Chapter 6 Drug Absorption, Distribution, Metabolism and Excretion
To meet the knowledge challenge created by the numerous drugs already marketed, combined with the many new drugs released annually, health-care professionals must develop an understanding of the fundamental principles of drug absorption, distribution, metabolism and excretion. From these processes stems the theoretical framework that provides the basis for the design of drug dosage regimens.
Key abbreviations
FOR a drug to produce an effect, it must reach its molecular target (see Chapter 5). The concentration of drug that finally interacts with the molecular target is influenced by the absorption, distribution, metabolism (into inactive or active metabolites) and excretion of the drug from the body. The interrelationship of these dynamic processes is shown in Figure 6.1. The study of the kinetics of a drug during these processes is collectively described by the term pharmacokinetics or simply ‘what the body does to the drug’. As there are many important definitions and concepts in the field of pharmacokinetics that you need to be familiar with to ensure safe clinical practice, these are introduced in the following relevant sections and in other chapters as ‘Key pharmacokinetic definitions’ and ‘Key pharmacokinetic concepts’.
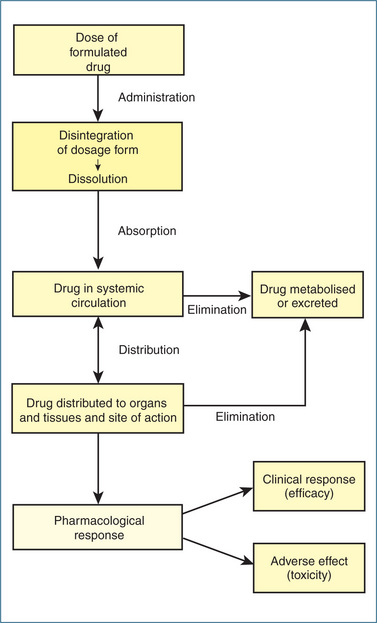
Figure 6-1 Interrelationship between drug absorption, distribution, metabolism and elimination. Note: For some drugs the site of action is the vascular system.
Drug absorption
Absorption (Box 6-1) is an important factor for all routes of administration with the exception of the intravenous route, where the drug is administered directly into the systemic circulation and does not require absorption from the site of administration.
Box 6-1 Key pharmacokinetic definition: absorption
Absorption is the process by which an unchanged drug proceeds from the site of administration into the blood.
With the exception of some drugs that are used solely for a local effect (e.g. ointments used for skin rashes), most drugs are administered outside the vascular system (extravascularly). Before they can be distributed to their site of action, drugs must be absorbed from the point of application into the systemic circulation. An oral drug may be in a solid form (tablet, capsule or powder) or in liquid form (solution or suspension). Disintegration of solid dosage forms must occur before dissolution, a process by which a drug goes into solution and becomes available for absorption (Figure 6.2). The drug dosage form is important because the faster the rate of dissolution, the more rapidly the drug is presented to the membrane for absorption. Basically, for oral drugs absorption is greatest for liquids, elixirs and syrups > suspension solutions > powders > capsules > tablets > coated tablets > enteric-coated tablets (slowest absorption).
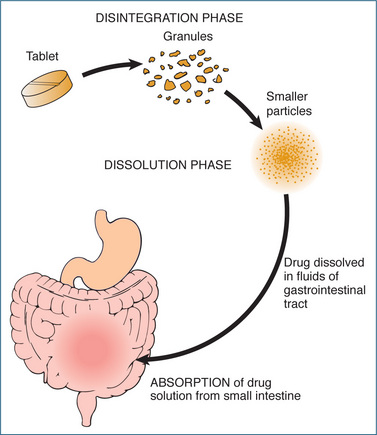
Absorption across biological membranes
For absorption to occur, it is necessary for a drug to cross a membrane and enter the blood vessels on the other side. The membrane typically consists of a lipid bilayer that contains protein molecules irregularly dispersed throughout it. These protein molecules themselves may act as carriers, enzymes, receptors or antigenic sites. Lipid (fat)-soluble drugs can easily pass through the lipid membrane, while ionised (charged) or water-soluble drugs have difficulty crossing cell membranes. The membrane, which also contains aqueous channels (pores), permits the passage of small water-soluble substances such as urea, alcohol and electrolytes as well as water itself.
When free to move to their sites of action, drug molecules are transported from one body compartment to another by way of the plasma; however, free movement can be somewhat limited because membranes also enclose these various sites. Whether the barrier to drug transport consists of a single layer of cells, such as the intestinal epithelium of the villi, or several layers of cells, such as skin, in order for a drug to gain access to the interior of a cell or a body compartment it has to penetrate cell membranes. All the physiological processes mediating absorption, distribution, metabolism and excretion are predicated on two main processes: passive transport and active or carrier-mediated transport.
Passive transport—diffusion
Most drugs cross membranes by a process of diffusion, which is the passive transfer of drug from a region of higher concentration to a region of lower concentration until equilibrium is established on either side of the membrane. Passive diffusion is influenced by the surface area of the membrane exposed to the drug (e.g. the large surface area of the small intestine), the concentration gradient of the drug and the lipid–water partition coefficient of the drug (i.e. the more lipid soluble the drug the faster it will diffuse across the membrane). For ionic drugs diffusion is also influenced by the ionisation state, which is discussed in the section ‘Variables that affect drug absorption’.
Carrier-mediated transport
Although passive diffusion is the dominant process, carrier-mediated transport is important for amino acids, glucose, some vitamins, neurotransmitters, metal ions and some drugs. In the case of active transport this requires an energy source: active transport involves the movement of drug molecules against the concentration gradient (from areas of low concentration to areas of high concentration) or, in the case of ions, against the electrochemical gradient. An example of an active transport mechanism is the sodium–potassium pump.
Carrier-mediated transport of drugs is important in the kidney, the gastrointestinal tract, biliary tract and the blood–brain barrier. Often referred to as ‘drug transporters’, these membrane proteins can function as either uptake transporters or efflux transporters. When we consider drugs there are two major families of transporters that are important: the ATP binding cassette (ABC) and solute carrier (SLC) transporters. The ABC transporters are primarily transporters that rely on the hydrolysis of ATP to provide the energy to pump substrates across membranes. A well known transporter in the ABC family is the efflux transporter P-glycoprotein (P-gp; encoded by the ABCB1 gene also known as MDR1, which stands for ‘multi-drug resistance’) that was first discovered in tumour cells. It is associated with the multi-drug resistance phenomenon observed clinically in patients treated with cancer chemotherapeutic drugs for long periods of time. This drug resistance results from over-expression of P-gp, which leads to an increased efflux of the cytotoxic drug from the cancer cell, thus lowering the intracellular concentration of drugs such as paclitaxel, vincristine and doxorubicin. Another transporter called breast cancer resistance protein (BRCP) has also been implicated in resistance to anticancer drugs (Chapter 42). P-gp is found in the intestine, kidney, liver, blood–brain barrier, placenta and testes. Drugs transported by P-gp include: digoxin; the immunosuppressants cyclosporin and tacrolimus; the anticancer drugs etoposide, doxorubicin and vincristine; the calcium channel blockers diltiazem and verapamil; the protease inhibitors indinavir and ritonavir; the antibiotic erythromycin and the antifungal ketoconazole.
The major family of uptake transporters is the organic anion transporting polypeptides (OATP). The first human OATP (OATP1A2) was isolated from liver but it is also found in brain, lung, kidney and testes. OATP1A2 transports a diverse range of compounds including bile acids, thyroid hormones, steroid sulfates, the antihistamine fexofenadine and opioid peptides. OATP1A2 may be important for regulating permeability of the blood–brain barrier to solutes. In the liver OATP1B1 appears to play a major role in the hepatic uptake of bile acids, sulfate and glucuronide conjugates and of drugs such as methotrexate and the lipid-lowering drug pravastatin. For example, pravastatin is taken up by the OATP1B1 transporter, which contributes to the efficient first-pass hepatic uptake of pravastatin. This enhances the pharmacological effect of pravastatin in the liver and minimises escape of the drug into the systemic circulation and hence limits the adverse effect in muscle tissue (see Chapter 24).
Within the ABC family there are seven subclasses, within the SLC superfamily there are 43 families and currently there are in excess of 300 known transporters. Specific transporters will be discussed in the following chapters where they are of pharmacological relevance in terms of pharmacokinetics, pharmacodynamics or adverse effects.
As knowledge of drug transporters continues to grow it is becoming increasingly apparent that drug transporters are subject to induction and inhibition by coadministered drugs (see the ‘Drug Interactions’ section in Chapter 10).
Variables that affect drug absorption
The rate and extent to which a drug is absorbed are influenced by the following variables.
Nature of the absorbing surface (cell membrane) that the drug must traverse
The drug molecule may pass through a single layer of cells (e.g. intestinal epithelium), in which case transport is faster than if it traverses several layers of cells (e.g. skin). The size of the surface area of the absorbing site is also an important determinant of drug absorption. Generally, the more extensive the absorbing surface, the greater the drug absorption and the more rapid its effects. Anaesthetics are absorbed immediately from the pulmonary epithelium because of the vast surface area of the lung. Absorption from the small intestine, which also offers a massive absorbing area, is more rapid than from an equivalent smaller absorbing surface, such as the stomach.
Blood flow
The circulation to the site of administration is a significant factor in the absorption of drugs. A rich blood supply (e.g. the sublingual route) enhances absorption, whereas a poor vascular site (e.g. the subcutaneous route) delays it. An individual in shock, for example, may not respond to intramuscularly administered drugs because of poor peripheral circulation. Drugs injected intravenously, on the other hand, are placed directly into the cardiovascular system and are available immediately. Food increases splanchnic blood flow and enhances absorption of orally administered drugs. Conversely, in hypovolaemic states absorption of drugs may be slowed due to decreased splanchnic blood flow.
Solubility of the drug
To be absorbed, a drug must be in solution; the more soluble the drug, the more rapidly it will be presented for absorption. Because cell membranes contain a fatty-acid layer, lipid solubility is a valuable attribute of drugs absorbed from certain areas (e.g. the gastrointestinal tract and the placenta). Chemicals and minerals that form insoluble precipitates in the gastrointestinal tract, such as barium salts, or drugs that are not soluble in water or lipids are not absorbed (e.g. the bile acid-binding resin cholestyramine).
Ionisation
In general, drugs exist as weak acids or weak bases and in body fluids they are either ionised or unionised. The ionised (charged polar) form is usually watersoluble (lipid-insoluble) and does not diffuse readily through the cell membranes of the body. By contrast, the un-ionised (non-polar) form is more lipid-soluble (less water-soluble) and is more apt to cross the cell membranes. In general, an acidic drug is relatively un-ionised in an acid environment such as the stomach but a basic drug tends to ionise in the same acid environment. In contrast, absorption of a basic drug is enhanced in a more basic site such as the small intestine, while the acidic drug tends to be more ionised (Figure 6-3). Despite the varying states of ionisation, little drug absorption occurs in the stomach (due to the small surface area and influence of the gastric emptying rate) and most occurs in the small intestine.
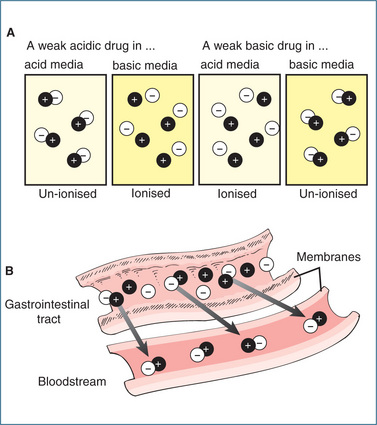
Figure 6-3 Effect of pH on drug ionisation and transport. A Effects of pH on the ionisation state of a drug that is either a weak acid or a weak base. B Effects of pH on the transport of drug molecules through membranes. Only un-ionised lipid-soluble drugs cross biological membranes.
The extent of ionisation is determined by the pH1 of the environment (see Clinical Interest Box 6-1). Remember that reference to a ‘weak’ or ‘strong’ acid refers to the tendency of the acid to dissociate (break up) into hydrogen ions (H+) and anions. This dissociation is often referred to as the pKa, which describes the strength of weak acids. In general, acids with lower pKa values (e.g. acetic acid, pKa 4.75) are ‘stronger’ acids that those with higher pKa values (e.g. carbonic acid, pKa 6.1). In simple terms the pKa is the pH at which half the chemical (this can be a drug or an endogenous chemical such as an electrolyte) is in its ionised form (pH – pKa = 0). To illustrate how a change in pH affects ionisation consider the following example. Drug X is a weak acid with a pKa of 5. In an acidic environment of pH 2, pH – pKa = −3 and drug X will be ∼0.1% ionised. However, in a more basic environment (pH 8), pH – pKa = 3 and drug X will ∼99.9% ionised (Table 6-1). Conversely, if a drug is a weak base (pKa ∼8), in an acidic environment of pH 3, pH – pKa = −5 and hence a basic drug will be >99.9% ionised.
Clinical interest Box 6-1 Incompatibility of drugs in solution
When either mixing or diluting drugs results in precipitation in the solution, this is most likely a result of formation of un-ionised (less water-soluble) drug. Most drugs reconstituted as injections are predominantly in the ionised or salt form. Consequently a change in pH is the most common cause of precipitation, for example diluting a drug such that the resulting pH generates more of the un-ionised (less water-soluble) form than was present in the original drug solution. An excellent reference source is Trissel’s Handbook of Injectable Drugs (2009).
Frusemide, the diuretic, is prepared as a mildly buffered alkaline solution to aid solubility. It should not be mixed with acidic solutions of pH<5.5 as it will precipitate. Precipitation can also result from formation of an insoluble salt. Dilution of the antibiotic ceftriaxone sodium in lactated Ringer’s solution will result in precipitation despite the alkaline pH of the Ringer’s solution maintaining the water-soluble (ionised) form of ceftriaxone. In this case, precipitation results from formation of an insoluble calcium salt of ceftriaxone. Knowledge of the fundamentals of acid–base balance and an understanding of ionisation are central to avoiding drug incompatibilities (Newton 2009).
Table 6-1 Guide for estimating the approximate percent ionisation for acids and bases
| % IONISATION | ||
| pH – pKa | Acid | Base |
| −3 | 0.1 | 99.9 |
| −2 | 1 | 99 |
| −1 | 10 | 90 |
| 0 | 50 | 50 |
| 1 | 90 | 10 |
| 2 | 99 | 1 |
| 3 | 99.9 | 0.1 |
Formulation
Drug formulation can be manipulated by pharmaceutical processing to achieve desirable absorption characteristics. An active drug can be combined with a resin or other substance from which it is slowly released, or prepared in a vehicle that offers relative resistance to the acidic environment of the stomach (e.g. enteric coating).
Routes of drug administration
The route of drug administration can affect both the rate of onset of action and the magnitude of the therapeutic response that results. Drugs are administered, in general, for either local or systemic effects. The local effect of a drug usually occurs at the immediate site of application, in which case absorption is a disadvantage. When a drug is given for a systemic effect, absorption is an essential first step before the drug appears in the circulation and is distributed to a location distant from the site of administration.
A drug may enter the circulation either by being injected there directly (intravenously) or by absorption from other extravascular sites. The traditional or standard routes of drug administration fall into the following major categories:
However, new technologies (Clinical Interest Box 6-2) continue to emerge with drugs delivered by drug-eluting stents in the field of cardiology, the application of nanoparticles targeting brain tumours, the use of nanocarriers for transdermal vaccine administration, and miniature micro electromechanical devices for passive and active drug delivery.
Clinical interest Box 6-2 Drug-eluting stents
Neointimal hyperplasia and restenosis in coronary vessels after insertion of metal stents led to the development of drug-eluting stents (DESs). These are designed to slowly release a drug that inhibits the proliferation of vascular smooth muscle cells, thus reducing restenosis of coronary vessels.
DESs were first approved by the US Food and Drug Administration (FDA) in 2003 and are widely used for coronary stent procedures. The major benefit of a DES is a reduction in the restenosis rate and the need for repeated revascularisation procedures. Basically the balloon-expandable DESs are made from either stainless steel, cobalt chromium or other metal alloys. The metal provides the platform for the polymer coating that is uniformly distributed along the stent and contains the drug, which is released at a controlled rate.
The sirolimus-eluting stent is made of a stainless steel platform coated with a polymer containing sirolimus at a concentration of 140 mcg/cm2, and 80% of the drug is released continuously over 30 days. Sirolimus blocks cell cycle transition and hence proliferation of smooth muscle cells and neointimal hyperplasia. The paclitaxel-eluting stent is also made of stainless steel and the polymer coating contains 100 mcg/cm2 paclitaxel, an antiproliferative agent. Unlike the former, the latter stent releases paclitaxel in a biphasic manner, with drug eluted initially within 48 hours and then over the following 2 weeks, but a large quantity of the drug remains bound to the polymer (Wykrzykowska et al 2009). The safety of DESs has been questioned and with the sirolimus-eluting stent there is evidence of increased mortality.
As stent thrombosis is reduced with dual antiplatelet therapy, the FDA has recommended the use of clopidogrel and aspirin for ≥12 months in patients with a DES (Maluenda et al 2009). Current research is focused more on developing fully biodegradable DESs that would provide a window for the vessel to heal itself while the biodegradable stent is slowly absorbed. Recent biodegradable DESs have included those made of magnesium and a small percentage of rare earth metals that degrade over 2 months to inorganic salts.
Oral route
Oral, or enteral, ingestion is the most commonly used method of administering drugs. It is safe, convenient and an economical route of administration. However, the frequent changes in the gastrointestinal environment produced by food, emotion, physical activity and other medications may make absorption unreliable and slow. Drugs may be absorbed from several sites along the gastrointestinal tract and they may also be metabolised by enzymes in the gastrointestinal mucosa before they are absorbed and enter the systemic circulation.
Absorption from the oral cavity
Although the oral cavity possesses a thin lining, a rich blood supply and a slightly acidic pH, little absorption occurs in the mouth. Despite its small surface area, the oral mucosa is capable of absorbing certain drugs as long as they dissolve rapidly in the salivary secretions (i.e. drugs given by the sublingual and buccal routes). In sublingual administration the drug is placed under the tongue to permit tablet dissolution in salivary secretions. Glyceryl trinitrate, used for treating angina, is administered in this manner and the person is advised to refrain as long as possible from swallowing saliva containing the tablet form of the drug. Because glyceryl trinitrate is un-ionised, with high lipid solubility, the drug readily diffuses through the lipid mucosal membranes. Drugs absorbed sublingually enter the systemic circulation directly without entering the portal system, thus bypassing the liver and escaping first-pass metabolism (see Chapter 8). Accordingly, absorption is rapid and the effects of the drug may become apparent within 2 minutes. In buccal administration the drug (tablet) is placed between the teeth and the mucous membrane of the cheek. Some hormones and enzyme preparations are administered by this route and are rapidly absorbed.
Absorption from the stomach
Although the stomach has a rich blood supply, it has a thick layer of mucus and a relatively small surface area and hence it is not an important site of drug absorption. The length of time a substance remains in the stomach is a significant variable in determining the extent of gastrointestinal absorption. Generally, a slow gastric emptying rate decreases the rate of drug absorption. This is why many drugs are administered on an empty stomach, with sufficient water to ensure dissolution and rapid passage into the small intestine. (Drugs that cause gastric irritation are usually given with food.) After solid-dose drug administration the recipient should be encouraged to sit upright for at least 30 minutes to shorten gastric emptying time (the time required for the drug to reach the small intestine) and also to reduce the potential for tablets or capsules to lodge in the oesophageal area. Prolongation of gastric emptying time increases the risk of destruction of acid-labile drugs (e.g. erythromycin base).
Absorption from the small intestine
The small intestine is highly vascularised and, with its many villi that have more permeable membranes, it presents a significantly larger absorption area than the stomach. It is the major site for absorption of orally administered drugs that pass from the stomach into this region and are absorbed primarily in the upper part of the small intestine. The intestinal fluid is alkaline (pH 7–8), which strongly influences the rate of absorption of the un-ionised basic drugs. Increased intestinal motility caused, for example, by diarrhoea or cathartics may decrease exposure to the intestinal membrane and thereby diminish absorption, leading to therapeutic failure from low systemic drug concentration. Prolonged exposure, on the other hand, allows more time for absorption, and hence the possibility of increased plasma concentration and adverse drug reactions.
Absorption from the rectum
The surface area of the rectum is not very large, but drug absorption does occur because of extensive vascularity. The veins of the rectum include the superior, middle and inferior veins. Only the superior rectal veins unite to form the inferior mesenteric vein, which is a tributary of the portal vein. Drug absorbed via the superior rectal veins flows to the liver via the portal vein and is metabolised, while the remainder of the drug that is absorbed (approximately 50%) escapes first-pass metabolism as it bypasses the liver. Rectal drug administration may be used for both local and systemic effects. This route is often used in unconscious individuals, in fasting patients, in those unable to swallow or when severe vomiting is present. Disadvantages to rectal drug administration include erratic absorption because of rectal contents, local drug irritation with some medications and uncertainty of drug retention.
Parenteral route
The parenteral route refers to the administration of drugs by injection. Intravenous administration is the most rapid route of drug administration, with high concentrations being achieved quickly in the systemic circulation. Absorption from subcutaneous or intramuscular injection sites is faster than via the oral route but is less reliable, as local blood flow and diffusion through the tissue influences the pattern of absorption.
Subcutaneous (SC)
A subcutaneous injection of a drug is given beneath the skin into the connective tissue or fat immediately underlying the dermis. This site can be used only for drugs that are not irritating to the tissue; otherwise severe pain, necrosis and sloughing of tissue may occur. The rate of absorption is slow and can provide a sustained effect.
Intramuscular (IM)
Intramuscular administration refers to the injection of a drug solution into muscle. Most often the drug is fully soluble in an aqueous solution and absorption occurs more rapidly than with subcutaneous injection because of greater tissue blood flow. However, not all drugs are formulated as aqueous solutions. Procaine penicillin is poorly soluble and is injected as an aqueous suspension that is slowly absorbed and hence has a prolonged duration of action. Some steroid hormones are synthesised as chemical esters, which increases their solubility in oil and slows the rate of absorption. Examples include testosterone propionate and the depot antipsychotic drug fluphenazine decanoate. Drug absorption via this route may not be ‘normal’ in obese or emaciated people because of differences in subcutaneous fat distribution.
Intravenous (IV)
This route of drug administration has both advantages and disadvantages. The intravenous route produces an immediate pharmacological response because the desired amount of drug is injected directly into the bloodstream, thereby circumventing the absorption process. However, adverse effects may occur as a result of the rapid attainment of a high plasma concentration. Intravenous drugs may be given as a small bolus dose or by constant infusion, which should generally be administered slowly to prevent adverse effects.
Intrathecal
Intrathecal drug administration means that the drug is injected directly into the spinal subarachnoid space, bypassing the blood–brain barrier. Many compounds cannot enter the cerebrospinal fluid or are absorbed in this region only very slowly. When rapid central nervous system (CNS) effects of drugs are desired, as with spinal anaesthesia or in treatment of acute infection of the CNS, this route may be used.
Epidural
Epidural drug administration refers to the injection of a drug within the spinal canal on or outside the dura mater that surrounds the spinal column. This is sometimes called extradural or peridural.
For other parenteral routes, see Box 6-2.
Inhalation
To ensure that normal gas exchange of oxygen and carbon dioxide is not interrupted in the lungs, drugs must be either gases or fine mists (aerosols) when they are administered by inhalation. The lungs provide a large surface area for absorption and the rich capillary network adjacent to the alveolar membrane promotes ready entry of drugs and toxic environmental chemicals into the bloodstream. Drugs such as bronchodilators are administered by various metered-dose inhalation devices (nebulisers, ‘puffers’) that deliver the drug during inhalation into the airway, producing primarily a local effect with reduced systemic adverse effects compared to oral administration.
Topical route
Depending on the site of application, absorption of drugs applied topically to the skin and mucous membranes is generally rapid. Examples include cutaneous application, nasal sprays and eye-drops.
Skin
Drugs applied to the skin are used to produce either a local or a systemic effect through the use of ointments or transdermal patches. Only lipid-soluble compounds are absorbed through the skin, which acts as a lipid barrier. To prevent significant systemic absorption only an intact skin surface should be used as absorption occurs more readily through abraded or burnt skin. Anything that enhances cutaneous blood flow or hydrates the skin will increase absorption e.g. massaging, warming the skin or covering with an occlusive dressing.
Transdermal administration is usually done with a patch that may contain a 1-, 3- or 7-day supply of medication, depending on the drug. Examples of drugs that are applied transdermally include oestrogen (hormone replacement therapy), glyceryl trinitrate (treatment of angina) and scopolamine for motion sickness.
Eyes
Topical administration of ophthalmic drugs produces a local effect on the conjunctiva or anterior chamber. Systemic absorption can occur through drainage from the nasolacrimal canal and, as this route bypasses the liver (no first-pass metabolism), adverse systemic effects may occur (e.g. unwanted effects due to the use of corticosteroids as eye drops). Suspensions and ointments are also used and eye/lid movements may promote the distribution of drug over the surface of the eye.
Key pharmacokinetic concept—drug bioavailability
After a drug crosses the membranes of the gastrointestinal tract, it enters blood in capillaries of the gut wall (the splanchnic circulation), finally ending up in the portal vein. The portal vein then carries the blood containing the drug to the liver, which is the main site of drug metabolism (discussed in a later section). The drug may pass through the liver and enter the systemic circulation as intact parent drug (unmetabolised) or may undergo metabolism in the liver. The extent to which a drug is metabolised (extracted) by the liver is highly variable.
The two factors that then determine the amount of drug reaching the systemic circulation are:
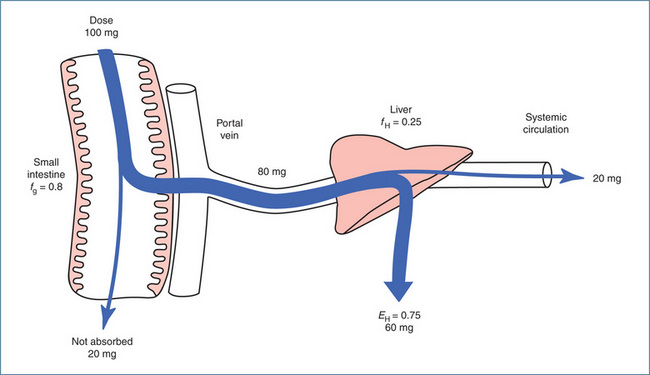
Figure 6-4 Factors affecting bioavailability. In this example, 80 mg of the original 100 mg dose is absorbed intact into the portal circulation (fraction absorbed is 0.8). The hepatic extraction ratio is 0.75, i.e. 60 mg is extracted in the first pass through the liver and 20 mg escapes extraction and is available for distribution via the systemic circulation. The bioavailability is F = fg × fH , which is 0.8 × 0.25 = 0.2 (20%).
Source: Birkett 2002, reproduced with permission.
Together fg and fH determine the bioavailability of a drug (Box 6-3).
Box 6-3 Key pharmacokinetic definition: bioavailability
Bioavailability is defined as the proportion of the administered dose that reaches the systemic circulation intact. It is usually expressed as a percentage. The symbol for bioavailability is F.

The approximate oral bioavailabilities of some commonly used drugs are alendronate 0.5%, amlodipine 70%, cefalexin 90%, digoxin 70%, quetiapine 9% and warfarin 90%.
Key pharmacokinetic concept—hepatic first-pass effect
As discussed, orally administered drugs that are absorbed travel first through the portal system and the liver before entering the systemic circulation. Depending on whether the drug is metabolised or not, a variable amount of drug can be extracted (fH) by the liver before the drug ever reaches the systemic circulation. In the example shown in Figure 6.4, 80 mg of the drug reaches the liver and 60 mg is extracted in the first pass through the liver. Consequently, the bioavailability of that drug is 20% and hence only 20 mg (a small fraction of the original 100-mg dose) is available for distribution and to produce a pharmacological effect. For such medications the oral drug dose is calculated to compensate for this first-pass effect. For example, morphine has a significant hepatic first-pass effect—30 mg oral morphine is equivalent to 10 mg morphine administered IM/IV/SC (Australian Medicines Handbook 2010). The hepatic first-pass effect explains why the oral doses of some drugs are much larger than the intravenous doses.
Drug bioequivalence
The term bioequivalence is used clinically when referring to two pharmaceutical formulations of the same drug that contain the identical concentration of the active ingredient in the same dosage form and administered by the same route. This is an important concept because, once the patent expires on a drug, other pharmaceutical companies can produce a generic equivalent of the original patented drug with a new proprietary (trade) name. The new generic product is then tested against the original market leader formulation (brand) to determine its relative bioavailability.2 The two pharmaceutical formulations (brand and the generic) are considered to be bioequivalent if their bioavailability is not significantly different under the test conditions (they attain similar concentrations in blood and tissues at similar times) and if there are no clinically important differences between their therapeutic or adverse effects (Birkett 2002). Regulatory authorities when granting a product licence for a generic equivalent place particular importance on evidence of bioequivalence.
Biosimilars
Pharmaceutical drugs are in general small low-molecular-weight medicinal chemicals and, once the patent has expired, the production of a generic drug is relatively easy. If the chemical and therapeutic bioequivalence of the generic product is established the generic drug is then marketed. However, more recently biopharmaceutical drugs have been introduced into clinical medicine. These drugs are large protein molecules, produced using ‘biotechnology’ methods, for example recombinant human technology, gene transfer methods and monoclonal antibody methods. Often the biopharmaceutical drug is produced using microbial cells and during the production process any small change (for example in post-translational modification) can cause a major impact on the biological activity of the protein drug. Once the patent expires a ‘biosimilar’ can be produced. A biosimilar is defined as ‘a biological product referring, but not identical, to an existing product, submitted for separate marketing approval following patent expiration’ (Schellekens et al 2008).
According to the EMEA Guidelines a biosimilar is not considered a generic medicinal product as it is often impossible to determine the bioequivalence of a protein. This is understandable when you consider that biopharmaceutical drugs are often manufactured from unique cell lines and it is virtually impossible to produce an ‘identical’ copy of the original patented (innovator) protein. For example, glycosylation of proteins (which is important for biological activity), ligand recognition and pharmacokinetics were found to differ between the original patented (innovator) epoetin and a number of biosimilar epoetins. These differences were thought to account for the altered clinical profile of the biosimilars. Similarly, differences in clinical activity and adverse effects have been reported for interferon-alpha-2a and enoxaparin biosimilars.
Many important issues remain unresolved with respect to biosimilars. These include the need for rigorous testing of the biosimilar against the innovator product, the standards of production and quality control, immunogenicity of the biosimilars and transparency of product labelling to provide clear guidance to health professionals on the interchangeability/substitution of a biosimilar with its innovator product. Current evidence indicates that some biosimilars are not interchangeable with the original innovator biopharmaceutical products.
Drug distribution
After a drug reaches the systemic circulation it can be distributed to various interstitial and intracellular compartments within the body including blood, bone, fat, total body water and extracellular water. Distribution is defined as the process of reversible transfer of a drug between one location and another (one of which is usually blood) in the body (see Figure 6-1). Some drugs remain almost exclusively in blood and these include heparin and warfarin. Other drugs are distributed to organs that are well perfused (e.g. heart, liver and kidneys) and the local drug concentration in these organs may be high initially. Drugs are also distributed more slowly to organs with poor blood supply, which include skeletal muscles and fat. Drugs that are widely distributed in the body include ethanol, digoxin, propranolol and morphine.
The rate at which a drug enters the different compartments of the body depends on the permeability of the capillaries, the partitioning of the drug between the vascular and tissue compartment and perfusion. As already discussed, lipid-soluble (un-ionised) drugs can readily cross capillary membranes to enter most tissues and fluid compartments, whereas ionised (lipid-insoluble) drugs do not diffuse readily across membranes. Cardiovascular function also affects the rate and extent of distribution of a drug, specifically cardiac output (the amount of blood pumped by the heart each minute) and regional blood flow (the amount of blood supplied to a specific organ or tissue).
Plasma protein binding
On entry into the circulatory system, a proportion of free drug molecules bind reversibly to proteins and lipoproteins to form drug–protein complexes. Plasma protein binding is commonly expressed as a percentage, which represents the proportion of the total drug bound, or as the fraction unbound (e.g. 75% bound corresponds to a fraction unbound of 0.25). The extent of drug binding depends on the affinity or attraction of the drug for the protein, the relative concentrations of the drug and the protein, and the number of drug binding sites on the protein. Drugs with a high affinity for the binding protein will be more ‘tightly’ but reversibly bound and the fraction of unbound drug will be low. Although plasma protein binding is a saturable process, the concentrations of most drugs in blood following therapeutic doses are generally lower than those of the binding proteins. For the majority of drugs in the therapeutic range the percentage bound and the unbound fraction are relatively constant. However, there are exceptions. For example, the high plasma concentrations of salicylate achieved with anti-inflammatory doses, used in the treatment of rheumatoid arthritis, can result in non-linear binding to albumin. Non-linear binding occurs when the concentration of the drug saturates the protein binding sites and adding more drug increases disproportionately the unbound concentration of the drug in plasma.
Protein binding is a reversible and dynamic process, with bound and unbound drug in equilibrium:
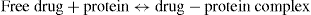
As free drug is removed from the circulation (e.g. by distribution, metabolism, excretion), the drug–protein complex dissociates very rapidly so that more free drug is released to replace what is ‘lost’. This is very important, as it is only the free or unbound drug that exerts a pharmacological effect. This is illustrated below; in this example the initial plasma drug concentration is 100 mg/L and the fraction of drug that is bound to plasma proteins is 0.8 (80%) and the unbound fraction is 0.2 (20%).

Acidic drugs (e.g. warfarin) bind mainly to plasma albumin, while basic drugs (e.g. quinine) bind to α1-acid glycoprotein. Among the highly protein-bound drugs is warfarin, which is about 99% protein-bound. This means that at any given time, 99% is bound to plasma proteins and only 1% of free drug is available for distribution (the drug–protein molecule is too large to diffuse through the blood vessel membrane) and to exert a pharmacological effect. Other examples of highly protein-bound drugs include NSAIDS >95%, alfentanil 92%, atorvastatin ≥98% and candesartan 99.8%. Drugs with low protein binding include cefalexin 14%, codeine 7%, fluconazole 11% and paracetamol 10%. Because albumin and (to a lesser extent) other plasma proteins provide a number of binding sites, two drugs can compete with one another for the same site and displace each other. Although this does occur it is now generally accepted that competition between drugs for plasma protein binding rarely leads to an increased drug effect provided clearance is not affected simultaneously.
Hypoalbuminaemia
Hypoalbuminaemia, or low levels of albumin in the blood, may be caused by hepatic dysfunction such as cirrhosis or by failure of the liver to synthesise sufficient plasma proteins. The decrease in albumin concentration results in an increase in the amount of free drug available for distribution to tissue sites. When an individual is given the usual dosage of a drug in the presence of decreased plasma protein binding, more of the free (unbound) drug is available to exert a pharmacological effect. This may result in toxicity, and the drug dosage should be reduced.
Phenytoin, an anticonvulsant drug, normally has an unbound fraction of 0.1, which increases to 0.2 in renal failure because of hypoalbuminaemia and the accumulation of competing endogenous compounds. The ‘normal’ therapeutic range for phenytoin based on total drug concentration is 10–20 mg/L and the therapeutic range of unbound drug concentration is 1–2 mg/L (i.e., 0.1 × 10 mg/L = 1 mg/L and 0.1 × 20 mg/L = 2 mg/L). In a renal failure patient when the unbound fraction is 0.2 the unbound concentration doubles to 2–4 mg/L (i.e., 0.2 × 10 mg/L = 2 mg/L, 0.2 × 20 mg/L = 4 mg/L), resulting in an enhanced pharmacological effect and toxicity. In this situation therapeutic drug monitoring is used to guide dosing and the therapeutic range based on total drug concentration is 5–10 mg/L, which maintains the unbound phenytoin drug concentration at 1–2 mg/L (i.e., 0.2 × 5 mg/L = 1 mg/L, 0.2 × 10 mg/L = 2 mg/L).
Tissue binding
Adipose tissue
Lipid-soluble drugs have a high affinity for adipose tissue, which is where these drugs are stored. Moreover, the relatively low blood flow in adipose tissue makes it a stable reservoir for a limited number of drugs and also for some environmental chemicals (e.g. DDT). For example, the lipid-soluble barbiturate anaesthetic thiopentone is initially rapidly distributed to brain, producing anaesthesia, but then redistributes to and accumulates in fatty tissue at levels 6–12 times those in the plasma. Continued administration of thiopentone causes a progressively longer period of anaesthesia as the drug accumulates in the body. This is one of the reasons why thiopentone is used for the induction of anaesthesia and not for surgical anaesthesia.
Bone
Some drugs have an unusual affinity for bone; for example, the antibiotic tetracycline accumulates in bone after being absorbed onto the bone-crystal surface. This serves as a storage site for tetracycline, which can depress bone growth in premature infants. Distribution of tetracycline to the teeth in a young child results in discolouration, which is thought to be due to formation of a tetracycline–calcium– orthophosphate complex. Brownish pigmentation of permanent teeth may also result if this drug is given during the prenatal period or early childhood. The bisphosphonates used for the treatment of osteoporosis are incorporated into the bone matrix and remain there until the bone is remodelled.
Barriers to drug distribution
Blood–brain barrier
The blood–brain barrier allows distribution of only lipid-soluble drugs (e.g. general anaesthetics and barbiturates) into the brain and cerebrospinal fluid. The barrier is made up of a layer of endothelial cells covered by a fatty sheath of glial cells joined by continuous tight intercellular junctions; normally drugs that are strongly ionised and poorly soluble in lipids cannot enter the brain. In some circumstances, such as meningitis, the blood–brain barrier can become ‘leaky’, and this allows access of drugs that would not normally be able to penetrate the brain. The use of penicillin systemically to treat bacterial meningitis is an example of taking advantage of the inflammatory disruption of the blood–brain barrier.
Placental barrier
The membrane layers that separate the blood vessels of the mother and the fetus constitute the placental barrier. In addition, enzymes in the placenta can metabolise some agents (e.g. catecholamines), inactivating them as they travel from the maternal circulation to the embryo. Despite the thickness of the structure, it does not afford complete protection to the fetus. Unlike the blood–brain barrier, the non-selective passage of drugs across the placenta to the fetus is a well-established fact. Although lipid-soluble substances preferentially diffuse across the placenta, the barrier is also permeable to a great number of water-soluble drugs. Consequently, many drugs intended to produce a therapeutic response in the mother may also cross the placental barrier and exert harmful effects on the developing embryo (see Chapter 9). Among the drugs easily transported across the placenta are steroids, narcotics, anaesthetics and some antibiotics.
Drug metabolism
Drug metabolism, or biotransformation, is the process of chemical modification of a drug and is almost invariably carried out by enzymes. The liver is the primary site of drug metabolism but, with certain drugs, other tissues (e.g. kidneys, lungs and intestinal mucosa) may also be involved to a limited extent in this process. Most drugs (around 70%) undergo metabolism to some extent, and in some but not all cases the products of metabolism have less biological activity than the parent drug. An exception to this is the use of prodrugs, which are inactive until converted to the active drug in the liver. Examples of prodrugs are the antihypertensive drug losartan, the anti-inflammatory drug sulindac and the antiplatelet agent clopidogrel. For the majority of therapeutic drugs, metabolism results in the formation of a more water-soluble compound or metabolite, which can then be excreted. Metabolism thus clears the parent compound and promotes urinary excretion. In terms of hepatic drug metabolism an important pharmacokinetic concept is ‘first-pass’ metabolism, which is discussed further in Chapter 8. To fully understand this concept it is important to have an appreciation of the types of drug metabolism reactions, the enzymes involved and those factors that can alter the extent of metabolism of drugs.
Classification of drug metabolism reactions
The vast majority of drugs are metabolised in the liver by either functionalisation and/or conjugation reactions. In many texts these are referred to as phase I and phase II reactions, respectively, which unfortunately in the 21st century is an inappropriate classification of drug metabolism reactions (Josephy et al 2005). Historically phase I reactions include oxidation and reduction reactions and hydrolyses or any combination of these three reactions. The phase II reactions are conjugation reactions typically involving the addition of glucuronic acid or sulfate. This terminology is now considered outmoded as it groups unrelated enzyme reactions and ignores our understanding of drug-metabolising enzymes. Indeed the term ‘functionalisation’ (the addition of a functional group) is not without its problems as some oxidation reactions remove functional groups. For simplicity we will use the terms ‘functionalisation’ and ‘conjugation’. It is important to recognise that a drug may:
These reactions are not necessarily sequential and can occur simultaneously (e.g. the metabolism of codeine by oxidation to morphine and by glucuronidation to codeine-6-glucuronide).
Functionalisation reactions
These reactions generally involve the introduction of a functional group into the molecule and include oxidation and reduction reactions or hydrolysis reactions (Table 6-2). These chemical reactions produce more water-soluble metabolites. In some cases the metabolites are more pharmacologically active than the parent compound and, uncommonly, may be more toxic (e.g. acrolein, the toxic metabolite of cyclophosphamide, and N-acetyl-pbenzoquinone imine, the toxic metabolite of paracetamol). The major family of enzymes associated with these reactions is the cytochrome P450 family.
Table 6-2 Functionalisation reactions
| ENZYME | CHEMICAL REACTION | DRUG SUBSTRATE → DRUG METABOLITE |
| CYP | Oxidation | Phenytoin → hydroxphenylhydantoin |
| S-warfarin → 7-hydroxywarfarin | ||
| Propranolol → 4-hydroxypropranolol | ||
| Esterases | Hydrolysis | Aspirin → salicylic acid |
| Clofibrate → clofibric acid | ||
| Succinylcholine → succinic acid | ||
| CYP | Reduction | Halothane → chlorotrifluoroethane |
Cytochrome P450 enzymes
The enzymes of greatest importance in functionalisation reactions are the superfamily of cytochrome P450 enzymes; the root symbol is CYP. CYPs are haem-containing enzymes found in the smooth endoplasmic reticulum of cells and are particularly abundant in liver cells (hepatocytes). CYPs catalyse the transfer of one atom of atmospheric oxygen to a substrate (drug or chemical), producing an oxidised substrate (metabolite) plus a molecule of water according to the following general reaction:

NADPH cytochrome P450 oxidoreductase (OR) is an enzyme essential for CYP activity since it provides the electrons (from NADPH/H+) necessary for the CYP oxidation/reduction cycle.
CYPs are involved not only in drug metabolism but also in the metabolism of environmental pollutants and dietary chemicals and in the synthesis and metabolism of bile acids, steroids, hormones and fatty acids. There are more than 50 individual human CYPs, which are grouped based on amino-acid sequence identity into families and subfamilies; of these, approximately 18 enzymes in families 1, 2 and 3 are able to metabolise drugs and foreign chemicals. In naming them, CYP is followed by a number designating the family, which is followed by a letter that denotes the subfamily and then another number that identifies the CYP form. For example, CYP2D6 is family 2, subfamily D and gene number 6. The human CYPs of greatest importance in hepatic drug metabolism are CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4 and CYP3A5. Table 6-3 lists CYPs involved in the metabolism of some common therapeutic drugs. For many common drugs it is important to be aware of which CYP is involved in metabolism so that administration of other drugs that may compete for or inhibit metabolism of the drug by the particular CYP can be avoided. Up-to-date information on CYP substrates can be found at the Indiana University Division of Clinical Pharmacology website, http://medicine.iupui.edu/clin pharm/ddis/.
Table 6-3 Common drugs metabolised by CYP enzymes
| CYP | DRUGS METABOLISED |
| CYP1A2 | Amitriptyline, caffeine, clozapine, ropivacaine, theophylline |
| CYP2A6 | Nicotine |
| CYP2C8 | Cerivastatin, paclitaxel, torsemide |
| CYP2C9 | Celecoxib, diclofenac, ibuprofen, irbesartan, phenytoin, S-warfarin |
| CYP2C19 | Citalopram, diazepam, omeprazole, phenytoin, R-warfarin |
| CYP2D6 | Amitriptyline, codeine, fluoxetine, haloperidol, metoprolol, perhexiline, nortriptyline, quetiapine |
| CYP2E1 | Alcohol, enflurane, halothane |
| CYP3A4 | Amiodarone, aprepitant, carbamazepine, cyclosporin, erythromycin, felodipine, hydrocortisone, saquinavir, tacrolimus, zolpidem |
Conjugation reactions
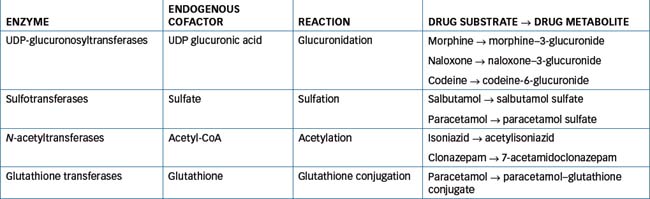
These involve the joining of a suitable functional group present in the drug molecule with the polar group of an endogenous substance in the body (e.g. glucuronic acid, sulfate, acetyl-coenzyme A or glutathione). The conjugated molecule is generally more polar or more water-soluble, which enhances urinary excretion. The relationship between drug metabolism and renal excretion is illustrated in Figure 6-5. Conjugation reactions are catalysed by a variety of different transferase enzymes, including the uridine diphosphate (UDP)-glucuronosyltransferases, sulfotransferases, N-acetyltransferases and glutathione transferases (Table 6-4). In humans there are 11 isoforms of sulfotransferases (SULT) classified into the SULT1, SULT2 and SULT4 families. The SULT1 family proteins play a major role in drug metabolism with SULT1A1 the most important enzyme. Two cytosolic N-acetyltransferases (NAT1 and NAT2) have been identified and of these only NAT2 seems to be important in drug metabolism. Over 20 glutathione transferases (GST) have been identified in humans. These enzymes are divided into two sub families: the cytosolic GSTs and the microsomal GSTs. The cytosolic GSTs play an important role in drug and xenobiotic metabolism while the microsomal GSTs have a greater role in the metabolism of leukotrienes and prostaglandins.
Figure 6-5 Relationship between drug metabolism and renal excretion. Metabolism via functionalisation and conjugation reactions results in decreasing lipid solubility, increasing water solubility and progressive enhancement of urinary excretion.
Source: Birkett et al 1979, reproduced with permission.
UDP-glucuronosyltransferases (UGT)
Like the CYPs, the UGT enzymes exist as a superfamily of enzymes that differ in terms of the drugs they metabolise. There are at least 12 UDP-glucuronosyltransferases that catalyse the conjugation of a substrate (drug) with glucuronic acid, which is derived from the co-factor UDP-glucuronic acid (UDPGA). Glucuronidation has been extensively studied in multiple species and, not surprisingly given their polar nature, glucuronide conjugates are excreted in urine and bile. Currently 18 catalytically active UGTs that primarily utilise UDPGA as cofactor have been identified, nine each from the UGT1 (1A1, 1A3, 1A4, 1A5, 1A6, 1A7, 1A8, 1A9 and 1A10) and UGT2 (2A1, 2A3, 2B4, 2B7, 2B10, 2B11, 2B15, 2B17 and 2B28) families. Individual UGTs exhibit distinct, but overlapping, substrate selectivity and differ in terms of regulation of expression. Age, diet, disease states, induction and inhibition by chemicals, ethnicity, genetic polymorphism and hormonal factors are known to influence UGT activity (Miners et al 2004).
Interindividual variability in drug metabolism
Large differences can occur between individuals in both the extent and the rate of metabolism of many drugs. Metabolism therefore becomes very important in determining the therapeutic and toxic responses to many drugs. This variability can be due to a range of factors, including:
Environmental factors – enzyme induction and inhibition
The basis for many drug–drug interactions during metabolism is related to either the induction or inhibition of enzyme activity. Induction or increased drug metabolism usually arises from increased synthesis of more of the enzyme protein via an effect on the genes that encode the specific drug-metabolising enzyme. The clinical impact of enzyme induction will then depend on the extent to which the plasma drug concentration is decreased (suboptimal) over the course of treatment with normal dosing. A drug may induce its own metabolism or its metabolism may be induced by coadministration of another drug. Cigarette smoke induces expression of CYP1A2 and thus increases the metabolism of caffeine, theophylline and imipramine. Other examples of the clinical consequences of enzyme induction include the need for higher doses of antiepileptic drugs in people treated with combinations of antiepileptic drugs, as many of these drugs are enzyme inducers, and the risk of contraceptive failure in women receiving phenobarbitone or rifampicin because of the increased metabolism of oral contraceptive steroids.
Inhibition of drug metabolising enzymes commonly occurs because of competition (i.e. two different drugs compete for metabolism by the same enzyme). This invariably results in a decrease in metabolism of one of the drugs. The clinical consequences of inhibition of drug metabolism include a decreased rate of elimination from the body, resulting in an increased plasma concentration and risk of toxicity. Examples include:
Disease states
In people with cardiac failure, liver perfusion and oxygenation may be decreased and this can reduce the activity of drug-metabolising enzymes. In liver disease the effects are harder to predict, as they depend on the disease type and severity, all of which can influence drug metabolism. In general, in severe cirrhosis and viral hepatitis the clearance of drugs metabolised by CYP is decreased.
Hormonal factors
Although gender-related differences have been observed for drug-metabolising enzymes in animal species, differences in humans appear to be minor and clinically insignificant. However, hormonal factors during pregnancy can have an important effect on drug metabolism, particularly during the third trimester. Induced activity of many CYP and UGT enzymes occurs, particularly during the third trimester. For example, it is well established that doses of the anticonvulsant drugs carbamazepine (metabolised by CYP3A4) and phenytoin (metabolised by CYP2C9) must be increased during pregnancy to maintain plasma concentrations in the therapeutic range. Following birth, doses decline to pre-pregnancy requirements. CYP2D6 activity is also induced during pregnancy. In contrast, there is evidence suggesting that the metabolism of caffeine (a CYP1A2 substrate) declines during pregnancy. Thus, although induction occurs most commonly, effects of pregnancy on drug metabolism are not always predictable.
Excretion of drugs and drug metabolites
A drug continues to exert a pharmacological effect (in some cases this may be an adverse effect) in the body until it is eliminated. In pharmacokinetic terms, elimination is defined as ‘the irreversible loss of drug from the site of measurement’ and occurs by the processes of metabolism and excretion (Rowland & Tozer 1995). For example, after administration, a drug may be metabolised by the liver but its metabolites may remain in the body. However, the parent drug is considered to have been eliminated from the site of measurement (the plasma). The terms ‘elimination’ and ‘excretion’ are often used interchangeably but excretion applies solely to the loss of (chemically) unchanged drug or metabolites in, for example, urine or bile. The term ‘unchanged’ in this context may appear confusing but it refers to the immediate chemical species that is being excreted, which can be either a parent molecule or a metabolite. In this regard, the liver, being the major site of drug metabolism, is the main organ of elimination, while the kidneys are the main organs of excretion.
Renal excretion
Some drugs are excreted unchanged in the urine, while other drugs are so extensively metabolised that only a small fraction of the parent drug is excreted unchanged. The process of excretion is accomplished primarily through glomerular filtration, tubular secretion and reabsorption (Figure 6-6). For example, free unbound drugs and watersoluble metabolites are filtered by the glomeruli (the glomerular filtration rate is around 120 mL/min), whereas protein-bound substances are not filtered. Most of the 120 mL of water from the plasma filtered at the glomerulus is reabsorbed during its passage through the renal tubule and only about 1–2 mL finally appears as urine. As the water is reabsorbed, a concentration gradient is established between the drug in the tubular fluid and the unbound drug in the blood (i.e. the drug in the urine is concentrated relative to that in the blood). If the drug is lipid-soluble enough to pass through the membranes it will be reabsorbed from the tubular fluid back into the systemic circulation. If the urine flow rate is high there is less of a concentration gradient and less drug is reabsorbed. Conversely, if the urine is more concentrated due to a low urine flow rate there is more of a concentration gradient and more drug is reabsorbed. The water-soluble compounds, on the other hand, are not reabsorbed and are excreted in the urine (Figure 6-5).
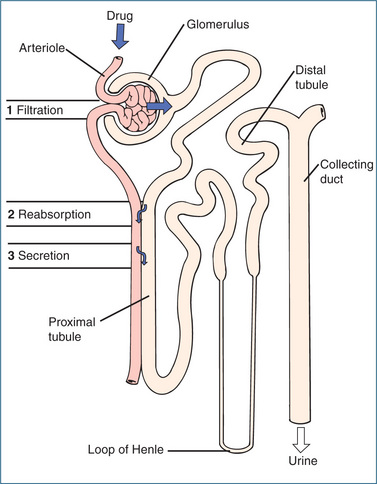
Figure 6-6 The drug excretion process, illustrating: 1 glomerular filtration, 2 tubular reabsorption and 3 active secretion.
Urinary pH varies between 4.6 and 8.2 and affects the amount of drug reabsorbed in the renal tubule by changing the degree of ionisation. Weak acids are excreted more readily in alkaline urine and more slowly in acidic urine; the reverse is true for weak bases. By altering the pH of urine, excretion of certain drugs can be increased, preventing prolonged action of a toxic compound, or decreased, prolonging the effect of a drug. In the case of drug overdoses involving weak organic acids such as aspirin or phenobarbitone, alkalinisation of the urine increases urine pH and the degree of ionisation (pH – pKa >1, refer to Table 6-1), resulting in decreased reabsorption and increased urinary drug excretion. Urine may be alkalinised by administering sodium bicarbonate. In contrast, high doses of vitamin C or ammonium chloride acidify the urine and promote the excretion of drugs that are weak bases.
The proximal tubule is the main site of active secretion, and both acidic and basic drugs are secreted into the lumen via their specific ‘acid’ and ‘base’ transporters. Examples of drugs secreted via the ‘acid’ (organic anion, OAT) transporters include penicillin, frusemide and probenecid; those secreted via the ‘base’ (organic cation) transporters include quinidine and procainamide. Another technique to reduce the rate of excretion of a drug is to competitively inhibit tubular secretion; for example, probenecid may be used to block tubular secretion and hence the renal excretion of penicillin. This prolongs the effect of the antibiotic by maintaining a higher therapeutic plasma concentration.
Another factor that affects renal excretion of drugs and drug metabolites is renal function, which is poorly developed in neonates and tends to decline in the elderly. Also, when a person has chronic renal failure, excretion of drugs is almost non-existent and, in people with cardiac failure, reduced blood flow to the kidneys may decrease renal excretion of unchanged drugs and drug metabolites.
Biliary excretion
After metabolism by the liver, drug metabolites may be transported into the bile, passed into the duodenum and excreted in faeces. Often, drug metabolites such as glucuronides are hydrolysed after the bile mixes with the intestinal fluid and the free drug is then reabsorbed and returned to the liver. This process is called enterohepatic cycling and for some drugs this ‘cycling’ produces a supply of recirculating drug that contributes to the overall pool of drug in the body. Examples of drugs that undergo significant enterohepatic cycling are morphine and ethinyloestradiol.
Pulmonary excretion
Gases and volatile drugs (e.g. general anaesthetics) are inhaled and excreted via the lungs. On inspiration, these agents enter the bloodstream and, after crossing the alveolar membrane, access the systemic circulation. Excretion from the lungs depends on the rate of respiration and exercise or deep breathing, which causes a rise in cardiac output and a subsequent increase in pulmonary blood flow, promoting excretion. In contrast, decreased cardiac output, such as that occurring in shock, decreases pulmonary drug excretion. Volatile substances, such as ethyl alcohol and paraldehyde, which are highly soluble in blood, are excreted in limited amounts by the lungs. In the case of ethyl alcohol pulmonary excretion is the basis of the alcohol breath test.
Excretion in sweat and saliva
Drug excretion through sweat and saliva is relatively unimportant because this process is slow relative to other forms of excretion and represents only a minor proportion of total excretion. The excretion of drugs and metabolites in sweat may be responsible for adverse effects such as dermatitis and skin reactions.
Excretion in breast milk
Many drugs or their metabolites cross the epithelium of the mammary glands and are excreted in breast milk. The risk to the infant of exposure to these drugs during breastfeeding depends on the maternal plasma drug concentration and the amount of milk ingested by the infant. Breast milk is acidic (pH 6.5); therefore basic compounds with low plasma protein binding and high lipid solubility such as narcotics (e.g. morphine and codeine) achieve high concentrations in this fluid. A major concern arises over the transfer of such drugs from mothers to their breastfed babies, which can result in adverse effects such as sedation and failure to thrive (Ilett et al 1997).
Key points

 The concentration that a drug attains at its site of action is influenced by the rate and extent to which the drug is absorbed into body fluids, distributed to the sites of action, metabolised into active or inactive metabolites and excreted from the body by various routes. The study of the kinetics of a drug during the processes of absorption, distribution, metabolism and excretion, or simply ‘what the body does to the drug’, is collectively described by the term ‘pharmacokinetics’. Absorption is defined as the process by which unchanged drug proceeds from the site of administration into the blood. It is an important factor for all routes of administration with the exception of intravenous administration. Variables that affect drug absorption include the nature of the absorbing membrane, blood flow, solubility of the drug, degree of ionisation and formulation characteristics. Bioavailability of a drug is defined as the proportion of the administered dose that reaches the systemic circulation intact. A biosimilar is defined as ‘a biological product referring, but not identical, to an existing product, submitted for separate marketing approval following patent expiration’. Current evidence indicates that some biosimilars are not interchangeable with the original innovator biopharmaceutical products. Distribution is defined as the process of reversible transfer of a drug between one location and another (one of which is usually blood) in the body. On entry into the circulatory system, free drug molecules bind to proteins to form drug–protein complexes. Protein binding decreases the free drug concentration and limits tissue distribution. As free drug is removed from the circulation, the protein–drug complex dissociates so that more free drug is released. Drug metabolism, or biotransformation, is the process of chemical modification of a drug and is almost invariably carried out by enzymes. The vast majority of drugs are metabolised in the liver by functionalisation and/or conjugation reactions. The major drug-metabolising enzymes are the cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT) families. Large differences may occur between individuals in the rate of metabolism of drugs. This variability may be due to genetic, environmental, age or disease-related factors.
The concentration that a drug attains at its site of action is influenced by the rate and extent to which the drug is absorbed into body fluids, distributed to the sites of action, metabolised into active or inactive metabolites and excreted from the body by various routes. The study of the kinetics of a drug during the processes of absorption, distribution, metabolism and excretion, or simply ‘what the body does to the drug’, is collectively described by the term ‘pharmacokinetics’. Absorption is defined as the process by which unchanged drug proceeds from the site of administration into the blood. It is an important factor for all routes of administration with the exception of intravenous administration. Variables that affect drug absorption include the nature of the absorbing membrane, blood flow, solubility of the drug, degree of ionisation and formulation characteristics. Bioavailability of a drug is defined as the proportion of the administered dose that reaches the systemic circulation intact. A biosimilar is defined as ‘a biological product referring, but not identical, to an existing product, submitted for separate marketing approval following patent expiration’. Current evidence indicates that some biosimilars are not interchangeable with the original innovator biopharmaceutical products. Distribution is defined as the process of reversible transfer of a drug between one location and another (one of which is usually blood) in the body. On entry into the circulatory system, free drug molecules bind to proteins to form drug–protein complexes. Protein binding decreases the free drug concentration and limits tissue distribution. As free drug is removed from the circulation, the protein–drug complex dissociates so that more free drug is released. Drug metabolism, or biotransformation, is the process of chemical modification of a drug and is almost invariably carried out by enzymes. The vast majority of drugs are metabolised in the liver by functionalisation and/or conjugation reactions. The major drug-metabolising enzymes are the cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT) families. Large differences may occur between individuals in the rate of metabolism of drugs. This variability may be due to genetic, environmental, age or disease-related factors.Review exercises
References and further reading
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Birkett D.J. Bioavailability and first-pass clearance. In: Birkett D.J., editor. Pharmacokinetics Made Easy. Sydney: McGraw-Hill, 2002. [ch 5]
Birkett D.J. Volume of distribution. In: Birkett D.J., editor. Pharmacokinetics Made Easy. Sydney: McGraw-Hill, 2002. [ch 2]
Birkett D.J., Grygiel J.J., Meffin P.J., Wing L.M.H. Fundamentals of Clinical Pharmacology; 4. Drug biotransformation. Current Therapeutics. 1979;6:129-138.
Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine 2009. http://medicine.iupui.edu/clinpharm/ddis/table.asp.[30 July 2009].
Holford N.H.G. Pharmacokinetics and pharmacodynamics: rational dosing and the time course of drug action. In Katzung B.G., editor: Basic and Clinical Pharmacology, 9th edn., The McGraw-Hill Companies, 2004. [ch 3]
Ilett K.F., Kristensen J.H., Wojnar-Horton R.E., Begg E.J. Drug distribution in human milk. Australian Prescriber. 1997;20:35-40.
Josephy P.D., Guengerich F.P., Miners J.O. “Phase I” and Phase II drug metabolism: terminology that we should phase out? Drug Metabolism Reviews. 2005;37:575-580.
Kim R.B. Transporters and drug discovery: why, when and how. Molecular Pharmaceutics. 2006;3(1):26-32.
Levy R.H., Thummel K.E., Trager W.F., et al, editors. Metabolic Drug Interactions. Philadelphia: Lippincott Williams & Wilkins, 2000.
Maluenda G., Lemesle G., Waksman R. A critical appraisal of the safety and efficacy of drug-eluting stents. Nature Medicine. 2009;85:474-479.
Miners J.O., Smith P.A., Sorich M.J., et al. Predicting human drug glucuronidation parameters: application of in vitro and in silico modeling approaches. Annual Reviews of Pharmacology and Toxicology. 2004;44:1-25.
Newton D.W. Drug incompatibility chemistry. American Journal of Health-System Pharmacy. 2009;66:348-357.
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007. [ch 7]
Rowland M., Tozer T.N. Clinical Pharmacokinetics: Concepts and Applications, 3rd edn. Philadelphia: Lea & Febiger; 1995.
Schellekens H., Lisman J., Bols T. Biosimilars in clinical practice—the challenges for hospital pharmacists. European Journal of Hospital Pharmacists Practice. 2008;14:32-33.
Trissel L.A. Handbook of Injectable Drugs, 15th edn. American Society of Health System Pharmacists; 2009.
Wykrzykowska J.J., Onuma Y., Serruys P.W. Advances in stent drug delivery: the future is in bioadsorbable stents. Expert Opinion Drug Delivery. 2009;6:113-126.