Shock
Shock reflects a state in which tissue perfusion is inadequate to meet tissue metabolic needs.268 Tissue perfusion may be inadequate because of low or unevenly distributed blood flow.269 Selection of the most appropriate therapy is facilitated by categorizing shock first as to the functional disturbance and second as to primary cause.
Classification Of Shock
Hypovolemic Shock
Hypovolemic shock is one of the more common causes of shock in small animals.268 Causes include but are not limited to hemorrhage, trauma, and severe dehydration such as that which accompanies renal dysfunction, vomiting, or hypoadrenocorticism.269 Physiologically, hypovolemic shock can present in three stages. The earliest stage is accompanied by compensatory mechanisms (e.g., increased heart rate, increased vascular resistance) designed to maintain blood pressure. As volume loss progresses, the second or middle stage is characterized by tachycardia with low systemic blood pressure and hypothermia. Capillary refill time is prolonged, and pulse pressure is poor. Blood is shunted away from less vital organs to the brain and heart, and blood clotting abnormalities may be accompanied by increased capillary permeability. Urine output decreases. If hypovolemia persists, the final stage of decompensation occurs. This stage is largely irreversible268 and is characterized by vascular dilation and pooling of blood in peripheral tissues. Poor cardiac filling leads to insufficient cardiac and brain perfusion. Death reflects myocardial failure, cardiac arrhythmias, respiratory failure associated with pulmonary edema, and cardiopulmonary arrest.268
Cardiogenic Shock
Cardiogenic shock is a state of low cardiac output associated with diastolic or systolic dysfunction. The heart is unable to function as a pump, and blood delivery to organs is insufficient. Causes include myocardial failure (acquired or congenital) and cardiac arrhythmias. Iatrogenic cardiogenic shock also can be drug induced.269 Clinically, because the underlying cause of shock is inadequate blood flow, cardiogenic shock presents similarly to hypovolemic shock, with the primary difference of increased atrial filling pressures accompanied by pulmonary edema.269
Distributive Shock
Distributive shock occurs when blood flow is distributed improperly to tissues. Improper distribution reflects a rapid, marked increase in peripheral vasodilation, vascular capacitance, and peripheral pooling of blood.269 Causes generally include those associated with the release of vasoactive mediators, most notably endotoxemia (see Chapter 8) or other causes of sepsis and anaphylaxis or anaphylactoid reactions. Injured and ischemic tissues (e.g., due to hypovolemic or cardiogenic shock) also lead to the release of vasoactive and procoagulant mediators. Vascular occlusive diseases such as saddle thrombi and pulmonary thromboembolism (e.g., dirofilariasis) also cause distributive shock.269 For example, with sepsis distributive shock might initially be “warm” in that blood flow is increased in peripheral tissues. As shock progresses and venous pooling continues, fluid is lost from the vascular space, venous return decreases, cardiac output decreases, and tissues become underperfused or “cool.”
Pathophysiology
Ideally, treatment of shock should focus on early reversal based on the underlying cause. As shock progresses, the underlying pathophysiology is the same, and treatment is oriented toward prevention and reversal of inadequate tissue perfusion. Some type of damage is likely to occur in any tissue subjected to a period of hypotension (mean arterial blood pressure <50 mm Hg). These include cell ischemia, inadequate oxygen delivery, and the generation of proinflammatory/procoagulant mediators.269
Sequelae of cellular ischemia
Tissues suffer damage from inadequate tissue oxygenation in 5 to 10 minutes, and the damage is irreversible at 15 to 20 minutes.269 Mitochondrial dysfunction accompanies ATP depletion, leading to anaerobic metabolism and lactic acid accumulation. Cell membrane (ATPase) pumps become disrupted, and intracellular destructive enzymes are released. Accumulation of intracellular calcium leads to the activation of enzymes that disrupt cellular homeostasis. Intracellular sodium and chloride increase, and magnesium and potassium decrease. ATP breakdown yields hypoxanthine and generation of xanthine oxidase (converted from xanthine dehydrogenase), which produces oxygen free radicals.269 With reperfusion, hyperemia occurs once blood flow is reestablished if the period of impaired oxygenation or poor tissue perfusion is short. The duration of hyperemia is determined by the extent of mediator release (potassium, hydrogen, NO, adenosine, adrenomedullin, the latter a hypotensive peptide first discovered in pheochromocytomas). On the other hand, if blood flow is less than 20% of normal for longer than 5 minutes, reperfusion after perfusion failure leads to reoxygenation injury. Injury reflects the production of self-destructive enzymes and metabolites and derangements in blood clotting.269 Together the consequences of reperfusion injury include uneven distribution of blood flow and focal ischemia (perhaps exacerbated by inappropriate thrombosis), swelling of capillary endothelial cells and subsequent plugging by leukocytes migrating to the area, and increased microvascular viscosity and interstitial edema.269
Oxygen free radicals
The generation of oxygen free radicals by mitochondria, macrophages, and neutrophils sets the stage for reperfusion injury should blood flow be reestablished after a sufficiently long period of poor perfusion, causing perhaps the most detrimental sequelae of shock.269 Xanthine oxidase metabolizes molecular oxygen into radicals such as superoxide anion, hydrogen peroxide, and the hydroxyl radical (see Chapter 29). Enzymes that normally scavenge oxygen free radicals (e.g., superoxide dismutase, catalase, and glutathione) are overwhelmed as tissues reperfuse. Production of oxygen free radicals is exacerbated by mediators released in response to oxygen free radicals, including cytokines (including TNF), interleukins (which also induce procoagulant activity), and prostaglandins.269
Role of nitric oxide
NO can be either protective or detrimental in the patient with sepsis and endotoxemia. Under basal physiologic conditions, NO serves as a free oxygen radical scavenger, limiting toxicity associated with superoxide and other radicals. Inhibition of platelet aggregation and leukocyte adhesion limits ischemia–reperfusion injuries.21 During shock large amounts of iNOS are formed; as such, NO becomes a major contributor to the pathophysiology of shock.21,269 Peroxynitrous acid, generated from the reaction of NO with oxygen free radicals, destroys cellular macromolecules, causing mitochondrial and cell membrane dysfunction, production of prostaglandins, and programmed cell death (apoptosis). The coagulation cascade is activated, ultimately leading to disseminated intravascular coagulation. Arteriovenous shunting (possibly caused by iNOS) contributes to maldistribution of blood flow, particularly in endotoxic and other septic shock, and may be a cause of irreversibility.269
Thus, although the initial responses of the body to iNOS might lead to important compensatory responses, ultimately the responses may prove to be detrimental. Nevertheless, inhibition of NOS is undesirable because systemic vascular resistance is improved only at the cost of loss of blood flow to vital organs. Platelet aggregation increases, along with the risk of thrombus formation and disseminated intravascular coagulation.269 Analogs of L-arginine, such as L-NAME (N-nitro-L-arginine methyl ester) competitively inhibit NO production by either cNOS or iNOS from L-arginine. Treatment of human patients in septic shock with L-NAME, however, led to pulmonary hypertension and reduced cardiac output.270 Drugs that selectively inhibit iNOS but not cNOS may be a more appropriate focus of investigation.
Gastrointestinal barrier
The sequelae of ischemia and hypoxia in the gut have profound clinical implications for the patient undergoing shock. Potent vasoconstrictors (endothelins), cytokines, and other mediators act in concert with leukocyte migration and epithelial necrosis to increase capillary permeability, transcapillary fluid filtration, and interstitial edema. Diarrhea is a common clinical complication of resuscitation from shock and may indicate the loss of the protective mucosal barrier in the gastrointestinal tract. Bacterial translocation and endotoxin absorption result in release of massive quantities of proinflammatory mediators, predisposing the patient to septicemia and, ultimately, to the systemic inflammatory response syndrome (see Chapter 29).269 This syndrome is characterized by multiorgan dysfunction.
The compensatory mechanisms implemented to counter the pathophysiologic sequelae (decreased tissue perfusion and oxygen delivery) of shock involve the neural, hormonal, and renal reflexes previously described for cardiovascular diseases. Vasoconstriction maintains arterial blood pressure and redistributes blood flow to vital organs (cerebral and coronary vessels). Cardiac output is increased by increasing heart rate and a fluid shift from interstitial to intravascular sites. Although compensatory mechanisms support the patient during the initial stages of shock, increased vascular resistance and myocardial oxygen demand ultimately will contribute to the demise of the patient.
Treatment
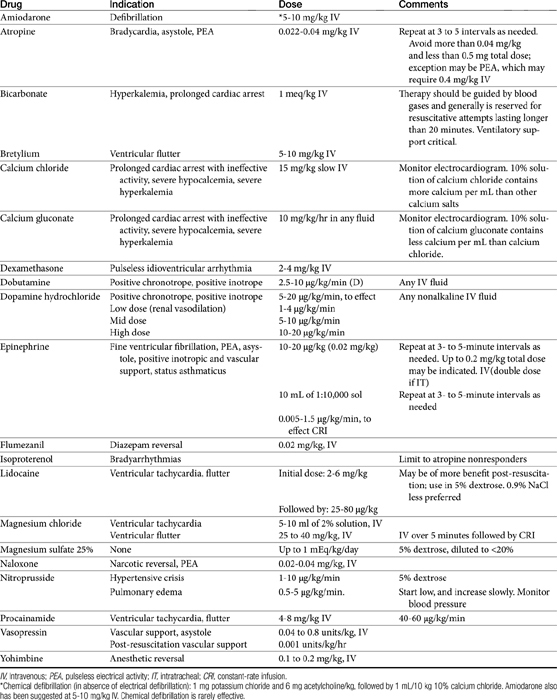
Successful therapy for shock focuses on reestablishing blood flow, blood pressure, and blood volume to normal or above normal (see Table 14-4).269 Monitoring response to therapy can, however, be difficult. Muir269 recommends that response to therapy and a good prognosis be based on frequent monitoring of behavior, level of consciousness, arterial blood pressure, tissue oxygenation, heart and respiratory rate and rhythm, mucous membrane color, capillary refill time, and urine output. Clinical pathology data should focus on packed cell volume, total protein, and serum lactate.
KEY POINT 14-59
Successful therapy for shock focuses on reestablishing blood flow, blood pressure, and blood volume to normal or above normal.
Treatment of shock should maximize tissue perfusion and oxygen delivery to and consumption by peripheral tissues and minimize the effects of proinflammatory and procoagulant mediators. For septic shock (discussed more extensively in Chapter 8) therapy also focuses more aggressively on prevention of endotoxin release and its effects. Regardless of the cause of shock, appropriate therapy is summarized by the acronym VIP: Ventilation to facilitate blood oxygenation, Infusion of fluids to restore blood volume, and support of the myocardial Pump to facilitate blood delivery (flow) to tissues.269
Fluid therapy
Fluid therapy should be aggressive but not overzealous. Care should be taken not to reduce packed cell volume and total protein to less than 20% and 3.5 g/dL, respectively, to minimize the risk of pulmonary or interstitial edema. Administration of hypertonic saline provides rapid but short-term (30 to 120 minutes) hemodynamic improvement in hypovolemic or endotoxic shock; duration of improvement can be extended if hypertonic saline is combined with a colloid.
Blood and blood substitutes
Treatment of hypotensive shock secondary to hemorrhage in which 25 mL/kg or more of blood is lost should include whole blood or packed red blood cells. Blood substitutes should be used when blood products are not available or for animals for which the risk of a transfusion reaction is too great.
Pressor drugs
Positive inotropic drugs are indicated to maintain arterial blood pressure and regional blood flow in patients whose myocardial function does not improve sufficiently after administration of fluids. Pressor drugs also are indicated for patients whose cardiac contractile activity is compromised. Dopamine and dobutamine have been the preferred pressor drugs; dopamine may be preferred for bradycardic animals. 229 Improved blood flow to the gastrointestinal tract will help minimize the risk of gastrointestinal mucosal damage and subsequent multiorgan failure.229 Volume replacement must occur before either drug can be used successfully. Refractory hypotension is often associated with septic shock. Underlying mechanisms include vasopressin deficiency, activation of ATP-sensitive K+ channels (in response to accumulation of lactate and hydrogen), and overproduction of NO (which precludes vascular smooth muscle contraction). Activation of K+ channels causes hyperpolarization, precluding interaction of vasoconstrictors with smooth muscle receptors. Among the current therapies is treatment with vasopressin. 269a In patients with septic or hypovolemic shock, vasopressin is released biphasically. Osmotic overstimulation may cause rapid release of endogenous vasopressin. However, only 20% of the total pool is available for rapid release. Slower release results in relative deficiency by 1 hour of sustained hypovolemic shock in humans. Whereas arterial pressure is minimally affected in healthy subjects, patients in vasodilatory shock express an exaggerated response to exogenous vasopressin therapy. Mechanisms include replacement of endogenous stores, inhibition of K+ channels, and decreased synthesis of nitric acid. Treatment at physiologic doses (0.01-0.04 units [70-kg human adult]) has been described as the best vasopressor drug; doses of first-line vasoproessors can be reduced, thus reducing the risk of adverse effects with these drugs. The drug can be given intratracheally.269b Yoo269c described a vasopressin dose determination study in dogs with experimentally-induced hemorrhagic shock. A dose of 0.4 IU/kg was the most effective dose, providing more hemodynamic response in decompensated shock compred to 1.6 IU/kg. Treatment ideally occurs when endogenous concentrations are lowest. Other benefits of vasopressin include increase urine output; improved cerebral, coronary, and pulmonary blood flow; and increased serum cortisol. Side effects occurring at nonphysiologic doses include platelet aggregation and renal, mesenteric, pulmonary, and coronary vasoconstriction. Vasopressin should be used only with caution in patients with cardiovascular disease. The use of glucocorticoids may be indicated in patients non-responsive to pressor agents as a result of functional adrenocorticodeficiency (See Chapter 30).
Miscellaneous drugs
The use of drugs intended to minimize the damage of oxygen free radicals has not been well established in animals such that a standard protocol can be followed. The use of glucocorticoids is controversial (see Chapter 30). Their potential benefits to the endotoxic shock patient have been delineated. In general, the efficacy of these products to limit vascular response to vasoactive compounds depends on the time of administration. Efficacy is greatest when administered before or within several hours of the onset of the pathophysiologic response to shock. Although survival (several hours) has been documented after use of glucocorticoids (compared with placebo) in human and animal clinical trials, long-term survival (beyond several days) has not been documented. Use of glucocorticoids in human patients with endotoxic or septic shock has been associated with an increased risk of infection in some studies but no increased risk in others. The use of glucocorticoids in veterinary medicine remains controversial. Of the drugs to be used, methylprednisolone may be preferred in most causes of shock because of its potential ability to scavenge oxygen free radicals. Administration should be short term.
KEY POINT 14-60
The use of drugs intended to minimize the damage of oxygen free radicals has not been well established in animals.
A number of NSAIDs have been studied for their ability to block response to mediators of endotoxic shock. Indomethacin and ibuprofen have shown efficacy in human patients. Flunixin meglumine has been studied in dogs. As with glucocorticoids, however, the effects of NSAIDs must be realized within the first 2 hours of the onset of endotoxic shock (i.e., before mediators have been able to stimulate response). Prolonged therapy with NSAIDs is not advised because of toxic effects. Although gastrointestinal toxicity is the major concern in most animals, the patient suffering from endotoxic shock may be more predisposed.
Despite the lack of scientific data to support clinical response to drugs that scavenge oxygen free radicals, their use should be strongly considered, particularly if there is little risk of toxicity (See Chapter 29).
The use of antimicrobials in patients suffering from or predisposed to endotoxic or septic shock is discussed elsewhere (see Chapter 8). Patients that have suffered vascular compromise are at risk of suffering the consequences of translocation of enteric pathogens. Prophylactic therapy should be oriented toward minimization of gastric erosion or ulceration (see Chapter 19) and selective decontamination of the digestive tract (targeting gram-negative aerobic pathogens). In humans oral antimicrobials that are not absorbed are recommended: a paste containing 2% polymyxin, 2% tobramycin, and 2% amphotericin (to target fungal organisms) for the oral cavity and a solution of polymixin (100 mg), tobramycin (80 mg), and amphotericin B (500 mg) for the gastrointestinal tract (about 0.1 mL/kg every 6 hours) have been recommended.271 However, this therapy is intended to reduce the incidence of nosocomial infections in the intensive care environment. The incidence of pneumonia, urinary tract infections, and catheter-related septicemia can be decreased. The role of selective digestive decontamination in patients subject to shock (with the exception of endotoxic shock) is less clear.
Cardiopulmonary Cerebrovascular Resuscitation
Generally, the goal of cardiopulmonary cerebrovascular resuscitation (CPCR) is to maintain or preserve neurologic function. It is beyond the scope of this chapter to discuss the causes of and recognition of the need for CPCR. Obviously, prevention is the key to success, and treatment of underlying diseases likely to cause cardiopulmonary arrest should be reviewed. Success is more likely if the cardiopulmonary arrest is associated with reversible conditions (e.g., anesthetic overdose, upper airway obstruction, hemorrhage, electrolyte imbalances). The focus of this discussion is on drugs used in CPCR. Which drugs are proper and when their use is indicated in the patient are controversial. Cardiac rather than respiratory arrest is discussed. The pharmacologic effects, side effects, and other pertinent clinical pharmacologic data for each of the drugs have been discussed elsewhere; discussion here is limited to the use of the drugs during or immediately after CPCR.
“Crash Cart” Drugs
Drugs that should be carried in a crash cart include epinephrine, atropine, magnesium chloride, naloxone, lidocaine, sodium bicarbonate, and bretylium tosylate.272
Epinephrine remains the mainstay of acute cardiac life support. It is intended to promote systemic vasoconstriction such that blood flow is diverted to the coronary and cerebral circulation. It is indicated for pulseless ventricular tachycardia, ventricular fibrillation, electromechanical dissociation (pulseless electrical activity), and ventricular asystole. The standard dose is 10 to 20 μg/kg (1 mg in humans) or 10 mL of a 1:10,000 solution repeated every 3 to 5 minutes. The optimal dose may be very high, ranging from 0.45 to 2 mg/kg (Table 14-4). For humans the American Heart Association has recommended a fivefold increase in the dose to 5 mg if there is no response to the initial 1 mg dose.267
Atropine has little indication in CPCR with the exception of bradycardia, pulseless electrical activity, and ventricular asystole. The dose for electromechanical dissociation and asystole is 1 mg intravenously, repeated every 3 to 5 minutes. In humans complete vagal blockade occurs at 0.04 mg/kg (3 mg); this dose is discouraged. Likewise, a total dose below 0.5 mg can cause parasympathomimetic effects and also is discouraged.267
Isoproterenol is a pure, nonselective β-agonist drug. As such, it is a positive inotrope but can also cause peripheral vasodilation. It will increase myocardial oxygen demand. Currently, its use is limited to bradyarrhythmias that do not respond to atropine.272
Bicarbonate provides little benefit and may in fact harm patients in metabolic acidosis. Acidosis associated with cardiac arrest is best treated with ventilatory and circulatory support. Potentially harmful effects of bicarbonate include arrhythmogenic alkalemia; increased generation of CO2; hyperosmolarity; hypokalemia; paradoxical central nervous system and myocardial intracellular acidosis; and a leftward shift in the oxyhemoglobin dissociation curve, limiting delivery of O2 to tissues.267 When used, bicarbonate therapy ideally should be guided by blood gas analysis (pH <7.15 to 7.2).267 Indications or situations in which bicarbonate may prove beneficial for humans requiring CPCR include hyperkalemia; tricyclic antidepressant overdose; prolonged cardiac arrest (protracted hypoperfusion-induced acidosis); and postresuscitation, bicarbonate-responsive, and anaerobic lactic acidoses.267 Sodium bicarbonate (1 mEq/kg intravenously) may be used after epinephrine in patients suffering from a prolonged cardiac arrest who have shown improvement in cardiovascular or cerebral recovery. It should be followed by correction of any deficit (monitored) that is greater than 5 mEq/kg. The bicarbonate-induced hypercarbia tends to be transient and generally harmless to the heart if used in conjunction with epinephrine.272
KEY POINT 14-62
Bicarbonate provides little benefit and may in fact harm patients in metabolic acidosis.
Calcium administration does not appear to enhance cardiac performance during CPCR. Ischemia associated with cardiac arrest causes intracellular accumulation of calcium, which can disrupt membranes and uncouple oxidative phosphorylation. Calcium can cause coronary vasospasm and will exacerbate the arrhythmic tendency of the unstable myocardium and impair relaxation.267,272 It will also exacerbate digoxin toxicity. Calcium causes precipitation when combined with sodium bicarbonate. Calcium is not recommended except in cases of prolonged cardiac arrest or absent or ineffective pump activity. Calcium chloride (10% solution contains 100 mg/mL Ca2+) is associated with the longest and most predictable increase in plasma ionized calcium.272 In human patients 1 g of calcium chloride (approximately 15 mg/kg) is generally sufficient, although toxicity may occur at this dose. Other indications for calcium include hyperkalemia, ionized hypocalcemia, and CCB overdose.267
Crystalloids (including hypertonic resuscitation and balanced electrolyte solutions) are indicated if the cause of cardiac arrest is hypovolemia. Inappropriate fluid load can, however, contribute to decreased cerebral blood flow and decreased coronary blood flow.272 The production of lactic acid will be enhanced in critically ill hyperglycemic patients, which can lead to or contribute to cell injury. Dextrose infusions are considered by the American Heart Association to be harmful to humans.267 As such, dextrose-containing fluids should be avoided. Isotonic saline or Ringer’s lactate is preferred as the resuscitation fluid.
KEY POINT 14-63
If crystalloid therapy is indicated in the shock patient, isotonic saline containing fluids free of dextrose is indicated.
Several routes of drug administration can be used to support resuscitation. Central venous catheter placement is ideal for immediate drug delivery to the heart. A sufficient bolus of a compatible isotonic fluid should follow any drug administered through peripheral tubing. Intracardiac injections probably offer no increased benefit compared with central intravenous administration. Potential complications include cardiac tamponade, coronary vessel laceration, and pneumothorax. Intracardiac bolus may destabilize the electrical properties of the heart.
Alternative routes of administration can be considered in the absence of venous access. Intratracheal administration is an effective alternative route to central intravenous administration for selected drugs during CPCR.272 In general, the dose is doubled and the drug is administered through a catheter in 10 to 20 mL of liquid for the drugs to reach the alveoli, where they will be subsequently absorbed. Insufflation will facilitate drug absorption. Doses of all drugs administered intratracheally should be increased by twofold to 2.5-fold. The duration of action of the drugs may be longer after intratracheal administration than after intravenous administration. Drugs shown to be effective after intratracheal administration include epinephrine, lidocaine, atropine, and naloxone. There are several drugs that should not be given via intratracheal administration because of the risk of tissue damage. Examples include sodium bicarbonate (depletes surfactant), norepinephrine, and calcium chloride.267 Drugs should not be mixed in the same syringe before intratracheal administration. Intraosseous administration is another alternative to intravenous use in small animals. The bone marrow provides a large venous access; the most common sites during CPCR are either the trochanteric fossa of the femur or distal cranial femur.272
Specific Conditions
Cardiac asystole refers to the complete absence of electrical activity. Therapy is oriented toward stimulating any electrical activity and then modifying the activity to generate a rhythm with a pulse.267 Epinephrine generally remains the drug of choice for cardiac arrest. Doses should be sufficient (> 0.01 mg/kg) to cause positive inotropic and peripheral vasoconstrictive effects yet low enough (<0.2 mg/kg) to avoid ventricular fibrillation. In an experimental model of cardiac arrest in dogs, declining renal function was positively correlated with the amount of epinephrine administered and the energy required for defibrillation.273 Because of its short duration of action, epinephrine should be administered every 3 minutes. Longer-term inotropic support should be provided with a less effective pressor drug such as dopamine or dobutamine. Electrolyte imbalance should be treated with the appropriate electrolyte. Bicarbonate is useful only in the previously described indications.
Ventricular fibrillation is best converted to a normal rhythm by electrical defibrillation. Potassium chloride, bretylium, and magnesium chloride have been used to pharmacologically treat defibrillation in the dog. Among these, magnesium chloride (5 to 10 mL of a 2% solution administered intravenously) may be best. Once an organized rhythm has been established, epinephrine may be beneficial for increasing vascular tone and improving blood flow to the brain. The initial dose should be low (0.02 mg/kg) and increased tenfold (0.2 mg/kg) in nonresponsive patients. Very high doses may, however, excessively increase myocardial oxygen demand. Lidocaine may prove useful for “coarsening” fibrillation, rendering it more amenable to electroconversion. In addition, it may increase vascular tone response to epinephrine.267 Bretylium has proved useful in some human cases of refractory ventricular fibrillation. The drug is administered immediately (1 minute) before electrical defibrillation. Refractory cases also may require correction of severe acidosis. Precaution is, however, taken to ensure that the pH is increased to no higher than 7.5 because of increased resistance to defibrillation.267
Ventricular tachycardia also is most amenable to electrical shock. Lidocaine is the drug of choice for control of ventricular tachycardia. Alternatives include procainamide (and, for humans, bretylium). Magnesium sulfate (25 to 40 mg/kg intravenously over 5 minutes) may be useful in refractory cases or in cases of ventricular flutter; constant-rate infusion over 4 to 8 hours is indicated if the patient responds.
Electromechanical dissociation (EMD; pulseless electrical activity) generally is fatal when caused by myocardial diseases. Treatable causes in humans include hypovolemia (e.g., acute blood loss, which should be treated with volume replacers), pericardial tamponade, and tension pneumothorax.267 Epinephrine or atropine (0.04 to 0.08 mg/kg intravenously) may be useful when EMD is associated with hypotension or pleural or pericardial disorders. Pulseless electrical activity is likewise accompanied by a poor prognosis, although naloxone, dexamethasone sodium phosphate, and calcium have been recommended. Calcium is most likely to be of benefit with hypocalcemia or extreme hyperkalemia.267
Bradyarrhythmias are most amenable to nondrug therapy. The slower the rate and the wider the ventricular complex on the electrocardiogram, the more ineffective will be the cardiac contractility.267 Atropine is most useful with narrow complex bradyarrhythmias. However, atropine should be used cautiously such that potentially lethal tachycardia might be prevented in hypoxic patients. A low dose (0.022 mg/kg) is indicated unless vagolytic arrest is present. Dopamine and epinephrine may be helpful for inotropic support. Isoproterenol is controversial because of peripheral vasodilation.
Anesthetic or narcotic overdoses should be reversed if posssible. Naloxone (0.02-0.04 mg/kg intravenously) is indicated for any narcotic. Yohimbine or atipamezole (0.1 to 0.2 mg/kg intravenously) is indicated in the presence of α-2 antagonists and may be effective for other chemical restraining agent, including ketamine or barbiturates.267a,b Flumazenil (0.02 mg/kg intravenously) is indicated for reversal of diazepine depressant effects.
Postresuscitation monitoring and care are critical to successful CPCR. Dobutamine is preferred to dopamine by many clinicians for postresuscitation inotropic support. SBP should be maintained above 90 mm Hg. Urine formation should be maintained at 1 to 2 mL/kg per hour. Furosemide may be indicated in the face of decreasing urine output. Neurologic function should be assessed along with the need for iron chelators, CCBs, or oxygen free radical scavengers. Vasopressin (0.04 to 0.8 units/kg intravenously or 0.001 units/kg/hr) may be indicated in patients whose vasculature remains nonresponsive to epinephrine.