated with this adverse drug event (ADE)(e.g., cyclosporine).
Clinical Use
Amlodipine, a congener of nifedipine, is often the drug of choice for treatment of feline hypertension.54 Because its actions are independent of the renin–angiotensin–converting enzyme system, the renal afferent (rather than efferent) artiole is preferentially dilated with amlodipine. Although renal perfusion and glomerular filtration are preserved in renal stressor states such as hypotension, increased glomerular perfusion may activate the RAAS, which may worsen renal disease in the impaired kidney. Accordingly, amlodipine should not be used as the sole afterload reducer in animals with myocardial failure associated with neural humoral compensatory mechanisms. As such, the use of amlodipine in dogs tends to be limited to hypertension associated with minimal compensatory mechanism. Mathur and coworkers55 have demonstrated an antihypertensive effect of amlodipine (0.25 mg/kg orally daily) in cats with surgically induced hypertension associated with renal insufficiency.
Other indications for amlodipine therapy include afterload reduction in dogs whose myocardial failure has not responded or for those that have developed an intolerance to enalapril and diuretics. In human patients a synergistic effect occurs when amlodipine or another peripherally acting CCB is combined with ACE inhibitors. The complementary actions of CCBs and ACE inhibitors may facilitate control of systemic hypertension while minimizing detrimental renal effects. In humans the co-administration of amlodipine and ACE inhibition at low doses has been suggested to provide superior renoprotective benefits compared with those of either drug alone.56 When combined with other therapies, a lower dose may be indicated (see Table 14-3). To guide therapy, clinicians should monitor patients with a targeted pressure of less than 150 mm Hg. Response should occur in 24 to 48 hours.
The use of amlodipine in veterinary cardiology has been reviewed.57 Using an open, uncontrolled study, Jepson and coworkers58 prospectively studied for 7 years (1998-2004) the efficacy of amlodipine (0.625 mg/cat/day) in hypertensive cats (n=141; median 15 years of age), with systolic blood pressure (SBP) 195 mm Hg; (184 and 214 mm Hg, 25th and 75th quartile, respectively). The dose of amlodipine was increased to 1.25 mg/cat if SBP remained above 160 mmHg. Phosphate-restricted diets were offered for azotemic cats, with aluminum hydroxide initiated if hyperphosphatemia became uncontrolled. Predictors of duration of survival in cats that did not survive to study end (n=89) were limited to urine protein: creatinine ratios (UPC), with the decline significant.
Helms59 studied transdermal absorption of amlodipine (0.625 mg once daily) using a nonrandomized (oral dosing occurred first), nonblinded design in client-owned cats (n=6) with systolic hypertension (SBP ≥180 to 220 mm Hg depending on underlying cause). Oral doses were titrated up until SBP < 180 mm Hg was achieved; the final dose was maintained for 7 days, and blood was collected at that point. Cats were then crossed over to transdermal amlodipine at the same oral dose established for each cat. Blood pressure was recorded at 3 and 7 days, and plasma amlodipine was determined 12 hours after the last dose at 7 days for both treatment groups. After oral dosing SBP decreased a median of 73.5 mm Hg below baseline; after 7 days of transdermal dosing, SBP remained 52 mm Hg below baseline. The relative decrease was greater after oral compared with transdermal administration. Plasma amlodipine concentrations ranged from 5.7 to 18.7 ng/mL after oral administration compared with 1.4 to 4.5 ng/mL after transdermal administration. The lack of a crossover design may be a limitation of the study because a time effect could not be considered. This may be particularly important for a drug with a half-life that is likely to exceed 30 hours (the canine half-life) in cats (resulting in time to steady state of 3 to 6 days) coupled with further delay related to drug receptor interactions. This could result in a substantial hold-over effect from the first treatment simply as a result of drug concentrations. Further, a cumulative effect for the sequential treatment groups would not be identified. Post transdermal SBP was not compared with post oral SBP, so it is not clear if the decline was significant.
Although diltiazem is not recognized for vasodilatory actions, it apparently can improve GFR and enhance urine production in dogs with leptospirosis. The presumed mechanism is renal arterial vasodilation and subsequent reversal of renovasoconstriction. Mathews and Monteith60 retrospectively studied the impact of diltiazem on acute renal failure in dogs (n = 11; seven dogs received standard care) associated with leptospirosis. Diltiazem was administered intravenously at 0.1 to 0.5 mg/kg slowly, followed by 1 to 5 μg/kg/min within 60 hours of admission and was continued until serum creatinine stabilized. Compared with standard therapy, renal recovery defined by reduction in serum creatinine occurred almost twice as fast in the diltiazem-treated group compared with the standard group, without a change in systemic blood pressure.
For cats with hyperthyroidism, the degree of hypertension associated with hyperthyroidism generally is mild, unless accompanied by renal insufficiency. Because hyperthyroidism-induced hypertension associated with hyperthyroidism alone is due to high-adrenergic output (causing increased cardiac inotropy and rate), β blockers are preferred to calcium channel blockers for control of hypertension. However, the addition of amlodipine may be indicated in hyperthyroid hypertensive cats that also exhibit renal insufficiency.
Angiotensin-Converting Enzyme Inhibitors
Structure–Activity Relationship
ACE inhibitors are carboxyalkyl dipeptide or tripeptide drugs whose chemistry yields three classes of drugs (see Figure 14-6): the sulfhydryl-containing drugs, which are structurally related to captopril; dicarboxyl-containing drugs related to enalapril (including lisinopril, benazepril, ramipril, and quinapril); and phosphorous-containing drugs related to fosinopril.18 At least nine drugs have been approved in the United States for use in humans. Those approved for veterinary use are in the dicarboxyl group, including enalapril (United States), and ramipril and imidapril (Canada and Europe). With exception of lisinopril, each is a prodrug, being metabolized, at least in part, by CYP 3A4, to their respective active form (enalaprilat, benazeprilat, ramiprilat, and imidaprilat, respectively). Although differences exist in potency for ACE, all appear clinically equal in the inhibition of RAAS, and no compelling reason exists to select one ACE inhibitor over another based on pharmacodynamic response.18
Pharmacodynamic Effects
The primary effect of ACE inhibition is prevention of the conversion of AGI, which is relatively inactive, to the active AGII. However, ACE inhibitors also inhibit bradykinin inactivation. Because bradykinin stimulates prostaglandin synthesis, pharmacodynamic actions of ACE inhibitors may reflect bradykinin or prostaglandin activity.18 However, of the two actions ACE inhibition is a more sensitive and measurable indicator of the complex dose–response relationship that characterizes the primary actions of ACE inhibitors (see the section on clinical pharmacology).61 Actions of ACE inhibition target the neurohumoral and renal compensatory responses associated with myocardial failure. The detrimental sequelae of neurohumoral compensatory effects involving the RAAS have been previously reviewed. Specific inhibition of ACE decreases circulating levels of AGII and aldosterone. Effects include vasodilation (arterial), decreased systemic blood pressure, increased cardiac output, and reduced heart rate. Aldosterone secretion is reduced (but not obliterated), and natriuresis (loss of sodium in urine) occurs. Mild venodilation reduces some preload. ACE inhibitors also target a variety of mediators associated with myocardial remodeling. In the normal feline kidney, ACE inhibitors mildly increase Na+ and Cl− urinary excretion and may increase K+ excretion. The ACE inhibitors have a positive inotropic effect on the heart but do not increase heart rate. The mechanism is unclear, but with enalapril it is associated with increased cardiac vasoactive intestinal peptide in rats.62 Primary cardiac effects increase their importance in the management of cardiac disease accompanied by CHF. Inhibition of ACE also increases circulating concentrations of the endogenous stem cell regulator Ac SDKP (N-acetyl-seryl-aspartyl-lysyl-proline), which may contribute a cardioprotective effect.18
Clinical Pharmacology
Pharmacokinetic–Pharmacodynamic Relationship
The use of ACE inhibitors has been reviewed in dogs and cats.12,61,63 The exact relationship among ACE inhibitors, ACE inhibition, impaired AGII production, pharmacodynamic responses (blood pressure and cardiovascular), and pharmacokinetics is complex.61 Most ACE inhibitors used in animals (e.g., enalapril and benazepril) are prodrugs, which are more easily absorbed than the active metabolite. Those that are not prodrugs include captopril and lisinopril.64 In addition to passive oral absorption, prodrugs are actively transported in the jejunum by peptide carrier-mediated transport (PEPT). Substrate specificity has been described for the different transport proteins.61 The importance of transport on drug absorption is not clear. Food does not appear to affect absorption. Parent compound oral bioavailability is low (20% to 40% in dogs); only a fraction of the low bioavailability appears to reflect first-pass metabolism by hepatic carboxyl esterases to active metabolites. Maximum drug concentrations occur approximately 30 minutes after administration in the dog. A small amount of drug not metabolized on first pass will be metabolized by esterases located in tissues.61
Tissue distribution and subsequent action of ACE inhibitors is among the most complex of drugs, being influenced by lipophilicity; systemic and local binding; receptor numbers and affinity; and, for prodrugs, presence of local metabolizing enzymes. Protein binding and its profound impact on pharmacokinetic–pharmacodynamic relationships are described later in this chapter. Lipophilicity determines distribution, particularly of the prodrug, into sanctuaries such as the brain, where local esterases produce the active compound. Accordingly, lipophilic prodrugs such as fosinopril are more likely to affect the central nervous system compared with less lipophilic drugs such as enalapril.61 Elimination of ACE inhibitors (i.e., active metabolite) is largely renal. However, the magnitude excreted renally varies with the drug.61 In dogs enalaprilat is largely (85% to 95%) eliminated by renal clearance, whereas benazeprilat and ramiprilat are cleared through both the kidney (45%) and bile. Fosinopril is also cleared (in humans) by liver and kidneys.
Protein binding of ACE contributes to complex pharmacokinetic–pharmacodynamic responses.61 Binding to proteins is both nonspecific, to albumin, yielding an inactive state, or specific, to ACE (located in circulation or tissue vascular endothelium), resulting in an active complex. Nonspecific binding is nonsaturable, whereas specific binding is saturable, with the latter resulting in nonlinear pharmacokinetics. Based on modeling, circulating ACE represents approximately 5%, 10%, and 30% of the total ACE pool in dogs, cats, and humans, respectively, with the balance represented by tissue ACE. Endothelial ACE occurs in many tissues but predominates in the lungs. Tissue endothelial ACE acts locally (paracrine) but also is released such that it can act systemically (hormonally). Bound, endothelial ACE is regularly cleaved to yield circulating ACE, which, is in turn, bound to circulating plasma proteins (in an inactive form) to ensure a steady supply of either ACE or its inhibitors. Endothelial ACE is accessible to ACE inhibitors in the extracellular space, facilitating an immediate response to ACE inhibitors. However, because the drug is bound to ACE, the (active) ACE inhibitor will not be detected (analytically). If drug is detectable, then all available tissue binding sites are saturated. Thus a decline in response that reflects a decline in drug concentrations occurs only after tissue sites are not saturated, which occurs only at drug concentrations that are not detectable. As such, the saturable nature of binding to ACE by the ACE inhibitor results in a nonlinear dose–response curve (detectable concentrations do not correlate well to response). Thus, in contrast to most drugs for which the terminal component of the plasma-concentration versus time is a reasonable guide to duration of effect, for ACE inhibitors the terminal curve reflects what is generally considered distribution for other drugs. The true terminal curve for ACE inhibitors reflects slower elimination at very low, but largely non-detectable concentrations. The slower elimination occurs as receptors become unsaturated, and free drug is eliminated; as such, it must take into account both elimination from the body and the relationship between drug and receptors—that is association–dissociation interactions. Consequently, the true terminal elimination phase of ACE inhibitors is physiologically (rather than pharmacokinetically) based, being influenced by receptor numbers (total binding capacity), the affinity between drug (metabolite) and receptor, and the amount of drug available for binding.61
Because bound, nondetectable drug is active drug, the pharmacodynamic (biologic) half-life of the ACE inhibitor might be anticipated to be much longer than the (detectable) plasma elimination half-life. Indeed, the complex pharmacokinetic–pharmacodynamic relationship of ACE inhibitors affects the design of a dosing regimen, particularly when adjusting doses for disease. This complexity reflects several characteristics. First, for most drugs, response correlates with the time course of drug concentrations. However, for ACE inhibitors response correlates better with tissue concentrations that are not detectable. Second, saturation of tissue receptors is characterized by a lag time. Therefore the slope of the true terminal phase of the plasma elimination curve is very flat, and maximum response may not be realized for several days.61 Third, once saturation occurs, further increases in dose are not likely to cause an increase in response. However, a very low dose (e.g., 0.03 mg/kg) might not be prudent because saturation will take longer and the maximal response may occur more slowly than with higher doses (e.g., 0.125 mg/kg). Simulation studies examining the pharmacokinetic–pharmacodynamic relationship of benzeprilat and ACE inhibition predict ACE inhibitor doses higher than 0.125 mg/kg every 24 hours will increase ACE inhibition very little,65 a finding supported by a study of ramilprat in cats.66 King and coworkers67 also demonstrated that increasing doses of 0.25, 0.5, and 2 mg/kg were well tolerated in cats but did not result in a comparable increase in ACE inhibition.
KEY POINT 14-18
The complex relationship between drug and tissue receptors includes a persistent effect in the absence of detectable drug and a saturable dose–response relationship.
For ACE inhibitors, neither circulating nor tissue concentrations significantly differ after 8 days of therapy. Consequently, the pharmacodynamic response tends to exceed the time course of the metabolite by several orders of magnitude based on measured elimination half-life of the metabolite. For example, the elimination half-life of enaliprat in dogs approximates 61 minutes, compared with a predicted pharmacodynamic half-life of 17 to 19 hours.61 For benazeprilat ACE inhibitory activity approximates 100% after multiple dosing at 0.25 mg/kg once daily and persists throughout each 24-hour dosing interval,68 despite the fact that the plasma elimination half-life of benazepril is 1.4 hours in dogs (39 ± 6 min for benazilprilat). This is consistent with the predicted pharmacokinetic–pharmacodynamic half-life of 17 to 19 hours.61,65 In cats receiving benazeprilat, plasma ACE was inhibited at 0.25 mg/kg, with 90% of activity persisting at 24 hours after a single dose.65 The modeled terminal elimination half-life of the active metabolite is approximately 28 hours compared with a measured half-life of approximately 60 minutes. Finally, in Beagles ramipril and ramiprilat elimination half-lives are 0.5 and 0.75 hours, respectively,69 with the pharmacokinetic–pharmacodynamic half-life predicted to be approximately 23 hours.61 In cats benazeprilat is excreted predominantly (about 85%) through the bile. Plasma half-life is approximately 1 hour, but the pharmacokinetic–pharmacodynamic half-life is characterized by an elimination half-life of 28 hours following single oral dosing.61
Contributing to the complex pharmacokinetic–pharmacodynamic relationship will be variability among normal animals resulting from differences in disposition, in receptor number and affinity. Further, concentrations accumulate with multiple dosing up to 50% in dogs.68,70 It is likely, however, if these differences in plasma disposition will translate to differences in pharmacodynamic response. Indeed, the pharmacokinetic–pharmacodynamic relationship between ACE and ACE inhibition is likely to mitigate differences in response among animals that reflect altered clearance because of disease, unless the disease also affects drug-receptor interaction. For example, a decrease in renal function that prolongs the half-life of an ACE inhibitor twofold is not likely to translate into a duration of effect that is twice as long. As such, benazepril (cleared predominantly by the liver) offers no advantage to enalapril (renally cleared) in the renal-compromised patient. The complex nature between pharmacokinetics and pharmacodynamics must be taken into account in studies that attempt to describe changes in response associated with disease, age, gender, drug interactions, and so forth. Such studies must address not only changes in pharmacokinetics but also the ways in which these changes influence (if at all) the physiologic response. Additionally, such studies must also address the impact of those factors on the interaction between drug and receptor (i.e., ACE), including the impact on the amount of ACE and its affinity for the drug. Finally, none of the discussion thus far has addressed the response of the body to inhibition of ACE. Because mechanisms other than ACE exist for formation of AGII, response to ACE inhibitors may decline within 24 hours after a maximum response has been realized despite maintenance of drug-receptor interactions.61
Dose modification in the diseased patient
Response to ACE inhibitors can be influenced by several factors. Sodium depletion in the dog shifts the ACE inhibitory mechanism of action from decreased renal AGII to enhanced kinin activity. Decreased circulating ACE associated with ACE inhibitors may correct after several days to weeks, a phenomenon referred to as angiotensin escape. Plasma renin activity may increase as negative feedback is lost. This effect may vary with the drug but is higher in hypertensive than in normotensive dogs.12 However, response to therapy (perhaps muted) may continue because of inhibition at the tissue level or alternative mechanisms of action. Variability in ACE inhibitory response also may reflect genotype differences in plasma and tissue ACE concentration or receptors. Genetic polymorphism appears to influence response in humans; ACE gene polymorphism has been demonstrated in dogs.12
The impact of liver disease on formation of active drug has not been determined for all prodrug ACE inhibitors. However, severe liver disease does not appear to impact metabolism of benazepril to benazeprilat in dogs.71 Dose modification in response to renal disease–induced altered drug disposition for drugs cleared by the kidney is complicated by the apparent lack of correlation of response to active drug concentration in plasma or vascular tissue.12,61 The impact of renal failure on ACE inhibitory pharmacokinetics (drug disposition) and pharmacodynamics (drug–receptor–response interaction) has been summarized by Lefebvre and Toutain.12 The impact on overall ACE inhibition reflects not only changes in drug concentration associated with altered clearance of drug from plasma but also distribution to tissue endothelial ACE and binding kinetics to ACE. Based on mathematical modeling in dogs with experimentally induced renal disease (GFR decreased by 50%), enapirilat clearance may decrease up to 45%, with changes in the area under the curve markedly varying from 30% to 40%. These changes are offset somewhat by decreased binding (decreased potency), such that enaliprilat inhibitory activity increased by about 65% in the first 24 hours. In contrast, neither benazeprilate nor ramiprilat response appears to be affected by renal disease in dogs.72 Dose adjustment for benazepril is not necessary in cats with renal insufficiency.73
In the cat the impact of increasing the dose of ACE inhibitors may vary with the intent (e.g., hypertension versus progressive renal disease). For example, feline hypertension in patients with low or undetectable plasma renin activity and aldosterone responded poorly to enalapril at 0.25 mg/kg once daily, but the response (decreased ACE activity) doubled when a second dose was added.74 In contrast, although benazepril was associated with a decrease in blood pressure and an increase in GFR in a surgical model of renal insufficiency, increasing the dose of benazepril (from 0.25 up to 2 mg/kg daily for 6.5 months) did not result in a proportional improvement in response in cats with chronic renal failure.12,75 Indeed, overzealous increase in dosing may put patients at risk for initiation of neurohumoral responses that may contribute to renal dysfunction. These studies highlight the complex nature of dose–response relationship of ACE inhibitors and underscore the need for monitoring response when changing doses regardless of species or disease state.
KEY POINT 14-19
Overzealous therapy with angiotensin-converting enzyme inhibitors may activate or potentiate the very neurohumoral endocrine responses targeted with therapy.
Ramipril and ramilprilat kinetics have been studied in both dogs72 and cats.66 In Beagles (n=10) the elimination half-life of free drug is 11.5 ± 6.0 minutes; this increased to 14.9 ± 7.2 minutes when renal impairment was surgically induced.72 After renal impairment, GFR was decreased by 58%. Although clearance of the free fraction of drug and metabolite was reduced, no pharmacodynamic changes occurred in renally impaired dogs, leading the authors to conclude that dose adjustment was not necessary in renal disease. In cats ramipril and its active metabolite ramiliprat have been reported after single and multiple oral dosing (0.125 to 1 mg/kg daily).66 Peak concentrations of ramiprilat occur in 1.5 hours, and the mean elimination half-life ranged from approximately 20 to 30 hours, depending on the route. Steady-state concentrations of metabolite were present after 2 days of dosing, and food did not affect oral absorption. Although a dose-dependent effect on ACE activity was present with single dosing, after multiple dosing the maximum inhibition of ACE was the same for all doses. Detectable drug was still present 72 hours after the last dose. Inhibition was still present 24 hours after the last dose. Based on their study, the authors concluded that the elimination of the active metabolite would not be altered in patients with renal disease; a dose of 0.125 mg/kg daily was suggested as a basis for further studies.
Adverse Events
Potential adverse drug events associated with ACE inhibitors have been reviewed12 and are largely limited, in dogs and cats, to the sequalae of hypotension (previously discussed) and to nephropathies induced by pharmacodynamic response to the drugs. In general, the risk is more likely in patients also receiving diuretics or in the face of impaired renal function. However, acute hypotension is a risk as therapy is begun, particularly in renin-dependent (sodium-depleted) patients. Clinical signs indicative of hypotension may include fatigue and apathy. Increased dietary salt or decreased diuretic therapy may be indicated initially as ACE inhibitor therapy is begun. Acute renal failure may occur as a result of altered renal perfusion. The RAAS, and particularly AGII, is the principal autoregulatory mechanism that maintains renal perfusion in the presence of low arterial pressures. In the kidney, angiotensin regulates both renal blood flow and GFR by modulating constriction of the postglomerular efferent arteriole in the presence of reduced GFR. In addition, tubular sodium is reabsorbed, and renin release is inhibited. Inhibition of ACE and AGII precludes constriction of the postglomerular efferent arteriole (Figure 14-7).76 As ACE inhibitor therapy is begun, total peripheral vascular resistance, renal vasoconstriction, and renal perfusion pressure decrease. Net glomerular filtration decreases in concert with decreased glomerular hydrostatic pressure. Decreased glomerular filtration after ACE inhibitor therapy is more likely to occur in the presence of excessive vasodilation, moderate to severe volume depletion, or minimal myocardial reserve. In patients with mild to moderate cardiac dysfunction, renal filtration generally is maintained despite initiation of ACE inhibitor therapy as long as afterload is decreased such that cardiac output (and thus GFR) can increase. As heart failure progresses, the risk of acute renal decompensation increases because reflex efferent arteriolar tone becomes increasingly important to the maintenance of glomerular filtration in these patients. All ACE inhibitors are equally likely to have a negative impact on renal function in patients at risk. This includes benazepril, for which clearance depends on liver metabolism.
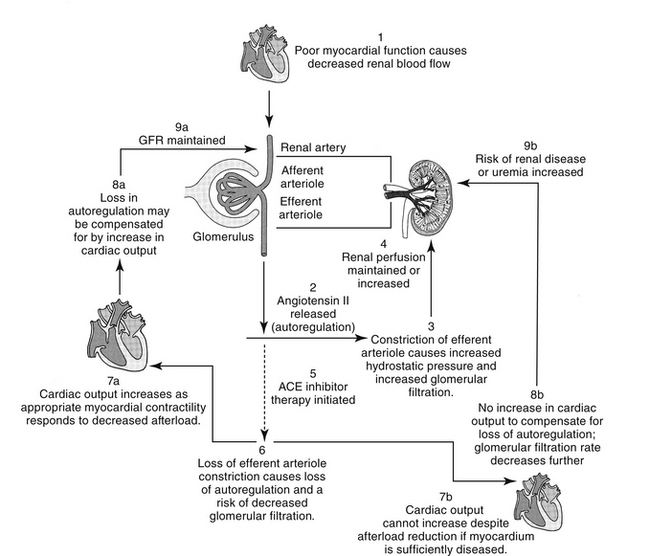
Figure 14-7 Despite their kidney-sparing effects in patients with glomerulonephritis, angiotensin-converting enzyme (ACE) inhibitors may contribute to renal disease in patients whose renal blood flow is threatened. In such patients maintenance of glomerular filtration may depend on increased efferent arteriolar tone (vasoconstriction). Use of ACE inhibitors reduces this tone. In patients whose cardiac function is sufficient (including those that respond to decreased afterload), increased cardiac output can compensate for decreased efferent arteriolar tone, thus maintaining glomerular filtration rate. In patients that are sodium depleted or have severe myocardial dysfunction, glomerular filtration rate may decline. Dashed line, inhibited.
KEY POINT 14-20
Renal adverse events associated with angiotensin-converting enzyme inhibitors are more likely if renal autoregulation is impaired by drug or disease.
Initial acute ACE inhibitor treatment of at-risk patients can decrease both creatinine clearance and GFR. Assuming that renal damage is not irreversible, however, chronic treatment generally causes GFR to return to baseline levels. An initial increase in serum urea nitrogen and creatinine associated with initiation of therapy may actually indicate a positive renal response; in human patients an increase from baseline of up to 30% in serum creatinine is acceptable as long as the GFR remains above the minimum of 20 mL/min. However, GFR is not routinely measured in animals and an ”acceptable” increase in GFR has not been established. Accordingly, an increase in either serum urea nitrogen or creatinine concentrations may indicate the need to decrease or temporarily discontinue therapy in dogs and cats. Although deterioration of renal function associated with ACE inhibitor therapy is generally reversible if therapy is discontinued, damage may become irreversible if decreased GFR persists. Therapy is contraindicated with preexisting hypotension, hypovolemia, hyponatremia, and acute renal failure. ACE inhibition may potentiate hypotensive episodes in surgical patients. However, withdrawal of therapy to prevent hypotension should be reconsidered in patients receiving ACE inhibitors for treatment of hypertension. Should it occur, hypotension can respond to fluid therapy, and as such, ACE inhibition probably should be continued up to the surgical procedure. Sodium depletion can activate the RAAS in normal dogs and cats. Experimentally, the use of ACE inhibitors and the subsequent loss of renal autoregulation in the presence of sodium restriction is accompanied by a decrease in renal blood flow, GFR, and filtration fraction. Accordingly, the use of ACE inhibitors in the presence of a low-sodium diet should be accompanied by frequent monitoring of renal function, particularly in patients predisposed to renal dysfunction.
The BENCH (BENazepril in Canine Heart Disease) study group reported the long-term tolerability of benazepril in dogs with CHF.77 Although the authors found plasma creatinine concentrations to be lower in the benazepril group than in the placebo group, a significant difference could not be demonstrated. However, serum creatinine levels tended to be higher than normal more often in the placebo group, leading the investigators to suggest that improved renal function was associated with benazepril. No abnormalities in electrolytes were associated with benazepril.
Although aldosterone production is impaired in the presence of ACE inhibitor therapy, hyperkalemia is unlikely even in the presence of high-potassium diets, unless the patient is otherwise predisposed to potassium retention. Potassium monitoring might be indicated in the presence of dietary sodium restriction78 or when combined with potassium-sparing diuretics such as spironolactone. Because the impact of aldosterone on sodium retention and potassium excretion is muted, the combination of ACE inhibitors with potassium-wasting diuretics decreases the risk of hypokalemia.78 The impact of ACE inhibitor therapy on erythropoiesis associated with renal disease is unclear, but studies suggest renal erythropoietin production probably will not normalize in response to ACE inhibitor therapy. As such, supplementation may be indicated.
The ACE inhibitors reportedly cause a dry cough in up to 20% of human, particularly female, patients. Onset ranges from 1 to 26 weeks of therapy, with clinical signs resolving within the first week of discontinuing therapy. The mechanism may reflect accumulation of pulmonary bradykinin or prostaglandins; interestingly, treatment with aspirin has reduced cough, suggesting a possible role for prostaglandins.18
Despite their ability to decrease proteinuria (discussed at more length later in this chapter), ACE inhibitors have been associated with the advent of proteinuria in human patients. Therapy need not be discontinued if it develops. Therapy also has been associated with skin rashes and angioedema. The latter is not dose related and generally occurs soon after dosing, within the first week of therapy.18 Neutropenia, glycosuria, and hepatotoxicity are rare side effects reported in humans.
The ACE inhibitors should not be used in pregnant animals. In human women therapy in the first trimester appears to be safe but results in multiple fetopathologies (potentially related to fetal hypotension) when administered in the second and third trimester.
Drug Interactions
Lefebvre and Toutain12 reviewed the literature addressing clinically relevant adverse reactions that might reflect interactions between ACE inhibitors and simultaneously administered drugs. Because AGII and aldosterone synthesis can occur in the absence of ACE, drugs that contribute to hypotension, sodium loss, or alteration of renal autoregulation should be avoided or used cautiously in patients receiving ACE inhibitors. The risks of combining ACE inhibitors and diuretics on renal dysfunction have been documented in humans and dogs. AGII and aldosterone may increase, despite the administration of furosemide in patients receiving ACE inhibitors. Further, the diuretic effect can decrease proportionately with GFR when combined with ACE inhibitors. However, the combined use of ACE inhibitors and sodium-wasting diuretics, and specifically furosemide, decreases the risk of hypokalemia.78 The impact of combined use of nonsteroidal antiinflammatory drugs (NSAIDs) and ACE inhibitors is less clear. Although presumably, NSAIDs that minimize cyclooxygenase-(COX-) 1 inhibition are less likely to be associated with nephropathy, in human patients the risk is equivalent when either traditional or newer NSAIDs are used. A number of studies have investigated the sequelae of combined NSAID and ACE inhibitor therapy in normal dogs and dogs with experimentally induced decreased renal function. In general, no significant detrimental impact on renal function has been reported. However, this should not be interpreted as proof of no effect, and the combined use should be avoided or undertaken cautiously, with additional attention to monitoring of renal function. Note that NSAIDs may attenuate antihypertensive actions of ACE inhibitors, although the impact may be less with those drugs that target principally COX-2. ACE inhibitors may increase the risk of nephrotoxocitiy induced by other agents, including contrast dye.79
The combination of ACE inhibitors with angiotensin receptor blockers (ARBs) might be considered for a beneficial therapeutic effect. However, a meta-analysis that studied the combined use of ACE inhibitors and ARBs found the addition of an ARB to ACE inhibitor therapy was not associated with a reduced mortality rate compared with ACE inhibitor treatment alone. The addition of an ARB did not affect exercise capacity, functional capacity, and quality of life. Of the outcome measures studied, only the rate of hospitalization was reduced with the combination of therapy.80
Beta blockers normalize β-receptor signaling and antagonize cytotoxic effects of circulating catecholamines, which increased with heart failure.80 Additionally, β blockers may also directly block AGII-mediated pathways. As such, β blocker therapy combined with ARBs and ACE inhibitors theoretically might completely remove the effects of angiotensin in cardiac patients. However, results of clinical trials regarding use of the combination in humans are controversial. A meta-analysis found that, compared with ACE inhibitors alone, the combination did not improve survival rates, although hospital stays did shorten.80 Further, subgroup analysis suggested that the addition of ARBs is redundant and combined use of ARBs with ACE inhibitors and β blockers potentially puts the patient at an increased risk of adverse drug events. A subsequent study using the ARB candesartan demonstrated that when it was combined with β blockers in patients also taking ACE inhibitors, it does tend to increase the risk of adverse events.81
Hypotension has been reported with the combined use of the β blocker atenolol, the ACE inhibitor quinapril, and an overdose of the selective serotonin reuptake inhibitor fluvoxamine, treatment with aminophylline was successful.82
Other reported drug interactions include decreased drug absorption when combined with antacids, although the clinical relevance of this finding in light of the relationship between plasma drug concentrations and responses is questionable.
Clinical Use in Canine Patients with Myocardial Disease
The role of ACE inhibitors in general for treatment of CHF associated with myocardial failure in dogs has been well established, with use emerging as part of cornerstone therapy. However, the timing of administration in the progression of diseases remains to be fully defined. The most extensively studied of the ACE inhibitors is enalapril, although captopril, benazepril, and lisinopril have also been studied. More recently, ramipril is being studied in dogs and cats. Captopril, the efficacy of which was demonstrated in an experimental model of canine mitral regurgitation (see BENCH study group)83 was the first generally accepted ACE inhibitor to be used in dogs and cats. Captopril can induce positive hemodynamic effects within 1 hour of therapy, although the effects are short lived (4 hours).84 Its short half-life led to dosing at 8-hour intervals. Subsequently, the safety and efficacy of quinipril were demonstrated when administered once daily (0.5 mg/kg) compared with captopril (0.5 mg/kg every 8 hours or 24 hours, respectively) in dogs (n=92) with CHF.85
Enalapril administered at 12-hour dosing intervals was also demonstrated to be of benefit in dogs with experimentally induced heart failure. The first placebo-controlled study demonstrating efficacy of ACE inhibitors in dogs with spontaneous myocardial disease (dilated cardiomyopathy [DCM] or mitral regurgitation) was the Cooperative Veterinary Enalapril (COVE) Study.86 This multicenter study involving 211 dogs demonstrated that enalapril therapy (for 28 days) was associated with improved longevity and quality of life.86 The IMPROVE87 study group reported in the same issue that enalapril (0.5 mg/kg twice daily) was associated with lower heart rate and mean systemic and pulmonary arterial pressures in dogs (35 with DCM, 22 with mitral regurgitation) compared with dogs in the placebo group. Dogs continued to receive traditional therapies (i.e., digoxin or diuretic). Ettinger and coworkers88 subsequently established the beneficial effects of enalapril for dogs (n=110; 15 locations throughout the United States) with either DCM or other chronic acquired heart disease (primarily mitral insufficiency). Although the study was not well controlled for stage of disease or differences in conventional therapy, results supported the importance of ACE inhibition in improving the quality of life and, particularly in dogs with DCM, prolonging life. The mean time to treatment failure (including treatment with traditional cardiovascular drugs) increased from 77 days in the placebo group to 156 days in the enalapril-treated group. Although more dogs died suddenly in the enalapril treatment group, all but one had preexisting ventricular arrhythmias for which they were receiving antiarrhythmic therapy. The BENCH study group83 focused on long-term survival afforded by the addition of benazepril to dogs (n=162) with class (International Small Animal Cardiac Health Council [ISACHC]) II or III heart failure associated with chronic valvular disease (n=125) or DCM (n=37). This prospective, randomized, placebo-controlled, double-blinded European study was multicenter (n=16) and multicountry (n=4). It studied benazepril (0.25 mg/kg once daily) as either sole therapy or as an add-to traditional therapy for up to 34 months. Mean survival significantly improved from 158 days in the placebo group to 428 days in the treatment group. Survival rates at 1 year were 20% and 49%, respectively, for the two groups. High-risk dogs had not been removed from the study group, suggesting that survival would improve in less severely afflicted animals. The risk of progression to a worsening class of heart failure was also reduced, whereas quality of life measures were improved in the benazepril group. Benefits could also be demonstrated in subgroup analysis for those dogs with cardiovascular disease, but not the smaller sample of dogs afflicted with DCM. Adverse events to benazepril were slightly less (not statistically so) than to placebo.
KEY POINT 14-21
Evidence of clinical efficacy of angiotensin-converting enzyme inhibitors is provided among clinical trials based on improved quality of life, improved survival, and objective measures of cardiac function.
Pouchelon and coworkers89 retrospectively addressed the efficacy of benazpril in dogs with Class II and III CHF (n=141). Dog were classified as untreated if they did not receive benazepril before the appearance of clinical signs of CHF; these dogs served as controls. For the treated dogs, the mean dose was 0.3 ± 0.13 mg/kg. Median survival times were greater (3.3 years) in the treatment group (n=34) compared with untreated dogs (1.9 years, n=59 dogs) for all breeds except Cavalier King Charles Spaniels.
Controversy still exists regarding when ACE inhibitor therapy should be initiated in the patient with CHF, if and when it is the preferred vasodilator, and its position in the sequence of drugs. For humans, ACE inhibitors are indicated for any patient with left ventricular systolic dysfunction even in the absence of clinical signs of overt cardiac failure.64 Studies in humans suggest that inhibition of ACE in patients with systolic dysfunction can prevent or delay the progression of heart failure, decrease the incidence of sudden death, and improve the quality of life.64 Although treatment plans have been offered for enalapril, based on severity of disease,88 the importance that ACE inhibitors may have in attenuating myocardial remodeling and the need for early intervention may supersede treatment plans based on severity.
KEY POINT 14-22
The timing of initiation of angiotensin-converting enzyme inhibitor therapy in relation to other drugs remains controversial.
To minimize negative sequelae of afterload reduction, particularly that leading to activation of the RAAS, treatment with enalapril or other ACE inhibitors might be initiated at a lower dose90 (see Table 14-3) with increased increments at weekly intervals until clinical signs indicate improvement or further increases cannot be tolerated. Renal function should be monitored weekly for the first month of therapy in patients predisposed to adverse renal effects.
The role of ACE inhibitors in cats with hypertrophic cardiomyopathy (HCM) is not clear. However, using an experimental feline banding model of pressure-overload left ventricular hypertrophy, 4 weeks of enalapril (0.5 mg/kg orally once daily) was associated with less intraventricular septum and free-wall diastolic thickness as well as decreased arterial (systolic and mean) pressure compared with placebo. Renal function and other physiologic parameters did not differ between placebo and treatment groups.91
Angiotensin-Converting Enzyme Inhibitors and Treatment of Progressive Renal Disease
The use of ACE inhibitors for the treatment and attenuation of progressive renal disease in humans and animals has been well reviewed.12 The evidence of efficacy is not clear, but therapy is generally accepted as beneficial. It is likely that a combination of therapies will be most beneficial.92 ACE inhibitors have repeatedly been demonstrated to provide superior renoprotective effects in patients with renal disease compared with other vasoactive drugs.93 The potential application of ACE inhibitors for this indication emerged as early as 1985, following a demonstrable beneficial effect on proteinuria and accompanying renal damage in a rat model of renal failure. Potential application to a variety of human renal diseases was demonstrated over the next decades, with use in animals lagging not far behind. A meta-analysis94 of clinical trials in humans with hypertension found a small benefit, probably reflecting reduction in blood pressure. This study was met with controversy, indicating that the role of ACE inhibitors in progressive renal disease has yet to be clearly defined. Benefits demonstrated in dogs and cats have resulted in the approval of benazepril in Europe and Australia for treatment of chronic progressive renal diseases in animals.
ACE inhibitors have three potential positive effects in the patient with renal disease: control of systemic and glomerular capillary hypertension, delayed progression of glomerulosclerosis and tubulointerstitial disease, and decreased proteinuria.12 In humans control of the progression of renal disease depends on control of systemic hypertension but this is not necessarily the mechanism whereby ACE inhibitors impart renoprotection. Systemic hypertension may accompany chronic renal disease in 50% to 93% of dogs and cats, and a similar dependency might be expected in animals. Hyperperfusion and hypertension accompanying renal diseases initially enhances the filtration capacity of remaining nephrons. However, this compensation for nephron loss occurs at a cost. AGII-mediated hypertension leads to distention of the glomerular capillary and mechanical strain on the glomerular mesangial cell. Podocyte coverage becomes inadequate, changing the size exclusion of the glomerulus such that permeability is increased. The exposed mesangial cells stimulate extracellular matrix proliferation and release of proinflammatory cytokines. Inflammatory and other proteins are increasingly filtered through the damaged glomerulus. The protein is toxic to tubular epithelials cells and stimulates further cytokine release. Included is transforming growth factor-β, which stimulates excessive renal deposition of extracellular matrix in both the glomerulus and tubulointerstitium. Persistent inflammation mediated by AGII leads to glomerular and tubulointerstitial fibrosis, a characteristic of end-state renal disease.12
In humans and experimental models, ACE inhibitors have demonstrated renoprotection in selected states of nephrotoxicosis associated with renal vascular hypertension. Examples include experimentally induced cyclosporine nephrotoxicity,95 in which ACE inhibitors decrease renal vascular resistance, increase GFR, and promote diuresis in experimentally induced doxorubicin nephprotoxicity.96 Beneficial effects of ACE inhibitors include decreased deposition of extracellular matrix. The effects of ACE inhibitors have been demonstrated in humans to be greater if therapy is begun early in a variety of renal diseases, with polycystic renal disease being the sole disease thus far that has not responded to therapy.12
KEY POINT 14-23
Efficacy of angiotensin-converting enzyme inhibitor therapy for progressive renal disease in dogs or cats depends on use early in disease.
In dogs a variety of studies have attempted to demonstrate renoprotective effects of ACE inhibitors. Studies in dogs have predominantly involved enalapril, whereas benazepril has been the drug of choice for cats for reasons that are not clear other than approval status of the drugs in the country of origin of each study. Results varied with the disease, the model (including experimental versus spontaneous nephropathies), and doses studied (enalapril at 0.5 mg/kg every 24 hours to 2 mg/kg every 12 hours). Outcome measures generally include objective measures such as azotemia, renal plasma flow, GFR, survival rates, weight gain or loss, and histopathology or more subjective measures such as appetite. In a canine experimental model, differences were limited to fewer histologic lesions after 6 months of treatment (1 mg/kg every 12 hours for 2 weeks followed by 0.5 mg/kg every 12 hours) compared with untreated controls. A controlled study in X-linked hereditary nephritis in Samoyeds demonstrated increased survival time in treated dogs (enalapril at 2 mg/kg every 12 hours from 4 weeks of age); therapy was begun before proteinuria could be detected. In the treatment group increased serum creatinine was delayed in onset and progression was slowed, although the magnitude of azotemia did not change. The life span of afflicted dogs increased 36%,97 an outcome measure that must be considered in the context of studies that found up to 53% increased survival time in dogs receiving a modified diet.98 In a third study, a difference in serum creatinine level could not be detected in untreated dogs versus enalapril-treated (0.5 mg/kg every 12 to 24 hours) dogs with spontaneous idiopathic renal disease. However, this lack of a difference probably reflected the small number of animals studied because serum creatitine levels had not increased more than 0.2 mg/dL in 13 of 16 treated dogs compared with 1 of 14 untreated dogs by the end of the 6-month study period.99
In cats benazepril has proved effective for treatment of hypertension associated with experimental feline renal disease75 and cats with HCM.100,100a Cats with experimentally induced chronic renal failure responded to benazepril (0.5 to 2 mg/kg once daily), although serum creatinine levels did not differ from those of the control group.75 In cats with polycystic renal disease, enalapril-treated (0.5 mg/kg once daily) cats had less azotemia compared with untreated cats, although GFR did not differ.101 Although the potential efficacy of ACE inhibition for treatment of progressive renal disease may be less clear in cats than in dogs, sufficient evidence exists to warrant its consideration as early in the disease as possible.
Angiotensin-Converting Enzyme Inhibitors and Treatment of Proteinuria
Among the specific renal indications of ACE inhibitors is proteinuria. Proteinuria appears to be correlated with renal disease, so much so that it is a marker of the progression of disease. Decreased proteinuria can be correlated with improved renal function, perhaps by preventing the inflammatory effects of protein in the tubule. Proteinuria is a demonstrated risk factor for the progression of chronic renal failure in cats.102
In humans, ACE inhibitors are considered superior to other vasoactive drugs in decreasing proteinuria, although the mechanism still is not understood and may depend on the cause of proteinuria. Use as part of combination therapy is likely to be the key.103 The mechanism may reflect maintenance of podocyte function in the face of glomerular hypertension; ACE inhibitors decrease podocyte hypertrophy. Reduced filtration pressure has been demonstrated in cats with experimental chronic renal disease.102 Whether control of systemic hypertension is sufficient to control podocyte function has yet to be demonstrated; additional direct effects on podocytes or mesangial cells may occur. Regardless of the mechanism, renal function improves in concert with reduction of proteinuria in human patients. Reduction of proteinuria appears to be dose dependent, occurs rapidly, and remains drug dependent in that proteinuria will recur once the drug is discontinued.
Response of canine proteinuria to ACE inhibitors is variable, but again this may reflect limitations in study design. Finco and coworkers104 were not able to demonstrate a renoprotective effect caused by decreased proteinuria in experimental renal disease in dogs.104 However, in another experimental model of renal failure, the urine protein to creatinine ratio was lowered in dogs receiving enalapril (0.5 mg/kg every 12 hours) for 6 months.105 In a prospective, blinded study of dogs with spontaneous glomerulonephritis, enalapril (0.5 mg/kg) twice daily (but not once daily) reduced the urinary protein to creatinine ratio to less than 50% of baseline, although response required 1 to 3 months. The ratio decreased by 4.2 in treated dogs, compared with a 1.9 increase in placebo-treated dogs.99 In cats benazepril also has proved effective for treatment of proteinuria associated with spontaneous renal diseases.106
Mizutani and coworkers107 prospectively studied the effect of benazepril (0.5 to 1 mg/kg) in cats (n=61) with chronic renal disease using a randomized, placebo, double-blinded parallel design. Cats were studied for up to 6 months. The urine protein to creatinine ratio was reduced in benazapril-treated cats; further, fewer cats progressed to stage 4 renal disease in the treatment group compared with the placebo group. Using a randomized, double-blinded parallel design, King and coworkers102 also prospectively studied the impact of benazepril (0.5-1 mg/kg daily) in cats (n=192; 96 treatment and placebo control each) with chronic renal disease (creatinine ≥ 2 mg/dL; urine specific gravity ≤ 1.025). Cats were treated for up to 1119 days. Proteinuria was significantly reduced based on the urine protein to creatinine ratio. Response was present at the first posttreatment sampling time (7 days) and was greatest in those with the highest ratios, although response was significant also in cats with low ratios (p ≤ 0.02). Survival time (which was inversely related to initial urine protein to creatinine ratio in the placebo group) did not differ between placebo and treatment groups (637 ± 480 for benazepril; 520 ± 323 placebo), although variability was marked for both treatment groups, limiting the power to detect a significant difference. Quality of life also did not differ between groups, although appetite improved more in treated cats with initial urine protein to creatinine ratio of 1 or higher. Therapy was well tolerated. Adverse events did not differ between treatment groups; creatinine concentrations did not differ between the groups, including as therapy began.
Angiotensin II Receptor Antagonists
The development of losartan, the first approved AGII receptor antagonist (ARA), represents a concerted pharmaceutical development endeavor to design a drug based on the core structure of the pharmacologic target. Currently, seven ARA drugs have been approved in the United States. Although each is devoid of agonistic activity, only very minor molecular changes can render the drugs as potent agonists.18 Generally, ARA receptors have a 10,000-fold greater affinity for AR-1 compared with AR-2 receptors. Potency varies among drugs, with candesartan being most and losartan being least potent for AR-1 blockade. However, losartan is metabolized to an active, more potent compound. Blockade of AR-1 is competitive, but inhibition of AGII response is described as insurmountable, perhaps because of the slow release of drug from the receptor.18 Regardless of the mechanism, maximal response to AGII is attenuated; blockade is effective despite increased concentrations of AGII or missed doses. Blockade generally attenuates most biologic effects of AGII. This includes peripheral vasoconstriction, pressor response (rapid and slow), the release of vasopressin, adrenal catecholamines or aldosterone, the advent of thirst, altered renal function, cellular hypertrophy, and hyperplasia.18
In humans oral bioavailability of the various ARA products is generally less than 50%, and protein binding is greater than 90%. Hepatic metabolism varies among the drugs: candesartan and olmesartan are prodrugs, losartan (14%) is metabolized (CYP2C9 and 3A4) to a metabolite more potent than the parent compound, and several others are metabolized to inactive metabolites. The half-life is variable among drugs, ranging from 2.5 hours for losartan (6-9 hours for its active metabolite) to 24 hours for telmisartan. Losartan is excreted both renally and by hepatic metabolism, with liver, but not renal, disease affecting disposition. Losartan is a competitive antagonist of thromboxane A2 and impairs platelet aggregations.18
The pharmacokinetics of ARAs have not been well established in dogs and cats. Several studies in dogs reflect preclinical studies for human products. Limited pharmacokinetic and pharmacodynamic data of losartan potassium were reported in dogs after oral administration of 5 to 20 mg/kg.107a Oral bioavailability ranged from 22% to 33%. The elimination half-life approximated 2 to 2.5 hours after oral administration but only 40 minutes after intravenous administration. The volume of distribution of 0.3 L/kg suggested limited tissue distribution; the drug is very highly (>95%) bound to plasma proteins. Clearance approximated hepatic blood flow; the proportion of drug converted to the active metabolite was not measured. Maximum drug concentration was dose dependent, with a low of 0.8 ± 4 μg/mL at 5 mg/kg to 2.9±1.6 at 20 mg/kg. Pharmacodynamically, an unbound plasma drug concentration of 3 ng/mL (total drug of 96 ng/mL) was determined to the 50th percentile concentration needed to block a pressor response. This concentration was almost achieved at the lowest oral dose of 5 mg/kg, suggesting a dose between 5 and 10 mg/kg. Irbesartan has been studied in dogs alone or in combination with hydrochlorthiazide.107b Dogs were treated with 30 mg/kg once daily for 8 days. Maximum drug concentrations at day 8 were 4.3±1.0 μg/mL, with an elimination half-life of 21±3.7 hours. Although pharmacodynamic response did not relate well with plasma drug concentrations, the effective concentration (EC50) associated with decreasing systemic or diastolic blood pressure was 3.3 ± 0.4 μg/mL, potentially supporting a dose of 30 mg/kg once daily. A lag time to efficacy of several days would be expected.
Indications for ARA treatment in humans include hypertension, diabetic nephropathy, stroke prophylaxis, and heart failure in patients intolerant to ACE inhibition. It is not clear whether antagonism of AGII receptors is more efficacious than therapy with ACE inhibitors. Compared with ACE inhibitors, ARAs differ in their impact on the RAAS in several aspects. Because non-ACE alternative pathways exist for generation of AGII, ACE inhibitors cannot completely attenuate its effects. In contrast, ARAs will effectively block all AGII effects. However, AGII receptors can still be activated. Because ARAs will result in a several-fold increase in renin, AGII concentrations will increase. Finally, inhibition of ACE also results in the inhibition of other pathways; this effect will not occur with ARAs, resulting in persistence of substrates (e.g., bradykinin, Ac-SDKP). Losartan has proved comparable in efficacy to enalapril, but efficacy compared with captopril was variable among studies. In human medicine, ACE inhibitors remain the first-line drugs of choice, with ARAs reserved for nonresponders. The combined use of ACE inhibitors and ARAs is controversial and requires further study.18 The ARAs have also been used alone or in combination with ACE inhibitors or amlodipine to treat proteinuria.
Arterial and Venous Dilators
Organic Nitrates
Organic nitrates activate cGMP, which ultimately decreases actin and myosin interaction, leading to relaxation of vascular smooth muscle and arterial and venous dilation (see Figure 14-4). At low concentrations venodilation predominates, and net systemic vascular resistance is usually not affected. Coronary vessels are directly dilated. Pharmacologic effects occur rapidly. First-pass metabolism precludes oral administration of nitrates; therapeutic routes include intravenous, sublingual, and topical (transdermal ointment).
Nitroglycerin is a member of the organic nitrate group. Although all vascular smooth muscle might be relaxed, the dose of nitroglycerin administered causes predominantly venous dilation and preload reduction. Pulmonary and systemic congestion and myocardial workload are reduced.108 Nitroglycerin is available for intravenous and sublingual use and as an ointment. The 2% ointment form has been the most commonly used preparation in veterinary medicine. It can be applied to the hairless portion of an animal’s skin (abdomen or ear). Gloves should be used by the caregiver to avoid percutaneous absorption of drug. The clinical indication for use is limited to acute (emergency) treatment of CHF.
Sodium Nitroprusside
Nitroprusside (see Figure 14-6)9 contains cyanide, which oxidizes intracellular sulfhydryl groups to produce methemoglobin; NO is simultaneously released. Activation of cGMP causes vasodilation (see Figure 14-3). The NO system that activates nitroglycerin is different from that activating nitroprusside, accounting for differences in vascular beds targeted by each drug.9 Both arterioles and venules are dilated by nitroprusside; systemic and pulmonary vasculatures are targeted. Nitroprusside is one of the most potent vasodilators available. The advantages of nitroprusside over other vasodilator drugs include its potency, its effect in both preload and afterload reduction, immediate hemodynamic effects, extremely short half-life, and low cost. Nitroprusside must be administered by constant intravenous (IV) infusion using an infusion pump; the potential for hypotension necessitates close monitoring. Free cyanide released during intracellular metabolism is cleared in part by transulfation of thiosulfate yielding thiocyanate, which itself is potentially toxic. Overdosing of nitroprusside may yield excessive cyanide, causing severe lactic acidosis; the risk is increased in patients with liver disease or those receiving diuretics (decreased thiosulfate stores). Co-administration of sodium thiosulfate may be indicated in at-risk patients. Clearance of the toxic end product, thiocyanate, may be decreased in patients with renal disease, particularly if infusion continues beyond 24 to 48 hours. Monitoring of thiocyanate should be considered in at-risk patients; concentrations should not exceed 0.1 mg/mL.9 Feline hemoglobin may be predisposed to oxidation and methemoglobin formation. The primary indication of sodium nitroprusside is treatment of severe (catastrophic) CHF. Nitroprusside is administered as a constant-rate infusion using a dedicated line and an infusion pump (1-10 μg/kg/min, starting at 1 μg/kg/min in dogs and 0.5 μg/kg/min in cats; increasing 0.5-1 μg/kg/min every 5 minutes to maintain systolic pressure at 90-1000 mm Hg). Nitroprusside is inherently unstable and will decompose under alkaline conditions and with exposure to light.
Prazosin
Prazosin is an α-adrenergic receptor blocker. However, it also is a venous dilator (perhaps owing to inhibition of cAMP) and thus might be considered as both a preload and an afterload reducer. Despite significant first-pass metabolism in humans, prazosin is effective after oral administration. Prazosin is an effective antihypertensive agent. However, tolerance develops rapidly. It is more effective when used in combination with other drugs. Clinical use of prazosin is limited because hydralazine affords a much better reduction in peripheral resistance and increase in cardiac output.
Phosphodiesterase Inhibitors
Several selective inhibitors of phosphodiesterase (PDE) V have been approved for treatment of penile erectile dysfunction in humans. These include sildenafil (Viagra), tadalafil (Cialis), and vardenafil (Levitra). In contrast to most other PDE inhibitors, PDE V inhibits breakdown of cGMP. Subsequent release of NO in nerve terminals of endothelial cells causes smooth muscle relaxation and increased blood flow. PDE V is located in corpus cavernosum smooth muscle, vascular and visceral smooth muscle, skeletal muscle, platelets, kidney, lung, cerebellum, and pancreas. Potency of tadalafil with PDE V is more than 9000-fold compared with that of other PDE isoforms except for PDE VI (retina) and PDE XI (in skeletal muscle), for which potency is 700- or 14-fold greater, respectively, indicating a relative selectivity for PDE V. Disposition of tadalafil in humans is characterized by oral absorption that is not impaired by food (bioavailability not reported on package insert), greater than 90% binding to plasma proteins, and a volume of distribution approximating 1 L/kg. In humans tadalafil is metabolized by CYP 3A4 to inactive catechols. In humans the elimination half-life is 17 hours. Drug interactions caused by inhibition or induction of CYP 3A4 may result in clinically relevant adverse drug events. For example, the dosing interval is prolonged from 12 hours to 72 hours in humans concomitantly receiving imidazole antifungals or other inhibitors of CYP 3A4. Despite hepatic metabolism, renal insufficiency may increase area under the curve and Cmax twofold to fourfold, whereas moderate hepatic disease does not appear to affect disposition. Although systemic hypotension is not a common side effect of tadalafil, PDE V inhibitors in general potentiate nitrate-induced hypotension and co-administration is contraindicated. Care is indicated when combined with α-blocker therapy. Visual disturbances in humans reflect temporary impairment of color vision, reflecting inhibition of PDE VI in the retina. The PDE V inhibitors have been used with variable success to treat pulmonary hypertension in dogs.