Chapter 45 Antifungal and Antiviral Drugs
Chapter focus
Over the past 20 years, the number of immunocompromised individuals has risen dramatically as a result of the spread of HIV and the use of immunosuppressant drugs in organ transplant recipients and chemotherapy for neoplastic diseases. These factors have contributed to a substantial rise in the incidence of severe fungal infections and the use of antifungal drugs. Similarly, viral infections (e.g. influenza and hepatitis) continue to exist globally, necessitating the continued development of effective antiviral (non-retroviral) drugs. The development of antiretroviral drugs has also evolved in concert with our knowledge of HIV and the need for multiple drug therapy to combat the devastating consequences of AIDS.
Key terms
Key abbreviations
AIDS acquired immunodeficiency syndrome
HIV human immunodeficiency virus
NNRTI non-nucleoside reverse transcriptase inhibitor
Antifungal drugs
HUMAN infections by fungi can be caused by any of about 50 species of plant-like, parasitic microorganisms. These simple organisms, lacking chlorophyll, are unable to make their own food and so are dependent on other life forms. Infections by fungi, termed mycoses, can range from mild and superficial to severe and life-threatening. Infecting organisms can be ingested orally, become implanted under the skin after injury, or be inhaled if the fungal spores are airborne. One species of fungi, Candida albicans, is usually part of the normal flora of the skin, mouth, intestines and vagina, and overgrowth and systemic infection from it can result from antibiotic, antineoplastic and corticosteroid drug therapy. This is often referred to as an opportunistic infection. Oral candidiasis (thrush) is common in newborn infants and immunocompromised individuals, whereas vaginal candidiasis is more common in pregnant women, women with diabetes mellitus and women who take oral contraceptives. The prevalence of mycoses as opportunistic infections in people with HIV is growing, whereas non-opportunistic fungal infections such as blastomycosis and histoplasmosis are usually rare.
The first antifungal agent was introduced in 1939, with others following throughout the 1940s. A lag in the further development of antifungal agents then occurred, primarily because after the discovery of penicillin the emphasis was on the development of antibiotics. With the increasing number of individuals with HIV and the use of immunosuppressant therapy during the 1980s and 1990s, severe fungal infections have again become a problem. This has resulted in renewed searches for antifungal agents with reduced toxicity to humans. A number of antifungal compounds (e.g. amphotericin B [Drug Monograph 45-1], the azole antifungals and nystatin) are available for use, and these are discussed below. The topical antifungal preparations are discussed in Chapter 48.
Drug monograph 45-1 Amphotericin B
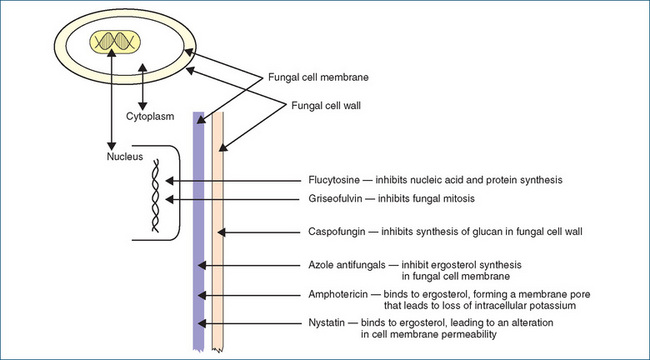
Amphotericin B was introduced in 1960 and is the premier drug for the treatment of severe systemic mycoses. It binds to ergosterol in the fungal cell membrane; this binding alters cell permeability and results in a loss of potassium and other elements from the cell (Figure Figure 45-1). Ergosterol is a major component of fungal cell membranes and not only provides the structure but is also involved in nutrient transport. Amphotericin B is effective for treating aspergillosis, blastomycosis, candidiasis (moniliasis), coccidioidomycosis, cryptococcosis, fungal endocarditis, histoplasmosis, cryptococcal meningitis, fungal septicaemia and many other severe systemic fungal infections.
Pharmacokinetics
Amphotericin B is widely distributed in the body after parenteral administration. After oral administration, little or no absorption occurs from the gastrointestinal tract. It has an initial half-life in adults of 24 hours and a terminal half-life of about 15 days. The routes of elimination are unknown, but excretion is via the kidneys. Around 40% of the drug is excreted over 7 days, but it can still be detected in the urine for at least 7 weeks after the drug is discontinued.
Drug interactions
The following effects can occur when amphotericin B is given with the drugs listed below:
Adverse reactions
Adverse reactions with the IV infusion of amphotericin B include headache, gastrointestinal (GI) distress, anaemia, hypokalaemia, infusion reaction (fever, chills, nausea, vomiting and hypotension), renal impairment, thrombophlebitis at the infusion site, blurred vision, cardiac dysrhythmias, rash, leucopenia, peripheral neuropathy, convulsions and thrombocytopenia. With oral administration (e.g. amphotericin lozenges), mild nausea and vomiting may be observed.
Warnings and contraindications
Caution should be exercised in people with renal impairment, as IV amphotericin alters renal function. The drug is contraindicated in people with amphotericin B hypersensitivity unless no alternative exists.
Dosage and administration
Amphotericin is available as a conventional IV formulation, as a lozenge, as a lipid complex and in a liposomal form. The lipid complex injection is a liposomal encapsulation of amphotericin used to treat aspergillosis and cryptococcosis in people who have infections that are refractory to, or who are unable to tolerate, standard amphotericin B therapy. The liposome formulation is effective but has significantly less nephrotoxicity than amphotericin B. The dosage of each formulation varies according to the infection, and specialist advice should be sought. In general, to avoid infusion-related reactions, the initial dose is either infused slowly over 2–6 hours, or a test dose of 1 mg is infused over the first 30 minutes, with the remainder infused over the next 2–4 hours. The commonest adverse reactions include fever, chills, nausea, hypotension, vomiting, dyspnoea and respiratory failure.
Clinical interest box 45-1 Australian medicinal plants
Antifungal compounds are present in several species. Researchers at the University of California found a compound, ‘polygodial’, from the leaves and fruits of the mountain pepper (Tasmannia sp.) that exhibits strong antifungal activity against Candida albicans and three strains of Trichophyton spp. The crushed fruits of the native cabbage (Scaevola taccada) are used to treat tinea but it is unclear whether the active ingredients are saponins or glycosidic compounds. An infusion of the inner bark of the green plum (Buchanania obovata) has been used for treating ringworm, and the leaves of the alpine cider gum (Eucalyptus gunnii) and other species are reported to contain at least three antifungal compounds.
Azole antifungals
Mechanism of action
The azole antifungals in clinical use are fluconazole, itraconazole, ketoconazole, miconazole, posaconazole and voriconazole. Those that contain two nitrogens in the azole ring are called imidazoles (e.g. ketoconazole and miconazole) and those with three nitrogens are called triazoles (e.g. itraconazole, fluconazole, posaconazole and voriconazole). These agents are fungistatic and affect the biosynthesis of fungal ergosterols by interfering with the cytochrome P450 dependent enzyme lanosterol demethylase (also called 14α-sterol demethylase) that catalyses ergosterol formation (Figure 45-1). Ergosterol is a major component of the fungal cell membrane and the azoles cause depletion of ergosterol and accumulation of 14α-methylated sterols. This results in inhibition of fungal growth, interference in nutrient transport and ultimately cell leakage and death. At therapeutic concentrations the azoles have a greater affinity for the fungal 14α-sterol demethylase than for the human liver equivalent cytochrome P450 14α-sterol demethylase (which is involved in cholesterol synthesis), and this improves their safety profile.
Indications
Fluconazole has good penetration in cerebrospinal fluid (CSF) and is used for treating cryptococcal meningitis, whereas itraconazole has poor CSF penetration but is widely distributed in the body and is indicated for treatment of aspergillosis, blastomycosis and histoplasmosis.
Ketoconazole is well distributed in body fluids (saliva, bile, urine, breast milk and inflamed joint fluid), tendons and other body tissues. It is indicated for the treatment of disseminated and mucocutaneous candidiasis, paracoccidioidomycosis and recalcitrant tinea infections. Miconazole is also widely distributed in body tissues, but neither ketoconazole nor miconazole adequately crosses the blood-brain barrier. Miconazole is primarily indicated for the treatment of disseminated and chronic mucocutaneous candidiasis. Voriconazole is indicated for serious fungal infections, including invasive aspergillosis and Candida, Scedosporium and Fusarium spp. infections. Currently posaconazole is indicated for invasive aspergillosis, fusariosis, zygomycosis, coccidioidomycosis, chromoblastomycosis and mycetoma in persons >13 years old when other antifungal therapy has failed or is inappropriate. The volume of distribution is large, suggesting that posaconazole distributes extensively into peripheral tissues. Fluconazole, itraconazole and voriconazole have replaced ketoconazole to a large extent because they have broader spectra of activity, fewer adverse reactions and reduced risk of drug interactions, compared with ketoconazole.
Pharmacokinetics
All of the azoles are administered orally; fluconazole and voriconazole may also be administered intravenously. Absorption rates are good if fluconazole is administered to a fasting person, while itraconazole and ketoconazole should be administered with food and voriconazole at least 1 hour before or 1 hour after a meal. Ketoconazole requires an acid medium for dissolution and absorption; therefore achlorhydria, hypochlorhydria or a rise in stomach pH caused by medications will impair its absorption. Absorption of posaconazole is enhanced with a high fat meal and this drug should be administered with food or a nutritional supplement containing ∼14 g of fat. Unlike the majority of the azoles posaconazole is not metabolised in the liver to any major extent, and the majority of the drug (77%) is excreted in faeces as the parent drug with ∼17% excreted in urine and faeces as glucuronide metabolites.
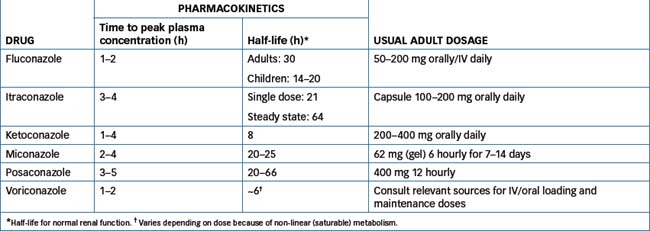
For pharmacokinetics and the usual adult dosages of azole antifungals, see Table 45-1.
Drug interactions
Ketoconazole inhibits human CYP3A4 and is subject to many drug interactions. The same interactions rarely occur with miconazole. The triazoles fluconazole and itraconazole inhibit a range of CYP enzymes and are also subject to many drug interactions. Voriconazole is metabolised by CYP2C9 and CYP3A4 and is also an inhibitor of the metabolism of other drugs metabolised by CYP3A4 (e.g.vincristine). Although not metabolised to a major extent, posaconazole is an inhibitor of CYP3A4 and hence drug interactions occur with other drugs metabolised by CYP3A4. Drug Interactions 45-1 lists the drugs that can interact with an azole antifungal agent, and the possible outcomes and management. Up-to-date drug interaction sources should always be consulted before administration of any of the azoles.
Drug interactions 45-1 Azole antifungals
| Drug | Possible outcomes | Management |
| Amphotericin | Antagonistic effect, reduced antifungal activity | Avoid concurrent use |
| Carbamazepine | Decreased metabolism of carbamazepine and increased risk of adverse reactions | Reduce dose of carbamazepine if required |
| Cyclosporin, tacrolimus | Increased plasma concentrations | Monitor renal function and cyclosporin/tacrolimus plasma concentrations |
| H2 antagonists, proton pump inhibitors, antacids | Reduced absorption, reduced antifungal effect | Avoid combination with H2 antagonists and proton pump inhibitors. Give 2 hours apart from antacids |
| Indinavir | Itraconazole and ketoconazole decrease metabolism of indinavir, which increases plasma concentration of indinavir | Reduce dose of indinavir |
| Phenytoin | Voriconazole increases phenytoin concentration and risk of toxicity | Monitor plasma concentration of phenytoin and reduce dose if necessary |
| Phenytoin increases metabolism of voriconazole, decreasing its efficacy | Increase voriconazole dose | |
| Rifamycins, isoniazid | Increased metabolism of ketoconazole, reduced antifungal effect | Avoid combination |
| Thiazolidinediones | Reduced metabolism increases risk of hypoglycaemia | Monitor blood glucose, reduce dose of thiazolidinediones if necessary |
| Triazolam | Itraconazole inhibits metabolism increasing the risk of sedation and adverse effects | Avoid combination or reduce dose of triazolam |
| Warfarin | Reduced metabolism increases risk of bleeding | Monitor INR and reduce warfarin dose if necessary |
Source: AMH 2010.
Adverse reactions
For azole antifungals, adverse reactions include nausea, vomiting, stomach distress, diarrhoea, flushing, drowsiness, dizziness, headache and hypersensitivity (fever, chills and rash). Miconazole has caused redness, swelling or pain at the injection site. Rarely, ketoconazole may cause photophobia, menstrual irregularities and gynaecomastia and impotence due to inhibition of adrenal steroid and testosterone synthesis. Other rare effects include liver toxicity, anaemia, agranulocytosis and exfoliative skin disorders such as Stevens–Johnson syndrome (for fluconazole) and thrombocytopenia (for fluconazole and miconazole). Voriconazole is associated with visual abnormalities including altered visual perception, blurred vision and colour changes. These effects are dose-related and are in general reversible. Data on the adverse effects of posaconazole are limited at present but are of a similar nature to the other azoles.
Caspofungin
Caspofungin is the first drug in a new class of antifungals, called the echiocandins, which have significant activity against Candida spp. and Aspergillus spp. It acts by inhibiting synthesis of glucan, a vital component of the fungal cell wall. Available only for intravenous administration, caspofungin is highly protein-bound (97%) and is slowly metabolised by hydrolysis and N-acetylation, with less than 2% excreted as unchanged drug in urine. Dosage adjustment is not necessary in patients with mild renal or mild hepatic impairment but dose reduction is required in those individuals who have moderate hepatic impairment, and this drug should not be used in the presence of severe hepatic impairment.
Although caspofungin does not induce or inhibit CYP, a number of drug interactions have been reported. The exact mechanisms are unclear, but when caspofungin is co-administered with known inducers (e.g. carbamazepine, dexamethasone, phenytoin and rifampicin), the plasma concentration of caspofungin is reduced substantially and an increase in dose is usually necessary. Cyclosporin increases caspofungin concentration, thereby increasing the risk of toxicity, and a reduction in the dose of caspofungin may be required. In contrast, caspofungin reduces tacrolimus concentration, and an increased dose of tacrolimus may be necessary.
As the site of action of caspofungin is unique to fungi, the drug is reasonably well tolerated. Adverse reactions include phlebitis at the injection site, rash, itch, fever, flushing, chills, nausea, vomiting and diarrhoea. Some elevations in liver enzymes have been reported and, rarely, resulting from possible histamine-mediated effects, facial swelling, bronchospasm and anaphylaxis. Hence, this drug is contraindicated in individuals allergic to caspofungin.
Flucytosine
Flucytosine enters fungal cells, where it is converted to fluorouracil, an antimetabolite (Figure 45-1). It interferes with pyrimidine metabolism, thus preventing nucleic acid and protein synthesis. It has selective toxicity against susceptible strains of fungi because human cells do not convert significant quantities of this drug into fluorouracil. Flucytosine is indicated for the treatment of fungal endocarditis (caused by Candida spp.), fungal meningitis (caused by Cryptococcus spp.) and fungal pneumonia, septicaemia or urinary infections caused by Candida or Cryptococcus spp. As resistance develops rapidly, combined use with another antifungal is recommended.
Flucytosine is administered IV and is widely distributed in the body, including CSF, the concentration in the latter being about 60%–90% of the plasma concentration. Flucytosine, with a half-life of 2.5–6 hours in individuals with normal renal function, is not significantly metabolised but is excreted via the kidneys, mostly as unchanged drug (80%). Capsules are not marketed but are available through the SAS.
Concurrent use in individuals who are receiving bone marrow suppressants or radiation therapy can result in an increased bone marrow-depressant effect. Monitor closely, as drug dosage reduction may be necessary. Antacids delay absorption, so administration should be separated by at least 2 hours. Adverse reactions include confusion, hallucinations, photosensitivity, headache, dizziness, sedation, gastric distress, anaemia, hepatitis, hypers ensitivity and bone marrow suppression (leucopenia and thrombocytopenia).
Griseofulvin
Griseofulvin is a fungistatic agent that inhibits fungal cell mitosis during metaphase. It is also deposited in the keratin precursor cells in skin, hair and nails, thus inhibiting fungal invasion of the keratin. When infested keratin is shed, healthy keratin will replace it. Griseofulvin is indicated for the treatment of susceptible organisms for onychomycosis (nail fungus), tinea barbae (fungal infection of the bearded section of face and neck), tinea capitis (fungal infection of the scalp; ringworm), tinea corporis (fungal infection of non-hairy skin), tinea cruris (fungal infection in the groin) and tinea pedis (fungal infection of the foot; athlete’s foot).
The oral absorption of griseofulvin varies from 25% to 70%. If griseofulvin is administered with or after a fatty meal, absorption is significantly enhanced. Griseofulvin is distributed in keratin layers in the skin, hair and nails, with very little being distributed in body tissues and fluids. It has a half-life of 24 hours and reaches peak plasma concentration in about 4 hours. The drug is metabolised by the liver to the primary metabolite 6-methylgriseofulvin which is excreted in urine (about 50%) and in faeces (about 30%).
Adverse reactions include headache, dizziness, gastric distress, insomnia, weakness, confusion, hypersensitivity (rash or hives), photosensitivity and, rarely, leucopenia, hepatitis and peripheral neuritis. Use griseofulvin with caution in individuals with lupus erythematosus or lupus-like syndromes. Avoid use in people with griseofulvin hypersensitivity, liver disease or porphyria.
The usual adult oral dose for tinea of the skin, hair and groin is 500 mg/day, and 1 g daily for tinea of the feet and nails. The duration of therapy depends on the infection site and severity of infection.
Nystatin
Nystatin is an antibiotic with antifungal activity and is used to treat cutaneous or mucocutaneous infections caused by the monilial organism Candida albicans. It has both fungistatic and fungicidal effects and is also used to suppress intestinal candidiasis. Nystatin adheres to sterols in the fungal cell membrane, altering cell membrane permeability, which results in loss of essential intercellular contents. Nystatin is poorly absorbed from the GI tract and is not absorbed when applied topically to skin or mucous membranes. It produces a local antifungal effect. Nystatin is not metabolised, and most of the unabsorbed nystatin is excreted in the faeces. No drug interaction has been documented.
Adverse reactions are uncommon and the drug is well tolerated by all ages. Large doses may cause gastric distress. Nystatin is contraindicated in people with nystatin hypersensitivity. The usual oral dose (tablet/capsule) for intestinal candidiasis in adults or children is 500,000–1,000,000 units 3–4 times daily. The dose of the oral liquid or lozenge for oral candidiasis is 100,000 units 4 times daily.
Terbinafine
Terbinafine is an orally and topically active allylamine antifungal agent and is indicated for severe ringworm unresponsive to topical treatment, onychomycoses (tinea unguium) and dermatophyte skin infections (tinea corporis, tinea cruris and tinea pedis). It inhibits squalene epoxidase thereby inhibiting formation of the lanosterol.
Terbinafine is distributed extensively and accumulates in sebum and hair within the first week of administration and by 3 weeks significant concentrations can be detected in stratum corneum and nails. Fungicidal concentrations persist in plasma and peripheral tissues for months after cessation of therapy (Kovarik et al 1995). Bioavailability is ∼40% due primarily to extensive first-pass metabolism. Terbinafine N-demethylation is mediated by CYP1A2, CYP2C8, CYP2C9, dihydrodiol formation by CYP1A2 and CYP2C9 and side chain oxidation by CYP1A2,CYP2C8, CYP2C9 and CYP2C19 while deamination is catalysed by CYP3A4. Terbinafine has minimal inhibitory activity towards a number of CYP substrate probes (e.g. tolbutamide 4-hydroxylation, S-mephenytoin 4’-hydroxylation, bufuralol l’-hydroxylation; Vickers et al 1999) but it is a potent inhibitor of CYP2D6 and hence interactions are likely with amitriptyline, nortriptyline and imipramine.
Adverse reactions include a variety of GI effects (e.g. nausea, vomiting, diarrhoea, abdominal pain) and skin reactions (e.g. itch, rash, urticaria). Rare adverse reactions include neutropenia, agranulocytosis, thrombocytopenia, pancytopenia and hepatotoxicity.
Antiviral drugs
Chemotherapy for viral diseases has been more limited than chemotherapy for bacterial diseases because the development and clinical application of antiviral drugs is more difficult. In many viral infections the replication of the virus in the body reaches its peak before any clinical symptoms appear. By the time signs and symptoms of illness appear, the multiplication of the virus is maximal and the subsequent course of the illness has been determined. To be clinically effective, antiviral drugs must be administered in a chemoprophylactic manner as preventive agents before disease appears. Development of antiviral drugs has also been impeded by the fact that viruses are simply either double-stranded DNA or single-stranded RNA contained within a capsid (viral protein coat), and they lack any metabolic capacity. Hence, viruses are true parasites; they replicate within the mammalian cell and use the host cells’ enzyme systems. Thus drugs that inhibit virus replication often disturb the host cells and in many instances are too toxic for use.
The antiviral drugs reviewed in this chapter are separated into two groups:
Antiviral (non-retroviral) drugs
The following drugs are used in the treatment of viral infections due to DNA and RNA viruses (excluding retroviruses, e.g. HIV):
DNA polymerase inhibitors
Aciclovir
Aciclovir (see Drug Monograph 45-2) is the prototypical drug and was the first to be approved (1982) for use in the treatment of herpes simplex virus (HSV) and varicella zoster virus (VZV). Other drugs similar to aciclovir (an acyclic guanine nucleoside analogue) include famciclovir, an oral prodrug that is metabolised to penciclovir; ganciclovir, structurally similar to aciclovir; valaciclovir, a prodrug of aciclovir; and valganciclovir, a prodrug of ganciclovir.
Aciclovir is selectively taken up by herpes simplex virus (HSV)-infected cells and is eventually converted via several cellular enzymes, including thymidine kinase, to an active triphosphate form. This active form (acyclo-GTP) inhibits viral DNA synthesis by two actions: (1) it inhibits the incorporation of the normal deoxyguanosine into viral DNA by the viral DNA polymerase and (2) in its place acyclo-GTP is incorporated into the growing DNA chain; this then causes termination of synthesis of the viral DNA.
Indications
Aciclovir is used in the prophylaxis and treatment of genital herpes infections and for the treatment of varicella (chickenpox) infections and HSV encephalitis. It is also used in people with AIDS.
Pharmacokinetics
The oral dose form is poorly absorbed (15%–30%), but plasma concentrations achieved are therapeutic. It is widely disseminated to various body fluids and tissues, including CSF and herpetic vesicular fluid. Concentrations in CSF are around 50% of the plasma drug concentration. The half-life is about 2.5 hours in individuals with normal renal function and about 20 hours in anuric patients. The drug is minimally metabolised by the liver (about 15%) and is excreted primarily as unchanged drug in urine.
Drug interactions
Concurrent use of aciclovir with probenecid will result in high plasma concentration of aciclovir and the risk of adverse neurological effects. During concomitant administration with theophylline, the plasma concentration of theophylline may increase and a decrease in theophylline dose may be necessary.
Adverse reactions
Adverse reactions with the oral dosage form include gastric distress, headache, dizziness, nausea, diarrhoea and vomiting. With the parenteral form, phlebitis at the injection site, acute renal failure with rapid injection or, rarely, encephalopathic alterations such as confusion, hallucinations, convulsions, tremors and coma can occur.
Warnings and contraindications
Use with caution in individuals with neurological abnormalities or kidney function impairment, or in dehydrated people (risk of precipitation of aciclovir crystals in the kidneys). Avoid use in people with aciclovir hypersensitivity.
Dosage and administration
Aciclovir is available as oral, topical and IV formulations. The usual oral adult dose for genital herpes infection is 400 mg orally every 8 hours during waking hours (three times daily) for 5–7 days. For prophylaxis of recurrent infection, the dose is 400 mg orally twice daily for up to 6 months. The parenteral adult dosage for HSV encephalitis and varicella zoster in immunocompromised individuals is 10 mg/kg IV 8-hourly. For other dosage recommendations, consult current drug information sources.
Famciclovir
Famciclovir has inhibitory actions against HSV (types 1 and 2) and VZV. It is indicated for the treatment of acute herpes zoster (shingles) infections and recurrent genital herpes. Administered orally, famciclovir is well absorbed and converted in the intestinal wall to the active antiviral metabolite penciclovir. Peak plasma concentration is achieved in about 1 hour; its half-life is 2–3 hours and it is excreted unchanged primarily in urine and faeces.
Adverse reactions include headaches, weakness, gastric distress (nausea, vomiting and diarrhoea) and fatigue. Exercise caution in people with kidney function impairment, as dosage reduction is required. Avoid use in lactating women. For recurrent genital herpes, the usual adult dose is 125 mg orally 12 hourly for 5 days and, as a prophylactic, 250 mg twice daily for up to 12 months.
Ganciclovir and valganciclovir
Ganciclovir is converted intracellularly to the triphosphate form, which is the active, antiviral agent. In the presence of the cytomegalovirus (CMV), ganciclovir is rapidly phosphorylated to ganciclovir triphosphate, which inhibits viral DNA polymerase, suppressing viral DNA synthesis. If ganciclovir is discontinued, viral replication will resume.
Ganciclovir is considered to be carcinogenic, and appropriate cytotoxic handling procedures should be adopted. It is used for CMV pneumonitis in bone marrow and renal transplant recipients and for sight-threatening CMV retinitis in severely immunocompromised individuals (AMH 2010). Valganciclovir is a prodrug of ganciclovir: the indications for its use include prophylaxis of CMV disease after organ transplant. As valganciclovir is quickly metabolised to the active component ganciclovir, the pharmacodynamics and pharmacokinetics of valganciclovir are the same as those of ganciclovir.
Ganciclovir is administered orally, by IV infusion or by ocular implant. The plasma half-life is 2.5–3.6 hours and the vitreous fluid half-life is about 13 hours. This drug is excreted unchanged, primarily by the kidneys, and the half-life is substantially prolonged in people with renal impairment.
Oral ganciclovir is indicated only for maintenance of CMV retinitis in people who have had resolution of active retinitis after induction therapy with parenteral ganciclovir. The plasma half-life following oral administration is 3–5.5 hours and peak plasma concentration occurs in 3 hours if administered with food. An ocular ganciclovir implant is used to treat sight-threatening CMV retinitis.
Drug Interactions 45-2 provides examples of interactions that can occur with ganciclovir or valganciclovir if given with the drugs listed.
Drug interactions 45-2 Ganciclovir/valganciclovir
| Drug | Possible effects and management |
| Bone marrow-depressant drugs | Concurrent use can result in increased bone marrow-suppressant effects. Monitor blood closely for neutropenia and thrombocytopenia |
| Foscarnet | Synergistic antiviral effects but increased haematological and renal toxicity. Monitor blood count and electrolytes |
| Imipenem | Avoid combined use because of increased risk of seizures |
| Probenecid | Inhibits renal secretion of ganciclovir, increasing the concentration and risk of toxicity. Avoid combination |
| Zidovudine (AZT) | Can result in severe haematological toxicity. Withhold zidovudine or change to another antiretroviral drug |
Adverse reactions with ganciclovir after IV or oral administration include granulocytopenia, thrombocytopenia, anaemia, gastric distress, central nervous system (CNS) effects (e.g. anxiety and tremors), hypersensitivity and phlebitis or pain at the injection site. After ocular implantation, a variety of ophthalmic disorders may result, such as retinal detachment, scleral induration, subconjunctival haemorrhage, conjunctival scarring and bacterial endophthalmitis. Use with caution in people with moderate to severe renal impairment, as dosage reduction will be required. Avoid use in people with aciclovir or ganciclovir hypersensitivity, an absolute neutrophil count <0.5 × 109 cells/L, a platelet count <25 × 109/L and in people with bone marrow suppression. Ganciclovir and valganciclovir are contraindicated in pregnancy and lactation because of potential embryotoxic and teratogenic effects. The adverse reactions of valganciclovir are similar to those of ganciclovir.
The usual adult dose of ganciclovir for CMV retinitis is 5 mg/kg by IV infusion every 12 hours for 1–2 weeks. For valganciclovir, the oral dose is 900 mg 12 hourly for 21 days, with a maintenance dose of 900 mg daily.
Valaciclovir
Valaciclovir is a prodrug that is converted to aciclovir by first-pass intestinal and liver metabolism. It is indicated for the treatment of varicella zoster infections and for primary and recurring genital HSV. Administered orally, valaciclovir is well absorbed and is converted to aciclovir, the active substance. It reaches peak plasma concentration in 1.6–2 hours; its half-life is 2.5–3.3 hours. Valaciclovir is converted to inactive metabolites by alcohol and aldehyde dehydrogenase, and excreted primarily in urine.
Adverse reactions include nausea, headache, weakness, gastric distress (constipation, diarrhoea, anorexia, abdominal pain and vomiting), dizziness, agitation and renal impairment. Rarely, valaciclovir can cause haematological toxicity, including neutropenia, leucopenia and thrombocytopenia. Use valaciclovir with caution in people with liver or renal function dysfunction. Avoid use in people with valaciclovir or aciclovir hypersensitivity, bone marrow or kidney transplant, advanced HIV infections or kidney function impairment.
The usual adult dose for genital herpes simplex is 500 mg 12 hourly for 5–10 days.
Neuraminidase inhibitors
Oseltamivir
Neuraminidase is an essential enzyme for replication of both influenza A and B strains because it plays an important role in releasing the virus from infected cells, enabling it to spread to other cells. Oseltamivir is a prodrug that undergoes ester hydrolysis in the GIT and liver to the active carboxylate metabolite, which inhibits the action of neuraminidase on the surface of the viral cell and prevents release of the viral particles (Robinson 2001). It is indicated for the treatment of influenza A and B in adults and children over 1 year.
Oseltamivir is well absorbed after oral administration and is metabolised (about 75%) by esterases in the liver and GIT to its active metabolite. Peak plasma concentration is reached 2–3 hours after dosing. The half-life is 6–10 hours in normal individuals but may be prolonged in people with renal impairment. Drug interactions are unlikely because oseltamivir is not metabolised by the hepatic cytochrome P450 system.
Adverse reactions include vomiting, nausea, headache, dizziness and vertigo. Use with caution in people with underlying respiratory or cardiac conditions and complicated influenza (e.g. pneumonia). The usual dose is 75 mg twice daily for 5 days. Oseltamivir should be started within 48 hours of the onset of symptoms and taken with food to reduce stomach discomfort.
Zanamivir
Similar to oseltamivir, zanamivir is a potent and selective inhibitor of the viral neuraminidase enzyme. It is indicated for the treatment of influenza A and B in adults and children >5 years old within 48 hours of the onset of symptoms. Administration is via oral inhalation, and approximately 10%–20% of the dose is systemically available. After inhalation, zanamivir is widely deposited within the respiratory tract. It is not metabolised and is excreted as unchanged drug in the urine.
Zanamivir is generally well tolerated and in clinical trials the frequency of adverse reactions is similar to that for placebo. Rarely, an allergic reaction has been reported (facial and oropharyngeal oedema), as have bronchospasm and dyspnoea.
Miscellaneous antiviral drugs
Adefovir
Adefovir dipivoxil is rapidly absorbed and metabolised by esterases to the active drug adefovir. The half-life after oral administration is 5–7 hours, and approximately 60% of the drug is eliminated unchanged by the kidney. To date, dose-related nephrotoxicity has been reported but other adverse reactions may not yet be fully known. Concomitant administration with other nephrotoxic drugs (e.g. NSAIDs) may increase the risk of nephrotoxicity. The dosage is 10 mg once daily, but in the presence of renal impairment a reduction in dose is necessary.
Adefovir dipivoxil is an oral prodrug that is effective in the treatment of chronic hepatitis B in adults. In the host cell, enzymes phosphorylate adefovir (an analogue of adenosine) to adefovir diphosphate, which then inhibits viral polymerase, resulting in termination of viral DNA synthesis.
Amantadine
Amantadine appears to block the uncoating of the influenza A virus and the release of viral nucleic acid into host respiratory epithelial cells. It also increases dopamine release and inhibits the reuptake of dopamine and noradrenaline centrally. Amantadine is indicated for the prevention and treatment of influenza A, for treatment of Parkinson’s disease and for the treatment of drug-induced extrapyramidal reactions.
Amantadine is rapidly absorbed orally; it is distributed to saliva and nasal secretions and crosses the blood-brain barrier. It has a half-life of 11–15 hours, reaching peak plasma concentration within 2–4 hours. It is excreted mostly unchanged by the kidneys, and the half-life is prolonged in people with renal impairment.
Adverse reactions include CNS toxicity, gastric distress and, with chronic therapy, livedo reticularis (a vasospastic disorder, worsened by exposure to cold, that is evidenced by a reddish-blue mottling of the legs and sometimes arms), anticholinergic effects (dry mouth, constipation, blurred vision, confusion and difficult urination), vomiting and orthostatic hypotension. Use amantadine with caution in patients with (or a history of) eczema rash or psychosis and in individuals with severe psychoneurosis. Avoid use in people with amantadine hypersensitivity, peripheral oedema, congestive heart failure, kidney impairment or epilepsy or other convulsive disorders.
The adult oral antiviral dose is 200 mg daily or 100 mg every 12 hours for ∼10 days. For those aged <10 years or >65 years, the once-daily dose is 100 mg for 10 days. Antiviral effects diminish within two days following the cessation of treatment.
Cidofovir
Cidofovir is used for treating CMV retinitis in AIDS. It selectively inhibits viral DNA polymerase and prevents DNA synthesis. It is administered IV with oral probenecid, which blocks active renal tubule secretion and hence the renal clearance of the drug. The drug is not metabolised, and 70%-85% of the dose is excreted unchanged in urine within 24 hours. Additive nephrotoxicity is observed with the aminoglycosides, amphotericin, foscarnet, vancomycin and NSAIDs. Reduction in dosage of drugs that undergo renal secretion (e.g. zidovudine) is required.
Adverse reactions include weakness, GI distress, alopecia, headache, nephrotoxicity, elevated plasma concentrations of liver enzymes, neutropenia, fever and, rarely, ocular hypotony. Cidofovir is contraindicated in individuals with cidofovir or probenecid hypersensitivity or severe kidney function impairment. Pretreatment with probenecid is used to reduce the nephrotoxicity. Administer 2 g probenecid orally 3 hours before cidofovir infusion and 1 g at 2 and 8 hours after completing the infusion. The induction dose of cidofovir is 5 mg/kg IV once weekly for 2 weeks and maintenance therapy is 5 mg/kg IV once every 2 weeks.
Entecavir
Entecavir is a novel deoxyguanosine nucleoside analogue with selective activity towards hepatitis B virus (HBV) with little or no activity against other viral pathogens. The active form of entecavir is the triphosphate, which inhibits HBV DNA replication at three stages: (1) priming of the HBV DNA polymerase, (2) at the reverse transcription stage and (3) at the synthesis stage of the positive strand of HBV DNA (Cada et al 2005). It is indicated for chronic hepatitis B in the presence of liver inflammation and especially if there is evidence of lamivudine resistance. Following oral administration peak plasma entecavir concentration occurs at ∼0.5–1.5 hours. Entecavir is well absorbed on an empty stomach and bioavailability is 100%. Entecavir does not undergo extensive metabolism and is not an inducer or inhibitor of CYP. It is primarily excreted as unchanged drug (62%–73%) in urine. The elimination half-life is 128-149 hours and dosage adjustment is required in individuals with creatinine clearance of <50 mL/minute.
Common adverse reactions include fatigue, headache, nausea, diarrhoea and dyspepsia. The usual dosage in individuals >16 years of age is 0.5 mg daily.
Foscarnet
Foscarnet is a virustatic agent that inhibits viral replication of all known herpes viruses in vitro, including CMV, HSV types 1 and 2, Epstein–Barr virus and varicella zoster virus. It acts by selective inhibition at the pyrophosphate binding site of viral DNA polymerase. If the drug is discontinued, viral replication will resume. It is currently used to treat CMV retinitis in patients with AIDS.
This drug is administered by intravenous infusion, has an elimination half-life of 3.3–6.8 hours, reaches peak plasma concentration at the end of the infusion, is not metabolised and is excreted primarily unchanged in the urine.
Adverse reactions include nephrotoxicity, gastric distress, neurotoxicity, anaemia, leucopenia and phlebitis or pain at injection site. Use with caution in people with anaemia, hypomagnesaemia, hypocalcaemia or a history of seizures. Avoid use in people with foscarnet hypersensitivity or kidney impairment and in dehydrated people. For CMV retinitis, the usual adult dose by IV infusion is 60 mg/kg administered over a minimum of 1 hour by infusion pump, every 8 hours for 2–3 weeks. The maintenance dosage is 90–120 mg/kg IV daily over 2 hours.
Palivizumab
Palivizumab is a humanised monoclonal antibody directed against a protein of the surface of respiratory syncytial virus (RSV). Once bound to the virus, palivizumab inhibits RSV replication. It is indicated for serious lower respiratory tract diseases caused by RSV in infants and children at high risk of RSV disease. These include infants born at 32–35 weeks’ gestation, or with bronchopulmonary dysplasia or with significant congenital heart disease (AMH 2010). Determination of the pharmacokinetics of monoclonal antibodies is difficult, and considerable individual variation in palivizumab plasma concentration has been reported. To date, the metabolism of palivizumab has not been determined and drug interactions have not been described.
Adverse reactions include fever, rash, wheeze, cough and, rarely, hypersensitivity including anaphylaxis. This drug is contraindicated in those with known sensitivity to palivizumab or any other humanised monoclonal antibody. Consult relevant sources for dosages for IM administration in infants.
Ribavirin aerosol
Ribavirin is virustatic, with a mechanism of action that is diverse and not completely understood. It rapidly penetrates virus-infected cells and is believed to reduce intracellular guanosine triphosphate (GTP) storage. It inhibits viral RNA and protein synthesis, thus inhibiting viral duplication, spread to other cells or both. It is indicated for serious viral pneumonia caused by RSV in children <2 years of age.
After oral inhalation, ribavirin is well absorbed and rapidly distributed to plasma, respiratory tract secretions and erythrocytes. Its half-life is 9.5 hours after oral inhalation and around 40 days in erythrocytes. Ribavirin is metabolised in the liver and excreted primarily by the kidneys. In-vitro studies indicate that ribavirin and zidovudine are antagonistic and should not be administered concurrently.
Adverse reactions are uncommon and can include skin rash or irritation (inhalation product), CNS effects (insomnia, headache and lethargy) with IV and oral dosages and gastric distress (anorexia and nausea) and anaemia (usually with higher doses). The health-care professional or provider should use caution in administering this medication, as headache, itching, swelling and red eyes have been reported. Use with caution in individuals with severe anaemia and avoid use in people with ribavirin hypersensitivity. In addition, worsening of lung function may occur in asthmatics, and ribavirin should be stopped if deterioration occurs.
The adult dose for ribavirin as an inhalation aerosol has not been established. For viral pneumonia in children, administer by oral inhalation via a small-particle aerosol generator, using a 20 mg/mL ribavirin concentration in the reservoir. Inhale for 12–18 hours/day for 3–7 days.
Ribavirin combined with peginterferon alfa (refer to Chapter 47) is used in the treatment of chronic hepatitis C (see Clinical Interest Box 45-2) in individuals over the age of 18 years with compensated liver complications (e.g. cirrhosis). Peginterferon alfa is administered SC once weekly combined with twice daily oral ribavirin dosed according to weight. This treatment is less effective in people who also have HIV.
Clinical interest box 45-2 Hepatitis C in new zealand
There are more than 30,000 people in New Zealand with hepatitis C, with approximately 25 new infections per week. It is estimated that there are 19,000 injecting drug users, and approximately 16,000 of these people have hepatitis C. Few patients are receiving treatment for the virus. Although hepatitis C medication is expensive, the courses are short and can prevent future health costs. For some of types of the disease, combination therapy has been shown to cure 80% of cases. Without adequate treatment and control, hepatitis C will spread among drug users and the wider New Zealand society. Costs have been predicted to rise by between $166 million and $400 million in the next 30 years. Hepatitis C has major effects on the liver and can be fatal.
Adapted from: I. Sheerin: www.otago.ac.nz/news/2005/01-04.05pressrelease.html/ [19 January 2006]. Hepatitis Resource Centre: www.hepc.org.nz/ [9 September 2009].
Antiretroviral drugs
The human immunodeficiency virus was discovered in 1983 and currently infects ∼40 million people worldwide. Since 1983 the development of antiviral drugs to combat the HIV epidemic has continued at a furious pace and continues to do because of the problem of HIV resistant strains. Vaccines have always been used to control viral infections but the prospect of an effective vaccine against HIV is still very remote (see Clinical Interest Box 45-3).
Clinical interest box 45-3 An AIDS vaccine
The development of a vaccine often proceeds not long after an infectious disease emerges. The search for an AIDS vaccine began in the early 1980s. There was considerable optimism that one would be found and that the problem could be controlled, as had smallpox. Progress on development of a vaccine has not been easy and the hurdles encountered have included identification of immunogens that will produce broad and long-lasting immunity, defining of structures and immunisation strategies that will elicit antibody production and development of strategies to deal with the different HIV strains and viral coats (Nabel 2001). According to Nabel in 2001: ‘The search for an HIV vaccine has been slow and at times frustrating, but the resolve of the biomedical research community to address this problem has grown. Although the solution is not yet at hand, progress is tangible and encouraging results now develop on a regular basis’.
In 2009 the future does not seem as rosy as the HIV Vaccine Trials Network and Merck-sponsored (HVTN502/Merck 018 or STEP) trial was terminated early in September 2007 because of preliminary data that suggested that the vaccine recipients with pre-existing adenovirus immunity were at risk of increased HIV acquisition. The dilemma is how we identify candidate vaccines when positive results from in vitro immunogenicity tests and evidence of protection in animal models fails to predict success of potential vaccines in humans (Weatherall et al 2009).
The following drugs are used in the treatment of HIV:
Drug monograph 45-3 Zidovudine
Zidovudine (azidothymidine [AZT]) was the first antiretroviral drug developed to target HIV infection. It was first used in 1987 and by the early 1990s it was evident that zidovudine slowed the rate of progression of AIDS. Survival rates, however, did not improve and it became apparent that resistant HIV strains had developed. By 1996, the use of combination therapy had emerged, with better results in retarding disease progression and decreasing mortality (Hovanessian 1999).
Zidovudine is an antiviral agent (virustatic) that intracellularly is converted by cellular enzymes to monophosphate, diphosphate and then to zidovudine triphosphate. The triphosphate form competitively inhibits the reverse transcriptase with respect to the incorporation of natural thymidine triphosphate in growing chains of viral DNA, thus inhibiting viral replication. It has a greater affinity for retroviral reverse transcriptase than for the human alpha-DNA polymerase; thus it selectively inhibits viral replication.
Indications
Zidovudine is indicated for the treatment of HIV infection and can be used in combination with abacavir and lamivudine. This drug is also used to prevent vertical transmission of HIV during pregnancy (2nd and 3rd trimesters) and to the neonate.
Pharmacokinetics
Administered orally, zidovudine is rapidly absorbed and distributed in plasma and CSF, reaches peak plasma concentration in 0.5–1.5 hours and has a half-life of around 1 hour in plasma (3.3 hours intracellularly). It is metabolised in the liver, and the major metabolite does not have antiviral activity. Both unchanged zidovudine (about 15%) and the main metabolite (about 75%) are excreted in urine.
Drug interactions
The following effects can occur when zidovudine is given with the drugs listed below:
Adverse reactions
These include nausea, myalgia, insomnia, severe headaches, bone marrow depression (anaemia and neutropenia), hepatotoxicity, myopathy, neurotoxicity and hyperpigmentation of nails.
The complete mechanism of action of indinavir is unknown. It appears to inhibit the replication of retroviruses (HIV types 1 and 2) by interfering with HIV protease, which cleaves inactive viral protein precursors into active proteins that make the HIV particle infectious. Indinavir affects the replication cycle of HIV. It is active both in acute infection and in chronically infected cells, which are generally not affected by the nucleoside analogue reverse transcriptase inhibitors, such as didanosine, lamivudine, stavudine, zalcitabine and zidovudine. Thus this product has a virustatic effect due to its HIV protease inhibitor effects.
Pharmacokinetics
Administered orally, indinavir reaches peak plasma concentration in about 1 hour. It is extensively metabolised in the liver by CYP3A4 and excreted primarily in faeces.
Drug interactions
The following effects can occur when indinavir is given with the drugs listed below:
| Drug | Possible effects and management |
| Cisapride, midazolam or triazolam | Indinavir and these drugs are metabolised in the liver by CYP3A4. Concurrent use can result in a decreased metabolism of these drugs and toxicity. Avoid, or a potentially serious drug interaction could occur |
| Didanosine | When given concurrently, take drugs at least 2 hours apart and take indinavir on an empty stomach. Indinavir needs an acidic pH for absorption, and didanosine needs a buffer to raise pH to prevent acid from destroying it |
| Ketoconazole | Concurrent use results in an increased plasma concentration of indinavir. Reduce the dose of indinavir to 600 mg every 8 hours if administered with ketoconazole |
| Rifabutin | Concurrent use results in an increased plasma concentration of rifabutin. Reduce the dose of rifabutin to 400 mg every 8 hours if administered concurrently with indinavir |
| Rifampicin | Rifampicin is a potent inducer of CYP3A4, which can decrease the plasma concentration of indinavir. Avoid concurrent use |
Adverse reactions
These include gastric distress, nausea, vomiting, diarrhoea, headache, dizziness, fatigue, insomnia, changes in taste sensation, kidney stones, fever, flu-like syndrome and, rarely, diabetes or hyperglycaemia and ketoacidosis.
Nucleoside reverse transcriptase inhibitors (NRTIs)
These drugs are indicated for the treatment of HIV infection (in adults and children) in combination with other retroviral drugs, for prophylaxis during pregnancy to prevent transmission to the fetus and for prophylaxis postexposure to HIV (see Clinical Interest Box 45-4). NRTIs are used in combination therapy with NNRTIs and PIs in HIV-infected patients. The most effective regimens continue to be combinations of three or more drugs that include at least two different classes of antiretrovirals. Unfortunately, the problems faced in the treatment of HIV continue to be rebound viral replication, development of resistance and inadequate drug potency.
Clinical interest box 45-4 HIV–AIDS in Australia
The cumulative number of HIV-infected people living in Australia at the end of 2008 was estimated at 17,444 (NCHECR 2009). New diagnoses of HIV infection increased by 38% over the past 10 years from 718 in 1999 to around 1000 cases per annum over the past 3 years. Between 2004 and 2008, 58% of newly diagnosed HIV infections occurred in people born in Australia and 11% of newly diagnosed cases had been previously diagnosed overseas. The majority of HIV transmission continues to occur primarily through sexual contact between men with relatively few new cases among injecting drug users or heterosexuals. In heterosexual partners, 59% of HIV infection cases over the past 5 years involved one or both partners from high prevalence countries. By 31 December 2008, a total of 10,348 cases of AIDS and 6765 AIDS-related deaths had been notified.
Source: National Centre in HIV Epidemiology and Clinical Research, 2009 Annual Surveillance Report, http://www.nchecr.unsw.edu.au/NCHECRweb.nsf/resources/SurvReports_3/$file/ASR2009.pdf [9 September 2009].
All of these drugs are substrates for the viral reverse transcriptase enzyme (Figure 45-2), which converts viral RNA into proviral DNA before it becomes incorporated in the host cell chromosome. In order to do this, the drugs must first be phosphorylated in the cytoplasm to enable them to effectively compete with the normal host cell triphosphates. The phosphorylated drug, once incorporated in the growing viral DNA chain, causes chain termination. These drugs are thus effective only in susceptible cells, but are ineffective in cells already infected with HIV.
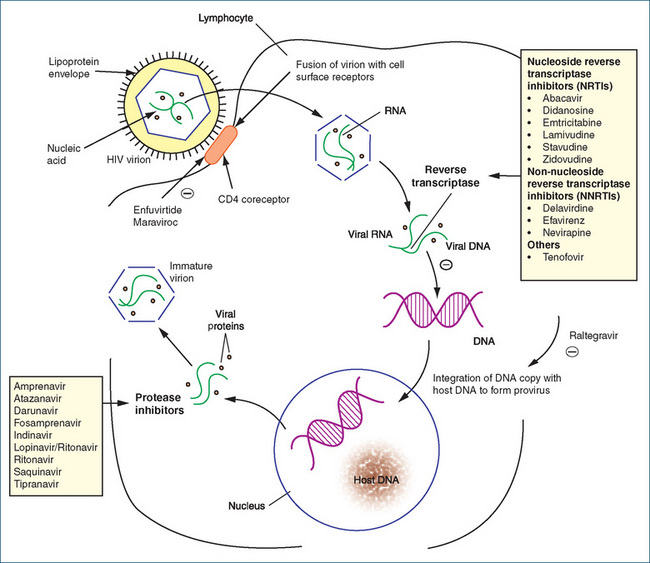
Figure 45-2 Inhibition sites for HIV replication. The HIV redundant genes are composed of RNA, which is translated to DNA by reverse transcriptase (RT) for viral reproduction. The RT inhibitors interfere with virus production at this site. When integrated DNA becomes part of the cell, the cell produces viral proteins requiring the protease enzyme for the production of new virions. The protease inhibitors block this enzyme to prevent the release of new viruses into the bloodstream. As a result, combination therapies can reduce the viral load of new HIV produced in the body.  = inhibition.
= inhibition.
Significant adverse reactions of the NRTIs include lipodystrophy (changes in cutaneous fat distribution: loss in the face, limbs and buttocks but accumulation in the abdominal, breast and dorsocervical region). Metabolic abnormalities may also occur, and these include insulin resistance and hyperlipidaemia. Many of the NRTIs are subject to numerous drug interactions, and current drug information sources should be consulted before commencing therapy.
Abacavir
Abacavir (also known as ABC) is selective against HIV-1 and HIV-2 and is indicated for the treatment of HIV infection. It is administered orally; bioavailability is ∼83%; the half-life is 1.5 hours; and the drug is widely distributed, including good penetration of the CSF. Abacavir is extensively metabolised by the liver, and less than 2% is excreted as unchanged drug in urine.
Adverse reactions include a hypersensitivity reaction in at least 5% of people within 6 weeks. The severity of this reaction is such that the drug should be stopped immediately and never reintroduced as therapy, as fatalities have occurred on rechallenge. Other adverse reactions include the whole range of GI symptoms, muscle problems (e.g. fatigue, myalgia and arthralgia) and respiratory symptoms (e.g. dyspnoea, sore throat and cough). The usual adult dose is 300 mg 12-hourly or 600 mg once daily.
Didanosine
Didanosine (also known as DDI) is converted intracellularly to its active form, ddA-triphosphate, which inhibits HIV DNA reverse transcriptase. This inhibition suppresses HIV replication.
This product, which is available in oral dosage forms, is acid-labile. The oral formulations are buffered to increase gastric pH to protect didanosine from gastric acid destruction. Didanosine crosses the blood-brain barrier, has a half-life of 1.5 hours in adults and reaches peak plasma concentration in 30–60 minutes. Excretion is primarily via the kidneys.
Drug Interactions 45-3 details the interactions that can occur when didanosine is given with the drugs listed.
Drug interactions 45-3 Didanosine
| Drug | Possible effects and management |
| Alcohol, azathioprine, oestrogens, frusemide, nitrofurantoin, pentamidine IV, sulfonamides, sulindac, stavudine, tetracyclines, thiazide diuretics, valproic acid or other drugs associated with pancreatitis | Concurrent drug use can result in pancreatitis. Avoid, or a potentially serious drug interaction could occur. If combination therapy is necessary, use extreme caution |
| Chloramphenicol, cisplatin, hydralazine, isoniazid, lithium, metronidazole, nitrofurantoin, nitrous oxide, phenytoin, stavudine, vincristine, zalcitabine or other drugs associated with peripheral neuropathy | Can increase the potential for peripheral neuropathy. Avoid, or a potentially serious drug interaction could occur. If one of these drugs must be used, monitor closely for numbness and tingling in the fingers and toes |
| Dapsone, itraconazole, ketoconazole, quinolone antibiotics, tetracyclines and indinavir | Buffered formulation of didanosine can result in decreased absorption of these drugs because they require an acidic medium. Administer these drugs at least 2 hours before didanosine |
Adverse reactions include peripheral neuropathy, CNS toxicity (e.g. headache, anxiety, increased irritability and insomnia), gastric distress (stomach pain, nausea and diarrhoea) and dry mouth. Less frequent and rare adverse reactions include pancreatitis, cardiomyopathy, blood disorders (anaemia, leucopenia or thrombocytopenia), hepatitis, convulsions, hypersensitivity and retinal depigmentation. Use with caution in individuals with gout, liver dysfunction, phenylketonuria (tablets contain phenylalanine) or impaired kidney function. Avoid use in people with hypertriglyceridaemia, pancreatitis, alcoholism or peripheral neuropathy. Didanosine tablets contain magnesium, which may present an unacceptable load in individuals with renal impairment, so dose reduction may be necessary.
The usual adult dose for patients weighing <60 kg is 250 mg once daily; for patients weighing >60 kg the dose is 400 mg daily. For dosing schedules in children, check current information sources. Capsules should be taken on an empty stomach at least 1 hour before or 2 hours after food.
Emtricitabine
Emtricitabine (also known as FTC) is an analogue of cytosine and is effective against HIV-1, HIV-2 and hepatitis B. It is rapidly absorbed after oral administration, with peak plasma concentrations occurring at 1–2 hours post-dose. Emtricitabine is excreted predominantly by the kidneys (86%) and in faeces (14%). Only 13% of the dose is hepatically metabolised, hence renal impairment significantly influences the plasma drug concentration and dose reduction may be necessary. The potential for drug interactions is low, and no clinically significant drug interactions have been noted with indinavir, zidovudine, stavudine or famciclovir.
Common adverse reactions include headache, diarrhoea, nausea and a rash. Predominantly in females, emtricitabine causes hyperpigmentation of the palms and/or soles of the feet. The dose in adults is 200 mg daily.
Lamivudine
Lamivudine (also known as 3TC) is converted in the body to an active metabolite, lamivudine triphosphate (L-TP), which inhibits HIV reverse transcription by terminating the viral DNA chain. It also inhibits RNA- and DNA-dependent DNA polymerase functions of reverse transcriptase. Synergism of antiviral activity is achieved with a combination dosage form containing 150 mg lamivudine and 300 mg zidovudine.
Rapidly absorbed after oral administration, lamivudine reaches peak plasma concentration in 1 hour (fasting state) or 3.2 hours (with food). L-TP has an intracellular half-life of 10–15 hours and is excreted unchanged by the kidneys. The potential for drug interactions exists with IV pentamidine, which increases the risk of pancreatitis, and trimethoprim, which competes with lamivudine for renal excretion. This latter combination can result in increased lamivudine concentration, and the combination should be avoided in people with renal impairment.
Adverse reactions with lamivudine include headaches, dizziness, fatigue, fever, nausea, vomiting, diarrhoea, gastric pain or distress, anorexia, neuropathy, insomnia, depression, cough, skeletal muscle pain, pancreatitis, paraesthesias, peripheral neuropathy and, rarely, anaemia, rash and neutropenia. Use lamivudine with caution in individuals with pancreatitis or peripheral neuropathy, including people with a history of these conditions, and in diabetics, as 5 mL of oral solution contains 1 g sucrose. Avoid use in people with lamivudine hypersensitivity and kidney function impairment.
For the treatment of HIV, the usual dose of lamivudine for children <12 years is 4 mg/kg 12-hourly to a maximum of 300 mg daily, and for adults 150 mg orally twice a day or 300 mg once daily.
Stavudine
Stavudine (also known as d4T) is converted to stavudine triphosphate, which competes with deoxythymidine triphosphate, resulting in inhibition of HIV replication and DNA synthesis. Oral stavudine is rapidly absorbed and reaches peak plasma concentration in 0.5–1.5 hours. It has a half-life of 1–1.6 hours and is excreted unchanged, primarily by the kidneys.
Drug interactions with stavudine include:
Adverse reactions include dose-related peripheral neuropathy, increased liver enzymes and anaemia. Other adverse reactions include arthralgia, hypersensitivity, myalgia, pancreatitis, weakness, GI distress, headache and insomnia. Use with caution in individuals with liver function impairment or alcoholism. Avoid use in people with peripheral neuropathy or kidney function impairment.
The usual adult oral dose of stavudine is 30 mg every 12 hours for people weighing <60 kg, or 40 mg every 12 hours for people weighing >60 kg.
Non-nucleoside reverse transcriptase inhibitors (NNRTIs)
The non-nucleoside reverse transcriptase inhibitors (NNRTIs) delavirdine, efavirenz and nevirapine block RNA-dependent and DNA-dependent DNA polymerases (Figure 45-2). These drugs are usually combined with two or more drugs to suppress HIV viral replication because use as sole agents results in rapid emergence of viral resistance (can occur within 1 week). Many interactions occur with these drugs because of the involvement of specific isoforms of CYP. Relevant drug information sources should be consulted for specific drug interactions, as some are life-threatening.
Delavirdine
Delavirdine is rapidly absorbed following oral administration, and peak plasma concentration occurs within 1 hour. The drug is extensively bound to plasma proteins and is metabolised in the liver by CYP3A4 and CYP2D6 to several inactive metabolites. The involvement of these CYPs makes delavirdine subject to multiple drug interactions.
Many significant drug interactions have been reported with drugs inhibiting metabolism (e.g. erythromycin, ketoconazole) and inducing metabolism (e.g. phenytoin, carbamazepine). Check a current reference source for full information on potential drug interactions. A decrease in gastric acidity due to H2-receptor antagonists (cimetidine and others) or concurrent antacids will decrease absorption of delavirdine. Avoid chronic use of these medications and separate antacid dosing by at least 1 hour.
Adverse reactions include diarrhoea, weakness, headache, nausea, vomiting, severe skin rash, conjunctivitis, fever, joint and muscle aches, oral lesions in mouth and, rarely, dyspnoea. Use with caution in people with renal impairment, as the metabolites are eliminated via the kidneys. Avoid use in individuals with known hypersensitivity to delavirdine and in people with hepatic impairment. Limited data are available on use during pregnancy, and specialist advice should be sought. The usual adult dose is 400 mg every 8 hours.
Efavirenz
Peak plasma concentration of efavirenz occurs 3–5 hours after oral administration. Metabolism of the drug is complex, as it both induces its own metabolism and inhibits the metabolism of other drugs by the hepatic cytochrome P450 system. Involvement of CYP3A4 in the metabolism of efavirenz increases the likelihood of drug interactions. The half-life is 52–76 hours, and less than 1% appears in urine as unchanged drug.
Many drug interactions occur and current information sources should be consulted. Drugs that should not be administered concurrently with efavirenz include the benzodiazepines, midazolam and triazolam, and cisapride. Other potentially significant drug interactions occur with warfarin, alcohol and protease inhibitors.
Individuals commonly complain of dizziness, drowsiness, insomnia, headache and abnormal dreaming. Allergic-type reactions and cardiovascular effects such as palpitations and tachycardia have been reported. Use with extreme caution in people with renal or hepatic impairment. Efavirenz is contraindicated in pregnancy, as it is embryotoxic and teratogenic in animals. The adult dose (capsule) is 600 mg daily and 720 mg (24 mL) of the oral liquid once daily. Refer to the manufacturer’s literature for other dosing information.
Nevirapine
Administered orally, nevirapine is well absorbed. It reaches peak plasma concentration in 4 hours and is distributed in CSF (45% of plasma concentration). Nevirapine is metabolised in the liver and is also an inducer of hepatic CYPs; thus autoinduction, or an increased clearance and a decrease in drug half-life, occurs within 2–4 weeks of therapy.
The effects that can occur when nevirapine is given with other drugs are listed in Drug Interactions 45-4.
Drug interactions 45-4 Nevirapine
| Drug | Possible effects and management |
| Oral contraceptives containing oestrogen; protease inhibitors such as indinavir, ritonavir and saquinavir | Nevirapine increases CYP3A activity, which will decrease the plasma concentrations of the oral contraceptives and the protease inhibitors. Concurrent use is not recommended |
| Methadone | Increased metabolism can cause methadone withdrawal or ineffective pain relief. Adjust dose if required |
| Rifabutin or rifampicin | Nevirapine induction of CYP3A can decrease plasma concentrations of these drugs. Monitor closely if used concurrently |
Adverse reactions include nausea, headache, diarrhoea, fever, hepatitis, ulcerative stomatitis and life-threatening skin reactions such as Stevens–Johnson syndrome. Nevirapine should be discontinued in individuals who develop a severe rash or a rash accompanied by symptoms such as fever, myalgia, fatigue, oral lesions and conjunctivitis. The rash usually occurs in the first 6 weeks, and women are at greater risk. Use with caution in individuals with kidney function impairment. Avoid use in people with nevirapine hypersensitivity or liver function impairment.
Initial therapy is 200 mg orally daily for 2 weeks. The maintenance dose is 200 mg twice daily in combination with another antiretroviral agent.
Protease inhibitors
The HIV protease, which is essential for viral infectivity, cleaves the viral precursor polypeptide into active viral enzymes and structural proteins. The protease inhibitors (PIs) prevent the protease from cleaving the polypeptide and hence block the subsequent maturation of the virus (Figure 45-2). The PIs include amprenavir, atazanavir, darunavir, fosamprenavir, indinavir (see Drug Monograph 45-4), lopinavir with ritonavir, ritonavir, saquinavir and tipranavir. These drugs are the most potent antiviral agents available and have suppressed viral replication for up to a year in clinical trials. Administering them in combination therapies has decreased viral loads and increased CD4 counts. Various combinations of these agents and their effects on HIV infection and AIDS complications and disease progression are continually under investigation.
Adverse reactions are common and include headache, diarrhoea, nausea, vomiting, elevated liver enzymes and a variety of metabolic disorders (e.g. diabetes, hypertriglyceridaemia and hypercholesterolaemia). Like many of the other antiretroviral drugs, PIs are subject to multiple drug interactions and, in particular, their use is contraindicated with cisapride, ergot alkaloids, pimozide, midazolam and triazolam.
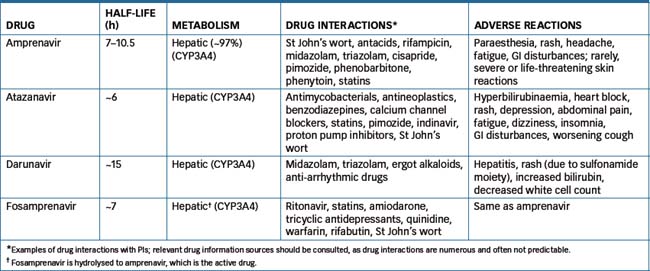
Comparative data for amprenavir, atazanavir, darunavir and fosamprenavir (the prodrug of amprenavir) are shown in Table 45-2.
Lopinavir with ritonavir
This drug is a combination of lopinavir, which is an inhibitor of HIV-1 and HIV-2 proteases, and ritonavir, which is also a PI but is in addition an inhibitor of the metabolism of lopinavir by CYP3A. The drug is formulated as soft capsules and as an oral liquid, and enhanced bioavailability occurs when both formulations are taken with food. Lopinavir is metabolised by hepatic CYP3A, and after multiple dosing <3% is excreted as unchanged drug in urine. Ritonavir is also metabolised by CYP3A and, although this was initially viewed as undesirable, it was later realised that this inhibitory action could be useful in allowing a lower dose of a PI to be given, as inhibition of metabolism by ritonavir would effectively increase the plasma concentration of the PI. This combination is now being investigated in numerous clinical trials.
Drug interactions are to some extent predictable, and include interactions with rifampicin, corticosteroids, statins, St John’s wort, azole antifungals, methadone, efavirenz and nevirapine. Adverse reactions include exacerbation of diabetes mellitus, increased bleeding, lipodystrophy (redistribution of fat including central obesity and ‘buffalo hump’), hyperlipidaemia and, rarely, pancreatitis.
Ritonavir
Administered orally, ritonavir reaches peak plasma concentration within 2 or 4 hours (fasting or non-fasting). Five metabolites have been identified with ritonavir, but only the M-2 metabolite has antiviral activity. It has a half-life of 3–5 hours and is excreted primarily in faeces.
Adverse reactions include weakness, nausea, vomiting, diarrhoea, stomach distress, allergic reactions, back or chest pain, chills, facial oedema, dizziness, headache, drowsiness, alterations in taste perception, numbness or tingling around mouth (circumoral paraesthesia), peripheral paraesthesia, flu symptoms and, rarely, diabetes, hyperglycaemia and ketoacidosis. Use with caution in patients with haemophilia and liver function impairment. Avoid use in people with ritonavir hypersensitivity. The usual dose is 600 mg twice a day, with meals. A lower starting dose (300 mg 12-hourly) may reduce the incidence of nausea, but the desirable dose of 600 mg 12-hourly should be achieved within 2 weeks.
Saquinavir
Administered orally, saquinavir has extensive first-pass metabolism, is highly protein-bound, is metabolised in the liver and is excreted primarily in the faeces.
Adverse reactions are usually mild and include diarrhoea, abdominal distress, headache, weakness and, rarely, paraesthesia, skin rash, confusion, ataxia, Stevens-Johnson syndrome, seizures, thrombocytopenia, anaemia and hepatotoxicity. Avoid use in people with saquinavir hypersensitivity. Dosage reduction may be required in individuals with hepatic dysfunction. The usual dose in combination with a nucleoside analogue (e.g. zalcitabine or zidovudine) is 1000 mg (with 100 mg ritonavir) twice a day, administered within 2 hours of a full meal.
Tipranavir
Tipranavir is a novel non-peptide PI that is used under specialist care in highly treatment-experienced people with HIV virus resistant to multiple PIs. Metabolised by CYP3A4, tipranavir is administered with low-dose ritonavir, which inhibits CYP3A and the intestinal P-gp efflux pump in order to achieve a therapeutic plasma concentration. Administration is contraindicated in the presence of moderate or severe hepatic impairment. Adverse effects include fatigue, myalgia and rash (due to the presence of a sulfonamide moiety).
Tipranavir (500 mg) is usually given twice daily with ritonavir (200 mg) with food.
Other antiretrovirals
Enfuvirtide
Enfuvirtide is a new and novel antiretroviral drug termed a fusion (HIV entry) inhibitor. It is a synthetic 36-amino-acid peptide that binds to the gp41 subunit of a glycoprotein found in the viral envelope. This blocks viral fusion with the CD4 receptor of the host cell and prevents the conformational changes required to allow fusion. Currently, this drug is indicated for HIV-1 infection in people where other treatments have failed.
The half-life is approximately 4 hours and pathways of elimination have not yet been determined. It is administered SC and, because it is a peptide, it is likely that it is catabolised to its constituent amino acids. Unlike many other antiretrovirals, enfuvirtide is not an inhibitor of CYP450 enzymes. Adverse reactions are common and include injection site reactions (e.g. pain, erythema, itch), which result in discontinuation in about 3% of people; peripheral neuropathy; insomnia; depression; respiratory symptoms (e.g. cough, dyspnoea) and loss of weight and appetite. The adult dose is 90 mg SC twice daily.
Maraviroc
It is essential for HIV to bind to the host cell in order to enter and then replicate and release new virions. Human CD4+ cells express chemokine receptors CCR5 and CXCR4. Maraviroc competitively and selectively binds to CCR5, blocking the interaction between the HIV glycoprotein gp120 and CCR5. gp120 is responsible for exposing coreceptor binding sites that then bind to CCR5 and CXCR4 on the membrane of the host cell. The drug is rapidly absorbed achieving maximum plasma concentrations in 1–4 hours. Like the majority of the PIs, maraviroc is also a substrate of CYP3A4 and P-gp and hence it is susceptible to multiple drug interactions.
Administered orally common adverse reactions include GI toxicity (e.g. abdominal pain, constipation), rash, bronchospasm, infections and neurological symptoms (e.g. myalgia, muscle spasms, paraesthesia, dysaesthesia and disturbed sleep).
Raltegravir
Raltegravir is an HIV integrase inhibitor that inhibits the strand transfer reaction, which is essential for integration of the viral DNA into the host DNA, and hence its viral expression and replication. It is indicated for treatment of multi-drug-resistant HIV infection in individuals whose current therapy is failing. Raltegravir is rapidly absorbed and is metabolised principally by UGT1A1 to raltegravir glucuronide, which is excreted in faeces (51%) and urine (32%). Clinical data are currently limited and specialist sources of drug information should be consulted. The usual dose is 400 mg twice daily swallowed whole.
Tenofovir
Tenofovir disoproxil fumarate is a prodrug that is converted to tenofovir, an analogue of adenosine. Similar to the NRTIs, tenofovir competitively inhibits the HIV reverse transcriptase and causes chain termination after its incorporation in DNA. Currently it is indicated for use in combination with other antiretrovirals. Administered orally, the bioavailability of tenofovir is enhanced if taken with a high-fat meal.
Similar to enfuvirtide, tenofovir is not an inhibitor of CYP and, as it is excreted predominantly by the kidneys (70%–80%), clinical CYP drug interactions are unlikely. However, tenofovir can cause nephrotoxicity, which may be exacerbated if administered concomitantly with other nephrotoxic drugs (e.g. lopinavir with ritonavir increases tenofovir concentration, increasing the risk of nephrotoxicity). Adverse reactions include GI disturbances, headache and hypophosphataemia. The usual adult dose is 300 mg daily of tenofovir disoproxil fumarate, which is equivalent to 136 mg of the active constituent tenofovir.
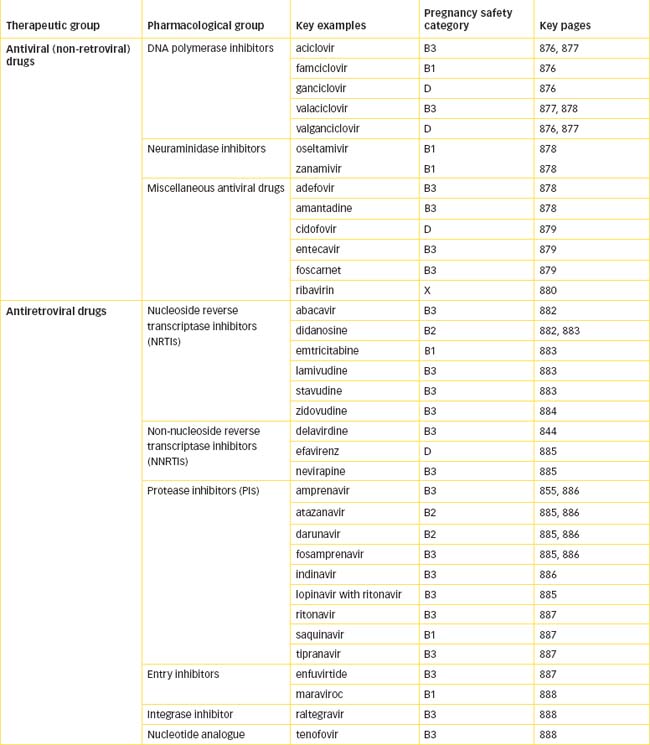
 The antifungal agents are potent and potentially toxic medications. They include amphotericin B, caspofungin, fluconazole, flucytosine, griseofulvin, itraconazole, ketoconazole, miconazole, nystatin, posaconazole, terbinafine and voriconazole. Drugs that are systemic fungistatic or fungicidal agents are used to treat a wide variety of mycotic infections. Chemotherapy for viral diseases has tended to be more limited than chemotherapy for bacterial diseases because the development and clinical application of antiviral drugs is more difficult. The antiviral and antiretroviral drugs discussed in this chapter include the guanine analogues (aciclovir, famciclovir, ganciclovir, valaciclovir, valganciclovir); the nucleoside analogue reverse transcriptase inhibitors (abacavir, didanosine, emtricitabine, lamivudine, stavudine and zidovudine); the non-nucleoside reverse transcriptase inhibitors (delavirdine, efavirenz and nevirapine); the protease inhibitors (amprenavir, atazanavir, durunavir, fosamprenavir, indinavir, lopinavir with ritonavir, ritonavir, saquinavir and tipranavir); the neuraminidase inhibitors (oseltamivir and zanamivir); and miscellaneous antiviral drugs (amantadine, cidofovir, foscarnet, palivizumab and ribavirin) and antiretrovirals (enfuvirtide, maraviroc, raltegravir and tenofovir). Many antiviral drugs are subject to multiple drug–drug interactions, and relevant sources should be consulted in relation to specific drugs.
The antifungal agents are potent and potentially toxic medications. They include amphotericin B, caspofungin, fluconazole, flucytosine, griseofulvin, itraconazole, ketoconazole, miconazole, nystatin, posaconazole, terbinafine and voriconazole. Drugs that are systemic fungistatic or fungicidal agents are used to treat a wide variety of mycotic infections. Chemotherapy for viral diseases has tended to be more limited than chemotherapy for bacterial diseases because the development and clinical application of antiviral drugs is more difficult. The antiviral and antiretroviral drugs discussed in this chapter include the guanine analogues (aciclovir, famciclovir, ganciclovir, valaciclovir, valganciclovir); the nucleoside analogue reverse transcriptase inhibitors (abacavir, didanosine, emtricitabine, lamivudine, stavudine and zidovudine); the non-nucleoside reverse transcriptase inhibitors (delavirdine, efavirenz and nevirapine); the protease inhibitors (amprenavir, atazanavir, durunavir, fosamprenavir, indinavir, lopinavir with ritonavir, ritonavir, saquinavir and tipranavir); the neuraminidase inhibitors (oseltamivir and zanamivir); and miscellaneous antiviral drugs (amantadine, cidofovir, foscarnet, palivizumab and ribavirin) and antiretrovirals (enfuvirtide, maraviroc, raltegravir and tenofovir). Many antiviral drugs are subject to multiple drug–drug interactions, and relevant sources should be consulted in relation to specific drugs.References and further reading
Australian Institute of Health and Welfare 2008. Australia’s Health 2008. Cat No AUS 99. Canberra: AIHW.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Bennett J.E. Antifungal agents. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 48]
Cada D.J., Levien T., Baker DE. Entecavir. Hospital Pharmacy. 2005;40:798-810.
De Clercq E. The design of drugs for HIV and HCV. Nature Reviews Drug Discovery. 2007;6:1001-1018.
Este J.A., Telenti A. HIV entry inhibitors. Lancetc. 2007;370:81-88.
Flexner C. Antiretroviral agents and treatment of HIV infection. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 50]
Hayden F.G. Antiviral agents (nonretroviral). In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 49]
Hovanessian H.C. New developments in the treatment of HIV disease: an overview. Annals of Emergency Medicine. 1999;33:546-555.
Kovarik J.M., Mueller E.A., Zehender H., et al. Multiple-dose pharmacokinetics and distribution in tissue of terbinafine and metabolites. Antimicrobial Agents and Chemotherapy. 1995;39:2738-2741.
Nabel G.J. Challenges and opportunities for development of an AIDS vaccine. Nature. 2001;410:1002-1007.
Robinson M Reining in influenza: the benefits of early treatment with oseltamivir. Australian Pharmacy Trade 2001. 19 April: 17–22.
Sheehan D.J., Hitchcock C.A., Sibley C.M. Current and emerging azole antifungal agents. Clinical Microbiology Reviews. 1999;12:40-79.
Vickers A.E.M., Sinclair J.R., Zollinger M., et al. Multiple cytochrome P-450s involved in the metabolism of terbinafine suggest a limited potential for drug–drug interactions. Drug Metabolism and Disposition. 1999;27:1029-1038.
Weatherall J., Kelleher A.D., Cooper D.A. Trials and tribulations of HIV vaccines. Future HIV Therapy. 2009;3:145-160.
Wong-Beringer A., Kriengkauykiat J. Systemic antifungal therapy: new options, new challenges. Pharmacotherapy. 2003;23:1441-1462.
Yost R., Pasquale T.R., Sahloff EG. Maraviroc a coreceptor CCR5 antagonist for management of HIV infection. American Journal of Health-System Pharmacy. 2009;66:715-726.