Chapter 29 Antiinflammatory Drugs
The Pathophysiology of Inflammation
Inflammation can occur in any vascularized tissue. Initiated as a protective mechanism intended to remove the underlying cause, be it chemical, physical, or biological, the acute inflammatory process facilitates return of the inflamed tissue to normal function.1,2 With time, however, its unfettered actions can lead to chronic inflammation, which can contribute to harm to the patient. The sequelae of inflammation are manifested as five cardinal signs: redness, heat, swelling or edema, pain, and loss of function. The initial response to vascular damage is vasoconstriction of small vessels in the area of injury, which serves to control hemorrhage within 5 to 10 minutes; however, vasodilation and increased vascular permeability of small venules are evident. Leukocytes, platelets, and erythrocytes in the injured vessels become “sticky,” adhering to the endothelium. Leakage of cells and plasma-derived protein-rich fluid is followed by platelet aggregation and fibrin formation. Initially, the predominant cell type infiltrating damaged tissues is the polymorphonuclear leukocyte (PMN), in part because it predominates in circulation. These cells provide a protective response by removing chemicals or other materials that may have initiated the inflammatory response. As the short-lived PMNs die, macrophages become the predominant cell type. The migration and concentration of PMNs to the site of injury is facilitated by chemical mediators that act as chemotactic agents. As PMNs die, the contents of the lysed cells accumulate to form the component of inflammatory exudate commonly referred to as pus.
The Role of Chemical Mediators in the Inflammatory Response
Released mediators perpetuate the inflammatory response (Table 29-1 and Figures 29-1 and 29-2) and are responsible for the clinical signs associated with inflammation, including pain and fever.3 Mediators (see Table 29-1) are derived from both the cells (both preformed and formed in situ) and fluid that reach the site of tissue damage by way of the bloodstream. Although quantitative differences between species and tissue concentrations of the mediators vary, the effect on and role in the pathophysiology of inflammation that each mediator has are predominantly the same. Leukocytes are a rich source of a variety of chemical mediators of inflammation. These cells, as well as cells of the injured tissues (either at the time of damage or after subsequent damage), perpetuate the inflammatory response and become potential targets of antiinflammatory drugs. Preformed mediators include those located in granules (e.g., histamine and serotonin) and lysosomes and other enzymes. Their release occurs in the earliest stages of inflammation, to be followed by the release of mediators formed in situ. This latter group includes products of arachidonic acid (AA) metabolism, including eicosanoids (prostaglandins [PGs], leukotrienes, and related compounds), and platelet-activating factor, as well as oxygen radicals, and cytokines. The role each of these mediators varies. Although most mediators are proinflammatory in action, antiinflammatory mediators also are formed. Examples include lipoxins, epoxyeicosatrienoic acids (EETs), hydroxyeicosatetranoic acids (HETEs), and epoxyoctadecenoic acids (EpOMEs), selected cytokines, and possibly cyclooxygenase-2 (COX-2)–generated PGs formed in later stages of inflammation (see Table 29-1).1,2 The induction of genes responsible for the generation of proinflammatory mediators is an emerging target of interest for antiinflammatory drugs. Among the inducible transcription factors that control inflammatory gene expression, nuclear factor NFκB is of particular interest because of its ability to coordinate soluble proinflammatory mediators such as cytokines and chemokines as well as leukocyte adhesion molecules. Normally, this factor is trapped in the cytosol by an inhibitory protein that hides the signal necessary for nuclear localization. However, in the face of proinflammatory cytokines (e.g., tumor necrosis factor-alpha [TNF-alpha], interleukin-1 [IL-1]), the inhibitory protein is phosphorylated and degraded, thus allowing movement of NFκβ to the nucleus.4 The role of matrix metalloproteinases (MMPs) in the inflammatory process is being increasingly recognized.5 The extraceullar matrix has long been recognized as a substrate for these proteolytic tissue-remodeling enzymes. However, other molecules serve as substrates, including cytokines and chemokines. As such, MMPs may act to control or perpetuate inflammation, and the sequelae of their inhibition may lead to adverse reactions as well as therapeutic benefits.
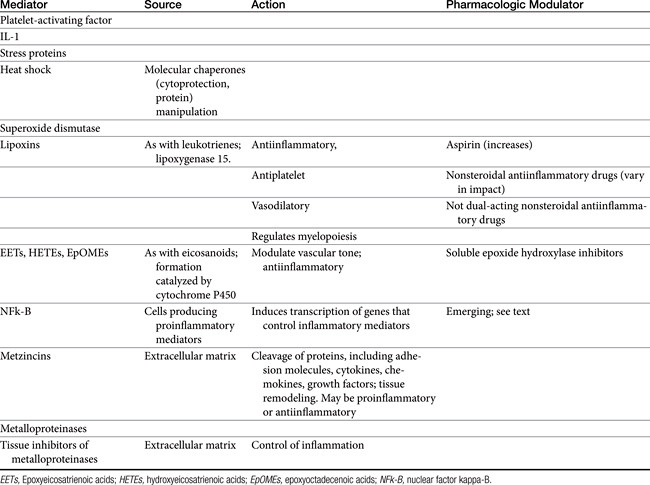
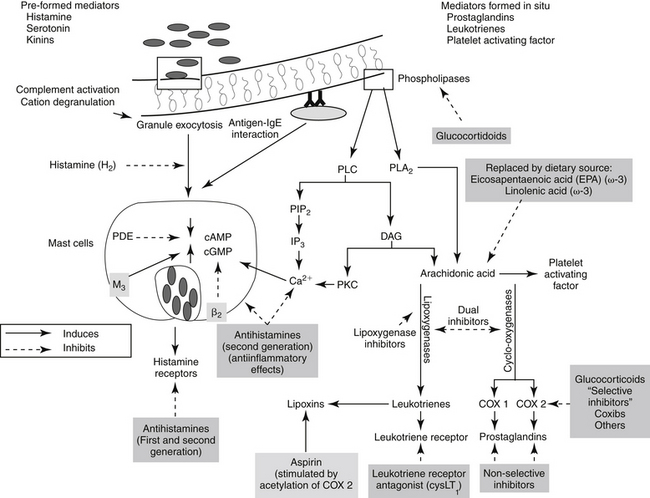
Figure 29-1 Leukocytes are an important source of inflammatory mediators that perpetuate the inflammatory response. Cellular mediators of inflammation include those preformed in granules or lysosomes, such as histamine and serotonin, and those formed in situ from arachidonic acid released by phospholipases in the cell membrane. Muscarinic receptors (M3) stimulate, and β-adrenergic receptors (2) inhibit, inflammatory mediator release. Although the phagocytic cell is intimately involved in the inflammatory reaction, it is not the only cell type capable of generating mediators of inflammation. The mediators of cellular origin interact with plasma-derived mediators, further compounding the response. Drugs used to control inflammation generally target specific mediators (see boxes) (see Table 29-1). cAMP, Cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; DAG, diacylglycerol; IP3, inositol triphosphate; PAF, platelet-activating factor; PDE, phosphodiesterase; PIP2, phosphatidylinositol; PLA2, phospholipase A2; PLC, phospholipase C.
Figure 29-2 The release of arachidonic acid from the phospholipids in cell membranes leads to a cascade of events resulting in the formation of inflammatory mediators. The family of cyclooxygenases (COX) results in the formation of constitutive prostaglandins (COX-1) and inducible prostaglandins (COX-2). The inducible prostaglandins contribute to all cardinal signs of inflammation. Selectivity of nonsteroiodal antiinflammatory drugs (NSAIDs) for COX-2 ultimately may result in marked safety of the NSAIDs. Lipoxygenases result in the formation of leukotrienes, which, along with their precursors, are potent inflammagens in a number of tissues. However, among these products are antiinflammatory lipoxins. The cysteinyl leukotrienes play a major role in type I hypersensitivity and potentially in chronic inflammatory diseases as well (see Chapter 19). Platelet-activating factor also is a potent inflammagen, although some of its actions may be mediated through leukotriene activity.
KEY POINT 29-1
The inappropriate inflammatory response reflects an imbalance between proinflammatory and antiinflammatory mediators.
Plasma-derived mediators also are important contributors to the inflammatory process. These include the kinins (e.g., bradykinin), released from their precursor form after appropriate physiologic or pathologic stimulation; complement and complement-derived peptides, released after activation of either the classic or the alternative pathway; and fibrinopeptides, released during the conversion of fibrinogen to fibrin during the clotting process and subsequent proteolysis of fibrin by plasmin.
Acute inflammation can cause severe organ or life-threatening damage. Monocytes, which follow neutrophils to the site of inflammation several hours later, release collagenases and elastases, softening local tissues. Interleukin release draws fibroblasts to the area, which in turn deposit collagen at the site. The collagen is gradually remodeled, and new blood vessels continue to form until oxygen tension is normal. If the cause of inflammation is removed, healing is complete. Failure to remove the inciting cause leads to persistent, chronic inflammation. Pharmacologic control of inflammation is oriented toward preventing the release of various chemical or plasma mediators, inhibiting their actions and treating pathophysiologic responses to them. Drugs useful for modulating the activity of chemical mediators derived from cells, plasma, or both are summarized in Table 29-1. Among the most important and frequently used drugs are the nonsteroidal antiinflammatory drugs (NSAIDs), all of which control fever; pain; and, to varying degrees, inflammation. A number of drugs and novel ingredients target inflammation particularly associated with specific conditions including osteoarthritis and septic shock; these are discussed separately. Glucocorticoids engage such a substantial role in the contribution, prevention, and treatment of inflammation that their discussion warrants a separate chapter.
Shared Pharmacology of Nonsteroidal Antiinflammatory Drugs
Chemistry
Although NSAIDs have been variably defined, the name is used to describe compounds that are not steroidal and that suppress inflammation. Generally, the classification is restricted to those drugs that inhibit one or more steps in the metabolism of AA, generally at the COX side of the cascade.6 The NSAIDs vary in their ability to influence inflammation. The mechanism of action of some of these drugs is not limited to inhibition of AA metabolism.7
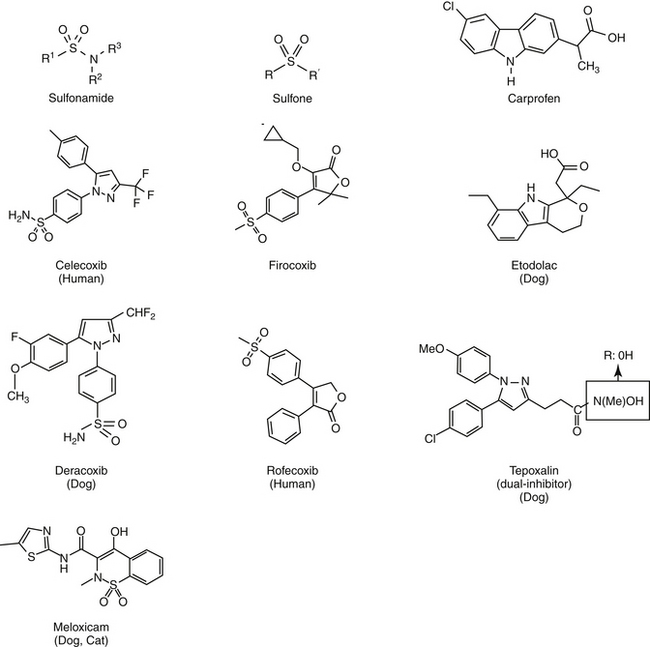
Aspirin, one of the earliest components of herbal therapy, is the progenitor NSAID, and terms such as aspirinlike and aspirin and related drugs have been used to refer to NSAIDs,6 although this older terminology may become obsolete with approval of newer NSAIDs. Structurally, NSAIDs can be broadly classified as either the salicylate or carboxylic acid derivatives, including the indoles (indomethacin), propionic acids (carprofen, ibuprofen, and naproxen), fenamates (mefenamic acid), oxicams (piroxicam), and pyrazolones or enolic acids (phenylbutazone and dipyrone)(Figures 29-3 and 29-4).6 Functionally, NSAIDs increasingly are being categorized by the enzymes they target: conventional (COX-1 and COX-2); newer COX-2–selective (COX-1 protective), which includes the coxibs as well as similarly acting drugs; more appropriate and dual inhibitors (tepoxalin) (see Figure 29-2). The coxibs include the sulfones and the sulfonamides. However, the term coxib refers to a chemical structure (see Figure 29-4) and not pharmacologic action and should not be used to refer to all drugs that might preferentially target COX-2 compared with COX-1. The coxibs are not arylamine sulfonamides (see Chapter 4), and therefore allergic reactions typical of sulfonamides antibiotics (see Chapter 7) may not occur. Nonetheless, several of these drugs are associated with keratitis sicca or suppression of thyroid gland activity.
Mechanism of Action
Prostaglandins
Prostaglandin Formation
Eicosanoids, such as PGs and leukotrienes, are 20-carbon chain derivatives of cell membranes. Eicosanoids are potent mediators of inflammation and are particularly important in the later stages.3,8 These compounds are synthesized when oxygen reacts with the polyunsaturated fatty acids of cell membrane phospholipids (see Figures 29-1 and 29-2). The most important of these fatty acids is AA, an omega-6 fatty acid, which is released into the cell from phospholipids of the damaged cell membranes. Release reflects activation of phospholipase A2 (dependent on calcium and calmodulin) located in the cell membrane.9 Once inside the cell, AA serves as a substrate for enzymes that generate intermediate and ultimately the final eicosanoid end product.8,10 The formation of lipoxygenases and their subsequent inhibition is discussed under “Miscellaneous Antiinflammtories.” COX PG synthase or prostaglandin H (PGH) synthase, located in all cells except mature red blood cells, add oxygen to AA, generating unstable PG endoperoxides (PGG2). Subsequent peroxidase reactions convert PGG2 to PGH2, the precursor of all PGs and thromboxane. The final PG end product depends on the presence of specific isomerase reductase or synthetase enzymes, some of which may be inducible.8,9
The direct effects of PGs are mediated through cell membrane–spanning G protein receptors located on target tissues and cells. At least nine subtypes have been identified, corresponding to each of the COX metabolites: DP1-2 (PGD2), EP1-4 (PGE2), FP A, B (PGF2α), IP (PGI2), and TP α, β (TXA2) (G&G);11 another four have been characterized for PGE2, opening the potential for selective drug activity.9 In addition to their receptor interactions, PGs also act indirectly by enhancing other mediators such as histamine and bradykinin.
The role of PGs in normal physiology as well as in disease might best be understood by considering them as protective in nature, even those associated with inflammation. Their formation is mediated by one of at least two isoforms of cyclooxygenases (see Figures 29-1 and 29-2), located on different genes in humans.12-14 COX-1, the “housekeeping” isoform,14 mediates the formation of constitutive PGs produced by many tissues, including gastrointestinal cells, platelets, endothelials cells, and renal cells. PGs generated from COX-1 are constantly present, providing homeostasis through a variety of normal physiologic effects. These include protection of the gastrointestinal mucosa, hemostasis, and the kidney when subjected to hypotensive insults. COX-2 is the product of an “immediate-early” gene that is rapidly inducible and tightly regulated.14 Regulation of COX expression is complex, with more sites present on the COX-2 compared with the COX-1 gene. Its expression is tightly restricted (but not absent) under basal conditions, but it is dramatically upregulated in the presence of inflammation or other diseases.9 Diseases associated with increases include (in humans) rheumatoid arthritis, seizures, ischemia, and (posttranslational) cancer.9 Proinflammatory cytokines such as TNFα and the interleukins stimulate the expression of COX-2 in many cell types, such as synovial cells, endothelial cells, chondrocytes, osteoblasts, and monocytes and macrophages.14 COX-2 catalyzes the formation of inducible PGs, which are needed only intermittently or under specific situations.12,13,15,16 A third COX isoform has been suggested, but it appears to be a variant of COX-1. Originally identified in high concentrations in canine cerebral cortex (hence the term used by some investigators, “canine COX-3”),17 it is inhibited by acetaminophen (and other NSAIDs). Its inhibition may influence central analgesic effects of NSAIDs.1,2 Differential or selective inhibition of COX-2 clearly offers potential some safety benefits by avoiding loss of homeostatic PGs; however, loss of COX-2 activity is also associated with adversities.14 It is important to note that COX-2 is ofttimes constitutively expressed: in the kidney and brain it mediates a cytoprotective effect in damaged or inflamed gastrointestinal mucosa.
Cyclooxygenases in health and disease
An appreciation of both efficacy and safety of COX inhibition (e.g., NSAIDs) is facilitated by an understanding of the role of COX in healthy and diseased tissues. COX has been described as the most common target of drug therapy with NSAIDs. In general, inhibition of COX-2 is responsible for efficacy, whereas inhibition of COX-1 is responsible for side effects.9 However, strict adherence to this simplistic approach will lead to therapeutic failure and increased morbidity with NSAID use. Although COX-1 does indeed appear to be the predominant constitutive enzyme responsible for housekeeping, both COX-1 and COX-2 are constitutively expressed in many tissues. Further, although COX-2 clearly is more active in the promotion of inflammation, COX-1 does appear to have some role.
Inflammation
PGs, and primarily PGE2, induce vasodilation, capillary permeability, and chemotaxis. As such, PGs cause the cardinal and clinical signs of inflammation, including pain and fever.8 PGE also modifies both T-cell and B-cell function, in part by inhibition of IL-2 secretion.8 Other inflammatory effects of PGE include its regulation IL-6, macrophage colony-stimulating factor, and vascular endothelial growth factor. In general, research consistently indicates that it is COX-2 that predominantly mediates formation of PGE associated with inflammation, pain and fever. For example, whereas COX-1 is largely absent in normal synovial cells, COX-2 is induced in most types of arthritis, including inflammatory arthritis in animals and rheumatoid arthritis in humans. In cartilage COX-2 is associated with IL-1 degradation of proteoglycan and apoptosis of synovial cells.18 However, not all of the inflammatory actions of COX-2 are undesirable in that COX-2 induction in response to inflammation contributes to tissue healing. For example, COX-2 is associated with healing in ligaments, bone, the gastrointestinal tract, and other tissues. The importance of COX-2 to dermal healing is not yet known.9
Central Nervous System
Both COX-1 and COX-2 are consitutively expressed in the brain and spinal cord. Constitutive COX is very responsive to ischemia, immunomodulation, cytokines, toxins, brain damage, and maturation processes.17 However, although both isoforms are present, COX-2 predominates. PGs of the central nervous system (CNS) play a major role in pain; howevever increasingly they are recognized for roles in other disorders. Among them is the role of COX in the pathogenesis of Alzheimer’s disease (AD). Extracellular deposition of fibrillar amyloid β (Aβ), intracellular accmumulation of abnormally phosphorylated tau protein, and subsequent formation of Aβ plaques mediating neurodegeneration and dementia in AD are associated with inflammation and COX-2.19 Mediators of inflammation are present throughout all stages of the disease, whereas COX-2 is absent in normal astrocytes or microglial cells. Further, COX-2 is upregulated in acute brain injury and in animal models of AD.9,19 Finally, the connection between AD and COX is supported by a lower incidence of AD in patients with rheumatoid arthritis, presumably because of the use of NSAIDs for its treatment.19 The protective effects of NSAIDs in the AD patient may reflect the antiplatelet properties of NSAIDs; aspirin in particular may decrease the risk of ischemic damage induced by blocked capillaries of the brain. Decreased formation of amyloid β protein also has been proposed,9 suggesting a possible role in other diseases associated with amyloid β protein deposition. Finally, COX-2 appears to be involved in the loss of glutamate-induced apoptotic cell death.19 Because N-methyl-d-aspartate (NMDA) receptors stimulate the arachidonic acid cascade, NSAIDs mute the role of NMDA receptors in pain. NSAIDs may also inhibit NMDA–mediated neuronal cell death by preventing increased extraceullar glutamate.19a
Pain
PGs have been implicated in causing increased pain perception (allodynia) in damaged compared with normal tissues.1,2 Induced COX-2 PGE as been associated with hyperalgesia (exaggerated response to pain) at the level of the spinal cord (primary hyperalgesia) or nociceptors (secondary hyperalgesia).17 Induction of COX-2 in the dorsal horn has been associated with central sensitization, manifested as a change in excitability threshold.9,17,18 Currently being descriped is the ability of PGs to contribute indirectly to neuropathic pain through influence on chemical mediators (e.g., histamine, bradykinin, substance P, nitric oxide [NO]), neurotransmitters (e.g., glycine inhibition or glutamate stimulation), or modulation of other receptors (e.g., NMDA).
Gastrointestinal and Other Healing
Both COX-1 and COX-2 are constitutively expressed in the gastrointestinal tract. However, constitutive expression of COX-1 has a predominant role in the protection of the gastrointestinal tract. COX-1 PGs decrease hydrochloric acid secretion, increase mucosal bicarbonate and mucus production, and increase epithelial cell proliferation and mucosal blood flow (Figure 29-5). The latter effect facilitates delivery of oxygen and nutrients to proliferating cells as well as rapid removal of damaging hydrogen ions that make their way into the mucosa. Drugs that express preferential potency for COX-2 PGs generally are associated with fewer gastrointestinal side effects compared to those targeting both COX isoforms, although the relative protection varies with the drugs. Protection appears to be more important in the presence of high drug concentrations (e.g., high doses).20 COX-2 is critically important to the gastrointestinal tract as well, being important for healing of gastrointestinal damage: COX-2 appears within 1 hour of gastrointestinal damage. Newer coxib PGs will inhibit gastrointestinal healing as has been demonstrated in dogs receiving firocoxib (see the discussion of tepoxalin).21 Interestingly, in the pancreas, constitutive COX-2 expression dominates, although the clinical relevance of this is not yet known.20 The impact of NSAIDS on bone healing is an emerging concern. COX influences osteoblast and osteoclast; experimental studies have demonstrated impaired bone healing.20a Their use in controlling pain and limiting ectopic bone growth must be balanced with the increased risk of impaired fracture healing; conventional NSAIDs might be preferred compared to newer NSAIDs. The impact of NSAID on soft tissue healing (or ligaments) remains to be confirmed; whereas short term use may have minimal negative (and potentially a positive) impact, long term used might be approached cautiously. A recent meta-analysis in humans found no advantage in terms of return to function for coxibs, but recommended caution with long term use.20b A strong association has been reported between NSAIDs and severe necrotizing soft tissue infections.20c
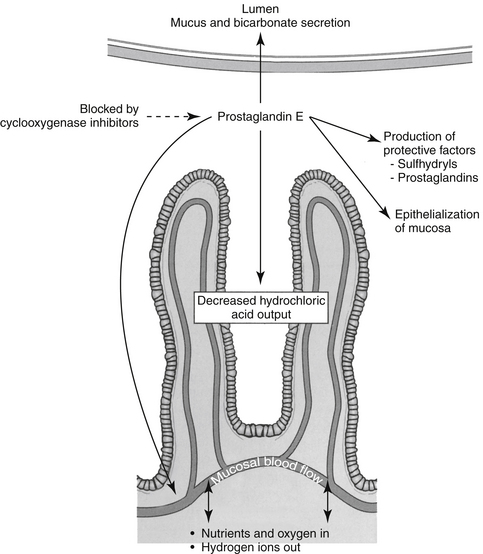
Figure 29-5 Among the many protective functions of constitutive prostaglandins is protection of the gastrointestinal mucosa from acid and other mediator-induced damage. Protective mechanisms include production of mucus and bicarbonate and inhibition of hydrogen ion secretion; epithelial turnover, which ensures rapid replacement of damaged cells; and increased mucosal blood flow, which ensures provision of oxygen and nutrients to the rapidly dividing epithelial cells as well as removal of hydrogen ions that are able to pass into the cells.
Cardiovascular Disease
The role of PGs in the cardiovascular system is largely beneficial, and the relationship between COX-1 and COX-2 and their respective PG end products exemplifies the complex “ying–yang” balance that characterizes them (Figure 29-6). Platelets contain thromboxane, a synthase, which catalyzes the formation of thromboxane from AA. Thrombosis reflects platelet aggregation and vasoconstriction. The formation of a thrombus is kept in check by the presence of prostacyclin synthase in vascular endothelial cells. This enzyme catalyzes metabolism of AA to prostacyclin (PGI2), a vasodilatory and platelet-inhibiting PG end product. However, whereas thromboxane A2 (TXA2) is associated with COX-2, prostacyclin synthase co-localizes with COX-1. Thus, whereas drugs that target both COX isoforms will potentially allow the balance to be maintained, drugs that preferentially target only one isoform risk disruption of the balance. Such may be the case with COX-2 selective NSAIDs. Their preferential inhibition of COX-2 may allow thrombus formation to go unchecked, increasing the risk of thromboembolic disorders. Indeed, in a meta-analysis of human clinical trials, two coxib drugs (rofecoxib and valdecoxib) were associated with an increased risk of stroke (see “Adverse Events”).20d
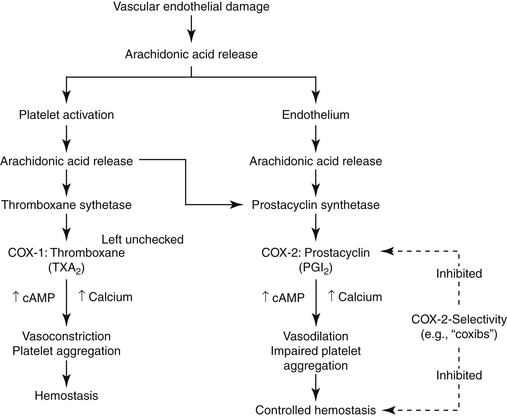
Figure 29-6 The role of constitutive prostaglandin products in hemostasis exemplifies the “ying-yang” relationship that often characterizes their action. Platelet activation in response to the damage is accompanied by the release of arachidonic acid, which is catalyzed by thromboxane synthetase, present in platelets, to thromboxane. Thromboxane causes platelet aggregation and local vasoconstriction. Excessive hemostasis is kept in check, however, by the simultaneous release of arachidonic acid from the damaged endothelial cell surface. Prostacyclin synthetase, located in the endothelial cells, results in the formation of prostacyclin, which is vasodilatory and inhibits platelet aggregation. cAMP, Cyclic adenosine monophosphate.
Kidney
In the kidney both COX-1 and -2 are constitutively expressed. Both are formed in the macula densa of humans and animals, but COX-2 may have a more important role than COX-1 (Figure 29-7). In animals, inhibition of COX-2 causes sodium and potassium retention in salt-depleted, but not normal, animals. However, in humans COX-2 appears to influence renal vasculature and podocytes. The role of COX in the kidney requires further elucidation before safety can be assumed for any NSAID; sparing COX-1 and targeting COX-2 can be expected to alter renal function. For example, kidneys do not develop in the embryos of COX-2–null knockout mice.22 The role of COX differs among tissues and species; their impact on adverse reactions are addressed under “Adverse Reactions.”9
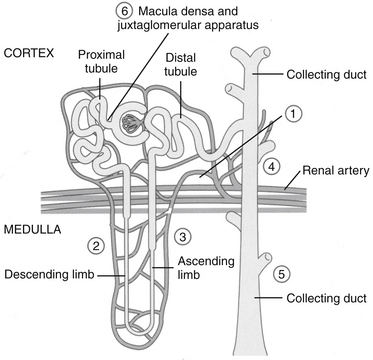
Figure 29-7 Both constitutive and inducible prostaglandins are important to renal blood flow, with the specific effect of each product varying among species. Renal prostaglandins ensure that intramedullary renal blood flow and urine formation will continue in the presence of decreased renal perfusion. Sites of action are noted by numbers and include shunting of blood from the cortices to the medullary intersitium, stimulation of natriuriesis, and inhibition of antidiuretic hormone. The nephrotoxicity of selected drugs reflects inhibition of renal prostaglandins.
Lungs
The role of PGs in the lungs does not appear to be as important as that of leukotrienes; the impact is primarily in disease, rather than health, for both eicosanoids. However, inflammatory diseases, such as asthma, are associated with smooth muscle proliferation, which is inhibited by COX-2. Thus, as in the gastrointestinal tract, COX-2 appears to have a protective role in the diseased lung. An interesting complication regarding eicosanoids in general is exemplified in the lungs: the increased formation of leukotrienes (LTs) in the presence of NSAIDs, presumably because of the increase availability of AA in the face of decreased PG synthesis.
Reproductive tract
Both COX-1 and -2 play a significant role in the normal reproductive tract. Induction of COX-2 is associated with ovulation, fertilization, implantation, and decidualization, as well as induction of labor.22 Female knockout mice devoid of COX-2 are largely infertile.22
Cancer
In the 1990s a reduced risk of colon cancer was associated with consistent aspirin use. Subsequent studies demonstrated a marked increase in COX-2 in a variety of soft-tissue tumors in humans and in transitional cell carcinoma in dogs.23 These studies suggest that benefits of NSAIDs in cancer may reflect inhibited COX-2.9 Mechanisms by which COX-2 may facilitate cancer growth or spread include impaired apoptosis, transactivation of epidermal growth factors or receptors and promotion of angiogenesis.9,17 Depending on the model, inhibition of COX-2 by NSAIDS reduces cell proliferation, increases apoptosis, and reduces metastasis. COX-2 inhibition may also enhance antitumor effects of radiation, although host toxicity also will be increased. Not surprisingly, gastrointestinal toxicity of anticancer drugs is also increased, presumably reflecting a combined toxic effect on the gastrointestinal tract.9 COX-1 may also have a role in cancer as is suggested by a decrease in colon cancer in COX-1 knockout mice.24
Cycloloxygenase and nonsteroidal antiinflammatory drugs
NSAIDs (Table 29-2) act to block PG synthesis by binding to and inhibiting cyclooxygenase (see Figures 29-1 and 29-2).8 This action is both dose and drug dependent. The planar form that characterizes these drugs is thought to facilitate their binding to COX.6,25 Several investigators have shown that some drugs (e.g., phenylbutazone and flunixin meglumine) also reduce later steps of the formation of PGE2 in inflammatory exudate at therapeutic doses.26 However, both the major therapeutic and toxic effects of NSAIDs have been correlated extensively with their ability to inhibit PG synthesis, with anti-inflammatory potency related to potency of impaired PG synthesis.8
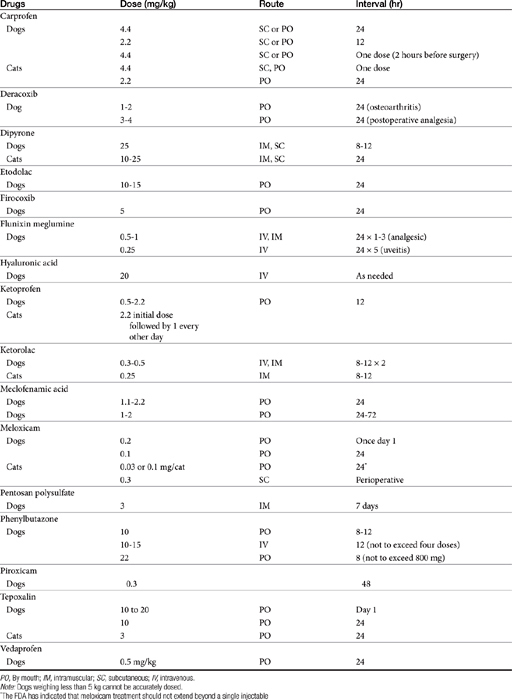
The differential effect of NSAIDs on the isoforms of COX presumably contribute to clinical differences in efficacy and safety, .17,27 Whereas as a class, NSAIDs appear to inhibit both COX-1 and COX-2, the ratio of the concentration of a NSAID necessary to inhibit the same level of COX-1 vesrus COX-2 activity (e.g., inhibition of 50% or 80% activivity [IC50 or IC80]) compares the potency each drug toward each isoform, and as such, provides a basis for screening predicted relative safety and efficacy of each. Thus, a COX-1 to COX-2 ratio greater than 1 (or a COX-2 to COX-1 ratio of less than 1) indicates greater potency for COX-2 which is desirable in that formation of inflammatory PGS should be inhibited preferentially to the housekeeping PGs.12,13,15,16 Inhibition of platelet activity is commonly used to measure in vitro COX-1 inhibition whereas inhibition of PGs released from macrophages stimulated with endotoxin is used to measure COX-2. A number of drugs associated with in vitro COX-2 selectivity in humans, including rofecoxib (Vioxx) and celecoxib (Celebrex); others include etodolac, meloxicam, and nimesulide; the newest drugs are valdecoxib, etoricoxib, and (soon to be approved) lumiracoxib.9 Drugs approved for use in animals include carprofen (the first) followed by etogesic; deracoxib; meloxicam; and, most recently, firocoxib.
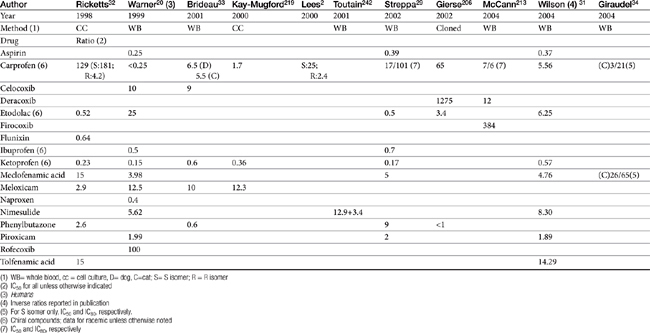
Although the gene for COX-1 is about 3 times as large as that for COX-2, the two COX proteins, which are membrane bound, share about 60% homology.9 Differences in NSAID selectivity for these two enzymes reflects smaller amino acids at two positions in COX-2 compared with larger amino acids at the same site in COX-1. The result is a larger and more flexible “pocket” into which the newer drugs insert and inhibit the COX-2 isoform.9 For example, ”coxib” drugs contain a sulfonamide group that interacts through hydrogen bonding to arginine of COX-2 but not histidine in the same position in COX-1.28 Despite the usefulness of the COX-1:COX-2 ratio in screening drugs for safety or efficacy, its applicability to clinical use is questionable. Further, comparing results among studies is difficult. For example, whereas some studies focus on the IC50, others determine the IC80, the latter probably being more clinically relevant. Assay methods contribute to variability in ratios among investigators for the same drug in the same species (Table 29-3). In vitro assasys based on cell culture or recombinant enzymes are easier to perform, but it is the ex vivo whole blood assays (as opposed to in vitro cell culture assays) that generally are recognized as the most representative of the clinical patient.1 However, even whole blood assays do not take into account different distribution patterns to different tissues.18,22 Extrapolation among species should be avoided.29 For example, wherease the COX-1 to COX-2 ratio of etodolac is much better than that for carprofen in humans,20,30 the opposite is true in dogs. Indeed, Wilson and coworkers31 have demonstrated that, wherease critical residues in the active sites of canine COXs are identical with human homologs, amino acids differ (up to 54) at in active sites and may contribute to differences in activity that preclude extrapolation among species (see Table 29-3). Further complicating extrapolation among and within species is the fact that NSAIDs generally exist and are largely marketed as enantiomers (see Chapter 1), each with a possible different ratios. For example, Ricketts and coworkers32 found the COX-1:COX-2 ratio of carprofen to be 129 for the racemic mixture but 181 for the S isomer and only 4.19 for the R isomer. Little information is available regarding COX selectivity in cats.33,34 Much of the information that is available is based on manufacturer-sponsored studies and as such might potentially reflect reporting bias. For example, in Table 29-3, drugs that generally performed best in each study tended to be the drug manufactured by the sponsor of the study. As such, COX ratios might be considered a screening procedure1,2
Table 29-3 COX-1 to COX-2 Ratios for Selected Nonsteroidal Antiinflammatory Drugs in the Dog by Different Investigators
Other Mechanisms of Action
Inhibition of eicosanoid formation is not the sole antiinflammatory mechanism of action of NSAIDs. The NSAIDs also appear to alter cellular and humoral immune responses and suppress inflammatory mediators other than PGs.7 As a group, all NSAIDs are planar and anionic, thus able to partition into lipid environments, including neutrophil cell membranes. As a result, cell membrane viscosity is altered, even at low concentrations.10 At higher concentrations, NSAIDs appear to uncouple protein–protein interactions within the plasma membrane and thus interfere with a variety of cell membrane processes. Examples include oxidative phosphorylation and cellular adhesion.10 Response of inflammatory cells to extracellular signals is impaired by affecting signal transduction proteins (G proteins).10 Neutrophil adherence and activation is inhibited, as is subsequent release of inflammatory cellular enzymes, including collagenase, elastase, hyaluronidase, and others.7 The extent of these effects varies with the drug. Whereas piroxicam inhibits superoxide ions and lysosomal enzymes release, ibuprofen does neither.10 All NSAIDs appear to inhibit neutrophil adhesion. Some NSAIDS directly block PG receptors. Inhibition of nuclear transcription factor NF- B, is responsible for expression of many genes linked to inflammation. Modulation of peroxisome proliferator-activated receptors (PPARs), heat shock proteins (HSPs) and mitogen-activated protein kinases (MAPK cascades), and impaired induction of nitric oxide synthase (iNOS) are among the COX-independent effects of the class of NSAIDs.2,28,35 Selected drugs (e.g., celecoxib but not rofecoxib) directly inhibit signaling needed to promote angiogensis, invasiveness, and proliferation of neoplasias.28 Interestingly, sulfone coxibs (see Figure 29-4) are pro-oxidant, whereas sulfonamide coxibs are not.28 Connective tissue metabolism may also be affected.7
NSAIDs are immunomodulators by virtue of their effect on several PGs and LTs as immunomodulators.8 Nonsteroidal antiinflammatory cells indirectly influence lymphocyte activity through altered PG formation.7 Selected NSAIDs appear to enhance cellular immunity by inhibiting PGE2, a mediator that dampens the immune response.7 This effect appears to be more important in the immunosuppressed animal.
Pharmacokinetics
Shared Pharmacokinetic Properties
Each drug will be discussed in depth; however, the NSAIDs share a number of pharmacokinetic properties as a class. Package insert data for dog and cat contains selected information for some drugs, although the information often is not comprehensive, with content tending to vary, in part based on the requirements implemented by the FDA at the time of drug approval. The United States Pharmacopeia has provided a series of monographs providing pharmacokinetic information regarding the use of NSAIDS in species; information will be addressed for individual drugs. As weak acids, the NSAIDs tend to be well absorbed after oral administration. Bioavailability can vary between animals but has not been established for many drugs because of the lack of intravenous preparations.36 Solutions of selected injectable preparations tend to be alkaline and can cause necrosis or pain if perivascular leakage occurs Food can impair the oral absorption of some NSAIDs or contribute to drug interactions.37,38 The drugs are lipid soluble. Whereas the volume of distribution tends to be small (approximating 10%) when based on total drug as a result of ≥ 90% binding to serum albumin39 the volume of distribution of unbound drug is consistent with distribution to extracellular fluid (Table 29-4). Only a small portion of pharmacologically active drug reaches peripheral tissues. Displacement from albumin (e.g., due to competition with other substrates for binding sites or from decreased serum albumin concentrations) may result in higher than expected concentrations of pharmacologically active drug and thus predispose the patient to drug-induced adverse effects. Although this increase is likely to be transient as the elimination of of unbound drug increases,36,40 adversities might yet emerge particularly in patients at risk (e.g, decreased hepatic or renal function). Clearance of unbound drug is likely to be reduced in geriatric patients. The volume of distribution of unbound drug in pediatric animals is twice that in adults, which will contribute potentially to longer half-life.36 For this and other reasons, (see metabolism), NSAIDs should not be used in the pediatric patient.
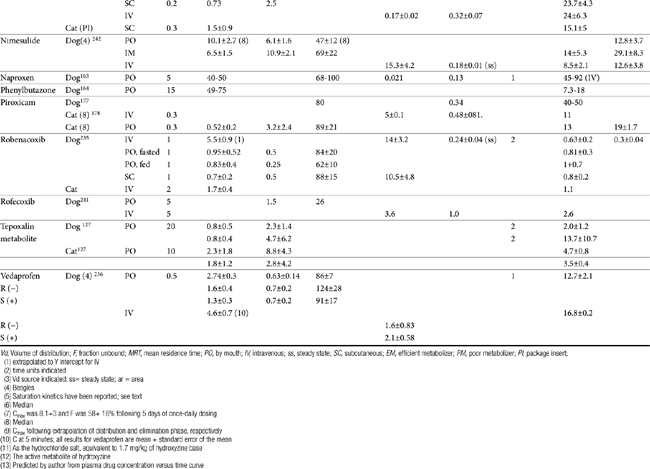
Most NSAIDs are eliminated primarily by both phase I and II hepatic drug-metabolizing enzymes. Clearance differs in both rate and extent among drugs and species. Differences in clearance are largely responsible for differences in drug half-life among animals.36 Failing to anticipate these differences may contribute to adverse reactions. Conjugated parent drug or metabolites are eliminated through the bile or urine, depending on the drug. Stereoselective metabolism plays a profound role in species, gender and age differences. Saturation of drug metabolizing enzymes may occur at doses higher than recommended clinically for some drugs as has been demonstrated for phenylbutazone and derocoxib (package insert). Genetic polymorphism has been described in dogs for at least one NSAID (celecoxib)41 and is likely to occur for others. Several drugs, or their enantiomers, undergo extensive enterohepatic circulation, again with differences among species (e.g., naproxen, meclofenamic acid, and the S enantiomer of carprofen in dogs), increasing the risk of gastrointestinal toxicity.42 Renal active tubular secretion occurs for some parent drugs. Naproxen, carprofen, ketoprofen, and celecoxib are among the drugs which exemplify pharmacodynamics and pharmacokinetics differences in safety and efficacy among species.36 Whereas allometric scaling generally can accurately predict systemic clearance and volume of distribution (based on body weight) of NSAIDs, allometric scaling generally cannot predict differences in pharmacodynamic indices (e.g., inhibition of TXB2 and PGE2 synthesis).43
Integration of Pharmacokinetics and Pharmacodynamics
Pharmacologic Effects
The pharmacologic effects of this class of drugs include analgesia, antipyresis, and control of inflammation. Antithrombosis also occurs, but this effect is variable, being the greatest for aspirin and the least to absent for newer COX-2 selective drugs. The effects are dose and drug dependent and include antithrombosis (when present), which occurs at the lowest concentrations (relatively lower for aspirin compared to all others); followed by antipyresis, analgesia and antiinflammation, with the latter occurring at the highest concentrations, although an exception appears to occur again for aspirin.10,43d Studies that compared efficacy and safety traditional or newer NSAIDs are generally are based on in vitro or ex vivo methods, with clinical trials limited. Yet, differences should be anticipated even among the newer NSAIDs. The mechanisms by which NSAIDs inhibit (interact with) COX are responsible, in part, for the variable antiinflammatory effects that characterize these drugs. Although several NSAIDs are characterized by a short plasma elimination half-life, the clinical response may last for over 24 hours after a single dose or up to 72 hours after multiple doses. These differential durations reflect, in part, drug-receptor interactions,with irreversible binding to COX most likely contribute to a longer biologic response.44 Lees and co-workers have reviewed the relationship of NSAID effect and dose, emphasizing the advantages of PK-PD modeling as a basis of dose design.44a Inhibition of COX-1 has been described as simple competitive inhibition, whereas that for COX-2 has been described as a slowly reversible, contributing to a longer biologic versus plasma half-life.45 Aspirin binds reversibly to the COX activity site on PGH synthase and then irreversibly inactivates the enzyme by acetylating a serine residue.10 Both thromboxane and prostacycline synthase are impacted. However, platelets cannot produce additional thromboxane synthase; as such, platelet activity depends on new platelet production in the absence of aspirin. In contrast, endothelial cells are able to synthesize more prostacyclin synthetase and are less susceptible to the inhibitory effects of low dose aspirin.10 In contrast to irreversible binders of COX, ibuprofen binds reversibly with COX and thus competes with AA for binding. In laboratory animals and humans, a relative potency (not to be confused with efficacy) has been established for selected conventional NSAIDs and COX: meclofenamic acid >indomethacin >naproxen >phenylbutazone >aspirin.44 However, differential potency for COX-1 versus COX-2 varies among the NSAIDs and their enantiomers. In addition to drug-receptor interaction, prolonged elimination of NSAIDs from inflammatory exudate compared with plasma may also result in differential effects among the drugs and species.37,44
Along with inhibition of PGs, disruption of cellular signaling is responsible for all three pharmacologic effects (antiypresis, analgesia, anti-inflammation) that characterize all NSAIDs.10 The antipyretic effect of NSAIDs may or may not be of benefit. Indeed, in human medicine little scientific support exists for the use of antipyretics for relief of discomfort or reduction of morbidity and mortality associated with fever.47 Fever may have a beneficial effect on the outcome of bacterial or viral infections and often inversely correlates with the severity of lesions or the time to resolution. Thus the antipyretic effects of NSAID may be of most benefit when increased metabolism induced by fever might prove detrimental, such as with septicemia.47 Central analgesic effects have been suggested for some but not all NSAIDs because NSAIDs can provide analgesia at very low intrathecal doses.15,16,46
The impact of COX on disease has led to a number of new applications for NSAIDs. The antiendotoxic effect of selected NSAIDs has been known (see the discussion of flunixin), but newer mechanisms are being examined. For example, several NSAIDs (carprofen, flunixin meglumine, and phenylbutazone) may specifically inhibit activation of the proinflammatory transcription factor nuclear factor kappa B (NF-κB) and on lipopolysaccharide (LPS) induction of iNOS. Using a mouse macrophages line, carprofen and flunixin, but not phenylbutazone, inhibited iNOS, whereas carprofen and, to a lesser degree, flunixin, inhibited NF-kappaB activation.35
Because NSAIDs are commonly first-choice treatments for arthritis, a focus on the effects of NSAIDs on cartilage is warranted. NSAIDs have been shown to inhibit proteoglycan synthesis in vitro, and for the salicylates this is supported by in vivo studies.48 This effect has been attributed to inhibition of uridine diphosphate glucose dehydrogenase, an enzyme important in proteoglycan synthesis.48 Hyaluronic acid synthesis, also dependent on this enzyme, does not appear to be as affected. The clinical effects of NSAIDs on cartilage are controversial. Some NSAIDs may, in fact, favorably modify the metabolism of proteoglycans, collagen, and matrix and may decrease the release of proteases or toxic oxygen metabolites.48 This may reflect, in part, control of inflammation. Thus, whereas several NSAIDs have documented adverse effects on normal cartilage, ranging from decreased proteoglycan synthesis (e.g., aspirin) to chondrocyte death (phenylbutazone), others (e.g., naproxen, piroxicam, ketoprofen, and possibly carprofen) are recognized for their chondroprotective effects. Dual inhibitors have been noted for their potential efficacy to slow the progression of arthritis.48b
KEY POINT 29-7
Newer nonsteroidal antiinflammatory drugs are less destructive to the cartilage at therapeutic concentrations than older nonsteroidal antiinflammatory drugs.
The use of NSAIDs in the control of acute pain is evolving. Preemptive use (just before and immediately after surgery) has been demonstrated in humans to be more effective than either placebo or control,17 and similar findings have been reported in the clinical use of COX-1–sparing drugs in animals. Drugs that target COX-2 (NSAIDs) offer a morphine-sparing effect for control of postoperative pain.17 Their use in multimodal analgesia is increasing.
NSAIDs are currently being investigated for their potential antitumor effects. Knapp and coworkers50 found piroxicam to be clinically useful in reducing tumor size and increasing survival time in dogs with transitional cell tumors of the urinary bladder. The result does not appear to reflect direct cytotoxic effects.49-51 NSAIDs may be beneficial when combined with other anticancer drugs.52 The effects of drugs inhibiting COX-2 cancerogenesis may be dose dependent: At least for aspirin, the maximum anticancer effect occurs at a dose less than that associated with control of inflammation but similar to that associated with antiplatelet effects.9 Mechanisms other than COX inhibition contribute to NSAID antitumor effects.51a In addition to the inhibition of cancer growth, the use of COX-1–sparing drugs may improve gastrointestinal tolerance of anticancer drugs.
Drug Interactions
The NSAIDs can be involved in a variety of drug interactions during any phase of drug disposition (see the discussion of individual drugs). Trepanier53 has reviewed some of these interactions. Displacement of only a small percentage of bound drug from albumin can increased the concentration of pharmacologically active drug in tissues. Few, if any, adverse reactions resulting from drug displacement have been reported, in part because of the failure to recognize the combination as problematic. In addition, the increase in pharmacologically active drug is only transient: Clearance of the unbound drug by both the liver and kidneys will increase.36,40 However, the impact of changes in protein binding in patients with altered liver function may be problematic; accordingly, attention to the possibility that protein-binding drug interactions may worsen risks associated with liver disease might be prudent. Several NSAIDs can induce or inhibit drug-metabolizing enzymes and thus the clearance and half-life of other drugs cleared by the liver.36 Phenylbutazone can both increase and inhibit selected drug-metabolizing enzymes depending on the second drug, whereas salicylates increase metabolism.36 Renal competition with other organic acids for active renal tubular secretion in the proximal tubule has been documented for aspirin and other drugs, although the clinical relevance of this is not clear.
KEY POINT 29-8
Among the less appreciated drug interactions involving nonsteroidal antiinflammatory drugs are pharmacodynamic interactions that impair normal physiologic responses.
KEY POINT 29-9
Nonsteroidal antiinflammatory drug–induced gastrointestinal adversities should be anticipated and clients counseled regarding their advent.
Drug interactions also occur at the level of pharmacodynamics and may increase the risk of adversities. Most notable is the combinations of NSAIDs or glucocorticoids, which increase the risk of gastrointestinal toxicity (discussed later). Also notable are those drugs that, like NSAIDs, alter renal blood flow, and potentially renal autoregulation, thus increasing the risk of nephrotoxicity (e.g., aminoglycosides, angiotensin-converting enzyme inhibitors, amphotericin B). NSAIDs, in turn, may blunt response to diuretics and hypertensive drugs. NSAIDs may also block endogenous responses to hypertensive drugs (e.g., alpha adrenergics such as phenylpropanolamine), thus increasing the risk of hypertension, although this is more likely a risk only in animals for which primary hypertension is a concern.54 The impact of combining NSAIDs with other drugs that affect platelet function or coagulation proteins is complicated by the differences in mechanisms of action between nonselective and preferentially selective drugs. Nonselective drugs would be expected to potentiate platelet function defects or deficiencies, whereas preferentially selective drugs would not. Prudence dictates that NSAID use ideally be avoided in patients with metabolic, hematologic, cardiovascular, or other disorders that put the patient at risk. NSAIDs are more likely to cause CNS effects if combined with fluorinated quinolones with unsubstituted piperazinyl rings (ciprofloxacin) at position 7.55 It is not clear if the interaction occurs at the level of CNS (at the level of the gamma-aminobutyric acid receptor) or as a result of altered clearance.56
Selective serotonin reuptake inhibitors increase the risk of upper gastrointestinal hemorrhage based on a meta-analysis of the human-medicine literature.57 The risk is greater in elderly patients; those drugs most selective for reuptake are associated with a higher risk. Among the proposed mechanisms is depletion of platelet serotonin and loss of platelet aggregation.
Adverse Reactions
All NSAIDs induce undesirable and potentially life-threatening side effects. In general, side effects tend to be predicted by toxicity studies implemented during the approval process,58 although the small number of animals tends to limit their prediction to only the most common side effects. The role of drug interactions in causing adverse events was previously described. Accidental poisoning has been described for several NSAIDs, with the most common among the conventional drugs being ibuprofen, acetaminophen, aspirin, and indomethacin;59 decreased use of these drugs as newer NSAIDs are used decreases relevancy. The most common clinical signs of toxicosis were vomiting and diarrhea followed by CNS depression, and circulatory manifestations.59 Most adverse reactions reflect the inhibitory effects of NSAIDs on PG activity. Acute intoxication by selected drugs can be fatal; the more common adverse drug events are discussed along with their prevention and treatment below.
The adverse event reporting site of the Food and Drug Administration (FDA) is publically acceptable and can be reviewed for adverse drug events associated with NSAIDs in animals.59a However, effective epidemiologic assessment is limited and cause and effect between drug and adverse drug event may not exist. Further, the FDA is not provided information regarding the number of units sold precludes standardizing incidence of adverse drug events among drugs. Among the greatest contributing limitations to the adverse event reporting site may be failure of veterinarians or clients to report adverse events.
Transitioning from one NSAID to another in an attempt to reduced toxicity (or improve efficacy) should be based on elimination half-life (plasma or biological, whichever is longer). For those that reversibly impair COX, 3 to 5 elimination half-lives of the current NSAID should elapse prior to initiating the second (see Table 29-4). Prudence dictates that prophylactic measures be implemented during transition for at-risk patients.
Toxicity as a result of overdosing should be treated as with other overdoses (i.e., removal of ingested drug, supportive therapy). Treatment generally should continue for at least 3 to 5 half-lives of the ingested NSAID; longer may be necessary, particularly if the intoxicating amount is sufficient to saturate drug metabolizing enzymes. The risk of saturation kinetics is greater in age extremes, for selected drugs (e.g., phenylbutazone, deracoxib), and in the presences of liver disease. For NSAIDs characterized by enterohepatic circulation, administration of cholestyramine should be considered. The drug binds to at least several NSAIDs, thus preventing enterohepatic circulation, decreasing half-life as well as gastrointestinal exposure.60 Treatment of other specific disorders is addressed with the adverse drug events.
Gastrointestinal
Mechanism of Gastrointestinal Adverse Events
Gastrointestinal damage is the most common and serious side effect of the NSAIDs. Dogs are described as being “exquisitively sensitive” (see package insert for meclofenamic acid, Fort Dodge) to NSAID-induced gastrointestinal ulceration.The incidence of gastrointestinal side effects associated with NSAID use in dogs (or cats) is not known. However, essentially every NSAIDs (conventional and new) used in the dog has been cited literature, at the FDA adverse event reporting site, in manufacturing package inserts, or other sources as causing gastrointestinal adverse drug events.61-66 Gastrointestinal ulceration should be anticipated in dogs receiving these drugs, and clients should be counseled regarding the side effects and potential treatments for ulcerative injury. The incidence of side effects associated with new NSAIDs approved for use in animals has led the inclusion of client information sheets (as part of package inserts) to be distributed to clients when NSAIDs are dispense to animals for at home therapy.
The mechanism of gastrointestinal damage is not completely understood. A review of the FDA-CVM adverse event website reveals gastrointestinal adversities to be the most common for most, if not all, NSAIDs for which adversities are reported. In the cat, gastrointestinal adversities are the first, or more commonly second, adverse event reported for most NSAIDs. It is likely that several mechanisms act in concert to cause adversities; the mechanisms and risks as they are perceived in human medicine have been reviewed.67-69 Several mechanisms offer a target for prevention and treatment. Gastroduodenal erosion and ulceration reflect, in part, inhibition of COX-1–stimulated PGE2-mediated bicarbonate and mucous secretion, epithelialization, and increased blood flow.70-71 Breakdown of small blood vessels resulting from a deficiency of mucus may be the initiating lesion.72 Enhanced LT synthesis from AA shunted from the COX pathway to the lipoxygenase pathway exacerbates damage: due to vasoconstriction-induced mucosal damage as well as platelet aggregation and neutrophil activation. Dual inhibitors which target LTs as well as PGs may be less ulcerogenic. Direct irritation contributes to gastrointestinal mucosal damage for acidic drugs: 70 ion trapping in the mucosa precipitates hydrogen ion diffusion from the lumen of the stomach to the mucosa.70,73 The lack of a sensitive indicator of gastrointestinal damage complicates assessment of gastrointestinal adverse drug events caused by NSAIDs. Boston and coworkers74 as others could not find a relationship appears between the presence of gastric lesions and positive fecal occult blood. A method based on sucrose absorption in the gastrointestinal tract may be successful for detecting NSAID-induced gastrointestinal damage.75 Some investigators have gone so far to suggest that endoscopic presence of erosive lesions is not necessarily indicative of ulcerogenic effects of NSAIDs.76 The lack of well-designed clinical trials assessing detection, risk, prevention or treatment is problematic. Some studies continue to inappropriately ascribe the lack of statistical differences among treatment groups as evidence of no treatment (diagnostic) effect. Yet, failure to detect a significant difference (type II statistical error) often reflects the small sample size characterizing these clinical trial and caution is recommended to not overinterpret “lack of significant difference” as “sample populations are similar”.
Prevention and Treatment of Gastrointestinal Adverse Events
Prevention of gastrointestinal toxicity is based on identifying those patients at greatest risk; avoiding NSAIDs drugs, if possible, in these patients; and if this is not possible, selecting the safest drug (i.e., COX-1–sparing drugs) and using it in a dosing regimen that results in the lowest exposure necessary for efficacy. Prevention also includes co-treatment with prophylactic drugs, including those that prevent gastric acid secretion or those that replace PGs.73 The use of other therapies that might decrease the need for NSAIDs (e.g., combination analgesic therapy, cartilage-supportive disease-modifying agents) also is indicated.
KEY POINT 29-10
Patients at risk for nonsteroidal antiinflammatory drug–induced gastrointestinal adversities should be identified before implementation of therapy, and preventive measures should be taken, if warranted.
Risk factors for NSAID-induced gastrointestinal adverse events
Several risk factors for NSAID-induced gastrointestinal adverse events identified in humans are applicable to animals. No chemical characteristic predicts the likelihood of gastrointestinal toxicity by a particular conventional NSAID.70,72 Drugs that undergo enterohepatic circulation (e.g., naproxen, carprofen, etodolac) may be associated with a greater incidence of gastrointestinal upset. Other risk factors include advanced age (altered disposition coupled with decreased ability to protect damaged mucosa),77 concurrent use of glucocorticoids (increasing the risk 4.4-fold in humans) or other NSAIDs (the exception might be low-dose aspirin, as discussed later), or anticoagulants. Comorbidity (renal, cardiovascular, or liver disase) is also a risk factor.73
The combination of steroids and NSAIDs causes worse lesions than either drug alone, as has been demonstrated for flunixin meglumine or dexamethasone.74,78,79 Further, the FDA adverse drug events site indicates that the risk of gastrointestinal toxicity is greater when drugs are given with glucocorticoids. In a restrospective study of gastrointestinal perforation associated with deracoxib, a risk factor was combination with glucocorticoids (or other NSAIDs) within the past 24 hours.80 Other NSAIDs appear to shift use of AA from the COX pathway to the production of cysteinyl LTs and LTB4, eicosonoids that promote leukocyte migration, break down the mucosal barrier, and stimulate gastric acid secretion. Inhibition of COX-2 may preclude angiogenesis critical to the healing ulcer.81 Persons with previous history of gastric ulcer disease also are predisposed to NSAID-induced gastrointestinal adverse drug events. Although peptic ulcer disease is unusual in animals, prudence dictates that evidence of any gastrointestinal disease associated with mucosal damage (e.g., inflammatory bowel disease) that might require COX-2–mediated healing be considered a potential risk factor.
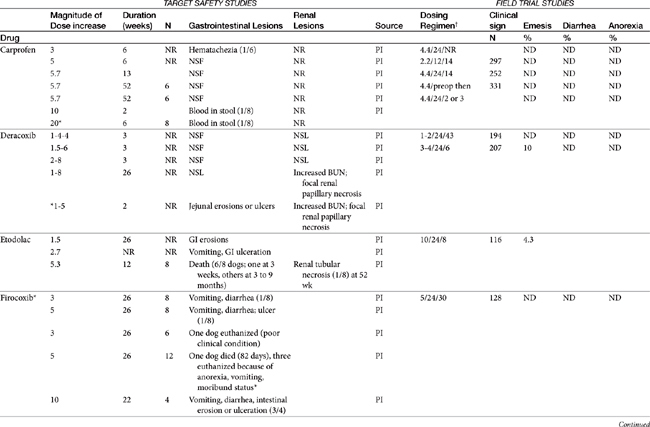
Dose-dependent toxicity has been demonstrated during the approval process essentially for all NSAIDs as is indicated by product package inserts (Table 29-5). The single most common cause of NSAID-induced toxicity found by a review of NSAID-induced adverse drug event reports by the FDA is overdosing, in particular failure to adhere to the recommended dosing regimen. Overdosing was associated with an increased risk of NSAID-induced gastrointestinal perforation in dogs.80 Drugs for which low-concentration preparations are not available increases the risk of toxicity because of inaccurate dosing. Use of compounded preparations (for which oral bioavailability is not known) should be implemented with extreme cautions; increasing bioavailability will result in a proportional increase in drug concentrations. The risk for overdosage is greater for selected drugs for which higher doses may result in zero-order elimination (e.g., deracoxib, phenylbutazone). For such drugs half-life is no longer germane, and the risk of drug accumulation dramatically increases.
Table 29-5 Safety Data for Selected NSAIDs in Dogs and Cat Based on Target Safety Studies and Clinical Trial Field Studies
The relationship between dose and risk of toxicity remains complex, even for NSAIDs, as is exemplified by aspirin. At low doses (concentrations), aspirin blocks COX activity and thus shunts AA into the lipoxygenase pathway. Although gastrotoxic LTs are produced (cysteinyl LTs and LTB4), aspirin also simultaneously triggers lipoxin (aspirin-triggered lipoxin [ATL]) formed by lipoxygenase-15. Lipoxin is antiinflammatory and thus inhibits the gastrotoxic leukocyte recruitment and activation caused by cysteinyl LTs, as well as mediates the antiadhesive platelet effects of aspirin. As such, low-dose aspirin tends to be both cardioprotective as well as safe to the gastrointestinal tract. However, at higher doses production of LTs is sufficient that the protective effects of lipoxin are masked and gastrotoxicity emerges. Interestingly, COX-1–sparing (COX-2 preferential drugs) inhibit the formation of lipoxin, contributing to the toxicity of the newer drugs when combined with low-dose aspirin.81 Theoretically, whereas COX-inhibiting drugs should not be combined with low-dose aspirin, dual-acting NSAIDs, which do not target lipoxygenase-15 (see the discussion of dual-acting NSAIDs), should not inhibit lipoxin and thus should maintain the low-dose aspirin gastroprotective and cardioprotective effects.81
In general, conventional NSAIDs are associated with a greater risk of gastrointestinal adverse drug events in humans, but exceptions may occur. For example, in humans (but not dogs) ibuprofen, which is essentially equivalent in its COX-1 versus -2 selectivity, is ranked as low risk, along with the COX-1–sparing drugs celecoxib, rofecoxib, meloxicam, and etodolac.73 Comparison in dogs of gastrointestinal safety among newer NSAIDs, including both COX-1–sparing drugs and dual-acting NSAIDs, might be based on toxicity data generated during the approval process and available on package inserts. This latter source includes both targeted safety studies (the dose is multiplied by 3, 5, or 10) and field studies (clinical trials at recommended doses under conditions of anticipated use) (see Table 29-5).
Comparison of clinical safety among NSAIDs ideally should reflect nonmanufacturer-sponsored, well-designed clinical trials involving a sufficient number of animals such that the power is sufficient to detect a significant difference. Placebo controls are imperative; positive responses have been reported in 40% or more of placebo-treated animals.82 However, such studies in veterinary medicine are few and far between. Data that do exist thus far support the safety of the newer drugs compared with conventional NSAIDs. For example, using a placebo-controlled, parallel, randomly assigned design in dogs (n = 6/group), Reimer and coworkers83 found that buffered aspirin caused endoscopic gastric lesions within 5 days of starting therapy compared with no or minimal lesions in animals treated with carprofen or etodolac (sample size may have limited the ability to discern a difference between the two groups); all drugs were administered at mid-recommended doses. Endoscopic lesions followed the same relative pattern among drugs throughout the 28-day study period, indicating that aspirin is more commonly associated with lesions in the canine gastrointestinal tract compared with the COX-1–sparing drugs. A separate study found no differences in gastrointestinal lesions in animals receiving ketoprofen (nonselective), carprofen, meloxicam, or placebo. However, the power of this latter study to detect a significant difference was not reported.
Pharmacologic prevention and treatment
Future pharmaceutical manipulations designed to deliver NSAIDs by alternative routes may decrease risk of gastrointestinal adverse drug effects, although their application to veterinary patients should not necessarily be assumed. For example, formation of nitroso derivatives of conventional NSAIDs may improve the safety margin of these drugs as a result of in vivo release of NO that provides gastroprotection while improving antiinflammatory and analgesic potency.2 However, NO also has been associated with the pathogenesis of osteoarthritis, thus exemplifying the continuously complicated nature of designing safe NSAIDs (see later discussion).22 In contrast to current drugs that only slowly dissociate with COX-1, newer NSAIDs appear to be weak and rapidly reversible binders of COX-1, thus enhancing safety.17,45
Topical (including transdermal) NSAID administration is appealing because it avoids direct contact between the drug and target tissue. Indeed, a meta-analysis of clinical trials comparing topical NSAIDs to placebos and oral NSAIDs in humans found this route to be effective, with no difference in response compared with oral. Topical administration was safe, although not necessarily safer than oral. Of the NSAIDs reviewed, ketoprofen was described as the best.84 Thus far, topical NSAID administration has not proved to be a vital means of avoiding gastrointestinal adverse drug events in small animals, primarily because of failed drug delivery. Because the amount of drug delivered cannot be predicted, administration of NSAID in novel drug delivery systems offered by compounding pharmacists is not recommended unless the amount of drug delivered by that system has been scientifically demonstrated, as would occur for an approved drug. Enteric coatings, combination drugs (e.g., with gastroprotectants), and other approaches may be of some benefit for some drugs (e.g., aspirin) and, if currently available, are discussed with the individual drugs.
Both the prevention and treatment for gastrointestinal toxicity focus on, in order of priority, control of gastric acid secretion, replacement of the missing PGs, and (for treatment) protection of the damaged mucosa.64,85 The two major categories included antisecretory drugs and the PG analog misoprostol; cytoprotectants, such as sucralfate, also play a role in treatment.
Among the antisecretory drugs, proton pump inhibitors (PPIs) generally have proved more effective than H2 receptor blockers for prevention and treatment of NSAID-induced gastrointestinal adverse drug events. An exception has been demonstrated in humans for famotidine, but only when it is administered at higher than recommended doses (40 mg twice rather than once daily).73 Boulay and coworkers86 demonstrated that cimetidine did not protect the gastric mucosa from developing lesions in dogs receiving nonbuffered aspirin (35 mg/kg every 8 hours). PPIs appeared to be more effective, as well as better tolerated, compared with misoprostol. In one human study, close to 30% of persons reciving misoprostol could not tolerate the full dose.76 Omeprazole generally is the PPI of choice, although lansoprazole may also be a reasonable choice. Although lansoprazole was not found to be superior to misoprostol for prevention of gastrointestinal adverse drug events in humans, improved compliance and better tolerance made it the preferred PPI drug in humans. However, the use of PPI will not prevent ulcers in all patients, as has been demonstrated in humans. Symptoms of GI side effects still occur in 20% of human patients receiving omeprazole.73 Although routine use of PPI in nonrisk animals receiving NSAIDs is not indicated, strong consideration should be given to their use in at-risk animals. As a class, the PPIs are inhibitors of drug-metabolizing enzymes, and care should be taken to review drug interactions before their use. Among them, esomeprazole may be the least likely to impact metabolizing enzymes.86a However, PPI also have been shown to induce selected CYP45O enzymes (P45OIA1 and A2) but this impact appears to be most clinically relevant to (human) poor metabolizers that are deficient in enzymes.86b
KEY POINT 29-11
Among the antisecretory drugs, proton pump inhibitors generally have proved more effective than H2 receptor blockers for prevention and treatment of nonsteroial antiinflammatory drug–induced gastrointestinal adversities.
Misoprostol is a synthetic PPGE analog that both prevents and helps heal gastrointestinal ulceration caused by NSAIDs.87 The efficacy of misoprostol has been well established in human patients suffering from NSAID-induced ulceration,71 and studies support similar benefits in dogs.88,89 Combination NSAID–misoprostol products have been approved for use by human patients,90 supporting its their combined use. Interestingly, misoprostol, when combined with an NSAID, also appears to enhance the antiinflammatory effect of the NSAID.90 Indeed, misoprostol inhibits IL-1, TNF, and thromboxane release from macrophages, and it is the most potent inhibitor of histamine release from human mast cells.90 Misoprostol has potentiated the antiinflammatory effect of a variety of compounds in animalsand appears to have analgesic effects, although at high concentrations, acting synergistically with other NSAIDs.90 Finally, misoprostol may be more effective in the presence of agents that decrease gastric acid secretion.64
Gastroprotective drugs include sucralfate and potentially glucosamine–chondroitin products (Figure 29-8). The benefits of sucralfate in the treatment of NSAID-induced adverse drug events include binding to and thus protecting damaged mucosa, as well as increasing PG synthesis, angiogenesis, and sulfhydryl (oxygen radical scavenger) production at the site of damage. Despite the fact that sucralfate binds only to damaged mucosa, it nonetheless consistently performs better than placebo in the prevention of gastric or duodenal ulcers in human patients receiving NSAIDs. Sucralfate is minimally effective than antisecretory drugs in preventing stress ulcers (e.g., critical care patients).90a The combined use of NSAIDs and disease-modifying agents (glucosamine and chondroitin sulfates) for treatment of osteoarthritis is discussed later. An added advantage of their combined use is the potential gastroprotection that might be realized because of enhanced mucopolysaccharide production.91 Interestingly, metronidazole has been ascribed a protective effect on the gastrointestinal tract when combined with NSAIDs, presumably through removal of the microbial impact on neutrophil chemoattractant.92
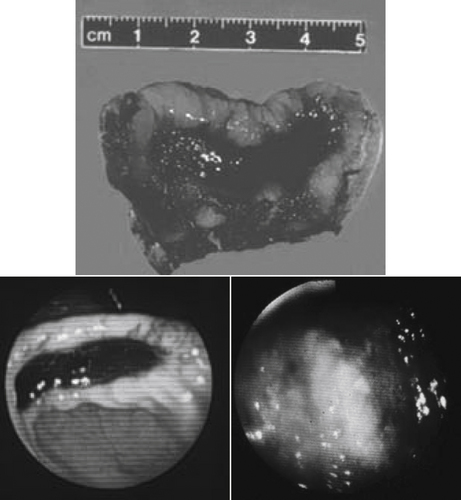
Figure 29-8 A gross (top) and endoscopic view of two gastric ulcers in dogs treated with aspirin and glucocorticoids. The bottom ulcer responded to treatment with misoprostol, sucralfate, and ranitidine.
KEY POINT 29-12
Although sucralfate will not prevent gastrointestinal ulceration, it can protect and facilitate healing of damaged mucosa once it emerges.
The Canadian Agency for Drugs and Technologies in Health (CADTH) sponsored an in-depth analysis of the literature in an attempt to identify the most cost-effective gastroprotective strategies in persons receiving NSAIDs.93 In their report, based on Monte-Carlo modeling, investigators considered patients receiving a nonselective NSAID (diclofenac) alone, a COX-2 preferentnial NSAID alone (celexocib); or either group of the NSAIDS with misoprostol, an H2 receptor blocker (ranitidine), or a proton pump inhibitor (omeprazole). In their 2007 report, they found that no prophylaxis and treatment with a traditional NSAID; but no gastroprotection was least costly but least effective in avoiding adverse events. Of the combinations, PPI consistently was more effective than no prophylaxis; further, the most cost-effective strategy was the combination of nonselective NSAIDs with PPIs. Combination of nonselective NSAIDs with H2 receptor antagonists also reduced the risk of gastrointestinal complications, but at a higher cost (25%). Use of Cox-2 selective drugs or combination with misoprotol were effective (but more costly) alternatives, although combination with misoprostol was associated with increased adversity because of its PG effects. The CADTH investigators further observed the need for a large, prospective, multicentered clinical outcome study that directly compares the efficacy of PPIs with H2 receptor antagonists to prevent NSAID-associated gastrointestinal adversities.
The use of 5-lipoxygenase inhibitors for prevention of NSAIDs gastrointestinal toxicity might be considered. Lis facilitate ulcer formation by virtue of their effect on platelets and blood vessels; these effects and others also might be inhibited by LT receptor antagonists (see later discussion). Among the approaches in reducing gastrointestinal toxicity associated with NSAIDs is the development of NO-NSAIDs.69 NO has been recognized as an important gastroprotectant. Its mechanism appears, in part, to reflect inhibition of neutrophil adherence to vascular endothelium, which is necessary for mucosal damage to occur. Whereas adverse reactions are likely to preclude use of drugs that promote NO release systemically, NSAIDs that facilitate local release of NO (COX-inhibiting NO donors; NO-NSAIDs) are currently being developed. The drugs contain a NO-releasing moiety; their continuing development has been reviewed.69 For example, the addition of the moiety to naproxen has yielded naproxcinod. The gastric mucosa also is protected by the formation of hydrogen sulfide (H2S) when produced in appropriate quantities. Accordingly, NSAIDs are being designed with a moiety that allows local, limited release of H2S. In either case (NO- or H2S-releasing moiety), the antiinflammatory potency of the drug may also be improved as gastric adversity is decreased.
Liver disease should be an anticipated sequela of long-term use of any drug extensively concentrated or metabolized by the liver, including NSAIDs (see Chapter 4).94 All NSAIDs approved for use in dogs have been associated with increased liver enzymes; most have been reported scientifically or anecdotally to cause hepatitis. Although their occurrence is discussed with individual NSAIDs, hepatic function tests (e.g., serum bile acids, albumin, urea nitrogen) should be implemented throughout drug exposure in patients at risk (discussed previously). The use of hepatoprotectant drugs (e.g., N-acetylcysteine, S-adenosylmethionine [SAMe]) should be considered for both prevention and treatment.
Clinical signs indicative of gastrointestinal ulceration may be exacerbated by an increased risk of bleeding induced by NSAIDs. In addition to their antiplatelet effects, selected NSAIDs (e.g., phenylbutazone) have also been associated with bone marrow dyscrasias.95-98 Gastrointestinal bleeding is probably the most common sign of bleeding dyscrasias, in part because of the ulcerogenic properties of these drugs. Epistaxis has also been reported. Because prostacyclin is mediated largely by COX-2, use of COX-2–selective drugs may increase the risk of thrombosis.99
Renal
Analgesic nephropathy is a relatively common adverse effect of NSAIDs in human beings.100 In the kidney both COX-1 and COX-2 mediate renal effects of PGs. Vasodilatory and tubuloactive PGs are protective, ensuring that medullary vasodilation and urinary output continue during states of renal arterial vasoconstriction (see Figure 29-7). The loss of this protective effect becomes important in patients with compromised renal function.100 NSAIDs inhibit the synthesis of renal PGs and may lead to deterioration of renal function in patients whose kidneys are physiologically stressed.100,101 However, some side effects reflect mechanisms other than PG inhibition. Renal side effects include both acute and chronic renal disease, nephrotic syndrome, interstitial nephritis, hyperkalemia, and disturbances in water and sodium movement.
Two different forms of renal disease associated with NSAIDs are generally described: hemodynamically mediated ischemic nephropathy and acute interstitial nephritis.102 Nephrotoxicity is more common in human patients than in veterinary patients, probably because therapy with NSAIDs is prolonged in the human patient and often occurs without physician supervision. NSAID therapy is also more common in geriatric patients, which is more likely to have reduced renal function as well decreased nephroprotective function. As such, animals that are likely to be predisposed to developing analgesic nephropathy are those that are geriatric, afflicted with conditions that impair renal blood flow (e.g., cardiac, renal, or cirrhotic liver disease), subjected to a hypotensive state (e.g., prolonged anesthesia without fluid support), and receiving nephroactive or nephrotoxic drugs in addition to the NSAID. Patients receiving more than one NSAID, aminoglycosides, amphotericin B, and possibly angiotensin-converting enzyme inhibitors are potential candidates for analgesic nephropathy.103 Interstitial nephritis, a less common syndrome associated with NSAID use in human patients, apparently has not been reported in dogs or cats. The cause of this syndrome appears to be a cell-mediated allergic response. Loss of renal PGs may potentiate the disease as inflammation progresses unchecked.
In contrast to gastrointestinal adverse drug events, the risk of renal adverse drug events may not necessarily be reduced with use of newer COX-2–selective drugs compared with conventional NSAIDs. In human medicine clinical trials have demonstrated that the renal effects of COX-1–protective (COX-2–selective) drugs are similar to those of conventional NSAIDs. A low-salt diet may increase the risk: Elderly human patients on a low-sodium diet receiving either single or multiple doses developed reductions in glomerular filtration rate, creatinine clearance, and sodium renal clearance.104 Acute changes in renal function in humans generally occur within 24 hours, with return to normal by 48 hours.105
KEY POINT 29-13
The newer nonsteroial antiinflammatory drugs may offer no advantage to older nonsteroial antinflammatory drugs in preventing nephrotoxicity; cats in particularly may be predisposed to adverse renal effects.
A number of studies have focused on the impact of perioperative NSAID use on renal function in dogs. Lobetti and Joubert106 studied the effects of ketoprofen (1 mg/kg) and carprofen (4 mg/kg) as well as ketorolac (0.5 mg/kg) and morphine (0.1 mg/kg; “control group”) on renal function as assessed by serum urea and creatinine concentrations, urine γ-glutamyltransferase, fractional renal clearance of sodium, and urinalysis. Measurements were collected before and 24 and 48 hours after ovariohysterectomy in dogs (n=40; four per group, with no placebo control). Fluids were not administered during the surgical procedure, which ranged between 1 and 5 hours in duration. Although all dogs remained clinically normal, transient azotemia was detected in two dogs each (2/4) in the ketoprofen and ketorolac groups, and changes in fractional clearance were detected in all three NSAID groups. Urine specific gravity increased in carprofen-treated dogs, although the clinical relavence of this is not clear.
In their study of carprofen safety in cats, Steagall and coworkers107 cite a report from the European Union that indicates 50% of the surveyed cases of acute renal failure in cats were associated with NSAID therapy. Gunew and coworkers108 prospectively studied the effects of long-term (approximately 6 months) treatment of cats with osteoarthritis (n = 46) meloxicam at 0.01 to 0.03 mg/kg using a case-controlled design. Cats were approximately 13 ± 4 years for both groups. Unfortunately, indices of renal dysfunction were compared between groups only at 1 month; no differences were detected at that time. The author has summarized adverse events reported for carprofen and meloxicam in dogs versus cats at the FDA adverse event reporting site for reported after 2006 but before 2009. (Figure 29-9). Although direct numbers of adverse events cannot be compared (no information on units or doses sold for each drug), the proportion of each advent might be compared among drugs. The percent of adverse events related to the kidney suggest that the incidence is clinically relevant in both the dog and cat. However, the data also suggests that the incidence of renal adverse events is generally higher for the cat compared to the dog. Further, the data suggests that the risk of increased BUN and serum creatinine is greater for meloxicam (45 and 46%, respectively) compared to carprofen (24 and 22%, respectively). However, assessment of the more recent data suggests a broader implication. For more recent data, the number of animals for which each specific adverse event occurs has been removed from the site. As such, current comparisons must be limited to ranking of adversities. The rank of increased BUN or serum creatinine is higher in cats for each NSAID (approved or not approved) compared to dogs. This includes both meloxicam and ketoprofen (Table 29-6). Accordingly, caution is recommended to not over estimate renal safety of these products in either dogs or cats; further, monitoring of renal function might be indicated in cats receiving NSAIDs long term, particularly if other risk factors for renal disease are present.
Figure 29-9 The proportion of renal-associated adverse drug events drug for nonsteroidal antiinflammatory drugs (NSAIDs) reported in either dogs or cats. The proportion comes from the total number of adverse events reported for that drug. The total number of adverse events should not be directly compared among drugs because the data cannot be adjusted for the number of units sold or animals treated. For example, a greater number of adverse events in dogs should be expected for carprofen compared with other NSAIDs because carprofen has been approved for a longer time. Further, veterinarians may be less inclined to report adverse events for newly approved NSAIDs if the event is one that is expected of the class. The cause-and-effect relationship for the adverse event and the NSAID was not confirmed but reflects cumulative adverse events reported to manufacturers or directly reported to the Food and Drug Administration (FDA) at the time the data were studied (2007–2008). The data were collected from the FDA’s adverse event reporting site from 2006 to 2008 (data were no longer available at time of publication).
Table 29-6 Post-Market Surveillance Data Ranking for Selected Renal-Related Adverse Events Reported for Orally Administered NSAIDs in Dogs and Cats∗
Studies have attempted to identify methods to prevent or treat NSAID-induced analgesic nephropathies. Despite some studies supporting its role,109 misoprostol does not have a recognized role in the prevention or treatment of NSAID-induced nephrotoxicity. Its impact on other drug-induced nephropathies associated with vasoconstriction has been variable (e.g., cyclosporine110,111). Misoprostol has been used clincally in human patients suffering from clinical conditions associated with peripheral or renal vasoconstriction; however, its effects may be dose related, with natriuresis, diuresis, and vasodilation occurring at low doses and vasoconstriction and impaired salt and water excretion occurring at high doses. Currently, preventive measures, including therapy with sodium-containing fluids, minimizing of drug interactions, and avoidance of use in patients at risk, appears to be the best method of avoiding the nephrotoxic effects of NSAIDS.
N-acetylcysteine has been studied experimentally for the prevention or treatment of NSAID-induced nephrotoxicity.102,112 Proposed mechanisms include vasodilation, although other mechanisms may be relevant. However, although animal models have indicated potential efficacy, clinical trials, including meta-analyses, have thus far failed to provide conclusive evidence of efficacy.113 Nonetheless, in the absence of effective therapies, N-acetylcysteine is generally recommended (approximately 10 mg/kg or more orally twice daily) preventively for drug-induced toxicity.
Cardiovascular
All conventional NSAIDs are able to impair platelet activity as a result of impaired PG (thromboxane) synthesis, a COX-1–selective action (see Figure 29-6). At pharmacologic doses aspirin selectively and irreversibly acetylates a serine residue of a platelet COX.114 The platelet form of this enzyme is up to 250-fold more sensitive to acetylation by aspirin compared with COX (prostacyclin synthetase) in vascular endothelial cells. Although platelets cannot regenerate more COX, endothelial cells apparently are able to rapidly synthesize and replace impaired COX.114 Platelet aggregation defects caused by aspirin will last until platelets can be replaced, generally 1 to 2 weeks.
KEY POINT 29-14
Cyclooxygenase-2–preferential drugs should be expected to increase the risk of thrombosis rather than hemostasis.
The effect of newer NSAIDs differs from that of conventional NSAIDs. The loss of COX-2–mediated formation of prostacyclin and its antithrombotic effects in the vascular endothelium predisposes (human) patients to COX-1–mediated platelet aggregation, leukocyte activation, and adhesion and accumulation of cholesterol in vascular cells. The potential association of COX-2 inhibitors and an increased risk of thromboembolic disease evolved in the analysis of side effects in treatment groups receiving selected COX-2 preferential drugs (e.g., rofecoxib or celecoxib compared with placebos or conventional NSAIDs) as part of clinical trials evaluating their use for treatment of cancer, treatment of AD, or association with gastrointestinal side effects. The trials initially were not designed to compare cardiovascular events, and initial evaluation could not discern whether it was treatment with rofecoxib or lack of treatment with naproxen (and thus loss of a cardioprotective effect) that led to the increased risk.115 Subsequent studies that have focused specifically on the risk of thromboembolic events have yielded conflicting results, ranging from a clear increased risk to no detectable increase in risk. The results of these studies have been reviewed,115 but conclusions are complicated by the presence of aspirin. However, neither conventional nor COX-selective NSAIDs are as likely to impair platelet aggregation as well as aspirin does. For platelet aggregation to be impaired, TXA2 must be inhibited by at least 95%, an impact that competitive TXA2 inhibitors cannot achieve; the possible exception is naproxen because of its long half-life. In contrast, aspirin irreversibly acetylates TXA2. Accordingly, both aspirin and naproxen tend to be associated with cardioprotective effects. However, aspirin has the added advantage of stimulating lipoxin, which provides antiplatelet effects.20 A number of NSAIDs, including naproxen, block aspirin’s cardioprotective effect, in part by physically binding the serine site of TXA2 acetylation by aspirin. Although this is less likely for newer NSAIDs, they may impair lipoxin, thus impairing antiplatelet effects through a different mechanism. This complicates interpretation of these studies, particularly if groups are not controlled for aspirin dose, duration, and timing. However, in general, review of these clinical trials indicate that COX-2–selective drugs do increase the risk of cardiovascular adverse events, particularly in patients already suffering from disease.115
The impact of COX-2 selective drugs on hemostasis in veterinary medicine is not clear, but preferential COX-2 inhibition by endogenous glucocorticoids may help explain the increased risk of thrombogenesis in dogs suffering from hyperadrenocorticism. In a canine model of coronary vessel occlusion, celecoxib decreased the vasodilatory response to AA, leading the authors to conclude that a potential risk exists for acute vascular events in animals suffering from inflammatory disorders and receiving COX-2–selective drugs.116 However, celecoxib also prevents the cardioprotective effects provided by low dose aspirin, contributing to the increased risk of thromboembolic events in some human patients.116a The lack of unequivocal information regarding COX selectivity of the various NSAIDs in either dogs or cats complicates assessment of risk. As such, NSAIDs should be used only with great caution in animals with comorbidity influencing hemostasis (e.g., heart worm thromboembolis). This may be particularly problematic in the cat (e.g., hypertrophic cardiomyopathy) for which little information regarding COX selectivity among drugs is available.
Miscellaneous
Thyroid
A number of NSAIDs adversely affect thyroid function, primarily through competition for or impairment of binding to carrier proteins. Displacement appears to cause plasma concentrations to rapidly increase, leading to decreased thyroid-stimulating hormone; however, with time, increased clearance of the hormone should alter the balance in the opposite direction. The impact of NSAIDs on the thyroid gland may reflect the sulfur moiety present on many of the drugs (see Figures 29-3 and 29-4); it is possible that displacement from protein-binding sites might increase clearance. Using a randomized crossover design, Daminet and coworkers117 reported that aspirin reduced total T4 within 24 hours of administration of 25 mg/kg twice daily; thyroid-stimulating hormone and free T4 were not affected. Ketoprofen at 1 mg/kg once daily had no effect compared with placebo. Whereas etodolac appears to have minimal impact on thyroid function.118 Ferguson (as reviewed by Sauve)119 reported slightly decreased serum T4 and cTSH in dogs receiving carprofen for 2 to 5 weeks. Sauve and coworkers119 prospectively studied in osteoarthritic dogs (n=62) the effect of meloxicam (n=14; 0.2 mg/kg load followed by 0.1 mg/kg daily for 60 days total), carprofen (n=14; 1.7 to 2.3 mg/kg twice daily for 60 days) and chondroitin sulfate–glucosamine–manganese combinations (n=18; 1 to 3 capsules daily for 60 days) compared with placebo on thyroid function. The study was randomized and placebo (n=16) controlled. Thyroid function did not significantly differ among any treatment group across time or between groups; however, free T4 declined 20% in the meloxicam group. Sample size may have limited the ability to detect differences in other groups.
Lungs
The “allergy” to aspirin described for human asthmatics is generally recognized to reflect increased production of LTs, although the loss of the inhibitory effect on LT production by PGs also has been suggested. Drugs that are COX-1 sparing do not appear to precipitate asthma as do conventional NSAIDs and aspirin in particular, suggesting that the newer drugs are safe in asthmatic patients.20 The impact of low-dose aspirin (which would allow the production of aspirin-induced lipoxin) on pulmonary inflammatory diseases has not been addressed.
Conventional Nonsteroidal Antiinflammatory Drugs
Aspirin: The Prototypic Nonsteroidal Antiinflammatory Drug
The discovery of the mechanisms of aspirin and its elucidation of COX enzyme isoforms was recently reviewed.122a
Structure–Activity and Preparations
Aspirin, the salicylic acid ester of acetic acid, is the prototype of the salicylate drugs, which includes sodium salicylate, bismuth subsalicylate, and others. In addition to inhibition of COX enzyme activity, salicylates inhibit the formation and release of kinins, stabilize lysosomes, and remove energy necessary for inflammation by uncoupling oxidative phosphorylation. Among the traditional NSAIDs, aspirin stands out for its impact on the AA cascade. Its effect on thromboxane synthetase was previously described. Additionally, at low doses, aspirin triggers the formation of lipoxins and their 15-epimers, epoxins.123,123a The latter are antiinflammatory mediators that counter the proinflammatoroy mediators of the cascade (see Figures 29-1 and 29-2). Aspirin is available in several different preparations, including plain, film-coated, buffered, time-release, and enteric-coated tablets. Capsules and suppositories are also available.70 Oral salicylates are also available in preparations intended to treat inflammatory bowel disease.
Disposition
The oral bioavailability of aspirin products may vary because of differences in disintegration, drug formulation, stomach content, and gastric pH.124 Although buffered aspirin is more soluble than plain aspirin, a larger proportion is ionized and less rapidly absorbed. The rate of absorption of both products is the same.70 Aspirin undergoes rapid metabolism to the hydrolyzed active product salicylic acid by plasma esterases. This metabolite is not as potent an analgesic or antiinflammatory drug because of the loss of the acetyl group, which is able to acetylate key proteins.70 Salicylic acid is between 50% and 70% bound to serum albumin among species.125 Hypoalbuminemia may result in transient increases in plasma drug concentrations associated with adverse effects. Distribution of salicylic acid into extracellular fluid is rapid and includes synovial and peritoneal fluids, saliva, and milk. Salicylic acid is eliminated by hepatic conjugation with glucuronide and glycine, renal excretion by glomerular filtration, and tubular secretion.125,126 Species differences in the biotransformation and elimination of salicylates are dramatic. Excretion is more rapid in alkaline urine, which might be used therapeutically to treat acute aspirin intoxication. Salicylate can achieve substantial concentrations in milk. Elimination occurs more slowly in pediatric patients.127
The disposition of acetyl salicylate appears to be the only salicylate for which data is available in dogs or cats.127,128 Other oral salicylates such as sulfasalazine have been used to treat chronic inflammatory conditions of the bowel (see Chapter 19). Although their mechanism of action is unclear, splitting of the diazo bond by colonic bacteria to yield sulfapyridine and 5-aminosalicylic acid (5-ASA) may be involved. The 5-ASA is considered to be the active moiety.129 Both the sulfapyridine and sulfasalazine (up to 25%) are absorbed from the small intestine, but most of the 5-ASA remains in the colon. That absorbed (approximately 20% in humans) is rapidly acetylated and inactivated by either the colonic mucosa or liver. Newer products composed principally of 5-ASA are being investigated for the treatment of chronic inflammatory bowel diseases.129 Alternative methods of delivery of these compounds such that side effects are minimized are also being studied.130
Adverse reactions
Aspirin is theoretically characterized by a wide safety margin in most species. The recommended therapeutic range in humans ranges from 100 to 250 μg/mL,131 with higher concentrations necessary to control inflammation, particularly that associated with immune-mediated disease. 5 Analgesia and antipyresis generally require concentrations of 20 to 50 μg/mL,70 whereas control of inflammation may require concentrations that exceed 50 μg/mL. Response of rheumatoid arthritis in human beings requires concentrations approximating 200 μg/mL. Although the drug concentration necessary to achieve an antithrombotic effect has not been established for aspirin in animals, smaller doses have proved efficacious (e.g., 3 mg/kg). These studies did not effectively evaluate safety (gastrointestinal changes). Studies based on gastrointestinal permeability demonstrate that aspirin alters gastrointestinal permability.7
Clinical signs of aspirin toxicity may be present at concentrations necessary for clinical response, but they worsen as serum salicylate concentrations exceed 300 μg/mL. The effects of NSAIDs on cartilage are largely detrimental. Hepatotoxicity and decreased renal function have been reported at 250 μg/mL. Decreased prothrombin time, deafness, and hyperventilation occur at 300 to 350 μg/mL, with severe toxicity, including metabolic acidosis, occurring at doses above 400 μg/mL.127
Treatment of acute aspirin toxicity is largely supportive, including increasing elimination. Toxicity (acute) is usually manifested as depression, vomiting, hyperthermia, electrolyte imbalances, convulsions, coma, and death. Toxicity is more likely in cats because of slow metabolism with accumulation. Acute toxicity includes serious acid–base disturbances resulting from uncoupling of oxidative phosphorylation. Hyperventilation resulting from direct stimulation of the respiratory center may be followed by depression at high doses. Bleeding disorders may also be evident,70,132 as might dose-dependent hepatotoxicity. Salicylate markedly suppresses augmented proteoglycan synthesis in osteoarthritic cartilage and permeates the damage joint more than the undamaged joint.133
Dogs
In dogs aspirin is distributed to a volume ranging from 0.4 to 0.6 L/kg. Bioavailability probably varies with the manufacturer as well as the preparation and ranges from 68% to 76%.134 The bioavailabilities of plain, buffered, and enteric-coated aspirin (25 mg/kg) do not appear to vary markedly, although plasma salicylate concentrations were most variable for the enteric-coated preparation.135 Concentrations of 91 to 120 μg/mL are achieved at 25 mg/kg administered at 8-hour intervals.127 After several doses of 25 mg/kg at 12-hour intervals, the biological half-life of aspirin is 7.5 hours in dogs. This time increased to a mean of 12.2 hours, however, when the dosing interval was decreased to 8 hours.136 In another study the elimination half-life of aspirin varied after intravenous injection of 36 to 60 mg/kg, ranging from 2.2 to 8.7 hours.
One study in clinical patients found that plasma salicylate concentrations correlated with response.134 The dose necessary to maintain clinical control of various lamenesses in dogs in one study ranged from 23 to 86 mg/kg twice daily, resulting in plasma drug concentrations ranging from 71 to 281 μg/mL.137 Marked individual variability in drug elimination among animals suggests that therapeutic drug monitoring may be useful to ensure that therapeutic drug concentrations have been achieved and that toxic concentrations (>300 μg/mL) are avoided.134 However, when 25 mg/kg is administered at 8-hour intervals, therapeutic concentrations can be expected to be maintained throughout the dosing interval. Gastrointestinal side effects of aspirin in dogs also appear to be dose and preparation related62,135 and may be decreased by using special preparations. Doses of 25 mg/kg of plain aspirin caused mucosal erosions in 50% of dogs that received plain aspirin, whereas minimal damage occurred in animals receiving buffered and coated preparations.135
Cats
As a phenol, aspirin is a compound for which glucuronidation is generally deficient in cats compared with other species.132,138 Its plasma elimination half-life may be dose dependent.139 The half-life is 22 to 27 hours after doses of 5 to 12 mg/kg but 45 hours after administration of 25 mg/kg.7,128 No clinical signs of toxicosis occurred in one study in which cats were treated with 25 mg/kg every 48 hours.138 Clinical signs of aspirin toxicity in cats are similar to those seen in human patients.140 Subtle changes in liver function may reflect nonspecific hepatitis, the primary histologic lesion.
Flunixin Meglumine
Structure–activity relationship
Flunixin meglumine is a nicotinic acid derivative approved for use in the horse. Described as a potent analgesic agent, it has been used to control pain that might otherwise respond only to opioids. It is particularly useful for visceral pain. In addition to its analgesic effects, flunixin meglumine has been studied and cited for its antiendotoxic effects in experimental models of septic shock in several species.141-145
Disposition
The disposition of flunixn has been studied in dogs and cats (see Table 29-4).60,146 Flunixin appears to undergo enterohepatic circulation in cats. Its half-life differs considerably depending on the study, with more recent studies perhaps being more accurate because of a lower limit of detection that allows description of the true elimination phase.60 However, because therapeutic concentrations are not known, the true elimination phase may reflect concentrations that are subtherapeutic and may not be clinically relevant. At least two transport systems appear to be involved in the disposition of flunixin in cats (one being organic anion transporter polypeptide-2 in the liver, the other renal in origin),60 which may subject the drug to drug interactions.
Therapeutic use
Although the mode of action has not been documented, flunixin is specifically recommended as an analgesic in the treatment of colic in horses and has been useful for control of visceral pain in dogs (e.g., parvovirus) or postoperative pain.66 It also has been useful for the treatment (and especially pretreatment) of endotoxic shock.44 It prevents many of the adverse effects caused by administration of endotoxin, TXA2, and PGI2.146 Flunixin meglumine appears to modulate response to septic shock in dogs.141,145,147 In dogs a dose of 1.1 mg/kg flunixin meglumine blocks PGI2 production, and 2.2 mg/kg improves survival times of septic dogs.146 Newer COX-2–targeting NSAIDs might be similarly effective in the treatment or prevention of endotoxemia. Pharmacokinetics of flunixin in septic dogs does not appear to differ from that of control dogs.146 Toxicity, most commonly manifested as gastrointestinal upset, limits use of this drug in dogs to 2 to 3 days. Doses at three to five times those recommended caused gastrointestinal disturbances in one study.
Ibuprofen
Ibuprofen is a propionic acid derivative that has been used in dogs. Ibuprofen may be less effective as an analgesic than aspirin, perhaps on account of differences in binding of COX (reversible for ibuprofen and irreversible for aspirin). Ibuprofen remains a popular and effective antiinflammatory drug in humans. The disposition of ibuprofen has been studied in dogs (see Table 29-4). Pharmacokinetics are similar at doses of 5 and 10 mg/kg.148 A dose of 12 to 15 mg/kg is, however, necessary to achieve therapeutic concentrations, as reported in humans.148 After repetitive administration of this dose, plasma drug concentrations decrease despite no change in drug half-life.148
KEY POINT 29-15
Ibuprofen and naproxen should be considered as contraindicated in the dog just as acetaminophen is contraindicated in the cat.
In humans ibuprofen is associated with a low incidence of gastrointestinal side effects and has been compared favorably with even the newer COX-2–selective drugs.73 This may lead clients to assume the same is true for dogs or cats. However, ibuprofen is among the least safe NSAIDs in dogs. Vomiting commonly occurs after several (2 to 6) days of ibuprofen therapy in dogs with either the gelatin- or enteric-coated capsules.148 Gastrointestinal inflammation and gastric erosions have been documented after administration of 8 mg/kg daily despite the lack of clinical signs of toxicity.148,149 Death associated with gastrointestinal hemorrhage occurred in one dog given 3 mg/kg every other day for 6 weeks.149 These effects occur despite a short half-life for ibuprofen in dogs (less than 5 hours). The COX-1 to COX-2 ratio apparently has not been determined for ibuprofen in dogs. Because gastric lesions occur at doses less than those necessary to achieve therapeutic concentrations, ibuprofen is not recommended for use in dogs.
Ketoprofen
Structure–activity relationship and mechanism of action
Ketoprofen is a propionic acid NSAID approved for use in humans and horses. Because ketoprofen is a strong inhibitor of COX, it has been ascribed powerful antiinflammatory, analgesic, and antipyretic properties. Although not firmly established, the efficacy of ketoprofen has been attributed to its ability to inhibit some lipoxygenases and thus formation of LTs.150 Ketoprofen is also a powerful inhibitor of bradykinin.150
Disposition
Ketoprofen is rapidly absorbed from the gastrointestinal tract. Although peak plasma drug concentrations are lower in dogs after oral than intravenous administration, mean residence times (4.59 versus 3.81 hours, respectively) were not different.151 Bioavailability does not seem to be impaired by food. As with other NSAIDs, ketoprofen is approximately 99% protein bound, principally to albumin. Elimination reflects metabolism to inactive metabolites by the liver and excretion as the glucuronide conjugate in the urine.150 Ketoprofen has a slightly shorter half-life in cats compared with dogs (see Table 29-4). Ketoprofen is sold as a racemic mixture. The R and S isomer are handled differently by the body and induce different pharmacodynamic and pharmacokinetic responses, with variability expected among and within species. Disposition is further complicated by the potential conversion that occurs between isomers, with the extent and sequelae also varying among and within species.127 Conversion of the R to S isomer, but not the S to R isomer, has been documented in dogs and cats. Cats may convert up to 37% of the R to the S isomer. Not surprisingly, species differ as to which isomer predominates after administration of the racemic mixture. In dogs the S isomer represented 91% of the peak plasma drug concentration 3 hours after administration of the R isomer.
Drugs that are similar in structure may be markedly different in their pharmacokinetic and pharmacodynamic description, as is exemplified by ketoprofen and a similar drug, fenoprofen, in cats.152 For ketoprofen the R and S isomers behave similarly: Respective clearance was 235 and 216 mL/hr/kg, and half-life of each was 0.5 hour. For the structurally similar fenoprofen, clearance for the R isomer was ninefold greater compared with that for the S isomer (980 versus 112 mL/hr/kg), leading to a shorter half-life (0.53 hour) for the R isomer compared with the S isomer (3 hours). Further, 93% of R-fenoprofen was converted to the S-isomer, compared with only 37% of R-ketoprofen.152 As such, at least for the cat, exposure to either isomer of ketoprofen may be largely equivalent (based on area under the curve) but is approximately sixfold higher for S-fenoprofen (the COX-active isomer) compared with R-fenoprofen.152 As with disposition, the pharmacodynamic impact of each ketoprofen isomer on inflammation and analgesia is variable among species. The anti-COX effect of ketoprofen (and fenoprofen) reflects the S-enantiomer in cats.
Drug interactions specific for ketoprofen have not yet been documented.153 However, in humans adverse reactions to ketoprofen occur in approximately 30% of the patients studied.154,155 The most frequent complaint was upper gastrointestinal upset. Other commonly encountered side effects include CNS reactions, such as headaches and dizziness, and nephritis. Side effects were severe enough in one report that therapy was discontinued in approximately 13% of patients.153 Alternative preparations, such as rectal suppositories, have been formulated for ketoprofen to reduce the incidence of gastrointestinal toxicity.151
KEY POINT 29-16
Ideally, the preferred nonsteroidal antiinflammatory drug for perioperative use will be one approved for use in the target species and known to be cyclooxygenase-2 preferential in action.
Ketoprofen is not approved for use in small animals in the United States but is approved for both dogs and cats in Canada and Europe. It has proved to be an efficacious NSAID in humans and animals. In human patients suffering from rheumatoid arthritis, ketoprofen has been shown to be as efficacious as aspirin, naproxen, aspirin, indomethacin, ibuprofen, diclofenac, and piroxicam.156 Similar results occurred in cancer patients receiving either aspirin–codeine combinations or ketoprofen.155 For control of postoperative pain, ketoprofen has proved as effective as pentazocine and meperidine156 and equally effective but longer in duration than acetaminophen–codeine combinations.157 In dogs analgesia provided by ketoprofen has been reported to last between 12 and 20 hours.127
The use of ketoprofen as an analgesic and antipyretic has been studied in cats. The antipyretic effect of ketoprofen (2 mg/kg subcutaneously followed by 1 mg/kg once daily orally) in febrile cats was rapid, being evident in 4 hours with temperatures normalized at that time.158 Temperatures did not change in the cats treated with antibiotics only. The use of ketoprofen as an analgesic is variable.159 In cats subjected to ovariohysterectomy, ketoprofen (2 mg/kg subcutaneously) compared favorably with buprenorphine (0.006 mg/kg or 6 μg/kg intramuscularly) and meperidine as gas anesthesia was discontinued. Response was based on visual analog scores and overall clinical assessment. Response was equal to that of buprenorphine at 4 hours and better at 8 hours. Response was better for both drugs compared with the control at both 4 and 8 hours but still present for buprenorphine only compared with control at 18 hours.
Ketorolac
Ketorolac is an NSAID approved for use in human patients. In contrast to many NSAIDs, ketorolac is only moderately effective as an antiinflammatory.160 It is a potent analgesic, however, that has been described as equivalent to morphine. As such, it has been used as a perioperative analgesic in dogs.161 Ketorolac appears to be superior to butorphanol and equal to flunixin meglumine for control of postoperative pain. The disposition of ketoralac is quite variable in in dogs (see Table 29-4) after single intravenous or oral dosing (0.34 mg/kg), although investigators indicate that the disposition was simlar to humans.162 In humans ketorolac causes gastrointestinal upset, and similar side effects occur in dogs. In one study 1 of 21 dogs developed gastrointestinal ulceration after a single dose.66 Therefore recommendations are to limit use to one to two treatments, or 3 days. Other side effects reported in human patients include dizziness, headache, nausea, and pain at the site of injection.160 Caution should be taken particularly in the perioperative patient, which is more likely to be dependent on renal PGs during the surgical procedure.
Meclofenamic Acid
Meclofenamate is an anthranilic NSAID available as a palatable granular preparation intended to be mixed with food for large animals and as a tablet. It is approved for use in dogs in the United States. Among the NSAIDs, it is noted for its slow onset of action. The package insert associated with the label of this drug describes dogs as being exquisitely sensitive to the gastrointestinal ulcerogenic effects of these drugs. Meclofenamic acid appears to have no clear advantage to other drugs for treatment of osteoarthritis in dogs and may be more likely to cause gastrointestinal upset.
Naproxen
Naproxen is approved for use in humans and is available in over the counter preparations. Although it contains a chiral carbon, it is sold as the pure S isomer. Its disposition has been studied in dogs and, notably, it varies from that in humans. Compared with 12 to 15 hours in humans and 5 hours in horses, the elimination half-life of naproxen after intravenous administration in dogs ranges from 45 to 92 hours.163 Peak concentrations in dogs after oral administration of 5 mg/kg were 40 to 50 μg/mL. Tissue concentrations paralleled plasma concentrations, with a peak of 20 to 30 μg/mL; concentrations declined over a period of 200 hours.164 Extensive enterohepatic circulation has been credited as the cause for prolonged elimination in dogs. Because of its long half-life, naproxen need be given only once daily to every other day. Although a loading dose has been recommended, the gastrointestinal toxicity of this drug in dogs suggests that a loading dose be avoided to minimize the risk of toxicity.
The dog has been described as the animal most sensitive to naproxen.163 Its use in dogs does not seem prudent. Gastrointestinal toxicity occurs at doses of 5 mg/kg daily.165 Toxicity appears most likely when plasma drug concentrations exceed 50 μg/mL. If this NSAID must be used in dogs, doses initially should be low (1 to 2 mg/kg) and subsequently titrated to the animal’s need. Animals should be watched closely for evidence of gastrointestinal upset. Bleeding dyscrasias have also been reported in dogs receiving large doses of naproxen.163,165,166
Naproxen has been cited for a positive protective effect on articular cartilage. In a canine experimental model of osteoarthritis, naproxen decreased the loss of proteoglycans and suppressed metalloproteinase activity.167
Phenylbutazone
Phenylbutazone is a weakly acidic, lipophilic NSAID approved for use in dogs. Inhibition of the AA cascade by phenylbutazone occurs after conversion to reactive intermediates at the level of PGH synthase and prostacyclin synthase. Prostanoid-dependent swelling, edema, erythema, and associated pain are reduced by phenylbutazone.37 Phenylbutazone has been associated with some attenuation of some clinical signs associated with endotoxic shock in experimental models.142,144
Bioavailability after intramuscular administration of phenylbutazone is less than that after oral administration in most species studied because of precipitation in the neutral pH of muscle.37,168 Phenylbutazone is metabolized by the liver, with less than 2% of the drug being excreted as a parent compound in the urine in some species. Its major metabolites are oxyphenylbutazone, which is less active than phenylbutazone, and inactive γ-hydroxyphenylbutazone.37,169,170 Dose-dependent zero-order kinetics has been reported in dogs.127 Reported adverse reactions caused by phenylbutazone include bleeding dyscrasias, hepatopathy, and nephropathy (primarily in horses).37,96,171,172 Phenylbutazone has chondrodestructive effects.173,174
Dogs
Despite approval of the oral preparation for use in dogs, there is little information regarding the use of phenylbutazone in dogs. The half-life in plasma (7.3 to 18 hours) is shorter than that in tissues (20 hours), although peak concentrations in tissues (13 to 20 μg/mL) were approximately one third of those in plasma (49 to 75 μg/mL)164 after a dose of 15 mg/kg orally. The elimination half-life (in Greyhounds) is 6 to 7 hours.169,170 Dogs apparently are more tolerant of phenylbutazone than are humans. When used to treat racing greyhounds, phenylbutazone should be used with caution because of routine drug testing. One study175 has documented that phenylbutazone can be detected in the urine of Greyhounds after topical administration in a commercially available cream.
Toxicity manifested as hemorrhage, biliary stasis, and renal failure has been reported in one dog receiving close to recommended doses.97,172,176 For reasons not explained, the package insert notes a total maximum dose and requires the drug to be discontinued slowly. Bone marrow dyscrasias (including neutropenia) also have been reported.
Cats
Although phenylbutazone has been used in cats, a high incidence of toxicity suggests extreme caution. One hundred percent of cats treated with 44 mg/kg daily became anorectic at 2 to 3 days, with 80% mortality at 2 to 3 weeks. Toxicity occurs primarily in the bone marrow and is characterized by decreased erythroblastic activity and possible interference with myeloid maturation. Gastrointestinal damage, nephrotoxicity, and hepatotoxicity also occur.96
Piroxicam
Piroxicam is an oxicam NSAID approved for humans that has been used to treat osteoarthritis in dogs. More recently, it has received attention for its ability to reduce the size of tumors (transitional cell tumors and others) in dogs.49,50-52 Piroxicam may interact by an additive or synergistic action with anticancer drugs to cause tumor cell death. Piroxicam is a potent antiinflammatory in musculoskeletal conditions.The disposition of piroxicam has been studied in both the dog177 and cat.178 Notably, the half-life of the drug is much shorter in cats (12 hours) compared with dogs (40 to 50 hours). Although the LD50 of piroxicam is greater than 700 mg/kg in dogs, gastric lesions and renal papillary necrosis have occurred in dogs receiving 0.3 to 1 mg/kg daily.49,177 The ratio of COX-2 to COX-1 suggests that gastrointestinal toxicity occurs. Little evidence of toxicity (gastrointestinal or bleeding), however, was noted after administration of 0.3 mg/kg every other day.49,177 Extrapolation of human use to dogs should be done cautiously because of possible differences in volume of distribution, therapeutic concentrations, or safety margin.
Cyclooxygenase-1–Sparing Nonsteroidal Antiinflammatory Drugs
Because the extent of COX-1 and COX-2 inhibition for many of the newer NSAIDs is not clear, particularly in dogs and cats, the term preferential COX-2 inhibitors is preferred in this text to selective COX-2 inhibitors, with the former reflecting the potential for inhibition of both isoforms. COX-1 sparing is intended to mean the same. Bergh and Budsberg179 reviewed the use of coxib NSAIDs in veterinary medicine.
Carprofen
Dogs
Carprofen is a proprionic acid–derived NSAID approved in the United States for use in dogs for the treatment of osteoarthritis.180 The drug is approved for use in dogs and cats in selected countries outside of the United States. Like other NSAIDs, carprofen has antipyretic, analgesic, and antiinflammatory effects.180 Its potency is equal to that of indomethacin and surpasses that of aspirin or phenylbutazone,180 and doses consequently are smaller for carprofen than for these NSAIDs.
The mechanism of action of carprofen is not certain, but, unlike other members of its class (e.g., ibuprofen, ketoprofen, naproxen), it may be relatively selective for inhibition of COX-2. McKellar and coworkers181 reported that it does not inhibit thromboxane activitiy in platelets, PGE2 (isoform not identified) nor 12-HETE in inflammatory cells. With canine platelet-derived COX-1 and macrophage-like cell COX-2, the ratio of COX-1 to COX-2 IC50 (concentration that caused 50% inhibition) for the racemic mixture (that available in the commercial preparation) was 129; the ratio was 181 for the S isomer (thus the “antiinflammatory” enantiomer) but only 4.19 for the R isomer (see Table 29-3).32 These differences in COX inhibition may explain the apparent safety of carprofen compared with conventional NSAIDs in dogs and with safety in the dog compared to that in humans, for whom the ratio is less than 1.
Pharmacokinetics
Carprofen has been studied in dogs after oral and subcutaneous administration (see Table 29-4). Increases in oral doses generally result in proportional increases in Cmax, with peak concentrations occurring at approximately 1 hour. Peak concentrations appear to occur more slowly with subcutaneous than with oral administration; the former route may prolong onset of efficacy. Although Cmax differed between oral and subcutaneous administration, the area under the curve did not, indicating bioequivalence among the two routes after both single and multiple dosing.182 Like other NSAIDs, carprofen is highly protein bound. Carprofen is metabolized by the liver and in dogs is characterized by a half-life of approximately 10 hours, which is sufficiently long that it has been approved for once-daily administration. Between 70% and 80% of carprofen metabolites are excreted in the feces, with the remainder in the urine.
The dispositions of the two carprofen enantiomers have been studied in dogs.181,183 Administration as a racemic mixture yields a Cmax of the R isomer at 18 μg/mL versus 14 μg/mL for the S isomer. Conversion apparently does not occur between the R or S isomer in dogs. Both the R isomer (72%) and the S isomer (92%) are extensively excreted in the bile, but enterohepatic circulation appears to be relatively specific for the S isomer (the isomer characterized by a very favorable COX-2 specificity), probably owing to greater glucuronidase-resistant isoglucuronides for the R isomer.183 Up to 34% of the S isomer is recirculated.127 Because both the clearance and the volume of distribution of the S isomer are greater than those of the R isomer, the mean residence times and half-lives of the two isomers do not differ.183 After a single subcutaneous dose of 4 mg/kg either preoperatively or postoperatively, duration of analgesia ranged from 18 to 24 hours.127
Adverse reactions
On the basis of toxicity data, carprofen appears to be among the safest of the new NSAIDs approved for use in dogs (see Table 29-5). According to the package insert, dogs dosed with more than 10 times the amount necessary to achieve therapeutic concentrations did not develop gastrointestinal side effects when dosed for 2 weeks or when dosed at almost six times the recommended dose for 52 weeks. In a clinical trial of 70 dogs, 6 of 36 carprofen-treated dogs (2.2 mg/kg orally every 12 hr) developed clinical signs indicative of gastrointestinal upset; three dogs that received placebo also developed gastrointestinal signs.184
KEY POINT 29-17
On the basis of toxicity and field trial data on package inserts, carprofen appears to be among the safest of the newer nonsteroidal antiinflammatory drugs approved for use in dogs.
Forsyth and coworkers185 compared the gastrointestinal side effects of carprofen (2 mg/kg orally twice daily for 7 days followed by 2 mg/kg once daily), meloxicam (0.2 mg/kg orally once daily), and ketoprofen (1 mg/kg orally every 24 hours) with those of placebo after 28 days of therapy in dogs. The fewest and least severe gastroduodenal lesions apparent endoscopically were in the carprofen-treated and control group, but there was no statistical significance between the three NSAIDs and the control group, making interpretation difficult. No animals revealed clinical signs associated with gastrointestinal upset. Reimer and coworkers83 found etodolac and carprofen to not differ from the placebo and all three to cause fewer gastric lesions than aspirin when dosed at labeled doses for 28 days. Craven and coworkers186 also prospectively compared the effects of carprofen (n = 10 dogs; 4 mg/kg days 1 and 2, 2 mg/kg daily thereafter) to meloxicam (n = 10 dogs; 0.2 mg/kg daily, 1 then 0.1 mg/kg daily) on gastrointestinal epithelium as measured by sugar permeability tests. Dogs were dosed for at least 7 days and studied for 8. Significant changes across time occurred within the carprofen-treated group but resolved by study end; further, although not significant, changes also appeared to have occurred for at least one sugar test for meloxicam. No significant changes were detected in urinary recovery of any sugar between the two treatment groups; placebo controls were not studied. The impact of this comparison is somewhat limited by the absence of untreated controls, large number of comparisons made (two treatment groups, 3 times, seven sugars; potentially, 42 comparisons), and the small number of dogs studied in the face of the variability of the data; the power of the study to detect significant differences was not reported.
Hepatotoxicity reflecting acute hepatic necrosis has been reported as an unexpected adverse effect of carprofen in dogs.187 Toxicity studies supporting carprofen approval found only mild changes in liver enzymes when the drug was administered at 25 mg/kg for 13 to 52 weeks (package insert data); serum alanine transaminase (SALT) increased with a dose of 80 to 160 mg/kg per day. However, approximately 1 year after its approval, reports of gastrointestinal toxicity led Pfizer to address concerns regarding side effects in a technical report.188 Of the 1 million dogs receiving carprofen, an incidence of 0.2% suspected side effects was reported, with 0.02% involving the liver. Although 33% of animals affected in initial studies were Labrador Retrievers, this number was not corrected for the prevalance of this species. Hepatopathy has been diagnosed in all breeds of dogs receiving carprofen clinically. At least 70% of afflicted animals were considered geriatric, suggesting that this age group is predisposed, perhaps because of decreased hepatoprotective function (e.g., glutathione scavenging of metabolites). Although death has occurred in some animals, timely discontinuation of the drug can lead to complete resolution of biochemical abnormalities. Animals with liver disease in one study also had evidence of renal tubular disease.187 MacPhail and coworkers187 studied 21 animals and reported clinical signs of anorexia, vomiting, lethargy, diarrhea, polyuria, polydipsia, and hematuria occurring between 5 and 30 days; however, clinical signs did not occur until as long as 60 and 180 days for two dogs. In this study 13 of the 21 dogs were Labrador Retrievers, with dogs ranging in age from 4 to 15 years. The most common clinical laboratory abnormalities included increased activities of SALT, aspartate transaminase (SAST), alkaline phosphatase (SAP), and bilirubin. Histologic lesions in the liver ranged from mild to severe and consisted of hepatocellular necrosis. Four of the 21 dogs died; those remaining that were hospitalized were treated with supportive therapy, including gastrointestinal protectants.
Hepatic disease associated with carprofen might be minimized by pretreatment evaluation because lesions appear to occur within the first several weeks of therapy. Consequently, monitoring (clinical laboratory tests) for hepatic damage and hepatic function (bile acids) at weekly or biweekly intervals for the first month is recommended, particularly in predisposed (e.g., geriatric) animals. Monitoring should continue at intervals of 2 to 4 weeks for 3 months and perhaps longer in patients at risk for liver disease. Animals receiving phenobarbital have been anecdotally reported to be more susceptible to hepatotoxicity. Induction of hepatic drug-metabolizing enzymes (e.g., phenobarbital, others) may increase the risk of toxicity if associated with toxic metabolites. Use of hepatoprotective agents such as N-acetylcysteine (a glutathione precursor) or SAMe may be beneficial during initial or continued hepatic damage.
The potential for carprofen-induced nephrotoxicity also has been studied in dogs. Crandell and coworkers105 found no changes compared with saline placebo in renal function (glomerular filtration rate, serum urea, and creatinine) in young healthy dogs (n = 12) receiving placebo, meloxicam (0.2 mg/kg), or carprofen (4 mg/kg) after 30 minutes of electrically stimulated pain. Dogs were studied using a randomized crossover design; anesthesia consisted of butorphanol and acepromazine as preanesthetics, ketamine and diazepam for induction, and isoflurane for maintenance.105 The study was designed such that power was sufficient to detect a change in glomerular filtration rate of 0.5 mL/kg/min, suggesting that the two treatment groups were the same. This is in contrast to the results of Forsyth and coworkers,189 who found that creatinine clearance was significantly less in dogs 24 hours after undergoing routine castration and receiving either carprofen (4 mg/kg intravenously), ketoprofen (2 mg/kg intravenously) compared to placebo or saline (0.2 mL/kg intravenously) at induction of anaesthesia. The anesthetic protocol is this study differed among animals (drugs included morphine, thiopental, and halothane).189 Bostrom and coworkers190 investigated the effect of carprofen (4 mg/kg intravenously) administered either 30 minutes before or 30 minutes after induction of anesthesia (acepromazine–thiopentone–isoflurane) on renal function (glomerular filtration rate) in dogs (n = 6) after experimentally induced decrease in blood pressure to 65 mm Hg. Significant adverse effects were not detected, although the power of the study to detect a significant difference was not assessed.
The impact of carprofen on hemostasis has been studied. Although carprofen was associated with decreased platelet aggregation and increased partial thromboplastin time in one study, all values were within normal limits. Neither buccal mucosal bleeding time nor complete blood count parameters changed after 12 days of dosing at 2.2 mg/kg twice daily.191 Other investigators have found no changes in in vitro indices of platelet function.
The effect of carprofen on cartilage physiology appears to be biphasic.192 In vitro studies with canine chondrocyte cell cultures revealed that carprofen increases the rate of polysulfated glycosaminoglycans (PGAG) synthesis at synovial fluid concentrations (≤10 μg/mL) achieved in human patients receiving a therapeutic dose of carprofen. The S-isomer stimulated PGAG synthesis at a tenfold higher rate compared with the R isomer (United States Pharmacopeia [USP]).127 Inhibition of PGAG synthesis, however, occurs at concentrations of 20 μg/mL or more.192 Concentrations that occur in dog synovial fluid after administration of a therapeutic dose of carprofen have not been determined.
Carprofen is approved for use in dogs for control of perioperative pain and treatment of osteoarthritis. It is available as either an oral preparation or an injectable solution intended for subcutaneous injection. Carprofen solution is stable for 1 month if not refrigerated. A chewable table is available, although the risk of accidental overdose may be greater with this product. Although approved for subcutaneous use, injectable carprofen has been given intravenously (4.4 mg/kg) as a single dose with no adverse effects.190
Carprofen appears to be equally or more effective than many other NSAIDs studied for the control of inflammation, and it has proved effective for control of the pain associated with the inflammation of osteoarthritis184 and postoperative pain193-195 when administered preoperatively. Because carprofen was the first COX-1–protective NSAID approved for use in dogs, approval of subsequent similar-acting NSAIDs generally used it as a positive control. As such, several studies have compared the efficacy of carprofen with that of other NSAIDs, although these studies tend to be manufacturer sponsored. Using a non–placebo-controlled randomized crossover design, Borer and coworkers196 compared single-dose carprofen (0.2 mg/kg intravenously or 4 mg/kg, orally), etodolac (17 mg/kg, orally), or meloxicam (0.2 mg/kg, orally); (n = 12; studied in groups of four at 3-week intervals) in dogs with experimentally induced acute synovitis (monosodium urate injection). Outcome measures included kinetic gait analysis (force plate), orthopedic evaluation, and serum C-reactive protein (CRP) (n = 6). Lameness was assessed on a biomechanical force platform and by orthopedic evaluations of the stifle joints; blood was collected to monitor serum CRP concentration. All dogs in the treatment groups had improved indices of lameness compared with control animals. Although greatest improvement occurred in carprofen-treated dogs, onset was fastest in etodolac-treated animals. Both carprofen and etodolac were associated with lower pain compared with butorphanol, although the authors concluded that meloxicam also was more effective than butorphanol. Serum CRP was not different among groups.196
Carprofen or meloxicam also were compared with a combination glucosamine–chondroitin sulfate (Cosequin) product using a prospective, double-blinded study in dogs (n = 71) with osteoarthritis; the study was supported by the manufacturers of meloxicam. Treatment continued for 60 days, and response was based on force plate analysis and subjective evaluation by owners. Although animals responded to both meloxicam and carprofen, only meloxicam was associated with a return to baseline function. Side effects were minimal, although one dog receiving carprofen developed hepatopathy and one dog receiving meloxicam withdrew from the study because of vomiting.197
Finally, Aragon and coworkers198 evaluated the quality of evidence of NSAIDs, including carprofen, or supplements used to treat osteoarthritis in dogs. The FDA’s evidence-based ranking was applied to scientific data collected from a review of the literature before 2006. Studies were ranked on the basis of study design, quality, and total body of evidence (quantity of studies or study subjects, consistency among the different reports, and the likelihood that the magnitude of the response was physiologically meaningful). The evidence was then ranked according to strength (high, moderate, low, or extremely low level of comfort). Only 16 clinical trials met the inclusion criteria. Of the compounds studied, the evidence for meloxicam was accorded a high level of comfort (i.e., that efficacy is scientifically valid), and the evidence for carprofen, etodolac, PGAG, and glucosamine –chondroitin–manganese was ranked as moderate. Hyaluronan was ranked extremely low. Care must be taken not to overinterpret this data because of the limited number of studies reviewed
Cats
Carprofen is approved for use in cats in certain countries outside the United States. The disposition of the drug and its enantiomers has been studied in cats at the dose associated with control of inflammation (4 mg/kg; Table 29.4).128,199-201 The smaller clearance for carprofen in cats results in an elimination half-life that is at least twice as long in cats as in dogs. The S isomer is cleared almost 3 times as rapidly as the R isomer, resulting in a shorter half-life for S compared with R in the cat
The relative COX-2 selectivity of carprofen that occurs in dogs has not been well documented in cats, although data from Brideau and coworkers33 suggests relative selectivity similar to that in dogs (see Table 29-3). Clinically, however, this may not be true. The gastrointestinal effects of single-dose carprofen (4 mg/kg intravenously) or aspirin (20 mg/kg intravenously) were studied in cats (n = 5) using endoscopy and a randomized crossover design. Lesions in the stomach and duodenum 8 hours after injection were limited to minor pinpoint erosion in one cat. Clinical laboratory tests were not affected by either drug.128 Duodenal perforation has been reported in cats receiving oral carprofen (2.2 mg/kg twice daily for 7 days) after ovariohysterectomy.202 The ulceration may have been exacerbated by the flunixin meglumine and dexamethasone with which the cat was treated before referral.
Parton and coworkers128 prospectively compared the gastrointestinal effects of carprofen (4 mg/kg intravenously) and salicylate (20 mg/kg intravenously) in cats using a crossover design. Only a single dose was studied; gastric lesions were scored endoscopically 8 hours after dosing. No differences were found between treatment groups or times, with one exception in the salicylate group for which pinpint erosive lesions were found. Steagall and coworkers107 also prospectively studied the adverse effects of carprofen, after multiple doses (6 days) of decreasing doses (4 mg/kg on day 1 to 1 mg/kg on day 6). A randomized, blinded crossover, placebo-controlled design was used, with a 4-week washout. Endoscopic lesions were scored before and 7 days after treatment. Changes in biochemistry profiles included decreased albumin but no other abnormalities, including gastrointestinal lesions or changes in renal dysfunction.
As a postoperative analgesic in cats, carprofen compares favorably with pethidine (meperidine), providing equal but longer analgesia (at least 24 hours) when administered at 4 mg/kg subcutaneously postoperatively.201 In a clinical trial of cats undergoing ovariohysterectomy, the analgesic effects of carprofen, ketoprofen, meloxicam, or tolfenamic acid were compared. Outcome was based on the visual analog scale and a nociceptive threshold at the incision site, as well as clinical response. No difference was found between treatment groups in providing analgesia. With nine of ten cats responding, all responses were described as good, although none prevented wound tenderness.203 Taylor and coworkers204 prospectively compared the efficacy of carpofen (4 mg/kg) and buprenorphine (0.01 mg/kg) as preventives for hyperalgesia in an experimental model of inflammatory pain in cats. Cats (n = 8) were studied using a randomized, crossover, placebo-controlled design. Drugs were administered before (3 hours for carprofen, 1 hour for buprenorphine) anesthesia. Carprofen completely prevented inflammation, and buprenorphine almost completely did so, although hypoalgesia was not realized.
Deracoxib
Deracoxib is approved for use as an oral chewable preparation in dogs. As a coxib, deracoxib would be expected to be COX-1 sparing, which appears to be true on the basis of studies using cloned canine COX enzymes (see Table 29-3). However, specificity is lost at higher concentrations. Deracoxib undergoes extensive hepatic metabolism. At 8 mg/kg (approximately 2 to 8 times the recommended dose [1 to 4 mg/kg]), nonlinear kinetics emerge, increasing the risk of toxicity. As with other NSAIDs, gastrointestinal toxicity occurs with increased doses. The package insert indicates that toxicity occurs at 3 times the dose when a micronized (which differs from the approved product) compound was administered (in a rapidly dissolving capsule) but not when the commercial preparation was administered at the same dose (see Table 29-5). Whether or not derocoxib is more likely than other NSAIDs to reach saturation kinetics is not known. The difference in preparations may have resulted in much more rapid exposure of the gastrointestinal mucosa to drug. The gastrointestinal lesions associated with the micronized material were attributed to a local effect (osmotic, pH, pharmacologic, others) rather than a systemic effect.205 Dose-dependent increases in blood urea nitrogen were reported when dogs received deracoxib at 3, 4, and 5 times the recommended dose (2 mg/kg); variable renal histologic lesions were reported. The disposition of deracoxib has been described in cats after a single oral dose (see Table 29-4). 206 Safety information in cats was not available at the time of this publication other than at the FDA CVM site (Table 29-6).
As a sulfonamide, deracoxib might be expected to be associated with adverse reactions (but not those typical of antimicrobial sulfonamides), including keratitis sicca.
In a study at least partially funded by the manufacturer, efficacy of deracoxib administered as a single dose (0.3, 1, 3, or 10 mg/kg) was compared with that of carprofen (2.2 mg/kg) in 24 hound dogs. Drugs were administered 30 minutes before induction of chemically induced (urate crystal) synovitis, and lameness indices (including clinical scores, force plate analysis, and joint fluid analysis) were measured for 24 hours after induction. Deracoxib was found to be somewhat more effective at 3 mg/kg (determined to be the minimum effective dose) and more effective than carprofen at 10 mg/kg (not a recommended dose) on the basis of outcome measures for lameness which included lameness scores, pain joint effusions, and pain threshold response207; no significant differences were found between carprofen or the placebo group.
Lascelles and coworkers80 described a series of cases of gastrointestinal perforation associated with deraxocib administration in dogs (n = 29). The mean age was 6.4 ± 3.2 years. Among the more important points of the report was that 55% of the dogs received a dose higher than recommended on the label; and 59% had received, within the previous 24 hours, either a different NSAID or a glucocorticoid. A total of 90% of the dogs had received one or the other and a total of 20% had received both. Of the dogs, 68% either died or were euthanized. The mean dose in dogs receiving the drug for long-term pain management was 3± 1.1 mg/kg/day, ranging from 2.3 to 6.2 mg/kg.
The safety of deracoxib (1.5 mgkg daily) was prospectively compared with that of buffered aspirin and placebo (25 mg/kg orally every 8 hours) in dogs (placebo controlled) based on gastroscopy and clinical scores assessing vomiting and diarrhea.208 Aspirin-related gastric lesions and vomiting were higher than similar lesions in the deracoxib-treated and placebo-treated groups, beginning with the first endoscopic exam at 6 days.
Etodolac
Etodolac is a pyranocarboxylic acid that has shown potent NSAID activity by inhibiting chondrocyte and synoviocyte biosynthesis of PGE2 COX-1 to COX-2 ratios vary among studies. The ratio using human cells appears to favor safety,209 although studies using canine cells32 reveal the opposite. Ricketts and coworkers32 reported a COX-1 to COX-2 ratio of only 0.517 compared with 129 for carprofen (study sponsored by carprofen manufacturer). In dogs the drug is rapidly absorbed, with food slowing absorption. Its elimination half-life is approximately 14 hours,210 which is sufficiently long to allow once-daily dosing. Food may prolong elimination (see Table 29-4). The drug is extensively metabolized, with the majority eliminated in the bile and up to 10% excreted in the urine; enterohepatic elimination is extensive, potentially increasing the risk of gastrointestinal toxicity.127 Etodolac is marketed as a racemic mixture, although the disposition of the enantiomers has not been well described in animals. Because it is approved for use in humans, much information is available regarding possible side effects in humans. However, in vitro studies indicate pharmacodynamic effects may differ in animals, and extrapolation of information may be inappropriate. Because of its longer half-life and enterohepatic elimination, etodolac may be associated with a greater risk of gastroduodenal ulceration than other COX-2 preferntial drugs. Toxicity studies also suggest that etodolac may not be as safe as other newer NSAIDs, which might be supported by COX ratios in the dog (see Table 29-3). According to the package insert, six of eight dogs developed gastrointestinal erosions and subsequently died after etodolac was administered at five times the recommended dose for 6 to 9 months (see Table 29-5). Five of six dogs developed excessive bleeding when receiving etodolac at the recommended dose, although the duration of therapy was not clear from the package insert. However, clinically, based on field studies, etodolac does not appear to be less safe than the other COX-2 preferential drugs.
Liver disease in dogs such as that associated with carprofen has not been reported in the literature but has been anecdotally reported and has been seen by the author. Etodolac (mean dose of 13.7 mg/kg orally every 24 hours for 28 days) administration did not significantly affect serum T4, T3, fT4, or cTSH concentrations or serum osmolality in healthy random-source mixed breed dogs (n = 19). However, plasma total protein, albumin, and globulin concentrations were decreased by day 14 of administration; decreases were still evident by day 28 of administration, although all concentrations were within normal limits.118 The effect of etodolac on cartilage metabolism has not been well documented, although one study found it to be associated with more cartilage damage than carprofen.211 Etodolac has been associated with keratitis sicca. 211a In a series of cases collected from the manufacturer or surveyed veterinary ophthalmologists, lesions were commonly considered severe, occurred in both eyes more than 50% of the time, with remission occurring in 10 to 15% of animals. Remission was 4 times more likely if treatment duration was less than 6 months.
Etodolac is available of an oral capsule prepration for use in dogs. Despite potential gastrointestinal adverse drug events, when dosed at 10 to 15 mg/kg once daily, etodolac was effective yet safe for controlling lameness associated with hip dysplasia.212 A clinical trial comparing carprofen and etodolac was previously discussed (see carprofen).196 Aragon and coworkers198 ranked etodolac as moderate with regard to scientific evidence of efficacy claims.
Firocoxib
Firocoxib has recently been approved for use in dogs in a chewable tablet form. Its disposition was reported before its approval by McCann and coworkers,213 who reported on COX-2 selectivity (canine whole blood), pharmacokinetic properties (2 mg/kg intravenously and 8 mg/kg orally) in healthy male and female mixed-breed dogs (n = 21). The IC90 for COX-2, according to the monograph, is 0.3 μg/mL, compared with a Cmax of about 0.5μg/mL at the recommended dose in dogs (Table 29-4). In the manufacturer sponsored study, the COX-1 to COX-2 ratio using canine blood was 384, compared with 6 for carprofen and 12 for deracoxib. Although the volume of distribution is larger than for most NSAIDs, clearance also is rapid, resulting in a relatively short half-life compared with that of other NSAIDs (see Table 29-4). Nonetheless, the drug is administered once a day. The oral bioavailability of firoxocib as listed on the package insert at 5 mg/kg is 38% (variability not reported); as such, care may be indicated in anticipating potential differences in oral bioavailability among animals.∗
Firocoxib is approved for use in dogs greater than 3.2 kg (smaller dogs cannot be accurately dosed) as a chewable tablet. The package insert overdose data and its impact on gastssrointestinal toxicity for firocoxib are indicated in Table 29-5. The manufacturer of firocoxib was interested in avoiding an age limit to drug administration, and accordingly, studied firoxocib in juvenile Beagle dogs (Table 29-5). Fatty livers were consistent findings in drug-treated juvenile dogs. In addition to the gastrointestinal effects, one juvenile dog given a dose 3 times that recommended developed juvenile polyarthritis, thrombocytopenia, decreased albumin, and elevated liver enzymes. In a separate study, one dog receiving 3 times the dose was euthanized because of poor clinical condition, although no indication was given as to whether adversity was related to the drug. However, 4 of 12 juvenile dogs receiving 5 times the dose either died (n = 1) or were euthanized (on account of a moribund status) as early as day 38 and as late as day 79; two of the dogs had gastrointestinal ulceration. This data should not be interpreted as a greater risk of toxicity to firocoxib as much as a potential indication that NSAIDs should be avoided in animals 6 months or younger.
For the approval process, efficacy of firocoxib was compared at 8 mg/kg with carprofen (2.2 mg/kg) as the positive control in healthy male Beagles (n = 24; eight per group) using chemically induced (urate) synovitis. The drugs were administered either prophylactically (administration 2 hours before induction of synovitis) or therapeutically (administration 1 hour after induction). Both drugs were effective in controlling pain and inflammation when administered either prophylactically or therapeutically; failure to find a difference between the two NSAIDs was attributed to the variability in the data compared to animal numbers studied. Although a treatment effect was found across time for firocoxib compared with carprofen, superiority of firocoxib compared with carprofen was not sufficient to allow label claims as such. Pollmeier and coworkers,214 in a manufacturer-sponsored study, reported the results of a multicenter field trial that compared firocoxib (5 mg/kg/day) to carprofen (4 mg/kg/day) using a double-blinded, randomized design in dogs (n = 218) with osteoarthritis. Efficacy was assessed by both owners and veterinarians. Reponse was slightly better for firocoxib at 96% compared with 92% for carprofen.
Meloxicam
Dogs
Structure–activity relationship
Meloxicam, like piroxicam, is a member of the oxicam group of NSAIDs. It is approved for use in tablet and oral suspension forms in the United States, Canada, and Europe in dogs and cats (the injectable form [subcutaneous] only is approved for one time use in cats in the US). The COX-2 to COX-1 ratio for meloxicam, unlike that for piroxicam, favors selective COX-2 inhibition in humans, suggesting that it has a wider margin of safety than most other NSAIDs.215-218 The drug is more potent (although not necessarily more efficacious) than aspirin, indomethacin, and piroxicam; hence its dose is smaller. In a study partially funded by the manufacturer of meloxicam, using in vitro methods, Kay-Mugford and coworkers219 determined the ratio of COX-1 and COX-2 activity of meloxicam, tolfenamic acid, carprofen, and ketoprofen using a canine monocyte–macrophage cell line. In this cell line, expression of COX-1 is constitutive, but COX-2 is induced in response to LPS. The COX-1 to COX-2 ratios were more favorable for meloxicam than carprofen (see Table 29-3), which suggests that meloxicam is COX-1 protective in the dog.
Disposition
The plasma concentration–time profile for meloxicam in dogs was described as being comparable to that in humans, in contrast to that of selected other species.219 Although oral bioavailability is close to 100% in dogs (according to package insert), time to peak concentration is approximately 7 to 8 hours, one of the longest of NSAIDs in dogs (see Table 29-4). Likewise, the elimination half-life is long compared with that of other NSAIDs (both conventional and new) in the dog, with a minimum of 3 days expected before steady state is achieved. A loading dose of 0.2 mg/kg is suggested in dogs, followed by a maintenance dose of 0.1 mg/kg. Cautious clinicians might consider administering the loading dose themselves rather than allowing the client to do so; this reduces the risk that the client will continue to administer the drug at a dose that is higher than appropriate. The package insert also indicates that disposition of meloxicam changes with chronic dosing, with elimination half-life becoming longer. As with most drugs, the selectivity enjoyed with the mechanism of action of this or any selective NSAID is likely to be lost at high doses.220
Adverse reactions
The safety profile of meloxicam, as indicated on the package insert, supports its safe use in dogs, although direct comparison with other newer NSAIDs is limited by differences in doses studied (see Table 29-5). Interestingly, in field studies the incidence of vomiting in dogs receiving meloxicam was among the highest of the newer NSAIDs. The safety of meloxicam has been compared with that of carprofen (see the discussion of carprofen). Meloxicam-induced hepatotoxicity has been reported in humans.221
The impact of meloxicam (0.1 mg/kg subcutaneously once daily), dexamethasone (0.25 mg/kg subcutaneously twice daily), or the combination of the two on the gastrointestinal mucosa was prospectively compared with that of saline after 3 days of therapy in Beagles (n = 20). Outcome measures included scores collected from five regions of the gastroduodenal region. Scores were greater for the combination-treated group than for the other groups; additionally, dexamethasone was associated with higher scores than either meloxicam or saline. No significant differences in histologic findings were found among groups, although this may reflect a low power for the study. However, the lack of correlation between gross and histologic gastroduodenal lesions is not unusual. Lesions associated with NSAIDs were greater in the pylorus and pyloric antrum compared with other regions, a finding also consistent with other studies.74
Gastrointestinal perforation associated with the administration of meloxicam was reported in a series of cases (n = 5).222 Ages ranged from 2.5 to 11 years. Doses associated with vomiting ranged from a single postoperative dose of 0.1 mg/kg (on day 8) to a high dose of 0.2 mg/kg orally as a single or divided daily dose. Onset of vomiting ranged from as early as day 2 to as late as day 8. Predisposing factors such as glucocorticoid or NSAID administration were not evident. Unidentified underlying gastrointestinal disorders were postulated as possible risk factors.
A single case report has described cutaneous and ocular lesions in a 10-year-old 25-kg mixed-breed dog with a history of atopy receiving meloxicam.223 Meloxicam (0.1 mg/kg orally once daily) was administered in anticipation of surgery. The patient returned after 3 days of therapy with cutaneous clinical signs consistent with a potential drug reaction and corneal edema. Skin biopsies were collected several days after initial lesions emerged; the patient was treated with glucocorticoids. Skin lesions had resolved by 6 weeks, and ocular lesions persisted.
Therapeutic Use
Meloxicam has been demonstrated to be both safe and effective for treatment of osteoarthritis in dogs.82,224 The efficacy of meloxicam in treatment of locomotor disorders in dogs compares favorably with that of other NSAIDs in studies supported by the manufacturer.218,225 In their evidence-based review of clinical trials addressing treatment of osteoarthritis, Aragon and coworkers198 found meloxicam to be the only drug worthy of the highest level of confidence reported in the study in regards to scientific validity of efficacy claims. Several studies have demonstrated efficacy of meloxicam compared to placebo. Doig and coworkers224 studied 40 dogs (manufacturer sponsored), and Peterson and Keefe82 studied 217 dogs with spontaneous osteoarthritis. Duration of therapy was 7 to 14 days, respectively. Placebo effects were reported in up to 38% of animals, underscoring the importance of placebo controls.82 Meloxicam (0.2 mg/kg every 24 hours) and aspirin (25 mg/kg every 12 hours) were compared in a clinical trial using a crossover (randomization not indicated) design in dogs (n = 12) with experimentally induced (cranial cruciate rupture) osteoarthritis of the stifle. Animals were treated for 21 days; outcome measures included measurement of PGE2, thromboxane B2 (TXB2) in blood and synovial fluid, and endoscopically collected gastric mucosa. Both aspirin and meloxicam administration significantly suppressed PGE2 concentrations in blood and synovial fluid. Aspirin, but not meloxicam, suppressed TXB2 concentration in blood and PGE2 in gastric mucosa, leading the authors to conclude that meloxicam behaves in vivo as COX-1 sparing.226
KEY POINT 29-18
The Food and Drug Administration has indicated that meloxicam should not be administered in cats more than as a single dose.
Meloxicam also has been compared with opioid analgesics for control of perioperative pain. Mathews and coworkers229 compared the safety and efficacy of preoperative administration of a single dose of meloxicam, ketoprofen, and butorphanol in dogs (n = 36; 12 per group) undergoing abdominal surgery. The butorphanol-treated group received a second dose immediately after surgery. Overall efficacy was rated as good or excellent in 9 of 12 dogs that received either meloxicam or ketoprofen compared with only 1 of 12 that received butorphanol. Analgesic effects of meloxicam were maintained for 20 hours postoperatively. Budsberg and coworkers230 compared the analgesic effects of meloxicam (0.2 mg/kg intravenously) and butorphanol (0.1 mg/kg) administered immediately after surgery (at incision closure) for control of postoperative pain in client-owned dogs (n = 40) presenting with rupture of the cranial cruciate ligament. Outcome was based on evidence of pain (visual analog scale) and serum cortisol concentrations for a 24-hour period. Differences in pain response did not emerge between the treatment groups until 8 hours later, with the meloxicam-treated group exhibiting less pain 8 to 11 hours after extubation. Failure to detect a significant difference between treatment groups earlier than 8 hrs may have reflected low overall scores for both groups coupled with low animal numbers (i.e., poor power of the study). Overall serum cortisol was less in the meloxicam-treated group compared with butorphanol group. Otherwise, significant differences could not be demonstrated. Using a model of acute inflammation (intraarticular injection of calcium pyrophosphate dehydrate), the effects of meloxicam (0.2 mg/kg intravenously for 3 doses) on proteoglycan biosynthesis in vitro and ex vivo were compared with indomethacin (0.5 mg/kg intravenously for 3 doses). Indomethacin is known to contribute to joint injury in humans in part by inhibiting sulphated proteoglycans., Neither drug contributed to joint damage, but pain and exudates were decreased more by meloxicam than indomethacin.231
Cats
The disposition of meloxicam has been studied in cats. Meloxicam is among the NSAIDs characterized by a shorter half-life in cats than in dogs (see Table 29-4). It is one of the few NSAIDs that appear to be well tolerated in cats. Its use in Canada for several years predated its approval for use in cats in the United States. Meloxicam is the only NSAID approved for use in cats in the United States, with the approved use limited to perioperative single dosing. Meloxicam is approved in cats outside the United States for single dose administration as both an injectable and oral product.
Despite its apparent safety compared with other NSAIDs, the therapeutic margin of meloxicam is relatively narrow. The association between renal disease and NSAIDs, including meloxicam, in cats was discussed previously. Cats do not tolerate multiple doses greater than or equal to 0.3 mg/kg, and gastric ulceration and death have occurred at three to six times the normal dose for 10 days. Several studies support the efficacy of meloxicam in cats. In one study the optimal dose of meloxicam to prevent endotoxin-induced fever in cats was 0.3 mg/kg.232 The need for a long-term safe yet effective analgesic in cats was supported by a symposium that focused on osteoarthritis in cats.233 The safety of meloxicam in cats in regards to renal disease was previously addressed (Table 29-6). A prevalence of degenerative joint disease was found in 34% of cats (n = 218), with previous reports placing the prevalence as high as 90%.
A number of clinical trials have addressed efficacy of meloxicam in cats. In cats subjected to ovariohysterectomy, meloxicam, carprofen, ketoprofen, and tolfenamic acid all were effective in controlling postoperative pain, with 10% of cats in each group requiring opioid rescue.203 In a comparison of carprofen (4.4 mg/kg subcutaneously) and meloxicam (0.3 mg/kg subcutaneously) in 80 cats undergoing ovariohysterectomy, no difference was found in control of postoperative pain; one cat in the carprofen group and two in the meloxicam group required opioid rescue.228
The efficacy of meloxicam for treatment of osteoarthritis in cats was reported in abstract form233 on the basis of a prospective crossover pilot study (n = 7; mean age 14 years). Cats were treated with either 0.05 mg/kg meloxicam orally for 5 days (first dose 0.1 mg/kg) or placebo (neither washout period nor randomization described). A tendency toward improvement has been described. Lascelles and coworkers227 prospectively compared the efficacy of meloxicam (0.3 mg/kg orally, followed by 0.1 mg/kg for 4 days orally) to ketoprofen (1 mg/kg orally once daily for 5 days) for treatment of acute or chronic pain associated with locomotor disease in cats (n =69 ). Although both drugs were effective in reducing clinical signs associated with pain, meloxicam was considered more palatable.
Robenacoxib
Robenacoxib is approved for use in companion animals outside the United States.45,234 The disposition of robenacoxib has been described in dogs after single-dose administration of 1 mg/kg either orally (fasted or fed), intravenously, or subcutaneously235 (see Table 29-4). Isoform preference in studies supported by the manufacturer indicate a COX-1 to COX-2 ratio (95% inhibition) of 450 using whole blood assays in cats.45 The disposition of robenacoxib in cats at 2 mg/kg intravenously has been reported.45 Data from the ex vivo and pharmacokinetic studies were subsequently integrated with a model of inflammation in cats (n=10), resulting in a recommended dose 2 mg/kg dose every 12 hours.
Vedaprofen
Vedaprofen is marketed outside the United States for use in dogs as an oral gel preparation. A document provided by the manufacturer indicates that the racemic mixture is approximately 8 times more potent toward COX-2 than COX-1, with the majority of activity exihibited by the S enantiomer. The pharmacokinetics of vedaprofen, including its enantiomers, are available in the dog (see Table 29-4)236 after administration either intravenously or as an oral gel preparation. Although volume of distribution was not provided, the elimination (disappearance) half-life was 12 to 17 hours, depending on oral versus intravenous administration. Bioavailability of the gel preparation was near complete. The S and R enantiomers are present generally at a ratio of 1:1, with variability ranging as low as 0.5 (toward the end of the 24-hour dosing interval) and as high as 3.10 (at 2 to 3 hours) after single dosing. After oral multiple dosing for 14 days, the R:S ratio averaged about 1.8 throughout the 24-hour dosing interval. The higher concentration of the R enantiomer appears to reflect more rapid clearance of the S enantiomer (see Table 29-4). The dispositionof vedaprofen did not appear to change substantially with 14 days of dosing.236 The efficacy and safety of vedaprofen were compared with that of meloxicam, which was used as the control during the approval process. Differences could not be detected for either safety or efficacy. Gastrointestinal side effects were reported in approximately 11 to 12% for both groups despite treatment for approximately 15 days (acute) and 40 days (chronic). Both drugs were effective in both acute (approximately 88%) and chronic (approximately 67%).237
Cyclooxygenase-1–Sparing Drugs Approved For Use In Human Medicine
Celecoxib
Celecoxib appears to be COX-1 sparing in the dog (see Table 29-3).Its disposition has been studied in Beagles after single and multiple oral dosing, using different (a solution and solid) dosing forms, with and without food.238 Celecoxib metabolism in dogs is characterized by polymorphism: Paulson and coworkers41 described dogs that metabolize the drug poorly (poor metabolizers [PMs]) compared with efficient metabolizers (EMs). The elimination half-life of the drug in PMs is approximately 4 times as long as that in EMs (see Table 29-4). The drug is normally extensively metabolized (principally hydroxylation followed by oxidation). In both EMs and PMs, approximately 80% of the drug is eliminated in the feces. In EMs disposition does not appear to change with chronic (1-year) dosing. Administration of the drug with a fatty meal increased both the Cmax and the bioavailability. The disposition of celecoxib also has been studied in normal Greyhounds after single (12.5 mg/kg) and multiple dosing. Decreases occurred in Cmax (22%), elimination half-life (shorter), and area under the curve (40%) after 10 days of dosing at 12.5 mg/kg per day.239 The pharmacodynamic and toxic effects of celecoxib have not been well studied in the dog.
Rofecoxib
Rofecoxib is described as a COX-2–selective drug in humans, but no information is available regarding its selectivity in dogs. Its disposition has been studied in dogs (see Table 29-4).241 The drug is cleared after oral administration primarily by biliary excretion after extensive hepatic metabolism to hydroxyl and glucuronide metabolites, with some urinary excretion.241 The effects of rofecoxib (0.5 mg/kg/day orally) on the canine gastrointestinal tract were compared endoscopically to those of the dual-acting NSAID licofelone (2.5 mg/kg orally twice daily) and placebo after 56 days of dosing.240 Whereas no endoscopic lesions were evident for either the placebo or licofelone, rofecoxib was associated with lesions in both the gastric (six of seven) and duodenal (four of seven) areas. This occurred despite a progressive decline in mean plasma drug concentrations for rofecoxib across time: Baseline concentrations were 110.1 ± 85.8 (2 days) compared with study end concentrations of 65.3 ± 49.1 ng/mL (56 days). Concentrations of licofelone also decreased: 761.0 ± 413.8 at baseline compared with 307 ± 106.5 ng/mL at study end.240
Nimesulide
Nimesulide is a COX-2–selective drug approved for use in humans that also appears to act as a COX-1–sparing drug in dogs.31,242 The disposition of nimesulide has been studied in dogs (n = 8-10) after single-dose administration of 5 mg/kg intravenously, intramuscularly, and orally and after 5 days of once-daily administration of the same dose (see Table 29-4).242 The terminal elimination half-life after intramuscular administration was longer than that after intravenous administration, indicating a flip-flop model for this route. On the basis of the integration of pharmacokinetics and pharmacodynamics, the authors determined an effective concentration (EC50) to be 2 to 6 μg/mL, which would be achieved at the studied dose, with some inhibition of COX-1 also occurring.243 However, the study was performed on Beagles, which may not be generally applicable to the canine population at large. The safety of nimesulide has not apparently been studied in dogs.
Miscellaneous Nonsteroidal Antiinflammatory Drugs
Indomethacin
Indomethacin is an NSAID that was developed specifically to abate the inflammatory response to the indolic hormones serotonin and tryptophan.6 As a powerful antiinflammatory, it became a standard for comparison with other drugs. In humans toxicities are not serious, but CNS side effects are undesirable.6 The incidence of gastrointestinal hemorrhage after administration of indomethacin at doses of 2 to 5 mg/kg precludes its clinical utility in dogs. In one study all dogs developed melena within 1 week of receiving 2 mg/kg daily; 60% of these animals had gastric ulcers.244 Interestingly, pretreatment with pure 5-lipoxygenase inhibitors prevents the ulcerogenic effects of indomethacin.2
Acetaminophen
Acetaminophen (paracetamol) is a coal tar analgesic used in human medicine as an effective alternative to aspirin for control of fever and pain. Classically, it is recognized to have poor antiinflammatory activity, although this view has become more controversial.245 Although often classified as an NSAID, its mechanism does not involve inhibition of COX. Rather, acetaminophen interferes with the endoperoxide intermediates (PGG2, PGH2) of AA conversion. Inhibition of a new isoenzym, COX-3, has been suggested, but studies suggest that this isoform is actually a variant of COX-1.9 Concentrations necessary to inhibit this variant are high, leading some investigators to suggest that its efficacy as an analgesic and antipyretic (but not antiinflammatory) reflect its effects on PGH synthases and glutathione-dependent PGE2 synthases.17 The relatively weak antiinflammatory activity of acetaminophen has been attributed to the high concentrations of peroxides that occur in peripheral inflammatory lesions but may reflect limited COX enzyme activity. Acetaminophen may be more effective against inflammatory conditions in the CNS.
The major disadvantage of acetaminophen use in veterinary patients is the narrow safety margin in cats.140 The drug is normally conjugated with glucuronide and to a lesser degree with sulfate. Drug that is not conjugated is metabolized by phase I microsomal enzymes to cytotoxic oxidative metabolites (see Chapter 4). Intracellular glutathione normally scavenges the metabolites, but in the case of overdose or glucuronide deficiency (as with the cat), the formation of toxic metabolites overwhelms the glutathione-scavenging system. In cats methemoglobinemia is the most common indication of toxicity, although centrolobular hepatic necrosis may also occur. Treatment of acetaminophen toxicity includes administration of antioxidants, including N-acetylcysteine, a precursor of glutathione, and ascorbic acid (vitamin C).246-248 The administration of cimetidine, a microsomal enzyme inhibitor, will reduce the formation of toxic metabolites and will result in clinical improvement if given within 48 hours of acetaminophen administration.249,250 The use of SAMe has been demonstrated scientifically and has been reported as clinically effective in a case report.251 A potential advantage of SAMe is response despite late administration.
Acetaminophen toxicity in cats has been retrospectively described in a series (n = 17) of cases.252 Treatments consisted of emesis induced by apomorphine or xylazine, activated charcoal, intravenous balanced crystalloids, intravenous N-acetylcysteine (140 mg/kg, followed by 70 mg/kg every 3 to 6 hours), and ascorbic acid (30 to 100 mg/kg orally or parenterally every 4 to 6 hours). Cimetidine was administered to three cats and methylene blue with dexamethasone to one. The survival rate was 82%. Responding cats were normal within 48 hours of treatment. Median acetaminophen dose in surviving animals was 170 mg/kg (highest 400 mg/kg) versus 100 mg/kg in nonsurvivors (highest dose 170 mg/kg), suggesting dose was not a determinant of outcome.Ten of 12 surviving cats were treated within 14 hours of ingestion. In four of the five deaths, N-acetylcysteine was not begun until 17 hours after acetaminophen exposure, emphasizing the importance of early treatment.
Acetaminophen can interfere (false increase) with glucose determination on certain bedside glucometers.253
In humans, according to the American College of Rheumatology, acetaminophen remains the first choice for treatment of patients with osteoarthritis because of its efficacy, safety, and cost. NSAIDs are recommended only after acetaminophen has failed. Among the NSAIDs, it is characterized as among the most able to penetrate the CNS, thus potentially providing better analgesia. Acetaminophen (15 mg/kg every 8 hours) may be as effective as aspirin for the control of postoperative pain and inflammation in dogs. An extended-relief formulation has proved useful (20 to 30 mg/kg every 8 to 12 hours) at a longer interval.254 Acetaminophen also can be combined with opioids,with a commercial preparation available with codeine (dose based on codeine at 1 to 2 mg/kg orally every 8 to 12 hours). Acetaminophen appears to be safe in dogs. At daily doses of 0.5 g every 8 hours (average weight 18 kg, or 28 mg/kg), acetaminophen causes no clinical signs of adverse drug effects.245 Other studies, however, have shown that adverse reactions (e.g., depression, methemoglobinemia, vomiting) can occur at higher (100 mg/kg) doses.255,256 In another study, 900 mg/kg intravenously caused fulminant hepatic failure in dogs.257
Leukotrienes and Their Inhibition
Leukotriene Formation
Although COX has been the primary target of NSAIDs, the complex role of lipoxygenases in the inflammatory process increasingly is being recognized and targeted. Further, improved gastrointestinal safety as a result of inhibition of leukotrienes supports their potential use. Goodman and coworkers258 recently reviewed the role of LTs and their inhibition in dogs and cats.
Lipoxygenase enzymes located within cells can also metabolize AA to inflammatory mediators, with lipoxygnases 5, 12, and 17 apparently being the most clinically relevant.7,8,17,259 Lipoxygenase enzymes are not as ubiquitous in the body as the COX enzymes, but the enzymes do vary in tissues location. Lipoxygenase-5 enzymes are found primarily in cells of myeloid origin, including macrophages (and all tissues in which they are found) as well as B-lymphocytes. The initial product formed from its action on AA is HPETE, followed by LT A4 (Figure 29-10; see also Figures 29-1 and 29-2). Further conversion (by way of hydroxylase) of LTA4 produces LTB4, a very potent chemotactant, and LTC4 (by way of reduced glutathione) or LTD4 (γ-glutamyltranspeptidase). From these, LTD4 is further converted to LTE4. LTs C, D, and E are referred to as cysteinyl-LTs (see Figure 29-2).22 A second lipoxygenase enzyme, lipoxygenase-12, occurs in platelets, where it catalyzes the formation of 12-HPETE.7,22 A third enzyme, lipoxygenase-15, catalyzes the formation of lipoxins. In contrast to LTs, lipoxins (LXA4 and LXB4) are characterized by anti-inflammatory activity.260 Lipoxin formation can occur when 5-lipoxygenase products interact with 12- lipoxygenase or 15- lipoxygenase.261 Lipoxins are described as counterregulatory, with a particular focus on those inflammatory responses that resolve inflammation.260 LTs act through membrane receptors located on inflammatory and other cells. Receptors are linked to G proteins, increasing intracellular calcium and decreasing cAMP (see Figure 29-1). For example, LTB4 binds to BLT1 and 2 subreceptor types located in inflammatory cells, whereas cysteinyl LTs (C-E) bind to CysLT1 and 2 subreceptor types located in eosinophils, macrophages, and bronchial smooth muscle.
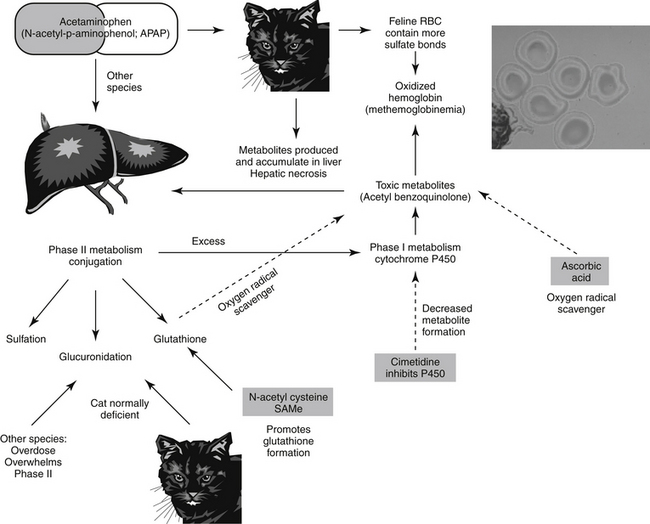
Figure 29-10 Acetaminophen toxicity reflects a cytotoxic type A adverse reaction. Because cats are deficient in glucuronidation, phase II metabolism is easily overwhelmed, and drug is shunted more aggressively back into phase I metabolism. The same process occurs in dogs after an overdose. The products of phase I metabolism are reactive and cause destruction of tissues (liver and red blood cells). Glutathione, an important phase II scavenger, prevents damage but is easily depleted in cats. Supplementation in the form of N-acetylcysteine can decrease damage. Cimetidine is useful because it decreases phase I metabolism and thus the formation of phase I metabolites.
Leukotrienes in Health and Disease
With the exception of lipoxins, each of the lipoxygenase-catalyzed products is a potent mediator of inflammation. At picomolar concentrations, they are effective in the activation, recruitment, migration, and adhesion of immune cells.22 LTs and other selected lipoxygenase products modulate lymphocyte function.8 LTB4 increases production and release of inflammatory cytokines, including activation of NF-kB (transcription factor responsible for expression of proinflammatory cytokins), and expression of TNF-alpha and IL-1β. In the cartilage these cytokines increase expression of MMPs.18 Inhibition of 5-lipoxygenase reduces expression of the cytokines, including TNF-alpha.22 In addition to their effect on leukocytes, the cysteinyl LTs are extremely spasmogenic, causing bronchoconstriction (being 100 times more potent than histamine) in the lungs and exacerbation of ulcerogenic effects in the gastrointestinal tract. The cysteinyl LTs are important mediators in hypersensitivity reactions. They are particularly effective chemoattractants for eosinophils; as such, they play important roles in a number of inflammatory and allergic diseases.258 LTB4 most commony is associated with inflammation associated with inflammatory bowel lesions; colonic epithelials cells are able to produce LTB4. A potential role for lipoxygenase in cancer also is emerging: Lipoxygenase-5 is overexpressed in cancer cells; upregulation in pancreatic cancer suggests a potential therapeutic role for lipoxygenase inhibitors.24,258,262 The role of LTs in eosinophil signaling and chronic (allergic) inflammatory diseases is discussed in Chapter 31.
KEY POINT 29-19
Although less ubiquitous in the body, leukotrienes also represent a balance of proinflammatory and antiinflammatory signals.
A potential sequela of COX blockade by NSAIDs is shunting of unused AA to the lipoxygenase pathway, resulting in increased production of LTs from AA that would have otherwise been metabolized to PG products.8 For example, aspirin hypersensitivity has been associated with the diversion of AA from the PG to the LT (5-lipoxygenase) pathway and the production of mediators that are more inflammatory than the products of PGH synthetase.10,22 Aspirin hypersensitivity also has been attributed to the loss of PGs that might otherwise have specifically inhibited the formation of LTs. The adverse reactions of NSAIDs thus reflect not only loss of PGs but also augmentation of LT synthesis.8 Examples include exacerbation of bronchoconstriction and increasing ulcerogenicity of NSAIDs. Newer NSAIDs that preferentially target COX-2 are not characterized by aspirin hypersensitivity, presumably because AA can yet be metabolized to PGs by way of COX-1.
Lipoxygenase Inhibition
Originally, studies indicated that NSAIDs were not capable of inhibiting LT synthesis. However, the antiinflammatory efficacy of some of the NSAIDs (e.g., ketoprofen)263 has been attributed, in part, to inhibition of lipoxygenase and thus prevention of LT formation.6 However, the ability of NSAIDs to inhibit 5-lipoxygenase is controversial.8 More recently, dual-acting NSAIDs, which impair COX-1 and -2 and lipoxygenases, have been approved. These include tepoxalin, which was initially studied in humans, but approved only for use in dogs. The class of di-tert-butylphenol antiinflammatory agents have the added advantage of antioxidant (phenolic properties) and radical-scavenging properties.22 These include tebufelone, darbufelone, and licofelone. Finally, the effects of LTs can be inhibited at the level of the receptor. LT-receptor antagonists include zafirlukast and montelukast (Figure 29-11).
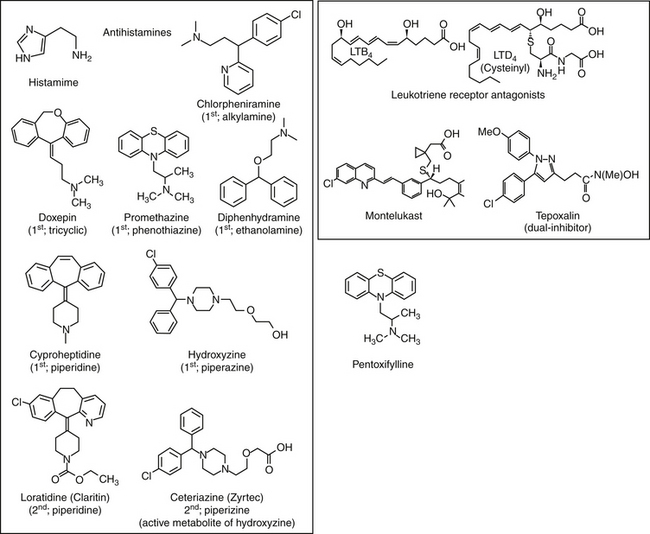
Lipoxygenase Inhibitors
Zileuton is a potent, selective 5-lipoxygenase inhibitor. It is the only drug of this class approved for use in the United States. Use of these drugs has largely been limited by hepatotoxicity; further, efficacy has been limited.
Dual-Acting Nonsteroidal Antiinflammatory Drugs
Despite the advantages of NSAIDs that COX-1–sparing drugs offer, the need for improved gastrointestinal tolerance continues. Because the formation of LTs from AA that might otherwise have been metabolized to PGs appears to contribute to the toxicity (gastrointestinal, repiratory, and other) of NSAIDs (conventional more so than newer drugs), a drug with both COX and lipoxygenase inhibitory effects has been the target of the pharmaceutical industry for some time. However, the presence of an iron atom in the structure of COX or lipoxygenase required that such drugs be redox active in their mechanism. Therefore these drugs tended to inhibit other redox-active enzyme systems, including those in the liver. Consequently, hepatotoxicy has limited the use of dual-acting drugs. Two drugs have been studied in human medicine for their dual actions: tepoxalin and licofelone. The latter is an AA substrate and as such acts as a competitive inhibitor of COX and lipoxygenase without redox activity;81 it is not clear whether tepoxalin similarly avoids redox activity. Neither drugs target lipoxygenase-12 or -15, and thus minimally affect lipoxin formation, further improving potential gastrointestinal tolerance. Further, in the cardiovascular system, because both COX-1 and COX-2 (prostacyclin and thromboxane formation) are inhibited, thrombembolic balance should not be disrupted.
In addition to enhanced safety, enhanced efficacy of dual-acting NSAIDs toward inflammatory diseases should be anticipated. As such has been demonstrated for licofelone in experimentally induced osteoarthritis in dogs. Not only are LT and PG formation decreased, but also formation of proinflammatory cytokines is decreased. Their ability to inhibit both COX and lipoxygenase may result in synergistic activity in the control of inflammation.264 Preliminary data suggest that dual inhibitors may slow the progression of osteoarthritis.265 Because of their potential enhanced efficacy and safety, dual inhibiting NSAIDs have been referred to as a class of breakthrough NSAIDs.22 Indications for dual inhibitors include osteoarthritis, the chronic inflammatory allergic diseases asthma and atopy, and chronic inflammatory bowel disease.
Tepoxalin
Among the dual-acting NSAIDs, only tepoxalin has been approved for use in animals. This drug was being investigated in humans, but pharmacokinetics (not safety) precluded further development.17 It is a potent antiinflammatory and analgesic pyrazole derivative that also inhibits production of IL-1 and suppresses NFkB activation and dependent gene expression.17 Its dual inhibitory effect has been demonstrated in dogs. For example, tepoxalin inhibited COX-1, COX-2, and lipoxygenase-5 in synovial fluids (stifle) of dogs with osteoarthritis.266,267 Tepoxalin, but not firocoxib or meloxicam, inhibited TXA2 in canine blood compared with baseline.267 Its disposition has been reported in both dogs and cats (see Table 29-4). In dogs tepoxalin is approved as a tablet that rapidly disintegrates in the oral cavity, with absorption nonetheless occurring in the intestinal tract. Food enhances oral absorption.127 Tepoxalin undergoes hepatic metabolism. It is converted to at least one major metabolite whose activity contributes substantially to its clinical effect in part because its half-life is substantially longer than that of the parent compound. Both the parent compound and its active metabolite are highly protein bound. Chronic administration does not appear to alter either the Cmax or the elimination half-life of either the parent compound or the active metabolite (USP).127 Essentially all drugs or metabolites, either as parent compound or metabolites, are eliminated in the feces. Feeding appears to increase absorption. The toxicity data generated for the approval of tepoxalin suggest that it should be among the safest of the NSAIDs recently approved for use in dogs (see Table 29-4), although gastrointestinal toxicity can occur. Its safety is supported by the study of Goodman and coworkers.21 Gastric and pyloric ulcers were monitored endoscopically in dogs (n = 6) treated with tepoxalin, firocoxib, or placebo for 7 days. Animals were studied using a randomized crossover design. Eicosanoids were determined in plasma and lesion margins, and lesions were scored. The firocoxib lesions were larger than placebo or tepoxalin lesions despite the fact that tepoxalin prostaglandin concentrations in the mucosa were lower than in either group. Safety appears to reflect blockade of LTs whose vasoconstrictive and neutrophil chemotaxis, adhesion, and degranulation effects are necessary for mucosal erosion.258 Agnello and coworkers266 demonstrated tepoxalin inhibition of PGE2 and LTB4 in the gastric mucosa of dogs. Despite its impact on both COX pathways, in a manufacturer-sponsored study, tepoxalin did not cause significant changes in hemostatic or renal function tests when administered (10 mg/kg orally) to young, healthy dogs (n = 8; 4 placebo) before surgery. Tests of renal function (serum blood urea nitrogen, creatinine and urine gamma glutamyl transferase [GGT] creatinine ratios) were studied for 48 hrs. Hemostatic tests included buccal mucosal bleeding time, as well as bleeding at the site of (experimental) surgical incision. The power of the study was not addressed. In a different study, tepoxalin did not alter renal function when administered with with angiotensin-converting enzyme inhibitors for up to 28 days.258
KEY POINT 29-20
Dual-acting inhibitors might be expected to be more efficacious toward some inflammatory conditions, particularly those associated with allergies, compared with nonsteroidal antiinflammatory drugs.
Tepoxalin also has been studied in cats. Whereas tepoxalin was well tolerated in cats when administered at 100 mg/kg once daily for 3 consecutive days, saturation kinetics occurred with two doses of 60 mg/kg 4 hours apart. Signs suggestive of CNS adverse drug events occurred (drunkenlike state), a response not recorded in any other species. Tepoxalin is a potent antipyretic agent in cats at doses between 5 and 10 mg/kg and provides analgesia at least equivalent to that of butorphanol at 10 mg/kg for onychectomy.268
The action of tepoxalin as a dual inhibitor might render it useful for control of pain or inflammation typical of other NSAIDs. This includes a potential added advantage for treatment of chronic allergic diseases for which LTs play a role in cellular signaling. However, preoperative use of tepoxalin may not be prudent, particularly in animals at risk for hemostasis problems. Its postoperative use might be considered. Bosmans and coworkers269 compared the combination of tepoxaline and buprenorphine to buprenorphine alone during the 24-hour period after cruciate repair in dogs (n = 20; 10 per group). Animals were studied using a parallel randomized, blinded design. Pain was assessed using visual analog scales and a multifactorial pain scale. No statistical differences could be demonstrated for either control of pain or side effects.
Licofelone
Licofelone impairs PGE production in a similar manner to conventional NSAIDs, but unlike other NSAIDs, it also prevents the formation of LTB4. Although licofelone has not been approved for use in dogs, its pharmacodynamic effects have been studied in dogs.240 Licofelone appears to have potent antioxidative properties.269a Its effects on the canine gastrointestinal tract (2.5 mg/kg orally twice daily) as measured endoscopically were negligible to those of rofecoxib (see the discussion of rofecoxib) after 56 days of treatment with either drug. In that study, as with rofecoxib, concentrations of licofelone also decreased across time. Baseline concentrations were 761.0 ± 413.8 compared with 307 ± 106.5 ng/mL at study end.240 The drug’s safety was explained in part because of decreased bioavailability at high doses but also because of prevention of lipoxygenase.2 Licofelone has been found effective in the treatment of experimentally induced arthritis in the dog.264
Leukotriene Receptor Antagonists
Zafirlukast and montelukast are competitive cysLT-1 receptor antagonists (LAR) approved for use to treat human asthma. However, their use increasingly is being expanded to treatment of a variety of chronic allergic inflammatory diseases. Interestingly, their use might be considered in combination with NSAIDs to reduce the risk of gastrointestinal toxicity. The currently available LARs are available only for oral administration. Disposition information is not available for dogs or cats. In humans the drugs are rapidly and nearly completely absorbed. Metabolism of zafirlukast is by CYP2C9 and for montelukast by CYP3A4 and CYP29C. Metabolites are not active. Half-lives in humans are 10 hours (zafirlukast) and 3 to 6 hours (montelukast). Maximum efficacy may require 2 to 4 weeks, although an initial response may be evident within several days. The drugs are generally very safe, probably reflecting the limited effects of LTs in the body.270
Pentoxifylline
Pentoxifylline is a methylxanthine derivative of theobromine with minimal bronchodilator activity but with clinically apparent rheologic effects.271 It is used to treat human patients with claudication associated with chronic occlusive arterial disease. Mechanisms do not include vasodilation or cardiac stimulatory effects. Its rheologic effects appear to reflect increased flexibility of red blood cells and reduced blood viscosity.272
The disposition of pentoxifylline is complex, with hepatic metabolism to at least seven metabolites, two of which (I and V) are responsible for most of the pharmacologic effects.273 Pentoxifylline inhibits the complement cascade, neutrophil degranulation, and cytokine production. Among the cytokines, inhibition of TNF-alpha may be particularly important to its efficacy.274 IL-1 and IL-6 also are inhibited, as is expression of adhesions molecules. Pentoxifylline also increases fibroblast collagenases, decreases collagen, fibronectin, and glycosaminoglycan production.274 Inhibition of phosphodiesterase contributes to its antinflammatory effects.
The disposition of the drug has been studied in normal dogs.275 After a dose of 30 mg/kg orally and 8 mg/kg intravenously (n = 5 dogs), pentoxifylline and its metabolites are characterized by an elimination half-life of 125 ± 192 and 450 ± 533 minutes, respectively, suggesting slow absorption. Peak concentrations were 41 ± 46 and 40 ± 32 μg/mL for intravenous and oral administration, respectively. Bioavailability was 76 ± 78%. No side effects occurred in any dog at this dose. Although therapeutic concentrations have been recommended in humans, this is based on the parent compound. However, therapeutic ranges should be based on both the parent drug and its most active metabolites.
In human patients inflammatory conditions for which pentoxifylline has been used include contact dermatitis, systemic vasculitis, and sepsis syndromes.271 However, for the latter indication (septic shock), one study demonstrated increased risk of mortality in septic dogs treated with pentoxifylline as a constant intravenous infusion, presumably because of decreased endotoxin clearance.276 Pentoxifylline was able to decrease the extent of esophageal stricture induced by erosive esophagitis.277 Pentoxifylline increasingly is being used to treat dermatolgic disorders in humans.274 The drug has been used to treat Collie dermatomyositis, although animals probably benefit as much from the antiinflammatory effects as from the rheologic effects.
Antihistamines
Histamine in Health and Disease
Histamine is a low-molecular-weight amine, synthesized from histidine by its decarboxylation. Histidine decarboxylase is ubiquitous in the body, occuring in the CNS, gastric parietal cells, mast cells, and basophils. Upon its formation histamine is stored. Most histamine is stored bound to heparin in granules located in either mast cells or their circulatory counterparts, basophils. Histamine is not the sole mediator of clinical relevance located in mast cells. Because histamine release occurs by mast cell degranulation, it is accompanied by the release of other mediators, including those preformed and stored in granules (e.g., serotonin) and those synthesized in situ (e.g., AA products). Currently, four histamine subreceptors have been identified. The H1 receptors couple to G proteins, activating the PLC–IP3–Ca2+ pathway [G&G].278 H2 receptors link to Gs, activating the adenylyl cyclase cAMP pathway. Both H3 and H4 receptors are inhibitory toward adenylyl cyclase; 35% or more homology between the two complicates pharmacologic distinction.
The impact of antihistamines is complex but somewhat predictable based on the location and impact of each receptor. For example, H1 receptors on vascular endothelium activate calcium mobilization and nitric oxide production (eNOS), causing local vascular smooth muscle relaxation. In contrast, activation of H1 receptors in smooth muscle will cause contraction (e.g., bronchoconstriction), which is balanced by stimulation of H2 receptors in the same cells that mediate relaxation. Both H1 and H2 receptors are distributed throughout the peripheral nervous system and CNS, whereas H3 receptors are limited to the CNS. The H3 receptors serve as autoreceptors. In general, their stimulation promotes sleep, and antagonism is manifested as wakefulness. H4 receptors are located primarily in cells of hematopoietic origin.278 These include granulocytes. Stimulation of H4 receptors on eosinophils induces a change in the shape of the cell, chemotaxis, and upregulation of adhesion molecules; antagonists therefore may be particularly useful for treatment of chronic allergic diseases.
The effects of histamine on H1 and H2 receptors vary with the site and extent of release. In general, histamine affinity for H1 receptors is probably greater than that for H2 receptors. In the vasculature both H1 and H2 receptor activation causes relaxation of arteriolar smooth muscle (resistance vessels), resulting in vasodilation. Whereas H1-mediated responses are rapid, H2- mediated response are slow and prolonged. In contrast to its effect on smaller vessels, histamine may cause contraction of the smooth muscle, causing hypertension in some species. In the heart contractility and automaticity increase, primarily through H2 receptors. Endothelial cells of venules contain more histamine receptors than other tissues. In postcapillary venules, H1 receptors cause endothelial cells to contract and separate, regulating cell-to-cell cytoskeleton interactions.278a The loss of the endothelial barrier results in increased permeability, typical of edema. Passage of inflammatory cells into tissues is facilitated. In nonvascular smooth muscle, H1 receptors cause contraction, whereas H2 receptor activation causes relaxation. Thus in the bronchi the predominant effect of smooth muscle contraction is bronchoconstriction; in the gastrointestinal tract, diarrhea may occur. Species sensitivity to these effects vary; for example, histamine does not appear to play a major role in bronchoconstriction associated with asthma in the cat. Release of histamine from mast cells and basophils causes H2-mediated inhibition of further histamine release. Gastric acid secretion is mediated by H2 receptors. The H1 receptors also stimulate sensory nerve endings; for example, pruritis reflects, in part, stimulation of type C nerve fibers.
The manifestation of the vascular effects of histamine varies depending on the magnitude of response. Local effects in the skin are manifested as the typical wheal and flare response; in contrast, systemic release, if sufficient, may be manifested as hypotensive shock, typical of anaphylactic or anaphylactoid response. The clinical signs vary with the species, depending on the “shock” organ, which in turn generally reflects the tissue with the greatest number of mast cells (lungs in cats, gastrointestinal tract in dogs).
Prevention or treatment of the effects of histamine can be accomplished in several ways: preventing histamine release (e.g., inhibition of mast cell degranulation), targeting the histamine receptor itself, or antagonizing (modulating) tissue response to histamine (e.g., the use of beta antagonists to blunt histamine-induced bronchoconstriction). Of these, the most comprehensive response is likely to occur if mast cell degranulation is prevented. This reflects the fact that histamine is not the only mediator released with degranulation. Others simultaneously released include those stored with histamine, such as serotonin (to which feline bronchial smooth muscle has been described as very sensitive), and those mediators formed in situ (e.g., eicosanoid mediators such as PGs, LTs [LTB4], or slow-reactive substance of anaphylaxis), platelet activating factor, and related compounds (see Figure 29-1)
Inhibitors of histamine release (mast cell degranulation) (see Figure 29-1), are exemplified by cromolyn sodium. It prevents calcium-dependent release of histamine from mast cells (but not basophils). Its mechanism of action may involve inhibition of calcium flux into the mast cell. Because degranulation is prevented, the release of other autacoids released with histamine is also prevented. However, the drug is poorly absorbed orally and can be given only by inhalation. Thus its use is limited to respiratory disease associated with mast cells. Because it prevents degranulation, it is useful only when administered prophylactically.
Structure–Activity Relationship
Anthistamines historically refer to those drugs specific for H1 receptors. Some of the drugs also inhibit muscarinic, serotonin, or alpha-adrenergic receptors. All H1 receptor blockers or antagonists are structurally similar to histamine; however, the primary amine of histamine is replaced by a tertiary amine (see Figure 29-11). Previously referred to as competitive antagonists at histamine receptors, they are now described as inverse agonists. They preferentially bind to the receptor in its inactive state, maintaining the conformation of the inactive state, thus prolonging the state of inactivity.205 Antihistamines are among the most used drugs in human medicine, with over 40 different drugs available worldwide, many of them nonprescription. Traditional categorization results in six chemical groups: ethanolamines, ethylenediamines, alkylamines, piperazines, piperidines, and phenothiazines. Alternatively, another classification is based on sedative potential, with first-generation drugs being sedating and second-generation drugs being nonsedating.
Pharmacologic Effects
The effects of the H1 receptor blockers are similar and predictable regardless of the preparation. Specific effects include smooth muscle inhibition in the gastrointestinal tract and respiratory tract; inhibition of histamine-induced vasodilation of resistance vessels in the vasculature (residual vasodilation may require H2 blockers); and strong antagonism of capillary and venule permeability. In the CNS both stimulation and depression may occur, depending on the drug. However, second-generation drugs (particularly cetirizine or fexofenadine, but less so loratadine) do not penetrate the blood–brain barrier and are less likely to be associated with CNS side effects.279 The difference appears to reflect, in part, whether the drugs are substrates for P-glycoprotein, with second-generation drugs more likely. Effects on anaphylactic (or anaphylactoid) shock vary with species and tissues. Inhibition of motion sickness may actually reflect anticholinergic activity (promethazine). Antiallergic effects of the newer drugs in particular may reflect inhibition of mediator release,205 probably by way of direct inhibition of inward calcium flux through calcium ion channels.
KEY POINT 29-22
The central nervous system–related side effects of antihistamines reflect their ability to penetrate and remain in the brain, a characteristic more consistent among the first-generation drugs than the newer second-generation drugs.
Example drugs include doxepin (marketed as a tricyclic antidepressant), diphenhydramine (antimuscarinic and sedating ethanolamine), chlorpheniramine (first-generation alkylamine; such drugs as a class are very potent and less sedating), hydroxyzine, cyclizine, and meclizine (a first generation piperazines), cetirizine (a second-generation piperazine and active metabolite of hydroxyzine), promethazine (a phenothiazine), cyproheptadine (a first-generation piperazine that also has antiserotonergic effects), and terfenadine (a second-generation piperazine). Dimenhydrinate (Dramamine) and diphenhydramine (Benadryl) are characterized by marked sedation and significant antimuscarinic effects. Tripelennamine is one of the most specific H1 antagonists and is associated with less sedation but more gastric side effects. Chlorpheniramine is one of the most potent H1 blockers and also is associated with less sedation but more CNS stimulation. Promethazine is the most potent inhibitor of motion sickness. Piperazines are very effective for motion sickness and may have more prolonged action (cyclizine and meclizine). Phenothiazines (promethazine as the prototype) are characterized by considerable anticholinergic activity and prominent sedation but are the most effective for motion sickness.
Pharmacokinetics
Few antihistamines have been studied in animals. Species differences are likely to preclude extrapolation of dosing regimens among species, and particularly between humans and dogs. This reflects, in part, differences in pH of the human skin (4.8) versus canine skin (similar to plasma). As weak bases, antihistamines are likely to accumulate in skin.280 In the ionized form, the accumulated drug will be inactive in human skin. However, the skin may serve as a depot, allowing slow release as the unionized, active form. This may either prolong elimination half-life from plasma or, if concentrations are too low to be detected in plasma, may allow a local pharmacodynamic effect that exceeds the duration indicated by pharmacokinetics. These benefits may not be realized in dogs.
Hydroxyzine is a piperazine antihistaminergic drug that is further metabolized to cetirizine, the active ingredient in Zyrtec. Its disposition has been described in dogs (n = 6) after oral and intravenous administration of 2 mg/kg hydroxyzine, using a randomized crossover design (14-day washout between routes).281 The pharmacokinetic study was accompanied by pharmacodynamic response based on immunoglobulin E (IgE) cutaneous wheal formation. Dogs tolerated each dose well. The maximum response to hydroxyzine occurred during the first 8 hours, correlating with plasma cetirizine concentrations that exceeded 1.5 μg/mL, with response attributed almost exclusively to cetirazine. Accordingly, the authors recommended cetirizine at 2 mg/kg twice daily.
In cats the disposition of cetrizine has been described after an oral dose of 0.9 ± 0.2 mg/kg (see Table 29-4).282 Despite the high proportion of bound drug, the effective concentration in humans of 10.5 to 27.3 ng/mL was achieved (based on calculation of free drug) and maintained during a 24-hour dosing period. Cats tolerated the single dose well, despite drug concentrations that are higher than those generally achieved in humans (human dose 10 mg/person).
Clemastine is an ethanolamine antihistaminergic drug. Its disposition and pharmacodynamic response have been described in dogs (Table 29-7).280 Target concentrations in human approximate 0.7 ng/mL. After oral administration bioavailability of clemastine was less than 5%. Although not reported, plasma drug concentrations versus time curves indicate that 0.7 ng/mL is achieved but maintained for less than 3 hours in dogs after oral absorption. Poor oral absorption contributed to poor response to antigen challenge, whereas intravenous administration inhibited wheal formation entirely for 7 hours. Accordingly, the oral dose of the drug should be at least 1 mg/kg every 12 hours or more; in contrast, the intravenous dose of 0.1 mg/kg would be effective twice daily.
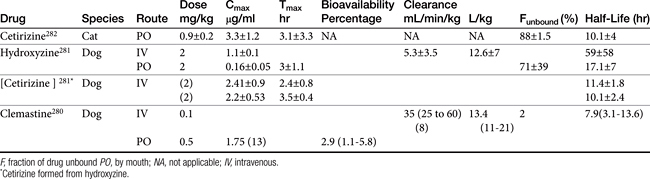
Drug Interactions
Antihistamines generally are substrates for P-glycoprotein and other drug transport systems; therefore competition should be expected when they are used in concert with other P-glycoprotein substrates. Selected drugs may also cause the induction or inhibition of drug-metabolizing enzymes.
Adverse Drug Reactions and Side Effects
Because side effects to first-generation drugs are problematic, alternative drugs were developed. For example, diphenhydramine adversely affects learning in children and alters cognition and mood. Several drugs cause a level of intoxication similar to that produced by alcohol; chlorpheniramine causes a hangover effect. Gastrointestinal side effects include anorexia, nausea and vomiting, constipation, and diarrhea. Other effects, such as a dry respiratory tract and urinary retention or dysuria, can be attributed to atropine-like (anticholinergic), antiadrenergic, and antiserotonin effects. Newer antihistamines (H1) do not interact with muscarinic receptors as effectively as did the first-generation drugs, thus avoiding many of these side effects. Side effects in the CNS are not unusual at therapeutic doses. However, they are rarely serious, and adaptation occurs with chronic use. Sedation is affected by pH, lipophilicity, and activity of transport proteins such as P-glycoprotein.283 Lack of sedation in the newer drugs has been attributed to transport proteins that mediate active efflux from the CNS and thus limit CNS distribution. Collies and related breeds deficient in P-glycoprotein might be expected to manifest more CNS reactions with newer drugs compared with other dog breeds.
Two early second-generation H1 antihistamines, astemizole and terfenadine, were associated with cardiotoxicity (prolongation of the QT interval: torsades de pointes) as a result of blockade of the rapid component of the delayed rectifier potassium current. More recent drugs, including loratadine, desloratadine (a metabolite of loratadine), cetirizine, and fexofenadine (a metabolite of terfenadine), are not associated with this effect.
Acute poisoning is not uncommon in humans; its occurrence in dogs and cats is not known. There is no specific therapy. Signs include ataxia, incoordination, convulsions, dry mouth, and fever, with treatment being supportive.
Therapeutic Use
Indications for antihistamines include the prevention or treatment of anaphylactic shock (IgE-mediated histamine release) or anaphylactoid response (e.g., cationic drug-induced histamine release), motion sickness (see the discussion of the gastrointestinal tract), particularly dimenhydrinate, piperazines, and promethazine, and allergic disease, including that affecting the skin. These indications are addressed in the relevant chapters. Antihistaminergic drugs have proved variably useful in the control of small animal allergic diseases.284-286 This no doubt reflects differences in receptors and drug receptor interactions. However, profound differences also are likely in the disposition of the drugs, including oral bioavailability (e.g., clemastine in dogs). Part of the lack of efficacy also reflects limited actions: Although the effects of histamine at H1 receptors may be blocked, limited to no effects on mast cell degranulation will result in inflammatory response to other mediators.