Chapter 26 Drugs Affecting Thrombosis and Haemostasis
Each year thousands of Australians and New Zealanders die from coronary heart disease and stroke, and many more are diagnosed with deep vein thrombosis, pulmonary embolus, transient cerebral ischaemic attacks and other non-fatal thrombotic events. The formation of thrombi or acute thromboembolic disorders requires the use of anticoagulant, thrombolytic and antiplatelet agents. In contrast, in some instances excessive bleeding needs to be counterbalanced by the use of specifi c haemostatic and antifi brinolytic drugs that hasten clot formation and reduce bleeding. All of these drugs are potent effective medications requiring a thorough knowledge of their pharmacology for safe usage.
Key abbreviations
aPTT activated partial thromboplastin time
DIC disseminated intravascular coagulation
HITS heparin-induced thrombocytopenic syndrome
INR international normalised ratio
Key background
THIS chapter reviews anticoagulant, antiplatelet, thrombolytic, haemostatic and antifibrinolytic drugs. Although blood clotting is a normal defence mechanism available for protection against excessive haemorrhage, the development of a thrombus (an aggregation of platelets, fibrin, clotting factors and the cellular elements of blood) that becomes attached to the inner wall of a blood vessel can obstruct blood flow and cause ischaemia. An embolus, a mass of undissolved matter that breaks off from the thrombus, can travel in the vascular system and lodge in a vital area of the body, causing death. In contrast, a defect in the blood coagulation cascade can lead to excessive bleeding or haemorrhage, even after a minor injury.
Arterial or venous thrombus formation is associated with significant morbidity and mortality and the risk of thrombosis increases with age. Thousands of people die each year from coronary heart disease and stroke, and many more are diagnosed with deep vein thrombosis, pulmonary embolus, transient cerebral ischaemic attacks and other non-fatal thrombotic events. Local trauma, vascular stasis and systemic alterations in the coagulability of blood are considered the main factors in the initiation of a thrombosis in an unbroken vessel. Basically, coagulation mechanisms are responsible for forming two kinds of thrombi: arterial thrombi and venous thrombi. The pathogenesis of arterial thrombosis is complex and involves genetic and environmental factors. Arterial thrombi are most frequently associated with ruptured atherosclerotic plaques, high blood pressure and turbulent blood flow that damage the endothelial lining of the blood vessel and cause platelets to stick and aggregate in the arterial system.
Venous thrombosis is also a multifactorial problem that occurs in areas where blood flow is reduced or static or from changes in total blood coagulability that initiate clotting and produce a thrombus in the venous system. Risk factors for venous thrombosis include inherited thrombophilia disorders (e.g. antithrombin deficiency), prolonged immobilisation (e.g. long-haul flights, major trauma), operative procedures (e.g. orthopaedic surgery), certain types of cancer (e.g. myeloproliferative disorders), pregnancy, use of the oral contraceptive pill and hormone replacement therapy. In many cases the first episode of thrombosis can be prevented by prophylactic drug therapy and recurrences prevented by a secondary drug management strategy (Bick 2000).
The haemostatic mechanism
Haemostasis is a process that spontaneously stops bleeding from damaged blood vessels. After any injury to a blood vessel, haemostasis is achieved by three sequential steps:
Platelet plug formation
After injury to a blood vessel, interruption of the continuity of its endothelial lining exposes collagen (a fibrous protein) in the underlying connective tissue. Platelets become activated changing their shape (refer to Chapter 27) and becoming spherical with long dendritic extensions that facilitate adhesion to the exposed collagen forming a dense aggregate, a process known as platelet adhesion. This attachment triggers the release of adenosine diphosphate (ADP) and thromboxane A2 from the dense granules in the platelet cytoplasm. Liberation of these factors causes further activation of nearby platelets and vasoconstriction through the action of thromboxane A2, which further limits blood flow through the damaged vessel. Platelet– membrane glycoprotein receptors mediate adhesion to the subendothelial tissue and the outer surfaces of the platelets become extremely sticky. The surface protein glycoprotein IIb-IIIa undergoes a conformational change during platelet activation expressing receptor function for fibrinogen. Binding of fibrinogen to glycoprotein IIb-IIIa mediates platelet aggregation and eventually the mass forms the haemostatic plug (see Figure 26-4). Because this plug is relatively unstable, it can stop the bleeding quickly as long as the damage to the vessel is minute. However, for long-term effectiveness the platelet plug must be reinforced with fibrin. This involves a series of chemical coagulation or clotting reactions. Blood coagulation ultimately results in the formation of a stable fibrin clot, which comprises a meshwork of fibrin threads that entraps platelets, blood cells and plasma. Thus the physical formation of a blood clot or thrombus plays a key role in haemostasis by permanently closing the hole in the injured vessel, preventing further bleeding.
Blood coagulation
The chemical events in blood coagulation involve two distinct pathways: the intrinsic pathway and the extrinsic pathway.
The intrinsic pathway
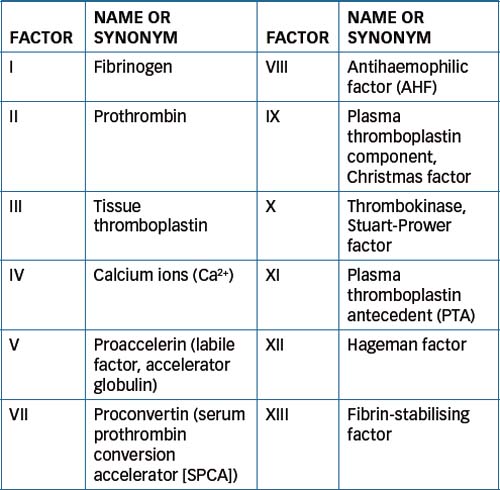
As all the chemical substances involved in coagulation are normally found in the circulating blood, this pathway is referred to as the intrinsic pathway of coagulation. In this complex pathway, activation by proteolysis of the first inactive blood coagulation factor causes activation of the next factor, and this cascading process continues through the whole pathway. Refer to Figure 26-1 for a summary of the main events of the intrinsic pathway and Table 26-1 for a list of the blood coagulation factors.
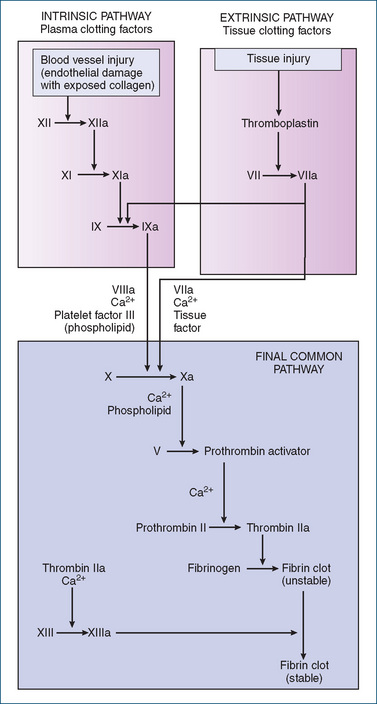
Figure 26-1 Coagulation mechanisms for intrinsic and extrinsic pathways for blood clotting. Final pathway (activation of factor X) is common to both the intrinsic and extrinsic coagulation systems.
The process occurs over several minutes and is initiated by injury to the endothelial lining of the blood vessel wall. Briefly:
The extrinsic pathway
The extrinsic pathway is activated within seconds by trauma to the vascular wall or to tissue external to the blood vessels. In this pathway, clotting occurs when the tissue protein thromboplastin is released from the damaged tissue, leaks into the bloodstream (hence the name extrinsic) and becomes part of a complex with factor VII and calcium ions. This combination of components activates factor X, which is the step at which the extrinsic pathway converges with the intrinsic pathway; coagulation then continues through a common route with the resultant formation of a stable clot. See Figure 26-1 for the extrinsic pathway.
The final pathway common to both the intrinsic and the extrinsic coagulation systems begins with the activation of factor X and ends in the formation of fibrin. Both systems function simultaneously in the body, and lack of a normal factor in either system will usually result in a blood coagulation disorder.
Anticoagulant drugs
Anticoagulant drug therapy (Figure 26-2) is primarily prophylactic because these agents act by preventing fibrin deposits, extension of a thrombus and thromboembolic complications. Although long-term anticoagulant therapy remains controversial, there is evidence that anticoagulant therapy reduces the incidence of thrombosis and therefore prolongs life. These drugs have no direct effect on a blood clot that has already formed or on ischaemic tissue injured by an inadequate blood supply because of the clot.
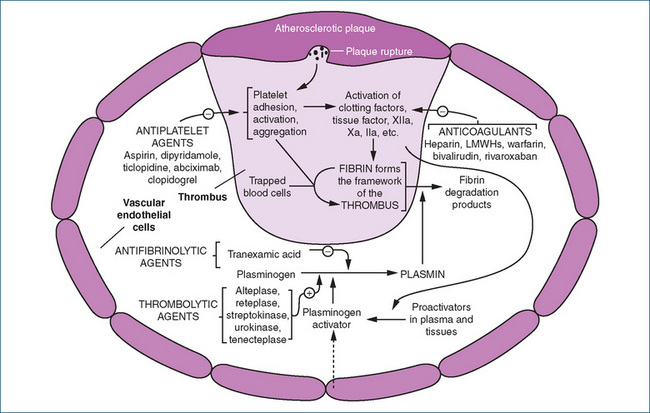
Figure 26-2 Sites of action of drugs interacting with the coagulation cascade and the fibrinolytic and platelet activation systems.
Source: Adapted from Rang, Dale, Ritter & Flower 2007, with permission.
The main groups of anticoagulant drugs include
Drug monograph 26-1 Heparin (unfractionated)
Heparin was discovered in 1916 and is a complex substance (a proteoglycan) consisting of repeating disaccharide (sugar) units attached to a core protein. As a consequence, it has a wide range of molecular weights (5000–40,000). This form of heparin is often referred to as ‘unfractionated’, or ‘standard’, heparin. It is formed in especially large amounts in the mast cells of the liver, lungs and intestinal mucosa and in general the source of heparin for injection is pig mucosa or bovine lung. As heparin is obtained from animal tissue, the potency of each preparation varies. This variability in preparation necessitates that the activity of each preparation (batch) is determined using a biological assay. The activity determined for each batch is then measured against an international standard and finally reported as units of activity.
MECHANISM OF ACTION Heparin produces its anticoagulant effect by combining with antithrombin III (heparin co-factor also simply called antithrombin or AT), a naturally occurring anticlotting factor in the plasma. The binding of heparin with AT III forms a complex that acts rapidly at multiple sites in the normal coagulation system, inactivating thrombin factor IIa and factors IXa, Xa, XIa and XIIa. Thrombin and factor Xa are most affected, and human thrombin is about 10-fold more sensitive to inhibition by the heparin-ATIII complex than factor Xa. By inactivating thrombin, heparin prevents fibrin formation and inhibits thrombin-induced activation of factor V and factor VIII. Inactivation of factor Xa of the intrinsic and extrinsic pathways prevents the conversion of prothrombin to thrombin, thereby inhibiting the formation of fibrin from fibrinogen. Furthermore, by preventing the activation of factor XIII (fibrin stabilising factor), heparin prevents the formation of a stable fibrin clot. As fibrin is associated with venous thrombi, heparin is useful in preventing venous thrombosis. The drug does not have fibrinolytic activity: it does not dissolve existing clots but can prevent their extension.
INDICATIONS Heparin is used to prevent and treat all types of venous thromboembolism in surgical and high-risk medical patients. It is used prophylactically to prevent blood clotting during surgery of the heart or blood vessels, during blood transfusion, in individuals with disseminated intravascular coagulation (DIC) and during haemodialysis. It is considered the drug of choice for acute arterial occlusion (including acute coronary syndromes) because its action is immediate and can be readily reversed if surgery is necessary. When rapid anticoagulation is necessary, it is used before the oral anticoagulant warfarin (Table 26-2).
PHARMACOKINETICS Heparin is administered parenterally because its large molecular size and polarity prevent any gastrointestinal absorption. These characteristics also prevent heparin from crossing the placenta and it is the drug of choice in pregnancy. Unlike warfarin, heparin does not cause fetal malformations. Its onset of action is immediate after intravenous injection. Subcutaneous injection usually results in an onset of action within 60–120 minutes. Clearance of heparin involves two mechanisms, one which is rapid and exhibits saturation and a second renal pathway that is non-saturable. At therapeutic doses heparin is cleared by the rapid saturable process, which occurs principally via binding to receptors on macrophages and endothelial cells. Saturation of this process accounts for the disproportionate increase in the anticoagulant effect with increasing dose. As a consequence the half-life of heparin increases from ∼30 minutes with a bolus dose of 25 U/kg to ∼150 minutes with a bolus dose of 400 U/kg (Hirsch et al 2001). Hence, the half-life is dose-dependent and averages 1.5 hours (range 1–6 hours). Heparin is highly protein-bound and only a small amount of unchanged heparin appears in urine. Heparin dosage is closely monitored with coagulation tests such as the activated partial thromboplastin time (aPTT) (see Clinical Interest Box 26-1). In some individuals higher doses of heparin are required to prolong the aPTT. Heparin resistance has been associated with antithrombin III deficiency, increased heparin clearance, elevations in heparin binding proteins and elevations of factor VIII and fibrinogen (Hirsh et al 2001).
DRUG INTERACTIONS The following interactions can occur when heparin is given with the drugs listed.
ADVERSE REACTIONS Bleeding, or haemorrhage, is the commonest adverse reaction. Early signs of heparin overdose include increased bruising, nosebleeds or excessive bleeding from minor cuts, wounds, brushing of teeth or menstrual period. Internal signs of bleeding include stomach pain; bloody or black stools; dizziness; persistent headaches; swollen, stiff or painful joints; and vomiting or coughing up of blood. Rarely, asthma-like symptoms, pruritus, urticaria and anaphylaxis have been reported. At the site of injection, a haematoma or blood accumulation under the skin, pain, or a local skin reaction such as irritation, peeling or sloughing can occur. If observed, immediate discontinuation of therapy is warranted. In 1%–3% of individuals, severe immune-mediated thrombocytopenia (heparin-induced thrombocytopenic syndrome, HITS) can occur, causing complications such as limb ischaemia, stroke, bleeding or death. A substantial drop in the baseline platelet count of 30%–50% indicates the need to withhold heparin.
Hyperkalaemia has been reported in patients administered 5000 units of heparin or more twice daily. Although reported in ‘normal’ patients it occurs more frequently in patients (8%–19%) with pre-existing perturbations of potassium homeostasis. The mechanism relates to inhibition of the final step in the biosynthetic pathway for the production of aldosterone in the adrenal gland. With reduced aldosterone synthesis a reduction in renal potassium excretion occurs, which ultimately causes hyperkalaemia. In addition, heparin also reduces the number and affinity of receptors for angiotensin II, which causes a decrease in the ability of angiotensin II to stimulate aldosterone synthesis (Oster et al 1995).
Administration of heparin for >1 month (e.g. in pregnancy to treat/prevent venous thromboembolism) is associated with a significant reduction in bone density in ∼30% of patients and symptomatic vertebral fractures occur in 2%–3% of patients receiving heparin. The mechanism underlying heparin-induced osteoporosis is unclear but may be related to decreased numbers of osteoblasts (decreased rate of bone formation) and increased activity of osteoclasts (increased bone resorption). LMWH may carry a lower risk of osteoporosis as current evidence suggests that >50-fold higher concentrations than those used clinically are required to produce the same effect on bone as heparin (Hirsch et al 2001).
WARNINGS AND CONTRAINDICATIONS Heparin should be used with caution in individuals with asthma and/or a history of allergies (increased risk of allergic reaction to animal proteins) or mild to moderate liver impairment (altered coagulation factors). Avoid heparin use in people with heparin hypersensitivity and those with an increased risk of bleeding (e.g. cerebral aneurysm, cerebrovascular bleeding, haemorrhage, severe hypertension, haemophilia, peptic ulcer disease, severe liver disease or blood dyscrasias; in women after recent childbirth; and in people who have recently had surgery or anaesthesia).
Table 26-2 Anticoagulant drugs: comparison of heparin and warfarin
| HEPARIN | WARFARIN | |
| Onset of action | Immediate | Slow (24–48 hours) |
| Route of administration | Parenteral | Oral |
| Duration of action | Short (< 4 hours) | Long (2–5 days) |
| Laboratory test for dosage control | Activated partial thromboplastin time (aPPT) | Prothrombin time (PT) |
| Antidote | Protamine sulfate | Vitamin K, whole blood or plasma |
| Pregnancy | Heparin and LMWH used. LMWH not recommended in pregnant women with prosthetic heart valves due to evidence of inadequate anticoagulation | Generally contraindicated (may be used in 2nd trimester if indicated) |
| Lactation | Safe to use | Safe to use |
Table 26-3 Comparison of regular heparin and low-molecular-weight heparins
| PROPERTY | REGULAR HEPARIN | LOW-MOLECULAR-WEIGHT HEPARIN |
| Molecular weight range | 3000–30,000 | 1000–10,000 |
| Average molecular weight | 12,000–15,000 | 4000–6000 |
| Mechanism of action | Inactivates factor Xa and IIa (thrombin) | Inactivates factor Xa |
| aPTT monitoring required | Yes | No |
| Inhibits platelet function | ++++ (high) | ++ (medium) |
| Route of administration | IV, SC | SC only |
| Protein binding | ++++ (high) | + (low) |
| Vascular permeability increased | Yes | No |
| Treatment of bleeding | Protamine | Protamine (partially effective) |
Clinical interest Box 26-1 Coagulation tests
Activated partial thromboplastin time (aPTT)
The aPTT test measures the overall activity of the intrinsic coagulation pathway. It is performed by warming a thromboplastin reagent that contains an activator with an aliquot of the patient’s plasma and then recalcifying this mixture after a set incubation period.
The length of time taken for the mixture to clot is called the aPTT and the normal range is usually about 26–39 seconds, depending on the analyser and the brand of reagent used.
A prolonged test can indicate:
The test is used to monitor heparin administration to a patient and the therapeutic range for the aPTT in a patient on heparin is about 50–90 seconds. The aPTT test cannot be used to monitor the effect of LMWH.
The PT test measures the overall activity of the extrinsic coagulation pathway (Figure 26-1). It is performed by adding thromboplastin to an aliquot of the patient’s plasma and measuring the time it takes for the mixture to clot.
This clotting time is used to determine the INR (international normalised ratio). The same test is performed using normal control plasma, and the prothrombin ratio is then derived by dividing the patient clotting time by the mean normal control clotting time.
Finally the INR is calculated using the ‘international sensitivity index’ of the thromboplastin reagent, so that the ratio obtained is independent of the reagent brand or testing laboratory. The normal range for the INR is 1.0–1.3.
A prolonged test can indicate:
The test is used to monitor warfarin administration to a patient and the therapeutic range for the INR in a patient on warfarin (for DVT) is about 2.0–4.0.
Courtesy of: SA Pathology, Flinders Medical Centre, Adelaide, Australia.
Warfarin interferes with hepatic synthesis of the vitamin K-dependent clotting factors through inhibition of the vitamin K epoxide reductase complex 1 (VKORC1). As a consequence, interference in the γ-carboxylation of the glutamic acid residues on factors II, VII, IX and X and on various anticoagulant proteins by γ-glutamyl carboxylase leads to the production of nonfunctional coagulation factors (Figure 26-3). Factor VII is depleted quickly; the sequential depletion of factors IX, X and II follows. Warfarin does not affect established clots but prevents further extension of formed clots, thereby diminishing the potential for secondary thromboembolic complications. The major advantage of warfarin is that it is effective orally and can be given once daily after the maintenance dose has been established.
INDICATIONS Warfarin is indicated for the prophylaxis and treatment of DVT and pulmonary thromboembolism. It is also used for the prophylaxis of thromboembolism associated with chronic atrial fibrillation, myocardial infarction or in individuals with prosthetic heart valves.
PHARMACOKINETICS Warfarin is a racemic drug comprising equal concentrations of S- and R-warfarin. It is well absorbed from the gastrointestinal tract and has a systemic bioavailability of >95%. Peak plasma concentration occurs in 3–9 hours and its duration of action is 2–5 days. Warfarin is highly protein-bound (99%) and the plasma half-life varies from 25 to 60 hours with an average of 40 hours. Warfarin crosses the placenta and fetal plasma attains a similar plasma concentration to that of the maternal circulation.
S-Warfarin is metabolised in the liver by CYP2C9 with a minor contribution from CYP2C8, CYP2C18 and CYP2C19. The predominant role of CYP2C9 (which exhibits significant pharmacogenetic variation) in S-warfarin metabolism accounts for the large variability observed in warfarin dose requirements. Some individuals with certain CYP2C9 variant alleles have reduced metabolism of S-warfarin and thus increased warfarin plasma concentration. These individuals are more prevalent in European, African and Asian populations; however, there is limited evidence currently to support that genotyping improves anticoagulation control or reduces the risk of haemorrhage (Limdi et al 2008). In contrast R-Warfarin is metabolised by CYP1A2 and CYP3A4 and to a minor extent by CYP1A1, CYP2C8, CYP2C18, CYP2C19 and CYP3A5.
DRUG INTERACTIONS Numerous drugs interact with warfarin and the INR should be monitored more often when instituting, ceasing or altering other drug therapy. An increase in the anticoagulant effect of warfarin has been reported with the herbal medicines dong quai, garlic, papaya and St John’s wort, and a decrease in anticoagulant effect with ginseng (Campbell et al 2001).
Drugs that may increase the anticoagulant effect ( ↑ INR) of warfarin, necessitating a dosage reduction
| Allopurinol | Dipyridamole* | Propranolol |
| Amiodarone | Disulfiram | Propylthiouracil |
| Anabolic steroids | Erythromycins | Salicylates |
| Aspirin | Fluconazole | SSRIs |
| Azithromycin | Fluoxetine | Streptokinase |
| Capecitabine | Gemcitabine | Sulfinpyrazone |
| Cefamandole | Gemfibrozil | Sulfonamides |
| Cefazolin | Indomethacin | Sulindac |
| Ceftriaxone | Ketoprofen | Tamoxifen |
| Celecoxib | Ketorolac | Thyroid hormone |
| Chloral hydrate† | Leflunomide | Ticarcillin |
| Chloramphenicol | Mefenamic acid | Quinidine |
| Cimetidine | Metronidazole | Urokinase |
| Ciprofloxacin | Miconazole | Valproate |
| Danazol | Phenytoin‡ | Zafirlukast |
| Dextran | Piperacillin | |
| Dextrothyroxine | Piroxicam |
Agents that may decrease the anticoagulant effect (↓ INR), often necessitating an increase in anticoagulant dosage
| Aminoglutethimide | Dicloxacillin |
| Aprepitant | Haloperidol |
| Azathioprine | Primidone |
| Barbiturates | Rifampicin |
| Carbamazepine | Sucralfate |
| Colestipol | Vitamin K |
| Cortisone |
ADVERSE REACTIONS These include bleeding (common), alopecia, anorexia, abdominal cramps or distress, leucopenia, nausea, vomiting, diarrhoea, purple toes syndrome (rare) and kidney damage (rare). Risk of abortion and teratogenicity is high and fetal abnormalities and facial anomalies have been reported if warfarin is administered during the first trimester. Administration during the second and third trimesters is associated with CNS abnormalities. If an anticoagulant is necessary during pregnancy, LMWH is usually the drug of choice because it does not cross the placenta.
The elderly may be more susceptible (see Clinical Interest Box 26-2). Risk factors for bleeding include age >70 years, previous history of stroke and falls, liver disease, chronic renal failure, drug interactions and evidence of gastrointestinal bleeding in the previous 18 months (Campbell et al 2001).
WARNINGS AND CONTRAINDICATIONS Use with caution in individuals with a history of severe allergic or anaphylactic reactions, oedema, elevated cholesterol or lipid concentrations or hypothyroidism, in the elderly and in unsupervised individuals who are alcoholics, psychotic, senile or mentally unstable.
Avoid use in people with known anticoagulant drug hypersensitivity, any medical or surgical condition associ ated with bleeding (aneurysm, cerebrovascular bleeding, surgery and severe trauma), blood disorders, severe uncontrolled hypertension, pericarditis, severe diabetics, ulcers, visceral cancer, vitamin C or vitamin K deficiencies, endocarditis or severe liver or kidney impairment. Brands of warfarin should not be interchanged due to a lack of bioequivalent data.
DOSAGE AND ADMINISTRATION The usual dose is 5 mg daily for 2 days and then adjusted according to the INR. The maintenance dose is in the range 1–10 mg daily and is taken at the same time each day. The INR range varies with specific indications, and local guidelines should be consulted.
* With doses of dipyridamole >400 mg/day.
† Usually occurs during first 2 weeks of therapy. With chronic concurrent therapy, the anticoagulant effect may return to normal or be decreased.
‡ Increased anticoagulant effect occurs initially. With chronic concurrent therapy, decreased activity may occur. May also see a decrease in metabolism of phenytoin, possibly leading to increased plasma concentrations and toxicity.
Heparin and the LMWHs are the drugs of first choice if a rapid anticoagulant effect is required because their onset is immediate if administered intravenously.
Low-molecular-weight heparins
Currently there are two LMWHs on the market, dalteparin and enoxaparin, and one heparinoid, danaparoid. The low-molecular-weight heparins are fragments approximately one-third the size of standard heparin and are prepared by enzymatic or chemical cleavage. The resulting fragments have a molecular weight range of 4000–6000 and hence are called ‘low-molecular-weight’ heparins. This difference in molecular weight produces an anticoagulant with properties considerably different from those of heparin.
Mechanism of action
Both types of heparin can inactivate factor Xa. Heparin (unfractionated) also inactivates thrombin (IIa) because it binds both antithrombin III and thrombin at the same time. In contrast, LMWHs increase the action of antithrombin III on factor Xa but, because of their small size, LMWHs cannot bind antithrombin III and thrombin at the same time. Hence, they have both an enhanced capacity to inhibit factor Xa, which contributes to their improved antithrombotic effect, and a relatively minor effect on aPTT. Danaparoid is a more selective inhibitor of factor Xa than the LMWHs. See Table 26-3 for a comparison of heparin and LMWHs.
Indications
The LMWHs are administered subcutaneously and are considered to be safer and require less monitoring. The indications are the same as for standard heparin, with the exception that dalteparin and enoxaparin are used during pregnancy and danaparoid is indicated only for the prevention of venous thromboembolism in surgical patients.
Pharmacokinetics
In comparison with standard heparin, LMWHs have a lower affinity for endothelial cells, macrophages and plasma proteins; an increased bioavailability; and a more predictable clearance that is independent of dose. Hepatic clearance plays a minor role and elimination is principally via the kidneys and hence their biological half-life is prolonged in patients with renal failure. For enoxaparin, consider dose reduction in severe renal impairment and avoid completely in end-stage renal disease. LMWHs have a longer half-life than heparin (2–4 times greater) when given subcutaneously and their anticoagulant effect also lasts longer. The elimination half-life for dalteparin is 3–5 hours and for enoxaparin 3–6 hours.
Drug interactions and adverse reactions
Drug interactions for the LMWHs are the same as those for heparin. In addition, if enoxaparin is given concurrently with ticlopidine, inhibition of platelet aggregation causes an increased risk of bleeding. Monitor closely.
Bleeding is a well-known complication of heparin therapy and the LMWHs have a similar risk. Common adverse reactions include local irritation effects such as erythema, haematomas, urticaria and pain at the injection site. The incidence of thrombocytopenia is less (around 0.6%) and data on osteoporosis also indicates a decreased incidence. Danaparoid may be used as an alternative to heparin or LMWH in people with heparin-induced thrombocytopenia, as cross-reactivity occurs in fewer than 10% of individuals.
Use LHWH with caution in people undergoing any medical procedure that increases the potential of bleeding. Avoid use in people with LMWH or heparin hypersensitivity, bleeding disorders, severe hypertension, stroke, thrombocytopenia, severe liver or kidney disease, endocarditis or retinopathy and in those who have recently had surgery. In persons with renal impairment, the risk of bleeding with LMWHs is greater, as they are eliminated by renal excretion.
Warfarin—Vitamin K antagonist
These drugs were discovered following an outbreak of a haemorrhagic disorder in cattle eating spoiled sweet clover in 1929. The active constituent was later identified as bishydroxy-coumarin in 1939. Synthesised analogues, including warfarin (the name comes from the Wisconsin Alumni Research Foundation and arin from coumarin), were originally thought to be too toxic and were used as rodenticides. Following survival of a man in 1951 after repeated high doses of the rat poison in an attempt to commit suicide, warfarin was introduced as an anticoagulant for humans in 1959 (see Drug Monograph 26-2). Warfarin is one of the most widely prescribed oral anticoagulants in Australia.
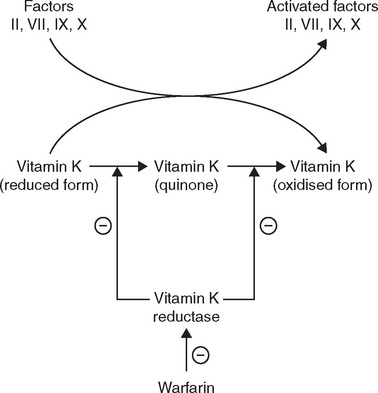
Figure 26-3 Site of action of warfarin and the role of Vitamin K. The oxidation of the reduced form of vitamin K is coupled to the carboxylation of the inactive factors II, VII, IX and X. The vitamin K reductase complex 1 (VKORC1) then regenerates the reduced form of vitamin K. The regeneration step is inhibited by warfarin and hence the formation of the activated clotting factors is inhibited.
Antithrombin-III-dependent anticoagulant
The synthetic antithrombin-III-dependent anticoagulant is fondaparinux, which binds ATIII potentiating the neutralisation of factor Xa by antithrombin, inhibiting both thrombin formation and thrombus development. It does not inhibit thrombin (activated factor IIa) and has no antiplatelet activity. It is as effective and as safe as the LMWHs. It is administered SC and is used in the prevention of venous thromboembolism in high-risk surgery such as hip fracture or replacement or knee replacement. The long half-life of 17 hours permits once-daily administration. As with the heparins, this drug is contraindicated in coexisting bleeding disorders and in cases of renal impairment. The latter is important because fondaparinux is excreted unchanged in urine. At present there are no data in pregnancy and breastfeeding.
Direct thrombin inhibitors
The use of medicinal leeches (Hirudo medicinalis) has its origins more than 2500 years ago. The discovery in 1884 by John Haycraft that blood in the leech gut did not coagulate finally led to the isolation of the anticoagulant hirudin from leech pharyngeal glands by Markwardt in the late 1950s. Hirudin is a direct irreversible non-covalent inhibitor of thrombin but the extent of anticoagulation was unpredictable. The modern anticoagulants are bivalirudin, which is a 20-amino-acid synthetic polypeptide analogue of hirudin, while lepirudin is a recombinant form of hirudin. Both drugs bind directly and reversibly to thrombin independently of ATIII and hence block the thrombogenic activity of thrombin. Relatively quick dissociation from thrombin leaves a small amount of active thrombin free for the control of haemostasis.
Bivalirudin has a plasma half-life of 25 minutes and lepirudin approximately 1.5 hours; both are administered by IV infusion. Bleeding disorders and significant reduction in renal function are factors for consideration prior to use of either of these drugs. As would be expected, common adverse reactions include bleeding and, in the case of lepirudin, a significant number of people (∼40%) develop antibodies. Fatal anaphylaxis has been reported on re-exposure to the drug.
A recently approved oral direct thrombin inhibitor is dabigatran etexilate, which is available only through hospitals or by private prescription. Dabigatran has low bioavailability and dabigatran etexilate, the prodrug of dabigatran, was developed to aid gastrointestinal absorption. Following hydrolysis of dabigatran etexilate by esterases, the active metabolite dabigatran, specifically and competitively inhibits both free and clot-bound thrombin by binding to the active site of the thrombin molecule. The plasma half-life is in the order of 12–14 hours and approximately 80% of the drug is excreted unchanged by the kidney. The remainder is excreted as acylglucuronides in the bile. In the absence of metabolism by CYP, dabigatran is not subject to major drug–drug interactions. The drug doubles the aPTT and the PT. It is indicated for the prevention of thromboemboli after major lower limb surgery (e.g. knee replacement) and total hip replacement. Unfractionated heparins, heparin derivatives, LMWHs, fondaparinux, thrombolytic drugs, GPIIb/IIIa receptor antagonists, clopidogrel, ticlopidine, dextran, sulfinpyrazone and vitamin K antagonists should not be administered concomitantly with dabigatran. Close observations should be carried out when there is an increased haemorrhagic risk, e.g. major trauma or recent biopsy.
Direct factor Xa inhibitor
Rivaroxaban is the second oral anticoagulant (after dabigatran etexilate) to become available in Australia since the introduction of warfarin. Listed on the PBS in August 2009 rivaroxaban is a direct reversible dose-dependent competitive inhibitor of factor Xa that binds directly to the active site thereby attenuating thrombin generation and preventing conversion of fibrinogen to fibrin. The drug prolongs both PT and aPTT and prolongation of PT correlates with the plasma drug concentration.
Rivaroxaban is absorbed rapidly and bioavailability ranges from 60%–80%. Approximately 30% of the dose is excreted unchanged in urine while the remainder is metabolised by CYP3A4, CYP2C8 and CYP-independent mechanisms. There are limited data on drug interactions but the incidence appears low. As rivaroxaban is a substrate for P-gp and CYP3A4, concomitant administration of the P-gp and CYP3A4 inhibitors ketoconazole, itraconazole, voriconazole, posaconazole and ritonavir are contraindicated because of increased risk of bleeding. Clinical evidence-based guidelines report similar efficacy after hip or knee replacement surgery for dabigatran, LMWHs, fondaparinux or rivaroxaban (NPS RADAR, August 2009). Recommended duration of therapy is 14 days after knee replacement and 35 days after hip replacement. Monitoring is not required as there is currently no way to relate PT to either therapeutic or adverse effects and the effect of rivaroxaban cannot be monitored by using the INR.
Antiplatelet agents
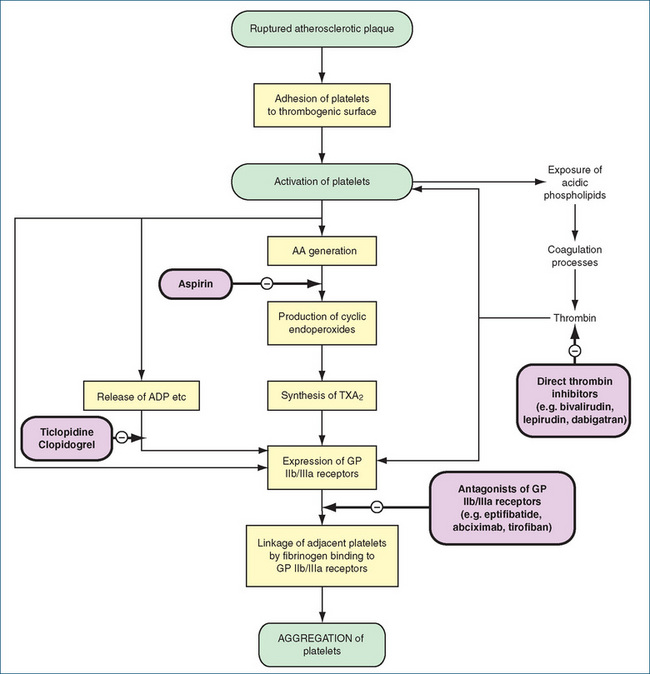
Platelets play a critical role in the production of thrombi following vascular damage. After adhesion of platelets to the thrombogenic surface, they become activated by mediators such as platelet activating factor, thromboxane A2, (which binds to Tx receptors), ADP, (which binds to P2Y12 and P2Y1 receptors) and thrombin, resulting in platelet aggregation. The latter occurs because the platelets stick to one another via fibrinogen bridges that link between the specific glycoprotein receptors, expressed on the surfaces of the platelets. ADP binding to the P2Y12 receptor stimulates the activation of the glycoprotein IIb/IIIa receptor. Glycoprotein IIb/IIIa is the most abundant surface protein (∼8000 molecules/platelet) and during platelet activation it undergoes a conformational change to express receptor function, which results in the binding of fibrinogen that mediates prolonged platelet aggregation. This is an autocatalytic process, as exposure of certain lipids on the surface of the platelets promotes further thrombin formation, platelet aggregation and fibrin formation. Although this process is desirable when forming a haemostatic plug, it is undesirable when triggered intravascularly. Our knowledge of the role of platelets in thromboembolic disease and our understanding of the pharmacology of aspirin has led to considerable development of drugs with ‘antiplatelet activity’.
Antiplatelet drugs are used in the treatment of arterial thrombosis and include aspirin (see Clinical Interest Box 26-3), the P2Y12 inhibitors clopidogrel and ticlopidine, dipyridamole and the glycoprotein IIb/IIIa receptor inhibitors abciximab, eptifibatide and tirofiban (Figure 26-4). (The role of aspirin as an analgesic is discussed in Chapter 15 and as an anti-inflammatory agent in Chapter 47.)
Clinical interest Box 26-3 Aspirin
Aspirin causes a long-lasting functional deficit in platelets by irreversibly inhibiting cyclooxygenase, an enzyme necessary for thromboxane A2 synthesis. Thromboxane A2 promotes plate let aggregation and vasoconstriction, and thus aspirin suppresses these actions. Platelets lack the metabolic capacity to synthesise new cyclooxygenase, and the deficit induced by aspirin lasts 8–10 days until new platelets are synthesised. This effect on platelet function explains both the effectiveness of aspirin as an antiplatelet agent and why it prolongs bleeding time.
Numerous studies have established the effectiveness of aspirin therapy in people with acute myocardial infarction and demonstrated conclusively a significant reduction in mortality. Follow-up studies in the Second International Study of Infarct Survival (ISIS-2) found that the benefit of early aspirin therapy persisted for several years, and further reductions in the incidence of death, reinfarction and strokes have been reported (Collins et al 1997). Aspirin is standard treatment for both cardiovascular and cerebrovascular diseases.
The antiplatelet effect of aspirin is achieved at a dose of 75–300 mg daily and no additional benefit has been observed at higher doses. Low-dose aspirin (100 mg/day) does not cause changes in bleeding time.
P2Y12 inhibitors
Clopidogrel is a second-generation thienopyridine derivative structurally related to ticlopidine. It is a prodrug that is predominantly metabolised (85%) to an inactive carboxylic acid metabolite. The remaining 15% is meta bolised to an active metabolite in a two-step process involving CYP2C19, CYP1A2 and CYP2B6 in the first step and CYP2C9, CYP2C19, CYP2B6 and CYP3A in the second step. Maximum concentration of the active metabolite is reached ∼1 hour after dosing. The active thiol metabolite inhibits ADP-induced platelet aggregation within 1–2 hours by irreversibly binding to the P2Y12 platelet receptor. This prevents ADP-mediated activation of the glycoprotein IIb/ IIIa complex and hence platelet aggregation. The P2Y12 receptors lose their ability to bind ADP for the life of the platelet. On cessation of treatment platelet function returns within ∼1 week. Unlike aspirin it has no effect on prostaglandin synthesis. Variability in clopidogrel response has been observed in multiple studies and current evidence suggests a combination of genetic (e.g. CYP2C19 polymorphism) and clinical causes (e.g. poor adherence to therapy). This variability in response has led to the development of more potent third-generation P2Y12 inhibitors (e.g. prasugrel [now available in Australia], cangrelor, ticagrelor and elinogrel), which are still at the clinical trial stage.
The current recommendation for patients with acute coronary syndrome and in those undergoing stent implantation is a combination of aspirin and clopidogrel. Clopidogrel is an established antiplatelet drug used to prevent thromboembolism in individuals with ischaemic heart disease. As would be anticipated in those with pre-existing bleeding problems, caution should be exercised, and in patients with planned surgery or dental procedures continued use of clopidogrel should be reviewed and possibly ceased before the procedure. Bleeding is a common adverse reaction and may be quite severe. Use in pregnancy should be avoided, as there are limited data available.
Ticlopidine is a first-generation thienopyridine and is believed to irreversibly inhibit ADP-induced activation of platelet–platelet aggregation in the same manner as clopidogrel.
This drug is used to decrease the risk of stroke in individuals who have had ischaemic attacks or in those who have had a thrombotic stroke. Recently, it has been used short-term in combination with aspirin to prevent thromboembolic events in people following placement of a coronary stent (Clinical Interest Box 6-2). Administered orally, the onset of action is slow, taking 3–7 days before a maximal effect is observed. Ticlopidine is metabolised extensively by the liver and no unchanged drug is detected in urine. Binding to plasma proteins occurs for both the parent drug (98%) and metabolites (40%–50%). Coadministration of ticlopidine with aspirin or other anticoagulants increases the risk of bleeding. With theophylline or phenytoin, coadministration increases their respective plasma concentrations, and monitoring of plasma theophylline and phenytoin concentration is recommended.
Clinical interest Box 26-2 Geriatric implications of anticoagulants
The elderly may be more susceptible to the effects of anticoagulants such as warfarin, so a lower maintenance dose is usually recommended for the geriatric patient, along with very close supervision and monitoring.
The primary adverse effects of excessive drug usage are prolonged bleeding from gums when brushing teeth or from small shaving cuts, excessive or easy skin bruising, blood in urine or stools and unexplained nosebleeds. These may be early signs of overdose that indicate the need for medical intervention.
Caution individuals to carry an identification card indicating the use of an anticoagulant. Also, remind patients to always consult their prescriber before starting any new drug, including over-the-counter medications and vitamins, or if changing a medication dose or when any drug administration is discontinued. Be aware that administration of concurrent drug therapy that can induce gastric irritation increases the risk of gastrointestinal bleeding. Drugs such as the non-steroidal antiinflammatory agents (e.g. ibuprofen, indomethacin) that are commonly prescribed for elderly people often cause gastrointestinal effects.
Individuals should be instructed to avoid alcohol or at least limit their daily alcohol intake to one alcoholic drink a day. Alcohol may cause liver damage, which increases the individual’s sensitivity to anticoagulants. Alcohol intoxication or heavy drinking may predispose to falls, poor compliance and poor nutritional habits, all of which can increase the risk of bleeding (Campbell et al 2001).
Health-care professionals should be aware that diet can interfere with the anticoagulant effect. In a previously stabilised person, vitamin C deficiency, chronic malnutrition, diarrhoea or other illnesses can result in an increased anticoagulant effect, and higher intake of green leafy vegetables (e.g. broccoli, cabbage, silver beet, lettuce and spinach) or consumption of a nutritional supplement or multiple vitamin containing vitamin K can result in decreased anticoagulant effectiveness.
Adverse reactions include nausea, stomach cramps, bloating, dizziness, skin rash, diarrhoea, tinnitus, bleeding, pruritus, neutropenia and, rarely, agranulocytosis, thrombocytopenia, purpura, hepatitis and Stevens–Johnson syndrome. In people planning dental or elective surgery, continued drug administration should be reviewed and possibly ceased before the procedure. Avoid use in people with ticlopidine hypersensitivity, haemophilia, bleeding disorders, current bleeding or severe liver function impairment.
Dipyridamole
The mechanism of action of dipyridamole is unclear but is thought to include inhibition of thromboxane A2 formation; inhibition of phosphodiesterase activity, which results in an increase in platelet cAMP; and inhibition of red blood cell uptake of adenosine, a platelet aggregation inhibitor.
Dipyridamole is used in combination with warfarin for the prevention of postsurgical thromboembolic complications after cardiac valve replacement, in combination with aspirin for the secondary prevention of ischaemic stroke and transient ischaemic attacks and IV for cardiac stress testing.
After an oral dose, dipyridamole is rapidly absorbed and reaches peak plasma concentrations within 45–75 minutes. Bioavailability ranges from 40% to 70% and is limited by hepatic first-pass metabolism. It is highly protein-bound, metabolised in the liver, and excreted principally as glucuronides in bile. Drug interactions include:
Adverse reactions include headache, dizziness, abdom inal upset, rash, allergic reaction, angina pectoris, blood pressure lability (hypertension, hypotension) and tachycardia. Use dipyridamole cautiously in individuals with unstable angina or recent myocardial infarction and in the presence of aortic stenosis. Avoid use in people with dipyridamole hypersensitivity.
Glycoprotein IIb/IIIa receptor inhibitors
As can be seen in Figure 26-4, inhibition of glycoprotein IIb/IIIa receptors will inhibit all pathways of platelet activation, as they constitute the point at which the pathways converge. Abciximab is a hybrid murine/human monoclonal antibody while eptifibatide and tirofiban are peptides based on a common sequence that occurs in glycoprotein IIa/IIIb receptors. These drugs are administered parenterally and are used in combination with heparin or aspirin (low-dose) to prevent ischaemic cardiac complications in people undergoing percutaneous transluminal coronary angioplasty or intracoronary stenting. To date, development of oral formulations has not been successful.
Abciximab has a longer duration of action than eptifibatide and tirofiban (it remains bound to platelets for about 15 days; platelet function recovers in ∼48 hours). Drug interactions include:
Adverse reactions include bleeding (minor and major), thrombocytopenia, visual changes, confusion, nausea, vomiting and hypotension. Monitor PT, aPTT, creatinine clearance, platelet count, haemoglobin and haematocrit before and during treatment.
Thrombolytic drugs
Thrombolytic (fibrinolytic) drugs are used to treat acute thromboembolic disorders. Unlike anticoagulants, they dissolve clots and are used in a hospital setting by experienced health-care professionals. These agents alter haemostatic capability more profoundly than does anticoagulant therapy. Consequently, when bleeding occurs, it is more severe and very difficult to control. The main drugs in this class include alteplase (also known as recombinant tissue plasminogen activator, rt-PA), reteplase, streptokinase, tenecteplase and urokinase. Streptokinase was the first thrombolytic agent released and is produced from cultures of β-haemolytic streptococci, urokinase is a product isolated from human urine, and alteplase, reteplase and tenecteplase are produced using recombinant DNA technology.
All five drugs have similar biochemical mechanisms of action on the fibrinolytic system, converting plasminogen in the blood to plasmin. Plasmin, a fibrinolytic enzyme, digests or dissolves fibrin clots wherever they exist and can be reached by plasmin. Alteplase, reteplase, tenecteplase and streptokinase are indicated for ST segment elevation myocardial infarction (STEMI); additional uses include in pulmonary embolism, acute ischaemic stroke (alteplase), peripheral arterial embolism and thrombosed cannulae (e.g. IV cannulae, central venous cannulae, haemodialysis shunts). Urokinase is not marketed in Australia but may be available via the Special Access Scheme (SAS).
These agents are administered intravenously. Alteplase, streptokinase and urokinase have elimination half-lives of 35 minutes, 23 minutes and up to 20 minutes, respectively. The time to peak effect after IV injection is from 20 minutes to 2 hours. Duration of the thrombolytic effect is about 4 hours for alteplase, streptokinase and urokinase. Reteplase has an elimination half-life of 13–16 minutes and usually has a peak effect within 2 hours. Tenecteplase shows biphasic elimination kinetics, with an initial half-life of ∼25 minutes and a terminal half-life of 130 minutes. The exact mechanisms of elimination for many of these drugs are not fully established.
The commonest adverse reaction is bleeding, including intracerebral haemorrhage. Others occurring less often include fever, headache, nausea, vomiting, hypotension, arrhythmias, allergic reaction, facial flushing, arthralgia and bronchospasm. With streptokinase and urokinase, adverse reactions include stomach pain or swelling, backache, bloody urine and stools, constipation, severe headaches, dizziness, arthralgia, tachycardia, bradycardia and fever.
For all these drugs, the absolute contraindications include:
Use of streptokinase results in the production of antistreptokinase antibodies. This drug may be ineffective if given between 5 days and up to 12 months after either a previous streptokinase treatment or an acute streptococcal infection.
In an acute coronary artery thrombosis evolving into a transmural myocardial infarction, thrombolytic therapy is most effective when started as early as possible or within 6–12 hours of the onset of symptoms. In general, bolus administration followed by an intravenous infusion is used. The drug regimens for acute myocardial infarction, pulmonary embolism, deep venous throm boembolism and arterial thromboembolism vary, and local institutional or manufacturer’s guidelines should be consulted.
Haemostatic and antifibrinolytic drugs
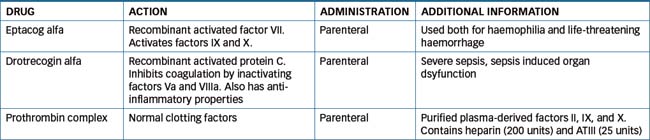
Haemophilia is a hereditary disorder caused by a deficiency of one or more plasma protein clotting factors. This condition usually leads to persistent and uncontrollable haemorrhage after even minor injury. The symptoms include excessive bleeding from wounds and haemorrhage into joints, the urinary tract and, on occasion, the CNS. There are two types of haemophilia: haemophilia A, the classic type in which factor VIII activity is deficient, and haemophilia B, or Christmas disease, in which factor IX complex activity is deficient. In recent years, a correct diagnosis of the coagulation disorder has led to specific factor replacement therapy, and this medical advance has resulted in effective management of patients at home. Haemostatic and antifibrinolytic drugs are compounds used to hasten clot formation and reduce bleeding. This group of drugs include eptacog alfa (factor VII), drotrecogin alfa, factor VIII, factor IX, protamine, tranexamic acid and vitamin K. The purpose of these agents is to control rapid loss of blood.
Table 26-4 provides a summary of three available drugs. Those not listed are discussed in more detail in following sections.
Factor VIII
Factor VIII, or the antihaemophilic factor, is a glycoprotein necessary for haemostasis and blood clotting. In the intrinsic pathway of the coagulation mechanism, the antihaemophilic factor is required for the transformation of prothrombin to thrombin. In the treatment or prevention of haemophilia A, factor VIII administration is based on replacing the missing plasma clotting factor to control and prevent bleeding.
When administered intravenously, factor VIII has a distribution half-life of 2.4–8 hours and an elimination half-life of 8.4–19.3 hours. The time to peak effect is 1–2 hours after IV administration. No significant drug interactions have been reported with factor VIII, but anticoagulants and antiplatelet drugs should not be administered to haemophiliacs. Mild to severe allergic reactions have been reported, such as bronchospasm, elevated temperature, chills or rash. Other adverse reactions, which might be related to the rate of infusion, include headache, increased heart rate, tingling of fingers, fainting, lethargy, sedation, hypotension, back pain, nausea or vomiting, visual disturbances and chest constriction.
Use factor VIII with caution in individuals with sensitivity to mouse, hamster or bovine proteins. Avoid use in people with antihaemophilic factor hypersensitivity. Individuals who develop antibodies to factor VIII might not respond to factor VIII therapy.
Factor IX complex
Factor IX complex is a purified plasma fraction prepared from pooled units of plasma. It contains factors II, VII, IX and X, which are known as the vitamin K coagulation factors. This agent is used for therapy in individuals with a deficiency of these factors during haemorrhage or before surgery. It is also indicated for patients with haemophilia B in whom factor IX is deficient (Christmas disease). Factor IX complex is used to prevent or control bleeding in individuals with factor IX deficiency. It is also used to treat patients with bleeding problems who have antibodies to factor VIII, and it will reverse haemorrhage induced by warfarin.
Factor IX has an elimination half-life of 18–32 hours and the time to peak effect after IV administration is 10–30 minutes. Interactions with other drugs have not been established.
Adverse reactions include chills and fever, especially when large doses are given. Also, if the intravenous infusion is given too rapidly, headache, flushing, rash, nausea, vomiting, sedation, lethargy, elevated temperature and tingling have been reported. The infusion should be stopped and in most people it can be resumed at a much slower rate.
Thrombosis and DIC have occurred as a result of the administration of factor IX. Myocardial infarction, pulmonary embolism and anaphylaxis have also been reported. It should not be used in individuals undergoing elective surgery, as they are at a greater risk of thrombosis.
Use factor IX with caution in individuals with trauma injuries and severe liver impairment, and in those who have recently had surgery. Avoid use in people with factor IX, hamster protein or mouse protein hypersensitivity, DIC and those with a history of thromboembolism. Factor IX should be administered slowly by intravenous injection or by intravenous infusion. The dosage is individualised according to the patient’s coagulation assay, which is performed before treatment. Check current references for specific dosing recommendations.
Protamine–heparin antagonist
Protamine, a protein-like substance derived from the sperm and mature testes of salmon and other fish, is a heparin antagonist and is used in over-anticoagulation. Protamine is a very weak anticoagulant alone, but when the sulfate form is given in conjunction with heparin, a complex is formed that dissociates the heparin–ATIII complex, thus reducing the anticoagulant action of heparin. Protamine is a basic protein (containing many free amino groups) and is able to combine with heparin to form an inactive complex.
Protamine is indicated for the treatment of an overdose of LMWH or standard heparin that has resulted in haemorrhaging. Blood transfusions may be necessary. It is also used to neutralise the effects of heparin administered during dialysis or cardiac or arterial surgery. It is administered intravenously and has an onset of action within 1 minute. Its duration of action is approximately 2 hours.
Adverse reactions include back pain, a feeling of warmth or tiredness, flushing, nausea and vomiting. Less often reported are bradycardia, sudden hypotension, shock and dyspnoea (all related to the too-rapid administration of protamine), bleeding (caused by protamine overdose or a rebound of heparin activity), hypertension and anaphylaxis.
Use protamine with caution in individuals who have been exposed to either protamine or protamine insulin in the past. Antibodies to protamine may have developed, which increases the risk of an allergic reaction. Avoid use in people with protamine hypersensitivity.
Protamine is administered by slow intravenous injection over 10 minutes. One milligram of protamine is necessary to neutralise around 100 units of standard heparin, if injected within 15 minutes of heparin administration. As heparin is cleared quite rapidly, a reduction in the dose of protamine is necessary if it is administered more than 15 minutes after the heparin dose. The standard dose of protamine (1 mg) will partially neutralise 100 units of dalteparin and 1 mg enoxaparin. Close monitoring with blood coagulation tests is required.
Tranexamic acid–antifibrinolytic drug
Tranexamic acid is a competitive inhibitor of plasminogen activation; at high doses, it is a non-competitive inhibitor of plasmin. It is indicated for use in a number of situations including heavy menstrual bleeding, to prevent haemorrhage in patients with mild-to-moderate coagulopathies undergoing minor surgical procedures e.g. cervical conisation, dental surgery and to prevent hereditary angiooedema. No significant drug interactions have been reported.
Adverse reactions include nausea, vomiting, diarrhoea, visual disturbances, thrombosis, hypotension, thromboembolism and menstrual discomfort.
Use with caution in women who are breastfeeding. Avoid use in people with tranexamic acid hypersensitivity, colour vision defects, haematuria, subarachnoid haemorrhage, a history of thrombosis or renal impairment.
Vitamin K—oral anticoagulant antagonist
Vitamin K (phytomenadione) is essential to the hepatic synthesis of prothrombin (factor II) and factors VII, IX and X. It acts as a co-factor for the carboxylase enzyme, which is necessary for the formation of prothrombin. A deficiency of vitamin K leads to hypoprothrombinaemia and haemorrhage.
Vitamin K is used to prevent and treat hypoprothrombinaemia. Prothrombin deficiency can occur because of inadequate absorption of vitamin K from the intestine (usually caused by biliary disease in which bile fails to enter the intestine) or because of destruction of intestinal organisms, which might occur with antibiotic therapy. It is also seen in the newborn because of a lack of establishment of intestinal organisms. Vitamin K is routinely administered to newborns to help prevent haemorrhage. Although prothrombin levels may be normal at birth, they decline until about day 6–8, when the liver becomes able to form prothrombin.
Vitamin K is also indicated in the preoperative preparation of individuals with deficient prothrombin, particularly those with obstructive jaundice. In addition, it may be given as an antidote for excessive anticoagulation with warfarin if simple cessation of warfarin therapy is not sufficient. When vitamin K is given concurrently with warfarin, a decrease in the anticoagulant effect is reported.
The onset of action for oral phytomenadione is 6–12 hours, and for the injectable form it is 1–2 hours. Vitamin K is metabolised in the liver and excreted via the kidneys and in the bile. Hence it is used with caution in people with biliary atresia, pancreatic insufficiency or fat malabsorption syndromes. Adverse reactions include facial flushing, taste alterations and redness or pain at the injection site.
Key points



 Anticoagulant therapy is primarily prophylactic, whereas thrombolytic drugs are used to dissolve already formed clots in the treatment of acute thromboembolic disease states. Heparin produces its anticoagulant effect by combining with antithrombin III to form a complex that acts at multiple sites in the normal coagulation system, inactivating factors IXa, Xa, XIa and XIIa. Inactivation of factor Xa of the intrinsic and extrinsic pathways prevents the conversion of prothrombin to thrombin, thereby inhibiting the formation of fibrin from fibrinogen. The low-molecular-weight heparins (LMWHs) are fragments of standard heparin prepared by enzymatic or chemical cleavage. They inactivate factor Xa more potently than factor IIa and hence have a relatively minor effect on aPTT. LMWHs are considered to be safer, easier to administer and require less monitoring than standard heparin. Bleeding is a well-known complication of heparin therapy, and the LMWHs have a similar risk. Warfarin is an orally administered anticoagulant and is indicated for the prophylaxis and treatment of deep venous thrombosis and pulmonary thromboembolism. Many drugs interact with warfarin, and the INR should be monitored more often when instituting, ceasing or altering other drug therapy. The synthetic antithrombin-III-dependent anticoagulant is fondaparinux, which binds ATIII potentiating the neutralisation of factor Xa by antithrombin, inhibiting both thrombin formation and thrombus development. The direct thrombin inhibitors include bivalirudin, lepirudin and the new oral anticoagulant dabigatran etexilate. Rivaroxaban is a new oral anticoagulant, which is a direct reversible dose-dependent competitive inhibitor of factor Xa. It binds directly to the active site, thereby attenuating thrombin generation and preventing conversion of fibrinogen to fibrin. Selected antiplatelet drugs inhibit platelet aggregation and can thus be used to reduce the risk of stroke. The antiplatelet drugs include aspirin, the P2Y12 inhibitors clopidogrel and ticlopidine, dipyridamole and the glycoprotein IIb/IIIa receptor inhibitors abciximab, eptifibatide and tirofiban Thrombolytic (fibrinolytic) drugs are used to treat acute thromboembolic disorders. Unlike anticoagulants, they dissolve clots and are used in a hospital setting by experienced health-care professionals.
Anticoagulant therapy is primarily prophylactic, whereas thrombolytic drugs are used to dissolve already formed clots in the treatment of acute thromboembolic disease states. Heparin produces its anticoagulant effect by combining with antithrombin III to form a complex that acts at multiple sites in the normal coagulation system, inactivating factors IXa, Xa, XIa and XIIa. Inactivation of factor Xa of the intrinsic and extrinsic pathways prevents the conversion of prothrombin to thrombin, thereby inhibiting the formation of fibrin from fibrinogen. The low-molecular-weight heparins (LMWHs) are fragments of standard heparin prepared by enzymatic or chemical cleavage. They inactivate factor Xa more potently than factor IIa and hence have a relatively minor effect on aPTT. LMWHs are considered to be safer, easier to administer and require less monitoring than standard heparin. Bleeding is a well-known complication of heparin therapy, and the LMWHs have a similar risk. Warfarin is an orally administered anticoagulant and is indicated for the prophylaxis and treatment of deep venous thrombosis and pulmonary thromboembolism. Many drugs interact with warfarin, and the INR should be monitored more often when instituting, ceasing or altering other drug therapy. The synthetic antithrombin-III-dependent anticoagulant is fondaparinux, which binds ATIII potentiating the neutralisation of factor Xa by antithrombin, inhibiting both thrombin formation and thrombus development. The direct thrombin inhibitors include bivalirudin, lepirudin and the new oral anticoagulant dabigatran etexilate. Rivaroxaban is a new oral anticoagulant, which is a direct reversible dose-dependent competitive inhibitor of factor Xa. It binds directly to the active site, thereby attenuating thrombin generation and preventing conversion of fibrinogen to fibrin. Selected antiplatelet drugs inhibit platelet aggregation and can thus be used to reduce the risk of stroke. The antiplatelet drugs include aspirin, the P2Y12 inhibitors clopidogrel and ticlopidine, dipyridamole and the glycoprotein IIb/IIIa receptor inhibitors abciximab, eptifibatide and tirofiban Thrombolytic (fibrinolytic) drugs are used to treat acute thromboembolic disorders. Unlike anticoagulants, they dissolve clots and are used in a hospital setting by experienced health-care professionals.
Review exercises
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Bick R.L. Proficient and cost-effective approaches for the prevention and treatment of venous thrombosis and thromboembolism. Drugs. 2000;60:575-595.
Campbell P., Roberts G., Eaton V., Coghlan D., Gallus A. Managing warfarin therapy in the community. Australian Prescriber. 2001;24:86-89.
Collins R., Peto R., Baigent C., Sleight P. Aspirin, heparin and fibrinolytic therapy in suspected acute myocardial infarction. New England Journal of Medicine. 1997;336:847-860.
Eriksson B.I., Quinlan D.J., Weitz J.I. Comparative pharmacodynamics and pharmacokinetics of oral direct thrombin and factor Xa inhibitors in development. Clinical Pharmacokinetics. 2009;48:1-22.
Gulseth M.P., Michaud J., Nutescu E.A. Rivaroxaban: an oral direct inhibitor of factor Xa. American Journal of Health-System Pharmacy. 2008;65:1520-1529.
Gurbel P.A., Antonino M.J., Tantry U.S. Recent developments in clopidogrel pharmacology and their relation to clinical outcomes. Expert Opinion in Drug Metabolism and Toxicology. 2009;5:989-1004.
Hirsh J., Dalen J.E., Anderson D.R., et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 2001;119:8S-21S.
Hirsh J., Warkentin T.E., Shaughnessy S.G., et al. Heparin and low-molecular-weight heparin mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest. 2001;119:64S-94S.
Holbrook A.M., Pereira J.A., Labiris R., et al. Systematic overview of warfarin and its drug and food interactions. Archives of Internal Medicine. 2005;165:1095-1106.
Limdi N.A., Veenstra D.L. Warfarin pharmacogenetics. Pharmacotherapy. 2008;28:1084-1097.
Majerus P., Tollenfsen D.M. Blood coagulation and anticoagulant, thrombolytic, and antiplatelet drugs. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw-Hill, 2006. [ch 54]
Mannuccio P. Hemostatic drugs. New England Journal of Medicine. 1998;339:245-253.
NPS RADAR. Rivaroxaban (Xarelto) for preventing venous thromboembolism after hip and knee replacement surgery. NPS RADAR August 2009.
Oster J.R., Singer I., Fishman L.M. Heparin-induced aldosterone suppression and hyperkalemia. American Journal of Medicine. 1995;98:575-586.
Rang H.P., Dale M.M., Ritter J.M., Flower R.J. Pharmacology, 6th edn. Edinburgh: Churchill Livingstone; 2007. [ch 21]
Reiner A.P., Siscovick D.S., Rosendaal F.R. Hemostatic risk factors and arterial thrombotic disease. Thrombosis Haemostasis. 2001;85:584-595.
Thomas M.D., Chauhan A., More R.S. Pulmonary embolism: an update on thrombolytic therapy. Quarterly Journal of Medicine. 2000;93:261-267.