Developmental Disturbances in Number of Teeth
Anodontia
True anodontia, or congenital absence of teeth, may be of two types, total and partial. Total anodontia, in which all teeth are missing, may involve both deciduous and permanent dentition (Fig. 1-43). This is a rare condition; when it occurs, it is frequently associated with a more generalized disturbance, hereditary ectodermal dysplasia (q.v.).

Figure 1-43 Anodontia.
There is congenital absence of nearly all permanent teeth with retention of many deciduous teeth
Induced or false anodontia occurs as a result of extraction of all teeth, while the term pseudo anodontia is sometimes applied to multiple unerupted teeth. The condition under discussion here is a true failure of odontogenesis and should not be confused with false or pseudoanodontia.
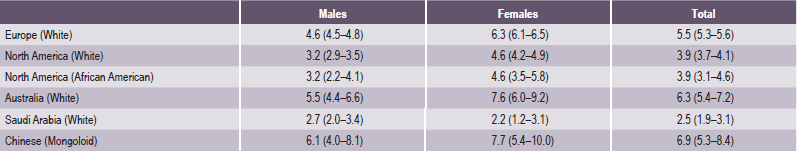
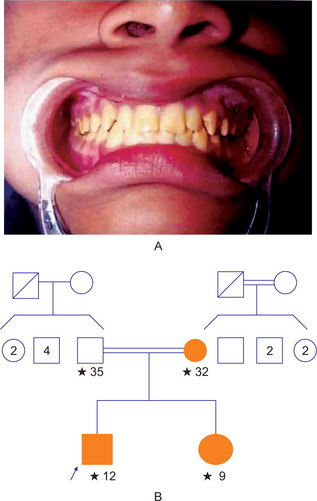
True partial anodontia (hypodontia or oligodontia) involves one or more teeth and is a rather common condition (Fig. 1-42). Although any tooth may be congenitally missing, there is a tendency for certain teeth to be missing more frequently than others (Table 1-11). Studies on the frequency of missing third molars have shown this tooth to be congenitally absent in as many as 35% of all subjects examined, with a frequent absence of all four third molars in the same person (Table 1-12). Other studies have shown that the maxillary lateral incisors and maxillary or mandibular second premolars are commonly missing, often bilaterally (Fig. 1-44). In severe partial anodontia, the bilateral absence of corresponding teeth may be striking. An outstanding review of this subject has been reported by Graber, who showed that the overall frequency of patients with congenitally missing teeth (excluding third molars) has ranged from 1.6–9.6% in various series of studies in different countries.
Table 1-11
Prevalence of dental agenesis by continent, race and gender in percentages (and 95% CI)
Source: Polder BJ et al. Community Dent Oral Epidemiol 32: 217–26, 2004.
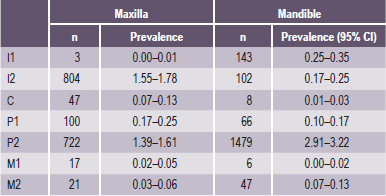
Table 1-12
Prevalence in percentages and 95% CI of dental agenesis of individual teeth derived from 48, 274 persons
Source: Polder BJ et al. Community Dent Oral Epidemiol 32: 217–26, 2004.
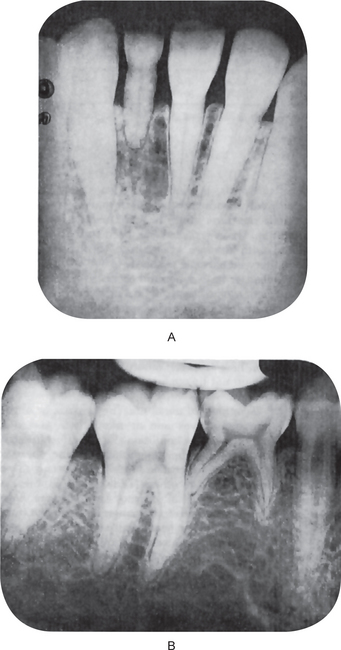
Figure 1-44 Partial anodontia.
The mandibular permanent left central incisor is congenitally missing. The deciduous incisor is retained (A). There is congenital absence of the mandibular second bicuspid with retention of the deciduous molar (B).
Congenitally missing deciduous teeth are uncommon but, when occurring, usually involve the maxillary lateral incisor. Mandibular lateral incisors and mandibular cuspids may also be missing, according to the study of Grahnen and Granath. Their studies also showed a close correlation between congenitally missing deciduous teeth and their permanent successors; suggesting, at least in some instances, a genetic factor.
Although the etiology of a single missing tooth is unknown, a familial tendency for this defect is present in many instances. Graber, in reviewing congenital absence of teeth, reported the accumulating evidence that it is actually the result of one or more point mutations in a closely linked polygenic system, most often transmitted in an autosomal dominant pattern with incomplete penetrance and variable expressivity. Some investigators believe cases of missing third molars to be an evidence of an evolutionary trend towards fewer teeth. Hereditary ectodermal dysplasia may be associated with partial anodontia, and in these instances the few teeth that are present may be deformed or misshapen, frequently cone-shaped.
Occasionally one sees children with teeth of one quadrant or both quadrants on the same side missing owing to X-ray radiation of the face at an early age. Tooth buds are extremely sensitive to X-ray radiation and may be destroyed completely by relatively low dosages. Teeth already forming and partially calcified may be stunted by X-ray radiation.
Supernumerary Teeth
A supernumerary tooth may closely resemble the teeth of the group to which it belongs, i.e. molars, premolars, or anterior teeth, or it may bear little resemblance in size or shape to the teeth with which it is associated. It has been suggested that supernumerary teeth develop from a third tooth bud arising from the dental lamina near the permanent tooth bud, or possibly from splitting of the permanent bud itself. Another theory, well supported in the literature, is the hyperactivity theory, which suggests that supernumeraries are formed as a result of local, independent, conditioned hyperactivity of the dental lamina. In some cases there appears to be a hereditary tendency for the development of supernumerary teeth.
A supernumerary tooth is an additional entity to the normal series and is seen in all the quadrants of the jaw. The etiology of supernumerary teeth is not completely understood. The anomaly does not follow a simple mendelian pattern. In a survey of 2,000 schoolchildren, Brook found that supernumerary teeth were present in 0.8% of primary dentition and in 2.1% of permanent dentition. Occurrence may be single or multiple, unilateral or bilateral, erupted or impacted, and in one or both jaws. Multiple supernumerary teeth are rare in individuals with no other associated diseases or syndromes. The conditions commonly associated with an increased prevalence of supernumerary teeth include cleft lip and palate, cleidocranial dysplasia, and Gardner syndrome. Supernumerary teeth associated with cleft lip and palate result from fragmentation of the dental lamina during cleft formation. The frequency of supernumerary permanent teeth in the cleft area in children with unilateral cleft lip or palate or both was found to be 22.2%. The frequency of supernumeraries in patients with cleidocranial dysplasia ranged from 22% in the maxillary incisor region to 5% in the molar region. While there is no significant gender predilection in primary supernumerary teeth, males are affected approximately twice as frequently as females in the permanent dentition.
Classification
Supernumerary teeth are classified according to morphology and location. In the primary dentition, morphology is usually normal or conical. There is a greater variety of forms presenting in the permanent dentition.
Four different morphological types of supernumerary teeth have been described:
This small peg-shaped conical tooth is supernumerary and most commonly found in the permanent dentition. It develops with root formation ahead of or at an equivalent stage to that of permanent incisors and usually presents as a mesiodens. It may occasionally be found high and inverted into the palate or in a horizontal position. In most cases; however, the long axis of the tooth is normally inclined. The conical supernumerary can result in rotation or displacement of the permanent incisor, but rarely delays eruption.
The tuberculate type of supernumerary possesses have more than one cusp or tubercle. It is frequently described as barrel-shaped and may be invaginated. Root formation is delayed compared to that of the permanent incisors. Tuberculate supernumeraries are often paired and are commonly located on the palatal aspect of the central incisors. They rarely erupt and are frequently associated with delayed eruption of the incisors.
The supplemental supernumerary refers to a duplication of teeth in the normal series and is found at the end of a tooth series. The most common supplemental tooth is the permanent maxillary lateral incisor, but supplemental premolars and molars also occur. The majority of supernumeraries found in the primary dentition are of the supplemental type and seldom remain impacted.
Odontome has been listed as the fourth category of supernumerary teeth by Howard. However, this category is not universally accepted. The term ‘odontoma’ refers to any tumor of odontogenic origin. Most authorities; however, accept the view that the odontoma represents a hamartomatous malformation rather than a neoplasm. The lesion is composed of more than one type of tissue and consequently has been called a composite odontoma. Two separate types have been described, the diffuse mass of dental tissue which is totally disorganized is known as a complex composite odontoma, whereas the malformation which bears some superficial anatomical similarity to a normal tooth is referred to as a compound composite odontoma.
Gardner syndrome is an interesting disease complex, reviewed by Fader and his associates and by Duncan and his associates. It is also characterized by the occurrence of multiple impacted supernumerary teeth. This syndrome consists of: (1) multiple polyposis of the large intestine, (2) osteomas of the bones, including long bones, skull, and jaws, (3) multiple epidermoid or sebaceous cysts of the skin, particularly on the scalp and back, (4) occasional occurrence of desmoid tumors, and (5) the impacted supernumerary and permanent teeth (Figs. 1-45, 1-46).
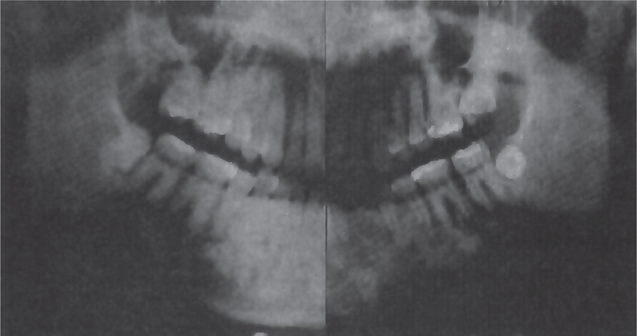
Figure 1-45 Gardner syndrome.
Multiple diffuse osteomas of the maxilla and mandible are present, although the patient does not have supernumerary teeth (Courtesy of Dr Wesley P Titterington, Dr William M Wade, Jr.
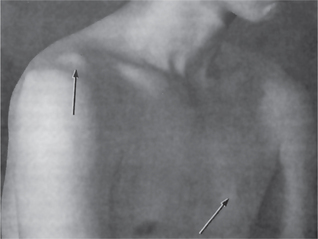
Figure 1-46 Gardner syndrome.
Sebaceous cysts of the skin are present over the shoulder and chest (Courtesy of Dr Wesley P Titterington, Dr William M Wade, Jr.
It is due to a single pleiotropic gene and has an autosomal dominant pattern of inheritance, with complete penetrance and variable expression. Indicative of its inheritability is a report by Watne and his associates, who studied 280 patients from 11 families with Gardner syndrome. They found that 126 of the 280, or 45% of the patients at risk, exhibited some part of the syndrome. Significantly, the intestinal polyps in this disease were premalignant, and polyps were found to be present in 85 of the 126 patients. In 41 of the patients, carcinoma of the intestine subsequently developed and only 27% survived. This disease is of interest to the dental profession, since the impacted teeth and osteomas of the jaws may lead to early diagnosis of the entire syndrome.
Predeciduous Dentition (Premature eruption, natal teeth, neonatal teeth)
Infants occasionally are born with structures which appear to be erupted teeth, usually in the mandibular incisor area. These structures must be distinguished from true deciduous teeth, or the so-called natal teeth described by Massler, which may have erupted by the time of birth. The predeciduous teeth have been described as hornified epithelial structures without roots, occurring on the gingiva over the crest of the ridge, which may be easily removed. Prematurely erupted true deciduous teeth, of course, are not to be extracted. These predeciduous teeth have been thought to arise either from an accessory bud of the dental lamina ahead of the deciduous bud or from the bud of an accessory dental lamina.
The concept of predeciduous teeth has been questioned by Spouge and Feasby; however. They are probably correct in believing that considering predeciduous teeth as an entity is a misinterpretation and that such structures, present at birth, undoubtedly represent only the dental lamina cyst of the newborn (q.v.). This cyst does commonly project above the crest of the ridge, is white in color and is packed within keratin, so that it appears ‘hornified’ and can be easily removed.
Post Permanent Dentition
A few cases are recorded of persons who have had all their permanent teeth extracted and yet have subsequently erupted several more teeth, particularly after the insertion of a full denture. The majority of such cases are the result of delayed eruption of retained or embedded teeth. A small number of cases; however, do appear to represent examples of a post permanent or third dentition, although it probably would be better to classify these as simply multiple supernumerary unerupted teeth, since they probably develop from a bud of the dental lamina beyond the permanent tooth germ.
Developmental Disturbances in Structure of Teeth
Amelogenesis Imperfecta (Hereditary enamel dysplasia, hereditary brown enamel, hereditary brown opalescent teeth)
A complex inheritance pattern gives rise to amelogenesis imperfecta (AI), a structural defect of the tooth enamel. It may be differentiated into three main groups: hypoplastic (HP), hypocalcified (HC), and hypomature (HM), depending on the clinical presentation of the defects and the likely stage of enamel formation that is primarily affected (Table 1-13). Each main clinical group of AI may be further divided into several subgroups depending on the mode of inheritance, as well as the clinical appearance of the defective enamel, although in some cases, overlapping clinical features may make distinction difficult. The prevalence of this condition has been estimated to range from (1 in 718) to (1 in 14,000), depending on the population studied. Hypoplastic AI represents 60–73% of all cases, hypomaturation AI represents 20–40%, and hypocalcification AI represents 7%. Disorders of the enamel epithelium also can cause alterations in the eruption mechanism, resulting in the anterior open bite.
Table 1-13
Classification of amelogenesis imperfecta according to Witkop (1989)
| Type I | Hypoplastic |
| IA | Hypoplastic, pitted autosomal dominant |
| IB | Hypoplastic, local autosomal dominant |
| IC | Hypoplastic, local autosomal recessive |
| ID | Hypoplastic, smooth, autosomal dominant |
| IE | Hypoplastic, smooth X-linked dominant |
| IF | Hypoplastic, rough autosomal dominant |
| IG | Enamel agenesis, autosomal recessive |
| Type II | Hypomaturation |
| IIA | Hypomaturation, pigmented autosomal recessive |
| IIB | Hypomaturation, X-linked recessive |
| IIC | Snow-capped teeth, autosomal dominant |
| Type III | Hypocalcified |
| IIIA | Autosomal dominant |
| IIIB | Autosomal recessive |
| Type IV | Hypomaturation-hypoplastic with tauro dontism |
| IVA | Hypomaturation-hypoplastic with taurodontism, autosomal dominant |
| IVB | Hypoplastic-hypomaturation with taurodontism, autosomal dominant |
Molecular Genetic Studies
Molecular genetic studies have shown that the etiology of AI is related to the alteration of genes involved in the process of formation and maturation of the enamel (Table 1-14). Although the genetic origin of the autosomal forms is less understood, analysis of X-linked AI has shown the defective gene for this specific AI type to be closely linked to the locus DXS85 at Xp22. Interestingly, this also has been identified as the general location of the human gene for amelogenin, the principal protein in developing enamel. Information from molecular genetic studies will ultimately lead to identification of the genes involved in normal and pathological enamel formation and provide a basis for definitive diagnostic tests.
Table 1-14
Gene(s) mutated in amelogenesis imperfecta (known and unknown)
| Type I | Hypoplastic AI |
| IA | Pitted hypoplastic AD |
| The gene defect for this AI type is unknown at this time | |
| IB | Local hypoplastic AD |
| A mutation in the enamelin gene (ENAM) has been identified as causing this AI type | |
| IC | Local hypoplastic AR |
| The gene defect for this AI type is unknown at this time | |
| ID | Smooth hypoplastic AD |
| Multiple mutations in the enamelin gene (ENAM); have been identified as causing this AI type | |
| IE | Smooth hypoplastic X-linked dominant |
| A variety of different mutations in the gene coding for the amelogenin protein (AMELX); cause this AI type | |
| IF | Rough hypoplastic AD |
| The gene defect for this AI type is unknown at this time | |
| IG | Enamel agenesis AR (rough hypoplastic AR) |
| The gene defect for this AI type is unknown at this time | |
| Type II | Hypomaturation AI |
| IIA | Pigmented hypomaturation AR |
| The gene defect for this AI type is unknown at this time | |
| IIB | Hypomaturation AI X-linked recessive |
| Multiple mutations in the AMELX gene have been identified in this AI type | |
| IIC | Snow capped teeth AD |
| The gene defect for this AI type is unknown at this time | |
| Type III | Hypocalcified AI |
| IIIA | Hypocalcified AI AD |
| The gene defect for this AI type is unknown at this time | |
| IIIB | Hypocalcified AI AR |
| The gene defect for this AI type is unknown at this time | |
| Type IV | Hypomaturation-Hypoplastic with taurodontism |
| IVA | AD Hypomaturation-hypoplastic with taurodontism |
| The gene defect for this AI type is unknown at this time | |
| IVB | AD Hypoplastic-hypomaturation with taurodontism |
| The gene defect for this AI type is unknown at this time |
Source: Modified from the website data of School of Dentistry, University of North Carolina, 2005.
Radiographic Features
The overall shape of the tooth may or may not be normal, depending upon the amount of enamel present on the tooth and the amount of occlusal and incisal wear. The enamel may appear totally absent on the radiograph, or when present, may appear as a very thin layer, chiefly over the tips of the cusps and on the interproximal surfaces (Fig. 1-47). In other cases the calcification of the enamel may be so affected that it appears to have the same approximate radiodensity as the dentin, making differentiation between the two difficult (Fig. 1-48).
Histologic Features
The general histologic features of the enamel also parallel the general type of amelogenesis imperfecta that has been diagnosed. There is a disturbance in the differentiation or viability of ameloblasts in the hypoplastic type, and this is reflected in defects in matrix formation up to and including total absence of matrix. In the hypocalcification types there are defects of matrix structure and of mineral deposition. Finally, in the hypomaturation types there are alterations in enamel rod and rod sheath structures.
Treatment
There is no treatment except for improvement of cosmetic appearance. However, in some cases, these teeth do not appear markedly abnormal to the casual observer.
Environmental Enamel Hypoplasia
Enamel hypoplasia may be defined as an incomplete or defective formation of the organic enamel matrix of teeth. Two basic types of enamel hypoplasia exist: (1) a hereditary type, described previously under amelogenesis imperfecta, and (2) a type caused by environmental factors. In the hereditary type, both the deciduous and permanent dentitions usually are involved and generally only the enamel is affected. In contrast, when the defect is caused by environmental factors, either dentition may be involved and sometimes only a single tooth; both enamel and dentin are usually affected, at least to some degree.
Many studies, both experimental and clinical, have been carried out in an attempt to determine the cause and nature of environmental enamel hypoplasia. It is known that a number of different factors, each capable of producing injury to the ameloblasts, may give rise to the condition, including: (1) nutritional deficiency (vitamins A, C, and D); (2) exanthematous diseases (e.g. measles, chickenpox, scarlet fever); (3) congenital syphilis; (4) hypocalcemia; (5) birth injury, prematurity, Rh hemolytic disease; (6) local infection or trauma; (7) ingestion of chemicals (chiefly fluoride); and (8) idiopathic causes.
In mild environmental hypoplasia, there may be only a few small grooves, pits, or fissures on the enamel surface (Fig. 1-49). If the condition is more severe, the enamel may exhibit rows of deep pits arranged horizontally across the surface of the tooth (Fig. 1-50). There may be only a single row of such pits or several rows indicating a series of injuries. In the most severe cases, a considerable portion of enamel may be absent, suggesting a prolonged disturbance in the function of the ameloblasts.
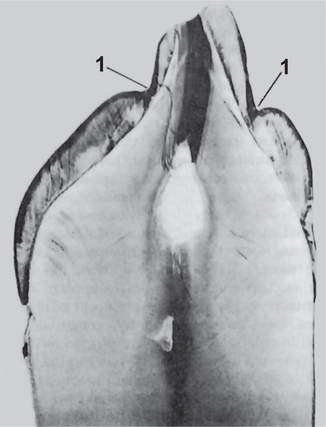
Figure 1-49 Enamel hypoplasia, environmental type.
The ground section of tooth shows a pit like defect on both the labial and lingual surfaces (1).
Figure 1-50 Enamel hypoplasia, environmental type.
Several rows of irregular, stained pits cover much of the labial surfaces of the teeth.
Hypoplasia results only if the injury occurs during the time the teeth are developing, or more specifically, during the formative stage of enamel development. Once the enamel has calcified, no such defect can be produced. Thus, knowing the chronologic development of the deciduous and permanent teeth, it is possible to determine from the location of the defect on the teeth the approximate time at which the injury occurred.
Hypoplasia due to Nutritional Deficiency and Exanthematous Fevers
Some studies have shown that rickets during the time of tooth formation is the most common known cause of enamel hypoplasia. For example, in a series of rachitic children reported by Shelling and Anderson, 43% of teeth showed hypoplasia. At present; however, rickets is not a prevalent disease. Deficiencies of vitamin A and C have also been named as causes.
Some studies have indicated that the exanthematous diseases, including measles, chickenpox and scarlet fever, are etiologic factors, but other investigators have been unable to confirm this finding. In general, it might be stated that any serious nutritional deficiency or systemic disease is potentially capable of producing enamel hypoplasia, since the ameloblasts are one of the most sensitive groups of cells in the body in terms of metabolic function.
The type of hypoplasia occurring from these deficiency or disease states is usually of the pitting variety described above. Since the pits tend to stain, the clinical appearance of the teeth may be very unsightly.
Clinical studies indicate that most cases of enamel hypoplasia involve those teeth that form within the first year after birth, although teeth that form somewhat later may be affected. Thus the teeth most frequently involved are the central and lateral incisors, cuspids, and first molars. Since the tip of the cuspid begins formation before the lateral incisor, some cases involve only the central incisor, cuspid, and first molar. Premolars and second and third molars are seldom affected, since their formation does not begin until about the age of three years or later.
There has been considerable controversy as to whether there is any relation between enamel hypoplasia and dental caries experience, and clinical reports have given conflicting results. It is most reasonable to assume that the two are not related, although hypoplastic teeth do appear to decay at a somewhat more rapid rate once caries has been initiated.
Enamel Hypoplasia due to Congenital Syphilis
The hypoplasia due to congenital syphilis is most frequently not of the pitting variety previously described but instead presents a characteristic, almost pathognomonic, appearance. This hypoplasia involves the maxillary and mandibular permanent incisors and the first molars. The anterior teeth affected are sometimes called ‘Hutchinson’s teeth,’ while the molars have been referred to as ‘mulberry molars’ (Moon’s molars, Fournier’s molars).
Characteristically, the upper central incisor is ‘screw-driver’ shaped, the mesial and distal surfaces of the crown tapering and converging toward the incisal edge of the tooth rather than toward the cervical margin (Fig. 1-51). In addition, the incisal edge is usually notched. The mandibular central and lateral incisors may be similarly involved, although the maxillary lateral incisor may be normal. The cause of the tapering and notching of the maxillary incisor has been explained on the basis of the absence of the central tubercle or calcification center. The crowns of the first molars in congenital syphilis are irregular and the enamel of the occlusal surface and occlusal third of the tooth appears to be arranged in an agglomerate mass of globules rather than in well-formed cusps (Fig. 1-52). The crown is narrower on the occlusal surface than at the cervical margin.
Figure 1-51 Enamel hypoplasia of congenital syphilis (mulberry molars).
The mandibular first molars show many small globular masses of enamel on the occlusal portion of the tooth.
Figure 1-52 Enamel hypoplasia of congenital syphilis (Hutchinson’s incisors).
There is characteristic notching of the incisal edges of the maxillary central incisors as well as tapering of the mesial and distal surfaces toward the incisal portion.
It has been reported by Fiumara and Lessell that between 1958 and 1969 there has been over a 200% increase in reported cases of primary and secondary syphilis in the United States and that, consequently, congenital syphilis in children under one year of age increased 117% during the 10–year period from 1960 to 1969. Investigating 271 patients with congenital syphilis, they found that over 63% had Hutchinson’s teeth but they pointed out that this may not be the true incidence, since some of the patients had their teeth extracted. In addition, approximately 65% of this group of patients with congenital syphilis also had the characteristic ‘mulberry molars.’
Not all patients with congenital syphilis will exhibit these dental findings. Also, occasional patients will appear to have Hutchinson’s teeth without having a history of congenital syphilis. Therefore, one must not be hasty in making the diagnosis of syphilis, particularly in the absence of the other conditions of Hutchinson’s triad (q.v.).
Enamel Hypoplasia due to Hypocalcemia
Tetany, induced by a decreased level of calcium in the blood, may result from several conditions, the most common being vitamin D deficiency and parathyroid deficiency (parathyroprivic tetany). In tetany the serum calcium level may fall as low as 6–8 mg per 100 ml, and at this level enamel hypoplasia is frequently produced in teeth developing concomitantly. This type of enamel hypoplasia is usually of the pitting variety and thus does not differ from that resulting from a nutritional disturbance or exanthematous disease.
Hypoplasia due to Birth Injuries
The neonatal line or ring, described by Schour in 1936 and present in deciduous teeth and first permanent molars, may be thought of as a type of hypoplasia because there is a disturbance produced in the enamel and dentin, which is indicative of trauma or change of environment at the time of birth. In traumatic births the formation of enamel may even cease at this time. In addition, Miller and Forrester have reported a clinical study with evidence that enamel hypoplasia is far more common in prematurely born children than in normal term infants. In this same study they not only drew attention to the widely recognized staining of teeth in children who had suffered from Rh hemolytic disease at birth (q.v.) but also reported enamel hypoplasia in these cases. Grahnen and Larsson have also shown an increased incidence of enamel hypoplasia in premature children, but interestingly no differences in caries incidence between this group and a control group of children.
Although the literature indicates that most cases of enamel hypoplasia of deciduous teeth involve enamel formed after birth, it is seen also in prenatal enamel. In such instances a gastrointestinal disturbance or some other illness in the mother may be responsible.
Enamel Hypoplasia due to Local Infection or Trauma
A type of hypoplasia occasionally seen is unusual in that only a single tooth is involved, most commonly one of the permanent maxillary incisors or a maxillary or mandibular premolar. There may be any degree of hypoplasia, ranging from a mild, brownish discoloration of the enamel to a severe pitting and irregularity of the tooth crown (Fig. 1-53). These single teeth are frequently referred to as ‘Turner’s teeth,’ and the condition is called ‘Turner’s hypoplasia.’
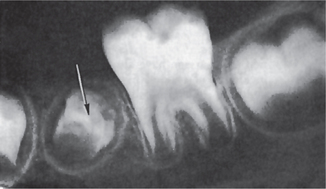
Figure 1-53 Enamel hypoplasia due to local infection (Turner’s hypoplasia).
The crown of the unerupted bicuspid is extremely irregular, owing to disruption of the forming tooth by infection through the preceding deciduous tooth (Courtesy of Dr Ralph E McDonald.
If a deciduous tooth becomes carious during the period when the crown of the succeeding permanent tooth is being formed, a bacterial infection involving the periapical tissue of this deciduous tooth may disturb the ameloblastic layer of the permanent tooth and result in a hypoplastic crown. The severity of this hypoplasia will depend upon the severity of the infection, the degree of tissue involvement, and the stage of permanent tooth formation during which the infection occurred.
A similar type of hypoplasia may follow trauma to a deciduous tooth, particularly when the deciduous tooth has been driven into the alveolus and has disturbed the permanent tooth bud. If this permanent tooth crown is still being formed, the resulting injury may be manifested as a yellowish or brownish stain or pigmentation of the enamel, usually on the labial surface, or as a true hypoplastic pitting defect or deformity. This form of dental injury has been discussed by Via, who has pointed out that a disturbance either in matrix formation or in calcification can occur, depending chiefly upon the stage of tooth formation at the time of injury.
Enamel Hypoplasia due to Fluoride: Mottled Enamel
Mottled enamel is a type of enamel hypoplasia that was first described under that term in this country by GV Black and Frederick S McKay in 1916. Earlier reference to the condition is known in the foreign literature; however. Black and McKay recognized that this lesion exhibited a geographic distribution and even suggested that it was a result of some substance in the water supply, although it was not until some years later that fluorine was shown to be the causative agent.
Etiology
It is now recognized that the ingestion of fluoridecontaining drinking water during the time of tooth formation may result in mottled enamel. The severity of the mottling increases with an increasing amount of fluoride in the water. Thus there is little mottling of any clinical significance at a level below 0.9–1.0 part per million of fluoride in the water, whereas it becomes progressively evident above this level.
Pathogenesis
This type of hypoplasia is due to a disturbance of the ameloblasts during the formative stage of tooth development. The exact nature of the injury is not known, but since there is histologic evidence of cell damage, it is likely that the cell product, the enamel matrix, is defective or deficient. It also has been shown that, with somewhat higher levels of fluoride, there is interference with the calcification process of the matrix.
Epidemiologic studies have reported that not all children born and reared in an area of endemic fluorosis exhibit the same degree of mottling even though they all have used the same water supply. Furthermore, a few persons may exhibit mild mottling even when exposed to a very low concentration of fluoride (Table 1-15). These findings may be related to individual variation in total water consumption and thus to total fluoride intake.
Clinical Features
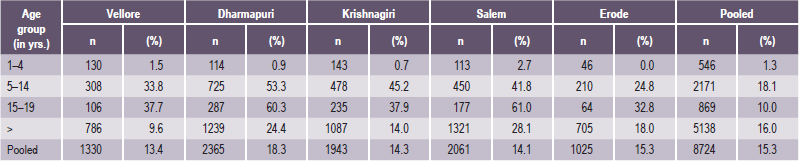
Depending upon the level of fluoride in the water supply, there is a wide range of severity in the appearance of mottled teeth, varying from: (1) questionable changes characterized by occasional white flecking or spotting of the enamel (Fig. 1-54A), through (2) mild changes manifested by white opaque areas involving more of the tooth surface area (Fig. 1-54B), to (3) moderate and severe changes showing pitting and brownish staining of the surface (Fig. 1-54C), and even (4) a corroded appearance of the teeth (Fig. 1-54D). Those teeth which are moderately or severely affected may show a tendency for wear and even fracture of the enamel. Early studies actually noted the difficulty of retaining restorations in such teeth.
Geographic Distribution
Mottled enamel has been reported in many parts of the world, including Europe, Africa, and Asia as well as the United States. In this country, persons in at least 400 areas in 28 states have shown dental evidence of endemic fluorosis, and undoubtedly more communities remain to be reported. Most of the areas affected are west of the Mississippi River, the largest single area being the Texas Panhandle, but there are numerous communities east of the Mississippi River in which fluorosis is present, the most notable being in Illinois.
The well-known relation between mottled enamel (or actually fluoride intake) and dental caries will be discussed in Chapter 9 on Dental Caries.
Treatment
Mottled enamel frequently becomes stained an unsightly brown color. For cosmetic reasons, it has become the practice to bleach the affected teeth with an agent such as hydrogen peroxide. This is frequently effective, but the procedure must be carried out periodically, since the teeth continue to stain.
Hypoplasia due to Idiopathic Factors
Although numerous factors have been reported as being possibly responsible for causing enamel hypoplasia, clinical studies have shown that, even with careful histories, the majority of cases are of unknown origin. Since the ameloblast is a sensitive type of cell and easily damaged, it is likely that in those cases in which the etiology cannot be determined, the causative agent may have been some illness or systemic disturbance so mild that it made no impression on the patient and was not remembered. Even relatively severe cases of enamel hypoplasia arise with no pertinent past medical history to account for their occurrence.
Dentinogenesis Imperfecta
Dentinogenesis imperfecta is an autosomal dominant condition affecting both deciduous and permanent teeth. Affected teeth are gray to yellowish-brown and have broad crowns with constriction of the cervical area resulting in a ‘tulip’ shape. Radiographically, the teeth appear solid, lacking pulp chambers and root canals. Enamel is easily broken leading to exposure of dentin that undergoes accelerated attrition. The gene maps to chromosome number 4. It encodes a protein called dentin sialophosphoprotein (DSPP). This protein constitutes about 50% of the noncollagenous component of dentin matrix. It is not known how the mutant protein causes near obliteration of the pulp. A clinically and radiographically indistinguishable dental condition is seen sometimes in patients with osteogenesis imperfecta. Dentin defect associated with osteogenesis imperfecta was earlier listed as dentinogenesis imperfecta type I (Sheilds classification). Extensive studies have proven that dentinogenesis imperfecta is clearly a disorder distinct from osteogenesis imperfecta hence the following revised classification is proposed.
Dentinogenesis imperfecta I: Dentinogenesis imperfecta without osteogenesis imperfecta (opalescent dentin): this corresponds to dentinogenesis imperfecta type II of Shields classification.
Dentinogenesis imperfecta II: Brandywine type dentinogenesis imperfecta: this corresponds to dentinogenesis imperfecta type III of Shields classification.
There is no substitute in the present classification for the category designated as DI type I of the previous classification (Shields).
Dentinogenesis imperfecta II is even more rare and paradoxically characterized by too little rather than too much dentin resulting in ‘shell teeth.’ Dentinogenesis imperfecta II may be an allelic variant of dentinogenesis imperfecta I (a different mutation in the same gene), both genes map to the same region on chromosome number 4.
Dentinogenesis Imperfecta I (Opalescent dentin, dentinogenesis imperfecta without osteogenesis imperfecta, opalescent teeth without osteogenesis imperfecta, dentinogenesis imperfecta, Shields type II, Capdepont teeth)
Dentinogenesis imperfecta II is caused by mutation in the DSPP gene (gene map locus 4q21.3), encoding dentin phosphoprotein and dentin sialoprotein. Dentinogenesis imperfecta is an entity clearly distinct from osteogenesis imperfecta with opalescent teeth, and affects only the teeth. There is no increased frequency of bone fractures in this disorder. The frequency may be 1 in 6,000–8,000 children. Witkop and Rao (1971) preferred the term opalescent dentin for this condition as an isolated trait, reserving dentinogenesis imperfecta for the trait when it is combined with osteogenesis imperfecta. The teeth are blue-gray or amber brown and opalescent. On dental radio graphs, the teeth have bulbous crowns, roots that are narrower than normal, and pulp chambers and root canals that are smaller than normal or completely obliterated. The enamel may split readily from the dentin when subjected to occlusal stress. Sauk et al (1976) noted an increase in glycosaminoglycans in EDTA soluble dentin in the teeth from patients with this disorder as compared to controls, and less glycosaminoglycan in EDTA insoluble residue.
Shields et al (1973) proposed that the variety of dentinogenesis imperfecta (dentinogenesis imperfecta type III) described in the Brandywine isolate by Hursey et al (1956) was distinct from dentinogenesis imperfecta type II.
A deficiency of dentin sialophosphoprotein had been suggested as a causative factor in dentinogenesis imperfecta. Zhanget al (2001) studied a Chinese family with dentinogenesis imperfecta Shields type II. Affected members in three generations showed discoloration and severe attrition of their teeth, with obliterated pulp chambers.
Dentinogenesis Imperfecta II (Shields type III, Brandywine type dentinogenesis imperfecta)
This disorder was found in the Brandywine triracial isolate in southern Maryland. The crowns of the deciduous and permanent teeth wear rapidly after eruption and multiple pulp exposures may occur. The dentin is amber and smooth (Fig. 1-55). Radiographs of the deciduous dentition show very large pulp chambers and root canals, at least during the first few years, although they may become reduced in size with age. The permanent teeth have pulpal spaces that are either smaller than normal or completely obliterated. Patients with Shields type III, or the Brandywine type, do not have stigmata of osteogenesis imperfecta. This disorder may be a separate mutation from dentinogenesis imperfecta I. Shields et al (1973) stated that multiple pulp exposures and markedly enlarged pulp chambers in the deciduous teeth do not occur in DGI-1; however, Witkop (1975) suggested that the two disorders are the same. Recent studies are consistent with the hypothesis that DGI-1 and DGI-2 are allelic or the result of mutations in two tightly linked genes. MacDougall (1998) provided information on the intervals separating four genes that map to this same region, all four of which are involved in dental development: DSPP, DMP-1, IBSP, and SPP1. MacDougall et al (1999) stated that the manifestations of DGI-2 can differ from those of DGI-1 by the presence of multiple pulp exposures, normal nonmineralized pulp chambers and canals, and a general appearance of ‘shell teeth.’ They illustrated the amber discoloration of the teeth, attrition, and fractured enamel, as well as the classic ‘shell teeth’ appearance on radiographs.
Histologic Features
The histologic appearance of the teeth in dentinogenesis imperfecta I emphasizes the fact that this is purely a mesodermal disturbance. The appearance of the enamel is essentially normal except for its peculiar shade, which is actually a manifestation of the dentinal disturbance. The dentin, on the other hand, is composed of irregular tubules, often with large areas of uncalcified matrix (Fig. 1-56). The tubules tend to be larger in diameter and thus less numerous than normal in a given volume of dentin (Fig. 1-57). In some areas there may be complete absence of tubules. Cellular inclusions, probably odontoblasts, in the dentin are not uncommon, and as pointed out previously, the pulp chamber is usually almost obliterated by the continued deposition of dentin. The odontoblasts have only limited ability to form well-organized dentinal matrix, and they appear to degenerate readily, becoming entrapped in this matrix.
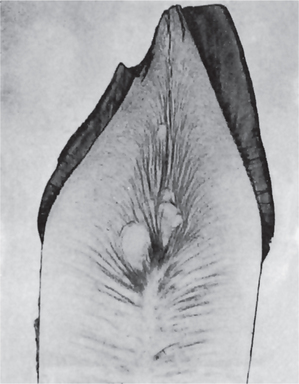
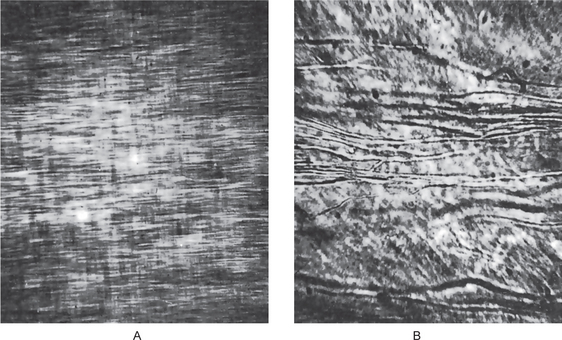
Figure 1-57 Dentinogenesis imperfecta.
(A) Normal dentin showing regular dentinal tubules. (B) large irregular dentinal tubules in dentinogenesis imperfecta. Both photomicrographs taken at same magnification.
The histopathology of the teeth in type III has not been adequately documented.
Chemical and Physical Features
Chemical analysis explains many of the abnormal features of the teeth of dentinogenesis imperfecta I. Their water content is greatly increased, as much as 60% above normal, while the inorganic content is less than that of normal dentin. As might be expected, the density, X-ray absorption, and hardness of the dentin are also low. In fact, the micro-hardness of the dentin closely approximates that of cementum, thus explaining the rapid attrition of affected teeth. There is no significant information available on teeth in type III.
Treatment
The treatment of patients with dentinogenesis imperfecta is directed primarily towards preventing the loss of enamel and subsequent loss of dentin through attrition. Cast metal crowns on the posterior teeth and jacket crowns on the anterior teeth have been used with considerable success, although care must be taken in the preparation of the teeth for such restorations. Caution must also be exercised in the use of partial appliances which exert stress on the teeth, because the roots are easily fractured. Experience has further shown that fillings are not usually permanent because of the softness of the dentin.
Dentin Dysplasia (Rootless teeth)
Dentin dysplasia is a rare disturbance of dentin formation characterized by normal enamel but atypical dentin formation with abnormal pulpal morphology.
At one time this was thought to be a single disease entity, but it now has been separated by Shields and his associates into type I (dentin dysplasia) and type II (anomalous dysplasia of dentin). However, Witkop has suggested that as a guide to the clinician, these conditions be referred to as radicular dentin dysplasia (type I) and coronal dentin dysplasia (type II). It has been found that type I is by far the more common.
The first description of the disease was that of Ballschmiede, who in 1920 reported the spontaneous exfoliation of multiple teeth in seven children of one family and called this phenomenon ‘rootless teeth.’ The first concise description of the disease was published in 1939 by Rushton, who was also the first to designate it as ‘dentin dysplasia.’
Etiology
Dentin dysplasia, both type I and type II, appears to be a hereditary disease, transmitted as an autosomal dominant characteristics. Nothing is known of the mutation rate, but it must be extremely low.
Clinical Features
Both dentitions are affected, although the teeth appear clinically normal in morphologic appearance and color. Occasionally there may be a slight amber translucency. The teeth generally exhibit a normal eruption pattern, although delayed eruption has been reported in a few cases. However, the teeth characteristically exhibit extreme mobility and are commonly exfoliated prematurely or after only minor trauma as a result of their abnormally short roots.
Both dentitions are also affected in this form of dentin dysplasia, although the involvement of each dentition is different clinically, radiographically, and histologically. The deciduous teeth have the same yellow, brown, or bluish-gray opalescent appearance as seen in dentinogenesis imperfecta. However, the clinical appearance of the permanent dentition is normal.
Radiographic Features
In both dentitions, the roots are short, blunt, conical, or similarly malformed (Fig. 1-58). In the deciduous teeth, the pulp chambers and root canals are usually completely obliterated, while in the permanent dentition, a crescent-shaped pulpal remnant may still be seen in the pulp chamber. This obliteration in the permanent teeth commonly occurs pre-eruptively. Of significant interest is the discovery of periapical radiolucencies representing granulomas, cysts, or abscesses involving apparently otherwise intact teeth.
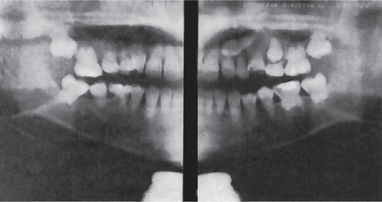
The pulp chambers of the deciduous teeth become obliterated as in type I and in dentinogenesis imperfecta. This does not occur before eruption. The permanent teeth; however, exhibit an abnormally large pulp chamber in the coronal portion of the tooth, often described as ‘thistle-tube’ in shape, and within such areas radiopaque foci resembling pulp stones may be found. Periapical radiolucencies do not occur unless for an obvious reason.
Histologic Features
A portion of the coronal dentin is usually normal. Apical to this may be areas of tubular dentin, but most of that which obliterates the pulp is calcified tubular dentin, osteodentin, and fused denticles. Normal dentinal tubule formation appears to have been blocked so that new dentin forms around obstacles and takes on the characteristic appearance described as ‘lava flowing around boulders’ (Fig. 1-59). Electron microscopic studies by Sauk and his coworkers have suggested that this pattern of ‘cascades of dentin’ results from repetitive attempts to form root structure. Interestingly, the dentin itself is histologically normal but is simply disoriented.
The deciduous teeth exhibit amorphous and atubular dentin in the radicular portion, while coronal dentin is relatively normal. The permanent teeth also show relatively normal coronal dentin, but the pulp has multiple pulp stones or denticles.
Excellent scanning electron microscopic studies of both types I and II dentin dysplasia have been reported by Melnick and his associates, and these have added appreciably to our knowledge of the atypical structure of this dentin.
Regional Odontodysplasia (Odontodysplasia, odontogenic dysplasia, odontogenesis imperfecta, ghost teeth)
This is an unusual dental anomaly in which one or several teeth in a localized area are affected in an unusual manner. Apparently the maxillary teeth are involved more frequently than the mandibular, the most frequently affected teeth being the maxillary permanent central incisor, lateral incisor, and cuspid. In the mandible, the same three anterior teeth are most often affected. The deciduous teeth as well as the permanent may be involved.
The etiology of this disease is unknown inasmuch as there is no history of trauma or systemic illness. It has been suggested that the condition may represent a somatic mutation, although the possibility has also been raised that it could be due to a latent virus residing in the odontogenic epithelium, which subsequently becomes active during the development of the tooth. In an excellent review of cases in the literature and a discussion of the condition, Walton and his coworkers observed that in three cases of regional odontodysplasia that they reported, all three patients had vascular nevi of the overlying facial skin as infants. They reported similar involvement in three additional cases in the literature, and these findings suggested to them that local vascular defects are involved in the pathogenesis of the condition. An early comprehensive account was also reported by Rushton.
Clinical Features
The teeth affected by odontodysplasia exhibit either a delay or a total failure in eruption. Their shape is markedly altered, being generally very irregular in appearance, often with evidence of defective mineralization.
Radiographic Features
The radiographs are uniquely characteristic, showing a marked reduction in radiodensity so that the teeth assume a ‘ghost’ appearance (Fig. 1-60A,B). Both the enamel and dentin appear very thin and the pulp chamber is exceedingly large. The enamel layer often is not evident.
Histologic Features
The most characteristic features of the disease are the marked reduction in the amount of dentin, the widening of the predentin layer, the presence of large areas of interglobular dentin, and an irregular tubular pattern of dentin. Characteristically, the reduced enamel epithelium around nonerupted teeth shows many irregular calcified bodies. Ultrastructural studies by Sapp and Gardner have proved enlightening in detailing some of the fine components of both the soft and calcified tissues in regional odontodysplasia.
Dentin Hypocalcification
Normal dentin is calcified by deposition of calcium salts in the organic matrix in the form of globules, which increase in size by further peripheral deposition of salts until all the globules are finally united into a homogeneous structure. In dentinal hypocalcification there is failure of union of many of these globules, leaving interglobular areas of uncalcified matrix. This globular dentin is easily detected in both ground sections and decalcified histologic sections of teeth, but there is no alteration in the clinical appearance.
Many clinicians believe that they can detect areas of globular dentin by the softness of the dental structure. Although this remains to be proved, it is logical that hypocalcified dentin would be softer than well-calcified dentin.
The causes of dentin hypocalcification are similar to those of environmental enamel hypocalcification and enamel hypoplasia. Obviously, any factor which interferes with normal calcification, such as parathyroid deficiency or rickets, could produce hypocalcification.
Disturbances of Growth (Eruption) of Teeth
It is recognized that a broad range of variation exists in the normal eruption times of the deciduous and permanent teeth in different persons. A valuable modification of the usually accepted chronology of the calcification and eruption times of the deciduous teeth has been published by Lunt and Law. Because of this inherent biologic variation, which is particularly notable in the human being, it is difficult to determine when the eruption dates of the teeth of a given person are outside the limits of the normal range. Nevertheless, certain cases do occur in which the eruption time is grossly beyond the extremes of normality and may be considered a pathologic state. The significance of this is frequently not apparent.
Premature Eruption
Deciduous teeth that have erupted into the oral cavity are occasionally seen in infants at birth. These are called natal teeth in contrast with neonatal teeth, which have been defined as those teeth erupting prematurely in the first 30 days of life. Usually only one or two teeth erupt early, most often the deciduous and mandibular central incisors. The etiology of this phenomenon is unknown, although in some instances it follows a familial pattern. It is well recognized in experimental animals that the secretion of several endocrine organs (e.g. thyroid, adrenals, and gonads) may alter the eruption rate of teeth, and it has been suggested that in some cases of early eruption in humans a poorly defined endocrine disturbance may be present. In cases of the adrenogenital syndrome (q.v.) developing early in life, premature eruption of teeth is sometimes seen. Most cases; however, defy explanation.
Spouge and Feasby have pointed out that prematurely erupted teeth are often well formed and normal in all respects except that they may be somewhat mobile. These teeth should be retained even though nursing difficulties may be experienced. A series of 18 cases of natal and neonatal teeth has been studied clinically and histologically by Berman and Silverstone, who reported that these were essentially normal teeth compatible with their chronologic age of development.
The premature eruption of permanent teeth is usually a sequela of the premature loss of deciduous teeth. This is best demonstrated in the situation in which only a single deciduous tooth has been lost, with subsequent eruption of the succedaneous tooth. Occasionally, cases occur involving the entire dentition, and here again the possibility of an endocrine dysfunction (e.g. hyperthyroidism) must be considered.
Eruption Sequestrum
The eruption sequestrum, an anomaly associated with the eruption of teeth in children, was first described by Starkey and Shafer.
Clinical Features
The eruption sequestrum is a tiny irregular spicule of bone overlying the crown of an erupting permanent molar, found just prior to or immediately following the emergence of the tips of the cusps through the oral mucosa. The spicule directly overlies the central occlusal fossa but is contained within the soft tissue. As the tooth continues to erupt and the cusps emerge, the fragment of bone completely sequestrates through the mucosa and is lost. For a few days, the fragment of bone may be seen lying on the crest of the ridge in a tiny depression from which it may easily be removed (Fig. 1-61A).
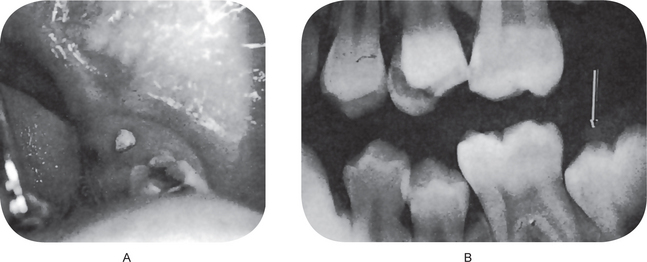
Radiographic Features
It is possible to recognize the eruption sequestrum radiographically even before the teeth begin to erupt into the oral cavity or before the bony spicule perforates the mucosa. It appears as a tiny irregular opacity overlying the central occlusal fossa but separated from the tooth itself (Fig. 1-61B).
Etiology
The explanation of this phenomenon is relatively simple. As the molar teeth erupt through the bone, they will occasionally separate a small osseous fragment from the surrounding contiguous bone, much in the fashion of a corkscrew. In most cases, this fragment probably undergoes total resorption prior to eruption. If the bony spicule is larger or eruption is fast, complete resorption cannot occur and the eruption sequestrum is observed.
Clinical Significance and Treatment
The clinical significance associated with this condition is that, occasionally, a child may complain of a slight soreness in the area, probably produced by compression of the soft tissue over the spicule during eating and just prior to its breaking through the mucosa, or by the movement of the spicule in the soft tissue crypt during mastication and following eruption through the mucosa. No treatment is necessary, since the condition corrects by itself.
Delayed Eruption
Retarded or delayed eruption of the deciduous teeth is difficult to establish unless the eruption is grossly overdue. In many cases the etiology is unknown, although in some instances it may be associated with certain systemic conditions, including rickets, cretinism, and cleidocranial dysplasia (q.v.). Local factors or circumstances may also delay eruption, as in the case of fibromatosis gingivae, in which the dense connective tissue will not permit eruption.
When the local factors can be established as the cause, their treatment may alleviate the condition. In cases of generalized or systemic disturbances in which the dental problem is of secondary importance, treatment of the primary condition, if possible, will frequently bring about tooth eruption.
Delayed eruption of the permanent dentition as a whole may be associated with the same local or systemic conditions causing the retardation of deciduous tooth eruption. Since there is a wider range of variation in the time of eruption of the permanent teeth, it is frequently difficult to state exactly when a case of retardation exists.
Multiple Unerupted Teeth
There is an uncommon condition in which there is a more or less permanently delayed eruption of teeth. The person affected may have retained his/her deciduous teeth, or more commonly the deciduous teeth may have been shed but the permanent teeth have failed to erupt. The term ‘pseudoanodontia’ is sometimes applied to this latter circumstance. In many instances, the clinical and radiographic examinations reveal apparently normal jaws and teeth. What seems to be lacking is eruptive force.
If this condition is due to an endocrine dysfunction, proper treatment may result in the eruption of teeth; if it is associated with cleidocranial dysplasia (q.v.), there is no known therapy.
Embedded and Impacted Teeth
Embedded teeth are individual teeth which are unerupted usually because of a lack of eruptive force. Impacted teeth are those prevented from erupting by some physical barrier in the eruption path. Some writers do not differentiate between the two terms and call all unerupted teeth impacted.
Lack of space due to crowding of the dental arches or to the premature loss of deciduous teeth with subsequent partial closure of the area they occupied is a common factor in the etiology of partially or completely impacted teeth (Fig. 1-62). Even more common; however, is the rotation of tooth buds resulting in teeth which are ‘aimed’ in the wrong direction because their long axis is not parallel to a normal eruption path.
Any tooth may be impacted, but certain ones are more commonly affected than others. Thus the maxillary and mandibular third molars and the maxillary cuspids are most frequently impacted, followed by the premolars and supernumerary teeth. Of the third molars, the mandibular teeth are more apt to exhibit severe impaction than the maxillary teeth.
Dachi and Howell have published the results of a study of 3,874 routine full-mouth radiographs of patients over 20 years of age. They found that 17% of these persons had at least one impacted tooth. The incidence of impaction of maxillary and mandibular third molars was 22% and 18%, respectively, while the incidence of impacted maxillary cuspids was 0.9%.
Impacted mandibular third molars
may exhibit a great variety of positions (Fig. 1-63). A simple classification of the types of impactions of mandibular third molars, based upon position, has been devised by Winter as follows:
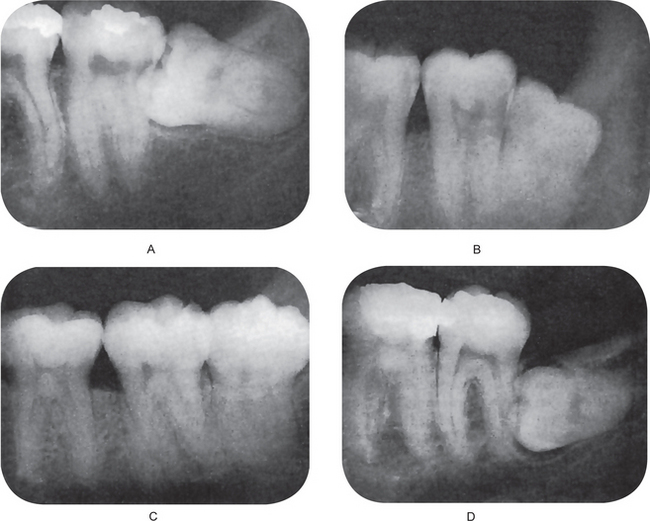
Mesioangular impaction
The third molar lies obliquely in the bone, the crown pointing in a mesial direction, usually in contact with the distal surface of the root or crown of the second molar. This is the most common type of impaction.
Distoangular impaction
The third molar lies obliquely in the bone, the crown of the tooth pointing distally toward the ramus, the roots approximating the distal root of the second molar.
Vertical impaction
The third molar is in its normal vertical position, but is prevented from erupting by impingement on the distal surface of the second molar or the anterior border of the ramus. Thus, in most cases of this type, there is simply lack of space for eruption.
Horizontal impaction
The third molar is in a horizontal position with respect to the body of the mandible, and the crown may or may not be in contact with the distal surface of the second molar crown or roots. In this type of impaction, the third molar may lie at any level within the bone from the crest of the ridge to the inferior border of the mandible.
In addition to these types of impaction in which there is variation of angulation in the sagittal plane, the impacted third molars may also be deflected either buccally or lingually in any case of the foregoing circumstances. Cases also have been recorded of complicated impactions in which the third molar is inverted, the crown pointing toward the inferior border of the mandible, or in which the third molar has been situated completely within the ramus of the mandible.
In the case of impaction of any tooth, but particularly of the mandibular third molar, it is important to determine whether the tooth is completely or only partially impacted. By definition, a completely impacted tooth is one which lies completely within the bone and has no communication with the oral cavity. A partially impacted tooth is not completely encased in bone but lies partially in soft tissue. Although there may be no obvious communication of the tooth with the oral cavity, one may exist (e.g. through a periodontal pocket on the distal of the second molar) and create an ideal situation for infection and even dental caries of the impacted tooth crown. A completely embedded or impacted tooth cannot become infected or carious.
Impacted maxillary third molars may be impacted in a manner similar to the mandibular third molar (Fig. 1-64). Thus they may show a mesioangular, distoangular, vertical, or even a horizontal position and may be deflected buccally or lingually.
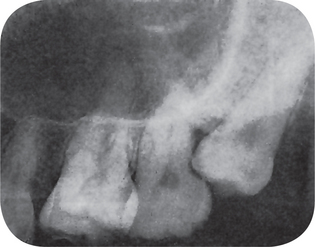
Impacted maxillary cuspids also assume a variety of positions ranging from horizontal to vertical (Fig. 1-65). In horizontally impacted cuspids the crown usually points in an anterior direction and may impinge on the roots of any of the incisors or premolars. The horizontal tooth may lie either labial or lingual to the associated teeth. The vertically impacted cuspid is usually situated between the roots of the lateral incisor and first premolar and is prevented from eruption simply by lack of space.
The treatment of an impacted tooth depends to a great extent upon the type of tooth involved and the individual circumstances. In some cases, such as the impacted cuspid, it is possible by a suitable orthodontic appliance to bring the tooth into normal occlusion. The majority of impacted teeth; however, must be surgically removed. Because of their location, impacted teeth frequently cause resorption of the roots of adjacent teeth. They may also cause periodic pain and even trismus, particularly when infection occurs around partially impacted teeth. Referred pain from impacted teeth has also been described.
A dentigerous cyst may develop around the coronal portion of an impacted tooth and may cause displacement of the tooth and destruction of bone. In the study of Dachi and Howell, 37% of impacted mandibular third molars and 15% of impacted maxillary third molars exhibited an area of radiolucency about the crown. About 10% of these radiolucencies were of such a size that they could be considered dentigerous cysts. Further more, cases of ameloblastoma have been reported developing in the wall of such a cyst (dentigerous cysts are discussed in more detail in Chapter 4 on Cysts and Tumors of Odontogenic Origin).
Occasionally, impacted teeth allowed to remain in situ may undergo resorption. The reason that some teeth are resorbed whereas others are not is unknown. The process usually begins on the crown of the tooth and results in destruction of the enamel and dentin, a well as of the cementum, with subsequent replacement by bone. Radiographically, early resorption resembles a carious lesion of the crown and has often been mistakenly called caries of an impacted tooth. Obviously, caries is impossible in a tooth that is completely impacted.
Ankylosed Deciduous Teeth (Submerged teeth)
‘Submerged’ teeth are deciduous teeth, most commonly mandibular second molars, that have undergone a variable degree of root resorption and then have become ankylosed to the bone. This process prevents their exfoliation and subsequent replacement by permanent teeth (Fig. 1-66). After the adjacent permanent teeth have erupted, the ankylosed tooth appears to have submerged below the level of occlusion. This illusion is explained by the fact that there has been continued growth of the alveolar process and also that the crown height of the deciduous tooth is less than that of the adjacent permanent teeth, so that the relative level of occlusion has been changed, not the position of the deciduous tooth. This relation between submersion and ankylosis of human deciduous molars has been studied in detail by Darling and Levers.
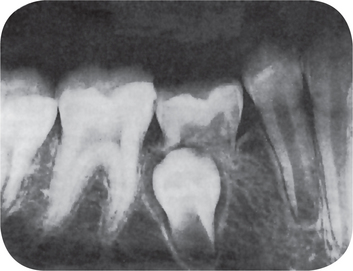
Figure 1-66 Ankylosed (submerged) deciduous second molar.
The ankylosed deciduous tooth appears submerged because of the difference in crown height between the deciduous and permanent teeth and continued growth of the alveolar process.
The diagnosis of ankylosis of a tooth is usually suspected clinically and confirmed by radiographic examination. The teeth affected lack mobility even though root resorption is far advanced. Upon percussion, an ankylosed tooth imparts a characteristic solid sound in contrast to the dull, cushioned sound of a normal tooth. Radiographically, at least partial absence of the periodontal ligament is seen, with areas of apparent blending between the tooth root and bone. The process is basically one of resorption of tooth substance and bony repair with the result that the tooth is locked in bone.
The cause of ankylosis is not known, although in some cases trauma, infection, disturbed local metabolism, or a genetic influence has been considered an important etiologic factor. These influences have been discussed by Henderson, who also emphasized that a patient who has had one or two ankylosed teeth is very likely to have other teeth ankylosed over a period of time. This condition is usually treated by the surgical removal of the ankylosed tooth to prevent the development of a malocclusion, a local periodontal disturbance, or dental caries.
Fissural (Inclusion, Developmental) Cysts of Oral Region
A number of different types of fissural (or inclusion) cysts of bone occur in the jaws and have generally been considered arising, as the name would indicate, along the lines of fusion of various bones or embryonic processes. These are true cysts (i.e. pathologic cavities lined by epithelium, usually containing fluid or semisolid material), the epithelium being derived from epithelial cells which are entrapped between embryonic processes of bones at union lines. These fissural cysts may be classified: (1) median anterior maxillary cyst, (2) median palatal cyst, (3) globulomaxillary cyst, and (4) median mandibular cyst.
There are several additional developmental cysts derived from embryologic structures or faults which involve the oral or adjacent soft tissue structures. These may be listed as: (1) nasoalveolar cyst, (2) palatal cysts of the neonate, (3) thyroglossal tract cyst, (4) benign cervical lymphoepithelial cyst, (5) epidermoid and dermoid cyst, and (6) heterotopic oral gastrointestinal cyst.
Nasopalatine Duct Cyst (Nasopalatine canal cyst, incisive canal cyst)
The most common of the nonodontogenic cyst, the nasopalatine duct cyst (NPDC) is a developmental cyst, non-neoplastic in nature. Its location is peculiar and specific in that it affects the midline anterior maxilla.
The nasopalatine ducts ordinarily undergo progressive degeneration; however, the persistence of epithelial remnants may later become the source of epithelia that gives rise to NPDC, from either spontaneous proliferation or proliferation following trauma (e.g. removable dentures), bacterial infection, or mucous retention. Genetic factors have also been suggested. The mucous glands present among the proliferating epithelium can contribute to secondary cyst formation by secreting mucin within the enclosed structure. NPDC can form within the incisive canal, which is located in the palatine bone and behind the alveolar process of the maxillary central incisors, or in the soft tissue of the palate that overlies the foramen, called the cyst of the incisive papilla.
Etiology
The cause of NPDC is essentially unknown. Trauma, infection, and mucous retention within associated salivary gland ducts have all been suggested as possible pathogenic factors; however, most believe that spontaneous cystic degeneration of residual ductal epithelium is the most likely etiology.
Clinical Features
No data currently available on the international frequency of this type of cyst. No racial predilection is known. Males are affected 18–20 times more often than females. NPDCs occur over a wide age range (8–84 year), although they also occur in fetuses. Most patients who are affected are aged 40–60 years.
Small cysts in the early stages of their development are frequently asymptomatic. Large cysts can be responsible for a variety of symptoms, including swelling (52–88%), discharge (25%), and pain (20–23%). About 70% of patients experience a combination of these symptoms. Paradoxically, patients with small cysts may have disproportionately severe symptoms, whereas patients with large ones may experience few or no symptoms. A salty taste in the mouth and devitalization of the pulps of associated teeth have been reported. Large and more destructive cysts that have perforated the labial and palatal bony plates may present as expansile, fluctuant swellings of the anterior palate and the palate. Extrabony cysts that develop within the soft tissues of the incisive papilla area of the anterior hard palate (called the cyst of the incisive papilla) may present as a translucent or bluish colored, dome-shaped swelling. The clinically apparent discoloration is due to the accumulation of fluid contents within the cyst. NPDCs clinically demonstrate slow and progressive growth, sometimes exceeding 60 mm in diameter. Tooth displacement is common finding, having been reported to occur in 78% of patients, whereas bony expansion is noted in only 1.4% of patients.
Histologic Features
Histopathologic examination discloses a cavity lined by epithelium and surrounded by a connective tissue wall. A reported 71.8% of NPDCs have squamous, columnar, cuboidal, or some combination of these epithelial types; respiratory epithelium is seen in 9.8%. The type of epithelium depends on the localization of the cyst, and it may also be reflective of the pluripotential character of the embryonic epithelial remnants. Rarely, dendritic melanocytes have been reported within the epithelium. Malignant transformation of the lining epithelium has not been reported. Often (81% of cases), a chronic inflammatory reaction consisting of lymphocytes and plasma cells is observed in the cyst wall; hemorrhage has been noted in 71% of the cases. Also helpful in the microscopic diagnosis of NPDC are the presence of structural elements in the cyst wall that are native to the nasopalatine canal (e.g. moderately sized peripheral nerves, arteries and veins, mucous glands, adipose tissue). A clear or straw-colored fluid aspirate is suggestive of NPDC; however, other cystic processes (e.g. lateral radicular cyst, cystic ameloblastoma) cannot be excluded on the basis of this finding alone. The cyst fluid has been reported to contain erythrocytes, leukocytes, desquamated epithelial cells, tissue debris, and bacteria.
Median Palatal Cyst
The median palatal cyst arises from epithelium entrapped along the line of fusion of the palatal processes of the maxilla.
Clinical Features
The median palatal cyst is located in the midline of the hard palate between the lateral palatal processes. It may become large over a prolonged period of time and produce a definite palatal swelling that is visible clinically (Fig. 1-67A). The cause the epithelial proliferation and subsequent cyst formation is unknown.
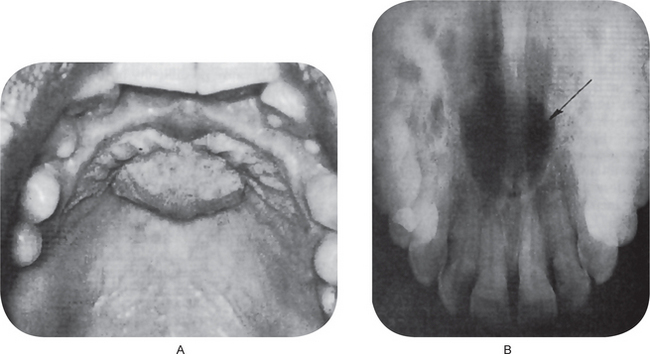
Radiographic Features
On the palatal radiograph a well-circumscribed radiolucent area is seen opposite the bicuspid and molar region, frequently bordered by a sclerotic layer of bone (Fig. 1-67B).
Histologic Features
The lining of such a cyst usually consists of stratified squamous epithelium overlying a relatively dense fibrous connective tissue band which may show chronic inflammatory cell infiltration. However, occasional cases have been reported to be lined by pseudostratified ciliated columnar epithelium or even a ‘modified’ squamous epithelium, as pointed out by Courage and his associates in a review of this lesion.
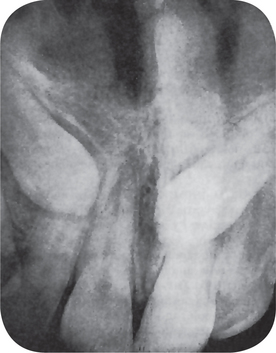
Globulomaxillary Cyst
The globulomaxillary cyst has traditionally been described as a fissural cyst found within the bone between the maxillary lateral incisor and canine teeth. Radiographically, it is a well-defined radiolucency which frequently causes the roots of the adjacent teeth to diverge. While there can be no doubt that cyst do occur in this region and that the pulps of the adjacent teeth may give positive vitality responses, there is now a considerable body of opinion against the idea that they are fissural cysts. The evidence against their being fissural cysts is, in fact, more substantial than the evidence in favor (Shear, 1996).
The WHO classification of cyst of the jaws (1992) considered this entity under the rubric ‘of debatable origin.’ We have included here a description of this cyst due to the cited reason.
The globulomaxillary cyst is found within the bone at the junction of the globular portion of the medial nasal process and the maxillary process, the globulomaxillary fissure, usually between the maxillary lateral incisor and cuspid teeth. However, there are reports of evidence that the cyst actually forms in the bone suture between the premaxilla and maxilla, the incisive suture, so that its location may be different from the cleft ridge and palate. Because of this, Ferenczy has suggested the term ‘premaxilla-maxillary cyst’ as more accurately describing its origin. The cause of the proliferation of epithelium entrapped along this line of fusion is unknown. Virtanen and Laine have carried out an extensive review and discussion of the globulomaxillary cyst.
Christ has also thoroughly evaluated the literature dealing with globulomaxillary cysts and has concluded that, embryologically, facial processes per se do not exist, and therefore, ectoderm does not become entrapped in the facial fissures of the nasomaxillary complex. Thus, he believes that this cyst should be removed from the category of orofacial fissural cysts, since modern embryologic concepts do not support such a view. Instead, he suggests that an odontogenic origin for this cyst is far more likely, the clinical and radiographic appearance being entirely compatible with a lateral periodontal, lateral dentigerous, or primordial cyst. In addition, numerous reported cases have had the histologic features of the odontogenic keratocyst (q.v.), while nests of odontogenic epithelium in the wall of globulomaxillary cysts are not rare. Furthermore, there is at least one case, reported by Aisenberg and Inman, of an ameloblastoma developing in a globulomaxillary cyst, which suggests an odontogenic origin.
Clinical Features
The globulomaxillary cyst seldom if ever presents clinical manifestations. Nearly every recorded case has been discovered accidentally during routine radiographic examination. Rarely, the cyst does become infected, and the patient may complain of local discomfort or pain in the area.
Radiographic Features
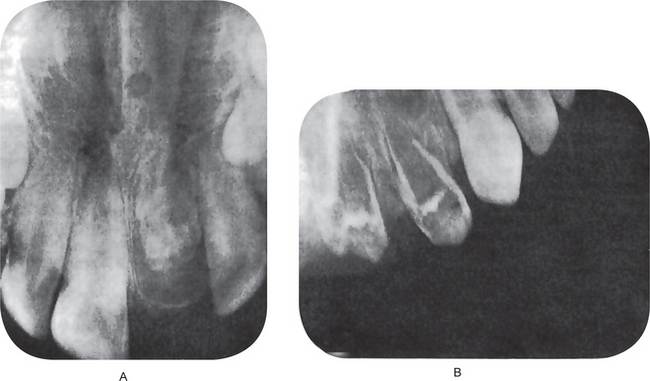
This cyst, on the intraoral radiograph, characteristically appears as an inverted, pear-shaped radiolucent area between the roots of the lateral incisor and cuspid, usually causing divergence of the roots of these teeth (Fig. 1-68A). Interestingly, there are several known cases of bilateral globulomaxillary cyst (Fig. 1-68B).
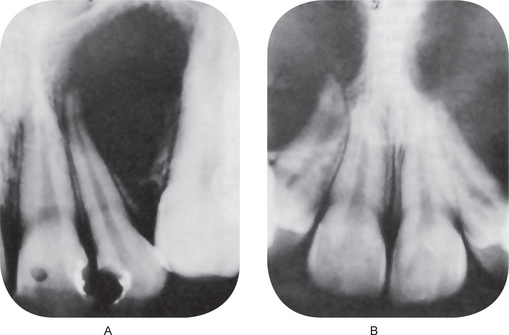
Figure 1-68 Globulomaxillary cyst.
There is a large cyst between the maxillary lateral incisor and cuspid teeth with a characteristic inverted pear shape (A). The same type of cyst may occur bilaterally (B). Note the divergence of the roots of these teeth (Courtesy of Dr Michael J Freeman and Richard Oliver)
Care must be exercised not to confuse this lesion with an apical periodontal cyst arising as a result of pulp involvement or trauma to one of the adjacent teeth. The teeth associated with a globulomaxillary cyst are vital unless coincidentally infected.
It has been emphasized recently by Wysocki, reviewing 37 cases of ‘globulomaxillary radiolucencies’ that many different types of lesions may present radiographically with features characteristic of a globulomaxillary cyst and that these must be included in any differential diagnosis of such a radiolucency in this area. He cited as examples such lesions as the periapical granuloma, apical periodontal cyst, lateral periodontal cyst, odontogenic keratocyst, central giant cell granuloma, calcifying odontogenic cyst, and odontogenic myxoma. He also concluded, in agreement with Christ, that cysts in the globulomaxillary region are odontogenic rather than fissural in origin.
Histologic Features
The globulomaxillary cyst classically has been described as being lined by either stratified squamous or ciliated columnar epithelium. However, Christ has emphasized that, in the literature, there is no accepted case of globulomaxillary cyst that is lined by pseudostratified ciliated columnar epithelium. The remainder of the wall is made up of fibrous connective tissue, usually showing inflammatory cell infiltration.
Median Mandibular Cyst
The median mandibular developmental cyst is an extremely rare lesion occurring in the midline of the mandible. It is of disputed origin. Some authorities consider it a true developmental condition originating from proliferation of epithelial remnants entrapped in the median mandibular fissure during fusion of the bilateral mandibular fissure during fusion of the bilateral mandibular arches. The possibility does exist; however, that the lesion may represent some type of odontogenic cyst such as a primordial cyst originating from a supernumerary enamel organ in the anterior mandibular segment, particularly since the bones uniting at the mandibular symphysis originate deep within the mesenchyme and thereby provide little opportunity for inclusion and subsequent proliferation of epithelial rests deep within the bone. It is also conceivable that this lesion represents a lateral periodontal cyst occurring in the midline, although the origin of this latter lesion is also obscure. The pertinent literature about this unusual type of cyst has been reviewed by White and his coworkers.
Clinical Features
Most of the median mandibular developmental cysts are clinically asymptomatic and are discovered only during routine radiographic examination. They seldom produce obvious expansion of the cortical plates of bone, and the associated teeth; unless otherwise involved, they react normally to pulp vitality tests.
Radiographic Features
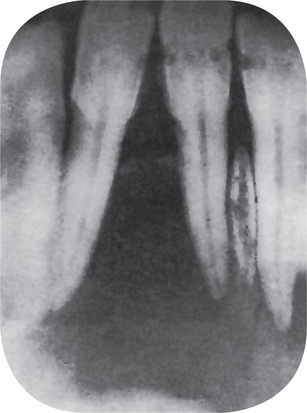
The radiographic appearance of the cyst is generally that of a unilocular, well-circumscribed radiolucency, although it may also appear multilocular (Fig. 1-69).
Nasoalveolar Cyst (Nasolabial cyst, Klestadt’s cyst)
The nasoalveolar cyst is not found within bone, but is usually described as a rare fissural cyst that may involve bone secondarily. It has been thought to arise at the junction of the globular process, the lateral nasal process, and the maxillary process as a result of proliferation of entrapped epithelium along the fusion line.
Clinical Features
The nasoalveolar cyst may cause a swelling in the mucolabial fold as well as in the floor of the nose, being located near the attachment of the ala over the maxilla. Superficial erosion of the outer surface of the maxilla may be produced by pressure of the nasoalveolar cyst, but it should be noted that they are not primarily central lesions and therefore may not be visible on the radiograph. Bilateral cases, such as reported by Brandao and his associates, are very rare.
Roed-Petersen, in a discussion of this lesion, reviewed 160 reported cases and noted that slightly over 75% of cases occurred in women. The mean age of occurrence was between 41 and 46 years, although cases have been reported in persons from 12–75 years of age.
Excellent reviews of the nasoalveolar cyst, with recapitulations of its etiology and pathogenesis, have been published by Moeller and Philipsen and by Campbell and Burkes. It has been suggested by Roed-Petersen and emphasized by Christ that this cyst probably originates from the lower anterior part of the nasolacrimal duct rather than from epithelium entrapped in the naso-optic furrow.
Palatal and Alveolar Cysts of Newborns (Epstein’s pearls, Bohn’s nodules, gingival cysts of the newborn)
A special form of odontogenic cyst is found in as many as 80% of newborn infants. Although this gingival cyst of the newborn has the microscopic appearance of an epidermoid cyst, it arises from epithelial remnants of the deeply budding dental lamina during tooth development, after the fourth month in utero, and is, therefore, discussed with the odontogenic lesions in the present text. A similar palatal cyst of the newborn is commonly found in the posterior midline of the hard palate, where it arises from epithelial remnants remaining in the stroma after fusion of the palatal processes which meet medially to form the palate. As originally described, the cysts along the median raphe of the palate were called Epstein’s pearls and the term Bohn’s nodules was used for cysts which originated from palatal gland structures and were scattered more widely over the hard and soft palates. Today these two terms are used interchangeably for both palatal and gingival cysts of newborns.
Clinical Features
Palatal cysts of the newborn typically present as multiple (usually less than six) 1–4 mm yellow-white, sessile mucosal papules of the posterior hard palate, and occasionally of the anterior soft palate. Occasional cysts are located at some distance from the midline. The cysts are usually somewhat larger and less numerous than the gingival cysts of the alveolar processes in newborns, but the two entities are, otherwise, clinically identical. Both types of cysts are so superficial that several may be ruptured at the time of examination (Fig. 1-70).
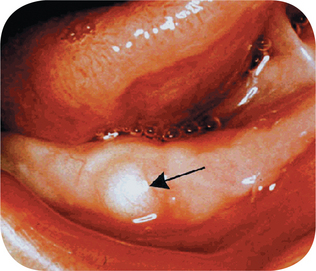
Histologic Features
Both gingival and palatal cysts of the newborn show a thin, stratified squamous epithelium cyst lining with a routine fibrovascular connective tissue stroma, usually without an inflammatory cell infiltrate. The cystic lumen is filled with degenerated keratin, usually formed into concentric layers or onion rings and the epithelium lacks rete processes. Occasional cysts will demonstrate a communication with the surface.
Treatment and Prognosis
No treatment is required for gingival or palatal cysts of the newborn. The cysts are very superficial and within weeks will rupture to harmlessly spill their contents into the oral or pharyngeal environment. The cyst lining then fuses with the overlying mucosa and becomes part of it. Occasionally, a larger cyst or a cyst situated more deeply in the submucosal stroma will remain for six to eight months before rupturing.
Thyroglossal Duct Cyst
The thyroglossal duct cyst is a rare but occasional cause of a benign midline neck mass. The cyst is usually located at the midline of the neck. The duct extends upward from the cyst with varying numbers of branches and secretory glands. These ducts or branches merge into a single duct at the level of the hyoid bone.
Thyroglossal duct cysts result from the dilatation of a remnant at the site where the primitive thyroid descended from its origin at the base of the tongue to its permanent location, low in the neck. Failure of subsequent closure and obliteration of this tract predisposes to thyroglossal cyst formation.
It most often occurs before age 20, but may be found in the older population as well.
Clinical Features
Thyroglossal duct cysts most often present with a palpable (able to be felt) asymptomatic midline neck mass at or below the level of the hyoid bone. The neck mass moves with swallowing. Some patients will have neck or throat pain, or dysphagia (difficulty in swallowing). The spectrum of clinical symptoms may be as varied.
Since the persistent duct or sinus can promote oral secretions, such cysts can become infected. Up to one half of thyroglossal cysts are not diagnosed until adult life. The tract may lie dormant for years or decades until some stimulus leads to cystic dilatation. Infection sometimes causes transient appearance of a mass or enlargement of the cyst, at times with periodic recurrences. Spontaneous drainage occurs in some instances.
Histologic Features
The thyroglossal tract cyst may be lined by stratified squamous epithelium, ciliated columnar epithelium, or intermediate transition type, since it is actually derived from cells originating from the embryonic pharyngeal floor (Fig. 1-71 A–D). With increased intracystic pressure, the cells may become flattened. The connective tissue wall of the cyst will frequently contain small patches of lymphoid tissue, thyroid tissue, and mucous glands. Interestingly, nodular collections of sebaceous glands in association with thyroglossal duct like structures of the tongue occasionally have been reported. Leider and his associates have described such a case as a sebaceous choristoma of the lingual thyroglossal duct.
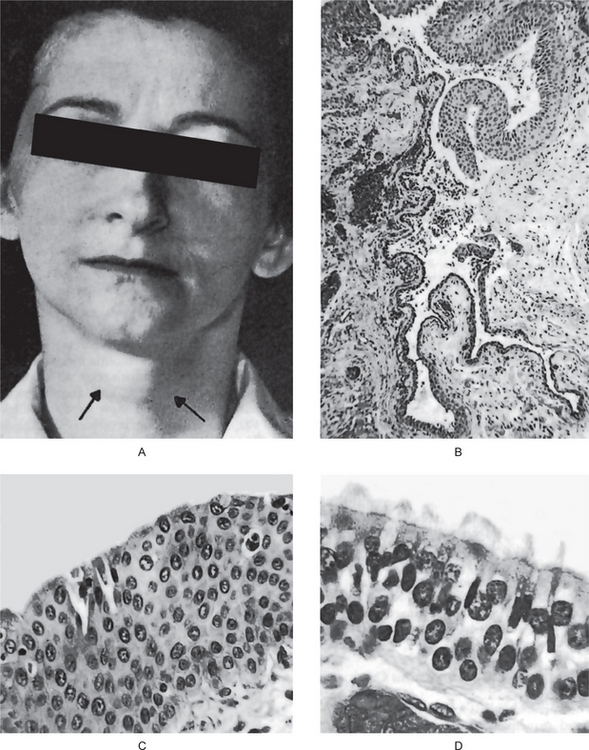
Treatment and Prognosis
Antibiotics are indicated if there is infection. Definitive surgical management requires excision not only of the cyst but also of the path’s tract and branches. The intimate association of the tract with hyoid bone mandates simultaneous removal of the central portion of the hyoid bone to ensure complete removal of the tract (Sistrunk procedure). Recurrence is unlikely after such an operation except with skin involvement and intraoperative cyst rupture.
Before thyroglossal duct cysts are excised, it is important to demonstrate that normally functioning thyroid tissue is in its usual location. Thyroid scans and thyroid function studies are ordered preoperatively.
Epidermal Inclusion Cyst (Epidermal cyst, epidermoid cyst, epithelial cyst, keratin cyst, sebaceous cyst, milia)
The terminology and nomenclature of jaw cysts varies. Epidermal inclusion cysts are the result of implantation of epidermal elements and its subsequent cystic transformation. The term epidermoid cysts is used in a general context in that, irrespective of the source of the epithelium, the term persists. Milia merely represent miniature epidermoid cysts. Sebaceous cyst is a misnomer, and the term should not be used at all because these cysts are not of sebaceous origin.
Etiology
The origin of epidermoid cyst is varied. They may form by the sequestration and implantation of epidermal rest during embryonal period, occlusion of the pilosebaceous unit, or iatrogenic or surgical implantation of epithelium into the jaw mesenchyme. HPV infection and eccrine duct occlusion may be additional factors in the development of palmoplantar epidermoid cysts. Epidermoid cysts result from the proliferation of epidermal cells within a circumscribed space of the dermis. They have been shown not to be of sebaceous origin based on the analysis of their lipid pattern, which demonstrates similarities to the epidermis. In addition, epidermoid cysts express cytokeratins 1 and 10, which are constituents of the suprabasilar layers of the epidermis. The source of this epidermis is often the infundibulum of the hair follicle. Inflammation is in part mediated by the horny material contained in epidermoid cysts. Extracts of this material have been shown to be chemotactic for polymorphonucleocytes.
Clinical Features
The epidermoid cysts are indolent in nature, slow to progress and remains asymptomatic until or unless secondarily infected. The occurrence of secondary malignancies of the types basal cell carcinoma, Bowen disease, SCC and even mycosis fungoides have been reported albeit rarely. In one study, epidermoid cysts were approximately twice as common in men as in women. Epidermoid cysts may occur at any time in life, but they are most common in the third and fourth decades of life. Gardner syndrome is an exception; the average patient age at onset is 13 years. Discharge of a foul-smelling cheese-like material is a common complaint. Less frequently, the cysts can become inflamed or infected, resulting in pain and tenderness. In the uncommon event of malignancy, rapid growth, friability, and bleeding have been reported. When located orally, the cysts can cause difficulty in feeding, swallowing, or even speaking (Fig. 1-72). Epidermoid cysts appear as firm, round, mobile, flesh-colored to yellow or white subcutaneous nodules of variable size. A central pore or punctum is an inconsistent finding that may adhere the cyst to the overlying epidermis and from which a thick cheesy material can sometimes be expressed. In individuals with dark pigmentation, epidermoid cysts may also be pigmented. In a study of Indian patients with epidermoid cysts, 63% of the cysts contained melanin pigment.
Figure 1-72 Epidermoid cyst.
Lesion appears as a symmetrical swelling in the floor of the mouth (Courtesy of Dr N Gururaj, CSI Dental College, Madurai, Tamil Nadu.
This cyst is mainly reported from sites of face, the trunk, the neck, the extremities, and the scalp. While facial involvement is also frequent in Gardner syndrome, the extremities tend to be affected more than the trunk. The ocular and oral mucosae can also be affected, and cysts have been reported on the palpebral conjunctivae, on the lips, on the buccal mucosa, under the tongue, and even on the uvula. The anterior fontanelle is an unusual location where epidermoid cysts have been found.
Certain hereditary syndromes have epidermoid cysts as part of their features. Examples include Gardner syndrome, basal cell nevus syndrome, and pachyonychia congenita. An epidermoid cyst may be a feature of pachyonychia congenita.
Histologic Features
The cystic lining is comprised of stratified squamous epithelium with glandular differentiation and is filled with desquamated keratin disposed in a laminar pattern. Dystrophic calcification and reactive foreign body reaction are seen associated with the cystic capsule. Malignant transformation of the cystic lining is noticed not infrequently.
Pigmented epidermoid cysts may demonstrate melanin pigment in the wall and a keratin mass. A surrounding infiltrate of melanocytes and melanophages may also be observed. Palmoplantar epidermoid cysts that are infected with HPV show characteristic histologic changes. The findings include intracytoplasmic eosinophilic inclusion bodies in the cyst wall, vacuolated cells and cells with condensed keratohyalin granules in the granular layer, elongated rete ridges, and vacuolated structures and parakeratotic nuclei in the keratinous mass. Structures resembling eccrine ducts are also observed in some lesions.
Treatment and Prognosis
Surgical removal seems to be the mainstay in the management protocol and recurrence is seldom noticed. Malignancies have been identified in epidermoid cysts. An unusual complication reported from an oral epidermoid cyst was sialadenitis due to pressure on the submandibular salivary duct.
Dermoid Cyst (Dermoid cystic tumor, cystic teratoma, ovarian cystic teratoma, cystic tumor of ovary, cystic tumors of omentum, congenital cyst of spine, spinal dermoid cysts)
A hamartomatous tumor containing multiple sebaceous glands and almost all skin adnexa, this may contain substances such as nails and dental, cartilage like, and bone like structures. If limited to the skin or subcutaneous tissue, dermoid cysts are thin-walled tumors that contain different amounts of fatty masses; occasionally, they contain horny masses and hairs. The origin of this cyst is probably by sequestration of skin and subsequent implantation of it along the lines of embryonic closure.
Clinical Features
Dermoid cyst occurs mostly on the face, neck, or scalp. In addition to the skin, dermoid cysts can be intracranial, intraspinal, or perispinal. Intra-abdominal cysts, such as cystic tumors of the ovary or omentum, occur as well. No racial predilection appears to exist; however, most cases of dermoid cysts in the literature occurred in whites. Dermoid cysts of the ovary or omentum are gender restricted, that is, they occur only in the female population. In other dermoid cysts, no gender predilection has been found. Dermoid cysts are described in persons of all ages. Intracranial or perispinal dermoid cysts are most often found in infants, children, or young adolescents. Intra-abdominal dermoid cysts are described in females aged 15–40 years. For example, cystic teratoma is a relatively rare tumor that most often occurs in females aged 15–40 years. Most dermoid cysts on the floor of the mouth occur in individuals aged 10–30 years. Only a few cases describe oral dermoid cysts in newborns or children. Dermoid cysts that are congenital and localized on the neck, head, or trunk are usually visible at birth. Occasionally, they occur on the neck or in a midline region. When on the head, dermoid cysts are often adherent to the periosteum. The usual diameter of the lesions is 1–4 cm. In many patients, dermoid cysts occur on the floor of the mouth or elsewhere in the mouth. Three subclasses of congenital mouth cysts are described in the literature: epidermoid (simple) cysts, dermoid (complex) cysts, and teratoid (complex) cysts. Most of these lesions occur in individuals aged 10–30 years. Other rare dermoid cysts in the oral cavity are those on the tongue. As of early 2000, 17 patients with intralingual dermoid cysts are described in the English-language literature. All cases occurred in young patients.
Histologic Features
In contrast to epidermal cysts, dermoid cysts in the skin are lined by an epidermis that possesses various epidermal appen dages. As a rule, these appendages are fully mature. Hair follicles containing hairs that project into the lumen of the cyst are often present. The dermis of dermoid cysts usually contains sebaceous glands; eccrine glands; and, in many patients, apocrine glands. Occasionally, the lining epithelium may proliferate as papillary boundaries extend externally or inward toward the lumen of the cyst. This proliferation may have some superficial resemblance to epidermal carcinomatous proliferation, and the growth may be misdiagnosed as a cancer.
Heterotopic Oral Gastrointestinal Cyst
Heterotopic islands of gastric mucosa have been found in the esophagus, small intestine, thoracic cysts, omphalomesenteric cysts, pancreas, gall bladder, and Meckel’s diverticulum, according to the review by Gorlin and Jirasek. In addition, Harris and Courtemanche cited at least 17 cases of cysts lined by gastric or intestinal mucosa occurring in the oral cavity, in the tongue, or in the floor of the mouth, probably from misplaced embryonal rests.
Clinical Features
This choristomatic cyst can be found in patients at any age, although the majority have been infants or young children. It may be significant that the lesion occurs overwhelmingly in males.
The cyst presents as a small nodule entirely within the body of the tongue, either anterior or posterior, or in the floor of the mouth, in the neck, or adjacent to the submaxillary gland. It may be asymptomatic or may cause difficulty in eating or speaking. Some cysts communicate with the surface mucosa by a tube or duct like structure.
Histologic Features
This cyst is usually lined partly by stratified squamous epithelium and partly by gastric mucosa similar to that found in the body and fundus of the stomach, with both parietal and chief cells. Sometimes intestinal epithelium is found, including Paneth, goblet and argentaffin cells. A muscularis mucosa may or may not be present.
References
Aagaard, A., Godiksen, G., Teglers, P.T. Comparison between new saliva stimulants in patients with dry mouth: a placebo-controlled double blind crossover study. J Oral Pathol Med. 1992; 21:376–380.
Abe, H., Ohnishi, T., Watanabe, S. Does plantar epidermoid cyst with human papillomavirus infection originate from the eccrine dermal duct? Br J Dermatol. 1999; 141(1):161–162. [Jul].
Abell, M.R. Lymphoid hamartoma. Radiol Clin North Am. 1968; 6:15.
Aberg, T., Wozney, J., Thesleff, I. Expression patterns of bone morphogenetic proteins (BMPs) in the developing mouse tooth suggest roles in morphogenesis and cell differentiation. Developmental Dynamics. 1997; 210:383–396.
Abrams, A.M., Howell, F.V., Bullock, W.K. Nasopalatine cysts. Oral Surg. 1963; 16:306–332.
Acree, T., Abreo, F., Smith, B.R. Diagnosis of dermoid cyst of the floor of the mouth by fine-needle aspiration cytology: a case report. Diagn Cytopathol. 1999; 20(2):78–81.
Adebajo, A.O., Crisp, A.J., Nicholls, A., Hazleman, B.L. Localized scleroderma and hemiatrophy in association with antibodies to double-stranded DNA. Postgrad Med J. 1992; 68:216–218.
Agris, J. Autologous fat transplantation: a 3-year study. Am J Cosm Surg. 1987; 4:95–102.
Aisenberg, M.S., Inman, B.W. Ameloblastoma arising within a globulomaxillary cyst. Oral Surg. 1960; 13:1352.
AJR. Castleman’s disease of the lung: radiographic, high-resolution CT, pathologic findings. 1996; 167:1055–1056. [(No abstract available)].
Akasaka, T., Imamura, Y., Kon, S. Pigmented epidermal cyst. J Dermatol. 1997; 24(7):475–478. [Jul].
Akira, K., Kitamura, J. Clinical report of a case of globulomaxillary cyst. Oral Surg. 1952; 5:705.
Akita, S., Hirano, A., Fujii, T. Recurrent, discharging congenital frontotemporal dermoid cyst. Ann Plast Surg. 2000; 44(4):465–466. [Apr].
Alexander, R.W., James, R.B. Melkersson-Rosenthal syndrome: review of literature and report of case. J Oral Surg. 1972; 30:599.
Alioglu, Z., Caylan, R., Adanir, M., Ozmenoglu, M. Melkersson-Rosenthal syndrome: report of three cases. Neurol Sci. 2000; 21(1):57–60. [Feb].
Allard, R.H., van der Kwast, W.A., van der Waal, I. Nasopalatine duct cyst: review of the literature and report of 22 cases. Int J Oral Surg. 1981; 10(6):447–461. [Dec].
Allen, C.M., Camisa, C., Hamzeh, S., Stephens, L. Cheilitis granulomatosa: report of six cases and review of the literature. J Am Acad Dermatol. 1990; 23(3 Pt 1):444–450. [Sep].
Allen, C.M. Animal models of oral candidiasis: a review. Oral Surg Oral Med Oral Pathol. 1994; 78:216–221.
Aloi, F., Tomasini, C., Pippione, M. Mycosis fungoides and eruptive epidermoid cysts: a unique response of follicular and eccrine structures. Dermatology. 1993; 187(4):273–277.
Aloi, F.G., Pippione, M. Molluscum contagiosum occurring in an epidermoid cyst. J Cutan Pathol. 1985; 12(2):163–165. [Apr].
Alvares, L.C., de Souza Freitas, J.A. Hypoplasia and hypocalcification of enamel: report of a case. Oral Surg Oral Med Oral Pathol. 1969; 28(1):73–75.
Amaral, W.J., Jacobs, D.S. Aberrant salivary gland defect in the mandible: report of a case. Oral Surg Oral Med Oral Pathol. 1961; 14:748–752.
American Journal of Human Genetic. 1990; 46:120–125.
Amos, E.R. Incidence of the small dens in dente. J Am Dent Assoc. 1955; 51:31.
Andersen, W.K., Rao, B.K., Bhawan, J. The hybrid epidermoid and apocrine cyst: a combination of apocrine hidrocystoma and epidermal inclusion cyst. Am J Dermatopathol. 1996; 18(4):364–366. [Aug].
Andlaw, R.J., Rock, W.P. A Manual of Paediatric Dentistry, 4, Edinburg: Churchill Livingstone; 1996:141–148.
Angle, E.H. Classification of malocclusion. Dent Cosmos. 1899; 41:284.
Ankyloglossia, Pediatr Surg Update. 1996;6(1):1.
Anneroth, G., Hall, G., Stuge, U. Nasopalatine duct cyst. Int J Oral Maxillofac Surg. 1986; 15(5):572–580.
Anonymous. Saliva: its role in health and disease. Working Group 10 of the commission on oral health, research and epidemiology. Int Dent J. 1992; 42:287–304.
Arafat, A., Brannon, R.B., Ellis, G.L. Adenomatoid hyperplasia of mucous salivary glands. Oral Surg. 1981; 52:51.
Archard, H.O., Heck, J.W., Stanley, H.R. Focal epithelial hyperplasia: an unusual and mucosal lesion found in Indian children. Oral Surg Oral Med Oral Pathol. 1965; 20:201–212.
Ariji, E., Fujiwara, N., Tabata, O. Nakayama E et al. Stafne’s bone cavity. Oral Surg Oral Med Oral Pathol. 1993; 76:375–380.
Ariji, E., Tabata, O., Kanda, S. CT imaging of the so-called Stafne’s bone cavity. J Jap Stomatol Soc. 1987; 37:303–315.
Arnold, M., Geilen, C.C., Coupland, S.E. Unilateral angiolymphoid hyperplasia with eosinophilia involving the left arm and hand. J Cutan Pathol. 1999; 26(9):436–440. [Oct].
Arons, M.S., Solitaire, G.B., Grunt, J.A. The macroglossia of Beckwith’s syndrome. J Plast Reconstr Surg. 1970; 45:341.
Ascher, K.W. Das syndrom blepharochalasis, Struma and Doppellippe Klin Wochenschr. 1922; 1:2287.
Ashley, F.L., Braley, S., McNall, E.G. The current status of silicone injection therapy. Surg Clin North Am. 1971; 51:501–509.
Ashley, F.L., Thompson, D.P., Henderson, T. Augmentation of surface contour by subcutaneous injections of silicone fluid. Plast Reconstr Surg. 1973; 57:8–13.
Asken, S. A Manual of Liposuction Surgery and Autologous Fat Transplantation under Local Anesthesia. In Terry and Associates, 2, Irvine: Keith C; 1986:119–157.
Awang, M.N., Siar, C.H. Dentigerous cyst due to mesiodens: report of two cases. J Dent Assoc. 1989; 35:117–118.
Ayhan A, Tuncer ZS, Bilgin F, Kucukali T. Squamous cell carcinoma arising in dermoid cyst. Eur J Gynaecol Oncol, (2): 144–47, Oct, 7.
Bäckman, B., Anneroth, G., Horstedt, P. Amelogenesis imperfecta: a scanning electron microscopic and microradiographic study. J Oral Pathol Med. 1989; 18(3):140–145.
Bäckman, B., Anneroth, G., Horstedt, P. Microradiographic study of amelogenesis imperfecta. Scand J Dental Res. 1989; 97(4):316–329.
Bäckman, M.D., Singer, A. Demonstration of the lyon hypothesis in X-linked dominant hypoplastic amelogenesis imperfecta. Birth Defects: Original Article Series. 1971; 7(7):204–209.
Balchsmiede G. Disseration (1920). In Steidler NE, Radden BG, Reade PC. Dentinal dysplasia; a clinicopathological study of eight cases and review of literature. Br J Oral Maxillofac Surg, 22: 274–86, 1984.
Ball, S.P., Cook, P.J.L., Mars, M., Buckton, K.E. Linkage between dentinogenesis imperfecta and Gc. Ann Hum Genet. 1982; 46:35–40.
Ballschmiede. Dissertation, Berlin, 1920. In: Herbet E.H., Apffelstaedt M., eds. Malformations of the Jaws and Teeth. New York: Oxford University Press, 1930.
Banoczy, J., Szabo, L., Csiba, A. Migratory glossitis. Oral Surg. 1975; 39:113.
Barrett, A.W. Oral melanotic macules that develop after radiation therapy. Oral Surg Oral Med Oral Pathol. 1994; 77:431–434.
Bart, R.S., Kopf, A.W. Hemifacial atrophy. J Dermatol Surg Oncol. 1978; 4:12–14.
Bartels, H.A., Maillard, E.R. Hairy tongue. Oral Surg. 1955; 8:659.
Bartholomew, L.G., Moore, C.E., Dahlin, D.C., Waugh, J.M. Intestinal polyposis associated with mucocutaneous pigmentation. Surg Gynecol Obstet. 1962; 115:1.
Baughman, R.A. Median rhomboid glossitis: a developmental anomaly? Oral Surg Oral Med Oral Pathol. 1971; 31:56–65.
Baughman, R.A., Heidrich, P.D., Jr. The oral hair: an extremely rare phenomenon. Oral Surg. 1980; 49:530.
Baughman, R.A. Median rhomboid glossitis: a developmental anomaly? Oral Surg. 1971; 31:56.
Beighton, P.The Ehlers-Danlos syndrome. London: Heinemann, 1970.
Beighton, P., Thomas, M.L. The radiology of the Ehlers Danlos syndrome. Clin Radiol. 1969; 20:354–361.
Benjamin, B., Weiner, H. Syndrome of cutaneous fragility and hyper elasticity and articular hyper laxity. Am J Dis Child. 1943; 65:246–257.
Berendt, H.C. Report of seven cases of anodontia partialis. Oral Surg. 1950; 3:1435.
Berman, D.S., Silverstone, L.M. Natal and neonatal teeth. Br Dent J. 1975; 139:361.
Berman, F.R., Fay, J.T. The retrocuspid papilla: a clinical survey. Oral Surg. 1976; 42:80.
Bertram, U.. Xerostomia: Acta Odontol Scand. 1967;25(Suppl 4).
Bhaskar, S.N., Bernier, J.L. Histogenesis of branchial cysts: a report of 468 cases. Am J Pathol. 1959; 35:407.
Bhaskar, S.N. Lymphoepithelial cysts of the oral cavity. Oral Surg. 1966; 21:120.
Bhatia, S.N. Genetics of cleft lip and palate. Br Dent J. 1972; 132:95.
Bhatt, A.P., Dholakia, H.M. Radicular variety of double dens invaginatus. Oral Surg. 1975; 39:284.
Biesecker, L.G., Peters, K.F., Darling, T.N., Hill, C.P. Clinical differentiation between Proteus syndrome and hemihyperplasia: description of a distinct form of hemihyperplasia. Am J Med Genet. 1998; 79:311–318.
Billings, R.J. Studies on the prevalence of xerostomia: preliminary results. Caries Res. 23, 1989. [Abstract 124, 35th ORCA Congress].
Bixler, D., Conneally, P.M., Cristen, A.G. Dentinogenesis imperfecta: genetic variations in a six-generation kindred. J Dent Res. 1969; 48:1196–1199.
Bixler, D. Heritability of clefts of the lip and palate. J Prosthet Dent. 1975; 33:100.
Bjornstrom, M., Axell, T., Birkhed, D. Comparison between saliva stimulants and saliva substitutes in patients with symptoms related to dry mouth: a multi-centre study. Swed Dent J. 1990; 14:153–161.
Black, G.V., McKay, F.S. Mottled teeth: an endemic developmental imperfection of the enamel of the teeth heretofore unknown in the literature of dentistry. Dent Cosmos. 1916; 58:129.
Blattner, R.J., Heys, F., Robinson, H.B.G. Osteogenesis imperfecta and odontogenesis imperfecta. J Dent Res. 1942; 21:325.
Blinder, D., Yahatom, R., Taicher, S. Oral manifestations of sarcoidosis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997; 83(4):458–461.
Blumberg, J.E., Hylander, W.L., Goepp, R.A. Taurodontism: a biometric study. Am J Phys Anthropol. 1971; 34:243.
Boardman, L.A., Thibodeau, S.N., Schaid, D.J. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med. 1998; 128(11):896–899.
Bodecker, C.F. Enamel hypoplasia. J Dent Res. 1941; 20:447.
Bodin, I., Isacsson, G., Julin, P. Cysts of the nasopalatine duct. Int J Oral Maxillofac Surg. 1986; 15(6):696–706.
Boerger, W.G., Waite, D.E., Carroll, G.W. Idiopathic bone cavities of the mandible: a review of the literature and report of case. J Oral Surg. 1972; 30:506.
Bohn H. Die Mundkrankheiten der Kinder Leipzig, W Engelmann, 1866.
Bonilla, J.A., Szeremeta, W., Yellon, R.F., Nazif, M.M. Teratoid cyst of the floor of the mouth. Int J Pediatr Otorhinolaryngol. 1996; 38(1):71–75. [Dec].
Boone, C.G. Nasoalveolar cyst. Oral Surg. 1955; 8:40.
Bouquot, J.E., Gundlach, K.K.H. Odd tongues: the prevalence of common tongue lesions in 23, 616 white Americans over 35 years of age. Quint Internat. 1986; 17:719–730.
Bouquot, J.E. Common oral lesions found during a mass screening examination. J Am Dent Assoc. 1986; 112(1):50–57.
Brandao, G.S., Ebling, H., Faria e Souza, I. Bilateral nasolabial cyst. Oral Surg. 1974; 37:480.
Braunschweig, I.J., Stein, I.H., Dodwad, M.I.M., Rangwala, A.F. Case report: 751: spindle cell lipoma causing marked bone erosion. Skeletal Radiol. 1992; 21:414–417.
Brekhus, P.J., Oliver, C.P., Montelius, G. A study of the pattern and combinations of congenitally missing teeth in man. J Dent Res. 1944; 23:117.
Brocq, L., Pautrier, L.M. Glossite losangue mediane de la face dorsale de la langue. Ann Derm Syph (Paris). 1914; 5:1–18.
Brondsted, K., Liisberg, W.B., Orsted, A. Surgical and speech results following palatopharyngoplasty operations in Denmark, 1959–77. Cleft Palate J. 1984; 21(3):170–179.
Bronstein, I.P., Abelson, S.M., Jaffe, R.H., von Bonin, G. Macroglossia in children. Am J Dis Child. 1937; 54:1328.
Brook, A.H. Dental anomalies of number, form and size: their prevalence in British school children. J Int Assoc Dent Child. 1974; 5:37–53.
Brown, F.H., Houston, G.D. Smoker’s melanosis: a case report. J Periodontol. 1991; 62:524–527.
Brown, R.S., Krakow, A.M. Median rhomboid glossitis and a kissing lesion of the palate. Oral Surg Oral Med Oral Pathol Oral Radiol Endodont. 1996; 82:472–473.
Brucker, M. Studies on the incidence and cause of dental defects in children II: Hypoplasia. J Dent Res. 1943; 22:115.
Buchner, A., Hansen, L.S. Lymphoepithelial cysts of the oral mucosa. Oral Surg Oral Med Oral Pathol. 1980; 50:441–449.
Buchner, A., Hansen, L.S. Melanotic macule of the oral mucosa: a clinicopathologic study of 105 cases. Oral Surg Oral Med Oral Pathol. 1979; 48:244–249.
Buchner, A., Silverman, S., Wara, W.M., Jr., Hansen, L.S. Angiolymphoid hyperplasia with eosinophilia (Kimura’s disease). Oral Surg. 1980; 49:309.
Buckerfield, J.B., Edwards, M.B. Angiolymphoid hyperplasia with eosinophils in oral mucosa. Oral Surg. 1979; 47:539.
Burdick, D., Prior, J.T., Scanlon, G.T. Peutz-Jeghers syndrome: a clinical-pathological study of a large family with a 10-year follow-up. Cancer. 1963; 16:854.
Burket, L.W. Nasopalatine duct structures and peculiar bony pattern observed in the anterior maxillary region. Arch Pathol. 1937; 23:793–800.
Burket, L.W. Histopathologic studies in congenital syphilis. Int J Orthod Oral Surg. 1937; 23:1016.
Cabrini, R.L., Barros, R.E., Albano, H. Cysts of the jaws—a statistical analysis. J Oral Surg. 1970; 28(7):485–489.
Calabro, F., Capellini, C., Jinkins, J.R. Rupture of spinal dermoid tumors with spread of fatty droplets in the cerebrospinal fluid pathways. Neuroradiology. 2000; 42(8):572–579.
Calista, D. Congential lower lip pits. Pediatr Dermatol. 2002; 19(4):363–364.
Calman, H.I. Nasoalveolar cyst. New York Dent J. 1954; 20:320.
Cam, Y., Neumann, M.R., Oliver, L., Janet, R.D. Immunolocalization of acidic and basic growth factors during mouse odontogenesis. Intern J Develop Biolo. 1992; 36:381–398.
Campbell, R.L., Burkes, E.J., Jr. Nasolabial cyst: report of case. J Am Dent Assoc. 1975; 91:1210.
Carlos, R., Sedano, H.O. Multifocal papilloma virus epithelial hyperplasia. Oral Surg Oral Med Oral Pathol. 1994; 77:631–635.
Carrol, M.K.O., Duncan, W.K., Perkins, T.M. Dentin dysplasia: review of literature and a proposed subclassification based on radiographic findings. Oral Surg. 1991; 72:119–125.
Carroll, C.M., Gaffney, R., McShane, D. Congenital nasal dermoids in children. J Med Sci. 1997; 166(3):149–151. [Jul-Sep].
Cataldo, E., Berkman, M.D. Cysts of the oral mucosa in newborns. Am J Dis Child. 1968; 116:44.
Cervenka, J., Gorlin, R.J., Anderson, V.E. The syndrome of pits of the lower lip and/or cleft palate. Am J Hum Genet. 1967; 19:416.
Chan, J.K.C., Hui, P.K., Ng, C.S. Epithelioid haemangioma (angiolymphoid hyperplasia with eosinophilia) and Kimura’s disease in Chinese. Histopathology. 1989; 15:557–574.
Chaudhry, A.P. A clinicopathologic study of intraoral lymphoepithelial cysts. J Oral Med. 1984; 39:79–84.
Chaudhry, A.P., ON, Johnson, Mitchell, D.F., Gorlin, R.J. Hereditary enamel dysplasia. J Pediatr. 1959; 54:776.
Choukas, N.C. Developmental submandibular gland defect of the mandible: review of the literature and report of two cases. J Oral Surg. 1973; 31:209–211.
Christ, T.F. The globulomaxillary cyst: an embryologic misconception. Oral Surg. 1970; 30:515.
Christen, A.G., Swanson, B.Z., Jr. Oral hygiene: a history of tongue scraping and brushing. J Am Dent Assoc. 1978; 96(2):215–219. [Feb].
Cohen, D.M., Green, J.G., Diekmann, S.L. Concurrent anomalies: cheilitis glandularis and double lip: report of a case. Oral Surg Oral Med Oral Pathol. 1988; 66(3):397–399. [Sep].
Cohen, M.M., Jr. Syndromes with cleft lip and cleft palate. Cleft Palate J. 1978; 15(4):306–328. [Oct].
Cohlon, S.Q. Excessive intake of vitamin A as a cause of congenntal anomalies in the rat. Science. 1953; 117:535.
Colby, R.A., Kerr, D.A.Pathologic Physiology of Oral Disease. St Louis: CV Mosby, 1959.
Colby, Ra, Kerr, D.A.Color Atlas of Oral Pathology. Phidelphia: JB Lippincott, 1956.
Collins, N.P., Edgerton, M.T. Primary branchiogenic carcinoma. Cancer. 1959; 12:235.
Compagno, J., Hyams, V.J., Safavian, M. Does branchiogenic carcinoma really exist? Arch Pathol Lab Med. 1976; 100:311.
Conneally, P.M., Bixler, D., Horton-Kelly, S., Daugherty, L. Confirmation of linkage between dentinogenesis imperfecta and GC. (Abstract) Cytogenet. Cell Genet. 1984; 37:438.
Connor, M.S. Anterior lingual mandibular bone concavity. Oral Surg. 1976; 48:113.
Conway, H., Jerome, A.P. The surgical treatment of branchial cysts and fistuals. Surg Gynecol Obstat. 1955; 101:621.
Conway, H.C., Wagner, K.J. Congenital anomalies of the head and neck. Plast Reconstr Surg. 1965; 36:71.
Cooke, B.E.D. Median rhomboid glossitis and benign glossitis migrans (geographical tongue). Br Dent J. 1962; 112:389.
Corney, G., Ball, S., Noades, J.E. Linkage studies on dentinogenesis imperfecta (DGI1). (Abstract) Cytogenet. Cell Genet. 1984; 37:439.
Correll, R.W., Jensen, J.L., Rhyne, R.R. Lingual cortical mandibular defects. Oral Surg. 1980; 50:287.
Courage, G.R., North, A.F., Hansen, L.S. Median palatine cysts. Oral Surg. 1974; 37:745.
Crawford, J.L. Concomitant taurodontism and amelogenesis imperfecta in the American caucasian. J Dent Child. 1970; 37:171.
Crosby, A.H., Edwards, S.J., Murray, J.C., Dixon, M.J. Genomic organization of the human osteopontin gene: exclusion of the locus from a causative role in the pathogenesis of dentinogenesis imperfecta type II. Genomics. 1995; 27:155–160.
Crosby, A.H., Scherpbier-Heddema, T., Wijmenga, C., Altherr, M.R. Genetic mapping of the dentinogenesis imperfecta type II locus. Am J Hum Genet. 1995; 57:832–839.
Crovetto de la Torre, M., Santolaya Jimenez, J., Urunuela Bernado, J. Agenesis of the major salivary glands and ectodermal dysplasia. Revue Laryngol Otol Rhinol. 1985; 106:91–93.
Dachi, S.F., Howell, F.V. A survey of 3,874 routine full-mouth radiographs II: a study of impacted teeth. Oral Surg. 1961; 14:1165.
Dado, D.V. Experience with the functional cleft lip repair. Plast Reconstr Surg. 1990; 86(5):872–881.
Damante, J.H., Camarini, E.T., Silver, M.S. Lingual mandibular bone defect: a developmental entity. Dentomaxillofac Radiol. 1998; 27:58.
Darling, A.I., Levers, B.G.H. Submerged human deciduous molars ankylosis. Arch Oral Biod. 1973; 18:1021.
Darling, A.I. Some observations on amelogenesis imperfecta and calcification of the dental enamel. Proc R Soc Med. 1956; 49:759.
Darwazeh, A.M., Pillai, K., Prevalence of tongue lesions in 1013 Jordanian dental outpatients. Community Dent Oral Epidemiol 1993;(5):232–324. [Oct, 21].
Davis, W.B. Reconstruction of hemiatrophy of the face: case report. Plastic Reconstr Surg. 1968; 42:489–491.
Davis, W.B. Congenital deformities of the face. Surg Gynecol Obstet. 1935; 61:201.
Dawes, C. Factors influencing salivary flow rate and composition. In: Edgar W.M., O’Mullane D.M., eds. Saliva and Oral Health. London: Br Dent Assoc; 1996:27–41. [(eds)].
Dawes, c, Macpherson, L.M.D. Effects of nine different chewing gums and lozenges on salivary flow rate and pH. Caries Res. 1992; 26:176–182.
de Jong, A.L., Park, A., Taylar, G., Forte, V. Lipoma of the head and neck in children. Int J Pediatr Otolaryngol. 1998; 43:53–60.
Dean, H.T. Chronic endemic dental flourosis. J Am Med Assoc. 1936; 107:1269.
Delbalso, A.M. Maxillofacial imaging, 1, Philadelphia: WB Saunders; 1990:323–324.
Di Biase, D.D. The effects of variations in tooth morphology and position on eruption. Dent Pract Dent Rec. 1971; 22:95–108.
Diamond, M., Weinmann, J.P.The Enamel of Human Teeth. New York: Columbia University Press, 1940.
Dilley, D.C., Siegel, M.A., Budnick, S. Diagnosing and treating common oral pathologies. Pediatr Clin North Am. 1991; 38(5):1227–1264.
Dinnerman, M. Vitamin A deficiency in unerupted teeth of infants. Oral Surg. 1951; 4:2014.
Dionisopoulos, T., Williams, H.B. Congenital anomalies of the Ear, Nose and Throat. New York: Oxford University Press, 1997; 243–262.
Dobrin, P.B., Schwarcz, T.H., Baker, W.H. Mechanisms of arterial and aneurysmal tortuosity. Surgery. 1988; 104:568–571.
Doku, H.C., Shklar, G., McCarthy, P.L. Cheilitis glandularis. Oral Surg. 1951; 4:1024.
Dolder, E. Deficient dentition. Dent Record. 1937; 57:142.
Donta, A.N., Lampadakis, J., Pilalitos, P., Spyropoulos, N.D. Findings from the clinical examination of the oral cavity of one hundred drug addicts. Hell Stomatol Chron. 1989; 33(2):101–105. [Apr-Jun].
Downs, W.G. Studies in the causes of dental anomalies. J Dent Res. 1928; 8:367.
Doyle, J.L., Weisinger, E., Manhold, J.H., Jr. Benign lymphoid lesions of the oral mucosa. Oral Surg. 1970; 29:31.
Dummer, P.M.H., Kingdon, A., Kingdon, R. Distribution of developmental defects of tooth enamel by tooth-type in 11–12-year-old children in South Wales. Community Dent Oral Epidemiol. 1986; 14:341–344.
Dummett, C.O., Barens, G. Oromucosal pigmentation: an updated literary review. J Periodontol. 1971; 42:726.
Duncan, B.R., Dohner, V.A., Priest, J.H. The Gardner syndrome: need for early diagnosis. J Pediatr. 1968; 72:497.
Durham, D.G. Cutis hyperelastica (Ehlers-Danlos syndrome). Arch Ophtalmol. 1952; 49:220–221.
Ehlers, F.E., Bundick, W.R. Cutaneous elasticity and hyper elasticity. Dermatology. 1956; 74:22–32.
Eidelman, E., Chosack, A., Cohen, T. Scrotal tongue and geographic tongue: polygenic and associated traits. Oral Surg Oral Med Oral Pathol. 1976; 42(5):591–596. [Nov].
el-Bardaie, A., Nikai, H., Takata, T. Pigmented nasopalatine duct cyst: report of 2 cases. Int J Oral Maxillofac Surg. 1989; 18(3):138–139. [Jun].
Elder, D., Elenitsas, R., Jaworsky, C., Johnson, B., Jr., Lever’s Histopathology of the skin. 8. Lippincott-Raven, Philadelphia, 1997.
Ellenbogen, R. Free autogenous pearl fat grafts in face a preliminary report of a rediscovered technique. Ann Plast Surg. 1986; 16:179–185.
El-Najjar, M.Y., DeSanti, M.V., Ozebek, L. Prevalence and possible etiology of dental enamel hypoplasia. Am J Phys Anthropol. 1978; 48(2):185–192.
Emerson, T.G. Hereditary gingival hyperplasia: a family pedigree of four generations. Oral Surg. 1965; 19:1.
Epstein, A. Ueber Epithelperlen in der Mundhohle Neugeborener Kinder Ztsch fur Heilkunde. 1880; 1:59.
Espelid, M. Geographic stomatitis: report of 6 cases. J Oral Pathol Med. 1991; 20:425–428.
Ettinger, R.L., Manderson, R.D. A clinical study of sublingual varices. Oral Surg. 1974; 38:540.
Everett, F.G., Holder, T.D. Cheilitis glandularis apostematosa. Oral Surg. 1955; 8:405.
Everett, F.G., Wescott, W.B. Commissural lip pits. Oral Surg. 1961; 14:202.
Eveson, J.W., Lucas, R.B. Angiolymphoid hyperplasia with eosinophilia. J Oral Path. 1979; 8:103.
Fader, M., Kline, S.N., Spatx, S.S., Zubrow, H.J. Gardner’s syndrome (intestinal polyposis, osteomas, sebaceous cysts) and a new dental discovery. Oral Surg. 1962; 15:153.
Fainstat, T. Cortisone-induced congential cleft palate in rabbits. Endocrinology. 1954; 55:502.
Fanibunda, K., Matthews, J.N.S. The relationship between accessory foramina and tumor spread on the medial mandibular surface. J Anat. 2001; 196:23–29.
Fara, M. The Musculature of Cleft Lip and Palate. In: McCarthy J.G., ed. Plastic Surgery. Philadelphia: WB Saunders; 1990:2598–2626. [(4)].
Farman, A.G. Hairy tongue (lingua villosa). J Oral Med. 1977; 32(3):85–91. [Jul-Sep].
Farman, A.G., van Wyk, C.W., Stax, J., Hugo, M. Central papillary atrophy of the tongue. Oral Surg. 1977; 43:48.
Farman, A.G. Atrophic lesions of the tongue: a prevalence study among 175 diabetic patients. J Oral Path. 1976; 5:255.
Ferenczy, K. The relationship of globulomaxillary cysts to the fusion of embryonal processes and to cleft plate. Oral Surg. 1958; 11:1388.
Fetsch, J.F., Weiss, S.W. Observations concerning the pathogenesis of epithelioid hemangioma (angiolymphoid hyperplasia). Mod Pathol. 1991; 4(4):449–455. [Jul].
Ficarra, G., Cicchi, P., Amorosi, A., Piluso, S. Oral Crohn’s disease and pyostomatitis vegetans: an unusual association. Oral Surg Oral Med Oral Pathol. 1993; 75(2):220–224. [Feb].
Finn, S.B. Hereditary opalescent dentin I: an analysis of literature of hereditary anomalies of tooth color. J Am Dent Assoc. 1938; 25:1240.
Fisher, A., Som, P.M., Mosemsson, R.E., Lidou, M., Liu, T.H. Giant intracranial aneurysms with skull base erosion and extracranial masses. CT and MR findings. J Comput Assist Tomogr. 1994; 18:939–942.
Fitzwilliams, C.D.L.The Tongue and its Diseases. New York: Oxford University Press, 1927.
Fiumara, N.J., Lessell, S. Manifestations of late congenital syphilis. Arch Dermatol. 1970; 102:78.
Fogh-Andersen, P. Inheritance of Harelip and Cleft Palate Copenhagen, Nyt, Nordisk Forlag Arnold Busck, 1942.
Foretich, E.A., Cardo, V.A., Jr., Zambito, R.F. Bilateral congenital absence of the submandibular duct orifices. J Oral Surg. 1973; 31:556.
Foster, T.D., Taylor, G.S. Characteristics of supernumerary teeth in the upper central incisor region. Dent Pract Dent Rec. 1969; 20:8–12.
Foster, T.D. The effects of hemifacial atrophy on dental growth. Br Dent J. 1979; 146:148–150.
Fox, P.C., van der Ven, P.F., Baum, B.J., Mandel, I.D. Pilocarpine for the treatment of xerostomia associated with salivary gland dysfunction. Oral Surg Oral Med Oral Pathol. 1979; 61:243–248.
Fraser, F. Workshop on embryology of cleft lip and cleft palate. Teratology. 1968; 1:353.
Fraser, F.C., Warburton, D. No association of emotional stress or vitamin supplement during pregnancy to cleft lip or palate in man. Plast Reconstr Surg. 1964; 33:395.
Fraumeni, J.F., Jr., Geiser, C.F., Manning, M.D. Wilms’ tumor and congenital hemihypertrophy: report of five new cases and review of literature. Pediatrics. 1967; 40:886–899.
Frerichs, D.W., Spooner, S.W. Median palatine cyst. Oral Surg. 1953; 6:1181.
Fromm, A. Epstein’s pearls, Bohn’s nodule and inclusion cysts of the oral cavity. J Dent Child. 1967; 34:275.
Frota-Pessoa, O.Personal Communication. Brazil: Sao Paulo, 1979. [12–10].
Fryns, J.P., Kleczowska, A., Kenis, H., Decock, P. Partial duplication of the short arm of chromosome, 2 (1321) associated with mental retardation and an Aarskog-like phenotype. Ann Genet. 1989; 32:174–176.
Fryns, J.P. Gingival fibromatosis and partial duplication of the short arm of chromosome 2 (dup(2)(p13p21)). Ann Genet. 1996; 39:54–55.
Gardner, D.G., Girgis, S.S. Taurodontism, shovelshaped incisors and the Klinefelter syndrome. J Can Dent Assoc. 1978; 8:372.
Gardner, D.G., Sapp, J.P. Ultrastructural, electronprobe, and microhardness studies of the controversial amorphous areas in the dentin of regional odontodysplasia. Oral Surg. 1977; 44:549.
Gardner, D.G. The dentinal changes in regional odontodysplasia. Oral Surg. 1974; 38:887.
Gelbier, M.J., Winter, G.B. Absence of salivary glands in children with rampant dental caries: report of seven cases. Int J Paediatr Dent. 1995; 5:253–257.
Giansanti, J.S., Budnick, S.D. Six generations of hereditary opalescent dentin: report of case. Dent Assoc. 1975; 90:439–443.
Giansanti, J.S., Allen, J.D. Dentin dysplasia, type II, or dentin dysplasia, coronal type. Oral Surg. 1974; 38:911.
Giansanti, J.S., Baker, G.O., Waldron, C.A. Intraoral mucinous, minor salivary gland lesions presenting clinically as tumors. Oral Surg. 1971; 32:918.
Giardiello, F.M., Brensinger, J.D., Tersmette, A.C. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000; 119(6):1447–1453. [Dec].
Giardiello, F.M., Welsh, S.B., Hamilton, S.R. Increased risk of cancer in the Peutz-Jeghers syndrome. New Engl J Med, 11. 1987; 316(24):1511–1514. [Jun].
Gibson, J. Geographic tongue. The clinical response to zinc supplementation. J Trace Elem Exp Med. 1990; 3:203–208.
Gier, R.E., Fast, B. Median maxillary anterior alveolar cleft. Oral Surg. 1967; 24:496.
Ginuta, J., Cataldo, E. Lymphoepithelial cysts of the oral mucosa. Oral Surg. 1973; 35:77.
Glenn, E.L., Miro, M. Stafne’s mandibular lingual cortical defect. J Maxillofac Surg. 1985; 13:172–176.
Glimcher, M.J., Friberg, U.A., Levine, P.T. The isolation and amino acid composition of the enamel proteins of erupted bovine teeth. Biochemi J. 1964; 93:202–210.
Global registry and database on craniofacial anomalies: report of a WHO registry meeting on craniofacial anomalies, 2001.
Gnanasekhar, J.D., Walvekar, S.V., al-Kandari, A.M., al-Duwairi, Y. Misdiagnosis and mismanagement of a nasopalatine duct cyst and its corrective therapy: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995; 80(4):465–470.
Gnepp, D.R., Sporck, F.T. Benign lymphoepithelial parotid cyst with sebaceous differentiation-cystic sebaceous lymphadenoma. Am J Clin Pathol. 1980; 74:683–687.
Godley, F.A. Frenulopasty with a buccal mucosal graft. Laryngoscope. 1994; 104(3):378–381.
Goebel, W.M. Bilateral patent nasopalatine ducts. J Oral Med. 1975; 30:96.
Goetsch, E. Lingual goiter; report of three cases. Ann Surg. 1948; 127:291.
Goldberg, H., Goldhaber, P. Hereditary intestinal polyposis with oral pigmentation (Jeghers’ syndrome). Oral Surg. 1954; 7:378.
Goldstein, E., Gottlieb, M.A. Taurodontism: familial tendencies demonstrated in eleven of fourteen case reports. Oral Surg. 1973; 36:131.
Googe, P.B., Harris, N.L., Mihm, M.C., Jr. Kimura’s disease and angiolymphoid hyperplasia with eosinophilia: two distinct histopathological entities. J Cutan Pathol. 1987; 14:263–271.
Gorlin, R.J., Cohen, M.M., Jr., Levin, L.S. Syndromes of the Head and Neck, 3rd, Oxford: Oxford University Press; 1990:868–897.
Gorlin, R.J., Jue, K.L., Jacobsen, U., Goldschmidt, E. Oculoauriculovertebral dysplasia. J Pediatr. 1963; 63:991–996.
Gorlin, R.J., Pindborg, J.J., Cohen, M.M.Syndromes of the Head and Neck. New York: McGrawHill, 1976.
RJ, Gorlin, HM, Goldman. Thomas’ Oral Pathology, 6. St Louis: CV Mosby, 1970.
Gorlin, R.J., Jirasek, J.E. Oral cysts containing gastric or intestinal mucosa: unusual embryologic accident or heterotopia. J Oral Surg. 1970; 28:9.
Gorlin, R.J., Meskin, L.H., St Geme, J W. Oculodentodigital dysplasia. J Pediatr. 1963; 63:69.
Graber, L.W. Congential absence of teeth: a review with emphasis on inheritance patterns. J Am Dent Assoc. 1978; 96:266.
Grace, L.G. Frequency of occurrence of cleft palates and harelips. J Dent Res. 1943; 22:495.
Grahnén, H., Granath, L.E. Numerical variations in primary dentition and their correlation with the permanent dentition. Odontol Recy. 1961; 12:348.
Grahnén, H., Larsson, P.G. Enamel defects in deciduous dentition of prematurely born children. Odontol Recy. 1958; 9:143.
Greenspan, D., Daniels, T.E. Effectiveness of pilocarpine in postradiation xerostomia. Cancer. 1987; 59:1123–1125.
Guiducci, A.A., Hyman, A.B. Ectopic sebaceous glands. Dermatologica. 1962; 125:44.
Gunter, G.S. Nasomaxillary cleft. Plast Reconstr Surg. 1963; 32:637.
Halperin, V., Kolas, S., Jefferis, K.R., Huddleston, S.D. The occurrence of Fordyce spots, beingn migratory glossitis, median rhomboid glossitis, and fissured tongue in 2, 478 dental patients. Oral Surg. 1953; 6:1072.
Hamilton, T.K., Baughman, R.D., Perry, A.E. Persistent pruritic plaque of the ear. Arch Dermatol. 1999; 135(4):464–465. [467–68, Apr].
Hamner, J.E., III., Witkop, C.J., Metro, P.S. Taurodontism Report of a case. Oral Surg. 1964; 18:409.
Hansson, L.G. Development of a lingual mandibular bone cavity in an 11-year-old boy. Oral Surg Oral Med Oral Pathol. 1980; 49:376–378.
Harada, K., Enomoto, S. A new method of tongue reduction for macroglossia. J Oral Maxillofac Surg. 1995; 53(1):91–92. [Jan].
Haring, O.M., Lewis, F.J. The etiology of congenital developmental anomalies. Surg, Gynecol Obstet (Int Abst Surg). 1961; 113:1.
Harris, A.M., van., Wyk, C.W. Heck’s disease (focal epithelial hyperplasia): a longitudinal study. Community Dent Oral Epidemiol. 1993; 21:82–85.
Harris, I.R., Brown, J.E. Application of cross-sectional imaging in the differential diagnosis of apical radiolucency. Int Endod J. 1997; 30(4):288–290. [Jul].
Harris, C.N., Courtemanche, A.D. Gastric mucosal cyst of the tongue. Plast Reconstr Surg. 1974; 54:612.
Hart, T.C., Pallos, D., Bowden, D.W., Boylard, J., Pettenati, M.J. Genetic linkage of hereditary gingival fibromatosis to chromosome 2p21. Am J Hum Genet. 1998; 62:876–883.
Harvey, W., Noble, H.W. Defects on the lingual surface of the mandible near the angle. Br J Oral Surg. 1968; 6:75.
Hasler, J.F., Standish, S.M. Podophyllin treatment of hairy tongue: a warning. J Am Dent Assoc. 1969; 78(3):563–567. [Mar].
Hassell, T.M., Hefti, A.F. Drug-induced gingival overgrowth: old problem, new problem. Crit Rev Oral Biol Med. 1991; 2:103–237.
Hayes, H. Aberrant submaxillary gland tissue presenting as a cyst of the jaw: report of a case. Oral Surg Oral Med Oral Pathol. 1961; 14:313–316.
Hedin, C.A. Disappearance of smoker’s melanosis after reducing smoking. J Oral Pathol Med. 1993; 22:228–230.
Hedin, M., Klamfeldt, A., Persson, G. Surgical treatment of nasopalatine duct cysts: a follow-up study. Int J Oral Surg. 1978; 7(5):427–433. [Oct].
Hellman, M. Our third molar teeth: their eruption, presence and absence. Dent Cosmos. 1936; 78:750.
Hemminki, A., Markie, D. Tomlinson I: a serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998; 392(6663):184–187. [Jan].
Henderson, H.Z. Ankylosis of primary molars: a clinical, radiographic, and histolgic study. J Dent Child. 1979; 46:117.
Henoch, E., Romberg, H.M. Klinische Ergebnisse. Berlin: A Forstner, 1846; 75–81. [(pub)].
Henzel, J.H., Pories, W.J., DeWeese, M.S. Etiology of lateral cervical cysts. Surg Gynecol Obstel. 1967; 125:87.
Hertzanu, Y., Cohen, M., Mendelsohn, D.B. Nasopalatine duct cyst. Clin Radiol. 1985; 36(2):153–158. [Mar].
Heymann. WR: Psychotropic agent-induced black hairy tongue. Cutis. 2000; 66(1):25–26. [Jul].
Heys, F.M., Hlattner, R.J., Robinson, H.B.C. Osteogenesis imperfecta and odontogenesis imperfecta: clinical and genetic aspects in eighteen families. J Pediatr. 1960; 56:234.
Hickman, J.W., Sheils, W.S. Progressive facial hemiatrophy: report of a case with marked homolateral involvement. Arch Intern Med. 1964; 113:716–720.
Hirshfeld, I. The retrocuspid papilla. Am J Orthod. 1947; 33:447.
Ho, K.H. Hemifacial atrophy. Br Dent J. 1987; 162(5):182–184. [Romberg’s disease].
Ho, K.K., Dervan, P., O’Loughlin, S., Powell, F.C. Labial melanotic macule: a clinical, histopathologic, and ultrastructural study. J Am Acad Dermatol. 1993; 28:33–39.
Hodge, H.C., Finn, S.B., Lose, G.B., Gachet, F.S. Hereditary opalescent dentin II: general and oral clinical studies. J Am Dent Assoc. 1939; 26:1663.
Hodge, H.C., Finn, S.B., Robinson, H.B.G., Manly, R.S. Hereditary opalescent dentin III: histological, chemical and physical studies. J Dent Res. 1940; 19:521.
Hogstrom, A., Andersson, L. Complications related to surgical removal of anterior supernumerary teeth in children. ASDC J Dent Child. 1987; 54:341–343.
Houston, W.J.B., Stephens, C.D., Tulley, W.J. A Textbook of Orthodontics, 2nd, Wright Publications; 1992:174–175.
Howard, R.D. The unerupted incisor. A study of the postoperative eruptive history of incisors delayed in their eruption by supernumerary teeth. Dent Pract Dent Rec. 1967; 17:332–341.
Hoyme, H.E., Seaver, L.H., Jones, K.L., Procopio, F. Isolated hemihyperplasia : report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet. 1998; 79:274–278. [hemihypertrophy].
Hung, W., Randolph, J.G., Sabatini, D., Winship, T. Lingual and sublingual thyroid glands in euthyroid children. Pediatrics. 1966; 38:647.
Hursey, R.J., Witkop, C.J., Jr., Miklashek, D., Sackett, L.M. Dentinogenesis imperfecta in a racial isolate with multiple hereditary defects. Oral Surg. 1956; 9:641–658.
Ingalls, T.H., Taube, I.E., Klingberg, M.A. Cleft lip and cleft palate: epidemiologic considerations. Plast Reconstr Surg. 1964; 34:1.
Thorac, J. Multicentric Castleman’s disease and POEMS syndrome. CT findings. 1997; 12:75–77. [(No abstract available)].
Janicke, S., Kettner, R., Kuffner, H.D. A possible inflammatory reaction in a lateral neck cyst because of odontogenic infection. Internat J Oral Maxillofac Surg. 1994; 23:369–371. [(branchial cyst)].
Janku, P., Robinow, M., Kelly, T., Bralley, R. The van der Woude syndrome in a large kindred: variability, penetrance, genetic risks. Am J Med Genet. 1980; 5:117–123.
Jarvinen, J., Kullaa-Mikkonen, A., Kotilainen, R. Some local and systemic factors related to tongue inflammation. Proc Finn Dent Soc. 1989; 85(3):199–209.
Jeghers, H., McKusick, V.A., Katz, K.H. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits: a syndrome of diagnostic significance. New Engl J Med. 1949; 241:993–1005.
Jenne, D.E., Reimann, H., Nezu, J. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998; 18(1):38–43.
Jensen, B.L., Kreiborg, S. Development of the dentition in cleidocranial dysplasia. J Oral Pathol Med. 1990; 19:89–93.
Jensen, J.L., Idiopathic diseasesEllis, G.L., AuClair, P.L., Gnepp, D.R., eds. Major Problems in Pathology: Surgical Pathology of the Salivary Glands; 25. WB Saunders, Philadelphial, 1991:79. [(eds)].
Johkoh, T. Intrathoracic multicentric Castleman’s disease: CT findings. Radiology. 1998; 209:477–481.
Johnson, O.N., Chaudhry, A.P., Gorlin, R.J., Mitchell, D.F. Hereditary dentinogenesis imperfecta. J Pediat. 1959; 54:786–792.
Jorgensen, R.J. Intraoral findings and anomalies in neonates. Pediatr. 1982; 69(5):577–582.
Jowett, A.K., Vainio, S., Ferguson, M.W., Sharpe, P.T. Epithelial-mesenchymal interactions are required for MSX-1 and MSX-2 gene expression in the developing murine molar tooth. Development. 1993; 117:461–470.
Jurkiewicz, M.J. Nahai F The use of free revascularized grafts in the amelioration of hemifacial atrophy. Plast Reconstr Surg. 1985; 76:44–55.
Kacker, A., Honrado, C., Martin, D., Ward, R. Tongue reduction in Beckwith-Weidemann syndrome. Int J Pediatr Otorhinolaryngol. 2000; 53(1):1–7. [Jun 9].
Kano, Y., Shiohara, T., Yagita, A., Nagashima, M. Treatment of recalcitrant cheilitis granulomatosa with metronidazole. J Am Acad Dermatol. 1992; 24(4):629–630. [Oct].
Kaplan, I., Moskona, D. A clinical survey of oral soft tissue lesions in institutionalized geriatric patients in Israel. Gerodontology, Summer. 1990; 9(2):59–62.
Karmiol, M., Walsh, R.F. Incidence of static bone defect of the mandible. Oral Surg. 1968; 26:225.
Kaugars, G.E., Heise, A.P., Riley, W.T. Oral melanotic macules: a review of 353 cases. Oral Surg Oral Med Oral Pathol. 1993; 76:59–61.
Kaugars, G.E., Pillion, T., Svirsky, J.A. Actinic cheilitis: a review of 152 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999; 88(2):181–186. [Aug].
Kennedy, J.M., Thompson, E.C. Hypoplasia of the mandible (Pierre Robin syndrome) with complete cleft palate: report of a case. Oral Surg. 1950; 3:421.
Kernahan, D.A. The striped Y—a symbolic classification for cleft lip and palate. Plast Reconstr Surg. 1971; 47(5):469–470. [May].
Kettunen, P., Thesleff, I. Expression and function of FGFs-4, -8, and -9 suggest functional redundancy and repetitive use as epithelial signals during tooth Morphogenesis. Developmental Dynamics. 1998; 211:256–268.
Killey, H.C., Kay, L.W. Benign Cystic Lesions of the Jaws, their Diagnosis and Treatment, 2. Edinburg: Churchill Livingstone, 1977.
Kinirons, M.J. Unerupted premaxillary supernumerary teeth: a study of their occurrence in males and females. Br Dent J. 1982; 153:110.
Kleinman, H.Z. Lingual varicosities. Oral Surg. 1967; 23:546.
Knapp, M.J. Lingual sebaceous glands and a possible thyroglossal duct. Oral Surg. 1971; 31:70.
Koillinen, H., Wong, F.K., Rautio, J., Rautio, coll. Mapping of the second locus for the Van der Woude syndrome to chromosome 1p34. Eur J Hum Genet. 2001; 9(10):747–752.
Kondo, S., Schutte, B.C., Richardson, R.J., Bjork, B.C. Mutations in IRF6 cause van der Woude and popliteal pterygium syndromes. Nat Genet. 2002; 32(2):285–289. [Epub Sep, 03, 2002].
Kovac-Kovacic, M., Skaleric, U. The prevalence of oral mucosal lesions in a population in Ljubljana, Slovenia. J Oral Pathol Med. 2000; 29(7):331–335.
Krassikoff, N., Sekhon, G.S. Familial agnathia-holoprosencephaly caused by an inherited unbalanced translocation and not autosomal recessive inheritance. Am J Med Genet. 1989; 34:255–257. [(Letter)].
Kreshover, S.J. The pathogenesis of enamel hypoplasia: an experimental study. J Dent Res. 1944; 23:231.
Kullaa-Mikkonen, A., Mikkonen, M., Kotilainen, R. Prevalence of different morphologic forms of the human tongue in young Finns. Oral Surg Oral Med Oral Pathol. 1982; 53(2):152–156. [Feb].
Kullaa-Mikkonen, A., Sorvari, T. Lingua fissurata: a clinical, stereomicroscopic and histopathological study. Int J Oral Maxillofac Surg. 1986; 15(5):525–533. [Oct].
Lacombe, D., Pedespan, J.M., Fontan, D. Phenotypic variability in van der Woude syndrome. Genet Couns. 1995; 6(3):221–226.
Lakhani, P.K., David, T.J. Progressive hemifacial atrophy with scleroderma and ipsilateral limb wasting (Parry-Romberg syndrome). J Royal Soc Med. 1984; 77:138–139.
Lamey, P.Z., Lewis, M.A. Oral medicine in practice: salivary gland disease. Br Dent J. 1990; 168:237–243.
Langlais, R.P., Cottone, J., Kasle, M.J. Anterior and posterior lingual depressions of the mandible. J Oral Surg. 1976; 34:502.
Langtry, J.A., Carr, M.M., Steele, M.C., Ive, F.A. Topical tretinoin: a new treatment for black hairy tongue (lingua villosa nigra). Clin Exp Dermatol. 1992; 17(3):163–164. May,
Lanthrop, G.M., Lalouel, J.M. Fast calculations of LOD scores and genetic risks on small computers. Am J Hum Genet. 1984; 6:460–465.
Lapage, C.P. Micrognathia in the newborn. Lancet. 1937; 1:323.
Larner, A.J., Bennison, D.P. Some observations on the aetiology of progressive hemifacial atrophy (‘Parry-Romberg syndrome’). (Letter). J Neurol Neurosurg Psychiat. 1993; 56:1035–1039.
Lau, E.C., Mohandas, T.K., Shapiro, L.J., Slavkin, H.C. Human and mouse amelogenin gene loci are on the sex chromosomes. Genomics. 1989; 4(2):162–168.
Lavelle, C.L.B. Applied Oral Physiology, 2. London: Wright, 1988.
Laymon, C.W. Cheilitis granulomatosa and Melkersson-Rosenthal syndrome. Arch Dermatol. 1961; 83:112.
Lederman, D.A. Suppurative stomatitis glandularis. Oral Surg Oral Med Oral Pathol. 1994; 78(3):319–322. [Sep].
Lederman, R.J. Progressive facial and cerebral hemiatrophy. Cleve Clin Q. 1984; 51:545–548.
Leider, A.S., Lucas, J.W., Eversole, L.R. Sebaceous choristoma of the thyroglossal duct. Oral Surg. 1977; 41:261.
Levine, N. The clinical management of supernumerary teeth. J Can Dent Assoc. 1961; 28:297–303.
Levine, N. Dark discoloration of the tongue. Geriatrics. 1996; 51(6):20. [Jun].
Levine, R.S., Saliva: 3. Xerostomia-aetiology and management. Dental Update. 1989. [Jun]. Levine RS. Saliva: 3. Xerostomia-aetiology and management. Dental Update, Jun, 1989.
TC, T.C. Gemination, fusion, twinning, and concrescence. J Dent Child. 1965; 32:93.
Lewkonia, R.M., Lowry, R.B. Progressive hemifacial atrophy (Parry-Romberg syndrome) report with review of genetics and nosology. Am J Med Genet. 1983; 14:385–390.
Li, R.W. Adhesive solutions: report of a case using multiple adhesive techniques in the management of enamel hypoplasia. Dental Update. 1999; 26(7):277–282. [284, 287–97].
Limbrock, G.J., Fischer-Brandies, H., Avalle, C. Castillo-Morales’ orofacial therapy: treatment of 67 children with Down syndrome. Dev Med Child Neurol. 1991; 33(4):296–303. [Apr].
Little, J.W., Rickles, N.H. The histogenesis of the branchial cyst. Am J Pathol. 1967; 50:533.
Littner, M., Dayan, D., Gorsky, M. Migratory stomatitis. Oral Surg Oral Med Oral Pathol. 1987; 63:555–559.
Liu, J.F. Characteristics of premaxillary supernumerary teeth: a survey of 112 cases. ASDC J Dent Child. 1995; 62:262–265.
Livolsi, V.A., Perzin, K.H., Savetsky, L. Carcinoma arising in median ectopic thyroid (including thyroglossal duct tissue). Cancer. 1974; 34:1303.
Lo, L.J., Noordhoff, M.S. Median cleft of the lower lip associated with lip pits and cleft of the lip and palate. Cleft Palate Craniofac J. 1999; 36(1):86–87. [Jan].
Logan, J., Becks, H., Silverman, S., Jr., Pindborg, J.J. Dentinal dysplasia, Oral Surg. 1962; 15:317.
Lunt, R.C., Law, D.B. A review of the chronology of calcification of deciduous teeth. J Am Dent Assoc. 1974; 89:599.
Lyons, D.C. Biochemical factors in the possible developments of malformations of the oral tissues, especially cleft palate and harelip. Oral Surg. 1950; 3:12.
MacKenzie, A., Ferguson, M.W. Sharpe PT. Hox-7 expression during murine craniofacial development. Development. 1991; 113:601–611.
MacMahon, B., McKeown, T. The incidence of harelip and cleft palate related to birth rank and maternal age. Am J Hum Genet. 1953; 5:176.
Mader, C.L. Fusion of teeth. J Am Dent Assoc. 1979; 98:62.
Mahoney, E.K. The treatment of localised hypoplastic and hypomineralised defects in first permanent molars. New Zealand Dent J. 2001; 97(429):101–105.
Manabe, M., Lim, H.W., Winzer, M., Loomis, C.A. Architectural organization of filiform papillae in normal and black hairy tongue epithelium: dissection of differentiation pathways in a complex human epithelium according to their patterns of keratin expression. Arch Dermatol. 1999; 135(2):177–181. [Feb].
Mandel, L., Baurmash, H. Thyroglossal tract abnormalities. Oral Surg Path. 1957; 10:113.
Mangion, J.J. Two cases of taurodontism in modern human jaws. Br Dent J. 1962; 113:309.
Mannens, M., Slater, R.M., Heyting, C., Bliek, J. Chromosome 11, Wilms’ tumour and associated congenital diseases. (Abstract) Cytogenet. Cell Genet. 1987; 46:655.
Marks, R., Radden, B.G. Geographic tongue: a clinico-pathological review. Australas J Dermatol. 1981; 22:75–79.
Mars, M., Farrant, S., Roberts, G.J. Dentinogenesis imperfecta: report of a fivegeneration family. Br Dent J. 1976; 140:206–209.
Martin, H.E., Howe, M.E. Glossitis rhombica mediana. Ann Surg. 1938; 107:39.
Maskow, B.S. Bilateral congenital nasopalatine communication. Oral Surg. 1982; 53:458.
Mason, D.K., Chisholm, D.M.Salivary Glands in Health and Disease. Phidelphial: WB Saunders, 1975.
Massler, M., Savara, B.S. Natal and neonatal teeth. J Pediatr. 1950; 36:349.
Mathewson, R.J., Siegel, M.J., McCanna, D.L. Ankyloglossia: a review of the literature and a case report. J Dent Child. 1966; 33:238.
McAdams, H.P. Castleman disease of thorax: radiologic features with clinical and histopathologic correlation. Radiology. 1998; 209:221–228.
McCarthy, F.P. A clinical and pathologic study of oral disease. J Am Med Assoc. 1941; 116:16.
McComb, H. Primary repair of the bilateral cleft lip nose: a 4–year review. Plast Reconstr Surg. 1994; 94(1):37–47. [48–50, Jul].
McConnel, F.M.S., Zellweger, H., Lawrence, R.A. Labial pits: cleft lip and/or palate syndrome. Arch Otolaryngol. 1970; 91:407.
McDonald, F.G., Mantas, J., McEwen, C.G., Ferguson, M.M. Salivary gland disorder: an ectodermal disorder? J Oral Pathol. 1986; 15:115–117.
McDonald, R.E., Avery, D.R. Dentistry for the Child and Adolescent, 7, St Louis: CV Mosby; 2000:115–122.
McEvitt, W.G. Cleft lip and palate and parental age: a statistical study of etiology. Plast Reconstr Surg. 1952; 10:77.
McGregor, J.M., Hay, R.J. Oral retinoids to treat black hairy tongue. Clin Exp Dermatol. 1993; 18(3):291. [May].
McKay, F.S., Black, G.V. An investigation of mottled teeth. Dent Cosmos. 1916; 58:477. [627, 781, 894].
McKay, F.S. Investigation of mottled enmel and brown stain. J Natl Dent Assoc. 1917; 4:273.
McKenna, K.E., Walsh, M.Y., Burrows, D. The Melkersson-Rosenthal syndrome and food additive hypersensitivity. Br J Dermatol. 1994; 131(6):921–922. [Dec].
Meadows, A.T., Lichtenfeld, J.L., Koop, C.E. Wilms’ tumor in three children of a woman with congenital hemihypertrophy. N Engl J Med. 1974; 291:23–24.
Mealey, B.L., Rasch, M.S., Braun, J.C., Fowler, C.B. Incisive canal cysts related to periodontal osseous defects: case reports. J Periodontol. 1993; 64(6):571–654. [Jun].
Megarbane, A., Souraty, N., Prieur, M. Interstitial duplication of the short arm of chromosome 2: report of a new case and review. J Med Genet. 1997; 34:783–786.
Mehregan, A.H., Shapiro, L. Angiolymphoid hyperplasia with eosinophilia. Arch Dermatol. 1971; 103:50–57.
Mellor, J.K., Ripa, L.W. Talon cusp: a clinically significant anomaly. Oral Surg. 1970; 29:224.
Melnick, M., Eastman, J.R., Goldblatt, L.I., Melnick, M. Dentin dysplasia, type II: a rare autosomal dominant disorder. Oral Surg. 1977; 44:592.
Melnick, M., Levin, L.S., Brady, J. Dentin dysplasia, type I: a scanning electron microscopic analysis of the primary dentition. Oral Surg. 1980; 50:335.
Menko, F.H., Koedijk, P.H., Baart, J.A. Van der Woude syndrome—recognition of lesser expressions: case report. Cleft Palate J. 1988; 25(3):318–321. [Jul].
Merchant, H.W., Haynes, L.E., Ellison, L.T. Soft-palate pigmentation in lung disease, including cancer. Oral Surg Oral Med Oral Pathol. 1976; 41:726–733.
Mermer, R.W., Rider, C.A., Clevel, D.B. Nasopalatine canal cyst: a rare sequelae of surgical rapid palatal expansion. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995; 80(6):620. [Dec].
Meskin, L.H., Redman, R.S., Gorlin, R.J. Incidence of geographic tongue among 3,668 students at the University of Minnesota. J Dent Res. 1963; 42:895.
Meyer, A.W. Median anterior maxillary cysts. J Am Dent Assoc. 1931; 18:1851.
Meyer, I. Dermoid cysts (dermoids) of the floor of the mouth. Oral Surg. 1955; 8:1149.
Michalowski, R. Munchausen’s syndrome: a new variety of bleeding type-selfinflicted cheilorrhagia and cheilitis glandularis. Dermatologica. 1985; 170(2):93–97.
Mignogna, M.D., Fedele, S., Lo Russo, L., Lo Muzio, L. The multiform and variable patterns of onset of orofacial granulomatosis. J Oral Pathol Med. 32(4), 2005.
Miles, A.E.W. Sebaceous glands in the lip and cheek mucosa of man. Br Dent J. 1958; 105:235.
Miller, A.S., McCrea, M.W. Sebaceous gland adenoma of the buccal mucosa. J Oral Surg. 1968; 26:593.
Miller, A.S., Winnick, M. Salivary gland inclusion in the anterior mandible. Oral Surg. 1971; 31:790.
Miller, A.S., Grelley, J.N., Catena, D.L. Median maxillary anterior alveolar cleft: report of three cases. J Am Dent Assoc. 1969; 79:896.
Miller, J., Forrester, R.M. Neonatal enamel hypoplasia associated with haemolytic disease and with prematurity. Br Dent J. 1959; 106:93.
Miller, J.B., Moore, P.M., Jr. Nasoalveolar cysts. Ann Otol Rhinol Laryngol. 1949; 58:200.
Miller, W.A., Seymour, R.H. Odontodysplasia. Br Dent J. 1968; 125:56.
Mitchell, L., Bennett, T.G. Supernumerary teeth causing delayed eruption— a retrospective study. Br J Orthod. 1992; 19:41–46.
Mitchell, L. An Introduction to Orthodontics. Oxford University Press, Oxford, London, 1996:23–25. [(1st ed)].
Mixter, R.C., Ewanowski, S.J., Carson, L.V. Central tongue reduction for macroglossia. Plast Reconstr Surg. 1993; 91(6):1959–1962. [May].
Moeller, J.F., Philipsen, H.P. A case of nasoalveolar cyst (Klestadt’s cyst) Tandlaegebladet. 1958; 62:659.
Monroe, C.W., Ogo, K. Treatment of micrognathia in the neonatal period: report of 65 cases. Plast Reconstr Surg. 1972; 50:317.
Monteleone, L., McLellan, M.S. Epstein’s perals and Bohn’s nodules of the palate. J Oral Surg. 1964; 22:301.
Montgomery, M.L. Lingual thyroid: a comprehensive review. West J Surg Obstet Gynecol. 1935; 43:661. [1936].
Morales, C., Peñarrocha, M., Bagan, J.V. Immunological study of MelkerssonRosenthal syndrome: lack of response to food additive challenge. Clin Exp Allergy. 1995; 25(3):260–264. [Mar].
Morgan, W.E., Friedman, E.M., Duncan, N.O., Sulek, M. Surgical management of macroglossia in children. Arch Otolaryngol Head Neck Surg. 1996; 122(3):326–329. [Mar].
Morgan WE. Macroglossia. Available at: http://www.bcm.tmc.edu/oto/, 1992.
Morris, L.F. Oral lesions in patients with psoriasis: a controlled study. Cutis. 1992; 49:339–344.
Moulton, F.R. Fluorine and Dental Health. American Association for the Advancement of Science, Washington DC, 1942.
Muchnick, R.S., Aston, S.J., Rees, T.D. Ocular manifestations and treatment of hemifacial atrophy. Am J Ophthalmol. 1979; 88:889–897.
Murray, J. Personal Communication. Iowa City, Iowa, 9/8/1987.
Mutch, J. Congenital absence of weeping associated with absence of salivation. Br J Ophthalmol. 1944; 28:333–334.
Nally, F. Diseases of the tongue. Practitioner. 1991; 235(1498):65–71. [Jan].
Nathanson, I. Macroglossia. Oral Surg. 1948; 1:547.
Nelson, D.L., Ledbetter, S.A., Corbo, L. Alu polymerase chain reaction: a method for rapid isolation of human-specific sequences from complex DNA sources. Proc Natl Acad Sci, USA. 1989; 86:6686–6690.
Neubuser, A., Peters, H., Balling, R., Martin, G.R. Antagonistic interactions between FGF and BMP signaling pathways: a mechanism for positioning the sites of tooth formation. Cell. 1997; 90:247–255.
Neville, B.W., Damm, D.D., Allen, C.M., Bouquot, J.E.Oral and Maxillofacial Pathology. Philadelphia: WB Saunders, 1995.
New, E.B., Erich, J.B. Dermoid cysts of the head and neck. Surg Gynecol Obstet. 1937; 65:48.
Newman, C.C., Wagner, R.F. Images in clinical medicine: black hairy tongue. N Engl J Med. 1997; 337(13):897. [Sep 25].
Nora, J.J., Fraser, F.C., Bear, J. Medical Genetics: Principle and Practice. In Lea and Febiger, 4th, Philadelphia: Lea and Febiger; 1994:270–279.
Nortje, C.J., Farman, A.G. Nasopalatine duct cyst: an aggressive condition in adolescent Negroes from South Africa? Int J Oral Surg. 1978; 7(2):65–72. [Apr].
Notestine, G.E. The importance of the indentification of ankyloglossia (short lingual frenulum) as a cause of breastfeeding problems. J Human Lactat. 1990; 6(3):113–115.
Nudleman, K., Andermann, E., Andermann, F., Bertrand, G. The hemi 3 syndrome: hemihypertrophy, hemihypaesthesia, hemiareflexia and scoliosis. Brain. 1984; 107:533–546.
Odell, E.W., Morgan, P.R.Biopsy Pathology of the Oral Tissues. London: Chapman and Hall Medical, 1998.
Oehlers, F.A.C. A case of multiple supernumerary teeth. Br Dent J. 1951; 90:211.
Oikarinen, V.J., Julku, M. An orthopantomographic study of developmental mandibular bone defects. Int J Oral Surg. 1974; 3:71.
Olason, H., Axell, T. Objective and subjective efficacy of saliva substitutes containing mucin and carboxymethylcellulose. Scand J Dent Res. 1991; 99:316–319.
Oliver, I.D., Pickett, A.B. Cheilitis glandularis. Oral Surg. 1980; 49:526.
Olsen, T.G., Helwig, E.B. Angiolymphoid hyperplasia with eosinophilia: a clinicopathologic study of 116 patients. J Am Acad Dermatol. 1985; 12:781–796.
Onofre, M.A., Brosco, H.B., Taga, R. Relationship between lower-lip fistulae and cleft lip and/or palate in von der Woude syndrome. Cleft Palate Craniofac J. 1997; 34(3):261–265. [May].
Oshima, H., Rochat, A., Kedzia, C., Kobayashi, K., Barrandon, Y. Morphogenesis and renewal of hair follicles from adult multipotent stem cells. Cell. 2001; 104:233–245.
Ozden, S., Ficicioglu, C., Kara, M., Oral, O et al. Agnathia-holoprosencephaly-situs inversus (Letter). Am J Med Genet. 2000; 91:235–236.
Mossey, P.A. The Heritability of malocclusion: Part 1: genetics, principles and terminology. Br J Ortho. 1999; 26:103–113.
Padayachee, A., van Wyk, C.W. Human papillomavirus (HPV) DNA in focal epithelial hyperplasia by in situ hybridization. J Oral Pathol Med. 1991; 20:210–214.
Page, L.R., Corio, R.L., Crawford, B.E., Giansanti, J.S. The oral melanotic macule. Oral Surg. 1977; 44:219.
Palmer, M.E. Case reports of evaginated odontomes in Caucasinas. Oral Surg. 1973; 35:772.
Papanayotou, P.H., Hatziotis, J. CH. Ascher’s syndrome. Oral Surg. 1973; 35:467.
Parry, C.H. Collections from the unpublished medical writings of the late Caleb Hillier Parry, 1, London: Underwoods (pub.); 1825:478.
Pauli, R.M., Hall, J.G. Lip pits, cleft lip and/or palate, and congenital heart disease. Am J Dis Child. 1980; 134(3):293–295. [Mar].
Pauli, R.M., Pettersen, J.C., Arya, S., Gilbert, E.F. Familial agnathia-holoprosencephaly. Am J Med Genet. 1983; 13(5):677–698.
Pavlin, J.E., O’Gorman, A., Williams, H.B. Epignathus: a report of two cases. Ann Plast Surg. 1984; 13(5):452–456. [Nov].
Pegum, J.S. Urea in the treatment of black hairy tongue. Br J Dermatol. 1971; 84(6):602. [Jun].
Pernu, H.E., Knuuttila, M.L.E., Huttunen, K.R.H., Tiiliksinen, A.S.K. Drug-induced gingival overgrowth and class II major histocompatibility antigens. Transplantation. 1994; 57:1811–1823.
Peter, M.S., Hugh, D.C. Head and Neck Imaging, 3, St. Louis: CV Mosby; 1996:516–518.
Pettenati, M.J., Rao, P.N., Hayworth-Hodge, R. Brewer G. Gene mapping by fluorescence in situ hybridization. In: Ross J., ed. Nucleic Acid Hybridization, Essential Techniques. New York: Wiley; 1998:81–109.
Pindborg, J.J.Pathology of the Dental Hard Tissues. Philadelphia: WB Saunders, 1970.
Poskanzer, D.C., Hemiatrophies and hemihypertrophiesVinken, P.J., Bruyn, G.W., eds. Handbook of Clinical Neurolgy; 1975;22:545–554. [Amsterdam, North-Holland].
Poswillo, D. The pathogenesis of the first and second branchial arch syndrome. Oral Surg Oral Med Oral Pathol. 1973; 35:302–328.
Powell, F.C. Glossodynia and other disorders of the tongue. Dermatol Clin. 1987; 5(4):687–693.
Praetorius-Clausen, F., Willisn, J.M. Papova virus-like particles in focal epithelial hyperplasia. Scand J Dent Res. 1971; 79:362.
Praetorius-Clausen, F.M., Ogeltoft, M., Roed-Petersen, B., Pindborg, J.J. Focal epithelial hyperplasia of the oral mucosa in a south-west Greenlandic population. Scand J Dent Res. 1970; 78:287.
Praetorius-Clausen, F. Geographical aspects of oral focal epithelial hyperplasia. Pathol Microbiol. 1973; 39:204.
Primosch, R.E. Anterior supernumerary teeth—assessment and surgical intervention in children. Pediatr Dent. 1981; 3:204–215.
Prinz, H., Greenbaum, S.S. Diseases of the Mouth and Their Treatment, 2. Philadelphia: Lea and Febiger, 1939.
Prowler, J.R., Glassman, S. Agenesis of the mandibular condyles. Oral Surg. 1954; 7:133.
Puvabanditsin, S., Garrow, E., Sitburana, O. Syngnathia and van der Woude syndrome: a case report and literature review. Cleft Palate Craniofac J. 2003; 40(1):104–106.
Ray, G.E. Congential absence of permanent teeth. Br Dent J. 1951; 90:213.
Redman, R.S. Prevalence of geographic tongue, fissured tongue, median rhomboid glossitis, and hairy tongue among 3,611 Minnesota schoolchildren. Oral Surg Oral Med Oral Pathol. 1970; 30(3):390–395.
Rees, T.D., Ashley, F.L., Delgado, J.P. Silicone fluid injections for facial atrophy. Plast Reconstr Surg. 1973; 52:118–127.
Rees, T.D., Coburn, R.J. Silicone treatment of partial lipodystrophy. J Am Med Assoc. 1974; 230:868–873.
Rees, T.D. Facial atrophy. Clin Plast Surg. 1976; 3:637–647.
Regezi, J.A., Sciubba, J.J., Oral Pathology. Clinical Pathologic Correlations. 3. WB Saunders, Philadelphia, 1999.
Rhodes, E.L., Stirling, G.A. Granulomatous cheilitis. Arch Dermatol. 1965; 92:40.
Richardson, E.R. Incidence of geographic tongue and median rhomboid glossitis in 3, 319 Negro college students. Oral Surg. 1968; 26:623.
Rieke, J.W., Hafermann, M.D., Johnson, J.T. Oral pilocarpine for radiation-induced xerostomia: integrated efficacy and safety results from two prospective randomised clinical trials. Int J Rad Onc Biol Phys. 1965; 31:661–669.
Ring, M.E. The treatment of macroglossia before the 20th century. Am J Otolaryngol. 1999; 20(1):28–36.
Ringrose, R.E., Jabbour, J.T., Keele, D.K. Hemihypertrophy Pediatrics. 1965; 36:434.
Risheim, H., Arneberg, P. Salivary stimulation by chewing gum and lozenges in rheumatic patients with xerostomia. Scand J Dent Res. 1993; 181:40–43.
Roa, R., Venkata. Naso-labial cyst. J Laryngol Otol. 1955; 69:352.
Roberts, E., Schour, I. Hereditary opalescent dentine—dentinogenesis imperfecta. Am J Orthodont. 1939; 25:267–276.
Robinson, C., Briggs, H.D., Kirkham, J., Atkinson, P.J. Changes in the protein components of rat incisor enamel during tooth development. Arch Oral Biolo. 1983; 28(11):993–1000.
Robinson, L.K., Hoyme, H.E., Edwards, D.K., Jones, K.L. Vascular pathogenesis of unilateral craniofacial defects. J Pediatr. 1987; 111:236–239.
Robinson, H.B.G., Koch, W.E. Diagnosis of cysts of the jaw. J Mo Dent Assoc. 1941; 21:187.
Robinson, H.B.G., Koch, W.E., Jr., Jasper, L.H. Infected globulomaxillary cyst. Am J Orthod Oral Surg. 1943; 29:608.
Robinson, H.B.G. Classification of cysts of the jaws. Am J Orthod Oral Surg. 1945; 31:370.
Roed-Petersen, B. Nasolabial cysts: a presentation of five patients with a review of the literature. Br J Oral Surg. 1969; 7:84.
Rogers, B.O. Embryology of the face and introduction to craniofacial anomalies. In: Converse J.M., ed. Reconstrucfive Plastic Surgery. Philadelphia: WB Saunders; 1977:2296–2358. [(2nd)].
Rogers, B.O. Progressive facial hemiatrophy: Romberg’s disease: a review of 772 cases. Proc 3d Int Cong Plast Surg. Excerpta Medica ICS. 1964; 66:681–689.
Rogers, R.S., III., Bekic, M. Diseases of the lips. Semin Cutan Med Surg. 1997; 16(4):328–336. [Dec].
Rogers, R.S., III., Melkersson-Rosenthal syndrome and orofacial granulomatosisMiles, D.A., Rogers, R.S., III., eds. Dermatologic Clinics; 14. WB Saunders, Philadelphia, 1996:371–379. [(2)].
Rosenthaler, H., Randel, H. Rotary reduction, enamel microabrasion, and dental bleaching for tooth color improvement. Compendium of Continuing Education in Dentistry. 1998; 19(1):62–67.
Roulston, D., Schwartz, S., Cohen, M.M., Suzuki, J.B. Linkage analysis of dentinogenesis imperfecta and juvenile periodontitis: creating a 5 point map of 4q. (Abstract). Am J Hum Genet. 1985; 37:A206.
Rowe, N.H. Hemifacial hypertrophy: review of the literature and addition of four cases. Oral Surg. 1962; 15:572.
Royer, R.W., Bruce, K.W. Median rhomboid glossitis: report of a case. Oral Surg. 1952; 5:1287.
Rushton, M.A. A case of dentinal dysplasia. Guys Hosp Rep. 1939; 89:369.
Rutrick, R., Black, P.W., Jurkiewicz, M.J. Bilateral cleft lip and palate: presurgical treatment. Ann Plast Surg. 1984; 12(2):105–117. [Feb].
Saarenmaa, L. The origin of supernumerary teeth. Acta Odontol Scand. 1951; 9:293.
Saito, T., Hida, C., Tsunoda, I. Melkersson-Rosenthal syndrome: distal facial nerve branch palsies, masseter myopathy and corticosteroid treatment. Fukushima J Med Sci. 1994; 40(1):39–44. [Jun].
Salonen, L., Axell, T., Hellden, L. Occurrence of oral mucosal lesions, the influence of tobacco habits and an estimate of treatment time in an adult. Swedish population. J Oral Pathol Med. 1990; 19(4):170–176. [Apr].
Sammett, J.F. Median rhomboid glossitis. Radiology. 1939; 32:215.
Sapp, J.P., Eversole, L.R., Wysocki, G.P.Contemporary oral and maxillofacial pathology. St. Louis: CV Mosby, 1997.
Sapp, J.P., Gardner, D.G. Regional odontodysplasia: an ultrastructural and histochemical study of the softtissue calcifications. Oral Surg. 1973; 36:383.
Sarnat, B.G., Shaw, N.G. Dental development in congenital syphilis. Am J Dis Child. 1942; 64:771. [Am J Orthod Oral Surg, 29: 270, 1943].
Sarnat, B.G., Schour, I. Enamel hypoplasia (chronochronologic, morphologic and etiologic classification. J Am Dent Assoc. 1941; 28:1989. [29: 67, 1942].
Sarti, G.M., Haddy, R.I., Schaffer, D. Black hairy tongue. Am Fam Physician. 1990; 41(6):1751–1755.
Satokata, I., Maas, R. MSX-1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nature Genetics. 1994; 6:348–356.
Sauk, J.J., Jr., Witkop, C.J., Jr., Brown, D.M., Corbin, K.W. Glycosaminoglycans of EDTA soluble and insoluble dentin in dentinogenesis imperfecta type I. Oral Surg. 1976; 41:753–757.
Sauk, J.J., Jr. Ectopic lingual thyroid. J Pathol. 1970; 102:239.
Sauk, J.J., Trowbridge, H.O., Witkop, C.J. An electron optic analysis and explanation for the etiology of dentin dysplasia. Oral Surg. 1972; 33:763.
Scheiner, M.A., Sampson, W.J. Scheiner MA, Sampson WJ. Supernumerary teeth: a review of the literature and four case reports. Aust Dent J. 1997; 42:160–165.
Schimmelpfennig, C.B., McDonald, R.E. Enamel and dentine aplasia. Oral Surg. 1953; 6:1444.
Schinkenickl, D.A., Muller, M.F. Lymphoepithelial cyst of the pancreas. Br J Radiol. 1996; 69:876–878.
Schneider, W., Reiter, E.H. Median cleft lip: report of a case. J Oral Surg. 1951; 9:329.
Schorff, J. Unusual cysts in the maxilla; cysts of nasopalatine duct and fissural cysts. Dent Items Interest. 1929; 51:107.
Schott, T.R., Correll, R.W., Wescott, W.B. Well-defined radiolucent area involving the anterior maxilla. J Am Dent Assoc. 1985; 110(1):86–88.
Schour, I., Smith, M.C. Mottled teeth: an experimental and histologic analysis. J Am Dent Assoc. 1935; 22:796.
Schour, I. The neonatal line in the enamel and dentin of the human deciduous teeth and first permanent molar. J Am Dent Assoc. 1936; 23:1946.
Schuller, D.E. Schleuning AJ. DeWeese and Saunders’ Otolaryngology-Head and Neck Surgery, 8. St. Louis: CV Mosby; 1994.
Schutte, B.C., Basart, A.M., Watanabe, Y., Laffin, J.J. Microdeletions at chromosome bands 1q32–q41 as a cause of van der Woude syndrome. Am J Med Genet, 21. 1999; 84(2):145–150.
Sciubba, J.J., Said-Al-Naief, N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003; 32(10):576–585. [Nov].
Scully, C., Cochran, K.M., Russell, R.I. Crohn’s disease of the mouth: an indicator of intestinal involvement. Gut. 1982; 23(3):198–201. [Mar].
Sedano, H.O., Gorlin, R.J. Familial occurrence of mesiodens. Oral Surg. 1969; 27:360.
Sedano, H.O., Sauk, J.J., Gorlin, R.J. Oral Manifestations of Inherited Disorders. Woburn: Butterworth; 1977.
Senia, E.S., Regezi, J.A. Dens evaginatus in the etiology of bilateral periapical pathologic involvement in caries-free premolars. Oral Surg. 1974; 38:465.
Seow, W.K. Taurodontism of the mandibular first permanent molar distinguishes between the tricho-dento-osseous (TDO) syndrome and amelogenesis imperfecta. Clini Genet. 1993; 43(5):240–246.
Sertie, A.L., Sousa, A.V., Steman, S. Linkage analysis in a large Brazilian family with van der Woude syndrome suggests the existence of a susceptibility locus for cleft palate at 17 p.11.2–11.1. Am J Hum Genet. 1999; 65(2):433–440. [Aug].
Severtson, M., Petruzzelli, G.J. Macroglossia. Otolaryngol Head Neck Surg. 1996; 114(3):501–502. [Mar].
Seward, G.R. Salivary gland inclusions in the mandible. Br Dent J. 1960; 108:321–325.
Sewerin, I. Praetorius-Clausen, F. Keratin-filled pseudocysts of ducts of sebaceous glands of the vermilion border of the lip. J Oral Path. 1975; 3:279.
Sewerin, I. The sebaceous glands in the vermilion border of the lips and in the oral mucosa of man. Acta Odontol Scand. 1975; 33:68. [(suppl)].
Shafer, W.G., Hine, M.K., Levy, B.M., A Textbook of Oral Pathology. 4. WB Saunders, Philadelphia, 1983:308–311.
Shafer, W.G. Dens in dente. New York Dent J. 1953; 19:279.
Shapiro, M., Peters, S., Spinelli, H.M. Melkersson-Rosenthal syndrome in the periocular area: a review of the literature and case report. Ann Plast Surg. 2003; 50(6):644–648. [Jun].
Shapiro, P.E. Noninfectious granulomas. In: Elder D., Elenitsas R., Jaworsky C., Johnson B., eds. Lever’s Histopathology of the Skin. 8. Philadelphia: Lippincott-Raven; 1997:327–328.
Sharma, L.K. Median cleft of the upper lip. Plast Reconstr Surg. 1974; 53:155.
Shear, M. Cysts of the Oral Region. Bristol: John Wright; 1983.
Shear, M. Hereditary hypocalcification of enamel. J Dent Assoc S Afr. 1954; 9:262.
Shelling, D.H., Anderson, G.M. Relation of rickets and vitamin D to the incidence of dental caries, enamel hypoplasia and malocclusion in children. J Am Dent Assoc. 1936; 23:840.
Shields, E.D., Bixler, D. El-Kafrawy. AMA proposed classification for heritable human dentine defect with a description of a new entity. Arch Oral Biol. 1973; 18:543–553.
Shiratsuchi, Y., Tashiro, H., Yasuda, K., Kanda, S. Posterior lingual mandibular bone depression. Int J Oral Maxillofac Surg. 1986; 155:98–101.
Shokeir, M.H.K. Dentinogenesis imperfecta: severe expression in a probable homozygote. Clin Genet. 1972; 3:442–447.
Shprintzen, R.J., Goldberg, R.B., Sidoti, E.J. The penetrance and variable expression of the Van der Woude syndrome: implications for genetic counseling. Cleft Palate J. 1980; 17(1):52–57.
Siddiqui, A., Pensler, J.M. The efficacy of tongue resection in treatment of symptomatic macroglossia in the child. Ann Plast Surg. 1990; 25(1):14–17. [Jul].
Siegal, M.J., Mock, D. Symptomatic benign migratory glossitis: report of two cases and literature review. Pediatr Dent. 1992; 14:392–396.
Simonsen, R.J., Kanca, J. Surface hardness of posterior composite resins using supplemental polymerisation after simulated occlusal adjustment. Quintessence International. 1986; 17(10):631–633.
Sklavounou, A., Laskaris, G. Oral psoriasis: report of a case and review of the literature. Dermatological. 1990; 180:157–159.
Skouteris, C.A., Patterson, G.T., Sotereanos, G.C. Benign cervical lymphoepithelial cyst: report of cases. J Oral Maxillofac Surg. 1989; 47:1106–1112.
Slavotinek, A.M., Collins, M.T., Muenke, M. Non-syndromic hemihyperplasia in a male and his mother. Am J Med Genet. 2003; 121A:47–51.
Smiley, G.R. A possible genesis for cleft plate formation. Plast Reconstr Surg. 1972; 50:390.
Smith, D.W. Dysmorphology (teratology). J Pediatr. 1966; 69:1150.
Smith, F.B. Benign lymphoepithelial lesion and lymphoepithelial cyst of the parotid gland in HIV infection. Prog AIDS Pathol. 1990; 2:61–72.
Smith, A.A., Farbman, A., Dancis, J. Tongue in familial dysautonomia: a diagnostic sign. Am J Dis Child. 1965; 110:152.
Smith, M.C., Smith, H.V. Mottled enamel of deciduous teeth. J Am Dent Assoc. 1935; 22:814.
Smith, M.C., Lanz, E., Smith, H.V. The cause of mottled enamel. J Dent Res. 1932; 12:149.
Smith, N.J.D., Smith, P.B. Congential absence of major salivary glands. Br Dent J. 1977; 142:259.
Snead, M.L., Lau, E.C., Fincham, A.G., Zeichner-David, M. Of mice and men: anatomy of the amelogenin gene. Connect Tissue Res. 1989; 22(1–4):101–109.
Som, P.M. Head and neck imaging, 2nd, St. Louis: CV Mosby; 1991:389.
Spouge, J.D., Feasby, W.H. WH. Erupted teeth in the newborn. Oral Surg. 1966; 22:198.
Spriestersbach, D.C., Spriestersbach, B.R., Moll, K.L. Incidence of clefts of the lip and palate in families with children with clefts and families with children without clefts. Plast Reconstr Surg. 1962; 29:392.
Stafne, E.C., Gibilisco, J.A. Calcifications of the dentinal papilla that may cause anomalies of the roots of the teeth. Oral Surg Oral Med Oral Pathol. 1961; 14:683–686.
Stafne, E.C. Bone cavities situated near the angle of the mandible. J Am Dent Assoc. 1969; 29:1942.
Stafne, E.C., Austin, L.T., Gardner, B. Median anterior maxillary cysts. J Am Dent Assoc. 1936; 23:801.
Stafne, E.C. Supernumerary teeth. Dent Cosmos. 1932; 74:653.
Starink, T.M. Focal epithelial hyperplasia of the oral mucosa: report of two cases from the Netherlands and review of the literature. Br J Dermatol. 1977; 96:375–380.
Starkey, P.E., Shafer, W.G. Eruption sequestra in children. J Dent Child. 1963; 30:84.
Stein, S.L., Mancini, A.J. Melkersson-Rosenthal syndrome in childhood: successful management with combination steroid and minocycline therapy. J Am Acad Dermatol. 1999; 41(5):746–748. [Pt1 Nov].
Stein, G. Enamel damage of systemic origin in premature birth and disease of early infancy. Am J Orthod Oral Surg. 1947; 33:831.
Stewart, R.E., Prescott, G.H. Oral Facial Genetics. St Louis: CV Mosby, 1976;. [(eds)].
Stout, F.W., Collett, W.K. Etiology and incidence of the median maxillary anterior alveolar cleft. Oral Surg. 1969; 28:66.
Strean, L.P., Peer, L.A. Stress as an etiologic factor in the development of cleft palate. Plastic Reconstr Surg. 1956; 18:1.
Suda, Y., Nakabayashi, J., Matsuo, I., Aizawa, S. Functional equivalency between Otx2 and Otx1 in development of the rostral head. Development. 1999; 126:743–757.
Sujansky, E., Smith, A.C.M., Prescott, K.E., Freehauf, C.L. Natural history of therecombinant (8) syndrome. Am J Med Genet. 1993; 47:512–525.
Sussman, G.L., Yang, W.H., Steinberg, S. Melkersson-Rosenthal syndrome: clinical, pathologic, and therapeutic considerations. Ann Allergy. 1992; 69(3):187–194.
Sutton, R.L. Cheilitis glandularis apostematosa (with case report). J Cutan Dis. 1909; 27:151–154.
Sutton, R.L. The symptomatology and treatment of three common diseases of the vermilion border of the lip. Int Clin. 1914; 3(24):123–128.
Swanson, K.S., Kaugars, G.E., Gunsolley, J.C. Nasopalatine duct cyst: an analysis of 334cases. J Oral Maxillofac Surg,. 1991; 49(3):268–271. [Mar].
Swerlick, R.A., Cooper, P.H. Cheilitis glandularis: a re-evaluation. J Am Acad Dermatol. 1984; 10(3):466–472. [Mar].
Takagi, R., Ohashi, Y., Suzuki, M. Squamous cell carcinoma in the maxilla probably originating from a nasopalatine duct cyst: report of case. J Oral Maxillofac Surg. 1996; 54(1):112–115. [Jan].
Takagi, Y., Sasaki, S.A. Probable common disturbance in the early stage of odontoblast differentiation in dentinogenesis imperfecta type I and type II. J Oral Path. 1988; 17:208–212.
Takeda, Y. Intra-osseous squamous cell carcinoma of the maxilla: probably arisen from non-odontogenic epithelium. Br J Oral Maxillofac Surg. 1991; 29(6):392–394. [Dec].
Taki, W., Picard, L., Kikuchi, H. Advances in International Neuroradiology and Intravascular Neuroradiology,. Amsterdam: Elsevier Science, 1996; 281–284.
Taylor, W.B., Lane, D.J. Congential fistulas of the lower lip. Arch Dermatol. 1966; 94:421.
Terry, B.R., Bolanos, O.R. A diagnostic case involving an incisive canal cyst. J Endod. 1989; 15(11):559–562.
Thesleff, I. Homeobox genes and growth factors in the regulation of craniofacial and tooth morphogenesis. Acta Odontol Scand. 1995; 53:129–134.
Thoma, K.H., Goldman, H.M. Oral Pathology, 5th. St. Louis: CV Mosby, 1960.
Thoma, K.H. Case report of a so-called latent bone cyst. Oral Med Oral Pathol. 1955; 8:963–966.
Thoma, K.H. Facial cleft or fissural cysts. J Orthod Oral Surg. 1937; 23:83.
Thomas, V.L., David, J.P. Erosion of the scapula by a benign lipoma: computed tomography diagnosis. J Comput Assist Tomogr. 1979; 3:679–680.
Tobias, N. Inheritance of scrota tongue. Arch Dermatol Syph. 1946; 52:266.
Toller, P.A. A clinical report on six cases of amelogenesis imperfecta. Oral Surg. 1959; 12:325.
Tolman, D.E., Stafne, E.C. Developmental bone defects of the mandible. Oral Surg Oral Med Oral Pathol. 1967; 24:488–490.
Treco, D.A. Preparation of yeast DNA, RNA and proteins. In: Dracopoli N.C., Haines J.L., Korf B.R., eds. Current Protocols in Human Genetics. New York: Wiley-Liss; 1994:11.1–13.11.5.
Trelles, M.A. Treatment of melanotic spots in the gingiva by argon laser. J Oral Maxillofac Surg. 1993; 51:759–761.
Tsang, W.Y., Chan, J.K. The family of epithelioid vascular tumors. Histol Histopathol. 1993; 8(1):187–212. [Jan].
Ueyama, Y., Mano, T., Nishiyama, A. Effects of surgical reduction of the tongue. Br J Oral Maxillofac Surg. 1999; 37(6):490–495. [Dec].
van der Waal, I., Beemster, G., van der Kwast, A.M. Median rhomboid glossitis caused by Candida. Oral Surg. 1979; 47:31.
Van der Woude, A. Fistula labii inferioris congenita and its association with cleft lip and palate. Am J Hum Genet. 1954; 6:224–256.
van Gool, A.V. Injury to the permanent tooth germ after trauma to the deciduous predecessor. Oral Surg. 1973; 35:2.
Vasconcelos, R., de Aguiar, M.F., Castro, W. Retrospective analysis of 31 cases of nasopalatine duct cyst. Oral Dis. 1999; 5(4):325–328.
Veau V. Division Palatine; Anatomie, Chirurgie, Phonetique Paris, Masson et Cie, 1931.
Velez, A., Alamillos, F.J., Dean, A. Congenital lower lip pits (Van der Woude syndrome). J Am Acad Dermatol. 1995; 32(3):520–521.
Via, W.F., Jr. Enamel defects induced by trauma during tooth formation. Oral Surg. 1968; 25:49.
Vichi, M., Franchi, L. Abnormalities of the maxillary incisors in children with cleft lip and palate. ADSC J Dent Child. 1995; 62:412–417.
Vickers, R.A., Gorlin, R.J., Smark, E.A. Lymphoepithelial lesions of the oral cavity: report of four cases. Oral Surg. 1963; 16:1214.
Vickey, I.M. Hemifacial atrophy. Br J Oral Surg. 1972; 9:102–109.
Vignale, R., Araujo, J., Pascal, G. Van der Woude syndrome: a case report. Pediatr Dermatol. 1998; 15(6):459–463. [Nov–Dec].
Viljoen, D., Pearn, J., Beighton, P. Manifestations and natural history of idiopathic hemihypertrophy: a review of eleven cases. Clin Genet. 1984; 26(##):81–86.
Viraben, R. Focal epithelial hyperplasia (Neck disease) associated with AIDS. Dermatology. 1996; 193:261–262.
Virtanen, I., Laine, P.A. Study on the relative frequency of the globulomaxillary cyst. Suom Hammaslaak Toim. 1961; 57:191.
Von Volkman, R. Einege Falle von Cheilitis Glandularis Apostematosa (Myxadenitis Labialis). Virchows Arch Pathol Anat. 1870; 142–144. [A],
Vorhies, J.M., Gregory, G.T., McDonald, R.E. Ankylosed deciduous molars. J Am Dent Assoc. 1952; 44:68.
Walsh, F.B. Facial hemiatrophy: report of 2 cases. Am J Ophthal. 1939; 22:1–10.
Walton, J.L., Witkop, C.J., Jr., Walker, P.O. Odontodysplasia. Oral Surg. 1978; 46:476.
Ward, G.E., Hendrick, J.W., Chambers, R.G. Thyroglossal tract abnormalities: cysts and fistulas. Surg Gynecol Obstet. 1949; 89:727.
Warkany, J., Deuschle, F.M. Congenital malformations induced in rats by maternal riboflavin deficiency: dentofacial changes. J Am Dent Assoc. 1955; 51:139.
Wartenberg, R. Progressive facial hemiatrophy. Arch Neurol Psychiat. 1945; 54:75–96.
Watkins, K.V., Chaudhry, A.P., Yamane, G.M. Benign focal melanotic lesions of the oral mucosa. J Oral Med. 1984; 39:91–96.
Watne, A.L., Core, S.K., Carrier, J.M. Gardner’s syndrome. Sure Gynecol Obstet. 1975; 141:53.
Weatherell, J.A., Weidmann, S.M., Eyre, D.R. Histological appearance and chemical composition of enamel proteins from mature human molars. Caries Research. 1968; 2(4):281–293.
Weathers, D.R., Baker, G., Archard, H.O., Burkes, E.J., Jr. Psoriasiform lesions of the oral mucosa (with emphasis on ‘ectopic geographic tongue’). Oral Surg. 1974; 37:872.
Weathers, D.R., Fine, R.M. Thrombosed varix of oral cavity. Arch Dermatol. 1971; 104:427.
Weathers, D.R., Corio, R.L., Crawford, B.E., Giansanti, J.S. The labial melanotic macule. Oral Surg. 1976; 42:196.
Webster, R.C. Cleft palate. Oral Surg. 1948; 1:647. [943, 2: 99, 485, 1949].
Weidmann, S.M., Eyre, D.R. Amino acid composition of enamel protein in the fully developed human tooth. Caries Research. 1967; 1(4):349–355.
Weinberg, S., Moncarx, V., Van de Mark, T.B. Midline cleft of the mandible: review of literature and report of case. J Oral Surg. 1972; 30:143.
Weinmann, J.P., Svoboda, J.F., Woods, R.W. Hereditary disturbances of enamel formation and calcification. J Am Dent Assoc. 1945; 32:397.
Weir, T.W., Johnson, W.C. Cheilitis glandularis. Arch Dermatol. 1971; 103:433.
Weissenbach, J., Gyapay, G., Dib, C. A second-generation linkage map of the human genome. Nature. 1992; 395:794–801.
Weitzner, S. Lymphoepithelial (branchial) cyst of parotid gland. Oral Surg. 1973; 35:85.
Wesley, R.K., Delaney, J.R., Pansler, L. Mucocutaneous melanosis and gastrointestinal polyposis (Peutz-Jeghers syndrome): clinical considerations and report of a case. J Dent Child. 1977; 44:131.
Wesley, R.K., Wysocki, G.P., Mintx, S.M., Jackson, J. Dentin dysplasia type I. Oral Surg. 1976; 41:516.
West, P.M.H., Love, D.R., Stapleton, P.M., Winship, I.M. Paternal uniparental disomy in monozygotic twins discordant for hemihypertrophy. J Med Genet. 2003; 40:223–226.
Westerman, A.M., Entius, M.M. de Baar E: Peutz-Jeghers syndrome: 78-year follow-up of the original family. Lancet 10. 1999; 353(9160):1211–1215.
Whelton, H. The anatomy and physiology of salivary glands. In: Edgar W.M., O’Mullane D.M., eds. Saliva and Oral Health. London: Br Dent Assoc; 1996:1–8.
White, D.K., Lucas, R.M., Miller, A.S. Median mandibular cyst: review of the literature and report of two cases. J Oral Surg. 1975; 33:372.
Wiesenfeld, D., Ferguson, M.M., Mitchell, D.N. Oro-facial granulomatosis—a clinical and pathological analysis. Q J Med. 1985; 54(213):101–113.
Wiesenfeld, D., Iverson, E.S., Ferguson, M.M., Hardman, F.G. Familial parotid gland aplasia. J Oral Med. 1985; 40:84–85.
Wiessinger, D., Miller, M. Breastfeeding difficulties as a result of tight lingual and labial frena: a case report. J Human Lactat. 1995; 11(4):313–316.
Williams, A.J., Wray, D., Ferguson, A. The clinical entity of orofacial Crohn’s disease. Q J Med. 1991; 79(289):451–458.
Williams, P.M., Greenberg, M.S. Management of cheilitis granulomatosa. Oral Surg Oral Med Oral Pathol. 1991; 72(4):436–491.
Wilson, G.W., Steinbrecher, M. Hereditary hypoplasia of the dentin. J Am Dent Assoc. 1929; 16:866.
Winchester, L., Scully, C., Prime, S.S., Eveson, J.W. Cheilitis glandularis: a case affecting the upper lip. Oral Surg Oral Med Oral Pathol. 1986; 62(6):654–656. [Dec].
Winter, G.B.Principles of Exodontia as Applied to the Impacted Mandibular Third Molar. St Louis: American Medical Book Company, 1926.
Winter, G.R., Maiocco, P.D. Osteogenesis imperfecta and odontogensis imperfecta. Oral Surg. 1949; 2:782.
Winzer, M., Gilliar, U., Ackerman, A.B. Hairy lesions of the oral cavity: clinical and histopathologic differentiation of hairy leukoplakia from hairy tongue. Am J Dermatopathol,. 1988; 10(2):155–159. [Apr].
Winzer, M., Gilliar, U. Hairy tongue and hairy oral leukoplakia—a differential histopathologic diagnosis. Z Hautkr. 1988; 15:517–520. [63 6 Jun].
Witkop, C.J., Jr., Niswander, J.D. Focal epithelial hyperplasia in Central and South American Indians and Latinos. Oral Surg Oral Med Oral Pathol. 1965; 20:213–217.
Witkop, C.J., Jr. Partial expression of sex-linked recessive amelogenesis imperfect in females compatible with the Lyon hypothesis. Oral Surg Oral Med Oral Pathol. 1967; 23(2):174–182.
Witkop, C.J., Jr. Hereditary defects in enamel and dentin. Acta Genet Statist Med. 1957; 7:236–239.
Witkop, C.J., Jr. Hereditary defects of dentin. Dent Clin North Am. 1975; 19:25–45.
Witkop, C.J., Jr., Rao, S. R. Inherited defects in tooth structure: birth defects. Orig Art Ser. 1971; VII(7):153–184.
Witkop, C.J., Jr.Genetics and Dental Health. New York: McGraw-Hill, 1962. [(ed)].
Witkop, C.J., Jr., Niswander, J.D. Focal epithelial hyperplasia in Central and South American Indians and Ladinos. Oral Surg. 1965; 20:213.
Witkop, C.J., Jr., Sauk, J.J., Jr., Heritable defects of enamel: in RE Stewart and GH Prescott (eds)Oral Facial Genetics. St Louis: Mosby, 1976.
Witsenburg, B., Boering, G. Eruption of impacted permanent upper incisors after removal of supernumerary teeth. Int J Oral Surg. 1981; 10:423–431#.
Wolf, J., Mattila, K., Ankkuriniemi, O. Development of a Stafne mandibular bone cavity. Surg Oral Med Oral Pathol. 1986; 61:519–521.
Wolford, L.M., Cottrell, D.A. Diagnosis of macroglossia and indications for reduction glossectomy. Am J Orthod Dentofacial Orthop. 1996; 110(2):170–177. [Aug].
Wong, F.K., Gustafsson, B. Popliteal pterygium syndrome in a Swedish family: clinical findings and genetic analysis with the Van der Woude syndrome locus at 1q32– q41. Acta Odontol Scand. 2000; 58:85–88.
Wong, G.A., Shear, N.H. Melkersson-Rosenthal syndrome associated with allergic contact dermatitis from octyl and dodecyl gallates. Contact Dermatitis. 2003; 49(5):266–267. [Nov].
Woodbourne, A.R., Philpott, O.S. Cheilitis glandularis: a manifestation of emotional disturbance. Arch Dermatol Syph. 1950; 62:820.
Woolf, C.M., Woolf, R.M., Broadbent, T.R. Genetic and nongenetic variables related to cleft lip and palate. Plast Reconstr Surg. 1963; 32:65.
Worsase, N., Pindborg, J. Granulomatous gingival manifestations of Melkersson-Rosenthal syndrome. Oral Surg. 1980; 49:131.
Wright, J.T., Aldred, M.J., Crawford, P.J., Kirkham, J. Enamel ultrastructure and protein content in X-linked amelogenesis imperfecta. Oral Surg Oral Med Oral Patholo. 1993; 76(2):192–199.
Wright, J.T., Robinson, C., Kirkham, J. Enamel protein in smooth hypoplastic amelogenesis imperfecta. Pediatric Dentistry. 1992; 14(5):331–337.
Wright, J.T. Analysis of kindred with amelogenesis imperfecta. J Oral Patholo. 1985; 14(5):366–374.
Wright, B.A. Median rhomboid glossitis: not a misnomer. Oral Surg. 1978; 46:806.
Wright, S. On the genetics of subnormal development of the head (otocephaly) in the guinea pig. Genetics. 1934; 19:471–505.
Wysocki, G.P. The differential diagnosis of globulomaxillary radiolucencies. Oral Surg. 1981; 51:281.
Yacobi, R., Brown, D.A. Cheilitis glandularis: a pediatric case report. J Am Dent Assoc. 1989; 118(3):317–318. [Mar].
Yip, W.K. The prevalence of dens evaginatus. Oral Surg. 1974; 38:80.
Zackin, S.J., Weisberger, D. Hereditary gingival fibromatosis. Oral Surg. 1961; 14:828.
Zaus, E., Teuscher, G.W. Report on three cases of congential dysfunction of the major salivary glands. J Dent Res. 1940; 19:326.
Zegarelli, E.V., Kesten, B.M., Kutscher, A.H. Melanin spots of the oral mucosa and skin associated with polyps. Oral Surg. 1954; 7:972.
Zimmer, W.M., Rogers, R.S., III., Reeve, C.M., Sheridan, P.J. Orofacial manifestations of Melkersson-Rosenthal syndrome: a study of 42 patients and review of 220 cases from the literature. Oral Surg Oral Med Oral Pathol. 1992; 74(5):610–619.
Zunt, S.L., Tomich, C.E. Erythema migrans–a psoriasiform lesion of the oral mucosa. J Dermatol Surg Oncol. 1989; 15:1067–1070.