CHAPTER 53 Disorders of the Adrenal Gland
HYPERADRENOCORTICISM IN DOGS
PITUITARY-DEPENDENT HYPERADRENOCORTICISM
Pituitary-dependent hyperadrenocorticism (PDH) is the most common cause of spontaneous hyperadrenocorticism, accounting for approximately 80% to 85% of cases. A functional adrenocorticotropic hormone (ACTH)–secreting pituitary tumor is found at necropsy in approximately 85% of dogs with PDH. Adenoma of the pars distalis is the most common histologic finding, with a smaller percentage of dogs diagnosed with adenoma of the pars intermedia and a few dogs diagnosed with functional pituitary carcinoma. Approximately 50% of dogs with PDH have pituitary tumors less than 3 mm in diameter, and most of the remaining dogs, specifically those without central nervous system (CNS) signs, have tumors 3 to 10 mm in diameter at the time PDH is diagnosed. Approximately 10% to 20% of dogs have pituitary tumors (i.e., macrotumors) exceeding 10 mm in diameter at the time PDH is diagnosed. These tumors have the potential to compress or invade adjacent structures and cause neurologic signs as they expand dorsally into the hypothalamus and thalamus (Fig. 53-1).

FIG 53-1 A, A 10-year-old male castrated mixed-breed dog with pituitary-dependent hyperadrenocorticism. Initial clinical signs of polyuria, polydipsia, and endocrine alopecia progressed to severe stupor, anorexia, adipsia, weight loss, and loss of body temperature regulation. B, Cross-section of the brain from the dog in A showing a pituitary macroadenoma that is severely compressing the surrounding brain structures.
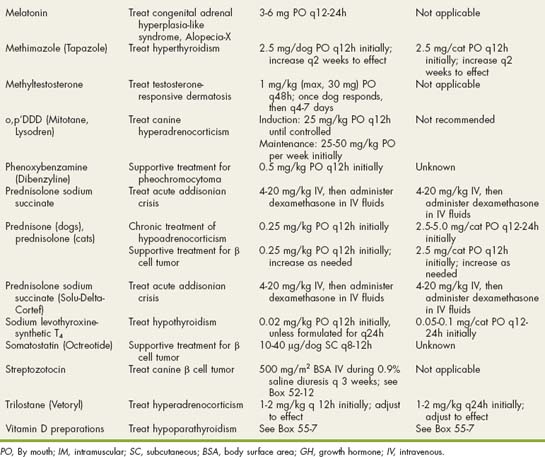
Excessive secretion of ACTH causes bilateral adrenocortical hyperplasia and excess cortisol secretion from the adrenal cortex (Fig. 53-2). Because normal feedback inhibition of ACTH secretion by cortisol is missing, excessive ACTH secretion persists despite increased adrenocortical secretion of cortisol. Episodic secretion of ACTH and cortisol is common and results in fluctuating plasma concentrations that may at times be within the reference range.
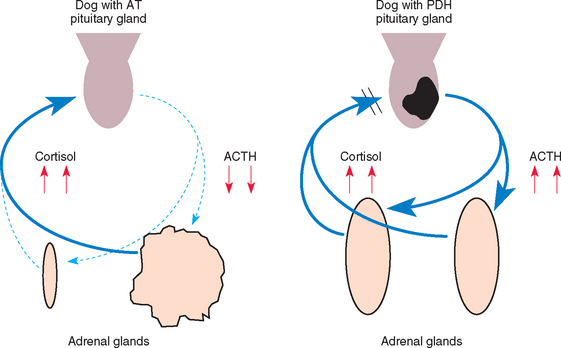
FIG 53-2 The pituitary-adrenocortical axis in dogs with a functioning adrenocortical tumor (AT; lef t) and in dogs with pituitary-dependent hyperadrenocorticism (PDH; righ t). Excess cortisol secretion from an AT causes pituitary suppression, decreased plasma adrenocorticotropic hormone (ACTH) concentration, and atrophy of the contralateral adrenal gland. Dogs with PDH have excess ACTH secretion, usually from a functional pituitary adenoma, which causes bilateral adrenomegaly and excess plasma cortisol concentrations.
ADRENOCORTICAL TUMORS
Adrenocortical tumors (ATs) account for the remaining 15% to 20% of dogs with spontaneous hyperadrenocorticism. Adrenocortical adenoma and carcinoma occur with equal frequency. There are no consistent clinical or biochemical features that help distinguish dogs with functional adrenal adenomas from those with adrenal carcinomas, although carcinomas tend to be larger than adenomas on abdominal ultrasound. Adrenocortical carcinomas may invade adjacent structures (e.g., phrenicoabdominal vein, caudal vena cava, kidney) or metastasize to the liver and lung.
Bilateral ATs can occur in dogs but are rare. A nonfunctional AT or an AT causing hyperadrenocorticism and a pheochromocytoma in the contralateral gland is a more common cause of bilateral adrenal masses in dogs. Macronodular hyperplasia of the adrenals has also been identified in dogs. The adrenals in such animals are usually grossly enlarged, with multiple nodules of varying sizes within the adrenal cortex. The exact pathogenesis of this latter syn drome is unclear, although most cases in dogs are presumed to represent an anatomic variant of PDH. Increased plasma 17-OH-progesterone concentrations have also been documented in dogs with an adrenal mass and clinical manifestations of hyperadrenocorticism but normal plasma cortisol concentrations after administration of ACTH or dexamethasone (see the section on atypical Cushing’s syndrome, p. 830).
Adrenocortical tumors causing hyperadrenocorticism (ATHs) are autonomous and functional and randomly secrete excessive amounts of cortisol independent of pituitary control. The cortisol produced by these tumors suppresses circulating plasma ACTH concentrations, causing cortical atrophy of the uninvolved adrenal and atrophy of all normal cells in the involved adrenal (see Fig. 53-2). This atrophy creates asymmetry in the size of the adrenal glands, which can be identified by abdominal ultrasonography. Most, if not all, of these tumors appear to retain ACTH receptors and respond to administration of exogenous ACTH. ATHs are typically unresponsive to manipulation of the hypothalamic-pituitary axis with glucocorticoids such as dexamethasone.
IATROGENIC HYPERADRENOCORTICISM
Iatrogenic hyperadrenocorticism typically results from the excessive administration of glucocorticoids to control allergic or immune-mediated disorders. It can also develop as a result of the administration of eye, ear, or skin medications containing glucocorticoids, especially in small dogs (weight less than 10 kg) receiving them long term. Because the hypothalamic-pituitary-adrenocortical axis is normal, the prolonged excessive administration of glucocorticoids suppresses circulating plasma ACTH concentrations, causing bilateral adrenocortical atrophy. In these animals ACTH stimulation test results are consistent with spontaneous hypoadrenocorticism despite clinical signs of hyperadrenocorticism.
SIGNALMENT
Hyperadrenocorticism typically develops in dogs 6 years of age and older (median age 10 years) but has been documented in dogs as young as 1 year. There is no apparent sex-related predisposition, although AT appears to be diagnosed more commonly in female dogs. PDH and ATH have been diagnosed in numerous breeds. All Poodle breeds, Dachshunds, various Terrier breeds, German Shepherd Dogs, Beagles, and Labrador Retrievers are commonly represented, and Boxers and Boston Terriers appear to be at increased risk for PDH. PDH tends to occur more frequently in smaller dogs; 75% of dogs with PDH weigh less than 20 kg. Approximately 50% of dogs with functional ATH weigh more than 20 kg.
CLINICAL SIGNS
The most common clinical signs are polyuria, polydipsia, polyphagia, panting, abdominal enlargement, endocrine alopecia, mild muscle weakness, and lethargy (Fig. 53-3; Table 53-1). Most dogs exhibit several, but not all, of these clinical signs. The more signs evident in the history, the greater the index of suspicion for hyperadrenocorticism. Additional findings on physical examination (see Table 53-1) help establish the diagnosis.
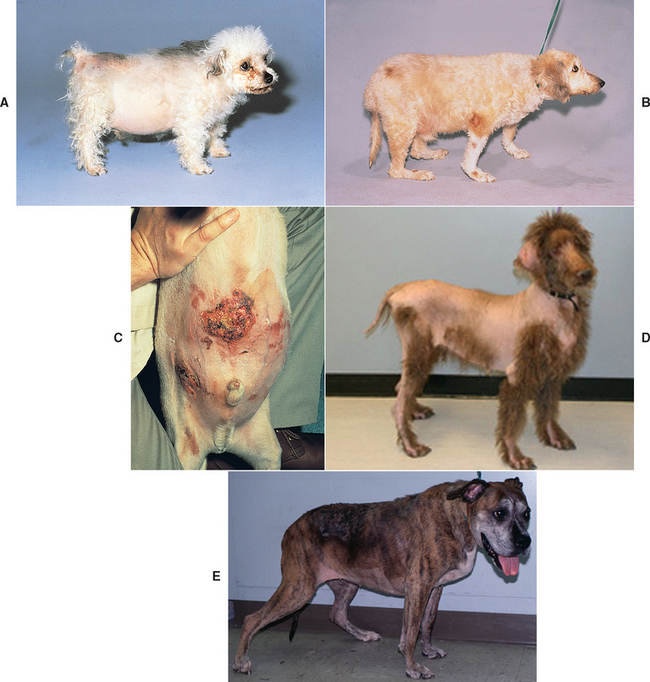
FIG 53-3 A, A 1-year-old male Miniature Poodle with pituitary-dependent hyperadrenocorticism (PDH). Note the truncal distribution of the endocrine alopecia with the pot-bellied appearance. B, A 9-year-old male castrated mixed-breed dog with PDH. Note the severe laxity of the ligaments, resulting in hyperextension of the carpal ligaments and ambulation on the hocks. A “rat tail” has also developed and is a finding also associated with hypothyroidism. C, An 8-year-old male castrated Chihuahua with PDH. Note the pot-bellied appearance and severe calcinosis cutis. D, A 7-year-old Standard Poodle with PDH. The primary owner complaints at presentation were polyuria, polydipsia, and progressively worsening symmetric endocrine alopecia. E, An adult mixed-breed dog with PDH. The primary owner complaints were polyuria, polydipsia, excessive panting, and severe weakness of the rear limbs. Note the absence of hair growth on the ventral abdomen, which had been shaved for an abdominal ultrasound 2 months before presentation.
 TABLE 53-1 Clinical Signs and Physical Examination Findings in Dogs with Hyperadrenocorticism
TABLE 53-1 Clinical Signs and Physical Examination Findings in Dogs with Hyperadrenocorticism
| CLINICAL SIGNS | PHYSICAL EXAMINATION FINDINGS |
|---|---|
| Polyuria, polydipsia* | Endocrine alopecia* |
| Polyphagia* | Epidermal atrophy* |
| Panting* | Comedones* |
| Abdominal enlargement* | Cutaneous |
| Endocrine alopecia* | hyperpigmentation* |
| Weakness* | Calcinosis cutis |
| Lethargy | Abdominal enlargement* |
| Calcinosis cutis | Hepatomegaly* |
| Cutaneous | Muscle wasting* |
| hyperpigmentation | Bruising |
| Neurologic signs (PMA) | Testicular atrophy |
| Stupor | Failure to cycle (intact female) |
| Ataxia | |
| Circling | Neurologic signs (PMA) |
| Aimless wandering | Dyspnea (pulmonary thromboemboli) |
| Pacing | |
| Behavioral alterations | Facial nerve paralysis |
| Respiratory distress-dyspnea (pulmonary thromboemboli) | Myotonia |
| Stiff gait (myotonia) |
PMA, Pituitary macroadenoma.
Dogs are occasionally seen because of isolated polyuria and polydipsia, bilaterally symmetric endocrine alopecia, or panting. There may be no other historic or physical examination findings consistent with hyperadrenocorticism. The diagnosis of hyperadrenocorticism is not readily apparent in these dogs. Fortunately, hyperadrenocorticism is a differential diagnosis for polyuria and polydipsia, endocrine alopecia, and panting and will be identified as the clinician works through the differentials for these problems. Similarly, hyperadrenocorticism causes insulin resistance and can lead to the development of diabetes mellitus. Clinical signs (other than polyuria and polydipsia) and physical examination findings suggestive of hyperadrenocorticism are often missing in diabetic dogs with concurrent hyperadrenocorticism. A clinical suspicion for hyperadrenocorticism develops after critical evaluation of routine blood test results (e.g., increased serum alkaline phosphatase [ALP] activity, isosthenuric urine) or after resistance to insulin treatment is identified.
PITUITARY MACROTUMOR SYNDROME
Neurologic signs may develop in dogs with PDH as a result of expansion of the pituitary tumor into the hypothalamus and thalamus (see Fig. 53-1). Neurologic signs may be present at the time PDH is diagnosed but usually develop 6 months or longer after PDH is identified. The most common neurologic sign is a dull, listless attitude (i.e., stupor). Additional signs of pituitary macroadenoma include inappetence, aimless wandering, pacing, ataxia, head pressing, circling, and behavioral alterations. In the event of severe compression of the hypothalamus, abnormalities related to dysfunction of the autonomic nervous system develop, including adipsia, loss of temperature regulation, erratic heart rate, and inability to be roused from a sleeplike state. Identification of a pituitary macrotumor requires computed tomography (CT) or magnetic resonance imaging (MRI; Fig. 53-4). There are no biochemical or endocrine test results that reliably correlate with the size of the pituitary tumor.
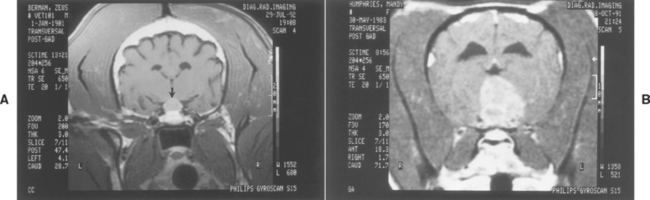
FIG 53-4 A, Postgadolinium administration magnetic resonance imaging (MRI) scan of a 9-year-old male castrated German Shepherd Dog with pituitary-dependent hyperadrenocorticism (PDH) and a pituitary mass (arrow). There were no neurologic signs present at the time the MRI scan was performed. B, Postgadolinium administration MRI scan of an 8-year-old Boston Terrier with PDH, a large pituitary mass invading the brainstem, and signs of disorientation, ataxia, and circling.
(From Feldman EC, Nelson RW: Canine and feline endocrinology and reproduction, ed 3, St Louis, 2004, WB Saunders.)
MEDICAL COMPLICATIONS: PULMONARY THROMBOEMBOLISM
Several medical complications can develop secondary to prolonged cortisol excess (Box 53-1). The most worrisome is pulmonary thromboembolism (PTE), which generally occurs in dogs undergoing adrenalectomy for AT. Thromboemboli may also affect the kidney, gastrointestinal tract, heart, and CNS. There is no apparent correlation between control of hyperadrenocorticism and development of thromboemboli. Factors predisposing to the development of PTE in dogs with hyperadrenocorticism include inhibition of fibrinolysis (corticosteroids stimulate the release of plasminogen activator inhibitors), systemic hypertension, protein-losing glomerulonephropathy, decreased serum antithrombin III concentrations, increased concentrations of several coagulationfactors, and an increased hematocrit value. Clinical signs of PTE include acute respiratory distress; orthopnea; and, less commonly, a jugular pulse. Thoracic radiographs may reveal no abnormalities, or they may show hypoperfusion, alveolar pulmonary infiltrates, or a pleural effusion. There may be an increased diameter and blunting of the pulmonary arteries, absence of perfusion of the obstructed pulmonary vasculature, and overperfusion of the unobstructed pulmonary vasculature. Normal thoracic radiograph findings in a dyspneic dog that does not have a large airway obstruction suggest a diagnosis of PTE. Arterial blood gas analysis typically reveals a decrease in the partial pressures of arterial oxygen and carbon dioxide, and mild metabolic acidosis. Thrombosis may be confirmed by angiography of the lungs or by radionuclear lung scanning. Therapy consists of general supportive care, oxygen, anticoagulants, and time (see Chapter 12). The prognosis for dogs with PTE is guarded to grave. If dogs do recover, it typically takes 5 to 10 days before they can be safely removed from oxygen support.
Diagnosis
A thorough evaluation should be done in any dog suspected of having hyperadrenocorticism and should include a complete blood count (CBC); serum biochemistry panel; urinalysis with bacterial culture; and, if available, abdominal ultrasonography. Results of these tests will increase or decrease the index of suspicion for hyperadrenocorticism; identify common concurrent problems (e.g., urinary tract infection); and, in the case of ultrasonography, provide valuable information for localizing the cause of the disorder (i.e., PDH versus AT). Endocrine studies required to confirm the diagnosis and localize the cause of the disorder can then be performed.
CLINICAL PATHOLOGY
Common clinicopathologic alterations caused by hyperadrenocorticism are listed in Box 53-2. An increase in ALP activity and cholesterol concentration is the most reliable indicator of hyperadrenocorticism. The major contributor to increased serum ALP is the corticosteroid-induced isoenzyme of ALP derived from the bile canalicular membrane of hepatocytes. Approximately 85% of dogs with hyperadrenocorticism have ALP activities that exceed 150 IU/L; values in excess of 1000 IU/L are common, and values in excess of 10,000 IU/L are occasionally identified. There is no correlation between the magnitude of increase in serum ALP activity and the severity of hyperadrenocorticism, response to therapy, or prognosis. There is also no correlation between the magnitude of increase in serum ALP activity and hepatocellular death or hepatic failure. The ALP activity can be normal in some dogs with hyperadrenocorticism, and an increase in ALP activity by itself is not diagnostic for hyperadrenocorticism. Similarly, an increase in the activity of the corticosteroid-induced isoenzyme of alkaline phosphatase (SIAP) is not a finding specific to hyperadrenocorticism or exogenous glucocorticoid administration; an increase in SIAP activity occurs commonly with many disorders, including diabetes mellitus, primary hepatopathies, pancreatitis, congestive heart failure, and neoplasia as well as in dogs receiving certain drugs (e.g., anticonvulsants). However, finding no SIAP in the serum may be of diagnostic value in ruling out hyperadrenocorticism.
Urine specific gravity is typically less than 1.020 in dogs with hyperadrenocorticism that have free access to water. Water-deprived hyperadrenal dogs maintain the ability to concentrate urine, although usually the concentrating ability remains less than normal. As such, urine specific gravities of 1.025 to 1.035 may be identified if urine is obtained after water has been withheld from the dog.
Proteinuria is a common finding in dogs with untreated hyperadrenocorticism. Proteinuria may be caused by glucocorticoid-induced systemic and glomerular hypertension, glomerulonephritis, or glomerulosclerosis. Urine protein : creatinine ratios are usually less than 4, although values in excess of 8 have been identified. Proteinuria decreases and often resolves in response to treatment of hyperadrenocorticism.
Urinary tract infection is a common sequela of hyperadrenocorticism. Hyposthenuria and the antiinflammatory effects of glucocorticoids commonly interfere with the identification of bacteria or inflammatory cells in the urine. Whenever hyperadrenocorticism is suspected, antepubic cystocentesis with bacterial culture of the urine and antibiotic sensitivity testing is strongly recommended, regardless of the urinalysis findings.
DIAGNOSTIC IMAGING
Abnormalities identified by thoracic and abdominal radiography and by abdominal ultrasonography are listed in Box 53-3. The most consistent radiographic findings in dogs with hyperadrenocorticism are enhanced abdominal contrast secondary to increased fat distribution in the abdomen; hepatomegaly caused by steroid hepatopathy; an enlarged urinary bladder secondary to the polyuric state; and dystrophic calcification of the trachea, bronchi, and occasionally the skin and abdominal blood vessels. The most important but least common radiographic finding is a soft-tissue mass or calcification in the area of an adrenal gland (Fig. 53-5). These findings are suggestive of an adrenal tumor. Approximately 50% of ATH are calcified; the frequency of calcification is equally distributed between adenoma and carcinoma. Metastasis of an adrenocortical carcinoma to the pulmonary parenchyma is occasionally evident on thoracic radiographs.
BOX 53-3 Abnormalities Identified by Abdominal and Thoracic Radiography and Abdominal Ultrasonography in Dogs with Hyperadrenocorticism
PDH, Pituitary-dependent hyperadrenocorticism; ATH, adrenocortical tumor causing hyperadrenocorticism.
* Common findings.
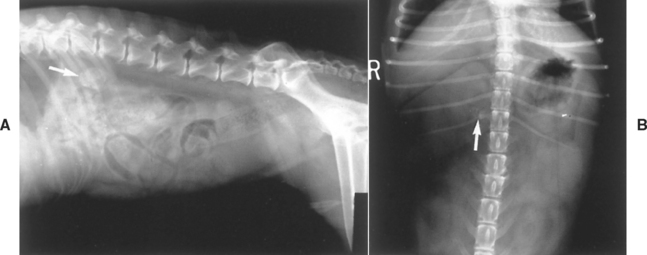
FIG 53-5 A, Lateral radiograph from a dog with adrenal-dependent hyperadrenocorticism showing a calcified adrenal mass cranial to the kidney (arrow). B, Ventrodorsal radiograph from a dog with adrenal-dependent hyperadrenocorticism showing a calcified adrenal mass craniomedial to the kidney and lateral to the spine (arrow). Compression of the abdomen in the region of the adrenal gland with a paddle has enhanced radiographic contrast, allowing better visualization of the adrenal mass.
Abdominal ultrasonography is used to evaluate the size and shape of the adrenals and to search for additional abnormalities in the abdomen (e.g., cystic calculi, tumor thrombus; Fig. 53-6). Finding bilaterally symmetric normal-size or large adrenals (defined as having a maximum width greater than 0.8 cm) in a dog with hyperadrenocorticism is evidence for adrenal hyperplasia caused by PDH. The adrenal glands in dogs with PDH are similar but not exactly the same in size and shape; should have smooth, not irregular borders; can exceed 2 cm in maximum width; may have a bulbous cranial or caudal pole; and do not invade surrounding blood vessels or organs (see Fig. 53-6). An AT is typically identified as an adrenal mass (Fig. 53-7). Size is quite variable, ranging from 1.5 to greater than 8 cm in maximum width. Small adrenal masses (i.e., less than 3 cm in maximum width) often maintain a smooth contour and may distort only a portion of the adrenal gland; one or both poles of the adrenal gland may still appear normal. With large adrenal masses (typically greater than 3 cm in maximum width), the adrenal gland usually becomes distorted and unrecognizable, the contour of the gland becomes irregular, and compression and/or invasion into adjacent blood vessels and organs may occur (Fig. 53-8). These changes suggest adrenocortical carcinoma. Identification of calcification within the mass does not differentiate adenoma from carcinoma. Generally, the larger the mass, the more likely it is carcinoma. Asymmetry in the size of the adrenal glands is evident (see Fig. 53-2). Ideally, the contralateral unaffected adrenal should be small or undetectable (maximum width typically less than 0.3 cm) as a result of AT-induced adrenocortical atrophy (see Fig. 53-7), although a normal-size contralateral adrenal gland does not rule out hyperadrenocorticism caused by AT. Identification of an adrenal mass and a normal-to-large contralateral adrenal gland in a dog with clinical signs supportive of hyperadrenocorticism suggests the possibility of PDH and a concurrent adrenal mass that may be a pheochromocytoma, a functional adrenocortical tumor, or a nonfunctional AT (Fig. 53-9). Finding normal-size adrenal glands in a dog with confirmed hyperadrenocorticism is most consistent with a diagnosis of PDH. Finding bilateral adrenomegaly with the appearance of multiple nodules of varying size is suggestive of macronodular hyperplasia (Fig. 53-10). Bilateral adrenal macronodular hyperplasia is believed to represent an anatomic variant of PDH. Failure to identify either adrenal is considered an inconclusive finding, and ultrasonography should be repeated at a later time.
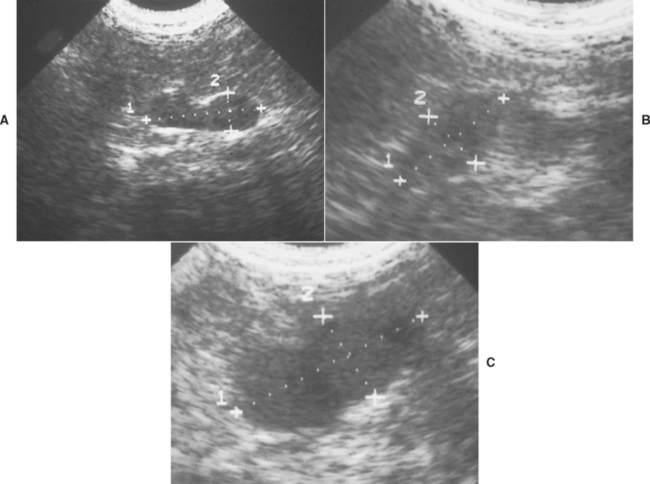
FIG 53-6 Ultrasound images of the adrenal gland in three dogs with pituitary-dependent hyperadrenocorticism (PDH) illustrating the differences in size and shape of the adrenal gland that can occur with PDH. A, The adrenal gland in this dog has maintained the typical kidney-bean shape often identified in normal dogs. However, the maximum diameter of the gland was enlarged at 0.85 cm. The contralateral adrenal gland was similar in size and shape. B, The adrenal gland in this dog is uniformly thickened and appears plump rather than kidney-bean shaped. The maximum diameter of the gland was 1.2 cm. The contralateral adrenal gland was similar in size and shape. C, Although the adrenal gland has maintained some semblance of a kidney-bean shape in this dog, the gland has undergone marked enlargement, with a maximum diameter of 2.4 cm. The contralateral adrenal gland was similar in size and shape.
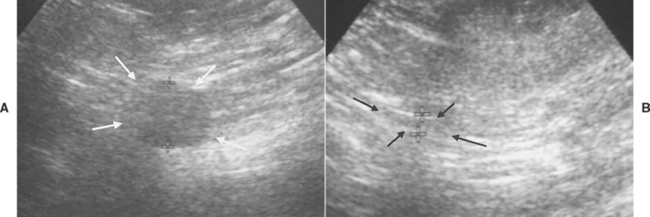
FIG 53-7 Ultrasound images of the adrenal glands in an 11-year-old male castrated Golden Retriever with adrenal-dependent hyperadrenocorticism. A, Cortisol-secreting tumor affecting the right adrenal gland (arrows). The maximum diameter of the adrenal mass was 1.6 cm. B, The left adrenal gland has undergone marked atrophy (arrows and crosses) as a result of suppression of pituitary adrenocorticotropic hormone secretion after negative feedback inhibition caused by the adrenocortical tumor. The maximum diameter of the left adrenal gland was less than 0.2 cm.
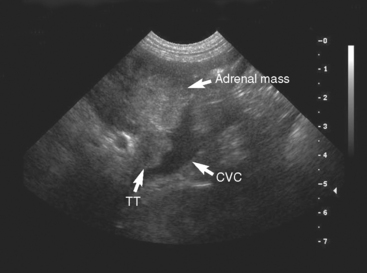
FIG 53-8 Ultrasound image of a mass affecting the left adrenal gland (adrenal mass) and extending into the lumen of the caudal vena cava (CVC) creating a tumor thrombus (TT) in a 9-year-old male Standard Poodle. The maximum width of the adrenal mass was 3.8 cm. The histopathologic diagnosis was pheochromocytoma.
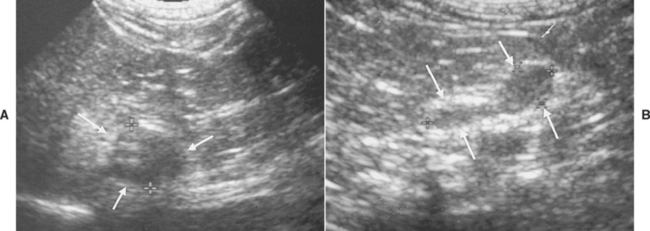
FIG 53-9 Ultrasound images of the adrenal glands in a 10-year-old female spayed Bichon Frise presented for acute onset of vomiting. A, An unexpected mass involving the right adrenal gland, measuring 1.4 cm in maximum diameter, was identified (arrows). B, The left adrenal gland was normal in size and shape (arrows); the maximum diameter was 0.6 cm. The normal-size left adrenal gland suggests that the right adrenal mass is either a pheochromocytoma or is nonfunctional. Results of routine blood work and tests for hyperadrenocorticism were normal.
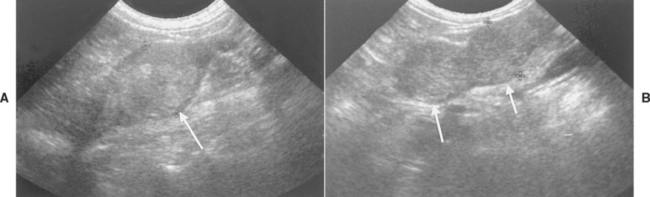
FIG 53-10 Ultrasound images of the adrenal glands (arrows) in an 11-year-old female spayed Shih Tzu. The right adrenal gland (A) measured 1.8 cm in maximum diameter and had a nodular echogenic pattern. In contrast, the left adrenal gland (B) had a large nodule located in each pole of the gland; each measured approximately 1.4 cm in maximum diameter. Tests of the pituitary-adrenocortical axis were diagnostic for pituitary-dependent hyperadrenocorticism; this finding, in conjunction with the findings on ultrasound, suggests macronodular hyperplasia of the adrenal glands.
CT and MRI can be used to evaluate the pituitary gland for a macroadenoma and assess the size and symmetry of the adrenal glands. Contrast enhancement using an iodinated contrast agent (CT) or gadolinium (MRI) given by continuous intravenous (IV) infusion during the imaging procedure aids in the identification of a pituitary macroad enoma and the adrenal glands during CT and MRI examination, respectively (see Fig. 53-4). The primary indications for CT or MRI are to confirm the presence of a visible pituitary tumor in a dog with clinical signs suggestive of macrotumor (see the section on pituitary macrotumor syndrome, p. 814) or in dogs diagnosed with PDH in which the client is willing to consider radiation treatment should a pituitary mass be identified (see the section on radiation therapy, p. 829) and to assess the size of an adrenal mass and extent of infiltration of the mass into surrounding blood vessels and organs before adrenalectomy. MRI is superior to CT in detecting small pituitary tumors; in detecting associated tumor features such as edema, cysts, hemorrhage, and necrosis; and in imaging the adrenal glands.
TESTS OF THE PITUITARY-ADRENOCORTICAL AXIS
The clinical signs, physical examination findings, and clinicopathologic alterations usually establish a presumptive diagnosis of hyperadrenocorticism, and results of an abdominal ultrasound provide valuable information regarding probable location of the lesion. Tests to establish the diagnosis of hyperadrenocorticism include the urine cortisol : creatinine ratio (UCCR), the ACTH stimulation test, the low-dose dexamethasone suppression (LDDS) test, and the oral dexamethasone suppression test (Table 53-2). Baseline serum cortisol measurement by itself is of no diagnostic value in diagnosing hyperadrenocorticism. Discriminatory tests are used to identify the etiology (i.e., PDH versus AT) in dogs with confirmed hyperadrenocorticism and include the low- and high-dose dexamethasone suppression test and baseline endogenous ACTH concentration. The most commonly used tests in our hospital are the UCCR, LDDS test, and abdominal ultrasound. An endogenous ACTH concentration is evaluated when abdominal ultrasound suggests an adrenal mass but results of the LDDS test are inconclusive or suggest PDH and when an adrenal mass is identified with contralateral adrenomegaly.
TABLE 53-2 Diagnostic Tests to Assess the Pituitary-Adrenocortical Axis in Dogs with Suspected Hyperadrenocorticism
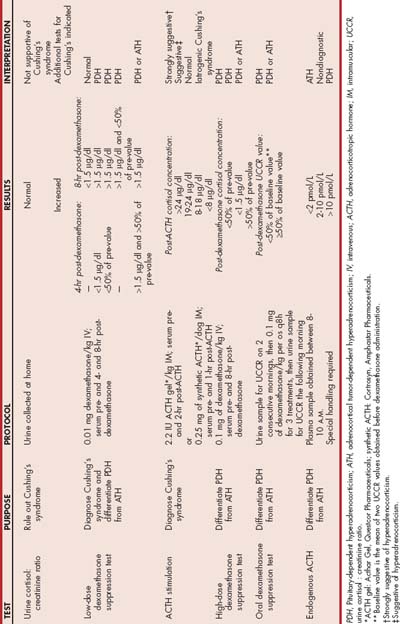
False-positive and false-negative test results occur with all of the diagnostic tests for hyperadrenocorticism. When the results are unexpected or questionable, another diagnostic test can be performed or the same diagnostic test repeated, preferably after waiting several months. Occasionally, results of different diagnostic tests performed in the same dog are contradictory. The decision to perform discriminatory tests or to initiate therapy should depend on the clinician’s index of suspicion for the disease formulated from a review of the history, findings on physical examination, and results of diagnostic tests. If there is doubt or uncertainty about the diagnosis, therapy for hyperadrenocorticism should be withheld and the dog reevaluated several months later.
Urine Cortisol : Creatinine Ratio
The UCCR is an excellent initial screening test for hyperadrenocorticism in dogs. Ideally, the UCCR should be determined from free-catch urine samples obtained by the client in the nonstressful home environment. The stress associated with driving the dog to the veterinary hospital and having the dog undergo a physical examination before collecting urine can increase the test results (Fig. 53-11). The UCCR is increased in dogs with hyperadrenocorticism compared with healthy dogs. Normal UCCR test results can occur in dogs with hyperadrenocorticism but are uncommon. Unfortunately, the specificity of the UCCR is only 20% in dogs. The UCCR is often increased in dogs with nonadrenal illness and in dogs with clinical signs consistent with hyperadrenocorticism but with a normal pituitary-adrenocortical axis (Fig. 53-12). A normal UCCR is a strong finding against hyperadrenocorticism and can be used as a screening test for normalcy; however, an increased UCCR is not diagnostic of hyperadrenocorticism. Additional tests are indicated when the UCCR is increased or when the UCCR is normal but the clinical picture strongly suggests hyperadrenocorticism.
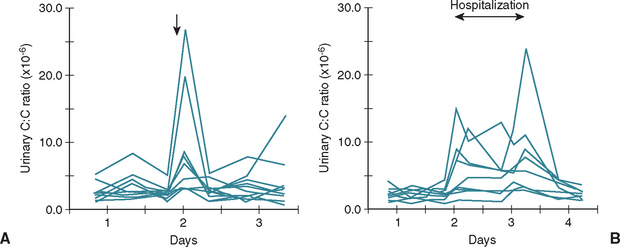
FIG 53-11 Urinary corticoid : creatinine (C : C) ratio measured in 12 pet dogs before and after a visit to a referral clinic for orthopedic examination (A) and in 9 healthy pet dogs before, during, and after a 1.5-day hospitalization at a referral clinic (B). The arrows indicate time of visit to the referral clinic. Note the increase in the urinary C : C ratio in a few dogs affiliated with a visit to a veterinary practice.
(From van Vonderen IK et al: Influence of veterinary care on the urinary corticoid : creatinine ratio in dogs, J Vet Intern Med 12:431, 1998.)
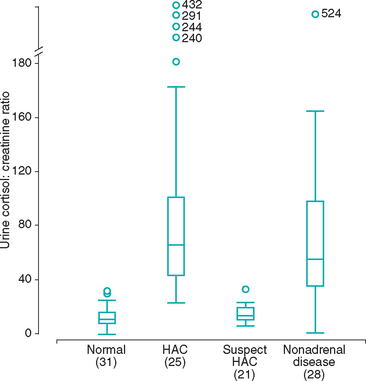
FIG 53-12 Box plots of the urine cortisol : creatinine ratios found in normal dogs, dogs with hyperadrenocorticism (HAC), dogs in which hyperadrenocorticism was initially suspected but that did not have the disease (suspect HAC), and dogs with a variety of severe, nonadrenal diseases. For each box plot, T-bars represent the main body of data, which in most instances are equal to the range. Each box represents an interquartile range (twenty-fifth to seventy-fifth percentile). The horizontal bar in each box is the median. Open circles represent outlying data points. Numbers in parentheses indicate the numbers of dogs in each group.
(From Smiley LE et al: Evaluation of a urine cortisol : creatinine ratio as a screening test for hyperadrenocorticism in dogs, J Vet Intern Med 7:163, 1993.)
Low-Dose Dexamethasone Suppression Test
In the normal dog relatively small doses of dexamethasone given intravenously can inhibit pituitary secretion of ACTH, causing a prolonged decline in the serum cortisol concentration (Fig. 53-13). Dexamethasone is used because it does not interfere with the radioimmunoassays used to measure cortisol. The abnormal pituitary in dogs with PDH is somewhat resistant to the negative feedback action of dexamethasone, and the metabolic clearance of dexamethasone may be abnormally accelerated as well. The administration of a small dose of dexamethasone to a dog with PDH causes the serum cortisol concentration to be variably suppressed; however, it is no longer suppressed by 8 hours after dexamethasone administration, compared with the response seen in normal dogs. ATH function independently of ACTH control, and dexamethasone does not affect the serum cortisol concentration, regardless of the dose or time of blood sampling because pituitary corticotrophs are already suppressed and blood ACTH concentration is undetectable.
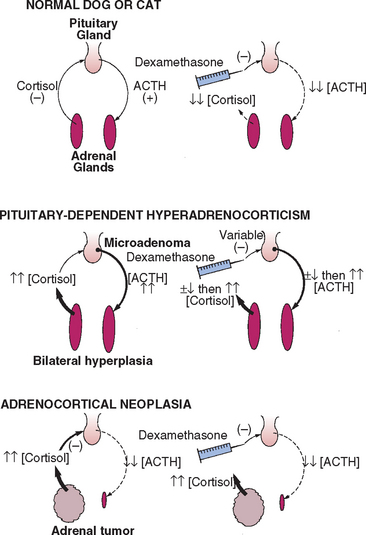
FIG 53-13 Effects of dexamethasone administration on the pituitary-adrenocortical axis in healthy dogs or cats and in dogs or cats with either pituitary-dependent hyperadrenocorticism (PDH) or adrenocortical neoplasia. In PDH dexamethasone may initially suppress pituitary adrenocorticotropic hormone (ACTH) secretion, but the suppression is short-lived. The plasma cortisol concentrations initially decline but increase above normal within 2 to 6 hours of dexamethasone administration. In adrenocortical neoplasia pituitary ACTH secretion is already suppressed; thus dexamethasone has no effect.
The LDDS test is a reliable diagnostic test for differentiating normal dogs from those with hyperadrenocorticism and may identify PDH. Sensitivity and specificity are approximately 90%. The LDDS does not identify iatrogenic hyperadrenocorticism, nor is it used to assess a dog’s response to mitotane (lysodren) or trilostane therapy. A normal or inconclusive LDDS test result does not by itself rule out hyperadrenocorticism. If hyperadrenocorticism is suspected, additional tests of the pituitary-adrenocortical axis should be performed. Similarly, an abnormal LDDS test result does not by itself confirm hyperadrenocorticism. Results of the LDDS test may be affected by concurrently administered anticonvulsant drugs, stress, excitement, exogenous glucocorticoids, and nonadrenal disease; the more severe the nonadrenal disease, the more likely the LDDS test result will be falsely positive. When performing the LDDS test, the clinician must ensure that all stressors are kept to a minimum; other procedures should not be performed until the test is completed, and the effect of concurrent clinical problems should be considered when interpreting results.
The protocol for the LDDS test and interpretation of results are described in Table 53-2. The clinician may use either dexamethasone sodium phosphate or dexamethasone in polyethylene glycol. The 8-hour postdexamethasone serum cortisol concentration is used to confirm hyperadrenocorticism. Normal dogs typically have serum cortisol values less than 1.0 μg/dl, whereas dogs with PDH and AT have serum cortisol concentrations greater than 1.5 μg/dl 8 hours after dexamethasone administration. In general, the higher the 8-hour postdexamethasone serum cortisol concentration is above 1.5 μg/dl, the more supportive the test result is for hyperadrenocorticism. Cortisol concentrations between 1.0 and 1.5 μg/dl are nondiagnostic. If results are in the nondiagnostic range, the clinician must rely on other information, including other tests of the pituitary-adrenocortical axis, to determine if hyperadrenocorticism is the correct diagnosis.
If the 8-hour postdexamethasone serum cortisol value supports a diagnosis of hyperadrenocorticism, the 4-hour serum cortisol value may then be of value in identifying PDH. Low doses of dexamethasone suppress pituitary ACTH secretion and serum cortisol concentrations in approximately 60% of dogs with PDH. Suppression does not occur in dogs with AT, nor does it occur in approximately 40% of dogs with PDH. Suppression is defined as a 4-hour postdexamethasone serum cortisol concentration of less than 1.5 μg/dl, a 4-hour postdexamethasone serum cortisol concentration less than 50% of the baseline concentration, or an 8-hour postdexamethasone serum cortisol concentration less than 50% of the baseline concentration. Any dog with hyperadrenocorticism that meets one or more of these criteria most likely has PDH. If none of these criteria is met, then results of the LDDS test are consistent with lack of suppression but not informative in terms of whether it is pituitary or adrenal in origin. Differentiation between PDH and AT must rely on results of abdominal ultrasound, the HDDS test, or plasma endogenous ACTH concentration.
Oral Dexamethasone Suppression Test
An alternative at-home oral dexamethasone suppression test has been used for years at the University of Utrecht, The Netherlands. This test relies entirely on results of UCCRs to establish the diagnosis of hyperadrenocorticism and to identify PDH. The client is instructed to collect two urine samples from the dog on 2 consecutive mornings and store them in the refrigerator. After collection of the second urine sample, the client should administer 3 doses of dexamethasone (0.1 mg/kg/dose) to the dog orally at 8-hour intervals. Urine is collected on the morning of the third day, and all three samples are delivered to the veterinarian for measurement of UCCRs. The first two urine samples are the screening test to diagnose hyperadrenocorticism. Abnormal values support hyperadrenocorticism; normal values rule out the disease. If both values are abnormal, then the average of the two values is used as the baseline value and compared with the third value obtained after dexamethasone administration. The dog is described as having responded to dexamethasone (suppressed) if the UCCR result from the third urine sample is less than 50% of the baseline value. Dogs meeting this criteria have results consistent with PDH, whereas those failing to demonstrate suppression could have either AT or PDH.
Adrenocorticotropic Hormone Stimulation Test
The ACTH stimulation test is used to establish the diagnosis of hyperadrenocorticism and hypoadrenocorticism, identify iatrogenic hyperadrenocorticism, identify atypical hyperadrenocorticism (see p. 830), and monitor mitotane and trilostane treatment. ACTH stimulation test results do not distinguish between PDH and AT. In our experience ACTH stimulation test results are clearly abnormal in approximately 30%, in the borderline range in another 30% and within the reference range in approximately 40% of dogs with PDH. Identification of ACTH stimulation test results in the borderline range is common, and clearly abnormal test results occur in dogs that do not have hyperadrenocorticism. Because of problems with sensitivity and specificity combined with the high cost of ACTH, I do not routinely use the ACTH stimulation test when evaluating dogs for hyperadrenocorticism.
The protocol for the ACTH stimulation test is given in Table 53-2. When synthetic ACTH is being used, a lower dose (5 μg/kg, administered intravenously or intramuscularly) is also effective and the unused reconstituted ACTH can be stored frozen at −20°C in plastic syringes for 6 months with no adverse effects on bioactivity of the ACTH. Four ranges of values are used in the interpretation of the ACTH stimulation test (Fig. 53-14). Post-ACTH serum cortisol values between 6 and 18 μg/dl are within the normal reference range, values of 5 μg/dl and below are suggestive of iatrogenic hyperadrenocorticism or hypoadrenocorticism, values between 18 and 24 μg/dl are considered borderline for hyperadrenocorticism, and values greater than 24 μg/dl are supportive of hyperadrenocorticism, assuming the clinical findings and clinicopathologic data are consistent with the disease. An increased post-ACTH serum cortisol value, especially one between 18 and 24 μg/dl, does not by itself confirm a diagnosis of hyperadrenocorticism, especially if the clinical features and clinicopathologic data are not consistent with the diagnosis.
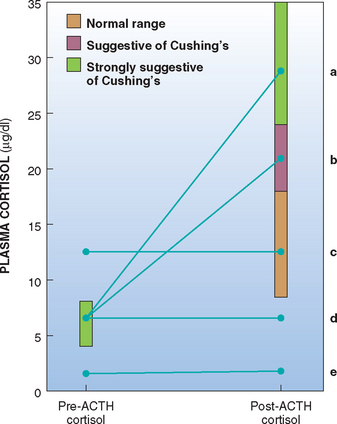
FIG 53-14 Interpretation of the adrenocorticotropic hormone (ACTH) stimulation test in dogs. Ideally, dogs with Cushing’s syndrome have an increased post-ACTH administration cortisol concentration (line a). Post-ACTH cortisol values that fall into the “gray zone” (line b) could be consistent with Cushing’s syndrome or result from the effects of concurrent illness or chronic stress. Post-ACTH cortisol values may also fall into the normal range in dogs with Cushing’s syndrome. The absence of a response to ACTH stimulation is suggestive of adrenocortical neoplasia (lines c and d) or iatrogenic hyperadrenocorticism (lines d and e). History and physical examination findings should differentiate between these possibilities.
Post-ACTH serum cortisol concentrations that do not increase above the preadministration value suggest iatrogenic hyperadrenocorticism or spontaneous hypoadrenocorticism, especially if the cortisol values are below the normal baseline range (i.e., less than 5 μg/dl; see Fig. 53-14). A history of recent glucocorticoid administration and the clinical presentation of the dog can help differentiate iatrogenic hyperadrenocorticism from spontaneous hypoadrenocorticism. In rare instances a dog with AT will have a minimal cortisol response to ACTH; however, its pre-ACTH and post-ACTH administration serum cortisol concentrations are within or above the reference range.
High-Dose Dexamethasone Suppression Test
ATs function independently of pituitary ACTH; therefore, regardless of the dose, dexamethasone should never suppress the serum cortisol concentration if the source of the cortisol is an AT. In contrast, dexamethasone-induced suppression of ACTH secretion from a pituitary tumor is variable and may depend on the dexamethasone dose. The administration of increased amounts of dexamethasone should eventually suppress pituitary ACTH secretion in most dogs with PDH. The protocol for the high-dose dexamethasone suppression (HDDS) test is similar to that for the LDDS test protocol, except that a higher dose (i.e., 0.1 mg/kg of body weight) of dexamethasone is used in an attempt to suppress pituitary ACTH secretion (see Table 53-2). Obtaining a 4-hour postdexamethasone blood sample is optional; in our experience, this has been informative in only 2% of dogs tested with both the LDDS and HDDS tests. Suppression is defined as a 4-hour or 8-hour postdexamethasone serum cortisol concentration less than 1.5 μg/dl and a 4-hour or 8-hour postdexamethasone serum cortisol concentration less than 50% of the baseline concentration. Any dog with hyperadrenocorticism that meets one or more of these criteria most likely has PDH. If a dog does not meet any of these criteria, this is consistent with lack of suppression. Approximately 25% of dogs with PDH and essentially 100% of dogs with ATH do not show suppression with the HDDS test. Higher doses of dexamethasone (e.g., 1.0 mg/kg) could be administered in an attempt to suppress pituitary ACTH secretion in dogs with dexamethasone-resistant PDH. However, the percentage of dogs with PDH that show suppression at higher doses of dexamethasone is similar to that observed for the 0.1 mg/kg protocol.
Endogenous Adrenocorticotropic Hormone Concentration
I do not routinely measure plasma ACTH concentrations because the LDDS test and abdominal ultrasound are very effective in differentiating between PDH and AT. I use plasma ACTH concentrations to provide clarity in confusing cases in which test results for hyperadrenocorticism and findings on abdominal ultrasound conflict (e.g., a dog with an adrenal mass but suppression on the LDDS test or a dog with an adrenal mass, enlargement of the contralateral adrenal gland, and lack of suppression on the LDDS test). Determination of a baseline plasma ACTH concentration is not used to diagnose hyperadrenocorticism because many of the concentrations in dogs with hyperadrenocorticism are within the reference range (2 to 25 pmol/L). However, determination of a single baseline plasma ACTH concentration may aid in distinguishing dogs with ATH from those with PDH once the diagnosis of hyperadrenocorticism is established. Adrenocortical tumors and iatrogenic hyperadrenocorticism should suppress ACTH secretion, and PDH is the result of excessive ACTH secretion (see Fig. 53-2). Approximately 60% of dogs with ATH have undetectable plasma ACTH concentrations, whereas 85% to 90% of dogs with PDH have plasma ACTH concentrations greater than 10 pmol/L and 35% have ACTH concentrations greater than 25 pmol/L. Plasma ACTH concentrations of 2 to 10 pmol/L are nondiagnostic. Several commercial veterinary endocrine laboratories perform endogenous ACTH assays for dogs. The laboratory should be consulted for information on sample collection and handling; results should be interpreted on the basis of the reference range established for the laboratory being used.
Medical Treatment
Medical options for treating hyperadrenocorticism are listed in Table 53-3. The most viable treatment options for dogs are mitotane and trilostane.
 TABLE 53-3
TABLE 53-3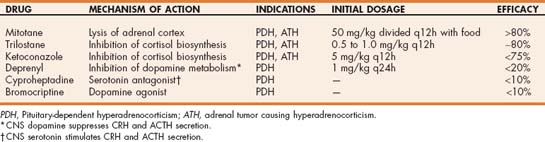
MITOTANE
Chemotherapy using mitotane (o,o’ DDD; Lysodren; Bristol Myers Oncology) is the most commonly used treatment for PDH and is a viable alternative to adrenalectomy for treatment of ATs causing hyperadrenocorticisim. There are two treatment protocols: the traditional approach, the goal of which is to control the hyperadrenal state without causing clinical signs of hypoadrenocorticism, and medical adrenalectomy, the goal of which is to destroy the adrenal cortex and create hypoadrenocorticism. I prefer the traditional approach initially and consider medical adrenalectomy in dogs that fail to respond to the traditional approach or that become nonresponsive to mitotane after months or years of maintenance therapy.
Traditional Approach to Mitotane Treatment
For the traditional approach, there are two phases of mitotane therapy: an initial induction phase designed to gain control of the disorder, and a lifelong maintenance phase designed to prevent recurrence of the signs of the disease.
Induction Therapy
The mitotane dosage during induction therapy is 40 to 50 mg/kg, divided into two doses. The daily dosage is reduced to 25 to 35 mg/kg in dogs without polydipsia or with concurrent diabetes mellitus. Gastrointestinal absorption of mitotane is enhanced in the presence of fat. Mitotane is more effective when each dose is ground up, mixed with a small amount of vegetable oil, and administered with food. Concurrent prednisone administration (0.25 mg/kg q24h) during induction therapy is a matter of personal preference. If prednisone is not used during induction therapy, it should always be dispensed before beginning induction therapy so that the client has glucocorticoids on hand should adverse reactions to mitotane develop.
The induction phase of mitotane treatment is typically done with the dog in the home environment. Client awareness of their dog’s activity, mental awareness, appetite, water consumption, and overall well-being is imperative for success. The usual amount of food offered to the dog can be decreased by approximately 25% during the induction phase to ensure that the dog remains hungry. Clients are instructed to stop mitotane treatment and contact their veterinarian if they observe lethargy, inappetence, vomiting, weakness, decreased water intake, or any other change in their dog that does not seem right. The veterinarian or a technician should call the client every day, beginning with the second day of therapy, to check on the health of the dog. The induction phase of therapy is usually complete once any reduction in appetite is noted or once daily water consumption decreases into the normal range (i.e., 80 ml/kg or less). Control is confirmed with the ACTH stimulation test. The first ACTH stimulation test should be performed 5 to 7 days after starting induction therapy, even if clinical signs of hyperadrenocorticism persist. Dogs that have responded clinically to the medication (or if the client is not certain about response) should not receive further therapy until results of the ACTH stimulation test are known. Dogs that have not yet responded clinically should undergo an ACTH stimulation test but should also remain on daily mitotane therapy pending results of the ACTH stimulation test.
The goal of therapy is to achieve a post-ACTH serum cortisol concentration of 2 to 5 μg/dl. Daily mitotane therapy and weekly ACTH stimulation tests should be continued until a post-ACTH serum cortisol concentration falls within the desired range or signs of hypocortisolism develop. In most dogs clinical signs resolve and a post-ACTH serum cortisol concentration of less than 5 μg/dl is achieved within 5 to 10 days of the start of the daily administration of 40 to 50 mg of mitotane/kg. A small number of dogs respond in less than 5 days, and an equally small number of dogs show minimal improvement after 20 to 30 consecutive days of therapy.
Reasons for a prolonged or poor response to mitotane treatment include inadequate dose, inadequate absorption from the gastrointestinal tract, concurrent administration of drugs (e.g., phenobarbital) that stimulate hepatic microsomal drug-metabolizing enzymes and could accelerate the metabolism of mitotane and decrease its serum concentration, existence of an AT rather than PDH, and client compliance issues. The absorption of mitotane is improved if it is given with food, especially a fatty meal, and if the tablet is crushed, mixed with a small amount of vegetable oil, and mixed with food. Typically, dogs with AT are more resistant to the adrenocorticolytic effects of mitotane than dogs with PDH. If tests to differentiate PDH from AT were not performed, dogs that are shown to be resistant to therapy, defined as showing little or no reduction in the post-ACTH plasma cortisol concentration after 20 or more days of therapy, should undergo further evaluation (i.e., abdominal ultrasound) to determine whether an AT is an explanation for the resistance. Rarely, dogs with PDH require more than 30 consecutive days of mitotane therapy before the desired response is seen.
Maintenance Therapy
Mitotane must be administered periodically to prevent recurrence of clinical signs. The maintenance phase of mitotane therapy should be initiated once the post-ACTH serum cortisol concentration is less than 5 μg/dl and the dog appears healthy. The maintenance dose is defined as the weekly amount of mitotane administered, regardless of whether the weekly dose is given once per week or divided into multiple doses and given on several days. Adverse reactions caused by sensitivity to the drug are less likely to occur when the weekly dose is divided and given on several days of the week. The typical initial weekly maintenance dosage of mitotane is 50 mg/kg administered orally, divided into two or three doses, and administered on 2 or 3 days of each week (e.g., Monday and Thursday or Monday, Wednesday, and Friday). The maintenance dose of mitotane is decreased from 50 mg/kg/week to 25 mg/kg/week if the post-ACTH serum cortisol concentration is less than 2 μg/dl and the dog appears healthy. Mitotane treatment is discontinued and prednisone treatment initiated if the post-ACTH serum cortisol concentration is less than 2 μg/dl and the dog is exhibiting clinical signs of hypoadrenocorticism (i.e., lethargy, inappetence, vomiting).
The initial dose of lysodren during maintenance therapy is arbitrary, and subsequent adjustments are made on the basis of results of ACTH stimulation tests; the first test is performed 3 to 4 weeks after the start of maintenance therapy. The goal of maintenance therapy is to maintain the post-ACTH serum cortisol concentration between 2 and 5 μg/dl in an otherwise healthy dog. The dose and frequency of administration of mitotane are adjusted, as needed, to maintain a hypoadrenal response to ACTH administration. If the post-ACTH serum cortisol is between 2 and 5 μg/dl, a change in treatment is not indicated and the ACTH stimulation test should be repeated in 6 to 8 weeks. If the post-ACTH serum cortisol concentration is greater than 5 μg/dl, the amount of mitotane per administration or the frequency of administration is increased; if the post-ACTH serum cortisol concentration is less than 2 μg/dl, the mitotane dose or frequency of administration is decreased; mitotane therapy is tempo rarily discontinued if clinical signs of hypoadrenocorticism are present. An ACTH stimulation test is performed 3 to 4 weeks after changing the dose or frequency of administration of mitotane. Once the post-ACTH serum cortisol concentration is stable and in the range of 2 to 5 μg/dl, the ACTH stimulation test should be repeated every 3 to 6 months thereafter unless clinical signs of hyperadrenocorticism or hypoadrenocorticism develop. In most dogs an initially effective maintenance dose of mitotane becomes inadequate as the compensatory sustained increase in plasma ACTH concentration counters the adrenocorticolytic effects of mitotane. With time (i.e., months to years), the dose and frequency of administration of mitotane must usually be increased to compensate for this effect. Periodic ACTH stimulation testing will identify an increase in the post-ACTH serum cortisol concentration above 5 μg/dl, allowing the clinician to adjust the mitotane treatment protocol before clinical signs of hyperadrenocorticism develop and another round of induction therapy is needed. In some dogs this can ultimately necessitate daily mitotane administration, sometimes with poor control of the disorder. Alternative therapy (i.e., medical adrenalectomy using mitotane, trilostane) should be considered for dogs that become insensitive to mitotane.
Adverse Reactions to Mitotane Treatment
Adverse reactions to mitotane treatment result from sensitivity to the drug or from excessive administration and the subsequent development of glucocorticoid and, if severe, mineralocorticoid deficiency (Box 53-4). The most common reactions to mitotane are gastric irritation and vomiting occurring shortly after its administration. If the gastric upset is the result of drug sensitivity and not hypoadrenocorticism, dividing the dose further, increasing the interval between administrations, or both can help minimize vomiting.
BOX 53-4 Adverse Effects of Mitotane in Dogs
PMA, Pituitary macroadenoma.
Direct Effect*
Secondary to Overdosage*
* Adrenocorticotropic hormone stimulation test, serum electrolytes, response to discontinuation of mitotane, and response to glucoco
The excessive administration of mitotane results in clinical signs of hypocortisolism, including weakness, lethargy, anorexia, vomiting, and diarrhea. Clinical improvement is usually seen within hours of the administration of prednisone (0.25 to 0.5 mg/kg, administered orally). If the dog responds, the initial dosage of glucocorticoids should be continued for 3 to 5 days and then gradually decreased and stopped over the ensuing 1 to 2 weeks. Mitotane therapy should be stopped until the dog is normal when it is not receiving glucocorticoids. An ACTH stimulation test performed once the dog is healthy and not receiving glucocorticoids can help determine when to start mitotane treatment. Ideally, mitotane treatment should be started when the post-ACTH serum cortisol concentration is 2 μg/dl or greater. The weekly dose of mitotane should be reduced when therapy is reinitiated.
Excessive administration of mitotane ultimately causes hypoaldosteronism. Mineralocorticoid deficiency should be considered in any dog with signs of hypocortisolism that does not respond to glucocorticoid therapy. Finding hyponatremia and hyperkalemia supports a diagnosis of hypoaldosteronism, and mineralocorticoid therapy is indicated in such dogs (see p. 840). Hypoaldosteronism can develop within days of the start of mitotane therapy in some dogs. Hypoaldosteronism can resolve and hyperadrenocorticism recur spontaneously, but this is unpredictable. Some dogs remain mineralocorticoid deficient for the remainder of their lives.
Mitotane may induce the development of neurologic signs, including stupor, head pressing, pacing, circling, seizures, ataxia, and blindness. Neurologic signs are usually transient, typically last 24 to 48 hours after mitotane administration, and usually occur in dogs that have been receiving the drug for more than 6 months. The primary differential diagnoses in such animals are pituitary macrotumor syndrome (see p. 814), hypoadrenocorticism, and thromboemboli. Adjustments in the dose or frequency of mitotane administration or temporary discontinuation of the therapy may alleviate the neurologic signs. An alternative mode of therapy should be considered if neurologic signs persist (discussed in more detail later).
Management of Concurrent Diabetes Mellitus
Hyperadrenocorticism and diabetes mellitus are common concurrent diseases in dogs. Presumably, hyperadrenocorticism develops initially and subclinical diabetes mellitus becomes clinically apparent as a result of the insulin resistance caused by the hyperadrenal state. For most of these dogs, glycemic control remains poor despite insulin therapy, and good glycemic control is generally not possible until the hyperadrenocorticism is controlled. Occasionally, diabetic dogs presumably in the early stages of hyperadrenocorticism (often identified while pursuing the cause for an increased ALP) will be responsive to insulin and have good control of glycemia. Because the diabetes is well controlled, the decision to treat or not treat the hyperadrenocorticism in these dogs should be based on other factors, such as the presence of additional clinical signs or physical examination findings and the clinician’s index of suspicion for the disease. The clinician should adopt a wait-and-see approach in the absence of strong evidence for hyperadrenocorticism in these dogs. Poor control of the diabetic state will eventually occur if hyperadrenocorticism is present.
The initial focus should be on treating the hyperadrenal state in a poorly controlled diabetic dog diagnosed with hyperadrenocorticism. Insulin therapy is indicated during induction therapy; however, aggressive efforts to control the blood glucose concentration should not be attempted. Rather, a conservative dose (0.5 to 1.0 U/kg) of intermediate-acting insulin (i.e., lente or NPH) is administered twice a day to prevent ketoacidosis and severe hyperglycemia (blood glucose concentration greater than 500 mg/dl). Monitoring induction therapy in the hyperadrenal dog with concurrent diabetes mellitus is similar to that used for the hyperadrenal dog (see the section on monitoring induction therapy) with one exception. Monitoring water consumption is not reliable when concurrent diabetes mellitus is present because both diseases cause polyuria and polydipsia and because polyuria and polydipsia may persist if poor control of glycemia persists despite the fact that the hyperadrenocorticism is under control. As control of the hyperadrenocorticism is achieved, insulin antagonism caused by the hyperadrenocorticism resolves and tissue sensitivity to insulin improves. To help prevent hypoglycemic reactions, clients are asked to test urine for the presence of glucose, preferably two or three times each day. Any urine sample found to be negative for glucose should be followed by a 20% to 25% reduction in the insulin dose and performance of an ACTH stimulation test. Critical assessment of glycemic control and adjustments in insulin therapy, if indicated, should be initiated once hyperadrenocorticism is controlled and maintenance mitotane therapy initiated.
Medical Adrenalectomy Using Mitotane
An alternative to the traditional mitotane treatment protocol is to intentionally cause complete destruction of the adrenal cortices by administering an excessive amount of mitotane. In theory, therapy for the ensuing adrenocortical insufficiency would then be necessary for the life of the dog. The protocol consists of administering mitotane at a dosage of 75 to 100 mg/kg daily for 25 consecutive days, given in three or four doses per day, with food, to minimize neurologic complications and ensure good intestinal absorption of the drug. Lifelong prednisone (0.1 to 0.5 mg/kg q12h initially) and mineralocorticoid (see p. 840) therapy is begun at the start of mitotane administration. The prednisone dose is tapered after completion of the 25-day protocol. Unfortunately, relapse with signs of hyperadrenocorticism occurs within the first year alone in approximately 33% of dogs so treated, indicating the need for periodic ACTH stimulation testing similar to that done in animals treated with the traditional mode of therapy. In addition, this treatment can be considerably more expensive than long-term treatment with mitotane because of the expense of treating addisonian dogs. For these reasons, medical adrenalectomy is reserved for dogs that show a poor response to the traditional form of treatment.
TRILOSTANE
Trilostane (Vetoryl, Arnolds Veterinary Products) is a competitive inhibitor of 3-β-hydroxysteroid dehydrogenase, which mediates the conversion of pregnenolone to progesterone in the adrenal gland. The net effect is inhibition of cortisol production (Fig. 53-15). Trilostane is currently the preferred enzyme blocker for treating hyperadrenocorticism. The clinical efficacy of trilostane is excellent (approximately 80%), and trilostane can control clinical signs of hyperadrenocorticism in dogs for prolonged periods of time (longer than 1 year). Trilostane is used as the primary treatment modality for PDH in dogs, as an alternative in dogs in which mitotane is ineffective or not usable because of problems with drug sensitivity, and as a way to reverse the metabolic derangements of hyperadrenocorticism before adrenalectomy. Trilostane is currently available as 30-, 60-, and 120-mg capsules. Compounding of capsules to different strengths (e.g., 10 or 20 mg) may be required. The published initial treatment protocol is 30 mg once a day for dogs weighing less than 5 kg, 60 mg once a day for dogs weighing 5 to 20 kg, 120 mg once a day for dogs weighing 20 to 40 kg, and 180 mg once a day for dogs weighing more than 40 kg. However, in our experience, twice-daily dosing using a lower dose provides better control than once-daily dosing using the aforementioned dosing schedule and the occurrence and severity of adverse reactions are less frequent. Our approach is to begin treatment using a trilostane dosage between 0.5 and 1 mg/kg twice daily.
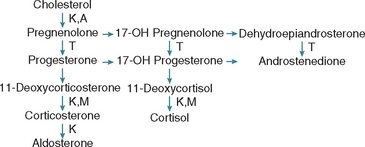
FIG 53-15 Steroid biosynthetic pathways in the adrenal cortex. The branching pathways for glucocorticoids, mineralocorticoids, and adrenal androgens are shown. The site of blockade in the steroid biosynthetic pathways by the enzyme inhibitors trilostane (T), ketoconazole (K), metyrapone (M), and aminoglutethimide (A) are also shown.
The dosage and frequency of administration of trilostane are adjusted, as needed, until clinical signs are controlled. An ACTH stimulation test and serum electrolytes should be performed 10 to 14 days after initiation of treatment and 4 to 6 hours after trilostane administration. In addition, the client should bring in a urine sample collected at home the morning of the ACTH stimulation test for a UCCR. The goals of therapy are the same as those of mitotane therapy: clinical improvement without the development of illness, lack of an adrenocortical response to ACTH, and a normal UCCR. Results of the ACTH stimulation test are used to adjust the dosage of trilostane; the goal is a post-ACTH cortisol concentration between 2 and 5 μg/dl. The UCCR is used to determine frequency of trilostane administration in dogs receiving the drug once daily. If clinical signs persist, results of the ACTH stimulation test are indicative of control of the disease, and the UCCR is increased, then the frequency of trilostane administration should be increased to twice a day. Serum electrolytes are monitored for changes consistent with the onset of hypoaldosteronism. Once control of the hyperadrenal state is attained, an ACTH stimulation test, serum electrolytes, and UCCR should be evaluated every 3 to 4 months.
Adverse effects of trilostane include lethargy, vomiting, and electrolyte shifts compatible with hypoadrenocorticism. Permanent hypoadrenocorticism has been reported in a small number of dogs. Histopathologic examination of the adrenal gland in dogs treated with trilostane has revealed adrenocortical necrosis of variable severity in some dogs—findings that, if severe, could explain persistent hypoadrenocorticism in affected dogs. Acute death has been reported in a small number of dogs shortly after the initiation of trilostane treatment.
KETOCONAZOLE
Ketoconazole reversibly inhibits adrenal steroidogenesis (see Fig. 53-15). The initial dosage of ketoconazole is 5 mg/kg q12h, and subsequent increases in the dosage are based on results of an ACTH stimulation test performed 10 to 14 days later and while the dog is still receiving ketoconazole. The goals of therapy are similar to those discussed for trilostane. Approximately 20% to 25% of dogs do not respond to the drug as a result of poor intestinal absorption. Adverse reactions are primarily a result of hypocortisolism and include lethargy, inappetence, vomiting, and diarrhea. Unfortunately, it is difficult to control the clinical signs of hyperadrenocorticism without creating problems with hypocortisolism.
l-DEPRENYL
l-Deprenyl (Anipryl, Deprenyl Animal Health) inhibits dopamine metabolism and increases hypothalamic and pituitary concentrations of dopamine, which in turn inhibits corticotropin-releasing hormone (CRH) and ACTH secretion. The current dosage recommendation for l-Deprenyl is 1 mg/kg once daily initially, with an increase to 2 mg/kg once daily if there is no response after 2 months. The efficacy of this drug for the treatment of PDH is, at best, 20%. The vast majority of dogs with PDH have a pituitary tumor, not alterations in neurotransmitter control of hypothalamic-pituitary gland function. Concentrations of an endogenous amphetamine, phenylethylamine, increase in the brains of dogs treated with l-Deprenyl, which may improve the dog’s level of activity and its interactions with family members independent of any improvement in the hyperadrenal state.
ADRENALECTOMY
Adrenalectomy is the treatment of choice for an AT unless metastatic lesions or invasion of surrounding organs or blood vessels is identified during the preoperative evaluation; the dog is considered a poor anesthetic risk because it has a concurrent disease (e.g., heart failure) or is debilitated as a result of its hyperadrenal state; or the probability of perioperative thromboembolism is considered high because of systemic hypertension, an increased urine protein : creatinine ratio, or a decreased serum antithrombin III concentration. The probability of successful adrenalectomy is lower and the likelihood of perioperative complications is greater the larger the adrenal mass. Removal of an adrenal mass that has a diameter in excess of 6 cm can be difficult even when the surgery is performed by an experienced surgeon. The larger the adrenal mass, the greater the probability that the adrenal mass is a carcinoma and that metastasis has occurred, regardless of findings during the preoperative evaluation. Treatment with mitotane or trilostane offers a viable alternative to adrenalectomy, especially for aged dogs or dogs at increased risk for anesthetic, surgical, or postsurgical problems. (See Suggested Readings for detailed information on surgical techniques.)
The most worrisome complication of adrenalectomy is thromboembolism, which typically develops during or within 72 hours of surgery and carries a high mortality rate (see p. 814). Several steps help minimize this complication. Trilostane treatment for 3 to 4 weeks before surgery can reverse the metabolic derangements of hyperadrenocorticism and minimize many of the complications associated with adrenalectomy. Plasma is a source of antithrombin III and should be administered during surgery. Heparin or other anticoagulant therapy should be administered during and for several days after adrenalectomy (see Chapter 12). Dogs should go for frequent short walks within hours of the surgery to promote blood flow and minimize clot formation. Anesthetic drugs and pain medications should be administered at dosages that allow the dog to be ambulatory within 4 hours of the surgery. Despite these measures, thromboembolism remains a common perioperative complication that should be thoroughly discussed with clients who are considering adrenalectomy.
Glucocorticoid therapy is not indicated before adrenalectomy because it may worsen hypertension, cause overhydration, and increase the risk of thromboembolic episodes. Beginning with anesthesia, IV fluids should be administered at a surgical maintenance rate. Acute hypocortisolism uniformly occurs after adrenalectomy. After the surgeon identi fies the adrenal tumor, dexamethasone (0.05 to 0.1 mg/kg) should be placed in the IV infusion bottle. This dose should be given over a 6-hour period. A tapering dose (e.g., decreasing the dose by 0.02 mg/kg/24 h) of dexamethasone should continue to be administered intravenously at 12-hour intervals until the dog can safely be given oral medication without the danger of vomiting (typically 24 to 72 hours postoperatively). At that point, the glucocorticoid supplement should be switched to oral prednisone (0.25 to 0.5 mg/kg q12h). Once the dog is eating and drinking on its own, the frequency of prednisone administration should be decreased to once a day and given in the morning. The prednisone dosage is then gradually reduced during the ensuing 3 to 4 months. If a unilateral adrenalectomy has been performed, prednisone supplementation can eventually be discontinued once the contralateral normal adrenocortical tissue becomes functional. Lifelong prednisone at a dosage of 0.1 to 0.2 mg/kg administered once or twice daily is usually required for dogs that undergo bilateral adrenalectomy.
Serum electrolyte concentrations should be closely monitored postoperatively. Development of mild hyponatremia and hyperkalemia is common within 72 hours of surgery and usually resolves in a day or two as exogenous glucocorticoid doses are reduced and the dog begins to eat. Mineralocortioid treatment is recommended if the serum sodium concentration decreases to less than 135 mEq/L or serum potassium concentration increases to greater than 6.5 mEq/L. An injection of desoxycorticosterone pivalate (DOCP; Percorten-V; Novartis Pharmaceuticals) is recommended, with measurement of serum electrolytes performed 25 days after the injection (see p. 840). If the dog is healthy and serum electrolytes are normal on day 25, the dog should be reevaluated 7 days later. If serum electrolytes are still normal, additional DOCP treatment is not needed. If hyponatremia or hyperkalemia is identified on day 25, another injection of DOCP should be administered but with the dosage reduced by 50% and serum electrolytes evaluated 25 days later.
RADIATION THERAPY
Approximately 50% of dogs have a pituitary mass identified on CT or MRI at the time PDH is diagnosed. In approximately 50% of these dogs, the pituitary mass grows over the ensuing 1 to 2 years, eventually causing pituitary macrotumor syndrome (see p. 814). Pituitary macroadenoma is tentatively diagnosed by ruling out other causes of the neurologic disturbances and is confirmed by CT or MRI findings (see Fig. 53-4). Development of neurologic signs from a pituitary macrotumor is a common reason for clients to request euthanasia of dogs with PDH. Irradiation has successfully reduced the tumor size and lessened or eliminated neurologic signs in dogs with pituitary macrotumor syndrome (Fig. 53-16). The primary mode of radiation treatment is cobalt 60 photon irradiation or linear accelerator photon irradiation. Treatment usually involves the delivery of a predetermined total dose of radiation given in fractions over a period of several weeks. Currently a total dose of 48 Gy, given in 4 Gy doses 3 to 5 days per week for 3 to 4 weeks, is typically administered to hyperadrenal dogs with pituitary macroadenoma at our hospital.
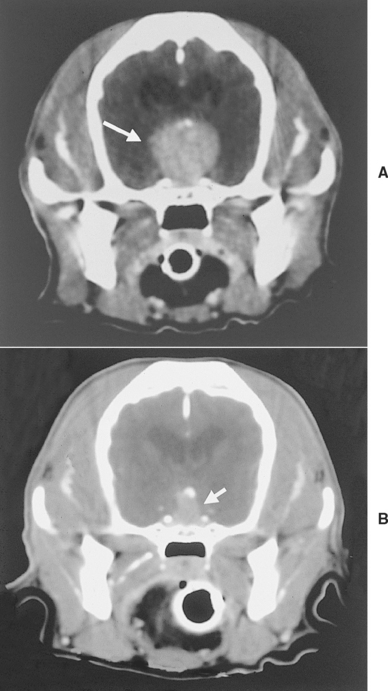
FIG 53-16 A, Computed tomography (CT) image of the pituitary region of a 9-year-old, female spayed Cocker Spaniel with pituitary-dependent hyperadrenocorticism (PDH). The PDH had been treated with mitotane for 2 years, at which time the dog developed lethargy, inappetence, and weight loss. A large mass measuring approximately 2.0 cm in diameter is evident in the hypothalamic-pituitary region (arrow). B, CT image of the pituitary region 18 months after completion of radiation therapy. The volume of the mass decreased by approximately 75%, compared with the volume before treatment. Clinical signs related to the pituitary macrotumor resolved, and mitotane treatment was discontinued after radiation treatment.
Prognostic factors that affect survival time after radiation therapy include the severity of neurologic signs and the relative size of the tumor. Generally, dogs with subtle or mild neurologic clinical signs and the smallest tumors show the best response to treatment. Theon et al. (1998) found a mean survival time after radiation of 25 months in dogs with mild neurologic signs, 17 months in dogs with severe neurologic signs, and only 5 months in untreated dogs with neurologic signs. Because of the high prevalence of a pituitary mass at the time PDH is diagnosed and the potential for future growth and development of neurologic signs, examination of the pituitary gland using CT or MRI and radiation therapy if a mass is identified should be discussed with the client at the time PDH is diagnosed. The goal of radiation therapy is to shrink the mass and prevent development of macrotumor syndrome; mitotane or trilostane therapy may still be needed to control clinical signs of hyperadrenocorticism.
Prognosis
The average life expectancy in dogs with adrenal-dependent hyperadrenocorticism that survive the initial postadrenalectomy month is approximately 36 months. Dogs with adrenocortical adenoma and adrenocortical carcinoma that has not metastasized (uncommon) have a good prognosis, whereas dogs with metastatic adrenocortical carcinoma (common) have a poor prognosis, with these dogs typically succumbing to the disease within a year of diagnosis. Although clinical signs can be controlled with trilostane and mitotane, death ultimately results from the debilitating effects of the tumor, complications of hyperadrenocorticism (e.g., pulmonary thromboembolism), or other geriatric disorders (e.g., renal insufficiency, congestive heart failure).
The prognosis for dogs with PDH depends in part on the age and overall health of the dog and on the client’s commitment to therapy. The mean life span of affected dogs after diagnosis of PDH is approximately 30 months. Younger dogs may live considerably longer (i.e., 5 years or longer). Many dogs ultimately die or are euthanized because of complications related to hyperadrenocorticism (e.g., pituitary macrotumor syndrome) or other geriatric disorders.
ATYPICAL CUSHING’S SYNDROME IN DOGS
Dogs with atypical Cushing’s syndrome have clinical features suggestive of hyperadrenocorticism but persistently normal or equivocal endocrine test results. An imbalance of one or more of the adrenocortical steroid hormone intermediates required for synthesis of cortisol (see Fig. 53-15) is believed to be the cause. It has been hypothesized that a relative deficiency in enzymes required for cortisol synthesis (such as 21-β-hydroxylase or 11-β-hydroxylase) cause accumulation of steroid precursors proximal to the blockade in the synthetic pathway. High concentrations of one or more steroid precursors may cause clinical signs or may be shunted into alternative metabolic pathways and cause excesses in other steroid hormones, such as androstenedione. Increased serum adrenocortical steroid hormone intermediates often occur in conjunction with cortisol in dogs with PDH and cortisol-secreting ATs. In contrast, dogs with atypical Cushing’s syndrome have normal or inconclusive serum cortisol concentrations and an increase in one or more adrenocortical steroid hormone intermediates, most notably 17-hydroxyprogesterone.
Adrenal tumors that secrete progesterone and 17-hydroxyprogesterone cause a clinical syndrome that mimics hyperadrenocorticism in dogs and cats. Clinical signs presumably result from intrinsic glucocorticoid activity of progestins, progestin-induced displacement of cortisol from cortisol-binding protein in the circulation, or both. An atypical form of PDH has also been described in which clinical features mimic hyperadrenocorticism, abdominal ultrasound reveals adrenal glands that are normal or mildly increased in size, tests of the pituitary-adrenocortical axis are normal or inconclusive, pre- and post-ACTH serum 17-hydroxyprogesterone concentrations are increased, and clinical signs improve with mitotane treatment. Diagnosis requires evaluation of serum and plasma adrenocortical steroid hormone intermediates and sex hormones before and 1 hour after the IV administration of 5 μg/kg of synthetic ACTH (Cosyntropin). The most common abnormality is an increase in serum 17-hydroxyprogesterone concentration. Currently, the only laboratory with established normal values for precursor and sex steroids is the Endocrinology Laboratory at the University of Tennessee, College of Veterinary Medicine, Knoxville, TN 37901-1071. Treatment recommendations have included low dosages of mitotane (10 mg/kg/day initially) and trilostane, although Sieber-Ruckstuhl et al. (2006) failed to document a decrease in 17-hydroxyprogesterone concentrations in dogs with PDH treated with trilostane.
I do not routinely measure serum adrenocortical steroid hormone intermediates or sex hormones when initially evaluating dogs for hyperadrenocorticism. I reserve measurement of these hormones for those dogs with clinical features suggestive of hyperadrenocorticism but persistently normal or equivocal test results for hyperadrenocorticism.
HYPERADRENOCORTICISM IN CATS
Hyperadrenocorticism is uncommon in cats. Although many of the clinical characteristics of feline hyperadrenocorticism are similar to those seen in dogs, there are some important differences that should be emphasized. Most notable is the very strong association with diabetes mellitus; the progressive, relentless weight loss leading to cachexia; and dermal and epidermal atrophy leading to extremely fragile, thin, easily torn and ulcerated skin (i.e., feline fragile skin syndrome) in cats with hyperadrenocorticism. Establishing the diagnosis can be difficult, and effective medical treatment for hyperadrenocorticism in cats has yet to be identified.
Etiology
Hyperadrenocorticism in cats is classified as either pituitary dependent (PDH) or adrenocortical tumor dependent (ATH). Approximately 80% of cats with hyperadrenocorti cism have PDH and 20% have ATH, with 50% of ATHs being adenomas and 50% carcinomas. Cats with PDH have a pituitary microadenoma, macroadenoma, or carcinoma identified at necropsy. Iatrogenic hyperadrenocorticism is uncommon in cats and typically takes months of prednisone or prednisolone administration before clinical signs occur.
CLINICAL SIGNS AND PHYSICAL EXAMINATION FINDINGS
Hyperadrenocorticism is a disease of older (average age 10 years) mixed-breed cats. There is a strong correlation between hyperadrenocorticism and diabetes mellitus, and the most common initial clinical signs of feline hyperadrenocorticism (i.e., polyuria, polydipsia, polyphagia) are more likely caused by diabetes than by hyperadrenocorticism. Other clinical signs and physical examination findings are not as frequently observed in cats as in dogs and tend to be very subtle in the early stages of the disease (Box 53-5; Fig. 53-17).
BOX 53-5 Clinical Features of Hyperadrenocorticism in Cats
* Common.
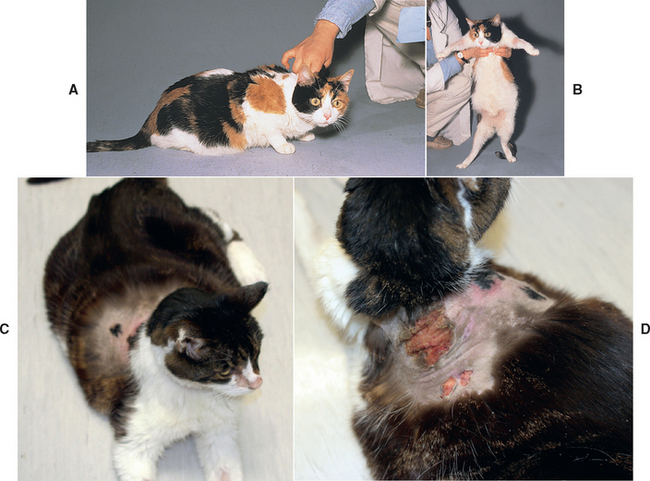
FIG 53-17 A and B, A 9-year-old cat with pituitary-dependent hyperadrenocorticism (PDH) and insulin-resistant diabetes mellitus. Note the relatively normal physical appearance of the cat in its normal posture (A). Abdominal enlargement and inguinal alopecia are evident on physical examination (B). C and D, A 16-year-old cat with PDH and insulin-resistant diabetes mellitus. Note the relatively normal appearance of the cat and the alopecia and ulceration in the dorsal cervical and anterior thoracic region in the area of a collar worn by the cat. Alopecia was also present in the ventral region of the neck.
A frequent clue to the existence of hyperadrenocorticism in cats is the presence of diabetes mellitus that is difficult to control and ultimately progresses to severe insulin resistance. Initially, clinical signs of hyperadrenocorticism are mild and tests of the pituitary-adrenocortical axis are often inconclusive and difficult to interpret in the presence of poorly controlled diabetes. With time, hyperadrenocorticism becomes more apparent as affected cats become progressively more debilitated despite aggressive insulin therapy; weight loss leads to cachexia; and dermal and epidermal atrophy result in extremely fragile, thin, easily torn, and ulcerated skin (Fig. 53-18). Dermal and epidermal lesions often occur when the cat is groomed or when the cat is handled during the physical examination. Insulin resistance is usually quite severe bythe time cachexia and skin fragility develop. The primary differential diagnosis for insulin resistance, cachexia, and feline fragile skin syndrome is excess progestins such as that which occurs with progesterone-secreting adrenal tumors (see p. 844).
FIG 53-18 A, A 15-year-old cat with pituitary-dependent hyperadrenocorticism (PDH), insulin-resistant diabetes mellitus, and feline fragile skin syndrome. Note the torn skin over the back of the neck that occurred while the cat was being restrained during a physical examination. B, A 12-year-old cat with hyperadrenocorticism and severe insulin-resistant diabetes mellitus. This cat weighed 2.2 kg and was receiving 25 units of regular insulin three times a day with no glucose-lowering effect. Note the emaciated appearance, presumably resulting from protracted poor glycemic control, alopecia, severe dermal and epidermal atrophy, and lesions resulting from easily torn skin (arrow). C, A 17-year-old cat with PDH and insulin-resistant diabetes mellitus. Note the emaciated appearance of the cat, the enlarged abdomen (pot-belly appearance), and absence of hair growth on the ventral abdomen, which had been shaved for an abdominal ultrasound 10 months before presentation.
CLINICAL PATHOLOGY
The classic clinicopathologic alterations seen in dogs with hyperadrenocorticism are infrequently found in cats. The most frequently observed abnormalities in cats are hyperglycemia, glycosuria, hypercholesterolemia, and a mild increase in alanine aminotransferase activity. These alterations can be explained by concurrent, poorly regulated diabetes mellitus. A stress leukogram, an increase in ALP activity, and isosthenuric-hyposthenuric urine are not common findings in hyperadrenal cats. An inability to document histologic changes in the liver consistent with steroid-induced hepatopathy, an absence of the steroid-induced alkaline phosphatase isoenzyme activity, and the relatively short half-life of ALP activity in cats may account for the absence of an observed increase in ALP activity. Urine abnormalities frequently identified in dogs with hyperadrenocorticism are not common in cats.
DIAGNOSTIC IMAGING
Abdominal ultrasonography is used to identify adrenal masses and to clarify the clinician’s index of suspicion for PDH. The interpretation of results of adrenal imaging in cats is similar to that in dogs (see p. 815). The maximum width of the adrenal gland in healthy cats is typically less than 0.5 cm. Adrenomegaly should be suspected when the maximum width is greater than 0.5 cm; a maximum width greater than 0.8 cm is strongly suggestive of adrenomegaly. The finding of easily visualized, bilaterally large adrenals in a cat with appropriate clinical signs, physical examination findings, and abnormal test results of the pituitary-adrenocortical axis is strong evidence for PDH. CT and MRI can be used to look for pituitary macroadenoma and to assess the size of an adrenal mass and extent of infiltration of the mass into surrounding blood vessels and organs before adrenalectomy.
TESTS OF THE PITUITARY-ADRENOCORTICAL AXIS
Although the tests used to diagnose hyperadrenocorticism in cats and dogs are similar (see p. 818), there are some important differences in the testing protocol and in the interpretation of results (Table 53-4). I rely most heavily on the UCCR, dexamethasone suppression test, and abdominal ultrasonography to establish the diagnosis of hyperadrenocorticism in cats. The ACTH stimulation test lacks sensitivity in the cat and is not recommended. I also rely on abdominal ultrasound rather than endogenous plasma ACTH concentration to differentiate PDH from ATH.
TABLE 53-4 Diagnostic Tests to Assess the Pituitary-Adrenocortical Axis in Cats with Suspected Hyperadrenocorticism
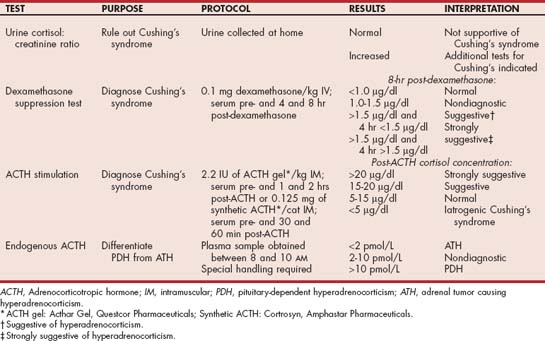
Urine Cortisol/Creatinine Ratio
The theory behind and the specifics regarding the UCCR are similar for dogs and cats and discussed on p. 819. The cat reference range for the UCCR performed on urine collected at home is less than 3.6 × 10-5 (often listed as less than 36); this value may vary among laboratories. I use the UCCR as the initial screening test for hyperadrenocorticism in cats. A normal UCCR is a strong finding against the diagnosis; an increased ratio does not establish the diagnosis by itself but supports performing the dexamethasone suppression test.
Dexamethasone Suppression Test
The duration of the suppressive effects of intravenously administered dexamethasone on serum cortisol concentrations is more variable in cats than dogs. Approximately 20% of healthy cats do not experience the suppressive effects of dexamethasone, and their serum cortisol concentrations are greater than 1.4 μg/dl 8 hours after dexamethasone administration. This “escape phenomenon” is more likely to occur in cats receiving lower doses of dexamethasone. Because of potential misinterpretation caused by the escape phenomenon and the fragile state of many diabetic hyperadrenal cats, I typically use only one dexamethasone suppression test protocol (0.1 mg/kg dexamethasone administered intravenously; blood obtained before and 4 and 8 hours after dexamethasone administration) when evaluating the pituitary-adrenocortical axis in cats. An 8-hour postdexamethasone serum cortisol concentration less than 1.0 μg/dl is suggestive of a normal pituitary-adrenocortical axis, values between 1.0 and 1.4 μg/dl are inconclusive, and values greater than 1.4 μg/dl are supportive of the diagnosis of hyperadrenocorticism. The higher the 8-hour post-dexamethsone serum cortisol concentration above 1.4 μg/dl, the more supportive the test is for the diagnosis of hyperadrenocorticism. Similarly, a serum cortisol concentration greater than 1.4 μg/dl at the 4-hour postdexamethasone blood sampling time adds further support for the diagnosis of hyperadrenocorticism (Fig. 53-19). Whenever the 4-hour post-dexamethasone cortisol value is less than 1.4 μg/dl (especially less than 1.0 μg/dl), the test results should be considered consistent with, but not definitively diagnostic of, hyperadrenocorticism and the clinician must rely on the clinical signs, physical examination findings, and results of other diagnostic tests to help establish the diagnosis. Results of the dexamethasone suppression test should never constitute the sole evidence for hyperadrenocorticism in cats.
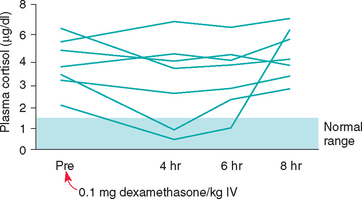
FIG 53-19 Dexamethasone suppression test results in seven cats with histologically confirmed hyperadrenocorticism. Blood for the cortisol determination was drawn before and 4, 6, and 8 hours after the intravenous administration of 0.1 mg of dexamethasone/kg body weight. In most cats the plasma cortisol concentration remained more than 1.4 μg/dl throughout the test—results that are very consistent with a diagnosis of hyperadrenocorticism.
Adrenocorticotropic Hormone Stimulation Test
The peak increase in the post-ACTH serum cortisol concentration occurs earlier in cats than in dogs, and serum cortisol concentrations can approach baseline values by 1 or 2 hours after the administration of synthetic or porcine ACTH, respectively. Whenever porcine ACTH gel is used, blood samples for cortisol determination should be obtained 1 and 2 hours after its administration. Whenever synthetic ACTH is used, blood samples should be obtained 30 minutes and 1 hour after its administration. The reference range for peak post-ACTH serum cortisol concentration is 5 to 15 μg/dl. Post-ACTH serum cortisol concentrations greater than 15 μg/dl are suggestive of hyperadrenocorticism. The sensitivity of the ACTH stimulation test in identifying hyperadrenocorticism is low in cats. Fewer than 50% of cats with hyperadrenocorticism confirmed at necropsy have abnormal ACTH stimulation test results consistent with the disease.
Endogenous Plasma Adrenocorticotropic Hormone Concentration
The endogenous plasma ACTH concentration test is discussed on p. 824. The reference range for baseline plasma ACTH concentrations in cats is 2 to 13 pmol/l. Undetectable plasma endogenous ACTH concentrations (less than 2 pmol/L) are consistent with ATH, endogenous ACTH concentrations greater than 10 pmol/l are consistent with PDH, and endogenous ACTH concentrations between 2 and 10 are nondiagnostic.
Diagnosis
Hyperadrenocorticism is diagnosed on the basis of history; findings on physical examination; results of routine blood and urine tests, abdominal ultrasonography, and tests of the pituitary-adrenocortical axis; and the clinician’s index of suspicion for the disease. Ideally, all diagnostic tests performed in the assessment of a cat with suspected hyperadrenocorticism should be abnormal. Discordant test results raise doubt regarding the diagnosis. False-positive and false-negative results occur with all of the diagnostic tests used to assess the pituitary-adrenocortical axis. Although normal UCCR and dexamethasone suppression test results are inconsistent with a diagnosis of hyperadrenocorticism, abnormal results of these tests do not by themselves confirm the diagnosis. If there is doubt or uncertainty about the diagnosis, therapy for hyperadrenocorticism should be withheld and the cat reevaluated 1 to 2 months later.
Treatment
Treatment of hyperadrenocorticism is problematic in cats, primarily because a reliable medical treatment for PDH has not been identified. Trilostane is the current treatment of choice because other treatments, such as mitotane, ketoconazole, and the enzyme inhibitor metyrapone, are ineffective or only transiently effective. The trilostane treatment and monitoring protocols are similar for dogs and cats (see p. 827). The initial dose is 1 to 2 mg/kg of body weight once daily. Adjustments in the dose and frequency of administration are based on clinical response and results of the ACTH stimulation test, UCCR, and serum electrolyte concentrations. In general, twice-daily dosing provides better control than once-daily dosing and should be the initial adjustment in cats that remain symptomatic at the starting dose given once daily. In one study by Neiger et al. (2004), three cats with PDH were still alive 6, 11, and 20 months after starting trilostane therapy at 30 mg/cat once a day.
Aminoglutethimide (Cytadren, 30 mg/cat, administered orally q12h; Ciba-Geigy Pharmaceuticals) inhibits the conversion of cholesterol to pregnenolone, thereby reducing cortisol hypersecretion. Aminoglutethimide has been used successfully in controlling clinical signs of hyperadrenocorticism and hyperprogesteronemia in cats with progesterone-secreting tumors (Fig. 53-20) and appears to maintain its efficacy for a relatively prolonged period of time (i.e., months). Cobalt irradiation may be tried in cats with pituitary macrotumor, although clinical signs of hypercortisolemia may persist despite shrinkage of the tumor.
FIG 53-20 A, A 9-year-old male castrated domestic long-haired cat with a 2-year history of poorly controlled diabetes mellitus, failure of hair to regrow after clipping 1 year before presentation, and recent development of feline fragile skin syndrome. Diagnostic evaluation revealed an adrenocortical tumor, increased serum progesterone concentration, and suppression of the pituitary-adrenocortical axis on adrenocorticotropic hormone stimulation and dexamethasone suppression testing. A progesterone-secreting adrenocortical tumor was suspected. B, Five weeks after initiating treatment with aminoglutethemide. Feline fragile skin syndrome was resolving, hair was growing, and gynecomastia had developed. The serum progesterone concentration had decreased from a pretreatment value of 4.7 ng/ml to less than 1 ng/ml. C, Four months after adrenalectomy. Insulin-requiring diabetes mellitus had resolved.
Adrenalectomy is the treatment of choice for ATH, and bilateral adrenolectomy is also an effective treatment for PDH. Medical treatment with trilostane is usually necessary for 4 to 6 weeks before adrenalectomy to reverse the catabolic state of the cat, improve skin fragility and wound healing, and decrease the potential for perioperative complications. The surgical approach and medical management during and after surgery are similar to those used in dogs (see p. 828). Treatment for hypoadrenocorticism should begin immediately after bilateral adrenalectomy and include injectable desoxycorticosterone pivalate (DOCP, 2.2 mg/kg, administered subcutaneously every 25 days initially; Percoten-V; Novartis Pharmaceuticals) or fludrocortisone acetate (Florinef, 0.05 mg/cat, administered orally q12h initially; ER Squibb & Sons) and prednisolone (1.0 to 2.5 mg once daily). Subsequent adjustments in the dose of DOCP or fludrocortisone acetate should be based on periodically measured serum electrolyte concentrations (see p. 840). Insulin therapy can be discontinued in approximately 50% of cats once hyperadrenocorticism is eliminated, and diabetes is easier to control using less insulin in the remaining cats.
Prognosis
The prognosis is guarded to poor. Untreated hyperadrenal cats die within months after the diagnosis has been established because of the deleterious effects of chronic hypercortisolism and insulin-resistant diabetes mellitus on skin integrity and on immune and cardiovascular function and as a result of progressive weight loss leading to severe cachexia. The effectiveness of trilostane remains to be determined. Unilateral (ATH) or bilateral (PDH) adrenalectomy has the potential for excellent success; however, success depends, in part, on correction of the debilitated state and skin fragility with medical treatment before surgery, involvement of a surgeon with expertise in adrenal surgery, avoidance of perioperative complications, and the client’s commitment to managing the iatrogenic adrenal insufficiency after bilateral adrenalectomy. Periodic evaluation of serum electrolytes and review of the treatment protocol are important. An addisonian crisis occurred months after surgery in several cats treated in our clinic and was believed to be responsible for the death of some.
HYPOADRENOCORTICISM
Etiology
Hypoadrenocorticism is a deficiency of mineralocorticoids, glucocorticoids, or both. Primary adrenocortical insufficiency (Addison’s disease) involving a deficiency of both mineralocorticoid and glucocorticoid secretion is the most common. The etiology of primary hypoadrenocorticism is usually classified as idiopathic because the cause of the disease is not obvious and necropsies are usually done years after the diagnosis is established, at which time idiopathic atrophy of all layers of the adrenal cortex is the most frequent histopathologic finding. Immune-mediated destruction of the adrenal cortices is believed to occur in most dogs and cats with idiopathic adrenal insufficiency; lymphocytes, plasma cells, and fibrosis are common findings in animals that undergo necropsy near the time of diagnosis. Bilateral destruction of the adrenal cortex by neoplasia (e.g., lymphoma), granulomatous disease, hemorrhagic infarction, arterial thrombosis, or drugs such as mitotane and trilostane can also cause primary adrenocortical insufficiency. For clinical signs to develop, it is believed that at least 90% of the adrenal cortices must be destroyed. The zones of the adrenal cortices are usually damaged at about the same rate, with aldosterone and glucocorticoid deficiency typically occurring in tandem. Destruction is progressive, ultimately leading to complete loss of adrenocortical function. Dogs and cats typically have complete loss of adrenocortical function at the time hypoadrenocorticism is diagnosed. A partial deficiency syndrome characterized by inadequate adrenal reserve may occur initially, with clinical signs manifested only during times of stress such as boarding, travel, and surgery. As destruction of the adrenal cortex progresses, hormone secretion becomes inadequate even under nonstressful conditions and a true metabolic crisis occurs without any obvious inciting event.
Mineralocorticoids (i.e., aldosterone) control sodium, potassium, and water homeostasis. In the setting of primary adrenocortical insufficiency, a loss of aldosterone secretion results in impaired renal conservation of sodium and chloride and the excretion of potassium, leading to the development of hyponatremia, hypochloremia, and hyperkalemia. The inability to retain sodium and chloride reduces extracellular fluid volume, leading to progressive development of hypovolemia, hypotension, a reduced cardiac output, and decreased perfusion of the kidneys and other tissues. Hyperkalemia has a deleterious effect on cardiac function, causing decreased myocardial excitability, an increased myocardial refractory period, and slowed conduction. A concurrent glucocorticoid deficiency typically results in gastrointestinal tract signs (e.g., anorexia, vomiting, diarrhea, weight loss) and changes in mental status (e.g., lethargy). One of the hallmark signs of hypocortisolism is impaired tolerance to stress, and clinical signs often become more pronounced when the animal is placed in stressful situations.
Some dogs and cats with hypoadrenocorticism present to the veterinarian with clinical signs of glucocorticoid deficiency but serum electrolyte concentrations that are within the reference range at initial presentation. A deficiency in glucocorticoid but not mineralocorticoid secretion is referred to as atypical hypoadrenocorticism and is discussed on p. 841. Glucocorticoid deficiency resulting from pituitary dysfunction is also called secondary hypoadrenocorticism. Destructive lesions in the pituitary gland or hypothalamus, the long-term administration of exogenous glucocorticoids, and idiopathic loss of function are the most common causes of secondary adrenal insufficiency. Naturally occurring, isolated hypoaldosteronism is rare in dogs and cats.
SIGNALMENT
Hypoadrenocorticism is typically a disease of young to middle-aged female dogs, with a median age of 4 to 6 years and a range of 2 months to 12 years. Dogs with glucocorti coid-deficient hypoadrenocorticism tend to be older at the time of diagnosis than dogs with mineralocorticoid and glucocorticoid deficient hypoadrenocorticism. Breeds reported to be at increased risk for hypoadrenocorticism are listed in Box 53-6. Hypoadrenocorticism is rare in cats. There is no apparent sex-related predisposition in cats, although it tends to occur in young to middle-aged cats (average age 6 years). Hypoadrenocorticism can, however, occur in aged dogs and cats as well.
CLINICAL SIGNS AND PHYSICAL EXAMINATION FINDINGS
Clinical signs and physical examination findings are listed in Box 53-7. The most common clinical manifestations are related to alterations in the gastrointestinal tract and mental status and include lethargy, anorexia, vomiting, and weight loss. Weakness is also a common client complaint. Additional physical examination findings may include dehydration, bradycardia, weak femoral pulses, and abdominal pain. Hyperkalemia and hypoadrenocorticism should be suspected in an animal with bradycardia and signs consistent with hypovolemia. Bradycardia by itself, however, is not pathognomonic for hypoadrenocorticism, especially in an otherwise healthy dog. Similarly, dogs with hypoadrenocorticism can have normal heart rates. Polyuria and polydipsia are rarely presenting signs, although they may surface during the taking of a complete history.
Clinical signs are often vague and easily ascribed to more common disorders involving the gastrointestinal and urinary tracts. Observant clients may occasionally describe an illness with a waxing-waning or episodic course; however, this bit of historic information is the exception rather than the rule. Most dogs with hypoadrenocorticism are first seen because of progressive problems that vary in severity, depending on the degree of stress and the adrenocortical reserve.
If hyponatremia and hyperkalemia become severe, the resultant hypovolemia, prerenal azotemia, and cardiac arrhythmias may result in an addisonian crisis. The clinical manifestations are as previously described; the only difference is in the severity of signs. In severe cases the animal may be presented in shock and be moribund. An addisonian crisis must be differentiated from other life-threatening disorders, such as diabetic ketoacidosis, necrotizing pancreatitis, acute hepatitis, septic peritonitis, and acute renal failure.
CLINICAL PATHOLOGY
Several abnormalities may be identified on a CBC, serum biochemistry panel, and urinalysis (Box 53-8). Hyperkalemia, hyponatremia, and hypochloremia are the classic electrolyte alterations in animals with adrenal insufficiency and are perhaps the most important evidence ultimately used to establish a diagnosis of hypoadrenocorticism. Serum sodium concentrations vary from normal to as low as 105 mEq/L (mean 128 mEq/L), and serum potassium concentrations vary from normal to greater than 10 mEq/L (mean 7.2 mEq/L). The sodium : potassium ratio reflects changes in these electrolyte concentrations in serum and has been frequently used as a diagnostic tool to identify adrenal insufficiency. The normal ratio varies between 27 : 1 and 40 : 1. Values are often less than 27 and may be less than 20 in animals with primary adrenal insufficiency.
BOX 53-8 Clinicopathologic Abnormalities Associated with Primary Hypoadrenocorticism in Dogs and Cats
Electrolyte alterations by themselves can be misleading. Normal serum electrolyte concentrations do not rule out adrenal insufficiency. Electrolyte abnormalities may not be evident in the early stages of the disorder, when clinical signs result from glucocorticoid deficiency, and do not develop with secondary adrenal insufficiency caused by pituitary failure. Alternatively, other disorders can cause alterations in serum electrolyte concentrations that mimic adrenal insufficiency, most notably disorders involving the hepatic, gastrointestinal, and urinary systems (see Boxes 55-2 and 55-3). For most disorders a thorough history and physical examination, together with a critical evaluation of results of the CBC, serum biochemistry panel, and urinalysis, allow the clinician to prioritize the potential differential diagnoses. Important clues for hypoadrenocorticism include lack of a stress leukogram in a sick dog or cat and identification of hypoalbuminemia, hypocholesterolemia, hypoglycemia, or a combination of these on the serum biochemistry panel.
The most challenging aspect of diagnosis is the differentiation between acute renal failure and primary adrenal insufficiency. The azotemia of adrenal insufficiency occurs secondary to reduced renal perfusion and an associated decrease in glomerular filtration rate after the onset of hypovolemia and hypotension. A compensatory increase in urine specific gravity to greater than 1.030 allows prerenal azotemia to be differentiated from primary renal azotemia and therefore adrenal insufficiency to be differentiated from acute renal failure, respectively.
Unfortunately, many hypoadrenal dogs and cats have an impaired ability to concentrate urine because of chronic urinary sodium loss, depletion of the renal medullary sodium content, loss of the normal medullary concentration gradient, and impaired water resorption by the renal collecting tubules. As a result, some hypoadrenal dogs and cats with prerenal azotemia have urine specific gravities in the isosthenuric range (i.e., 1.007 to 1.015). Fortunately, the initial therapy for acute renal failure is similar to that used for adrenal insufficiency. Ultimately, the differentiation between these two disorders must rely on testing of the pituitary-adrenocortical axis and the animal’s response to initial fluid and other supportive therapy.
ELECTROCARDIOGRAPHY
Hyperkalemia depresses cardiac conduction and causes characteristic alterations on an electrocardiogram (ECG; see Box 55-4). The severity of the ECG abnormalities correlates with the severity of hyperkalemia. The ECG can be used as a diagnostic tool to identify and estimate the severity of hyperkalemia and as a therapeutic tool to monitor changes in the blood potassium concentration during therapy.
DIAGNOSTIC IMAGING
Hypoadrenal dogs and cats with severe hypovolemia often have microcardia, a descending aortic arch that is flattened and has a decreased diameter, and a narrow caudal vena cava, as seen on lateral thoracic radiographs. These findings are a crude means of evaluating the degree of hypovolemia and hypotension. Concurrent generalized megaesophagus may be evident and may resolve in response to treatment for the hypoadrenocorticism. Abdominal ultrasonography may reveal small adrenal glands (i.e., maximum width less than 0.3 cm), a finding suggestive of adrenocortical atrophy. However, finding normal-size adrenal glands does not rule out hypoadrenocorticism.
Diagnosis
Hypoadrenocorticism is often tentatively diagnosed on the basis of the history; physical examination findings; clinicopathologic findings; and, in the case of primary adrenal insufficiency, identification of appropriate electrolyte abnormalities. Results of an ACTH stimulation test confirm the diagnosis (see Table 53-2). Baseline serum cortisol concentrations of greater than 2 μg/dl are inconsistent with the diagnosis of hypoadrenocorticism, but baseline serum cortisol concentrations of 2 μg/dl or less do not confirm the diagnosis. UCCRs are not reliable for confirming the diagnosis. One major criterion is used in confirming the diagnosis of adrenal insufficiency: a post-ACTH serum cortisol concentration less than 2 μg/dl (Fig. 53-14). A post-ACTH serum cortisol concentration of 4 μg/dl or greater is inconsistent with the diagnosis of adrenal insufficiency. Post-ACTH serum cortisol values between 2 and 4 μg/dl are equivocal and may occur with relative adrenal insufficiency—a syndrome defined as inadequate production of cortisol in relation to increased demand during periods of critical illness such as sepsis. Prolonged or excessive inflammatory cytokine activity suppresses pituitary and adrenal function in humans and possibly in dogs as well. In a recent study by Burkitt et al. (2007), dogs with severe sepsis had a suppressed response of the adrenal cortex to exogenously administered ACTH, an increase in serum cortisol concentration of less than 3 μg/dl after ACTH administration, and resolution of the relative adrenal insufficiency after resolution of the illness.
Results of the ACTH stimulation test do not distinguish dogs and cats with naturally occurring primary adrenal insufficiency from those with secondary insufficiency resulting from pituitary failure, dogs and cats with secondary insufficiency resulting from prolonged iatrogenic glucocorticoid administration, or dogs with primary adrenocortical destruction caused by mitotane or trilostane overdosing. Concurrent abnormal serum electrolyte concentrations imply the existence of primary adrenal insufficiency and the need for mineralocorticoid and glucocorticoid replacement therapy. Normal serum electrolyte concentrations do not differentiate between primary hypoadrenocorticism that progresses and primary hypoadrenocorticism that does not progress to mineralocorticoid deficiency or between primary hypoadrenocorticism and secondary hypoadrenocorticism (see the section on atypical hypoadrenocorticism). If secondary hypoadrenocorticism can be documented, only glucocorticoid replacement therapy is indicated. Primary and atypical or secondary hypoadrenocorticism can be differentiated prospectively by periodically measuring serum electrolyte concentrations, by measuring baseline endogenous ACTH concentration, or by measuring plasma aldosterone concentrations during the ACTH stimulation test (Table 53-5). In theory, measurement of plasma aldosterone concentration should be helpful in distinguishing between the various forms of adrenal insufficiency. Unfortunately, there is no clear demarcation in plasma aldosterone concentrations between these groups of dogs.
 TABLE 53-5
TABLE 53-5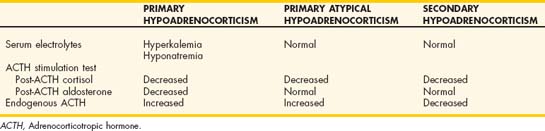
Treatment
The aggressiveness of therapy depends on the clinical status of the animal and the nature of the insufficiency (i.e., glucocorticoid or mineralocorticoid or both). Many dogs and cats with primary adrenal insufficiency are presented in varying stages of an acute addisonian crisis, requiring immediate, aggressive therapy. In contrast, dogs and cats with isolated glucocorticoid deficiency often have a chronic course that poses more of a diagnostic than a therapeutic challenge.
THERAPY FOR ACUTE ADDISONIAN CRISIS
An acute addisonian crisis involves both a mineralocorticoid and a glucocorticoid deficiency. The treatment of acute primary adrenal insufficiency is directed toward correcting hypotension, hypovolemia, electrolyte imbalances, and metabolic acidosis; improving vascular integrity; and providing an immediate source of glucocorticoids (Box 53-9). Because death resulting from hypoadrenocorticism is often attributed to vascular collapse and shock, rapid correction of hypovolemia is the first and most important therapeutic priority. Physiologic saline solution is the IV fluid of choice because it aids in correcting hypovolemia, hyponatremia, and hypochloremia. Hyperkalemia is reduced by simple dilution and by improved renal perfusion. Potassiumcontaining fluids (see Table 55-2) are relatively contraindicated but should be used in lieu of not giving IV fluids at all.
 BOX 53-9 Initial Treatment for Acute Addisonian Crisis
BOX 53-9 Initial Treatment for Acute Addisonian Crisis
IV, Intravenous; IM, intramuscular.
Fluid Therapy
Rate: 40 to 80 ml/kg/h IV initially
Potassium supplementation: contraindicated
Dextrose: 5% dextrose infusion (100 ml of 50% dextrose per liter of fluids)
Glucocorticoid Therapy
Dexamethasone sodium phosphate, 0.5 to 1.0 mg/kg IV, repeat q12h at dosage of 0.05 to 0.1 mg/kg in IV fluids until oral prednisone can be administered.†
Alternatively, hydrocortisone hemisuccinate or hydrocortisone phosphate,* 2 to 4 mg/kg IV or prednisolone sodium succinate,* 4 to 20 mg/kg IV, then dexamethasone sodium phosphate, 0.05 to 0.1 mg/kg in IV fluids q12h.
Mineralocorticoid Therapy
Desoxycorticosterone pivalate (DOCP; Novartis), 2.2 mg/kg IM q25 days initially.
Bicarbonate Therapy
Indicated if HCO3 <12 mEq/L or total venous CO2 <12 mmol/L or animal is severely ill. mEq HCO3 = body weight (kg) × 0.5 × base deficit (mEq/L); if base deficit unknown, use 10 mEq/L. Add one quarter of calculated HCO3 dose to IV fluids and administer over 6 hours. Repeat only if plasma HCO3 remains <12 mEq/L.
If hypoglycemia is suspected or known to be present, 50% dextrose should be added to the IV fluids to produce a 5% dextrose solution (i.e., 100 ml of 50% dextrose per liter of fluids). The addition of dextrose to isotonic solutions produces a hypertonic solution that ideally should be administered through a central vein to minimize phlebitis.
Dogs and cats with acute adrenal insufficiency usually have a mild metabolic acidosis that does not require therapy. Fluid therapy alone corrects the mild acidosis as hypovolemia lessens and tissue perfusion and glomerular filtration rate improve. If the total venous carbon dioxide or the serum bicarbonate concentration is less than 12 mmol/L or 12 mEq/L, respectively, conservative bicarbonate therapy is indicated. In a severely ill animal in which laboratory results are not yet known, a base deficit of 10 mEq/L can be assumed to be present. The milliequivalents of bicarbonate needed to correct the acidosis can be determined from the following equation:
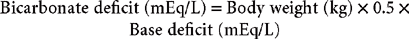
One fourth of the calculated bicarbonate dose should be administered in the IV fluids during the initial 6 to 8 hours of therapy. The acid-base status of the animal should be reassessed at the end of this time. Rarely, a dog or cat may require additional parenterally administered sodium bicarbonate.
Sodium bicarbonate therapy helps correct the metabolic acidosis and also decreases the serum potassium concentration. The intracellular movement of potassium ions after bicarbonate administration, in conjunction with the dilutional effects of saline fluid therapy and improved renal perfusion, is quite effective in lowering the serum potassium concentration and returning any ECG abnormalities toward normal. Additional therapy to rapidly correct life-threatening hyperkalemia is rarely needed (see Box 55-3).
Glucocorticoid and mineralocorticoid therapy is also indicated in the initial management of an acute addisonian crisis. Ideally, glucocorticoids should not be given until after completion of the ACTH stimulation test. IV infusion of saline is usually sufficient therapy during the initial 1 or 2 hours while the ACTH stimulation test is being completed. Dexamethasone does not interfere with the cortisol assay and can be used if glucocorticoid therapy cannot be delayed. Our glucocorticoid of choice for treating an acute addisonian crisis is dexamethasone sodium phosphate, given intravenously at an initial dosage of 0.5 to 1.0 mg/kg and repeated q12h at a dose of 0.05 to 0.1 mg/kg in the IV solution until oral medication can be safely given. Rapid-acting, water-soluble glucocorticoids such as hydrocortisone sodium succinate, hydrocortisone hemisuccinate, hydrocortisone phosphate, and prednisolone sodium succinate may be measured by the cortisol assay, causing falsely increased cortisol results, and should not be administered until after the ACTH stimulation test is completed. We do not routinely use these glucocorticoids for treating acute adrenal insufficiency.
Currently available mineralocorticoid supplements include DOCP (Percorten-V; Novartis Pharmaceuticals) and fludrocortisone acetate (Florinef; ER Squibb & Sons). Both are intended for the long-term maintenance therapy of primary adrenal insufficiency. Injectable DOCP is the preferred mineralocorticoid for the treatment of a sick dog or cat suspected of having adrenal insufficiency. The drug is initially administered at a dose of 2.2 mg/kg intramuscularly or subcutaneously every 25 days initially. In an animal in an emergency hypoadrenal crisis, the drug should be administered intramuscularly. The IV administration of saline solution and the intramuscular administration of DOCP correct electrolyte abnormalities in most hypoadrenal animals within 24 hours. There are no adverse reactions to a single injection of DOCP administered to dogs subsequently shown to have normal adrenocortical function. Atrial natriuretic peptide provides natural protection against hypernatremia. Fludrocortisone acetate is also an effective treatment. However, it is available only in tablet form, and most dogs and cats are too ill to receive oral therapy initially.
Most dogs and cats with acute adrenal insufficiency show dramatic clinical and biochemical improvement within 24 to 48 hours. Over the ensuing 2 to 4 days the animal should be gradually switched from IV fluids to oral water and food. Maintenance mineralocorticoid and glucocorticoid therapy should be initiated. If the animal fails to make this transition smoothly, persistent electrolyte imbalance, insufficient glucocorticoid supplementation, a concurrent endocrinopathy (e.g., hypothyroidism), or concurrent illness (most notably renal damage or pancreatitis resulting from poor perfusion and hypoxia caused by adrenal insufficiency) should be suspected.
MAINTENANCE THERAPY FOR PRIMARY ADRENAL INSUFFICIENCY
Mineralocorticoids and usually glucocorticoids are required for maintenance of the dog or cat with primary adrenal insufficiency. The preferred mineralocorticoid supplementation is injectable DOCP, which slowly releases the hormone at a rate of 1 mg/day/25 mg suspension. The initial dosage is 2.2 mg/kg body weight, given intramuscularly or subcutaneously every 25 days. Subsequent adjustments are based on results of serum electrolyte concentrations, which are initially measured 12 and 25 days after each of the first two or three DOCP injections. If the dog or cat has hyponatremia or hyperkalemia (or both), on day 12 the next dose should be increased by approximately 10%. If the day 12 electrolyte profile is normal but the day 25 profile is abnormal, the interval between injections should be decreased by 48 hours. DOCP is very effective in normalizing serum electrolyte concentrations. The only adverse reaction is polyuria and polydipsia that improve after reduction of the DOCP dose. Most dogs (and presumably cats) receiving DOCP also require a low dose of glucocorticoids (prednisone, 0.25 mg/kg q12h initially).
Drawbacks to DOCP are problems with availability and the inconvenience and expense associated with the need to make monthly visits to the veterinarian for the injection. To minimize the inconvenience and expense, the client is routinely taught to give the injection subcutaneously at home. Every third or fourth treatment, the client should bring the dog or cat into the clinic for a complete physical examination, measurement of serum electrolyte concentrations, and administration of DOCP to ensure that problems with the administration of DOCP have not developed. Once the dog or cat is healthy and serum electrolyte concentrations are stable, the amount of DOCP administered can be decreased by 10% increments initially and the frequency of DOCP administration can be shortened to every 21 days to allow lower doses of DOCP to be administered (typically about 1.5 mg/kg/injection), thereby decreasing the expense of treatment. The goal is to identify the lowest dosage of DOCP that maintains the health of the dog or cat and keeps serum electrolyte concentrations in the reference range.
Fludrocortisone acetate (Florinef) is another commonly used mineralocorticoid supplement. The initial dose is 0.02 mg/kg/day, divided into two doses, and administered orally. Subsequent adjustments in the dose are based on serum electrolyte concentrations, which are initially assessed every 1 to 2 weeks. The goal is to reestablish normal serum sodium and potassium concentrations. The dose of fludrocortisone acetate must typically be increased during the first 6 to 18 months of therapy. This increasing need may reflect the continuing destruction of the adrenal cortices. After this time the dose usually plateaus and remains relatively stable.
The major drawbacks to oral therapy with fludrocortisone acetate are the wide range in the doses required to control serum electrolyte concentrations; the development of polyuria, polydipsia, and incontinence in some dogs (presumably caused by the potent glucocorticoid activity of this drug); resistance to the effects of the drug, which has been observed in some animals; and persistent mild hyperkalemia and hyponatremia in some animals. Ineffectiveness of fludrocortisone acetate should be suspected when clients report that their pet is “just not right” and hyponatremia and hyperkalemia persist despite high dosages of the mineralocorticoid supplement. The concurrent administration of hydrocortisone hemisuccinate or oral salt may help alleviate the electrolyte derangements in dogs and cats in which fludrocortisone acetate by itself is not completely effective. Alternatively, switching to DOCP should be considered.
Glucocorticoid supplementation is initially indicated for all dogs and cats with primary adrenal insufficiency. Prednisone (dogs) and prednisolone (cats) is given at an initial dose of 0.25 mg/kg twice a day orally. Over the ensuing 1 to 2 months the dose of prednisone or prednisolone should gradually be reduced to the lowest amount given once a day that still prevents signs of hypocortisolism. Approximately 50% and fewer than 10% of dogs receiving fludrocortisone and DOCP, respectively, ultimately do not require glucocorticoid medication, except during times of stress. All clients should have glucocorticoids available to administer to their dogs and cats in times of stress. Veterinarians should also be aware of the increased glucocorticoid requirements of hypoadrenal dogs and cats undergoing surgery or during times of illness with a nonadrenal-related disease. The glucocorticoid dose being administered should be doubled on days when increased stress is anticipated.
The most common reason for persistence of clinical signs despite appropriate treatment is inadequate glucocorticoid supplementation. When healthy and in a nonstressed environment, dogs and cats with adrenal insufficiency typically require small amounts of prednisone or prednisolone, if any. However, when stressed or ill, these same animals may require large amounts of prednisone or prednisolone (i.e., 0.25 to 0.5 mg/kg) given twice a day. Failure to provide adequate amounts of glucocorticoids can lead to persistent and worsening lethargy, inappetence, and vomiting. The amount of prednisone or prednisolone required to offset the deleterious effects of stress and illness is variable and unpredictable. As such, it is always better to err on the high end of the dosage range and then gradually decrease the dosage over the ensuing weeks.
Prognosis
The prognosis in dogs and cats with adrenal insufficiency is usually excellent. The most important factors in determining an animal’s long-term response to therapy are client education about the disease and client dedication to treatment. If there is good client–veterinarian communication, if frequent rechecks are performed, and if clients are conscientious about carrying out therapy, dogs and cats with adrenal insufficiency can have a normal life expectancy.
ATYPICAL HYPOADRENOCORTICISM
Some dogs and cats with hypoadrenocorticism present to the veterinarian with clinical signs of glucocorticoid deficiency but with serum electrolyte concentrations that are within the reference range at initial presentation. A deficiency in glucocorticoid but not mineralocorticoid secretion is referred to as atypical hypoadrenocorticism. Glucocorticoid deficiency may be adrenocortical in origin (primary atypical hypoadrenocorticism; most common) or may result from impaired secretion of ACTH by the pituitary gland (secondary hypoadrenocorticism). Baseline endogenous plasma ACTH concentrations are normal or increased when the primary problem is adrenal in origin and decreased when the primary problem is pituitary in origin (Table 53-5). Glucocorticoid but not mineralocorticoid deficiency of adrenal origin may represent a dog or cat in the early stages of development of typical primary hypoadrenocorticism with destruction of the zona fasciculata more advanced than destruction of the zona glomerulosa. Mineralocorticoid deficiency and abnormal serum electrolyte concentrations develop weeks to months later. In some dogs and cats glucocorticoid deficiency does not progress to mineralocorticoid deficiency. The etiology for this form of hypoadrenocorticism is not known, although drugs such as megesterol acetate, mitotane, and trilostane are recognized causes.
Glucocorticoid deficiency resulting from pituitary dysfunction is called secondary hypoadrenocorticism. Destructive lesions (e.g., neoplasia, inflammation) in the pituitary gland or hypothalamus and the long-term administration of exogenous glucocorticoids are the most common recognized causes of secondary adrenal insufficiency. Adrenocortical atrophy may develop after the injectable, oral, or topical administration of glucocorticoids. Adrenal function usually returns within 2 to 4 weeks after the medication is discontinued, unless long-acting depot forms of glucocorticoids are used.
Glucocorticoid-deficient hypoadrenocorticism is usually identified during the diagnostic evaluation of dogs and cats with chronic, vague gastrointestinal clinical signs such as lethargy, anorexia, vomiting, diarrhea, and weight loss. Results of routine blood and urine tests are typically normal. Diagnosis requires an ACTH stimulation test (see p. 822). Therapy involves the administration of glucocorticoids, as previously described for the treatment of primary hypoadrenocorticism. The exception is secondary adrenal insufficiency induced by the overzealous administration of glucocorticoids, in which case therapy revolves around a gradual reduction in the dose and frequency of administration, with eventual discontinuation of the medication. Dogs and cats with secondary adrenal insufficiency should not have mineralocorticoid deficiency. The periodic measurement of serum electrolytes is advisable because primary glucocorticoid-deficient adrenal insufficiency and dogs and cats believed to have secondary adrenal insufficiency may progress to mineralocorticoid deficiency months after glucocorticoid-deficient hypoadrenocorticism is diagnosed.
PHEOCHROMOCYTOMA
Etiology
Pheochromocytoma is a catecholamine-producing tumor derived from the chromaffin cells of the adrenal medulla. Pheochromocytomas are uncommon in dogs and rare in cats. Pheochromocytomas are usually solitary, slow-growing tumors ranging in size from nodules of less than 0.5 cm in diameter to masses greater than 10 cm in diameter. Pheochromocytoma involving both adrenal glands has been reported. Pheochromocytoma should be considered a malignant tumor in dogs and cats. Pheochromocytomas commonly invade into the lumen of the adjacent phrenicoabdominal vein and caudal vena cava, entrap and compress the caudal vena cava and phrenicoabdominal vein, or both (see Fig. 53-8). Mural invasion or luminal narrowing of the aorta, renal vessels, adrenal vessels, and hepatic veins and infiltration into the adjacent kidney and body wall may also occur. Distant sites of metastasis include the liver, lung, regional lymph nodes, bone, and CNS. Paragangliomas are tumors arising from chromaffin cells located outside of the adrenal medulla, most commonly near the sympathetic ganglia, and are rare in dogs and cats.
Clinical Features
Pheochromocytomas occur most commonly in older dogs and cats, with a median age of 11 years at the time of diagnosis in dogs. There is no apparent sex- or breed-related predisposition.
Clinical signs and physical examination findings develop as a result of the space-occupying nature of the tumor and its metastatic lesions or as a result of excessive secretion of catecholamines (Table 53-6). The most common clinical signs are generalized weakness and episodic collapse. The most common abnormalities identified during physical examination involve the respiratory, cardiovascular, and musculoskeletal systems and include excessive panting, tachypnea, tachycardia, weakness, and muscle wasting. Excess catecholamine secretion may also cause severe systemic hypertension, resulting in nasal and retinal hemorrhage and retinal detachment. Because catecholamine secretion is sporadic and unpredictable, clinical manifestations and systemic hypertension tend to be paroxysmal and are usually not evident at the time the dog is examined. Because clinical signs and physical examination findings are often vague, nonspecific, and easily associated with other disorders, pheochromocytoma is often not considered a possible differential diagnosis until an adrenal mass is identified with abdominal ultrasound.
TABLE 53-6 Clinical Signs and Physical Examination Findings Associated with Pheochromocytoma in Dogs
| CLINICAL SIGNS | PHYSICAL EXAMINATION FINDINGS |
|---|---|
Diagnosis
Pheochromocytoma should be on the list of differential diagnoses for dogs presenting with clinical signs suggestive of catecholamine excess, dogs with an unexpected adrenal mass identified by abdominal ultrasound, and dogs that develop unexpected problems with systemic hypertension or cardiac arrythmias during anesthesia. Pheochromocytoma may also be an unexpected or incidental finding at necropsy or may cause sudden collapse and death from a sudden, massive, and sustained release of catecholamines by the tumor.
There are no consistent abnormalities identified in the CBC, serum biochemistry panel, or urinalysis that would raise suspicion for pheochromocytoma. A history of acute or episodic collapse, the identification of appropriate respiratory and cardiac abnormalities during physical examination, the documentation of systemic hypertension, and identification of an adrenal mass by abdominal ultrasonography are most helpful in establishing a tentative diagnosis of pheochromocytoma. Systemic hypertension may be sustained or episodic. Failure to document systemic hypertension in a dog with appropriate clinical signs does not rule out a diagnosis of pheochromocytoma.
The ultrasound identification of an adrenal mass with a normal-size contralateral adrenal gland is perhaps the most important clue for pheochromocytoma. Pheochromocytoma is one of several differentials for an adrenal mass (Table 53-7; see also the discussion of incidental adrenal mass). The primary differential diagnosis is adrenal-dependent hyperadrenocorticism. Interestingly, many of the clinical signs (e.g., panting, weakness) and blood pressure alterations seen in dogs with hyperadrenocorticism (common) are similar to those seen in dogs with pheochromocytoma (uncommon). In addition, pheochromocytoma and adrenocortical carcinoma both invade adjacent structures and cause tumor thrombi in the phrenicoabdominal vein and caudal vena cava. Kyles et al. (2003) found 6 of 11 dogs with pheochromocytoma and 6 of 28 dogs with an adrenocortical tumor had tumor thrombi. It is important to rule out adrenal-dependent hyperadrenocorticism before focusing on pheochromocytoma in a dog with an adrenal mass.
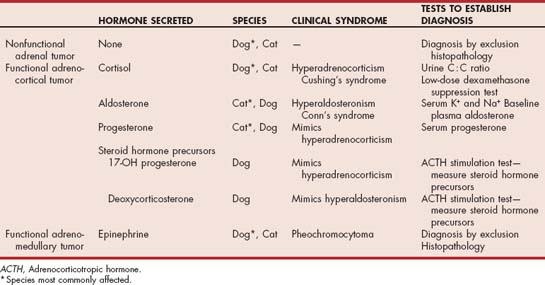
Measurement of urinary catecholamine concentrations or their metabolites can strengthen the tentative diagnosis of a pheochromocytoma. Unfortunately, these tests are not commonly performed in dogs and cats. As a result, the antemortem definitive diagnosis of a pheochromocytoma ulti mately relies on histologic evaluation of the surgically excised adrenal mass.
Treatment
A period of medical therapy to reverse the effects of excessive adrenergic stimulation, followed by surgical removal of the tumor, is the treatment of choice for pheochromocytoma. The success of chemotherapy and radiation therapy in humans with pheochromocytoma has been limited, and results of chemotherapy or radiation therapy for the treatment of pheochromocytoma in dogs or cats has not been reported. Mitotane is ineffective for treating tumors arising from the adrenal medulla. Long-term medical therapy is primarily designed to control excessive catecholamine secretion.
Potentially life-threatening complications are common during the perioperative period, especially during induction of anesthesia and manipulation of the tumor during surgery. The most worrisome complications include episodes of acute, severe hypertension (systolic arterial blood pressure of more than 300 mm Hg), episodes of severe tachycardia (heart rate of more than 250 beats/min) and arrhythmias, and hemorrhage. Preoperative α-adrenergic blockade is indicated to prevent severe clinical manifestations of hypertension in the preoperative period, to reverse the hypovolemia that is frequently present, and to promote a smooth anesthetic induction. Phenoxybenzamine is the drug of choice for α-adrenergic blockade. Our current protocol for the management of hypertension in dogs with pheochromocytoma includes preoperative phenoxybenzamine and intraoperative phentolamine. Our initial dosage of phenoxybenzamine is 0.5 mg/kg q12h. Unfortunately, many dogs with pheochromocytoma have episodic clinical signs and hypertension, making it difficult to adjust dosage on the basis of improvement in clinical signs and blood pressure. In addition, this dosage is often ineffective in preventing severe hypertension during surgery. Therefore we gradually increase the phenoxybenzamine dosage every few days until clinical signs of hypotension (e.g., lethargy, weakness, syncope), adverse drug reactions (e.g., vomiting), or a maximum dosage of 2.5 mg/kg q12h is attained. Surgery is recommended 1 to 2 weeks later. The drug should be continued until the time of surgery. If severe persistent tachycardia is identified, β-adrenergic antagonist therapy (e.g., propranolol: 0.2 to 1.0 mg/kg, per os, q8h; atenolol: 0.2 to 1.0 mg/kg, PO, q24h to q12h) should be used during the preoperative period but only after α-adrenergic blockade has been initiated. Complications may still occur despite prior treatment with α-adrenergic blocking drugs; close monitoring of the dog during the perioperative period is critical for a successful outcome after adrenalectomy. (See Suggested Readings for more information on the perioperative and surgical management of dogs with a pheochromocytoma.)
Long-term medical management is designed to control excessive catecholamine secretion. The α-adrenergic blocking drug phenoxybenzamine (0.50 mg/kg, administered orally q12h initially) is used to prevent severe clinical manifestations of hypertension. Propranolol or atenolol may also be necessary to control tachycardia and cardiac arrhythmias. However, propranolol and atenolol should be given only after α-adrenergic blockade has been initiated because severe hypertension may develop after blockade of β-receptor–mediated vasodilation in skeletal muscle.
Prognosis
The prognosis depends in part on the size of the adrenal mass, presence of metastasis or local invasion of the tumor into adjacent blood vessels or organs (e.g., kidney), avoidance of perioperative complications if adrenalectomy is performed (i.e., hypertension, cardiac arrhythmias, respiratory distress, and hemorrhage), and the presence and nature of concurrent disease. Surgically excisable tumors carry a guarded to good prognosis. Survival time in our dogs that underwent adrenalectomy and survived the immediate postoperative period ranged from 2 months to longer than 3 years. If metastatic disease is not present, perioperative complications are prevented, and serious concurrent disease not present, the dog has the potential to live a significant length of time (i.e., more than a year). Pretreatment with an α-adrenergic blocking drug before surgery and the involvement of an experienced anesthesiologist and surgeon with expertise in adrenal surgery help minimize potentially serious perioperative complications associated with anesthesia and digital manipulation of the tumor. Medically treated dogs can live longer than 1 year from the time of diagnosis if the tumor is relatively small (less than 3 cm diameter), vascular invasion is not present, and treatment with an α-adrenergic blocking drug is effective in minimizing the deleterious effects of episodic excessive catecholamine secretion by the tumor. Most dogs die or are euthanized because of complications caused by excessive catecholamine secretion, complications caused by tumor-induced venous thrombosis, or complications caused by invasion of the tumor or its metastases into surrounding organs.
INCIDENTAL ADRENAL MASS
Ultrasonography has become a routine diagnostic tool for the evaluation of soft tissue structures in the abdominal cavity. One consequence of abdominal ultrasonography is the unexpected finding of a seemingly incidental adrenal mass. Many factors determine the aggressiveness of the diagnostic and therapeutic approach to an adrenal mass, including the severity of concurrent problems, the original reason for performing abdominal ultrasound, the age of the dog or cat, the likelihood that the mass is hormonally active, the likelihood that the mass is a malignant or benign tumor, the size and invasiveness of the mass, and the client’s desires and willingness to pursue the problem. The first consideration is to be certain that an adrenal mass exists. Abdominal ultrasound should always be repeated to confirm that the mass is a repeatable finding. An adrenal mass is suspected when the maximum width of the adrenal gland exceeds 1.5 cm, there is loss of the typical kidney-bean shape of the gland, and there is asymmetry in shape and size between the affected adrenal gland and the contralateral adrenal gland. Bulbous enlargement of the cranial or caudal pole of the adrenal gland is common in dogs with normal adrenal glands and can be misinterpreted as an adrenal mass.
An adrenal mass is not always neoplastic or producing and secreting a hormone. The mass may be normal tissue, granuloma, cyst, hemorrhage, or an inflammatory nodule. Adrenalectomy is the treatment of choice if the mass is malignant and has not spread, but adrenalectomy may not be indicated if the mass is benign, small, hormonally inactive, and not invading surrounding structures. Unfortunately, it is not easy to determine whether an adrenal mass is neoplastic and malignant or benign before surgical removal and histopathologic evaluation. Guidelines to suggest malignancy include size of the mass, invasion of the mass into surrounding organs and blood vessels, and identification of additional mass lesions with abdominal ultrasound and thoracic radiographs. The bigger the mass, the more likely it is to be malignant and the more likely metastasis has occurred, regardless of findings on abdominal ultrasound and thoracic radiographs. Cytologic evaluation of specimens obtained by ultrasound-guided fine-needle aspiration of the adrenal mass may provide guidance regarding malignancy and origin of the mass (i.e., adrenal cortex versus medulla).
An adrenal tumor may secrete a hormone or be nonfunctional. Excess secretion of cortisol, catecholamines, aldosterone, progesterone, and steroid hormone precursors have been documented in dogs and cats (see Table 53-7). The most common functional adrenal tumors secrete cortisol or catecholamines. Aldosterone-secreting adrenal tumors causing primary hyperaldosteronism (Conn’s syndrome) are uncommon in dogs and cats. Excessive secretion of aldosterone causes sodium retention and potassium depletion, which is manifested as increased serum sodium (greater than 155 mEq/L) and decreased serum potassium (less than 3.0 mEq/L) concentrations. Hypokalemia causes lethargy and weakness, which are the most common clinical signs of primary hyperaldosteronism. Hypernatremia causes systemic hypertension. An adrenal mass should be identified on abdominal ultrasound, and the contralateral adrenal gland should be normal in size and shape. Documenting an increased baseline plasma aldosterone concentration is used to confirm the diagnosis.
Adrenal tumors secreting progesterone, 17-hydroxyprogesterone (see the section on atypical Cushing’s syndrome, p. 830), and other adrenocortical steroid precursors have also been documented in dogs and cats. Progesterone-secreting adrenal tumors are identified most commonly in cats. Excessive progesterone secretion in affected cats caused diabetes mellitus and feline fragile skin syndrome, characterized by progressively worsening dermal and epidermal atrophy, patchy endocrine alopecia, and easily torn skin (see Fig. 53-20). Clinical features mimicked feline hyperadrenocorticism, which is the primary differential diagnosis. Results of tests of the pituitary-adrenocortical axis are normal to suppressed in cats with progesterone-secreting adrenal tumors, and the contralateral adrenal gland is normal in size and shape on abdominal ultrasound. Diagnosis requires documenting an increased plasma progesterone concentration.
After discovering an incidental mass, the clinician should review the history, physical examination, and results of routine blood and urine tests for evidence of hyperadrenocorticism, hyperaldosteronism, or pheochromocytoma and should perform the appropriate tests to confirm the diagnosis. Whenever surgical removal of an adrenal mass is planned, a UCCR and an LDDS test should be evaluated and the perioperative management adjusted accordingly if test results are consistent with hyperadrenocorticism. If hormonal tests for hyperadrenocorticism are normal and clinical signs suggestive of pheochromocytoma are present, the clinician should assume that the adrenal mass is a pheochromocytoma and treat with an α-adrenergic antagonist before adrenalectomy (see p. 843). If the diagnostic evaluation does not support hyperadrenocorticism or pheochromocytoma, the anesthesiologist should be pre-pared to manage intraoperative blood pressure and cardiac rhythm disturbances should the mass turn out to be a pheochromocytoma.
An aggressive diagnostic and therapeutic approach is often not warranted for a small adrenal mass (less than 2 cm in maximum width), especially if the dog or cat is healthy and there are no clinical signs related to adrenal dysfunction. In these cases, it may be preferable to determine the rate of growth of the mass by repeating abdominal ultrasound initially at 2, 4, and 6 months. If the adrenal mass has not changed in size during this time, the clinician can increase the time interval between ultrasound evaluations to every 4 to 6 months (Fig. 53-21). However, if the adrenal mass is increasing in size and/or clinical signs develop, the clinician should consider adrenalectomy.
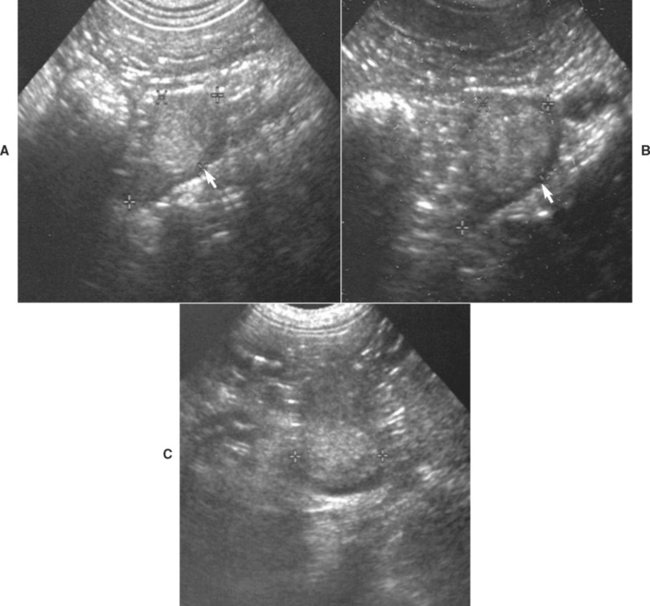
FIG 53-21 A, An 11-year-old male castrated Doberman Pinscher mix presented for clinical signs consistent with acute gastroenteritis. Abdominal ultrasound identified a 1.4-cm diameter adrenal mass (arrow) and a normal-size contralateral adrenal gland. The history, physical examination, and results of routine blood and urine tests were not supportive of adrenal disease, and the dog responded to symptomatic therapy for acute gastroenteritis. The adrenal mass was periodically evaluated with ultrasound. Over the ensuing 2 years the dog remained healthy and there was minimal growth or change in the echogenicity of the adrenal mass. B, The adrenal mass 1 year after presentation; maximum diameter was 1.8 cm. C, The adrenal mass 2 years after presentation; maximum diameter was 2.0 cm.
Feldman EC, Nelson RW. Canine and feline endocrinology and reproduction, ed 3. St Louis: WB Saunders, 2004.
Fossum TW. Small animal surgery, ed 3. St Louis: Mosby, 2007.
Slatter D. Textbook of small animal surgery, ed 3. Philadelphia: WB Saunders, 2003.
Barker EN, et al. A comparison of the survival times of dogs treated with mitotane or trilostane for pituitary-dependent hyperadrenocorticism. J Vet Intern Med. 2005;19:810.
Behrend EN, et al. Intramuscular administration of a low dose of ACTH for ACTH stimulation testing in dogs. J Am Vet Med Assoc. 2005;229:528.
Bell R, et al. Study of the effects of once daily doses of trilostane on cortisol concentrations and responsiveness to adrenocorticotrophic hormone in hyperadrenocorticoid dogs. Vet Rec. 2006;159:277.
Hoerauf A, et al. Ultrasonographic characteristics of both adrenal glands in 15 dogs with functional adrenocortical tumors. J Am Anim Hosp Assoc. 1999;35:193.
Huang H, et al. Iatrogenic hyperadrenocorticism in 28 dogs. J Am Anim Hosp Assoc. 1999;35:200.
Kintzer PP, et al. Treatment and long-term follow-up of 205 dogs with hyperadrenocorticism. J Vet Intern Med. 1997;11:43.
Meij BP, et al. Results of transsphenoidal hypophysectomy in 52 dogs with pituitary-dependent hyperadrenocorticism. Vet Surg. 1998;27:246.
Neiger R, et al. Trilostane treatment of 78 dogs with pituitary-dependent hyperadrenocorticism. Vet Rec. 2002;150:799.
Reusch CE, et al. The efficacy of l-Deprenyl in dogs with pituitary-dependent hyperadrenocorticism. J Vet Intern Med. 1999;13:291.
Ruckstuhl NS, et al. Results of clinical examinations, laboratory tests, and ultrasonography in dog’s with pituitary-dependent hyperadrenocorticism treated with trilostane. Am J Vet Res. 2002;63:506.
Theon AP, et al. Megavoltage irradiation of pituitary macrotumors in dogs with neurologic signs. J Am Vet Med Assoc. 1998;213:225.
Vaessen MMAR, et al. Urinary corticoid : creatinine ratios in healthy pet dogs after oral low-dose dexamethasone suppression tests. Vet Rec. 2004;155:518.
Van Sluijs FJ, et al. Results of adrenalectomy in 36 dogs with hyperadrenocorticism caused by adrenocortical tumor. Vet Q. 1995;17:113.
Vaughn MA, et al. Evaluation of twice-daily, low-dose trilostane treatment administered orally in dogs with naturally occurring hyperadrenocorticism. J Am Vet Med Assoc. 2008;232:1321.
Wenger M, et al. Effect of trilostane on serum concentrations of aldosterone, cortisol, and potassium in dogs with pituitary-dependent hyperadrenocorticism. Am J Vet Res. 2004;65:1245.
Zimmer C, et al. Ultrasonographic examination of the adrenal gland and evaluation of the hypophyseal-adrenal axis in 20 cats. J Small Anim Pract. 2000;41:156.
Atypical Cushing’s Syndrome in Dogs
Behrend EN, et al. Serum 17-β-hydroxyprogesterone and corticosterone concentrations in dogs with nonadrenal neoplasia and dogs with suspected hyperadrenocorticism. J Am Vet Med Assoc. 2005;227:1762.
Benitah N, et al. Evaluation of serum 17-hydroxyprogesterone concentration after administration of ACTH in dogs with hyperadrenocorticism. J Am Vet Med Assoc. 2005;227:1095.
Chapman PS, et al. Evaluation of the basal and postadrenocorticotrophic hormone serum concentrations of 17-hydroxyprogesterone for the diagnosis of hyperadrenocorticism in dogs. Vet Rec. 2003;153:771.
Frank LA, et al. Steroid hormone concentration profiles in healthy intact and neutered dogs before and after cosyntropin administration. Dom Anim Endocr. 2003;24:43.
Hill KE, et al. Secretion of sex hormones in dogs with adrenal dysfunction. J Am Vet Med Assoc. 2005;226:556.
Ristic JME, et al. The use of 17-hydroxyprogesterone in the diagnosis of canine hyperadrenocorticism. J Vet Intern Med. 2002;16:433.
Cauvin AL, et al. The urinary corticoid : creatinine ratio (UCCR) in healthy cats undergoing hospitalization. J Fel Med Surg. 2003;5:329.
Duesberg CA, et al. Adrenalectomy for treatment of hyperadrenocorticism in cats: 10 cases (1988–1992). J Am Vet Med Assoc. 1995;207:1066.
Meij BP, et al. Transsphenoidal hypophysectomy for treatment of pituitary-dependent hyperadrenocorticism in 7 cats. Vet Surg. 2001;30:72.
Neiger R, et al. Trilostane therapy for treatment of pituitary-dependent hyperadrenocorticism in 5 cats. J Vet Intern Med. 2004;18:160.
Burkitt JM, et al. Relative adrenal insufficiency in dogs with sepsis. J Vet Intern Med. 2007;21:226.
Hughes AM, et al. Clinical features and heritability of hypoadrenocorticism in Nova Scotia Duck Tolling Retrievers: 25 cases (1994-2006). J Am Vet Med Assoc. 2007;231:407.
Lennon EM, et al. Use of basal serum or plasma cortisol concentrations to rule out a diagnosis of hypoadrenocorticism in dogs: 123 cases (2000-20005). Am Vet Med Assoc. 2007;231:413.
Sieber-Ruckstuhl NS, et al. Cortisol, aldosterone, cortisol precursor, androgen and endogenous ACTH concentrations in dogs with pituitary-dependent hyperadrenocorticism treated with trilostane. Dom Anim Endocr. 2006;31:63.
Thompson AL, et al. Comparison of classic hypoadrenocorticism with glucocorticoid-deficient hypoadrenocorticism in dogs: 46 cases (1985-2005). J Am Vet Med Assoc. 2007;230:1190.
Kook PH, et al. Urinary catecholamine and metanephrine to creatinine ratios in healthy dogs at home and in a hospital environment and in 2 dogs with pheochromocytoma. J Vet Intern Med. 2007;21:388.
Kyles AE, et al. Surgical management of adrenal gland tumors with and without associated tumor thrombi in dogs: 40 cases (1994-2001). J Am Vet Med Assoc. 2003;223:654.
Ash RA, et al. Primary hyperaldosteronism in the cat: a series of 13 cases. J Fel Med Surg. 2005;7:173.
Rossmeisl JH, et al. Hyperadrenocorticism and hyperprogesteronemia in a cat with an adrenocortical adenocarcinoma. J Am Anim Hosp Assoc. 2000;36:512.
Syme HM, et al. Hyperadrenocorticism associated with excessive sex hormone production by an adrenocortical tumor in two dogs. J Am Vet Med Assoc. 2001;219:1725.
 Drugs Used in Endocrine Disorders
Drugs Used in Endocrine Disorders