Antiarrhythmic Drugs
Cardiac Arrhythmias
Underlying molecular causes of arrhythmias have been reviewed in humans.1 Arrhythmias result from a combination of factors, including genetic predisposition, external stressors and subsequent myocardial remodeling, as well as iatrogenic contributions. Ischemia, electrolyte imbalances, activation of the RAAS and other systems, and pharmacologic therapy are among the triggers for arrhythmias.1 Inherited causes of arrhythmias are rare, but generally pathophysiology is straightforward, particularly if associated with mutations in ion channel genes. In contrast, arrhythmias associated with acquired heart disease are complex. Structural and electrical remodeling, hemodynamic changes, and neuroendocrine signals each influence ion channel function, intracellular calcium response, and intercellular communication and matrix composition.
Regardless of the source, all cardiac arrhythmias arise from two primary abnormalities: impulse initiation, which includes spontaneous automaticity and triggered activity, and impulse propagation—that is, conduction (generally manifested as reentrant arrhythmias). Impulse initiation establishes heart rate and is determined primarily by the rate of diastolic depolarization—that is, the slope of phase 4. In the normal heart the slope (heart rate) is autonomically controlled, being decreased by acetylcholine released from parasympathetic nerves and increased by norepinephrine released from the adrenal cortex. Stroke volume decrementally decreases as heart rate increases; accordingly, cardiac output may ultimately decrease with faster rates. The slope of phase 4 can be affected by a number of abnormal conditions. Enhanced automaticity occurs when the rate of spontaneous diastolic depolarization increases sufficiently to allow emergence of pathologically slowed or increased rates (e.g., sinus tachycardia). Ectopic foci (pacemakers that normally are latent) may emerge and may cause tachycardia if the frequency exceeds that of the sinoatrial node. Arrhythmias of initiation may also be triggered by an abnormal depolarization (phase 0), resulting in secondary upstrokes in the action potential. Two types of triggered arrhythmias occur. Delayed after-depolarization (DAD) occurs after a normal action potential and is followed by an overload of intracellular calcium. Examples include arrhythmias associated with myocardial failure, myocardial ischemia, adrenergic stress, and digoxin toxicity. Early after-depolarization (EAD) upstrokes occur during phase 3 repolarization and follow abnormally long cardiac action potentials. They generally result from abnormal inward sodium or calcium channel currents or exchange pumps and are associated with very slow heart rates or low extracellular K+ or in association with drugs that cause prolonged action potential duration.
KEY POINT 14-25
Arrhythmias reflect altered impulse initiation (automaticity) or impulse propagation (conduction).
Drugs that decrease automaticity do so through several mechanisms. Some decrease the rate (slope) of phase 4 spontaneous depolarization, suppressing the ectopic focus such that the sinoatrial node is allowed to resume its dominance. Automaticity might also be reduced by lengthening the time needed to attain threshold potential by increasing either the excitation threshold (more positive; e.g., Na+ or Ca2+ channel blockers) or the diastolic membrane potential (i.e., hyperpolarization, or more negative). Finally, spontaneous discharge might be reduced by prolonging the action potential duration, such as might occur with prolonged effective or absolute refractory periods.5 Triggered automaticity can be impaired by inhibiting the development of after-depolarizations or interfering with inward sodium or calcium channels. Shortening (rather than prolongation) of the action potential duration will inhibit EADs. Although the mechanism is not well known, magnesium will also inhibit EADs.5
KEY POINT 14-26
Automaticy can be reduced by prolonging the effective refractory period or increasing the time it takes for the membrane potential to reach threshold (phase 4).
Arrhythmias associated with conduction abnormalities are exemplified by reentrant arrhythmias. Reentrant arrhythmias may be either anatomic or functional. Functional reentrant arrhythmias are exemplified by pathologies such as ischemia that markedly slow conduction. Anatomic arrhythmias involve two or more pathways that travel to the same region of the heart but differ in electrophysiology (e.g., refractory period or conduction speed). Emergence of reentrant arrhythmias reflects the unique histologic make up of myocardial muscle. The geometric connection of myocardial cells to one another facilitates rapid movement of impulses throughout the myocardium such that the heart acts as a single large cell (Figure 14-8). One region might receive signals from several different pathways. Coordination of impulse conduction ensures that myocardial fibers are excited in a sequence that maximizes pump efficiency. Normally, retransmission of an impulse received in a specific region from an alternative (i.e., second) direction is prevented because either the signals arrive simultaneously, thus canceling one another, or the fiber remains refractory from the first impulse and cannot respond to the second. However, if the refractory period is abnormally short, or conduction of the reentering, alternative impulse is markedly slowed, the fiber will no longer be refractory from the first impulse. The impulse can be reinitiated and travels in the opposite direction, until it returns to the region again, generating a (circus) reentrant arrhythmia (see Figure 14-8). Altered membrane channel and ion movements with injured cardiac tissue channels slow movement through fast Na+ channels (phase 0) and slow Ca2+ channels. Conduction is accordingly slowed, potentially generating reentrant arrhythmias.
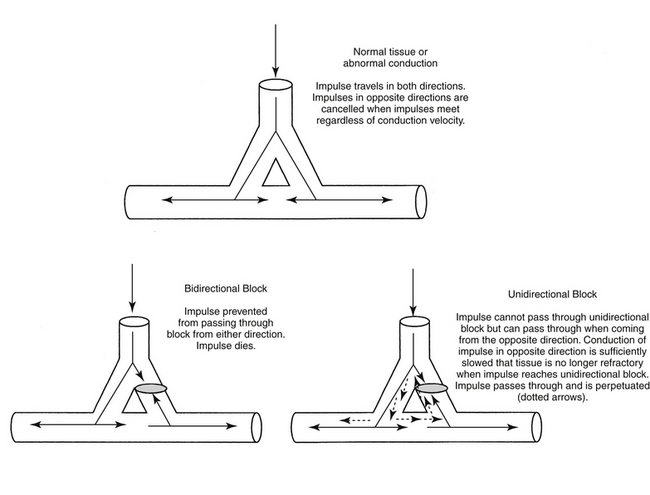
Figure 14-8 In the normal myocardium, electrical impulses travel down one or more paths of a bifurcated myocardial cell. Bifurcations allow coordination of contractions in all directions such that myocardial emptying is efficient and complete. Several mechanisms exist to ensure that impulses travel only in the proper direction. Those impulses that travel in opposite directions will be canceled by one another, precluding their transmission in the wrong direction. In the presence of a unidirectional block (i.e., resulting from damaged myocardium), transmission of one of the impulses will be prevented. However, although the impulse that normally would be canceled is now able to pass through the damaged tissue in the other direction, it will meet tissue that remains refractory from the first impulse. Consequently, it will not be transmitted any further. However, if conduction of the second unimpeded impulse is slowed, the myocardium will no longer be refractory when the impulse passes through the unidirectional block. Therefore the myocardium is receptive to the electrical impulse of this misdirected impulse, and the signal will be transmitted. Because of the combined effects of the unidirectional block and slowed conduction, the impulse may be perpetuated, resulting in a circus or reentrant arrhythmia. Drugs can reduce the arrhythmia by causing a bidirectional block, which prevents the second impulse from traveling through the damaged site, or by increasing the rate of conduction of the second impulse such that it reaches myocardial tissue while it is still in its refractory state from the first impulse.
Clinically relevant antiarrhythmics act to prevent reentrant arrhythmias by blocking specific ion channels or by targeting autonomic function, thus altering initiation, or conduction or action potential duration (thus refractory periods) of cardiac fibers. Initiation and thus automaticity can be suppressed through blockade of ion channels or drugs that facilitate adenosine or acetylcholine (thus increasing the maximum diastolic or resting potential) or antagonize adrenergic receptors (thus decreasing the slope of phase 4). Acceleration of conduction or prolongation of the ERP or APD causes the second impulse to reach tissue still refractory from the first impulse. Examples include Ca2+ channel blockers, β-adrenergic receptor antagonists, and digitalis glycosides. Slowed conduction caused by these drugs may increase the risk of functional reentrant arrhythmias. However, because reentrant arrhythmias respond best to drugs that prolong the refractory period, such drugs nonetheless remain effective for treatment of reentrant arrhythmias if the effective refractory period is prolonged. Examples of drugs that prolong ERP or APD might include drugs that delay recovery of sodium channels (particularly those that target inactivated channels) or potassium channel blockade (the delayed rectifier channels is particularly amenable to drug blockade), or drugs that prolong the APD. However, potassium channel blockade is also particularly conducive to causing arrhythmias, perhaps in part because of the importance of potassium channels in multiple phases of the action potential cycle.
In general, the complex action of antiarrhythmic drugs renders antiarrhythmic therapy generally inefficient and risky. All antiarrhythmic drugs are proarrhythmogenic. A major contributing factor to both inefficiency and risk is lack of knowledge regarding the particular electrophysiologic mechanism that underlies each arrhythmia or the drug. For example, the number of potassium channels, their differences in location, control, and role in the action potential complicate understanding, let alone predicting the impact of potassium channel blockade on cardiac function or rhythm. Increasingly, the arrhythmogenicity of drugs is likely to be understood as the differences in potassium channel impact by these drugs are understood. For example, blockade of delayed rectifier potassium current (Ik5) in the presence of high (β) adrenergic output predictably causes torsades de pointes in dogs.110a
Differences in clinical actions of antiarrhythmic drugs reflect in part different affinities of ion channel–blocking drugs for target receptors on the ion channel proteins, with some drugs targeting specific receptor subtypes. Affinity may change with conformation of the protein channel. As such, affinities are often state dependent (i.e., occurring only in the open, conducting state; in the closed, resting state; or during the inactivated, recovering state). Further, cardiac drugs often target more than one channel, and changes induced by a drug in one current generally influences the other currents. Direct and indirect (through the autonomic system) effects on cardiac contractility contribute to adverse effects.5 Finally, most antiarrhythmic drug target channels in both normal and abnormal tissues. Electrolyte abnormalities and hypokalemia in particular often predispose arrhythmogenicity and may increase adverse effects to antiarrhythmics.109,110 Indeed, a randomized human clinical trial that focused on the use of antiarrhythmic drugs for prevention of sudden death found that sudden death actually increased, primarily because of the proarrhythmic effects of drugs that target ion channels.111 Accordingly, antiarrhythmics should be considered dangerous,109,111 and their use should be pursued, whenever possible, under the guidance of clinicians with appropriate expertise (i.e., cardiologists).
KEY POINT 14-27
Reentrant arrhythmias can be decreased by increasing the rate of conduction or prolonging the effective refractory period.
Antiarrhythmic cardiac drugs fall into four main classes according to their dominant electrophysiologic effect on myocardial cells5,112 (Figure 14-9). Although this classification serves to couple electrophysiologic actions with antiarrhythmic effects, increasing emergence of the complex mechanisms of actions of these drugs complicates the classification, and care should be taken not to assume that drugs within classes behave the same way; indeed, most drugs have multiple effects that cross into multiple classes.
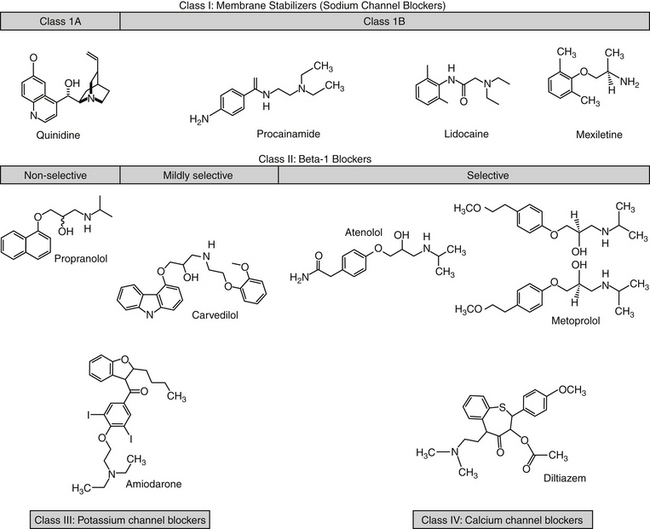
Figure 14-9 Structures of selected antiarrhythmic drugs. The drugs are classified by their mechanism of action, although much overlap occurs, particularly with potassium channel effects. Metoprolol exemplifies the chiral carbon present on most β blockers (some have two). Rotation of the four groups around the carbon yields enantiomers (stereoisomers that are non-superimposablemirror images). Although similar in structure, the enantiomers are likely to be different in both pharmacokinetic and pharmacodynamic properties. Most products are sold as racemic mixtures of both isomers, although only one may be active. The body is likely to handle the isomers differently. Class 1C drugs are not shown.
Class I Antiarrhythmic Drugs
Class I agents comprise the standard membrane-stabilizing drugs such as lidocaine, quinidine, and procainamide. These agents work by selectively blocking the fast Na+ channels and depressing phase 0 of the action potential through the direct membrane-stabilizing or “local anesthetic” effect. Accordingly, class I drugs increase the threshold of excitability and decrease the rate of spontaneous phase 4 depolarization, thus reducing the emergence of ectopic foci (decreased automaticity). Although decreased phase 0 depolarization also decreases conduction velocity (thus prolonging conduction and increasing the risk of re-entrant arrhythmias), effects on automaticity and generation of ectopic foci appear to predominate over effects on conduction velocity. Some class I drugs also prolong action potential duration, especially the effective refractory period, and as such are particularly useful in treating reentrant arrhythmias.113 Class I agents can be further subdivided according to their effects on the refractory period and the rate of repolarization. Class 1A are intermediate blockers, including quinidine and procainamide. Class 1 B are rapid blockers, including lidocaine and mexiletine. Class 1C blockers are characterized by slow blocking kinetics and include flecainide, propafenone, and moricizine.
The risk of proarrhythmogenicity of antiarrhythmics is increased in dogs and cats by virtue of the dearth of scientifically based data supporting their use. However, regarding the data that do exist, when comparisons are made between dogs and humans, the concentrations at which antiarrhythmic effects of class I drugs are realized generally are similar. However, marked differences in pharmacokinetics exist; indeed, an exception to the generalization occurs for procainamide because of differences in its metabolism between dogs and humans.114
KEY POINT 14-28
All antiarrhythmics are proarrhythmic, and their use should be initiated cautiously and only if proper monitoring tools are available.
KEY POINT 14-30
Blockade of sodium channels might be expected to affect nondepolarizing cells more than depolarizing cells.
KEY POINT 14-31
The anticholinergic effects of quinidine –which initially result in an increased heart rate –contribute to its efficacy in the treatment of atrial fibrillation.
Class IA Drugs
Class 1 A drugs increase the threshold for excitability and decrease automaticity, reduce the rate of phase 0 depolarization and thus slow conduction, prolong both the effective refractory period and the action potential duration, and delay repolarization. A shared arrythmogenic effect of sodium channel blockers with atrial flutter reflects slowed conduction. Subsequent slowing of atrial flutter may allow more signals to be transmitted through the atrioventricular node, causing the heart rate to increase.
Quinidine
Quinidine, derived from the bark of the cinchona plant, is a diastereomer of the antimalarial drug quinine. Quindine affects most types of cardiac muscles.5,112 Its efficacy against supraventricular and ventricular arrhythmias facilitates its classification as a broad-spectrum antiarrhythmic. Quinidine has both direct and indirect effects. For direct effects, quinidine blocks open Na+ channels, increasing the threshold for excitability and decreasing automaticity, thus suppressing ectopic pacemakers. Blockade of multiple K+ currents prolongs action potentials, particularly at slow heart rates. Electrophysiologically, quinidine increases the QRS complex; the QT interval may also be prolonged, causing torsades de pointes at therapeutic or subtherapeutic concentrations in some patients.5 Because it also prolongs the effective refractory period, especially in the atria, quinidine is particularly useful for treatment of reentrant arrhythmias such as atrial fibrillation.112,113,2,115 In the atria quinidine also has an indirect effect through antivagal (“atropine-like”) actions, contributing to undesirable side effects, and specifically, tachycardia.
Although given intravenously (which markedly increases the risk of cardiotoxicity) and intramuscularly, quinidine is most practically administered orally. Intramuscular injection is painful. Quninidine has been prepared as different salts to manipulate (prolong) oral absorption. Quinidine sulfate is absorbed rapidly after oral administration,5,112 whereas the gluconate form is absorbed more slowly. Despite marked (90%) binding to α glycoproteins, quinidine distributes rapidly to most tissues, resulting in a large volume of distribution. In states of high stress, it may be necessary to increase doses of quinidine to overcome increased binding associated with increased inflammatory and other α glycoproteins, although this may be an issue more commonly encountered in humans with myocardial infarction. Quinidine binds to tissue, including cardiac proteins. Hepatic metabolism is mediated by CYP3A and is extensive, with excretion of parent compound or metabolites in the urine. Variability in metabolism is marked among patients and can be influenced by other drugs, necessitating individualized therapy. The duration of action of quinidine may be shortened or lengthened by drugs that target CYP3A4. The half-life is about 6 hours, but the dosing interval may be prolonged with slow-release preparations.
Quindine disposition has been studied in Beagle dogs (n=4). An intravenous dose of 1 mg/kg distributed to a volume of 1.17 ± 0.40 L/kg and was cleared at a rate of 5.90 ± 0.40 mL/min/kg, yielding an elimination half-life of 3.46 ± 1.44 h and mean residence time (MRT) of 3.34 ± 1.25 h. After oral administration of the sulfate salt (100 mg orally), Cmax was 2162 ± 598 ng/mL and oral bioavailability was 73%.116
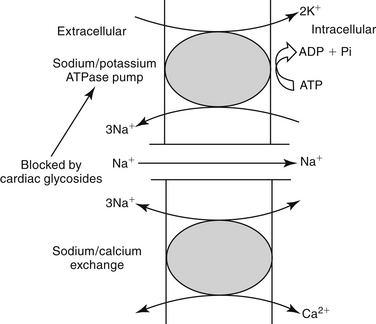
Quindine can cause or be affected by drug interactions, particularly by drugs that affect CYP 2D6 and 3A. It is a potent inhibitor of CYP 2D6, prolonging the elimination of selected drugs or preventing the formation of active drug (e.g., morphine from codeine).5 Its clearance is affected by other drugs that impair (e.g., cimetidine, verapamil) or induce (e.g., phenobarbital, phenytoin, rifampin) CYP 2D6. Clearance of quinidine was decreased in normal Beagles (n=4) by 50% after treatment with the CYP3A substrate inhibitor ketoconazole. The elimination half-life (hr) increased from 3.46 ± 1.44 to 6.78 ± 1.98; Cmax increased from 2162 ± 598 to 3295 ± 636 ng/mL.116 Quindine clearance also is prolonged by drugs that alkalinize the urine (e.g., carbonic anhydrase inhibitors, thiazide diuretics). Quindine decreases digoxin clearance and may compete with it for P-glycoprotein–mediated efflux. Competition at cardiac binding sites also may displace digoxin, further increasing plasma digoxin concentrations, which may exacerbate digoxin-induced cardiac arrhythmias.117
Quinidine is associated with cardiotoxicity-induced arrhythmias such as atrioventricular blockade or ventricular arrhythmias. Sudden death caused by syncope has been reported. The atropine-like (antivagal) effects of quinidine probably account for some potentially serious side effects. Loss of vagal tone, important to the control of conduction in the atrioventricular node, may increase impulse conduction to the ventricles, resulting in paradoxical acceleration, an undesirable increase in heart rate in the patient with supraventricular tachycardias (including atrial fibrillation). This loss of vagal tone also impacts drugs whose cardiac effect is based on enhanced vagal tone. In humans, digitalization prior to therapy is indicated. Quinidine is also an α-adrenergic blocking agent and can cause vasodilation and potential hypotension. Gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea) may occur, particularly with the sulfate form.
As a broad-spectrum drug, quinidine may be effective for acute or chronic management of supraventricular and ventricular arrhythmias. However, its efficacy particularly targets atrial arrhythmias. Quinidine is contraindicated in the presence of complete A-V heart block. Daily doses range from 5 to 15 mg/kg; therapeutic concentrations are 2 to 6 μg/mL. The drug can be given intravenously, but only with extreme caution.
Procainamide
Procainamide differs from the local anesthetic procaine only by replacement of an ester with an amide (see Figure 14-9). Like quinidine, procainamide blocks open Na+ channels and outward K+ channels. Thus it decreases automaticity, increases refractory periods, and slows conduction as well as prolongs the action potential duration. Its N-acetyl metabolite (in which the dog is deficient) is active and accounts for a major proportion of activity in humans. Although the metabolite does not block Na+ channels, it does prolong the action potential duration (a K+-blocking effect).5 The metabolite is equal in efficacy but less potent than the parent compound in the control of ventricular arrhythmias.118 The effects of procainamide on automaticity, excitability, responsiveness, and conduction are similar to those produced by quinidine. Indirect effects (those affecting the autonomic nervous system) are significantly weaker, with no α-adrenergic blockade or paradoxical acceleration.5
KEY POINT 14-32
Because the acetyl metabolite of procainamide targets potassium channels, decreased efficacy can be anticipated in dogs compared to humans.
Procainamide is rapidly and almost completely absorbed after oral administration, although the rate varies with the preparation. Peak concentrations occur within 45 to 75 minutes with a capsule but take longer with tablets. Bioavailability in the dog approximates 85%.119 Only about 20% of the drug is protein bound to glycoproteins in humans.5 Distribution occurs to most tissues except the brain,5 yielding a large (1.44 L/kg in dogs) volume of distribution.119 Procainamide is extensively biotransformed by the liver to metabolites. In humans, N-acetylprocainamide is a major active metabolite, contributing significantly to antiarrhythmic effects. In dogs acetylation is deficient and therapeutic concentrations reflect principally the parent drug whereas for humans, it reflects both parent and metabolite. In dogs the mean concentration necessary to control arrhythmias in ouabain (a cardiac glycoside)-intoxicated dogs was 33.8 μg/mL, with a range of 25 to 48.5 μg/mL.120 This compares with a therapeutic range of concentration of 5 to 30 μg/mL for the combined parent and metabolite in humans. The elimination half-life of procainamide in the normal dog is 2.5 to 2.8 hours.
The fluoroquinolones decrease clearance of procainamide or its N-acetyl metabolite in humans. The extent and target (parent versus metabolite) varies with the fluoroquionlone drug. In humans ciprofloxacin decreases clearance of both by nearly 20%; competition appears to be occurring for renal tubular transport proteins.121 Toxicities of procainamide include cardiotoxicity, similar to that induced by quinidine; hypotension with rapid intravenous administration (bolus); and gastrointestinal signs (anorexia, nausea, vomiting, diarrhea). Cardiac toxicity is indicated by a 50% widening of the QRS complex or by bradyarrhythmias or tachyarrhythmias.
Procainamide is available as oral capsules, tablets, and sustained-release tablets. Intravenous preparations are available for acute or unstable situations. Intravenous infusion is a reasonable approach for the treatment of the acute patient and may be preferred to rapid bolus, which may be associated with hypotension. Procainamide also can be administered intramuscularly (15 to 20 mg every 2 hours [dog] or 8 to 16 mg every 3 to 6 hours [cat]). An advantage to procainamide over other antiarrhythmics is ease of converting from an intravenous to an oral preparation. The transition is best accomplished by stopping an infusion, and, at one elimination half-life, administering the first oral dose (125 to 500 mg every 6 to 8 hours, for a total of 33 mg/kg per day).
Procainamide is a broad-spectrum antiarrhythmic drug. In general, its effectiveness as a ventricular antiarrhythmic drug parallels or exceeds that of quinidine, and it is useful for patients who have failed quinidine therapy. Arrhythmias for which procainamide have proved useful include ventricular122 and, to a lesser degree, supraventricular types. Procainamide can suppress digitalis-induced toxicity but fatalities may occur.
Disopyramide
The pharmacologic effects and spectrum of disopyramide are similar to those of procainamide and quinidine. Although it is effective in controlling supraventricular arrhythmias, its primary use is for ventricular tachyarrhythmias. Disopyramide has been studied in the dog.123 It is quickly absorbed after oral administration, but it undergoes rapid metabolism and clearance. Its half-life is less than 2 hours in the dog, necessitating multiple daily administrations. Like quinidine, disopyramide has potent antivagal effects and can therefore increase the ventricular rate dramatically in patients with supraventricular tachycardias. More important, disopyramide also has a negative inotropic effect on the heart and can be lethal for patients with preexisting myocardial disease. Clinical indications for disopyramide are probably limited by its potential adverse affects on the heart.124
Class IB Drugs
Class IB drugs include agents such as lidocaine and its congeners and phenytoin. Lidocaine (see Figure 14-9), widely used as a local anesthetic, is the prototypic class IB drug. Increasingly, it is being used for a variety of conditions (discussed later). Lidocaine preferentially binds to open (phase 0) and inactivated (phases 1 through 3) channels. Therefore cardiac tissues with long action potential duration or tissues that are rapidly firing (because more time is spent in the inactivated state), such as ischemic tissues, are more affected by lidocaine. Because atrial tissues have a very short action potential compared with ventricles, lidocaine preferentially targets arrhythmias in the abnormal Purkinje system and ventricles. In contrast, it minimally affects the sinoatrial node, atria, or atrioventricular node, although it has been used to successfully treat supraventricular arrhythmias in a limited number of dogs.125 Additionally, abnormal ventricular tissues are preferentially affected compared to normal myocardial tissues. Lidocaine also minimally affects the autonomic system.
Lidocaine suppresses automaticity, increases the threshold, and hyperpolarizes (increases the resting membrane potential) of Purkinje fibers. Increased conduction velocity may facilitate inhibition of reentrant arrhythmias. The action potential duration is either unaffected or shortened. Lidocaine efficacy is dependent on potassium; hypokalemia will minimize its efficacy.
KEY POINT 14-33
Lidocaine targets inactive sodium channels, which are more prevalent in diseased, rapidly beating tissues, thus minimizing binding to normal or atrial myocardial tissues.
Lidocaine is well absorbed orally, but it is subject to first-pass metabolism, with only one third of the drug reaching systemic circulation. Rectal bioavailability ranges from 32% to 56%, depending on the preparation.126 Peak concentrations in dogs after intravenous administration of 6 mg/kg approximate 10 μg/mL, declining to 0.1 μg/mL by 3 hours, with the elimination half-life approximating 50 minutes.127 After intramuscular administration, absorption is complete, with peak concentrations at the same dose approximating 1.8 μg/mL at 30 minutes. Distribution is rapid to a volume of approximately 1.4 L/kg.127,128 About 70% of the drug is bound to protein (glycoprotein). Lidocaine is cleared by hepatic metabolism to active and inactive metabolites. Clearance in female dogs (n=4; dose of 2 mg/kg infused over 5 minutes) was 27.5 ± 6.0 mL/min/kg. As a flow-limited drug, systemic clearance of lidocaine should decrease in proportion with hepatic blood flow. Portosystemic shunting will increase oral bioavailability; experimentally induced portosystemic shunting in dogs increases it from 15% to 80%.129 Lidocaine is prepared for intravenous administration. Lidocaine can be administered intravenously as a rapid bolus or as a continuous intravenous infusion. It can also be given intramuscularly in emergency situations. Topical creams, salves, or gels and patches are intended for local anesthetic use only and will not provide antiarrhythmic efficacy.
As an antiarrhythmic, lidocaine is indicated for emergency treatment of ventricular arrhythmias. Pharmacologic effects occur rapidly. Lidocaine has several undesirable effects. Although lidocaine may decrease the risk of acute death associated with ventricular fibrillation, one study demonstrated decreased hospital survival, mitigating its routine use. The primary toxicity in the dog occurs in the central nervous system, with symptoms that range from drowsiness or agitation to muscle twitching and convulsions at higher plasma concentrations. Therapeutic and toxic lidocaine concentrations have been established experimentally. In an study of canine myocardial infarction, inhibition of electrically stimulated ventricular tachycardia responded to mean concentrations of 3.5 μg/mL (1 mg/kg intravenously followed by 80 μg/kg/min).130 Wilcke and coworkers131 compared effective and toxic concentrations in six dogs. Minimum effective concentrations for experimentally induced ouabain toxicity (outcome measure was eradication of ventricular tachycardia) ranged from 3.8 to 7.65 μg/mL (mean 6.25 μg/mL). The time necessary to eradicate the arrhythmia ranged from 0.3 to 1 hour after infusion of 1480 mg/hr. However, neurologic manifestations of toxicity appeared at 6.3 to 10.4 μg/mL (mean 8.21 μg/mL) (tonic extension) and became more exaggerated (cortical seizures) at 7.3 to 11.2 μg/mL (mean 9.58 μg/mL). Other neurologic manifestations of lidocaine toxicity include anxiety, sedation, and disorientation. The risk of toxicity with intravenous administration is greater in the cat compared with the dog; however, the cat s prone to cardiac toxicity. Cardiac suppression will occur at higher doses; exacerbation of heart failure has been reported in human patients with poor left ventricular function.5 Lidocaine can worsen first-degree or second-degree atrioventricular block and is contraindicated for patients with third-degree heart block because of suppression of ventricular automaticity. A large intravenous bolus may cause sinus arrest; cats are more prone to this adversity.
Drug interactions involving lidocaine are limited. Care should be taken when mixing lidocaine solution with other drugs to avoid pharmaceutic direct drug-drug interactions. Selected drug interactions reflect competition with other basic drugs for binding sites on glycoproteins, although their clinical relevance is not clear. Lidocaine increases hepatic blood flow: a 12-hour infusion of 76 μg/kg/min increased hepatic arterial blood flow 1.6- to 9.2-fold.128 Despite its flow-limited behavior, lidocaine appears to decrease its own clearance, with the impact being demonstrable following longer infusions compared with bolus administration.128,132 Changes were more profound in one study following a 24-hour compared with a 90-minute infusion period.132 Another study found that multiple administration of lidocaine (once daily) was associated with a decrease in hepatic intrinsic clearance from 1224 ± 859 to 285 ± 104 mL/min/kg.128 Lidocaine metabolism is inhibited by a number of drugs, including midazolam and thiamylal.133
Tocainide (no longer marketed in the United States) and mexiletine are class IB antiarrhythmic drugs that are similar in chemistry and mechanism of action to lidocaine but modified to reduce first-pass metabolism, thus allowing oral administration. For example, tocainide is 100% orally bioavailable. Using a model of canine myocardial infarction, tocainide prolonged conduction intervals and the effective refractory period by 26% to 31% in damaged tissue compared with 6% to 8% in the noninfarcted zone.134 Tocainamide has been used for long-term management of ventricular arrhythmias, particularly those that respond to lidocaine or fail to respond to procainamide. Arrhythmias that are refractory to lidocaine are not likely to respond to tocainide. Tocainide is prepared as a racemic mixture. Both the R and S enantiomers appear to have greater antiarrhythmic activity alone compared with the racemic mixture, whereas the racemic mixture appears more toxic than either enantiomer alone. Consequently, safety and efficacy might be improved if single enantiomer preparations become available.135 Tocainaide may be more likely than lidocaine to cause CNS and gastrointestinal adverse reactions. Bone marrow dyscrasias and pulmonary fibrosis have limited use of tocainide. Blood dyscrasias have occurred in dogs.110 Contraindications to lidocaine should be followed for tocainide.
After intravenous administration of tocainide, spontaneous premature ventricular complexes were reduced in dogs with experimentally induced myocardial infarction.136 In Doberman Pinschers (n=23) with cardiomyopathy, tocainide at 15 to 25 mg/kg every 8 hours reduced the number of ventricular premature complexes short term by at least 70% in 80% of treated dogs; ventricular tachycardia was corrected in 90% of affected dogs. Serum concentrations at 2 hours and 8 hours were 6.2 to 19.1 mg/L and 2.3 to 11.1 mg/L, respectively. Long-term control was more difficult in animals whose left ventricular shortening fraction was less than 17%. Although efficacious, tocainide therapy may be limited by side effects. Anorexia and gastrointestinal disturbances occurred in 35% of dogs, with peak concentrations being higher in afflicted dogs (14 μg/mL) compared with dogs not exhibiting toxicity (11 μg/mL). Serious adverse effects occurred in 58% of dogs treated longer than 4 consecutive months and included progressive corneal endothelial dystrophy; renal dysfunction occurred in 25% of the dogs.137
Mexiletine, like tocainide, is a structural analog of lidocaine, indicated for oral management of life-threatening ventricular arrhythmias. Like lidocaine, it decreases the the APD more than the ERP such that the ratio of ERP/APD increases (package insert). Normal cardiac tissue is minimally impacted. In humans and dogs, mexiletine is characterized by high oral bioavailability, low first pass metabolism, 50-60% bound to plasma protein, and a volume of distribution of 5-7 liters/kg. Clearance in humans is primarily hepatic via CYP2D6 metabolism (hence, major differences in metabolizer phenotypes may occur), with some CYP1A2; metabolites are largely inactive. extensive metabolizer phenotypes. In dogs, most of the drug may be renally excreted, although its plasma elimination half-life approximates 10-12 hours; liver disease may prolong clearance. Mexiletine exists as a racemic mixture; in humans, the S isomer is characterized by a higher area under the curve (mean R/S ration = 0.8). 137a In dogs, mexiletine has demonstrated stereoselective effects, with R-(-)-mexiletine being more potent in the prevention of ventricular tachycardia than S-(+)- mexiletine. 137b
In human clinical trials involving mexiletine, approximately 40% of study participants withdrew from studies because of adverse effects; this rate was similar to treatment groups receiving quinidine or procainamide. Rarely, mexiletine has been associated with liver disease, warranting monitoring in patients with abnormal tests indicative of liver disease. Although mexiletine does not cause heart block in normal animals, it occasionally exacerbates pre-existing conduction disturbances; its use is contraindicated in the presence of 2nd or 3rd degree heart block. Mexiletine is arrhythmogenic: sustained ventricular arrhythmias worsened in approximately 10% of human patients receiving mexiletine. Minimum therapeutic plasma concentrations appear to approximate 0.5 mcg/ml in humans (range 0.05 to 2 mcg/ml). Dosing intervals are designed to minimize fluctuation.
Studies of mexiletine in dogs are largely experimental in nature, supporting use in humans. Experimentally, including studies using canine models, it suppresses induced ventricular arrhythmias. Mexiletine (5 mg/kg) (or esmolol at 1.25 mg/kg or verapamil at 0.4 mg/kg) was limited in its success in preventing torsades de pointes in a canine model involving blockade of delayed rectifying current and β-adrenergic stimulation. 110a However, mexiletine (4.5 mg/kg load followed by 1.5 mg/kg/hr infusion) was able to partially prevent the proarrhythmic effects of β-adrenergic A-V blockade (sotalol 4.5 mg/kg load followed by 1.5 mg/kg/hr infusion) in diuretic (furosemide and hydrochlorthiazide)-induced hyopkalemic dogs. Mexiletine prevented sotalol-associated torsades de pointes that was electrically induced in the dogs. 110b Pharmacodynamic effects of melixetine were determined in three canine models of arrhythmias. The minimum effective plasma concentrations (μg/ml) of mexiletine necessary to suppress arrhythmias were 1.8 ± 0.6 when induced by digitalis, 3.7+0.9 for adrenaline, and 2.2 ± 0.4 for coronary ligation μg/mL.110c The plasma elimination half-life in dogs is 3 to 4 hr. Mexiletine may be more effective when used in combination with other drugs. For example, it was more effective at in preventing experimentally induced ventricular arrhythmias in dog myocardium when combined with quinidine. 110d
Like tocainide, mexiletine is more likely than lidocaine to cause central nervous system and gastrointestinal toxicity. Mexiletine induces seizures in dogs at 25 mg/kg; vomiting occurs at 15 to 30 mg/kg. However, the drug was well tolerated at 15 mg/kg for 13 weeks in normal dogs. 138b
Contraindications to lidocaine should be followed for mexiletine.
Phenytoin is an anticonvulsant with a limited spectrum of cardiac antiarrhythmic activity. Its mechanism and impact are similar to those of lidocaine. Its primary use in veterinary medicine has historically been management of digitalis-induced arrhythmias; phenytoin will shorten atrioventricular nodal and Purkinje refractory period in digitalized patients.
Class IC Drugs
Class IC drugs cause effects similar to those of the class IB drugs except that they do not prolong the refractory period. Examples include encainide, flecainide, lorcainide, and propafenone. Conduction velocity is depressed. In addition to sodium channel blockade, flecainide also blocks (in vitro) delayed rectifier potassium channels and calcium channels. Action potential duration is shortened in Purkinje cells (as a result of blockade of late-opening sodium channels) but prolonged in ventricular tissues (because of potassium channel blockade). In contrast to most class 1B drugs, flecainamide affects atrial tissues, prolonging the action potentials proportionately with rates, rendering it effective for atrial arrhythmias. However, it affects normal as well as abnormal tissues. Flecainide studies in dogs have been largely experimental. In one study concentrations of 610 ± 111 ng/mL increased the electrically induced defibrillation threshold in dogs.138 The arrhythmogenicity of flecainamide can be lethal. Reentrant ventricular tachycardia may emerge when treating atrial arrhythmias, heart block may occur in conduction disturbances, and congestive heart failure can be exacerbated if ventricular performance is poor.5
Propafenone is a Na+ channel blocker that is similar to flecainide, including K+ channel blockade. It is prepared as a racemic mixture with the S-(+) isomer also exhibiting β-adrenergic antagonism in some (human) patients at concentrations at or above 1 μg/mL. Its efficacy is greater with atrial than with ventricular arrhythmias. It is eliminated through CYP2D6 metabolism as well as renal excretion, generally undergoing first-pass metabolism to an equipotent active metabolite. At least one other metabolite is active. Metabolism can be saturated with only small dose increases, resulting in disproportionately high plasma drug concentrations. Inhibitors of CYPD26 will contribute to high drug concentrations. It is available in slow-release preparations. Dosing is modified in human patients with liver disease.
Class II Antiarrhythmic Drugs
Class II antiarrhythmic drugs are β-adrenergic receptor blocking agents. Although discussed here because they affect cardiac arrhythmias, β-blockers have a number of potentially positive effects, particularly on the failing heart. Abbott26 has reviewed β blockade in the management of systolic dysfunction. β-adrenergic stimulation causes the following effects, which are antagonized by β blockers: decreased magnitude and inactivation of calcium current, increased magnitude of potassium and chloride repolarizing currents, increased pacemaker activity, and increased DAD- and EAD-mediated arrhythmias.5,9 Adrenergic signals stimulate atrial chronotropic β receptors more so than ventricular receptors. As such, β-blocking drugs (propranolol, alprenolol, and metoprolol) are most effective in slowing atrial compared with ventricular rates and are more effective during states of adrenergic stress.139 Beta blockers decrease pacemaker current and thus sinus rate.5 Because β blockers increase atrioventricular nodal conduction and prolong atrioventricular nodal refractoriness, they are useful for reentrant arrhythmias associated with the atrioventricula node.5 In acutely ischemic myocardial tissue, β blockers increase the energy necessary to fibrillate the heart and thus may decrease mortality (in humans) if used in the first couple of weeks after myocardial infarct.5
A number of clinical trials have revealed the efficacy of β-blockade in the treatment of heart failure, reflecting a major effect on remodeling associated with the failing heart.140 The effects of β blockade in chronic heart failure have been described as protective. Effects considered protective include decreased heart rate, decreased energy consumption, and antifibrillatory effects. Prevention of adrenergic overactivation decreases myocardial cell necrosis. Beta blockers that induce an upregulation of β receptors improve contractility. These benefits tend to outweigh negative inotropic effects that might lead to deterioration of hemodynamics and decompensation, although care must be taken in patients experiencing acute failure. The success of the “paradoxical intervention” may not be obvious until 2 to 3 months after initiation of additional β blocker therapy.
KEY POINT 14-34
In addition to slowing the heart rate—particularly in the presence of high sympathetic tone—β blockers also provide cardioprotection and slowing of progressive myocardial failure.
Drug therapy with β blockers presents pharmacokinetic and pharmacodynamic challenges generally not encountered by many other drugs. Among the challenges are the potential sequelae of interactions between drugs and receptors. Pharmacodynamically, chronic exposure of receptors to agonists or antagonists may cause receptors to be internalized and destroyed, resulting in decreased response or desensitization. Desensitization of β receptors may accompany long-term β blockade therapy. However, initiation of therapy at low doses may attenuate desensitization, an approach that is recommended in human patients with CHF.7 Patients are monitored closely for acute adversities, and the dose is gradually increased only as low doses are tolerated. Low-dose metoprolol therapy simultaneously reduces vascular resistance and avoids reflex tachycardia, an advantage previously recognized only for carvedilol.141 Use of low doses may also decrease the rebound effect that has been documented in humans once β blockade is discontinued. Rebound is associated with worsening heart failure and arrhythmias. Down-titration (decreasing the dose as the drug is discontinued over a 2-week period) reduces the rebound effects.142 Use of β blockers in states of excessive adrenergic stimulation may allow unopposed α-adrenergic stimulation–mediated hypertension. Beta-blockers should be tapered rather than suddenly decreased to avoid the negative sequelae of receptor up regulation.
Beta blockers are not equal in efficacy. Protective effects of carvedilol and metoprolol were compared in progressive CHF in humans.143 Carvedilol performed better in preventing cardiovascular-related illness in a clinical trial (COMET) involving human patients with heart failure associated with ischemia or idiopathic cardiomyopathy (see later discussion). Pharmacokinetically, distribution into the central nervous system—which may be important for maximal response—varies with lipophilicity, with effects of water-soluble drugs (atenolol, nadolol) potentially muted compared with those of more lipophilic drugs (e.g., metoprolol). Distribution is complicated by binding to both albumin and α-glycoproteins, the latter a protein whose concentration increases with inflammation; this may be more relevant to acute myocardial infarction. Binding decreases concentration of active drug; however, because clearance of flow-limited drugs generally is not affected by protein binding, a compensatory increase in drug clearance should not be anticipated and the risk of drug interactions at the level of protein binding increases. Lipophilic β-adrenergic drugs generally are characterized by hepatic clearance that is flow-limited (exceptions are the water-soluble atenolol and nadolol, which are predominantly renally excreted). For flow-limited drugs, the rate of substrate delivery (hepatic blood flow) determines clearance. As such, oral administration is characterized by significant (if not total) hepatic extraction and first-pass metabolism. Beta blockers interact with P-glycoprotein, with carvedilol being an example; its impact is sufficient to warrant its therapeutic use for inhibition of drug transport, which otherwise would result in multidrug resistance. 143a The impact of disease and its successful treatment may impact response. Progressive cardiac disease or improvement thereof might profoundly alter clearance; disposition will be markedly altered in patients with liver disease associated with portosystemic shunting. Pharmacokinetics are further complicated by production of metabolites that are variably active. For example, for carvedilol, M4 and M5 are equipotent to the parent drug; M14 appears to be responsible for greater antioxidant effects compared with the parent. The hydroxymetabolite of metoprolol has 20% of the parent compound in dogs.144
Finally, most β-adrenergic blockers contain at least one chiral carbon, and thus are present as two isomers, each differing both kinetically and dynamically from the other.141 The stereometric character of β-blockers markedly complicates their use. Complicating interpretation, the isomers can be described according to their spatial orientation (S versus R) or the direction in which they rotate light (+ or −); the two are not necessarily related. Blockade of β-1 receptors is achieved predominantly by the S isomer for most drugs. An exception occurs for sotalol, for which the R isomer exhibits more β blocker activity. In the dog the (−) isomer (based on rotation of polarized light; not related to R or S terminology is almost twofold more potent than the (+) isomer of metoprolol.145 In contrast, non–β-1-blockade effects (e.g., blockade of α receptors, antiarrhythmic activity) generally are not stereoselective. All β blockers are marketed as a racemic mixture; exceptions include imolol (marketed as the S isomer), labetolol (which contains two chiral carbons), and nadolol (which contains three chiral carbons); all these are marketed as a mixture of four isomers. Absorption of β blockers does not appear to be stereoselective, although timolol may be stereoselectively metabolized by intestinal epithelial cells.141 Binding to proteins may be steroselective, with selectivity varying with the binding protein. Distribution to tissues does not appear to be stereoselective beyond that determined by differences in protein binding. However, β blockers appear to be stored at terminal nerve endings in a stereoselective manner, with preferential storage of the (−) isomer, particularly for water-soluble drugs such as atenolol. Metabolism of β blockers is very complicated. Each isomer may be stereoselectively metabolized to active metabolites, which maintain the chiral carbon; as such, metabolites may be stereoselectively active or further metabolized. For metoprolol and carvedilol (but not significantly for other lipid-soluble drugs), metabolism is stereoselective, resulting in stereoselective plasma drug concentrations. In humans concentrations of the (+) carvedilol isomer (the less active isomer) are approximately twice that of the (−) isomer with regard to Cmax and area under the curve; for metoprolol the concentrations of the isomers are almost equivalent, with the (–) isomer up to 30% higher than the (+) isomer. Renal clearance of water-soluble drugs (atenolol, nadolol) does not appear to be stereoselective. Drug interactions also may be stereoselective. In humans CCBs (verapamil and diltiazem) decrease first-pass metabolism. However, the inhibition is stereoselective for verapamil (but not diltiazem) toward the (+) R-isomer of propranolol. Cimetidine and quinidine also have exhibited stereoselective inhibition of β blockers. Finally, genetic polymorphism has been demonstrated for enzymes responsible for CYP-mediated drug metabolism in humans (and should be anticipated in dogs), although differences do not appear to be stereoselective.141
KEY POINT 14-35
Like many cardioactive drugs, enantiomers and active metabolites contribute to variability in disposition among patients.
Nonselective B Blockers
Propranolol is the prototype β blocker. It is a competitive, nonselective β blocker of both β-1 and β-2 receptors. Like all β blockers, propranolol is most effective in the presence of elevated sympathetic tone. Its negative chronotropic effect is less likely in conditions not associated with elevated levels of catecholamines (e.g., less effective if associated with hypokalemia, fever, some heart diseases). It will slow ventricular rates in patients suffering from supraventricular arrhythmias, including those induced by digitalis toxicity, but is rarely able to convert a supraventricular arrhythmia to a normal sinus rhythm. As a β-1 blocker, propranolol is also a negative inotrope. This pharmacologic effect might be detrimental in the patient with small cardiac reserve (e.g., the patient with decompensated CHF) during acute treatment. Propranolol has been studied in euthryoid and hyperthyroid cats.146 Changes in disposition induced by hyperthyroidism suggest that a lower dose is indicated for oral administration because of increased bioavailability. Clinical indications of propranolol (β blockers) as an antiarrhythmic include reduction of ventricular rate in cases of supraventricular tachycardias, hypertrophic and other forms of obstructive heart disease, and hyperthyroidism. The effects of propranolol are dose, time, duration and route dependent; although oral administration may be associated with decreased bioavailability of the parent drug, formation of a more effective metabolite may cause better response with oral administration.146b
The toxic effects of propranolol are the result of nonselective β blockade and include bradyarrhythmias, hypotension, heart failure, bronchospasm, and hypoglycemia, particularly in diabetics. In an intact experimental canine model, insulin (4 IU/min intravenously with glucose) was shown to be superior to epinephrine for treatment of acute propranolol toxicity.147
Nadolol (5 to 10 mg orally every 6 to 12 hours [cat]; 40 to 60 mg orally every 6 to 12 hours [dog]) also is a nonspecific β blocker that is renally excreted. Side effects and contraindications typical of propranolol occur for nadolol.110 Sotalol is another non-specific β-blocker that also prolongs the APD and ERP of atrial and ventricular fibers (Class III).
Selective Beta Blockers
Metoprolol is a relatively selective, lipophilic β-1 blocker. It is marketed as a racemic mixture, although the (−) isomer provides the predominant β blockade effect. The clinical pharmacology of metoprolol exemplifies the complexities associated with therapeutic use. Metoprolol undergoes hepatic clearance to several metabolites, of which the α-hydroxyl metabolite, the product of metabolism steroselective for the S(−) isomer is active. However, the proportion of this metabolite formed from the parent compound may not be clinically significant. O-Demethylation and N-dealkylation appear to be the major metabolites formed in dogs.148
The β-blocking effect of α-hydroxymetoprolol has been compared with that of metoprolol after intravenous administration in the dog. The dose–response relationship was linear for both compounds, but the metabolite required 10 times the plasma concentration of the metoprolol (5 times the dose) for therapeutic equivalence. The volume of distribution for the metabolite was 2 liters/kg compared with 3.5 liters/kg for metoprolol, whereas clearance was 3.5 mL/kg/min for the metabolite compared with 20 mL/kg/min for metoprolol. The net effect of these differences resulted in an elimination half-life for the α-hydroxy metabolite of 7 hours compared with 2 hours for the parent compound. Approximately 5% of an intravenous dose of metoprolol was metabolized to the active metabolite.144 In Beagles (n=4, 8 years old, all female) receiving 1.37 mg/kg metoprolol orally, the area under the curve (0-48 hours; μg/mL/hr) was 2 for α-hydroxymetoprolol, compared with 4.39 for metoprolol acid and 1.89 for metoprolol. Peak plasma concentrations (estimated from concentration versus time curve) of metoprolol and its metabolites were 400 ng/mL at 0.5 hour for metoprolol, compared with 100 ng/mL at 3 hours for α-hydroxymetoprolol. An intravenous dose of 2 mg/kg resulted in a decrease in heart rate in normal dogs by 35% at a concentration of 379 ± 24 ng/mL; the concentration of the α-hydroxyl metabolite was approximately 38 ng/mL. A similar intravenous dose of the metabolite (2 mg/kg) resulted in a 25% reduction in heart rate. These studies suggest that the active metabolite of metoprolol may not play a major role in cardiac response, although the impact on other effects (i.e., antiarrhythmic, cardioprotective) is not clear.
Stereoselective pharmacodynamic effects of metoprolol also have been described in dogs. The concentration (μg/mL) necessary to achieve 50% of the inhibitory effect for each isomer were as follows: for Vmax 250 ± 80 (R) versus 70 ± 30 (S), dP/dtmax: 450 ± 210 (R) versus 70 ± 40 (S) and heart rate, 520 ± 210 (R) versus 82 ± 27 (S). As such, the (S) isomer was more potent than the (R) isomer at a ratio of 3.7, 6.8, and 6.3 for Vmax, dp/dtmax, and heart rate, respectively.145 Stereoselectivity has also been reported for the disposition of metoprolol in anesthetized dogs, (although applicability to awake dogs is not clear). The peak times to maximum inhibitory effect of either metoprolol isomer occurred at 90 to 120 minutesBecause the isomers were not given intravenously, volume of distribution(Vd) and CLs could not be corrected for bioavailability (F); however, differences occurred in Vd/F, CLs/F, and area under the curve between the isomers;.145 Another investigator149 documented that hepatic clearance of metoprolol in dogs is slightly selective for the (S) isomer.
Metoprolol is prepared as an oral tartrate (Lopressor®), and slow-release succinate salt (Toprol XI®); the latter allows once-daily dosing in humans. The slow-release preparation does not appear to have been studied in dogs or cats. Indications of the slow-release preparation in humans include hypertension as well as treatment of stable CHF.
Verapamil (3 mg/kg) inhibited clearance of metoprolol in dogs approximately 50% to 70%. The effect is profound after oral administration, abolishing first-pass metabolism, with inhibition selective toward O-demethylation of the (S) isomer.149
A large multicenter clinical trial studied the use of metoprolol for treatment of acquired heart disease and particularly DCM compared with a placebo in dog.149a The drug did not appear to decrease mortality rates but did improve ventricular function as well as quality of life.
Carvedilol represents one of the more recently (third) generation of β blockers associated with potential antiarrhythmic, antihypertensive, and antiremodeling effects.150-152 Carvedilol is specifically approved to reduce cardiovascular mortality in human patients. Although it is characterized by β1 and β2 as well as α-adrenergic blockade, it is relatively (mildly) β1-selective in human patients. Vascular endothelium contains β1 and β2 receptors as well as α-1 receptors, each targeted by carvedilol. As such, it decreases total peripheral resistance and preload without compromise of cardiac output or causing reflex tachycardia. This advantage however, might be minimized by low-dose therapy of other (i.e., not carvedilol) β blockers.153Advantages compared with traditional selective β blockers such as metoprolol include reduced mortality in human patients with left ventricular failure, perhaps resulting from a more complete antagonism of sympathetic activation.140,142,154,155 Carvedilol benefits do not reflect a reduction in heart rate as much as improvement in left ventricular function. Additional advantages may include antioxidant and antiproliferative properties and inhibition of apoptosis in the heart.150-152,156 Finally, carvedilol may inhibit the synthesis of endothelin in coronary arteries.157 Carvedilol appears to protect against doxorubicin-induced cardiomyopathy.158 In a rabbit model of ischemia, carvedilol provided superior cardioprotection, probably because of antioxidant and antineutrophil effects.159 Similar effects were reported in a human patient receiving doxorubicin.160
KEY POINT 14-36
An advantage of carvedilol compared with other selective β blockers is α-adrenergic blockade, which may decrease afterload.
Carvedilol has been relatively well studied in dogs, It is well absorbed and undergoes extensive hepatic metabolism, including glucuronidation and subsequent biliary excretion.161 The kinetics and selected pharmacodynamics have been studied in anesthetized162 and awake163,164 dogs. In anesthetized dogs the elimination half-life was 54 minutes (compared with 2.4 hours in humans), and the volume of distribution was 2 L/kg. When studied at doses ranging from 10 μg to 630 μg/kg, heart rate did not decrease, although reports in awake dogs indicate otherwise. Pulmonary and systemic pressures decreased in treated animals but increased in control animals, consistent with the β blockade effect of the drug. The authors recommend an optimal plasma drug concentration of 100 ng/mL, achieved after intravenous infusion of 150 to 310 μg/mL. Disposition was characterized by marked variability. The median peak concentration (extrapolated) of carvedilol after intravenous administration of 1.75 μg/mL was 476 ng/mL (range 203 to 1920 ng/mL), elimination half-life (t1/2) was 282 minutes (range 19 to 1021 minutes), and MRT was 360 minutes (range 19 to 819 minutes). Volume of distribution at steady state was 2 L/kg (range 0.7 to 4.3 L/kg). After oral administration of 1.5 mg/kg, the median peak concentration was 24 μg/mL (range 9 to 173 μg/mL), time to maximum concentration was 90 minutes (range 60 to 180 minutes), t1/2 was 82 minutes (range 64 to 138 minutes), and MRT was 182 minutes (range 112 to 254 minutes). Median bioavailability after oral administration of carvedilol was 2.1% (range 0.4% to 54%). However, the bioavailability of active metabolites (M4 and M5, which are equipotent to parent drug in humans) was not determined, and it is not clear whether these metabolites are produced in dogs. On the basis of these data, monitoring should be considered to adjust dose. The half-life of 3 hours suggests 8-hour dosing. However, pharmacodynamic studies were also performed.164 Normal dogs were studied at baseline and after multiple-dose (>5 days) oral administration of carvedilol (1.5 mg/kg of body weight orally every 12 hours). Dogs were challenged with isoproterenol. Carvedilol had no effect on heart rate or blood pressure in six of eight dogs at baseline or study end, but heart rate reduced after multiple dosing in two of eight dogs. Carvedilol attenuated isoproterenol-induced changes in heart rate by 54% to 76% through 12 hours and by 30% at 24 hours. The effects of isoproterenol on blood pressure was attenuated by 80% to 100% through 12 hours. Based on normal dogs, an oral dose of 1.5 mg/kg was recommended every 12 hours. The magnitude of β-blockade response correlated strongly to peak plasma carvedilol concentration, suggesting that therapeutic drug monitoring may be clinically useful. Carvedilol also has been compared to bisopropol (see later discussion).
The efficacy of carvedilol and metoprolol for the treatment of chronic heart failure has been compared in humans. The efficacy of these drugs has also been compared with that of standard therapy in humans.154,165 No difference could be demonstrated in most outcome measures between carvedilol versus metoprolol treatment, although blood pressures were lower in carvedilol compared with metoprolol patients. Patients receiving either carvedilol or metoprolol significantly improved compared with those receiving standard therapy.154 More recently, the results of the COMET study, which compared carvedilol with metoprolol, have been reported. The COMET study was a randomized, double-blind, parallel comparison of carvedilol at approximately 0.3 mg/kg twice a day and metoprolol tartrate at approximately 0.7 mg/kg twice a day. Patients (n=3000) were studied for 58 weeks and had stable chronic heart failure, New York Heart Association (NYHA) functional class II to IV, with left ventricular dysfunction. Patients continued ACE inhibition and diuretics. Patients were randomly assigned to receive either carvedilol or metoprolol and followed for 58 months. Endpoints were cardiovascular events (which may be less relevant in dogs or cats), and the proportion of such events in each group (584 for carvedilol versus 667 for metoprolol), although statistically significant, may not be as clinically relevant.
In a study of myocardial perfusion in dogs receiving either carvedilol (2 mg/kg) or metoprolol (4 mg/kg) orally, carvedilol was associated with greater increase in myocardial perfusion and decrease in blood pressure, whereas metoprolol was associated with greater decrease in heart rate.166 Carvedilol appears to be an inhibitor of P-glycoprotein, at least as was demonstrated in vitro in cancer cells: the LD50 of doxorubicin in breast cancer cells increased by twentyfold (200 to 10 ng/mL)167 despite doxorubicin cardioprotection.160
Oyama and coworkers168prospectively failed to find a significant impact of carvedilol (0.3 mg/kg twice daily) in dogs with DCM (n=16; n=7 placebo) using a placebo-controlled, double-blinded randomized design. Endpoints for which significant differences were not documented included changes in ventricular function, activation of neurohumoral compensatory responses, or owner-perceived quality of life. However, animal death reduced sample size and the power of the study to detect a significant difference was not reported. Marcondes-Santos and coworkers169 found some beneficial effect of carvedilol when added to traditional therapy (digoxin, benazepril, codeine) in dogs (n=13) with chronic mitral valvular disease; 12 control dogs with disease did not receive carvedilol. Dogs were studied using a prospective blinded parallel study. A tendency for improvement occurred for quality of life and a reduction in SBP, and improved disease classification during the 3-month study period.
Atenolol (see Figure 14-9) is a selective β2 blocker. In humans its use is associated with greater mortality compared to non-atenolol β blockers. The difference presumably reflects a risk of ventricular fibrillation that is greater with atenolol compared to others. Compared to most other clinically used selective β blockers, atenolol is much less lipid soluble and thus less likely to penetrate the central nervous system; increased mortality may be related to the lack of centrally mediated vagal tone, which would otherwise counteract the risk of fibrillation.170 The implications of this difference among β blockers is not clear for dogs or cats. Little information is available regarding active metabolites or stereoisomers.
Atenolol is 90% bioavailable after oral administration in normal adult cats.171 Elimination half-life in normal adult cats is 3.44 ± 0.5 and 3.65 ± 0.39 hours after intravenous and oral administration, respectively. A dose of 3 mg/kg orally generates a peak plasma concentration of 0.48 ± 0.16 μg/mL and will block cardioresponsiveness to isoproterenol for 12, but not 24, hours, suggesting a 12-hour dosing interval. Using a prospective, randomized, crossover, blinded study, atenolol (6.25 mg every 12 hours) was studied in healthy cats as either an oral or transdermal preparation.172 Peak (2-hour) and trough (12-hour) concentrations were measured after 1 week of administration.Therapeutic concentrations (250 ng/mL) were reached in six of seven cats (579 ± 212 ng/mL) 2 hours after oral administration, but in only two of seven cats (177 ± 123 ng/mL) following transdermal administration. Trough plasma atenolol concentrations were 258 ± 142 ng/mL following oral administration and 62.4 ± 17 ng/mL following transdermal administration. The authors concluded Monitoring might be considered in animals that must receive atenolol transdermally. Atenolol is indicated for cats with HCM associated with outflow obstruction and respiratory distress. Henik and coworkers173 reported the efficacy of atenolol (1 to 2 mg/kg PO every 12 hours) for control of hypertension in hyperthyroid cats (n=20). Although heart rate was decreased, SPB was not well controlled, indicating the need for an additional vasodilator. Crandell and Ware174 described the successful use of atenolol for treatment of cardiac toxicity associated with phenylpropanolamine overdose in a dog.
KEY POINT 14-37
Transdermal delivery of atenolol in cats is not predictable and should be implemented only if monitoring is available.
Esmolol is a β1-selective blocker (S enantiomer) characterized by a very (ultra) short half-life owing to metabolism by erythrocyte esterases. Methanol is a metabolite of esmolol in humans, but its formation does not appear to be clinically relevant. Duration of effect is about 10 minutes; therefore its effects will rapidly dissipate once the drug is discontinued.5 It is administered intravenously and has proved useful in dogs for acute ventricular arrhythmias associated with inhalation anesthesia and surgical removal of hyperactive thyroid glands.110 In anesthetized dogs receiving a constant-rate infusion, steady-state concentrations occurred in 10 minutes, with duration of β blockade paralleling drug concentrations.175 Peak β blockade occurred within 15 seconds after loading with a 500 μg/kg constant-rate infusion and at 30 to 45 seconds after switching to a maintenance dose of 12.5, 25, or 50 μg/kg/min. Duration of β blockade was less than 15 minutes once drug was discontinued. Esmolol has been proved to be effective for treatment of cats with HCM and left ventricular outflow tract obstruction.176
Bisoprolol is among the β-1 selective blockers that have prolonged the life span of humans with cardiac disease. Among its distinguishing characteristics is less lipophilicity than other drugs; consequently, the parent drug is eliminated by both hepatic metabolism and renal excretion (approximately 50% each) in dogs.177 Bisoprolol is less lipophilic than other β blockers, including carvedilol; the implication is not clear, but pharmacodynamic data for bisoprolol apparently are not yet available in dogs. However, Beddies and coworkers177 compared the pharmacokinetics of carvedilol and bisoprolol (1 mg/kg either drug) after both intravenous and oral administration in 12 Beagles using a parallel nonrandomized study design (six dogs per group; oral drug was followed by intravenous drug). Intravenous administration of bisoprolol resulted in a Cmax (presumed to be extrapolated) of 408 ± 75 (presumed to be ng/mL) with a 3.9 ± 0.3 hour half-life. Volume of distribution was 2.4 ± 0.6 L/Kg, and clearance 0.42 ± 0.08 L/h/kg). After oral administration of bisoprolol, the Cmax was 322 ± 261 at 1.1 ± 0.7 hr. Oral bioavailability of bisoprolol was 91.4% compared with 14.3% for carvedilol. For carvedilol, after intravenous administration, Cmax (presumed to be extrapolated) for carvedilol was 788 ± 348 (presumed to be ng/mL) and half-life 1 ± 0.2 hour. Volume of distribution was 2.9 ± 0.6, and clearance 2.1 ± 0.5 L/hr/kg. After oral administration the Cmax of carvediolol was 51 ± 42 at 1.1 ± 07 hours.
Class III Antiarrhythmic Drugs
Class III drugs prolong the cardiac action potential and refractory period by selective potassium channel blockade. As such, they have no effect on the fast Na+ conductance and prolong APD without slowing conduction velocity. They generally do not cause β-blockade; sotalol is an example exception. There are two members of this class: bretylium and amiodarone. Bretylium is used as an antifibrillatory drug in humans. It accumulates in sympathetic nerve terminals, where it blocks norepinephrine release, but only after an initial release of stored neurotransmitter. Bretylium is minimally effective in dogs, in part because it affects primarily the Purkinje fibers and ventricles, limiting its spectrum of activity. It is not used clinically in veterinary medicine but is used in human medicine for ventricular arrhythmias. It reportedly can cause defibrillation in cases of ventricular fibrillation in humans and has been investigated for similar effects in dogs.178 Because it causes the release of norepinephrine from adrenergic neurons, it may be associated with untenable, undesirable side effects.
Amiodarone is a structural analog of thyroid hormone; its mechanism may involve interaction with nuclear thyroid hormone receptors.5 It blocks activated sodium channels, calcium channels, and multiple potassium channels. Conduction velocity is slowed, the action potential duration is prolonged, and repolarization is delayed. Further, it noncompetitively blocks adrenergic receptors. It also inhibits cell-to-cell coupling, which may be important to its effects in diseased tissues. It is a powerful antiarrhythmic drug useful for both atrial and ventricular arrhythmias.179 In the normal canine heart, however, amiodarone causes negative inotropic effects.180 It also causes both α-blocking and nonselective β-blocking effects. Proarrhythmogenic effects are more likely in the presence of hypokalemia. Amiodarone is metabolized via CYP 3A4 to an active metabolite in dogs. Amiodarone, however, shows only moderate efficacy for the treatment of arrhythmias (supraventricular or ventricular) in dogs and cats,110 although it was more effective than bretylium in preventing sustained ventricular tachycardia or fibrillation in a canine model of reperfusion arrhythmia.178 Therapeutic concentrations of 0.5 to 2 μg/mL (1 to 2.5 μg/mL)181 have been recommended.5 Because it is very lipophilic, with lipid to plasma ratios may be as high as 300:1, amiodarone accumulates in cells (myocardial concentrations exceed plasma by 15 fold), and is slowly released, resulting in a lag time to onset and long duration of maximum effect. Generally, a loading dose is administered for several weeks, followed by a maintenance dose. Adverse effects tend to persist as the drug is eliminated over a period of weeks to months; one half- the peak effect is reached only after 21 days following drug discontinuation. Lipophilicity also limits oral absorption (bioavailability in humans is 30%). Adverse effects require long-term therapy, with the most serious being pulmonary fibrosis, which can be rapidly fatal. Other side effects include corneal deposits, hepatic dysfunction, neurologic dysfunctions (up to 40% in humans), including peripheral neuropathy, muscular weakness, and altered thyroid function (hyperthyroidism and hypothyroidism). Photosensitivity and blue discoloration of the skin have been reported. Amiodarone is associated with drug interactions.
Saunders and coworkers182 retrospectively studied the effect of amiodarone (median loading and following maintenance doses of 16.5 and 9 mg/kg/day) for treatment of atrial fibrillation in dogs (n=17). A variety of cardiac diseases were studied. Cardioconversion to normal sinus rhythm occurred in six dogs, and heart rate was decreased by at least 20% in 13 dogs. The drug was discontinued in five dogs because of adversities, including bradycardia and increased liver enzymes. Kraus and coworkers181 retrospectively studied amiodarone toxicity in Doberman Pinschers (n=22) with occult DCM. All dogs were simultaneously receiving either tocainamide or mexiletine; some dogs also were receiving other antiarrhythmics. All dogs had been dosed with a loading dose (9.0-12.1 mg/kg) followed by a maintenance dose (range 4.3 to 6.3 mg/kg). Serum amiodarone concentrations ranged within and between animals, in part because of dose changes, but roughly ranged from 1.5 to 3.7 μg/mL at doses that ranged from 200 to 400 mg/kg once (for higher concentrations) to (for lower concentrations) twice daily. Adverse events that emerged during loading or maintenance included anorexia and vomiting associated with increased liver enzymes in up to 45% of dogs. Dogs often responded to temporary discontinuation of the drug, but persistent hepatic involvement necessitated discontinuation of therapy for some dogs.
Hepatopathy associated with amiodarone therapy has been reported in a series of 4 cases (3 of which were Doberman Pinchers).182a Doses ranged from 8 to 10 mg/kg per day with duration as short as 6 weeks and as long as 8 months. Patients were receiving other drugs, including melixetine. Concentrations of amiodarone approximated 1.7 mcg/ml at the time of toxicity. Hepatopathy included increased serum liver enzymes in all dogs, and hyperbilirubinerma in 2 of the dogs. No risk factors were identified other than left ventricular dysfunction. Because the drug had been used by the authors in only 10 dogs, the incidence of hepatopathy was described as 40% in this small population of dogs. Hepatopathy resolved in 3 cases after amiodarone was discontinued despite continuation of other cardiac drugs (including melixetine); one dog died suddently.
Sotalol is a class III potassium blocking antiarrhythmic drug with nonselective β-blocking properties. Studies in dogs appear to be limited to experimental studies supporting its use in humans. For example, sotalol disposition was described in dogs (n=3). Oral absorption was rapid and oral bioavaiability 75 to 90%; elimination half-life was 4.5 ± 1 h.177a the volume of distribution is larger (by 4 fold) than total body water. 177b Excretion appears to be renal. Plasma concentrations necessary to achieve 50% and 100% of β-blockade after isoproterenol administration were 1-2 and 2.3 to 3.4 μg/ml, respectively.177b In an early canine model, the relative efficacy of β-blockade following isoproterenol stimulation was l-propranolol > propranolol >>sotalol >bunolol. 146b Sotalol is largely cleared in dogs by renal excretion; volume of distribution ranges from 1.1 to 1.6 L/kg. The half-life is shorter in dogs at 4 to 5 hrs compared to 7 to 18 hrs in humans. Experimentally, sotalol has been used to induce torsades de pointes in hyokalemic dogs (2.5 meg/L).
Class IV Antiarrhythmic Drugs
Class IV antiarrhythmic drugs are the CCBs. Those blockers particularly effective on the vasculature are discussed with the vasodilator drugs. Diltiazem and verapamil have been used in both dogs and cats for their effects on heart rate. Both drugs are also, however, characterized by negative inotropic effects, with verapamil being a more effective but less safe negative inotrope.
Because calcium entry into myocardial cells is regulated primarily by slow channels, the CCBs also affect the heart. Specialized tissues capable of automaticity and atrioventricular conduction tissues are particularly affected by CCBs. The differences in pharmacologic effects induced by these drugs often reflect their impact on recovery of slow calcium channel.2 CCBs that do not alter the rate of recovery (e.g., nifedipine and its congener amlodipine) will have little effect on conducting tissues. Drugs that do delay recovery of the channels can also delay conduction. For example, verapamil and diltiazem decrease the rate of recovery of calcium channels and thus not only decrease the magnitude of the cardiac action potential but also slow conduction through the atrioventricular node. The faster that atrioventricular nodal stimulation occurs, the more effective the atrioventricular nodal blockade is. Both drugs are useful for supraventricular arrhythmias. However, verapamil also has been shown to be useful in the treatment of experimentally induced ventricular tachycardias in dogs.183 Because of their effect on slow calcium channels, CCBs also decrease myocardial contractility.2 Verapamil also appears to provide cardioprotective effects in dogs with acute Chagas disease, which is characterized by destruction of sympathetic nerve terminals and alterations of β-receptor density.184 A proposed mechanism is increased adrenergic adenylyl cyclase activity.
KEY POINT 14-38
Calcium channel blockers active in the heart target cells normally capable of automaticity and thus are more useful for supraventricular arrhythmias.
Hypotension, bradycardia, and tachycardia (generally reflex) are the predominant clinical indicators of CCB overdose. In patients with poor myocardial reserve, exacerbation of CHF may result in peripheral or pulmonary edema
Diltiazem
Diltiazem (see Figure 14-9) is the most commonly used CCB in veterinary medicine, in part because it has been studied in both dogs and cats. It exerts its greatest effects in the sinoatrial and atrioventricular nodes, tissues in which slow Ca2+ influx is largely responsible for phase 0 depolarization. Diltiazem slows sinus rate and atrioventricular conduction. The ventricular rate is reduced in patients with atrial fibrillation or flutter, but primary ventricular arrhythmias are generally unresponsive to diltiazem. Myocardial oxygen demand decreases in response to the effects of diltiazem. Cardiac side effects include hypotension, bradycardia, and various degrees of heart block. In human patients a therapeutic range of 50 to 300 ng/mL has been identified and can be used as a target for clinical response in animals.
The magnitude of the hemodynamic affects of the CCB reflects the route of administration. Bioavailability is reduced as a result of first-pass metabolism for nifedipine > verapamil > diltiazem, with bioavailability of diltiazem only 50% after oral administration. However, the extent of metabolism decreases with chronic administration, facilitating chronic oral therapy. Diltiazem is metabolized by acetylation, which is deficient in the dog but not in the cat; the impact of acetylation deficiencies in the dog on diltiazem disposition has not been studied.
In an attempt to identify a product that allows convenient (once to twice daily) dosing intervals, the disposition of several diltiazem products has been studied in cats. Comparison of intravenous, standard, and slow-release (CD) diltiazem preparations reveals a disappearance half-life of 120 minutes for the intravenous and oral preparations, but 460 minutes (7 hours) for the CD preparation. The bioavailabilities of the oral preparations are 71% (standard) and 36% (CD). The higher bioavailability of the standard preparation for cats compared with humans (30%) may reflect less first-pass metabolism, despite the fact that cats are efficient acetylators. Peak plasma concentrations for the standard preparation occurred at 45 minutes; peak steady-state concentrations of CD should occur at 1 to 2 days. Diltiazem is approximately 55% to 65% bound to serum proteins in cats. Maximum prolongation of the PR interval is less than 20% for either preparation, occurring at approximately 18 hours, when plasma diltiazem concentrations are between 50 and 100 ng/mL. The pharmacodynamic effects have not been studied in cats with HCM. However, on the basis of the pharmacokinetic data, the standard diltiazem product is administered at 1 mg/kg every 8 hours, but the CD product (prepared in gelatin as either 60-mg or 90-mg capsules) can be administered at 10 mg/kg every 24 hours. An extended-release diltiazem (Dilacor XR) also was studied in cats (n=13; 10 normal and 3 with HCM) after oral administration of either 30 or 60 mg (9.3 to 14.8 mg/kg, obtained as 60-mg tablets in 120-mg capsules; tablets were halved [with difficulty] to yield 30 mg fractions). Peak serum concentrations following 60 mg (8 cats; concentrations not measured until 6 hours) were markedly variable, ranging from approximately 71 to 1500 ng/mL (mean 787 ± 488 ng/mL); by 24 hours, concentrations were 196 ± 232 (range 5 to 920) ng/mL. At 30 mg (5 cats), peak concentrations were 448 ± 370 (20 to 800) at 6 hours and 43 ± 24 (20 to 88 ng/mL) at 24 hours. Variability in absorption indicates that monitoring, if available, would be a useful tool for targeting effective concentrations.185 The recommended dose is one half of one of the pellets given once or twice daily.
Diltiazem appears to be relatively safe in cats. A retrospective study of client-owned cats (n=25) with HCM treated with 60 mg diltiazem once daily, reported side effects evident at 60 mg per cat included lethargy, gastrointestinal disturbances (vomiting, diarrhea), and weight loss (36%). Clinical signs appeared in 1 to 7 days.185 Diltiazem has been studied in cats after single-dose transdermal administration. Therapeutic concentrations (50 to 200 ng/mL) were not acheived in any cat (n = 6).185a Administration as a transdermal gel is not recommended until multiple dosing has been documented to predictably achieve therapeutic concentrations.
Diltiazem has been studied in dogs.186,187 The PR interval is prolonged approximately 20% at plasma concentrations of 60 ng/mL. In dogs suffering from atrial fibrillation or CHF, diltiazem (0.5 to 1 mg/kg every 8 hours) can be used for its negative chronotropic effects to reduce heart rate in the presence of sustained supraventricular arrhythmias. Diltiazem can be used either alone or in combination with digoxin. Because of its negative inotropic effects, however, it must be used cautiously in patients with myocardial failure, and digitalization may be indicated before diltiazem therapy is begun. The drug can be used intravenously (0.2 to 0.4 mg/kg, followed by infusion of 4 to 8 μg/kg per minute), although extreme caution is recommended.
A single case of diltiazem toxicity has been reported in a dog that accidentally ingested between 95 and 109 mg/kg of a slow-release product.188 Clinical signs included cardiac arrhythmias, bradycardia, hypotension, mental depression, and gastrointestinal upset. The patient was treated with a temporary pacemaker. Costello and Syring189 reviewed the toxicity associated with CCBs in general. Therapeutic strategies discussed for treatment included calcium as the initial therapy to increase availability to cells. Response should be monitored by electrocardiogram and heart rate. Glucagon may improve or reverse bradycardia and hypotension through unclear mechanisms that increase cAMP. Experimental doses used in dogs were 0.2 to 0.25 mg/kg bolus followed by 150 μg/kg/min constant-rate infusion. Because calcium blockade may cause hypoinsulinemia (and hyperglycemia) at the level of insulin release from pancreatic β cells, insulin and dextrose may be indicated (4 U/min in 20% dextrose and potassium supplementation as needed), as is a pacemaker. Finally, drugs that act as direct agonists at calcium channels may be beneficial, although no drug is clinically available or described with this effect. Among the drugs studied is 4-aminopyridine.
Verapamil
Verapamil is similar to diltiazem in its actions. It undergoes first-pass metabolism after oral administration in humans. Although it is available in both intravenous and oral preparations, caution is recommended with intravenous use. It has been studied for the treatment of acute supraventricular tachycardia in the dog at a dose of 0.05 to 0.15 mg/kg up to 0.2 to 5 mg/kg.110,190 Supraventricular arrhythmias that do not respond to class IA drugs often respond to oral verapamil therapy.110 Among the CCBs used in small animals, verapamil is associated with the greatest negative inotropic effects and as such should be used cautiously in animals with ventricular myocardial dysfunction. Further, verapamil may be associated with more drug interactions than diltiazem.
Miscellaneous Antiarrhythmic Drugs
Digoxin, a cardiac glycoside traditionally recognized as a positive inotrope used to improve cardiac muscle contractility in the failing heart, is also a negative chronotrope as a result of both its direct (inhibition of Na+,K+-ATPase pump) and indirect (cholinergic-like) effects. In fact, its most common use in the treatment of canine CHF is probably as a negative chronotrope rather than a positive inotrope. Vagomimetic effects inhibit atrioventricular nodal calcium currents and activate acetylcholine-mediated atrial potassium channels. Cardiac glycosides increase the slope of phase 4, particularly in the presence of low extracellular potassium. Indirectly, glycosides hyperpolarize the RMP and shorten atrial action potential durations but increase atrioventricular nodal refractory periods, thus enhancing activity against atrial reentrant arrhythmias.6 Although digoxin is less effective in slowing the heart rate in the presence of high catecholamine output compared with nonadrenergic stress conditions, only moderate decreases in heart rate are necessary to improve cardiac function. Use of digoxin as a negative chronotrope is discussed in greater depth along with its positive inotropic effects.
Dofetilide is a pure delayed rectifier K+ channel blocker. As such, it has no extracardiac adverse events. Its efficacy is in the maintenance of normal sinus rhythm in the patient with atrial fibrillation. It is renally excreted, with dose modification necessary with renal disease. The drug is available only through limited distribution. Ibutilide also is a pure delayed rectifier K+ channel blocker whose use is limited to intravenous administration because of extensive first-pass metabolism. It is metabolized by the liver, with a duration in humans ranging from 2 to 12 hours. Among the more common side effects is torsades de pointes.
Moricizine is a phenothiazine analog that blocks Na+ channels. It has been used to chronically treat ventricular arrhythmias but has been associated with increased mortality when used acutely to treat myocardial infection. Characterized by first-pass metabolism, its long duration of action in humans (many hours) probably reflects active metabolites.
Atropine is considered an antiarrhythmic by virtue of its blockade of vagally mediated cardiac slowing in some bradyarrhythmias. It is useful, however, only for short-term management. Longer-acting orally administered anticholinergics (e.g., propantheline) are indicated only rarely for bradyarrhythmias, with pacemaker placement being the preferred treatment.
Adenosine is an endogenous nucleoside. It must be administered as a rapid intravenous bolus. Adenosine can acutely block reentrant supraventricular arrhythmias and (rare) ventricular tachycardia associated with DAD events. Transient asystole is a not-uncommon adverse event that lasts less than 5 seconds because of rapid intracranial domination. Atrial fibrillation and bronchospasm are rare events. The effects of adenosine are ameliorated by methylxanthine derivatives.
Magnesium is an antiarrhythmic indicated in patients with torsades de pointes, probably by inhibition of ion flow responsible for EAD. When given intravenously, it has also been used for treating arrhythmias associated with digoxin toxicity. Chronic oral magnesium therapy has not been demonstrated to decrease arrhythmias.