Chapter 15 Drugs Acting on Blood or Blood-Forming Organs
Drugs Stimulating Medullary Poeisis
Bone Marrow Regulation
The bone marrow contains nonhematopoietic (osteoblasts, of pluripotential stromal origin) and hematopoietic (osteoclasts, of macrophage/monocyte origin) stem cells as well as nonosteogenic cells such as platelets and lymphocytes. Interaction is critical to hematopoiesis, with both nonhematopoietic and nonosteogenic cells contributing to regulation of hematopoietic cells and medullary poiesis. Multiple factors control the commitment, proliferation, and differentiation of bone cell precursors. These include but are not limited to cytokines, growth factors, systemic hormones, and transcriptional regulators. Cell–cell and cell–matrix interactions maintain contact between osteoblastic cells and osteoclast marrow precursors and between the stroma and hematopoietic cells. Among the interacting signals are adhesion molecules, including but not limited to integrins, selectins, and cadherins. Osteoclastogenesis is stimulated by interleukin (IL)-1 and tumor necrosis factor (TNF). The latter is released from marrow mononuclear cell lines in response to NFκB ligand (RANKL), IL-6, IL-11, macrophage colony-stimulating factor (M-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) released from the stroma. Megakaryocytes appear to play a major role in the regulation of hematopoiesis through expression of a number of mediators such as RANKL, n-methyl d aspartate-type glutamate receptors, calcium-sensing receptors, osteonectin, and osteocalcin. Another example is thrombopoietin, which simultaneously regulates megakaryopoeisis and inhibits osteoclastogenesis.1
Red Blood Cell Formation
Hematopoiesis
Hematopoiesis occurs through differentiation of stem cells that are formed early in embryonic life. Differentiation occurs in a series of steps in which burst-forming units (BFUs) and colony-forming units (CFUs) are formed for each of the major cell lines. These undifferentiated cells continue to proliferate and differentiate under the influence of a number of cellular and humoral factors that are produced by bone marrow and peripheral tissues.
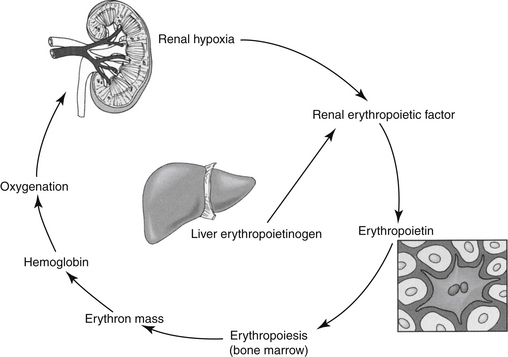
Erythropoietin (EPO) is the most important regulator of the proliferation of committed erythroid cells (see the discussion of erythropoiesis-stimulating agents [ESAs]) (Figure 15-1).2 The kidney is the major site of EPO production, where it is released in response to anemia or hypoxia. Among the more important regulators of the myeloid series are granulocyte colony-stimulating factor (G-CSF) and granulocyte/macrophage colony-stimulating factor (GM-CSF). Several vitamins are needed for red and white blood cell formation (hematopoiesis and granulopoiesis, respectively).2,3
Drugs Affecting Red Blood Cell Formation
Pharmacologic therapy (Table 15-1) for anemia is oriented toward (1) providing components needed for red blood cell (RBC) production (e.g., proteins, vitamin B12, and folic acid), including hemoglobin synthesis (iron and other minerals); and (2) stimulating bone marrow formation of RBCs.
Components Needed for Red Blood Cell Production
Vitamin B12
Vitamin B12 (cyanocobalamin), the “maturation factor,” is essential for DNA synthesis, and its deficiency inhibits nuclear maturation and division. Because cells that rapidly multiply are affected first, reduced RBC proliferation and a “maturation arrest” in the bone marrow are among the first indications of a vitamin B12 deficiency. The erythroblast cannot continue to divide, becomes very large, and is referred to as a megaloblast (megaloblastic anemia). The mature erythrocyte is also large and is referred to as a macrocyte. Although its oxygen-carrying capacity is adequate, the enlarged cell is very fragile because of its large size, and it has a reduced life span.2,3
Vitamin B12 is a porphyrin-like compound with a ring containing a centrally located cobalt.2 It can be acquired from both the diet and microbes in the gastrointestinal tract. Most microbial production is in the large intestine, however, where B12 is not readily absorbed because it is dependent upon receptor mediated uptake. Dietary deficiency of B12 is unlikely and usually results from poor absorption (including pancreatic enzyme deficiency) from the gastrointestinal tract. Absorption of B12 is complicated (Figure 15-2) and depends on several factors.2 Gastric acid and pancreatic enzymes are needed to release B12 from dietary and salivary binding proteins.2 To avoid being digested, B12 is protected by binding to intrinsic factor (excreted by the exocrine pancreas in cats4) and R protein, which are secreted by the parietal cells. The bound B12 complex is carried to the ileum, where B12 is adsorbed to highly specific receptor sites on the brush border. Vitamin B12 enters the cell by pinocytosis and then enters the blood, where it is bound with transcobalamin, the plasma carrier. Excessive vitamin B12 is stored in large quantities in the liver and is slowly released as needed. Vitamin B12 is excreted into the bile but undergoes enterohepatic cycling. Interference with absorption by the ileum will result in continuous depletion of B12. Many months of defective vitamin B12 absorption are necessary, however, before vitamin B12 deficiency occurs. The anemia resulting from B12 deficiency is also referred to as pernicious anemia. Exclusive uptake of cobalamine in the ileum as led to its detection in serum as a tool for diagnosing disease of the ileum.4 Deficiency has been described (in cats) by increased serum methylmalonic acid (MMA) coupled with serum cobalamine at or below 100 ng/mL (the lowest detectable concentration). However, the description of clinical and biochemical signs associated with deficiency focused on the gastrointestinal tract and did not address tests indicative of anemia.
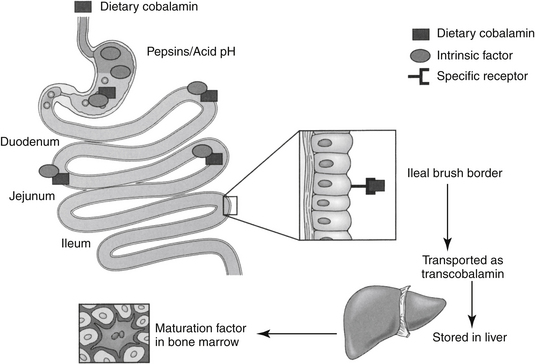
Figure 15-2 The absorption of vitamin B12 from the gastrointestinal tract is complicated and depends on the release of intrinsic factor and hydrochloric acid from the stomach. The drug complexes with a receptor. This complex is protected as it passes to the ileum, where it is absorbed by receptor-mediated pinocytosis.
Vitamin B12 is available as a parenteral preparation in the pure form of cyanocobalamin or the more highly protein bound hydroxocobalamin. Hydroxocobalamin may provide more sustained effects than cyanocobalamin when given by injection.2 Vitamin B12 is also available for oral administration in the pure form or in combination with other vitamins and minerals. Methylcobalamin is another congener of vitamin B12 and represents one of the two intracellular active forms of the vitamin (the other being deoxyadenosylcobalamin).2 Foods high in vitamin B12 include selected microbial sources and animal (meat) products. There are no significant toxicities associated with therapy. Indications for B12 therapy are limited to situations of B12 malabsorption such as ileectomy, gastrectomy, malabsorption syndromes, or chronic administration of cimetidine or other antisecretory drugs because an acid environment is necessary for release of B12 from the diet and for intrinsic factor activity. In one study4 response to cobalamin treatment (250 μg subcutaneously once weekly in cats (n = 19) with gastrointestinal disease was prospectively studied, with a focus on indicators of gastrointestinal health. Indicators of cobalamin deficiency (serum MMA, cysteine) improved and were correlated with weight gain and folic acid use, however, improvements in indices of red blood cell health were not addressed.
Folic Acid
Folic acid (pteroylglutamic acid) is a cofactor needed for DNA synthesis because it promotes the formation of a nucleotide necessary for DNA formation. Folic acid is also necessary for RNA synthesis, and it serves as a methyl donor for the formation of vitamin B12.2 Folic acid is acquired from the diet, although it can also be formed by microbes. Dietary sources include yeast, liver, kidney, and green vegetables. Folic acid is also stored in the liver but not as avidly as is vitamin B12. It undergoes enterohepatic circulation but is destroyed daily by catabolic processes. Daily requirements are high, and serum levels will fall rapidly several days after dietary deficiency. Gastrointestinal absorption of folic acid is not as complex as that of vitamin B12, although it requires protein digestion and the presence of dihydrofolate reductase in the small intestine. Jejunal pathology can result in folate deficiency. The degree of folic acid binding to plasma proteins is not well understood.
Folic acid is available as both a parenteral and an oral (pure or combined product) form. The minimum daily requirement in humans is 50 μg/day, but this can increase to 100 to 200 or more in patients with high cell turnover rates (e.g., hemolytic anemia).2 In humans the most popular form of folic acid supplementation is as part of a multivitamin preparation containing 400 to 500 μg of pteroylglutamic acid. In high disease states, 1 to 2 1-mg tablets are consumed. This dose may be the source of the recommended dose for cats and dogs. Compared with commercially available products, the dose recommended for small animals seems excessive, and it is not clear that such a high dose is necessary. There are, however, no apparent significant toxicities associated with therapy.
Indications for therapy are inadequate intake as a result of the administration of several drugs (methotrexate, potentiated sulfonamide antibiotics, some anticonvulsants such as phenytoin), liver diseases, malabsorption, or other chronic debilitating diseases. Folinic acid (leucovorin) is a formyl derivative of tetrahydrofolic acid and as such does not require the action of dihydrofolate reductase in order to act as a folate by contributing a carbon moiety. In humans, administration of folinic acid increases serum folate activity owing to 5-methyltetrahydrofolate. Folinic acid apparently serves as a substrate for inhibitors of dihydrofolate reductase such as methotrexate, or diaminopyrimidines such as trimethoprim or ormetoprim, and as such, can resolve some deficiencies associated with folic acid deficiency. However, it will not replace deficient folic acid or any of its derivatives prior to the tetrahydrofolate step in folic acid use.2 As such, leucovorin can be used clinically to circumvent the actions of dihydrofolate reductase (e.g., methotrexate) and replace materials in the folic acid pathway beyond tetrahydrofolate, but can not prevent folic acid deficiency. Leucovoran also inhibits thymidylate synthtase and as such, facilitates anticancer efficacy of 5-fluorouracil.
Hemoglobin Synthesis: Iron
Hemoglobin consists of a heme portion and a globin portion. The heme portion is formed from a pyrrole ring, four of which combine to form a protoporphyrin compound. This in turn combines with iron to form heme. Four hemes combine with the globulin globin to form hemoglobin. Several factors are necessary for hemoglobin formation.
Iron is a component of hemoglobin, myoglobin, and other substances, such as those found in cytochrome and electron transport systems.2,3 Approximately 65% of total body iron is present in hemoglobin, 4% as myoglobin, and 1% in cytochromes and electron transport systems. The remaining 15% to 30% is stored as either ferritin, the soluble form of iron stores, or hemosiderin, the insoluble stores. Oral absorption of iron is slow, complicated, and not well understood (Figure 15-3). It is available in the diet in either a heme form, which makes up a small percentage of the total but readily absorbed form, or in a nonheme (ferric oxide) form. The nonheme form represents the largest dietary fraction, but its absorption is profoundly affected by dietary factors. Nonheme iron must be converted to the ferrous form for absorption to occur; conversion depends on an acidic environment. Absorption of iron is increased by hydrochloric acid and decreased in situations that decrease acid production in the stomach (e.g., chronic use of antisecretory drugs).
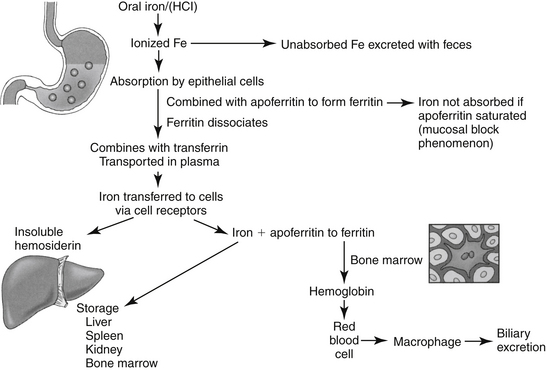
Figure 15-3 Absorption of usable iron from the gastrointestinal tract is maximized in an acidic environment. Iron combines with apoferritin in cells to form ferritin, the soluble form of iron. Iron is bound to transferrin in plasma. Saturation of apoferritin leads to saturation of transferrin. Excessive iron is stored as hemosiderin, a nonsoluble form of iron. In the presence of saturation, iron is eliminated in the gastrointestinal tract.
Iron is absorbed primarily from the proximal jejunum, where it immediately combines in the enterocyte with apoferritin to form ferritin. When transferred out of the enterocyte, ferritin dissociates, and iron combines with the globulin transferrin. Iron is transported in the plasma in this form, but the binding is loose so iron can be easily transferred to tissues. Iron is transferred to cells by way of specific receptors that interact with transferrin, especially in the liver. In cells, iron again combines with apoferritin to become ferritin. Small quantities are also stored as the very insoluble hemosiderin; the quantity of this storage form increases when the total quantity of iron in the body is much more than apoferritin can accommodate. When apoferritin is saturated, transferrin cannot release iron, and it thus becomes close to 100% bound (estimated iron stores).
There is no mechanism for the excretion of iron other than by the gastrointestinal tract. Gastrointestinal tract elimination occurs by (1) exfoliation of enterocytes containing iron, (2) biliary elimination, and (3) elimination in diet of iron not absorbed. Total body iron is regulated by altering the rate of absorption. If all the apoferritin in the enterocyte is combined with iron, the amount of iron in the enterocyte is high, and absorption from the diet is slowed. Absorption is faster if iron stores are depleted. This mechanism has been referred to as the mucosal block phenomenon.
Preparations
Iron is available in both oral and parenteral preparations. Oral preparations are prepared as ferrous (bivalent) or ferric (trivalent) salts. Ferrous salts tend to be the treatment of choice for oral supplementation and are dosed according to their iron content.2 Examples of bivalent ferrous salts include sulfate (20% iron), gluconate (12%), and fumarate (33%). The ferrous bivalent salts are more soluble in the gastric environment and are absorbed three times faster than the trivalent salts. The efficacy of polysaccharide oral iron products approximates that of ferrous products. Slow-release iron products have not been well studied. These products may be continued for several months; toxicities and side effects are dose related.
KEY POINT 15-2
Response of anemia to iron supplementation should be anticipated only if the cause is iron deficiency. Excessive iron therapy should be avoided because excessive iron cannot be easily excreted.
Parenteral preparations are indicated if oral preparations cannot be tolerated or are not feasible. Iron dextran administered by intramuscular injection generally is preferred. Parenteral administration results in a more rapid accumulation of iron stores, which may take months (in humans) with oral therapy. Other indications for parenteral therapy include diseases of the gastrointestinal tract that preclude iron absorption or that will exacerbate another disease (e.g., inflammatory bowel disease) or intolerance of oral supplementation.2 Much of the iron given intramuscularly remains at the site of injection for several months. The remaining iron enters the plasma but must first be phagocytized by reticuloendothelial cells for processing. This may take several months, and evaluation of total body iron may be difficult until all of the iron is processed.2
In human patients iron injection is preceded by a test dose (approximately 0.5 mL). It is given only in a large muscle mass and is associated with long-term discomfort, local skin discoloration, and a perceived risk of malignant change at the site of injection. Selected iron dextran preparations also can be administered intravenously. Dosing is based on a conversion of body weight (0.66 × weight in kilograms) and on the patient’s hemoglobin level compared with the desired hemoglobin level (14.8 g/dL).2 After an initial 0.5-mL test dose, the calculated dose is given in 2-mL increments each day until completed. Side effects associated with intravenous administration include malaise, fever, arthralgias, urticaria, and generalized lymphadenopathy.2
Drug interactions
Several drugs, including tetracyclines and antacids, and several foods bind to and precipitate iron when given orally. Absorption is enhanced in the presence of ascorbic acid because it reduces ferric iron to its ferrous state and prevents formation of insoluble and unabsorbable iron compounds.5
Clinical use
Indications for iron therapy are limited to treatment or prevention of iron deficiency such as occurs with blood loss or successful therapy with anabolic steroids or recombinant EPO in a patient suffering from chronic anemia. The efficacy of “shotgun” products is questionable. As with any hematinic preparation, provision of these compounds will be ineffective if the nutritional status of the animal is poor. Shotgun preparations are products that contain a combination of hematinic agents such as vitamin B12, folic acid, pyridoxine, riboflavin, nicotinic acid, pantothenic acid, thiamine, biotin, ascorbic acid, and vitamin E. The need for inclusion of all of these products is not clear. An exception might be made for ascorbic acid, which enhances oral absorption of iron, and pyridoxine, which is useful in human patients with selected anemias. One human patient with pure red cell aplasia has responded to riboflavin. Occasionally, these products might be considered dangerous because some additives may be sufficiently high to mask clinical signs of other nutrient deficiencies that ultimately may become life threatening.2
Bone Marrow Stimulation
Erythropoiesis-Stimulating Agents
EPO, an endogenous glycosylated protein hormone (MW 30,000 daltons), is the most important regulator of committed erythroid progenitor proliferation.2 Extensive glycosylation is responsible for the activity of the molecule.6 The kidney (peritubular cell) is the primary source of endogenous EPO, although the liver produces a small amount in some species. Insufficient oxygen delivery to tissues is the primary stimulus for promotion of the transcription and thus production and secretion of EPO2 (see Figure 15-1). Hypoxia or anemia will increase synthesis by a hundredfold or more. However, renal disease, damage to the bone marrow, or deficiencies of iron or other essential vitamins can impair synthesis. Inflammatory cytokines also impair secretion (as well as iron delivery). Prostaglandins appear to increase and nonsteroidal anti-inflammatory drugs appear to decrease EPO production.
KEY POINT 15-3
Formation of antibodies to foreign erythropoietin may lead to therapeutic failure as long as 12 months after the beginning of therapy. As such, treatment should be reserved for states of erythropoietin deficiency or for those limited indications for which efficacy has been demonstrated through well-designed clinical trials. Iron deficiency might be anticipated in some animals.
Mechanism of Action
Once released from the renal cell, EPO travels to the bone marrow, where it binds to receptors on the surface of committed erythroid progenitors. The influence of EPO (including ESAs) on RBC production occurs at several steps. EPO binding initiates changes in intracellular phosphorylation.2 Additionally, EPO stimulates proliferation and differentiation of erythroid precursors, including BFU–erythroid CFUs: erythroid, erythroblasts, and reticulocytes.2 Recombinant human EPO also stimulates the release of reticulocytes from the bone marrow into the blood, where they subsequently mature.7-11
Preparations
Human EPO has been isolated and cloned and can be synthesized in large quantities with recombinant techniques using a mammalian (hamster cell line). ESAs include two recombinant EPO (rEPO) products. Epoetin is a recombinant human product (rhEPO), available in two products that differ in their glycosylated carbohydrate patterns: alpha (Epogen, Eprex) and beta (Procrit). A second ESA is darbepoetin, a recombinant hyperglycosylated analog of the human protein, often referred to as a second-generation EPO. Hyperglycosylation increases stability. Darbepoetin is dosed in μg/kg, whereas epoetin is dosed as units/kg for epoetin: a dose of 6.25 to 12.5 μg/kg of darbepoeitin is equivalent in humans to a dose of 2500 to 5000 units/kg of rhEPO (0.0025 μg darbepoetin to 1 unit epoetin).12 Albumin is included as a vehicle in both epoetin alpha and darbepoetin. However, because of the concern regarding potential transmission of disease carried by albumin, alternative vehicles such as polysorbate 80 have been used to formulate epoetin beta.13 Products are intended for intravenous or subcutaneous administration. Both canine (rcEPO)14 and feline (rfEPO)15 recombinant products have been developed but are not yet commercially available. Canine EPO is more closely homologous to feline EPO compared with rhEPO and will be preferred in cats should rcEPO approval precede the feline product.
Disposition
The disposition of ESAs has not been well characterized in either humans or animals. Glycosylation (addition of carbohydrates) prolongs elimination compared with the endogenous proteins. In humans the elimination half-life of rh-EPO is about 6 to 9 hours after intravenous administration, with a very small volume of distribution (0.055 L/kg). As with most endocrine products, plasma half-life may not necessarily reflect biological half-life.9,11 Glycosylation of darbepoetin is greater than that of epoetin, leading to an accordingly longer (threefold) biological half-life in humans. Dosing subsequently can be reduced to every other day (3 times weekly).2
Adverse Effects
Antibody formation
Use of ESAs is associated with immunogenicity, and relevant side effects should be anticipated when administering foreign protein products intended to mimic endogenous protein products.16 Pure red cell aplasia (PRCA) is a recognized, albeit rare, sequela of rEPO in humans.17 In 1998 a formulation was approved internationally that varied from the U.S. product by vehicle, storage, and handling. The use of the product was associated with an increased incidence of PRCA, leading to the assumption that the differences increased the immunogenicity of the product.18 The increased frequency of disease facilitated its characterization.Clinically manifested as a rapid onset of severe anemia, refractoriness to rhEPO therapy, and a low reticulocyte count, PRCA is associated with high concentration of EPO-neutralizing antibodies, probably of IgG origin.6,19 Diagnosis is confirmed by the presences of EPO neutralizing antibodies in circulation, absence of bone marrow red cells and normal to elevated transferrin saturation. Testing no longer appears to be available for either eryropoietin or antibodies for dogs or cats. In one study, greater than 95% of human cases afflicted with chronic renal disease received the drug subcutaneously for a mean of nine months before the onset of PRCA. Antibodies resolved in 80% of patients when the drug was discontinued but only in the presence of immunosuppressive therapy.13,17,19 A lower incidence of PRCA was found in patients with cancer chemotherapy–related anemia, probably because anticancer therapy led to immunosuppression.13 The incidence of PRCA has been reduced by methods intended to reduce immunogenicity. These included changes in processing that contributed to immunogenicity (such as freeze drying), changes in packaging (replacement of rubber stoppers with Teflon, removal of silicone lubricant in prefilled syringes), and a shift from subcutaneous to intravenous administration.13 Darbepoetin may be associated with less immunogenicity compared with rhEPO, as is shown by response in one human patient to darbepoetin despite development of rhEPO-induced PRCA.20
Because of the foreign nature of EPO, adverse effects reported in humans also should occur with rhEPO administration in animals. Local and systemic allergic reactions include cellulitis, fever, arthralgia, and mucocutaneous ulcerations, which occurred in 12% of animals treated in a pilot study.21-23 Signs resolve with drug withdrawal but may reappear when treatment is resumed. Because antibodies directed toward the foreign rhEPO may stimulate production of antibodies targeted toward endogenous EPO, RBCs, hematocrit, and hemoglobin may progressively decrease starting as early as 2 weeks of therapy.21-23 Antibodies are indicated by a bone marrow myeloid-to-erythroid cell ratio of less than 8. Discontinuation of rhEPO will result in resolution of antibody formation and, to some degree, the anemia (i.e., that caused by antibodies), but drug therapy cannot be reinstituted.22 The incidence of antibody formation is high, developing in 63% to 100% of healthy dogs receiving EPO.24 Red cell aplasia was associated with anti-Epogen antibodies in two out of three dogs treated longer than 90 days and seven out of eight cats treated for 180 days or more. For the cats, 70% were refractory to further EPO therapy, requiring transfusion therapy until anti-epoetin antibody concentrations decreased (2 to 4 months after rhEPO was discontinued. Because both exogenous and native EPO may be impaired, aplastic anemia resulting from bone marrow failure may ultimately occur. Immunosuppressive therapy directed toward antibody formation, although apparently successful in humans, has not been addressed in veterinary medicine. Antibodies generally require 2 to 12 months after therapy is discontinued for eradication. Species specificity of rEPO should minimize the advent of anti-EPO antibodies as has been demonstrated for both the dog24,25 and cat.15 Randolph and coworkers15 have demonstrated that the feline recombinant product not only does not induce antibodies but also will re-establish erythropoiesis in most cases of rhEPO-induced red cell aplasia. Unfortunately, the same does not appear to be true for the canine recombinant preparation25 in dogs, suggesting that even species-specific origin of rEPO may be associated with antibodies.
Miscellaneous adverse effects
Other adverse effects that may require monitoring in patients receiving EPO include systemic hypertension, iron deficiency, hyperkalemia, and polycythemia.8 Flulike symptoms occur in some human patients receiving the drug intravenously. Hypertension occurs in many human patients with renal disease as the hematocrit normalizes. Patients who begin the use of rhEPO when in a state of hypertension are likely to experience a further increase in blood pressure. The mechanism of hypertension is not known but may include increased blood viscosity or peripheral vasoconstriction. Blood pressure increases within as little as 2 weeks of therapy and tends to stabilize by month 4. Increased blood pressure has been reported in 40% to 50% of dogs or cats treated with ESAs.26 It may be necesary to adjust drug therapy for hypertension (e.g., increase amlodipine dose).27 Polycythemia is another likely sequela of ESAs if response to therapy is not monitored on the basis of packed cell volume (PCV).27 Other miscellaneous adverse effects of rhEPO in dogs and cats include seizures (one in six dogs, two of eleven cats26), particularly in animals with moderate to severe azotemia and as a terminal clinical sign.27 Depleted iron stores should be anticipated in patients with iron deficiency (discussed later).22 Pain on subcutaneous injection (a route being used less frequently in humans in order to minimize PRCA) varies with products, with the alpha (e.g., Eprex) being more painful in humans than the beta (Procrit). In an animal model involving renal ablation, rhEPO actually hastened the progression of chronic renal disease. However, the clinical relevance of this finding has not been documented.
Indications and Clinical Use
The primary indication for ESAs in humans is anemia of chronic renal disease (CRD).7,8,21,23,28 Most human patients receiving renal dialysis also receive ESAs. Indications in animals are likely to be similar to those in humans, particularly for renal disease, because clinical signs of weakness, somnolence, depression, and poor appetite associated with CRD in cats (and presumably dogs) is due to anemia. Decisions must be made regarding the product (epoetin, alpha or beta, versus darbepoetin), route (intravenous versus subcutaneous), and dosing regimen (dose and interval). Subcutaneous administration of epoetin—which can cause pain on administration—has been associated with better response in humans, although it appears to be more immunogenic. In contrast, darbepoetin appears to be equally efficacious regardless of the route.29 Preference among the ESA products is not yet clear; evaluation of darbopoetin currently is ongoing.30 Advantages of darbopoetin may include faster response time, particularly with front end loading, decreased dosing intervals, and decreased risk of immunogenicity. The average dose of darbepoetin used in human patients with CRD or cancer ranged from 3.5 to 5 μg/kg 1 to 3 times per week; preliminary studies indicate that administration as infrequent as once every 3 weeks may be sufficient.
Because it has been available longer, more information is available regarding rhEPO use in both humans and animals. In humans with CRD, rhEPO normalizes the hematocrit, hemoglobin concentration, and RBCs after administration of 15 to 300 U/kg rhEPO 3 times weekly. Some human patients have required 500 U/kg. A stair-step approach has been used for nonresponding CRD in human patients in that the dosage continues to be increased until the desired response is achieved.8 When the desired response is achieved (a hematocrit of 30% to 40%), a smaller maintenance dose (25 to 100 U/kg three times a week) is given to maintain the hematocrit above 30%.8 Normalizing the PCV in patients with underlying renal disease may be associated with a greater risk of cardiovascular side effects.31 Because of convenience and cost, in humans the maintenance dose generally has been given subcutaneously, whereas the induction dose is often given intravenously. However, intravenous administration increasingly is being used for maintenance dosing to minimize the risk of immunogenicity (in human patients).18 The intravenous route in humans has the added advantage of convenience in those individuals with chronic access ports (i.e., human patients undergoing dialysis), a situation that is less likely in animals.
Erythropoetin therapy is indicated in dogs or cats with a PCV of 20% to 25% or less owing to renal disease (see Table 15-1). To date, of the commercially available products, reports (whether scientific or anecdotal) of use in animals are limited to alpha EPO. Recombinant human EPO has been used in small animals with chronic anemia.21,22,23 When administered in uremic animals with CRD, the hematocrit of most patients normalizes within 3 to 4 weeks of therapy, and the clinical well-being of patients improves. Response to therapy is indicated by reticulocytosis, and an increase in hematocrit of 0.5% to 1% each day. Hypokalemia associated with uremia in cats should also resolve, in part because of improved appetite.22 White blood cells and platelets do not seem to be affected. Caution is indicated for patients that are hypertensive before rhEPO therapy. Informed consent should be obtained before use because of the high incidence of antibody formation. Initial treatment should begin at 100 U/kg (or, if a slower increase is acceptable, 50 to 75 U/kg) subcutaneously 3 times a week until the target hematocrit (originally 37% to 45% in dogs or 30% to 40% in cats; more recently 30% to 35% for dogs and 25% to 30% in cats) has been reached.27 Generally, response requires approximately 4 weeks. The PCV should be measured twice weekly for the first several months of therapy. The dose is decreased to a maintenance dose of 50 to 100 U/kg and the frequency of administration to twice weekly once the target PCV has been achieved. The dose should be titrated to maintain the PCV in the mid-target range. If the PCV does not increase sufficiently, the dose of rhEPO can be increased in 25- to 50-U/kg increments. The maintenance dose will vary for each patient. A maximum dose has not been established in animals, although weekly doses of 300 to 1050 U/kg have been reported.22
Among the more common causes of ESA therapeutic failure in humans is iron deficiency. Iron therapy generally is begun along with rhEPO. Iron therapy is not easily monitored. In humans serum ferritin and transferrin iron saturation are the most common tests used.32 An increasing percentage of hypochromic red blood cells and decreasing content of hemoglobin in reticulocytes may also reveal iron deficiency but will not reveal iron overload. Iron gluconate and iron sucrose are considered the safest of the intravenous iron medications.32 However, because intravenous iron dextran therapy is more expensive and is associated with anaphylactic therapy, oral iron therapy is preferred.2,8 Further, the body can control iron content with oral but not intravenous administration. Differences in the gastrointestinal absorption of iron may alter response to rhEPO therapy. Addition of 200 mg of oral iron, however, appears to successfully maintain adequate iron stores in human patients with CRD receiving rhEPO. Oral iron supplementation in dogs is recommended at 100 to 300 mg of ferrous sulfate daily (20- to 60-mg elemental iron) and for cats 50 to 100 mg (10 to 20 elemental).27 Iron dextran (10 to 20 mg/kg for dogs and 50 mg total dose for cats) is recommended by intramuscular administration (deep) for animals that cannot tolerate oral supplementation. Note that oral absorption of iron may not occur until serum ferritin is below 30 to 50 ng/mL (human patients). Erythropoiesis improves, however, even in human patients whose transferrin saturation is above that diagnostic of iron deficiency (16%).33
Anemias other than that associated with CRD also may be associated with decreased levels of endogenous EPO in animals and thus might respond to supplementation. However, the potential application of these indications in veterinary medicine must be balanced by the lack of scientific support and the adversities that are more likely in animals compared with humans if rh-EPO is used (e.g., antibody production). These have been reviewed by Langston and coworkers27 and others.2,34 An enzyme-linked immunosorbent assay (ELISA) kit developed to detect canine EPO may be helpful in identifying the need for supplementation although access at the time of publication is not known.35
The most common nonrenal use of ESAs in humans is anticipation of autologous blood transfusion; use of rhEPO increased the number of autologous units collected, and pretreatment may decrease the need for allogenic transfusion therapy. Although the need is less clear, a similar approach should be of benefit in animals; experimentally, pretreatment with iron and rhEPO in dogs resulted in higher hematocrit levels after transfusion than those in pretreatment with iron and saline.27 Selected (nonmyeloid) cancer anemias associated with chemotherapy may respond to EPO supplementation, including multiple myeloma and myelodysplastic diseases (the latter defined on the basis of cytopenia, dysplastic hematopoietic precursor cells and less than 30% blasts in the bone marrow).2,27,34 Anemia associated with cancer may reflect an inadequate EPO response; in humans the ratio of measured to predicted EPO (the latter based on population studies) of less than 0.9 is interpreted as an indication of potential response. Response may be related to dose: One retrospective study of multiple myeloma in humans reported a higher overall survival in patients who received a high dose (greater than 60,000 U/week) rather than a lower routine dose.36 The use of ESAs in human cancer patients has been reviewed,38 and guidelines for such use have been published.38,39
A combination of rhEPO and immunosuppressive therapy apparently was not successful in cats with pure red cell aplasia (not associated with previous rhEPO therapy) but may have prolonged survival rates of dogs with myelofibrosis.27 An interesting and potentially relevant application of ESA is for patients in the intensive care unit. In human patients in the intensive care unit, blood loss, including that associated with repetitive phlebotomy for diagnostic testing, can be significant. The veterinary patient—particularly the smaller one—may be similarly predisposed. Relative EPO deficiencies in these patients further predispose to anemias, as is indicated by transferrin saturations of less than 20% in the majority of patients. The use of rhEPO decreases the number of transfusions needed in these patients. The use of exogenous EPO in viral immunosuppressive diseases is tempting but controversial and might best be based on demonstration of endogenous EPO deficiency. Whereas endogenous EPO appears to be low and thus exogenous therapy is of benefit in human patients with HIV-associated anemia, EPO concentrations are increased in feline patients with feline leukemia virus.27 However, the PCV increased after 2 weeks of rhEPO in nonanemic cats positive for feline immunodeficiency virus; antibodies did not appear with this short-course therapy. Other uses of rhEPO in dogs have been characterized by variable success. The use of rHEPO in racing animals is likely to be associated with the production of antibodies and therefore is discouraged not only for ethical and legal reasons but also for health reasons. Human growth factors (granulopoietin and EPO) were associated with bone marrow recovery in a dog with ehrlichiosis.40 However, therapy took several months before response in peripheral counts were realized. The use of EPO in patients with pancytopenia associated with parvovirus is controversial, particularly in light of one case of fulminant infection in a human patient with immunodeficiency who was subsequently treated with rhEPO.41 Evaluating response of drug-induced bone marrow depression to rHEPO is complicated by the impact of discontinuing the inciting drug.
Therapeutic Failure
The most likely reason for a patient not to respond to rhEPO is inappropriate therapy. If the anemia is not associated with CRD, rhEPO therapy may be ineffective if endogenous rhEPO concentrations are maximally increased. Failure in a patient with low endogenous rhEPO may occur for several reasons. The dosage may be insufficient. Failure of an anemic animal to respond to rhEPO may indicate the development of antibodies directed toward rhEPO, as previously discussed.22 Animals that develop antibodies should recover, with antibodies potentially resolving within 3 to 4 weeks in dogs. Recovery may depend in part on the severity of renal disease, with recovery less likely in those animals with severe renal disease. In humans pure red cell aplasia as a result of antibody formation may be treated with a low dose (3.3 mg/kg) of cyclosporine.6,42 Differences in vehicles, stabilizers, and handling were identified as possible contributors to autoantibodies in human patients with renal disease who were receiving recombinant EPO; changes in response to these problems has dramatically reduced the incidence of antibody formation.16 Response to ESAs is likely to require iron supplementation.22 Chronic inflammatory disease decreases response to rhEPO, as do myelophthisic diseases that replace bone marrow with fibrous tissue. Alternatively, anemia may persist because patient nutrition is not sufficient to support increased EPO.
Granulopoietin and Related Products
At least six growth factors influence granulopoiesis.2 Formation of both granulocytes and macrophages from multipotential stem cells is stimulated by stem cell factor, a glycoprotein produced by bone marrow stromal cells; IL-1 and IL-6, inflammatory cytokines that mediate many of the systemic manifestations of acute inflammation; and IL-3. GM-CSF, along with G-CSF, is produced by a variety of tissues in response to cytokines such as IL-1, TNF, and endotoxin.2,5 Supplementation with exogenous factors stimulates the proliferation of various cell types, with the type affected depending on the factor or combination of factors. IL-3 increases both platelets and granulocytes, and GM-CSF administered by itself stimulates granulocyte and macrophage proliferation. When given in combination with IL-3, however, GM-CSF stimulates thrombopoiesis, and when combined with EPO and IL-3, erythropoiesis is stimulated as well.43
As with EPO, recombinant products have been developed for therapeutic stimulation of granulopoiesis. Both recombinant canine (rcGSF) and recombinant feline (rfGSF) G-CSF have been cloned, but neither is commercially available at this time. Feline GSF has much closer homology with rcGSF than with rhGSF and thus is the preferred product. Despite their lack of availability, the factors are increasingly being studied for treatment of neutrophil disorders in dogs and cats.5,44
In normal dogs G-CSF (5 μg/kg per day) will increase neutrophil counts over fourfold within 24 hours, reaching a maximum of approximately 72,000/mL by 19 days, with counts returning to normal within 5 days after discontinuation of therapy.44 In dogs afflicted with cyclic neutropenia, rcGSF (2.5 μg/kg every 12 hours) prevents neutropenia and associated clinical signs, although cycling of neutrophils is not prevented. Chemotherapy-induced neutropenia (induced by mitoxantrone) was minimized in dogs receiving rcGSF for 20 days. Studies in normal cats receiving rcCSF reveal an approximately threefold increase in neutrophil count that persists until the drug is discontinued. No adverse effects occur.44 Cohn et al.45 demonstrated that plasma G-CSF concentrations increased just after the onset of neutropenia in puppies (n = 8) experimentally exposed (oronasal) to parvovirus. Neutrophil counts rebounded within 2 to 3 days, and G-CSF concentrations decreased before neutropenia resolved, questioning the benefits of exogenous therapy. However, recombinant human G-CSF did correct canine cyclic hematopoiesis in one study.46
Other compounds have been studied for their effect on granulopoiesis. An extract of Serratia marcescens activates interferon- α and interferon-γ, as well as IL-1, IL-6, and GM-CSF, and induces myeloproliferation either directly or through release of other cytokines.44
Vincristine
Vincristine has demonstrated efficacy in the treatment of thrombocytopenia. Its mechanism is not clear, but several have been proposed. These include decreased upregulation of glycoprotein receptors on platelets, thereby reducing interaction with von Willebrand factor (vWF) and platetet aggregation, or reduction in immunglobulin G (IgG) autoantibodies that damage endothelial cells or suppress von Willebrand factor–cleaving protease activity.47 Other mechanisms suggested in the veterinary literature have included impaired macrophage phagocytosis of platelets and effects at the bone marrow (increased megakaryocyte fragmentation and stimulation of thrombopoiesis).48 For acute episodes of thrombocytopenia purpura in humans, vincristine treatment has been considered a salvage treatment (1.4 mg/m2 followed by 1 mg on days 4 and 7).49 However, its success in these patients has led to more frequent use as first-choice therapy. A retrospective study in humans found patient outcome (survival) to be better when vincristine therapy was administered initially as opposed to 3 days into treatment.50 The authors subsequently encourage the early, initial use of vincristine as part of combination therapy, including traditional modalities. Dogs with severe immune-mediated thrombocytopenia (IMT; platelet count less than 15,000/μL) appear to be respond similarly. In a prospective study, Rozanski and coworkers48 found that platetet counts in dogs with IMT increased more rapidly in patients that received prednisone (1.5 to 2 mg/kg/day) and vincristine (0.02 mg/kg) as opposed to prednisone alone. Further, animals refractory to prednisone alone for 7 days responded rapidly when vincristine was added to therapy. However, all dogs also were treated with doxycycline (10 mg/kg/day) for 7 days. Because IMT in dogs is characterized by increased antiplatelet antibodies on platelet surface, response to vincristine might be expected in this syndrome more than other causes of thrombocytopenia. Because vincristine is a relatively safe drug, initial therapy with vincristine should be considered in dogs with moderate to severe IMT.
Treatment of Disorders of Hemostastis
Anabolic or Androgenic Steroids
Chemistry
Anabolic steroids are synthetic compounds structurally related to testosterone. They have protein-anabolic activity similar to that of testosterone but ideally minimal androgenic effects (i.e., minimal masculinization) (Table 15-2).51,52 Testosterone is the sole endogenous androgen in most mammals. Its androgenic effects and rapid elimination have led to the manufacture of synthetic compounds. Chemical manipulations that have produced clinically useful anabolic steroids include (Figure 15-4) (1) alkylation (addition of a methyl [CH3] group) at the 17β position (e.g., methyl-testosterone, stanozolol), which impairs hepatic metabolism, thus prolonging elimination half-life; (2) esterification of the 17β hydroxyl group (e.g., Deca-Durabolin) such that absorption from parenteral sites is prolonged; and (3) modification of the steroidal ring structure. The sequelae of changes in the steroidal structure vary with the modification and include prolonged absorption or elimination.
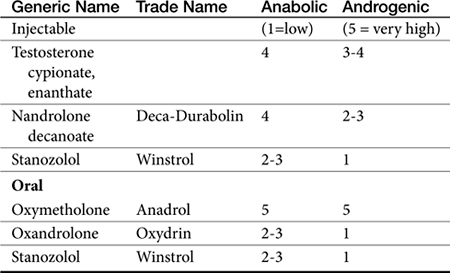
Figure 15-4 Chemical structures of several anabolic steroids. Rings and their positions are indicated for testosterone. Esters on R-groups also alter absorption characteristics. Methylation (methyltestosterone, stanozolol) increases anabolic activity as well as toxicity. Nandrolone is a nonmethylated anabolic steroid. Mibolerone has little anabolic activity, whereas the double bond at C1 and C2 contributes to marked activity with boldenone. Nonmethylated products tend to be less efficacious but less hepatotoxic.
Mechanism of Action
The mechanism of hematinic action of anabolic steroids on the cellular level is typical of the steroidal compounds.52-55 Anabolic steroids enter the RBCs, where they enhance glycolysis and the formation of steroidal 17-keto metabolites. These metabolites are delivered to tissues, including the bone marrow, where they interact with a cytosolic receptor in the appropriate cell and are transferred to the cell nucleus. In the nucleus they induce the formation of RNA and synthesis of an effector protein that brings about the pharmacologic effect.
The sequelae of steroid-induced nuclear transcription are manifold.54,55 The proposed sequelae on RBC formation include (1) stimulation of EPO production by way of EPO-stimulating factor, (2) differentiation of stem cells into EPO-stimulating factor–sensitive cells (e.g., hemocytoblasts), and (3) direct stimulation of erythroid-progenitor cells. Anabolic steroids also increase intracellular concentrations of 2,3-bisphosphoglycerate in erythrocytes; oxygen release into tissues is subsequently increased.
Efficacy of the anabolic steroids on the RBC mass depends on the presence of supportive materials.54,55 This includes adequate concentrations of androgen dehydrogenase in the RBC, adequate EPO concentrations, and sufficient bone marrow cellularity. Thus the effectiveness of anabolic steroids in treating anemia may be limited depending on the cause (i.e., renal disease accompanied by low EPO levels). Administration of high doses of these steroids may cause a negative feedback inhibition.
Pharmacologic Effects
The difference in the pharmacologic effects of these drugs among the various tissues and, specifically, whether an androgenic versus an anabolic effect will predominate cannot be attributed to differences in target tissue receptor structure because there appears to be only one androgen receptor type. Differences in response may be concentration dependent (e.g., reproductive tissues requiring higher concentrations than nonreproductive tissues), with drugs differing in their binding to the androgen receptor. However, mibolerone has little anabolic effect despite tight receptor binding and methylation; in contrast, boldenone, which is not methylated, has marked anabolic activity, attributed to a double bond at positions 1 and 2 (see Figure 15-4).56
The effect of several anabolic steroids in renal EPO was studied in the perfused canine kidney exposed to 4 hours of hypoxia.57 Activities important to secretion of EPO included a double bond at position 4 and 5 and the absence of a methyl group at position 10 (see Figure 15-4). Alternatively, differences in response may reflect conversion of the drug by target tissues to an active metabolite that subsequently causes the pharmacologic effect.54,55 In the presence of continued administration, anabolic steroids initiate and maintain a positive nitrogen balance, although the response is short-lived in intact males.52,54,55 The anabolic effects of stanozolol have been studied in dogs. Based on urine excretion of urea, retention of intravenously administered radiolabeled amino acid was increased in sled dogs (n = 10) receiving stanozolol either 2 mg/dog orally twice daily (increased from a baseline of 30% to posttreatment retention of 50%), or 25 mg weekly for 4 weeks (increased from 27% to 67%). The authors58 concluded that such an effect might be beneficial with acute or chronic conditions characterized by protein loss. Anabolic steroids antagonize glucocorticoid-induced protein catabolism by competitively inhibiting glucocorticoid binding to glucocorticoid receptors. Although anabolic steroids appear to vary in their ability to antagonize the effects of glucocorticoids, protein anabolism may be induced in patients experiencing glucocorticoid-induced protein catabolism. Anabolic steroids also reduce urinary excretion of nitrogen, sodium, potassium, chloride, and calcium.
As part of their anabolic activity, these compounds increase the circulating RBC mass (and possibly granulocytic mass). Red blood cell indices, hemoglobin, and hematocrit increase in various types of anemia. White blood cell mass may increase in cases of pancytopenia, although white blood cell response takes longer than the RBC response. Response by thrombocytes is slower and less predictable.52,54,55
Pharmacokinetics
The absorption and disposition of anabolic steroids depend on the type of preparation, the presence of specific receptors, and the species to which it is administered.52,54,55 Most anabolic steroids depend on hepatic metabolism for elimination, with those metabolites that are anaologs of the parent compound accompanied by metabolic activity.56 The risk of abuse in athletes (including the racing animal) has led to metabolic profiling of some steroids. Among the steroids, Beagle hepatic microsomal metabolism of steroids yielded hydroxylated and oxidized metabolites testosterone, methyltestosterone, mibolerone, and boldenone, with testosterone a metabolite of boldenone and androstenedione a major metabolite of testosterone (see Figure 15-4).
Preparations
Anabolic steroids have been divided into two categories depending on the presence or absence of an alkyl (CH3) group at the 17-carbon position, although the relevance of this division is probably less important than the impact of the individual drugs (see Figure 15-4).54,55 Oral and parenteral preparations are available, with the alkylated products being better absorbed orally. Oil-based parenteral preparations are intended for slow release. Examples of alkylated anabolic steroids include methyltestosterone, (oral), oxymetholone (oral), stanozolol (oral and parenteral), methandrostenolone, ethylestrenol, and norethandrolone (see Table 15-1). Examples of nonalkylated anabolic steroids include testosterone cypionate or enanthate, oxymetholone, and nandrolone in its decanoate form (parenteral).
Toxicity Versus Efficacy
A review of risks in human athletes includes cardiovascular adversities, increased risk of cancer, behavioral disorders, and increased risk of tendon damage.59 Hepatotoxicity is the most common serious adverse effect associated with androgenic or anabolic steroids.54,55 Toxicity ranges from mild increases in clinical laboratory tests to hepatocellular carcinoma (in humans). Drugs alkylated at the 17-carbon position are more likely to cause hepatotoxicity (see Figure 15-4). Although the mechanism is not known, the drugs or their metabolites may be carcinogenic or may increase the metabolism of other drugs to carcinogenic or hepatotoxic compounds. Cholestatic liver damage occurs early and can be significant but is apparently reversible if the drug is discontinued before irreversible hepatic lesions develop. It is not clear if the hepatotoxic effects that occur in humans are likely in dogs or cats.
Masculinization is a major undesirable (or desirable) side effect of anabolic steroids in humans.52,54,55,59 Virilization can occur but is seldom objectionable in dogs or cats. In dogs and cats, anabolic steroids increase libido in males, interfere with the female reproductive cycle, and cause masculinization of fetuses if administered during pregnancy. Other undesirable consequences of anabolic steroid therapy in dogs include hyperplasia of the perineal glands and stimulation of androgen-dependent tumors, such as anal carcinomas and prostatic carcinomas. In humans edema resulting from water retention occurs.52,54,55
Clinical Indications
The use of anabolic steroids for treatment of renal disease and catabolic effects associated with cachexia are addressed in their respective chapters. Human recombinant EPO has largely replaced anabolic steroids in the treatment of chronic anemias, particularly those associated with renal disease. Anabolic steroids are, however, indicated for treatment of chronic, nonregenerative anemias that fail to respond to EPO; for animals that react adversely to EPO; or for hematopoietic diseases that are not typically associated with decreased concentrations of EPO.54,55 Of human patients with aplastic anemias who have failed conventional therapy of anemia, 50% or more have responded to anabolic steroids.54,55 Anabolic steroids have been used in conjunction with anticancer chemotherapeutic drugs to protect bone marrow production of RBCs, thus allowing longer and perhaps more toxic chemotherapy.52,54,55 Anabolic steroids presumably help decrease the catabolic effects of cancer. Treatment several weeks before cancer chemotherapy may enhance response to anabolic steroids. Use of anabolic steroids for cancer has not been established in dogs and cats.
Hematopoietic response to anabolic therapy is variable, and the time to clinical improvement is long, frequently 3 or more months. Cellularity of the bone marrow appears to determine rate of response to anabolic steroids. Cessation of therapy may result in recurrence of the underlying diseases. Among the anabolic steroids, nandrolone decanoate has the greatest hematopoietic effect.54,55 Leukocyte and thrombocyte counts may also increase after therapy with anabolic steroids, although these effects are slower in onset and less likely, particularly for thrombocytes. Danazol is the anabolic steroid of choice in the treatment of thrombocytopenia, especially that which is immune mediated. Beneficial effects probably include impaired clearing of IgG-labeled platelets and decreased antibody formation. Response to therapy may take several months, and relapse may occur when danazol therapy is discontinued. Anabolic steroids and, in particular, danazol, have been used with some benefit for patients with hemolytic anemia. Benefits include not only expansion of the RBC mass but also impaired complement activation and binding to RBCs. Human patients suffering from anemia caused by renal disease respond to continuous administration of anabolic steroids. RBC indices improve, as well as appetite, muscle mass, and strength.
In general, the following approaches should be used with anabolic steroids in dogs and cats (see Table 15-1). State restrictions regarding the use of these drugs should be observed. Nandrolone decanoate is the drug of choice except in cases of thrombocytopenia, for which danazol is the drug of choice. Several months of therapy may be necessary before a clinical response is observed. The dose should be decreased once the maximum therapeutic effect occurs. Relapse of disease may occur once the drug is discontinued, and hepatotoxicity is a potential contraindication for use of these drugs.
A final consideration regarding anabolic steroids is use in racing dogs. In addition to potential performance-enhancing abuse, testosterone, methyltestosterone, and mibolerone are used to prevent estrus.56 Detection of urinary metabolites in racing animals is an ongoing field of study.56
Oxygen-Carrying Solutions
Oxygen-carrying solutions (often referred to as blood substitutes) offer the advantages of oxygen delivery to tissues similar to that of blood, without the concerns of transmission of disease. Two types of blood substitutes have been developed to both carry and deliver substantial amounts of oxygen to tissues: free hemoglobin–based oxygen-carrying solutions (HBOCs) and fluorocarbons.60,61 Fluorocarbons are chemicals that are miscible with blood and can carry as much as 5.25 mL of oxygen per 100 mL of blood, depending on oxygen tension. Although the oxygen-carrying capacity of these compounds is not adequate for total blood replacement, their low viscosity makes them potentially useful in disorders characterized by abnormalities in microcirculation. In addition, use of these agents in nonvascular tissue (e.g., the peritoneal cavity) may enhance supplementing oxygen exchange in tissues during respiratory failure.
Of the two blood substitutes pursued for clinical use, HBOCs (e.g., oxyglobin, approved for use in dogs) have proved most useful. Free, unmodified hemoglobin (purification involves washing, lysing, ultrafiltering, and pasteurizing) is not an effective blood substitute. Free hemoglobin decreases the amount of oxygen released from tissues by reducing concentrations of 2,3-diphosphoglycerate such that the oxyhemoglobin dissociation curve shifts to the left. As a result, the Pa50 of oxygen in HBOCs is reduced from 28 mm HG to 10 mm Hg. In addition, the free smaller dimers of hemoglobin are more readily cleared by the kidneys, resulting in a short half-life (20 minutes), as well as an increased risk of nephrotoxicity. Smaller molecules also may be responsible for the increase in vascular (systemic and pulmonary) resistance that may accompany use of HBOCs, including Oxyglobin use. Enlarging the size of the hemoglobin molecules by cross-linking (e.g., with fumarate or dasprin [HemAssist]), polymerization techniques (e.g., using oxidized raffinose [Hemolink] or gluteraldehyde [Oxyglobin and Hemopure]) or surface conjugation reduces the negative sequelae of free smaller molecules.62 Surface conjugation techniques have the added advantage of blocking the hemoglobin receptor site that interacts with and blocks nitric oxide, thus reducing the risk of vasoconstriction. Of the HBOC products, Oxyglobin, the first approved for use by the Food and Drug Administration, is a bovine hemoglobin–based product indicated for the treatment of canine anemia. Human hemoglobin–based products also have been developed (PolyHeme, a surface conjugated product); recombinant products are currently being studied. Intended indications for newer products extend beyond supplementation of oxygen in critically anemic patients, including sensitizing of cancer cells or scavenging of nitric oxide.62
Manipulation of the hemoglobin tetramer by cross-linking, polymerization, and other methods yields products with variable molecular weights: for Oxyglobin the average MW is 200 kDa (range 64-500 kDa) whereas conjugation of surface proteins (e.g., polyethylene glycol [Enzon] yields lower molecular weight products (117-125); recombinant products may be as small as 64 kDa. Among the advantages of the larger molecular weight (discussed later) is the ability of the compounds to act as colloids, increasing preload, stroke volume, and cardiac output (similar to hetastarch, plasma, and dextran-70), with the added advantage of increased oxygen delivery.62 In general, using experimental canine models of acute hemorrhagic hypovolemia, use of a bovine HBOC either increases oxygen delivery or arterial oxygen content, although studies generally have not been able to consistently demonstrate both occurring.62 One study concluded that treatment of acute anemia in dogs with 15 mL/kg of a bovine HBOC was able to increase oxygen content equivalent to packed RBCs.63 In canine models of hemorrhagic hypovolemia, intestinal perfusion and oxygenation appear to be maintained; however, increased systemic vascular resistance and decreased cardiac output have also been demonstrated.
Oxyglobin (Biopure Corp., www.oxyglobin.com; availability at the time of publication is not clear) is a polymerized bovine HBOC fluid. Oxyglobin increases plasma and total hemoglobin concentrations and thus increases arterial oxygen content. Its molecular weight imparts colloidal properties similar to those of dextran 70 and hetastarch. Its polymerized nature precludes renal filtration and renal side effects of hemoglobinuria. Because the solution is free of RBC walls, antigens associated with transfusion reactions are largely absent. Likewise, the absence of plasma reduces reactions associated with isoantibodies. However, antibodies may develop to bovine hemoglobin, and caution is recommended with repeated administration 10 or more days apart. As a foreign protein, anaphylactic reactions are possible.
Oxyglobin is eliminated similarly to hemoglobin by reticuloendothelial cells. Its elimination half-life in dogs is estimated to range between 30 and 40 hours, with 90% of the drug eliminated within 5 to 7 days. Conditions for HBOCs studied in controlled canine clinical trials included immune-mediated hemolysis (n = 30), blood loss (gastrointestinal, traumatic, surgical, rodenticide intoxication; n = 25), and ineffective erythropoiesis (idiopathic, RBC aplasia, ehrlichiosis; n = 9). Relative to pretreatment, plasma hemoglobin concentration significantly increased (P = 0.001) and clinical signs associated with anemia (lethargy/depression, exercise intolerance, and increased heart rate) significantly improved (P = 0.001) after treatment with Oxyglobin. Treatment success was defined as the lack of need for additional oxygen-carrying support (i.e., blood transfusion) for 24 hours after the completion of infusion with the HBOC. Success in the treatment group was 95% compared with 32% in untreated control dogs.64
Side effects of HBOCs include circulatory overload and its negative sequelae (e.g., pulmonary edema, pleural effusion, increased central venous pressure, dyspnea, coughing), particularly in patients already suffering from volume overload (e.g., congestive heart failure) or in cases of accidental overdose (>10 mL/kg/hr). Hypertension is a recognized side effect of HBOC use. Smaller molecules are potentially more likely to reduce nitric oxide and subsequent increased vascular resistance.62 Species are likely to differ in their vasoactive response. Oxyglobin caused a 30% increase in peripheral vascular resistance in isovolemic dogs; hypertension also has been induced in hypovolemic cats.62
Vasoconstriction may be sufficiently profound that tissue perfusion is compromised. Further, standard methods of assessing adequate volume resuscitation may be ineffective in the presence of HBOCs.65 Oxyglobin mildly decreases PCV immediately after infusion and increases total and plasma hemoglobin concentrations for at least 24 hours. PCV and RBC counts are not accurate measures of anemia for 24 hours after administration. Overhydration should be avoided with the use of an HBOC because of its plasma-expanding properties; likewise, administration of other colloidal solutions is discouraged. Central venous pressure or clinical signs indicative of circulatory overload should be monitored during and immediately after administration of Oxyglobin, particularly in normovolemic patients or patients with cardiac disease. Cats also may be predisposed to volume overload: One retrospective study of Oxyglobin use in cats with anemia found that 29 of 72 developed volume overload compared with 37 of 72 that responded to therapy. Hypothermia appeared to increase the risk once body temperature was returned to normal.66
Transient changes or side effects after administration of Oxyglobin reported by Biopure Corp. include yellow-orange discoloration of the skin, sclera, and gums; red to dark green discoloration of feces; brown-black discoloration of urine; vomiting; diarrhea; and decreased skin elasticity within 48 hours of dosing. The frequency and intensity of these clinical signs are dose dependent. HBOC solutions may interfere with several laboratory tests, particularly those based on colorimetric procedures (see package insert) or hemostatic tests based on optical methods. Pulse oximetry, blood gas analysis, and related procedures generally are not affected.
Oxyglobin may be warmed to 37° C before administration. As long as it is not frozen, it will remain stable for up to 24 months. Care should be taken to avoid temperatures that are too cold in the refrigerator. Oxyglobin is intended as a one-time use only at a recommended dose of 30 mL/kg intravenously at a rate of up to 10 mL/kg/hr. Although animals may not need the full 30 mL/kg; according to the manufacturer, 10 mL/kg may be sufficient in some cases. The foil bag in which the product is contained is oxygen impermeable. Once the bag is opened, oxygen can penetrate the plastic bag, resulting in the oxidation of the hemoglobin, as evidenced by the formation of methemoglobinemia. Unlike intact RBCs, which can reduce methemoglobinemia, HBOC solutions cannot. A brown discoloration will appear when approximately 25% of the hemoglobin has been oxidized. The product also offers an excellent environment for bacterial growth, even with refrigeration. The product should be used within 4 days of opening. Retrospective studies using HBOCs have been performed in both dogs67 and cats.66 However, neither study was accompanied by a non–HBOC-treated group, and the retrospective nature of the study precluded implementation of methods to reduce bias. A retrospective study of immune-mediated hemolytic anemia found that all dogs treated with Oxyglobin died, but the study did not address bias introduced by selection of only those most severely afflicted animals for treatment with HBOCs.67 Gibson and coworkers66 retrospectively studied HBOC use in 72 cats treated for anemia, including hemolytic anemia associated with acetaminophen toxicosis. Although 37 cats improved after a mean dose of 14.6 mL/kg (4.8 mL/hr), adverse events (not necessarily associated with HBOC) occurred in 44, including pleural effusions (n = 21) and pulmonary edema (n = 8). The anemic crisis was survived by 23 cats; euthanasia or death of the remaining cats was attributed to the underlying disease rather than administration of the HBOC.66 Oxyglobin also has been used successfully in a variety of other species.62
The use of Oxyglobin was reviewed retrospectively in cats (Germany) (n = 53, treated, 48 studied).68 The drug was generally administered by a fluid pump at 5 mL/kg/hr. Bags had been stored for 24 hours or less at 4° C. Some cats were treated more than once in a 24-hour period. The bases of treatment varied but included the general condition of the patient, the current hematocrit or hemoglobin level, and underlying disease. Cats receiving blood products were excluded from the study. Domestic Shorthair represented the predominant breed (n = 37); ages ranged from 0.3 to 14 years. Indications were blood low anemia (n = 25), hemolysis (n = 13), and ineffective erythropoiesis (n = 8). Reasons for administration of Oxyglobin rather than blood included lack of availability of feline blood products (including compatible donor) or to reduce loss of previously opened (within 24 hours) product coupled with reduced availability of feline donors. All patients also received Ringer’s lactate. An overall survival rate was not offered, but a survival rate was offered for each group. Hemoglobin changes ranged from −0.8 to 3.3 in the blood loss group (decreased in 5 cats) with a 24-hour survival rate of 72%. In the hemolytic group, hemoglobin changed from 0.2 to 5.2 gm/dL for a 24-hour survival rate of 84%. In the group with ineffective erythropoiesis, hemoblogin changed from −0.5 to 2.4 g/dL, with an 88% survival rate. Of the cats treated, 11 of the 48 had some type of cardiac disease; in this subgroup (which included cats from each of the preceding treatment groups), adverse reactions that might be related to treatment occurred in seven cats. These included pulmonary edema, pleural effusion, or respiratory distress; one of the cats died within 6 hours, and four died within 28 hours as a result of volume overload after receiving a range of 6.7 to 20 mL/kg in the 24-hour period.
Drugs Acting on Coagulation or Clotting
Hemostastis has been reviewed69 elsewhere. Briefly, it occurs in three steps (Figure 15-5), with the role players varying somewhat with the site or rate of flow: high shear vessels include the smaller to medium arteries, whereas low shear vessels include the larger arteries or veins. Platelet adherence occurs in the presence of damaged tissue or disrupted endothelium; platelets also will adhere to one another. Platelet adherence is mediated by laminin, collagen,or fibrinonectin in low shear vessels and adenosine diphosphate (ADP) and vWF in high shear stress vessels. Adherence is followed by platelet activation, during which granular contents of platelets are released. The contents recruit more platelets but also integrate platelet activation with the inflammatory process through release of mediators such as IL-1, platelet-activating factor, and serotonin. In high shear stress vessels, ADP and vWF are the primary mediators of activation, whereas thromboxane is the primary activator in low shear stress vessels. Thrombin will activate platelets (by way of protease-activated receptors) in any vessel. Because thrombin is not inhibited by cyclooxygenase inhibition, it is considered the strongest of the activators. Platelet adherence and subsequent activation are then followed by assembly of glycoproteins, causing aggregation. Again, the role players vary with the site, with vWF and glycoprotein Ib-IX-V mediating the response in high shear flow rates and fibrinogen in low shear flow vessels. The final common pathway to platelet aggregation is the glycopeptides IIb through IIIa receptors; this protein is the target of several investigative antithrombotic drugs in humans.69
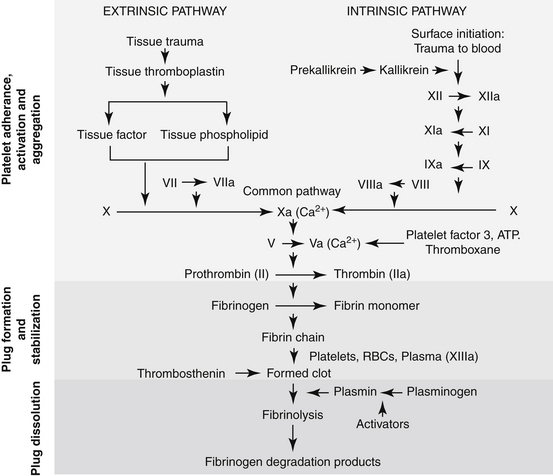
Figure 15-5 Hemostasis occurs in several phases: vascular and platelet, coagulation, and fibrinolysis. Each phase must remain in equilibrium to maintain a normal state of hemostasis. Endogenous procoagulants and anticoagulants function in each phase. The vascular and platelet phase is composed of vascular wall contraction and platelet adherence. Damage to the vessel exposes subendothelial collagen, which stimulates platelet adherence, which, in turn, depends on von Willebrand factor. Activation of the coagulation phase relies on a complex series of interdependent events. Injury results in activation of procoagulant substances in a cascade manner. Activation (designated by a lower case a, e.g., Xa) generally occurs as a result of proteolysis of a small molecule from the inactive factor. The major events in the coagulation cascade include formation of thromboplastin as a result of tissue trauma (designated as intrinsic if the blood is damaged and extrinsic if the vascular structures are damaged); transformation of prothrombin to thrombin; and rapid conversion of fibrinogen into fibrin. Calcium is involved as a factor at several steps. The fibrinolytic phase is initiated by the conversion of plasminogen to plasmin, which degrades fibrin.
Although a number of endogenous compounds play a role in platelet aggregation, a number of exogenous substances do as well (e.g., infectious organisms, complement factors). Further, endogenous factors provide balance, preventing coagulation. Among the most important antiplatelet mediators are nitric oxide and prostacyclin, both released from the endothelium. These two mediators appear to act synergistically to inhibit platelet adhesion, activation, and aggregation. Aggregation leads to the organized deposition of coagulation proteins with the formation of a temporary platelet plug. It ultimately should be stabilized in a cross-linked fibrin network. The final step in hemostasis is plug removal, which is accomplished through the initiation of fibrinolysis following activation of plasminogen to plasmin (see Figure 15-5). Disorders reflect not only failed hemostasis but increased risk of thrombosis as a result of a hypercoagulable state induced by disease or, less commonly, drugs.
Hemostatic Drugs
Hemostatic drugs are used to prevent or attenuate bleeding. They can be administered either topically or systemically. In general, topical agents either act as a factor in the coagulation cascade or stimulate some aspect of the coagulation cascade (see Figure 15-5).
Topical
Lyophilized concentrates of one or more clotting factors are available as topical preparations. These preparations usually provide an artificial factor or structural matrix that facilitates clotting (Figure 15-6). An intact hemostatic mechanism is necessary for their efficacy. These are absorbable products and are indicated for capillary oozing from small, superficial vessels. Examples of concentrated factors include thromboplastin (prothrombin is converted to thrombin, used locally in surgery), thrombin (converts fibrinogen to fibrin; available as powder, solution, or sponge), and fibrinogen. Examples of artificial matrices include absorbable gelatin sponge and oxidized cellulose (treated surgical gauze that promotes clotting) (see Figure 15-6).

Figure 15-6 Examples of coagulants (A).Oxidized cellulose (Surgicel shown here) provides a matrix for clotting (B). Gelfoam also provides an artificial structural matrix that initiates the clotting process (C). Products that contain silver nitrate (shown here), tannic acid, or salicylic acid precipitate proteins and cause coagulation by destroying tissue. Their use is limited to small, topical lesions.
Astringents act locally by precipitating proteins. These agents do not penetrate tissues and thus are restricted to surface cells. They can be damaging to surrounding tissues. Examples include ferric sulfate, silver nitrate (e.g., sticks; see Figure 15-6), and combinations that include tannic acid (e.g., STA).
Epinephrine and norepinephrine are hemostatic drugs only by virtue of their vasoconstrictive effects. They may be included in topical medications or injectable local anesthetics to decrease blood flow to the tissues.
Systemic
Fresh blood or blood components are hemostatic drugs only in states of (coagulation) factor deficiency. Examples include plasma (contains electrolytes, albumin, and some coagulation factors), fresh frozen plasma (plasma in which factors V and VII are stable), cryoprecipitate, and platelet-rich plasma.
Vitamin K
Vitamin K is a hemostatic drug only in instances of vitamin K deficiency. It is necessary for hepatic synthesis of coagulation factors II, VII, IX, and X (Figure 15-7). Several forms of vitamin K are available for therapeutic use. Vitamin K1 (phytonadione), the plant form, can be given parenterally and orally. It is more effective and more rapid in onset compared with other vitamin K analogs. However, anaphylactic reactions have been reported with intravenous administration, particularly with those preparations containing polysorbate 80, a known releaser of histamine in dogs. The drug can also be given intramuscularly. The effects of vitamin K1 occur within 1 hour of administration,3,70 although several hours are needed to resolve bleeding tendencies as coagulation proteins are replaced.
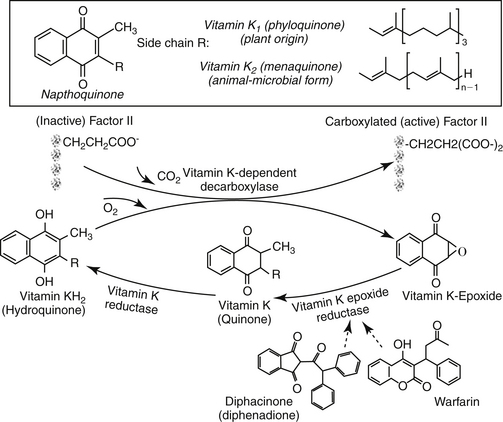
Figure 15-7 Vitamin K catalyzes the carboxylation of factors II, VII, IX, and X. As part of the reaction, the oxidized vitamin must be reduced to continue activation of coagulation factors. Coumarin derivatives block the reduction of vitamin K, rendering it incapable of activating coagulation factors. The arrow on warfarin identifies the chiral carbon that leads to S and R enantiomers. Both isomers are highly (>95%) bound to plasma proteins. Enterohepatic circulation was evident in cats 4 hours after administration.
Vitamin K2 (menaquinone) is the natural animal and microbial form of vitamin K. Vitamin K3 (menadione) is one of several synthetic forms. It must be metabolized to the active state. Vitamin K3 usually is absorbed too slowly to be used effectively in acute conditions, but it can be used for chronic therapy once the acute crisis has been resolved. In states of hypocoagulation, vitamin K1 is the preferred form, administered either subcutaneously or orally (3 mg/kg subcutaneously in multiple sites followed by 1 mg/kg every 24 hours parenterally or orally).71 Fat will enhance the oral absorption of vitamin K.
The most common use of vitamin K is treatment of rodenticide toxicity (see later discussion of anticoagulants). Duration of treatment for rodenticide poisoning will vary according to the toxicant. Toxicities from rodenticides containing coumarin or warfarin should be treated for 4 to 6 days; intoxication with diphacinone or brodifacoum requires treatment for at least 14 days.71 For toxicity with longer-acting rodenticides (i.e., indandiones), the dose of vitamin K is often reduced for subsequent weeks:71 0.5 mg/kg, week 2; 0.25 mg/kg, weeks 3 and 4. Prothrombin time should be monitored for 2 days after vitamin K is discontinued to detect residual rodenticide toxicity. Screening tests should remain normal for 3 to 4 days after therapy has been discontinued.
Protamine sulfate
Protamine sulfate is a low-molecular-weight, positively charged drug that binds to heparin, forming a salt and neutralizing its anticoagulant effects. It is used as a procoagulant only in instances of heparin overdosing. Protamine should be used cautiously because it also has anticoagulant activity, probably by impairing thrombin and fibrinogen. It is difficult to dose accurately because the dose is based on the amount of heparin to be antagonized (1 to 1.5 mg protamine for each 1 mg of heparin). In addition, the dose decreases as time elapses after heparin was administered.72 No more than 50 mg should be given in a 10-minute period.
Desmopressin
Desmopressin acetate (deamino 8-D-arginine vasopressin) is a synthetic analog of vasopressin and antidiuretic hormone that is used to treat central diabetes insipidus However, the drug also transiently increases serum concentrations of vWF in part through the release of preformed vWF from endothelial cells and macrophages as well as release of preformed Factor VIII.73 Increased release was demonstrated 1 hour after infusion in normal and afflicted dogs, although increase in afflicted dogs was greater.74 Kraus and coworkers75 have demonstrated that desmopressin decreases buccal mucosal bleeding time in Doberman Pinschers afflicted with vWF A similar response has been demonstrated for Greyhounds with vWF deficiency treated with 1 mg/kg desmopressin diluted in 1 mL of saline.76 The maximum response occurs 1 to 2 hours after treatment, although it may occur after as little as 10 minutes of infusion, with effects lasting 2 hours after intravenous administration.69 The increase is sufficient to provide improved coagulation activity in animals suffering from von Willebrand disease that undergo surgery.71 Desmopressin is generally administered by slow intravenous infusion over 20 to 30 minutes in humans after dilution in a minimum of 100 mL of physiologic saline. However, the intranasal (human) preparation also can be given subcutaneously at 1 U/kg.69 Rapid infusion may result in tachycardia, flushing, tremor, and abdominal discomfort. Repetitive administration results in depletion of the storage pools and loss of procoagulant effect and may increase the risk of tachyphylaxis.71,73
Recombinant human factor VIIa
The role of tissue factor (TF), a transmembrane glycoprotein receptor located in multiple extravascular tissues, is key to the initiation of hemostasis. It is through activation of Factor VII (FVII) bound to receptors located on TF that Factors IX and X are activated, leading to platelet activation and subsequent coagulation. A recombinant FVII product, derived from transfected baby hamster kidney cells, is chemically identical to activated FVII. The drug product was developed for treatment of human hemophiliacs (A and B) who had developed antibodies (following multiple blood transfusions) to Factors VIII or IX.77 Theoretically, although FVIIa should effect hemostasis through a TF factor, afflicted hemophiliacs are not able to mount an effective coagulative response primarily through loss of an amplified response by FVIII and IX. However, supraphysiologic doses appear to stimulate hemostasis (thrombin formation) by way of TF-independent pathways. Although activation of Factor X is not as effectively amplified as in the normal patient, more effective platelet involvement yields a more stable fibrin clot than would be formed in Factor-deficient patients. The fibrin clot is actually denser than that in normal patients. Because rhFVII requires exposed TF and activated platelets, its effects tend to be limited locally. The drug has been used on a compassionate-use basis for treatment of hemophiliacs undergoing procedures associated with a risk of bleeding or in patients suffering from refractory bleeding. The use of rhFVII has been described in dogs, including those suffering from either A or B hemophilia. Use in homozygote von Willebrand’s disease was not as successful. However, hypersensitivity reactions developed in several animals within 30 minutes of treatment, and all animals developed antibodies within 2 weeks of therapy. Nonetheless, should safety be improved, suggested potential applications of rhFVII therapy in animals, beyond treatment of factor deficiencies, have included disseminated intravascular coagulation (DIC), thrombocytopenia or thrombocytopathia, anticoagulant rodenticide toxicosis, overzealous anticoagulant therapy, high-risk surgeries, and liver and gastrointestinal diseases.77
Fibrinolysin inhibitors
Fibrinolysin inhibitors prevent the activity of plasmin (fibrinolysin) and therefore promote the persistence of clots (see Figure 15-5). The lysine analog aminocaproic acid is one of the few examples. Its therapeutic use is limited to treatment after an overdose of a fibrinolytic agent. It theoretically acts by substituting for lysine-binding sites, forming a complex with plasminogen, precluding its conversion to plasmin (e.g., inhibits streptokinase).78 It was studied after intravenous administration (1750 mg of 250 mg/mL solution followed by 250 mg/mL every hour for 9 hours) in a canine (n = 4; 10-21 kg) model of urokinase and venous injury–induced thrombosis. Aminocaproic acid inhibited plasmin formation completed, compared with heparin (20,000 U intravenously every 3 hours), which inhibited it by approximately 80%.79
Antihemostatic Drugs
Feline aortic thromboembolism (FATE) is a useful model for the discussion of treatment of thromboembolic disease. With FATE the goals of therapy are to reduce the formation of the thrombus, prevent embolization, improve collateral blood flow, control pain, and provide supportive care. Because thrombus formation in the heart (atria) is facilitated by blood stasis associated with atrial enlargement and endothelial damage, supportive therapy may include cardiogenic support. Drugs used to limit the formation of thrombi include anticoagulants, thrombolytics, and antithrombotics.
Anticoagulants
Anticoagulants interfere either directly or indirectly with the clotting cascade (see Figure 15-5). Several in vitro anticoagulants are used for blood collection intended for transfusion therapy and should be considered as drugs. Examples include citrate phosphate dextrose, acid citrate dextrose, sodium citrate, and heparin. All except heparin (discussed later) act by effectively removing Ca2+ from the cascade system. In vivo anticoagulants include heparin and the vitamin K antagonists.
Heparins
Heparin is a heterogeneous mixture of sulfated (anionic) polysulfated glycosaminoglycans (PSGAG) (Figure 15-8). Glycosaminoglycans are a family of structurally diverse and distinct polyanionic complex carbohydrates. Included in this group are heparin, heparan sulfate, chondroitin 4-sulfate, chondroitin 6-sulfate, dermatan sulfate, keratin sulfate, and hyaluronic acid. Each of the glycosaminoglycans is composed of repeating polysaccharides consisting of an amino sugar (e.g., glucosamine or galactosamine) and uronic (e.g., glucuronic or iduronic) acid or a hexose (galactose; keratin sulfate only). Each is ubiquitous in location. Among them, heparan sulfate proteoglycans are diverse in location with those that are endothelial derived serving as the primary endogenous anticoagulant. They are an integral part of stromal matrices, basement membranes, and almost all cell surfaces where they provide cohesion between vessels and vascular stroma.43 Endogenous synthesis results in multiple compounds that vary in the lengths of carbohydrate chains Heparin and heparan sulfate are the most complex members of this class of compounds and are characterized by the greatest biologic diversity.43 Proteoglycans are formed when proteins, which serve as the core for the PGAG, are posttranslationally (in the Golgi apparatus) modified with the addition of glycosaminoglycan chains. Each is sulfated to variable degrees, depending on the repeating subunits of which the disaccharide is composed.
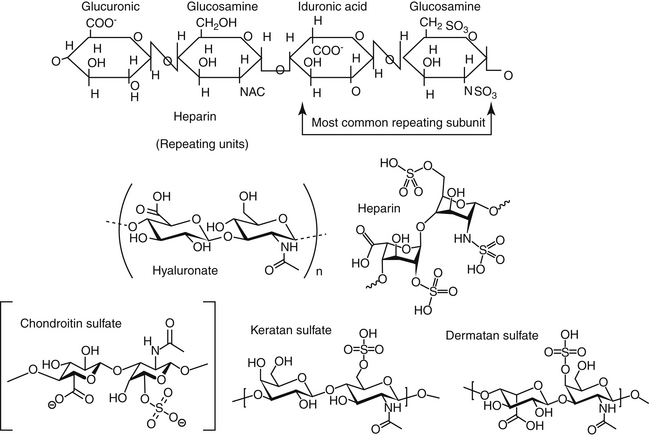
Figure 15-8 Heparin is a heterogeneous mixture of sulfated, anionic, and polysulfated glycosaminoglycans. The subunits include uronic acid, sulfonaminoglycosamine, N-acetylglucosamine, and a pentasaccharide sequence. Heparin is one of many polysulfated glycosaminoglycans (PGAGs) found throughout the body. Other structurally similar PGAGS include hyaluronate, heparin sulfates, chondroitin sulfates, keratan sulfates, and dermatan sulfates.
Heparin was so named because of its initial discovery in high concentrations in the liver. It is often located attached to serine residues of core proteins (e.g., coagulation factors). Commercially available heparin is prepared from porcine intestinal mucosa and bovine lung. It is stored in mast cells, along with other PGAGs, including chondroitin and dermatan sulfate. Heparin also is stored in the body in basophils and, to a lesser extent, in vascular endothelium. Tissues with high concentrations of mast cells serve as a source of heparin. Heparin is a highly sulfated (generally 2 to 3 per subunit) glycosaminoglycan composed of alternating sequences of sulfoaminoglycosamine and uronic acid units and smaller amounts of N-acetylglucosamine (see Figure 15-8). Its structure also includes a unique and specific sulfated glucosamine pentasaccharide sequence that binds to antithrombin (AT) III and thus serves as the active site of the molecule. The number of active sites in a molecule of heparin is variable, but it is present in only about 30% of the molecules. In its native state, the molecular weight is quite variable, ranging from 3 to 50 kDa. The molecular weight of commercial unfractionated heparin (UFH) generally varies from 1800 to 30,000 daltons (mean 15,000, representing about 45 monosaccharide chains).80 However, heparin also has been prepared through filtration methods as fractionated or low-molecular-weight (LMW) heparins, which normally make up less than 5% of endogenous heparin. The LMW heparins generally are only 1000 to 10,000 daltons in size and are specifically defined as an average weight of 8000 daltons or less, with at least 60% of the product being less than 8000 daltons in size (see Figure 15-8). A variety of LMW heparins have been made, with each varying by the method of fractionation or depolymerization. The LMW heparins have much higher affinity for AT (by way of Xa) compared with UFH and thus are more potent, providing 70 units/mg of antifactor Xa and a ratio of antifactor Xa to AT activity of 1.5 or more.80 Doses of fractionated LMW preparations are thus lower than unfractionated products (Table 15-3). Preparations of UFH also contain contaminants such as dermatan sulfate; these contaminants may be present in LMW heparin preparations but in diluted form. Intentional contamination of heparins with oversulfated chondroitin sulfates is addressed with adverse reactions. Some of these compounds also have anticoagulant activity, albeit less than heparin.81
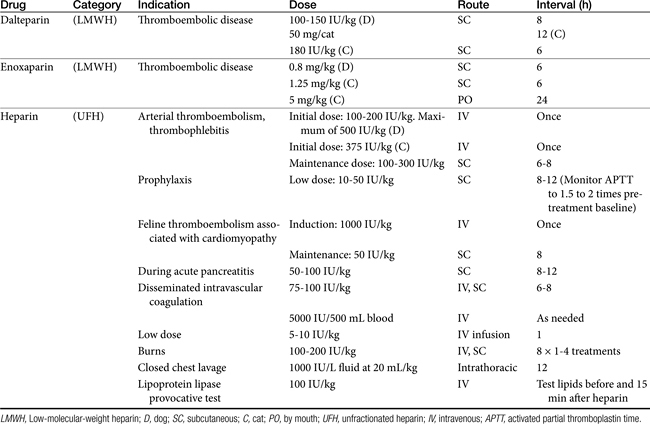
Mechanism of action
Heparin interacts with many proteins, probably by electrostatic forces between the polyionic groups of the glycosaminoglycan and the cationic groups of proteins. Example proteins include proinflammatory chemokines, growth factor, extracellular matrix proteins, and leukocyte proteins. Intracellular effects reflect interaction of heparin or heparan sulfates with cell surface-binding proteins (which are not necessarily true receptors) followed by endocytotic internalization.
The most recognized sequelae of protein–heparin interaction are the anticoagulant actions of heparin.43 However, this effect is indirect, reflecting facilitation of the actions of endogenous anticoagulant proteins. The most notable is AT, one of several endogenous serine proteinase inhibitors (serpins) whose thrombin inhibition occurs only in the presence of heparin. Long chains of heparin bind to specific lysine residues on AT. At low heparin concentrations (0.1 to 1 U/mL), AT rapidly inhibits the activity of the clotting factors IIa (thrombin), Xa, and IXa, resulting in prolongation of activated partial thromboplastin time (APTT) and thrombin time (TT); prothrombin time (PT) is only mildly affected. At high heparin concentrations (>5 U/mL), heparin interacts with heparin cofactor II (HCII), which binds only to thrombin, causing further inhibition. Binding of heparin to AT or HCII results in a stable complex (Figure 15-9). Binding induces conformational change in the anticoagulant complex such that the active sites of the inhibitor is exposed, facilitating binding to the clotting factors. As such, heparin increases the velocity, but not magnitude, of interaction between the endogenous anticoagulants and clotting factors. The rate of interaction can increase 10,000-fold in the presence of heparin. The anticoagulant and clotting factor complex will remain intact after activity is inhibited; as such AT is a “suicide” substrate.54 In contrast, heparin will be released to interact with other molecules. Other inhibitors, whose concentrations are less than one hundredth of that of AT are also inhibited at high concentrations; these include plasminogen activator inhibitor, protein C inhibitor, and tissue factor pathway factor inhibition of Xa. Heparin also inhibits factors IXa and to some degree Xia; the kallikrein–kinin system and, through binding to complement C1, the complement system are inhibited. Factor VIIa is minimally inhibited.
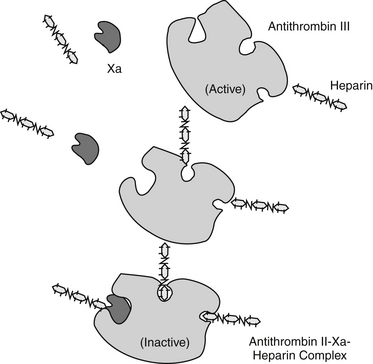
Figure 15-9 The pentasaccharide sequence of heparin interacts with the amino residues of antithrombin III, changing the conformation of antithrombin II and its ability to interact with factors IXa, XIa, XIIa, and, in particular, II and Xa. Heparin may directly interfere with factor X.
Because the specific AT-binding site of heparin is generally absent in other glycosaminglycans (limited in heparin sulfates and absent in chondroitin and dermatan sulfates), these latter compounds are characterized by minimal anticoagulant activity.
Simultaneous inhibition of both AT and thrombin requires molecules greater than 5400 MW. LMW heparins generally act through AT inhibition of Xa rather than IIa. Thus LMW heparin fractions are more potent anticoagulants (a lower concentration inhibits Xa) compared with high-molecular-weight (HMW) fractions,81 but HMW heparins may be more effective anticoagulants because their large structure facilitates simultaneous binding of both AT and thrombin simultaneously.72 Whereas the ratio of antiXa:IIa activity for UFH is generally 1:1, the same ratios range from 2 to 4:1 up to 9:1 for LMW heparins in humans.82 Lower ratios might be expected for dogs because LMW heparin appears to have less effect on AT.
KEY POINT 15-5
Although fractionated low-molecular-weight heparins are more potent inhibitors of Xa, products that contain high-molecular-weight fractions may actually be more effective anticoagulants because their large structure facilitates simultaneous binding of both antithrombin and thrombin.
The effect of heparin on thrombin and Xa occurs only when the factors are not bound to platelets or fibrin; platetet factor IV blocks the interaction between heparin and AT. However, heparins have an antiplatelet effect because of a high affinity for platelet factor IV. Most heparin preparations also inhibit platelet aggregation, negating effects of platelet-derived growth factor on vascular smooth muscle and leading to their descriptor as platelet-active anticoagulants. vWF (platelet factor VIII) is among the factors bound and inhibited by heparin.80 Heparins also bind to vascular endothelium, causing release of two other endogenous PGAGs and altering vascular endothelial permeability.81
Heparin has other effects not related to its anticoagulant activity. In human patients heparin may prove useful for prevention of atherosclerosis and for accelerated formation of collateral circulation in the presence of thrombosis (angiogenesis).81,83 Heparin can inhibit or modulate selected targets of an allergic inflammatory response, including neutrophils and T cells. Heparins appear to decrease leukocyte adherence to vascular endothelium and facilitate leukocyte migration along a chemoattractant gradient to sites of inflammation. Derivatives of low anticoagulant activity block superoxide anion generation, probably by interacting with superoxide dismutase. Heparin appears to impair mast cell degranulation by altering intracellular calcium release through blockade of inositol 1,4,5-triphosphate receptors.43 Heparin also appears able to influence tumor cell metastasis. Tumor cell invasion from the vasculature probably involves host degradative enzymes, including heparinases, which target both heparin and heparan sulfate. Exogenous heparin inhibits the hydrolysis of heparan sulfate. Heparins with low anticoagulant activity have reduced the metastasis of several tumor types in various experimental models.43 Smooth muscle cell proliferation such as that which accompanies atherosclerosis and bronchial asthma also is influenced by heparin and heparan sulfate.84 Heparin inhibits capillary endothelial growth but binds with high affinity to fibroblast growth factors and potentiates their growth-promoting effects.54 As such, heparin facilities growth of smooth muscle of the vasculature (including pulmonary), airways, intestine, and contractile cells such as fibroblasts and renal glomerular mesangial cells. Effects can be correlated with molecular size and sulfation of heparin; heparin and other heparans may serve as reservoir for growth factors. Endogenous heparin has been implicated in diseases characterized by smooth muscle proliferation; manipulation of selected members of the heparins may ultimately prove to be a therapeutic alternative to treatment.43 Finally, heparin liberates lipoprotein lipase and can decrease serum triglyceride concentrations, thus “clearing” lipemia.72,54
Drug disposition
The complex chemistry of heparin and its binding to proteins and cells leads to complex disposition. Absorption and distribution of heparin are limited by the large size and polarity. Studies characterizing heparin elimination are generally based on response to therapy (i.e., changes in the APTT). However, more recent studies in animals have detected the drug chemical in blood.85Absorption of heparin after oral administration or by aerosolization is negligible; as such, heparin is a parenteral anticoagulant. Subcutaneous absorption is rapid; intramuscular administration is generally contraindicated because of the risk of hemorrhage. Heparin is metabolized by heparinase in the liver and by reticuloendothelial cells. Metabolites of heparinase activity are excreted in the urine. The anticoagulant activity of the metabolites apparently has not been addressed. The elimination half-life of heparin appears to depend on molecular weight. The LMW fractions are cleared less rapidly compared with larger molecules, leading to a half-life two to three times longer than that of endogenous heparin. As such, molecules with a higher affinity for AT activity will be cleared more rapidly than those responsible for anti-Xa activity.86 It is perhaps for this reason that UFH remains the preferred heparin in situations in which a rapid resolution of response upon discontinuation of therapy is desired (e.g., surgical patients or other patients at risk for bleeding).
The pharmacodynamics and elimination half-time of response (based on anti Xa activity) of dalteparin (Fragmin; anti-Xa activity of 2500 U/mL) have been reported in dogs (Table 15-4). Targeted therapeutic ranges based on anti-Xa activity approximates 0.3 U/mL for UFH and 0.5 U/mL for LMW heparin.87 Dose response indicates increasing effects with increasing dose, with response more rapid in onset and of greater magnitude with intravenous compared with subcutaneous administration. Response occurs in 2 minutes with intravenous administration, compared with 2 hours with subcutaneous administration. Peak APTT and TT at 2 hours after subcutaneous administration of 200 IU/kg were similar to that measured at 1 minute after 25 IU administered intravenously. Peak effects after intravenous administration approximated 1.75 times baseline for APTT and up to 2.5 times baseline (TT); return to baseline occurred at approximately 1 hour. The impact of subcutaneous administration on APTT and TT was muted: peak effect was no more than 1.25 times baseline for either APTT or TT at all doses. Return to baseline appeared to be dose dependent for both routes but generally was faster (1 to 4 hours, but up to 8 hours) after intravenous compared with SC administration (6 to 10 hours). The authors noted that significant anticoagulant activity lasted up to 3 hours, but even the highest subcutaneous doses failed to affect either APTT or TT to a clinically significant degree. The authors also concluded that monitoring LMW heparin administration in dogs would best be accomplished by measuring anti-FXa activity with chromogenic substrates.88 The influence of LMW heparin on APTT and TT in dogs appears to be less than that in humans, suggesting species differences in coagulation response. However, this may reflect a shorter APTT in normal dogs compared with normal humans.
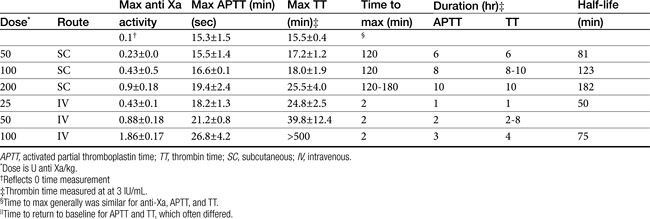
The pharmacodynamics (based on anti-Xa activity) of 5 days’ treatment of dalteparin (100 IU/kg subcutaneously twice daily) or enoxaparin (1 mg/kg subcutaneously twice daily) was compared with that of UFH (250 IU/kg subcutaneously every 6 hours) in cats (n = 5) using a randomized (for the first treatment) crossover design with a 14-day washout between treatments. Peak activity for all three products occurred at approximately 2 to 4 hours, with the maximum effect of each product in anti-Xa (U/mL) approximating 0.6 (UFH), 0.45 (enoxaparin) and 0.3 (dalteparin). Concentrations reached the human therapeutic range of 0.5 to 1 U/mL anti-Xa activity in only one cat receiving dalteparin. For each drug mean activity was not detectable before the next dose (trough). These data led the investigators to conclude that higher doses than those studied are indicated,87 Vargo and coworkers drew similar conclusions in their study of rhe effect of dalteparin (100 IU/kg SC, b.i.d, for 13 days) in healthy cats (n = 8) (based on anti-Xa activity). Activity was measurable in only 4 cats; activity present by 4 hr after a single dose (the first sampling time) and returned to baseline by 6 hr; in these cats. Prothrombin time and APTT were not significantly altered by dalteparine administration; although sample size was small, clinical values were essentially unchanged in the cats.88a The half-life of heparin is prolonged in renal or liver failure.81 Bioavailability (subcutaneously) of the LMW heparins appears to be similar in humans, approximating 80% to 90%,82 and approximates 100% in dogs.89
Preparations
Because heparin is a heterogeneous mixture, only about 33% of the molecules i the drug preparation inhibit coagulation, and correlation between heparin plasma concentration and anticoagulant activity is not possible. Correlation between dose and response is further complicated by differences in effects based on molecular weight. The concentration of heparin is standardized by bioassay as units of activity, with 1 unit of heparin (referred to as a Howell unit) being equivalent to approximately 2 μg of pure heparin, which in turn (according to the Online Medical Dictionary; http://www.online-medical-dictionary.org/) is the amount of heparin that will maintain anticoagulant activity in 1 mL of feline blood for 24 hours at 0o C.90 Heparin is available as a sodium or lithium salt. The sodium salt is usually the preferred preparation for in vivo use. LMW heparins currently available for prevention of acute deep vein thrombosis or pulmonary thromboembolism in the United States include dalteparin, enoxaparin (also approved for extended use in prevention of deep vein thrombosis), and ardeparin. The cost for treating a cat with enoxaparin for 1 month has been estimated at $80 (US). Storage of diluted drug (20 mg/mL) and storage in either glass vial or individual tuberculin syringes for up to 4 weeks does not result in statistical differences of activity. A generic LMW heparain has recently been approved.
Drug interactions and side effects
Heparin stands out as a drug that will interact with many other drugs. Heparin should not be mixed with any other drug. Hemorrhage is the major complication of heparin therapy, occurring in 18% to 22% of human patients receiving heparin.81 Hemorrhage is less likely to occur with low dosages and constant intravenous infusion (as opposed to intermittent intravenous administration). Hypersensitivity may play a role in hematoma formation.91 The incidence of hemorrhage can be reduced by (1) confirmation of the need for therapy; (2) use of the appropriate dose and frequency; (3) avoidance of combination therapy with other antihemostatic drugs, including aspirin and other salicylates; and (4) monitoring of the effects of therapy with clotting or coagulation tests. Use of LMW heparin should also be considered. At higher doses the APTT can be useful for assessing the likelihood of hemorrhage. Monitoring APTT is less useful when low doses of heparin are given. Heparin is contraindicated for the bleeding patient and for DIC unless replacement blood or plasma therapy is given (see later discussion of clinical indications). Excessive therapy (theoretically) might be treated with protamine sulfate, a compound that complexes with heparin. It is dosed according to the amount of heparin to be neutralized (see previous discussion of protamine). The risk of bleeding in surgical patients receiving LMW heparin has not been well assessed; however, the longer duration of action of these products may contribute to an increased risk of bleeding. In human patients undergoing cardiovascular surgery, although all antithrombotic drugs studied increased the risk of bleeding, the use of the LMW product enoxaparin was associated with the greatest amount of blood loss and need for blood transfusions.92
Heparin-induced thrombocytopenia has been reported in 5% to 30% of human patients receiving heparin and is more likely to occur with bovine as opposed to porcine preparations.81,93,94 Two types (type I and type II, similar to type A and type B adverse reactions) of thrombocytopenia have been described. Several days of therapy are necessary for type I thrombocytopenia to occur; it resolves once therapy is discontinued. Type I thrombocytopenia occurs less frequently with LMW heparins. Type II thrombocytopenia occurs in fewer people, is more severe and rapidly fulminating, and is characterized by a longer time to onset (6 to 10 days). Type II thrombocytopenia may be an allergic response to the secondary and tertiary structures of heparin and has caused paradoxical thrombosis.81Antibody formation toward heparin bound to platelet factor IV has been demonstrated.80 The syndrome is recognized by a greater than 50% decrease in platelets that cannot be explained by other causes and/or skin lesions at the site of injection.80 As with type I thrombocytopenia, the incidence of type II thrombocytopenia is likely to be less with LMW heparins. However, cross-reactivity of 20% or more toward heparin antiplatelet antibodies has been reported for LMW heparins; heparanioids such as danaparoid have been suggested for human patients demonstrated to develop heparin-induced thrombocytopenia.
A small number of human patients have developed skin necrosis with both UFH and LMW heparin. Lesions are similar to those caused by toxic epidermal necrolysis and can be lethal. The cause is unknown.
In human patients receiving long-term therapy, heparin has induced osteoporosis. The mechanism is not known but does not seem to involve prostaglandin E2. The effect is more dramatic with UFHs. LMW heparins have an osteopenic or calcium-sparing effect, although the sequelae and nature of this effect are not characterized.81
Liver leakage enzymes increase in up to 93% of human patients receiving heparin. The increases peak approximately 7 days into therapy and then return to normal, with no obvious detrimental clinical sequelae. Heparin also interferes with and falsely increases bile acids. When used as an in vitro anticoagulant, heparin falsely increases blood urea nitrogen, bile acids, and sodium and potassium salts. Finally, heparin can interfere with several hormones. It interferes with thyroid hormones, causing decreases in both thyronine and thyroxine. It is a predictable and potent inhibitor of aldosterone secretion in human patients, causing natriuresis and the potential for hyperkalemia, particularly in predisposed patients.95,96 The effects of heparin on the renin–angiotensin–aldosterone system may be responsible for its antihypertensive effects.97
A unique side effect of heparin recently identified reflects adulteration of heparin ingredients by the addition of synthetic or animal origin oversulfated (4 sulfates per disaccharide unit) chondroitin sulfates (OSCS). Acute reactions in humans (which are potentially lethal) reflect two mechanisms: direct interaction with kallikrein and release of C3a and C5a anaphylatoxins.98 Hypotensive patients (e.g., those undergoing renal dialysis) and patients receiving angiotensin-converting enzyme inhibitors (which inhibit degradation of bradykinin) were at risk of reacting adversely. Because LMW heparins are processed from UFHs, contamination is also possible, although contaminants will be diluted compared with unfractionated preparations. The dog, rat, and pig were studied as models for toxicologic evaluation (as reported by manufacturers Sanofi–Aventis) of contaminated heparins; however, the dog was not selected because cardiovascular toxicity did not occur after administration of doses (0.83, 2.09, 10 mg/kg) considered clinically relevant (although coagulation disorders precluded dosing higher than 10 mg/kg). Consequently, the dog probably is less sensitive to the effects of OSCS, which is consistent with the relative safe use of polysulfated aminoglycans (i.e., Adequan) in dogs compared with humans.
N-acetylcystiene (NAC) manifests some actions similar to heparin. It has been used as a renoprotectant and hepatoprotectant agent in humans undergoing surgery. Compared with placebo controls, human patients receiving 150 mg/kg NAC followed by 150 mg/kg as a 24-hour infusion had lower postoperative PTs and prolonged coagulation times (based on thromboelastometry tracings); further, platelet aggregation also was decreased. Blood loss increased in these patients, leading the investigators to warn that patients receiving NAC preoperatively had an increased risk of bleeding.99
Clinical indications and use
Clinical indications for heparin therapy include the prevention or treatment of venous or pulmonary embolism (e.g., nephrotic syndrome, autoimmune hemolytic anemia, hyperadrenocorticism, or heartworm disease) and atrial fibrillation with embolization (e.g., feline cardiomyopathy) and as an anticoagulant for diagnostic use and blood transfusion.
Thromboembolism
Heparin remains the drug of choice when rapid anticoagulant activity is necessary in acute thrombosis; in humans heparin is continued for 4 to 5 days to allow time to onset of oral anticoagulants. Generally, heparin is effective only on the fluid phases of thrombin-dependent proteins and is ineffective toward clot-bound factors. However, heparin not only prevents further thrombosis but also appears to facilitate resolution of the thrombus, promoting recanalization through activation of tissue plasminogen activator81 and stimulating angiogenesis. Efficacy of therapy does not seem to be related to the anatomic location of the thrombus. Heparin and other antithrombotic agents have not been well studied in dogs and cats. Many studies that focus on pharmacokinetics and pharmacodynamics are performed in normal animals; response to drugs may be much different in the face of diseases requiring therapy. In human patients (and possibly dogs), the dose of heparin necessary to control coagulation may be greater than that in the normal patient who is not suffering from a coagulopathy.100 For thrombosis, heparin is administered parenterally; deep subcutaneous or intrafat injection prolongs persistence of therapeutic concentrations. Large hematomas can occur with deep intramuscular injection. Heparin is also administered intravenously, either intermittently or as a constant infusion. In human patients the use of weight-based nomograms to determine initial and maintenance heparin infusion rates results in a higher percentage of patients reaching the targeted activated thromboplastin time (APTT) range earlier in the course of therapy.101 Monitoring is particularly important to establish effective doses. Doses are variable with author, route, species, and intent. Generally, subcutaneous doses range from 150 to 250 U/kg in dogs and from 250 to 375 U/kg in cats every 8 hours. The dose of heparin in normal cats necessary to maintain therapeutic concentrations of heparin (as established in humans, 0.35 to 0.7 U/mL) in one study was 300 U/kg of heparin every 8 hours.85
For most causes of thromboembolism, therapy is more likely to be successful in patients if heparin is administered at a rate to cause the APTT time to be 1.5 to 2.5 times baseline or the activated clotting time (ACT) to be 1.2 to 1.5 times baseline (prolongation by 15 to 20 seconds).80,101-103 However, monitoring APTT generally is ineffectual if low doses of heparin (or LMW heparin) are given. Rather, anti-Xa activity should be measured.88 A baseline should be established before therapy is begun, and monitoring should occur 2 hours after a subcutaneous dose; measurements might be immediate. Alternatively, targeting anti-Xa activity of 0.35 to 0.7 IU/mL has been recommended in human patients requiring higher doses of UFH to maintain targeted APTT; less heparin was needed in one study, decreasing the risk of bleeding.80 Replacement of AT should accompany heparin therapy when indicated (e.g., nephrotic syndrome, DIC). Gradual discontinuation of heparin therapy has been recommended to prevent a hypercoagulable state.102,104 Long-term heparin therapy can be accomplished at home with subcutaneous injections, or warfarin therapy can be implemented (see later discussion of vitamin K antagonists).
LMW heparins (e.g., enoxaparin) have been studied in humans and to some degree in dogs and cats. Advantages when compared with HMW heparins include specificity of action, better absorption after subcutaneous injection, more predictable dose–response relationship because of improved prediction of pharmacokinetics, and prolonged elimination half-life. Reduced incidence of bleeding (in nonsurgical patients) because of more specific targeting of Xa compared with thrombin and other factors, as well as reduced thrombocytopenia, has also been suggested as an advantage of LMW heparins.81,105 Because LMW heparin does not effectively affect thrombin, APTT and TT are minimally affected and therefore should not be used to monitor response. Rather, anti-Xa activity should be measured. According to Mischke and Grebe,88 in humans an anti-Xa activity target of 0.3 to 0.6 U/mL at 4 hours is suggested for patients at high risk of thrombosis (the lower target intended to minimize the risk of bleeding complications; a target of 0.14 to 0.34 has been recommended for patients with acute myocardial infection already receiving thrombolytic therapy) and 0.4 to 0.9 U/mL for low-risk patients.80,88 Whether the targets appropriate for dogs are also appropriate for cats has yet to be established. However, if the same targets are relevant, a dose of 100 U/kg of LMW heparin (dalteparin) would achieve the targeted anti-Xa activity in dogs at 2 to 4 hours (see Table 15-4)
A retrospective study of cats with FATE and treated with dalteparin found the drug, when administered at 99 U/kg either once or twice daily, to be both convenient and effective. Mean follow-up time of 172 days was associated with recurrence in 8 of 43 cats, with complications related to bleeding described as infrequent.103 The use of LMW heparin (dalteparin, starting daily dose of 102 IU/kg subcutaneously) has been compared to warfarin (0.08 mg/kgl using PT and an international normalized ratio target of 2 to 3) for treatment of arterial thrombosis in cats.106 Dalteparin increased mean survival time to 255 days compared with 69 days for warfarin-treated cats, although the differences were not statistically significant, probably due, in part, to small sample size. Further, 17% of the warfarin-treated cats exhibited bleeding adverse events, whereas none of the LMW heparin–treated cats did.
Supportive therapy for treatment of FATE includes the use of analgesics to control pain. The use of newer nonsteroidal antiinflammatories that are more potent for cyclooxygenase 2 (responsible for prostacycline formation) than cyclooxygenase 1 (responsible for thromboxane formation) might prudently be avoided. Their use may shift the balance toward thrombogenesis (see Chapter 29). Use of opioid analgesics, including a fentanyl patch and/or constant-rate infusion, should be considered. Acepromazine should be avoided for either treatment or prevention of thromboemblism; its vasodilatory properties are not likely to facilitate collateral circulation and hypotension-induced decreased blood flow may increase the risk of thrombus growth.103
For prevention of pulmonary thromboembolism, heparin can be administered at a lower dose (30 to 75 U/kg). Monitoring ACT or APTT may not be useful at these doses. Hemorrhage, however, remains a potential side effect in at-risk patients (i.e., those undergoing surgery).
A Cochrane review in human medicine found insufficient evidence to recommend use of LMW heparin instead of vitamin K antagonists.107
In patients suffering from severe heartworm disease, low-dose heparin given 1 to 2 weeks before treatment and 3 to 6 weeks after treatment improves survival rates compared with no treatment or with aspirin therapy.108 In the event of thrombosis associated with adulticide therapy, heparin should be administered (50 to 70 U/kg every 8 hours) for at least 7 days (assuming the platelet count increases to above 150,000/mm3).
LMW heparins have been recommended as treatment or prevention of thrombosis associated with immune-mediated hemolytic anemia.
Disseminated Intravascular Coagulaopathy
Heparin is used with blood and plasma for the treatment of DIC. The use of heparin for treatment of DIC remains controversial.71 Its efficacy for DIC depends on adequate concentrations of AT (≥40% of normal). Replacement therapy with either whole fresh blood or fresh or fresh frozen plasma is indicated if AT levels are not normal or for actively bleeding patients. A loading dose is generally followed by a maintenance dose; the loading dose can be preincubated with blood or plasma in order to maximize effects on AT Maintenance dosing should be based on changes in APTT rather than on a fixed dose. This is particularly true in patients suffering from DIC because the synthesis of cofactors varies among patients with DIC. Although normal dogs respond rapidly to heparin therapy, identification of changes in APTT in DIC patients may be difficult. Neutralization of heparin with polybrene (hexadimethrine bromide)71 can be used to distinguish changes in the APTT due to heparin from those due to DIC once a baseline APTT has been established. The recommended dose of heparin is markedly variable, ranging from a low of 75 to 100 U/kg administered subcutaneously (a dose unlikely to change APTT but likely to be sufficient to target AT) to a high of 750 to 1000 U/kg (severe disease with organ damage caused by microthrombosis) every 8 hours. An intravenous bolus of 5000 U or 80 U/kg followed by constant-rate infusion of 5 to 18 U/kg/hr also has been recommended. For a hypercoagulable state, a subcutaneous dose of 200 to 300 U/kg every 6 hours is recommended. The APTT should be monitored every 6 hours. In general, if higher doses are used, the APTT should be prolonged by 1.5-fold to twofold. Once a therapeutic response is established, the APTT and platelet count should be monitored daily. Heparin should not be abruptly discontinued in patients with DIC because of the risk of rebound hypercoagulability associated with AT deficiency. Nonanticoagulant uses of heparin are increasing in human medicine. Anecdotal reports in humans indicate that heparin can be beneficial for the treatment of asthma and allergic inflammation.
DIC has been reviewed in cats,109 including its treatment. Of the 46 cats studied retrospectively, 43 died or were euthanized, precluding conclusions regarding the most effective therapies.
Vitamin K Antagonists (Oral Anticoagulants)
The oral anticoagulants differ primarily in their duration and magnitude of effect. Studies of their importance in veterinary medicine have focused primarily on their toxic rather than their therapeutic indications,11 although these drugs are being used increasingly to treat thromboembolic diseases.
Chemistry
The vitamin K antagonists consist of two groups: the coumarin derivatives (dicoumarol and warfarin) and the indandione anticoagulants. Both interfere with the hepatic synthesis of vitamin K–dependent clotting factors II, VII, IX, and X and anticoagulant proteins C and S (see Figure 15-7).95 They block the reduction of vitamin K by vitamin K epoxide after its use in factor synthesis, thus effectively reducing the concentration of vitamin K. Two points related to this mechanism are important. First, the anticoagulant activity occurs only in vivo; second, there is a delay in anticoagulant activity (and therefore therapeutic or toxic effect) for 8 to 12 hours because of the persistence of factors synthesized before administration. Factor VII has the shortest half-life and therefore is the first factor to become deficient. Antithrombotic effects occur in 4 to 6 days after therapy is begun, however, as serum concentrations of factors IX and X decrease. Serum concentrations of the anticoagulant protein C also decrease but more rapidly than clotting factors, thus possibly rendering the patient hypercoagulable. Other factors that might contribute to the hypercoagulable state increased thrombin generation as a result of the release of procoagulant platelet-derived factors, expression of tissue factor in response to damaged endothelium, and release of platelet factor IV by activated platelets with subsequent neutralization of heparin.80 Heparin therapy might be used for the first 2 to 5 days after warfarin therapy is begun to prevent this hypercoagulable state.
Disposition
Warfarin contains a chiral carbon (see Figure 15-7) and is marketed as a racemic mixture. The S isomer is approximately 5.5 times more potent than the R isomer with regard to anticoagulant activity in all species pharmacodynamically studied.110 The vitamin K antagonists are rapidly and completely orally absorbed. Warfarin is 75,000 times more soluble in water than is dicoumarol and is characterized by much better oral bioavailability.72 Peak levels occur in 1 hour. There are, however, marked differences in product bioavailability, and products should be interchanged with caution. For products used as rodenticides, warfarin derivatives often have a drug half-life up to 7 days, whereas the indandione diphacinone has an elimination half-life of 30 days.11 All coumarin derivatives are highly bound to serum albumin, limiting the distribution volume to plasma volume. Vitamin K antagonists are metabolized by the liver to inactive metabolites by the cytochrome P450 system and subsequently conjugated to glucuronide. They undergo an enterohepatic cycle.
Although response does not relate in a dose-dependent manner to plasma drug concentrations, the disposition of warfarin, including its enantiomers, has been reported in cats (n = 10) after single intravenous administration of 0.5 mg/kg and then at 0.1, 0.25, and 0.5 mg/kg (n = 4) orally of the racemic mixture.110 The disposition of the S isomer resulted in significantly higher area under the curve compared with the R isomer, in part because of longer mean residence time and half-life, which were approximately twice as long in the S (approximately 24 hours) compared with the R (11 hours) isomers. Variability was great, with a range that varied almost threefold (11 to 38 hours) for both half-life and mean residence time for the S isomer. A dose-dependent effect was evident. Problematically, distribution of warfarin throughout the study tablets was even. Anticoagulant potency of the two isomers was not studied in cats.
Preparations
Vitamin K antagonists are prepared for therapeutic use as tablets and solutions (e.g., warfarin, dicoumarol). They are, however, more commonly used as oral rodenticides.
Drug interactions
A variety of factors can increase the activity of warfarin anticoagulants. Hypoproteinemia, antimicrobial therapy, hepatic disease, hypermetabolic states, pregnancy, and the nephrotic syndromes are some examples. Drug interactions are most significant when used therapeutically for chronic treatment. Because they are highly protein bound, they will be displaced by (or will displace) other drugs that are protein bound, and their anticoagulant effects may be increased to the point of toxicity. Examples include acetylsalicylic acid and other nonsteroidal antiinflammatory drugs. Drug interactions occur with other antihemostatic agents.
Clinical use
Clinical use of the coumarin derivatives in dogs and cats is increasing as monitoring techniques improve and doses are refined. The high incidence of recurrent thromboembolism in cats receiving aspirin has led to renewed interest in the use of warfarin for management of arterial thromboembolism. Currently, cardiologists are recommending use of warfarin even in the absence of prior thromboembolic events once adequate left atrial enlargement (left atrial to aortic ratio <2) has been confirmed by echocardiography.111 The drug is discontinued if atrial size should normalize (ratio of <1.25).
Warfarin therapy for humans is characterized by doses that vary as much as twentyfold. Variability should be anticipated as a result of species differences in drug disposition, warfarin preparations, the target disease, and the presence of other drugs that may interact with warfarin. Accurate dosing should be based on drug-response monitoring. For long-term anticoagulant needs, or in combination with heparin (100 U/kg every 8 hours) for acute treatment of thromboembolism, warfarin can be administered at 0.1 to 0.2 mg/kg (or one fourth to one half of a 1-mg tablet in cats) orally every 24 hours.70,111
The PT should be measured at baseline before initiation of therapy. Heparin therapy (when also being used) may prolong the prothrombin (as well as ACT and APTT), but a baseline should still be established. Warfarin therapy should be monitored by 4 to 5 days of therapy. Beginning monitoring earlier may help detect changes more easily. Monitoring techniques should be standardized. An optimum sampling time of 2 hours has been recommended.111 The target response to warfarin therapy varies. Recommendations have included an increase in baseline prothrombin of 1.3 to 1.6 times baseline to 1.5 to 2.5 times baseline.111
A standardized approach to interpreting prothrombin response has been recommended based on an international reference preparation of standardized human brain thromboplastin. The international normalized ratio (INR) is determined for the patient (INR = patient prothrombin/control PTISI), where ISI is the international sensitivity index of the thromboplastin control used to determine the prothrombin. The ISI is provided by the manufacturer of the thromboplastin control. Standardization is important because controls can vary by more than twofold. A target INR of 2 to 3 has been recommended for prevention of feline thromboembolism while minimizing the risk of bleeding. Either the dose or the interval can be manipulated to achieve the target INR (or prothrombin). Tablet size restrictions may, however, make it difficult to fine-tune warfarin therapy. The dosing interval might be prolonged to as long as 48 hours for some patients.102 Heparin therapy can be discontinued approximately 3 days after warfarin therapy is begun; prothrombin should be expected to decrease as a result. After the patient is sent home, monitoring can be decreased to 2-week intervals.
Warfarin therapy has been combined with aspirin to treat or prevent recurrent thromboembolism or increased risk of thromboembolism. Although the combination is rational, the risk of bleeding is intensified. Not only are both platelets and coagulation factors targeted with this combination, but drug interactions between these two highly protein-bound drugs can also complicate safe therapy.
Toxicity
Toxicity manifested as hemorrhage has been the major veterinary medical concern with vitamin K antagonists. Secondary poisoning resulting from the ingestion of a rodent that has eaten treated bait is the most common cause of toxicity. Toxicity can also occur, however, by overdosing warfarin for treatment of thromboembolism. Treatment for anticoagulant rodenticide toxicity is symptomatic and specific. Specific therapy is vitamin K. Vitamin K1 is a very effective antidote, but it must be given as long as the anticoagulant is present in the body at toxic levels. This time varies, depending on the drug, from several days to several weeks after ingestion. Vitamin K3 (menadione) is much less expensive but is also far less effective and never should be used as the sole antidote in cases of severe coagulopathy. Treatment with vitamin K will preclude the therapeutic benefits of warfarin for up to 3 weeks.
Fibrinolytics (Thrombolytics)
Fibrinolytic agents increase the activity of plasmin (fibrinolysin), the endogenous compound that is responsible for dissolving clots.112
Streptokinase and Streptodornase
Streptokinase and streptodornase are synthesized by streptococcal organisms. Urokinase is prepared from cultures of human renal cells. Streptokinase, streptodornase, and urokinase are used in the treatment of wounds that do not respond to antibacterial therapy, burns, ulcers, chronic eczemas, ear hematomas, otitis externa, osteomyelitis, chronic sinusitis, or other chronic lesions. They are available as powders for local or systemic administration. Streptokinase may be useful for the treatment of feline thromboembolic disease. In an experimental model, streptokinase reduced mean thrombus weight after administration of a loading dose (90,000 IU/cat) and a constant maintenance infusion of 45,000 IU (studied for only 3 hours). Clinical use of streptokinase has not been reported.70
Tissue Plasminogen Activator
Tissue-type plasminogen activator (tPA) activates plasminogen bound to fibrin. Its use should be limited to cases of thrombosis (rather than prevention). Some studies have indicated a potential use for tPA for dissolution of thrombi in cats, but side effects should be minimized before general use of the drug can be accepted. Although 50% of cats with spontaneous aortic thrombosis treated with tPA (0.25 to 1 mg/kg per hour for a total dose of 1 to 10 mg/kg) had resolution of clinical signs, 70% of the cats treated died from reperfusion injury, heart failure, or other side effects.70
Antithrombotics
Platelet activity is controlled by substances generated both outside and inside the platelet. Adenophosphate, prostaglandins, thromboxane, serotonin, cAMP, and GMP are generated within the platelet, where they interact with platelet receptors or with the platelet itself. Modulation of all platelet activity can be achieved by interaction with these compounds. Among platelet receptors are two P2YP type purinergic receptors that are activated by ADP and coupled with G-protein (Gq). Activation causes platelet shape to change, induces phosphatidylinositol turnover and platelet aggretation. This response is offset by a second receptor type coupled to Gi. Activation of this receptor by ADP inhibits adenylyl cyclase and decreases cAMP, resulting in muted platelet activation. Both receptors must be stimulated for platelets to activate.
The thienopyridines ticlopidine and clopidogrel are ADP antagonists with antiplatelet activity. Their inhibitory effects on P2Y12 G1-coupled adenosine receptors are irreversible, targeting both primary and secondary aggregation to a variety of agonists. Ticlopidine has been extensively studied in human patients with thromboembolic disease. Inhibition occurs in 2 to 5 days. The thienopyridines also inhibit the conformation change of glycoprotein IIb/IIa induced by ADP. Because binding of fibrinogen and vWF is prevented, thrombrus progression is inhibited. Because both ticlopidine and clopidrogel are prodrugs, pharmacodynamic tests must occur in vivo and pharmacokinetics must be based on the active metabolite. The enzyme responsible for metabolism has not yet been identified.54 Maximum effects are delayed up to 8 to 11 days; a loading dose (twice maintenance) is suggested in humans. Effects persist for several days after discontinuation of therapy; this may reflect an irreversible nature of inhibition and the need for new platelets. Serious side effects are rare in humans but include neutropenia and thrombocytopenia. The drug has proved equal to aspirin in the prevention of human stroke. Ticlopidine has been studied in dogs. A dose of 62 mg/kg every 24 hours inhibits platelet aggregation in normal dogs, but higher doses may be necessary in the presence of thromboembolic disease.3,113 It is not clear if the cat metabolizes ticlopidine to the active drug. However, ticlopidine also has been studied in cats (n = 8) at 50, 100, and 200 mg/cat once daily orally and 250 mg/cat every 12 hours for 10 days using (apparently) a nonrandomized crossover design with at least 14 days between treatments.114 Outcome measures included oral mucosal bleeding times and platelet aggregation (blood aggregometry). Significant reduction in platelet aggregation occurred with doses at 100 mg and above, although effects were present on all three test days (3, 7, and 10) only for the 250-mg twice-daily dose. Ticlopidine was associated with dose-dependent anorexia and vomiting, which occurred shortly after dosing, causing the authors to conclude that clinical utility of ticlopidine was limited. However, cats developed tolerance to these side effects. One of the eight cats was removed from the study because of sudden death, although the death could not be attributed to the drug.
Clopidogrel is related to ticlopidine and is less likely to cause neutropenia. Also a prodrug, its onset of action also is slow, with the loading dose in humans being 4 times the maintenance dose. Clopidogrel has been studied for its effect on thrombolysis, based on the observation that platelet activation may decrease thrombolysis or facilitate rethrombosis.115 Clopidogrel had no impact on thrombolysis in cats with experimentally induced (tPA) thrombosis after 3 days of oral therapy (75 mg), although only three cats were studied. However, a follow-up study in blood collected from cats (n = 9) found no effect.115 The pharmacokinetic-pharmacodynamic relationship of clopidregel was described in dogs (n = 8) receiving approximately 1 mg/kg PO every 24 hr for 3 days followed by either 0.5 or 1 mg/kg every 24 hr for 3 days (n = 5).115a Indices of platelet inhibition (e.g, ADP-induced platelet aggregation decreased by 80 to 93% from baseline) were present by day 3 in either treatment group, with the duration of effect lasting 3 to 7 days. Peak metabolite concentration 210 + 200 ng/ml occurred 1 hour after administration; the rapid disappearance and low (below detectable) concentrations of the metabolite precluded complete pharmacokinetic description. Experimentally, clopidogrel (5 mg/kg) proved more effective than aspirin (5 mg/kg) when combined with t-PA (80 μg/kg) or heparin (200 U/kg) for prevention of experimentally induced coronary artery occlusion.115b
Dipyridamole is a vasodilator that inhibits cAMP phosphodiesterase and increases cAMP in the platelet. When used alone, it minimally affects platelet activity but acts synergistically with aspirin to inhibit platelet activity.116
Aspirin causes an irreversible and thus long-lasting negative effect on platelet activity, which is clinically manifested as prolonged bleeding time. Aspirin irreversibly inactivates prostaglandin G and H synthetase, the enzyme that catalyzes the initial conversion of arachidonic acid to thromboxane A2. Two types of this enzyme are present: the constitutive form responsible for conversion of arachidonic acid to prostaglandin H (and ultimately thromboxane) and an inducible form that activates cells in response to growth factors during inflammation. The antiplatelet effects of aspirin result from acetylation of the constitutive form of the enzyme.117 Platelets are not able to synthesize proteins and thus cannot regenerate the enzyme necessary for thromboxane formation. The irreversible nature of platelet inhibition allows aspirin administration to occur at 3-day or longer intervals. Reduced formation of the various eicosanoids responsible for platelet aggregation and coagulation accounts for the variety of different pharmacologic (therapeutic and toxic) responses to aspirin. Side effects of aspirin can, however, be minimized by taking advantage of the dose–response relationship between aspirin and its many pharmacologic effects.
Prospective, controlled studies that support the efficacy of aspirin as an antithrombotic either as a preventive or treatment for FATE are limited. In a retrospective study of cats with acute FATE, survival did not differ between cats receiving 5 mg (n = 12) versus ≥ 40 mg/cat (n = 34) every 72 hours.118 An advantage of the lower dose might be inhibition of thromboxane synthetase (causing platelet aggregation) without inhibition of prostacyclin synthetase (inhibition of platelet aggregation). However, the power of the study to detect a treatment difference was not described, and failure to detect a significant difference should not be interpreted as equal efficacy between the two doses. On the other hand, the incidence of side effects was lower in the low-dose aspirin group.
One study reported that a very high dose of aspirin (650 mg/cat) in cats before initiation of thrombus formation improved collateral circulation compared with controls that did not receive aspirin,119 suggesting that aspirin prophylaxis might help reduce recurrence and that therapy should be initiated as soon as possible in the acute event.
The antiplatelet effect of aspirin can be separated to a large degree from its other actions by administration of a low dose (0.5 mg/kg every 12 hours in a normal dog). One retrospective study of the use of an ultralow dose of aspirin (0.5 mg/kg) versus (unfractionated) heparin (75-125 U/kg every 6 to 8 hours) in 151 dogs with immune-mediated hemolytic anemia being treated with prednisolone and azathioprine found the percentage of animals surviving the hospital stay to be 84% for the aspirin group versus approximately 54% for either the heparin-treated or no-antihemostatic-drug–treated group.120 Survival at 1 year was 67% for the aspirin-treated group versus 46% for the heparin-treated group and 34% for the no-antihemostatic-drug–treated group. The study was biased by the assignment of treatments: Coagulopathy was worse in the heparin group compared with the aspirin group. Nonetheless, the study supports the potential efficacy of aspirin at very low doses compared with no antihemostatic drug; its inclusion in the treatment regimen is made more prudent because of the potential for glucocorticoid therapy to worsen the risk of thromboemblism (see Chapter 30).
Other “low doses” suggested for aspirin include 10 mg/kg in heartworm-infested dogs and 25 mg/kg twice weekly in cats.72,117,121 Although experimental studies reveal antiplatelet activity for aspirin, clinical response is variable.122,123 Aspirin, however, remains a component of therapy directed toward prevention and treatment of arterial thrombosis,2,117,124 including severe pulmonary arterial thrombosis associated with heartworm disease.108 Coupled with cage confinement, aspirin (4 to 6 mg/kg per day) administered for 2 to 3 weeks before caparsolate therapy may improve survival after treatment.108 Aspirin is still the most commonly used drug for prevention of arterial thrombosis in cats (25 mg/kg orally every 72 hours) despite the lack of clinical proof of efficacy.70 Recurrence of thrombosis may, however, be as high as 75%.
Interestingly, one report in a canine model of coronary thrombosis describes a synergistic antiplatelet effect when administered with metoclopramide.125 Aspirin was administered at 0.03 to 0.1 mg/kg intravenously and metoclopramide at 0.1 or 0.3 mg/kg intravenously. The effect of metoclopramide was attributed to either s5HT2 or antagonism of alpha2.
Other drugs with antiplatelet activity are available but have not been sufficiently studied in animals. These include the tirofiban and abciximab, inhibitors of glycoprotein (IIb/IIIa), a platelet surface integrin that serves as a receptor for fibrinogen and vWF. For example, abciximab is the Fab fragment of a human monoclonal antibody directed toward the receptor used to treat coronary thromboembolism. Propranolol has been found to be ineffective as an inhibitor of platelet activity.71,126
Homocysteine
The recognition of arterial damage in two infants with inborn errors of metabolism resulting in an increase of homocysteine led to the investigation of the possible role of homocysteine in thromboembolic disease.127 However, homocysteine accumulation generally is accompanied by an increase of its precursor, S-adenosylhomocysteine (SAH). This demethylated product of numerous S-adenosylmethionine (SAM)-dependent transmethylation reactions may cause a potent feedback inhibition, resulting in hypomethylation of reactions critical to thrombogenic control. In homocystinuria, lowered plasma homocysteine is accompanied by lowered SAH concentrations and restored transmethylation reactions. It is not clear if increased homocysteine (or SAH) is a cause or effect (and thus a marker) of thromboembolic disorders. Because the kidneys are responsible for clearing up to 70% of homocysteine, concentrations increase in concert with creatinine in patients with renal disease, reaching increases of threefold to fivefold above baseline. Other factors that increase homocysteine include decreased folate and vitamin B concentrations and cardiovascular disease itself.
1. Compston J.E. Bone marrow and bone: a functional unit. J Endocrinol. 2002;173:387-394.
2. Kanshansky K., Kipps T.J. Hematopoietic agents: growth factors, minerals, and vitamins. In Brunton L., Lazo J.S., Parker K.L., editors: Goodman and Gilma’s the pharmacologic basis of therapeutics, ed 11, New York: McGraw-Hill, 2006.
3. Adams H.R. Drugs acting on blood and blood elements. In: Adams H.R., editor. Veterinary pharmacology and therapeutics. ed 7. Ames, Iowa: Iowa State University Press; 1995:531-543.
4. Ruaux C.G., Steiner J.M., Williams D.A. Early biochemical and clinical responses to cobalamin supplementation in cats with signs of gastrointestinal disease and severe hypocobalaminemia. J Vet Intern Med. 2005;19:155-160.
5. Weiss D.J. Leucocyte disorders and their treatment. In: Kirk R.W., editor. Current veterinary therapy XI. Philadelphia: Saunders; 1995:452-456.
6. Locatelli F., Aljama P., Barany P. Erythropoiesis-stimulating agents and antibody-mediated pure red-cell aplasia: where are we now and where do we go from here? Nephrol Dial Transplant. 2004;19:288-293.
7. Eschbach J.W., Adamson J.W. Recombinant human erythropoietin: implications for nephrology. Am J Kidney Dis. 1988;11:203-209.
8. Eschbach J.W., Adamson J.W. Guidelines for recombinant human erythropoietin therapy. Am J Kidney Dis. 1989;16(Supp l):2-8.
9. Faulds D., Sorkin E.M. Epoetin (recombinant human erythropoietin); a review of its pharmacodynamic and pharmacokinetic properties. Drugs. 1989;38(6):863-899.
10. Schwenk M.H., Halstenson C.E. Recombinant human erythropoietin. DICP. 1989;23:528-535.
11. Schulman A., Lusk R., Lippincott C.L., et al. Diphacinone-induced coagulopathy in the dog. J Am Vet Med Assoc. 1986;188:402-405.
12. Patton J., Reeves T., Wallace J. Effectiveness of darbepoetin alfa versus epoetin alfa in patients with chemotherapy-induced anemia treated in clinical practice. Oncologist. 2004;9(4):451-458.
13. Bennett C.L., Luminari S., Nissenson A.R. Pure red-cell aplasia and epoetin therapy. N Engl J Med. 2004;351(14):1403-1408.
14. MacLeod J., Tetreault J., et al. Expression and bioactivity of recombinant canine erythropoietin. Am J Vet Res. 1998;59(9):1144.
15. Randolph J.F., Scarlett J.M., Stokol T., et al. Expression, bioactivity and clinical assessment of recombinant feline erythropoietin. Am J Vet Res. 2004;65:1355-1366.
16. Casadevall N., Rossert J. Importance of biologic follow-ons: experience with EPO. Best Pract Res Clin Haematol. 2005;18(3):381-387.
17. Evens A.M., Bennett C.L., Luminari S. Epoetin-induced pure red-cell aplasia (PRCA): preliminary results from the research on adverse drug events and reports (RADAR) group. Best Pract Res Clin Haematol. 2005;18(3):481-489.
18. Schellekens H. Immunologic mechanisms of EPO-associated pure red cell aplasia. Best Pract Res Clin Haematol. 2005;18(3):473-480.
19. Carson K.R., Evens A.M., Bennett C.L., et al. Clinical characteristics of erythropoietin-associated pure red cell aplasia. Best Pract Res Clin Haematol. 2005;18(3):467-472.
20. Asari A., Gokal R. Pure red cell aplasia secondary to Epoetin alpha responding to darbepoetin alpha in a patient on peritoneal dialysis. J Am Soc Nephrol. 2004;15:2204-2207.
21. Cowgill L. Application of recombinant human erythropoietin (r-Hu-EPO) in dogs and cats. In: Kirk R.W., editor. Current Veterinary Therapy XI. Philadelphia: Saunders; 1992:484.
22. Cowgill L. CVT update: use of recombinant human erythropoietin. In: Bonagura J., editor. Current Veterinary Therapy XII. Philadelphia: Saunders; 1995:961-966.
23. Cowgill L.D., Polzin D.J., Osborne C.A., et al. Results of recombinant human erythropoietin clinical trial. Proc 12th ACVIM Forum. 1994;12:490-492.
24. Randolph J., Stokol T., Scarlett J.M., et al. Comparison of biological activity and safety of recombinant canine erythropoietin with that of recombinant human erythropoietin in clinically normal cogs. Am J Vet Res. 1999;60(5):636.
25. Randolph J.F., Scarlett J., Stokol T., et al. Clinical efficacy and safety of recombinant canine erythropoietin in dogs with anemia of chronic renal failure and dogs with recombinant human erythropoietin-induced red cell aplasia. J Vet Intern Med. 2004;18:81-91.
26. Cowgill L.D., James K.M., Levy J.K., et al. Use of recombinant human erythropoietin for management of anemia in dogs and cats with renal failure. J Am Vet Med Assoc. 1998;212(4):521-528.
27. Langston C.E., Reine N.J., Kittrell D. The use of erythropoietin. Vet Clin North Am Small Anim Pract. 2003;33(6):1245-1260.
28. Adamson J.W., Eschbach J.W. Treatment of the anemia of chronic renal failure with recombinant human erythropoietin. Proc 9th ACVIM Forum. 1991;9:603-610.
29. Cervelli M., Gray N., McDonald S., et al. Randomized cross-over comparison of intravenous and subcutaneous darbepoetin dosing efficiency in haemodialysis patients. Nephrology. 2005;10(2):129.
30. Glaspy J. Phase III clinical trials with darbepoetin: implications for clinicians. Best Pract Res Clin Haematol. 2005;18(3):407-416.
31. Besarab A., Bolton W.K., Browne J.K., et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998;339:584-590.
32. Eschbach J.W. Iron requirements in erythropoietin therapy. Best Pract Res Clin Haematol. 2005;18(2):347-361.
33. Wingard R.L., Parker R.A., Ismail N., et al. Efficacy of oral iron therapy in patients receiving recombinant human erythropoietin. Am J Kidney Dis. 1995;25:433-439.
34. Garton J.O., Gertz M.A., Witzig T.E., et al. Epoetin alfa for the treatment of anemia of multiple myeloma. Arch Intern Med. 1995;155:2069-2074.
35. Giampaoli S., Facciabene A., Mennuni C. A protocol to guide development of a sensitive ELISA for canine erythropoietin. Vet Clin Pathol. 2003;32(4):199-201.
36. Baz R, Brand C, McGowan B, et al: High dose recombinant human erythropoietin use is associated with increased overall survival in patients with multiple myeloma, 2005 American Society of Clinical Oncology Annual Meeting, abst 6621.
37. Bohlius J.F., Langensiepen S., Engert A. Effectiveness of erythropoietin in the treatment of patients with malignancies: methods and preliminary results of a Cochrane review. Best Pract Res Clin Haematol. 2005;18(3):449-454.
38. Djulbegovic B. Erythropoietin use in oncology: a summary of the evidence and practice guidelines comparing efforts of the Cochrane Review group and Blue Cross/Blue Shield to set up the ASCO/ASH guidelines. Best Pract Res Clin Haematol. 2005;18(3):455-466.
39. Lichtin A. The ASH/ASCO clinical guidelines on the use of erythropoietin. Best Pract Res Clin Haematol. 2005;18(3):433-438.
40. Aroch I., Harrus S. The use of recombinant human granulocyte colony stimulating factor and recombinant human erythropoietin in the treatment of severe pancytopenia due to canine monocyte ehrlichiolsis. Israel J Vet Med. 6, 2001.
41. Borwkoski J., Amrikachi M., Hudnall S.D. Fulminant parvovirus infection following erythropoietin treatment in a patient with acquired immunodeficiency syndrome. Arch Pathol Lab Med. 2001;124(3):441-445.
42. Duffield J.S., Mann S., Horn L., et al. Low-dose cyclosporin therapy for recombinant erythropoietin-induced pure red-cell aplasia. Nephrol Dial Transplant. 2004;19:479-481.
43. Tyrrell D.J., Kilfeather S., Page C.P. Therapeutic uses of heparin beyond its traditional role as an anticoagulant. Trends in Pharmaceutical Science. 1995;16:198-204.
44. Kruth S.A. Cytokines and biological response modifiers in small animal practice. In: Kirk R.W., editor. Current veterinary therapy XI. Philadelphia: Saunders; 1995:452-456.
45. Cohn L.A., Rewerts J.M., McCaw D., et al. Plasma granulocyte colony-stimulating factor concentrations in neutropenic, parvoviral enteritis-infected puppies. J Vet Intern Med. 1999;13:581-586.
46. Lothrop C.D., Warrren D.J., Souza L.M., et al. Correction of canine cycline hematopoiesis with recombinant human granulocyte colony stimulating factors. Blood. 1988;72:1324-1328.
47. Ferrara F., Annunziata M., Pollio F., et al. Vincristine as treatment for recurrent episodes of thrombotic thrombocytopenic purpura. Ann Hematol. 2002;81(1):7-10.
48. Rozanski E.A., Callan M.B., Hughes D. Comparison of platelet count recovery with use of vincristine and prednisone or prednisone alone for treatment for severe immune-mediated thrombocytopenia in dogs. J Am Vet Med Assoc. 2002;220(4):477-481.
49. Chamouni P., Lenain P., Buchonnet G., et al. Difficulties in the management of an incomplete form of refractory thrombotic thrombocytopenic purpura, the usefulness of vincristine. Transfus Sci. 2000;23:101-106.
50. Ziman A., Mitri M., Klapper E., et al. Combination vincristine and plasma exchange as initial therapy in patients with thrombotic thrombocytopenic purpura: one institution’s experience and review of the literature. Transfusion. 2005;45:41-49.
51. Dennis J.S. Anabolic steroids: their potential in small animals. Compend Contin Educ Pract Vet. 1990;12:1403-1419.
52. Bagatelle C.J., Brenner W.J. Androgens in men—uses and abuses. N Engl J Med. 1996;334:707-714.
53. Molinari P.F. Erythropoietic mechanism of androgens: a critical review and clinical implications. Haematology. 1982;67(3):442-460.
54. Majerus P.W., Tollefsen D.M. Blood coagulation and anticoagulant, thrombolytic, and antiplatelet drugs. In Brunton L., Lazo J.S., Parker K.L., editors: Goodman and Gilman’s the pharmacologic basis of therapeutics, ed 11, New York: McGraw-Hill, 2006.
55. Snyder P.J. Androgens. In Brunton L., Lazo J.S., Parker K.L., editors: Goodman and Gilman’s the pharmacologic basis of therapeutics, ed 11, New York: McGraw-Hill, 2006.
56. Williams T.M., Kind A.J., Hill D.W. Drug metabolism: in vitro biotransformation of anabolic steroids in canines. J Vet Pharmacol Ther. 2000;23(2):57-66.
57. Paulo L.G., Fink G.D., Roh B.L., et al. Effects of several androgens and steroid metabolites on erythropoietin production in the isolated perfused dog kidney. Blood. 1974;43:39-47.
58. Olson M.E., Morck D.W., Quinn K.B. The effect of stanozolol on 15nitrogen retention in the dog. Can J Vet Res. 2000;64(4):246-248.
59. Maravelias C., Dona A., Stefanidou M., et al. Adverse effects of anabolic steroids in athletes: a constant threat. Toxicol Lett. 2005;158(3):167-176.
60. Lowe K.C. Blood transfusion or blood substitution. Vox Sang. 1986;51:257-263.
61. Gould S.A., Sehgal L.R., Rosen A.L., et al. Red cell substitutes: an update. Ann Emerg Med. 1985;14:798-803.
62. Day T.K. Current development and use of hemoglobin-based oxygen-carrying (HBOC) solutions. J Vet Emerg Crit Care. 2003;13(2):77-93.
63. Kelly N., Rentko V.T. Comparative cardiovascular response by Oxyglobin versus packed red blood cells in acute anemia. J Vet Emerg Crit Care. 1998;8(3):252.
64. Rentko V.A., Wohl J., Murtaugh R., et al. Clinical trial of a hemoglobin based oxygen-carrying (HBOC) fluid in the treatment of anemia in dogs. J Vet Intern Med. 1996;10:177-178.
65. Driessen B., Jahr J.S., Lurie F., et al. Inadequacy of low-volume resuscitation with hemoglobin-based oxygen carrier hemoglobin glutamer-200 (bovine) in canine hypovolemia. J Vet Pharmacol Therap. 2001;24:61-71.
66. Gibson G.R., Callan M.B., Hoffman V., et al. Use of a hemoglobin-based oxygen-carrying solution in cats: 72 cases (1998–2002). J Am Vet Assoc. 2002;221(1):96-102.
67. Grundy S.A., Barton C. Influence of drug therapy on survival of dogs with immune-mediated hemolytic anemia: 88 cases (1989–1999). J Am Vet Med Assoc. 2001;218(4):543-548.
68. Weingart C., Kohn B. Clinical use of a haemoglobin-based oxygen carrying solution (Oxyglobin®) in cats (2002-2006). J Feline Med Surg. 2008;10:431-438.
69. McMichael M. Primary hemostasis. J Vet Emerg Crit Care. 2005;15:1-8.
70. Meurs K.M. Primary myocardial disease. In Ettinger S.J., Feldman E.C., editors: Textbook of Veterinary Internal Medicine, ed 6, St Louis: Saunders, 2005.
71. Greene R.A., Thomas J.S. Hemostatic disorders: coagulopathies and thrombosis. In: Ettinger S.J., Feldman E.C., editors. Textbook of veterinary internal medicine. ed 4. Philadelphia: Saunders; 1995:1946-1963.
72. Adams H.R., Carr A.P. Hemostatic and anticoagulant drugs. In: Adams H.R., editor. Veterinary pharmacology and therapeutics. ed 7. Ames, Iowa: Iowa State University Press; 1995:544-559.
73. McMichael M. Primary hemostasis. J Vet Emer Crit Care. 2005;15(2):1-8.
74. Johnstone I.B. Desmopressin enhances the binding of plasma von Willebrand factor to collagen in plasmas from normal dogs and dogs with type I von Willebrand’s disease. Can Vet J. 1999;40(9):645-648.
75. Kraus K.H., Turrentine M.A., Jergens A.E. Effect of desmopressin acetate on bleeding times and plasma von Willebrand factor in Doberman pinscher dogs with von Willebrand’s disease. Vet Surg. 1989;18(2):103-109.
76. Sato I., Parry B.W. Effect of desmopressin on plasma factor VIII and von Willebrand factor concentrations in Greyhounds. Aust Vet J. 1998;76:809-812.
77. Kristensen A.T., Edwards M.L., Devey J. Potential uses of recombinant human factor VIIa in veterinary medicine. Vet Clin North Am Small Anim Pract. 2003;33(6):1437-1451.
78. Brockway W.J., Castellino F.J. The mechanism of the inhibition of plasmin activity by ε-aminocaproic acid. J Biol Chem. 1971;246(14):4641-4647.
79. Takeda Y., Parkhill T.R., Nakabayashi M. Effects of heparin and ε-aminocaproic acid in dogs on plasmin-125 I generation in response to urokinase injections and venous injury. J Clin Invest. 1972;51:2678-2685.
80. Hirsh J., Warkentin T.E., et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest. 2001;119(Suppl):64S-94S.
81. Freedman M.D. Pharmacodynamics, clinical indications, and adverse effects of heparin. J Clin Pharmacol. 1992;32:584-596.
82. Weitz J.I. Low-molecular-weight heparins. N Engl J Med. 1997;337(10):688-698.
83. Folkman J., Shing S. Control of angiogenesis by heparin and other sulfated polysaccharides. Adv Exp Med Biol. 1992;313:355-364.
84. Burden T.S., Buhler F.R. Regulation of smooth muscle proliferative phenotype by heparinoid-matrix interactions. Trends in Pharmaceutical Science. 1988;9:94-98.
85. Kellerman D.L., Lewis D.C., Myers N.C., et al. Determination of a therapeutic heparin dosage in the cat. Proc Am Coll Vet Intern Med. 1996;14:748.
86. Brieger D., Dawes J. Production method affects the pharmacokinetic and ex vivo biological properties of low molecular weight heparins. Thromb Haemost. 1997;77(2):317-322.
87. Alwood A.J., Downend A.B., Brooks M.B., et al. Anticoagulant effects of low-molecular-weight heparins in healthy cats. J Vet Intern Med. 2007;21:378-387.
88. Mischke R., Grebe S. The correlation between plasma anti-factor Xa activity and haemostatic tests in healthy dogs, following the administration of a low molecular weight heparin. Res Vet Sci.. 2000;69(3):241-247.
88a. Vargo C.L., Taylor S.M., Carr A., et al. The effect of a low molecular weight heparin on coagulation parameters in healthy cats. Can J Vet Res. 2009;73(2):132-136.
89. Grebe S., Jacobs C., Kietzmann M., et al. Phamacokinetics of low-molecular-weight heparins Fragmin D in dogs. Berl Munch Tierarztl Wochenschr. 2000;113(3):103-107.
91. Grodman-Gross C.A., Sastri S.V. Heparin-associated hematomas: possible allergic reaction. DICP Ann Pharmacother. 1987;21:180-183.
92. McDonald S.B., Renna M., Spitznagel E.L., et al. Preoperative use of enoxaparin increases the risk of postoperative bleeding and re-exploration in cardiac surgery patients. J Cardiothorac Vasc Anesth. 2005;19(1):4-10.
93. Greinacher A., Michels I., Schäfer M., et al. Heparin-associated thrombocytopenia in a patient treated with polysulphated chondroitin sulphate: evidence for immunological crossreactivity between heparin and polysulphated glycosaminoglycan. Br J Haematol. 1992;81:252-254.
94. Shumate M.J. Heparin-induced thrombocytopenia. N Engl J Med. 1995;333:1006.
95. Aull L., Chao H., Coy K. Heparin-induced hyperkalemia. DICP. 1990;24:244-246.
96. Oster J.R., Singer I., Fishman L.M. Heparin-induced aldosterone suppression and hyperkalemia. Am J Med. 1995;98:575-586.
97. Susic D., Mandal A.K., Jovovic D., et al. Antihypertensive action of heparin: role of the renin-angiotensin aldosterone system and prostaglandins. J Clin Pharmacol. 1993;33:342-347.
98. Kishimoto T.K., Viswanathan K., Ganguly T., et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N Engl J Med. 2008;358(23):2457-2467.
99. Niemi T.T., Munsterhjelm E., Pöyhiä R., et al. The effect of N-acetylcysteine on blood coagulation and platelet function in patients undergoing open repair of abdominal aortic aneurysm. Blood Coagul Fibrinolysis. 2006;17(1):29-34.
100. Mungall D., Raskob G., Coleman R., et al. Pharmacokinetics and dynamics of heparin in patients with proximal vein thrombosis. J Clin Pharmacol. 1989;29:896-900.
101. Gunnarsson P.S., Sawyer W.T., Montague D., et al. Appropriate use of heparin. Arch Intern Med. 1995;155:526-532.
102. Hawkins E.C. Diseases of the lower respiratory system. In: Ettinger S.J., Feldman E.C., editors. Textbook of veterinary internal medicine. ed 4. Philadelphia: Saunders; 1995:767-811.
103. Smith S.A., Tobias A.H. Feline arterial thromboembolism: an update. Vet Clin Small Anim. 2004;34:1245-1271.
104. Lauer M.A., Houghtaling P.L., Peterson J.G. Attenuation of rebound ischemia after discontinuation of heparin therapy by glycoprotein IIb/IIIa inhibition with eptifibatide in patients with acute coronary syndromes observations from the platelet IIb/IIIa in unstable angina: receptor suppression using integrilin therapy (PURSUIT) trial. Circulation. 2001;104:2772.
105. Frydman A.M., Bara L., Woler M., et al. The antithrombotic activity and pharmacokinetics of enoxaparin, a low molecular weight heparin, in humans given single subcutaneous doses of 20 to 80 mg. J Clin Pharmacol. 1988;28:609-618.
106. DeFrancesco T.C., Moore R.R., Atkins C.L., et al. Comparison of dalteparin and warfarin in the longterm management of feline arterial thromboembolism. Proc ACVIM: #281. 2005.
107. Van der Heijden J.F., Hutten B.A., Büller H.R., et al. Vitamin K antagonists or low-molecular-weight heparin for the long term treatment of symptomatic venous thromboembolism. Cochrane Database Syst Rev. 2001. Issue 3, Art. No.: CD002001. doi:10.1002/14651856.CD002001, 2008
108. Rawlings C.A., Calvert C.A. Heartworm disease. In: Ettinger S.J., Feldman E.C., editors. Textbook of veterinary internal medicine. ed 4. Philadelphia: Saunders; 1995:1046-1068.
109. Estrin M.A., Wehausen C.E., Jessen C.R., et al. Disseminated intravascular coagulation in cats. J Vet Intern Med. 2006;20:1334-1339.
110. Smith C.E., Rozanski E.A., Freeman L.M., et al. Use of low molecular weight heparin in cats: 57 cases (1999-2003). J Am Vet Med Assoc. 2005;225(8):1237-1241.
111. Harpster N.K., Baty C. Warfarin therapy of the cat at risk of thromboembolism. In: Bongura J., editor. Current veterinary therapy XII. Philadelphia: Saunders; 1995:868-873.
112. Witty L.A., Krichman A., Tapson V.F. Thrombolytic therapy for venous thromboembolism: utilization by practicing pulmonologists. Arch Intern Med. 1994;154:1601-1604.
113. Boudreaux M.K., Dillin A.R., Ravis W.R. Effects of treatment with aspirin or aspirin/dipyrimadole combination in heartworm negative, heartworm infected and embolized heartworm-infected dogs. Am J Vet Res. 1991;52:1992-1999.
114. Hogan D.F., Ward M.P. Effect of clopidogrel on tissue-plasminogen activator-induced in vitro thrombolysis of feline whole blood thrombi. Am J Vet Res. 2004;65:715-719.
115. Hogan D.F., Andrews D.A., Talbott K.K., et al. Evaluation of antiplatelet effects of ticlopidine in cats. Am J Vet Res. 2004;65:327-332.
115a. Brainard B.M., Kleine S.A., Papich M.G., et al. Pharmacodynamic and pharmacokinetic evaluation of clopidogrel and the carboxylic acid metabolite SR 26334 in healthy dogs. Am J Vet Res. 2010;71(7):822-830.
115b. Yao S.K., Ober J.C., Ferguson J.J., et al. Clopidogrel is more effective than aspirin in preventing coronary artery reocclusion after thrombolysis. Trans Assoc Am Physicians. 1993;106:110-119.
116. Boudreaux M.K., Dillin A.R., Sartin E.A., et al. Effects of treatment with ticlopidine in heartworm negative, heartworm infected and embolized heartworm-infected dogs. Am J Vet Res. 1991;52:2000-2006.
117. Patrono C. Aspirin as an antiplatelet drug. N Engl J Med. 1994;330:1287-1294.
118. Smith S.A., Tobias A.H., Jacob K.A., et al. Arterial thromboembolism in cats: acute crisis in 127 cases (1992–2001) and long-term management with low-dose aspirin in 24 cases. J Vet Intern Med. 2003;17:73-83.
119. Schaub R.G., Gates K.A., Roberts R.E. Effect of aspirin on collateral blood flow after experimental thrombosis of the feline aorta. Am J Vet Res. 1982;43(9):1647-1650.
120. Weinkle T.K., Center S.A., Randolph J.F., et al. Azathioprine and ultra-low-dose aspirin therapy for canine immune-mediated hemolytic anemia. Proc ACVIM. 2004.
121. Jackson M.L. Platelet physiology and platelet function: inhibition by aspirin. Compend Contin Educ Pract Vet. 1987;9:627-638.
122. Rawlings C.A., Keith J.C., Lewis R.E., et al. Aspirin and prednisolone modification of radiographic changes caused by adulticide treatment in dogs with heartworm infection. J Am Vet Med Assoc. 1983;183:131-132.
123. Rawlings C.A., Keith J.C., Losonsky J.M., et al. Aspirin and prednisolone modification of postadulticide pulmonary arterial disease in heartworm-infected dogs: arteriographic study. J Am Vet Med Assoc. 1983;44:821-827.
124. Chastain C.B. Aspirin: new indications for an old drug. Compend Contin Educ Pract Vet. 1987;9:165-170.
125. Duval N., Grosset A., O’Connor S.E. Combination of aspirin and metoclopramide produces a synergistic antithrombotic effect in a canine model of coronary artery thrombosis. Fundam Clin Pharmacol. 1997;11:57-62.
126. Allen D.G., Johnstone I.B., Crane S. Effects of aspirin and propranolol alone and in combination on hemostatic determinants in the healthy cat. Am J Vet Res. 1985;46:660-663.
127. Brattström L., Wilcken D. Homocysteine and cardiovascular disease: cause or effect? Am J Clin Nutr. 2000;72:315-323.