Developmental Disturbances of Oral and Paraoral Structures
 . Developmental Disturbances of Jaws
. Developmental Disturbances of Jaws
. Abnormalities of Dental Arch Relations
. Developmental Disturbances of Lips and Palate
. Hereditary Intestinal Polyposis Syndrome
. Developmental Disturbances of Oral Mucosa
. Developmental Disturbances of Gingiva
. Developmental Disturbances of Tongue
. Developmental Disturbances of Oral Lymphoid Tissue
. Developmental Disturbances of Salivary Glands
. Developmental Disturbances in Size of Teeth
. Developmental Disturbances in Shape of Teeth
. Developmental Disturbances in Number of Teeth
. Developmental Disturbances in Structure of Teeth
Craniofacial Anomalies
Craniofacial anomalies (CFA) are a diverse group of deformities in the growth of the head and facial bones. Anomaly is a medical term meaning ‘irregularity’ or ‘different from normal’. These abnormalities are congenital (present at birth) and have numerous variations: some are mild, others are severe and require surgery.
There is no single factor that causes these abnormalities. Instead, there are many factors that may contribute to their development, including the following:
• Combination of genes. A child may receive a particular combination of gene(s) from one or both parents, or there may be a change in the genes at the time of conception, which results in a craniofacial anomaly.
• Environmental. There is no data that shows a direct correlation between any specific drug or chemical exposure causing a craniofacial anomaly. However, any prenatal exposure should be evaluated.
• Folic acid deficiency. Folic acid is a B vitamin found in orange juice, fortified breakfast cereals, enriched grain products, and green, leafy vegetables. Studies have shown that women who do not take sufficient folic acid during pregnancy, or have a diet lacking in folic acid, may have a higher risk of having a baby with certain congenital anomalies, including cleft lip and/or cleft palate.
Some of the most common types of craniofacial anomalies (Table 1-1) include:
Table 1-1
Examples of most common craniofacial anomalies
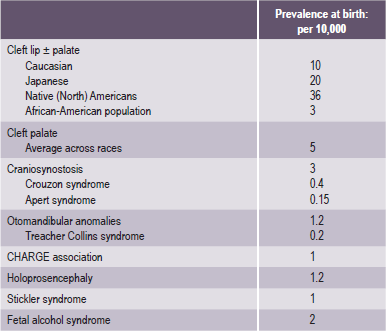
Source: Rovin et al, 1964; Temple, 1989; Cohen et al, 1992; Lewanda et al, 1992; Croen et al, 1996; Derijcle et al, 1996; Sampson et al, 1997; Blate et al, 1998.
• Cleft lip and/or cleft palate. A separation that occurs in the lip or the palate or both. Cleft lip and cleft palate are the most common congenital craniofacial anomalies seen at birth.
Cleft lip. An abnormality in which the lip does not completely form. The degree of the cleft lip can vary greatly, from mild (notching of the lip) to severe (large opening from the lip up through the nose).
Cleft palate. Occurs when the roof of the mouth does not completely close, leaving an opening that can extend into the nasal cavity. The cleft may involve either side of the palate. It can extend from the front of the mouth (hard palate) to the throat (soft palate). The cleft may also include the lip.
• Craniosynostosis. A condition in which the sutures in the skull of an infant close too early, causing problems with normal brain and skull growth. Premature closure of the sutures may also cause the pressure inside the head to increase and the skull or facial bones to change from a normal, symmetrical appearance.
• Hemifacial microsomia. A condition in which the tissues on one side of the face are underdeveloped, affecting primarily the ear (aural), mouth (oral), and jaw (mandibular) areas. Sometimes, both sides of the face can be affected and may involve the skull as well as the face. Hemifacial microsomia is also known as Goldenhar syndrome, brachial arch syndrome, facio-auriculovertebral syndrome (FAV), oculo-auriculovertebral spectrum (OAV), or lateral facial dysplasia.
• Vascular malformation. A birthmark or a growth, present at birth, which is composed of blood vessels that can cause functional or esthetic problems. Vascular malformations may involve multiple body systems. There are several different types of malformations, named after the type of blood vessel that is predominantly affected. Vascular malformations are also known as lymphangiomas, arteriovenous malformations, and vascular gigantism.
• Hemangioma. A type of birthmark; the most common benign (noncancerous) tumor of the skin. Hemangiomas may be present at birth (faint red mark) or appear in the first month after birth. A hemangioma is also known as a port wine stain, strawberry hemangioma, and salmon patch.
• Deformational (or positional) plagiocephaly. A misshapen (asymmetrical) shape of the head (cranium) from repeated pressure to the same area of the head. Plagiocephaly literally means ‘oblique head’ (from the Greek ‘plagio’ for oblique and ‘cephale’ for head).
Collectively they affect a significant proportion of the global society (Table 1-1).
Global Epidemiology
The frequency of occurrence of cleft lip, with or without cleft palate, has been computed on a global scale and is estimated to be 1 in every 800 newborn babies (Tables 1-2 and 1-3). A child is therefore born with a cleft somewhere in the world approximately every two-and-half minutes. Accurate data on the frequency of occurrence of these disorders is relevant for implementing strategies aimed at primary prevention and effective management of these disabled children (Table 1-2). Like anywhere else, the epidemiological data in this situation is also inherently handicapped by:
Table 1-2
Cleft lip with or without cleft palate
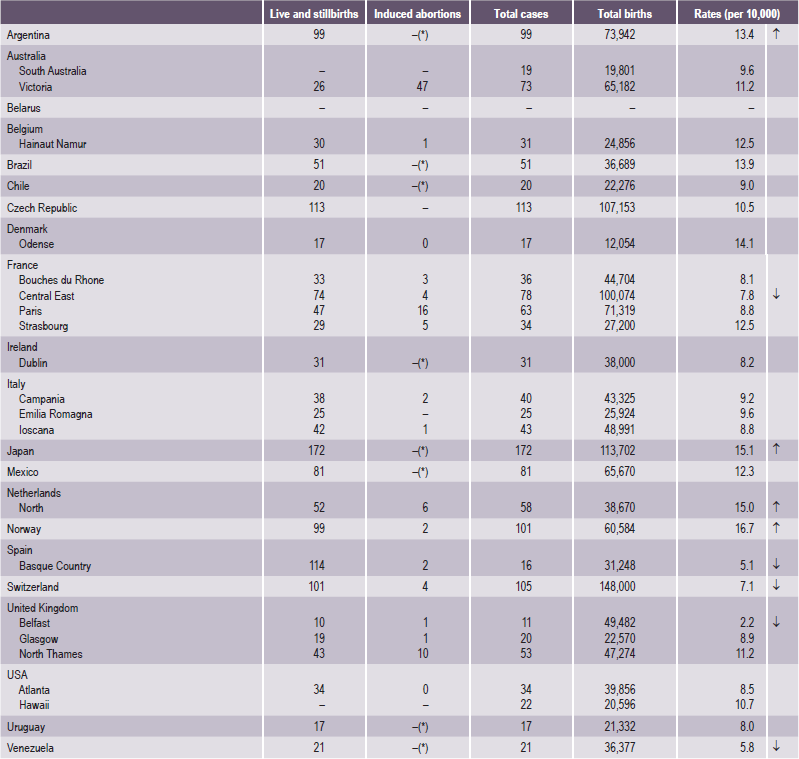
Source: WHO (1998), World Atlas of Birth Defects (1st Edition).
*Abortion for birth defect not permitted.
↑= 99% significantly higher than the mean.
↓= 99% significantly lower than the mean.
Table 1-3
Cleft palate without cleft lip
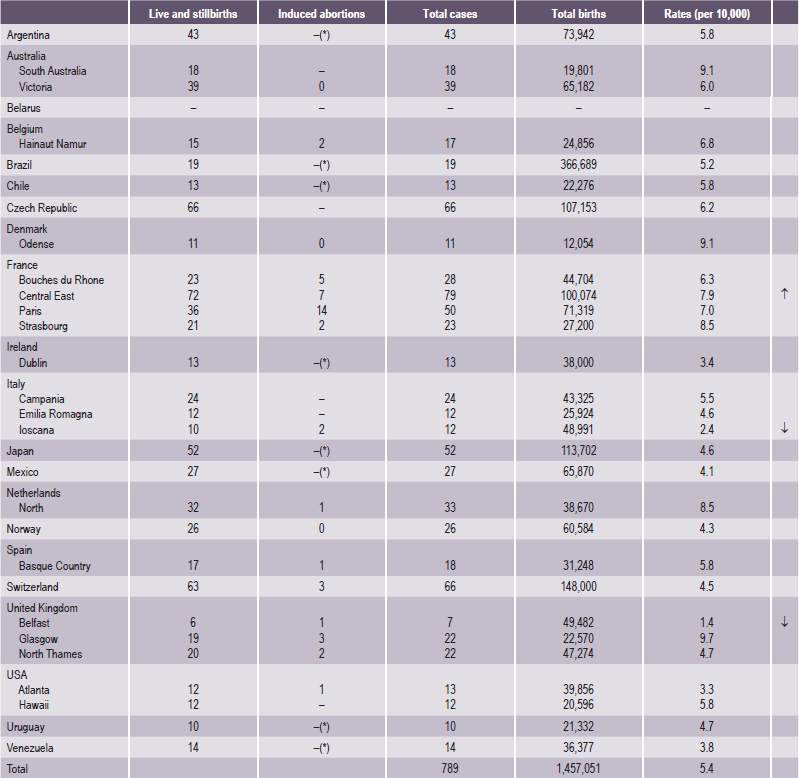
Source: WHO (1998), World Atlas of Birth Defects (1st Edition).
*Abortion for birth defect not permitted.
↑= 99% significantly higher than the mean.
↓= 99% significantly lower than the mean.
• The heterogeneity of orofacial clefting,
• The lack of standard criteria for collection of data, and
• In particular, the lack of and/or failure to apply an internationally comparable classification for orofacial clefting.
Defining the affected population is also problematic because, on many occasions, the terminology is so vague that it is not clear whether it denotes all birth, or all live births. The word ‘births’ is again somewhat ambiguous because it usually includes stillbirths, a term which lacks clarity.
Registration of Targeted Craniofacial Anomalies in India
1. Three multicenter studies in India have provided almost similar frequency of CFA: meta-analysis of 25 early studies from 1960–1979, involving 407,025 births, showed:
CL/P = 440 cases, 1.08 per 1,000 births,
CP = 95 cases, 0.23 per 1,000 births.
2. A prospective national study of malformations in 17 centers from all over India from September 1989 to September 1990 involving 47,787 births showed:
CL/P = 64 cases, 1.3 per 1,000 births,
CP = 6 cases, 0.12 per 1,000 births.
3. The latest three-center study, conducted in 1994–1996, involved 94,610 births in Baroda, Delhi and Mumbai, and showed a frequency of:
This was the most rigorously conducted study and it found the number of infants born every year with CLP to be 28,600; this means 78 affected infants are born every day, or three infants with clefts are born every hour (Table 1-4)!

CFA are not lethal but they are disfiguring, and thus cause a tremendous social burden. However, these disorders have an excellent outcome if surgical repair is carried out competently. Recent information regarding the etiology of CFA provides the means to carry out primary or secondary prevention. Maintaining a registry would be very useful as a benefit to the community and in reducing the burden of these anomalies, either by prevention or surgical repair.
Another reason why a registry would be desirable is the changing pattern of morbidity and mortality in India emerging as a result of the achievements in immunization, the success in providing primary health care and the existence of a well-developed health infrastructure. In many university and city hospitals congenital malformations and genetic disorders have become important causes of illness. All these reasons show that starting a registry of these disorders deserves high priority in India.
Existing epidemiological data on CFA
The epidemiological information that exists on CFA anomalies in India needs to be examined to decide what data should be collected for the registry:
• Higher frequency of CL+CP among Indian males is similar to that observed among Caucasians. The ratio is more than that observed in Africans and Japanese.
• The higher prevalence of CL+CP as compared with CL among Indians is like that observed in Africans, and is more than that observed in Caucasians.
• Children born prematurely are more frequently affected in India, as elsewhere.
• About 10.9% of 459 cases of all clefts are syndromic in Chennai. Of these, about 50 % are due to single-gene disorders, about 18% due to chromosomal disorders, and the rest due to undetermined causes.
• Chromosomal studies would be desirable in cases with associated abnormalities.
• Syndromes are more commonly associated with CP than with CL, as elsewhere.
• Lateralization (more clefts on the left side) in India is similar to that observed in other races.
• In one study in India, the intake of drugs was observed in 18% of parents — mostly steroidal compounds (progestogens as tests for pregnancy).
• A greater history of terminated pregnancies has been observed among cases, as compared with controls.
• History of severe vomiting has been observed to be about six times more common among case mothers than among controls.
• There is some difference in the frequency of orofacial clefts in different states in India; however, this needs verification. The state of origin (or mother tongue) of the parents should be recorded.
• Clefts are more commonly found in certain caste groups among Hindus.
• In India CP has less frequency in those with blood group A.
• CL occurs more in those with group O and AB.
• Association of clefts with certain HLA types has been documented in India.
• In a study in Chennai, significantly more consanguinity was observed among couples having children with clefts as compared with controls.
Genetics: Principles And Terminology
Genotype and Phenotype
The science of genetics is concerned with the inheritance of traits, whether normal or abnormal, and with the interaction of genes and the environment. This latter concept is of particular relevance to medical genetics, since the effects of genes can be modified by the environment.
Table 1-5
Genes involved in craniofacial and dental disorders, sorted by acronym gene name
| Acronym | Full name |
| ACTC | Actin, Alpha, Cardiac Muscle |
| APP | Amyloid Beta A4 Precursor Protein (APP) |
| ARVCF | Armadillo Repeat Gene Deleted in VCFS |
| ATP&E | ATPhase, H+ Transporting, Lysosomal, Subunit E |
| CA1 | Carbonic Anhydrase 1 |
| CLTCL1 | Clathrin, Heavy Polypeptide-Like 1 |
| COL01A1 | Collagen, Type I, Alpha-1 |
| COL01A2 | Collagen, Type I, Alpha-2 |
| COL02A1 | Collagen, Type II, Alpha-1 |
| COL04A4 | Collagen, Type IV, Alpha-4 |
| COL04A5 | Collagen, Type IV, Alpha-5 |
| COL05A1 | Collagen, Type V, Alpha-1 |
| COL06A1 | Collagen, Type VI, Alpha-1 |
| COL10A1 | Collagen, Type X, Alpha-1 |
| COMT | Catechol-O-Methyltransferase |
| CYLN2 | Cytoplasmic Linker 2 |
| DCN | Decorin |
| DGSI | DiGeorge Syndrome Critical Region Gene |
| ELN | Elastin |
| FBLN2 | Fibulin 2 |
| FBN1 | Fibrillin 1 |
| FGF8 | Fibroblast Growth Factor 8 |
| FGFR1 | Fibroblast Growth Factor Receptor 1 |
| FGFR2 | Fibroblast Growth Factor Receptor 2 |
| FGFR3 | Fibroblast Growth Factor Receptor 3 |
| GNAS1 | Guanine Nucleotide-Binding Protein, Alpha-Stimulating Activity Polypeptide |
| GOLGA1 | Golgi Autoantigen, Golgin Subfamily A, 1 |
| GP1BB | Glycoprotein Ib, Platelet, Beta Polypeptide |
| GPC3 | Glypican 3 |
| GPC4 | Glypican 4 |
| GPR1 | G Protein-Coupled Receptor 1 |
| GTF2I | General Transcription Factor II-I |
| HIP1 | Huntingtin-interacting Protein 1 |
| HIRA | Histone Cell Cycle Regulation Defective, S. Cerevisiae, Homolog of, A |
| HOXD10 | Homeo Box D10 |
| ITGB2 | Integrin, Beta-2 |
| KRT04 | Keratin 4 |
| KRT06A | Keratin 6A |
| KRT06B | Keratin 6B |
| KRT13 | Keratin 13 |
| KRT16 | Keratin 16 |
| KRT17 | Keratin 17 |
| LAMB1 | Laminin, Beta-1 |
| MEOX2 | Mesenchyme Homeo Box 2 |
| MSX1 | MSH, Drosophila, Homeo Box, Homolog of, 1 |
| MSX2 | MSH (Drosophila) Homeo Box Homolog 2 |
| PNUTL1 | Peanut-Like 1 |
| PTHR1 | Parathyroid Hormone Receptor 1 |
| RO60 | Autoantigen Ro/SSA, 60–KD |
| SCZD | Schizophrenia |
| SCZD4 | Schizophrenia 4 |
| SCZD8 | Schizophrenia 8 |
| SRC | V-SRC Avian Sarcoma (Schmidt-Ruppin A-2) Viral Oncogene |
| SRY | Sex-Determining Region Y |
| SSA1 | Sjögren Syndrome Antigen A1 |
| SSB | Sjögren Syndrome Antigen B |
| TNFRSF11B | Tumor Necrosis Factor Receptor Superfamily, Member 11B |
| TTPA | Tocopherol Transfer Protein, Alpha |
| TWIST | Twist, Drosophila, Homolog of |
| WBSCR1 | Williams-Beuren Syndrome Chromosome Region 1 |
Source: Report of the National Institutes of Dental and Craniofacial Research Genetics Workgroup, Meeting held November 14–16, 1999.
Consideration of the heritability of a particular feature or trait requires a consideration of the relationship between genotype and phenotype. Genotype is defined as the genetic constitution of an individual, and may refer to specified gene loci or to all loci in general. An individual’s phenotype is the final product of a combination of genetic and environmental influences. Phenotype may refer to a specified character or to all the observable characteristics of the individual. The proportion of the phenotypic variance attributable to the genotype is referred to as heritability.
Genetic variation in man may be observed at two levels:
In specific traits, individual genotypes are readily identified and differences are qualitative (discrete), for example, the ABO blood antigen system. Gene frequencies can be estimated and the Mendelian type of analysis can be applied.
In continuous traits such as height, weight or tooth size, differences are characterized quantitatively between individuals. These quantitative traits in man are more elusive to study because they are determined by the alleles of many gene loci, and therefore, the Mendelian type of analysis is not appropriate. They are further modified by environmental conditions which obscure the genetic picture. If the genetic variation of a particular phenotypic trait is dependent on the simultaneous segregation of many genes and affected by environment it is referred to as being subject to multifactorial inheritance. Genetic differences caused by the segregation of many genes is referred to as polygenic variation and the genes concerned are referred to as polygenes. These genes are, of course, subject to the same laws of transmission and have the same general properties as the single genes involved in qualitative traits, but segregation of genes is translated into genetic variations seen in continuous traits through polygenes.
Different types of genetic ‘product’ can be thought of as being different distances from the fundamental level of gene activity. Enzymes, for instance, are almost direct products of gene action, and in most cases where genetic variation of enzyme structure has been demonstrated, it has been shown that a single locus is responsible for the structure of a single enzyme. The structure, and consequently, the activity of an enzyme is therefore usually simply and directly related to allele substitutions at a single locus.
Morphological characters, on the other hand, such as the numerous dimensions used to describe the shape of the face and jaws, are furthest removed from the fundamental genetic level and are the end results of a vast complexity of interacting, hierarchical, biochemical, and developmental processes. Each gene is therefore likely to influence many morphological characters so that a deleterious mutation, although producing a unitary effect at the molecular level, almost always results in a syndrome of morphological abnormalities. When a gene is known to affect a number of different characters in this way its action is said to be pleiotropic. A reverse hierarchy also exists, making each morphological character dependent on many different genes (Mossey, 1999).
Modes of Inheritance
Population genetics deals with the study of the mode of inheritance of traits and the distribution of genes in populations.
All chromosomes exist in pairs so our cells contain two copies of each gene, which may be alike or may differ in their substructure and their product. Different forms of genes at the same locus or position on the chromosome are called alleles. If both copies of the gene are identical, the individual is described as homozygous, while if they differ, the term used is heterozygous.
The exception to the rule that cells contain pairs of chromosomes applies to the gametes, sperm and ovum, which contain only single representatives of each pair of chromosomes, and therefore, of each pair of genes. When the two gametes join at fertilization, the new individual produced again has paired genes, one from the father and one from the mother. If a trait or disease manifests itself when the affected person carries only one copy of the gene responsible, along with one normal allele, the mode of inheritance of the trait is called dominant (Fig. 1-1A). If two copies of the defective gene are required for expression of the trait, the mode of inheritance is called recessive (Fig. 1-1B).
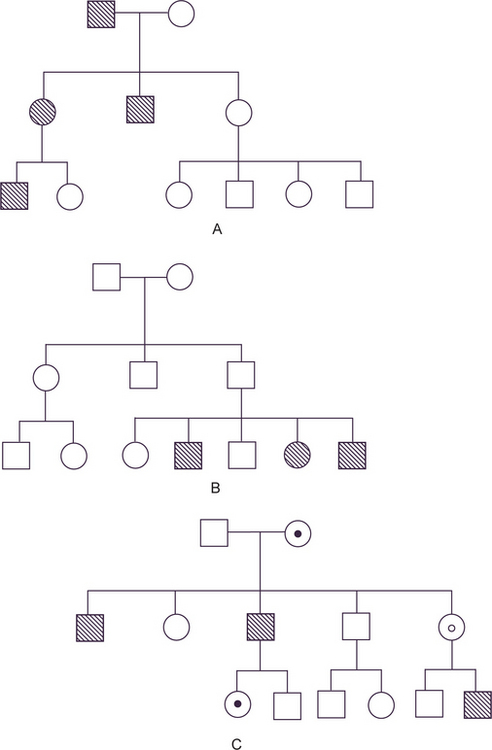
Figure 1-1 (A) Autosomal dominant pedigree. (B) Autosomal recessive pedigree. (C) X-linked recessive pedigree. Courtesy of PA Mossey, 1999.
The special case of genes carried on the X chromosome produces yet different pedigrees. Since male-to-male transmission is impossible and since females do not express the disease when they carry only one copy of the diseased gene (since it is modified by the homologous X chromosome), the usual pedigree consists of an affected male with clinically normal parents and children, but with affected brothers, maternal uncles, and other maternal male relatives (Fig. 1-1C). This mode of inheritance is described as X-linked recessive.
It has been long appreciated that many normal traits, such as height, intelligence, and birth weight, have a significant genetic component, as do a number of common diseases, such as diabetes mellitus, schizophrenia, hypertension, and cleft lip and palate. However, the pattern of inheritance of these traits does not follow the simple modes just described. Mathematical analysis of many of these has led to the conclusion that they follow the rules of polygenic inheritance, i.e. determined by a constellation of several genes, some derived from each parent.
The determination of heritability for polygenic or multifactorial characters is difficult, as a feature of continuous variation is that different individuals may occupy the same position on the continuous scale for different reasons. Using mandibular length as an example, micrognathia can occur in chromosomal disorders, such as Turner’s syndrome, in monogenic disorders such as Treacher Collins syndrome or Stickler syndrome, or due to an intrauterine environmental problem, such as fetal alcohol syndrome. Combined with this the concept of etiological heterogeneity encompasses the principle of the same gene defect producing different phenotypic anomalies, and syndromes can be due to defective gene activity in different cells. Conversely, different gene defects or combinations of defective genes can produce a similar phenotypic abnormality. Genetic lethality or reduced reproductive fitness can also complicate the diagnostic picture and genomic imprinting can result in a gene defect ‘skipping’ a generation. These complexities serve to hamper progress in the understanding of polygenic or multifactorial disorders such as orofacial clefting (Mossey, 1999).
Multifactorial Inheritance
In contrast to single-gene inheritance, either autosomal or sex-linked, the pedigree pattern does not afford a diagnosis of multifactorial inheritance. In multifactorial traits, the trait is determined by the interaction of a number of genes at different loci, each with a small, but additive effect, together with environmental factors (i.e., the genes are rendering the individual unduly susceptible to the environmental agents). Many congenital malformations (Table 1-6) and common diseases of adult life are inherited as multifactorial traits and these are categorized as either continuous or discontinuous.
Table 1-6
Extrinsic
Mechanical
Unstretched uterine and abdominal muscles
Small maternal size
Amniotic tear
Unusual implantation site
Uterine leiomyomas
Unicornuate uterus
Bicornuate uterus
Twin fetuses
Intrinsic
Malformational
Spina bifida
Other central nervous system malformations
Bilateral renal agenesis
Severe hypoplastic kidneys
Severe polycystic kidneys
Urethral atresia
Functional
Neurologic disturbances
Muscular disturbances
Connective tissue defects
Source: MM Cohen Jr, The Child with Multiple Birth Defects. Raven Press, New York, 1982, p 10. Cited by RJ Gorlin, MM Cohen, LS Levin. Syndromes of the Head and Neck, 3rd Ed, Oxford University Press; 1990, New York.
Molecular Genetics in Dental Development
The first sign of tooth development is a local thickening of oral epithelium, which subsequently invaginates into neural crest derived mesenchyme and forms a tooth bud. Subsequent epithelial folding and rapid cell proliferation result in first the cap, and then the bell stage of tooth morphogenesis. During the bell stage, the dentine producing odontoblasts and enamel secreting ameloblasts differentiate. Tooth development, like the development of all epithelial appendages, is regulated by inductive tissue interactions between the epithelium and mesenchyme (Thesleff, 1995).
There is now increasing evidence that a number of different mesenchymal molecules and their receptors act as mediators of the epithelial-mesenchymal interactions during tooth development. Of the bone morphogenetic proteins (BMPs) 2, 4, and 7 mRNAs shift between the epithelium and mesenchyme in the regulation of tooth morphogenesis (Aberg et al, 1997). The fibroblast growth factor (FGF) family have also been localized in epithelial and mesenchymal components of the tooth by immunohistochemistry (Cam et al, 1992); and in dental mesenchyme tooth development and shape is regulated by FGF8 and FGF9 via downstream factors MSX1 and PAX9 (Kettunen and Thesleff, 1998).
Control of Tooth Development
Homeobox genes have particular implications in tooth development. Muscle specific homeobox genes Msx-1 and Msx-2 appear to be involved in epithelial mesenchymal interactions, and are implicated in craniofacial development, and in particular, in the initiation developmental position (Msx-1) and further development (Msx-2) of the tooth buds (MacKenzie et al, 1991; Jowett et al, 1993). Further evidence of the role of Msx-1 comes from gene knock-out experiments which results in disruption of tooth morphogenesis among other defects (Satokata and Maas, 1994). Pax-9 is also transcription factor necessary for tooth morphogenesis (Neubuser et al, 1997). BMPs are members of the growth factor family (TGF) and they function in many aspects of craniofacial development with tissue specific functions. BMPs have been found to have multiple roles not only in bone morphogenesis, (BMP 5, for example, induces endochondral osteogenes is in vivo), but BMP 7 appears to induce dentinogenesis (Thesleff, 1995).
Congenital Deformations Of Head And Neck
These are common, and mostly resolve spontaneously within the first few days of postnatal life. When they do not, further evaluation may be necessary to plan therapeutic interventions that may prevent long-term consequences. Approximately 2% of infants are born with extrinsically caused deformations that usually arise during late fetal life from intrauterine causes. Approximately 30% of deformed infants have two or more deformations. Deformed infants tend to show catch-up growth during the first few postnatal months after release from the intrauterine environment. The common deformations considered under the craniofacial category are nasal, auricular and mandibular deformities.
Teratogenic Agents
Teratogens are agents that may cause birth defects when present in the fetal environment. Included under such a definition are a wide array of drugs, chemicals, and infectious, physical, and metabolic agents that may adversely affect the intrauterine environment of the developing fetus. Such factors may operate by exceedingly heterogeneous pathogenetic mechanisms to produce alterations of form and function as well as embryonic and/or fetal death (Gorlin et al, 1990).
The mechanisms of teratogenesis are selective in terms of the target and effect. Thus, characteristic patterns of abnormalities can be expected to be associated with particular teratogenic agents. However, the extent to which an individual may be adversely affected by exposure to a given teratogen varies widely. This depends on the following four factors:
It is of considerable interest to the dental profession that experimental studies of teratogenic agents have almost invariably revealed a variety of head, neck and oral malformations. Conway and Wagner have compiled a list of the most common malformations of the head and neck as shown in Table 1-7.
Table 1-7
| Type | Occurrence (%) |
| Unilateral incomplete | 33 |
| Unilateral complete | 48 |
| Bilateral incomplete | 7 |
| Bilateral complete | 12 |
Source: V Veau: Division palatine; anatomie, chirurgie, phonetique. Paris, Masson et Cie, 1931.
Stem cells are clonogenic cells that have the capacity for self-renewal and multilineage differentiation. The microenvironment in which stem cells reside is called a stem cell niche and is composed of heterologous cell types, extracellular matrix and soluble factors to support the maintenance and self-renewal of the stem cells. Stem cells have been isolated and characterized from a wide variety of sources. Adult progenitor cellular populations have been isolated from distinctive tissues and fluids, such as bone marrow, peripheral blood, umbilical cord blood, Wharton’s jelly, placenta, amniotic fluid and membrane, adipose tissue, dermis, hair follicles, synovial membrane, skeletal muscle, central nervous system, olfactory bulb, retina, and liver among others. From these biological sources, progenitors of the mesenchymal, epithelial, hematopoetic (discussed elsewhere), neural, endothelial and trophoblastic lineages have been identified (Fig. 1-2).
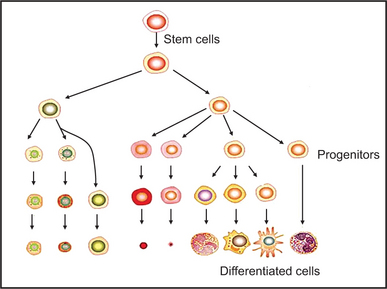
Figure 1-2 Mandibular micrognathia. Courtesy: Prof Anil Sukumaran, Prof R Rajendran. King Saud University, Riyadh, KSA.
In tissue engineering applications, mesenchymal stem cells are among the most used populations because of the wide variety of sources from which they can be harvested, their ability to self-renew, and their multilineage potential following adequate induction. Moreover, the vast majority of the tissues relevant in surgical repair / regeneration are of mesenchymal origin. This is of particular relevance in the craniofacial area because during development cells originating from the neural crest are known to migrate, differentiate, and participate in the morphogenesis of virtually all structures of the region including muscle, ligament, bone, cartilage, periodontal tissues and teeth.
There are two major categories of stem cells, which are of relevance to dentistry:
Totipotent embryonic stem cells are derived from the inner cell mass of mammalian blastocyst and can be maintained indefinitely in culture. Somatic stem cells have a limitation in their potential of differentiation. The differentiation potential of dental stem cells lies in the formation of dentin or periodontium associated tissues, whether these cells are derived from pulp, periodontal ligament or dental follicle. It is obvious that dental ectomesenchymal stem cells can be classified into two different groups with respect to their differentiation potential. The first group is associated with the dental pulp, consisting of dental pulp stem cells (DPSCs), stem cells from exfoliated deciduous teeth (SCEDs), and stem cells from apical papilla (SCAPs); the second group contains periodontal ligament stem cells (PLSCs) and dental follicle progenitor cells (DFPCs), related to the periodontium.
Embryonic dental epithelium is capable of inducing the development of a tooth germ in combination with non-dental ectomesenchymal tissue. However, oral ectomesenchyme in combination with non-dental epithelium did not induce tooth development. Later steps in tooth development are driven by a complex interaction of both ectodermal and ectomesenchymal tissues along with growth promoting factors of bone morphogenic protein-4 (BMP-4) and fibroblast growth factor-8 (FGF-8), produced by the oral ectoderm for the initiation of tooth development.
Oral ectoderm derived ameloblasts are unable to proliferate or regenerate once they have reached the maturation stage of development. Continuously growing mouse incisors and molars in some mammalian species show replenishing populations of enamel organs composed of stellate reticulum, stratum intermedium and enamel epithelial cells which provide models for dental epithelial stem cells. Mice have an epithelial stem cell niche located at their incisor labial apical ends, known as the cervical loop. The cervical loop has been considered to be a determinative region in odontogenesis due to its ability to produce enamel and dentin. One specialized structure found at the apical region of the labial cervical loop in mouse incisors, “apical bud” (Ohshima H et al, 2003), is suggested as stem cell compartment that could differentiate into ameloblast through interaction with mesenchymal cells and growth factors.
In humans, these dental epithelial stem cells are lost after tooth eruption; therefore, are not available for studies on dental development. In contrast to dental epithelial stem cells, undifferentiated cells of the oral ectomesenchyme are not entirely lost after tooth eruption in humans. It became possible to isolate precursor cells from the dental pulp and ectomesenchymal stem cells were isolated from the dental pulp of extracted wisdom teeth.
They are a heterogeneous population of multipotent self-renewal cells that possess clonogenic competence and the capacity to differentiate into all cell lineages of the mesenchymal and connective tissue elements. They also seem to be able to differentiate into epithelial cells and lineages derived from neuroectoderm. Mesenchymal stem cells have been shown to produce a wide range of bioactive molecules capable of immunomodulatory functions along with the ability to modulate the regenerative processes in the human body (Ohshima H et al, 2001).
Developmental Disturbances of Jaws
Agnathia (Otocephaly, holoprosencephaly agnathia)
Agnathia is a lethal anomaly characterized by hypoplasia or absence of the mandible with abnormally positioned ears having an autosomal recessive mode of inheritance. More commonly, only a portion of one jaw is missing. In the case of the maxilla, this may be one maxillary process or even the premaxilla. Partial absence of the mandible is even more common. The entire mandible on one side may be missing, or more frequently, only the condyle or the entire ramus, although bilateral agenesis of the condyles and of the rami also has been reported. In cases of unilateral absence of the mandibular ramus, it is not unusual for the ear to be deformed or absent as well. It is probably due to failure of migration of neural crest mesenchyme into the maxillary prominence at the fourth to fifth week of gestation (postconception). The prevalence is unknown and less than 10 cases are described. The prognosis of this condition is very poor and it is considered to be lethal.
Micrognathia
Micrognathia literally means a small jaw, and either the maxilla or the mandible may be affected. Many cases of apparent micrognathia are not due to an abnormally small jaw in terms of absolute size, but rather to an abnormal positioning or an abnormal relation of one jaw to the other or to the skull, which produces the illusion of micrognathia.
True micrognathia may be classified as either congenital, or acquired. The etiology of the congenital type is unknown, although in many instances it is associated with other congenital abnormalities, including congenital heart disease and the Pierre Robin syndrome (q.v.) This form of the disease has been discussed by Monroe and Ogo. It occasionally follows a hereditary pattern. Micrognathia of the maxilla frequently occurs due to a deficiency in the premaxillary area, and patients with this deformity appear to have the middle third of the face retracted. Although it has been suggested that mouth-breathing is a cause of maxillary micrognathia, it is more likely that the micrognathia may be one of the predisposing factors in mouth-breathing, owing to the associated maldevelopment of the nasal and nasopharyngeal structures.
True mandibular micrognathia of the congenital type is often difficult to explain. Some patients appear clinically to have a severe retrusion of the chin but, by actual measurements, the mandible may be found to be within the normal limits of variation. Such cases may be due to a posterior positioning of the mandible with regard to the skull or to a steep mandibular angle resulting in an apparent retrusion of the jaw. Agenesis of the condyles also results in a true mandibular micrognathia.
The acquired type of micrognathia is of postnatal origin and usually results from a disturbance in the area of the temporomandibular joint. Ankylosis of the joint, for example, may be caused by trauma or by infection of the mastoid, of the middle ear, or of the joint itself. Since the normal growth of the mandible depends to a considerable extent on normally developing condyles as well as on muscle function, it is not difficult to understand how condylar ankylosis may result in a deficient mandible.
The clinical appearance of mandibular micrognathia is characterized by severe retrusion of the chin, a steep mandibular angle, and a deficient chin button (Fig. 1-3).
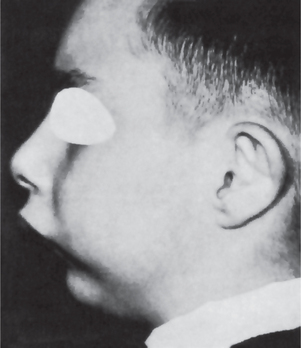
Micrognathia may be caused by or may be a feature of several conditions (Table 1-8).
Table 1-8
Congenital conditions
• Catel-Manzke syndrome
• Cerebrocostomandibular syndrome
• Cornelia de Lange syndrome
• Femoral hypoplasia—unusual facies syndrome
• Fetal aminopterin-like syndrome
• Miller-Dieker syndrome
• Nager acrofacial dysostosis
• Pierre Robin syndrome
• Schwartz-Jampel-Aberfeld syndrome
• van Bogaert-Hozay syndrome
Intrauterine acquired conditions
• Syphilis, congenital
Chromosomal abnormalities
• 49, XXXXX syndrome
• Chromosome 8 recombinant syndrome
• Cri du chat syndrome 5p
• Trisomy 18
• Turner’s syndrome
• Wolf-Hirschhorn syndrome
Mendelian inherited conditions
• CODAS (cerebral, ocular, dental, auricular, skeletal) syndrome
• Diamond-Blackfan anemia
• Noonan’s syndrome
• Opitz-Frias syndrome
Autosomal dominant conditions
• Camptomelic dysplasia
• Cardiofaciocutaneous syndrome
• CHARGE syndrome
• DiGeorge’s syndrome
• Micrognathia with peromelia
• Pallister-Hall syndrome
• Treacher Collins-Franceschetti syndrome
• Trichorhinophalangeal syndrome type 1
• Trichorhinophalangeal syndrome type 3
• Wagner vitreoretinal degeneration syndrome
Autosomal recessive conditions
• Bowen-Conradi syndrome
• Carey-Fineman-Ziter syndrome
• Cerebrohepatorenal syndrome
• Cohen syndrome
• Craniomandibular dermatodysostosis
• De la Chapelle dysplasia
• Dubowitz syndrome
• Fetal akinesia-hypokinesia sequence
• Hurst’s microtia-absent patellae-micrognathia syndrome
• Kyphomelic dysplasia
• Lathosterolosis
• Lethal congenital contracture syndrome
• Lethal restrictive dermopathy
• Marden-Walker syndrome
• Orofaciodigital syndrome type 4
• Postaxial acrofacial dysostosis syndrome
• Rothmund-Thomson syndrome
• Smith-Lemli-Opitz syndrome
• ter Haar syndrome
• Toriello-Carey syndrome
• Weissenbacher-Zweymuller syndrome
• Yunis-Varon syndrome
X-linked inherited conditions
• Atkin-Flaitz-Patil syndrome
• Coffin-Lowry syndrome
• Lujan-Fryns syndrome
• Otopalatodigital syndrome type 2
Autoimmune conditions
• Juvenile chronic arthritis
Source: www.diseasesdatabase.com, Health on the Net Foundation, 2005.
Macrognathia
Macrognathia refers to the condition of abnormally large jaws. An increase in size of both jaws is frequently proportional to a generalized increase in size of the entire skeleton, e.g. in pituitary gigantism. More commonly only the jaws are affected, but macrognathia may be associated with certain other conditions, such as:
• Paget’s disease of bone, in which overgrowth of the cranium and maxilla or occasionally the mandible occurs,
• Acromegaly, in which there is progressive enlargement of the mandible owing to hyperpituitarism in the adult, or
• Leontiasis ossea, a form of fibrous dysplasia in which there is enlargement of the maxilla.
Cases of mandibular protrusion or prognathism, uncomplicated by any systemic condition, are a rather common clinical occurrence (Fig. 1-4). The etiology of this protrusion is unknown, although some cases follow hereditary patterns. In many instances the prognathism is due to a disparity in the size of the maxilla in relation to the mandible. In other cases the mandible is measur-ably larger than normal. The angle between the ramus and the body also appears to influence the relation of the mandible to the maxilla, as does the actual height of the ramus. Thus prognathic patients tend to have long rami which form a less steep angle with the body of the mandible. The length of the ramus, in turn, may be associated with the growth of the condyle. It may be reasoned, therefore, that excessive condylar growth predisposes to mandibular prognathism.
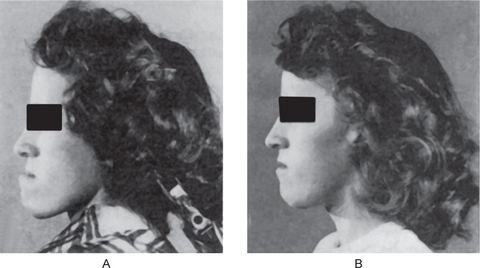
Figure 1-4 Macrognathia (prognathia) of the mandible. (A) The protrusion of the mandible is obvious. (B) The same patient after surgical correction (ostectomy) (Courtesy of Dr G Thaddeus Gregory and Dr J William Adams.
General factors which conceivably would influence and tend to favor mandibular prognathism are as follows:
• Increased height of the ramus
• Increased mandibular body length
• Anterior positioning of the glenoid fossa
• Posterior positioning of the maxilla in relation to the cranium
Surgical correction of such cases is feasible. Ostectomy, or resection of a portion of the mandible to decrease its length, is now an established procedure, and the results are usually excellent from both a functional and a cosmetic standpoint.
Facial Hemihypertrophy (Hyperplasia)
Hemihyperplasia is a rare developmental anomaly characterized by asymmetric overgrowth of one or more body parts. Although the condition is known more commonly as hemihypertrophy, it actually represents a hyperplasia of the tissues rather than a hypertrophy. Hemihyperplasia can be an isolated finding, but it also may be associated with a variety of malformation syndromes (Table 1-9). Almost all cases of isolated hemihyperplasia are sporadic.
Table 1-9
Malformation syndromes associated with hemihyperplasia
• Beckwith-Wiedemann syndrome
• Neurofi bromatosis
• Klippel-Trenaunay-Weber syndrome
• Proteus syndrome
• McCune-Albright syndrome
• Epidermal nevus syndrome
• Triploid/diploid mixoploidy
• Langer-Giedion syndrome
• Multiple exostoses syndrome
• Maffucci’s syndrome
• Ollier syndrome
• Segmental odontomaxillary dysplasia
Source: HE Hoyme et al, 1998.
Hoyme et al (1998) provided an anatomic classification of hemihyperplasia:
• Complex hemihyperplasia is the involvement of half of the body (at least one arm and one leg); affected parts may be contralateral or ipsilateral,
• Simple hemihyperplasia is the involvement of a single limb, and
• Hemifacial hyperplasia is the involvement of one side of the face.
Etiology
The cause is unknown, but the condition has been variously ascribed to vascular or lymphatic abnormalities; CNS disturbances; and chromosomal abnormalities.
Clinical Features
Patients affected by facial hemihypertrophy exhibit an enlargement which is confined to one side of the body, unilateral macroglossia and premature development, and eruption as well as an increased size of dentition. Familial occurrence has been reported on a few occasions, according to the excellent review of the condition by Rowe, who described four additional cases. Of all reported cases, females are affected somewhat more frequently than males (63% versus 37%), according to the review by Ringrose. There is an almost equal involvement of the right and left sides.
Oral Manifestations
The dentition of the hypertrophic side, according to Rowe, is abnormal in three respects: crown size, root size and shape, and rate of development. Rowe has also pointed out that not all teeth in the enlarged area are necessarily affected in a similar fashion. There is little information about the effects on the deciduous dentition, but the permanent teeth on the affected side are often enlarged, although not exceeding a 50% increase in size. This enlargement may involve any tooth, but seems to occur most frequently in the cuspid, premolars, and first molar. The roots of the teeth are sometimes proportionately enlarged but may be short.
Characteristically, the permanent teeth on the affected side develop more rapidly and erupt before their counterparts on the uninvolved side (Fig. 1-5). Coincident to this phenomenon is premature shedding of the deciduous teeth. The bone of the maxilla and mandible is also enlarged, being wider and thicker, sometimes with an altered trabecular pattern.
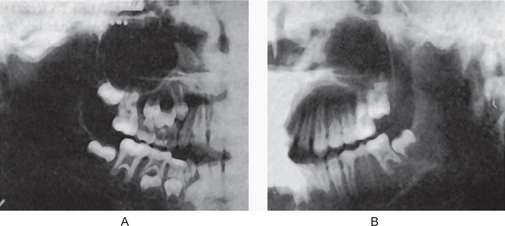
Figure 1-5 Facial hemihypertrophy.
The difference in the eruption pattern of the teeth on each side is apparent. The teeth on the affected side, where all deciduous teeth have already been lost, are not appreciably larger than those on the unaffected side (Courtesy of Dr John B Wittgen.
The tongue is commonly involved by the hemihypertrophy and may show a bizarre picture of enlargement of lingual papillae in addition to the general unilateral enlargement and contralateral displacement. In addition, the buccal mucosa frequently appears velvety and may seem to hang in soft, pendulous folds on the affected side.
Histologic Features
Tissue examination has been infrequently reported but is generally uninformative. In those cases reported, true muscular hypertrophy was not found.
Treatment and Prognosis
There is no specific treatment for this condition other than attempts at cosmetic repair. Cosmetic surgery is advised after cessation of growth. Effect on life expectancy is not certain, but in some cases patients have lived a normal life span. Periodic abdominal ultrasound/ MRI is recommended to rule out tumors.
Facial Hemiatrophy (Parry-Romberg syndrome, Romberg-Parry syndrome, progressive facial hemiatrophy, progressive hemifacial atrophy)
Hemifacial atrophy remains almost as much an enigma today as it was when first reported by Romberg in 1846. Hemifacial atrophy, originally described by Parry and Henoch and Romberg consists of slowly progressive atrophy of the soft tissues of essentially half the face, which is characterized by progressive wasting of subcutaneous fat, sometimes accompanied by atrophy of skin, cartilage, bone and muscle. Although the atrophy is usually confined to one side of the face and cranium, it may occasionally spread to the neck and one side of the body and it is accompanied usually by contralateral Jacksonian epilepsy, trigeminal neuralgia, and changes in the eyes and hair. Evidence of a mendelian basis is lacking. Lewkonia and Lowry reported the case of a 16-year-old boy who developed facial changes at age seven and had localized scleroderma on one leg and the trunk. The presence of antinuclear antibodies in his serum suggested that the Parry-Romberg syndrome may be a form of localized scleroderma. Hemifacial atrophy is a form of localized scleroderma and is supported by its concurrence with scleroderma.
Hemifacial atrophy is a rare condition that occurs sporadically although some familial distribution has been found. The majority of cases are sporadic with no definite inheritance being proven in the literature.
Etiology
The etiology has been the subject of considerable debate. Wartenburg considered the primary factor to be a cerebral disturbance leading to increased and unregulated activity of sympathetic nervous system, which in turn produced the localized atrophy through its trophic functions conducted by way of sensory trunks of the trigeminal nerve. Other workers suggested extraction of teeth, local trauma, infection and genetic factors could also be a cause. In a paper published in 1973, Poswillo attributed the development of facial deformities to the disruption of the stapedial artery. Poswillo fed pregnant rats with triazine and pregnant monkeys with thalidomide and showed the consistent maldevelopment of first and second branchial arch structures. Robinson, in 1987, supported Poswillo’s theory by demonstrating carotid flow abnormalities in two and defects related to vascular disruption in a third child with craniofacial microsomia.
Clinical Features
Hemifacial atrophy is a syndrome with diverse presentation. The most common early sign is a painless cleft, the ‘coup de sabre,’ near the midline of the face or forehead. This marks the boundary between normal and atrophic tissue. A bluish hue may appear in the skin overlying atrophic fat.
The affected area extends progressively with the atrophy of the skin, subcutaneous tissue, muscles, bones, cartilages, alveolar bone and soft palate on that side of the face. In addition to facial wasting that may include the ipsilateral salivary glands and hemiatrophy of the tongue, unilateral involvement of the ear, larynx, esophagus, diaphragm, kidney and brain have been reported.
It starts in the first decade and lasts for about three years before it becomes quiescent. The final deformity varies widely, burning itself out in some patients with minimal atrophy, while in others progressing to marked atrophy.
Neurological disorders are found in 15% of patients, while ocular findings occur in 10–40%, the most common being enophthalmos.
Rarely, one half of the body may be affected. This condition may be accompanied by pigmentation disorders, vitiligo, pigmented facial nevi, contralateral Jacksonian epilepsy, contralateral trigeminal neuralgia and ocular complications.
The disease occurs more frequently in women; female to male ratio is 3 : 2. It has a slight predilection for the left side and appears in the first or second decades of life. It progresses over a period of two and 10 years, and atrophy appears to follow the distribution of one or more divisions of the trigeminal nerve. The resulting facial flattening may be mistaken for Bell’s palsy (Fig. 1-6).
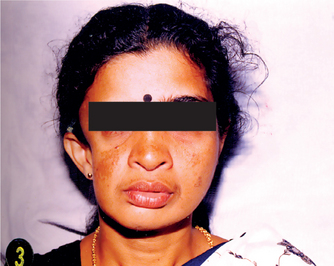
Oral Manifestations
Dental abnormalities include incomplete root formation, delayed eruption and severe facial asymmetry, resulting in facial deformation and difficulty with mastication. Hemiatrophy of the lips and the tongue is reported, as are dental effects. Foster has reported that growth of the teeth may be affected just as other tissues are involved. Eruption of teeth on the affected side may also be retarded.
Abnormalities of Dental Arch Relations
In the preceding sections the conditions discussed are those in which there is an actual or apparent abnormal variation in size of one or both jaws. Of far greater importance than a simple disparity in size is the disparity in relation of one jaw to the other and the difficulties in occlusion and function that result.
A great many different types of malocclusion exist, and many classifications have been evolved in an attempt to unify methods of treatment. The classification of Angle, proposed in 1899, is the most universally known and used. That classification, with the approximate percentage occurrence as determined by Angle in a large group of orthodontic patients, is as follows:
Class I. Arches in normal mesiodistal relations 69.0%.
Class II. Mandibular arch distal to normal in its relation to the maxillary arch.
Division 1. Bilaterally distal, protruding maxillary incisors 9.0.
Subdivision. Unilaterally distal, protruding maxillary incisors 3.5.
Division 2. Bilaterally distal, retruding maxillary incisors 4.0.
Subdivision. Unilaterally distal, retruding maxillary incisors 10.0.
Class III. Mandibular arch mesial to normal in its relation to the maxillary arch.
Since these abnormal jaw relations constitute a separate course of study, no further allusion to this subject will be made here.
Developmental Disturbances of Lips And Palate
Congenital Lip and Commissural Pits and Fistulas
Congenital lip pits and fistulas are malformations of the lips, often following a hereditary pattern, that may occur alone or in association with other developmental anomalies such as various oral clefts. Both Taylor and Lane and McConnel and his associates have emphasized that in 75–80% of all cases of congenital labial fistulas, there is an associated cleft lip or cleft palate, or both. The association of pits of the lower lip and cleft lip and/or cleft palate, termed van der Woude’s syndrome, has been reviewed by Cervenka and his associates.
Commissural pits are an entity probably very closely related to lip pits, but occur at the lip commissures, lateral to the typical lip pits. Everett and Wescott have described this entity and noted that it is also frequently hereditary, possibly a dominant characteristic following a Mendelian pattern, and may be associated with other congenital defects.
Etiology
Many theories of the etiology of congenital lip pits have been offered, but none has been universally accepted. Pits may result from notching of the lip at an early stage of development, with fixation of the tissue at the base of the notch, or from failure of complete union of the embryonic lateral sulci of the lip, which persist and ultimately develop into the typical pits.
Commissural pits are also difficult to explain, but they occur at the site of the horizontal facial cleft and may represent defective development of this embryonic fissure.
Clinical Features
The lip pit or fistula is a unilateral or bilateral depression or pit that occurs on the vermilion surface of either lip but far more commonly on the lower lip (Fig. 1-7A). In some cases a sparse mucous secretion may exude from the base of this pit. The lip sometimes appears swollen, accentuating the appearance of the pits.
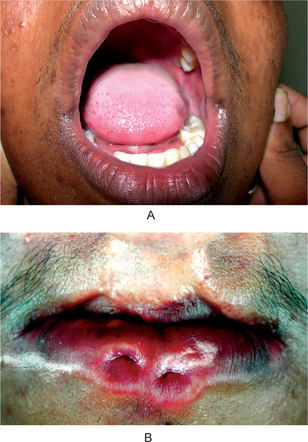
Figure 1-7 (A) Congenital lip pits. (B) Congenital commissural pits. Courtesy of Dr Spencer Lilly, Meenakshi Ammal Dental College, Chennai.
Commissural pits appear as unilateral or bilateral pits at the corners of the mouth on the vermilion surface (Fig. 1-7B). An actual fistula may be present from which fluid may be expressed. Whether this tract, either in lip or commissural fistulas, represents a true duct is not clear. Interestingly, in several cases preauricular pits have been reported in association with commissural pits.
van der Woude Syndrome (Cleft lip syndrome, lip pit syndrome, dimpled papillae of the lip)
van der Woude syndrome is an autosomal dominant syndrome typically consisting of a cleft lip or cleft palate and distinctive pits of the lower lips. The degree to which individuals carrying the gene are affected is widely variable, even within families. These variable manifestations include lip pits alone, missing teeth, or isolated cleft lip and palate of varying degrees of severity. Other associated anomalies have also been described.
Etiology
The most prominent and consistent feature of van der Woude syndrome is orofacial anomalies. They are due to an abnormal fusion of the palate and lips, at days 30–50 postconception. The van der Woude syndrome can be caused by deletions in chromosome band 1q32, and linkage analysis has confirmed this chromosomal locus as the disease gene site. The gene has been localized to chromosome band 1q32. Further studies have raised the possibility that the degree of phenotypic expression of a gene defect at this locus may be influenced by a second modifying gene that has been mapped to chromosome band 17p11.
Clinical Features
In general, van der Woude syndrome affects about 1 in 100,000–200,000 people. About 1–2% of patients with cleft lip or palate have van der Woude syndrome. The van der Woude syndrome affects both genders equally and no difference among them have been reported. The severity of the van der Woude syndrome varies widely, even within families. About 25% of individuals with the van der Woude syndrome have no findings or minimal ones, such as missing teeth or trivial indentations in the lower lips. Others have severe clefting of the lip or palate. The hallmark of the van der Woude syndrome is the association of cleft lip and/or palate with distinctive lower lip pits. This combination is seen in about 70% of those who are overtly affected but in less than half of those who carry the gene. The cleft lip and palate may be isolated. They may take any degree of severity and may be unilateral or bilateral. Submucous cleft palate is common and may be easily missed on physical examination. Hypernasal voice and cleft or bifid uvula are clues to this diagnosis. It is possible as well that a bifid uvula is an isolated finding in certain individuals with the van der Woude syndrome. The lower lip pits seen in this syndrome are fairly distinctive (Fig. 1-8). The pits are usually medial, on the vermilion portion of the lower lip. They tend to be centered on small elevations in infancy, but are simple depressions in adults. These pits are often associated with accessory salivary glands that empty into the pits, sometimes leading to embarrassing visible discharge. Occasionally lip pits may be the only manifestation of the syndrome. Affected individuals may have maxillary hypodontia; missing maxillary incisors or missing premolars. Again, this may be the only manifestation of the syndrome. Although infrequently reported, other oral manifestations include syngnathia (congenital adhesion of the jaws); narrow, high, arched palate; and ankyloglossia (short glossal frenulum or tongue-tie).
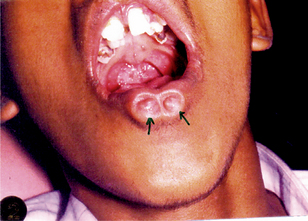
Extraoral Manifestations
The reported incidence of extraoral manifestations are rare but include limb anomalies, popliteal webs, and brain abnormalities. Accessory nipples, congenital heart defects, and Hirschsprung disease have also been reported. It is uncertain whether these extraoral manifestations are unassociated additional anomalies or infrequently expressed aspects of van der Woude syndrome.
Treatment
Along with a thorough orofacial examination, a thorough general physical examination helps to determine if there are other associated anomalies of the cardiovascular system, genitourinary system, limbs, or other organ systems. Examination and genetic counseling by a pediatric geneticist (dysmorphologist) is suggested for families that may be affected by the van der Woude syndrome. This should include an examination of as many potentially affected family members (probands) as possible. Surgical repair of the cleft lip and palate or other anomalies may be required, when planning surgical intervention, imaging studies of affected areas, such as CT scanning of the oropharynx, may be appropriate. Even among those less severely affected, surgical excision of lip pits is often performed, either to alleviate discomfort or for cosmetic reasons (e.g. improving the appearance of lip pits or reducing mucous discharge).
Cleft Lip and Cleft Palate
The term cleft lip and palate is commonly used to represent two types of malformation, i.e., cleft lip with or without cleft palate (CL/P) and cleft palate (CP). Cleft lip and palate are common congenital malformations. The reported incidence of clefts of the lip and palate varies from 1 in 500 to 1 in 2500 live births depending on geographic origin, racial and ethnic background and socioeconomic status. In general, Asian population have the highest frequencies, often at 1 in 500 or higher, with Caucasian population intermediate, and Africanderived population the lowest at 1 in 2500.
An understanding of the normal human maxillofacial development is necessary before a discussion of cleft lip and palate. During the fourth week of gestation, the maxillary processes emerge from the first branchial arch on each side and the nasal placodes form from the frontal prominence. By the fifth week, all the primordia for the lip and palate are present. The medial, lateral nasal and the frontonasal processes are formed from the nasal placodes and the maxillary processes continue to enlarge. During the seventh week the medial nasal, frontonasal and maxillary processes fuse to form the primary palate, which becomes the medial portion of the upper lip, alveolus and the anterior part of hard palate up to the incisive foramen. When the primary palate is completely formed, the maxillary processes enlarge intraorally to form the palatine processes. During the 8th week of gestation, the palatal shelves fill up the space on both sides of the tongue. During the 9th and 10th weeks, the mandibular arch enlarges and the tongue drops. The palatal shelves transpose horizontally and fuse with each other and with the anterior part of the palate. Palatal fusion occurs anteroposteriorly and the process is completed by the 11th to 12th weeks (Shapiro, 1976).
Failure in the fusion of the nasal and maxillary processes leads to the cleft of the primary palate, which can be unilateral or bilateral. The degree of cleft can vary from a slight notch on the lip to complete cleft of the primary palate. Cleft of the secondary palate is medial. It varies from bifid uvula to complete cleft palate up to the incisive foramen. When it is associated with the primary palate, a complete uni- or bilateral cleft lip and palate is formed.
Etiology
It has been clearly established by Fogh-Andersen and confirmed by numerous other investigators that two separate and distinct entities exist:
Heredity is undoubtedly one of the most important factors to be considered in the etiology of these malformations. However, there is increasing evidence that environmental factors are important as well. According to Fogh-Andersen, slightly less than 40% of the cases of cleft lip with or without cleft palate are genetic in origin, whereas slightly less than 20% of the cases of isolated cleft palate appear to be genetically derived. Most investigations indicate that the inheritance pattern in cleft lip with or without cleft palate is different from that in isolated cleft palate. The mode of transmission of the defect is uncertain. This has been discussed by Bhatia, who pointed out that the possible main modes of transmission are either by a single mutant gene, producing a large effect, or by a number of genes (polygenic inheritance), each producing a small effect which together create this condition. It should be pointed out that cytogenetic studies have failed to reveal visible alterations in chromosomal morphology of the affected individuals.
Bixler more recently has expanded upon this concept and reiterated that there are two forms of clefts. The most common is hereditary, its nature being most probably polygenic (determined by several different genes acting together). In other words, when the total genetic liability of an individual reaches a certain minimum level, the threshold for expression is reached and a cleft occurs. Actually, it is presumed that every individual carries some genetic liability for clefting, but if this is less than the threshold level, there is no cleft. When the individual liabilities of two parents are added together in their offspring, a cleft occurs if the threshold value is exceeded. However, even though this is the most common form of cleft, the threshold value is sufficiently high that it is a low-risk type. The second form of cleft is monogenic or syndromic and is associated with a variety of other congenital anomalies. Since these are monogenic, they are of a high-risk type. Bixler has pointed out that, fortunately, the clefting syndromes are rare and probably make up only 5% of all cleft cases even though, according to Cohen, there are now over 300 clefting syndromes reported in the literature.
Although there is insufficient evidence that nutritional disturbances cause cleft palates in human beings, abnormal dietary regimens have caused developmental clefts in animals. Cleft palate has been experimentally produced in newborn rats by feeding diets either deficient or excessive in vitamin A to maternal rats during pregnancy. Riboflavin-deficient diets fed to pregnant rats have also produced offspring with a high incidence of cleft palate. The administration of cortisone to pregnant rabbits has induced similar clefts in their young.
Strean and Peer reported that physiologic, emotional, or traumatic stress may play a significant role in the etiology of human cleft palate, since stress induces increased function of the adrenal cortex and secretion of hydrocortisone. Their study, based on histories of 228 mothers of children with cleft palate, confirms the experimental findings of cleft palate in animals due to the action of stressor agents or the administration of cortisone. However, Fraser and Warburton have reported data which indicate that neither maternal emotional stress nor the lack of a prenatal nutritional supplement was causally related to the occurrence of cleft lip or cleft palate.
Other factors that have been suggested as possible causes of cleft palate include:
• A defective vascular supply to the area involved
• A mechanical disturbance in which the size of the tongue may prevent the union of parts
• Circulating substances, such as alcohol and certain drugs and toxins
Despite the numerous clinical and experimental investigations, the etiology of cleft palate in the human being is still largely unknown. It must be concluded; however, that heredity is probably the most important single factor.
Clinical Features
Cleft lip with or without palate is more common in males than in females. Males have also been reported to have more severe defects, whereas the isolated cleft palate is more common in females.
Clefts can be divided into nonsyndromic and syndromic forms. Syndromic forms of clefts include those cases that have additional birth defects like lip pits or other malformations, whereas nonsyndromic clefts are those cases wherein the affected individual has no other physical or developmental anomalies and no recognized maternal environmental exposures. At present, most studies suggest that about 70% of cases of cleft lip with or without palate and 50% of isolated cleft palate are nonsyndromic. The remaining syndromic cases can be subdivided into chromosomal anomalies, teratogens and uncategorized syndromes.
Cleft lip can occur as a unilateral (on the left or right side) or as a bilateral anomaly. The line of cleft always starts on the lateral part of the upper lip and continues through the philtrum to the alveolus between the lateral incisor and the canine tooth. The clefting anterior to the incisive foramen (i.e. lip and alveolus) is also defined as a cleft of primary palate. Cleft lip may occur with a wide range of severity, from a notch located on the left or right side of the lip to the most severe form, bilateral cleft lip and alveolus that separates the philtrum of the upper lip and premaxilla from the rest of the maxillary arch. When cleft lip continues from the incisive foramen further through the palatal suture in the middle of the palate, a cleft lip with palate (either unilateral or bilateral) is present (Figs. 1-9, 1-10). A wide range of severity may be observed. The cleft line may be interrupted by skin or mucosa bridges, hard (bone) bridges, or both, corresponding to a diagnosis of an incomplete cleft. This occurs in unilateral and bilateral CLP.
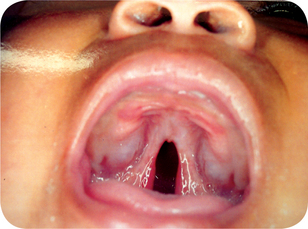
Figure 1-9 Cleft involving both the hard and the soft palates. Courtesy of Dr R Manikandan, Meenakshi Ammal Dental College, Chennai)
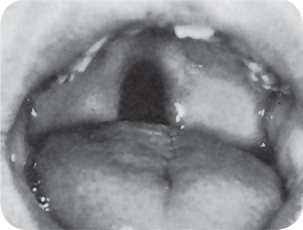
Figure 1-10 Cleft involving the soft palate only. Courtesy of Dr John M Tondra and Harold M Trusler.
Isolated cleft palate is etiologically and embryologically different from cleft lip with or without cleft palate. Several subtypes of isolated cleft palate can be diagnosed based on severity. The uvula is the place where the minimal form of clefting of the palate is observed (Fig. 1-11). A more severe form is a cleft of the soft palate. A complete cleft palate constitutes a cleft of the hard palate, soft palate, and cleft uvula. The clefting posterior to the incisive foramen is defined as a cleft of secondary palate.
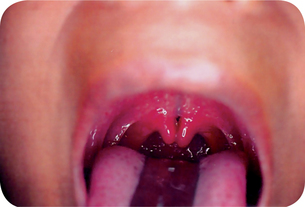
Figure 1-11 Cleft or biid uvula. Courtesy of Dr Manikandan R, Meenakshi Ammal Dental College, Chennai.
A median maxillary anterior alveolar cleft is a relatively common defect, occurring in approximately 1% of the population, according to Stout and Collett, but this is unrelated to cleft lip or cleft palate. This type of cleft has been discussed by Gier and Fast, who suggested that it might be due to precocious limitation of the growth of the primary ossification centers on either side of the midline at the primary palate, or to their subsequent failure to fuse. In addition, Miller and his coworkers have suggested that at least some cases may represent an incomplete manifestation of the median cleft-face syndrome (hypertelorism, median cleft of the premaxilla and palate, and cranium bifidum occultum). This syndrome has no clinical manifestations and is usually detected only on routine intraoral radiographic examination (Fig. 1-12).
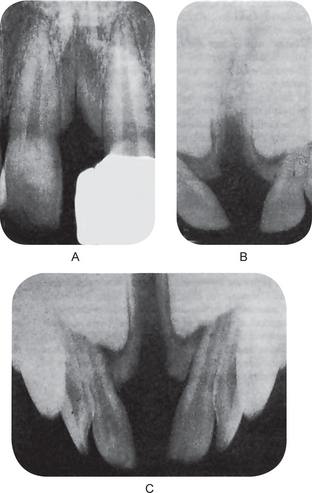
Figure 1-12 Median maxillary anterior alveolar cleft.
All degrees of severity of the cleft may occur (Copyright by the American Dental Association. Reprinted by permission and by courtesy of Dr Arthur S Miller. From AS Miller, JN Greeley and DL Catena: Median maxillary anterior alveolar cleft: report of three cases. J Am Dent Assoc, 79: 896, 1969.
Clinical Significance
Most cases of cleft lip can be surgically repaired with excellent cosmetic and functional results. It is customary to operate before the patient is one month old or when he has regained his original birth weight and is still gaining.
Both the physical and psychologic effects of cleft palate on the patient are of considerable concern. Eating and drinking are difficult because of regurgitation of food and liquid through the nose. The speech problem is also serious and tends to increase the mental trauma suffered by the patient.
Most individuals with CL, CP, or both, require the coordinated care of providers in many fields of medicine and dentistry, as well as those in speech pathology, otolaryngology, audiology, genetics, nursing, mental health, and social medicine.
Treatment
Treatment of CLP anomalies requires years of specialized care. Although successful treatment of the cosmetic and functional aspects of orofacial cleft anomalies is now possible, it is still challenging, lengthy, costly, and dependent on the skills and experience of a medical team. This especially applies to surgical, dental, and speech therapies. Undoubtedly, closure of the CL is the first major procedure that tremendously changes children’s future development and ability to thrive. Variations occur in timing of the first lip surgery; however, the most usual time occurs at approximately three months of age. Pediatricians used to strictly follow a rule of ‘three 10s’ as a necessary requirement for identifying the child’s status as suitable for surgery (i.e. 10 lb, 10 mg/L of hemoglobin, and age 10 weeks). Although pediatricians are presently much more flexible, and some surgeons may well justify a neonatal lip closure, the rule of three 10s is still very useful.
Anatomical differences predispose children with CLP and with isolated CP to ear infections. Therefore, ventilation tubes are placed to ventilate the middle ear and prevent hearing loss secondary to otitis media with effusion (OME). In multidisciplinary teams with significant participation of an otolaryngologist, the tubes are placed at the initial surgery and at the second surgery routinely. The hearing is tested after the first placement when ears are clear with tubes. If no cleft surgery is planned early, placing the tubes by age six months and monitoring hearing with repeated testing is recommended. Complications include eardrum perforation and otorrhea, particularly in patients with open secondary palates in which closure is planned for a later date.
Cheilitis Glandularis (Actinic cheilitis, squamous cell carcinoma)
Cheilitis glandularis (CG) is a clinical diagnosis that refers to an uncommon and poorly understood inflammatory disorder of the lip. The condition is characterized by progressive enlargement and eversion of the lower labial mucosa that results in obliteration of the mucosal-vermilion interface. With externalization and chronic exposure, the delicate labial mucous membrane is secondarily altered by environmental influences, leading to erosion, ulceration, and crusting. Most significantly, susceptibility to actinic damage is increased. Therefore, CG can be considered a potential predisposing factor for the development of actinic cheilitis and squamous cell carcinoma.
Etiology
CG is an unusual clinical manifestation of cheilitis that evolves in response to one or more diverse sources of chronic irritation. Lip enlargement is attributable to inflammation, hyperemia, edema, and fibrosis. Surface keratosis, erosion, and crusting develop consequent to longstanding actinic exposure, unusual repeated manipulations that include self-inflicted biting or other factitial trauma, excessive wetting from compulsive licking, drying (sometimes associated with mouth-breathing, atopy, eczema, and asthma), and any other repeated stimulus that could serve as a chronic aggravating factor.
Clinical Features
In 1870, von Volkman introduced the term cheilitis glandularis. He described a clinically distinct, deeply suppurative, chronic inflammatory condition of the lower lip characterized by mucopurulent exudates from the ductal orifices of the labial minor salivary glands. In 1914, Sutton proposed that the characteristic lip swelling was attributable to a congenital adenomatous enlargement of the labial salivary glands. This remained the prevailing hypothesis until 1984 when Swerlick and Cooper reported five new cases and a retrospective analysis of all cases of CG reported until that time. Their studies revealed no evidence to support the assertion that salivary gland hyperplasia is responsible for CG.
Cheilitis glandularis is a chronic progressive condition. Patients typically present for diagnostic consultation within 3–12 months of onset. Complaints vary according to the nature and the degree of pain, the enlargement and the loss of elasticity of the lip, and the extent of evident surface change. Asymptomatic lip swelling initially occurs with clear viscous secretion expressible from dilated ductal openings on the mucosal surface. Some patients report periods of relative quiescence interrupted by transient or persistent painful episodes associated with suppurative discharge. A burning discomfort or a sensation of rawness referable to the vermilion border may be reported. This is associated with atrophy, speckled leukoplakic change, erosion, or frank ulceration with crusting. CG affects the lower lip almost exclusively. In more suppurative cases, application of gentle pressure can elicit mucopurulent exudate. Prolonged exposure to the external environment results in desiccation and disruption of the labial mucous membrane, predisposing it to inflammatory, infectious, and actinic influences. This is an uncommon condition. CG has been associated with a heightened risk for the development of squamous cell carcinoma. In many cases, dysplastic (premalignant) surface epithelial change is evident, and frank carcinomas have been reported in 18–35% of cases. The disorder appears to favor adult males; however, cases have been reported in both genders. The condition most frequently occurs between the fourth and seventh decades of life; however, the age range is wide. The risk of dysplasia and carcinoma increases with age, especially in fair-skinned individuals with sun-damaged skin. This is because the characteristic eversion of the lower lip results in long-term chronic exposure of the thinner, more vulnerable labial mucosa to actinic influence.
Classification
CG had historically been subclassified into three types, now believed to represent evolving stages in the severity of a single progressive disorder.
• In the simple type, multiple, painless, papular surface lesions with central depressions and dilated canals are seen.
• The superficial (suppurative) type (also referred to as Baelz disease) consists of painless, indurated swelling of the lip with shallow ulceration and crusting.
• CG of the deep suppurative type (CG apostematosa, CG suppurativa profunda, myxadenitis labialis) comprises a deep-seated infection with formation of abscesses, sinus tracts and fistulas, and potential for scarring.
The latter two types of CG have the highest association with dysplasia and carcinoma, respectively.
Histologic Findings
Lip biopsy is indicated to rule out specific granulomatous diseases that predispose to lip enlargement and aid in establishing a definitive diagnosis. A representative incisional biopsy specimen should consist of a wedge (or punch) of lip tissue that includes surface epithelium and is of adequate depth to ensure inclusion of several submucosal salivary glands. The term cheilitis glandularis is a provisional descriptive designation rather than a definitive diagnosis. It refers to a constellation of clinical findings that can reflect a broad scope of possible histologic changes; therefore, no consistent or pathognomonic features of this disorder are seen at the microscopic level. Instead, a diverse array of possible alterations can be seen in both the surface epithelium and the submucosal tissues. These findings best enable the clinician to presumptively determine the etiology and the nature of individual cases.
The minor salivary glands may appear normal under the microscope, or they may exhibit various changes indicative of nonspecific sialadenitis. These changes can include atrophy or distention of acini, ductal ectasia with or without squamous metaplasia, chronic inflammatory infiltration and replacement of glandular parenchyma, and interstitial fibrosis. Suppuration and sinus tracts may be present in cases that involve bacterial infection. Other possible histologic findings include stromal edema, hyperemia, surface hyperkeratosis, erosion, or ulceration.
Differential Diagnosis
Differential diagnoses of this condition include actinic keratosis, atopic dermatitis, cheilitis granulomatosa (Miescher-Melkersson-Rosenthal syndrome), sarcoidosis and squamous cell carcinoma.
Treatment
The approach to treatment is based on diagnostic information obtained from histopathologic analysis, the identification of likely etiologic factors responsible for the condition, and attempts to alleviate or eradicate those causes. In cases with acute or chronic suppuration, bacterial culture and sensitivity testing is indicated for selection of appropriate antibiotic therapy. Given the relatively small number of reported cases of CG, neither sufficient nor reliable data exist with regard to medical approaches to the condition. Therefore, treatment varies accordingly for each patient. In cases where a history of chronic sun exposure exists (especially if the patient is fair skinned or the everted lip surface is chronically eroded, ulcerated, or crusted), biopsy is strongly recommended to rule out actinic cheilitis or carcinoma.
Cheilitis Granulomatosa (Miescher-Melkersson-Rosenthal syndrome)
Cheilitis granulomatosa is a chronic swelling of the lip due to granulomatous inflammation. Miescher cheilitis is the term used when the granulomatous changes are confined to the lip. Miescher cheilitis is generally regarded as a monosymptomatic form of the Melkersson-Rosenthal syndrome, although the possibility remains that these may be two separate diseases. Melkersson-Rosenthal syndrome is the term used when cheilitis occurs with facial palsy and plicated tongue. Melkersson-Rosenthal syndrome is occasionally a manifestation of Crohn’s disease or orofacial granulomatosis.
Etiology
The cause is unknown. A genetic predisposition may exist in Melkersson-Rosenthal syndrome; siblings have been affected, and a plicated tongue may be present in otherwise unaffected relatives. Crohn’s disease, sarcoidosis, and orofacial granulomatosis may present in a similar clinical fashion, and with identical histologic findings. Dietary or other antigens are the most common identified causes of orofacial granulomatosis. Contact antigens are sometimes implicated.
Clinical Features
Cheilitis granulomatosa is episodic with nontender swelling and enlargement of one or both lips (Fig. 1-13). Occasionally, similar swellings involve other areas, including the periocular region. The first episode of edema typically subsides completely in hours or days. After recurrent attacks, swelling may persist and slowly increase in degree, eventually becoming permanent. Recurrences can range from days to years. Attacks sometimes are accompanied by fever and mild constitutional symptoms (e.g. headache, visual disturbance).
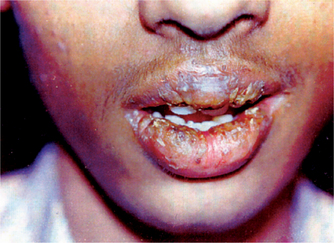
The earliest manifestation is sudden diffuse or occasionally nodular swellings of the lip or the face involving (in decreasing order of frequency) the upper lip, the lower lip, and one or both cheeks. The forehead, the eyelids, or one side of the scalp may be involved (less common). The upper lip is involved slightly more often than the lower lip, and it may feel soft, firm, or nodular on palpation. Once chronicity is established, the enlarged lip appears cracked and fissured with reddish brown discoloration and scaling. The fissured lip becomes painful and eventually acquires the consistency of firm rubber. Swelling may regress very slowly after some years. Regional lymph nodes are enlarged (usually minimally) in 50% of patients. A fissured or plicated tongue is seen in 20–40% of patients. Its presence from birth (in some patients) may indicate a genetic susceptibility. Patients may lose the sense of taste and have decreased salivary gland secretion. Facial palsy of the lower motor-neuron type occurs in about 30% of patients. Facial palsy may precede attacks of edema by months or years, but it more commonly develops later. Facial palsy is intermittent at first, but it may become permanent. It can be unilateral or bilateral, partial or complete. Other cranial nerves (e.g. olfactory, auditory, glossopharyngeal, hypoglossal) are occasionally affected. Poorly defined association of psychiatric and neurologic features are reported. Autonomic disturbances may occur. In granulomatous cheilitis, normal lip architecture is eventually altered by the presence of lymphedema and noncaseating granulomas in the lamina propria. The frequency is unknown; the condition is rare. Morbidity depends on whether underlying organic disease, such as Crohn’s disease or sarcoidosis, is present. There is no racial and sexual predilection. The age of onset is usually young adulthood.
Differential Diagnosis
Differential diagnoses include insect bites and sarcoidosis. Serum angiotensin-converting enzyme test, chest radiography or gallium or positron emission tomography (PET) scanning may be performed to help exclude sarcoidosis. Gastrointestinal tract endoscopy and radiography may be used to help exclude Crohn’s disease.
Histologic Features
The histologic features of cheilitis granulomatosa and the swellings of the syndrome are rather characteristic, consisting of a chronic inflammatory cell infiltrate—particularly peri- and para-vascular aggregations of lymphocytes, plasma cells, and histiocytes—and focal noncaseating granuloma formation with epithelioid cells and Langhans type giant cells. The microscopic findings are suggestive of sarcoidosis, but as yet there is insufficient evidence to relate cheilitis granulomatosa with sarcoid.
Treatment
Patch tests may be used to help exclude reactions to metals, food additives, or other oral antigens. Some cases may be associated with such sensitivities. If found, avoidance of the implicated allergen is recommended. This condition can be conservatively managed by intralesional corticosteroid injections, nonsteroidal anti-inflammatory agents, mast cell stabilizers, clofazimine and tetracycline (used for antiinflammatory activity). Surgery and radiation have been reported to be used.
Hereditary Intestinal Polyposis Syndromen
(Peutz-Jeghers syndrome, intestinal hamartomatous polyps in association with mucocutaneous melanocytic macules)
The Peutz-Jeghers syndrome is an autosomal dominantly inherited disorder characterized by intestinal hamartomatous polyps in association with mucocutaneous melanocytic macules. A 15-fold elevated relative risk of developing cancer exists in this syndrome over that of the general population; cancer primarily is of the GI tract, including the pancreas and luminal organs, and of the female and male reproductive tracts and the lung. The syndrome was named after Peutz, who noted a relationship between the intestinal polyps and the mucocutaneous macules in 1921, and after Jeghers, who is credited with the definitive descriptive reports in 1944 and later in 1949.
Etiology
The cause of the Peutz-Jeghers syndrome appears to be a germline mutation of the STK11 (serine threonine kinase 11) gene in most cases, located on band 19p13.3. Penetrance of the gene is variable, causing varied phenotypic manifestations among patients with Peutz-Jeghers syndrome (e.g. inconsistent number of polyps, differing presentation of the macules) and allowing for a variable presentation of cancer. Because the signaling pathway of the STK11 gene product currently is not identified, the mechanism of hamartomatous polyp formation and mucocutaneous pigmentation is not known. In cancer formation, STK11 inactivation appears to occur early and might be followed by interruption of the APC/β catenin and p53 pathways, but this has not been fully elucidated. STK11 may be a tumor suppressor gene in that its overexpression can induce a growth arrest of a cell at the G1 phase of the cell cycle and that somatic inactivation of the unaffected allele of STK11 often is observed in polyps and cancers from patients with the Peutz-Jeghers syndrome.
Clinical Features
The Peutz-Jeghers syndrome has been described in all races. The occurrence of cases in males and females is about equal. The average age at diagnosis is 23 years in men and 26 years in women. The affected individuals usually have a positive family history of the Peutz-Jeghers syndrome. The principal causes of morbidity stem from the intestinal location of the polyps (i.e. small intestine, colon, stomach). Morbidity includes small intestinal obstruction and intussusception (43%), abdominal pain (23%), hematochezia (14%), and prolapse of a colonic polyp (7%), and these typically occur in the second and third decades of life.
The presenting complaints include repeated bouts of abdominal pain in patients younger than 25 years, unexplained intestinal bleeding in a young patient, or menstrual irregularities in females (due to hyperestrogenism from sex cord tumors with annular tubules). Cutaneous pigmentation (1–5 mm macules) of the perioral region crossing the vermilion border (94%), perinasal, and perioral areas is seen; pigmentation may also be present on the fingers and toes, on the dorsal and ventral aspects of the hands and feet, and around the anus and genitalia. This pigmentation may fade after puberty. Mucous membrane pigmentation primarily affects the buccal mucosa (66%) and the intestinal mucosa rarely. Other manifestations of this syndrome include precocious puberty, prolapse of tissue from the rectum, rectal mass (rectal polyp), testicular mass, gynecomastia and growth acceleration (due to Sertoli cell tumor).
Labial and Oral Melanotic Macule
The oral mucosa is usually not pigmented despite the fact that it has the same density of melanocytes as the skin. Occasional patients; however, will show a focal area of melanin deposition, either as a response to local chronic conditions (mechanical trauma, tobacco smoking, chronic autoimmune mucositis), racial background (the darker a person’s skin color the more likely they are to have oral pigmentation), or systemic medications, especially chloroquine. Moreover, certain syndromes and systemic diseases have oral pigmentation as part of their spectrum.
Most focal melanin deposits of the oral mucosa which are not associated with race or an appropriate syndrome are innocuous surface discolorations called oral melanotic macule (focal melanosis). This entity represents not only a focal increase in melanin deposition but a concomitant increase in the number of melanocytes. Unlike the cutaneous ephelis (freckle), the oral melanotic macule is neither dependent on sun exposure, nor does it show the elongated rete ridges of actinic lentigo. Some authorities have questioned the purported lack of an association with actinic irradiation for melanotic macule located on the vermilion border, preferring to consider the lesion at this site to be a distinct entity called labial melanotic macule. Melanotic macules are found in the mouths of 1 of every 1,000 adults.
Clinical Features
The oral melanotic macule has a 2:1 female predilection with an average age of 43 years at the time of diagnosis, although it can develop at any age. One-third of lesions occur on the vermilion border of the lower lip, but the buccal mucosa, gingiva and palate are other sites of common occurrence. Almost one fifth of the lesions are multiple.
The typical macule is a well-demarcated, uniformly tan to dark brown, asymptomatic, round or oval discoloration less than 7 mm in diameter. The lesion is not thickened and has the same consistency as surrounding mucosa. It tends to have an abrupt onset and seldom enlarges after diagnosis.
A special case of oral melanosis, called smoker’s melanosis is found on the gingival or buccal mucosa in heavy smokers. It has an adult onset and is often associated with a concomitant superficial white/gray keratosis. The keratosis may become thick enough to mimic leukoplakia, although it is not known whether or not it is a true precancer. Both the pigmentation and the keratosis diminish or disappear once the tobacco habit is stopped.
While the melanotic macule is an innocuous lesion, it must be remembered that focal oral and oropharyngeal pigmentation might represent an internal malignancy (usually lung), an oral manifestation of a systemic disease or one facet of a genetic syndrome.
Histologic Features
The oral melanotic macule is characterized by an otherwise normal stratified squamous epithelium with abundant melanin deposits within the keratinocytes of the basal and parabasal layers. Deposits may also be seen within subepithelial stroma (melanin incontinence), perhaps within macrophages or melanophages. There is no underlying inflammatory response. The melanin can be distinguished from iron deposits with melanin stains or by the loss of brown color after bleaching. Brown formalin deposits can be differentiated by their association with erythrocytes rather than with basal layer epithelial cells.
Treatment and Prognosis
No treatment is required for oral melanotic macule except for esthetic considerations. The intraoral melanotic macule has no malignant transformation potential, but an early melanoma could have a similar clinical appearance. For this reason, pigmented macular lesions of recent onset, large size, irregular pigmentation, unknown duration, or with a history of recent enlargement should be excised and examined histopathologically.
Developmental Disturbances of Oral Mucosa
Fordyce’s Granules (Fordyce’s disease)
This is not a disease of the oral mucosa, as the name might indicate, but rather a developmental anomaly characterized by heterotopic collections of sebaceous glands at various sites in the oral cavity. It has been postulated that the occurrence of sebaceous glands in the mouth may result from inclusion in the oral cavity of ectoderm having some of the potentialities of skin in the course of development of the maxillary and mandibular processes during embryonic life. A complete review of Fordyce’s granules was published by Miles, and a superb investigation of the sebaceous glands of the lips and oral cavity was carried out by Sewerin.
Clinical Features
Fordyce’s granules appear as small yellow spots, either discretely separated or forming relatively large plaques, often projecting slightly above the surface of the tissue (Fig. 1-14). They are found most frequently in a bilaterally symmetrical pattern on the mucosa of the cheeks opposite the molar teeth but also occur on the inner surfaces of the lips, in the retromolar region lateral to the anterior faucial pillar, and occasionally on the tongue, gingiva, frenum, and palate. Ectopic sebaceous glands have been discussed in an excellent review by Guiducci and Hyman and are recognized to occur, besides in the oral cavity, in the esophagus, the female genitalia including the cervix uteri, the male genitalia, the nipples, the palms and soles, the parotid gland, the larynx, and the orbit.

Studies by Halperin and coworkers, confirmed by Miles, have indicated that the oral condition is present in approximately 80% of the population, with apparently no significant differences in occurrence between the genders or races. Fewer children than adults exhibit Fordyce’s granules, probably because the sebaceous glands and hair system do not reach maximal development until puberty. Nevertheless, Miles has reported that large numbers of sebaceous glands in the cheeks and lips may sometimes be found in children long before the age of puberty. Because of the high incidence of these glands in the oral cavity, Knapp has suggested that they be regarded as sebaceous nevi rather than ectopic glandular tissue.
Histologic Features
These heterotopic collections of sebaceous glands are identical with those seen normally in the skin, but are unassociated with hair follicles, although a single hair follicle and hair shaft growing from the gingiva—an extremely rare occurrence—has been reported recently by Baughman (Fig. 1-15). The glands are usually superficial and may consist of only a few or a great many lobules, all grouped around one or more ducts which open on the surface of the mucosa. These ducts may show keratin plugging.
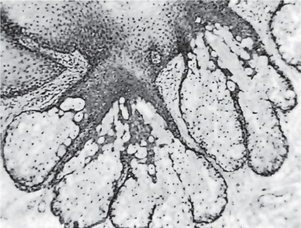
Treatment
These glands are innocuous, have no clinical or functional significance, and require no treatment. However, very rarely a benign sebaceous gland adenoma may develop from these intraoral structures, such as in the case involving the buccal mucosa reported by Miller and McCrea. Sewerin has also reported the occasional development of keratin-filled pseudocysts from the ducts of these sebaceous glands.
Focal Epithelial Hyperplasia (Heck’s disease)
One of the most contagious of the oral papillary lesions is focal epithelial hyperplasia or Heck’s disease, another HPV-induced epithelial proliferation first described in 1965 in Native Americans. The level of contagion is exemplified by the fact that in some isolated population up to 40% of children have been affected. Today, it is known to exist in numerous populations and ethnic groups and to be produced by one of the subtypes of the human papillomavirus, HPV-13, and possibly HPV-32. Where the infection is endemic among children, adults seem to have minimal evidence of residual oral lesions and so the lesions are presumed to eventually disappear on their own.
Focal epithelial hyperplasia is somewhat different from other HPV infections in that it is able to produce extreme acanthosis or hyperplasia of the prickle cell layer of the epithelium with minimal production of surface projections or induction of connective tissue proliferation. The mucosa may be 8–10 times thicker than normal.
Clinical Features
Heck’s disease primarily occurs in children, but lesions may occur in young and middle-aged adults. There is no gender predilection. Sites of greatest involvement include the labial, buccal and lingual mucosa, but gingival and tonsillar lesions have also been reported.
Individual lesions are broad-based or so slightly elevated as to present as well demarcated plaques. Lesions are frequently papillary in nature, but relatively smooth-surfaced, flat-topped lesions are more commonly seen. Papules and plaques are usually the color of normal mucosa, but may be pale or, rarely, white. Hyperplastic lesions are small (0.3–1.0 cm), discrete, and well-demarcated, but they frequently cluster so closely together that the entire mucosal area takes on a cobblestone or fissured appearance.
Histologic Features
Epithelial hyperplasia in this disease presents microscopically as an abrupt and sometimes considerable focal acanthosis of the oral epithelium. The thickened mucosa extends upward, not down into underlying connective tissues, hence the lesional rete ridges are at the same depth as the adjacent normal rete ridges. The ridges themselves are widened, often confluent and sometimes club-shaped; they are not long and thin as in psoriasis and other diseases. Some superficial keratinocytes show a koilocytic change similar to that seen in other HPV infections, while occasionally others demonstrate a collapsed nucleus which resembles a mitotic figure (mitosoid cell). These presumably result from viral alteration of the cells. Virus-like particles have been noted ultrastructurally within both cytoplasm and nuclei of cells within the spinous layer, and this layer is positive for HPV antigen with in situ hybridization.
The lesion is usually easily differentiated from squamous papilloma, verruca vulgaris and condyloma by its lack of pronounced surface projections; the presence of mitosoid cells, and the lack of connective tissue cores in the surface projections, when present. The sessile nature of focal epithelial hyperplasia also serves to separate it from the former two lesions, although this is not a guaranteed distinction.
Focal epithelial hyperplasia also tends to lack the pronounced elongation of thin rete ridges seen in keratoacanthoma and pseudoepitheliomatous hyperplasia, and it lacks the central keratin-filled core of the keratoacanthoma. It also lacks the subepithelial foamy or granular histiocyte-like cells required for the diagnosis of verruciform xanthoma.
Treatment and Prognosis
Conservative excisional biopsy may be required to establish the proper diagnosis, but additional treatment is unnecessary, except perhaps for esthetic reasons relating to visible labial lesions. Spontaneous regression has been reported after months or years, and the disease is rather rare in adults. No case of focal epithelial hyperplasia has been reported to transform into carcinoma. It should be remembered that focal epithelial hyperplasia may be an oral manifestation of AIDS.
Developmental Disturbances of Gingiva
Hereditary gingival fibromatosis is a benign, idiopathic condition affecting both arches. It affects males and females equally and is usually autosomal dominant. The gingiva are markedly enlarged, asymptomatic, nonhemorrhagic, nonexudative. It may be an isolated finding or associated with other syndromes (see below). A relationship with growth hormone deficiency has been suggested. The condition is either of a nodular form or, more commonly, a symmetric form. Onset of the condition usually begins with eruption of the permanent teeth. This condition predisposes one to malpositioning of the teeth, retention of deciduous teeth, esthetic and functional problems. Treatment is gingivectomy in one or several appointments. Recurrence is possible after some years.
Fibromatosis Gingivae (Elephantiasis gingivae, hereditary gingival ibromatosis, congenital macrogingivae)
Fibromatosis gingivae is a diffuse fibrous overgrowth of the gingival tissues, described for many years under a variety of terms. In the majority of reported cases, the condition was hereditary, being transmitted through a dominant autosomal gene. Zackin and Weisberger have reviewed this condition and presented a family of 11 affected children and 10 normal children from six marriages over four generations, while Emerson has reported the pedigree of a family over four generations in which nine marriages between affected and unaffected persons resulted in seven affected and all 11 normal offsprings. But many cases have appeared to be sporadic, with no familial background. Occasionally other abnormalities have been reported in association with fibromatosis gingivae, but of these, only hypertrichosis has been noted more than a few times. Even this association, in terms of total number of reported cases, is rare.
Clinical Features
This condition is manifested as a dense, diffuse, smooth, or nodular overgrowth of the gingival tissues of one or both arches, usually appearing about the time of eruption of the permanent incisors. It has been reported; however, in even very young children and, in a few instances, at birth (Fig. 1-16A). The tissue is usually not inflamed, but is of normal or even pale color, and it is often so firm and dense that it may prevent the normal eruption of teeth. It is not painful and shows no tendency for hemorrhage. The extent of the tissue overgrowth may be such that the crowns of the teeth are nearly hidden even though they are fully erupted with respect to the alveolar bone (Fig. 1-17).
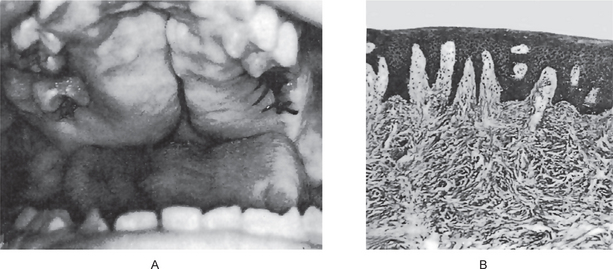
Figure 1-16 Fibromatosis gingivae. (A) The palatal vault is nearly filled with a dense fibrous mass of tissue. The apparent midline cleft extends only to the bone and is not a true cleft. The maxillary gingiva is also involved. (B) The photomicrograph reveals that the mass is made up only of a dense mass of fibrous connective tissue covered by normal epithelium.
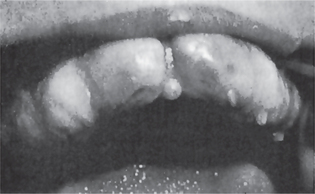
Histologic Features
The microscopic picture of the tissue in fibromatosis gingivae is similar to that of any fibrous hyperplasia. The epithelium may be somewhat thickened with elongated rete pegs, although the bulk of the tissue is composed of dense fibrous connective tissue. The bundles of collagen fibers are coarse and show few interspersed fibroblasts or blood vessels. Inflammation is an unrelated and variable finding (Fig. 1-16B).
Treatment and Prognosis
When tooth eruption is impeded, surgical removal of the excessive tissue and exposure of the teeth are indicated. The cosmetic appearance may also require surgical excision. The lesion sometimes recurs. It has been reported that tooth extraction alone will cause the tissues to shrink almost to normal and that recurrences can be prevented by this means.
Retrocuspid Papilla
The retrocuspid papilla, first described by Hirshfeld in 1933 but not reported until 1957, is a small, elevated nodule located on the lingual mucosa of the mandibular cuspids.
Clinical Features
This soft, well-circumscribed, sessile, mucosal nodule, commonly bilateral, is located lingual to the mandibular cuspid, between the free gingival margin and the mucogingival junction.
It is exceedingly common in children, occurring in 99% of those between the ages of eight and 16 years, according to the original report of Hirshfeld, but decreases in incidence with age, occurring in 38% of those between the ages of 25 and 39 years and in 19% of those between the ages of 60 and 80 years. Thus there appears to be regression of the structure with maturity. In addition, most studies have found a greater occurrence bilaterally than unilaterally. A study by Berman and Fay, who have reviewed the studies dealing with the retrocuspid papilla, reported from their own data that the structure was consistently more common in females than in males.
Histologic Features
The structure appears as an elevated mucosal tag often showing mild hyperorthokeratosis or hyperparakeratosis, with or without acanthosis. The underlying connective tissue is sometimes highly vascularized and may exhibit large stellate fibroblasts as well as occasional epithelial rests.
Developmental Disturbances of Tongue
Aglossia and Microglossia Syndrome
This malformation is very rare, since the first publication which was attributed by Gaillard and Nogué Antoine De Jussieu in 1718, to present till date there have been less than 35 cases reported (Grinspan, 1976). This anomaly is almost always associated to malformations in the extremities, especially the hands and feet, cleft palate and dental agenesia. Aglosia syndrome is, in reality, a microglossia with extreme glossoptosis. What is commonly observed is a rudimentary, small tongue. As a consequence of the lack of muscular stimulus between the alveolar arches, these do not develop transversely and the mandible does not grow in an anterior direction, producing as a result a severe dentoskeletal malocclusion (Fig. 1-18). This syndrome shows no predilection for gender and has no genetic implications. Its etiology must be searched for in some sort of fetal cell traumatism in the first few weeks of gestation. Neither language nor swallowing are sensibly affected by this condition.
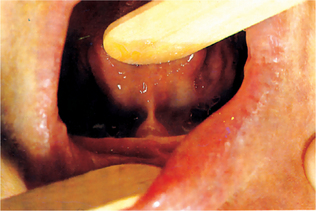
Macroglossia (Tongue hypertrophy, prolapsus of the tongue, enlarged tongue, pseudomacroglossia)
Macroglossia, meaning large tongue, has been a documented anatomical anomaly for several centuries. The earliest known written description of tongue lesions comes from the Egyptian Papyrus Ebers, originally thought to be from around 1550 BC. Obviously, tongue lesions have since been categorized by their etiologies. Macroglossia has an extensive list of possible causes. Its treatment has been largely surgical in the modern era.
Although the exact incidence of macroglossia is unknown (because the etiologies are too numerous to quantify), some congenital syndromes often express macroglossia in their phenotypes, most commonly Down syndrome (1 per 700 live births) and Beckwith-Wiedemann syndrome (0.07 per 1,000 live births). In Beckwith-Wiedemann syndrome, 97.5% of patients have macroglossia.
Reports on the etiology of macroglossia are extensive. Historically, Virchow described it as a form of elephantiasis. In the last 100 years, Butlin and Spencer attributed it to the dilation of lymphatics, muscle hypertrophy, or inflammation. Because of the large number of possible etiologies, multiple classification schemes have been used to list the causes.
The two broadest categories under the heading of macroglossia are true macroglossia and pseudomacroglossia.
Physical examination of the oral cavity and head morphology is helpful to deduce true macroglossia from pseudomacroglossia. Severe retrognathia and unusually small maxillary and/or mandibular size may indicate the latter. In addition, check tongue tone and mobility to rule out simple atonia or hypotonia indicating poor posturing of the tongue—as is commonly observed in Down syndrome (Fig. 1-19).
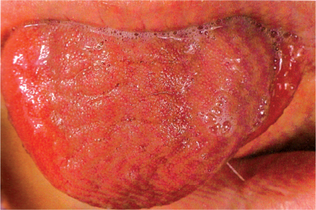
In addition to the oral cavity and airway, assess other features in the patient that may indicate congenital or systemic syndromes. Certain vitamin deficiencies may present with angular stomatitis, nonpitting edema of the lower extremities may indicate hypothyroidism, and unusual body morphologies may indicate the early signs of diseases-like acromegaly.
Treatment
The goal of nearly all surgery is to return the patient to an anatomically and physiologically normal condition; and this applies to macroglossia as well. The goal is to reduce tongue size and thereby improve function. Those main functions include articulation, mastication, deglutition, protection of the airway, and gustation. Of these, only gustation is not often improved with surgical intervention.
Ankyloglossia or Tongue-tie
Ankyloglossia, or tongue-tie as it is more commonly known, is said to exist when the inferior frenulum attaches to the bottom of the tongue and subsequently restricts free movement of the tongue. At one time, such restriction was believed to cause speech problems and it was routine to clip the membranous frenulum (frenulectomy) to free the tongue tip. Ankyloglossia occurs in approximately 1.7% of all neonates without preference for either gender and is reported to be transitory. With growth, the frenulum lengthens so normal tongue function is established. The criterion for diagnosis is based upon observation of lingual mobility; no current specific indications for surgery are emphasized in either the dental or medical literature reviewed. Simple incision of the frenulum may result in the development of scar tissue and further restriction of tongue movement (Schuller and Schleuning, 1994). Some authors indicate that if the tongue is able to touch the lower incisor teeth or just beyond the lower teeth, articulation will not be adversely affected. In some cases the frenulum is reported to tear spontaneously during infancy. Tongue-tie can cause feeding problems in infants; if this is the case, feeding difficulties are usually noticed early in an infant’s life. Feeding difficulties may be a reason to consider early surgery to cut the lingual frenulum and loosen the tongue. In some children, tongue-tie may also cause speech defects, especially articulation of the sounds: l, r, t, d, n, th, sh, and z. Preventing speech defects or improving a child’s articulation may be another reason to consider surgical intervention. The tongue is remarkably able to compensate; however, many children have no speech impediments due to ankyloglossia. Tongue-tie may contribute to dental problems as well, causing a persistent gap between the mandibular incisors.
Cleft Tongue
A completely cleft or bifid tongue is a rare condition that is apparently due to lack of merging of the lateral lingual swellings of this organ. A partially cleft tongue is considerably more common and is manifested simply as a deep groove in the midline of the dorsal surface (Figs. 1-20, 1-21). The partial cleft results because of incomplete merging and failure of groove obliteration by underlying mesenchymal proliferation. Interestingly, it is often found as one feature of the oral-facial-digital syndrome in association with thick, fibrous bands in the lower anterior mucobuccal fold eliminating the sulcus and with clefting of the hypoplastic mandibular alveolar process.
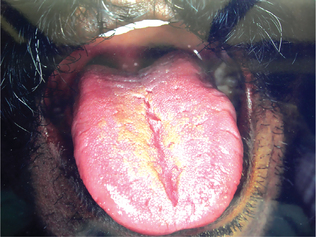
It is of little clinical significance except that food debris and microorganisms may collect in the base of the cleft and cause irritation.
Fissured Tongue (Scrotal tongue, lingua plicata)
Fissured tongue is a condition frequently seen in the general population and it is characterized by grooves that vary in depth and are noted along the dorsal and lateral aspects of the tongue (Fig. 1-22). Although a definitive etiology is unknown, a polygenic mode of inheritance is suspected because the condition is seen clustering in families who are affected. Patients are usually asymptomatic, and the condition is initially noted on routine intraoral examination as an incidental finding. Fissured tongue is also seen in Melkersson-Rosenthal syndrome and Down syndrome and in frequent association with benign migratory glossitis (geographic tongue).
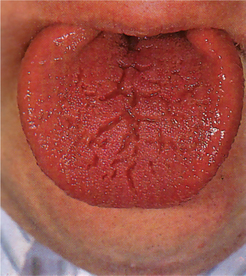
Melkersson-Rosenthal syndrome is a rare condition consisting of a triad of persistent or recurring lip or facial swelling, intermittent seventh (facial) nerve paralysis (Bell’s palsy), and a fissured tongue. The etiology of this condition is also unknown. The orofacial swelling usually manifests as pronounced lip enlargement. It may or may not affect both lips, and it may be tender or erythematous. Histologic examination of this tissue exhibits characteristic noncaseating granulomatous inflammation. Therapy for these lesions is often intralesional steroid injections. The facial paralysis is indistinguishable from Bell’s palsy, and it may be an inconsistent and intermittent finding with spontaneous resolution. The presence of fissured tongue in association with these other features is diagnostic of the condition.
This condition affects only the tongue and is a finding in Melkersson-Rosenthal syndrome, which consists of a triad of fissured tongue, cheilitis granulomatosa, and cranial nerve VII paralysis (Bell’s palsy).
Clinical Features
The prevalence worldwide varies by geographic location and has been reported to be as high as 21%. Fissured tongue is a totally benign condition and is considered by most to be a variant of normal tongue architecture. No predilection for any particular race appears to exist. Some reports have shown a slight male predilection. Although fissured tongue may be diagnosed initially during childhood, it is diagnosed more frequently in adulthood. The prominence of the condition appears to increase with increasing age.
The lesions are usually asymptomatic unless debris is entrapped within the fissure or when it occurs in association with geographic tongue (a common finding). On clinical examination, fissured tongue affects the dorsum and often extends to the lateral borders of the tongue. The depth of the fissures varies but has been noted to be up to 6 mm in diameter. When particularly prominent, the fissures or grooves may be interconnected, separating the tongue dorsum into what may appear to be several lobules. Although a specific etiology has not been elicited, a polygenic or autosomal dominant mode of inheritance is suspected because this condition is seen with increased frequency in families with an affected proband.
Histologic Features
A biopsy is rarely performed on a fissured tongue because of its characteristic diagnostic clinical appearance; however, histologic examination has shown an increase in the thickness of the lamina propria, loss of filiform papillae of the surface mucosa, hyperplasia of the rete pegs, neutrophilic microabscesses within the epithelium, and a mixed inflammatory infiltrate in the lamina propria.
Median Rhomboid Glossitis
Embryologically the tongue is formed by two lateral processes (lingual tubercles) meeting in the midline and fusing above a central structure from the first and second branchial arches, the tuberculum impar. The posterior dorsal point of fusion is occasionally defective, leaving a rhomboid-shaped, smooth erythematous mucosa lacking in papillae or taste buds. This median rhomboid glossitis (central papillary atrophy, posterior lingual papillary atrophy) is a focal area of susceptibility to recurring or chronic atrophic candidiasis, prompting a recent shift towards the use of posterior midline atrophic candidiasis as a more appropriate diagnostic term.
The latter term has certain difficulties; however, because not all cases improve with antifungal therapy or show initial evidence of fungal infection. The erythematous clinical appearance; moreover, is due primarily to the absence of filiform papillae, rather than to local inflammatory changes, as first suggested in 1914 by Brocq and Pautrier. The lesion is found in one of every 300–2,000 adults, depending on the rigor of the clinical examinations. It is seldom biopsied unless the red discoloration is confused with precancerous erythroplakia or its surface shows pronounced nodularity.
Clinical Features
Median rhomboid glossitis presents in the posterior midline of the dorsum of the tongue, just anterior to the V-shaped grouping of the circumvallate papillae (Fig. 1.23A, B). The long axis of the rhomboid or oval area of red depapillation is in the anteroposterior direction. Most cases are not diagnosed until the middle age of the affected patient, but the entity is, of course, present in childhood. There appears to be a 3 : 1 male predilection.
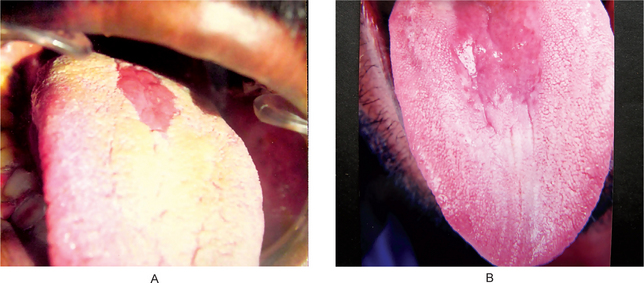
Those lesions with atrophic candidiasis are usually more erythematous but some respond with excess keratin production, and therefore, show a white surface change. Infected cases may also demonstrate a midline soft palate erythema in the area of routine contact with the underlying tongue involvement; this is commonly referred to as a kissing lesion.
Lesions are typically less than 2 cm in greatest dimension and most demonstrate a smooth, flat surface, although it is not unusual for the surface to be lobulated. Occasional lesions are located somewhat anterior to the usual location. None have been reported posterior to the circumvallate papillae.
Prior to biopsy, the clinician should be certain that the midline lesion does not represent a lingual thyroid, as it may be the only thyroid tissue present in the patient’s body. Differential diagnoses include the gumma of tertiary syphilis, the granuloma of tuberculosis, deep fungal infections, and granular cell tumor.
Histologic Features
Median rhomboid glossitis shows a smooth or nodular surface covered by atrophic stratified squamous epithelium overlying a moderately fibrosed stroma with somewhat dilated capillaries. Fungiform and filiform papillae are not seen, although surface nodules may mimic or perhaps represent anlage of these structures. A mild to moderately intense chronic inflammatory cell infiltrate may be seen within subepithelial and deeper fibrovascular tissues.
Chronic candida infection may result in excess surface keratin or extreme elongation of rete processes and premature keratin production with individual cells or as epithelial pearls (dyskeratosis) deep in the processes. Silver staining for fungus will often reveal candida hyphae and spores in the superficial layers of the epithelium. This pseudoepitheliomatous hyperplasia may be quite pronounced, and the tangential cutting of such a specimen may result in the artifactual appearance of cut rete processes as unconnected islands of squamous epithelium, leading to a mistaken diagnosis of well differentiated squamous cell carcinoma. Because of this difficulty, it is recommended that the patient be treated with topical antifungals prior to biopsy of a suspected median rhomboid glossitis.
Treatment and Prognosis
No treatment is necessary for median rhomboid glossitis, but nodular cases are often removed for microscopic evaluation. Recurrence after removal is not expected, although those cases with pseudoepitheliomatous hyperplasia should be followed closely for at least a year after biopsy to be certain of the benign diagnosis. Antifungal therapy (topical troches or systemic medication) will reduce clinical erythema and inflammation due to candida infection. This therapy, as stated earlier, should ideally be given prior to the biopsy, in order to reduce the candida-induced pseudoepitheliomatous hyperplasia features. Some lesions will disappear entirely with antifungal therapy.
Benign Migratory Glossitis (Geographic tongue)
Benign migratory glossitis is a psoriasiform mucositis of the dorsum of the tongue. Its dominant characteristics is a constantly changing pattern of serpiginous white lines surrounding areas of smooth, depapillated mucosa. The changing appearance has led some to call this the wandering rash of the tongue, with the depapillated areas have reminded others of continental outlines on a globe, hence the use of the popular term geographic tongue (Fig. 1-24). As with psoriasis, the etiology of benign migratory glossitis is unknown, but it does seem to become more prominent during conditions of psychological stress and it is found with increased frequency (10%) in persons with psoriasis of the skin. The great majority of those with oral involvement; however, lack psoriatic skin involvement. Approximately 1–2% of the population are affected, although most cases are so mild that they are never formally diagnosed.
Figure 1-24 Benign migratory glossitis. Courtesy of Dr Spencer Lilly, Meenakshi Ammal Dental College, Chennai.
Histologic Features
All of the microscopic features of psoriasis are present in benign migratory glossitis and migratory stomatitis, but these will not be obvious unless the biopsy is taken from a prominent serpiginous line at the periphery of a depapillated patch. A thickened layer of keratin is infiltrated with neutrophils, as are lower portions of the epithelium to a lesser extent. These inflammatory cells often produce small microabscesses, called Monro's abscesses, in the keratin and spinous layers. Rete ridges are typically thin and considerably elongated, with only a thin layer of epithelium overlying connective tissue papillae. When rete ridges are not elongated, the pathologist should consider Reiter's syndrome as a diagnostic possibility. Chronic inflammatory cells can be seen in variable numbers within the stroma and silver or PAS staining will often demonstrate candida hyphae or spores in the superficial layers of the epithelium. There is no liquefactive degeneration of basal cells, as seen in lichenoid lesions, and there is no ulceration except in cases of Reiter’s syndrome.
Treatment and Prognosis
No treatment is usually necessary for benign migratory glossitis and stomatitis. Symptomatic lesions can be treated with topical prednisolone and a topical or systemic antifungal medication can be tried if a secondary candidiasis is suspected. Occasional symptomatic cases respond well to topical tetracycline or systemic, broadspectrum antibiotics, but this should not be expected.
Hairy Tongue (Lingua nigra, lingua villosa, lingua villosa nigra, black hairy tongue)
Hairy tongue (lingua villosa) is a commonly observed condition of defective desquamation of the filiform papillae that results from a variety of precipitating factors (Fig. 1-25). The condition is most frequently referred to as black hairy tongue (lingua villosa nigra); however, hairy tongue may also appear brown, white, green, pink, or any of a variety of hues depending on the specific etiology and secondary factors (e.g. use of colored mouthwashes, breath mints, candies).
Etiology
The basic defect in hairy tongue is the hypertrophy of filiform papillae on the dorsal surface of the tongue, usually due to a lack of mechanical stimulation and debridement. This condition often occurs in individuals with poor oral hygiene (e.g. lack of toothbrushing, eating a soft diet with no roughage that would otherwise mechanically debride the dorsal surface of the tongue). Contributory factors for hairy tongue are numerous and include tobacco use and coffee or tea drinking. These factors account for the various colors associated with the condition.
Clinical Features
Normal filiform papillae are approximately 1 mm in length, whereas filiform papillae in hairy tongue are more than 15 mm in length. Hairy tongue has been reported with greater frequency in males, patients infected with human immunodeficiency virus (HIV), and those who are HIV negative and use intravenous drugs. Hairy tongue is rarely symptomatic, although overgrowth of Candida albicans may result in glossopyrosis (burning tongue). Patients frequently complain of a tickling sensation in the soft palate and the oropharynx during swallowing. In more severe cases, patients may actually complain of a gagging sensation. Retention of oral debris between the elongated papillae may result in halitosis. No racial predilection is associated with hairy tongue. Because hairy tongue is usually asymptomatic, the history is often irrelevant. In most cases, lesions are noted as part of an intraoral examination, although patients may complain of a tickling or gagging sensation. The tongue has a thick coating in the middle, with a greater accentuation towards the back. Bacterial and fungal overgrowth play a role in the color of the tongue. The only complication associated with hairy tongue is an occasional candidal overgrowth, which often results in an uncomfortable glossopyrosis (burning tongue). Altered taste sensation is a rare complication.
Differential Diagnosis
This condition has to be differentiated from candidiasis, leukoplakia, oral lichen planus and oral hairy leukoplakia. Culture of the tongue’s dorsal surface may be taken if a superimposed oral candidiasis or other specific oral infection is suspected. Distinguishing between oral hairy leukoplakia and hairy tongue is important if patients are found or suspected to be HIV positive. This can be accomplished by a simple mucosal punch biopsy and appropriate immunostaining of the specimen for the presence of Epstein-Barr virus, the causative agent of oral hairy leukoplakia. However, in most cases, the diagnosis is made retrospectively on the basis of the clinical response to mechanical debridement.
Histologic Features
Histopathologic findings in hairy tongue consist of elongated filiform papillae, with mild hyperkeratosis and occasional inflammatory cells. Debris accumulation among the papillae and candidal pseudohyphae is not unusual finding. No other specific microscopic findings are associated with this entity.
Treatment
The treatment of hairy tongue is variable. In many cases, brushing of the tongue with a toothbrush or using a commercially available tongue scraper is sufficient to remove elongated filiform papillae and retard the growth of additional ones. Surgical removal of the papillae by using electrodesiccation, carbon dioxide laser, or even scissors is the treatment of last resort when less complicated therapies prove ineffective. The prognosis for hairy tongue is excellent. If the precipitating factors cannot be adequately controlled or compensated for, patients may have to make tongue brushing or scraping part of their daily oral hygiene regimen.
Lingual Varices (Lingual or sublingual varicosities)
A varix is a dilated, tortuous vein, most commonly a vein which is subjected to increased hydrostatic pressure but poorly supported by surrounding tissue. Varices involving the lingual ranine veins are relatively common, appearing as red or purple shotlike clusters of vessels on the ventral surface and lateral borders of the tongue as well as in the floor of the mouth. However, varices also do occur in other oral sites such as the upper and lower lip, buccal mucosa, and buccal commissure.
There has been no direct association established between these varicosities and other specific organic diseases. However, Kleinman has concluded that these varicosities represent an aging process and that, when they occur prior to 50 years of age, they may indicate premature aging. In his study, lingual varicosities were not related to cardiac pulmonary disease. In an investigation of 1,751 persons (755 males and 996 females) ranging in age from 7 to 99 years, Ettinger and Manderson found that 68% of the persons over 60 years of age had sublingual varices. In this study, there appeared to be a significant relationship between the presence of leg varicosities and sublingual varices.
Thrombosis of any of these varices is a relatively frequent occurrence, as indicated in a report of 12 such cases by Weathers and Fine, but is apparently of little clinical significance.
Lingual Thyroid Nodule
The thyroid gland develops in the embryo from the ventral floor of the pharynx by means of an endodermal invagination or diverticulum. The tongue forms at the same time from this pharyngeal floor and is anatomically associated with the thyroid gland by connection through the thyroglossal tract, the lingual remnant of which is known as the foramen caecum.
The lingual thyroid is an anomalous condition in which follicles of thyroid tissue are found in the substance of the tongue, possibly arising from a thyroid anlage that failed to ‘migrate’ to its predestined position or from anlage remnants that became detached and were left behind. Baughman has reviewed and discussed in detail the various theories on the development of lingual thyroglossal remnants and the lingual thyroid nodule.
Etiology
The benign enlargement of lingual thyroid tissue is thought to be due in some cases to functional insufficiency of the chief thyroid gland in the neck, since some patients with such a lingual lesion are without a demonstrable main thyroid gland. Other cases of lingual thyroid nodules occur in patients residing in goitrous areas, but it is not certain that the condition is a form of goiter. In addition, it has been suggested that the failure of the primitive thyroid anlage to descend is the cause of the majority of cases of nongoitrous sporadic cretinism.
Clinical Features
The incidence of this benign condition is not known, since a routine autopsy seldom includes an examination of the base of the tongue. Montgomery, in an exceptionally complete and thorough review of the entire subject of the lingual thyroid, analyzed 144 acceptable previously reported cases, pointing out that these represented only cases showing hypertrophy of the lingual thyroid tissue. Though information is inadequate about racial and geographic distribution of lingual thyroid nodules, a difference in gender incidence does appear to be significant. Of 135 cases in which the gender was recorded, 118 patients were female and only 17 were male. The majority of patients had their onset of symptoms relatively early in life, chiefly during puberty, adolescence, and early maturity, though cases have been recorded as early as birth and as late as the seventh decade.
In contrast, Sauk carried out a study to determine the frequency with which ectopic thyroid tissue occurred in the tongue of ‘normal’ persons, i.e. those without a definite symptomatic mass for which treatment was sought. In a series of 200 consecutive necropsies, ectopic thyroid tissue was found in 10% of the cases, with an equal distribution between the sexes. Nearly identical data have been reported by Baughman in his own investigation of 184 tongues from human cadavers. Since the condition is more often clinically apparent in females, Sauk suggested that hormonal factors may be involved in the genesis of symptoms. Most cases appear to arise in females during puberty, adolescence, pregnancy, or menopause.
The lingual thyroid may be manifested clinically as a nodular mass in or near the base of the tongue in the general vicinity of the foramen caecum and often, but not always, in the midline (Fig. 1-26). This mass, which more commonly appears as deeply situated rather than as a superficial exophytic lesion, tends to have a smooth surface. In some cases it may appear vascular, while in others the color of the mucosa is not atypical. The size of the lesion in many of the reported cases has been approximated 2–3 cm in diameter. The chief symptoms of the condition may vary, but the presenting complaint is often dysphagia, dysphonia, dyspnea, hemorrhage with pain, or a feeling of tightness or fullness in the throat.
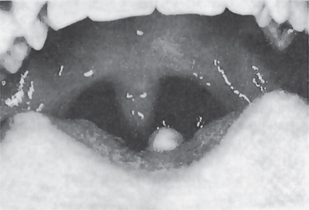
Histologic Features
The benign lingual thyroid nodules may present a variety of microscopic patterns, but in the majority of cases they resemble either normal thyroid tissue or thyroid tissue of an embryonal or fetal type. In some instances the nodules exhibit colloid degeneration or goiter.
Great care must be taken to distinguish these lesions from lesions derived from accessory salivary glands in the same location. Both the lingual thyroid and these salivary glands may give rise to adenomas and adenocarcinomas in the tongue.
Treatment
Care must be exercised in handling the lingual thyroid lesion. It has been emphasized by Hung and his associates that a careful physical examination should be performed to demonstrate the presence of a normally located thyroid gland in patients presenting with midline masses in the lingual or sublingual area. If the thyroid gland cannot be palpated, a scintiscan with a tracer dose of radioactive iodine, 131I, should be carried out to determine whether there is a normally located thyroid gland and if the lingual mass is ectopic thyroid. It is usually recommended that a patient with an ectopic thyroid gland should have a trial of replacement thyroid hormone therapy before excision is contemplated, since this will often decrease the size of the lesion and make surgery unnecessary. Occasionally, the clinical manifestations of the lesion and its size necessitates surgical excision.
Developmental Disturbances of Oral Lymphoid Tissue
Reactive Lymphoid Aggregate (Reactive lymphoid hyperplasia)
The lingual tonsil, one of the largest oral lymphoid aggregates, is located on the posterior portion of the tongue on the dorsolateral aspect. It is typically surrounded by a crypt lined by stratified squamous epithelium. It frequently becomes inflamed and enlarged so that it is clinically evident. Such enlargement is usually bilateral but, if unilateral, may easily be mistaken clinically for early carcinoma. This reactive lingual tonsil has often been called ‘foliate papillitis,’ referring to the vestigial foliate papilla in this area.
Similar reactive hyperplasia may occur in the lymphoid aggregates in the other locations mentioned previously, the buccal mucosa being especially common. This presents clinically as a firm nodular submucosal mass which may be tender. Since this lymphoid tissue may be the site for the development of lesions of the malignant lymphoma group, early microscopic diagnosis is essential whenever the lesions do not regress in a short period of time.
Hyperplastic lymphoid polyps have also been described as polypoid structures composed entirely of lymphoid tissue. They are reported to occur on the gingiva, buccal mucosa, tongue, and floor of the mouth.
Lymphoid Hamartoma (Angiofollicular lymph node hyperplasia, angiomatous lymphoid, Castleman tumor, giant benign lymphoma, hamartoma of lymphatics, giant lymph node hyperplasia)
Castleman’s disease is a rare disorder characterized by noncancerous benign growths that may develop in the lymph node tissue throughout the body (i.e. systemic disease of plasma cell type). Most often, they occur in the chest, stomach, and/or neck (i.e. localized disease of hyaline vascular type). Less common sites include the armpit (axilla), pelvis, and pancreas. Usually the growth represents abnormal enlargement of the lymph nodes normally found in these areas (lymphoid hamartoma). There are two main types of Castleman’s disease: hyaline vascular type and plasma cell type. The hyaline vascular type accounts for approximately 90% of the cases. Most individuals exhibit no symptoms of this form of the disorder (asymptomatic) or they may develop noncancerous growths in the lymph nodes. The plasma cell type of Castleman’s disease may be associated with fever, weight loss, skin rash, early destruction of red blood cells, leading to unusually low levels of circulating red blood cells (hemolytic anemia), and/ or abnormally increased amounts of certain immune factors in the blood (hypergammaglobulinemia).
A third type of Castleman’s disease has been reported in the medical literature. This type may affect more than one area of the body (multicentric or generalized Castleman’s disease). Many individuals with multicentric Castleman’s disease may exhibit an abnormally large liver and spleen (hepatosplenomegaly). Researchers’ opinions in the medical literature differ as to whether multicentric Castleman’s disease is a distinct entity or a multicentric form of the plasma cell type of Castleman’s disease. The exact cause of Castleman’s disease is not known. Some researchers speculate that increased production of interleukin-6 (IL-6) may be involved in the development of Castleman’s disease. IL-6 is a substance produced by structures within the lymph nodes.
Angiolymphoid Hyperplasia with Eosinophilia (Epithelioid hemangioma, histiocytoid hemangioma, pseudopyogenic granuloma, papular angioplasia, inlammatory angiomatous nodules)
Angiolymphoid hyperplasia with eosinophilia (ALHE) is an uncommon idiopathic condition that presents with isolated or grouped plaques or nodules in the skin of the head and neck. Most patients present with lesions in the periauricular region, forehead, or scalp.
A distinct pathologic entity, ALHE is marked by a proliferation of blood vessels with distinctive large endothelial cells. These blood vessels are accompanied by a characteristic inflammatory infiltrate that includes eosinophils. The lesion is benign but may be persistent and difficult to eradicate. Whether ALHE represents a benign neoplasm or an unusual reaction to varied stimuli, including trauma, remains unclear. While ALHE shows some similarity to Kimura disease, it is a distinct condition.

Figure 1-27 Angiolymphoid hyperplasia. H and E section showing irregular clusters of blood vessels and intermixed lymphocytes and eosinophils in this example of ALHE.
Etiology
ALHE is idiopathic. Whether this condition is a neoplastic or reactive state is uncertain; a reactive cause is favored.
Clinical Features
Although ALHE may be a benign tumor numerous factors suggest that it is an unusual reactive process. The condition may be multifocal. ALHE has occurred following various forms of trauma or infection. Hyperestrogenic states (e.g. pregnancy, oral contraceptive use) may foster lesion growth. Additionally, the distinctive inflammatory infiltrate in ALHE appears to be an intrinsic (not secondary) component of the lesion. Approximately 20% of patients have blood eosinophilia. Although frequency is unknown, cases have been reported worldwide. ALHE is uncommon but not rare; it may be more common in Japan than in other countries. ALHE can persist for years, but serious complications (e.g. malignant transformation) do not occur. ALHE is seen most commonly in Asians, followed by Caucasians. Although less commonly, blacks too can develop ALHE. ALHE is somewhat more common in females; however, a male predominance has been noted in selected Asian studies. ALHE presents most commonly in patients aged 20–50 years, with mean onset of 30–33 years. This condition is rare in elderly patients and in the non-Asian pediatric population.
Patients with ALHE typically present with an expanding nodule or group of nodules, usually in the vicinity of the ear. The lesion(s) may be associated with pain or pruritus. Uncommon symptoms include pulsation and spontaneous bleeding.
ALHE typically appears as dome-shaped, smooth-surfaced papules or nodules. Approximately 85% of lesions occur in the skin of the head and neck; most of them are on or near the ear or on the forehead or scalp. The lesions range from erythematous to brown in color and may be eroded or crusted. Approximately 80% of patients present with isolated lesions, while the remaining patients usually demonstrate grouped papules or nodules in a single region. Rarely, the lesions may be pulsatile. Most lesions are 0.5–2 cm in diameter, with a range of 0.2–8 cm. Larger nodules tend to be deeply centered within the subcutis.
Differential Diagnosis
Granuloma faciale, insect bites, pyogenic granuloma (lobular capillary hemangioma), angiosarcoma, hemangioendothelioma and hemangioma.
Histologic Features
The clinical presentation of papules around the ears may suggest ALHE, but a biopsy is required to establish the diagnosis. ALHE shows characteristic histologic features, including a proliferation of small blood vessels, many of which are lined by enlarged endothelial cells with uniform ovoid nuclei and intracytoplasmic vacuoles. These distinctive endothelial cells have been described as having a cobblestone appearance. In addition, a perivascular and interstitial infiltrate composed primarily of lymphocytes and eosinophils is present. Eosinophils typically comprise 5–15% of the infiltrate. Rarely, they can account for as much as 50% of the infiltrate. Occasionally, the infiltrate is devoid of eosinophils. Lymphoid aggregates with or without follicle formation are typical.
Lymphoepithelial Cyst
The oral lymphoepithelial cyst develops within a benign lymphoid aggregate or accessory tonsil of the oral or pharyngeal mucosa. The surface of such aggregates may be indented with tonsillar crypts, as are the much larger pharyngeal tonsils of the lateral pharyngeal walls. The crypts may become obstructed by keratin or other debris, or the surface opening may become constricted during episodes of inflammatory hyperplastic responses. Certain cases develop a complete disunion of the crypt epithelium from the surface epithelium, resulting in a subepithelial cyst lined by the old crypt epithelium. This cyst was first reported by Parmentier in 1857 as hydatid cyst. Outside of the head and neck region, lymphoepithelial cyst is found most frequently in the pancreas and testis.
A similar but much larger cervical lymphoepithelial cyst (branchial cleft cyst) most probably develops from entrapped salivary duct epithelium in the lymph nodes of the lateral neck, rather than from the branchial cleft. These are discussed in a separate section of the present book. Another similar cyst, the parotid cyst, is found in major salivary glands, especially in AIDS patients, although it often lacks a surrounding lymphoid aggregate in Chapter 3 on Tumors of the Salivary Gland.
Clinical Features
Oral lymphoepithelial cyst presents as a movable, painless submucosal nodule with a yellow or yellow-white discoloration. Occasional cysts are transparent. Almost all cases are less than 0.6 cm in diameter at the time of diagnosis, which is usually during the teen years or the third decade of life. Approximately half of all intraoral examples are found on the oral floor (Fig. 1-28A), but the lateral and ventral tongue are not uncommon sites of occurrence, nor is the soft palate, especially the mucosa above the pharyngeal tonsil. Of course, this cyst may also occur within the pharyngeal tonsils themselves. Occasional superficial cysts rupture to release a foul-tasting, cheesy, keratinaceous material.
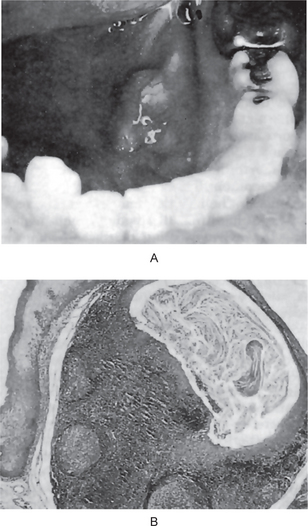
Figure 1-28 Oral lymphoepithelial cyst.
An elevated nodule in the floor of the mouth (A). Histologically exhibited a keratinfilled epithelium-lined cyst in a lymphoid aggregate (B). (Courtesy of Dr Joseph A Regezi.
This cyst has a clinical appearance similar to that of an epidermoid cyst or a dermoid cyst of the oral/pharyngeal mucosa, but its growth potential is much less than the other cysts. The lymphoepithelial cyst never occurs on the alveolar mucosa, hence, can easily be distinguished from a gingival cyst of adults or from an unruptured parulis or ‘pus pocket’ at the terminus of a fistula (extending from the apical or lateral region of an abscessed tooth).
Histologic Features
The lymphoepithelial cyst is lined by atrophic and often degenerated stratified squamous epithelium, usually lacking rete processes and usually demonstratinga minimal granular cell layer. Orthokeratin is seen to be sloughing from the epithelial surface into the cystic lumen, often completely filling the lumen and sometimes showing dystrophic calcification. Rarely, mucus-filled goblet cells may be seen within the superficial layers of the epithelium, and occasional cysts will demonstrate an epithelium-lined communication with the overlying mucosal surface. The cyst is entrapped within a well-demarcated aggregate of mature lymphocytes. The aggregate or ‘tonsil’ will have a variable number of germinal centers, sometimes none at all. The lymphoid aggregate may be hyperplastic.
This combination of epithelium-lined cyst with lymphoid aggregates is unique enough to make the diagnosis an easy one (Fig. 1-28B), but the pathologist must differentiate this lesion from the Warthin tumor (papillary cystadenoma lymphomatosum). The latter lesion is lined not by squamous epithelium but by a bilayered cuboidal, columnar or oncocytic ductal epithelium. It is almost always found in the parotid gland, but rare oral examples have been reported.
Occasional cysts have very small lumina with degenerated epithelial linings and may mimic metastatic deposits of well differentiated squamous cell carcinoma. Deeper sections will reveal the true nature of the benign lesion.
Treatment and Prognosis
No treatment is usually necessary for the oral lymphoepithelial cyst unless its location is such that it is constantly being traumatized. Most lesions are, however, removed by conservative surgical excision in order to arrive at a definitive diagnosis. There is no malignant potential to this lesion but the lymphoid stroma, as with all lymphoid tissues, can become involved with an extranodal lymphoma.
Developmental Disturbances of Salivary Glands
Aplasia (Agenesis)
Any one or group of salivary glands may be absent, unilaterally or bilaterally. Aplasia becomes manifest with the development of xerostomia and its sequelae. A diagnosis of salivary gland aplasia is made after exclusion of the common causes of xerostomia medications, Sjögren’s syndrome and radiation. The CT scan or MRI will indicate the gland’s absence and its replacement by fat and fibrous tissue. Scintiscanning with a radioisotope will confirm the initial diagnosis. The absence of the salivary duct orifice/papilla is an additional clue.
Aplasia occurs for unknown reasons as an isolated finding or in conjunction with other developmental defects such as hemifacial microsomia, the LADD syndrome (see below) and mandibulofacial dysostosis (Treacher Collins). In the more severe cases, the ensuing xerostomia causes clinical problems. Salivary loss leads to increased caries, burning sensations, oral infections, taste aberrations and difficulty with denture retention.
Hemifacial microsomia, a relatively frequent entity (1 in 3,500 births), is characterized by an asymmetric mild to severe underdevelopment of the craniofacial skeleton, the external ear, and facial soft tissues including the parotid gland. It involves structures derived from the first and second branchial arches. The majority of cases occur sporadically but rare familial cases have been reported.
LADD is a hereditary autosomal dominant syndrome. The lacrimal (L) apparatus usually demonstrates occlusion of the lacrimal puncta, nasolacrimal duct obstruction with overflow of tears (epiphora), lacrimal sac inflammation (dacrocystitis) and lacrimal gland aplasia. The auricles (A) are deformed with the ear having a cup-shaped appearance. There is some hearing loss. Dentally (D), peg-shaped teeth, hypodontia, and enamel hypoplasia are noted. Various combinations of salivary gland agenesis, with varying degrees of xerostomia, are present. Digital (D) deformities are manifested by deviation of the fingers medially or laterally (clinodactyly).
Mandibulofacial dysostosis is familial in origin. Patients have a symmetric notching of the lower eyelids with the eyes slanting downward at the lateral borders. Maldevelopment of the mandible and maxilla, a defective development of the malar bones results in bird-like appearance and often salivary aplasia are seen.
Xerostomia (Dry mouth)
Xerostomia is not a disease but can be a symptom of certain diseases. It can produce serious negative effects on the patient’s quality of life, affecting dietary habits, nutritional status, speech, taste, tolerance to dental prosthesis and increased susceptibility to dental caries. The increase in dental caries can be devastating in many patients and therefore special care must be made to control this condition.
Temporary Causes
Anxiety and depression are well recognized as causes of reduced salivary flow, as many students become aware at examination times! However, these psychological problems are often treated with drugs, which may be salivary inhibitors.
A blockage of the duct of a major salivary gland, commonly the submandibular, can produce dryness on the affected side, together with pain and swelling in the gland, especially on stimulation. If untreated, the obstruction may lead to progressive fibrosis of the gland and permanent xerostomia.
Inflammation of the salivary glands can cause reduced secretion. Acute infections include mumps and postoperative parotitis, while chronic conditions include swellings related to nutritional deficiency and hypersensitivity to iodine. However, many cases of intermittent swelling of the salivary glands are idiopathic and are described as ‘chronic nonspecific sialadenitis’ and may be associated with duct calculi.
A wide variety of drugs may cause xerostomia. Anticholinergic and sympathomimetic agents may be implicated and this group includes tricyclic antidepressants, bronchodilators and antihistamines. Diuresis produced by drugs or alcohol can result in dehydration and xerostomia. In most cases function recovers after the drug is withdrawn. Zyban, a newly introduced drug to aid smoking cessation can cause xerostomia and since the demand for the drug is likely to be high, its use may become a common etiological factor.
Permanent Causes
Congenital absence of one or more of the major salivary glands is a rare but recognized condition of unknown etiology.
This combination of dry mouth, dry eyes, and often rheumatoid arthritis, mainly affects women over 40 years of age and is often accompanied by a mild fever. About half of the patients with this syndrome also present with, or go on to develop swellings of the major salivary glands, which display similar histology to Mikulicz’s disease.
Xerostomia is associated with diabetes mellitus, probably as a consequence of polyurea, as well as Parkinson’s disease, cystic fibrosis and sarcoidosis. It has been reported in cases of vitamin A, riboflavin and nicotinic acid deficiencies and anemia.
One of the most dramatic and distressing causes of xerostomia is therapeutic radiography for head and neck tumors. The effect on the glands of the irradiated side is often rapid and profound. Postradiation glandular atrophy is partly due to a reduction in the vascularity of the gland and partly to the direct effect of the X-rays on the highly specialized and sensitive secretory epithelial cells. While recovery of function can occur after several months, in many cases a permanent xerostomia develops. Radiation does not appear to damage the teeth or periodontal tissues directly, the effect on the dentition resulting solely from the reduction in salivary flow.
Clinical Features
It is important to establish whether the dryness is continual or intermittent, whether it is accompanied by pain or swelling, if unilateral or bilateral and whether there is any relevant history of anxiety, stress or depression, a systemic disorder, irradiation, trauma, surgery or medication. The patient’s occupation, diet and domestic situation are often relevant.
Unilateral dryness with pain or discomfort and swelling in the affected gland on stimulation is often an indication of a duct calculus. Sjögren’s syndrome commonly produces bilateral swelling, often constant and accompanied by the other symptoms of the syndrome, and in many cases lymph node enlargement. A punch biopsy of labial glands and by serological tests may be needed to confirm the diagnosis. As well as looking for evidence of enlargement of salivary glands and lymph nodes and unilateral dryness, the palpation of the floor of the mouth for evidence of submandibular duct calculi and examination of the major duct openings, as inflammation or swelling of the orifice may indicate the presence of a distally placed calculus. These are often revealed by simple radiography.
While the more dramatic forms of xerostomia are not common, a more typical case of nonspecific xerostomia is a postmenopausal woman, living alone with few family or other social contacts, surviving on a low income and a marginally inadequate diet, and wearing old ill-fitting dentures. The combination of atrophy of the oral mucosa due to hormonal changes and a mild chronic candidosis and reduced salivary flow due to age and depression for which she might have medication, together with a marginal iron deficiency anemia, is sufficient to produce a degree of discomfort which a cursory oral examination will not reveal.
All degrees of xerostomia exist. In some cases, the patient complains of a dry or burning sensation but the mucosa appears normal. In other cases there is a complete lack of saliva.
When the deficiency of saliva is pronounced, there may be severe alterations in the mucous membranes, and the patient may have extreme discomfort. The mucosa will appear dry and atrophic, sometimes inflamed or, more often, pale and translucent. The tongue may manifest the deficiency by atrophy of the papillae, inflammation, fissuring, and cracking and in severe cases by areas of denudation. Soreness, burning, and pain of the mucous membrane and tongue are common symptoms. Xerostomia, in all its aspects, has been discussed by Bertram.
Clinical Significance
Aside from annoyance to the patient, there is one feature of the condition that is serious. In many cases, chronic xerostomia predisposes to rampant dental caries and subsequent loss of teeth. Moreover, patients with xerostomia have difficulty with artificial dentures. Dental appliances are extremely disagreeable against dry mucosa and cannot be tolerated by some patients.
Treatment
The basic principles of management are first to eliminate or address any etiological factors such as drugs, calculi and emotional problems. It is also advisable to promote salivary stimulation by using sugar-free chewing gum which is effective and convenient. Salivary substitutes can also be given. The relief that can be given to many sufferers is limited. However, patience and consideration, especially towards the elderly, is as important as clinical intervention. Regular review to monitor the condition of teeth, gingivae and mucosa and to give support and reinforcement of preventive measures is advised.
Hyperplasia of Palatal Glands
An unusual localized hyperplasia or hypertrophy of minor accessory salivary glands in the palate has been described by Giansanti and his associates, although they have also accepted the view that this lesion may represent a benign adenoma of these glands. The cause of this focal enlargement is unknown although, according to these investigators, the following have been reported to result in salivary gland enlargement: (1) endocrine disorders, (2) gout, (3) diabetes mellitus, (4) menopause, (5) hepatic disease, (6) starvation, (7) alcoholism, (8) inflammation, (9) benign lymphoepithelial lesion, (10) Sjögren’s syndrome, (11) adiposity, hyperthermia, oligomenorrhea, and parotid swelling syndrome, (12) aglossiaadactylia syndrome, (13) Waldenstrom’s macroglobulinemia, (14) uveoparotid fever, (15) Felty’s syndrome, (16) certain drugs, and (17) the aging process.
A series of 10 cases has been reported recently by Arafat and her associates. They could also find no associated abnormalities and had to consider the cases idiopathic. Interestingly, one of their cases involved the glands of the retromolar area rather than the palate.
Clinical Features
Palatal gland hyperplasia presents as a small localized swelling, measuring from several millimeters to 1 cm or more in diameter, usually on the hard palate or at the junction of the hard and soft palates. The lesion has an intact surface and is firm, sessile, and normal in color. It is usually asymptomatic and the patient may be unaware of the lesion. Too few cases have been reported to determine whether there is any age or gender predilection.
Atresia
Congenital occlusion or absence of one or more of the major salivary gland ducts is an exceedingly rare condition. When it does occur, it may result in the formation of a retention cyst or produce a relatively severe xerostomia. Such a case has been reported by Foretich and his associates.
Aberrancy
Because of the widespread distribution of normal accessory salivary glands in the oral cavity, it is difficult to define the condition of aberrancy. Since such accessory glands may be found in the lips, palate, buccal mucosa, floor of the mouth, tongue, and retromolar area, aberrancy may be construed as simply that situation in which these glands are found farther than normal from their usual location. In any event, there is no clinical significance to be attached to aberrant salivary glands other than that they may be the site of development of a retention cyst or neoplasm.
Occasional cases have been reported in the literature of salivary gland tissue present within the body of the mandible. It has been found, in many instances, that this glandular tissue anatomically communicated with the normal submaxillary or sublingual gland, generally through a stalk or pedicle of tissue which perforated the lingual cortical plate. For this reason, this aberrancy of salivary gland tissue probably represents only an extreme example of the condition known as the ‘developmental lingual mandibular salivary gland depression’ described next.
Developmental Lingual Mandibular Salivary Gland Depression (Static bone cavity or defect of the mandible, lingual mandibular bone cavity, static bone cyst, latent bone cyst, Stafne cyst or defect)
A Stafne bone cyst is an unusual form of slightly aberrant salivary gland tissue wherein a developmental inclusion of glandular tissue is found within or, more commonly, adjacent to the lingual surface of the body of the mandible within a deep and well-circumscribed depression. The oldest described occurrence of this phenomenon is in a skull dated to the sixth to fourth centuries BC. The phenomenon was first recognized by Stafne in 1942, hence the eponym. However, this cyst has been referred to by many other names, including static bone cavity, defect of the mandible, lingual mandibular bone cavity, static bone cyst, latent bone cyst, and Stafne bone defect. The incidence of occurrence has been reported as 0.1–1.3% in various studies. The general consensus is that this is a congenital defect, although it rarely has been observed in children. These lesions generally may be regarded as developmental rather than pathologic defects. A predilection for males over females seems to exist.
Radiographically, the lesion usually appears as an ovoid radiolucency located between the inferior alveolar canal and the inferior border of the mandible in the region of the second or third molars. It can be differentiated from the traumatic or hemorrhagic bone cyst, which by location almost invariably lies superior to the inferior alveolar canal.
Although the classic Stafne cyst is described in the posterior mandible, an anterior variant presenting as a round or ovoid radiolucency in the area between the central incisors and first premolars exists; however, it is far less common.
Anterior Lingual Depression
It has also been recognized that a similar asymptomatic round or ovoid radiolucency may occur in the anterior segment of the mandible, generally appearing as a rather poorly circumscribed lesion somewhere between the central incisor and the first premolar area. This anterior radiolucency also represents a cavity or depression on the lingual surface of the mandible. It has been reviewed by Miller and Winnick and more recently by Connor. Langlais and his coworkers examined 12 dried mandibles with such anterior depressions and concluded that these might represent either anatomic variants related to the digastric or sublingual fossa or developmental anomalies caused by impingement of the sublingual gland. These are far less common than the posterior lesion (Fig. 1-29).
Figure 1-29 Developmental lingual mandibular salivary gland depression of sublingual gland. Courtesy of Dr Michael J Freeman.
Complications
A complication occasionally reported in the literature is the development of a true central salivary gland neoplasm from the included salivary gland tissue, but this is rare. This has been discussed in Chapter 3 on Tumors of the Salivary Glands (q.v.) in the section on mucoepidermoid carcinoma.
Treatment
These lesions generally represent benign developmental anomalies that normally do not require any treatment. A complication occasionally reported in the literature is the development of a true salivary gland neoplasm in the tissue associated with one of the cortical defects. Therefore, re-cording the finding of these lesions and periodically observing them radiographically seem prudent. Clinical or radiographic changes may indicate the need for further investigation.
Developmental Disturbances in Size of Teeth
Microdontia
This term is used to describe teeth which are smaller than normal, i.e. outside the usual limits of variation. Three types of microdontia are recognized: (1) true generalized microdontia, (2) relative generalized microdontia, and (3) microdontia involving a single tooth.
In true generalized microdontia, all the teeth are smaller than normal. Aside from its occurrence in some cases of pituitary dwarfism, this condition is exceedingly rare. The teeth are reportedly well formed, merely small.
In relative generalized microdontia, normal or slightly smaller than normal teeth are present in jaws that are somewhat larger than normal, and there is an illusion of true microdontia. Since it is well recognized that a person may inherit the jaw size from one parent and the tooth size from the other parent, the role of hereditary factors in producing such a condition is obvious.
Microdontia involving only a single tooth is a rather common condition (Fig. 1-30). It affects most often the maxillary lateral incisor and the third molar. These two teeth are among those that are most often congenitally missing. It is of interest to note; however, that other teeth which are often congenitally absent, the maxillary and mandibular second premolars, seldom exhibit microdontia. Supernumerary teeth; however, are frequently small in size.
Figure 1-30 Microdontia. The third molar is small and incompletely formed. The second molar had been previously extracted.
One of the common forms of localized microdontia is that which affects the maxillary lateral incisor, a condition that has been called the ‘peg lateral’ (Fig. 1-31). Instead of exhibiting parallel or diverging mesial and distal surfaces, the sides converge or taper together incisally, forming a peg-shaped or cone-shaped crown. The root of such a tooth is frequently shorter than usual.
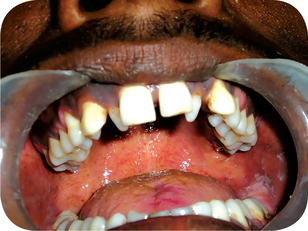
Macrodontia
Macrodontia is the opposite of microdontia and refers to teeth that are larger than normal. Such teeth may be classified in the same manner as microdontia.
True generalized macrodontia, the condition in which all teeth are larger than normal, has been associated with pituitary gigantism, but is extremely rare.
Relative generalized macrodontia is somewhat more common and is a result of the presence of normal or slightly larger than normal teeth in small jaws, the disparity in size giving the illusion of macrodontia. As in microdontia, the importance of heredity must be considered.
Macrodontia of single teeth is relatively uncommon, but is occasionally seen. It is of unknown etiology. The tooth may appear normal in every respect except for its size. True macrodontia of a single tooth should not be confused with fusion of teeth, in which, early in odontogenesis, the union of two or more teeth results in a single large tooth.
A variant of this localized macrodontia is the type that is occasionally seen in cases of hemihypertrophy of the face, in which the teeth of the involved side may be considerably larger than those of the unaffected side.
Developmental Disturbances in Shape of Teeth
Gemination
Geminated teeth are anomalies which arise from an attempt at division of a single tooth germ by an invagination, with resultant incomplete formation of two teeth. The structure is usually one with two completely or incompletely separated crowns that have a single root and root canal. It is seen in deciduous as well as permanent dentition, and in some reported cases, appears to exhibit a hereditary tendency. It is not always possible to differentiate between gemination and a case in which there has been fusion between a normal tooth and a supernumerary tooth (Fig. 1-32).
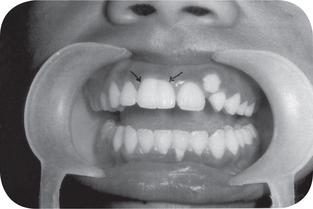
The term ‘twinning’ has sometimes been used to designate the production of equivalent structures by division resulting in one normal and one supernumerary tooth. These terms, as well as ‘fusion’ and ‘concrescence,’ have been discussed by Levitas.
Fusion
Fused teeth arise through union of two normally separated tooth germs. Depending upon the stage of development of the teeth at the time of the union, fusion may be either complete or incomplete. It has been thought that some physical force or pressure produces contact of the developing teeth and their subsequent fusion. If this contact occurs early, at least before calcification begins, the two teeth may be completely united to form a single large tooth (Fig. 1-33). If the contact of teeth occurs later, when a portion of the tooth crown has completed its formation, there may be union of the roots only. The dentin; however, is always confluent in cases of true fusion. The tooth may have separate or fused root canals, and the condition is common in the deciduous as well as in the permanent dentition. In fact, Grahnen and Granath have reported that fusion of teeth is more common in the deciduous than in the permanent dentition.
Figure 1-33 Fusion of teeth. (A) There has been complete fusion between the mandibular left central and lateralincisors and the right central and lateral incisors. (B) The intraoral radiograph showing a common pulp chamber and root canal in each pair of fused teeth.
In addition to affecting two normal teeth, fusion may also occur between a normal tooth and a supernumerary tooth such as the mesiodens or the distomolar (Fig. 1-34). In some cases the condition has been reported to show a hereditary tendency.
Figure 1-34 Fusion of right lower central and a supernumerary tooth. Courtesy of Dr Spencer Lilly, Meenakshi Ammal Dental College, Chennai.
The possible clinical problems related to appearance, spacing, and periodontal conditions brought about by fused teeth have been discussed by Mader, who has also reported several illustrative cases.
Concrescence
Concrescence of teeth is actually a form of fusion which occurs after root formation has been completed. In this condition, teeth are united by cementum only. It is thought to arise as a result of traumatic injury or crowding of teeth with resorption of the interdental bone so that the two roots are in approximate contact and become fused by the deposition of cementum between them. Concrescence may occur before or after the teeth have erupted, and although it usually involves only two teeth, there is at least one case on record of union of three teeth by cementum.
The diagnosis can frequently be established by radiographic examination. Since with fused teeth the extraction of one may result in the extraction of the other, it is desirable that the dentist be forewarned of the condition and advices the patient.
Dilaceration
The term ‘dilaceration’ refers to an angulation, or a sharp bend or curve, in the root or crown of a formed tooth (Fig. 1-35). The condition is thought to be due to trauma during the period in which the tooth is forming, with the result that the position of the calcified portion of the tooth is changed and the remainder of the tooth is formed at an angle. The curve or bend may occur anywhere along the length of the tooth, sometimes at the cervical portion, at other times midway along the root or even just at the apex of the root, depending upon the amount of root formed when the injury occurred. It has been emphasized by van Gool that such an injury to a permanent tooth, resulting in dilaceration, often follows traumatic injury to the deciduous predecessor in which that tooth is driven apically into the jaw. He has discussed this condition in detail, reporting 18 such cases.
Since dilacerated teeth frequently present difficult problems at the time of extraction if the operator is unaware of the condition, the need for preoperative radiographs before any surgical procedures are carried out is self-evident.
Talon Cusp
The talon cusp, an anomalous structure resembling an eagle’s talon, projects lingually from the cingulum areas of a maxillary or mandibular permanent incisor. This cusp blends smoothly with the tooth except that there is a deep developmental groove where the cusp blends with the sloping lingual tooth surface (Fig. 1-36). It is composed of normal enamel and dentin and contains a horn of pulp tissue.
This anomaly has been discussed by Mellor and Ripa, who have emphasized the problems it poses for the patient in terms of esthetics, caries control, and occlusal accommodation. They have recommended prophylactically restoring the groove to prevent caries. If there is occlusal interference, it should be removed but exposure of the pulp horn, necessitating endodontic therapy, is almost certain to occur. Fortunately, this anomaly is quite uncommon among the general population. However, it has been reported by Gardner and Girgis that it appears to be more prevalent in persons with the Rubinstein-Taybi syndrome (developmental retardation, broad thumbs and great toes, characteristic facial features, delayed or incomplete descent of testes in males, and stature, head circumference, and bone age below the fiftieth percentile). The talon cusp has not been reported as an integral part of any other syndrome, although Mader, in his thorough review, suggested that it may be associated with other somatic and odontogenic anomalies.
Dens in Dente (Dens invaginatus, dilated composite odontome)
The ‘dens in dente’ is a developmental variation which is thought to arise as a result of an invagination in the surface of tooth crown before calcification has occurred (Fig. 1-37). Several causes of this condition have been proposed. These include an increased localized external pressure, focal growth retardation, and focal growth stimulation in certain areas of the tooth bud.
Figure 1-37 Dens in dente.
(A) Dens invaginatus. (B) A slight invagination may be seen in the lingual pit area of the maxillary lateral incisor on the radiograph. (C) The ground section of tooth represents a severe form of ‘dens in dente’ and illustrates how the anomaly may resemble a tooth within a tooth (Courtesy of Dr Twinkle S Prasad.
The permanent maxillary lateral incisors are the teeth most frequently involved, and in the majority of cases the ‘dens in dente’ appears to represent simply an accentuation in the development of the lingual pit (Fig. 1-37A). The maxillary central incisors are sometimes involved, and the condition is frequently bilateral. Oehlers has presented an excellent discussion of this condition and emphasized that not only are posterior teeth sometimes affected but also an analogous form of invagination occasionally occurs in the roots of teeth. This radicular variety of ‘dens in dente’ has been discussed by Bhatt and Dholakia, who pointed out that the radicular invagination usually results from an infolding of Hertwig’s sheath and takes its origin within the root after development is complete.
The cases that have been reported in the literature indicate that the condition is fairly common and that an extremely wide range in degree of variation can exist. The term ‘dens in dente,’ originally applied to a severe invagination that gave the appearance of a tooth within a tooth, is actually a misnomer, but it has continued in usage (Fig. 1-37B). In the mild form, there is a deep invagination in the lingual pit area, which may not be evident clinically. Radiographically, it is recognized as a pear-shaped invagination of enamel and dentin with a narrow constriction at the opening on the surface of the tooth and closely approximating the pulp in its depth. Food debris may become packed in this area with resultant caries and infection of the pulp, occasionally even before the tooth has completely erupted. The more severe forms of ‘dens in dente’ may exhibit an invagination that extends nearly to the apex of the root, and these present a bizarre radiographic picture (Fig. 1-38), reflecting a severe disturbance in the normal anatomic and morphologic structure of the teeth.
It is important to realize that this condition, particularly in its mild form, is fairly common. The clinical studies of Amos have shown that if the minor invaginations are included, the incidence may be as high as 5% of all patients examined (Table 1-10). The more severe forms; however, are much less common.
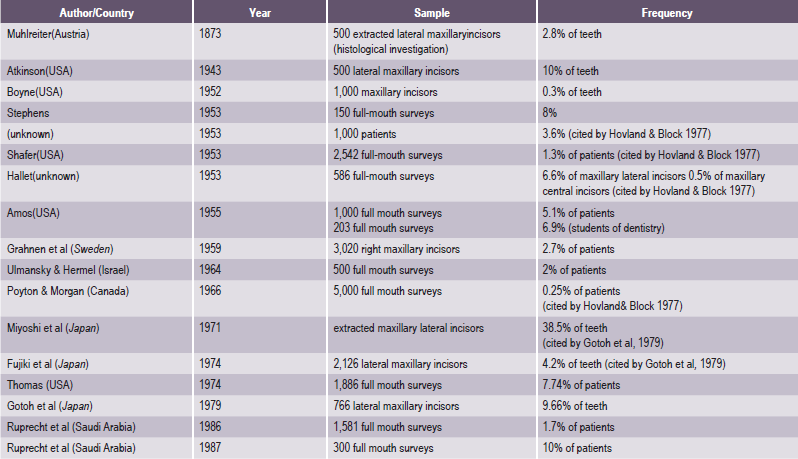
To prevent caries, pulp infection, and premature loss of the tooth, the condition must be recognized early and the tooth prophylactically restored. Fortunately, the defect may be recognized radiographically even before the teeth erupt.
Dens Evaginatus (Occlusal tuberculated premolar, Leong’s premolar, evaginated odontome, occlusal enamel pearl)
The dens evaginatus is a developmental condition that appears clinically as an accessory cusp or a globule of enamel on the occlusal surface between the buccal and lingual cusps of premolars, unilaterally or bilaterally, although it has been reported to occur rarely on molars, cuspids, and incisors (Fig. 1-39). It has been thought to develop only in persons of Mongoloid ancestry: Chinese, Japanese, Filipinos, Eskimos, and American Indians. Its prevalence in a group of 2,373 Chinese schoolchildren in Singapore, reported by Yip, was 2.2%. However, Palmer has reported a series of cases in Caucasians in England.
The pathogenesis of the lesion is thought to be the proliferation and evagination of an area of the inner enamel epithelium and subjacent odontogenic mesenchyme into the dental organ during early tooth development. Thus, it has been considered to be the antithesis of the mechanism of development of the dens in dente or dens invaginatus.
The clinical significance of the condition is similar to that of the talon cusp, which it may physically resemble. This ‘extra’ cusp may contribute to incomplete eruption, displacement of teeth and/or pulp exposure with subsequent infection following occlusal wear or fracture. This phenomenon has been discussed by Senia and Regezi, who have reported periapical infection of caries-free premolars affected by dens evaginatus.
Taurodontism
The term ‘taurodontism’ was originated by Sir Arthur Keith in 1913 to describe a peculiar dental anomaly in which the body of the tooth is enlarged at the expense of the roots. The term means ‘bull-like’ teeth and its usage is derived from the similarity of these teeth to those of ungulate or cudchewing animals. Shaw further classified taurodont teeth into hypotaurodont, mesotaurodont, and hypertaurodont forms, with hypertaurodontism being the extreme form in which the bifurcation or trifurcation occurs near the apices of the roots and hypotaurodontism being the mildest form.
A variety of possible causes of taurodontism have been enumerated by Mangion as follows: (1) a specialized or retrograde character, (2) a primitive pattern, (3) a mendelian recessive trait, (4) an atavistic feature, and (5) a mutation resulting from odontoblastic deficiency during dentinogenesis of the roots. Hammer and his associates believe that the taurodont is caused by failure of Hertwig’s epithelial sheath to invaginate at the proper horizontal level. The heritability of this condition requires further study, although after finding 11 cases of taurodontism among members of three families, Goldstein and Gottlieb have stated that the condition appears to be genetically controlled and familial in nature.
This condition is of anthropologic interest in as much as it has been found commonly in fossil hominids, especially in the Neanderthal man, with a very high prevalence during the neolithic period. At one time it was thought to be confined to these early populations, but it is now known to be widespread in many modern races. Hammer and his associates have discussed these anthropologic aspects in detail, while Blumberg and his coworkers have carried out a biometric study of the condition. A case of taurodontism occurring concomitantly with amelogenesis imperfecta has been reported by Crawford. In addition, it has been reported that many patients with the Klinefelter syndrome (males whose sex chromosome constitution includes one or more extra X chromosomes) exhibit taurodontism, but it is not a constant feature of this syndrome. For this reason, Gardner and Girgis have recommended that male patients exhibiting taurodontism should have chromosomal studies performed, especially if there is any nonspecific diagnosis of mental retardation and if the patient has a tall, thin appearance with long arms and legs and a prognathic jaw.
Figure 1-40 Normal tooth (A), taurodontism (B). Note increased pulpal width in B. Courtesy of Paulsen, Alvin C, Plymate SR. ‘Klinefelter’s syndrome.’ The Genetic Basis of Common Diseases. Editors: King et al. Oxford Monograph on Medical Genetics,Chapter 44,885:1992)
Clinical Features
Taurodontism may affect either the deciduous or permanent dentition, although permanent tooth involvement is more common. The teeth involved are almost invariably molars, sometimes only a single tooth, at other times several molars in the same quadrant. The condition may be unilateral or bilateral or may exhibit any combination of quadrant involvement. The teeth themselves have no remarkable or unusual morphologic clinical characteristics.
Radiographic Features
The unusual nature of this condition is best visualized on the radiograph. Involved teeth frequently tend to be rectangular in shape rather than taper toward the roots. The pulp chamber is extremely large with a much greater apico-occlusal height than normal. In addition, the pulp lacks the usual constriction at the cervical of the tooth and the roots are exceedingly short. The bifurcation o trifurcation may be only a few millimeters above the apices of the roots (Fig. 1-41). This radiographic picture is quite striking and characteristic.
Supernumerary Roots
This developmental condition is not uncommon and may involve any tooth (Fig. 1-42). Teeth that are normally singlerooted, particularly the mandibular bicuspids and cuspids, often have two roots. Both maxillary and mandibular molars, particularly third molars, also may exhibit one or more supernumerary roots. This phenomenon is of considerable significance in exodontia, for one of these roots may be broken off during extraction and, if unrecognized and allowed to remain in the alveolus, may be the source of future infection.