Molecular Biology I: DNA Structure, Genetic Role and Replication
Genetic information stored in deoxyribonucleic acid (DNA) is inherited and expressed. Inheritance involves transfer of genes from parent cells to daughter cells; a gene is the length of DNA that directs synthesis of a polypeptide. At molecular level, inheritance requires replication whereby the parent DNA directs synthesis of daughter DNAs. The latter then passes into the daughter cells during cell division.
Gene expression determines characteristics of an individual. At molecular level, it involves DNA directed synthesis of proteins. This process occurs in two stages: transcription and translation. During transcription, which takes place in the nucleus and is directed by DNA, messenger RNA (mRNA) is synthesized. Structure of mRNA mirrors the image of a segment of DNA. In translation, the structure of the mRNA specifies the amino acid sequence of a polypeptide. In prokaryotic cells, translation follows immediately on transcription, whereas in eukaryotic cells these processes are separated both in time and place; transcription occurs in the nucleus and translation in the cytoplasm.
This chapter deals with the structure and genetic role of DNA. Causes of DNA damage and repair mechanism are also explained in detail. After going through this chapter the student should be able to understand:
• Structure of DNA: polynucleotide chains; size of DNA; and double helix.
• Physical properties of DNA: denaturation; buoyant density and supercoiling.
• DNA as genetic material: extra-chromosomal DNA; gene sequencing; architecture of human genome.
• Replication: semiconservative; initiation, elongation and termination in DNA replication: DNA polymerases.
• Mutations: Types and causes of mutations; repair systems for mutated DNA.
I Structure of Deoxyribonucleic Acid (DNA)
DNA is the largest macromolecule in the body, consisting of millions of nucleotide units that are linked covalently. DNA is the storehouse of the genetic information which is translated into perceptible traits like the colour of skin, hair texture, height, etc.
A Basic Chemistry
DNA is an antiparallel dimer of nucleic acid strands. Each strand is a linear deoxyribose-phosphate chain with purine and pyrimidine bases attached to the 2’-deoxyribose subunits. The deoxyribose differs from the ribose in that it lacks the hydroxyl group at the 2’-position. Carbon-3’ and -5’ of the deoxyribose subunits are involved in ester linkages with an inorganic phosphate to form a 3’,5’-phosphodiester bond (Fig. 21.1 ). Thus, alternating phosphate and 2’-deoxyribose units form the backbone structure of a DNA strand. The C-1 of the deoxyribose forms a β-N-glycosidic bond with nitrogen-1 of a pyrimidine base or with nitrogen-9 of a purine base.
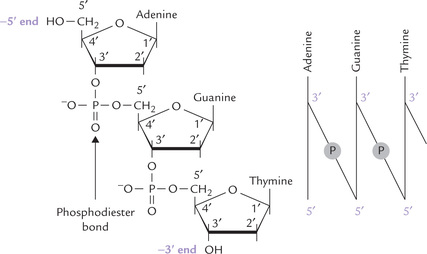
Fig. 21.1 Structure of a segment of DNA and formation of a phosphodiester bond between adjacent nucleotides.
The bases in DNA are adenine, guanine, cytosine and thymine; they lie flat in the interior of the dimer (Fig. 21.2 ). The phosphate groups are strongly acidic and, therefore, DNA molecule carries multiple negative charges at the physiological pH.
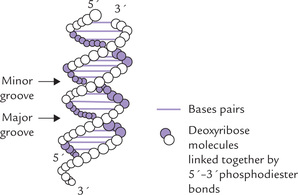
Antiparallel strands: By antiparallel it implies that the two chains of a double helix have opposite polarity, i.e. they run in opposite directions. The polarity of a DNA strand is defined by two distinct ends:
• The 5’-end: This end has a free 5’-OH group on the deoxyribose as shown in Figure 21.1. The 5’-OH group is not connected to another nucleotide, though sometimes it may have a phosphate group esterified to it.
• The 3’-end: This end has a 3’-OH group that is not connected to another nucleotide (though it may also have a phosphate group on it).
Figure 21.1 shows a segment of DNA with the 5’ end at the top and the 3’ end at the bottom.
Polarity of the DNA chain is analogous to that of the polypeptide chain with an amino terminus and a car-boxy terminus. Since the carbons of 2’-deoxyribose are numbered by a prime to distinguish them from the carbons and nitrogens of the bases, the ends are designated as 5’ and 3’ ends. A closer look at this simple oligonucleotide reveals that variability of DNA structure depends on sequence of bases. Since DNA consists of four bases, it is possible to construct 64 (43) different trinucleotides with them.
B Size of DNA
DNA molecules are extremely large. Contour length (the end-to-end length of a stretched-out native molecule) of a DNA molecule is several times more than the dimensions of the cell that accommodates it. For example, the DNA olecule of an Escherichia coli cell contains 4.7 million base pairs. Its contour length is 1.4 millimeter, which is 700 times greater than the diameter of the bacterial cell. Size of the genome of various other living systems and viruses is shown in Table 21.1 .
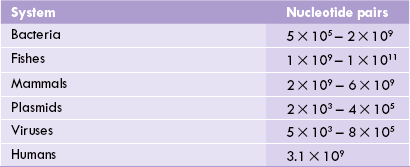
Viruses contain DNA which amounts to about 10% of DNA present in bacterial cells, which is consistent with the fact that viruses do not contain sufficient information for independent growth. On the other extreme are the eukaryoticcells which contain far more DNA than prokaryotes. An individual cell of a slime mold, one of the lowest eukaryotes, contains over 10 times more DNA than the E. colicell. Length of the human genome (all the chromosomes taken together) totals one to two meters; there are about 1013 cells in a human being and if we add up the total length of DNA in a person, it comes out to 2 × 1010 km: an astronomical length, of the order of the diameter of the solar system. Clearly, more complex the organism, greater would be the DNA content of its cell, but exceptions do exist. For example, a broad bean cell has more DNA than a human cell, and among vertebrates amphibia have the most. The apparent anomalies in DNA content are related to the fact that a vast majority of DNA is not in the form of functional genes, as described later.
C The Double Helix
Based on X-ray diffraction photographs of DNA taken by Rosalind Franklin, a two chain higher order structure was proposed by James Watson and Francis Crick in 1953 (Nobel Prize, 1962). The prominent features of this model of DNA, known as Watson-Crick’s double helix (now known as B-DNA) are as here.
1. DNA is composed of two poly-deoxyribonucleotide(or simply deoxynucleotides) strands of opposite polarity, wound around each other on a common axis in aright-handed, helical structure (Fig. 21.2). Each strandis composed of monomeric units, of deoxynucleoside 5’-monophosphates namely
2. The more hydrophilic deoxyribose subunits and phosphate groups are on the superhelix exterior, in contact with the aqueous environment; whereas the planar bases are stacked in the interior, where the environment is hydrophobic. The resulting helix has a spiral staircase appearance—the deoxyribosylphosphate residues act as the backbone, and the bases, which are oriented perpendicular to the superhelix axis, act as steps.
3. The bases on one strand interact with the bases on the other strand to form base pairs, which are planar and oriented nearly perpendicular to the axis of helix. (Fig. 21.3 ). The composite strength of the hydrogen bonds formed between the bases of the opposite strands is responsible for holding the two chains together and maintaining the double-helix structure.
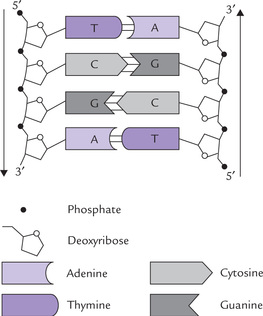
Fig. 21.3 DNA segment showing orientations of different components of the nucleotide subunits of DNA.
4. Each base pair is formed by hydrogen bonding between a purine and a pyrimidine. To form hydrogen bonds, the tautomeric forms of the bases must be of the keto type (Chapter 20).
5. Because of the specificity of interactions between purines on one strand and pyrimidines on the opposite strand, the two strands are said to possess structures complementary to each other. Complementary base pairing occurs between A and T (A = T), and between G and C (G = C). As a result, the double-stranded DNA molecules always contain equal (molar) amounts of A and T, and of G and C. Each A-T base pair is held together by two hydrogen bonds and each G-C by three. The complementary base pairing proves the Chargaff rule (refer to Box 21.1).
Alteration of base pairing is potentially a disastrous event for the cell; it may lead to abnormal cell growth or even cancer (Case 21.1).
6. The bases lie flat in the interior, stacked on top of one another, each base being turned about 35° with respect to the next. They interact through van der Waals forces and hydrophobic interactions. Collectively these two forces are known as base-stacking interactions because of their contribution to the stacked arrangement of bases in DNA.
7. Each strand is wound into a right-handed helix, and each turn accommodates 10 base pairs. Since adjacent base pairs are stacked 3.4 A° apart, pitch of the helix is 3.4 × 10 = 34 A°.
8. The ribosylphosphate backbones of the two strands are slightly offset from the centre of the helix and so two types of grooves of unequal width, the major groove and the minor groove, run the length of the DNA molecule. The major groove is more open (12 A° wide) and deep, and the minor groove is narrow (6 Å wide) and shallow. The unequal size is because the N-glycosidic bonds are not exactly opposite each other. Within these grooves, the bases are exposed and hense access is possible for molecules, which need to interact with base (e.g. the regulatory proteins). The latter interacts with DNA at these grooves without affecting properties of the double helix.
9. The double helix has a stiff, extended native conformation because of electrostatic repulsion between phosphate groups. However, it can be bent and twisted to a limited extent without major distortions of the regional structure.
 The double helical structure is stabilized by Watson-Crick base pairing and by base-stacking interactions (i.e. van der Waals forces and hydrophobic interactions) between stacked base pairs.
The double helical structure is stabilized by Watson-Crick base pairing and by base-stacking interactions (i.e. van der Waals forces and hydrophobic interactions) between stacked base pairs.
Sense and antisense DNA strands
DNA transcription is normally initiated at specific sites on the DNA template and involves small, single-strand portions of the genome. The strand of the duplex DNA that serves as template during transcription is known as the antisense- or non-coding-strand since its sequence is complementary to that of the transcribed RNA. The other DNA strand is its sense or coding-strand. It has the same nucleotide sequence and orientation as the transcribed RNA (except for the replacement of U with T). Two strands of DNA in an organism’s chromosome can therefore contain different genes.
D Alternate Higher Order Structures of DNA
The double helix may assume a number of shapes in three-dimension. Although the vast majority of the DNA in living cells exists in the form described above (B-form), five alternate forms have also been recognized (A-, C-, D-, E- and Z-DNA). Among these, A and Z forms are important and may help to regulate gene expression. Their comparative features are listed in Table 21.2 .
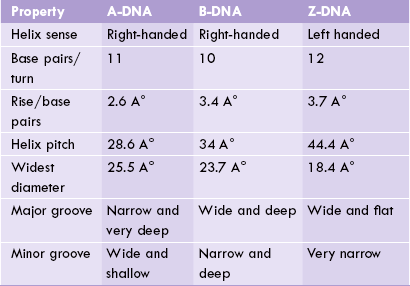
A-DNA
First reported by Richard Dickerson, this form is more squat in shape with its bases tilted. In the laboratory, at low salt concentrations and a maximum degree of hydration, the B-form predominates; but, when the salt concentration is increased and degree of hydration lowered, a reversible B- to A-DNA transition occurs. The A-helix is right-handed with 10.7-11 base pairs per helical turn and a rise of only 2.6 A° per base pair. The base pairs are tilted 19 A° relative to the helical axis. These features cause the helix to be thicker and more compact than B-DNA.
Small stretches of A-DNA are found within the conventional B-DNA strands. Double-stranded DNA-RNA hybrids also tend to assume the shape of A-DNA, because the -OH groups in the 2’ position prevent the B-conformation.
Z-DNA
The Z-form, first reported by Alexender Rich, is named so (Z stands for “zigzag”) because location of the phosphodiester groups on the outer surface of the Z-helix can be connected by a broken (zigzag) line, rather than a smooth spiral as in D-DNA. The helix is left-handed, and is thinner and more stretched out than B-DNA or A-DNA. It has 12 pairs per turn and a 3.7 A° rise per base pair. While formation of Z-DNA is favoured at high ionic concentration, it can also be induced at normal ionic concentration by DNA methylation. The control of gene expression caused by DNA methylation may be mediated by B- to Z-transition.
Base Composition Dictates Higher Order Structure
Formation of the alternate forms in a given segment of DNA is determined by its base composition:
(i) the polypurine segments become A-like,
(ii) the polypyrimidine segments are B-like, and
(iii) the segments in which purine and pyrimidine basesalternate (especially when G and C follow eachother and C is methylated in 5’ position), theZ-DNA is formed.
All DNAs have grooves, which can interact with proteins such as histones. Such grooves have different dimensions, and therefore different protein-binding affinity in various forms of DNA. It is possible that the presence of the A- or Z-DNA within the normal B-DNA strand is a means of regulating its biological activity through changes in binding of regulatory proteins. This may help to regulate the gene expression.
II Some Physical Properties of DNA
A Denaturation
The term denaturation refers to disruption of native conformation of a biomolecule, so that it does not retain its higher-order structure. In case of DNA, denaturation refers to separation of the double strands of the DNA into two component strands. When temperature of the medium containing a DNA molecule is raised, the hydrogen bonds linking the complementary base pairs tend to break. As a result, separation of the two polynucleotide chains occurs.
Melting Temperature
Denaturation, also called melting of DNA, occurs over a narrow range of temperature. Melting temperature (Tm indicates the temperature at which half of the double-stranded structure is lost. At the physiological pH and ionic strength, the melting temperature of DNA is between 85°C and 95°C. Since G and C are linked more strongly by three hydrogen bonds, in contrast to hydrogen bonds linking A and T, the DNA molecules having higher GC content have relatively higher melting temperature. For every 10% increase in GC content, the melting temperature increases by 5°C. One simple formula is Tm (° C) = 2 [number of AT base pairs] + 4 [number of GC base pairs].
Monitoring of Strand Separation
Denaturation affects properties of DNA and these can be used to monitor strand separation.
1. Hyperchromia effect: The heterocyclic DNA bases absorb ultraviolet light of wavelength 260 nm. Absorbance of this light increases by 40% upon denaturation. This is called hyperchromic effect. Thus, melting of DNA is monitored by the increase of UV absorbance: the melting temperature is characterized by a sharp increase in absorbance.
2. Viscosity: Viscosity of the DNA solution decreases on melting because the single strands are far more flexible than the stiff, resilient double helix.
Denatured DNA can Undergo Renaturation
Renaturation, also termed reannealing, is the process of formation of DNA double helix from two separated strands of DNA. For example, with gradual cooling to 5°-20°C below the melting temperature, the separated strands of the denatured DNA tend to join and form double-helix because of reformation of base pairs. This process, known as annealing (or renaturation), is useful in determining genetic similarity between DNAs derived from different organisms. It is a rapid process; even the large DNA molecules may take few seconds to minutes for renaturation.
The Watson-Crick double helix being unstable, the DNAs can undergo denaturation (into random coils), and can also reanneal under appropriate conditions.
In addition to heat, alkali treatment and decrease of salt concentration also cause denaturation. A variety of chemical agents that interfere with hydrogen bonding or base stacking are effective denaturants as well. Formamide, for example, is used as an alternative to heat dena-turation in the laboratory. However, acid and ethanol cause DNA precipitation.
B Buoyant Density
The maximum buoyant density of DNA is 1.70 ± 0.01. The DNA molecules having higher GC content have more compact structure and hence greater buoyant density. For every 10% increase in the GC content, the density rises by 0.12 units.
C Hybridization
This refers to the pairing between RNA and complementary base sequences of a strand of DNA to form DNA-RNA hybrids that are slightly less stable than the corresponding DNA double helices.
D DNA Supercoiling and Topoisomerases
Many naturally occurring prokaryotic DNA molecules are circular. The circular DNA is compacted by supercoiling which forms an important aspect of the DNA tertiary structure. In the relaxed form, the two polynucleotide chains of a DNA molecule are wound around a common central axis. Supercoiling implies twisting of the central axis of DNA upon itself (Fig. 21.4 ). It is not a random process but is precisely regulated by action of enzymes called topoisomerases. These enzymes cause under-winding of the double-helix by removing one or more helical turns in its relaxed state (10 base pairs per turn). If one turn is removed by these enzymes, only seven turns would remain for the 84 base pairs. Each turn now accommodates 12 base pairs instead of 10. This is a deviation from normal and, therefore, it induces a thermo-dynamic strain. This strain can be relieved either by partial strand separation or by supertwist of the duplex around its own axis—much as a telephone cord twists around itself. This kind of supertwist permits the neighbouring bases to assume positions which closely resemble position of bases in a double helix.
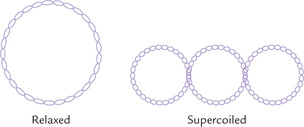
The supercoiling mentioned above arises due to under-winding and is called negative supercoiling. The opposite situation arises due to over-winding and is called positive supercoiling. Most bacterial DNAs are super-twisted negatively, having approximately 5% to 7% fewer turns than expected from the number of base pairs. This is helpful because:
(a) Negative supercoiling counteracts, to some extent, the positive supercoiling upstream of the unwinding DNA (Chapter 21).
(b) Supercoiling also aids in the process of DNA packaging, discussed later. The negative supercoiling shown in Figure 21.4 is introduced by an enzyme in prokaryotic cells, called gyrase, with the expenditure of ATP energy. Gyrase is a member of a group of topoisomerases.
Topoisomerases: The topoisomerases generate supercoils, but their more important role is to relieve the supercoils during the process of DNA replication and transcription. They perform this role by cleaving the phosphodiester bond on one or both DNA strands (nuclease activity) and then resealing the “nick” (ligase activity) after rotation of the DNA strand(s) to relieve tension. Various types of topoisomerases (Type I to IV) have been recognized. Topoisomerase I breaks only one strand, and topoisomerase II breaks both. Gyrase is topoisomerase II. Topoisomerases are thus integral components of DNA replication and transcription mechanisms (see Table 21.4).
III DNA as Genetic Material
DNA carries the blue-print of heritable traits in living systems. Simply stated, DNA is the genetic material in all prokaryotic and eukaryotic organisms. The genetic material must fulfill the following basic requirements:
1. It must serve as bearer of genetic information and be able to transmit this information with a high degree of fidelity to the daughter cells.
2. It must be stable so that the stored information remains protected.
3. A limited degree of variation must be allowed so as to provide raw material for natural evolution.
Miescher first isolated DNA in 1869, but the first experimental evidence to indicate that it is the genetic material came much later from the persuasive experiments of Avery, Macleod and McCarty (1944) on bacterial transformation. These workers demonstrated transformation of an uncapsulated strain of the bacterium Pneumococcus into a capsulated form by the addition of DNA extracted from a capsulated colony.
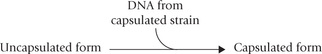
The DNA preparation used was shown to be free of proteins, and the DNA cleaving enzyme, deoxyribonuclease, was shown to destroy its activity. These observations provided evidence that the specific alteration of a heritable function (uncapsulated to capsulated transformation) was brought about by transfer of DNA.
Hershey and Chase in 1952 proved that viral DNA was infectious. These workers infected E. coli by T-2 bacterio-phage. DNA of the latter was labelled with P-32 and the protein coat was labelled with S-35. When E. coli was infected with this doubly labelled phage, only P-32 labelled nucleic acid entered the bacterial cell. It was thus observed that only DNA could enter the cell, none of the other viral constituents (e.g. proteins or carbohydrates) had this ability.
A Extrachromosomal DNA
In eukaryotic cells, small amount of DNA is present in mitochondria. The mitochondrial DNA (mt DNA) undergoes replication and directs protein synthesis, independent of chromosomal DNA. Precise role of the mt DNA is still unclear. Probably it represents vestiges of the foreign DNA (bacterial) that infected the cells early in evolution (Chapter 24).
In bacterial cells, extrachromosomal DNA is present in structures called plasmids. Each plasmid contains circular DNA material. The plasmid DNA codes for proteins that confer resistance to antibiotics. Plasmids can be transferred from one cell to another and are, therefore, called vector molecules (Chapter 25).
Gene Sequencing
Several techniques have been developed for determining base sequence of DNA. One of these, called Maxam-Gilbert method employs selective chemical cleavage of a single-stranded DNA sample. The sample is divided into four batches and subjected to selective chemical cleavage at specific bases. Batch 1 is cleaved on 5’ side of cytosine; batch 2 is cleaved on 5’ side of guanine; batch 3 cleaved on 5’side of purines (A or T); and batch 4 on 5’ side of pyrimidines (C or T).

Smaller sets of DNA fragments are obtained (one set from each batch), whose length identifies position of a particular base. For example, a 20-nucleotide fragment in the batch 2 (G only) reaction mixture identifies G at position 21. Similarly, a fragment containing 23 nucleotide in batch 4 (C or T) reaction, but not in batch 1 (C only) reaction, indicates that T is at position 24.
The length of DNA fragments is determined by polyacryl-amide gel electrophoresis, a technique that can separate fragments differing in length even by one nucleotide.
Another sequencing technique is the Sanger method (developed by Fred Sanger), which is based on controlled interruption of DNA synthesis by dideoxynucleoside tri-phosphates (ddNTP). The latter can be incorporated in the new strand by DNA polymerase, but halts further chain growth (Fig. 21.5 ). Enzymatic DNA synthesis is performed in four test tubes. Each tube contains all the four deoxyribonucleoside triphosphates (dATP, dGTP, dCTP, dTTP) but only one of the four ddNTP. For example, in tube 1 ddATP is added, in tube 2 ddGTP is added, etc. In each test tube a single-stranded, DNA template is copied.
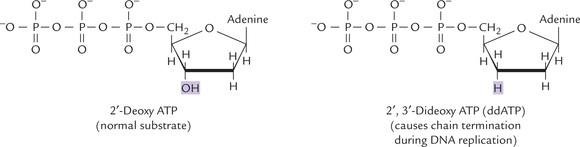
Fig. 21.5 Structure of a dideoxynucleoside triphosphate. DNA polymerases can incorporate a dideoxynucleotide into a new DNA strand, but further chain growth is prevented for lack of a free 3’-hydroxy group.
A series of radioactive DNA fragments are generated, differing in length. The length depending upon:
(a) Type of ddNTP added: in tube 1, chain termination occurs at A, in tube 2 the chain terminates at G.
These fragments are separated by polyacrylamide gel electrophoresis according to their size. The sequence of the DNA fragment can be directly deduced from the pattern of radioactive bands in the gel.
Determination of base sequence of the entire DNA of an organism, called gene mapping is now possible with tools of recombinant DNA technology (Chapter 25).
IV Architecture of the Human Genome
In human cell there are 46 chromosomes, as already mentioned, with a total length of DNA per cell of 1-2 meters. It has to be packed into a nucleus millions of times smaller in size (dimensions, 6 × 9 μm). DNA has responded to this challenge over the course of evolution through a very elaborate packing procedure by associating with a group of basic proteins called histones. The nucleoprotein complex so formed contains 50% DNA and 50% protein, and is called chromatin. It occurs in two forms: euchromatin, which has a loose structure and heterochromatin, which is more condensed and stains more deeply with basic dyes. Only euchromatin contains actively transcribed genes. Evidently, the access to information is not interfered with (due to tight DNA packaging) in chromatin.
Eukaryotic genomes are composed of several linear DNA duplexes that are organized into separate chromosomes. Each chromosome has a single DNA, stabilized by interaction with histones, the basic proteins.
Chromatin stains with basic dyes, such as haematoxylin and fuschin, and so is conspicuous in histological preparations. Chromatin from the resting interphase is usually studied, wherein it is visualized as amorphous and randomly distributed material. But in dividing cells (meta-phase), the chromatin condenses and assembles into specific number of well-defined chromosomes.
A Histones
There are five histones associated with DNA, essential features of which are listed in Table 21.3. They are rich in arginine and histidine, giving them a positive charge, that forms ionic bonds with the negative charges on the phosphate groups of the double helix. The amino acid sequences of eukaryote histones are highly conserved throughout the phylogenic tree. (This suggests that histones were invented early during evolution to precisely combine with an equally invariant structure, i.e. DNA). Thus, the histones, H3 and H4, from pea seedlings and calf thymus differ in only four and two amino acid positions respectively, and the changes are very conservative (valine for isoleucine, lysine for arginine). However, such extreme conservation is rarely seen in H1, which shows variation in structure in different species, and even in different tissues of the same organism.
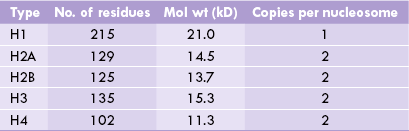
B Nucleosomes
Clusters of histones consisting of two molecules each of H2A, H2B, H3 and H4 form an octamer protein complex, called a nucleosome core, around each of which the DNA wraps two turns (about 146 base pairs) in a left-handed orientation. The octamer with its DNA is a nucleosome, a small disc-like structure with a diameter of 10 nm. The nucleosome is the structural unit of chro-matin: in electron microscopy it shows like beads on string (Fig. 21.6a ), where the beads are nucleosomes.
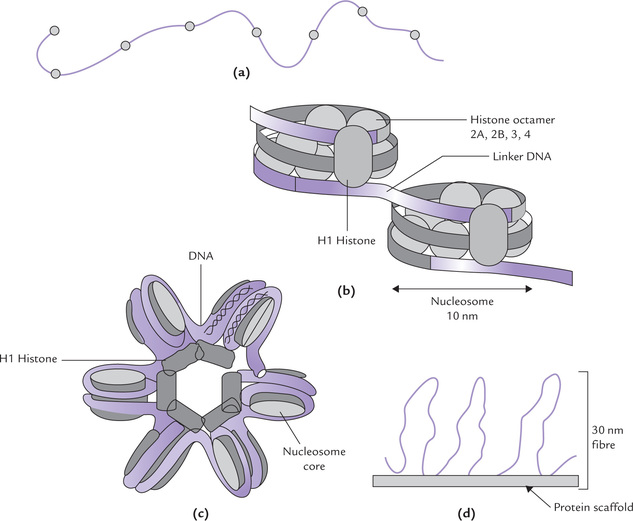
Fig. 21.6 Chromosome structure. (a) Swollen fibre of chromatin, (b) Nucleosome connected by linker DNA, (c) Formation of helix or solenoid: there are 6 nucleosomes per turn in the solenoid (30 nm fibre), (d) Fibres on central protein scaffold of chromosomes.
The nucleosomes are connected by an intervening stretch of DNA of a variable length of 50-60 base pairs. It is called linker DNA and is associated with the H1 histone (Fig. 21.6b).
C Packaging of DNA
There are several levels of organization that result in tight packaging of DNA into the nucleus.
1. Winding around nucleosomes: The wrapping of DNA around the nucleosome core results in 7-fold compaction of DNA. The nucleosomes, being of about 10 nm diameter each, form the 10 nm fibres shown in Figure 21.6b.
2. Nucleosomes packed into a 30 nm fibre about three nucleosomes wide: The 10 nm fibre is condensed further to form a 30 nm fibre illustrated in Figure 21.6c. The nucleosomes are packed in the 30 nm fibre in an as yet undetermined manner, though it is often depicted as a helical or solenoid arrangement of the nucleosomes. There are six nucleosomes per turn of the helix; this would yield a fiber three nucleosomes wide, which is indeed the diameter observed (30 nm).
The condensation of DNA achieved so far is about 40-fold, which may be sufficient for the dispersed chro-matin of the interphase nucleus. However, further 200fold compaction is required for the formation of fully condensed chromosome.
3. The 30 nm fibres form loops, thousands of nucleosomeslong attached to central scaffolding: The further compaction into the metaphase chromosome is achieved by attachment of the loops (of 30 nm fibre) to the central proteinscaffold of the chromosome (Fig. 21.6d). Each loop of30 nm fibre from one scaffold attachment to the next measures approximately 0.4-0.8 jum and contains 45,000-90,000 base pairs. It is believed that these loops arefurther condensed, forming yet other coils and/or foldsand achieve a 10,000-fold packing of the original DNA.
V Some Commonly Used Terms Related to Molecular Genetics
A Gene Families and Pseudogenes
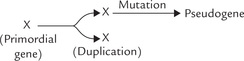
Most protein-coding genes are present in a single copy in the haploid genome. Occasionally, however, two identical or near identical copies of a gene are present together on the same chromosome. They are called duplicated genes. They originate by duplication of a primordial gene.
Some genes that code for very abundant RNAs or proteins are present in multiple copies. In most cases identical (or near identical) copies of the gene are arranged in tandem over long stretches of DNA separated by untranscribed sequences. Examples in humans include rRNA genes (200 copies), the 5S rRNA genes (2000 copies), the histone genes (20-50 copies) and most of the tRNA genes. In some other cases, gene duplication of a primordial gene is followed by divergent evolution of the duplication products. Consequently, we find that two or more similar but not identical genes are present in the genome, usually together on the same chromosome. Such structurally related genes with a common evolutionary origin constitutes a gene family. The α- and β-like globin gene clusters are the classic examples of such gene families.
Pseudogenes are unexpressed DNA sequences that exhibit remarkable sequence homology to some functional gene. They originate by gene duplication, followed by crippling mutation in one of the duplication products that renders it defunct. Pseudogenes are generally located close to their functional counterpart on the chromosome.
B Jumping Genes
These genes, described first by McClintock, do not appear to have a fixed position within the genome, but rather move from one location to another. They can jump to any place in the chromosome—there are no specific insertion sites for them. Such mobile DNA sequences in prokaryotes include insertion sequences and transposons.
Insertion Sequence
An insertion sequence is a mobile segment of DNA (750-1500 base-pair long) that contains a single gene and is framed by inverted repeats (IR) of 9-41 base pairs. The gene codes for a protein called transposase that catalyzes the movement of insertion sequence to the new genomic positions (i.e. transposition). From one point of view, insertion sequence (IS) might even be regarded as a futile element—doing nothing but code for its own movement from place to place. Most insertion sequences are present in 5-30 identical (or near identical) copies in the cell (Fig. 21.7 ).
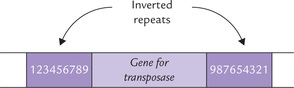
Fig. 21.7 The insertion sequence consisting of gene for transposase, flanked by inverted repeats of a base pairs (1-9 Indicate sequence of base pairs).
Transposition is a rare event, occurring once every 1000 to 10,000-cell generations. It can occur in two forms: conservation transposition and duplicative transposition.
Transposons
In common with insertion sequences, the transposons are flanked by inverted repeats and contain gene for their own transposase (the latter catalyzes their movement into new genomic locations). A distinctive feature of transposons is that (unlike IS) they contain useful genes, for example, antibiotic resistance genes.
C Silent Genes
These are mutant forms of genes which do not have a phenotypic effect. They may play a role in gene regulation.
D Complex Genes
These are transcribed together to yield more than one RNA transcripts. In prokaryotic genome such genes are frequently arranged in tandem along a single strand (eukaryotic genes, by contrast, are transcribed individually).
E Cell Interaction Genes
These genes play an important role in immunological recognition. They belong to major histocompatibility complex (Chapter 33).
Oncogenes, antioncogenes and protooncogenes are described in Chapter 32.
VI Replication
Every time a cell divides, the entire content of its chromosomal DNA must be duplicated so that a complete complement of DNA can be given to each daughter cell. DNA is able to duplicate, or as is more commonly stated, replicate, by separating and copying the parent strands to produce two daughter molecules that are identical to the parent DNA. Each separated parent strand serves as template for the synthesizing a new strand, so that each of the two daughter molecules contains a parent strand and a new daughter strand (Fig. 21.8 ). It is noteworthy that the template strand is copied in the complementary sense, which is dependent on Watson-Crick base pairing.
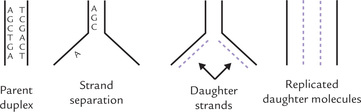
Fig. 21.8 Semiconservative nature of DNA replication. Each new daughter molecule consist of a parent strand and a daughter strand, which are complementary to each other.
Division of the cell into two daughter cells (mitosis) follows replication of chromosomal DNA into two daughter cells and each daughter cell gets an identical copy of the replicated DNA. In this way the genetic information is duplicated and transmitted to the next generation.
A Replication is Semiconservative
The method of replication shown in Figure 21.9 is a ‘semiconservative mechanism’ because each replicated duplex contains one parental strand and one newly synthesized strand. Messelon and Stahl confirmed this mechanism at the molecular level by the following experiment.
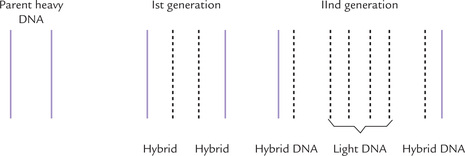
1. They grew E. coli for several generations in a medium containing 15 N-ammonium chloride as the only source of nitrogen so that any newly synthesized DNA incorporated this heavy isotope of nitrogen (i.e. 15 N-DNA). This is heavy DNA.
It is possible to separate (by density gradient centrifugation in cesium chloride) this 15N-DNA (heavy) from 14N-DNA (light) and from the hybrid (14N/15N-DNA) of intermediate density.
2. Fully 15N labelled cells were then abruptly transferred to a medium containing 14N-ammonium chloride, so that all subsequent DNA chains synthesized would be ‘light.
3. DNA was isolated at various generation times and its density monitored by density gradient centrifugation (Fig. 21.9).
Generation 1: There was a single band of DNA halfway between those of 14N-DNA and 15N-DNA, indicating its hybrid nature (14N/15N-DNA).
Generation 2: Half the DNA was hybrid DNA and the other 14N-DNA.
In succeeding generations: Proportion of light strands increases, but hybrid molecules persist.
These observations rule out the conservative mode. For the conservative model to be correct,
(a) all new DNAs must have normal density,
On the other hand, the semiconservative replication predicts all hybrid DNAs at the first generation and after two generations equal amounts of hybrid and 14N-DNAs; with no 15N-DNA ever being formed. In view of the results discussed above, the conservative mechanism can be ruled out, thereby confirming the semiconservative mechanism.
B Three Phases of Replication
The three phases of replication are initiation, elongation and termination. E. coli cells are mostly used for the study of replication. Fundamental mechanism and the participating enzymes of E. coli are similar to those of other living systems, including humans. Replication in prokaryotes is described in this Chapter; the comparative features of eukaryotic replication are discussed in Chapter 24.
Initiation
Initiation is a highly specific and precisely regulated stage of replication. It is at this stage that regulation of replication occurs; once the initiation phase gets started, replication of whole of the DNA must complete.
This phase starts with the unwinding of the chromosome at a single origin site, termed the oriC locus. It consists of a unique sequence of 245 base pairs that is very rich in A-T pairs, presumably to facilitate strand separation (A-T pairs having two hydrogen bonds are less tightly bound than G-C pairs which have triple hydrogen bonding). A complex of up to 30 subunits of the 52-kD DnaA protein binds to oriC and causes a segment of DNA to melt open. The melting process requires ATP hydrolysis, and is facilitated by the AT rich nature of the DNA segment. Further strand separation and subsequent unwinding of the duplex DNA are needed to produce the single-stranded DNA template required for DNA replication. A class of proteins termed helicases accomplishes separation.
Helicases
Strand separation by helicases is brought about through dissolution of hydrogen bonds holding the two DNA strands together. The process is ATP dependent; (it is likely that the hydrolysis of ATP could produce a cyclic change in shape of the helicase, which enables it to move unidirectionally along the DNA strand and disrupt the hydrogen bonds).
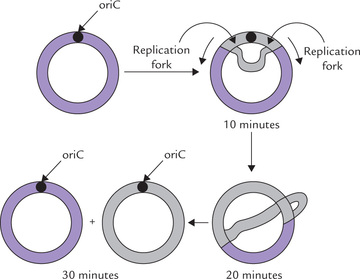
At least nine helicases have been described in E. coli. The DNA-binding proteins A, B and C (abbreviated as DnaA, DnaB and DnaC respectively) are the most important of these. Initial separation at the initiation site by DnaA protein is continued further by the hexameric DnaB protein, which is the major strand separating protein. It separates the DNA strands in both directions, resulting in the formation of two replication forks which move in opposite directions: one moves clockwise and the other counterclockwise, till they meet at the opposite side of chromosome (Fig. 21.10 ). DnaC is required for loading the DnaB at origin.
Single-stranded Binding Proteins (SSB Protein)
Once separated, the single-stranded regions associate with SSB protein, which prevents reannealing. This protein has high affinity for single-stranded DNA but has no base sequence specificity—it can bind anywhere on the separated strand. The regions of single-stranded DNA stabilized by SSB protein are rigid and semi-extended, which is an appropriate configuration for its role as a template for DNA replication. The SSB exists as tetramer, and binding of one tetramer facilitates binding of another to an adjacent region of single-stranded DNA (i.e. cooperative binding).
As the helicase moves in advance of the replication fork, the SSB proteins move on and off the DNA and are recycled for use on another site.
DNA Gyrase
The strand separation at the replication fork applies a turning force to the double helix, which may soon result in positive supercoiling (overwinding) of the remaining unseparated DNA and cessation of further separation. This is prevented by DNA gyrase, a type II DNA-topoisomerase that relaxes positive supercoils passively and introduces negative supercoils by an ATP-dependent mechanism.
Gyrase, SSB and helicases are collectively known as unwinding proteins; together they create single-stranded DNA for replication. A diagrammatic representation of their action is given in Figure 21.11 and summarized in Table 21.4.
Table 21.4
Escherichia coli DNA replication proteins
| Protein | Function |
| Dna A | Recognizes and binds specifically at replication origin (leading strand). |
| Dna B | Major strand separator (lagging strand). |
| Dna C | Required for loading Dna B at origin. |
| SSB | SSB protein prevents reannealing. |
| Primase | RNA primer synthesis. |
| DNAP III holenzyme | Progressive chain elongation. |
| DNAP I | Gap filling, primer excision. |
| Topoisomerase I | Removes negative supercoils. |
| Topoisomerase II | Forms negative supercoils, ATP dependent. |
| Ligase | Connects Okazaki pieces, ATP-dependent. |
Role of each of these proteins is of vital significance. Disrupted action of a protein may sometimes halt the prokaryotic replication altogether, thereby endangering growth and survival of the cell (Case 21.2).
To initiate replication, parental strands are first melted apart at a specific region and further separated in an ATP-dependent process that requires a helicase. SSB prevents reaggregation of the single strands and the gyrase relaxes positive supertwists.
Thus, a coordinated action of several proteins ensures specific initiation at the precise location. Regulation of initiation is important because replication occurs only once in a cell cycle.
The following points merit re-emphasis at this stage.
• Separation of the parental strands and synthesis of the new strands occurs simultaneously during replication. The site of simultaneous separation and synthesis is known as replication fork.
• Replication is bidirectional, which means that it moves simultaneously in both directions from the initiation site. In other words, there are two replication forks: one moves clockwise and the other anticlockwise.
Because of the small size of the bacterial genome and fast speed of replication (up to 3 billion base pairs may be accumulated in a few hours), the two replication forks are sufficient to duplicate the DNA in only about 30 minutes.
These concepts were first demonstrated by Cairns; using techniques, discussed in Box 21.2.
Elongation
The principal enzymes for the synthesis of new strands are DNA polymerases (DNAP) which catalyze polymerization of deoxyribonucleotides under direction of DNA template. These enzymes catalyze the stepwise addition of deoxyribo-nucleotide residues to the free 3’-hydroxyl end of a preexisting DNA or RNA primer strand; thus the replication is propagated in the 5’ → 3’ direction. The overall reaction is
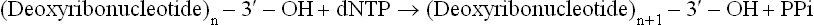
DNAP needs participation of the following:
DNA template
Each separated parental polynucleotide strand serves as template and copied by DNAP (in complementary sense) according to Watson-Crick base pairing rules.
Precursors
Any of the four deoxyribonucleotides-5’-tri-phosphate (dATP, dGTP, dCTP and dTTP) can serve as a precursor molecule for DNA synthesis. In the above equation, dNTP represents a precursor and PPi represents pyrophosphate cleaved from the dNTP.
Primer
DNAP is incapable of assembling the first nucleotide of a new chain. It needs a pre-existing oligonucleotide sequence called primer, containing a 3’-hydroxy group at end (Fig. 21.12 ). This group attacks the innermost phosphate of the incoming precursor molecule and forms a phosphodiester bond at the end of the growing chain. This complex reaction is driven forward by two factors:
(a) Pyrophosphate cleavage: The second and third phosphates of the precursor nucleotide form pyrophosphate, which is cleaved rapidly to inorganic phosphates by cellular pyrophosphatases. The hydrolysis of pyrophosphate increases the negative ΔG0′ value for the synthesis, thus helping to drive the reaction.
(b) New hydrogen bond: Incorporation of a nucleotide into the new strand of DNA involves the formation of new hydrogen bonds with its template partner. There occurs liberation of free energy in this process, which adds to the thermodynamic drive of the process.
At the cost of over-emphasizing a point (for it is important): DNA is always elongated in the 5’→ 3’ direction. The direction of elongation refers to polarity of the new strand and not of the template strand, which has an opposite polarity. The polarity of DNA strands has been explained earlier.
DNA polymerases bring about template-directed synthesis of the new DNA strand stepwise, nucleotide by nucleotide, by nucleotide in the 5’→3’ direction.
Processivity: DNA polymerase has capacity to remain associated with the template, catalyzing addition of several (nearly 100) nucleotide units to the growing daughter strand per binding event. This property, called processivity, enhances efficiency of replication several-fold.
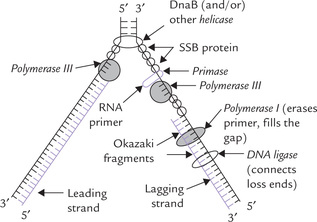
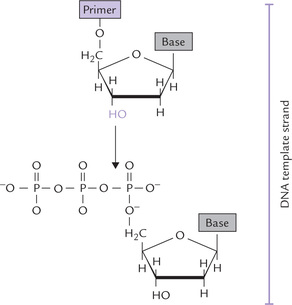
Role of primase: Despite the fact that DNA polymerase can read the base sequences of their template and incorporate “correct” bases with incredible accuracy, it cannot proceed until an RNA primer with a 3’-OH terminus is formed. A specific class of enzymes called primases accomplishes this task. (Table 21.4). A primase forms a short stretch (approximately 3-5 ribonucleotide units) of RNA in the replication fork. This small RNA, base paired with the template strand, is extended by the DNAP III (Fig. 21.13 ).
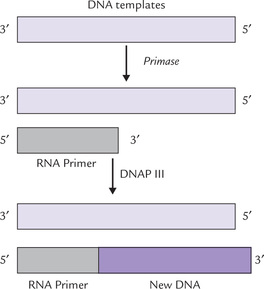
Fig. 21.13 DNA polymerase-III cannot assemble the first nucleotide of a new chain. This task is left to primase, which synthesizes a Short sequence of RNA that serves as the primer for DNAP III (DNAP = DNA polymerase).
The primer is hydrolyzed soon after by DNAP I (discussed later). The same enzyme then fills the gap so created with a new deoxyribonucleotide (DNA) sequence. DNA ligase then joins the loose ends.
Leading and lagging strands: A major problem, which remained unanswered for long, concerned the direction of DNA synthesis. It arises from the following observations: that DNAP can synthesize a new chain only in the 5’ → 3’ direction, reading the template in 3’ → 5’. If synthesis always proceeds in 5’ → 3’ direction, then how can simultaneous synthesis be possible in two strands of a DNA molecule which are antiparallel?
This problem was answered by Okazaki and colleagues in 1960s. These workers found that in only one of the strands, continuous synthesis takes place in 5’→ 3’ direction, which is also direction of movement of the replication fork. This strand is termed as the leading strand. In the other strand, known as the lagging strand, synthesis takes place in short DNA fragments, named Okazaki fragments. As the replication fork moves, the primase comes into action repeatedly, each time synthesizing short oligonucleotide sequences which serve as primers for DNAP III. DNA fragments, each approximately 1000 nucleotides long, are then added by DNAP III at the 3’ ends of these primers. The RNA primer is removed soon by 5’-exonuclease activity of DNAP I and the gap is filled by its polymerase activity: it synthesizes a fragment of DNA that replaces the RNA primer.
Finally, the enzyme DNA ligase catalyzes formation of a continuous strand of DNA by forming phosphodiester bonds between loose ends (Fig. 21.11). The loose ends refer to DNA synthesized by DNAP III and the small fragments of DNA synthesized by DNAP I. The bond formation requires energy provided by pyrophosphate cleavage of ATP.
At each replication fork, the leading strand is synthesized continuously in direction of unwinding while the lagging strand is synthesized in opposite direction as discontinuous segments (Okazaki fragments) that are later joined.
Primosomes for repeated initiation of Okazaki fragments: To function effectively, primase needs to be complexed with various other proteins to form a primosome (~600 kD). Various components of a primosome work cooperatively to prime for the repeated initiation of the Okazaki fragments. The complex includes a helicase (DnaB) and primase. It moves processively in the 5’ →3’-direction on the lagging strand to repeatedly displace SSBs, recognize start loci and synthesize RNAs.
A primase-containing-primosome synthesizes a RNA primer: a primosome is required to initiate each Okazaki fragment.
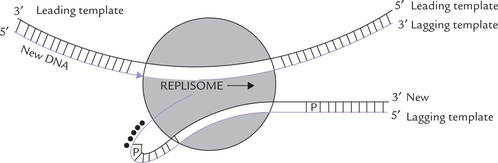
Replisomes for simultaneous synthesis of leading and lagging strands: Though the leading strand is synthesized continuously in the direction of unwinding, and the lagging strand in an opposite direction, it has been proposed that more or less simultaneous synthesis of both the strands occurs at replication fork. A dimeric DNA polymerase III holoenzyme, a primosome, a helicase, and replication proteins may form a complex termed replisome. Simultaneous synthesis of both the strands occurs by the action of a replisome if the DNA of the lagging strand template is looped backward so that it is in the same orientation as the leading strand template during polymerization.
As evident from Figure 21.14, the orientation of the lagging strand template (passing through the polymerase site in one subunit of the dimeric DNA polymerase III holoen-zyme) is in the same direction as the leading strand template in the other polymerase subunit. After the addition of about 1000 nucleotides, the polymerase would release the lagging strand template to allow formation of new loop. Primase would synthesize a short RNA primer, and the process would be repeated. In this way, the lagging strand would not be too far behind the leading strand synthesis.
Termination
The two replication forks moving in opposite directions from the origin meet at the opposite side of the circular DNA. This is the terminal event in DNA replication. However, it is still not clear what causes the ultimate separation of the daughter strands from the parent strand. In E. coli, possibly topoisomerase IV is involved in this process. Precise mechanism of the termination is still under investigation.
Replication forks move bidirectionally from the origin of replication until adjacent forks fuse at the opposite side of the chromosome, and thus the replication is completed.
Replication is a fast process. DNAP III, the principal enzyme of chain elongation, adds about 600-1000 nucleotide units per second. Replication of whole length of circular DNA of E. coli, which is 1.3 mm long—approximately 1000 times the diameter of the cell—takes just about 30 minutes.
Single-stranded organisms such as bacteriophages also undergo replication process, but by a different method, called rolling-circle replication mode. The details of this process are given in Box 21.3.
C Inhibitors of DNA Replication
Inhibitors of DNA replication are classified into the general categories:
1. Inhibitors that interact directly with DNA.
2. Inhibitors of deoxyribonucleotide synthesis.
3. Inhibitors that interact with replicative enzymes involved in DNA synthesis.
A large number of compounds in the first category bind between the stacked bases of the DNA duplex, disrupting the normal structure of DNA and causing some unwinding. This inhibits the DNA from serving as template for both replication and transcription. Examples of such compounds include acridine, ethidium and actinomycin. Mitomycin cross links adenine and guanine on DNA strands and prevents unwinding.
In the second category are compounds acting at various sites in the pathways for purine and pyrimidine biosynthesis. Actions of some of these, such as 6-mercaptopurine (inhibitor of purine synthesis) and 5-fluorouracil (inhibitor of thymidylate synthesis), and their role in treatment of cancer and infectious diseases have been discussed earlier in Chapter 20. Some compounds such as hydroxy-urea and sulfonamides inhibit the synthesis of both purine and pyrimidine nucleotides.
In the third category are such compounds that inhibit DNA polymerase III (e.g. arylhydrazinopyrimidines) or DNA gyrase, e.g. norfloxacin and ciprofloxacin that are used as antibiotics. Others, such as ethylmaleimide, aphidicolin, butyl-phenyl-dGTP, act on enzymes of eukaryotic replication (Chapter 24).
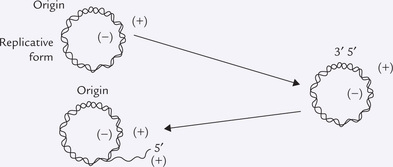
Box 21.3 Other Modes of DNA Replication
Several bacterial and viral genomes are replicated by the leading-lagging strand mechanisms. In addition, other modes of DNA replication for small circular chromosomes are also known. For example, some bacteriophages contain a single-stranded circular DNA, the ( + ) strand. A complementary strand to it, the (-) strand, is synthesized by a mechanism resembling conventional leading strand synthesis to form a circular duplex DNA. The latter is known as the replicative form. Additional copies of the ( + ) strand can be synthesized by the rolling-circle replication mode. This process initiates at a single strand break in the ( + ) strand and uses the (-) strand as a template. As the new ( + ) strand is synthesized, the existing ( + ) strand gets displaced by it. The replicating structure now resembles the Greek letter, sigma (σ), and therefore, this mechanism is commonly known as the replication mode σ. Multiple rounds of rolling-circle replication can generate a large number of tandemly linked ( + ) strands. These are later separated by an endonuclease for packaging into individual phage particles.
D Types of DNA Polymerases
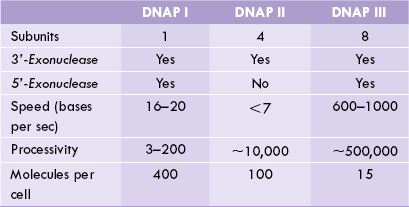
Prokaryotes have three different DNA polymerases (I, II and III), each designed for a different task.
DNA Polymerase I was the first DNAP enzyme to be isolated and was initially thought to be principal enzyme for replication. However, soon after its isolation in 1955, evidence began to accumulate that it alone cannot account for the observed speed of replication, and that other types of DNA polymerases must be present. The reason for this being, first, the rate at which this enzyme accumulates nucleotides (20 per second) is much slower than the observed rate of replication (3 billion base pairs added in a few hours). Second, it has lower processivity. Third and most importantly, when activity of this enzyme was altered in some bacterial strains, the affected bacterium still retained ability for an independent existence.
This observation by John Cairns intensified search for other DNA polymerases, which soon led to discovery of DNA polymerase II and DNA polymerase III in the early years of 1970s. Further, these studies helped to define the exact role of the DNAP I. Because of its low processivity, DNA I tends to fall off its template after polymerizing only a few dozen nucleotides. Its primary role is in DNA repair, playing an accessory role in DNA replication, where it erases primers and fills gaps (Fig. 21.11).
DNA Polymerase II has intermediate processivity. Though its exact role is not known, it is believed to participate in DNA repair.
DNP Polymerase III is the major enzyme of DNA replication. Its processivity is high: it can catalyze polymerization of thousands of nucleotides at a rate of almost 1000 nucleotides per second.
Comparative properties of the three DNA polymerases have been shown in Table 21.5 .
E Exonuclease Activities of DNA Polymerases
Nuclease refers to an enzyme that catalyzes hydrolysis of a phosphodiester bond in a nucleic acid. The nucleases that cleave the internal phosphodiester bonds are termed endonucleases and those that cleave bonds at ends are called exonucleases. Some exonucleases cleave only at the 3’ end (the 3’-exonuclease activity) while others cleave at the 5’ end (the 5-exonucleases activity).
The 3’-exonuclease activity: All the three types of bacterial DNAPs possess the 3’-exonuclease activity (Table 21.5), meaning that they can cleave the phosphodiester bond starting from the 3’ end of the chain. This activity provides a means for proofreading: any wrong base, mistakenly incorporated by DNAP, is promptly removed. It is important to correct such mistakes since wrongly incorporated base can lead to permanent change in the genetic information, which can have disastrous consequences. The 3’-exonuclease activity prevents any such eventuality by removing the mismatched base from the 3’ end (Fig. 21.15 ). The DNAP then replaces the mismatched base with the correct one, and the replication continues.
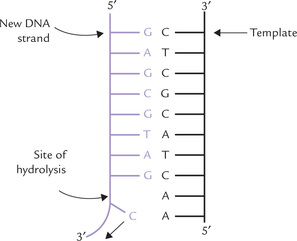
It is noteworthy that DNAPs are very accurate enzymes rarely making mistakes; the error rate being 1 in 104. The proofreading mechanism can reduce this rate to about 1 in 107.
The 5’-exonuclease activity: DNA polymerase I can cleave phosphodiester bond of DNA, starting from the 5’ end of a chain. This is 5’-exonuclease activity, which is different from the 3’-exonuclease activity. First, the cleavage can occur at a bond several residues from the 5’ end. Second, the active site for 5’ → 3’ hydrolysis is different from that for 3’→ 5’ hydrolysis.
The 5’-exonuclease is commonly referred to as the error correcting activity since it is required for removing the damaged DNA during DNA repair. For example, excision of pyrimidine dimers formed by exposure to ultraviolet light, as discussed later. The 5-exonuclease activity plays a key role in DNA replication by removing RNA primers.
VII Mutations
Mutations are permanent, heritable changes in the base sequence of genomic DNA. The most common changes are substitution, addition, rearrangement, or deletion of one or more bases. Mutations can either occur spontaneously or result from
(a) damage to the DNA by such external agents as radiation and chemicals,
Although mutations provide the raw material for natural evolution, they may lead to severe and debilitating diseases including cancers, and contribute to normal aging process. They lead to a genetic disease when they occur in the germline and to a defective cell clone when they occur in a somatic cell. The former (germline mutations) are perpetuated in a heritable manner in the genome of an organism, and the latter (somatic type) results in production of cells with reduced viability or impaired functions.
Mutations are rare and random events as far as individual organism is concerned. The probability of a mutation occurring in lifetime of an E. coli cell is only one chance in 109. For a human cell, the probability is greater (1 in 105). Mutations of more than 2500 human genes have been detected to date, many of them impair biological functions or are even lethal. For instance, a single base alteration produced by certain carcinogens in cigarette smoke may lead to cancerous transformation of the affected cell (Case 21.1). However, not all mutations give rise to a mutant phenotype; some remain silent and some are beneficial. Also, some mutations (Box 21.4) suppress harmful effects of some other mutations. In the following sections, the molecular basis of mutagenesis, and the DNA repair mechanisms that keep the mutation rate within tolerable limits are discussed.
A Types of Mutations
Following types of mutations are known to occur.
Point Mutations
These are changes of a single base pair of the DNA and are most common of all mutations. Transitions are point mutations in which one purine is substituted for another purine, or one pyrimidine is substituted for another pyrimidine. Transversions are point mutations in which a purine is replaced by a pyrimidine, or vice versa. When such base substitutions occur on the coding region of DNA, it can change the amino acid sequence of the encoded protein and the following manifestations are possible:
1. Missense mutation leads to a changed codon, whichencodes a different amino acid, so that a single aminoacid change appears in the encoded polypeptide. Forexample, replacement of A by C in the triplet UAUresults in replacement of tyrosine by serine:
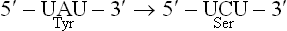
Depending on the amino acid that is changed, missense mutations can have either no effect or very serious consequences. Sickle cell anaemia, a severe haemolytic disease, is an example of a missense mutation with serious consequences.
2. Silent mutations arise due to degeneracy of geneticcode, discussed later (Chapter 22). In this instance,the point mutation produces a codon that still codesfor the same amino acid. For example, replacementof T by C in the first position of the triplet (TAA) doesnot produce any change in the amino acid encoded:

The point mutations occurring in the non-coding regions of DNA are also silent.
3. Nonsense mutations are the ones where single base substitution causes generation of a stop codon, UAA, UAG or UGA. As a result, premature termination of translation occurs with complete loss of function of the truncated protein.
4. Favourable mutations are the ones which may produce useful proteins. Such mutations are made use of in agriculture to yield better varieties, such as tryptophan-rich maize (normal maize is deficient in tryptophan).
Insertion or Deletion of Base Pairs
Insertion and deletion are mutations in which bases are added to the normal sequence or removed from it. Both may lead to frameshifts (if the number of base pairs is not a multiple of 3), changing the reading frame of all codons beyond the point of insertion or deletion (Fig. 21.16 ). This changes the amino acids beyond the insertion/deletion so that the encoded polypeptide chain is completely abnormal. Frameshifts not only change the amino acid sequence but also generally lead to premature termination (or rarely elongation) of the encoded polypeptide. This occurs when stop codons are generated or removed by the frameshift. The insertion or deletion of three base pairs, or any multiple of three, does not result in a frameshift, and the resulting protein is more likely to retain part or all of its biological activity.

Fig. 21.16 Frameshift mutations caused by a deletion or an insertion of a base. Note the change in amino acids from the site of deletion/insertion and onwards.
Frameshift mutations may be induced by some chemicals including acridine (Fig. 21.17 ) and proflavin, which have planar, fused ring structures that intercalate (insert themselves) between the adjacent stacked DNA bases. Figure 21.17 shows structure of acridine, which has some resemblance to purine base. When replication occurs in the region of an intercalated molecule, one or both daughter strands are synthesized that either lack one or more nucleotides or have additional ones.
Fig. 21.17 Acridine, a mutagenic agent, produces frameshift mutation by virtue of its flat, ring structure that resembles a purine base.
Frameshift mutations may also result from insertion of large chunks of genetic material, such as transposons.
Triplet Expansion
It is a newly discovered type of mutation that leads, by an unknown mechanism, to a great increase in the number of triplet repeats. Once triplet expansion reaches a critical size, they begin to interfere with gene function and then result in clinical syndrome. Some genetic diseases are caused by such trinucleotide amplification within the confines of an important gene (Chapter 24).
The site where a mutation occurs has a significant effect on the protein product. Mutations in a promoter or enhancer leaves the structure of polypeptide intact, but can decrease (or sometimes increase) its rate of synthesis. Mutations in an intron-exon junction results in abnormal splicing and hence production of a non-functional protein (Chapter 24).
Finally, translocation (transfer of genetic material from one chromosomal location to another) may disrupt an important gene to cause disease.
B Mutagens and Mutagenesis
Mutations may occur following exposure to physical agents or chemical reagents, collectively called mutagens. For example, hydroxylamine reacts with a DNA base, changing its chemistry and hydrogen bonding properties, and therefore, is a mutagen. Mutagenesis is the process of producing a mutation. It may occur spontaneously or may be induced by mutagens. If it occurs spontaneously in nature without the action of a known mutagen, it is called spontaneous mutagenesis and the resulting mutations are spontaneous mutations, also called basal mutations. If a mutagen is used, the process is called induced mutagenesis.
Basal Mutagenesis (or Spontaneous Mutations)
Several bases lose their exocyclic amino groups spontaneously, resulting in amino- to keto-group conversion. Thus cytosine is converted to uracil (U) which can form the base pair UA. In a later round of replication CG to TA mutation may follow.
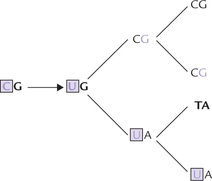
• Adenine is likewise converted spontaneously to hypoxanthine (H), which can form the base pair, HC. This may lead to error of replication (AT to GC mutation) in a later round.
• Likewise, spontaneous deamination of guanine to xanthine (X), followed by XC pairing may lead to mutation in a later round.
Presence of thymine (instead of uracil) in DNA minimizes deleterious effects of spontaneous mutations (Box 21.5).
Induced Mutagenesis
Radiations
Approximately 10% of all DNA damages caused by non-biological agents are because of ultraviolet or ionizing radiation.
(a) Ultraviolet (UV) radiations (wavelength 200400 nm) from sunlight or tanning lamps induce dimerization of adjacent pyrimidines, particularly adjacent thymines, along one strand of the DNA to form (cis-syn cyclobutane) pyrimidine dimers and other photo-products. This distorts the DNA structure, inhibits transcription and disrupts replication.
(b) Ionizing radiations, including X-rays and radioactive decay, possess sufficient energy to displace electrons from their orbitals, creating highly reactive intermediates that react with DNA bases to cause strand breaks. They also generate reactive oxygen species that damage DNA. Single-stranded breaks are more common, though rarely double-stranded breaks also occur. Ionizing radiations are capable of penetrating deep and therefore cause both somatic and germline mutations.
Chemicals
Several chemicals alter DNA bases or the structure of DNA, which often leads to mutations. Types of chemical mutagens are:
1. Deaminating agents, particularly nitrous acid and the compounds that can be metabolized to nitrous acid or nitrites (Fig. 21.18a ) convert amino groups to keto groups by oxidative deamination. Thus, cytosine, adenine and guanine are converted to uracil, hypoxanthine and xanthine respectively, which results in AT-to-GC or GC-to AT transitions, as discussed on page 458.
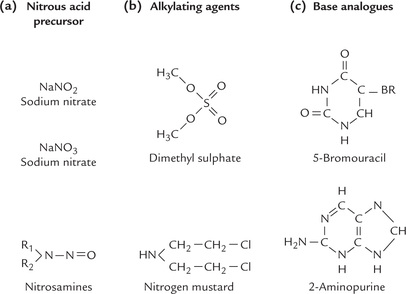
2. Alkylating agents (Fig. 21.18b) can cause chemical alterations in the bases so that the base pairing is changed. For example, dimethylsulphate can methylate a guanine residue to yield 6-methylguanine, which is unable to base pair with cytosine of the complementary strand. Alkylating agents are also capable of cross-linking the bases in the same (or opposite) strand.
3. Base analogues can erroneously get incorporated into DNA in place of a normal base. Some lead to inhibition of replication. Others, such as bromouracil (BU), are mutagenic because they lead to mispairing. BU (Fig. 21.18c), an analogue of thymine, usually pairs with adenine. But through the influence of its electronegative Br atom, it frequently assumes a tautomeric form that base pairs with guanine instead of adenine. This may induce an AT-to-GC transition in subsequent round of replication. In this way a mutation is caused by a spontaneous tautomeric shift (keto to enol). Occasionally, BU is incorporated into DNA in place of cytosine, which generates a GC-to-AT transition.
4. Intercalating agents, such as acridine and proflavin, tend to cause frameshift mutations, discussed earlier.
Hydroxylamine is an interesting chemical mutagen, which reacts specifically with cytosine and converts it into modified base that pairs with adenine. This ultimately leads to GC-to-AT mutation.
Oxidative Radicals
DNA within the cell suffers from many environmental insults, the most serious of which is caused by reactive oxygen metabolites. In presence of these metabolites, the metal ions such as iron and copper catalyze the oxidation of DNA, which may lead to mutations. The damage sustained by DNA may range from oxidation of sugar and base moieties of nucleotides to breaking of strands. Oxidative damage is increased in aging and in age-related degenerative diseases, including cancer. Approximately 20,000-60,000 modifications of DNA occur per cell per day.
C Mutagenesis and Carcinogenesis
Most mutagens can cause cancer, and therefore, they are carcinogenic as well. Some mutagens that exist in the carcinogenic form, directly damage the DNA, whereas other mutagens are pro-carcinogens. Pro-carcinogens in their native form do not damage DNA, but are converted to carcinogens in the liver or in other tissues via a variety of detoxification reactions (e.g. oxidation by cytochrome P450). This process is called metabolic activation (see benzo(a)pyrene; Case 21.1).
Ames test for estimating carcinogenicity: The test, developed by Bruce Ames, an American biochemist, is based on the high correlation between carcinogenesis and mutagenesis. Ames constructed special test-strains of the bacterium Salmonella typhimurium that are his-, meaning that they cannot synthesize histidine. Therefore, they cannot grow in absence of histidine in the medium. The DNA of these histidine auxotroph strains contain nucleotide substitutions or deletions that prevents the production of histidine biosynthetic enzymes. Mutagenesis in these strains is indicated by their reversion to the his + phenotype (can synthesize histidine).
About 109 of the his- bacteria are spread on a culture plate lacking histidine and the suspected mutagen is added to the plate. Occasionally, the mutagen causes reversal of the histidine mutation to the his + phenotype; the latter can now synthesize histidine, and therefore, grow in the absence of histidine. This is detected by growth into visible colonies after 2 days at 37°C. Counting the number of such colonies that have reverted to the his + phenotype scores the mutagenicity of a compound. There is a significant correlation between the results of the Ames test and direct tests of carcinogenic activity in animals.
Addition of a small amount of liver homogenate, (which contains the liver P450 detoxification enzymes), in the Ames’s test medium test may detect procarcinogens.
VIII Repair Systems for Mutated DNA
An elaborate system exists in the body, which can repair most damages to DNA, thus preventing alteration in the stored genetic information. Despite its inherent stability, a number of chemical changes are occurring in DNA all the time, which may be spontaneous or triggered by ‘insults’ by a variety of agents. These changes may add up to result in large number of mutations per day in each cell if there were not constant repair systems. Repair is possible because usually only one strand of DNA is damaged at a given place, and there is always the other strand to act as template and direct the repair of the damaged part.
High fidelity of DNA replication is achieved (in addition to 3’ → 5’ proofreading) by DNA repair mechanism. Some forms of DNA damages, e.g. alkylated bases and pyrimidine dimers may be repaired in a single step, others are more complex.
The principal classes of repair systems and their essential features are discussed here:
Base Excision Repair and AP Site Repair
Some chemical changes, such as deamination convert normal bases into abnormal ones. These are removed from the DNA and replaced with the correct base by the base excision repair mechanism. Deamination of cytosine, for example, creates uracil, which is hydrolyzed off by DNA glycosylase through cleavage of the N-glycosidic bond (between the base and 2’-deoxyribose). This leaves apurine or apyrimidine (AP) sites, in which the deoxyribose has no base attached to it. Repair of the AP site involves:
• nicking of the polynucleotide chain adjacent to the lesion by AP (apurinic)-endonuclease,
• replacement of the section containing the nick by DNA polymerase I, and
• final sealing by ligase (Fig. 21.19 ).
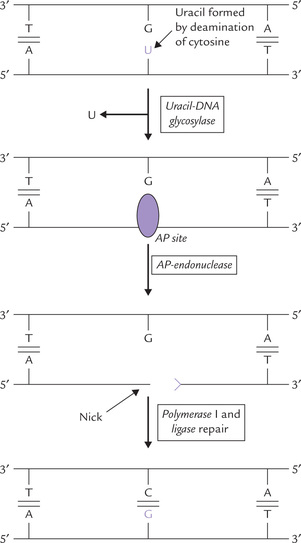
Spontaneous depurination: AP sites can also be formed by spontaneous hydrolysis of the purine-deoxyribose link, which is somewhat unstable. It is repaired as above by successive actions of AP endonuclease, DNAP-I, and DNA ligase.
Mismatch Repair
The proofreading mechanisms (discussed earlier in this chapter) ensure that DNA is replicated with the almost incredible degree of accuracy, but in a system involving such vast numbers of nucleotide additions, mistakes invariably occur. Such mistakes (mostly base mismatches) cause some distortion of the duplex chain, illustrated in Figure 21.20 .
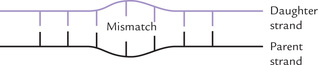
The mismatches are repaired by the mismatch repair system, which has been described here in prokaryotes (and appears equally important in eukaryotes).
Strand discrimination: An important point is that the repair system must be able to discriminate between the parent (template) strand and the new daughter strand because it is the base on the daughter strand that is incorrect and so needs to be excised. Clearly, excising the newly synthesized base would preserve the genetic information, whereas excising the base on the parental strand would permanently alter the DNA, producing a mutation. Therefore, the challenge is to recognize the newly synthesized strand and remove the mismatched base on that strand.
How is such strand discrimination made? The repair enzymes identify the parent strand because it is methylated at the palindrome GATC (Fig. 21.21 ); a cytoplasm enzyme, Dam-methylase, methylates the adenine of this palindromic sequence. This methyl group on the parent strand does not affect the base pairing or the DNA structure but serves as a tag by which the repair system identifies it as the parent strand. When a new strand is synthesized, it takes some time to methylate it, and so, for this brief period it remains unmethylated. The repair enzymes recognize the methylated parent strand and leave it, but they act on the not-yet methylated new strand to remove the mismatched portion. The gap created by its removal is then filled by synthesis of new DNA and ligation.
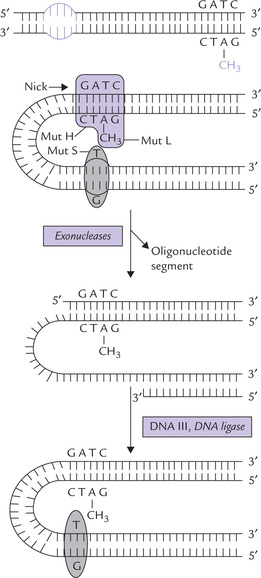
Fig. 21.21 Mechanism of mismatch repair. The mispaired base in newly synthesized DNA strand is shown by arrow.
Being guided by methylation, this system is referred to as methyl directed mismatch repair system. It correctly repairs mismatches that may be located as much as 1000 base pairs distant from a methylated palindromic (GATC) sequence. Further information on beneficial effects of methylation is given in Box 21.6.
Mechanism of repair
Exact mechanism of the mismatch repair is not clearly understood. Some proteins identified in this process are Mut S, Mut H and Mut L:
• Mut S protein recognizes the mismatch distortion in the helix.
• Mut H binds to an unmethylated GATC.
• Mut L, links the above two proteins to form a complex (Fig. 21.21).
Identifying the unmethylated strand as being newly synthesized, the Mut H nicks it. The entire section of DNA from the nick to beyond the error is removed by combined action of exonuclease, followed by the synthesis of new DNA (by DNAP III) and ligation (by DNA Ligase).
Nucleotide Excision Repair
This system repairs the bulky lesions that cause large distortions in the double helix, such as:
(a) T-dimers and other photoproducts,
(b) Adducts formed by covalent linking of some foreign molecules to DNA, and
A short stretch of nucleotides, including the lesion is excised, which is followed by its correct replacement, the opposite strand serving as template for this (Fig. 21.22 ).
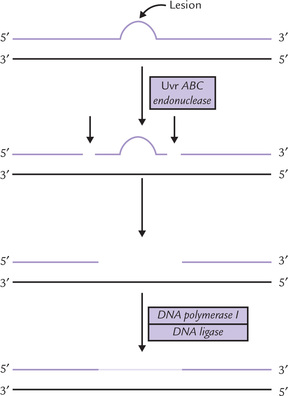
The key enzyme in E. coli, an unusual endonuclease is termed Uvr ABC endonuclease complex, after the genes coding for the enzyme. It binds at the site of lesion and makes two nicks on the damaged strand. It is an ATP-dependent reaction which cleaves the lesion-containing strand at the seventh and fourth phosphodiester bonds on the 5’ and 3’ sides of the lesion, respectively. The oligonucleotide segment between the two nicks is then excised by the same enzyme. The excision gap is then filled by action of DNA polymerase I and ligated by DNA ligase.
The ABC endonuclease scans the DNA, recognizes the lesion, and removes the portion of damaged DNA. The resulting gap is filled by DNA polymerase and DNA ligase.
Further information on action of the ABC endonuclease is given in Box 21.7.
Direct Repair
Direct repair refers to those types where the repair occurs without removal of a base or a nucleotide. For example, repair of the pyrimidine dimers (produced by dimerization of adjacent thymines by UV rays) is effected by reversing this damage by photoreactive photolyases. The photolyase binds to the dimer and cleaves it after exposure to visible light. In fact, light exposure elevates the two cofactors present in it (FADH and methenyl-tetrahydrofolate) to a higher energy state. The excited form of FADH is capable of transferring an electron to the pyrimidine dimer, which breaks it.
Another example of direct repair involves the alkyl groups on bases, for example, the O6 alkyl derivatives of guanine, (which are formed by some mutagenic agents). These may be directly repaired by transfer of alkyl group to cysteine side chain of a repair enzyme. In doing so the enzyme destroys its own action, hence acting as a suicide enzyme.
Recombination Repair
The recombination repair system may act when damaged DNA undergoes replication before the lesion has been repaired. It is an essential repair process for the rapidly dividing cells because a replication machinery promptly arrives at the damaged site, often before its repair (by nucleotide excision system).
The replication machinery halts when it encounters a lesion, such as pyrimidine dimer, and resumes polymerization at some point beyond the dimer block. The result of this process is that the daughter strand has large gap opposite the pyrimidine dimer (Fig. 21.23 a, b).
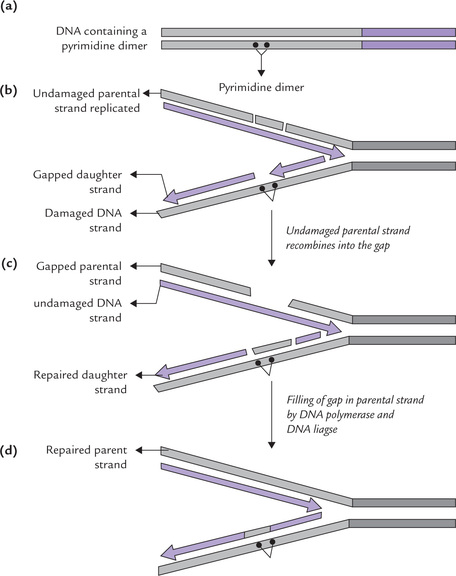
The gap is repaired by the process of recombination, which involves:
(i) Excision of an undamaged DNA segment from the parental DNA strand.
(ii) Insertion of this DNA into the gapped strand (Fig. 21.23c).
(iii) A combined action of polymerase I and ligase jointsthis inserted piece to adjacent regions, thus fillingin the gap.
(iv) This creates a gap in the parental strand, but it canbe easily repaired since it is placed opposite an undamaged strand, and so can be filled-in according to thetemplate, and then sealed (Fig. 21.23d).
(v) Finally, the pyrimidine dimer, associated with anintact complementary strand, can then be eliminated by nucleotide excision repair or photoreactive photolyase.
Since recombination repair occurs after DNA replication, it is often called post-replication repair. It, however, fails if two dimers in opposite strands are very near one another because no undamaged segments are available.
SOS Repair Response
Agents that extensively damage DNA induce a complex series of cellular changes in E. coli, known as SOS response. These changes result in production of proteins and enzymes that allow DNA chain growth across the damaged segments, though at the cost of fidelity of replication. The chain synthesis is possible because the editing system is relaxed, which means loss of the ability to remove incorrect bases added to the growing strand. As a result the process is error prone and, therefore, mutagenic.
The SOS response in prokaryotic cell induces a special set of survival enzymes that relax fidelity of replication, permitting the replication complex to synthesize DNA opposite the damaged template strand. The result is often mutations.
It may be mentioned that DNA polymerase II is one of the important proteins that are induced as a result of the SOS response. Unlike DNA polymerase III, which is stopped by such lesions, this enzyme can replicate past them. Precise function of the other proteins of this specialized system is unknown. In essence, special properties of DNAP II help the replication past the DNA lesions.
IX Defective DNA Repair and Human Diseases
Inability to carry out certain stages of DNA repair results in human diseases discussed below.
1. Xeroderma pigmentosum (XP; Greek: xeros, dry + derma,skin) is a group of recessively inherited disorders due tomutations in genes encoding polypeptide(s) of thenucleotide excision repair. This system is responsible forthe repair of bulky DNA lesions, including the pyrimidine dimers that are formed by UV radiation in sunexposed skin. At least seven subtypes of XP have beenidentified in different patients, each caused by the deficiency of a different polypeptide in the repair system.
Individuals with this disease are extremely sensitive to sunlight, developing freckles and ulcerative lesions on the sun-exposed skin. Moreover, they develop skin cancer at a 2000-fold greater rate than normal, usually during the first and second decades of life. In some subtypes, a variety of seemingly unrelated symptoms develop, such as progressive neurological degeneration, developmental deficits, and opacification or ulceration of the cornea. The only effective treatment for this otherwise fatal disease is complete avoidance of sunlight.
2. Cockayne’s syndrome (CS) is a rare recessively inherited disease, also associated with defective nucleotide excision repair. In addition to the genes that are defective in XP, two additional genes are mutated in CS. Individuals with CS are hypersensitive to UV radiation, show growth retardation and neurological degeneration, but do not have an increased incidence of skin cancer.
3. Bloom’s syndrome, Fanconi’s anaemia and ataxia-telangi-ectasis are collectively referred to as chromosome breakage syndromes as they are thought to be caused by defects in the repair of double-strand DNA breaks. Fanconi’s syndrome is a lethal aplastic anaemia, Bloom’s syndrome is a premature aging disorder, and ataxia-telengiectasia is characterized by severe abnormalities in various organ systems and a high incidence of lymphoreticular cancer. Patients are highly sensitive to radiations, so their exposure to diagnostic X-rays should be minimized.
4. Hereditary non-polyposis colon cancer (HNPCC), is a dominantly inherited cancer susceptibility syndrome in which the post-replication mismatch repair is defective. One of these mismatch repair genes (hMSH2) is located on the chromosome 3 and the other on the short arm of chromosome 2. HNPCC is responsible for approximately 6% of all cases of colorectal cancer; and risk of cancers of other tissues mildly increases, including the small bowel, stomach, biliary system and ovary.
X Site Specific Recombination
The exchange of polynucleotide strands between separate DNA segments is called genetic recombination. It requires cutting and covalent joining of DNA. There are two types of genetic recombination: site specific- and general-recombination.
A Site Specific Recombination
It occurs between two short, specific DNA sequences. The process of recombination is effected by an enzyme that can recognize specific sequences either on one or on both DNA molecules (donor and target sites). For example, the viral integrase, which effects integration of the × phage, recognizes both the phage DNA and the target site on the bacterial chromosome. Likewise, the retroviral integrase recognizes the LTRs of the retroviral cDNA. The transposase is likewise specific for the inverted terminal repeats of its own insertion sequence (see section on jumping genes).
B General Recombination
It occurs between DNA segments with extensive sequence homology, hence also known as homologous recombination. It neither requires any specific enzyme systems nor any specific DNA sequences; it depends only on sequence similarity between the recombining molecules. Foreign DNA, for example, can be installed in a host’s chromosome by this mechanism provided it has sufficient sequence homology with the chromosomal DNA.
A prototypic model for homologous recombination between homologous DNA duplexes was proposed by Robin Holliday in 1964. As shown in Figure 21.23, this type of recombination results in a reciprocal exchange between two duplex DNA molecules. The corresponding strands of two aligned homologous DNA duplexes are nicked, and the nicked strands cross over to pair with the nearly complementary strands on the homologous duplex, thereby forming a cross-shaped intermediate called the Holliday intermediate (Fig. 21.24 ). This intermediate is then resolved into two duplex DNAs by the successive action of an endonuclease (ruvC in E. coli).
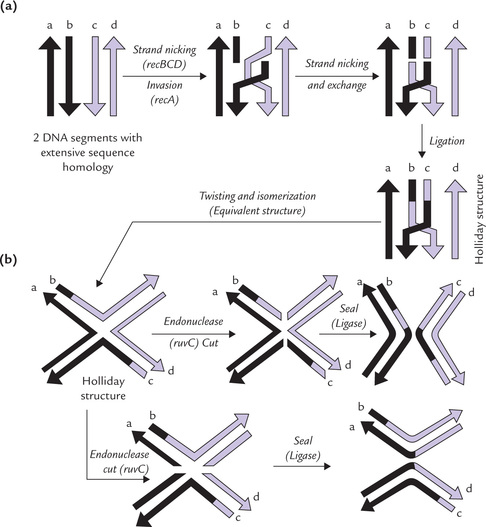
Fig. 21.24 Key steps in general recombination, (a) Formation of Holliday intermediate, (b) Resolution of Holliday ntermediate.
Several enzyme proteins act successively to bring about the recombination:
1. A complex of recB, recC and recD proteins, which act as endonuclease and helicase to generate single-stranded DNA.
2. The recA protein, which catalyzes the invasion of a DNA duplex by the single-stranded DNA.
General recombination involves exchange of the homologous DNA segments. A crossover structure (Holliday junction) is formed that can be subsequently resolved.
Homologous recombination is used for recombining genetic information in bacteria. In higher organisms, during prophase of meiosis, pairs of genes may exchange by crossing-over between homologous chromosomes, thereby promoting genetic diversity. Genes may also move from a given site to a non-homologous site by transposition; the mobile genetic elements are called transposons, discussed earlier in the chapter.
Though transposons shuttle among different locations, their movement does not seem to be of any advantage to the bacterial cell. Rather they can inflict deleterious effect by jumping into a useful gene and halting its expression. In view of these, mobile genes are, in general, thought to be selfish DNA elements.
Exercises
Essay type questions
1. Discuss the events which proved that DNA is the genetic material.
2. Outline the structural features of DNA double helix. What are the distinctive structural features of eukaryotic DNA?
3. What are the alternative higher order forms of DNA and how are they different from B- DNA? Discuss role of histones in DNA packing.
4. Outline the experiments that show that DNA replicates by semiconservative mechanism. Describe the mechanism of DNA replication.
5. Explain the basis of semi-discontinuous DNA synthesis. Discuss roles of primosomes and replisomes in replication.