CHAPTER 12 Thromboembolic Disease
GENERAL CONSIDERATIONS
Thromboembolic (TE) disease involves either a locally formed (in situ) clot, an aggregation of platelets and other blood elements (thrombus), or a clot or other aggregate that breaks away from its origination site (embolus) and is carried downstream by blood flow. Both thrombi and emboli can partially or completely obstruct blood flow, either in a vessel or in the heart. TE disease can occur whenever normal hemostatic mechanisms are disturbed. Most clinically recognized TE events involve the distal aorta, pulmonary arteries, heart, or cranial vena cava. (For additional information on the pathogenesis of TE, see Chapter 87.)
The clinical sequelae of TE disease depend mainly on the size and location of the clot(s). These factors determine how much functional compromise occurs and in which organs and tissues. Acute, profound clinical signs result from some thrombolemboli. Others cause subclinical tissue damage and varying degrees of pathology. TE disease is sometimes suspected antemortem; in other cases it is discovered at necropsy (or not at all).
There is normally an interplay among the different factors that promote coagulation, inhibit coagulation, and promote fibrinolysis. A proper balance of these factors maintains blood fluidity and minimizes loss when vessels are damaged. Platelets, the vascular endothelium, proteins of the coagulation cascade, and the fibrinolytic system are all involved in normal hemostasis. Injury to the vascular endothelium quickly induces several reactions that cause vasoconstriction, hemostatic plug formation, and attempts at vascular repair in order to prevent blood loss.
Intact endothelium normally produces factors with antiplatelet, anticoagulant, and also fibrinolytic effects. Antiplatelet substances include nitric oxide and prostacyclin. Nitric oxide inhibits platelet activation and promotes local vasodilation. Prostacyclin also inhibits platelet activation and aggregation, while mediating vascular smooth muscle relaxation. Anticoagulant substances synthesized by intact endothelium include thrombomodulin, protein S, and heparan sulfate. These substances inhibit the coagulation process in a number of ways.
Damaged endothelial cells promote thrombus formation. Although this reduces blood loss in the event of vascular laceration, in other settings TE disease results. Endothelial damage contributes to thrombus formation in several ways. For example, injured endothelial cells release endothelin, which promotes vasoconstriction and decreases local blood flow as well as tissue factor (thromboplastin), which activates the extrinsic pathway of the coagulation cascade.
Exposed subendothelial collagen and other substances stimulate platelet adherence and aggregation. This is followed by platelet activation. Activated platelets release a number of substances that further stimulate the process of platelet aggregation. Fibrinogen binds to surface glycoprotein (gp) IIb/IIIa receptors, which are expressed on activated platelets. Fibrinogen linkage forms a primary platelet plug, which then stabilizes as platelets contract and fibrinogen is converted to fibrin via the action of thrombin (factor IIa) produced by the coagulation cascade.
Both the intrinsic and extrinsic pathways of the coagulation cascade feed into the common pathway to produce thrombin (see Chapter 87). Tissue factor (released from monocytes and damaged cells) stimulates the extrinsic pathway by activating factor VII. The intrinsic pathway amplifies the process; it also modulates fibrinolysis. Thrombin converts fibrinogen into fibrin monomers. These polymerize to soluble fibrin, which then is cross-linked by the action of thrombin-activated factor XIII. This insoluble fibrin stabilizes the clot. Thrombin also stimulates further platelet aggregation as well as contributes to negative feedback inhibition of clotting by interacting with thrombomodulin, proteins C and S, and antithrombin (AT).
After a thrombus forms, several mechanisms limit its extent and promote its breakdown. Thrombolysis requires plasmin. Its inactive precursor, plasminogen, is converted to plasmin by tissue plasminogen activator (t-PA) when fibrin is present. During activation of the coagulation cascade, t-PA is simultaneously released by endothelial cells. Several other substances also can act as plasminogen activators. Plasmin degrades fibrinogen and soluble (noncross-linked) fibrin to yield fibrinogen/fibrin degradation products (FDPs). Plasmin also cleaves cross-linked fibrin in stabilized clots into large fragments (x-oligomers) that are further broken down into d-dimers and other fragments. d-dimers are produced only with active coagulation and subsequent fibrinolysis. There are also negative feedback constraints on fibrinolysis (e.g., plasminogen activator inhibitors, α2-antiplasmin, thrombin-activated fibrinolytic factor). Defective fibrinolysis is thought to play a role in pathologic thrombosis.
Inhibition of platelet adherence and activation is important in preventing primary platelet plug formation. In addition, there are three main mechanisms that limit thrombus formation: AT, protein C, and the fibrinolytic system. AT is a small protein produced by the liver, which is responsible for most of the anticoagulant effect of plasma. AT, with its co-factor heparan sulfate, binds and inactivates thrombin; factors IXa, Xa, Xia, and XIIa; and kallikrein. Protein C, a vitamin K–dependent glycoprotein, also is involved in countering thrombosis.
Malfunction of one or more of these systems promotes thrombosis.
Pathophysiology
TE disease is more likely when changes in normal hemostatic processes create conditions that favor clot formation or impair thrombolysis. Three general situations (so-called Virchow’s triad) promote pathologic thrombosis: abnormal endothelial structure or function, slowed or static blood flow, and a hypercoagulable state (either from increased procoagulant substances or decreased anticoagulant or fibrinolytic substances). A number of common diseases produce such conditions (Box 12-1).
 BOX 12-1 Diseases Potentially Associated with Thromboembolism
BOX 12-1 Diseases Potentially Associated with Thromboembolism
Diseases that induce severe or widespread endothelial injury also cause loss of normal endothelial antiplatelet, anticoagulant, and fibrinolytic functions. Increased coagulability and platelet activation favor pathologic thrombosis. Injured endothelium also releases tissue factor as well as antifibrinolytic factors. Subendothelial tissue, exposed because of endothelial cell damage, promotes thrombosis by acting as a substrate for clot formation and stimulating platelet adherence and aggregation.
Systemic release of inflammatory cytokines (e.g., tumor necrosis factor [TNF], various interleukins, platelet activating factor, nitric oxide) can cause widespread endothelial injury, induce tissue factor expression, and also inhibit anticoagulant mechanisms. This occurs with sepsis and likely other systemic inflammatory conditions as well. Neoplastic invasion, vascular disruption resulting from other disease, and postischemic injury also induce endothelial damage. Mechanical trauma to the vascular endothelium (as with catheterization) can also precipitate TE disease, especially when other predisposing conditions exist. Pulmonary artery endothelial injury resulting from heartworm disease (HWD) is well known (see Chapter 10). The inflammatory reaction to dead or dying worms and worm fragments exacerbates the endothelial damage and prothrombotic conditions.
Stagnant blood flow promotes thrombosis by impeding the dilution and clearance of coagulation factors. Poor flow can promote local tissue hypoxia and endothelial injury as well. Abnormal turbulence has also been associated with thrombus formation. Turbulence can mechanically injure the endothelial surface.
Hypercoagulability may develop secondary to various systemic diseases in dogs and cats; multiple mechanisms are thought to be involved. Nevertheless, thrombus formation in such cases may also depend on altered endothelial integrity or blood flow. AT deficiency is a common cause of hypercoagulability. Excessive loss, increased consumption, or possibly inadequate hepatic synthesis leads to AT deficiency. Decreased protein C activity and other mechanisms may also contribute to hypercoagulability.
Increased platelet aggregability has been associated with neoplasia, some heart diseases, diabetes mellitus, and nephrotic syndrome in some animals. Thrombocytosis alone, without an increase in platelet aggregability, is not thought to increase the risk for thrombosis.
Defective fibrinolysis can promote pathologic thrombosis by preventing efficient breakdown of physiologic clots. This can result from either reduced levels of fibrinolytic substances (e.g., t-PA, plasminogen, urokinase) or increased production of plasminogen activator inhibitors; the latter is a major mechanism of TE disease in humans with hypertension.
Pancreatitis, shock, trauma, sepsis, neoplasia, severe hepatopathy, heatstroke, immune-mediated disease, and other conditions can lead to gross thrombosis as well as disseminated intravascular coagulopathy (DIC). DIC involves massive activation of thrombin and plasmin, with generalized consumption of coagulation factors and platelets. DIC produces extensive thrombosis as well as hemorrhage in the microcirculation, resulting in widespread tissue ischemia and multiorgan failure.
Protein-losing nephropathy (resulting from glomerulonephritis, renal amyloid deposition, or hypertensive injury) can lead to marked AT deficiency. Because of its small size, AT is lost through damaged glomeruli more easily than most procoagulant proteins, which predisposes to thrombosis. Protein-losing enteropathies also cause AT deficiency, but concurrent loss of larger proteins tends to maintain a balance between procoagulant and anticoagulant factors. Other factors also may contribute to TE disease in animals with protein-losing nephropathies, such as increased platelet aggregation secondary to hypoalbuminemia.
Thrombosis associated with immune-mediated hemolytic anemia (IMHA) is also thought to be multifactorial, with the systemic inflammatory (immune-mediated) response playing a large role. Thrombocytopenia, hyperbilirubinemia, and hypoalbuminemia have been identified as risk factors for TE disease. The role of high-dose corticosteroid therapy in pathologic thrombosis is unclear. However, TE disease is relatively common in animals receiving exogenous corticosteroids and in those with hyperadrenocorticism (see next paragraph). Other predisposing factors are usually concurrent in these cases as well.
TE disease occurs in some dogs with spontaneous hyperadrenocorticism. This endocrinopathy has been associated with decreased fibrinolysis (resulting from increased plasminogen activator inhibitor [PAI] activity) and high levels of several coagulation factors. Diabetes mellitus is occasionally associated with TE disease in dogs. Platelet hyperaggregability and possibly hypofibrinolysis are thought to be involved. Occasionally, a patient with clinically relevant TE disease does not have any detectable abnormality that can result in hypercoagulability (e.g., Greyhounds with aortic TE disease not associated with detectable hemostatic or cardiovascular abnormalities). Cats with myocardial disease (see Chapter 8) are at risk for intracardiac thrombus formation and subsequent arterial embolization. The mechanisms involved probably relate to poor intracardiac blood flow (especially within the left atrium), altered blood coagulability, local tissue or blood vessel injury, or a combination of these. Increased platelet reactivity occurs in some of these cats. Abnormal turbulence may be a factor when mitral regurgitation occurs. DIC may accompany thromboembolism. Some cats with TE disease have decreased plasma arginine and vitamin B6 and B12 concentrations; hyperhomocysteinemia may be a factor in some cases. Hyperhomocysteinemia and low plasma vitamin B concentrations are risk factors for thromboembolism in people. It is not known if hypercoagulability induced by a genetic abnormality exists in some cats, as it does in people.
PULMONARY THROMBOEMBOLISM
Pulmonary thromboemboli in dogs are associated with HWD, other heart diseases, immune-mediated hemolytic anemia (IMHA), neoplasia, DIC, sepsis, hyperadrenocorticism, nephrotic syndrome, pancreatitis, trauma, hypothyroidism, and right atrial thrombus related to infection.
Pulmonary TE disease appears to be rare in cats compared with dogs, except in those with HWD. Nevertheless, pulmonary TE disease has been associated with a variety of systemic and inflammatory disorders in cats, including neoplasia, HWD, anemia (probably immune mediated), pancreatitis, glomerulonephritis, encephalitis, pneumonia, heart disease, sepsis, glucocorticoid administration, protein-losing enteropathy, and hepatic lipidosis.
Pulmonary TE disease that causes pulmonary hypertension variably produces right ventricular enlargement and hypertrophy, interventricular septal flattening, and high tricuspid regurgitation jet velocities. Sometimes a clot is identified within the pulmonary artery or right atrium.
SYSTEMIC ARTERIAL THROMBOEMBOLISM IN CATS
The most common cause for arterial TE disease in cats is cardiomyopathy (see Chapter 8). Thrombi initially form in the left heart and can become quite large. Although some remain in the heart (usually the left atrial [LA] appendage; see Figure 8-6), others embolize to the distal aorta or, less often, other sites. Marked LA enlargement may magnify the risk for thromboembolus formation, but this is controversial. Neoplastic and systemic inflammatory disease are sometimes associated with systemic thromboemboli in cats. Hyperthyroidism may be a risk factor for TE disease in cats independent of its cardiac effects. In some cases, no predisposing condition is identified.
Systemic arterial emboli usually lodge at the aortic trifurcation (so-called saddle thrombus; Figure 12-1), but iliac, femoral, renal, brachial, and other arteries can be affected depending on embolus size and flow path. Besides obstructing flow in the affected artery, thromboemboli release vasoactive substances that induce vasoconstriction and compromise collateral blood flow development around the obstructed vessel. Tissue ischemia results and causes further damage and inflammation. An ischemic neuromyopathy occurs in the affected limb(s), with peripheral nerve dysfunction and degeneration as well as pathologic changes in associated muscle tissue.
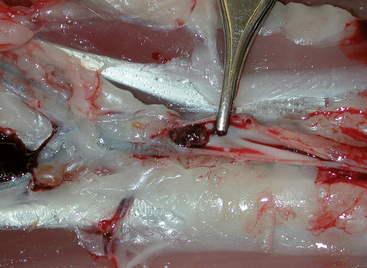
FIG 12-1 Postmortem image with opened distal aorta, from a cat with cardiomyopathy. A thromboembolus (just left of the forceps tip) is lodged at the aortic trifurcation. The rear limbs are to the left in the image; cranial is to the right.
Coronary thromboembolism with myocardial necrosis has occurred in cats with cardiac disease, especially severe hypertrophic cardiomyopathy or infective endocarditis, as well as from carcinoma emboli.
Clinical Features
Arterial TE disease in cats usually causes acute and dramatic clinical signs secondary to tissue ischemia (Fig. 12-2). Male cats are at higher risk for thromboembolism, but this gender bias appears to be related to the prevalence of hypertrophic cardiomyopathy. Distal aortic embolization occurs in most cases. However, the clinical findings depend on the area embolized as well as the extent and duration of arterial blockage.
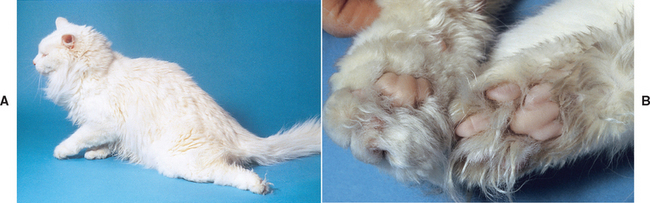
FIG 12-2 A, Cat with thromboembolism to the distal aorta. The left rear limb was dragged behind as the cat tried to walk; there was slightly better function in the right rear. B, The pads of the left rear paw (right side of image) in this cat were paler as well as cooler compared with the left forepaw (left side of image).
Signs of pain and poor systemic perfusion are usually present. Hypothermia and azotemia are common. A cardiac murmur, gallop sound, or arrhythmia is often identified, but these signs are not always evident even with underlying heart disease. Clinical signs of heart disease before the TE event are often absent. Tachypnea and open-mouth breathing are common in cats with acute arterial embolization, despite the absence of overt congestive heart failure (CHF). These signs may represent a pain response, although increased pulmonary venous pressure could be involved. Thoracic radiographs should be obtained as soon as possible because it is important to determine whether pulmonary edema underlies the respiratory signs.
Acute hindlimb paresis without palpable femoral pulses is typical. Common clinical findings are summarized in Box 12-2. Motor function in the rear limbs is minimal to absent in most cases, although the cat is usually able to flex and extend the hips. Sensation to the lower limbs is poor. One side may show greater deficits than the other. Emboli are occasionally small enough to lodge more distally in only one limb, which causes paresis of the lower limb alone. Embolization of a brachial artery produces (usually right) forelimb monoparesis. Intermittent claudication (see p. 201) occurs rarely. Thromboemboli within the renal, mesenteric, or pulmonary arterial circulation may result in failure of these organs and death. Emboli to the brain could induce seizures or various neurological deficits.
Diagnosis
Thoracic radiography is used to screen for cardiopulmonary abnormalities such as evidence for heart failure or other disease associated with thromboemboli (e.g., glomerulonephritis, neoplasia, HWD). Most cats with arterial TE disease have some degree of cardiomegaly (especially LA enlargement) when cardiomyopathy is the underlying cause. Signs of heart failure include dilated pulmonary veins, pulmonary edema, or pleural effusion. A few affected cats have no radiographic evidence of cardiomegaly.
Echocardiography delineates the type of myocardial disease and may reveal the presence of an intracardiac thrombus (see Figure 8-6). The most common site for intracardiac thrombi is the left auricular appendage. Most cats with arterial TE disease associated with cardiomyopathy have some degree of LA enlargement. An LA dimension of >20 mm (measured from the two-dimensional long-axis four-chamber view) may increase the risk for TE disease, although more than half of aortic TE disease cases in one study had a smaller left atrium (Smith, 2003). If echocardiography is unavailable, nonselective angiocardiography can help define the nature of underlying cardiac disease and determine the location and extent of the thromboembolism.
Cats with arterial thromboembolism often have azotemia. This can be prerenal, resulting from poor systemic perfusion or dehydration; primary renal, resulting from embolization of the renal arteries or preexisting kidney disease; or a combination of both. Metabolic acidosis, DIC, electrolyte abnormalities (especially low serum sodium, calcium, potassium, and elevated phosphorus), and stress hyperglycemia are common. Hyperkalemia may develop secondary to ischemic muscle damage and reperfusion. Skeletal muscle damage and necrosis are accompanied by elevations of alanine aminotransferase and aspartate aminotransferase activities, beginning within 12 hours of the TE event and peaking by 36 hours. Widespread muscle injury causes lactate dehydrogenase and creatine kinase activities to be increased soon after the event; elevations in these enzyme activities may persist for weeks. Metabolic acidosis, DIC, and hyperkalemia may also be present secondary to ischemic muscle damage and reperfusion. Cats with arterial TE disease usually have a normal coagulation profile.
Other causes of acute posterior paresis to be considered include intervertebral disk disease, spinal neoplasia (e.g., lymphoma), trauma, fibrocartilaginous infarction, diabetic neuropathy, and possibly myasthenia gravis.
Treatment and Prognosis
The goals of treatment are to manage concurrent CHF and arrhythmias (if present), prevent extension of the embolus and formation of additional thrombi, promote collateral circulation, and provide supportive care (Box 12-3). The treatment of heart failure is outlined in Chapter 8 and Box 8-1. Propranolol is discouraged in cats with cardiomyopathy and arterial thromboembolism because its nonselective β-blocking effect may contribute to peripheral vasoconstriction from unopposed α-receptors, and the drug has no antithrombotic effects at clinical doses.
 BOX 12-3 Therapy for Thromboembolic Disease
BOX 12-3 Therapy for Thromboembolic Disease
An analgesic is recommended, especially for the first 24 to 36 hours, because this is a painful condition. Butorphanol (0.15 to 0.5mg/kg, administered intramuscularly into the cranial lumbar area or subcutaneously q1-3h) has been recommended, especially for the first 24 to 36 hours after the embolic event. Low-dose morphine (0.1 to 0.3mg/kg q3-6h, administered intramuscularly or subcutaneously) could be considered, but some cats experience dysphoria. A fentanyl patch (25 μg/h size) applied to a shaved area of skin could be used to help alleviate pain for up to 3 days, but because it takes about 12 hours to become effective, another analgesic is used simultaneously during this initial period. Respiratory depression and reduced gastrointestinal (GI) motility are potential side effects.
Acepromazine is not recommended for animals with arterial TE disease, despite its α-adrenergic receptor–blocking effects. Improved collateral flow has not been documented, and hypotension and exacerbation of dynamic ventricular outflow obstruction (in cats with hypertrophic obstructive cardiomyopathy) are potential adverse effects. Other supportive care is given to improve and maintain adequate tissue perfusion, minimize further endothelial damage and blood stasis, and optimize organ function as well as to allow time for collateral circulation development.
Antiplatelet and anticoagulant therapies are used to reduce platelet aggregation and growth of existing thrombi. Although fibrinolytic therapy is used in some cases, dosage uncertainties, the need for intensive care, and the potential for serious complications stemming from reperfusion injury limit its use.
Aspirin (acetylsalicylic acid) is used commonly to block platelet activation and aggregation in patients with, or at risk for, TE disease. Aspirin irreversibly inhibits cyclooxygenase, which reduces prostaglandin and thromboxane A2 synthesis and therefore subsequent platelet aggregation, serotonin release, and vasoconstriction. Because platelets cannot synthesize additional cyclooxygenase, this reduction of procoagulant prostaglandins and thromboxane persists for the platelet’s life span (7 to 10 days). Endothelial production of prostacyclin (also via the cyclooxygenase pathway) is reduced by aspirin but only transiently as endothelial cells synthesize additional cyclooxygenase. Aspirin’s benefit may relate more to in situ thrombus formation; efficacy in acute arterial thromboembolism is unknown. Adverse effects of aspirin tend to be mild and uncommon, but the optimal dose is unclear. Cats lack an enzyme (glucuronyl transferase) that is needed to metabolize aspirin, so less frequent dosing is required compared with dogs. In cats with experimental aortic thrombosis, 10 to 25mg/kg (1.25 grains/cat) given by mouth once every (2 to) 3 days inhibited platelet aggregation and improved collateral circulation. However, low-dose aspirin (5mg/cat q72h) has also been used with fewer GI adverse effects, although its efficacy in preventing TE events is unknown. Aspirin therapy is started when the patient is able to take food and oral medications.
Other antiplatelet drugs are being studied. The thienopyridines inhibit adenosine diphosphate (ADP)-binding at platelet receptors and subsequent ADP-mediated platelet aggregation. Clopidogrel (Plavix; 18.75mg/cat PO q24h) appears to have significant antiplatelet effects; daily dosing may be possible.
Heparin is indicated to limit extension of existing thrombi and prevent further TE episodes; it does not promote thrombolysis. Unfractionated heparin and a number of low-molecular–weight heparin (LMWH) products are available. Heparin’s main anticoagulant effect is produced through AT activation, which in turn inhibits factors IX, X, XI, and XII and thrombin. Unfractionated heparin binds thrombin as well as AT. Heparin also stimulates release of tissue factor inhibitors from vascular sites, which helps reduce (extrinsic) coagulation cascade activation. Optimal dosing protocols for animals are not known. Unfractionated heparin is usually given as an initial intravenous (IV) bolus followed by subcutaneous (SC) injections (see Box 12-3). Heparin is not given IM because of the risk for hemorrhage at the injection site. Heparin doses (from 75 to 500U/kg) have been used with uncertain efficacy. An initial IV dose of 200IU/kg, followed by 150 to 200IU/kg administered subcutaneously q6-8h for 2 to 4 days is one protocol. Monitoring the patient’s activated partial thromboplastin time (aPTT) is recommended, although results may not accurately predict serum heparin concentrations. Pretreatment coagulation testing is done for comparison, and the goal is to prolong the aPTT to 1.5 to 2.5 times baseline. Activated clotting time is not recommended to monitor heparin therapy. Hemorrhage is the major complication. Protamine sulfate can be used to counteract heparin-induced bleeding. However, an overdose of protamine can paradoxically cause irreversible hemorrhage. Dosage guidelines for protamine sulfate are as follows: 1mg/100U of heparin is given if the heparin was given within the previous 60 minutes; 0.5mg/100U of heparin is given if the heparin was given more than 1 but less than 2 hours earlier; and 0.25mg/100U of heparin is given if more than 2 hours have elapsed since heparin was administered. Fresh frozen plasma may be needed to replenish AT. Heparin treatment is continued until the patient is stable and has been on antiplatelet therapy for a few days.
LMWH is a safer alternative to unfractionated heparin. LMWH products are a diverse group of depolymerized heparin that vary in size, structure, and pharmacokinetics. Their smaller size prevents simultaneous binding to thrombin and AT. LMWH products have more effect against factor Xa through their inactivation of AT. Because they have minimal ability to inhibit thrombin, they are less likely to cause bleeding. LMWH products have greater bioavailability and a longer half-life than unfractionated heparin when given subcutaneously because of lesser binding to plasma proteins as well as endothelial cells and macrophages. However, LMWH products do not markedly affect coagulation times, so monitoring aPTT is generally not necessary. LMWH effect can be monitored indirectly by anti-Xa activity. Optimal anti-Xa activity level in cats is not known; the target range in people is reported as 0.5 to 1.0U/ml, although 0.3 to 0.6U/ml has also been used. The LMWH products have differences in biological and clinical effects and are not interchangeable. The most effective dosage for the various LMWH products is not clearly established in dogs and cats. Commonly used dosages of dalteparin sodium (Fragmin; 100-150U/kg administereed subcutaneously q8-24h) and enoxaparin (Lovenox; 1mg/kg administered subcutaneously q12-24h) were extrapolated from human use. However, according to a recent study (Alwood et al., 2007), these doses do not produce a (human) target level of anti-Xa activity in cats. Although enoxaparin produced anti-Xa activity close to this level at 4 hours postdose, activity was undetectable 8 hours later. On the basis of this study, the predicted optimal dose and dosing interval to maintain anti-Xa activity within the (human) therapeutic range in normal cats are as follows: dalteparin, 150U/kg administered subcutaneously q4h; and enoxaparin, 1.5mg/kg administered subcutaneously q6h. The optimal therapeutic range in cats as well as the most effective dosage in sick cats are not yet known.
Drugs used to promote clot lysis include streptokinase and human recombinant tissue plasminogen activator (rt-PA). These agents increase conversion of plasminogen to plasmin to facilitate fibrinolysis. Veterinary experience with these agents is quite limited. Although they effectively break down clots, complications related to reperfusion injury and hemorrhage, the high mortality rate, the cost of therapy, the intensive care required, and the lack of clearly established dosing protocols have prevented their widespread use. Furthermore, a clear survival advantage has not been shown. If used, this therapy is best instituted within 3 to 4 hours of vascular occlusion. An intensive care setting, including continuous serum potassium concentration (or electrocardiographic [ECG]) monitoring to detect reperfusion-induced hyperkalemia, is recommended.
Streptokinase is a nonspecific plasminogen activator that promotes the breakdown of fibrin as well as fibrinogen. This action leads to the degradation of fibrin within thrombi and clot lysis but also potentially leads to systemic fibrinolysis, coagulopathy, and bleeding. Streptokinase also degrades factors V, VIII, and prothrombin. Although its half-life is about 30 minutes, fibrinogen depletion continues for much longer. Streptokinase has been used with variable success in a small number of dogs with arterial TE disease. The reported protocol is 90,000IU of IV streptokinase infused over 20 to 30 minutes, then at a rate of 45,000IU/hour for 3 (to 8) hours. Dilution of 250,000IU into 5ml saline, then into 50ml to yield 5000U/ml for infusion with a syringe pump has been suggested for cats. Adverse effects are minor in some cases, and bleeding may respond to discontinuing streptokinase. However, there is a risk for serious hemorrhage, and the mortality rate in clinical cases is high. Acute hyperkalemia (secondary to thrombolysis and reperfusion injury), metabolic acidosis, bleeding, and other complications are thought to be responsible for causing death. Streptokinase can increase platelet aggregability and induce platelet dysfunction. It is unclear if lower doses would be effective with fewer complications. Streptokinase combined with heparin therapy can increase the risk of hemorrhage, especially when coagulation times are increased. Streptokinase is potentially antigenic because it is produced by β-hemolytic streptococci. No survival benefit has been shown for streptokinase therapy compared with conventional (i.e., aspirin and heparin) treatment in cats.
rt-PA is a single-chain polypeptide serine protease with a higher specificity for fibrin within thrombi and a low affinity for circulating plasminogen. Although the risk of hemorrhage is less than with streptokinase, there is the potential for serious bleeding as well as other side effects. rt-PA is also potentially antigenic in animals because it is a human protein. Like streptokinase, rt-PA induces platelet dysfunction but not hyperaggregability. Experience with rt-PA is very limited, and the optimal dosage is not known. An IV dose of 0.25 to 1mg/kg/h up to a total of 1 to 10mg/kg was used in a small number of cats; although signs of reperfusion occurred, the mortality rate was high. The cause of death in most cats was attributed to reperfusion (hyperkalemia, metabolic acidosis) and hemorrhage, although CHF and arrhythmias were also involved.
Surgical clot removal is generally not advised in cats. The surgical risk is high, and significant neuromuscular ischemic injury is likely to have already occurred by the time of surgery. Clot removal using an embolectomy catheter has not been very effective in cats.
In general, the prognosis is poor in cats with arterial TE disease. Historically, only a third of cats survive the initial episode. However, survival statistics improve when cats euthanized without therapy are excluded or when only cases from recent years are analyzed. Survival is better if only one limb is involved and/or if some motor function is preserved at presentation. Hypothermia and CHF at presentation are both associated with poor survival in cats. Other negative factors may include hyperphosphatemia, progressive hyperkalemia or azotemia, progressive limb injury (continued muscle contracture after 2 to 3 days, necrosis), severe LA enlargement, presence of intracardiac thrombi or spontaneous contrast (“swirling smoke”) on echocardiogram, DIC, and history of thromboembolism.
Barring complications, limb function should begin to return within 1 to 2 weeks. Some cats become clinically normal within 1 to 2 months, although residual deficits may persist for a variable time. Tissue necrosis may require wound management and skin grafting. Permanent limb deformity develops in some cats, and amputation is occasionally necessary. Repeated events are common. Significant embolization of the kidneys, intestines, or other organs carries a grave prognosis.
PROPHYLAXIS AGAINST ARTERIAL THROMBOEMBOLISM
Prophylactic therapy with an antiplatelet or anticoagulant drug is commonly used in animals thought to be at increased risk for TE disease. These include cats with cardiomyopathy (especially those with marked LA enlargement, echocardiographic evidence for intracardiac spontaneous contrast or thrombus, or a previous TE event) and animals with sepsis, IMHA, severe pancreatitis, or other procoagulant conditions. However, the efficacy of TE prophylaxis is unknown, and a strategy that consistently prevents thromboembolism is not yet identified.
Drugs used for arterial TE prophylaxis include aspirin, clopidogrel, warfarin (coumadin), and LMWH. Aspirin and clopidogrel present a low risk for serious hemorrhage and require less monitoring compared with warfarin. Adverse GI effects (e.g., vomiting, inappetence, ulceration, hematemesis) occur in some animals. Buffered aspirin formulation or an aspirin-Maalox combination product may be helpful. Low-dose aspirin (5mg/cat every third day) has been advocated in cats. Although adverse effects are unlikely with this dose, it is not known whether antiplatelet effectiveness is compromised. Warfarin (discussed in more detail later) is associated with greater expense and a higher rate of fatal hemorrhage. No survival benefit has been shown for warfarin compared with aspirin in cats. In some reports, recurrent thromboembolism occurred in almost half of cats treated with warfarin. Clopidogrel or LMWH prophylaxis may be more efficacious, with less risk of hemorrhage, but more experience with this therapy is needed. Recurrent TE events occurred in 20% of cats in one study (Smith, 2004). LMWH is expensive and must be given by daily SC injection, but some owners are motivated to do this. In cats without thrombocytopenia, aspirin may be used concurrently. Diltiazem, at clinical doses, does not appear to have significant platelet-inhibiting effects.
Warfarin inhibits the enzyme (vitamin K epoxide reductase) responsible for activating the vitamin K–dependent factors (II, VII, IX, and X), as well as proteins C and S. Initial warfarin treatment causes transient hypercoagulability because anticoagulant proteins have a shorter half-life than most procoagulant factors. Therefore heparin (e.g., 100IU/kg administered subcutaneously q8h) is given for 2 to 4 days after warfarin is initiated. There is wide variability in dose response and potential for serious bleeding, even in cats that are monitored closely. Warfarin is highly protein-bound; concurrent use of other protein-bound drugs or change in serum protein concentration can markedly alter the anticoagulant effect. Bleeding may be manifested as weakness, lethargy, or pallor rather than overt hemorrhage. A baseline coagulation profile and platelet count are obtained, and aspirin discontinued, before beginning treatment. The usual initial warfarin dose is 0.25 to 0.5mg (total dose) administered orally q24-48h in cats. Uneven distribution of drug within the tablets is reported, so compounding rather than administering tablet fragments is recommended. Drug administration and blood sampling times should be consistent.
The dose is adjusted either on the basis of prothrombin time (PT) or the international normalization ratio (INR). The INR is a more precise method that has been recommended to prevent problems related to variation in commercial PT assays. The INR is calculated by dividing the animal’s PT by the control PT and raising the quotient to the power of the international sensitivity index (ISI) of the thromboplastin used in the assay, or INR = (animal PT/control PT)ISI. The ISI is provided with each batch of thromboplastin made. Extrapolation from human data suggests that an INR of 2 to 3 is as effective as higher values, with less chance for bleeding. Using a warfarin dose of 0.05 to 0.1mg/kg/day in the dog achieves this INR in about 5 to 7 days. Heparin overlap until the INR is >2 is recommended. When PT is used to monitor warfarin therapy, a goal of 1.25 to 1.5 (to 2) times pretreatment PT at 8 to 10 hours after dosing is advised; the animal is weaned off heparin when the INR is >1.25. The PT is evaluated (several hours after dosing) daily initially, then at progressively increasing time intervals (e.g., twice a week, then once a week, then every month to 2 months) as long as the cat’s condition appears stable.
If the PT or INR increases excessively, warfarin is discontinued and vitamin K1 administered (1 to 2mg/kg/day administered orally or subcutaneously) until the PT is normal and the packed cell volume (PCV) is stable. Transfusion with fresh frozen plasma, packed red blood cells, or whole fresh blood is sometimes necessary.
SYSTEMIC ARTERIAL THROMBOEMBOLISM IN DOGS
Arterial TE disease in dogs is relatively uncommon compared with cats. Nevertheless, it has been associated with many conditions, including protein-losing nephropathies, hyperadrenocorticism, neoplasia, chronic interstitial nephritis, HWD, hypothyroidism, gastric dilatation-volvulus, pancreatitis, and several cardiovascular diseases. Kidney disease was present in about half of the dogs with TE disease in one report (Van Winkle, 1993). Vegetative endocarditis is the most common cardiac disease associated with systemic thromboembolism. Other cardiovascular conditions that have been associated with canine TE disease include patent ductus arteriosus (surgical ligation site), dilated cardiomyopathy, myocardial infarction, arteritis, aortic intimal fibrosis, atherosclerosis, aortic dissection, granulomatous inflammatory erosion into the left atrium, and other thrombi in the left heart. TE disease is a rare complication of arteriovenous (A-V) fistulae; it may relate to venous stasis from distal venous hypertension. Aortic TE has occurred in Greyhounds without overt underlying abnormalities as well as in those with protein-losing nephropathy or intramuscular hemangiosarcoma in the thigh muscles. Affected dogs typically present for intermittent rear limb lameness (claudication) and have weak femoral pulses on the affected side, and the thrombi are obvious during abdominal ultrasonography.
Atherosclerosis is uncommon in dogs, but it has been associated with TE disease in this species, as it has in people. Endothelial disruption in areas of atherosclerotic plaque, hypercholesterolemia, increased PAI-1, and possibly other mechanisms may be involved in thrombus formation. Atherosclerosis may develop with profound hypothyroidism, hypercholesterolemia, or hyperlipidemia. The aorta as well as coronary and other medium to large arteries are affected. Myocardial and cerebral infarctions occur in some cases, and there is a high rate of interstitial myocardial fibrosis in affected dogs.
Vasculitis related to infectious, inflammatory, immune-mediated, or toxic disease occasionally underlies TE events. Arteritis of immune-mediated pathogenesis is described in some young Beagles and other dogs. Inflammation and necrosis that affect small to medium-sized arteries may be associated with thrombosis.
Coronary artery thromboembolism causes myocardial ischemia and infarction. Infective endocarditis, neoplasia that involves the heart directly or by neoplastic emboli, coronary atherosclerosis, dilated cardiomyopathy, degenerative mitral valve disease with CHF, and coronary vasculitis are reported causes. In other dogs coronary TE events have occurred with severe renal disease, IMHA, exogenous corticosteroids or hyperadrenocorticism, and acute pancreatic necrosis. These cases may have TE lesions in other locations as well.
Clinical Features
There appear to be no age, breed, or sex predilections for arterial TE disease in dogs. As in cats, the distal aorta is the most common location for clinically recognized thromboemboli. In contrast to cats, most dogs have some clinical signs from 1 to 8 weeks before presentation. Less than a quarter of cases have peracute paralysis without prior signs of lameness, as usually occurs in cats. Signs related to the TE event include pain, hindlimb paresis, lameness or weakness (which may be progressive or intermittent), and chewing or hypersensitivity of the affected limb(s) or lumbar area. Although about half of affected dogs present with sudden paralysis, this is often preceded by a variable period of lameness. Intermittent claudication, common in people with peripheral occlusive vascular disease, may be a manifestation of distal aortic TE disease. This involves pain, weakness, and lameness that develop during exercise. These signs intensify until walking becomes impossible, then disappear with rest. Inadequate perfusion during exercise leads to lactic acid accumulation and cramping.
Physical examination findings in dogs with aortic thromboembolism are similar to those in cats, including absent or weak femoral pulses, cool extremities, hindlimb pain, loss of sensation in the digits, hyperesthesia, cyanotic nailbeds, and neuromuscular dysfunction. Occasionally, a brachial or other artery is embolized. TE disease involving an abdominal organ causes abdominal pain, with clinical and laboratory evidence of damage to the affected organ.
Coronary artery thromboembolism is likely to be associated with arrhythmias, as well as ST segment and T wave changes on ECG. Ventricular (or other) tachyarrhythmias are common, but if the atrioventricular (AV) nodal area is injured, conduction block may result. Clinical signs of acute myocardial infarction/necrosis may mimic those of pulmonary TE disease; these include weakness, dyspnea, and collapse. Respiratory difficulty may develop as a result of pulmonary abnormalities or left heart failure (pulmonary edema) depending on the underlying disease and degree of myocardial dysfunction. Some animals with respiratory distress have no radiographically evident pulmonary infiltrates. Increased pulmonary venous pressure preceding overt edema (from acute myocardial dysfunction) or concurrent pulmonary emboli are potential causes. Other findings in animals with myocardial necrosis include sudden death, tachycardia, weak pulses, increased lung sounds or crackles, cough, cardiac murmur, hyperthermia or sometimes hypothermia, and (less commonly) GI signs. Signs of other systemic disease may be concurrent. Acute ischemic myocardial injury that causes sudden death may not be detectable on routine histopathology.
Diagnosis
Thoracic radiography is used to screen for cardiac abnormalities, especially in animals with systemic arterial TE disease and for pulmonary changes in animals suspected to have pulmonary thromboemboli. Evidence for CHF or other pulmonary disease associated with TE disease (e.g., neoplasia, HWD, other infections) may also be found.
A complete echocardiographic exam is important to define whether (and what type of) heart disease might be present. Thrombi within the left or right heart chambers and proximal great vessels can be readily seen with two-dimensional echocardiography. In dogs with coronary TE disease, the echocardiographic examination may indicate reduced myocardial contractility with or without regional dysfunction. Areas of myocardial fibrosis secondary to chronic ischemia or infarction appear hyperechoic compared with the surrounding myocardium. Thromboemboli in the distal aorta (or other vessel) may be visible by ultrasonography as well. Doppler studies can demonstrate partial or complete obstruction to blood flow in some cases.
Angiography may be used to document vascular occlusion when ultrasonography is inconclusive or unavailable. It also can show the extent of collateral circulation. The choice of selective or nonselective technique depends on patient size and the suspected location of the clot.
Routine laboratory test results depend largely on the disease process underlying the TE event(s). Systemic arterial TE disease also produces elevated muscle enzyme concentrations from skeletal muscle ischemia and necrosis. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activities rise soon after the TE event. Widespread muscle injury causes increased lactate dehydrogenase and creatine kinase (CK) activities as well.
Coagulation test results in patients with TE disease are variable. The concentration of FDPs or d-dimer may be increased, but this can occur in patients with inflammatory disease and is not specific for a TE event or DIC. Modestly increased d-dimer concentrations occur in diseases such as neoplasia, liver disease, and IMHA. This could reflect subclinical TE disease or another clot activation mechanism because these conditions are associated with a procoagulant state. Body cavity hemorrhage also causes a rise in d-dimer concentrations. Because this condition is associated with increased fibrin formation, elevated d-dimer levels may not indicate TE disease in such cases. The specificity of d-dimer testing for pathologic thromboembolism is lower at lower d-dimer concentrations, but the high sensitivity at lower concentrations provides an important screening tool. d-dimer testing appears to be as specific for DIC as FDP measurement. A number of assays have been developed to measure d-dimer concentrations in dogs; some are qualitative or semiquantitative (i.e., latex agglutination, immunochromatographic, and immunofiltration tests), others are more quantitative (i.e., immunoturbidity, enzymatic immunoassays). It is important to interpret d-dimer results in the context of other clinical and test findings. Assays for circulating AT and proteins C and S are also available for dogs and cats. Deficiencies of these proteins are associated with increased risk of thrombosis.
Thromboelastography (TEG) provides an easy point-of-care method of assessing global hemostasis and is quite valuable when evaluating patients with TE disease.
Treatment and Prognosis
The goals of therapy are the same as for cats with TE disease: Stabilize the patient by supportive treatment as indicated, prevent extension of the existing thrombus and additional TE events, and reduce the size of the thromboembolus and restore perfusion. Supportive care is given to improve and maintain adequate tissue perfusion, minimize further endothelial damage and blood stasis, and optimize organ function as well as to allow time for collateral circulation development. Correcting or managing underlying disease(s), to the extent possible, is also important. Antiplatelet and anticoagulant therapies are used to reduce platelet aggregation and growth of existing thrombi as in cats (see p. 199). The results of the TEG, if available, should be used to monitor response to anticoagulants in patients with TE disease.
Management strategies used for TE disease are outlined in Box 12-3. Although fibrinolytic therapy is used in some cases, dosage uncertainties, the need for intensive care, and the potential for serious complications limit its use. The reported streptokinase protocol for dogs is 90,000IU infused intravenously over 20 to 30 minutes, then continued at a rate of 45,000IU/hour for 3 (to 12) hours. In dogs, rt-PA has been used as 1mg/kg boluses administered intravenously q1h for 10 doses, with IV fluid, other supportive therapy, and close monitoring. The half-life of t-PA is about 2 to 3 minutes in dogs, but effects persist longer because of binding to fibrin. The consequences of reperfusion injury present serious complications to thrombolytic therapy. The iron chelator deferoxamine mesylate has been used in an attempt to reduce oxidative damage caused by free radicals involving iron. Allopurinol also has been used but with uncertain results. Clot removal using an embolectomy catheter has not been very effective in cats but might be more successful in dogs of larger size.
Fluid therapy is used to expand vascular volume, support blood pressure, and correct electrolyte and acid/base abnormalities depending on individual patient needs. However, for animals with heart disease and especially CHF, fluid therapy is given only with great caution (if at all). Hypothermia that persists after circulating volume is restored can be addressed with external warming. Specific treatment for heart disease, CHF, and arrhythmias is provided as indicated (see Chapters 3 and 4 and other relevant chapters). Acute respiratory signs may signal CHF, pain, or pulmonary thromboembolism. Differentiation is important because diuretic or vasodilator therapy could worsen perfusion in animals without CHF.
Because acute arterial embolization is particularly painful, analgesic therapy is important in such cases, especially for the first 24 to 36 hours (see Box 12-3). Loosely bandaging the affected limb(s) to prevent self-mutilation may be needed in some animals with aortic TE disease. Renal function and serum electrolyte concentrations are monitored daily or more frequently if fibrinolytic therapy is used. Continuous ECG monitoring during the first several days can help the clinician detect acute hyperkalemia associated with reperfusion (see Chapter 2, p. 31). In general, the prognosis is poor.
PROPHYLAXIS AGAINST ARTERIAL THROMBOEMBOLISM
Prophylactic strategies are the same as for cats. Aspirin, LMWH, warfarin, or possibly clopidogrel are agents to consider. If warfarin is used, the usual initial warfarin dose is 0.25 to 0.5mg (total dose) administered orally q24(to 48)h in cats; 0.1 to 0.2mg/kg administered orally q24h in dogs. A loading dose of ∼0.2mg/kg for 2 days appears to be safe in dogs.
VENOUS THROMBOSIS
Thrombosis in large veins is more likely to be clinically evident than thrombosis in small vessels. Cranial vena caval thrombosis has been associated with IMHA and/or immune-mediated thrombocytopenia, sepsis, neoplasia, protein-losing nephropathies, mycotic disease, heart disease, and glucocorticoid therapy (especially in patients with systemic inflammatory disease) in dogs. Most cases have more than one predisposing factor. An indwelling jugular catheter increases the risk for cranial caval thrombosis, probably by causing vascular endothelial damage or laminar flow disruption or by acting as a nidus for clot formation.
Portal vein thrombosis, along with DIC, has occurred in dogs with pancreatitis and pancreatic necrosis. Peritonitis, neoplasia, hepatitis, protein-losing nephropathy, IMHA, and vasculitis have also been diagnosed occasionally in dogs with portal thrombosis. A high proportion of dogs with incidental portal or splenic vein thrombosis are receiving corticosteroids.
Systemic venous thrombosis produces signs related to increased venous pressure upstream from the obstruction. Thrombosis of the cranial vena cava can lead to the cranial caval syndrome. The cranial caval syndrome is characterized by bilaterally symmetric subcutaneous edema of the head, neck, and forelimbs; another cause of this syndrome is external compression of the cranial cava, usually by a neoplastic mass. Pleural effusion occurs commonly. This effusion is often chylous because lymph flow from the thoracic duct into the cranial vena cava is also impaired. Palpable thrombosis extends into the jugular veins in some cases. Because vena caval obstruction reduces pulmonary blood flow and left heart filling, signs of poor cardiac output are common.
Vena caval thrombosis may be visible on ultrasound exam, especially when the clot extends into the right atrium. Portal vein thrombosis and thromboemboli in the aorta or other large peripheral vessels can also be documented on ultrasound examination.
Clinicopathic findings generally reflect underlying disease as well as tissue damage resulting from vascular obstruction. Cranial caval thrombosis has been associated with thrombocytopenia.
Alwood AJ, et al. Anticoagulant effects of low-molecular–weight heparins in healthy cats. J Vet Intern Med. 2007;21:378.
Boswood A, Lamb CR, White RN. Aortic and iliac thrombosis in six dogs. J Small Anim Pract. 2000;41:109.
Bright JM, Dowers K, Powers BE. Effects of the glycoprotein IIb/IIIa antagonist abciximab on thrombus formation and platelet function in cats with arterial injury. Vet Ther. 2003;4:35.
Buchanan JW, Beardow AW, Sammarco CD. Femoral artery occlusion in Cavalier King Charles Spaniels. J Am Vet Med Assoc. 1997;211:872.
Carr AP, Panciera DL, Kidd L. Prognostic factors for mortality and thromboembolism in canine immune-mediated hemolytic anemia: a retrospective study of 72 dogs. J Vet Intern Med. 2002;16:504.
Cook AK, Cowgill LD. Clinical and pathological features of protein-losing glomerular disease in the dog: a review of 137 cases (1985–1992). J Am Anim Hosp Assoc. 1999;32:313.
De Laforcade AM, et al. Hemostatic changes in dogs with naturally occurring sepsis. J Vet Intern Med. 2003;17:674.
Driehuys S, et al. Myocardial infarction in dogs and cats: 37 cases (1985–1994). J Am Vet Med Assoc. 1998;213:1444.
Fox PR, Petrie JP, Hohenhaus AE. Peripheral vascular disease. In: Ettinger SJ, Feldman EC, editors. Textbook of veterinary internal medicine. ed 6. Philadelphia: WB Saunders; 2005:1145-1165.
Good LI, Manning AM. Thromboembolic disease: physiology of hemostasis and pathophysiology of thrombosis. Compend Contin Educ Pract Vet. 2003;25:650.
Good LI, Manning AM. Thromboembolic disease: predispositions and clinical management. Compend Contin Educ Pract Vet. 2003;25:660.
Hogan DF, et al. Antiplatelet effects and pharmacodynamics of clopidogrel in cats. J Am Vet Med Assoc. 2004;225:1406.
Hogan DF, et al. Evaluation of antiplatelet effects of ticlopidine in cats. Am J Vet Res. 2004;65:327.
Kidd L, Stepien RL, Amrheiw DP. Clinical findings and coronary artery disease in dogs and cats with acute and subacute myocardial necrosis: 28 cases. J Am Anim Hosp Assoc. 2000;36:199.
Laste NJ, Harpster NK. A retrospective study of 100 cases of feline distal aortic thromboembolism: 1977-1993. J Am Anim Hosp Assoc. 1995;31:492.
McMichael MA, et al. Plasma homocysteine, B vitamins, and amino acid concentrations in cats with cardiomyopathy and arterial thromboembolism. J Vet Intern Med. 2000;14:507.
Moore KE, et al. Retrospective study of streptokinase administration in 46 cats with arterial thromboembolism. J Vet Emerg Crit Care. 2000;10:245.
Nelson OL, Andreasen C. The utility of plasma d-dimer to identify thromboembolic disease in dogs. J Vet Intern Med. 2003;17:830.
Olsen LH, et al. Increased platelet aggregation response in Cavalier King Charles Spaniels with mitral valve prolapse. J Vet Intern Med. 2001;15:209.
Palmer KG, King LG, Van Winkle TJ. Clinical manifestations and associated disease syndromes in dogs with cranial vena cava thrombosis: 17 cases (1989–1996). J Am Vet Med Assoc. 1998;213:220.
Schermerhorn TS, Pembleton-Corbett JR, Kornreich B. Pulmonary thromboembolism in cats. J Vet Intern Med. 2004;18:533.
Schoeman JP. Feline distal aortic thromboembolism: a review of 44 cases (1990–1998). J Feline Med Surg. 1999;1:221.
Smith CE, et al. Use of low molecular weight heparin in cats: 57 cases (1999-2003). J Am Vet Med Assoc. 2004;225:1237.
Smith SA, et al. Arterial thromboembolism in cats: acute crisis in 127 cases (1992-2001) and long-term management with low-dose aspirin in 24 cases. J Vet Intern Med. 2003;17:73.
Smith SA, Tobias AH. Feline arterial thromboembolism: an update. Vet Clin North Am: Small Anim Pract. 2004;34:1245.
Stokol T, et al. d-dimer concentrations in healthy dogs and dogs with disseminated intravascular coagulation. Am J Vet Res. 2000;61:393.
Thompson MF, Scott-Moncrieff JC, Hogan DF. Thrombolytic therapy in dogs and cats. J Vet Emerg Crit Care. 2001;11:111.
Van Winkle TJ, Hackner SG, Liu SM. Clinical and pathological features of aortic thromboembolism in 36 dogs. J Vet Emerg Crit Care. 1993;3:13.
 Drugs Used in Cardiovascular Disorders
Drugs Used in Cardiovascular Disorders