Cancer
Cellular proliferation is a regulated process, which ensures that cell division is coordinated with physiological demands of the tissue or the organ. Under normal conditions, cells of adult body divide only to replace those cells that die (apoptosis) after fulfilling their task. Derangements of the regulatory mechanisms result in uncontrolled and excessive cellular proliferation—the cells are said to be malignantly transformed, and the condition is known as cancer. Thus, cancer refers to a state of abnormal, excessive and uncontrolled proliferation of the cells of a tissue; the cellular proliferation far exceeds physiological requirements of the tissue. The proliferating cells may migrate beyond the tissue of origin and infiltrate the adjacent tissues. The word cancer is derived from the Greek word, karikoma meaning “crab”: the cells from a mass of cancer tissue move away like feet of a crab.
After going through this chapter, the student should be able to understand:
Terminology relating to cancer and distinctive features of cancer cells.
Factors implicated in aetiology of cancer: the somatic mutations causing activation of protooncogene or inactivation of antioncogenes; the oncogenic retroviruses and DNA viruses; chemical carcinogens; and ionizing radiations.
Mechanism of activation of protooncogenes by somatic mutations, gene rearrangements, gene amplification, promoter insertion and enhance insertion.
Role of growth factors in causing malignant transformation of cell, and cancer markers in diagnosis and follow up cancer patients.
I Tumours: Benign and Malignant
Excessive cellular proliferation leads to accumulation of an abnormal cell mass called tumour. If the growth of the tumour is self-limiting, the tumour is said to be benign. However, if the cells have high propensity for invasion and distant spread, the tumour is malignant (from the Latin, meaning “engendering harm”). Generally benign tumours are not life-threatening (e.g. moles, warts), and not even considered as cancers. The term cancer is reserved as for the diseases resulting from malignant tumours. The diseases resulting from the benign and the malignant tumours are collectively called neoplasia (Greek word for “new growth”).
 Cancer is characterized by derangements of cellular growth control, resulting in excessive proliferation and accumulation of an abnormal cell mass called tumour.
Cancer is characterized by derangements of cellular growth control, resulting in excessive proliferation and accumulation of an abnormal cell mass called tumour.
The malignant tumours are classified according to the type of somatic cells from which they are derived. For example, tumours of epithelial origin are known as carcinomas, and the tumours originating from connective tissue are called sarcomas. Although the transformed cells may retain some of the important characteristics of the cells of their origin (carcinomas produce keratin, a product of epithelial cells; and sarcomas produce certain constituents of extracellular matrix), these cells acquire some distinctive features not seen in the normal cells.
Incidence of cancers: More than 100 different types of human cancers are known, which are second only to heart disease as the major health issue in the developed world, accounting for about 23% deaths annually. Among children of 7-13 year age, cancer is the leading cause of death: leukaemia accounting for about 50% of cancer-related deaths. Among adults, the incidence of cancers increases with advancing age; about two-third of newly diagnosed cancers have been reported in persons over 60 years.
II Distinctive Features of Malignant Cells
Some of the distinctive features of cancer cells are:
De-differentiation:
The cancer cells assume features of the immature stem cells. For example, carcinoma cells lose the normal columnar or cuboidal shape of the progenitor cells and assume an undifferentiated appearance.
Moreover, the malignantly transformed cells often lose capability to synthesize specialized proteins which are synthesized by the normal cell. For example, the cancerous liver cells may stop producing the normal liver enzymes and instead start secreting certain proteins that are normally synthesized by the fetal liver; for instance, the a-fetoproteins (AFP). Such abnormal proteins are called tumour markers.
High mitotic rate and disordered growth:
The rapidly proliferating cancer cells have high mitotic rate. These cells are capable of growing in the absence of normal mitogenic stimuli.
The growth of cancer cells is disorganized and disordered. The cells spread in all directions, often crossing the anatomical boundaries of the affected tissue. Consequently, they infiltrate into the surrounding areas.
Dedifferentiation and disordered growth are together referred to as anaplasia. The greater the degree of anaplasia, higher the malignant behaviour of the cells and poorer is the prognosis for the patient.
Genetic abnormalities:
The cancer cells commonly show certain genetic abnormalities such as abnormal chromosomal number, deletion or translation of chromosomes, gene amplification or extrachromosomal genetic elements. These aspects of cancer cells are described later in this chapter.
Hereditary:
Cancer is a manifestation of some heritable alteration in the genome. This enables them to divide excessively without losing the distinctive neoplastic characteristics.
Monoclonal origin:
All malignant cells in a patient descend from a single aberrant somatic cell. In the other words, cancers are monoclonal in origin. For example, in patients with chronic myelogenous leukaemia (CML) the characteristic chromosomal translocation between chromosome 9 and chromosome 22 (called the Philadelphia chromosome) are present in all tumour cells, but not in the normal somatic cells. This observation confirms monoclonal origin of the cancer cells.
Loss of apoptosis:
Apoptosis refers to death of a cell after its function has been fulfilled (Box 32.1). Cancer cells lack this property. Even in extremely adverse circumstances such as lack of growth factor, DNA damage, or other environmentally induced injuries, the cancer cells retain ability to survive and proliferate.
Biochemical alterations:
The cancer cells show a number of biochemical changes such as:
(i) The glycolytic sequence is greatly enhanced, indicating higher energy demands of the multiplying cells.
(ii) The membrane permeability and cell-surface charge are altered.
(iii) Activities of certain enzymes, such as proteases, are decreased. The isoenzymes typical of fetal cells, make appearance in cancer cells.
(iv) The antigens present on cell membrane are altered; certain new antigens appear, while some antigens present on the normal cell surface are lost.
(v) Rates of synthesis of DNA and RNA are much higher than that in the normal cell.
(vi) Tumour cells can grow in the absence of normal mitogenic stimuli. In cell cultures they exhibit normal growth under conditions of growth-factor deprivation. However, the quantity of growth factors secreted by tumour cells is greater than normal cells.
Metastasis
The cancer cells can spread to distant tissues through blood or lymph. Such spread of the cancer cells and colonization of distinct tissues is called metastasis. On reaching the far tissues in this manner, the cancer cells form secondary tumour.
Metastasis is the major cause of cancer-related morbidity and mortality. Biochemical basis of metastatic spread are not clearly understood, but it is believed to require poor adherence to neighbouring cells in the tumour and loss of contact inhibition.
• Loss of contact inhibition: Normal cells growing in tissue culture show contact inhibition. When these cells are allowed to grow in tissue culture medium on a surface, such as that of a Petri-dish, they grow and divide until the surface is covered with a monolayer and further growth is inhibited. Cancer cells, however, show loss of contact inhibition and continue to grow beyond the monolayer to form multilayered masses of cells.
• Poor adherence to surface: Normal cells are firmly adhered to the surface, but cancer cells show loss of such anchorage properties. Alteration in structure of the protein, vinculin, is possibly responsible for this.
There are typical differences between malignant neoplastic cells and the normal progenitor cells: cancer cells are immortal, grow in absence of mitogenic stimuli; have abnormally high mitotic rate, gross genetic abnormalities, disordered growth patterns, loss of contact inhibition; and exhibit features of immature stem cells.
III Aetiology
Cancer is a multifactorial disease. A number of factors are implicated in its aetiology, viz. genetic, hormonal, metabolic, physical, chemical and environmental. However, most cancers are spontaneous and no specific cause could be held responsible for their development. Each day, a few cells may undergo spontaneous mutation and become cancerous, but such cells are promptly removed by the immune system. Cancer develops only when (a) the number of mutant cells is too large to be removed by immune system, and (b) immune system is not efficient enough to remove the mutant cells.
The factors that are implicated in cancer aetiology appear to be vastly diverse, but all of them converge at a single point. All produce certain permanent changes in DNA, which are transmissible at cell divisions. Thus, cancer is truly a disease of genome.
Most human tumours are non-viral in origin and arise from spontaneous or induced mutations. Certain viruses, chemicals and ionizing radiations are also capable of causing similar changes in DNA, and hence fostering cancerous transformation of the affected cell.
A Somatic Mutations
Somatic mutations can cause malignant transformation of the cell in two ways:
1. By activation of the growth promoting genes called protooncogenes; the activated protooncogene is called oncogene.
2. By inactivation of the growth inhibiting genes (i.e. antioncogenes; also called tumour suppressor genes).
Certain growth promoting protein-products are encoded by the (activated) protooncogenes; examples include growth factors, the receptors for these growth factors and the components of the mitogen activated protein (MAP) kinase pathway. These protein-products can drive the cell into malignancy. Such mutations, causing activation of the protooncogenes, are called “gain of function mutations”.
Growth inhibiting protein products are encoded by the antioncogenes. Cyclin-CDK inhibitors, the retinoblastoma protein pRb, and the p53 protein are some examples of inhibitory proteins. Mutations in antioncogenes, decrease production of these growth inhibiting substances, leading to malignant transformation. Therefore, these mutations are referred to as the “loss of function mutations”
Malignant transformation can be caused by activation of the genes that code for growth-promoting proteins; or by inactivation of genes that code for growth-inhibiting products.
Malignant transformation occurs even if only single copy of a growth-promoting protooncogene becomes activated (Fig. 32.1 ). The inactivation of an antioncogene, on the other hand, is effective in causing cancerous transformation only if both copies of the gene are inactivated: a single active gene can still produce enough of the tumour-suppressing protein products to permit normal growth. Most of the cancers result due to a combination of several mutations that include both activated protooncogenes and the inactivated antioncogenes. A combined effect of these two types of mutations mostly drives the cell into malignancy.
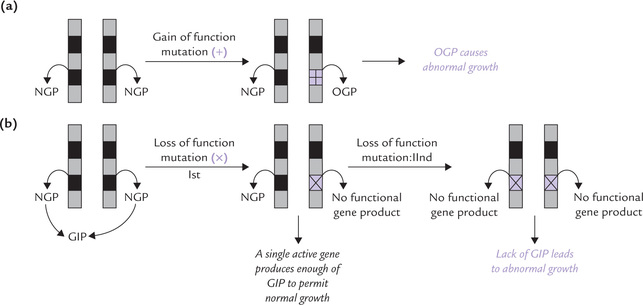
Fig. 32.1 Activation of growth promoting genes (proto-oncogenes) or inactivation of growth inhibiting genes (anti-oncogenes) cause malignant transformations, (a) A single mutation in protooncogene can cause malignant transformation, (b) In antioncogenes, a single mutation is insufficient to drive the cell into malignancy (NGP = normal gene product, OGP = oncogenic gene product, GIP = growth inhibiting product).
How does the Protooncogene Activation Cause Malignancy
Protooncogenes are important regulatory genes presen in normal cells that play a key role in cell growth an division. Their expression is meticulously controlled. I is only when the protein product encoded by the proto oncogene is required by the host cell, that these genes are expressed. Excessive production of such protein product provides excessive stimulation to the host cells, leadin to their malignant transformation.
The mutationally activated form of protooncogen (called oncogene) is also known to cause malignancy. It is generated by somatic mutations, of which the com monest are point mutations. Others are gene amplification, gene translocation, truncation, etc. These change in cellular genome, cause activation of a normal (proto onco-) gene to abnormal and overactive (onco-) gene which stimulates cellular growth and proliferation.
Thus, protooncogene-activation may cause malignanc in two ways:
1. By causing excessive and uncontrolled synthesis of a structurally normal gene product. (This usually follows a mutation in regulatory site of the gene).
2. By creating an abnormal protein product (oncoprotein) which is “superactive”, i.e. it causes rapid stimulation of the cell growth and proliferation.
Excessive amount of the normal-gene-product as also the overactive, oncogenic-gene-product provide excessive stimulation for cell growth, thus driving the cell into malignancy.
More than 80 protooncogenes are presently known, each one located on a specific chromosome. Some of these are listed in Table 32.1 .
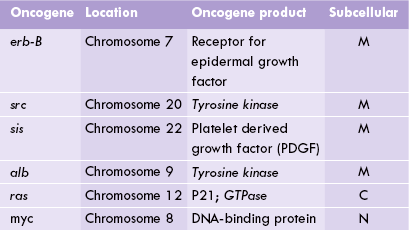
Antioncogene Inactivation — More Common Cause of Malignant Transformation
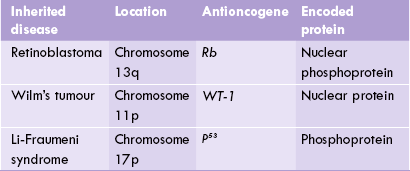
Antioncogenes offer protection against cancer since they code for proteins having inhibitory effect on cell division (Table 32.2 ). Mutation of antioncogenes, therefore, results in the loss of inhibition on cell division-a hallmark of cancerous growth (Fig. 32.1). In fact, inactivation of antioncogenes is more common than the formation of active oncogenes in spontaneous cancers.
The inactivating mutation can be a missense mutation, nonsense mutation, splice-site mutation, or a large deletion.
Two tumour suppressor genes, discussed as follows, have key roles in determining cell cycle progression and apoptosis, and are the genes most frequently mutated in cancer cells.
RB1 Gene
This antioncogene, also called retinoblastoma gene encodes a nuclear phosphoprotein with a molecular weight of 105,000. The protein, termed p110RB1, acts specifically in retinoblasts. It binds to DNA and inhibits the expression of some growth-promoting genes. This inhibits cell division. Deletion or loss-function mutations of the Rb gene removes this inhibitory effect in the neural progenitor cells in the retina, resulting in their cancerous transformation.
p53 Gene
It encodes a cell cycle regulator protein of 375 amino acids, with a molecular weight of 53,000. The p53 gene is expressed in almost all cells, and mutations in it are the most common genetic changes in spontaneous cancers. It plays a vital role during the cell cycle just before replication. It monitors for any signs of DNA damage, and if damage is detected, it causes arrest of the cell cycle in the G1-phase. This gives adequate time for repair of the damaged DNA, before the S-phase of the cell cycle begins. Alternatively, if the damage cannot be repaired in this time, it causes programmed cell death (apoptosis) of the cell. Thus, this gene plays a key role, not only in regulating G1/S-phase cell cycle progression, but also in monitoring DNA damage.
Individuals with inherited mutations in the p53 gene are predisposed to develop a wide range of tumours, including sarcomas, carcinomas of the lung, breast, larynx and colon; brain tumours; and leukaemias. This syndrome, called Li Fraumeni syndrome, is associated with a high incidence of various cancers at an early age in the affected families.
B Oncogenic Viruses
Some viruses, called oncogenic viruses, are capable of causing cancer by introducing a cancer-producing gene in the host cell genome (Fig. 32.2 ). Rous (Nobel Prize, 1966) was the first scientist to prove the role of viruses in aetiology of cancer by demonstrating that the cell-free filtrates from certain chicken sarcomas induced new sarcomas in chickens.
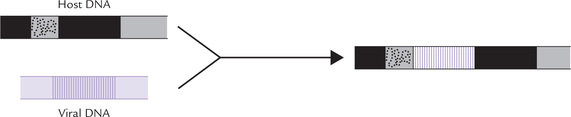
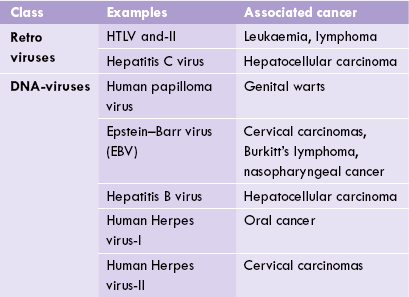
Viruses are nucleoproteins; their genomic material comprises either DNA (i.e. DNA viruses) or RNA (i.e. retroviruses). Certain viruses of either type are capable of producing cancer in the host cell. A selected list of cancer-causing viruses is given in Table 32.3 .
Oncogenic Retroviruses
Genomic material of these viruses is RNA. It directs synthesis of complementary DNA, known as retroviral DNA, by the enzyme reverse transcriptase. The retroviral DNA gets incorporated into the host cell genome, (and is now known as provirus). It fosters cancerous transformation of the host cell by either of the two mechanisms discussed here.
1. The retroviral DNA is expressed at a high rate under the directions of viral promoter and enhancer in the long terminal repeats (LTRs). This results in overexpression of the protein encoded by the viral oncogene (Fig. 32.3a ). The excessive production of the viral oncoprotein fosters malignant transformation of the cell.
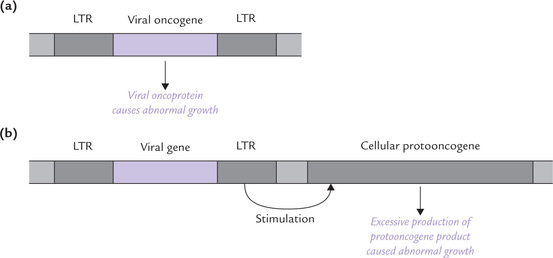
Fig. 32.3 Cancerous transformation of host cells following insertion of virion with long terminal repeats (LTR). (a) Excessive production of viral oncogene product causes abnormal growth, (b) The integrated retrovirus induces overproduction of the cellular protooncogene product which then leads to abnormal growth.
2. A second route to retroviral transformation arises when a virion with LTR is inserted into the host genome next to a cellular protooncogene. Transcription of the protooncogene is boosted up by the LTR (Fig. 32.3b). Thus, viral oncogene-insertion increases production of the protein product encoded by the neighbouring protooncogene. Excessive production of such protein, having growth enhancing action, results in uncontrolled cell division, and hence a cancerous growth follows.
Some retroviruses cause cancer by introducing an oncogene into the host cell genome or by inserting themselves next to a cellular protooncogene.
Viral Oncogenes Resemble Cellular Protooncogenes (Table 32.4)
In case of Rous sarcoma virus, the viral src oncogene (v-src) resembles a cellular oncogene (c-src) present in the normal cell (i.e. protooncogene). Apparently, the virus picked up this gene accidentally during a previous infection cycle. The v-src oncogene codes for a protein that possesses tyrosine kinase activity. This protein phos-phorylates several intracellular proteins, which in turn are involved in mitogenic signalling. Thus, production of the kinase protein by the v-src provides a potent stimulus for cell proliferation, which ultimately results in cancer.
Several retroviruses carry viral equivalents of cellular genes. Their oncogene product(s) foster cancerous transformation of the infected cell (Table 32.4 ).

DNA Oncogenic Viruses
Several groups of DNA viruses have been recognized which induce development of human cancers. In contrast to the retroviruses, however, the DNA viruses neither carry viral equivalent of cellular genes (v-onc) nor the LTRs. Instead their genomes encode proteins that are able to interact with, and inactivate, the regulatory proteins that control the cell cycle. The genomic material of these viruses gets incorporated in the host cell genome (Fig. 32.2). The viral genome is capable of overriding the regulatory checks and balances of the host cell and undergoes rapid replication. Along with it the rate of replication of the host cell genome is also greatly enhanced, being boosted up by the protein product encoded by the viral genome.
Some examples of DNA oncogenic viruses
These include human papilloma virus, Epstein-Barr virus (EBV) and hepatitis B virus (Table 32.3). Human papilloma virus causes genital and skin warts, and has a possible role in development of cancer cervix. Hepatitis B virus (HBV) is involved in development of hepatocellular carcinoma. Epstein-Barr virus is a herpes virus, which has been implicated in aetiology of Burkitt’s lymphoma. This cancer develops in children suffering from chronic malaria or in a transplant recipients.
C Chemical Carcinogenesis
Certain chemical agents, called chemical carcinogens, react with host cell DNA to induce mutations. Some of the mutant cells may die, some become normal through repair of mutation, but a few may be induced to undergo rapid multiplication. The latter action (i.e. proliferation-enhancing action) of these chemicals may result in rapid, uncontrolled cell division—a hallmark of malignant transformation.
Some of the carcinogenic chemical agents are enlisted in Figure 21.18. These may be organic or inorganic, and account for about 80% of all human cancers. They may enter the human body through diet, drugs or may be related to occupation (e.g. asbestos), or lifestyle (tobacco).
D Radiation Energy
X-Rays, gamma rays and electromagnetic radiations are mutagenic in nature, causing cancers. Similarly, particulate radiations (a-particles, (3-particles, protons and neutrons) are also capable of inducing cancer. These radiations can directly damage the DNA structure, or may produce free radicals, which in turn cause damage to DNA.
IV Molecular Basis of Protooncogene Activation
A central event in carcinogenesis is the protooncogene activation. This section deals with various ways by which the activation may occur.
A Point Mutation of Protooncogene
Certain protooncogenes, for instance the ras oncogene, are activated by point mutation (Fig. 32.4a ). The mutated ras oncogene produces a protein (GTPase) in which amino acid is substituted at the 12th position. This alteration greatly diminishes the GTPase activity. Because this activity plays a key role in control of all growth, its loss manifests as excessive proliferation of cell.
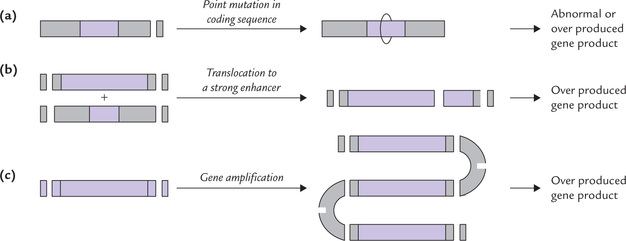
B Gene Rearrangements
A common type of gene rearrangement is chromosomal translocation which involves splitting of a piece of chromosome and joining it to some other chromosome. In this way, a gene is translocated to a transcriptionally active site where it is under the influence of some strong enhancer or promoter sequence (Fig. 32.4b). The translocated gene is overexpressed at the new site.
Chromosomal translocation has been described in the following conditions:
Burkitt’s Lymphoma
The c-myc gene locus on chromosome 8 is split off and joined to chromosome 14. At this site the c-myc gene is rapidly expressed under the influence of the local enhancer sequence. Consequently, the protein product of the c-myc gene is produced in large amounts, which makes the cell malignant.
Leukaemias
Gene rearrangements have been described in chronic myelogenous leukaemia and acute lymphoblastic leukaemia. In these diseases, recombination between the chromosome 9 and the chromosome 22 occurs with the following consequences. Shortening (or deletion) of the short arms of the chromosome 22 occurs (due to translocation of the genetic material to the chromosome 9). The abnormal chromosome 22 so produced is called philadelphia (Ph’) chromosome, which is seen in 80-85% patients of chronic myelogenous leukaemia (CML) and in 5-20% patients of acute lymphoblastic leukaemia (ALL). The abnormality persists throughout the course of the disease (in remission as well as in relapse), and is generally unaffected by therapy. In about 20% cases of CML, there is translocation of chromosomes 9-22, which results in activation of c-alb present on the chromosome 9.
C Gene Amplification
Gene amplification may cause overproduction of certain proteins because of several fold amplification of certain DNA sequences (Fig. 32.4c). Gene amplification is frequently seen in cancer cells, when the patient is treated with methotrexate. This drug inhibits the enzyme dihydrofolate reductase (DFR). In an attempt to develop resistance against this drug, the gene for the DFR gets amplified, resulting in increased production of the enzyme (up to 400-fold).
D Promoter Insertion
The retroviral DNA is inserted immediately upstream of the cellular protooncogene and enhances its expression as shown in Figure 32.5a . The retroviral DNA has two identical promoter regions contained in the long terminal repeats (LTRs). The promoter located in the downstream LTR is used for enhancing the transcription of protooncogene. The transcription occurs at an abnormally high rate, without any negative controls that normally regulate the rate of transcription.
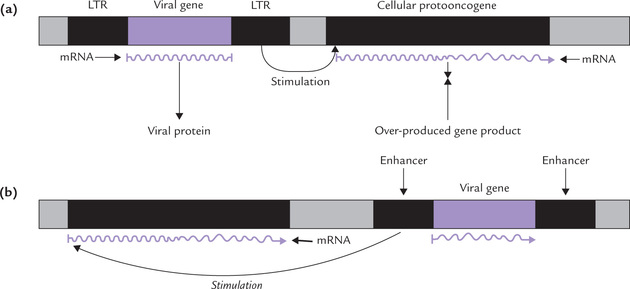
E Enhancer Insertion
The viral enhancer (in the LTR of retrovirus) may also stimulate transcription of the neighbouring protooncogene (Fig. 32.5b). The viral enhancer differs from the promoter as it can act over considerable distances; in some cases it is located 10,000 base pairs away from the protooncogene. The stimulation occurs even if the viral enhancer is:
• inserted downstream of the protooncogene
• inserted with opposite polarity (i.e. wrong 5’→3’ orientation).
Thus, the viral enhancers have similar actions as the normal cellular enhancers (Chapter 24).
V Some Oncoproteins and Associated Cancers
The protein products of oncogenes are involved in transmission of mitogenic signals, thereby causing transformation and multiplication of cells. They can be divided into four categories: growth factors, growth-factor receptors, GTP-binding protein and non-receptor tyrosine-kinase.
A Growth Factors
Several growth factors that allow the cell to respond to signals from distant sources are known. Most growth factors stimulate cell proliferation, but some others cause their target cell to migrate, to differentiate, or to grow to a large size without dividing. Growth factors even can induce opposite effects in different cell types causing some cells to proliferate and others to stop growing, or to undergo terminal differentiation. Some of them act on a wide variety of cell types; others have narrow specificities (Table 32.5 ).
Growth factors transmit the signal across the plasma membrane to the interior of the cell, thereby regulating cell division. They act by binding to a protein receptor on the plasma membrane, which contains tyrosine kinase domain within its covalent structure. The above binding activates the receptor by phosphorylation, triggering a signalling cascade that leads to phosphorylation of intracellular transcription factors. The latter in turn regulate expression of genes that affect cell growth and proliferation.
Insulin-like growth factor (IGF-1), for example, is released from the liver in response to growth hormone. The growth promoting effects of growth hormone is mediated, in part, by IGF-1. Pygmies are said to have just as much growth hormone as taller people, but they release less of IGF-1.
B Growth Factor Receptors
Growth factors bind to the extracellular domains of trans-membrane receptors that have tyrosine kinase domains present within their intracellular domain. These receptor-tyrosine-kinases are monomeric and enzymatically inactive in the absence of the growth factor. The binding of the corresponding growth factor causes receptor to dimerize and undergo autophosphorylation and activation (Fig. 32.6 ).
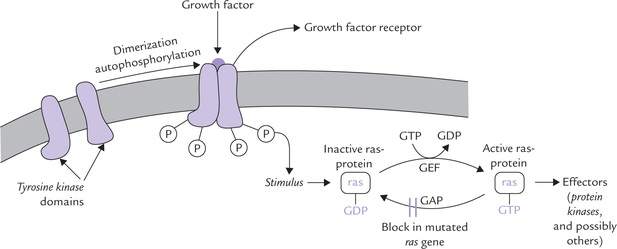
Fig. 32.6 Activation of ras-protein by growth factors. The sequence of events is: binding of GF → Dimerization of stimulated GF receptor → Receptor autophosphorylation →Activation of ras (stimulated by GEF) → Hydrolysis of the bound GTP by ras (stimulated by GAP) (GEF = guanine nucleotide exchange factor, GAP = GTPase activity promoter).
The phosphotyrosine residues in turn cause phosphorylation of cytoplasmic proteins, which act as intracellular messenger and stimulate cell division by still unknown mechanism.
C GTP-binding Proteins
These signal transducing proteins transfer the signal beyond tyrosine kinase. They have been detected in about 30% of the human cancers, and are believed to play a major role in cell growth and differentiation. An example is ras protein (or p21), encoded by ras-gene. It is a small GTPase (MW 21000), which controls cell division, but when mutated, leads to cancer, as discussed here.
The ras-protein cycles between an active GTP-bound form and an inactive GDP-bound form (Fig. 32.6). Upon stimulation by growth factors, the inactive form is activated by exchange of GTP for GDP. The exchange is facilitated by a guanine nucleotide exchange factor (GEF), which opens a nucleotide-binding site, allowing the GDP to escape and GTP to enter in its place. The activated ras-protein causes activation of effectors (protein kinases and others), which phosphorylate specific targets that promote cell growth and differentiation. This effect is, however, transient only because the activated ras gets inactivated quickly. This is because of intrinsic GTPase activity of the ras-protein. This activity serves to terminate the signal and return the activated rasGTP to its original inactivated state (ras-GDP). Although the activity is slow, it is augmented by helper protein termed GTPase activity promoter (GAP).
Point mutation in ras gene results in the production of altered ras-protein, which loses its GTPase activity. This leads to “locking” of the protein in its activated form (i.e. persistent activation), causing uncontrolled cell multiplication.
D Non-receptor Kinases
These proteins are located on the interior of the inner plasma membrane. They respond to external growth stimuli and cause phosphorylation of certain cytoplasmic proteins, which are involved in cell growth and differentiations. Mutations in the genes encoding these proteins cause increase in their kinase activity, and this in turn, increases phosphorylation of the cellular target protein, and this boosts up cell multiplication.
VI Tumour Markers
A tumour marker is any substance that is produced by the tumour cell, and which is related to the presence or progress of a tumour.
It is known that metabolism of tumour cells differs from that of the normal progenitor cells. Certain genes that are active during fetal period, but are suppressed in adult cells, are reactivated in the tumour cells. Therefore, tumour cells synthesize certain abnormal proteins, such as surface antigens, cytoplasmic proteins or enzymes, measurement of which (in plasma or serum) provides a useful marker for supporting cancer-diagnosis and follow up. Thus, these proteins serve as biochemical markers of cancer, and accordingly referred to as tumour markers.
The α-fetoprotein (AFP), for example, is secreted by fetal liver cells but the adult liver loses capability to synthesize this glycoprotein. In patients with hepatocellular carcinoma, the liver cells synthesize large amount of AFP and release it into the serum. Therefore, serum AFP levels increase markedly (Table 32.6 ). In malignant testicular teratoma also, elevation of AFP has been reported in about 90% of the patients. Thus, AFP estimation is a useful indicator of hepatic and testicular malignancies.
Table 32.6
| Cancer marker | Clinical utility |
| Tumour antigens | |
| α-Fetoprotein | Increases in tumours of liver and germ cells |
| Carcinoembryonic antigen | Increases in cancers of colon, pancreas, lung and breast |
| Enzymes | |
| Acid phosphatase | Increases in carcinoma prostate |
| Alkaline phosphatase | Increases in metastasis of liver, bone and carcinoma of head of pancreas |
| β-Glucuronidase (urinary) | Increases in cancers of pancreas and urinary bladder |
| Hormones | |
| Human chorionic gonadotropin | Increases in choriocarcinoma |
| Calcitonin | Increases in medullary carcinoma of thyroid |
| Others | |
| Prostate specific antigen (PSA) | Increases in prostate cancer |
| Paraprotein | Appears in multiple myeloma |
Characteristics of a Required Tumour Marker
1. Be clearly related to the malignant process.
2. Provide information on the type and location of the tumour and its stage.
3. Be of value in prognosis. For this the concentration of tumour a marker should correlate with tumour mass.
4. Be useful for monitoring treatment: the decline in concentration of a marker is an indication of the success of the treatment (surgery, chemotherapy, radiotherapy, or a combination of these).
However, in reality most tumour markers have certain limitations.
Tumour marker, the substance related to presence or progress of a tumour, may be hormone, enzyme, or oncofetal protein. Tumour markers are of most value in monitoring treatment and assessing follow up, but are also used in diagnosis, prognosis and screening for the presence of a disease.
A host of tumour markers has been described, which fall into one of the several groups: they may be tumour antigens, e.g. carcinoembryonic antigen (CEA) in colorectal carcinoma; or hormones, e.g. calcitonin secreted in medullary carcinoma of thyroid; or enzymes, e.g. acid phosphatase in carcinoma prostate. Some important ones are described here.
Carcinoembryonic antigen (CEA):
Measurement of serum level of CEA, a complex glycoprotein (MW 180,000) produced from embryonic tissues of liver and intestine, has been found useful in colorectal cancer. In about 65% of patients with this cancer, CEA can be detected in serum. In advanced stages of the disease, especially when the metastatic spread involves the liver, about 90-100% of the patients have elevated CEA levels.
However, lack of sensitivity and specificity limit utility of CEA for screening colorectal cancer. Elevated levels are also found in gastric, bronchial and ovarian carcinomas, as also in a number of non-malignant conditions, particularly liver diseases of various types, pancreatitis, and small bowel diseases. Heavy smokers (57%) also have high levels of CEA. Thus, CEA is not a reliable indicator of cancer detection and may lead to diagnostic ambiguities. Its utility in follow up of the known cases of colorectal cancer is also not established. The fact that it does not relate to the mass of the tumour limits its utility in monitoring the response to treatment.
α-Fetoprotein (AFP):
It is glycoprotein (MW 7000) that is synthesized in yolk sac in early fetal life. Normal serum level is 12 U/ml. It helps in the diagnosis of liver and germline carcinomas along with indicating the presence of spina bifida, amencepalia and atresia of oesophagus.
Human chorionic gonadotropin (hCG):
Normal serum level is 5 IU/ml and it is useful in mammary carcinoma and trophoblastic tumours.
Prostate specific antigen (PSA):
It is a 32 HD glycoprotein, produced by secretory epithelium of prostate gland. It is secreted into seminal fluid where it exists either in a free form, or complexed with α1-antitrypsin. PSA has protease activity, and is necessary for liquefaction of the seminal coagulum. The PSA level (especially of complexed form) is elevated in prostate cancers.
Presently, definition and scope of tumour markers has been expanded, and it includes not only those substances that are secreted in blood, but also those found on the surface of or within the cells. Such markers are not necessarily unique products of the malignant cells, but may simply be expressed by the tumours in greater amount than by normal cells.