Chapter 30 Glucocorticoids and Mineralocorticoids
Glucocorticoids (GLCs) are among the most frequently used and misused drugs in veterinary medicine. Optimal therapy with GLCs requires a thorough understanding of their actions on all body systems and knowledge of the pharmacodynamic and pharmacokinetic differences of the synthetic GLC derivatives. The physiologic and pharmacologic effects of GLCs, beyond suppression of the hypothalamic–pituitary–adrenal axis, have not been extensively studied in the dog or cat; therefore much of the information presented represents data extrapolated from human patients or rodent studies. Whenever possible, information specific to the dog or cat has been included and is indicated as such.
Physiology: Control of Endogenous Glucocorticoid Secretion
The adrenal cortex comprises three zones, each of which synthesizes steroidal hormones. From superficial to deep, these include the zonas glomerulosa, the source of mineralocorticoids (predominantly aldosterone but also corticosterone); reticularis, a source of (weak) androgens; and fasciculata, the source of GLCs (cortisol and cortisone). As with many hormones, the secretion of corticosteroids reflects a balance between positive and negative feedback pathways. Corticotropin-releasing hormone (previously corticotropin release factor, CRH) is secreted by the hypothalamus and travels through the hypophyseal portal system to the adenohypophysis, where it stimulates the synthesis and secretion of adrenocorticotropin (ACTH) from the basophilic cells of the adenohypophysis (Figure 30-1). In addition to its role in promoting ACTH secretion, CRF appears to be involved in the autonomic, immunologic, and behavioral response to stress independent of the hypothalamopituitary axis. Administration of CRF to dogs results in a decrease in gastric acid secretion by activation of the sympathetic nervous system, an immediate decrease in mean arterial blood pressure accompanied by reflex tachycardia, and a marked increase in plasma vasopressin concentrations.1 These actions of CRF are independent of GLCs.
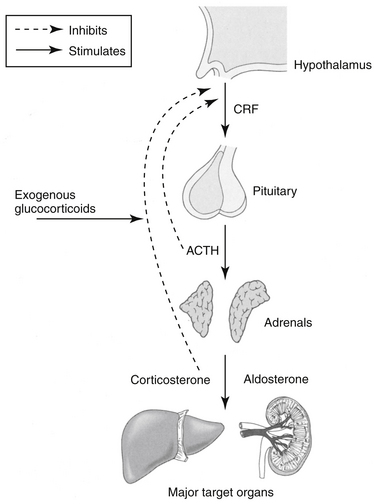
Figure 30-1 The relationship between the hypothalamus, pituitary, and adrenal glands. Release of corticotropin-releasing factor (CRF) is prevented by feedback inhibition of corticosteroids (exogenous or endogenous). Release of adrenocorticotropic hormone (ACTH) is subsequently inhibited, and further synthesis, primarily of glucocorticoids, is inhibited. The primary target organ of mineralocorticoids is the kidney; the liver and cardiovascular system are important targets of glucocorticoids.
In addition to CRF, ACTH also is stimulated by arginine vasopressor (AVP), which is a weak stimulator of ACTH but a strong stimulator of CRH and catecholamines; angiotensin II; serotonin; and vasoactive intestinal peptide. Other stimulators include the inflammatory cytokines interleukin (IL) -1, -2, and -6 and tumor necrosis factor-α(TNF-α).2 Most stimulators of ACTH also stimulate CRH.2 The primary short-term effects of ACTH are stimulation of the adrenal cortex synthesis and secretion of cortisol, corticosterone, aldosterone (the effect of ACTH on mineralocorticoid secretion is minimal), and weak androgenic substances. Long-term effects of ACTH increase the production of enzymes and cofactors necessary for cortisol production and cause an increase in adrenal receptors for low-density lipoprotein cholesterol.2
Cortisol and corticosterone concentrations in plasma subsequently influence CRF and ACTH secretion such that increased concentrations inhibit release of CRF and ACTH and reduced concentrations stimulate release of CRF and ACTH. Exogenous factors, such as trauma, heat, stress, surgery, and neural impulses, also mediate CRF and ACTH secretion. Exogenous corticosteroid administration can also suppress CRF and ACTH release. The degree of suppression depends on the particular drug used. For example, the synthetic drug dexamethasone is 50 to 100 times more potent in suppressing ACTH secretion than is the endogenous compound, cortisol.3 The diurnal variation in GLC secretion that occurs in humans has not been well documented in dogs or cats. Nonetheless, morning dosing (in dogs) is among the strategies used to minimize the risk of adrenocortical suppression that might result from exogenous glucocorticoid therapy. These approaches thus include: 1. Determining a minimum effective dose; 2. Alternate day dosing; 3. Use of a “short-acting” glucocorticoids whose effects are 24 hrs or shorter in duration; 4. Dosing in the morning (and potentially dosing at nights for cats); and 5. Tapering the dose over days to weeks as therapy is discontinued, thus facilitating readaptation of the adrenal gland to secretion.
Mechanism of Action
The myriad physiologic effects of GLCs result from interaction of the drugs with the glucocorticoid receptor (GR), one member of a nuclear hormone receptor superfamily that also includes receptors for thyroid hormone, mineralocorticoid, estrogen, and progesterone.4 Other activities may also reflect nonreceptor mechanisms.
Mineralocorticoid and Glucocorticoid Receptors
Two primary types of corticosteroid receptors exist. Type I, or mineralocorticoid receptors (MRs), bind both endogenous GLCs and aldosterone. Type II, or GRs, bind endogenous and exogenous GLCs but have a poor affinity for mineralocorticoids. The GRs and MRs are sufficiently similar that drugs may bind to both, causing similar responses. In human medicine, the cause and effect relationship between hypertension, heart failure, and MRs has led to a reassessment of the relationship between GLC and mineralocorticoid as well as androgen and progesterone receptors.5 The distinction among the receptors, including the sequence of their evolution, becomes important when assessing the selectivity or lack thereof of therapeutic agents on each receptor type. In human medicine this is particularly germane to the impact of GLCs on the MRs and its implication in patients with cardiovascular disease.
Every cell appears to have GRs, although the liver is the primary target. The type and concentration of GRs varies between species and tissue. Type II receptors are more ubiquitous in the brain. The degree to which GLCs bind to each receptor type varies with the circulating concentration. At basal levels type I receptors are preferred, but as cortisol concentrations increase (e.g., during stress), type II receptors increasingly are activated.6
Within a given tissue, GR numbers appear to fluctuate with changing cell cycles and age and in response to a variety of endogenous or exogenous compounds. More than 15 endogenous regulators have been identified for GLC receptors.7 Response to GLCs reflects receptor density or GLC concentration, depending on the tissue. GR density is autoregulated; increased receptor density associated with hypoadrenocorticism can be reversed with GLC replacement.8 Likewise, chronic GLC therapy will result in downregulation of receptor density. However, autoregulation is tissue specific, and in some tissues the concentration of GLC is a more important determinant of response than is the number – or even type - of receptors. For example, as noted, the MR acts similarly to the GR and is sufficiently similar in structure that responses to substrates are also likely to be similar.8,9 Any steroid with a ketone group at position 11 (Figure 30-2) will interact positively with either the MR or GR.2 However, GLCs generally are present in concentrations that exceed those of mineralocorticoids by 1000-fold or more,2 which suggests the potential for GLC response to predominate in any tissue exhibiting either GRs or MRs. The ability of receptors to respond to mineralocorticoids despite differences in concentration reflects a combination of conditions: (1) binding of glucorticoids by globulins, which effectively sequesters the hormone, precluding interaction with GRs or MRs; (2) activation of discrete sets of target genes, perhaps reflecting a difference in repression of transcriptional induction (e.g., aldosterone activates the expression of several genes, with that of Na+, K+-ATPase in the basolateral membranes of tubular cells being the best characterized); (3) restrictive expression of MRs only, with the principle sites occurring in the renal cortical distal tubules and collecting ducts, colon, salivary and sweat glands, and hippocampus8; (4) differing rates of GLC inactivation, which allows mineralocorticoids to predominate and stimulate response. For example, tissues differ in their expression of 11 β-hydroxysteroid dehydrogenase type 2 (11 β-HSD2), the enzyme that inactivates cortisol to corticosterone. Tissues with high concentrations of 11 beta-HSD2are those that typically respond to mineralocorticoids—kidney, colon, salivary gland—allowing mineralocorticoid effects to predominate.8,10 In the presence of 11 β-HSD2, GLC expression occurs only if present in concentrations several magnitudes higher than mineralocorticoids.
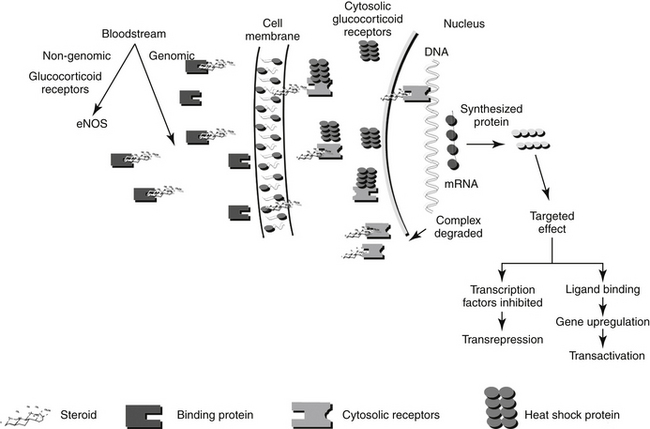
Figure 30-2 The intracellular mechanisms of a glucocorticoid include release from its binding protein in the bloodstream, passive diffusion of the drug through the cell membrane, movement through the cytosol in association with a receptor and chaperone heat shock protein, translocation to the nucleus, binding to DNA, transcription, either stimulation or inhibition of protein synthesis, and degradation of the protein–steroid complex.
KEY POINT 30-1
Similarities in the mineralocorticoid and glucocorticoid receptors and steroid structures contribute to the shared action of drugs.
Certain disease conditions may reflect aberrations in methods allowing differential substrate response such as an absence of 11 β-HSD2 such that tissues respond to the inappropriate steroid.9 The importance of renal metabolism of cortisol is implied in the impact renal failure has on prolongation of plasma cortisol half-life in human patients; half-life increased 30% in one study.10 Impaired 11 β-HSD2 accompanying renal disease contributes to the pathophysiology of salt and water retention and hypertension in renal failure and therefore could contribute to selected consequences of renal disease (hypertension, sodium retention, hyperkalemia, and decreased glomerular filtration rate). Inhibition of 11β HSD2by selected compounds (licorice, selected angiotensin-converting enzyme inhibitors [ramipril]) contributes to pharmacologic or adverse events as well as drug interactions. Ramipril causes decreased ACTH-induced hypertension in the rat.10 Inhibition allows GLCs to excessively stimulate MRs such that hyperaldosteronism emerges. Such interactions also can be favorable:
Nuclear and Non-nuclear Mechanisms of Action of Corticosteroids
The molecular description of the GR as it relates to its mechanism of interaction has been described.11-14 The GR consists of at least two isoforms.11 The α isoform binds to GLCs, DNA, and transcription factors and thus modulates transcription activity. The α isoform may also act through nongenomic mechanisms. In contrast, although the β isoform binds to DNA, it does not bind other ligands, fails to activate transcription, and appears to be able to interfere with the activities of α isoforms.12 Corticosteroid receptors are located in the cytoplasm of the target cell complexed with heat shock proteins (Hsp 70 and Hsp 90) that act as chaperones, proteins which in non-functional activities (e.g., folding, assembly) of macromolecules, and an immunophilin, an intracellular protein that binds other immunosuppressive compounds.9,11 The complexed receptor is inactive until bound to a steroid ligand.9 Steroids enter the cells by passive diffusion, although a rate-limited active transport mechanism may also exist (Figure 30-2) Once in the cell, the GLC binds to the receptor, causing the heat shock protein and other molecules to dissociate. Intracellular GLCs impart their effects in one of three ways: The steroid–receptor complex moves into the nucleus and (1) binds to specific DNA sequences causing the regulation of genes (for example, lipocortin or genes responsible for gluconeogenesis) (referred to as a “cis” or element, resulting in transactivation), (2) interacts with other transcription factors (e.g., AP-1, NFκB), generally at lower concentrations, that prevent the transcription of targeted genes (e.g., TNFα, IL-1B, IL-4 and 5; referred to as a “trans element” resulting in transrepression); or (3) through non-genomic mechanisms involving membrane-associated GR and secondary messenger systems.2,12a Non-genomic responses are rapid in onset, and includes vascular responses (eNOS activation), lymphocytolysis, and possibly, induction of inflammatory prostaglandins. “Permissive” effects of GLC may reflect non-genomic responses.
Because the effect of older, current and future GLC reflects the molecular relationship between drug and cellular macromolecules, a brief review or molecular interactions is warranted. Genomic effects of GLCs are initiated by translocation of the activated receptor–GLC complex to the nucleus (Figure 30-2). In the nucleus the complex binds to regulatory proteins of target genes. The short DNA sequences recognized by the activated GR are referred to as glucocorticoid responsive elements (GREs). It is through these GREs that specificity of GLC-modulated gene transcription is controlled.4 Transcription of the gene and subsequent formation of the targeted protein is either induced (transactivated) or inhibited (transrepressed). The receptor and GLC are eventually metabolized (the exact location or timing is not documented). The cellular half-life of the activated complex is about 10 hours.15 It is not known whether the rate of metabolism of the GLC–receptor complex is dependent on the specific GLC involved. Some of the proteins that are regulated by GLCs are listed in Table 30-1. The proteins encoded by these genes are responsible for some of the physiologic (pharmacologic) effects of GLCs.
Table 30-1 Immunomodulatory Actions of Glucocorticoids
| Actions | Mechanism |
|---|---|
| Interference with cytokine production | 1 |
| Interleukin-1 | |
| Interleukin-2 | |
| Interleukin-6 | |
| Interleukin-8 | |
| Tumor necrosis factor-alpha | |
| Interferon-gamma | |
| Downregulation of cell adhesion molecules | 1 |
| Intercellular Adhesion Molecule 1 (ICAM-1) | |
| E-selectin | |
| Inhibition of chemokine synthesis | |
| Interference with leukocyte function enzymes | 1 |
| Inducible nitric oxide synthase | |
| Granzyme B | |
| Upregulation of cytokine receptors | |
| Interleukin-1 | |
| Interleukin-6 | |
| Induction of lymphocyte apotosis | |
| CD4+CD8+ | |
| Inhibition of cyclooxygenase-2 expression | |
| Inhibition of wound healing | 2 |
| Epithelial cells | |
| Fibroblasts | |
| Induction of acute phase proteins (permissive effect) | 3 |
| Serum amyloid A | |
| Serum amyloid P | |
| AP-1 | |
| NFk-B | |
| Stat proteins |
1 = protein-protein interaction with transcription factors
KEY POINT 30-2
As with all steroids, corticosteroids cause their primary effects by inducing or inhibiting the synthesis of target proteins at the level of the gene.
Once activated, the activated ligand receptor influences gene expression by binding to specific GREs in the promoter regions of GLC-regulated genes. However, in addition to this classical transactivation mechanism, also referred to as positive GRE (pGRE), genes may also be negatively targeted, or transrepressed, through negative GRE (nGRE). The GR generally has a lower affinity for nGRE than pGRE. Transrepression involves protein–protein coupling to other transcription factors (e.g., nuclear factor kappa-B [NFκB], activator protein-1 [AP-1], STAT-5 (signal transducer and activator of transcription) or nuclear factor of activated T-cells [NFAT]) such that their activity is modulated. The importance of these different GR interactions results in differential physiologic (and pharmacologic) effects and ultimately provides a basis for directed drug therapy. The largely undesirable metabolic effects of long-term GLC therapy appear to be mediated by pGRE interaction (i.e., transactivation gene expression). In contrast, the desirable antiinflammatory effects appear to reflect modulation of transcription factors (i.e., transrepression through interaction with nGREs).16,17 Indeed, nGREs contribute to the regulation of the hypothalamic–pituitary–adrenal axis (targeting pro-opiomelanocortin [POMC] and CRH), bone metabolism (through osteocalcin), skin function, inflammation (IL-1β), angiogenesis (proliferin), and lactation (prolactin).17 Trans (protein-coupling) effects, and repression in particular, appear to occur at concentrations lower than that necessary for cis (transactivation) effects,18 providing another mechanism of directed therapy. “Designer” GLC might be developed such that potency for protein coupling (trans) repression is greater than DNA interaction, leading to cis or trans activation. Dexamethasone and prednisolone are examples of a “symmetric” GLC, characterized by equal binding affinity for both actions. Interestingly, medroxyprogesterone acetate exhibits a preponderance of transrepression activity rather than transactivation.16 Newer GLCs are likely to have a preponderance of activity for either activation or repression. The recognition of this dual mechanism of GLC action will also facilitate the development of GR antagonists, which might be useful for the treatment of Cushing’s disease.16 Another term that has emerged is “Dissociated GLCs” which target GLC-dependent transactivation or alternatively act as inhibitors of NFκB-dependent transcription, thus retaining their in vivo antiinflammatory activity but avoiding their undesirable metabolic effects.19
KEY POINT 30-3
The desired inhibitory effects of glucocorticoids reflect indirect repression of transcription factors, whereas undesirable effects reflect direct interaction with DNA.
Differences in GR affinity for nGREs versus pGREs may also offer a mechanism for differential targeted drug response based on dose (or concentration). Treatment with low, rather than high, doses of GLCs appears to minimize undesirable side effects.20 Using a retrospective analysis of published literature (meta analysis precluded by method variability) coupled with a prospective study, researchers found that most adverse effects to GLCs in humans were associated with high, rather than low, doses (7.5 mg prednisolone equivalent in humans [approximately 0.1 mg/kg]) of GLC (see the section on adverse events).
Glucocorticoid Resistance
Failure to respond to steroid therapy is observed in human patients being treated for a variety of diseases. Poor compliance and low bioavailability must be ruled out before other factors are considered. The documented phenomenon of GLC resistance (both familial and iatrogenic or drug induced forms have been reported in humans) may reflect, among other causes, reduced receptor numbers or affinity for GLC.21 Cellular response to GLCs has been directly correlated with receptor numbers. Reversible downregulation of receptor numbers and a subsequent decrease in biological effect are documented sequelae of GLC treatment.7 Such effects have been demonstrated on lymphocyte receptors.22 GLC failure in patients receiving long-term GLC therapy for treatment of host-versus-graft rejection has been attributed to downregulation of cytoplasmic GRs in T lymphocytes. GLC doses subsequently must be increased.23 Pulsing of high doses of GLCs may overcome this relative resistance (see the section on therapeutic use).24 The relative balance of the α and β isoforms of GR appear to influence cell sensitivity to GLCs; higher concentrations of β contribute to GLC resistance.12 Treatment of inflammatory bowel disease offers an example of potential mechanisms of GLC resistance. Some human patients with severe disease are nonresponsive to even high doses of GLCs. Lack of response has been associated with poor antiproliferative effects on blood T lymphocytes, in contrast to near-complete inhibition of responders. A similar relationship has been demonstrated for other chronic allergy-based diseases, such as asthma or rheumatoid arthritis, and renal allograft rejection. A relatively fast in vitro T lymphocyte proliferation assay has been useful in identifying nonresponders before treatment.25 Several molecular mechanisms have been proposed to account for poor control of T cell proliferation by GLCs. These include overexpression of the multidrug resistance gene (MDR1 polymorphism), resulting in increased P-glycoprotein–mediated efflux of cytoplasmic (see relevant discussion in the section on neoplasia). This mechanism might be reversible by co-administration of cyclosporine, an inhibitor of P-glycoprotein. Other mechanisms include impaired GLC signaling by the GR and increased activation by epithelial cells of proinflammatory mediators that directly inhibit GR transcription. This latter mechanism (postreceptor failure) is supported by the presence of clinical signs consistent with Cushing’s disease in humans with poor inflammatory control.25 Differences in activation patterns of NFκB have been associated with GLC resistance in human patients with chronic inflammatory bowel disease.19 Ultimately, differences in response at the molecular level may also be shown .
Physiologic Effects
Physiologic effects of GLCs required for the “normal” day-to-day function of the animal can be easily appreciated when GLCs are absent, as in the case of adrenocortical insufficiency (hypoadrenocorticism or Addison’s disease), or present in excessive concentrations, as in hyperadrenocorticism. The primary role of aldosterone, the major mineralocorticoid, is regulation of sodium homeostasis. Interestingly, the effects of GLCs and mineralocorticoids can be directly antagonistic at some sites. For example, in some tissues aldosterone stimulates and GLCs inhibit sodium resorption.8 Likewise, cortisone can have different and opposing effects, depending on whether it interacts with MRs or GRs. Cortisone inhibits microglial proliferation through GRs but stimulates proliferation through MRs.26 Dexamethasone also inhibits microglial cell proliferation at concentrations lower than those for cortisone.26
Effects on Intermediary Metabolism: Carbohydrates, Proteins, and Lipids
The natural function of GLCs is to protect glucose-dependent cerebral functions by stimulating the formation of glucose by the liver, decreasing its peripheral utilization and promoting its storage as glycogen.27 Teleologically, these effects protect glucose-dependent tissues, the brain and heart, from starvation.9 The hyperglycemic effect of GLCs, as seen in the stressed patient, is due to an increase in gluconeogenesis and insulin antagonism. Gluconeogenesis is the result of an increase in precursors necessary for gluconeogenesis as well as induction of hepatic enzymes that catalyze reactions of glucose synthesis. Increased breakdown of proteins, particularly skeletal muscle and collagen, provides gluconeogenic precursors (e.g., amino acids and glycerol). This effect is exhibited clinically as muscle wasting, delayed wound healing, and thinning of the skin. The anti-insulin effect of GLCs is a result of decreased peripheral tissue utilization of glucose and reduced affinity of cellular receptors for insulin. Utilization appears to be decreased by translocation of insulin receptors from the cell membrane to an intracellular location inaccessible by insulin.9
KEY POINT 30-5
The physiologic effects of glucocorticoids reflect their underlying physiologic intent: maintenance of the body, particularly during states of stress.
Metabolism of lipids is also affected by GLCs. Specifically, GLCs promote lipolysis, generating the free fatty acids that, along with amino acids, serve as substrates for hepatic glycogen synthesis and inhibit long-chain fatty acid synthesis. Effects of GLCs on lipid metabolism reflect, in part, a permissive effect of the steroids on other agents, including growth hormone and β-adrenergic receptors. One sequela of these effects is redistribution of body fat (such as is typified by Cushing’s disease). Differences in adipocyte sensitivity to insulin and the facilitating effects of GLCs may explain the redistribution phenomenon.9
Water and Electrolyte Balance
Cortisol appears to be essential for the maintenance of renal blood flow and glomerular filtration rate. Volume repletion alone is not sufficient to return renal function to normal in states of hypoaldosteronism. GLCs also generally increase the filtration fraction; impact on renal vascular resistance is not clear, varying with the method studied.10 Aldosterone is the most potent natural corticosteroid that affects fluid and electrolyte balance. Mineralocorticoids act to enhance sodium reabsorption in exchange for potassium (from the distal renal tubules and collecting ducts) or hydrogen (the intercalated cells), resulting in a positive sodium balance, expansion of extracellular fluid volume, and an increase in glomerular filtration rate. Sodium reabsorption is enhanced by increasing the number of open Na+ and K+ pores; Na+, K+-ATPase activity at the basolateral membrane also is increased, causing sodium to be returned to the circulation. The ratio of exchange of these mono-charge cations may be greater than than 11:1.
KEY POINT 30-6
Actions of glucocorticoids will maintain blood glucose, blood pressure, cardiac output, and vascular volume. Accordingly, their absence can result in life-threatening cardiovascular collapse.
Effects of mineralocorticoids are not limited to renal tissues but also include the colon, ileum, ciliary apparatus, salivary, and (in human beings) sweat glands. GLCs influence water and electrolyte balance through mineralocorticoid actions. Differences in tissue response to various corticosteroids was addressed previously. Cortisol and the synthetic GLCs possess varying degrees of mineralocorticoid activity, but all have less than 1% of the mineralocorticoid activity of aldosterone. GLCs also impart a permissive effect on tubular mechanisms that maintain the glomerular filtration rate. GLCs have an inhibitory effect on antidiuretic hormone and may decrease permeability of the distal renal tubules to water through a direct action. The polyuria and polydipsia commonly observed in dogs (but not cats) receiving GLCs may result from a combination of mineralocorticoid and GLC effects.
Other reported renal actions of GLC include downregulation of sodium–phosphate co-transport and sulfate co-transport, upregulation on sodium–bicarbonate co-transport, modulation of the effects of sodium on amino acid transport, and other effects on acid–base balance.10 GLCs also influence several aspects of calcium movement. In the renal tubular cells, calcium excretion is increased, and in the small intestine its absorption is impaired. GLCs also increase parathormone secretion, which in turn increases osteoclast-mediated bone resorption. The net effect of GLCs on calcium homeostasis is a decrease in total body calcium stores.
Hemolymphatic System
GLCs tend to increase the red blood cell content of the blood by retarding erythrophagocytosis. Lymphopenia, eosinopenia, and monocytopenia caused by cellular redistribution and neutrophilia caused by increased release from bone marrow, demargination, and a reduction of their removal from the circulation are all associated with GLC administration.9 This blood cell profile represents the “stress leukogram” seen clinically in patients with increased concentrations of endogenous GLCs. The acute effects of GLCs on circulating lymphocytes are due to sequestration from the blood rather than lymphocytolysis, although cells of lymphocytic malignancies are destroyed by GLCs. In addition to reducing the number of circulating lymphocytes, GLCs also alter the responses of lymphocytes to mitogens and antigens. T lymphocytes are inhibited to a greater degree than B lymphocytes (see following discussion of immunomodulation). GLCs also induce lymphocyte apoptosis.28 GLCs also have direct inhibitory effects on eosinophils, including eosinophil migration.29 GLCs directly induce eosinophil and lymphocyte apoptosis.30
Antiinflammatory and Immunosuppressive Effects
GLCs are most frequently used in clinical medicine for their antiinflammatory and immunosuppressive actions (see Table 30-1).31 Because the antiinflammatory and immunosuppressive effects of GLCs reflect specific actions on white blood cells, these effects are inextricably linked. They generally occur only when the amounts of steroid are present in concentrations greater than those found in the normal physiologic state (i.e., pharmacologic concentrations). The effects of GLCs on leukocyte numbers were discussed earlier. GLCs also profoundly affect white blood cell function. Ultimately, both the humoral and cell-mediated arms of the immune response are affected. The antiinflammatory effects of GLCs reflect all three mechanisms of GLC action (genomic transcription and transactivation; nongenomic membrane receptor signaling).12
GLCs upregulate or downregulate as many as 2000 genes involved in the regulation of the immune response (Table 30-4).2 GLCs inhibit early and late phases of the inflammation. Responses that are inhibited include edema formation, fibrin deposition, leukocyte migration, phagocytic activity, collagen deposition, and capillary and fibroblast proliferation. Many of these processes involve lymphokines and other soluble mediators of inflammation, and it is through these mediators that GLCs exert their antiinflammatory actions. GLCs induce annexin I (also known as lipocortin), which inhibits phospholipase 2, thus blocking the release of arachidonic acid and its subsequent conversion to eicosanoids (i.e., prostaglandins, thromboxanes, prostacyclins, and leukotrienes). (Figure 30-3). GLCs also preferentially inhibit transcription of cyclooxygenase-2 (COX-2), the inducible form of cyclooxygenase.12,32,33 Interestingly, recent evidence indicates that under some circumstances, GLCs may actually induce COX-2.34 The net effect of specificity to inhibit COX-2 may be inhibition of inflammatory prostaglandins without negatively affecting the protective effects of prostaglandins in other body systems (e.g., gastrointestinal, renal, hemostasis). GLCs induce the protein MAPK phosphatase 1, which through various actions inactivates a number of proteins important in the signaling of cytokines.12 Through physical interaction, transcription of NFκB is inhibited by GLCs. Many of the antiinflammatory effects of GLCs occur rapidly, apparently independently of changes in gene expression (nongenomic). Examples include activation of endothelial nitric oxide synthetase,12 offering an alternative mechanism of action of a new class of more selective antiinflammatory drugs.35
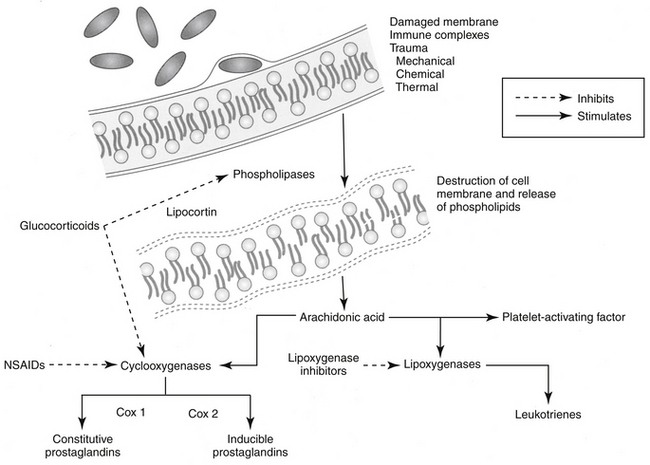
Figure 30-3 Site of glucocorticoid action in the arachidonic acid cascade. Glucocorticoids inhibit phospholipase and its subsequent degradation of cell membrane phospholipids to mediators of inflammation (platelet-activating factor, leukotrienes, and prostaglandins). Lipocortin is the effector protein whose synthesis is stimulated by glucocorticoids. In addition, glucocorticoids selectively inhibit cyclooxygenase-2. NSAIDs, Nonsteroidal antiinflammatory drugs.
KEY POINT 30-7
Glucocorticoids inhibit essentially every phase of the inflammatory response, including the immune response and wound healing.
Among the sequelae of these mechanisms, GLCs inhibit release of TNFα and IL-2 from activated macrophages. TNFα induces cytotoxicity and can enhance neutrophil and eosinophil function.27 The release of platelet-activating factor from leukocytes and mast cells is inhibited by GLCs. Platelet-activating factor induces vasodilation, platelet and leukocyte aggregation, smooth muscle contraction (especially in the bronchi), and increased vascular permeability.36
The immunosuppressive actions of GLCs, like their antiinflammatory actions, involve disruption of intercellular communication of leukocytes through interference with lymphokine production, biological action, or both. GLCs inhibit the synthesis and release of IL-1 by macrophages, thereby suppressing the activation of T cells. GLCs also inhibit IL-2 synthesis by activated T cells. IL-2 plays a critical role in amplification of cell-mediated immunity. GLCs block the effects of the migration inhibitory factor-γ (causing macrophages to migrate away from the affected area) and interferon-γ (IFN-γ) on macrophages.27 IFN-γ, which is released from activated T cells, plays an important role in facilitating antigen processing by macrophages. Additionally, GLCs suppress the bactericidal and fungicidal actions of macrophages. GLCs also alter synthesis of and biologic response to collagenase, lipase, and plasminogen activator. The antiinflammatory effects of GLCs also may reflect inhibition of the inducible form of nitric oxide synthase (iNOS).37 Synovial macrophage nitric oxide production and iNOS synthesis are inhibited by dexamethasone. The inhibitory effect appears to be mediated by lipocortin.The immunosuppressive actions of GLCs are more pronounced on the cellular arm than the humoral arm of the immune system (Figure 30-4). GLCs have minimal effects on plasma immunoglobulin concentrations but can modulate immunoglobulin function. For example, opsonization of bacteria is inhibited. Therapeutic doses of GLCs do not significantly decrease an animal’s antibody response to antigenic challenge (e.g., vaccinations). The immunosuppressive effects of GLCs may also reflect actions on the hypothalamic–pituitary–adrenal axis. Multiple cytokines appear to regulate this axis. Specifically, IL-1 appears to stimulate the release of CRH and directly increase the release of ACTH, and it may cause the adrenals to release GLCs. These interactions appear to be important to modulation of stress and thus maintenance of homeostasis.9
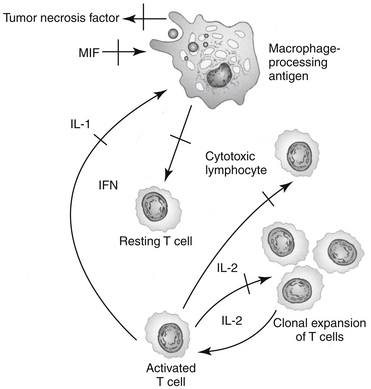
Cardiovascular System
Corticosteroids have two major effects on the cardiovascular system. Mineralocorticoids and, to a lesser extent, GLCs affect the maintenance of extracellular fluid volume, as described previously. Interestingly, mineralocorticoids also appear to have direct actions on cardiovascular tissues; increased cardiac fibrosis has been induced experimentally in rats by administration of excessive mineralocorticoids, suggesting an indication for spironolactone as a diuretic in congestive heart failure associated with myocardial disease.9 In addition, corticosteroids (predominantly GLCs) enhance vascular reactivity to other vasoactive substances (e.g., norepinephrine, angiotensin II).9 Mechanisms appear to include increased receptor numbers in the vascular wall or other tissues. Other proposed mechanisms of GLC-induced hypertension include reduced activity of depressor systems (e.g., kallikrein-kinin, prostaglandins, and nitric oxide) and increased responsiveness to angiotensin II and norepinephrine.38 In humans, Cushing’s syndrome or prolonged administration of synthetic GLCs is associated with hypertension characterized by sodium chloride retention and volume expansion. However, hypertension may be independent of these effects; rather, downregulation of nitric oxide synthetase may be a more plausible explanation.39 In contrast, in patients with insufficient concentrations of GLCs, negative sequelae include increased capillary permeability, decreased cardiac output, and inadequate vasomotor response of the smaller blood vessels to catecholamines. In humans, glucocorticoids are associated with worsening of cardiovascular disease. Hypertension appears to reflect increased glucocorticoid-mediated peripheral vascular resistance rather than aldosterone–mediated rather than sodium retention. The latter mechanism, along with myocardial remodeling, has been associated with worsening congestive heart failure.39b
Bone and Cartilage
GLCs antagonize the effects of vitamin D3, accelerate bone resorption, and decrease bone formation (through direct action on osteoblasts), resulting in osteoporosis. This phenomenon is well documented in human patients after chronic GLC therapy, but to the authors’ knowledge it has not been observed in animals.40 At physiologic doses, glucocorticoids stimulate collagen. At supraphysiologic doses GLCs inhibit collagen synthesis by fibroblasts; depress chondrocyte metabolism; and decrease the proteoglycan content of cartilage, resulting in morphologic changes in articular cartilage.41,42
Skeletal Muscle
The permissive effects of GLCs include their influence on the ability of skeletal muscle to function normally. Too little will result in muscle wasting (generally resulting from hypokalemia). Likewise, and paradoxically, too much also will result in muscle wasting. Increased use of amino acids from muscle proteins is likely to contribute to this effect. Although the exact mechanisms of muscle wasting are unknown, the term steroid myopathy has been coined to reflect this condition in human patients and is used to refer to similar manifestations in small animals with hyperadrenocorticism.9
Central Nervous System
In the central nervous system (CNS), the highest concentration of MRs is in the limbic system, whereas GRs are more ubiquitous in the brain.6 Indirectly, GLCs maintain adequate plasma concentrations of glucose for cerebral functions, maintain cerebral blood flow, and influence electrolyte balance in the CNS. GLCs decrease formation of cerebrospinal fluid, which results in a reduction of intracranial pressure. In human beings GLCs are believed to influence mood (e.g., producing euphoria), behavior, and brain excitability.9 The euphoric effect commonly recognized in dogs also is likely to reflect differences in GRs. Steroids, including GLCs, also appear to regulate neuronal excitation, with differential effects occurring for some drugs. For example, dexamethasone inhibits microglial cell proliferation; cortisone also will inhibit proliferation at higher doses through GRs, but stimulate proliferation through MRs.26 GLCs appear to have a protective effect in the CNS through induction of glutamine synthetase in both the CNS and peripheral nervous system; the enzyme adds an amine to and thus removes glutamate.43 Increased glutamate has been associated with CNS pathology, although the relationship between the two remains controversial.44,45 However, efficacy reflects regulation through transcription, which takes at least 24 hours. Glutamine has several roles in the CNS; in the presence of hyperammonia (e.g., liver disease) increases formation of glutamine in brain cells and the potenetial for cerebral edema.45b The use of glucocorticoids for treatment of cerebral edema might be avoided in situations in which increased glutamine increases the risk of cerebral edema.
Interestingly, GLC concentrations appear to be directly related to the severity of memory impairment in animals, including humans.6 Learning is impaired in animals exposed to prolonged reductions in circulating concentrations of GLCs; conversely, memory also appears to be impaired if GLC concentrations are too high.
Respiratory System
GLCs are reported to have “permissive” effects (increased receptor number or affinity have been proposed) on β2-receptors, promoting bronchodilation.46 Other mechanisms are likely to evolve as non-genomic effects are understood. The effects of GLCs on leukotrienes, platelet-activating factor, and other mediators important in the pathogenesis of respiratory inflammatory diseases were discussed earlier and also are addressed in Chapters 20 and 29.
Alimentary Tract
GLCs decrease the absorption of calcium and iron from the gastrointestinal tract and increase the absorption of fats. Secretion of gastric acid, pepsin, and trypsin are increased by GLCs. Gastric mucosal cell growth and renewal are reduced by GLCs, and mucus production is decreased, resulting in compromise of the protective barrier of the gastric mucosa. Collectively, these effects contribute to increased susceptibility to gastric ulceration. A retrospective study in humans found that 5% developed gastric mucosal lesions while receiving GLCs, particularly patients with rheumatoid arthritis and collagenosis.47 It is not clear whether the effects of GLCs on the gastric mucosa occur as a result of impaired mucosal prostaglandin synthesis, although failure of misoprostol (prostaglandin E) to protect against GLC-induced ulceration indicates that they do not. Indeed, some studies have shown that GLCs provide a gastroprotective effect during stress-induced ulceration.48 Deposition of glycogen in the liver, resulting in hepatomegaly in animals chronically treated with GLCs, is a consequence of increased glycogen synthesis, as described previously. Excessive cortisol levels can induce pancreatic ductule hyperplasia.
Reproduction
GLCs can induce parturition during the latter stages of pregnancy in ruminants and horses, but their effect in dogs and cats has not been determined. GLC administration in pregnant dogs has been associated with cleft palate and abortion. GLCs have been shown to inhibit cell division, DNA synthesis, or both in the developing liver, lung, brain, and thymocytes.
Structure–Activity Relationship
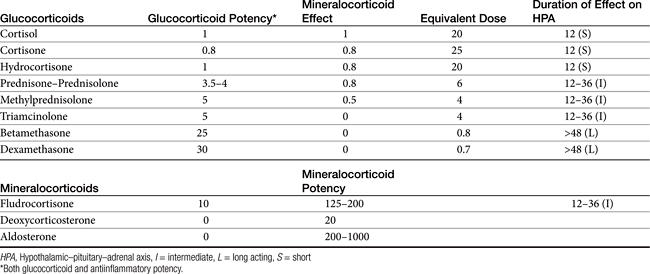
There are close to 50 generic corticosteroid products approved for human use and several approved for use in small animals. These drugs differ primarily in their duration of action, mineralocorticoid activity, and antiinflammatory potency (Table 30-2). As the antiinflammatory potency of a particular agent increases, its biological half-life and duration of action also tend to increase. Antiinflammatory properties also parallel the effects on metabolism, that is, carbohydrate and protein metabolism, but mineralocorticoid effects can be altered independently by changing the molecular structure of the steroid (Figure 30-5). The 4,5 double bond and the 3-ketone are necessary for mineralocorticoid and GLC effects. Synthetic modifications of cortisol increase the antiinflammatory activity, decrease protein binding, and decrease hepatic metabolism, thus prolonging activity. First-generation GLCs formed with the addition of a 1,2 double bond increased the ratio of GLC to mineralocorticoid effects (prednisolone, prednisone, and methylprednisone) and duration of action. The second-generation steroids were fluorinated at the C-9 position, increasing potency. Methylation at the C-16 position eliminates mineralocorticoid activity (dexamethasone, betamethasone, and triamcinolone).
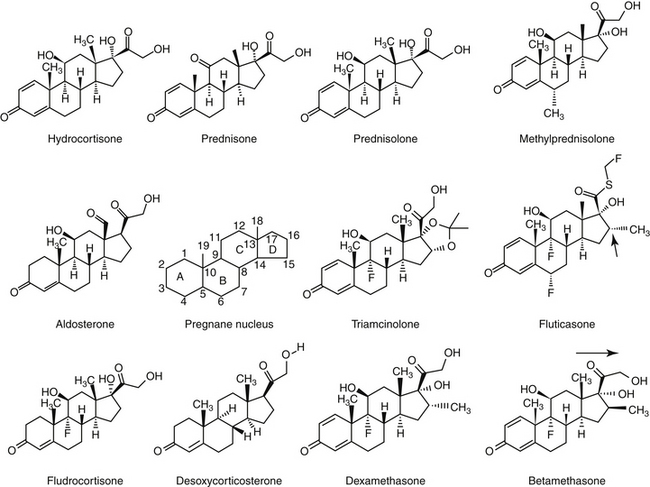
Figure 30-5 Chemical structures of selected adrenocorticosteroids. The pregnane nucleus provides the skeletal structure of corticosteroids. Corticosteroid activity depends on the double bond at position 4,5 and the keto group in the 3 position. Compounds with a double bond at position 1,2 have increased glucocorticoid activity (e.g., prednisone versus hydrocortisone) as well as a longer duration of action. Prednisolone differs from the prodrug prednisone only by the presence of a hydroxyl rather than a ketone group on the C ring. A methyl group at the C-16 position (arrow) eradicates mineralocorticoid activity (e.g., dexamethasone). Fluorination at the C-9 position enhances corticosteroid potency. The difference between betamethasone and dexamethasone is the orientation of the methyl group at position 16. The addition of acetate rather than succinate esters (not shown) to the various compounds also prolongs duration of action.
KEY POINT 30-8
In general, as exogenous glucocorticoid effects become more potent, mineralocorticoid effects become less potent.
The effects of GLCs are generally recognized to be dose dependent. Indeed, GLCs can be classified on the basis of their potency, generally relative to hydrocortisone. For example, dexamethasone is 30 times and prednisolone 4 times as potent as hydrocortisone in impairing glucose metabolism (Table 30-2).20 Daily doses of glucocorticoids used therapeutically have been variably referred to as physiologic, which generally reflects 0.1 to 0.25 mg/kg predniosolone or equivalent, and a pharmacologic or supraphysiologic, ranging, for prednisolone, 0.8 mg/kg IV, to 1 to 2 mg/kg/day, and a high dose of approximately 10 mg/kg orally (or 4 to 60 mg/m2/day). Some references refer to very high supraphysiologic doses (in dogs: hydrocortisone at 2-4 mg/kg, IV every 6 to 8 hrs, 4 to 20 mg/kg prednisolone IV every 2 to 6 hrs, 0.5 to 2.0 mg/kg dexamethasone, or 30 to 100 mg/kg methylprednisolone), which also might be referred to as a “shock” dose.
Clinical Pharmacology
Absorption
Several products are well absorbed orally. Differences between oral absorption occur, for example between prednisone and prednisolone in both the dog and cat (see below). For intramuscular or subcutaneous administration, the duration and onset of action of a particular GLC can be altered by the addition of an ester, usually bound to C-21. The GLC esters must be hydrolyzed – often by esterases located at the site of administration - to release the active, free form of the drug. However, the sodium phosphate and sodium succinate esters are water soluble, can be administered intravenously, and are rapidly hydrolyzed. These characteristics make them ideal for treatment of acute conditions. The acetate, acetonide, valerate, and dipropionate esters are water insoluble and release of the active steroid is very slow, providing GLC activity for days to weeks (i.e., repositol, or depot, products). The major advantage of these esters is convenience of administration. Administration at 2- to 6-week intervals, depending on the preparation used and disease being treated, has been recommended. Disadvantages include unpredictability of blood concentrations, chronic suppression of the hypothalamic–pituitary–adrenal axis (up to 12 weeks or more after administration of a single dose), possible induction of steroid resistance (mediated by receptor downregulation), and the fact that the drug cannot be withdrawn should adverse reactions develop. For these reasons the authors recommend the use of short- to intermediate-acting preparations administered daily or on alternate days over repositol steroid preparations.
KEY POINT 30-9
The slow-release salts of glucocorticoids preclude advantages of alternate-day dosing that occur with administration of short- or intermediate-acting base drugs.
There are many forms of GLCs available for topical use, including extensions of the skin such as the external ear canal and anal sacs. Once absorbed through the skin, topical corticosteroids are handled by the body in the same capacity as systemically administered GLCs. The extent of percutaneous absorption of topical GLCs depends on factors such as the vehicle, the ester form of the steroid (greater lipid solubility enhances percutaneous absorption), duration of exposure, surface area, and the integrity of the epidermal barrier. Ointment bases are occlusive and are therefore more likely to increase percutaneous absorption of the same GLC in a cream base. Highly potent preparations in any form should not be used on abraded skin.
GLCs are absorbed and can achieve physiologic and possibly pharmacologic concentrations after local administration. This includes the skin, as previously noted, synovial spaces, conjunctival sac, and the respiratory tract.9 Suppression of the hypothalamic–pituitary–adrenal axis has been documented after ocular,50 otic,51 and topical52 administration for several weeks. The impact of dexamethasone and betamethasone administered as a topical commercial otic preparation on adrenal function was studied in normal dogs (with normal ears). Drugs were administered twice daily for 2 weeks according to the package insert: dexamethasone at 10 drops (0.223 mg) and betamethasone at 4 drops (0.1193) per ear. For the betamethasone-treated group (n = 7), ACTH response tests were normal at the end of the treatment period, which was in contrast to the dexamethasone-treated group, for which the adrenal gland was suppressed in 71% (five of seven) of animals. Response normalized in 1 to 2 weeks for all animals. No animal exhibited signs of hyperadrenocorticism.53 Ginel and coworkers54 prospectively examined the effect of 2 weeks of topical betamethasone administration (4 to 8 drops, depending on body weight, of 0.88 mg betamethasone/mL, Otomax) compared with placebo using a randomized crossover design with a 4-week washout period. Although adrenal gland suppression did not occur, serum alkaline phosphatase increased (but remained within normal limits) and response to intradermal testing was muted by day 14 of treatment.
Using a prospective parallel approach, Aniya and Griffin55 recently compared the effect of vehicle and concentration of dexamethasone applied topically in the normal (not inflamed) ear of dogs (n = 21). Dexamethasone was applied as either 0.01% or 0.1% in saline (n = 7 each) and 0.1% in the commercially available product (Tresaderm; n = 7), for which propylene glycol is the carrier. Animals were treated for 2 weeks with 10 drops in each ear twice daily, resulting in a dose of approximately 0.89 mg dexamethasone for the 0.1% product (0.07 to 0.25 mg/kg). On the basis of ACTH stimulation, four of seven dogs in each 0.1% treatment group, but no dogs in the 0.01% treatment group, experienced adrenal gland suppression. Further, serum alkaline phosphatase was increased in one of seven dogs in the 0.1% propylene glycol treatment group. Finally, Reeder and coworkers56 compared four GLC-containing otic preparations in dogs with otitis externa using a randomized, double-blinded parallel design. Drugs studied included 0.007 to 0.02 mg/kg once daily of mometasone (Mometamax; n = 9), 0.002 to 0.03 mg/kg twice daily of triamcinolone (Panalog; n = 12), 0.004 to 0.024 mg/kg twice daily of dexamethasone (Tresaderm; n = 8), and 0.006 to 0.03 mg/kg twice daily of betamethasone (DVMax; n = 11). Dogs were treated for 7 days. Mometasone was described to have 1.5 times the potency of fluticasone and 22 times that of dexamethasone.56 Plasma cortisol was suppressed at 7 days in 0%, 9%, 17%, and 50% of the animals receiving Mometamax, betamethasone, triamcinolone, and dexamethasone, respectively.
KEY POINT 30-10
With few exceptions, it should be assumed that topical administration of glucocorticoids will result in drug absorption sufficient to affect the hypothalamic–pituitary–adrenal axis.
Selected steroids have been studied after transdermal administration in cats. Using a randomized crossover design, Willis-Goulet and coworkers57 measured dexamethasone in the serum of cats (n = 6) after single-dose oral or transdermal administration of 0.05 mg/kg. Although drug was detected after oral administration (peak 30 μg/mL), drug was not detected after transdermal administration. A similar finding was reported by other investigators.59 Oral absorption of GLCs may be affected by P-glycoprotein, depending on the specific drug.58 Cortisol, aldosterone, and dexamethasone are demonstrated substrates. Even in dogs, oral absorption of prednisone may not result in prednisolone concentrations comparable to that achieved with oral prednisolone.
Distribution, Metabolism, and Excretion
In humans (and presumably many animals), endogenous (and exogenous) cortisol is bound to corticosteroid-binding globulin (transcortin). The two main binding proteins are cortisol-binding globulin and albumin.2
Transcortin is an α-globulin secreted by the liver. Whereas it has a high affinity for steroids, it has a relatively low binding capacity. Albumin, which has a low affinity but large binding capacity, also binds GLCs. Corticosteroids compete with one another (endogenous and exogenous) for binding sites and at high concentrations will displace one another. Steroidal hormones tend to be eliminated by oxidation or reduction followed by conjugation (generally glucuronide or sulfate) and excretion (principally renal). Metabolism occurs at both hepatic and extrahepatic (including the kidney) sites. Prednisone is the prodrug form of prednisolone; it is assumed, but not proven, that no barrier exists to formation of the latter Cortisol is metabolized by the liver by phase I and II enzymes. Metabolism to inactive corticosterone is mediated by 11 beta-hydroxysteroid dehydrogenase, particularly in the kidney.2 Renal expression of this enzyme is one means whereby aldosterone response predominates; elimination of cortisol in human patients with renal disease may be prolonged as much as 30%.60 Cortisol is metabolized by the liver by phase I and II enzymes and in the kidney, where it is converted to the inactive metabolite cortisone by 11 beta-hydroxysteroid dehydrogenanse.2
Biliary and fecal elimination of corticosteroids do not appear to be that significant.9
Drugs and Preparations
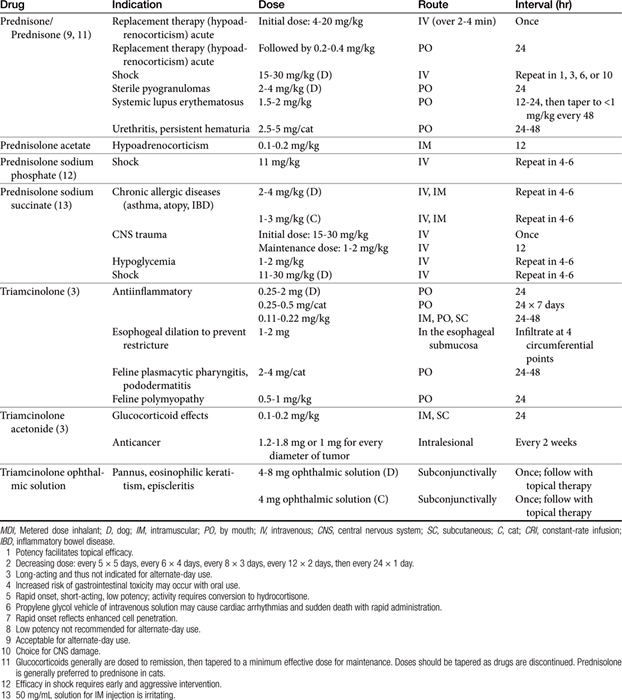
Knowledge of a few commercial preparations is sufficient for most clinical purposes (Tables 30-3 and 30-4). Selection is most commonly based on balancing the need for efficacy with the risk of adverse effects. Some distinct characteristics of selected steroids are presented in the following sections.


Hydrocortisone
Hydrocortisone is identical to cortisol, the most important endogenous GLC for most species. Extrahepatic elimination of hydrocortisone contributes substantially to clearance in the dog. Elimination is flow limited, with clearance being greatest in organs with greatest blood flow.61 Elimination of hydrocortisone from the plasma is biphasic in dogs, with an elimination half-life of the terminal phase of 50 minutes. The volume of distribution in dogs is greater than 1 L/kg. Because of its short duration of action (<12 hours) and low potency, hydrocortisone is not frequently used for systemic therapy. Hydrocortisone is available in creams and ointments for topical use.
Prednisolone and Prednisone
Prednisone is rapidly metabolized by the liver to prednisolone (C-11 ketol reduction). Liver disease probably has minimal effect on activation. Prednisone elimination in dogs is biphasic, with apparent half-lives of 15 and 82 minutes for each phase after intravenous administration.62 However, prednisolone has an intermediate duration of action (12 to 36 hours) and is therefore considered appropriate for alternate-day administration.
Prednisone and prednisolone generally are considered equivalent in terms of therapeutic dosing in both dogs and cats. However, the two are not necessarily equivalent, as has been demonstrated in dogs and cats. Colburn and coworkers63 demonstrated in Beagles (n = 16) that relative bioavailability is greater for prednisolone. After 5 mg orally of either drug, the Cmax was twice as high and the area under the curve 40% higher for prednisolone compared with prednisone. All dogs received either 5 mg prednisolone or prednisone orally using a randomized crossover design. Both steroids were measured with each study. The pertinent pharmacokinetic parameters differed between the two drugs and were as follows: AUC (area under the curve; ng∗mL/hr), Cmax, (ng/mL), Tmax (hour), and elimination half-life (hour). After administration of 5 mg prednisolone, the data for prednisolone was 986, 273, 0.9, and 1.8; and for prednisone, 855, 205, 1.3, and 1.6. After oral administration of 5 mg prednisone, the numbers were for prednisolone: 638, 111, 2, and 2.2; and for prednisone: 694, 132, 1.2, and 2.3. These data suggest rapid interconversion between both steroids, regardless of which is administered, in the dog but also about a 65% relative bioavailability for prednisolone (or prednisone) when prednisone is administered compared to prednisolone. Prednisolone also has been studied after IV administration but only in a single Beagle.63a The dog was dosed at 1.8 and 3.6 mg/kg. Clearance was 34 ml∗kg/min and volume of distribution was 0.7 L/kg. Elimination half-live was 0.3 hour. The oral bioavailability of prednisolone solution or tablet approximated 50% but this was increased to essentially 100% when administered as a slurry. This study, although in one dog, supports a dose-dependent disposition of prednisolone.
That the two are not equivalent in cats was demonstrated by Graham-Mize and Rosser.59 Disposition of prednisolone in cats after oral administration was as follows: area under the curve 3230.55 ng/mL/hr and Cmax 1400.81 ng/mL, with a half-life for excretion of 1 hour. This compares to an area under the curve of 672.63 ng/mL/hr and Cmax of 122.18 ng/mL after oral administration of prednisone. The half-life of prednisone was longer at 2.46 hours. According to these data, the dose of prednisone must be at threefold to fivefold higher than that of prednisolone to achieve equivalent activity. Boothe has studied the efficacy of drug delivery of prednisone and prednisolone after multiple (3 weeks) transdermal administration. Cats were studied using a random crossover design. Cats received prednisone and prednisolone (2 mg/kg) either orally or transdermally twice daily. At the end of each 3-week dosing period, and a 1-week washout, cats were rotated to the next randomly assigned treatment. Two-hour peak and 12-hour trough serum samples were assayed for the presence of prednisone and prednisolone. After 3 weeks of dosing (n = 6), neither prednisone nor prednisolone concentrations were detectable after oral or gel administration of prednisone. Prednisolone was not detectable after gel administration of prednisolone, but prednisolone reached therapeutic concentrations after oral administration of prednisolone. The use of prednisolone rather than prednisone might be more prudent for both species. Liver disease does not appear to impact conversion of prednisone to prednisolone as was demonstrated in human patients with chronic active liver disease (n = 10) compared to healthy volunteers (n = 7).63b
Methylprednisolone
Methylprednisolone possesses lipid antioxidant activity that has been shown to be beneficial in the treatment of experimental spinal cord trauma in cats and experimentally induced Escherichia coli bacteremia.65,66 In contrast, methylprednisolone did not appear clinically beneficial in a canine model of spinal trauma, although the model may have caused insufficient damage for effective evaluation.67 Although some other GLCs (dexamethasone, prednisolone) are efficacious as lipid antioxidants, methylprednisolone is the most potent. Methylprednisolone has an intermediate duration of action (12 to 36 hours) and is also a good candidate for alternate-day administration. The disposition of methylprednisolone succinate was described in normal dogs and then again after induction of hemorrhagic shock (n = 5).68 The drug is characterized by a clearance of 1.6 ± 0.5 L∗kg/hr and half-life 15.3 ± 3.8 minutes. During states of hemorrhagic shock, the clearance is reduced to 0.5 ± 0.2 L∗kg/hr and half-life was prolonged to 41 ± 23 minutes.
Dexamethasone
Dexamethasone is a highly potent GLC, but it has virtually no mineralocorticoid activity. It possesses some lipid antioxidant activity. The disposition of dexamethasone in dogs exhibits dose dependency. Greco and coworkers69 studied the disposition of dexamethasone after a low (0.01 mg/kg) and high (0.1 mg/kg) dose using a randomized crossover disease. The relevant pharmacokinetic parameters at each dose were A and B, (the Y intercept extrapolated from the initial and terminal components of the plasma drug concentration versus time curve, respectively; ng/mL), elimination half-life (hour), mean residence time (hour), and volume of distribution (L/kg). At 0.01 mg/kg, the data were: 22 ± 13.5, 5.3 ± 4.11, 192, 174 ± 42, and 1.9 ± 1.2. At 0.1 mg/kg, the data were 118 ± 74, 9.0 ± 5.7, 412, 324, and 6.41 ± 2. In addition to dose-dependent differences in clearance, the authors suggested that saturation of plasma protein-binding sites at the higher dose might explain the marked increase in volume of distribution. Elimination half-life is longer at a higher (0.1 mg/kg) compared with lower (0.01 mg/kg) dose, despite an increase in clearance; the differences presumably reflects the increase in the volume of distribution of dexamethasone at the higher dose.69 The disposition of dexamethasone did not differ in the presence of simultaneous administration of ACTH (0.5 U/kg intravenously), although the power of the study was not addressed. The prolonged duration of action (biological half-life approximately 48 hours) of dexamethasone makes it inappropriate for alternate-day administration.
Betamethasone
Betamethasone is a very potent GLC that varies from dexamethasone only in the orientation of a side chain. Thus, like dexamethasone, it has virtually no mineralocorticoid activity. It has a long duration of action (biological half-life approximately 48 hours) and is therefore not appropriate for alternate-day administration.
“Soft” Glucocorticoids
Among the mechanisms whereby undesirable side effects of GLCs can be minimized is topical administration of drugs that are potent for GRs but also rapidly metabolized should the drug be absorbed into systemic circulation via the oral route. These efforts generally reflect manipulation of chemical groups on the D ring of the GLC. Examples include beclomethasone, budesonide, and fluticasone propionate, steroids designed specifically for use in inhalant metered doses. Their potency when inhaled varies in clinical trials, with fluticasone propionate being most potent and budesonide and beclomethasone dipropionate approximately equipotent. Time of onset in humans to budesonide is approximately 10 hours based on evidence of clinical improvement at that time. Improvement can be expected over the next 1 to 2 days, with maximum effects potentially not being evident until 2 weeks after therapy has begun.
KEY POINT 30-11
The use of topical “soft” glucocorticoids for treatment of asthma or inflammatory bowel disease may reduce side effects in humans but not necessarily in dogs or cats.
Budesonide is rapidly metabolized in the liver by CYP3A4, with affinity of metabolites of the GCR being less than 1% of the parent. Essentially 100% of topically (inhalant) administered drug in humans appears as metabolites in the urine, indicating that systemic absorption of the drug does occur.70 Indeed, inhaled drug is generally considered to be absorbed, and as much as 25% of the inhaled dose in humans circumvents hepatic metabolism before entering systemic circulation. Because the potency that characterizes enhanced topical activity can similarly lead to peripheral effects, further development has focused on minimizing peripheral effects of topically administered GLCs that avoided hepatic metabolism.18 Budesonide represented one of the first successful products of those efforts, with local receptor binding of the drug being greater than peripheral binding. Intracellular fatty acid esterification inactivates the drug. As such, esterification serves as a storage site, with esterified drug being rereleased as the concentration of free drug declines with clearance. The ability to esterify varies among tissues, with pulmonary tissue apparently having a much higher capacity compared with other tissues, leading to greater storage in airways than in peripheral tissues.
Topical GLCs combine enhanced efficacy with some reduction in systemic exposure. Because lipophilicity increases transcutaneous movement, GLC esters are the most effective choices in human medicine for treatment of atopic dermatitis and other inflammatory skin diseases. The 17-esters, in particular, are characterized by high activity, presumably because of tighter GR binding (i.e., increased potency). However, greater potency may lead to irreversible skin atrophy. This contrasts with 21-esters, whose lower potency may decrease the risk. Combination 17, 21-double esters such as prednicarbate, a prednisolone double ester, offers the advantages and minimizes the disadvantages of both ester types and therefore is characterized by an improved benefit–risk ratio.71
The side effects of GLCs might be reduced but probably will not be eliminated by topical therapy. Drugs that impaired CYP3A4 may increase the plasma drug concentration of budesonide over sevenfold. Even by itself, budesonide appears to suppress the hypothalamic–adrenal–pituitary axis in dogs based on a clinical trial in dogs (n = 6) with inflammatory bowel disease. Endogenous ACTH and baseline and post-ACTH–stimulated cortisol were decreased after treatment with budesonide (30 days at 3 mg/m2) compared with baseline.72 Although no differences were described in serum alkaline phosphatase activity, urine specific gravity or the incidence of side effects typical of GLCs (polyphagia, polyuria), the power to detect these indicators of hyperadrenocorticism was not described. Anecdotally, dogs receiving budesonide for prolonged periods to treat inflammatory bowel disease do develop clinical signs associated with iatrogenic hyperadrenocorticism.
Tirilazad Mesylate
Tirilazad mesylate is a novel, non-GLC, 21-aminosteroid (lazaroid) that possesses potent antioxidant activity (i.e., it protects against oxygen-derived free radicals). Unlike the GLCs, this agent does not inhibit phospholipase A2, but it does inhibit lipid peroxidation-induced arachidonic acid release.73 The mechanism appears to reflect, in part, insertion into the cell membrane. These products are discussed in depth in Chapter 29.
Fludrocortisone
Fludrocortisone is a synthetic steroid hormone with mineralocorticoid activity. In humans, although it has tenfold activity at the GR compared with cortisone, it has no appreciable GLC effect at standard human doses. Its mineralocorticoid activity is 125-fold greater than cortisol and is used for mineralocorticoid replacement therapy.9 Fludrocortisone acetate is administered orally at 24- to 48-hour intervals for treatment of hypoadrenocorticism.
Desoxycorticosterone Pivalate
Desoxycorticosterone pivalate (DOCP) is an ester salt of a synthetic steroid hormone with mineralocorticoid (no GLC) activity. This form of desoxycorticosterone has a 20- to 30-day duration of action after intramuscular injection. Addisonian patients who have failed to completely respond to fludrocortisone may respond to therapy with DOCP.
Therapeutics
Unless GLCs are being administered for replacement therapy in a deficiency state (i.e., hypoadrenocorticism), GLC therapy is not directed at the inciting agent. GLC therapy is intended to reduce the physiologic processes that are activated in response to the disease. Despite the adverse events associated with their use, GLCs continue to be heavily used in veterinary medicine, potentially at doses that exceed those recommended. Indeed, in human medicine the use of GLCs clearly exceeds that recommended in textbooks and review papers.20 The advantages of low versus high doses have been previously discussed and are addressed again in the section on adverse reactions. In general, an antiinflammatory dose is considered to be 10 times the physiologic dose, and immunosuppressive doses are twice the antiinflammatory dose. Shock doses of GLCs have been reported at 5 to 10 times the immunosuppressive dose; however, the disadvantages of this high dose and the advantages of low-dose therapy in shock patients are discussed later. When treating a patient for an immediately life-threatening condition such as immune-mediated hemolytic anemia, the clinician should institute aggressive therapy, with a minimum effective dose determined after response has been achieved. Because high doses of GLCs are often required to adequately treat immune-mediated diseases, adverse effects are likely to occur and should be anticipated. Tapering of doses not only helps prevent side effects associated with long-term therapy but may also prevent the antibody rebound that has been associated with abrupt withdrawal of GLCs in human patients treated for prevention of graft-versus-host transplant rejection. Dose reduction in patients with autoimmune diseases should be conducted gradually. The reduced dose should be continued for at least 2 weeks before the next attempted dose reduction, and the actual dose should be decreased by no more than half. It is essential to assess the patient’s status frequently for recurrence of clinical signs. Concurrent administration of additional immunosuppressive (azathioprine, cyclophosphamide) or antiinflammatory drugs other than NSAIDs (antihistamines, omega fatty acids) may allow the GLC dose to be decreased (dose-sparing effect; see Chapters 29 and 31).74 High-dose pulse therapy has been reported in human patients with acute relapse of chronic graft-versus-host disease. In an open-design study, patients receiving no immunosuppressive therapy or patients that failed (a median of two failures) current therapy (mean prednisolone dose of 0.2 mg/kg per day, range of 0 to 2.5 mg/kg per day) were treated with methylprednisolone at 10 mg/kg intravenously or orally for 4 days. The rationale behind the high dose is based on the lympholytic properties of this dose, thus causing destruction of lymphocytes that otherwise would cause irreversible organ damage. The high dose is assumed to target the (nongenomic) metabolic processes necessary for sustained activity of lymphocytes, as opposed to the low (genomic) doses that target lymphocyte replication. Additionally, the high dose is considered to overcome GR saturation associated with GLC therapy, causing significant GLC downregulation. Induction of T lymphocyte apoptosis may also occur. Standard infection prophylactic therapy also was begun on day 1, consisting of fluconazole, levofloxacin, and valacyclovir (days 1 to 7) and a sulfadiazine–trimethoprim combination (continued until evidence of immunosuppression resolved). On day 5 a new immunosuppressive therapy (which varied with the hospital) was started using standard doses of standard immunosuppressive drugs. With use of this protocol, 75% of patients responded (48% classified as major response, indicating resolution of unequivocal improvement). During a 2-year follow-up, patients tolerated the therapy well, with no major life-threatening effects occurring in the first three months after treatment. However, three patients developed infections after completion of the therapy, suggesting profound immunosuppression. Yet the median time to progression of disease was 2 years after treatment, leading the authors to conclude that high-dose pulse steroid therapy is an effective and well-tolerated treatment for progressive graft-versus-host disease.24 The author (Boothe) has used this approach in a single case of lymphosarcoma in a dog whose sole treatment was GLC therapy. After the first relapse at 5 weeks, a high dose of dexamethasone (1 mg/kg intravenously) was followed by a second remission, with relapse occurring 3 weeks later. Should this approach be considered as a “rescue” scenario, antimicrobial (and possibly antifungal) treatment might be anticipated along with prophylactic antiulcer therapy.
Long-term use should strive to identify the smallest dose of GLC that will achieve the desired effect, particularly for relatively benign, chronic inflammatory conditions such as atopy or flea allergy dermatitis. Optimum doses (initial and maintenance) for the variety of syndromes responsive to GLCs are largely based on trial and error, with intermittent reevaluation. Eventually, anti-inflammatory GLCs with minimal metabolic effects will be associated with a lower risk. An every-other-day dosage regimen with a short- or intermediate-acting agent can achieve therapeutic effects without untoward effects in many patients. Agents that are ideal for alternate-day administration include prednisone, prednisolone, and methylprednisolone. The duration of action of hydrocortisone and cortisone may be too short for effective alternate-day therapy. Although triamcinolone’s duration of action is similar to those of prednisolone and methylprednisolone, its ability to suppress the hypothalamic–pituitary–adrenal axis is more typical of the long-acting agents such as dexamethasone and betamethasone. Side effects can occur if withdrawal of a GLC occurs too rapidly. In human patients receiving GLCs, the most frequent problem encountered with rapid withdrawals is recrudescence of the underlying condition for which the GLC was indicated.9 The most severe but rare complication, however, is acute adrenal insufficiency. Because of variability in GLC impact on the hypothalamic–pituitary–adrenal axis and variability within and between animals, predicting which animal is likely to develop insufficiency is difficult. In general, iatrogenic Addison’s disease is not common, but even this risk can be minimized by gradual withdrawal of the GLC. In human patients those who receive supraphysiologic doses for 2 weeks within the preceding year are considered to have some level of hypothalamic–pituitary–adrenal suppression.9
Therapeutic Indications∗
Dermatologic
GLCs are the cornerstone of therapy of many of the autoimmune diseases affecting the skin, including the pemphigus complex, systemic lupus erythematosus, and discoid lupus erythematosus. Optimal therapy for each of these diseases varies, insofar as some may respond to GLCs alone and some may require a combination of GLCs and alternate immunosuppressive drugs such as azathioprine or cyclophosphamide.
The long-term management of canine atopy (allergic inhalant dermatitis) frequently requires the use of GLCs. If at all possible, hyposensitization and avoidance therapy should be attempted before medical management. Although GLCs are the most effective antiinflammatory–antipruritic medication currently available for the atopic patient, alternate forms of therapy, as previously noted (e.g., antihistamines, misoprostol, omega fatty acids, and combinations thereof), should be attempted before sentencing a patient to chronic GLC therapy. Initial treatment with prednisolone at an antiinflammatory induction dose of 0.5 to 1 mg/kg per day is recommended. After resolution of clinical signs, the therapy should be switched to an alternate-day regimen. An effective alternate-day dose can be achieved by taking the lowest effective daily dose and increasing it by 50%.75 Some dogs may require medication every 3 to 4 days. The use of injectable repositol forms of GLCs is not recommended for treatment of canine atopy.
Atopy is also believed to exist in the cat. There is little information regarding the efficacy of antihistamines and fatty acid supplements as antipruritics in the cat. GLCs are the therapy of choice for treatment of atopic pruritis in the cat.76 As with dogs, short-acting to intermediate-acting compounds (prednisolone 1 to 2 mg/kg per day) are recommended, and alternate-day therapy should be the goal.
Otic
Otitis externa, often occurring as a component of atopy, is frequently responsive to topical GLC therapy (see Chapters 8 and 9 for discussion of antimicrobial drugs). Products usually contain an antibiotic and antifungal in addition to the GLC, and these agents can help resolve secondary infections. It is important to note that topically administered GLCs can be absorbed systemically.51 Efforts to remove or resolve the inciting factors should be made in cases of chronic otitis externa. More severe otitis externa, such as idiopathic hyperplastic otitis externa of Cocker Spaniels, may require systemic GLC treatment (prednisolone at 0.5 to 1 mg/kg per day).77 Follow-up examinations, usually at 2-week intervals, should include otoscopic and cytologic examinations to identify potential complications (otitis media, secondary yeast or bacterial infection, parasites, and so forth). It should be stressed that GLC therapy is not a substitute for thorough cleaning and drying of the ear.
Respiratory
GLCs have a pivotal role in the treatment of selected respiratory conditions (see Chapter 20). Their efficacy as bronchodilators (through their permissive effects on β2‑receptors) and as antiinflammatory agents has been well documented in patients with asthma. Among clinically used antiinflammatory drugs, GLCs alone inhibit prostaglandins, leukotrienes, and platelet-activating factor. Their effects on macrophage processing are well documented. These mediators have important roles in the pathophysiology of chronic bronchial disease. In human patients inhaled GLCs (beclomethasone, triamcinolone) tend to be first-line drugs. In cats suffering from bronchial asthma, GLCs are administered acutely and are the cornerstone of long-term therapy. Preference for the particular drug varies among clinicians. In patients suffering from status asthmaticus, water-soluble preparations of prednisolone tend to be used for their rapid effects, whereas dexamethasone may be preferred because it is more potent and provides a more prolonged effect. A combination of both products might be considered for immediate effects, followed by the more prolonged effects. Long-term therapy generally consists of oral forms of prednisolone, although some clinicians may prefer triamcinolone because of its enhanced potency. Alternate-day therapy may, however, be of no benefit for this intermediate- to long-acting GLC. As with other conditions requiring prolonged therapy, a higher dose is administered initially, with tapering of the dose to a minimal acceptable level. Lifelong therapy may be necessary for some cats. The use of repositol steroid preparations is controversial. Although they are convenient, the lack of predictability regarding retreatment may preclude their effective use. In cases of relapse, intravenous administration of dexamethasone or nebulized steroid therapy may be of benefit. Greater discretion is indicated for long-term use of GLCs in chronic respiratory diseases in dogs. Noninfectious diseases associated with eosinophilic or macrophage infiltrates are indications for GLC therapy, usually in conjunction with bronchodilator therapy. Short-acting GLCs can be used on a short-term basis (<48 hours) to break a cough cycle in patients with upper respiratory syndromes associated with inflammation (i.e., tracheobronchitis).
The design of topically administered inhalant GLCs and their use is also discussed with regard to “soft steroids” and adverse events, as well as in Chapter 20.
Musculoskeletal
Understanding the implications of GLC use for the treatment of osteoarthritis requires appreciation of the pathophysiology of osteoarthritis and the effects of GLCs on normal joint physiology. GLCs have variable and opposite effects on joint physiology, depending on the dose used.78 Low concentrations appear to be chondroprotective, whereas higher concentrations are chondrodestructive.
The term steroid arthropathy was coined to refer to the destructive condition that occurs in joints after multiple intraarticular injections of GLCs in human patients.78 In animal models (including dogs), the detrimental effects of GLCs can occur after administration of a single intraarticular injection or multiple systemic doses. In addition to the destruction induced by GLCs on cartilage, indirect damage may occur because of failure to rest an injured joint. Finally, GLCs negatively affect subchondral bone by inhibiting osteoblastic activity.
Despite the obvious role GLCs have in preventing cartilage catabolism and controlling inflammation, their use for the treatment of osteoarthritis is controversial. Although short-term use is generally accepted for acute conditions or trauma, long-term use is less acceptable. Much of the controversy might be resolved if a physiologic dose could be defined. In a canine model of osteoarthritis, an oral dose of 0.2 to 0.25 mg/kg per day of prednisone or an intraarticular dose of 5 mg per month of triamcinolone hexacetonide significantly reduced the incidence and severity of cartilage lesions and osteophyte formation79 The advantages of intraarticular administration include minimization (but not total elimination) of systemic effects. Disadvantages include the potential need for chemical restraint and the risk of sepsis.
Newer GLCs characterized by increased antiinflammatory potency yet decreased negative effects on cartilage may resolve the controversy regarding the use of GLCs in the treatment of osteoarthritis. Until these drugs are available or a physiologic dose has been established, the authors recommend reservation of GLCs in the treatment of osteoarthritis to animals that have failed to respond to chondroprotective agents or nonsteroidal antiinflammatory agents that are chondroprotective. Concurrent use of GLCs and nonsteroidal antiinflammatory agents enhances the risk for gastrointestinal ulceration and is not recommended. Finally, if GLCs are used, low doses (as noted previously) should be administered, and the simultaneous use of disease-modifying chondroprotective agents should be considered.
In humans suffering from osteoarthritis, intraarticular treatment with triamcinolone, methylprednisolone, and prednisolone are used to treat symptoms associated with osteoarthritis. Contraindications for such use include evidence of infection (e.g., bacteremia, adjacent osteomyelitis, or overlying skin) and joint prostheses. In humans corticosteroid injections are considered relatively safe, with long-term concerns focusing on the risk of long-term joint damage or infection.80 The use of GLCs for treatment of osteoarthritis reduces the number of GRs in treated patients, leading to a decrease in responsiveness. Subsequent activation of metalloproteinases may contribute to joint degeneration, although long-term (>1 year) therapy was associated with a trend toward improvement in humans with osteoarthritis of the knee. However, because the long-term effects of GLC therapy are not clear, their use is generally limited to patients who have not responded to other therapies.
Central Nervous System
GLCs are used for treatment of both brain and spinal cord disease. Their beneficial effects include protection from free radicals, reduction in intracranial pressure (decreased production of cerebrospinal fluid), and maintenance of normal microvasculature integrity. GLCs appear to be beneficial in reducing or preventing cerebral edema associated with neoplasia; however, cerebral edema caused by trauma is thought to be less responsive to GLC therapy.
There is increasing evidence that lipid peroxidation and resultant formation of oxygen-derived free radicals play an important role in tissue damage subsequent to brain or spinal cord trauma. Several GLCs, including methylprednisolone (most potent), dexamethasone, and prednisolone, are capable of inhibiting lipid peroxidation.73 Their ability to inhibit phospholipase A2 may also protect injured nervous tissue. For treatment of acute CNS injury, the water-soluble salts should be used. Recommended dosage regimens for these agents in the treatment of CNS injury in dogs and cats are methylprednisolone sodium succinate 30 mg/kg intravenously, followed by 15 mg/kg 2 and 6 hours later, and then 2.5 mg/kg per hour for the first 48 hours; prednisolone sodium succinate 60 mg/kg intravenously, followed by 30 mg/kg 2 and 6 hours later, and then 5 mg/kg per hour for the first 48 hours; and dexamethasone sodium phosphate 4 mg/kg intravenously every 6 hours for the first 48 hours.73 In studies involving cats with spinal cord injury, methylprednisolone sodium succinate–treated cats had significantly improved neurologic outcome, whereas the neurologic outcome of cats treated with dexamethasone was not significantly different than that of cats receiving placebo.81,82
Noninfectious, or so-called steroid-responsive, meningitis and granulomatous meningoencephalitis are diseases that frequently respond dramatically to GLC therapy. Prednisolone at a dose of 2 to 4 mg/kg per day has been recommended. The use of glucocorticoids for treatment of intervertebral disk disease is addressed in Chapter 28.
Inflammatory Bowel and Liver Disease
Inflammatory bowel disease in cats (especially those with lymphocytic–plasmacytic infiltrates) is frequently responsive to prednisolone at 2.2 mg/kg per day83 (see Chapter 19). For more severe cases, dexamethasone at 0.22 mg/kg per day plus metronidazole may be effective. In either case, the initial dose should be maintained for 2 weeks beyond the time that the cat’s clinical signs begin to resolve. Cats that respond immediately should receive the initial dose for at least 4 weeks before dose reduction is attempted.
Inflammatory bowel disease in dogs may be less responsive to GLC therapy than is the case in cats (canine patients with eosinophilic infiltrates are an exception). Initial treatment of lymphocytic–plasmacytic enteritis and colitis is with 2.2 mg/kg of prednisolone per day. Frequently, additional therapy is necessary (metronidazole, azathioprine). In all cases dietary modification should accompany pharmacologic therapy. Alimentary lymphosarcoma should be ruled out before steroid therapy is initiated (especially in cats). The use of GLC for treatment of inflammatory bowel disease is also discussed in Chapter 19.
Corticosteroid therapy for liver disease remains somewhat controversial. Clinical studies in human medicine have shown that steroid therapy in chronic active hepatitis improves survival rates. Nonsuppurative cholangitis–cholangiohepatitis may respond to immunosuppressive GLC therapy.
Gingivitis and Stomatitis
The eosinophilic granuloma complex in cats is usually responsive to high-dose GLC therapy such as methylprednisolone acetate 20 mg intramuscularly every 2 to 3 weeks for three treatments. Lymphocytic–plasmacytic gingivitis and stomatitis in cats may respond to GLC therapy (prednisone 2 to 4 mg/kg orally every 24 hours).
Septic Shock and Functional Adrenocortical Deficiency
The antibacterial treatment of septic shock is addressed in Chapter 8, and the antiinflammatory component also is addressed in Chapter 29. A number of bacterial and viral infections activate the HPAA such that GLC release increases. The mechanism probably reflects cytokines or other inflammatory mediators.4 It is likely that activation reflects both a direct effect on the adrenal glands or the hypothalamus or pituitary. Other proteins (e.g., Staphylococus, Clostridium, Mycoplasma) can also stimulated the response. A number of viruses have similar capability. However, although both bacterial and viral infections affect the hypothalamic–pituitary–adrenal axis such that GLC release is increased, other factors in the infected patient, such as pain, psychological stress, and the debilitating effects of infection, likewise activate the hypothalamic–pituitary–adrenal axis4 and will modulate response to infection.
The use of GLCs in septic shock has been controversial, although their potential benefits were recognized as early as 195184,85 (see Chapter 29). Evidence varies with experimental versus clinical studies. Earlier experimental models of septic shock showed GLCs to be beneficial if administered before or concurrently with endotoxin administration (i.e., within the first 2 hours). In canine models, severe mesenteric vasoconstriction within the first 15 minutes can lead to irreversible shock. Thus GLCs are unlikely to provide beneficial effects if they are administered 30 to 60 minutes after administration of the endotoxin.86 One of the differences between results of experimental versus clinical (spontaneous) models of septic shock may be the choice of outcome measures. Survival rates in experimental models are often based on short-term analysis (i.e., survival for several hours). In addition, the likelihood of knowing the time of onset of endotoxemia or sepsis in a clinical setting is extremely low.
The use of GLCs in human patients suffering from sepsis and septic shock was addressed in a large meta-analysis of human clinical trials.84 In this study 10 of 49 published reports investigating the effects of corticosteroids in human patients experiencing septic shock were considered to be superior on the basis of scientific design (e.g., controlled study, blinding techniques). Even within these 10 studies, however, differences in experimental design, definition of septic shock, drugs, dosing regimens, timing of steroid administration, and other design considerations limited the number of conclusions that could be made. Mortality rates often were the basis of analysis; however, mortality was not determined at the same time in all 10 studies. Of the 10 studies examined, only one study offered a significant positive effect for patients receiving GLCs. Most studies show a beneficial effect of steroids during the first few hours after the initiation of shock. Patients with sepsis associated with gram-negative infections demonstrated a slightly better outcome. The most common or severe side effects after steroid treatment was gastrointestinal bleeding (reported in five studies), generally associated with administration for 6 or more days or months. Superinfection (secondary infection) was reported in 7 of the 10 studies. The authors made the following conclusions: (1) The broad use of corticosteroids in patients with sepsis or septic shock is not beneficial, and the authors went so far as to suggest that a new trial would not be indicated. (2) Short-term, high-dose treatment did not increase the risk of complications. (3) Very early initiation (or prophylactic use) of steroid therapy in gram-negative infections might reduce the generalized inflammatory response to infection.84 The results of this and other studies led to a decline in the use of GLCs in septic shock in the early 1990s, only to be followed by a re-examination of the question in the late ‘90s and early 2000s. Renewed interest reflected the recent recognition that sepsis and septic shock are aggravated by transient adrenal failure and negative cardiovascular sequelae associated with adrenal insufficiency.2 Mortality rates are increased in acutely septic patients in which corticotropin stimulation is attenuated.87 In humans bilateral adrenal hemorrhage or necrosis is present in as many as 30% of patients dying as a result of septic shock, and peripheral ACTH resistance has been described in close to 20% of humans with septic shock. Predisposition to acute renal failure in septic shock may be facilitated by underlying pituitary or adrenal pathology, or co-treatment of drugs that impair cortisol synthesis (e.g., ketoconazole) or increase cortisol metabolism (e.g., phenobarbital).2
In contrast to early studies that focused on high doses of GLCs for treatment of septic shock, the realization of the potential role of adrenal insufficiency led to a physiologic approach to adrenal hormone replacement. A recent meta-analysis88 concluded that the dose of GLC used in the patient experiencing septic shock affected risks associated with increased mortality. Whereas clinical trials reported before 1989 (n = 8) found decreased survival in patients treated with GLCs, analysis studies after 1997 (n = 5) consistently found beneficial effects on survival if physiologic doses were used, even if longer than 6 days. The mean total hydrocortisone equivalent reported in the later trials was 1209 mg, or 300 mg per day (approximately 4 mg/kg/day), compared with a mean total dose of 20 times that amount in the earlier studies. The impact of secondary bacterial infections was considered a contributing factor to increased mortality rates in trials before 1989. Although the incidence of secondary bacterial infections did not differ between the two time periods, the time to resolution of the secondary infection was shorter after 1997, presumably because of less immunosuppression and fewer deaths associated with secondary infection. The authors concluded that whereas short courses of high-dose GLCs decreased sepsis survival, a 5- to 7-day course of physiologic hydrocortisone doses, with subsequent tapering of doses, increased survival. The impact of physiologic GLCs appears to reflect reversal of vasopressor-dependent septic shock, including improved response to vasoactive drugs.88 Other investigators have described decreased body temperature and heart rate when subjects are treated with low-dose GLCs. Improved survival rates in septic human patients treated with low-dose GLC therapy have led to the development of criteria intended to aid in the recognition of adrenal failure so that low-dose GLC therapy might be initiated. Basal cortisol concentration below 15 μg/dL are considered indicative of adrenal insufficiency in humans; for dogs, concentrations are < 2 μg/dL pre and post ACTH response (see also Chapter 21).2 Treatment for adrenal failure in humans focuses on replacement therapy with hydrocortisone (200 to 300 mg/kg per day [for a 70-kg patient]) combined with fludrocortisone (50 μg per day) for up to 7 days.
Septic shock is not the only acute illness for which functional ACTH deficiency may occur. Deficiency associated with acute illnesses in humans has been reviewed.89 Acute situations such as severe infection, trauma, burns, and surgery may cause up to a sixfold increase in cortisol secretion, with the increase proportional to the illness. Secretion may be impaired, however, for a variety of reasons, including preexisting disease affecting the hypothalamic-pituitary-adrenal axis, drug-impaired synthesis (e.g., imidazole antifungals, etomidate), adrenal necrosis, or hemorrhage as might accompany coagulopathies, or iatrogenic suppression such as that which may occur with exogenous GLC therapy. Cytokines associated with systemic inflammation may cause tissue GLC resistance; indeed, human patients with the most severe illness may have the highest cortisol concentrations, making “spot checks” of cortisol less helpful in the diagnosis of these patients.89 The deficiency is generally transient but is difficult to diagnosis. In humans, hyponatremia, hyperkalemia, hypoglycemia, and eosinophilia may be present. A corticotropin stimulation test might be used to detect an incremental response. Deficiency, once diagnosed, is treated in humans differentially, depending on the severity of illness. No treatment is indicated for mild illness. A moderate illness or condition may be treated with approximately 0.2 mg/kg per day, with discontinuation (or return to predeficiency doses in those patients receiving GLCs) within 24 hours of resolution. Severe illness or major surgery is treated with approximately 0.7 mg/kg cortisone every 6 hours; on resolution, the dose is tapered 50% every day. Septic shock is treated similarly with the addition of 0.7 μg/mL of fludrocortisone daily; treatment continues for 7 days.89
The final word on the role of GLCs in critical patients has yet to be determined. A meta-analysis of controlled clinical trials (n = 5) evaluating the efficacy of GLC therapy in patients experiencing septic shock. The analysis found that a short course of high-dose hydrocortisone decreased survival rates, whereas a longer course (5 to 7 days) of low-dose hydrocortisone increased survival rates.88 However, a recent multicenter clinical trial in humans (n = 251) with persistent hypotension despite fluid and resuscitation therapy found no significant response to corticotropin in patients that received low-dose hydrocortisone (approximately 0.72 mg/kg; n = 251) compared with saline placebo (n = 248) every 6 hours. Although time to reversal of shock was shorter in the treatment group, no difference in mortality rates was detected between groups or between responders and nonresponders within each treatment group.90 Factors that may have led to the lack of difference, as opposed to other studies that have reported treatment success, included lack of fludrocortisone treatment, patients that were less severely ill compared with those in other studies, and continued therapy for up to 11 days (as opposed to 7 or fewer days for other studies). The authors concluded that corticotropin response does not seem to be a viable test for predicting the need for steroid therapy. Interestingly, perioperative glucocorticoid supplementation in the surgical patient has been a point of controversy for more than half a century.90a The rationale is based on the recognition of surgery-associated adrenal insufficiency. The need for use and guidelines for administration were reviewed by Salem and coworkers in 1994. In 2008, Jung and Inder revisited the issue, recognizing that no consensus exist regarding dosing regimens or duration. However, high doses (>200 mg hydrocortisone equivalent, or approximately 2.75 mg/kg a day) beyond 2 to 3 days clearly are to be avoided in the uncomplicated case.90b
Neoplasia
Prednisolone is often included in combination chemotherapy protocols for treatment of lymphoma and multiple myeloma for their cytotoxic actions. The impact of GLC treatment alone in patients with GLC-responsive cancer, before implementation of combination chemotherapy, is controversial. In general, use of GLCs before chemotherapy is generally discouraged because of the clinical perception that such use is associated with a decreased duration of survival, which presumably reflects rapidly developing resistance to GLCs. Indeed, Price and coworkers91 demonstrated a significant decrease in the median duration of remission in dogs receiving GLCs before chemotherapy (134 days) compared with that of dogs not receiving GLCs (267 days). Proposed mechanisms whereby this might occur include the advent of multidrug resistance (e.g., by way of P-glycoprotein or related proteins) or a decrease in GLCs receptors. The use of GLCs as a single agent for treatment of lymphoma theoretically may rapidly induce multidrug resistance in neoplastic cells, causing the cell to be less responsive to not only GLCs but also other anticancer drugs, such as doxorubicin and vincristine. However, an in vitro study using canine cancer cells demonstrated dexamethasone resistance to cisplatin and methotrexate, but the resistance did not appear to reflect the multidrug resistant genes studied.92 Because of the controversy surrounding single-agent chemotherapy of lymphoma with prednisolone, this approach is generally not recommended except under certain circumstances, such as in the presence of life-threatening or organ (kidney) hypercalcemia.
The impact of GLCs on drug–receptor interaction may be a more likely cause for the development of resistance. Failure has been correlated to receptor density, with cells (e.g., T lymphocytes) characterized by low density being less responsive to chemotherapeutic drugs in general.
GLCs are often used in patients with mast cell tumors to decrease the inflammatory response associated with mast cell degranulation. Additionally, GLCs may induce a partial or complete remission in some canine patients with mast cell tumors.93
Ophthalmic
Topical GLC therapy is efficacious for treatment of noninfectious conjunctival, scleral, corneal, and anterior uveal inflammatory diseases.94 Before topical corticosteroid use, however, the corneal epithelium should be examined (using fluorescein dye) for signs of ulceration. The effects of glucocorticoids on corneal epithelial cells is biphasic, stimulating growth at low (physiologic) concentrations and inhibiting it at higher concentrations (supraphysiologic or pharmacologic).94a The presence of corneal ulceration precludes the use of topical corticosteroids. Prednisolone acetate penetrates the intact corneal epithelium and is therefore effective for treatment of intraocular inflammation.94 Systemic absorption of topical ophthalmic corticosteroids has been documented in dogs. Systemic GLC therapy is necessary for treatment of noninfectious posterior segment inflammatory conditions.
Hematopoietic
GLCs are the mainstay, first-line therapy for both immune-mediated hemolytic anemia and thrombocytopenia. Some anecdotal evidence suggests that initial treatment with dexamethasone may be superior to prednisone therapy, but no controlled studies have substantiated this claim. This may, reflect, however, poor bioavailability of prednisone. Prednisone (2 to 4 mg/kg per day) or dexamethasone (0.3 to 0.6 mg/kg per day) therapy should be continued until erythrocyte or platelet numbers steadily increase. Subsequently, the dose should be slowly (over 3 to 6 months) tapered. Addition of GLCs is also beneficial for treatment of immune-mediated neutropenia and for treatment and prevention of transfusion reactions.
Hypoadrenocorticism
Replacement corticosteroid therapy for treatment of hypoadrenocorticism is discussed in Chapter 21. An Addisonian crisis requires treatment with rapid-acting GLCs and mineralocorticoids. Not all synthetic GLCs are effective for replacement of mineralocorticoid deficiency. Dexamethasone, methylprednisolone, triamcinolone, and betamethasone are virtually devoid of mineralocorticoid activity. For treatment of acute hypoadrenocorticism, if a mineralocorticoid such as fludrocortisone (Florinef) or DOCP is unavailable, prednisolone or cortisone may provide adequate mineralocorticoid activity.
Adverse Effects
Adverse reactions resulting from GLC therapy can occur through either cessation of therapy or prolonged use. Acute adrenal insufficiency results from rapid withdrawal of GLCs after prolonged treatment (see earlier discussion of clinical use). Termination of GLC therapy in a chronically treated patient should be performed gradually over several months. Complete recovery of the hypothalamic–pituitary–adrenal axis may require 9 months. During this time, patients may need supplemental GLCs during “stressful” situations (e.g., surgery).
Complications of GLCs related to prolonged therapy are described later. In general, dogs are more susceptible to these complications than are cats. The incidence of adverse effects resulting from GLC administration is frequently related to the dose and duration of treatment. Alternate-day therapy with short-acting preparations can significantly reduce the incidence of adverse reactions.
The avoidance of GLC side effects is a focus of intense research in human medicine. The critical role that GLCs play in human asthma has directed much research in the design and development of the ideal GLC: one that combines marked efficacy and long-term safety, a convenient delivery device, and a reasonable price. Among the current methods by which risks are reduced is the design of drugs that target transrepression and through manipulation of the pharmacokinetic properties of GLC. This includes the administration of “soft” GLCs (previously discussed).18 Another mechanism by which systemic effects of GLC might be reduced is activation of a prodrug at the site of action. For example, ciclesonide is a novel, inhaled corticosteroid approved or treatment of asthma. The drug is activated in the lungs to the potent drug desisobutyryl–ciclesonide (des-CIC) in the lungs. The metabolite exhibits 100-fold affinity for GR in the lungs compared with the parent compound, and thus is comparable to fluticasone in potency. Further, the drug is characterized by low bioavailability, in part because of its high first-pass metabolism.95
The use of low rather than high doses may prevent some undesirable side effects, although the impact varies with the adverse event in human medicine.20 Osteoporosis persists, whereas osteonecrosis might be reduced. Myopathy is rare with low doses, whereas endocrinopathies, including glucose intolerance, fat redistribution, and weight gain, remain present with low doses; however, diabetes mellitus rarely occurs with low doses. Cardiovascular adverse events (e.g., atherosclerosis, sodium and water retention) tend to be minimized at low doses, whereas dermatologic events consistent with iatrogenic Cushing’s syndrome persist, albeit at a lower level. Psychologic disturbances, including steroid psychosis, are largely absent at low doses. Ophthalmic events such as glaucoma and cataracts are largely reduced with low doses, as are gastrointestinal events, including gastroduodenal ulceration (generally associated with concomitant use of nonsteroidal antiinflammatory drugs) and pancreatitis. Immunosuppression resulting in increased susceptibility to infectious disease also is largely reduced but still remains for some pathogens (e.g., Pneumocystis carinii) at low doses. The impact of dose dependency on the use of GLCs for treatment of septic shock was previously discussed.
Steffan and coworkers96 compared adverse events associated with cyclosporine A (n = 119; 5 mg/kg per day induction followed by tapering doses) versus methylprednisolone (n = 59; 0.5 to 1 mg/kg per day for 1 week, then every other day for 3 weeks, then tapered) in dogs with atopic dermatitis. The incidence of adverse events was similar between groups. The incidence of vomiting was significantly less in the methylprednisolone-treated group compared with the cyclosporine A–treated group (vomiting, 37% versus 5%). Whereas infections occurred with similar frequency in the two treatment groups, they were more likely to be classified as severe or very severe in the methylprednisolone-treated group, leading to more dropouts. Polyuria and polydipsia were more common in the methylprednisolone-treated group (25%) versus the cyclosporine A–treated group (4.3%); increased appetite and weight gain were also greater in the methylprednisolone-treated group (12% versus 3%). Serum chemistries indicative of renal function did not change from baseline levels. Potassium decreased approximately 0.7 mg/dL (and increased in the cyclosporine A–treated group) but was within normal values.
Levine and coworkers97 retrospectively compared the adverse events associated with dexamethasone (n = 49) compared with no treatment (n = 80) and other GLC therapy (primarily methylprednisolone sodium succinate) for treatment of acute thoracolumbar intervertebral disk herniation in dogs. The mean dose was 2.25 mg ± 4.28 m/kg (range 1 to 30 mg/kg). Adversities that were greater in the dexamethasone-treated group included vomiting, diarrhea, and anemia. Urinary tract infections also were more frequent in the dexamethasone-treated patients, although not all patients were tested for infection.
Central Nervous System
GLCs act as centrally active appetite stimulants to induce polyphagia in dogs and, to a lesser degree, in cats. This may contribute to the development of obesity in some patients, but it also may stimulate the appetite of an anorexic patient. Changes in mood or behavior have been noted in human patients treated with GLCs. Although these are more difficult to document in veterinary patients, most clinicians have noted positive attitude changes in patients being treated with GLCs.
Musculoskeletal
Muscle weakness and muscle atrophy are commonly observed in dogs with hyperadrenocorticism and dogs receiving large doses of GLCs (see previous discussion). These effects in part reflect muscle wasting that may result from the catabolic gluconeogenic effects of GLCs.
GLCs are associated with osteoporosis and vertebral compression in 30% to 50% of human patients receiving the drugs chronically. Mechanisms include direct inhibition of osteoblasts (decreased bone formation); inhibition of calcium absorption from the intestines; increased parathormone secretion, which in turn increases osteoclast-mediated bone resorption; and increased calcium excretion.9 Osteoporosis is not a clinically recognized entity in small animals receiving GLCs, in part because of differences in body posture as well as duration of therapy. Prudence is indicated, however, when GLCs are used in the face of conditions that facilitate or are facilitated by the negative sequelae of GLCs on bone formation. Aseptic necrosis has been reported in human patients after a short course of high doses of GLCs.9
Gastrointestinal and Liver
The likelihood of GLCs causing gastrointestinal ulceration is controversial. In general, patients that develop ulceration are predisposed to ulceration because of stress or are receiving other drugs that contribute to gastrointestinal damage, most notably nonsteroidal antiinflammatory drugs. In stressed animals peripheral effects of centrally or peripherally mediated norepinephrine may be important to the manifestation of gastrointestinal ulceration mediated by GLCs.98 Colonic perforation in dogs with spinal cord injuries has been associated with the use of dexamethasone, perhaps in part because of modulation of local blood flow. Pancreatitis has been associated with the use of GLCs alone and in combination with azathioprine. GLCs can induce hepatocellular swelling as a result of fat, perivascular glycogen, or water accumulation. Centrilobular vacuolization is common, and focal necrosis occasionally occurs. Clinically, these changes are seen as hepatomegaly and elevated liver enzyme activity. In dogs induction of a steroid-induced alkaline phosphatase isoenzyme occurs in patients receiving GLCs. The effects of GLCs on the liver are slowly reversible, persisting 1 to 1.5 months after therapy is discontinued.
Metabolic
Glucose intolerance, insulin resistance, overt diabetes mellitus, and hyperlipidemia can be induced in canine patients receiving GLC therapy.
Hepatic
GLCs consistently cause diffuse to centrilobular vacuolization and perivascular glycogen accumulation in hepatocytes.99,100 Focal necrosis has been occasionally described in clinical cases. Similar lesions result with hyperadrenocorticism. In addition to serum biochemical changes consistent with liver disease, GLCs also cause elevations in a steroid-specific alkaline phosphatase isoenzyme, which can be discerned with the levamisole test. This increase (induction) is not considered indicative of liver disease if other indicators of liver disease are absent. The pathologic changes associated with GLCs are slowly reversible over 1 to 1.5 months after discontinuation of therapy.
Endocrine
Exogenous administration of GLCs suppresses the hypothalamic–pituitary–adrenal axis through feedback inhibition on the anterior pituitary and the hypothalamus. Chronic suppression of the axis can result in iatrogenic hypoadrenocorticism on discontinuation of exogenous GLCs.
GLCs cause a decrease in plasma concentrations of total triiodothyronine, thyroxine, and thyroid-stimulating hormone, with a normal free triiodothyronine and thyroxine concentration.101 Therefore, when assessing thyroid function in animals receiving GLCs, the clinician should keep in mind their influence on total thyroid hormone concentrations. GLC administration does not result in clinical hypothyroidism because the free hormone concentration is minimally affected. GLCs increase parathormone secretion.
Renal
Polyuria and polydipsia are classically associated with excessive GLC (either endogenous or exogenous). Several factors probably contribute to this effect, including increased glomerular filtration rate resulting from increased vascular volume, increased renal calcium excretion, inhibitory actions on antidiuretic hormone, and direct actions decreasing permeability of the distal tubule. Glucocorticoids may increase the risk of azotemia in patients with renal disease because of their catabolic effects, particularly in the presence of metabolic acidosis.
Cardiovascular
Many human patients receiving GLCs develop hypertension as a result of sodium retention. Hypertension occurs in dogs with hyperadrenocorticism and presumably could also occur in veterinary patients receiving exogenous GLCs. Postulated mechanisms for hypertension in canine hyperadrenocorticism include increased activation of angiotensin I, increased vascular responsiveness to catecholamines, and reduction of vasodilator prostaglandins.
Immune Function
Although the intent of GLC therapy generally reflects suppression of some facet of immune function, such use generally is associated with an increased risk of bacterial, viral, and fungal infection, although this effect is dose dependent, as was previously discussed with regard to treatment of septic shock. It is generally recognized that a single dose of a short- to intermediate-acting GLC is unlikely to significantly suppress immune function, but caution with this approach is prudent, particularly for infectious diseases. Chronic administration of GLCs can result in recurrent cystitis, septicemia, and endocarditis. Causes for these effects are multiple, including immune suppression and, potentially, upregulation of P-glycoprotein (see the discussion of neoplasia). Ihrke and coworkers102 demonstrated as early as 1985 that close to 40% of dogs receiving immunosuppressive GLC therapy (mean of 0.8 mg/kg) also had a urinary tract infection. In a study of close to 200 dogs with pruritic skin disease, those receiving GLCs (either prednisone [70%] or methylprednisolone [30%]) for longer than 6 months had a higher incidence of urinary tract infections compared with dogs that did not receive GLCs.103 Doses ranged from 0.12 to 1 mg/kg, with a mean of 0.28 ± 0.14, generally administered every 48 hours. The overall incidence was lower than that of the earlier study, at 12% for treated dogs compared with 0% for untreated dogs, when based on the first urine sample collected for each dog. However, the number increased to 23% when subsequent samples were included in the analysis, indicating the need for repetitive culturing in animals receiving GLCs. This study provided evidence that clinicians should perform routine cultures for patients with hyperadrenocorticism rather than rely on clinical signs or urinalysis. No dog with bacteriuria manifested clinical signs of urinary tract infection. Whereas the presence of bacteriuria and pyuria consistently indicated the presence of a urinary tract infection, the absence of bacteruria did not rule out urinary tract infection: Bacteriuria was not detected in 24% of urine samples that subsequently yielded bacterial growth. Female, but not male, dogs receiving GLCs were predisposed to infection, although this finding may have been biased by the limited number of male dogs treated with GLCs. Although E. coli was the most common organism isolated (14 of 34), a total of 10 different organisms were isolated. This study might be interpreted as justification of the routine use of antimicrobials. However, only 6 of 52 urine samples collected in patients receiving antibiotics were positive on culture. Particularly problematic was the fact that isolates cultured in dogs receiving cephalexin were resistant to the drug. Indeed, 51% of all isolates were resistant to cephalexin (a pattern typical of E. coli in general) and 22% were resistant to enrofloxacin, emphasizing the need for selection of an antimicrobial on the basis of susceptibility testing (see Chapter 8). Presumably, achieving bactericidal concentrations of a drug at the site of the urinary tract infection is paramount, should the decision be made to treat the infection. Dogs with intervertebral disk disease have an increased risk of UTI when treated with glucocorticoids (see Chapter 27).
Wound Healing
The impact of glucocorticoids on wound healing has been generally recognized for decades.103a The impact of exogenous glucocorticoids largely reflects their antiinflammatory effects and down regulation of pro-inflammatory cytokines (as reviewed by Grose et al, 2002).103a However, these effects occur at supraphysiologic (i.e., pharmacologic) doses. The effects of endogenous glucocorticoids (physiologic concentrations) also appear to be suppressive, but these latter effects may actually be beneficial in the control of late would healing.103a
Immune Reactions
Paradoxically, despite their immunomodulatory effects, GLCs have been associated with allergic reactions in humans, albeit rarely. Types I, III, and IV reactions have been reported, with reactions more common in patients treated chronically (e.g., asthmatics, transplant recipients) and patients receiving either hydrocortisone or methylprednisolone intravenously.49,104 In addition to the active ingredient, reaction to (sulfate) excipients or to succinate salts has also been suspected. Vasculitis reactions have been reported after oral administration of either prednisone or prednisolone. Nakamura and coworkers105 reported anaphylaxis that occurred after intravenous administration of succinate-containing GLCs (hydrocortisone or methylprednisolone) in severely atopic asthmatic humans. In this retrospective study, phosphate-containing drugs appeared to be safe. The authors recommended slow intravenous drip administration in patients with severe allergic disease who have been previously exposed to GLCs. Erdmann and coworkers106 reported an anaphylactic reaction in an atopic person receiving oral prednisone; skin testing revealed reaction to prednisolone, prednisolone hydrogen succinate, prednisone, and betamethasone but not methylprednisolone or dexamethasone. Accordingly, anaphylaxis should be suspected with any GLC if clinical signs are supportive. Allergic reactions may not be limited to type 1. Finally, reactions may not be limited to active drug but may involve vehicles or salt forms. One case of anaphylaxis has been reported in a dog receiving dexamethasone.107 The dog had been previously treated with dexamethasone in propylene glycol as well as oral prednisone therapy for immune-mediated hemolytic anemia. The vehicle contained methylparaben and propylparaben as well as benzyl alcohol. No occurrences of anaphylaxis or anaphylactoid reactions were cited in a review of adversities associated with dexamethasone treatment in dogs with thoracolumbar disease (n = 161).97 In a review of the literature, Schaer and coworkers107 noted that anaphylactoid reactions also have been reported.
Respiratory
Pulmonary thromboembolism is a potential complication of hyperadrenocorticism and presumably also can occur with excessive exogenous GLC. The exact pathophysiologic mechanism is unknown but may be a result of obesity, hypertension, increased hematocrit level, or hypercoagulability. In addition, because of the myopathic effect of GLCs, muscles of respiratory function may be weakened in animals receiving high doses of GLCs.9
Growth Retardation
The administration of small doses of GLCs to children has been associated with retardation of growth. The mechanism may reflect changes in collagen synthesis but is likely to be much more complex. Prepartum exposure to GLCs can predispose experimental animals to malformations (e.g., cleft palate).
Contraindications
GLCs produce profound actions on virtually all systems of the body. Many of these effects can be tolerated by a healthy animal, but in certain disease states the use of GLCs may be deleterious. Diseases and conditions that the authors consider to be major, moderate, and minor contraindications to GLC therapy are mentioned in the subsequent sections.
Infectious Disease
Infectious diseases are a major contraindication. GLCs can exacerbate viral, bacterial, and fungal infections because of their immunosuppressive properties (although a single antiinflammatory dose of a short-acting agent is unlikely to be harmful).
Diabetes Mellitus
Diabetes mellitus is a major contraindication. GLC-mediated gluconeogenesis and general anti-insulin effects make regulating diabetic patients difficult.
Renal Failure
Renal failure is a moderate contraindication. Increased protein catabolism induced by GLCs results in increased quantities of nitrogenous waste products to be excreted. Additionally, patients with renal failure are already immunocompromised. Further immunosuppression would greatly increase their risk of developing pyelonephritis.
Corneal Ulceration
Corneal ulceration is a major contraindication. Topically administered GLCs delay healing of the cornea and may lead to corneal perforation.
Pancreatitis
Pancreatitis is a moderate contraindication. GLCs are believed to aggravate pancreatitis by increasing fatty acid circulation.
Gastrointestinal Ulceration
Gastrointestinal ulceration is a moderate contraindication. GLCs promote progression of ulcerative disease by delaying healing, increasing acid and pepsin secretion, and reducing the rate of mucosal cell proliferation in the gastrointestinal tract. Patients with eosinophilic enteritis, lymphocytic–plasmacytic enteritis, and certain ulcerative and granulomatous enteritides are potential exceptions. Colonic ulceration is a significant risk in animals with spinal trauma that are receiving dexamethasone. Concurrent administration of a nonsteroidal antiinflammatory drug greatly increases the likelihood of gastrointestinal ulceration and is not recommended. Misoprostol, a prostaglandin E2 analog, may provide protection against GLC-induced gastric ulceration.
Pregnancy
Pregnancy is a moderate contraindication. GLC administration has been associated with abortion and cleft palate in the dog. Additionally, GLC administration to a bitch within 24 hours of parturition reduces neonatal intestinal permeability of immunoglobulins, thereby reducing colostrum absorption.108
Epilepsy
Epilepsy is a moderate contraindication. Endogenous GLCs suppress neuronal excitability, perhaps by potentiating the inhibitory effects of the neurotransmitter γ-aminobutyric acid. Chronic GLC use has been reported to be associated with lowering the seizure threshold. The mechanism for this possible effect is not clear. Exogenous GLCs may; however, downregulate steroid receptors, resulting in increased neuronal excitability.
1. Mol J.A., van Wolferen M., et al. Predicted primary and antigenic structure of canine corticotropin-releasing hormone. Neuropeptides. 1994;27:7.
2. Prigent H., Maxime V., Annane D. Clinical review: corticotherapy in sepsis. Crit Care. 2004;8(2):122-129.
3. Feldman E.C., Nelson R.W. Hypoadrenocorticism (Addison’s disease). In: Feldman E.C., Nelson R.W., editors. Canine and feline endocrinology and reproduction. ed 3. St Louis: Saunders; 2004:394-439.
4. Webster J.I., Sternberg E.M. Role of the hypothalamic–pituitary–adrenal axis, glucocorticoids and glucocorticoid receptors in toxic sequelae of exposure to bacterial and viral products. J Endocrinol. 2004;181:207-221.
5. Funder J.W., Mihailidou A.S. Aldosterone and mineralocorticoid receptors: Clinical studies and basic biology. Mol Cell Endocrinol. 2009;301(1-2):2-6.
6. Alderson A.L., Novack T.A. Neurophysiological and clinical aspects of glucocorticoids and memory: a review. J Clin Exp Neuropsychol. 2002;24(3):335-355.
7. Burnstein K.L., Cidlowski J.A. The down side of glucocorticoid receptor regulation. Mol Cell Endocrinol. 1992;83:C1.
8. Rashid S., Lewis G.F. The mechanisms of differential glucocorticoid and mineralocorticoid action in the brain and peripheral tissues. Clin Biochem. 2005;38:401-409.
9. Schimmer B.P., Parker K.L. Adrenocorticotropic hormone: adrenocortical steroids and their synthetic analogs; inhibitors of the synthesis and actions of adrenocortical hormones. In: Brunton L.L., Lazo J.S., Parker K.L., editors. Goodman & Gilman’s the pharmacological basis of therapeutics. ed 11. New York: McGraw-Hill; 2006:1587-1612.
10. Mangos G.J., Whitworth J.A., Williamson P.M. Glucocorticoids and the kidney. Nephrology. 2003;8:267-273.
11. Gupa B.B.P., Lalchhandama K. Molecular mechanisms of glucocorticoid action. Curr Sci. 2002;83(9):1103-1111.
12. Rhen T., Cidlowsk J.A. Inflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med. 2005;353:1711-1723.
12a. Haller J., Mikics E., Makara G.B. The effects of non-genomic glucocorticoid mechanisms on bodily functions and the central neural system. A critical evaluation of findings. Front Neuroendocrinol. 2008;29(2):273-291.
13. Reutner W., Smith C.L. In vitro glucocorticoid receptor binding and transcriptional activation by topically active glucocorticoids. Arzneimittelforschung. 1998;48(9):956-960.
14. Kumar R., Thompson E.B. Gene regulation by the glucocorticoid receptor: structure: function relationship. J Steroid Biochem Mol Biol. 2005;94:383-394.
15. Bodine P.V., Litwack G. The glucocorticoid receptor and its endogenous regulators. In: Litwack G., editor. Receptor. Clifton, N.J: Humana; 1990:83.
16. Schulz M., Eggert M. Novel ligands: fine tuning the transcriptional activity of the glucocorticoid receptor. Curr Pharm Des. 2004;10:2817-2826.
17. Dostert A., Heinzel T. Negative glucocorticoid receptor response elements and their role in glucocorticoid action. Curr Pharm Des. 2004;10:2807-2816.
18. Brattsand R. The ideal steroid. Curr Pharm Des. 2003;9:956-1964.
19. Bacher S., Schmitz L.M. The NFκB pathway as a potential target for autoimmune disease therapy. Curr Pharm Des. 2004;10:2827-2837.
20. da Silva J.A.P., Jacobs J.W.G., Kinwan J.R., et al. Low-dose glucocorticoid therapy in rheumatoid arthritis. A revew on safety: published evidence and prospective trial data. Ann Rhem Dis online. Aug 26, 2005. Downloaded from ard.Bmjournals.com 10-10-2005
21. Lamberts S.W.J., Koper J.W., et al. Familial and iatrogenic cortisol receptor resistance. J Steroid Biochem Mol Biol. 1992;43:385.
22. Shipman G.F., Bloomfield C.D., Gajl-Peczalska K.J., et al. Glucocorticoids and lymphocytes. III. Effects of glucocorticoid administration on lymphocyte glucocorticoid receptors. Blood. 1983;61(6):1086-1090.
23. Reding R., Webber S.A., Fine R. Getting rid of steroids in pediatric solid-organ transplantation. Pediatr Transplant. 2004;8:526-530.
24. Akpek G., Lee S.M., Anders V., et al. A high dose pulse steroid regimen for controlling active chronic graft-versus-host diseases. Biol Blood Bone Marr Transpl. 2001;7:495-501.
25. Farrell R.J., Kelleher D. Mechanisms of steroid action and resistance in inflammation: glucocorticoid resistance in inflammatory bowel disease. J Endocrinol. 2003;178:339-346.
26. Tanaka J., Fujita H., Matsuda S., et al. Glucocorticoid and mineralocorticoid receptors in microglial cells: the two receptors mediate differential effects of corticosteroids. Glia. 1997;20(1):23-37.
27. Haynes R.C.Jr. Adrenocorticotropic hormone: adrenocortical steroids and their synthetic analogs; inhibitors of the synthesis and actions of adrenocortical hormones. In: Gilman A.G., Rall T.W., et al, editors. The pharmacological basis of therapeutics. ed 8. New York: Pergamon; 1990:1445.
28. Planey S.L., Litwack G. Glucocorticoid-induced apoptosis in lymphocytes. Biochem Biophys Res Commun. 2000;279(2):307.
29. Sugimoto Y., Ogawa M., Tai N., et al. Inhibitory effects of glucocorticoids on rat eosinophil superoxide generation and chemotaxis. Int Immunopharmacol. 2003;3(6):845-852.
30. Druilhe A., Létuvé S., Pretolani M. Glucocorticoid-induced apoptosis in human eosinophils: mechanisms of action. Apoptosis. 2003;8(5):481-495.
31. Reichardt H.M. Immunomodulatory activities of glucocorticoids: insights from transgenesis and gene targeting. Curr Pharm Des. 2004;10:2797-2805.
32. Ristimaki A., Narko K., Hla T. Down-regulation of cytokine-induced cyclo-oxygenase-2 transcript isoforms by dexamethasone: evidence for post-transcriptional regulation. Biochem J. 1996;318(Pt 1):325-331.
33. Crofford L.J. COX-1 and COX-2 tissue expression: implications and predictions. J Rheumatol Suppl. 1997;49:15-19.
34. Pujols L., Benitez P., Alobid I., et al. Glucocorticoid therapy increases COX-2 gene expression in nasal polyps in vivo. Eur Respir J. 2009;33:502-508.
35. Pfeilschifter J., Eberhardt W., Hummel R., et al. Therapeutic strategies for the inhibition of inducible nitric oxide synthase—potential for a novel class of anti-inflammatory agents. Cell Biol Int. 1996;20(1):51-58.
36. Campbell W.B. Lipid-derived autocoids: eicosanoids and platelet activating factor. In: Gilman A.G., Rall T.W., et al, editors. The pharmacological basis of therapeutics. ed 8. New York: Pergamon; 1990:600.
37. Yang Y.H., Hutchinson P., Santos L.L., et al. Glucocorticoid inhibition of adjuvant arthritis synovial macrophage nitric oxide production: role of lipocortin 1. Clin Exp Immunol. 1998;111:117-122.
38. Saruta T. Mechanism of glucocorticoid induced hypertension. Hypertens Res. 1996;19:1-8.
39. Wallerath T., Witte K., Schafer S.C., et al. Down-regulation of the expression of endothelial NO synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc Natl Acad Sci U S A. 1999;96(23):13357-13362.
39b. Ng M.K., Celermajer D.S. Glucocorticoid treatment and cardiovascular disease. Heart. 2004;90(8):829-830.
40. Seale J.P., Compton M.R. Side-effects of corticosteroid agents. Med J Aust. 1989;144:217.
41. Glade M.J., Krook L., et al. Morphologic and biochemical changes in cartilage of foals treated with dexamethasone. Cornell Vet. 1983;73:170.
42. Adams M.E. Cartilage research and treatment of osteoarthritis. Curr Opin Rheumatol. 1992;4:552.
43. Shirasawa N., Yamanouchi H. Glucocorticoids induce glutamine synthetase in folliculostellate cells of rat pituitary glands in vivo and in vitro. J Anat. 1999;194(Pt 4):567-577.
44. Vardimon L. Neuroprotection by glutamine synthetase. Isr Med Assoc J. 2000;2(Suppl):46-51.
45. Obrenovitch T.P. High extracellular glutamate and neuronal death in neurological disorders. Cause, contribution or consequence. Ann NY Acad Sci. 1999;890:273-286.
45b. Albrecht J., Sonnewald U., Waagepetersen H.S., et al. Glutamine in the central nervous system: function and dysfunction. Front Biosci. 2007;12:332-343.
46. Barnes P.J. Our changing understanding of asthma. Respir Med. 1989;83(Suppl):217.
47. Horcicka V., Linduskova M., Vykydal M. Injury to gastric mucosa due to cortisonoid therapy. Acta Univ Palacki Olomuc Fac Med. 1990;126:151-155.
48. Filaretova L., Mattcev N., Bogdanov A., et al. Role of gastric microcirculation in the gastroprotection by glucocorticoids released during water restraint stress in rats. Clin J Physiol. 1999;42:145-152.
49. Ventura M.T., Muratore L., Calogiuri G.F., et al. Allergic and pseudoallergic reactions induced by glucocorticoids: a review. Curr Pharm Des. 2003;9(24):1956-1964.
50. Roberts S.M., Lavach J.D., Macy D.W., et al. Effect of ophthalmic prednisolone acetate on the canine adrenal gland and hepatic function. Am J Vet Res. 1984;45:1711-1713.
51. Meyer D.J., Moriello K.A., et al. Effect of otic medications on liver function test results in healthy dogs. J Am Vet Med Assoc. 1990;196:743.
52. Zenoble R.D., Kemppainen R.J. Adrenocortical suppression by topically applied corticosteroids in healthy dogs. J Am Vet Assoc. 1987;191:685-688.
53. Ghubash R., Marsella R., Kunkle G. Evaluation of adrenal function in small-breed dogs receiving otic glucocorticoids. Vet Dermatol. 2004;15:363-368.
54. Ginel P.J., Garrido C., Lucena R. Effects of otic betamethasone on intradermal testing in normal dogs. Vet Dermatol. 2007;18:205-210.
55. Aniya J.S., Griffin C.E. The effect of otic vehicle and concentration of dexamethasone on liver enzyme activities and adrenal function in small breed healthy dogs. Vet Dermatol. 2008;19:226-231.
56. Reeder C.J., Griffin C.E., Polissar N.L., et al. Comparative adrenocortical suppression in dogs with otitis externa following topical otic administration of four different glucocorticoid-containingmedications. Vet Ther. 2008;9(2):111-121.
57. Willis-Goulet H.S., Schmidt B.A., Nicklin C.F., et al. Comparison of serum dexamethasone concentrations in cats after oral or transdermal administration using Pluronic Lecithin Organogel (PLO): a pilot study. Vet Dermatol. 2005;14:83-89.
58. Ueda K., Okamura N., Hirai M., et al. Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progresterone. J Biol Chem. 1992;267(34):24248-24252.
59. Graham-Mize C.A., Rosser E.J. Bioavailability and activity of prednisone and prednisolone in the feline patient, Plenary Session Abstracts. Vet Dermatol. 2004;25(Suppl 1):1-19.
60. Rohatag S., Appajosyula S., Derendorf H., et al. Risk-benefit value of inhaled glucocorticoids: a pharmacokinetic/pharmacodynamic perspective. J Clin Pharmacol. 2004;44:37-47.
61. McCormich J.R., Herman A.H., Lien W.M., et al. Hydrocortisoine metabolism in the adrenalactomized dogs: the quantitative significance of each organ system in the total metabolic clearance of hydrocortisone. Endocrinology. 1974;94:17-26.
62. El Dareer S.M., Struck R.F., White V.M., et al. Distribution and metabolism of prednisone in mice, dogs, and monkeys. Cancer Treat Rep. 1977;61(7):1279-1289.
63. Colburn W.A., Sibley C.R., Buller R.H. Comparative serum prednisone and prednisolone concentrations following prednisone or prednisolone administration to beagle dogs. J Pharm Sci. 1976;65:997-1001.
63a. Tse F.L.S., Welling P.G. Prednisolone bioavailability in the dog. J Pharm Sci. 1977;66:1751-1754.
64. Boothe DM, Bockelman A: Absorption of prednisolone and prednisone after transdermal versus oral administration in cats, In preparation, 2011.
64a. Uribe M., Summerskill W.H. Comparative serum prednisone and prednisolone following administration to patients with chronic active liver disease. Clin Pharmacokinet. 1982;7:452-459.
65. Arvidsson S., Falt K., Haglund U. Feline E. coli bacteremia—effects of misoprostol/scavengers or methylprednisolone on hemodynamic reactions and gastrointestinal mucosal injury. Acta Chir Scand. 1990;156:215-221.
66. Means E.D., Anderson D.K., Waters T.R., et al. Effect of methylprednisolone in compression trauma to the feline spinal cord. J Neurosurg. 1981;55:200-208.
67. Coates J.R., Simpson S., Wright J., et al. Clinicopathologic effects of a 21 aminosteroid compound (U743894) and high dose methylprednisolone on spinal cord function after simulated spinal cord trauma. Vet Surg. 1995;24:128-139.
68. Toutain P.L., Autefage A., Oukessou M., et al. Pharmacokinetics of methylprednisolone succinate, methylprednisolone, and lidocaine in the normal dog and during hemorrhagic shock. J Pharm Sci. 1987;76(7):528-534.
69. Greco D.S., Brown S.A., Gauze J.J., et al. Dexamethasone pharmacokinetics in clinically normal dogs during low- and high-dose dexamethasone suppression testing. Am J Vet Res. 1993;54(4):580-585.
70. Astra Zeneca, Budesonide (Rhinocort) package insert.
71. Hammer S., Spika I., Sippl W., et al. Glucocorticoid receptor interactions with glucocorticoids: evaluation by molecular modeling and functional analysis of glucocorticoid receptor mutants. Steroids. 2003;68:329-339.
72. Tumulty J., Broussard J., Steiner J.M., et al. The influence of budesonide on the pituitary-adrenal axis in dogs with inflammatory bowel disease (IBD). ACVIM. 2003.
73. Brown S.A., Hall E.D. Role of oxygen-derived free radicals in the pathogenesis of shock and trauma, with focus on central nervous system injuries. J Am Vet Med Assoc. 1992;200:1849.
74. Miller W.H., Scott D.W. Medical management of chronic pruritus. Compend Contin Educ Pract Vet. 1994;16(4):449.
75. Bevier D.E. Long term management of atopic disease in the dog. Vet Clin North Am. 1990;20:1486.
76. Gilbert S. Feline pruritis therapy. In: Bonagura J.D., Twedt D.C., editors. Kirk’s current veterinary therapy XIV: small animal practice. St Louis: Saunders, 2009.
77. Royschuk R.A. Otitis externa part II: an update on management. ACVIM Proc. 1994:145.
78. Chunekamrai S., Krook L.P., et al. Changes in articular cartilage after intra-articular injections of methylprednisolone acetate in horses. Am J Vet Res. 1989;50(10):1733.
79. Pelletier J.-P., Martel-Pelletier J. Protective effects of corticosteroids on cartilage lesions and osteophyte formation in the Pond-Nuki dog model of osteoarthritis. Arthritis Rheum. 1989;23:181.
80. Fajardo M., Di Cesare P.E. Disease-modifying therapies for osteoarthritis: current status. Drugs Aging. 2005;22(2):141-161.
81. Hoerlein B.F., Redding R.W., et al. Evaluation of dexamethasone, DMSO, mannitol, and solcoseryl in acute spinal cord trauma. J Am Anim Hosp Assoc. 1983;19:216.
82. Hoerlein B.F., Redding R.W., et al. Evaluation of naloxone crocetin, thyrotropin releasing hormone, methylprednisolone, partial myelotomy, and hemilaminectomy in the treatment of acute spinal cord trauma. J Am Anim Hosp Assoc. 1985;21:67.
83. Parnell N.K. Chronic colitis. In: Bonagura J.D., Twedt D.C., editors. Current veterinary therapy XIV. St Louis: Saunders; 2009:515-520.
84. Lefering R., Neugebauer E.A. Steroid controversy in sepsis and septic shock: a meta-analysis. Crit Care Med. 1995;23:1294-1303.
85. Hardie E.M., Kruse-Elliott K. Endotoxic shock. Part II: a review of treatment. J Vet Intern Med. 1990;4:306-314.
86. Haskins S.C. Management of septic shock. J Am Vet Med Assoc. 1992;200:1915.
87. Annane D., Sébille V., Trochée G., et al. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. J Am Med Assoc. 2000;283:1038-1045.
88. Minneci P.C., Deans K.J., Banks S.M., et al. Meta-analysis: the effect of steroids on survival and shock during sepsis depends on the dose. Ann Intern Med. 2004;141:47-56.
89. Cooper M.S., Stewart P.M. Corticosteroid insufficiency in acutely ill patients. N Eng J Med. 2003;348:727-734.
90. Sprung C.L., Annane D., Keh D., et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111-124.
90a. Salem M., Tainish R.E., Bromberg J., et al. Perioperative glucocorticoid coverage. A reassessment 42 years after emergence of a problem. Ann Surg. 1984;219:416-425.
90b. Jung C., Inder W.J. Management of adrenal insufficiency during the stress of medical illness and surgery. The Med J Aust. 2008;188:409-413.
91. Price G., Page R., Fischer B., et al. Efficacy and toxicity of doxorubicin/cyclophosphamide maintenance therapy in dogs with multicentric lymphosarcoma. J Vet Int Med. 1991;5:259-262.
92. Mealey K.L., Bentjen S.A., Gay J.M., et al. Dexamethasone treatment of a canine, but not human, tumour cell line increases chemoresistance independent of P‑glycoprotein and multidrug resistance-related protein expression. Vet Comp Oncol. 2003;1:67-75.
93. McCaw D.L., Miller M.A., et al. Response of canine mast cell tumors to treatment with oral prednisone. J Vet Intern Med. 1994;8(6):406.
94. Hollingsworth S.R. Ocular immunotherapy. In: Bonagura J.D., Twedt D.C., editors. Kirk’s current veterinary therapy XIV. St Louis: Saunders; 2009:1149-1153.
94a. Bourcier T., Forgez P., Bordene V., et al. Regulation of human corneal epithelial cell proliferation and apoptosis by dexamethasone. Invest Ophthamol Vis Sci. 2000;41:4133-4141.
95. Belvisi M.G., Bunchschuh D.S., Stoeck S., et al. Preclinical profile of ciclesonide, a novel corticosteroid for the treatment of asthma. J Pharmacol Exp Ther. 2005;314(2):568-574.
96. Steffan J., Alexander D., Brovedani F., et al. Comparison of cyclosporine A with methylprednisolone for treatment of canine atopic dermatitis: a parallel, blinded randomized controlled trial. Vet Dermatol. 2003;14:11-22.
97. Levine J.M., Levine G.J., Boozer L., et al. Adverse effects and outcome associated with dexamethasone administration in dogs with acute thoracolumbar intervertebral disk herniation: 161 cases (2000-2006). J Am Vet Med Assoc. 2008;232:411-417.
98. Bakke H.K., Murison R., Walther B. Effect of central noradrenaline depletion on corticosterone levels and gastric ulcerations in rats. Brain Res. 1986;368(2):256-261.
99. Badylak S.E., Van Fleet J.F. Sequential morphological and clinic pathologic alterations in dogs with experimentally induced glucocorticoid hepatopathy. Am J Vet Res. 1981;42:1310-1318.
100. Fittshen C., Bellamy J.E. Prednisone-induced morphologic and chemical changes in the liver of dogs. Vet Pathol. 1984;21:399-406.
101. Feldman E.C., Nelson R.W. Hypothyroidism. In: Feldman E.C., Nelson R.W., editors. Canine and feline endocrinology and reproduction. ed 3. St Louis: Saunders; 2004:86-151.
102. Ihrke P.J., Norton A.L., Ling G.V., et al. Urinary tract infection associated with long-term corticosteroid administration in dogs with chronic skin diseases. J Am Vet Med Assoc. 1985;186:43-46.
103. Torres S.M., Dias S.F., Nogueir S.A., et al. Frequency of urinary tract infection among dogs with pruritic disorders receiving long-term glucocorticoid treatment. J Am Vet Med Assoc. 2005;227:239-243.
103a. Grose R., Werner S., Kessler D., et al. A role for endogenous glucocorticoids in wound repair. EMBO Reports. 2002;3:575-582.
104. Calogiuri G.F., Muratore L., Nettis E., et al. Anaphylaxis to hydrocortisone hemisuccinate with cross-sensitivity to related compounds in a paediatric patient. Br J Dermatol. 2004;151(3):707-708.
105. Nakamura H., Matsuse H., Obase Y., et al. Clinical evaluation of anaphylactic reactions to intravenous corticosteroids in adult asthmatics. Respiration. 2002;69:309-313.
106. Erdmann S.M., Abuzabra F., Merk H.F., et al. Anaphylaxis induced by glucocorticoids. J Am Board Fam Pract. 2005;18:143-146.
107. Schaer M., Ginn P.E., Hanel R.M. A case of fatal anaphylaxis in a dog associated with a dexamethasone suppression test. J Vet Emerg Crit Care. 2005;15(3):213-216.
108. Gillette D.D., Filkins M. Factors affecting antibody transfer in the newborn puppy. Am J Physiol. 1966;210(2):419.