Chapter 24 Overview of the Heart and Drugs Affecting Cardiac Function
Dyslipidaemia, or increased plasma concentrations of cholesterol and triglycerides in the body, has been clinically associated with atherosclerosis. Atherosclerosis is a disorder of the arteries that is characterised by cholesterol deposits in the lining of the blood vessels, which eventually produce degenerative changes and obstruct blood flow. Atherosclerosis can result in angina, heart failure, myocardial infarction, cerebral artery disease and renal artery insufficiency. It is also a factor in hypertension. The treatment guidelines for the management of dyslipidaemia include dietary and lifestyle modifications and drug treatment. The main classes of lipid-lowering drugs are the ‘statins’, bile acid-binding resins and fibrates.
Key abbreviations
HMG-CoA 3-hydroxy-3-methylglutaryl coenzyme A
IDL intermediate-density lipoproteins
Key background
Dyslipidaemia
Dyslipidaemia is a metabolic disorder characterised by increased concentrations of lipids and lipoproteins. Lipidlowering drugs are used along with dietary modifications to treat dyslipidaemia. Clinical and experimental studies have provided evidence of an important relation between high levels of circulating triglycerides and cholesterol and atherosclerosis. Atherosclerosis, a disorder that involves large- and medium-sized arteries, is characterised by cholesterol deposits in the arterial wall, which eventually produce degenerative changes and obstruct blood flow.
Atherosclerosis is a causative factor in coronary artery disease (CAD), which can result in angina, heart failure and myocardial infarction; cerebral arterial disease that results in senility or cerebrovascular accidents; peripheral arterial occlusive disease, which can cause gangrene and loss of limb; and renal arterial insufficiency. It is also a factor in hypertension. Intensive research to develop effective and safer lipid-lowering drugs is ongoing.
Lipids do not circulate freely in the bloodstream. Instead, they are transported as complexes called lipoproteins. Lipoproteins are composed of an interior core, consisting of cholesteryl esters and triglycerides, which are covered by a layer of phospholipids, free cholesterol and apolipoproteins. Hyperlipo proteinaemias are always associated with an increased concentration of one or more lipoproteins.
Classification of lipoproteins
The three primary lipoproteins found in the blood of fasting individuals are very-low-density lipoproteins (VLDLs), low-density lipoproteins (LDLs) and highdensity lipoproteins (HDLs). The intermediate-density lipoproteins (IDLs) have short half-lives (minutes to a few hours) and their concentrations in plasma tend to be very low (Table 24-1).
Table 24-1 Lipoproteins: core lipids and lipid transported
| LIPOPROTEINS | CORE LIPID | LIPID TRANSPORTED |
| Chylomicrons/chylomicron remnants | Dietary triglycerides | Dietary triglyceride |
| VLDL | Endogenous triglyceride | Endogenous triglyceride |
| IDL | Endogenous cholesteryl esters and triglycerides | Endogenous cholesterol |
| LDL | Endogenous cholesteryl esters | Endogenous cholesterol |
| HDL | Endogenous cholesteryl esters | Endogenous cholesterol from periphery |
VLDLs contain a large amount of triglyceride (50%–65%) and 20%–30% cholesterol, and are formed in the liver from endogenously synthesised triglycerides, cholesterol and phospholipid. These lipoproteins contain 15%–20% of the total blood cholesterol and most of the triglyceride found in the body. The apolipoproteins apoB-100, apoE and apoC-I-III are synthesised in the liver and incorporated into VLDL. After VLDL particles are secreted from the liver into the circulation, their triglyceride content is released as a result of the action of the enzyme lipoprotein lipase (LPL), which is located in the endothelium of adipose, muscle and cardiac tissue capillaries. As the triglycerides are hydrolysed by LPL the resulting free fatty acids are taken up by adjacent tissues. Drugs that enhance the action of LPL will lower plasma triglyceride levels.
When triglyceride hydrolysis is almost complete the remnant VLDL (termed IDL) is released from the capillary endothelium and re-enters the circulation. Approximately 40%–50% of the IDL is cleared from plasma by the liver via LDL receptors, which recognise the apoB-100 and apoE components of the remnants. The remainder of the IDL is converted to the cholesterol-rich lipoprotein LDL, which contains 60%–70% of total blood cholesterol. LDL particles have a half-life of 1–2 days, which accounts for their high concentration in plasma in comparison to VLDL and IDL. The quantity and density of systemic LDL particles correlates with the risk of atherosclerosis, and elevated LDL levels indicate that an individual has a greater risk of developing atherosclerosis. LDL (∼75%) is cleared from plasma mainly via hepatic LDL receptors and defects in the LDL receptor gene are associated with high plasma concentrations of LDL and familial hypercholesterolaemia.
HDLs are the smallest and most dense lipoproteins and can be separated based on density into HDL2 (larger and more cholesterol-rich) and HDL3 particles (smaller, less cholesterol-rich). Their function is to transfer cholesterol from peripheral cells to the liver either directly or by exchanging cholesteryl esters for triglycerides from LDL and VLDL. This exchange is mediated by cholesteryl ester transfer protein and accounts for approximately 66% of the removal of cholesterol from HDL. The LDL particles are then cleared from plasma by LDL receptors principally in the liver, and the level of hepatic LDL receptors generally controls the level of circulating LDL in humans. High levels of HDL are considered beneficial and decrease the risk of coronary heart disease. This transport mechanism prevents the accumulation of cholesterol in the arterial walls, thereby providing protection against the development of atherosclerosis.
Chylomicrons are large particles that transport dietary cholesterol and fatty acids absorbed from the gastrointestinal tract to the liver. This is known as the exogenous pathway of lipid transport, whereas the lipoproteins transporting cholesterol between the liver and peripheral cells are part of the endogenous pathway. Chylomicrons consist mainly of triglycerides (85%–95%) and are produced in the small intestine during absorption of a fatty meal. They are cleared from the bloodstream by LPL after 12–14 hours. The chylomicron that remains following the removal of the triglyceride content is cleared rapidly by the liver and is not converted into LDL.
Apolipoproteins
Lipoproteins contain proteins on their surface called apolipoproteins (apo). These proteins have a variety of functions: they serve as ligands for cell receptors, activate enzymes involved in lipoprotein metabolism and provide structure for the lipoprotein. If apolipoprotein metabolism is impaired, an increased risk of atherosclerosis exists; thus plasma concentrations of apolipoproteins are important in evaluating lipid disorders. The apolipoproteins include apoA-I, apoA-II, apoA-IV, apoA-V, apoB-100, apoB-48, apoC-I, apoC-II, apoC-III, apoE and apo(a). The latter is associated with Lp(a) a lipoprotein (structurally related to plasminogen) that promotes thrombosis. Apolipoprotein A-I is thought to confer the beneficial effect of HDL; HDL particles that have both A-I and A-II appear not to be as atheroprotective. In contrast, a deficiency of the C-II apolipoprotein in VLDL particles results in impaired triglyceride metabolism and hypertriglyceridaemia.
Figure 24-1 illustrates cholesterol transport in tissues and indicates the sites of action of the lipid-lowering drugs discussed in the following sections. Dietary fats and cholesterol are transported into the system as chylomicrons via the exogenous pathway. In the endogenous pathway the liver synthesises cholesterol and triglycerides, which are transported from the liver to peripheral tissues as VLDL particles. The function of HDL is to carry about 25% of plasma cholesterol from the periphery back to the liver, where it is processed into bile acids. As the cholesterol HDL carries is ultimately for excretion, it is known as ‘good’ cholesterol. In contrast, LDL carries more than 50% by weight of cholesterol and its relation with the development of atherosclerosis has resulted in its label of ‘bad’ cholesterol.
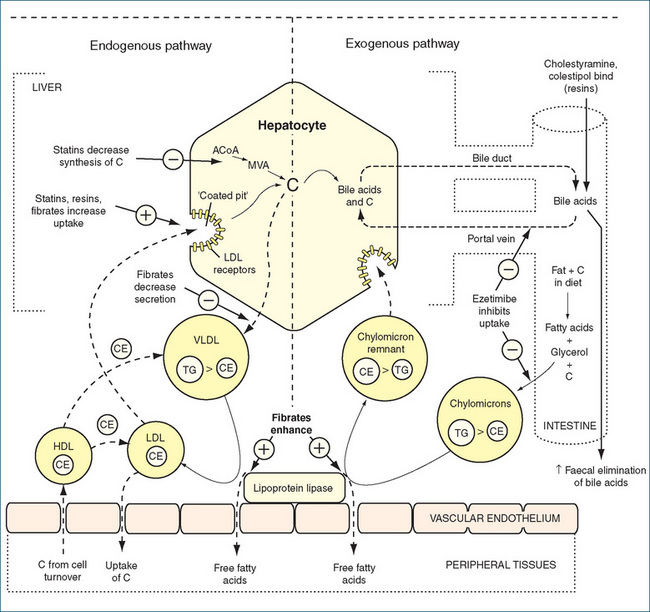
Figure 24-1 Schematic diagram of cholesterol transport in the tissues, with sites of action of the main drugs affecting lipoprotein metabolism. C = cholesterol; CE = cholesteryl ester; HDL = high-density lipoprotein; HMG-CoA reductase = 3-hydroxy-3-methyl-glutaryl- CoA reductase; LDL = low-density lipoprotein; MVA = mevalonate; TG = triglyceride; VLDL = very-low-density lipoprotein.
Source: adapted from Rang et al. 2007. Reproduced with permission.
Plasma lipoproteins are usually in a state of dynamic equilibrium. When the liver and tissues outside the liver need cholesterol they increase the synthesis of LDL receptors on their respective cell surfaces (Figure 24-1). These receptors are necessary for the binding of LDL, thus enabling the release of free fatty acids. When the cellular need for cholesterol is met, the synthesis of LDL receptors decreases, and this controls the plasma level of LDL. Modulation of the number of hepatic LDL receptors is an integral part of the therapeutic approach to the management of hypercholesterolaemia.
Hyperlipoproteinaemias
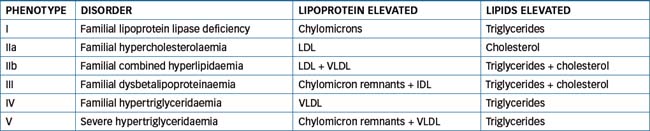
Dyslipidaemias can be classed as primary or secondary. The primary, or genetically determined, hyper lipoproteinaemia forms are classified into six phenotypes, depending on the lipoprotein particle elevated (Table 24-2). Factors such as diabetes mellitus, obesity, hypothyroidism, nephrotic syndrome, excess alcohol consumption and drug treatment (e.g. corticosteroids, thiazide diuretics) constitute the secondary causes of dyslipidaemia. In these cases, investigation of underlying disease pathology or current drug treatment is necessary before instituting lipid-lowering drug therapy.
 Management strategies for dyslipidaemia
Management strategies for dyslipidaemia
Treatment guidelines
Dietary modification is important in the treatment of high LDL-cholesterol (LDL-C) levels. The National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand Position Statement on Lipid Management 2005 recommends a dietary model based on the consumption of ‘a low saturated fat eating plan incorporating moderate amounts of polyunsaturated and monounsaturated fats and oils, marine omega-3s via two to three fish meals per week and at least 2 g of plant omega-3s (alpha-linolenic acid) per day, and a wide variety of fruits, vegetables and wholegrain cereal products’. In Australia, the Pharmaceutical Benefits Scheme (PBS, refer to Chapter 2) criteria for subsidy of lipid-modifying drugs now reflect treatment according to risk of future cardiovascular events. Persons eligible for subsidy at any cholesterol concentration include persons with symptomatic coronary, or cerebrovascular or peripheral vascular disease; or at high risk (e.g. diabetes mellitus or with a family history of symptomatic coronary heart disease). Identification of higher-risk individuals can be aided by a number of tools that can be found online (http://www.nps.org.au/health_professionals/tools/cardiovascular_risk_calculator).
A 6-week period of dietary modification is still required before a person considered at a lower risk is eligible for subsidised drugs available through the PBS (Table 24-3). The Australian PBS guidelines have been developed from data on levels of risk and controlled clinical trials.
Table 24-3 Australian pbs criteria for subsidy of lipid-lowering drugs
| LOWER RISK PATIENT GROUP | CHOLESTEROL THRESHOLDS FOR PBS SUBSIDY |
| Patients with diabetes (apart from high-risk groups) | Total-C > 5.5 mmol/L |
| Aboriginal and Torres Strait Islander people without diabetes or other high risk | Total-C > 6.5 mmol/L or |
| total-C > 5.5 mmol/L and HDL-C < 1 mmol/L | |
| Patients with hypertension | Total-C > 6.5 mmol/L or |
| total-C > 5.5 mmol/L and HDL-C < 1 mmol/L | |
| Patients with HDL-C < 1 mmol/L | Total-C > 6.5 mmol/L |
| Familial hypercholesterolaemia identified by: | ≤ 18 years of age |
| LDL-C > 4 mmol/L | |
| >18 years of age | |
| LDL-C > 5 mmol/L or | |
| total-C > 6.5 mmol/L or | |
| total-C > 5.5 mmol/L and HDL-C < 1 mmol/L | |
| Family history of symptomatic CHD: | ≤ 18 years of age |
| LDL-C > 4 mmol/L | |
| >18 years of age | |
| LDL-C > 5 mmol/L or | |
| total-C > 6.5 mmol/L or | |
| total-C > 5.5 mmol/L and HDL-C < 1 mmol/L | |
| High cholesterol in patients not eligible above who are: | Total-C > 7.5 mmol/L or |
| triglycerides > 4 mmol/L | |
| Patients not otherwise included | Total-C > 9 mmol/L or |
| triglycerides > 8 mmol/L |
Source: National Prescribing Service RADAR February 2007. Available: http://nps.org.au/health_professionals/publications/nps_radar [24 August 2009].
In the absence of satisfactory reduction of high plasma lipid levels through exercise, diet and lifestyle modification, lipid-lowering drugs offer health-care professionals a management strategy for the treatment of dyslipidaemia. This is of proven benefit in individuals with high cardiovascular risk factors. Current guidelines recommend target levels of LDL-C <2.0 mmol/L in high risk patients with existing coronary heart disease and HDL-C >1.0 mmol/L and triglycerides <1.5 mmol/L (National Heart Foundation 2005).
The main classes of lipid-lowering drugs used are:
The choice of drug depends on the individual’s plasma lipid profile and whether the aim is to reduce the level of LDL-C (hypercholesterolaemia) or triglyceride (hypertriglyceridaemia), or both LDL-C and triglycerides (hyperlipidaemia). Table 24-4 provides an indication of the current drug treatment of dyslipidaemia.
Table 24-4 Current drug treatment of dyslipidaemia
| DISORDER | LIPID-LOWERING DRUGS |
| Hypercholesterolaemia | Statins, bile acid-binding resin, nicotinic acid, ezetimibe, fibrate |
| Hypertriglyceridaemia | Fibrate, fish oil, nicotinic acid |
| Mixed hyperlipidaemia | Guided by predominant disorder and including statins, fibrate, nicotinic acid |
Source: AMH 2010.
HMG-CoA reductase inhibitors
Numerous studies (e.g. WOSCOPS (1995), CURVES (1998), PRINCE (2001), REVERSAL (2003), JUPITER (2003)) have now established that ‘statins’ significantly reduce the risk of coronary heart disease, stroke and death in individuals undergoing treatment for an average of >5 years. This group of drugs, which includes atorvastatin, fluvastatin, pravastatin, rosuvastatin and simvastatin, was first introduced into clinical practice in the late 1980s. They are reversible competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase), the rate-limiting enzyme necessary for cholesterol biosynthesis. HMG-CoA reductase catalyses the conversion of HMG-CoA to mevalonic acid, which is an essential precursor in the synthesis of cholesterol (Figure 24-1). HMG-CoA reductase inhibitors are indicated for the treatment of primary hypercholesterolaemia (types IIa and IIb) caused by an elevated LDL-C level that is not controlled by diet or other treatment measures.
Simvastatin and pravastatin are chemically modified derivatives of the original fungal metabolite lovastatin while atorvastatin, fluvastatin and rosuvastatin are synthetic compounds. These drugs are particularly effective, lowering total cholesterol by 10%–45% and raising HDL by 2%–13%. The decrease in cholesterol production in the liver leads to increased expression of the LDL receptor gene with subsequent increased synthesis of LDL receptors, resulting in a greater clearance of LDL-C from the circulation. A modest increase also occurs in HDL as well as a slight reduction in plasma triglycerides. The widespread use of these drugs worldwide is attributable to their proven efficacy in randomised clinical trials in reducing CAD, angina, strokes and the need for angioplasty and coronary artery bypass grafts.
In addition to beneficial effects on lipid profiles, the statins have a number of other ‘antiatherosclerotic’ properties (Corsini et al 1999). Clearly these antiatheromatous actions may contribute to the overall beneficial effects observed with statin therapy. These include:
Pharmacokinetics
At the pharmacokinetic level the statins currently available have some important differences, which are summarised in Table 24-5. Atorvastatin, fluvastatin, pravastatin and rosuvastatin are administered as the active β-hydroxy acid form, whereas simvastatin is administered as an inactive lactone (a prodrug) that requires metabolic activation by the liver to the active hydroxy acid form. Although rosuvastatin is the most hydrophilic statin and simvastatin is more lipophilic, all statins are absorbed rapidly following oral administration reaching peak concentrations within 5 hours. Food variably affects absorption: there is no apparent effect on the absorption of simvastatin and rosuvastatin while bioavailability of fluvastatin, pravastatin and atorvastatin is decreased. However, the overall lipid-lowering efficacy of statins is not affected by whether the statin is taken with an evening meal or at bedtime.
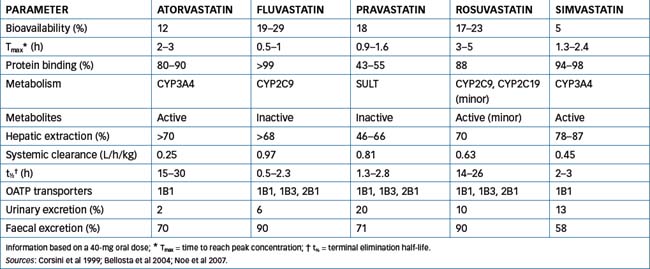
All of the statins have low systemic bioavailability, indicating extensive first-pass metabolism. With the exception of pravastatin, which is metabolised by cytosolic sulfotransferases (SULT), all statins are substrates for cytochrome P450 (CYP). Fluvastatin exhibits saturable first-pass metabolism and is metabolised by CYP2C9 and to a lesser extent by CYP3A4, atorvastatin and simvastatin are metabolised by CYP3A4 while rosuvastatin is metabolised to a minimal extent by CYP2C9 and CYP2C19.
Interaction with various drug transporters is complex. Atorvastatin is both a substrate and an inhibitor of the efflux transporter P-glycoprotein (refer to Chapter 6) and a substrate and an inhibitor of the sinusoidal uptake organic anion transporter OATP1B1. Pravastatin is a substrate of OATP1B1, OAT2B1 and OATP1B3 (all expressed on the basolateral membrane on human hepatocytes), which contributes to the efficient hepatic uptake of pravastatin. Hepatic uptake by the various transporters enhances the pharmacological effect of the statins by delivering the drugs directly to the liver as the target organ. Together, hepatic uptake and extensive first-pass metabolism minimise the ‘escape’ of the drug into the systemic circulation, hence limiting the adverse effect in muscle tissue.
The predominant route of excretion of the statins is via the faeces, with renal excretion accounting for <2% with atorvastatin, 6% with fluvastatin, 20% with pravastatin and 30% with rosuvastatin. An initial response is seen within 1–2 weeks, and the maximum therapeutic response occurs within 4–6 weeks of chronic drug administration. Bile acid-binding resins can impede absorption, so statins should be administered either 1 hour before or 4 hours after administration of the resin.
Drug interactions and adverse drug reactions
Potential drug interactions with the statins should always be considered, especially as these drugs are often one of multiple medications taken by people with cardiovascular disease. Interactions occur with drugs that can either inhibit or induce CYP and with other drugs that may be transported. Fluvastatin and rosuvastatin are metabolised by CYP2C9 and pravastatin by SULT and hence they are less subject to interactions than the other statins. With atorvastatin and simvastatin significant drug interactions occur with the macrolide antibiotics (azithromycin, erythromycin and clarithromycin), antifungal drugs (ketoconazole, itraconazole and fluconazole), HIV-protease inhibitors, calcium channel blockers (diltiazem and verapamil) and cyclosporin (see Drug Monograph 24-1). Grapefruit juice should be avoided if taking atorvastatin or simvastatin because grapefruit juice inhibits metabolism of both of these drugs by CYP3A4. Inhibition of metabolism leads to an increased plasma drug concentration and the likelihood of adverse effects.
Although these drugs are well tolerated, adverse reactions include stomach cramps or pain, rash, constipation or diarrhoea, nausea, headaches, myalgia and, rarely, myopathy, rhabdomyolysis, alopecia, impotence, gynaecomastia, anaphylaxis and angio-oedema. Muscle pain or weakness, severe tiredness or flu-like symptoms should be reported to the treating doctor. Long-term safety data with rosuvastatin is currently limited. Use with caution in situations of impaired hepatic and renal function. Avoid use in people with hypersensitivity to any HMG-CoA reductase inhibitor, organ transplant recipients receiving immunosuppressant drugs and people with any disease state or condition that may predispose them to renal failure.
Statins and adverse muscular effects
In general the statins are well tolerated. However, the most common adverse drug reaction relates to muscle toxicity (Table 24-6). This ranges from myalgia through to myopathy, myositis and rhabdomyolysis. The incidence of myalgia is ∼190 per 100,000 person-years while rhabdomyolysis has an incidence of 1.6–4.4 per 100,000 person-years (Law & Rudnicka 2006; Graham et al 2004). Currently, there is no consensus on the exact definition of statin-induced myopathy and the underlying mechanisms are poorly understood (Joy & Hegele 2009). Predisposing factors include older age, female, excessive alcohol intake and concomitant administration of fibrates, cyclosporin, protease inhibitors, macrolide antibiotics or amiodarone. Statin treatment should be ceased if persistent unexplained muscle pain occurs and the creatine kinase level is elevated. In the absence of any identified cause of the myopathy (e.g. hypothyroidism, neuromuscular diseases), statin therapy may be recommenced after a month if the creatine kinase level is within the normal range.
Table 24-6 Overview of the symptoms and progression of statin-induced muscle toxicity
| PROGRESSIVE SYMPTOMS | CHARACTERISTICS |
| Myalgia—diffuse muscle discomfort involving predominantly proximal muscles | |
| Myopathy—muscle pain with creatine kinase level >10 x upper limit of normal | |
| Myositis—characterised by muscle weakness with or without elevated creatine kinase levels | |
| Rhabdomyolysis—characterised by muscle destruction, presence of myoglobin in urine, muscle pain and swelling and elevated creatine kinase |
Adapted from: Evans 2004.
Hepatic cholesterol synthesis is maximal in the early hours of the morning (midnight to 2 am) and statins with short half-lives (fluvastatin, pravastatin and simvastatin) are taken in the evening to maximise efficacy. The recommended adult doses are: atorvastatin, 10 mg daily, titrated monthly as needed (range 10–80 mg/day); fluvastatin, 40 mg daily in the evening with or after food, titrated monthly as necessary (range 20–80 mg/day); pravastatin, 20–80 mg, with dosage adjustments at monthly intervals as needed; rosuvastatin, 5–10 mg once daily (range 5–20 mg once daily) to a maximum of 40 mg once daily under specialist supervision; simvastatin, 10–80 mg once daily, titrated monthly as necessary.
Bile acid-binding resins
Cholestyramine and colestipol are non-absorbable anion-exchange resins, also called bile acid sequestrants. These drugs are used for their cholesterol-lowering effects. Cholesterol is the major precursor of bile acids, which are secreted from the gallbladder into the small intestine. Bile acids perform two functions in the small intestine: they emulsify fat from food to facilitate chemical digestion and they are required for absorption of lipids (including fat-soluble vitamins, A, D, E and K). Bile acids are returned to the liver via enterohepatic recirculation.
The anion-exchange resins bind bile acids in the intestine, thereby decreasing absorption of exogenous cholesterol. To compensate for the loss of bile acids removed by the resins and excreted in the faeces, the liver increases the rate of endogenous metabolism of cholesterol into bile acids and increases the expression of hepatic LDL receptors, and hence uptake of LDL-C from plasma. Long-term increased faecal loss of bile acids causes a reduction in plasma cholesterol concentration that is blunted by the increased synthesis of cholesterol by the liver. An increase in plasma triglycerides limits the use of bile acid-binding resins in people with hypertri glyceridaemia. These drugs used to be the mainstay of lipid-lowering therapy but are now principally used as adjunct therapy to the statins.
Both cholestyramine and colestipol are used in treatment of hypercholesterolaemia and mixed hyperlipidaemia. They are also used to treat pruritus induced by bile acid deposits in dermal tissues (from partial biliary obstruction) and for diarrhoea following ileal resection.
Plasma cholesterol levels usually decrease within 1–2 weeks but in some individuals may increase or exceed previous levels with continued therapy. With cholestyramine, plasma cholesterol levels may continue to fall for up to 1 year. After withdrawal of colestipol and cholestyramine, plasma cholesterol levels tend to rise in around 2–4 weeks. Pruritus will return in about 1–2 weeks after discontinua tion of the drugs. Close monitoring for effectiveness is necessary.
The bile acid-binding resins are not absorbed from the gastrointestinal tract and hence there are no major systemic effects. They bind bile acids in the intestine and are excreted via the faeces. See Drug Interactions 24-1 for drug interactions with bile acid-binding resins and possible outcomes.
Adverse reactions include constipation, indigestion, abdominal pain, nausea, vomiting, flatulence, dizziness, headache and, rarely, gallstones, pancreatitis, bleeding ulcers and malabsorption syndrome. Use bile acid-binding resins with caution in people with gallstones, hypothyroidism, haemorrhoids, kidney disease or bleeding disorders. Avoid use in people with cholestyramine or colestipol hypersensitivity, biliary obstruction or constipation. Cholestyramine is contraindicated in the presence of phenylketonuria because of the presence of aspartame in the product (aspartame is metabolised to phenylalanine). Safety in pregnancy has not been established.
The adult dose of cholestyramine is 4 g once or twice daily before meals; the maintenance dose is 12–16 g daily in 2–3 divided doses. The colestipol adult dose is 10–30 g daily before meals in 2–4 divided doses. When used in combination with other lipid-lowering drugs, the doses are 4 g for cholestyramine and 5 g for colestipol, once or twice daily.
Fibrates
Although several fibric acid derivatives are available overseas (e.g. bezafibrate, ciprofibrate and clofibrate), only fenofibrate and gemfibrozil are available in Australia (see Drug Monograph 24-2). The results of several large studies raised specific concerns regarding the use of clofibrate. First, there is the possibility of an increased risk of inducing malignancy and cholelithiasis in humans with the use of this drug. Individuals taking clofibrate had twice the risk for cholelithiasis and cholecystitis requiring surgery of non-users. Second, there is no evidence of reduced cardiovascular mortality with its use. In fact, studies reported an increase in cardiac arrhythmias, angina and thromboembolic episodes. The clinical use of clofibrate has consequently declined tremendously.
Gemfibrozil and fenofibrate are more effective in reducing VLDL that is rich in triglycerides than in lowering LDL that is high in cholesterol. The increase in HDL observed with gemfibrozil has been reported to reduce the incidence of coronary disease and stroke and, in a follow-up period of 5 years, a reduction of 22% was observed in either fatal or non-fatal myocardial infarction (Rubins et al 1999). In contrast, the FIELD study (2005) found that fenofibrate did not reduce the risk of coronary events in people with type 2 diabetes. The mechanism of action is not completely understood but recent studies indicate that fibrates interact with the peroxisome proliferator activated receptors (PPARs), which regulate gene transcription. Specifically an interaction with PPAR may provide an explanation for the reduction of triglycerides via stimulation of fatty acid oxidation, increased lipoprotein lipase activity and the reduction in apoC-III synthesis. These drugs are indicated principally for the treatment of severe hypertriglyceridaemia, mixed hyperlipidaemia and as second-line treatment for hypercholesterolaemia.
Additional drugs
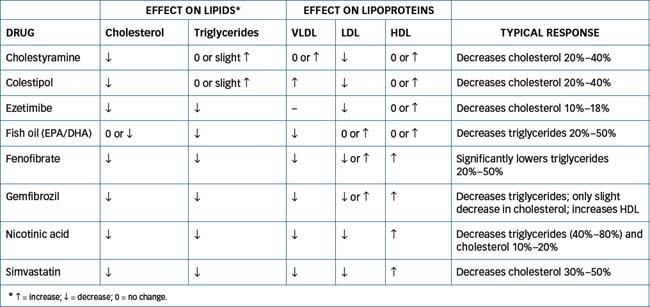
Several additional drugs are discussed below, and a comparison of their lipid-lowering effects is given in Table 24-7.
Nicotinic acid
The lipid-lowering effect of nicotinic acid (niacin) has been known since 1955. Nicotinic acid is a water-soluble vitamin that inhibits mobilisation of free fatty acids from peripheral tissue. This results in a reduction in the hepatic synthesis of triglycerides and the secretion of VLDL. Nicotinic acid also increases the plasma HDL concentration markedly (20%–30%). It is used principally as an adjunct to other therapies, such as the fibrates and bile acid-binding resins, in the treatment of severe and mixed hypertriglyceridaemia. It is also used to treat niacin (vitamin B3) deficiency and, because it increases blood flow through skin and muscle, it is used to treat peripheral vascular disease. Recent studies have investigated whether the combination of slow release nicotinic acid with simvastatin in patients with the metabolic syndrome would improve clinical outcomes. Over the three years of the study the beneficial antiatherogenic effect was offset by the adverse effect of nicotinic acid on glucose metabolism and insulin resistance (Vittone et al., 2007Vittone et al 2007).
Nicotinic acid is well absorbed orally and has a plasma half-life of about 45 minutes. Extensive metabolism occurs in the liver and about 35% of the dose is excreted unchanged in urine. Reduction in plasma triglyceride concentration occurs within several hours of the start of dosing.
Combination with HMG-CoA reductase inhibitors can result in myopathy and rhabdomyolysis. Potentiation of the effect of antihypertensive drugs has also been reported. Adverse reactions include increased feelings of warmth and flushing of the face and neck. Flushing can be very intense and can be reduced in severity by taking aspirin 30–60 minutes before each dose of nicotinic acid. Other common adverse effects include hypotension, nasal stuffiness, diarrhoea, vomiting and dyspepsia. Less frequently encountered are pruritus, skin rash, dry skin or eyes, hyperglycaemia, hyperuricaemia and jaundice. Liver toxicity has been reported with doses of 2 g per day and higher doses. Use with caution in people with gout, diabetes mellitus, peptic ulcer, liver disease and CAD. A reduction in dose may be necessary in situations of renal impairment. Nicotinic acid is contraindicated in people with a recent myocardial infarction or symptomatic hypotension.
The adult maintenance dose for hyperlipidaemia is 500 mg–1 g three times daily. The initial dose of 250 mg three times daily is increased by 250 mg every 4 days until the maintenance dose is achieved (maximum dose is 3.0–4.5 g daily).
Ezetimibe
Ezetimibe is a novel lipid-lowering drug released in Australia in 2004. It is the first of a new group of drugs that inhibits intestinal absorption of both cholesterol and phytosterols. Not only is it effective in inhibiting intestinal absorption of dietary cholesterol, it also inhibits reabsorption of cholesterol excreted in bile (see Figure 24-1). The exact mechanism of action is unknown but ezetimibe localises at the brush border of the small intestine and is thought to inhibit absorption of cholesterol by binding to a specific transport protein (NPC1L1) in the small intestine wall. Ezetimibe also inhibits intestinal absorption of plant sterols (see Clinical Interest Box 24-1). Despite inhibiting cholesterol absorption, ezetimibe does not alter absorption of fat-soluble vitamins and nutrients (Kosoglou et al 2005). The average reduction in LDL-C with ezetimibe is about 18% and it has minimal effect on HDL-C and triglycerides. It is primarily used as an adjunct to diet for the treatment of hypercholesterolaemia and homozygous phytosterolaemia.
Clinical interest Box 24-1 Plant sterols
There is substantial interest in the use of plant sterols as a component of dietary management of people with hyperlipidaemia. Plant sterols such as sitosterol are similar in structure to cholesterol and, although minimally absorbed, they compete with cholesterol for incorporation into micelles in the gastrointestinal tract. This results in less absorption of cholesterol. In turn, this stimulates hepatic LDL receptor formation, increasing uptake of LDL and so lowering the plasma cholesterol level. Plant sterols have been incorporated into margarine and mayonnaise. A daily intake of 2–3 g reduces LDL cholesterol by 5%–10%, but there is evidence that absorption of dietary carotenoids might be impaired. Toxicity appears not to be an issue in the short term, but long-term safety studies have yet to be undertaken.
Source: Heart Foundation of Australia, Guidelines & Publications (http://www.heartfoundation.org.au).
Drug monograph 24-1 Atorvastatin
Atorvastatin is a second-generation synthetic drug that resembles the natural substrate HMG-CoA and hence it is a reversible inhibitor of HMG-CoA reductase. Unlike simvastatin, atorvastatin is administered as the active hydroxy acid form.
INDICATIONS Atorvastatin is indicated for the treatment of hypercholesterolaemia and mixed hyperlipidaemia.
PHARMACOKINETICS Atorvastatin is well absorbed and maximum plasma concentrations occur within 1–2 hours. Bioavailability is low (∼12%), which may be accounted for by high hepatic first-pass metabolism and presystemic (gut wall) metabolism. Two active metabolites have been detected in plasma, 2-hydroxy-atorvastatin and 4-hydroxy-atorvastatin, and both of these metabolites are in equilibrium with their respective inactive lactone forms. CYP3A4 is the major enzyme responsible for formation of the two active metabolites, which are glucuronidated by UGT1A1 and UGT1A3. The biliary route is the major route of elimination of atorvastatin and its metabolites with <2% excreted as unchanged drug in urine, hence changes in renal function have no significant effect on the pharmacokinetics of atorvastatin. The half-life of atorvastatin is ∼20 hours, which may be increased in individuals with hepatic disease.
DRUG INTERACTIONS Inhibitors of CYP3A4, such as antibiotics (clarithromycin, erythromycin), antifungal drugs (fluconazole, itraconazole, ketoconazole), cyclosporin, verapamil and grapefruit juice, all elevate the plasma drug concentration. In contrast, inducers of CYP3A4, which include barbiturates, carbamazepine, phenytoin and St John’s wort, all lower the plasma drug concentration.
WARNINGS AND CONTRAINDICATIONS Use of atorvastatin is contraindicated in conditions of pre-existing liver disease, in women of childbearing age unless adequate contraceptive cover is assured (ADEC Category D), in people with severe intercurrent illness (infection, trauma) or prior to major surgery.
ADVERSE REACTIONS Common adverse reactions include gastrointestinal discomfort, headaches, insomnia and dizziness. An elevation of hepatic transaminase levels can occur within the first few weeks of treatment (dose-related, start at lower end of the dosage range). Of a more serious nature is the potential for the development of myopathy, which can progress to rhabdomyolysis and renal failure. The latter is more likely when the statins are combined with inhibitors of CYP3A4, but an increased incidence has also been observed in combination with the fibrate class of lipid-lowering drugs and nicotinic acid.
DOSAGE AND ADMINISTRATION The initial dose is 10 mg, increasing to a maximum of 80 mg daily. Effectiveness of the dose is determined by monitoring plasma lipids. Atorvastatin is also available in combination with amlodipine for patients stabilised on at least 5 mg of amlodipine daily. Use of the combination drug concomitantly with ritonavir will increase the plasma concentrations of both atorvastatin and amlodipine, increasing the risk of myopathy, rhabdomyolysis, headache, fatigue and dizziness.
Drug monograph 24-2 Gemfibrozil
Gemfibrozil primarily decreases plasma triglycerides found in VLDL and moderately increases HDL. The mechanism of this action has not been established, but it may involve an inhibition of peripheral lipolysis and a decrease in hepatic extraction of free fatty acids, resulting in reduction of triglyceride production. In addition, the drug may accelerate turnover and removal of cholesterol from the liver, to be excreted in the faeces.
PHARMACOKINETICS Gemfibrozil is well absorbed from the gastrointestinal tract and reaches peak levels in 1–2 hours. Its onset of action in reducing plasma VLDL levels occurs within 2–5 days, with the peak effect seen in 4 weeks. It is metabolised in the liver by glucuronidation, which accounts for around 70% of the metabolites excreted by the kidneys. A small proportion (2%) is excreted as unchanged drug and 6% is eliminated via the faeces.
DRUG INTERACTIONS When gemfibrozil is administered with warfarin or phenindione, an increased anticoagulant effect is reported. Monitor the INR closely because the anticoagulant dose may need to be decreased significantly. If administered with HMG-CoA reductase inhibitors, an increased risk of rhabdomyolysis, myoglobinuria and acute renal failure can occur. This has been reported within 3 weeks to several months of combined drug therapy, and concurrent drug administration should be avoided. The efficacy of cyclosporin may be reduced by concurrent administration of the fibrates; monitor and adjust dose of cyclosporin if necessary.
ADVERSE REACTIONS These include muscle aches and cramps, nausea, vomiting, rash, diarrhoea, flatulence and abdominal distress.
WARNINGS AND CONTRAINDICATIONS Use with caution in people with gallstones or gallbladder disease. Avoid use in people with gemfibrozil hypersensitivity, liver or kidney disease and especially primary biliary cirrhosis.
DOSAGE AND ADMINISTRATION The adult dose is 1.2 g daily in two divided doses, preferably 30 minutes before food.
Drug interactions 24-1 Bile acid-binding resins*
| Drug | Possible effects and management |
| Warfarin | Concurrent use significantly decreases absorption of warfarin thus the anticoagulant effect might be reduced. It is suggested that warfarin be given 1 hour before or 4–6 hours after cholestyramine. Also, monitor international normalised ratio (INR) and adjust warfarin dose as necessary |
| Digoxin | Reduced absorption occurs with bile acid-binding resins. It is recommended that drugs be administered at least 1 hour before or 4–6 hours after administration of resin. Monitor plasma digoxin concentration |
| Thyroxine | Decreased absorption of thyroxine. Separate doses of thyroxine and cholestyramine by at least 4 hours and monitor clinical response to thyroxine |
* Multiple interactions with other drugs affecting either absorption and/or enterohepatic recycling have been reported. Consult relevant drug information sources before administering bile acid-binding resins.
Ezetimibe is conjugated in the intestine, forming an active glucuronide, which accounts for approximately 90% of the drug in plasma after 30 minutes. Ezetimibe and ezetimibe glucuronide are then transported to the liver and subsequently secreted in bile back into the intestine (enterohepatic recycling). The half-life of both is approximately 22 hours and about 80% of the administered dose is excreted in faeces and about 11% in urine. As ezetimibe is not metabolised to any major extent in the liver, significant interactions with the majority of drugs used to treat dyslipidaemia are not a major issue. However, coadministration with cholestyramine reduces bioavailability, and hence these drugs should be administered several hours apart.
Ezetimibe is administered once daily as a 10-mg dose. As it is a relatively new drug, the full range of adverse effects is not yet known. Commonly, ezetimibe causes headache and diarrhoea and, as with the statins, muscle disorders (e.g. myalgia, muscle cramps, weakness and pain) have been reported. A combination of ezetimibe (10 mg) and simvastatin (40–80 mg) has been listed on the Australian PBS. This combination increases the lipidlowering effect of the statin by up to 20% and is a valuable combination for individuals who are unable to tolerate a higher dose of a statin.
Fish oils
Evidence indicates that consumption of omega-3 fatty acids found in fish and fish oil reduces the incidence of ischaemic heart disease and sudden death from myocardial infarction, and prolongs the life of myocardial infarction survivors. Other beneficial effects of omega-3 fatty acids include a small reduction in BP and heart rate and a decrease in platelet aggregation and anti-arrhythmic effects. However, the consumption of omega-3 fatty acids by humans has decreased as a result of the increased consumption of commercial livestock, which have lower levels of omega-3 fatty acids than their wild or free-range counterparts (Horrocks & Yeo 1999). One of the active components of fish oil is docosahexaenoic acid (DHA), which is present in oily fish such as mackerel, salmon and tuna. Consumption of a fish-rich diet is thought to account for the lower incidence of coronary heart disease in the Japanese and in Greenland Eskimos. In one trial, subjects treated with simvastatin but with persistent hypertriglyceridaemia were additionally prescribed fish oil (2 g twice daily) for 1 year. A significant reduction was observed in plasma triglycerides (20%–30%) and VLDL-C (30%–40%) (Durrington et al 2001).
In 2004 the FDA approved a prescription form of omega-3 fatty acids for the treatment of high triglycerides in adults. Each 1-g capsule (Lovaza®) contains ∼465 mg of eicosapentaenoic acid (EPA) and 375 mg of DHA and the recommended dose is two capsules twice a day or 4 capsules once a day (total dose 4 g, containing 1860 mg EPA and 1500 mg DHA). Omega-3 fatty acids are thought to exert their beneficial effects through reducing VLDL formation and accelerating VLDL metabolism to LDL particles. This ultimately reduces triglyceride levels but also potentially increases LDL-C levels. Similar to the fibrates, omega-3 fatty acids have a high affinity for PPAR and may upregulate the metabolism of fatty acids in the liver. Current evidence indicates that, when used at the recommended dosage (4 g/day) in patients with very high triglycerides, omega-3 fatty acids reduce triglycerides by ∼ 45% and VLDL-C by ∼50%.
Numerous studies are ongoing and, although fish oil supplements appear to have few adverse effects, high doses may increase bleeding time. Fish oil may be added to a lipid-lowering drug regimen but a daily dose needs to be calculated according to the EPA and DHA content of the individual fish oil supplements. Current guidelines from the Heart Foundation suggest eating a healthy diet rich in fruit and vegetables and consuming ∼500 mg per day of combined DHA and EPA through a combination of two or three serves (150 g serve) of oily fish per week or fish oil capsules or liquid food and drink enriched with marine omega-3 polyunsaturated fatty acids (http://www.heartfoundation.org.au/SiteCollectionDocuments/HW%20FS%20FishOil%20RevEv.pdf).Drugs at a glance 24: Lipid-lowering drugs

 Key points
Key points
 Dyslipidaemia is a metabolic disorder characterised by increased concentrations of plasma cholesterol and triglycerides, two of the major lipids in the body. High circulating levels of these lipids have been associated with atherosclerosis, a disorder in which lipids are deposited in the linings of medium- and large-sized arteries, eventually producing degenerative changes and obstruction of blood flow. Atherosclerosis is a causative factor in CAD, which in turn can result in angina, heart failure, myocardial infarction, cerebral artery disease, peripheral artery occlusive disease and renal arterial insufficiency. In the absence of satisfactory reduction of high plasma lipid levels through exercise, diet and lifestyle modification, lipid-lowering drugs offer health-care professionals a management strategy for the treatment of dyslipidaemia. This is of proven benefit in individuals with high cardiovascular risk factors. In Australia, the Pharmaceutical Benefits Scheme (PBS) criteria for subsidy of lipid-modifying drugs now reflect treatment according to risk of future cardiovascular events. Persons eligible for subsidy at any cholesterol concentration include persons with symptomatic coronary or cerebrovascular or peripheral vascular disease; or at high risk e.g. diabetes mellitus or with a family history of symptomatic coronary heart disease. The main classes of lipid-lowering drugs are the HMG-CoA reductase inhibitors (‘statins’), bile acid-binding resins and fibrates. These agents may be used either individually or in combination if not contraindicated. Numerous studies have now established that statins significantly reduce the risk of coronary heart disease, stroke and death in individuals undergoing treatment for an average of >5 years. Cholestyramine and colestipol are non-absorbable anion-exchange resins, also called bile acid sequestrants, that are used for their cholesterol-lowering effects. The HMG-CoA reductase inhibitors and bile acid-binding resins are subject to numerous drug interactions. Current drug use should be established before commencing a lipidlowering drug. Gemfibrozil and fenofibrate are more effective in reducing VLDL that is rich in triglycerides than in lowering LDL that is high in cholesterol. Ezetimibe is a novel lipid-lowering drug that inhibits intestinal absorption of both cholesterol and phytosterols. Not only is it effective in inhibiting intestinal absorption of dietary cholesterol, it also inhibits reabsorption of cholesterol excreted in bile. Effectiveness varies depending on the specific type of dyslipidaemia. See Tables 24-2, 24-4 and 24-7 for classifications of hyperlipoproteinaemia, drug treatment approaches and a comparison of lipid-lowering effects.
Dyslipidaemia is a metabolic disorder characterised by increased concentrations of plasma cholesterol and triglycerides, two of the major lipids in the body. High circulating levels of these lipids have been associated with atherosclerosis, a disorder in which lipids are deposited in the linings of medium- and large-sized arteries, eventually producing degenerative changes and obstruction of blood flow. Atherosclerosis is a causative factor in CAD, which in turn can result in angina, heart failure, myocardial infarction, cerebral artery disease, peripheral artery occlusive disease and renal arterial insufficiency. In the absence of satisfactory reduction of high plasma lipid levels through exercise, diet and lifestyle modification, lipid-lowering drugs offer health-care professionals a management strategy for the treatment of dyslipidaemia. This is of proven benefit in individuals with high cardiovascular risk factors. In Australia, the Pharmaceutical Benefits Scheme (PBS) criteria for subsidy of lipid-modifying drugs now reflect treatment according to risk of future cardiovascular events. Persons eligible for subsidy at any cholesterol concentration include persons with symptomatic coronary or cerebrovascular or peripheral vascular disease; or at high risk e.g. diabetes mellitus or with a family history of symptomatic coronary heart disease. The main classes of lipid-lowering drugs are the HMG-CoA reductase inhibitors (‘statins’), bile acid-binding resins and fibrates. These agents may be used either individually or in combination if not contraindicated. Numerous studies have now established that statins significantly reduce the risk of coronary heart disease, stroke and death in individuals undergoing treatment for an average of >5 years. Cholestyramine and colestipol are non-absorbable anion-exchange resins, also called bile acid sequestrants, that are used for their cholesterol-lowering effects. The HMG-CoA reductase inhibitors and bile acid-binding resins are subject to numerous drug interactions. Current drug use should be established before commencing a lipidlowering drug. Gemfibrozil and fenofibrate are more effective in reducing VLDL that is rich in triglycerides than in lowering LDL that is high in cholesterol. Ezetimibe is a novel lipid-lowering drug that inhibits intestinal absorption of both cholesterol and phytosterols. Not only is it effective in inhibiting intestinal absorption of dietary cholesterol, it also inhibits reabsorption of cholesterol excreted in bile. Effectiveness varies depending on the specific type of dyslipidaemia. See Tables 24-2, 24-4 and 24-7 for classifications of hyperlipoproteinaemia, drug treatment approaches and a comparison of lipid-lowering effects.Review exercises
References and further reading
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Beaumont J.L., Carlson L.A., Cooper G.R., Fejfar Z., Fredrickson D.S., Strasser T. Classification of hyperlipidaemias and hyperlipoproteinaemias. Bulletin of the World Health Organization. 1970;43:891-915.
Bellosta S., Paoletti R., Corsini A. Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation. 2004;109:III-50-III-57.
Blumenthal R.S. Statins: effective antiatherosclerotic therapy. American Heart Journal. 2000;139:577-583.
Chan K.Y., Boucher E.S., Gandhi P.J., et al. HMG-CoA reductase inhibitors for lowering elevated levels of C-reactive protein. American Journal of Health-System Pharmacy. 2004;61:1676-1681.
Colquhoun D, Ferreira-Jardim A, Udell T, Eden B. Review of evidence: fish, fish oils, n-3 polyunsaturated fatty acids and cardiovascular health. PRO-059, August 2008. Online. http://www.heartfoundation.org.au/SiteCollectionDocuments/HW%20FS%20FishOil%20RevEv.pdf [25 August 2009].
Corsini A., Bellosta S., Baetta R., et al. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacology & Therapeutics. 1999;84:413-428.
Durrington P.N., Bhatnagar D., Mackness M.I., et al. An omega-3 polyunsaturated fatty acid concentrate administered for one year decreased triglycerides in simvastatin treated patients with coronary heart disease and persisting hypertriglyceridaemia. Heart. 2001;85:544-548.
Evans M. Statin safety in perspective—maximising the risk:benefit. British Journal of Cardiology. 2004;11:449-454.
FIELD study investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849-1861.
Graham D.J., Staffa J.A., Shatin D., et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. Journal of the American Medical Association. 2004;292:2585-2590.
Horrocks L.A., Yeo Y.K. Health benefits of docosahexaenoic acid (DHA). Pharmacological Research. 1999;40:211-225.
Joy T.R., Hegele R.A. Narrative review: statin-related myopathy. Annals of Internal Medicine. 2009;150:858-868.
Knopp R.H. Drug treatment of lipid disorders. New England Journal of Medicine. 1999;341:498-511.
Kosoglou T., Statkevich P., Johnson-Levonas A.O., et al. Ezetimibe: a review of its metabolism, pharmacokinetics and drug interactions. Clinical Pharmacokinetics. 2005;44:467-494.
Law M., Rudnicka A.R. Statin safety: a systematic review. American Journal of Cardiology. 2006;97:52C-60C.
Lennernas H. Clinical pharmacokinetics of atorvastatin. Clinical Pharmacokinetics. 2003;42:1141-1160.
Mahley R.W., Bersot T.P. Drug therapy for hypercholesterolemia and dyslipidemia. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw-Hill, 2006. [ch 35]
McKenney J.M., Sica D. Prescription omega-3 fatty acids for the treatment of hypertriglyceridemia. American journal of Health-System Pharmacy. 2007;64:595-605.
National Heart Foundation of Australia and the Cardiac Society of Australia and New Zealand Position Statement on Lipid Management 2005. Heart Lung and Circulation 2005; 14: 275–291.
Noe J., Portmann R., Brun M.E., et al. Substrate-dependent drug–drug interactions between gemfibrozil, fluvastatin and other organic anion-transporting peptide (OATP) substrates on OATP1B1, OATP2B1 and OATP1B3. Drug Metabolism and Disposition. 2007;35:1308-1314.
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007. [ch 20]
Rubins H.B., Robins S.J., Collins D., et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol [VA-HIT]. New England Journal of Medicine. 1999;341:410-418.
Vittone F., Cait A., Morse J.S., et al. Niacin plus simvastatin reduces coronary stenosis progression among patients with metabolic syndrome despite a modest increase in insulin resistance: a subgroup analysis of the HDL-Atherosclerosis Treatment Study (HATS). Journal of Clinical Lipidology. 2007;1:203-210.
American Heart Association: www.americanheart.org
National Heart Foundation of Australia: www.heartfoundation.org.au