Chapter 44 Antibacterial Drugs
The discovery of sulfonamides and penicillin in the 1940s revolutionised the treatment of infectious diseases. The multiplicity of infectious organisms and the success of the early drugs spurred the development of numerous classes of antimicrobial drugs. Although many infections have been controlled with antimicrobial drugs, during the past 20 years drug-resistant strains of microorganisms have steadily increased, and this has necessitated the continued development of antimicrobial drugs. The challenge facing health-care professionals is the continued and prudent use of antimicrobial drugs in the face of increasing antibiotic resistance.
MRSA methicillin-resistant Staphylococcus aureus
Antibiotics
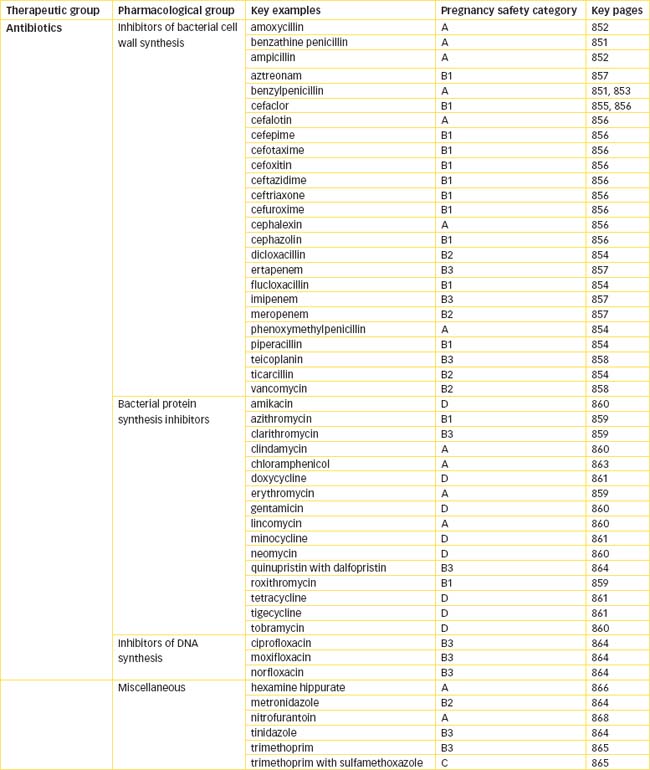
ANTIBIOTICS are chemical substances produced from various microorganisms (bacteria and fungi) that kill or suppress the growth of other microorganisms. This term is commonly used also to describe synthetic antimicrobial agents such as sulfonamides and quinolones that are not products of microorganisms. Hundreds of antibiotics are available that vary in antibacterial spectrum, mechanism of action, potency, toxicity and pharmacokinetic properties. For ease of understanding, this chapter is divided into:
Inhibitors of bacterial cell wall synthesis
Penicillins
Penicillins are antibiotics derived from several strains of common moulds often seen on bread or fruit (Figure 44-1). Introduced into clinical practice in the 1940s, penicillin and related antibiotics constitute a large group of antimicrobial agents that remain the most effective and least toxic of all available antimicrobial drugs. The common chemical feature of penicillins, cephalosporins, monobactams and carbapenems is a β-lactam ring that is essential to activity of the drug but is also the site of attack by resistant bacteria that possess β-lactamase enzymes that render the antibiotic inactive.
Mechanism of action
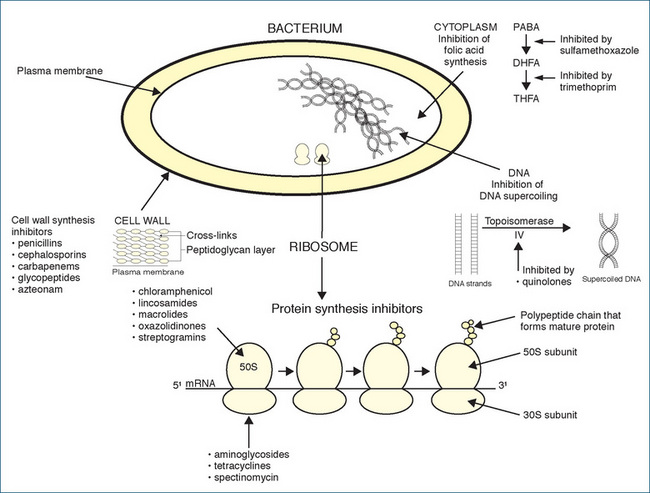
The bacterial cell wall is a rigid cross-linked structure of peptidoglycan, which is composed of crossed linked glycan chains comprised of alternating molecules of the amino sugars N-acetylglucosamine and N-acetylmuramic acid. Peptidoglycan is essential for the normal growth and development of bacteria. The thickness of the cell wall varies: in Gram-positive bacteria it is 50–100 molecules thick, whereas in Gram-negative bacteria it is 1–2 molecules thick. Penicillins weaken the cell wall by inhibiting the transpeptidase enzymes responsible for cross-linking the glycan strands; this results in cell lysis and death (Figure 44-2). Penicillins are considered to be bactericidal time-dependent drugs because they kill susceptible bacteria, but their effectiveness can be influenced by the presence of, or resistance to, certain β-lactamase enzymes and their combination with β-lactamase inhibitors.
To a limited extent, certain antibiotics are combined with β-lactamase inhibitors such as clavulanate or tazobactam. This extends the spectra of their antibacterial activity and improves their effectiveness but increases the cost. Although most penicillins are much more active against Gram-positive than Gram-negative bacteria, ticarcillin, aztreonam, imipenem and the combination of penicillins with β-lactamase inhibitors are more effective against Gram-negative bacteria (Escherichia coli, Klebsiella pneumoniae and others). Penicillins are divided into the following categories.
Narrow-spectrum penicillins
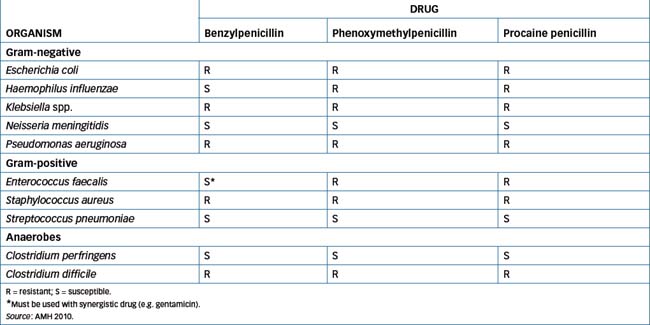
These include benzylpenicillin (also known as penicillin G) and phenoxymethylpenicillin (also called penicillin V). Benzylpenicillin and phenoxymethylpenicillin are comparable therapeutically but phenoxymethylpenicillin is more stable in stomach acid and is available as an oral preparation. Benzylpenicillin is available in various salt formulations—benzylpenicillin sodium, which may be administered IM/IV, while procaine penicillin (also known as procaine benzylpenicillin) and benzathine penicillin (also known as benzathine benzylpenicillin) are only administered IM. Accidental administration of either procaine penicillin or benzathine penicillin intravascularly ‘may result in severe neuromuscular damage; CNS effects including anxiety, agitation, fear of death and hallucinations (usually resolve in 15–30 minutes, but rarely last for up to 24 hours) may also occur’ (AMH 2010). The active substance in all these formulations is benzylpenicillin. Examples of susceptible and resistant bacteria are shown in Table 44-1.
Narrow-spectrum penicillinase-resistant penicillins
These are resistant penicillins with antistaphylococcal activity, and include dicloxacillin and flucloxacillin. A chemical alteration of the penicillin structure resulted in penicillins resistant to β-lactamase inactivation; thus they are used against penicillinase-producing staphylococci. These antibiotics are not, however, effective against methicillin-resistant bacteria.
Moderate-spectrum β-lactamase-sensitive aminopenicillins
These include amoxycillin and ampicillin. Although these antibiotics have a similar spectrum of activity to that of penicillin, they have greater efficacy against selected Gram-negative bacteria (e.g. Haemophilus influenzae) but are usually not very effective against Staphylococcus aureus and Escherichia coli (β-lactamase-producing bacteria) unless combined with clavulanic acid (e.g. amoxycillin with clavulanic acid).
Broad- and extended-spectrum (antipseudomonal) penicillins
This group includes piperacillin, piperacillin–tazobactam and ticarcillin–potassium clavulanate. These antibiotics have a broader spectrum of antimicrobial activity, but only piperacillin (as the single drug) is effective against Pseudomonas aeruginosa.
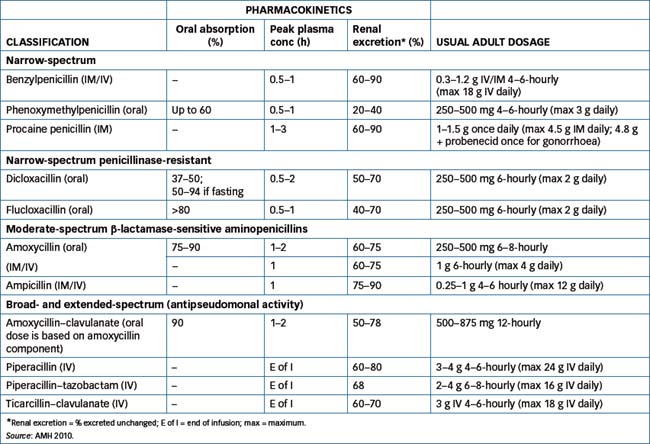
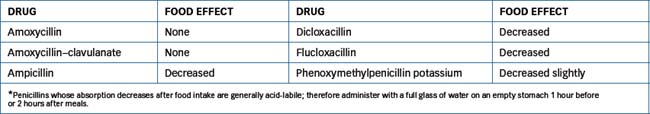
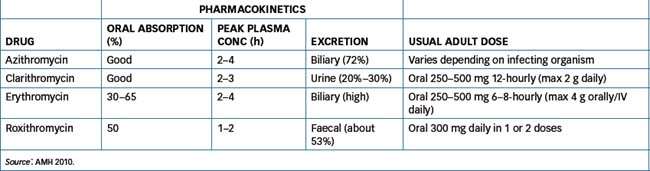
Health-care professionals should be aware that antibiotic therapy may provide an environment that is conducive to the unrestrained growth of undesirable microorganisms, such as bacteria or fungi that would ordinarily have been controlled by the normal body flora. This is a condition known as superinfection and may present as diarrhoea from altered gastrointestinal (GI) flora or a Candida infection vaginally. Table 44-2 lists penicillin pharmacokinetics and usual adult dosages and Table 44-3 the effect of food on oral penicillin absorption.
Drug interactions
Examples of interactions with penicillins include:
Adverse reactions
For penicillins these include diarrhoea, nausea, vomiting, headache, sore mouth or tongue, oral and vaginal candidiasis, allergic reactions, anaphylaxis, serum sickness-type reaction (rash, joint pain and fever), hives and pruritus. Rare adverse reactions include cholestatic hepatitis with some penicillins, especially dicloxacillin, flucloxacillin, amoxycillin–potassium clavulanate and ticarcillin–potassium clavulanate; leucopenia or neutropenia; mental disturbances (with large dosages of procaine penicillin); convulsions (with high dosages of penicillin and/or in people with advanced renal function impairment); and interstitial nephritis. Platelet dysfunction has been reported with piperacillin and ticarcillin, especially in people with renal impairment.
Warnings and contraindications
Use penicillins with caution in people with general allergies or on salt-restricted diets, and in those with poor cardiac reserve or renal or hepatic impairment. Many parenteral formulations of penicillins (e.g. benzylpenicillin) contain a high concentration of sodium that may precipitate or worsen heart failure in patients with cardiac reserve. Avoid use in people with penicillin hypersensitivity (see Figure 44-3, Clinical Interest Box 44-1, Yates 2008), bleeding disorders, congestive heart failure, cystic fibrosis, GI disease (especially antibiotic-associated colitis, as peni cillins may cause pseudomembranous colitis) and mononucleosis.
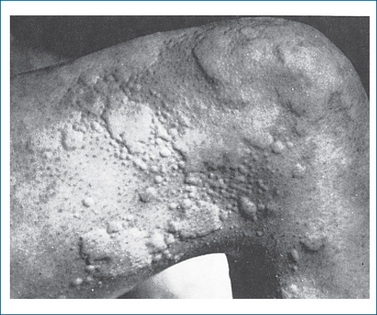
Clinical interest Box 41-1 Penicillin rash and anaphylaxis
A hypersensitivity reaction to penicillin is perhaps the one adverse drug reaction most people seem to have an awareness of, either through personal experience or through knowledge of someone who has experienced a reaction. In fact, a hypersensitivity reaction is the commonest adverse effect noted with penicillins (varying from 1% to 10% of people), and it appears that no one penicillin is the sole culprit. Manifestations of penicillin allergy range from the most common maculopapular or urticarial rash through to bronchospasm and the least common reaction, anaphylaxis. A reaction can occur with any dosage and in the absence of prior knowledge of exposure, which often occurs as a result of ingestion of food of animal origin where antibiotics may have been used in animal feed, or through ingestion of fungi producing penicillin. Anaphylaxis occurs in approximately 0.004%–0.04% of exposed people, and about 0.001% will die as a result. This reaction most often occurs after injection, but has been reported after ingestion of small doses and even after minute intradermal injection to test for penicillin sensitivity.
Cephalosporins
 Penicillins were discovered as a result of someone leaving a window open in Fleming’s laboratory (or so the story goes!), while cephalosporins were isolated from sea fungus found near a sewerage outlet off the Sardinian coast in 1948. Since then, chemical modification of the central active component, 7-aminocephalosporanic acid and the addition of side-chains have created compounds with different and greater microbiological and pharmacological activities. To classify easily the differences in antimicrobial activity, cephalosporins are divided into first, second, third and fourth generations.
Penicillins were discovered as a result of someone leaving a window open in Fleming’s laboratory (or so the story goes!), while cephalosporins were isolated from sea fungus found near a sewerage outlet off the Sardinian coast in 1948. Since then, chemical modification of the central active component, 7-aminocephalosporanic acid and the addition of side-chains have created compounds with different and greater microbiological and pharmacological activities. To classify easily the differences in antimicrobial activity, cephalosporins are divided into first, second, third and fourth generations.
Mechanism of action
Like penicillin, cephalosporins inhibit bacterial cell wall synthesis and are also bactericidal (Figure 44-2). Because they inhibit cell division and growth, rapidly dividing bacteria are affected most. They are effective in numerous situations, but with only a few exceptions they are rarely the drugs of first choice. The first-generation cephalosporins (e.g. cephazolin, cephalexin) are primarily active against Gram-positive bacteria, whereas the second-generation drugs (cefaclor and others) had increased activity against Gram-negative microorganisms. The third-generation drugs are more active against Gram-negative bacteria (ceftazidime is also effective against P. aeruginosa) and β-lactamase-producing microbial strains but less effective against Gram-positive cocci. Cefepime is a fourth-generation cephalosporin with antimicrobial effects comparable to those of third-generation cephalosporins and is also more resistant to some β-lactamases.
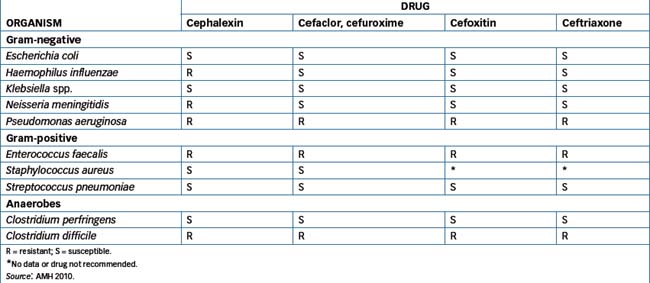
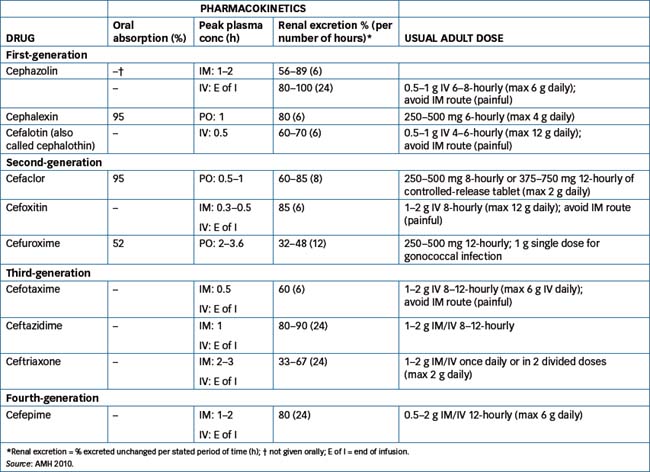
Initially, the advantage of cephalosporins over penicillins was their resistance to degradation by β-lactamase, but widespread use of cephalosporins has been linked to the emergence of resistant strains of K. pneumoniae and methicillin-resistant S. aureus (MRSA) and Clostridium difficile. Examples of susceptible and resistant bacteria are shown in Table 44-4 and Table 44-5 lists cephalosporin pharmacokinetics and the usual adult dosages.
Indications
Cephalosporin antibiotics may be prescribed for patients allergic to penicillins. However, the possibility of a crossreaction is 5%–15%, and cephalosporins should not be used if a person reports a history of a serious reaction or anaphylaxis to penicillin. If use is critical, specialist advice should always be sought prior to administration. Drugs in this group are indicated for the treatment of a variety of infections and are also used as prophylaxis in bowel and gynaecological surgery. Combinations of third-generation cephalosporins and aminoglycosides are used synergistically to treat P. aeruginosa, Serratia marcescens and other susceptible organisms.
Drug interactions
Examples of interactions with cephalosporins include:
Adverse reactions
These include diarrhoea, abdominal cramps or distress, oral and/or vaginal candidiasis, rash, pruritus, redness, oedema, allergic reaction, anaphylaxis, Stevens–Johnson syndrome, haemolytic anaemia, renal toxicity, convulsions, neurotoxicity (including seizures), blood dyscrasias, thrombocytopenia and bleeding.
Warnings and contraindications
Use cephalosporins with caution in individuals with impaired/low vitamin K synthesis because of increased risk of bleeding. Monitoring of INR (see Chapter 26) is recommended. Because parenteral cephalosporins have high sodium contents, their use should be avoided in sodium-restricted individuals. Avoid use in people with reports of anaphylaxis to penicillin, penicillin derivatives, penicil lamine or cephalosporins; bleeding disorders; GI disease or kidney function impairment.
Carbapenems
Ertapenem, imipenem and meropenem are members of a class of antibiotics called carbapenems, which are related to the β-lactam antibiotics but differ from them in having another 5-membered ring in their chemical structure. They bind to penicillin-binding proteins, thus inhibiting bacterial cell wall synthesis. Carbapenems have the broadest spectrum of activity of all the antimicrobials against Gram-positive and Gram-negative aerobic and anaerobic organisms. All of these drugs are inactive against MRSA and Enterococcus faecium, and ertapenem is inactive against P. aeruginosa and Acinetobacter. Imipenem is degraded by renal dipeptidase and is combined with the dipeptidase inhibitor cilastatin, which inhibits renal dihydropeptidase and blocks the tubular secretion of imipenem, thus preventing renal metabolism of this drug. Meropenem and ertapenem are more resistant to renal dipeptidase degradation and are given alone. These drugs are expensive and are generally reserved (with the exception of ertapenem) for nosocomial and life-threatening infections when other antibiotics are contraindicated or inappropriate. Meropenem is used for treating meningitis, whereas imipenem is contraindicated because of the high incidence of seizures (this is likely to apply also to ertapenem, which is likewise associated with seizures, but there are currently no data on ertapenem use in meningitis).
When carbapenems are administered intravenously, peak plasma concentration is achieved rapidly. The half-life of imipenem is 2–3 hours, about 1 hour for meropenem and about 4 hours for ertapenem. These drugs are excreted unchanged to varying degrees in urine within 10 hours. Dosage adjustment is required in people with significant renal impairment. Imipenem and meropenem should not be given with probenecid, as renal secretion of the carbapenems is inhibited, increas ing the risk of toxicity. Seizures have been reported in combination with ganciclovir, so this combination should be avoided.
Adverse reactions include gastric distress, diarrhoea, nausea, vomiting, allergic-type reactions, confusion, psychiatric disturbances, insomnia and raised liver enzyme levels. Pseudomembranous colitis has also been reported with these drugs. The risk of seizures is high in people with pre-existing central nervous system (CNS) disturbances and in renal impairment if the plasma concentration increases substantially. Avoid use in people with hypersensitivity to imipenem, cilastatin or other β-lactams (e.g. penicillin and cephalosporin), and in kidney impairment and CNS disorders. Renal and hepatic function and haematological parameters should be monitored during prolonged treatment.
Aztreonam
Aztreonam, the first drug in the monobactam class of antibiotics, is a synthetic bactericidal antibiotic with a similar mechanism of action to that of penicillin. It binds to penicillin-binding proteins, resulting in inhibition of bacterial cell wall synthesis, cell lysis and death. It is active only against Gram-negative aerobic organisms (e.g. P. aeruginosa). It is reserved for treatment of infections when other antibacterial drugs are contraindicated and for urinary tract, bronchitis, intra-abdominal, gynaecological and skin infections. Aztreonam is highly resistant to most β-lactamase enzymes.
Aztreonam is not given orally, as it is not absorbed from the GIT. After IM injection, peak plasma concentration occurs in 0.6–1.3 hours. The half-life in adults with normal renal function is 1.4–2.2 hours, and 60%–70% of the drug is eliminated in the urine within 8 hours. Concomitant administration of probenecid or frusemide results in a clinically insignificant increase in the plasma concentration of aztreonam. No significant drug interaction has been reported at this stage.
Adverse reactions include gastric distress, diarrhoea, nausea, vomiting, hypersensitivity and thrombophlebitis at the site of injection. Rarely, anaphylaxis, hepatitis, jaundice, thrombocytopenia and prolonged bleeding time have been reported. Use aztreonam with caution in people receiving anticoagulant therapy. A low risk of allergic reaction exists in those allergic to penicillins or cephalosporins. The drug is contraindicated in people with aztreonam hypersensitivity. Administered by IV infusion, the dose in adults with normal renal function is 0.5–2 g every 6–8 hours. The maximum dose is 8 g IV daily. Dose reduction is required in moderate and severe renal impairment.
Glycopeptides
Vancomycin and teicoplanin are both complex glycopeptide antibiotics. Vancomycin was isolated from the soil actinomycete Streptococcus orientalis, while teicoplanin was isolated from Actinoplanes teichomyceticus. Both drugs inhibit bacterial wall synthesis and are primarily active against gram-positive bacteria. Due to increasing problems with vancomycin resistance, Australia has adopted the guidelines recommended for vancomycin use by the Centers for Disease Control Hospital Infection Control Practices Advisory Committee (USA). Similar consideration applies to teicoplanin. These guidelines are as follows (Australian Medicines, 2010AMH 2010):
Vancomycin absorption from the intestinal tract is poor; hence it is usually administered intravenously and never intramuscularly. In contrast, teicoplanin can be administered IM. An oral formulation of vancomycin is available but is only ever used for the treatment of pseudomembranous colitis. The elimination half-life varies between the two drugs: parenteral vancomycin has an elimination half-life of 4–6 hours in adults and about 2–3 hours in children, whereas it is close to 100 hours for teicoplanin. Both are excreted primarily by the kidneys, and dosage adjustment is crucial in persons with compromised renal function. Routine therapeutic plasma drug concentration monitoring is undertaken for vancomycin in circumstances such as during concomitant aminoglycoside administration, in patients on haemodialysis and during high-dose prolonged treatment in patients with unstable or impaired renal function. Therapeutic plasma drug concentration monitoring is usually unnecessary with teicoplanin unless treating severe infections such as endocarditis. With both drugs the intention is to allow individualisation of dose to ensure effective plasma drug concentrations. Examples of drug interactions and possible outcomes are listed in Drug Interactions 44-1.
Drug interactions 44-1 Vancomycin or teicoplanin
| Drug | Possible effects and management |
| Aminoglycosides | Increased potential for ototoxicity and nephrotoxicity with vancomycin and teicoplanin. Monitor renal function and plasma drug concentrations, adjust dose if necessary |
| Bile acid-binding resins (cholestyramine or colestipol) | When given concurrently with the oral dosage form, a reduction in vancomycin antibacterial activity is reported. Avoid this combination if possible. If not, give oral vancomycin several hours apart from the other medications |
| Muscle relaxants and general anaesthetics | Vancomycin may potentiate the neuromuscular blockade produced by non-depolarising muscle relaxants and suxamethonium. Vancomycin infusion should be completed before induction of general anaesthesia because of increased risk of vancomycinrelated adverse reactions (e.g. hypotension) |
Adverse reactions are more common with rapid IV infusions and include rash, itching, chills and fever. Rarely, the ‘red-neck’ or ‘red-man’ syndrome is reported after bolus or too rapid drug injection with vancomycin (less often with teicoplanin), which results in histamine release and chills, fever, tachycardia, pruritus, rash or red face, neck, upper body, back and arms (AMH 2010). In combination with aminoglycosides, there is an increased risk of nephrotoxicity and ototoxicity, particularly in people with renal impairment. Avoid use in people with glycopeptide hypersensitivity (allergic cross-reactions occur between teicoplanin and vancomycin), deafness, history of hearing loss or kidney disease. The dosage of either drug depends on age, renal function and indication, and relevant drug information sources should be consulted prior to administration.
Bacterial protein synthesis inhibitors
Macrolide antibiotics
The macrolide antibiotics are so named because they contain a many-membered lactone ring that has one or more sugar molecules attached. They inhibit bacterial RNA-dependent protein synthesis by binding to the 50S ribosomal subunit (Figure 44-2). Macrolides are bacteriostatic: that is, they inhibit growth of microorganisms and, in high concentrations with selected organisms, may be bactericidal. The macrolide antibiotics include azithromycin, clarithromycin, erythromycin and roxithromycin. Erythromycin was the first macrolide and is the key drug from this classification.
These agents have similar antimicrobial action (against Gram-positive and some Gram-negative microorganisms) and are used for respiratory, GIT, skin and soft tissue infections when β-lactam antibiotics are contraindicated because of allergy. Clarithromycin is used in conjunction with amoxicillin and the proton pump inhibitor omeprazole for the eradication of Helicobacter pylori (see Chapter 30). For macrolide pharmacokinetics and usual adult dosages, see Table 44-6.
Macrolides inhibit hepatic CYP3A4 and as a consequence are subject to numerous drug interactions. In general, they inhibit the metabolism of other drugs, leading to increased plasma concentration and the risk of toxicity. The potential for drug interactions is greatest with erythromycin > clarithromycin > roxithromycin > azithromycin. Drug Interactions 44-2 lists some of the more significant drug interactions but is not complete, and reference should be made to appropriate sources if uncertain about a specific drug.
Drug interactions 44-2 Macrolide antibiotics
| Drug | Possible outcomes | Management |
| Benzodiazepines (triazolam, midazolam) | Increased/prolonged sedation | Avoid combined use with erythromycin. Exercise caution with other macrolides |
| Bromocriptine | Increased adverse reactions | Avoid combined use |
| Buspirone | Increased adverse reactions | Avoid combined use with erythromycin. Exercise caution with other macrolides |
| Carbamazepine | Neurotoxicity | Decrease dose of carbamazepine if necessary |
| Cisapride | Cardiotoxicity | Combination contraindicated |
| Cyclosporin, tacrolimus | Nephrotoxicity, neurotoxicity | Adjust doses of cyclosporin and tacrolimus if necessary |
| Digoxin | May cause digoxin toxicity | Monitor plasma digoxin concentration |
| Ergot alkaloids | Ergotism | Adjust dose of ergot alkaloid or stop administration |
| Pimozide | Cardiac arrhythmias | Combination contraindicated |
| Theophylline | Theophylline toxicity | Monitor plasma theophylline concentration. Use alternative agent |
| Warfarin | Increased risk of bleeding | Monitor INR, decrease or cease warfarin. Use alternative antibiotic |
INR = international normalised ratio
Source:AMH 2010.
Adverse reactions to these drugs are as follows:
Use macrolide antibiotics with caution in people with severe liver function impairment. In addition, use erythromycin cautiously in patients with hearing loss, and clarithromycin cautiously in persons with severe kidney function impairment. Avoid these drugs in people with individual drug hypersensitivity and avoid use of erythromycin in those with cardiac arrhythmias.
Lincosamides
The lincosamides include lincomycin and clindamycin. Lincomycin inhibits protein synthesis by binding to the bacterial 50S ribosomal subunit and preventing peptide bond formation. It is primarily bacteriostatic, although it may be bactericidal in high doses with selected organisms. It is used to treat serious streptococcal and staphylococcal infections but clindamycin is preferred, as it is has better oral absorption and is more potent than lincomycin.
Clindamycin, which is a semisynthetic derivative of lincomycin, has a similar mechanism of action to that of lincomycin but is more effective. It is indicated for the treatment of bone and joint, pelvic (female), intraabdominal and skin and soft tissue infections, bacterial septicaemia and pneumonia caused by susceptible bacteria.
Oral clindamycin is well absorbed and should be administered with food or with a full glass of water. It is rapidly distributed to most body fluids and tissues, with the exception of cerebrospinal fluid; the highest concentrations are noted in bone, bile and urine. The half-life of clindamycin in adults is 2–3 hours. It reaches peak blood concentrations within 0.75–1 hour of oral administration in adults, within 1 hour in children and within 3 hours of intramuscular injection. It is metabolised in the liver and excreted primarily in bile, with only a small proportion (6%–10%) excreted as unchanged drug in urine.
Examples of drug interactions with clindamycininclude:
Adverse reactions include GI distress (stomach pain, diarrhoea, nausea, vomiting), oral and/or vaginal candidiasis, hypersensitivity, neutropenia and thrombocytopenia. A significant adverse and limiting effect for both drugs is antibiotic-associated pseudomembranous colitis in which the normal bacterial flora within the colon is disturbed facilitating the growth of Clostridium difficile or other bacteria. With overgrowth of the bacteria potent bacterial toxins are released causing inflammation of the colon. Patients at risk include the elderly, those with a compromised immune system (e.g. resulting from cancer chemotherapy) and patients with pre-existing inflammatory bowel disease. Use these drugs with caution in people with GI disorders, especially ulcerative colitis, antibiotic-induced colitis and regional enteritis. Avoid use in those with clindamycin or lincomycin hypersensitivity.
The usual adult oral dose of clindamycin is 150–450 mg 6–8-hourly; by IV infusion, it is 600–2700 mg daily to a maximum of 4.8 g IV daily. In children, the IM/IV infusion dose is 5–10 mg/kg every 6–8 hours.
Aminoglycosides Aminoglycosides are potent bactericidal antibiotics usually reserved for serious or life-threatening infections. They are very effective against many Gram-negative bacteria but as monotherapy they have limited activity against Gram-positive bacteria. Safer and less toxic agents are available to treat most Gram-positive infections but combinations of aminoglycosides with β-lactam antibiotics provide a synergistic effect against Listeria spp and Staphylococcus aureus. Currently available drugs are the parenteral aminoglycosides, including amikacin, gentamicin and tobramycin; and the oral aminoglycoside neomycin.
The mechanism of action of aminoglycosides involves irreversible binding to the 30S ribosomal subunit of susceptible bacteria, thus inhibiting protein synthesis (interferes with the mRNA–ribosome complex), leading to eventual cell death (bactericidal). They are indicated for the treatment of serious or life-threatening infections when other agents are ineffective or contraindicated. They are used with penicillins, cephalosporins or vancomycin for their synergistic effects and are especially useful for the treatment of Gram-negative infections such as those caused by Pseudomonas spp., E. coli, Proteus spp., Klebsiella spp., Serratia spp. and others.
Aminoglycosides are poorly absorbed (<1%) from the intestinal tract but are rapidly absorbed intramuscularly, with peak plasma concentrations occurring 30–90 minutes after injection. As the aminoglycosides are strongly polar molecules, they do not distribute to the CNS, and tissue concentrations are low. They are almost entirely eliminated by the kidneys and, in people with normal renal function, the plasma half-life is in the range of 2–3 hours.
Determining the plasma concentration of aminoglycosides is essential to ensuring that therapeutic concentrations are achieved without the risk of adverse reactions due to high plasma concentrations. These drugs exhibit a significant post-antibiotic effect, inhibiting the growth of organisms after the plasma concentration has fallen below the minimal inhibitory concentration. Although aminoglycosides have in the main been administered 2–3 times daily, there is substantial literature evidence to indicate that once-daily dosing in adults and children is as effective and no more toxic than the 2–3 times daily dosing regimens. In the absence of any contraindications, dosing once daily is recommended in people with normal renal function. Monitoring the plasma concentration is generally not undertaken if the course of treatment is shorter than 48 hours. If it is longer, monitoring of plasma concentration and renal function determines the dosage regimen. Methods used for drug concentration monitoring vary among facilities and may rely on interpreting either the trough plasma concentration from a special graph or the area under the plasma concentration– time curve (AUC). Local diagnostic laboratory services should be contacted for specialist information.
Drug Interactions 44-3 lists those that may occur when aminoglycosides are given with the drugs listed.
Drug interactions 44-3 Aminoglycosides
Source:AMH 2010.
| Drug | Possible effects and management |
| Aminoglycosides (two or more concurrently) | Potential for ototoxicity, nephrotoxicity and neuromuscular blockade is enhanced. Hearing loss may progress to deafness even after the drug is stopped. In some cases, hearing loss may be reversed. Avoid, or a potentially serious drug interaction may occur |
| Loop diuretics | Increased risk of irreversible hearing loss. Avoid prolonged use of high doses of aminoglycosides |
| Muscle relaxants | Aminoglycosides may potentiate the neuromuscular-blocking effect of non-depolarising muscle relaxants and suxamethonium |
| NSAIDs | NSAID-induced reduction in renal function may lead to increased plasma concentration of aminoglycoside. Monitor drug concentration and renal function and adjust dose if necessary |
| Penicillins and cephalosporins | Antibacterial action of aminoglycoside may be enhanced as a result of greater penetration. Do not mix aminoglycosides with penicillins and cephalosporins, as the parenteral solutions are incompatible |
Adverse reactions include nausea, vomiting, tinnitus, increase or decrease in urinary frequency, ataxia, dizziness, nephrotoxicity, neurotoxicity, ototoxicity (auditory and vestibular), hypersensitivity, peripheral neuritis and, rarely, neuromuscular blockade (difficulty breathing, increased sedation, weakness). Use these drugs with extreme caution in dehydrated individuals (increased risk of toxicity) and in people with myasthenia gravis, parkinsonism and hearing impairment. Aminoglycosides are contraindicated in people with a known allergic reaction or hypersensitivity to these drugs.
The usual adult doses (<48 hours) of aminoglycosides in adults with normal renal function are: for amikacin, 16–24 mg/kg IM/IV once daily or in 2–3 divided doses; and for gentamicin or tobramycin, 4–7 mg/kg IM/IV once daily. For additional dosing recommendations, refer to current recommendations. For neomycin, the oral dose is 1 g hourly for 4 hours, then 1 g every 4 hours up to a maximum daily dose of 12 g.
Tetracyclines
Tetracyclines were the first broad-spectrum antibiotics developed after a systematic search for antibiotic-producing microorganisms in soil. The first drug produced was chlortetracycline, which was released in 1948. This group now includes a number of drugs that have a common basic structure and similar chemical activity: doxycycline, minocycline and tetracycline (available only through the SAS). Minocycline is not as well tolerated as doxycycline, producing vestibular and CNS adverse effects and hepatitis and serum-sickness during long-term use for acne. More recently tigecycline, the first glycylcycline antiobiotic (a tetracycline derivative), has become available and it is discussed in the following section.
Tetracyclines are bacteriostatic for many Gram-negative and Gram-positive organisms; they exhibit cross-sensitivity and cross-resistance. Tetracyclines inhibit protein synthesis by reversibly blocking the 30S subunit of the ribosome and preventing access of tRNA to the mRNA–ribosome complex. These drugs have been commonly used to treat many infections, such as acne vulgaris, actinomycosis, anthrax, bacterial urinary tract infections (UTIs), bronchitis and numerous systemic bacterial infections sensitive to this class of drug.
Oral tetracyclines are fairly well absorbed and are distributed in most body fluids. Concentration in cerebrospinal fluid varies and can range from 10% to 25% of the plasma drug concentration after parenteral administration. Tetracyclines localise in teeth, liver, spleen, tumours and bone. Doxycycline can reach clinical concentrations in the eye and prostate, whereas minocycline reaches high levels in saliva, sputum and tears. Doxycycline and minocycline are metabolised in the liver, but most tetracyclines are excreted via the kidneys. Table 44-7 lists details of tetracycline half-lives and usual adult dosages.
Table 44-7 Half-life and usual adult dosage of tetracyclines
| DRUG | HALF-LIFE (h) | USUAL ADULT DOSAGE |
| Doxycycline | 12–22 | 200 mg on day 1, then 100 mg daily (max 200 mg daily); syphilis: 300 mg daily for 10 days |
| Minocycline | 11–23 | 200 mg on day 1, then 100 mg twice daily (max 200 mg daily) |
| Tetracycline | 6–11 | 250–500 mg 6-hourly (max 2 g daily) |
| Tigecycline | 36 | 100 mg IV for the first dose then 50 mg IV every 12 hours |
Adverse reactions include dizziness (minocycline), oesophagitis (doxycycline), ataxia, GI distress, photosensitivity (doxycycline, see Clinical Interest Box 44-2, depends on dose and extent of sun exposure), discolouration of infants’ or children’s teeth (do not give to children under 8 years), skin and mucous membrane pigmentation (minocycline), dark or discoloured tongue, rectal or genital fungal overgrowth; rarely, hepatotoxicity, pancreatitis and benign intracranial hypertension. Minocycline and doxycycline only may be taken with food or milk to reduce gastric adverse effects as interactions with food and milk are less problematic in comparison totetracycline. Tetracyclines are not recommended in people with renal impairment, but doxycycline and minocycline may be used. These drugs are contraindicated in pregnant women after week 18 and in children less than 8 years of age, as they cause discolouration of teeth and enamel dysplasia.
Clinical interest Box 44-2 Photosensitivity reaction with doxycycline
The New Zealand Centre for Adverse Reactions Monitoring (CARM) has reported that, of the 10 most commonly reported drugs associated with photosensitivity reactions in New Zealand, doxycycline ranked first and trimethoprim/ sulfamethoxazole ranked sixth. The photosensitivity reaction presents as unexpected sunburn or as a dry or blistering rash on skin exposed to the sun. The face, neck, arms, backs of hands, lower legs and feet were the most affected sites and the reaction occurred either immediately or as long as 3 days after exposure to sunlight. With doxycycline all patients should be advised of the risk of a photosensitivity reaction. Precautions to take include avoiding direct sunlight or use of sunlamps and sunbeds; trying to stay in the shade or using an umbrella; wearing closely-woven clothes, a broad-brimmed hat; and liberal application of sunblock with a high sun protection factor (SPF) (http://dermnetnz.org/reactions/drug-photosensitivity.html).
If a photosensitivity reaction is suspected the drug culprit should be identified and if possible the drug should be withdrawn. Other drugs included in the CARM list were hydrochlorothiazide (second), amiodarone (third), piroxicam (fourth), chlorpromazine (fifth), captopril (seventh), enalapril (eighth), bendroflumethiazide (ninth) and carbamazepine (tenth). In cases where it is not possible to withdraw the drug patients should be advised to take the precautions as outlined above.
Adapted from Prescriber Update 2010; 31(1): 7-8 (http://www.medsafe.govt.nz/profs/puarticles/summer%20reminder%20-%20photosensitivity%20reactions.htm [8 March 2010]).
Tigecycline is the first of the glycylcyline antibiotics to be approved for use by the FDA (June 2005). It is structurally related to minocycline and, like minocycline, binds to the 30S ribosomal subunit blocking protein synthesis. However, in tigecycline the addition of an N,N,-dimethyglycylamido group to the minocycline molecule results in an increase (>5-fold) in the affinity of tigecycline for the ribosomal target when compared to minocycline or tetracycline. This increased affinity explains the expanded spectrum of activity against tetracyclineresistant organisms such as penicillin-resistant Streptococcus pneumoniae, methicillin-resistant Staphylococcus aureus (MRSA) and Staphylococcus epidermidis (MRSE) and vancomycin-resistant Enterococcus (VRE) species. Resistance has been documented in Australian isolates of Acinetobacter baumanni and acquired resistance has been reported in Klebsiella pneumoniae, Enterobacter aerogenes, Enterobacter cloacae and Enterococcus faecalis. Tigecycline is approved for use in the treatment of complicated skin and soft tissue infections and complicated intra-abdominal infections caused by susceptible organisms.
Tigecycline is administered intravenously and concentrations in the gallbladder, lung and colon are higher than plasma concentrations. It is not extensively metabolised and is primarily eliminated as unchanged drug in bile and faeces (∼59%) and urine (∼22%) (Greer 2006). Dosage adjustment is not usually necessary in mild–moderate hepatic impairment but is recommended in severe hepatic impairment because studies have demonstrated a reduced clearance of tigecycline (∼55%) and an increase (∼43%) in tigecycline half-life. Adverse effects are similar to those described for the tetracyclines.
Chloramphenicol
Chloramphenicol, a broad-spectrum antibiotic, potently inhibits bacterial protein synthesis by binding to the 50S subunit of the bacterial ribosome. It is a bacteriostatic agent used for a wide variety of Gram-negative and Grampositive organisms and anaerobes; however, because it has the potential to be seriously toxic to bone marrow (aplasia, leading to aplastic anaemia and possibly death), its use has declined but local application (ophthalmic use) is still prevalent. It is indicated for the treatment of Haemophilus influenzae, Streptococcus pneumoniae and Neisseria meningitidis, as it may be bactericidal to these organisms.
Chloramphenicol has good oral and parenteral bioavailability, with the highest concentrations reported in the liver and kidneys. Concentrations up to 50% of those in plasma have been noted in cerebrospinal fluid. Chloramphenicol is metabolised in the liver to an inactive glucuronide, 75%–90% of which is excreted in the urine over a 24-hour period. In neonates, chloramphenicol causes ‘grey baby syndrome’: a blue–grey skin discoloration, hypothermia, irregular breathing, coma and cardiovascular collapse. This occurs because of lack of maturation of glucuronide-conjugating enzymes in the liver during the first 3–4 weeks of life and inadequate renal capacity to excrete unchanged drug. The half-life of chloramphenicol in an adult is 1.5–3.5 hours; in neonates 24–48 hours old it is 1–2 days or more; in those 10–16 days of age it is approximately 10 hours. Peak plasma concentration is reached in 1–1.5 hours via the IV route.
The effects shown in Drug Interactions 44-4 may occur when chloramphenicol is given with the drugs listed.
Drug interactions 44-4 Chloramphenicol
| Drug | Possible effects and management |
| Barbiturates | Increased metabolism of chloramphenicol and reduced antibacterial activity. Increase chloramphenicol dose if necessary |
| Phenytoin | Impaired phenytoin metabolism and increased risk of toxicity. Monitor phenytoin concentration and reduce dose if required |
| Tacrolimus | Increased plasma concentration of tacrolimus when administered concurrently with chloramphenicol. Monitor plasma concentration of tacrolimus and decrease tacrolimus dose if necessary to avoid toxicity |
| Warfarin | Inhibition of warfarin metabolism may lead to bleeding. Monitor INR and reduce warfarin dose if necessary |
INR = international normalised ratio.
Adverse reactions include diarrhoea, nausea, vomiting, blood dyscrasias, grey baby syndrome in neonates, hypersensitivity, neurotoxic reactions (delirium, confusion and headaches), peripheral neuritis, optic neuritis and, possibly, irreversible bone marrow depression that may result in aplastic anaemia. Use chloramphenicol with extreme caution in people who have had antineoplastic chemotherapy orradiation therapy. Chloramphenicol is contraindicated in people with a history of hypersensitivity to the drug and in those with pre-existing bone marrow suppression and/or blood dyscrasias. Specialist advice should be sought before prescribing to infants and children.
Oxazolidinones
The only drug available in this class is linezolid, a novel compound with a broad spectrum of activity against community and nosocomial Gram-positive organisms (e.g. VRE and MRSA). Unlike chloramphenicol, which binds to the 50S ribosomal subunit and inhibits bacterial protein synthesis, linezolid’s site of action is proximal to the 50S subunit. At that specific site, linezolid inhibits protein synthesis by interfering with the formation of a complex that is essential for protein translation. The site and mechanism of action of linezolid were thought to reduce the likelihood of cross-resistance between Gram-positive bacterial strains; however, enter ococci and MRSA resistant to linezolid have now been isolated.
Linezolid is indicated for the treatment of serious infections due to Gram-positive organisms where other drugs are either contraindicated or not appropriate. It is available in both oral and IV formulations. After oral administration, linezolid is well absorbed (∼100% bioavailability) and peak plasma concentrations occur in 1–1.5 hours. It is metabolised by the liver to inactive metabolites, and approximately 30% of the drug is excreted unchanged in urine. The half-life is in the order of 4.5–5.5 hours and dose adjustment is generally not necessary in the elderly or in the presence of renal or hepatic dysfunction.
The predominant adverse reactions include nausea, diarrhoea and headache. As linezolid may cause thrombocytopenia, leucopenia, eosinophilia and neutro penia, full blood count should be monitored before treatment and weekly during therapy. Linezolid is a weak inhibitor of monoamine oxidase and hence has the potential to interact with adrenergic drugs (e.g. adrenaline, dopamine, ephedrine), serotonergic drugs (e.g. SSRIs) and tyramine-rich foods or drinks. Stop linezolid for at least 7 days before commencing any drugs that interact with MAOIs (to avoid hypertension due to accumulation of endogenous catecholamines) or to avoid serotonin syndrome. Counselling should be provided on foods to be avoided during linezolid treatment.
Streptogramins
Quinupristin with dalfopristin is a combination of 30 parts streptogramin B (quinupristin) with 70 parts streptogramin A (dalfopristin). These compounds are semisynthetic derivatives of a naturally occurring antibiotic produced by Streptomyces pristinaespiralis. The combination acts synergistically to inhibit bacterial protein synthesis through binding to the 50S ribosomal subunit. Quinupristin with dalfopristin is active against Gram-positive cocci and is indicated for the treatment of vancomycin-resistant Enterococcus faecium and severe MRSA when other antibiotics are inappropriate.
Quinupristin with dalfopristin is administered only via the IV route, and after reconstitution is further diluted with 5% glucose and infused over 60 minutes. This drug combination should never be given in sodium chloride solutions, nor the line flushed with sodium chloride or heparin solutions, because of incompatibility. The half-life is <1 hour for both drugs, and metabolism via conjugation is the main route of clearance. Approximately 80% of the administered dose is excreted in bile and hence no dosage adjustment is necessary in renal failure. Quinupristin with dalfopristin is an inhibitor of CYP3A4, and toxicity resulting from higher plasma concentrations of the interacting drugs may occur with drugs that are metabolised by CYP3A4. These include indinavir, midazolam, nifedipine and other calcium channel blockers and the immunosuppressant cyclosporin.
As would be anticipated, the commonest adverse reactions are infusion-related (e.g. pain and thrombophlebitis at the injection site). Nausea, vomiting and diarrhoea are also commonly observed, and arthralgias and myalgia may be related to accumulation of metabolites, particularly in people with hepatic dysfunction. Specialist advice should be sought prior to using this drug and it is extremely expensive.
Inhibitors of DNA synthesis
Fluoroquinolones
The quinolones (including fluoroquinolones) are synthetic, broad-spectrum agents with bactericidal activity. They interfere with bacterial topoisomerase II (DNAgyrase) and topoisomerase IV, the enzymes involved in the supercoiling of DNA that is necessary for the duplication, transcription and repair of bacterial DNA (see Figure 44-2). An equivalent topoisomerase II enzyme exists in eukaryotic cells, but this enzyme is inhibited by fluoroquinolones only at much higher concentrations. Examples of fluoroquinolones include ciprofloxacin, moxifloxacin and norfloxacin and ofloxacin (available on authority through the PBS). The quinolones are effective against Pseudomonas aeruginosa and are reserved for infections when alternative drugs are either contraindicated or ineffective. These include bone and joint infection, Legionella pneumonia, epididymo-orchitis, prostatitis and complicated UTIs. Individual quinolones may vary in their spectrum of activity. Unfortunately, bacterial resistance to the quinolones is increasing worldwide, and appropriate use is needed to extend their clinical life.
The oral bioavailability of ciprofloxacin is 50%–70%, 86% for moxifloxacin, 30%–40% for norfloxacin and 95%–100% for ofloxacin. They are widely distributed in the body with the following half-lives: ciprofloxacin, 4 hours; moxifloxacin ∼15 hours; norfloxacin, 3–4 hours; and ofloxacin, 4–7 hours. Ciprofloxacin and moxifloxacin are the only quinolones available for parenteral use. Qinolones are metabolised in the liver (minimally for ofloxacin) and excreted primarily by the kidneys.
Quinolones interact with a number of drugs (see Drug Interactions 44-5), and the effects shown may occur when quinolones are given with the drugs listed.
Drug interactions 44-5 Quinolones
| Drug | Possible effects and management |
| Antacids, ferrous sulfate or sucralfate | May decrease absorption of ciprofloxacin, reducing drug effectiveness. Administer quinolone at least 2 hours before these medications |
| Nitrofurantoin | Antibacterial effect of quinolones is antagonised. Avoid combination |
| Theophylline and other xanthines | Ciprofloxacin and norfloxacin may inhibit theophylline metabolism, resulting in increased theophylline plasma concentration and toxicity. Monitor theophylline plasma concentration closely, as dosage adjustments may be necessary |
| Warfarin | May result in increased anticoagulant effect and potential for bleeding. While not currently reported with all quinolones, it is recommended that the INR be monitored closely whenever these drugs are administered concurrently. Reduce warfarin dose if necessary |
INR = international normalised ratio.
Adverse reactions include dizziness, drowsiness, restlessness, stomach distress, diarrhoea, nausea, vomiting and photosensitivity; rarely, CNS stimulation (psychosis, confusion, hallucinations, tremors), hypersensitivity (skin rash, redness, Stevens–Johnson syndrome, face or neck swelling, shortness of breath) and interstitial nephritis. In addition, Achilles tendinitis and tendon rupture injuries have been reported and persons taking corticosteroids concurrently and >60 years old are at an increased risk. Additionally, young athletes participating in extensive training are also at risk of tendon adverse effects. These drugs should be ceased at the first sign of tendon pain or inflammation. Use quinolones with caution in people with CNS disorders, including epilepsy and seizures. Avoid use in those with quinolone hypersensitivity.
Miscellaneous antibiotics
Metronidazole and tinidazole
Metronidazole and tinidazole are reduced intracellularly in anaerobic microorganisms or anoxic or hypoxic cells to a short-acting cytotoxic agent that interacts with DNA, inhibiting bacterial synthesis and causing cell death. They are selectively toxic to many anaerobic bacteria and protozoa. Both are indicated for the treatment of amoebiasis (intestinal and extraintestinal), bone infections, brain abscesses, CNS infections, bacterial endocarditis, genitourinary tract infections, septicaemia, trichomoniasis and other infections caused by organisms susceptible to metronidazole.
Oral metronidazole is well absorbed and distributed throughout the body, penetrating many tissues, including vaginal secretions, seminal fluid, saliva and breast milk. It reaches peak plasma concentration within 1–2 hours and has a half-life of 8 hours. It is metabolised in the liver (about 50%), and both unchanged metronidazole and metabolites are excreted by the kidneys. Tinidazole has a longer half-life and is given once daily.
The effects shown in Drug Interactions 44-6 may occur when metronidazole is given with the drugs listed.
Drug interactions 44-6 Metronidazole
| Drug | Possible effects and management |
| Alcohol | Metronidazole interferes with the metabolism of alcohol, leading to an accumulation of acetaldehyde. This may result in disulfiram-type effects: flushing, headaches, nausea, vomiting and abdominal distress. Avoid, or a potentially serious drug interaction may occur |
| Anticoagulants (warfarin) | May enhance anticoagulant effects by inhibiting warfarin metabolism. Monitor INR closely, as dosage adjustments may be necessary. Consider using an alternative drug |
| Barbiturates | Induce metabolism of metronidazole, reducing its effectiveness. To avoid treatment failure an increased dose of metronidazole may be necessary |
| Disulfiram | Avoid concurrent use or use within 14 days of disulfiram administration in alcoholic patients. Adverse reactions such as confusion and psychosis have been reported |
INR = international normalised ratio.
Adverse reactions include dizziness, headache, gastric distress, diarrhoea, anorexia, nausea, vomiting, dry mouth, taste alterations, dark urine, peripheral neuropathy, CNS toxicity, hypersensitivity, leucopenia, thrombophlebitis, vaginal candidiasis and convulsions (with high drug doses). Use with caution in people with renal disease and hepatic impairment. Avoid use in those with metronidazole hypersensitivity, blood dyscrasias, severe liver disease or active organic CNS disease.
Dosage regimens vary according to the infection being treated, and relevant sources should be consulted for information.
Trimethoprim–sulfamethoxazole (co-trimoxazole)
The use of sulfonamides has declined substantially because of widespread bacterial resistance. These agents are primarily bacteriostatic, rather than bactericidal, in concentrations that are normally useful in controlling infections in humans. All the sulfonamides used therapeutically are synthetically produced and, because they are structurally similar to para-aminobenzoic acid (PABA), they competitively inhibit the bacterial enzyme dihydropteroate synthetase, necessary for incorporating PABA into dihydrofolic acid (see Figure 44-2). The blocking of dihydrofolic acid synthesis results in a decrease in tetrahydrofolic acid, which interferes with the synthesis of purines, thymidine, and DNA in the microorgan ism. Susceptible bacteria are particularly sensitive to sulfonamides because bacteria need to synthesise their own folic acid. The combination with trimethoprim (trimethoprim–sulfamethoxazole) is synergistic, as this agent blocks a further step in the synthesis of folic acid. The combination often has no advantage over the use of trimethoprim as monotherapy but is associated with a greater incidence of adverse reactions. Trimethoprim alone is indicated for uncomplicated UTIs, epididymo-orchitis and prostatitis.
After oral administration of trimethoprim–sulfamethoxazole the absorption of trimethoprim is more rapid than that of sulfamethoxazole. Peak concentration occurs within 2 hours for trimethoprim and 4 hours for sulfa-methoxazole. Trimethoprim distributes into tissues while sulfamethoxazole distributes in extracellular fluids. About 60% of the trimethoprim and 25%–50% of the sulfamethoxazole is excreted by the kidneys in 24 hours.
Examples of drug interactions include:
Adverse reactions include sore mouth, fever, nausea, vomiting and, in severe cases, phototoxicity, interstitial nephritis, hypoglycaemia and lowered mental acuity. Use with caution in HIV-infected people, because of the increased risk of allergic reactions, and in renal impairment, as the dose may need to be reduced. The combination is contraindicated in those with a history of previous allergic reaction to a sulfonamide (or related drugs, e.g. thiazide diuretics) or trimethoprim, and in severe renal or hepatic impairment.
The ratio of trimethoprim to sulfamethoxazole is 1:5 (i.e. 160/800 mg means 160 mg trimethoprim with 800 mg sulfamethoxazole). The usual adult oral dose is 80/400–160/800 mg every 12 hours and, in children, 4/20 mg/kg 12-hourly.
Urinary tract antimicrobials
 Urinary tract infections rank as one of the most commonly reported problems to general practitioners in Australia. UTIs are broadly classified as acute uncompli cated UTI, acute uncomplicated pyelonephritis, complicated UTI and acute complicated pyelonephritis. The term ‘uncomplicated’ usually applies to non-pregnant women without underlying abnormalities (functional or ana tomical) of the urinary tract. In the USA approximately 11% of 2000 women surveyed (age >18 years) reported at least one UTI in the past 12 months (Foxman et al 2000). The incidence of UTIs increases in institutional settings, up to as much as 50% of the population in extended-stay hospitals. See Table 44-8 for predisposing risk factors for UTIs.
Urinary tract infections rank as one of the most commonly reported problems to general practitioners in Australia. UTIs are broadly classified as acute uncompli cated UTI, acute uncomplicated pyelonephritis, complicated UTI and acute complicated pyelonephritis. The term ‘uncomplicated’ usually applies to non-pregnant women without underlying abnormalities (functional or ana tomical) of the urinary tract. In the USA approximately 11% of 2000 women surveyed (age >18 years) reported at least one UTI in the past 12 months (Foxman et al 2000). The incidence of UTIs increases in institutional settings, up to as much as 50% of the population in extended-stay hospitals. See Table 44-8 for predisposing risk factors for UTIs.
Table 44-8 Predisposing risk factors for utis
| Risk factors | Frequency reported |
| Urinary tract instrumentation (urethral and ureteral catheterisation)* | Up to 67% |
| Pregnant women | 4%–10% |
| Non-pregnant women | 2%–5% |
* After a week of indwelling catheterisation, up to 100% colonisation and bacteriuria (Ahronheim 1992).
UTIs are primarily caused by bacteria. In communityacquired infections, 75%–90% of uncomplicated UTIs are caused by E. coli. It has been reported that E. coli may cause up to 90% of all community-acquired uncomplicated UTIs. Hospital-acquired infections are often complicated and difficult to treat. Organisms involved include P. aeruginosa, Serratia spp., Enterobacter spp. and other Gram-negative microorganisms.
Drug therapies for lower UTIs are often started before culture and sensitivity reports are available. Treatment guidelines for the various types of UTIs can be found in the Therapeutic Guidelines: Antibiotic (Antibiotic Expert Group 2006); depending on the circumstances, drug regimens may involve monotherapy (e.g. trimethoprim or cephalexin or amoxicillin–clavulanate for the treatment of acute uncomplicated UTI) or combination therapy. Drugs that may be used include ampicillin, amoxycillin– clavulanate, cephalexin, ciprofloxacin, nitrofurantoin, norfloxacin, trimethoprim, trimethoprim–sulfamethoxazole or in severe infections gentamicin. Many of these drugs have been covered in preceding sections; the remaining drugs indicated for treatment of UTIs are discussed below. Current guidelines recommend treating febrile UTI or acute pyelonephritis with antimicrobials for at least 14 days. A current randomised clinical trial is investigating the efficacy of 7-day treatment in comparison to 14-day treatment (van Nieuwkoop et al 2009).
Hexamine hippurate
Hexamine hippurate, which is used to treat UTIs, combines the action of hexamine with hippurate. Its effectiveness depends on the release of formaldehyde, which requires an acid medium. The acids released from hippurate salts contribute to this acidity and, if sulfonamides are coadministered, this may result in crystalluria from precipitation of the formaldehyde with the sulfonamide. Formaldehyde may be bactericidal or bacteriostatic and its effects are believed to be the result of denaturation of bacterial protein. The drug is ineffective in alkaline urine as alkalinity inhibits the conversion of hexamine to formaldehyde. Because of its fairly wide bacterial spectrum, low toxicity and low incidence of resistance, hexamine has often been the drug of choice in the long-term suppression of infections.
Hexamine is absorbed orally and takes 0.5–2 hours to reach peak urinary formaldehyde concentration at a urinary pH of 5.6. Excretion is via the kidneys. Use
hexamine with caution in severely dehydrated patients. Avoid use in people with severe kidney impairment, as renal tubule concentration will be inadequate to achieve a response. For prophylaxis of recurrent UTIs, the adult oral dose is 1 g 12-hourly, and that for children aged 6–12 years is 500–1000 mg every 12 hours.
Nitrofurantoin
Nitrofurantoin is a broad-spectrum bactericidal agent. Its mechanism of action is not fully understood but it is reduced by bacteria to reactive substances that inactivate or alter cell wall synthesis, bacterial ribosomal proteins and DNA and RNA function. It is indicated for the treatment of acute UTIs caused by organisms such as E. coli, S. aureus and Klebsiella species, and for prophylaxis of recurrent UTIs.
After oral administration, nitrofurantoin is well absorbed and has a half-life of 20–60 minutes. About 65% of the drug is excreted, mainly as unchanged drug in the urine. Antacids decrease absorption while probenecid inhibits tubular secretion of nitrofurantoin, leading to increased plasma concentration and possible toxicity. Combination with quinolones is contraindicated as they antagonise the bacterial action of nitrofurantoin. Use nitrofurantoin with caution in people with G6PD deficiency. Acute allergic pneumonitis may occur within days of initiation of treatment and presents as fever, chills, cough, dyspnoea, chest pain and often a rash. It is more common in women aged 40–50 years. Rarely, chronic irreversible interstitial pulmonary fibrosis can occur in older patients following chronic treatment (>6 months). Avoid use in those with nitrofurantoin hypersensitivity, peripheral neuropathy, lung disease or moderate to severe renal impairment.
Key points


 Antibiotics are chemical substances, produced by microorganisms, that kill or suppress the growth of other microorganisms. The term antibiotic is now commonly used also to describe synthetically produced antimicrobial drugs. Penicillin was originally discovered in moulds and then found to be bactericidal by inhibiting the synthesis of the bacterial cell wall. Penicillins can be broadly divided into the following groups: narrow-spectrum penicillins; narrow-spectrum penicillinaseresistant penicillins with antistaphylococcal activity; moderate-spectrum β-lactamase-sensitive aminopenicillins; and the broad- and extended-spectrum (antipseudomonal activity) penicillins. There are now four generations of cephalosporins. Modification of the central β-lactam ring has resulted in many drugs with different microbiological and pharmacological activities. Ertapenem, imipenem and meropenem are members of the class of antibiotics called carbapenems, which are related to the β-lactam antibiotics. They have a wide spectrum of activity against Gram-positive and Gram-negative aerobic and anaerobic organisms. Vancomycin is reserved for treatment of serious infections that are resistant to penicillins—methicillin-resistant Staphylococcus aureus (MRSA) and multi-resistant Staphylococcus epidermis (MRSE). The macrolide antibiotics, typified by erythromycin, are bacteriostatic agents that inhibit protein synthesis and, at high concentrations, are bactericidal for selected microorganisms. The lincosamides, such as clindamycin, are primarily bacteriostatic and are used to treat serious streptococcal and staphylococcal infections. The aminoglycosides are very potent bactericidal antibiotics that are primarily indicated for serious or life-threatening infections. Therapeutic drug monitoring plays a major role in ensuring that a therapeutic plasma concentration is achieved and the risk of toxicity is minimised. Metronidazole is a short-acting cytotoxic agent that is selectively toxic to protozoa as well as anaerobic bacteria.
Antibiotics are chemical substances, produced by microorganisms, that kill or suppress the growth of other microorganisms. The term antibiotic is now commonly used also to describe synthetically produced antimicrobial drugs. Penicillin was originally discovered in moulds and then found to be bactericidal by inhibiting the synthesis of the bacterial cell wall. Penicillins can be broadly divided into the following groups: narrow-spectrum penicillins; narrow-spectrum penicillinaseresistant penicillins with antistaphylococcal activity; moderate-spectrum β-lactamase-sensitive aminopenicillins; and the broad- and extended-spectrum (antipseudomonal activity) penicillins. There are now four generations of cephalosporins. Modification of the central β-lactam ring has resulted in many drugs with different microbiological and pharmacological activities. Ertapenem, imipenem and meropenem are members of the class of antibiotics called carbapenems, which are related to the β-lactam antibiotics. They have a wide spectrum of activity against Gram-positive and Gram-negative aerobic and anaerobic organisms. Vancomycin is reserved for treatment of serious infections that are resistant to penicillins—methicillin-resistant Staphylococcus aureus (MRSA) and multi-resistant Staphylococcus epidermis (MRSE). The macrolide antibiotics, typified by erythromycin, are bacteriostatic agents that inhibit protein synthesis and, at high concentrations, are bactericidal for selected microorganisms. The lincosamides, such as clindamycin, are primarily bacteriostatic and are used to treat serious streptococcal and staphylococcal infections. The aminoglycosides are very potent bactericidal antibiotics that are primarily indicated for serious or life-threatening infections. Therapeutic drug monitoring plays a major role in ensuring that a therapeutic plasma concentration is achieved and the risk of toxicity is minimised. Metronidazole is a short-acting cytotoxic agent that is selectively toxic to protozoa as well as anaerobic bacteria.Review exercises
References and Further reading
Ahronheim J.C. Handbook of Prescribing Medications for Geriatric Patients. Boston: Little Brown; 1992.
Antibiotic Expert Group. Therapeutic Guidelines: Antibiotic version 13. Melbourne: Therapeutic Guidelines; 2006.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Chambers H.F. Aminoglycosides. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw–Hill, 2006. [ch 45]
Chambers H.F. Protein synthesis inhibitors and miscellaneous antibacterial agents. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw–Hill, 2006. [ch 46]
Fihn S.D. Acute uncomplicated urinary tract infection in women. New England Journal of Medicine. 2003;349:259-266.
Foxman B., Barlow R., D’Arcy H., et al. Urinary tract infection: self-reported incidence and associated costs. Annals of Epidemiology. 2000;10:509-515.
Greer ND. Tigecycline (Tygacil): the first in the glycylcycline class of antibiotics. Proceedings (Baylor University Medical Center) 2006; 19: 155–161.
Petri W.A.Jr. Sulfonamides, trimethoprim–sulfamethoxazole, quinolones, and agents for urinary tract infections. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw–Hill, 2006. [ch 43]
Petri W.A.Jr. Penicillins, cephalosporins, and other β-lactam antibiotics. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw–Hill, 2006. [ch 44]
Raper K.B., Alexander D.F., Penicillin V. Mycological aspects of penicillin production. Journal of the Elisha Mitchell Scientific Society. 1945;61:74.
van Nieuwkoop C., van’t Wout J.W., Assendelft W.J.J., et al. Treatment duration of febrile urinary tract infection (FUTIRST trial): a randomized placebo-controlled multicenter trial comparing short (7 days) antibiotic treatment with conventional treatment (14 days). BMC Infectious Diseases. 2009;9:131-150.
Yates A.B. Management of patients with a history of allergy to beta-lactam antibiotics. American Journal of Medicine. 2008;121:572-576.