Chapter 47 Anti-inflammatory and Immunomodulating Drugs
The body’s resistance to disease is both non-specific (e.g. physical and chemical barriers that offer immediate protection, such as the skin, mucous membranes and gastric acidity) and specific (i.e. an acquired specific resistance or immunity that develops slowly). Exposure to a chemical, foreign body or microorganism may elicit an inflammatory response and provoke a specific immune response. In some individuals who are sensitised to a particular antigen, further exposure can precipitate an allergic reaction. In humans, the immune system consists of lymph, lymphatic vessels, lymph nodes, red bone marrow, the spleen, tonsils, the thymus gland and billions of circulating cells, mainly lymphocytes and antibody-secreting plasma cells. During a lifetime, an individual may acquire immune capabilities through both natural and artificial means.
This chapter reviews the uses of anti-inflammatory and immunomodulating drugs. These include the non-steroidal anti-inflammatory drugs (NSAIDs), which are among the most widely prescribed drugs in the world; disease-modifying antirheumatic drugs (DMARDs), used as first-line treatment for rheumatoid arthritis; drugs used to treat gout, a disorder of uric acid metabolism; immunosuppressant drugs, crucial in the treatment of multiple diseases and to the success of organ transplantation; and antihistamines, which are widely used for motion sickness, vertigo and skin and allergic disorders.
Key abbreviations
AIDS acquired immune deficiency syndrome
DMARDs disease-modifying antirheumatic drugs
HIV human immunodeficiency virus
Key background
THE immune system is composed of cells and organs that mount defensive responses against pathogens (e.g. microbes, toxins and chemicals) and cancer cells and in some cases, unfortunately, against normal body tissues (autoimmune diseases). The body’s resistance to disease is both non-specific (e.g. physical and chemical barriers that offer immediate protection, such as the skin and gastric acidity) and specific (i.e. an acquired specific resistance or immunity that develops more slowly). The lymphatic system is responsible for eliciting highly specific immune responses and comprises lymph, lymphatic vessels, lymph nodes, red bone marrow (see Chapter 26) and the thymus gland, spleen and tonsils. These organs and tissues are collectively responsible for the production and maturation of the immunocompetent cells and for facilitating the immune response. Figure 47-1 illustrates the organs and tissues of the immune system.
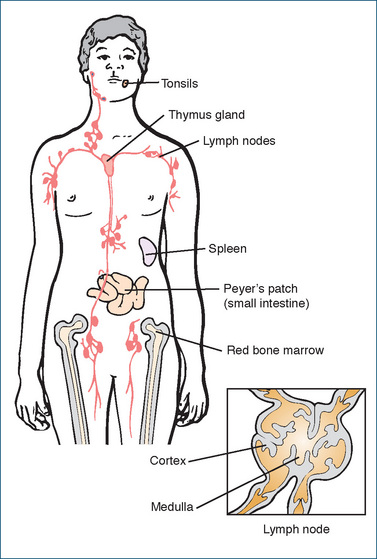
Figure 47-1 Location of organs and tissues of the immune system. The inset shows a cross-section of a lymph node.
Thymus gland
The thymus gland has two lobes and is located in the mediastinal area. The thymus gland is larger at birth than it is in an adult. By the time a person reaches puberty, the thymus has grown to nearly six times its original size. After puberty, this gland undergoes involution (a process of rolling inwards with a decrease in both cell size and organ size), and in the elderly it is usually a small mass (about 3 g) of reticular fibres with some lymphocytes and connective tissue. Although its importance has been largely discounted over the years, there are many unanswered questions about the thymus gland and its relation to the other tissues and organs in the immune system. The immune system develops when lymphoid stem cells produced in the red bone marrow migrate to the thymus, spleen and other lymphoid tissues and organs in the body. Thymic hormones produced in the thymus are thought to aid in the maturation of the prothymocytes, which ultimately develop into T lymphocytes (see following section on specific resistance).
Spleen
The spleen, the largest lymphatic organ in the body, is located on the left side of the body in the extreme superior, posterior corner of the abdominal cavity. Its main functions are to act as a storage site or reservoir for blood and a processing station for time-expired blood cells and platelets. Resident macrophages lining the white pulp (lymphatic tissue) and central arteries of the spleen remove pathogens from the blood, while the venous sinuses of the red pulp area of the spleen process worn-out blood cells and store platelets. The white pulp area of the spleen also contains lymphocytes and plasma cells that are involved in the immune process.
Tonsils
The tonsils are accumulations of lymphoid tissue (lymphatic nodules) named according to their location: lingual, palatine and pharyngeal tonsils. They intercept foreign substances that enter the body by way of the respiratory or gastrointestinal tracts. Similar lymphoid tissue located in the submucosal areas of the gastrointestinal (GI) tract (Peyer’s patches) mount immune responses against bacteria and viruses entering from the gut. Other lymphatic nodules are located in the urinary and reproductive tracts and help to intercept antigens in the blood and in the lymph nodes.
Lymph nodes
The lymph nodes are capsulated organs located throughout the body and are involved with lymph circulation. They are essentially a row of unidirectional in-line filters, which screen the lymph flowing through them. Foreign material is either engulfed by the resident macrophages or destroyed by immune responses elicited by the lymphocytes located throughout the lymph nodes. Lymph nodes are common secondary malignant tumour sites.
The outer portion of the lymph node is called the cortex and contains B lymphocytes (B cells), which become antibody-secreting plasma cells. The thymus-dependent zone exists in the deep area or inner cortex and contains mainly T lymphocytes (T cells), formed or seeded from the thymus gland, which, when exposed to an antigen, divide rapidly and produce large numbers of new T cells sensitised to that antigen. The inner portion is the medulla, which contains B lymphocytes and plasma cells.
Resistance to disease
Non-specific resistance—inflammation
The initial and immediate lines of defence for the body are the skin, the mucous membranes of the respiratory and gastrointestinal tracts and chemical secretions such as saliva and gastric acid. Penetration of these barriers by pathogens results in the mobilisation of internal defence mechanisms. These include natural killer (NK) cells, which are lymphocytes that are of neither B nor T cell type, and phagocytic neutrophils and macrophages. Phagocytosis involves several phases, including digestion and in some cases killing of the pathogen. If cells are damaged by bacteria or viruses, physical trauma (e.g. a cut), foreign bodies, chemical substances, surgery, radiation or electricity, the body will elicit an inflammatory response.
The four characteristic signs of inflammation are swelling (oedema), redness (erythema), pain and heat, which are accounted for by three basic events: (1) blood vessel vasodilation and increased capillary permeability, (2) cellular infiltration and (3) tissue repair. These processes involve a variety of chemical mediators that modify and contribute to the inflammatory response. After an injury occurs, for example, the body will release chemical substances such as histamine, prostaglandins, leukotrienes, kinins and complement into the tissue, forming a chemotactic gradient, and fluids and cells will begin to accumulate in the area. Blood vessels dilate (primarily because of the action of histamine and kinins) within 30 minutes of the insult, and this allows an increase in blood flow and exudation of fluid due to increased capillary permeability in the injured tissues. The exudate includes protein-rich fluids high in fibrinogen that will attract other substances to the area, such as complement, antibodies and leucocytes. Fluid collection in the area results in oedema, which generally occurs within 4 hours of the injury.
During the cellular phase, neutrophils and monocytes attracted by chemotactic agents such as kinins, leukotrienes and complement will migrate into the area from the dilated blood vessels. If the injury is due to a foreign substance or bacteria, the monocytes will transform into wandering macrophages, which are more powerful phagocytes than the neutrophils and engulf and destroy the foreign material (phagocytosis). The phagocytosis process tends to localise, or wall off, the foreign material to prevent its spread through the tissues. Large numbers of phagocytes can lead to pus accumulation and the eventual destruction and removal of the foreign material. The resulting debris is removed by the macrophages and neutrophils, thus resolving the inflammatory reaction.
Mediators of inflammation
The complement system is composed of plasma proteins (at least 18 distinct proteins and their cleavage products) present in the blood in the form of inactive proteases. Activation of the protein called complement 1 (C1) by proteolytic cleavage is the initial step in this cascading pathway that mediates destruction of invading pathogens.
The system is divided into two pathways: the ‘classical pathway’, which is activated by an antigen-antibody complex, and the ‘alternative pathway’, which is antibody-independent and involves protein factors B, D and P (properidin) and activation of the complement cascade at C3 (Figure 47-2). Complement is essential in the response to an acute inflammatory reaction caused by bacteria, some viruses and immune complex diseases. Complement enhances chemotaxis, increases blood vessel permeability and eventually causes cell lysis.
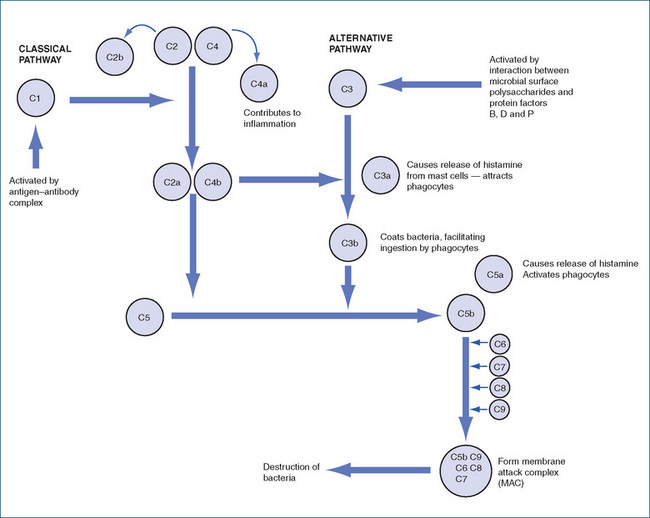
Figure 47-2 The complement system. Complement protein C1, activated by an antigen–antibody complex, initiates the classical pathway. The alternative pathway is initiated at C3 by an interaction between the microbe and factors B, D and P.
Histamine, a product of mast cells and basophils; prostaglandins generated from arachidonic acid; and cytokines released from inflammatory tissue are some of the mediators capable of producing local reactions, smooth muscle contraction, increased chemotaxis, blood vessel vasodilation and other inflammatory effects.
Specific resistance
Provocation of an immune response against specific pathogens or transplanted tissue is called specific resistance, or immunity. The primary types of immunity are cell-mediated immunity and humoral (antibody-mediated) immunity. The key features that differentiate immunity from non-specific resistance are the specificity of recognition of the antigen (e.g. pollen, bacteria and transplanted tissue) and memory evocation, i.e. a second encounter with the same antigen provokes an immediate and more substantial immune response. The part of an antigen that triggers an immune response, such as antibody production, or activates specific T cells is called the epitope. Sometimes the body’s ability to differentiate between true antigenic proteins and self-proteins fails and the immune system attacks the normal body cells, producing autoimmune disorders. The cells that carry out the immune responses are described as immunocompetent cells. T and B cells and the polymorphonuclear leucocytes (PMLs) are involved in the immune response, although only mononuclear T and B cells are immunocompetent cells. The PMLs are non-specific cells that interact with lymphocytes to produce an inflammatory response, whereas B and T cells are capable of recognising specific antigens and initiating the immune response.
Cell-mediated immunity
In humans, stem cells from the bone marrow are transformed into T cells in the thymus gland and B cells in red bone marrow (see Chapter 26). Before migrating to lymphoid tissue and organs, T and B cells acquire cell surface antigen receptors that are capable of recognising specific antigens. When in contact with an antigen, T cells proliferate to form specialised ‘killer’ T cells, thus providing cell-mediated immunity. The B cells transform into plasma cells, which form antibodies (immunoglobulins) that search out, identify and bind with specific antigens to provide humoral (antibody-mediated) immunity. T cells are generally long-lived. When they are not in their special areas, they circulate continuously through the body by way of the bloodstream and lymphatic system. When the T cells first recognise the foreign substance (antigen), they are stimulated and become activated. The activated lymphocytes then proliferate and differentiate into highly specialised cells that have the capacity to recognise and respond to the antigen. These specialised cells, or clones, constitute an identical population of lymphocytes that are capable of recognising one specific antigen. Three major groups of T cells identified are cytotoxic (killer) T cells, helper T cells and memory T cells.
Cytotoxic (killer) T cells
These cells display a protein called CD8+ on their membranes and can bind tightly to organisms or cells that contain their binding-specific antigen. Following stimulation by cytokines produced by helper T cells, the killer cells become cytolytic, i.e. capable of lysing (killing) cancer cells, tissue transplant cells and other cells that are foreign to the person’s body. Body tissue that contains viruses or foreign cells may also be attacked by the killer T cells.
Helper T cells
These make up the majority of the T cells that have the plasma membrane protein CD4+. They increase the activation of B cells, T cells and NK cells. Helper T cells are activated by antigen-presenting cells; once activated, they secrete various cytokines, which comprise a large group of molecules that are involved in signal pathways between cells during immune responses. These cytokines include interleukin-2 (IL-2, the major stimulus for T-cell proliferation), IL-4 and IL-5 (which stimulate B cells and cause plasma cells to secrete antibodies) and gamma interferon (which stimulates phagocytosis and enhances immune responses). Helper T cells also secrete macrophage migration inhibition factor, which slows or stops the migration of macrophages away from the affected area. The activated macrophages can attack and destroy a vastly increased number of the invading organism.
AIDS is the final outcome of an infection with HIV. This virus binds to protein on the cell membranes of the helper T cells (T4 cells) and, over the course of many years, the T4 cells are destroyed by the virus. This ultimately leads to the immunodeficiency syndrome known as AIDS (refer to Chapter 45).
Memory T cells
Cells remaining from a population of clones after a cell-mediated immune response are called memory T cells. The function of these cells is immediate recognition and vigorous handling of the same foreign antigen on presentation at a later date. In essence, they are the ‘memory’ of a previous response to a specific antigen.
Humoral (antibody-mediated) immunity
Humoral immunity is a response effective in the extracellular environment, in contrast to cell-mediated immunity, which deals with intracellular organisms. Humans possess B cells, which can produce an array of antibodies that recognise different antigens. Antibody response can be elicited by various antigenic proteins, such as the protein coat of a bacterium, or by intentional vaccination.
B lymphocytes
There are millions of B cells that reside in various parts of the body, e.g. lymph nodes, the spleen and GI tract. Following antigen recognition, B cells, assisted by helper T cells that secrete IL-2, become activated. Some of the activated B cells specific for the antigen will enlarge and differentiate to form plasmablasts, which are plasma cell precursors, and some will remain undifferentiated and become memory B cells (Figure 47-3). The plasma cells secrete an antibody specific to the antigen for around 4–5 days or until the plasma cell dies (primary response). Different antigens stimulate different B cells, and the respective clonal populations each produce only one type of antibody. Initially, the immunoglobulin is IgM (see following section), which increases in quantity for up to 2 weeks; production then declines so that very little IgM is present after a few weeks. After the initial IgM production, IgG antibodies start to appear at around day 10, peak in several weeks and maintain high levels for a much longer period.
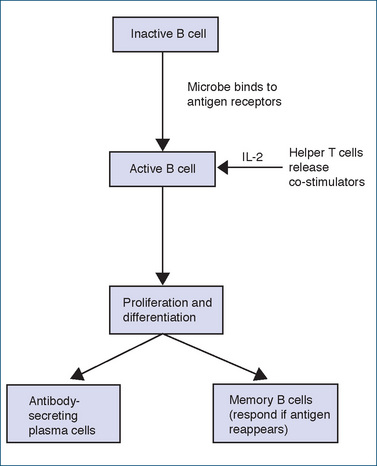
Figure 47-3 Schematic of B lymphocyte activation and differentiation into plasma cells (antibody-secreting) and memory B cells.
The first (primary) response to an antigen may be slow, weak and of short duration, but the memory B cells on second exposure to the same antigen will cause a more rapid and potent antibody response, and antibodies will be formed for months rather than for only a few weeks (secondary response). This is why vaccination using several doses given at periods of weeks or months apart is so effective.
Antibodies
Antibodies (immunoglobulins) are gamma globulins (a type of protein) that are specific for particular antigens. They are produced by lymphoid tissue in response to antigens and consist of four polypeptide chains: two heavy (H) and two light (L) chains that can form either a T or a Y shape. Differences in the constant region of the H chains provide the basis for the five classes of antibodies that have been identified: IgG, IgM, IgA, IgD and IgE. (‘Ig’ stands for immunoglobulin; the other letters designate the classes.)
IgG is the major immunoglobulin in the blood (about 75%–80% of the total antibodies in the normal person) and is capable of entering tissue spaces, coating microorganisms and activating the complement system, thus accelerating phagocytosis. It is the only immunoglobulin capable of crossing the placenta to provide the fetus with passive immunity until the infant can produce its own immune defence system.
IgM is the first immunoglobulin produced during an immune response. It is located primarily in the bloodstream and develops in response to an invasion of bacteria or viruses. IgM activates complement and can destroy foreign invaders during the initial antigen exposure. Its level decreases in about 2 weeks, while IgG levels are progressively increasing.
IgA is located primarily in external body secretions— saliva, sweat, tears, mucus, bile and colostrums—and it is found in respiratory tract mucosa and in plasma. It helps to provide a defence against antigens on exposed surfaces and antigens that enter the respiratory and gastrointestinal tracts. The plasma cells in the intestinal area secrete IgA and secretory component to defend the body against bacteria and viruses.
IgD is located in blood and on lymphocyte surfaces together with IgM. IgD is involved in activation of B cells. Levels are elevated in chronic infections.
IgE binds to histamine-containing mast cells and basophils. It is involved in allergic and hypersensitivity reactions and can mediate the release of histamine in immune response to parasites (helminths). Concentrations are low in the plasma because the antibody is firmly fixed on tissue surfaces. Once activated by an antigen, it will trigger the release of the mast cell granules, resulting in the signs and symptoms of allergy and anaphylaxis.
Allergic (hypersensitivity) reactions
Substances foreign to the body act as antigens to stimulate the production of antibodies or immunoglobulins (IgE, IgG, IgM). When a previously sensitised individual is again exposed to the foreign substance, the antigen reacts with the antibodies to release substances such as histamine, which then provoke allergic symptoms. There are four different types of allergic, or hypersensitivity, reactions.
Natural and acquired immunity
The body has certain inherited and innate abilities to resist antigens. This ability is known as natural resistance, or natural immunity, which is different from acquired immunity. Some general defences inherent in natural resistance come from factors familiar to the focus of health care: for example, adequate rest, nutrition, exercise and freedom from undue stress. Physiological factors, which discourage proliferation of microbes, include the acidity of gastric secretions, respiratory tract cilia and bactericidal lysozyme in tears. During a lifetime, an individual may also acquire further immune capabilities through both natural and artificial means. This type of acquired immunity is acquired either actively or passively (Figure 47-4).
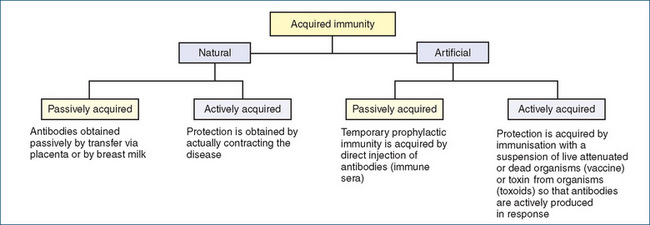
Antibodies activate cellular defences to engulf antigens. Custom-made immunoglobulins, or antibodies, provide acquired immunity to the specific type of antigen for varying lengths of time. Those antibodies gradually disappear from the plasma but the potential for their rapid replication in response to a repeat challenge by that specific antigen continues to exist after the initial exposure. The result, known as naturally acquired immunity, is a process of active immunity because of the body’s active involvement in creating the antibodies. Naturally acquired immunity can also result from a process of passive immunity, when antibodies made by the mother’s body are passively transferred by means of the placenta or by breast milk (especially colostrum, the breast milk produced shortly after delivery) to the fetus or infant.
On the other hand, artificial induction of the immune state, or artificially acquired immunity, is initiated purposefully for the protection of an individual (see Clinical Interest Box 47-1). It can also be induced either actively or passively. Artificially acquired active immunity is evoked by the deliberate administration of antigens, which may be live, partly modified organisms; killed organisms; or their toxins. The parenteral route is the predominant mode of administration. Periodic reactivation of actively acquired artificial immunity to certain organisms by booster doses (e.g. tetanus) is sometimes necessary. Artificially acquired passive immunity is conferred by the parenteral administration of antibody-containing immune plasma from immune humans or animals (Figure 47-4).
Clinical interest box 47-1 H1N1 infuenza
The 2009 pandemic ‘swine flu’ (2009 H1N1 virus) has a unique combination of genes from both North American and Eurasian swine lineages and this particular strain had not been identified previously in either pigs or humans. A free vaccine (produced by CSL Biotherapies) to protect, in the first instance, Australians from H1N1 influenza virus was registered by the Therapeutic Goods Administration on 18 September 2009 following a successful preliminary clinical trial. The vaccine is prepared in embryonated chicken eggs using the same procedure used for the preparation of seasonal influenza vaccines. The vaccine is a monovalent, unadjuvanted, inactivated, split-virus vaccine and is presented in multidose vials containing thimerosal as a preservative. An initial trial was conducted in 240 healthy non-pregnant adults (18–64 years) randomised to receive either 15 mcg or 30 mcg of haemagglutinin antigen in a 1:1 ratio. Three weeks after vaccination 96.7% of the subjects receiving the 15-mcg dose and 93.3% of those receiving the 30-mcg dose had antibody titres of 1:40 or more. No serious adverse reactions were reported but local injection site tenderness or pain and headache were reported in 46.3% and 45% of subjects, respectively (Greenberg et al 2009).
Artificially acquired active immunity generally secures protection for a longer duration than any kind of passive immunity, and is usually the prophylactic treatment of choice for populations at potential risk. Adverse effects include local pain at the injection site and headache with mild to moderate fever. Artificially acquired passive immunity is often chosen for susceptible individuals after a known exposure. A combination of active and passive approaches is also used occasionally. Various products used in artificial passive immunisation have caused adverse reactions because of individual hypersensitivities to animal products, especially horse plasma or eggs; to the preservative used in a medication; or to an antibiotic. The products of bacterial metabolism are the agents responsible for other adverse reactions. Table 47-1 compares the processes of active and passive immunity.
Table 47-1 Comparison of active and passive immunity
| Characteristic | Active immunity | Active immunity |
| Source | Individual | Other human or animal |
| Efficacy | High | Low to moderate |
| Method | Contracting disease Immunisation with vaccines or toxoids | Administer preformed antibody by injection, maternal transplacental ransfer or in breast milk |
| Time to develop | 5–21 days | Immediate effect |
| Duration | Long, up to years | Usually shorter than active immunity |
| Ease of reactivation | Easy with booster dose | Can be dangerous; anaphylaxis may occur, especially if animal sources are used |
| Purpose | Prophylaxis | Prophylaxis and therapeutic |
Non-Steroidal antiinflammatory drugs (NSAIDs)
Non-steroidal anti-inflammatory drugs (NSAIDs) are one of the most commonly administered groups of drugs worldwide, and new formulations and new NSAIDs continue to enter the market place. Generally, NSAIDs are either prescribed or purchased over the counter (OTC) for their analgesic, anti-inflammatory and antipyretic properties. In 2004–2005, 20% of Australians in the age group 45–54 years, 38.6% aged 55–64 years, 49% aged 65–74 years and 49.9% aged >75 years were reported to have chronic arthritis (Australia’s Health 2008).
Around 14 different NSAIDs are available in Australia and, although aspirin is also an NSAID, this term commonly refers to the aspirin-like substitutes on the market. Despite diversity in their chemical structures (Table 47-2), all the NSAIDs possess the same therapeutic properties—analgesic, antipyretic and anti-inflammatory effects. Unfortunately, they share to varying degrees the same adverse reactions. This is because inhibition of prostaglandin (PG) synthesis, which accounts for all the therapeutic effects, also causes renal and GI toxicity (Figure 47-5).
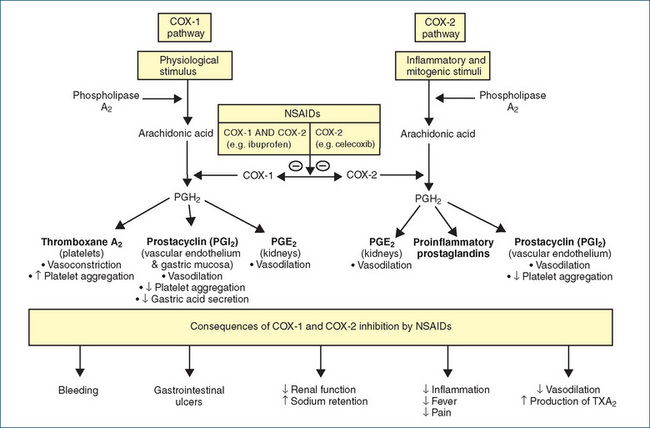
Figure 47-5 Sites of action of NSAIDs and synthesis of thromboxane A2, prostacyclin, prostaglandin E2 (PGE2) and proinflammatory prostaglandins from arachidonic acid, which is released from phospholipid membranes by the action of phospholipase A2. The cyclo-oxygenase isoforms (COX-1 and COX-2) then catalyse the metabolism of arachidonic acid to prostaglandin H2. Subsequent metabolism to the eicosanoids (e.g. thromboxane, and prostaglandins) differs in different cells.  = inhibition.
= inhibition.
Mechanism of action: inhibition of prostaglandin synthesis
Although aspirin was first introduced into clinical medicine in 1899, it was not until 1971 that Sir John Vane and his associates identified that the anti-inflammatory action of aspirin and indomethacin (introduced in 1965) was related to the inhibition of the enzyme cyclo-oxygenase (COX). COX catalyses the oxygenation of arachidonic acid, which is a 20-carbon fatty acid esterified to phospholipids of cell membranes. Arachidonic acid is released from the cell membrane by a variety of physical, chemical and hormonal stimuli through the action of acylhydrolases, principally phospholipase A2. Once released, arachidonic acid is metabolised principally to prostaglandins (PGs) and leukotrienes (LTs) (Figure 47-5). At the time, this single enzyme was thought to be responsible for the synthesis of all the PGs, which fall into several main classes, designated by letters and distinguished by substitutions on the cyclopentane ring (e.g. PGEb PGE2, PGI2) (Figure 47-5).
Prostaglandins are synthesised by most cells in the body and they bind to a number of PG receptors. With regard to PGE2 there are currently four subtypes of EP receptors, EP1-EP4. PGE2 is the major PG involved in inflammation and pain and contributes to inflammatory erythema and pain. It does not cause pain directly but appears to potentiate the pain induced by mediators such as bradykinin or histamine by sensitising nociceptors on sensory nerve terminals to painful stimulation. This property of hyperalgesia is also shared by PGI2. Both of these PGs are found in the synovial fluid of arthritic patients. PGE2 also causes fever. Following its release by inflammatory mediators from endothelial cells in blood vessels of the hypothalamus, PGE2 binds to EP3 receptors in a specialised region of the hypothalamus interfering with temperature control and thus producing a pyretic effect.
In the kidney PGE2 and PGI2 play an important role (especially in older people) in maintaining glomerular filtration, acting as vasodilators increasing renal blood flow, inhibiting sodium reabsorption and stimulating renin release. Within the GI tract PGE2, acting on EP3 receptors, and PGI2, acting on IP receptors, reduce the secretion of gastric acid and through a vasodilator action increase gastric mucosal blood flow. In addition, PGE2 stimulates production of a viscous mucous that plays a major role in protecting the gastric mucosa against gastric acid-induced damage.
It had been recognised in 1967 that the formation of ‘cytoprotective prostaglandins’ by the stomach and the intestine was necessary to maintain the integrity of the GI mucosa. With the exception of aspirin, which irreversibly acetylates the COX enzymes, the discovery that NSAIDs competitively inhibited PG synthesis established the link between the common occurrence of gastric ulcers and NSAID use. With the knowledge of a relationship between COX inhibition and GI toxicity, the following decades (1970s–1990s) saw the development of drugs intended specifically to have fewer GI and renal adverse effects. Unfortunately, the newer NSAIDs were not an advance in terms of toxicity, and the increasing popularity of these drugs was paralleled by increasing incidences of adverse reactions. Risks of serious GI tract reactions were shown to be very high with indomethacin; high with piroxicam, naproxen and most others, including aspirin; and lower with ibuprofen.
By 1990, a second COX enzyme (COX-2) had been identified. It was soon established that COX-1 (the original enzyme identified) is expressed in most tissues and, in particular, catalyses the synthesis of protective mucosal PGs in the GI tract and renal vasodilatory PGs. COX-2 is constitutively expressed in many tissues, e.g. brain, kidney, placenta, GIT (often at low levels), and is upregulated in cancer tissues and by inflammatory cytokines, laminar shear stress and various growth factors. As the older NSAIDs (e.g. the -profens) inhibit both COX-1 and COX-2, it was thought at that time that inhibition of COX-2 accounted for the anti-inflammatory actions of NSAIDs, whereas inhibition of COX-1 explained the GI and renal toxicity. This led to the search for drugs that would inhibit selectively COX-2. Celecoxib was the first COX-2 selective drug (coxib) released (in 1998), and was followed soon after by rofecoxib, which was marketed in 1999. Clinical trials of celecoxib and rofecoxib demonstrated some advantages over the older, non-selective NSAIDs in terms of GI tolerability but renal toxicity was still evident with the COX-2 inhibitors. An intense marketing campaign based on GI tolerability led to an unexpectedly high use of COX-2 inhibitors when they were made readily available in 2001 in Australia.
With the publication of the VIGOR (Vioxx Gastrointestinal Outcomes Research) study in 2000 (Bombardier et al 2000), warning bells were starting to ring about the risk of serious cardiovascular events with rofecoxib. In 2004, the US Food and Drug Administration (FDA) posted on its website an internal study of over 1.39 million patients who used rofecoxib, celecoxib or traditional NSAIDs. The FDA study found that, in comparison with other NSAIDs, rofecoxib increased the risk of heart attack and sudden cardiac death. In September 2004 it was recommended that the APPROVe trial be halted after 3 years because of the increased incidence of cardiovascular events with rofecoxib compared with the placebo group (Bresalier et al 2005). The increased CV events appear to relate to inhibition of endothelial cell prostacyclin production (which inhibits platelet aggregation) by COX-2 inhibitors, resulting in the unopposed actions of thromboxane A2, which in turn promotes platelet aggregation. On 28 September 2004, Merck voluntarily withdrew rofecoxib from the marketplace. In Australia in February 2005, a ‘black box’ warning was placed on the product information for celecoxib. A number of other coxibs have either not been approved for use or have been withdrawn. With the exception of celecoxib and parecoxib, which selectively inhibit COX-2, and meloxicam, which inhibits COX-2 at normal doses and at higher doses also COX-1, all of the other NSAIDs in current clinical use inhibit both COX-1and COX-2.
In addition to inhibition of COX, the non-selective NSAIDs also reduce synthesis of superoxide radicals, inhibit expression of adhesion molecules, reduce the activity of nitric oxide synthase, induce apoptosis, modify lymphocyte activity and cell membrane function and decrease activity of proinflammatory cytokines. All of these actions may contribute variously to the anti-inflammatory action of NSAIDs but this remains to be established.
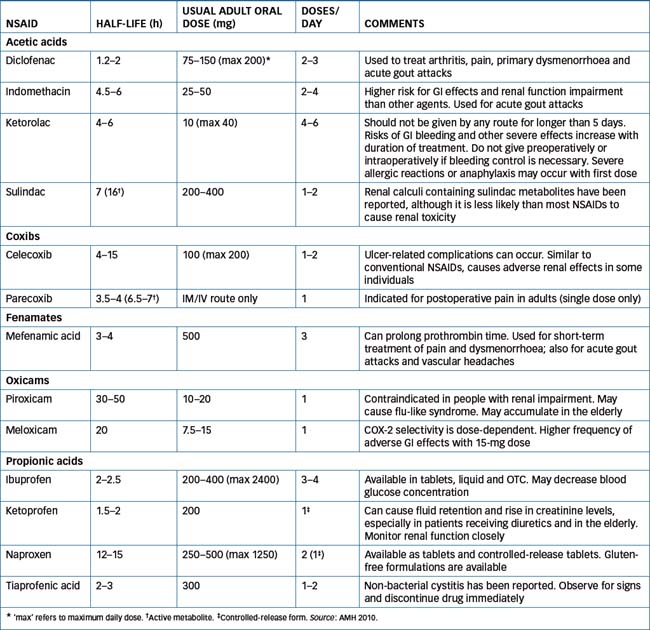
In general, by interfering with PG synthesis, NSAIDs tend to reduce the inflammatory process and ultimately provide pain relief. Hence NSAIDs are indicated for the treatment of acute or chronic rheumatoid arthritis, osteoarthritis, ankylosing spondylitis and other rheumatic diseases; mild to moderate pain, especially when the anti-inflammatory effect is also desirable (e.g. after dental procedures, obstetric and orthopaedic surgery and soft-tissue athletic injuries); gout; fever; non-rheumatic inflammation; and dysmenorrhoea. The choice between the various drugs in the group comes down to a clinical decision based on pharmacokinetic properties (especially short versus long half-life) and pharmacodynamic properties. Table 47-3 lists most of the commonly used NSAIDs, grouped together under chemical class where possible, and shows important properties. Some NSAIDs are available over the counter, as are aspirin and paracetamol. The differences between the prescription and OTC NSAIDs are usually in the strengths of the products and the indications for which they are recommended. Prescription strengths of naproxen (base), for example, for treatment of inflammatory pain are 250-mg and 500-mg tablets and controlled-release tablets of 750-mg and 1000-mg. In contrast, the OTC product specifically for dysmenorrhoea is marketed as 275-mg tablets.
Pharmacokinetics
For specific NSAID pharmacokinetics, usual adult dose and comments see Table 47-3. Oral absorption of these drugs is very good. Food may delay absorption but it has not been proven to significantly change the total amount of drug absorbed. Protein binding is high (greater than 90%). Most of these drugs are metabolised to varying degrees by the liver and excreted by the kidneys. Sulindac is inactive (a prodrug) until converted by the liver to an active sulfide metabolite. Similarly, parecoxib is converted to the active metabolite valdecoxib.
Adverse reactions
Although NSAIDs are commonly used and readily available, there has been a long history of adverse reactions. Indeed, it has been said that if aspirin had not been discovered until after the thalidomide disaster in the 1960s, when controls on drug testing were tightened, it would never have been approved for marketing. NSAIDs are responsible for almost one-quarter of all adverse drug reactions officially reported in the UK, and feature worldwide in reports of drug-related deaths.
Gastrointestinal
Adverse reactions of greatest concern are gastrointestinal (gastric pain, distress and/or ulceration, GI bleeding and perforation) and renal (nephrotoxicity, dysuria and haematuria). The risk of GIT effects appears to be higher with ketoprofen and piroxicam, and lower with ibuprofen and diclofenac. The beneficial effect of the latter two drugs is lost when the dose is increased. Virtually every person taking NSAIDs has some gastric damage, which may be unnoticeable or can develop into frank ulceration and haemorrhage. In attempts to lessen the risk, NSAIDs, including aspirin, have been formulated in enteric-coated forms to minimise the presence of drug in the stomach. This has been unsuccessful, however, because the effect is not only a local one but occurs also due to suppression of mucoprotective PGs by systemically absorbed NSAIDs. Treatment of NSAID-induced peptic ulcers is with misoprostol (a PG analogue) and proton pump inhibitors (see Chapter 30). The incidence of gastroduodenal ulceration appears to be lower with the COX-2-selective drugs, but ulcer healing rates may be impaired in individuals with pre-existing ulceration.
Renal
Adverse renal effects of NSAIDs occur consistently in a small percentage (1%–5%) of patients. The most common adverse effects include fluid retention, electrolyte disturbances (e.g. hyponatraemia, hyperkalaemia) and an increase in blood pressure. Additionally, as a consequence of fluid retention NSAIDs may worsen congestive heart failure and, in a small percentage (<1%) of at-risk patients (e.g. the elderly or those with volume depletion, renal insufficiency, heart failure, or diabetes), NSAIDs cause acute renal failure due to inhibition of synthesis of vasodilator PGs. Rarely, NSAIDs can cause acute tubulointerstitial nephritis or acute papillary necrosis. The adverse renal effects appear to be comparable for the selective and non-selective NSAIDs. Importantly, NSAIDs have been implicated in the ‘triple whammy’ (refer to Clinical Interest Box 23-4), a combination of an ACE inhibitor/angiotensin receptor antagonist, a diuretic and an NSAID that results in an adverse renal outcome.
Cardiovascular toxicity
Cardiovascular toxicity has been reported with both non-selective NSAIDs and COX-2 inhibitors (see Clinical Interest Box 47-2). Unlike aspirin, the COX-2 inhibitors do not reduce platelet aggregation, and in many studies prothrombotic activity manifests, leading to a higher risk of myocardial infarction and stroke.
Clinical interest box 47-2 NSAIDs and CV toxicity
Evidence of cardiovascular (CV) toxicity led to the withdrawal of rofecoxib (Vioxx) on 28 September 2004. Estimates of the relative risk of myocardial infarction while taking rofecoxib vary but, in general, meta-analyses suggest a near doubling of the risk with about 1 cardiovascular death per 1000 persons treated in 1 year. Approximately 80 million people worldwide took rofecoxib and the meta-analyses would suggest about 80,000 excess deaths. Further studies suggest that increased CV effects are not idiosyncratic and are shared with other NSAIDs including the non-selective NSAIDs. For example, high doses of ibuprofen and diclofenac, but not naproxen, have been found to be associated with excess risk, similar to that of coxibs (Patrono 2009). The mechanism has not been fully elucidated and a simplistic explanation involves an imbalance between the synthesis of thromboxane and prostacyclin, inhibition of the latter by COX-2 inhibitors predisposing to heart attack and stroke.
The availability of COX-2 inhibitors differs between Australia (only celecoxib and parecoxib are marketed) and New Zealand. In Australia, the Therapeutic Goods Administration (TGA) advised health professionals (doctors and pharmacists) in February 2005 that any individual taking more than 200 mg a day of celecoxib or more than 15 mg a day of meloxicam should have their drug therapy reviewed. Although the magnitude of the CV risk and the exact duration of therapy associated with increased risk are still unknown, it was recommended that COX-2 inhibitors be prescribed only when other treatments could not be tolerated or had caused serious adverse effects. It is currently recommended that celecoxib and meloxicam not be prescribed for those individuals with increased risks of CV events, such as heart attacks. In addition, treatment should be limited to the shortest possible time, and the dose during long-term therapy of celcoxib should not exceed 200 mg daily. Similar recommendations have been made by Medsafe in New Zealand (see Clinical Interest Box 47-3). The European Medicines Agency (EMEA) recommends the lowest possible dose of COX-2 inhibitors for the shortest duration and use is contraindicated in the presence of established ischaemic heart disease or stroke and in peripheral arterial disease. Caution should be exercised in people with increased cardiovascular risk factors (e.g.hypertension, hyperlipidaemia, diabetes and smoking) (EMEA 2005).
Clinical interest box 47-3 Cox-2 inhibitors in New Zealand
Medsafe has concluded the updating of the data sheets for the following COX-2 inhibitors: Arcoxia (etoricoxib), Celebrex (celecoxib), Dynastat (parecoxib), Mobic (meloxicam) and Prexige (lumiracoxib). These changes were recommended by the Medicines Adverse Reactions Committee (MARC), and are intended to best manage the cardiovascular risks of the COX-2 inhibitors and to identify those select patients in whom appropriate use may be warranted.
Key points for prescribers are:
Prescribers are additionally encouraged to continue reporting any suspect adverse reaction of clinical concern relating to the use of COX-2 inhibitors. These reports will assist Medsafe and MARC in the ongoing safety monitoring of these medicines.
Source: http://www.medsafe.govt.nz/profs/PUarticles/watchingbriefsDec05.htm#Update [11 September 2009].
Other adverse reactions
Other common adverse reactions to NSAIDs include skin reactions (rashes, urticaria). All NSAIDs, especially aspirin, can precipitate asthma attacks in sensitive persons, and in some individual NSAIDs may cause specific adverse reactions, e.g. salicylates generally can cause tinnitus, impaired haemostasis and acid-base imbalances.
Use NSAIDs with caution in the elderly, in debilitated patients and in people with compromised cardiac function and/or hypertension. Avoid use in persons with a history of hypersensitivity or a severe allergic reaction to aspirin (or to other NSAIDs), asthma, severe renal or liver disease, active ulcer disease or GI bleeding.
Drug interactions with NSAIDs
In view of the widespread use of NSAIDs, both prescribed and OTC preparations, it is important to know the most common and serious drug interactions that can occur with this drug group. Generally speaking, NSAIDs have the potential to interact with other drugs that affect inflammatory responses, kidney or liver function, blood pressure, blood coagulation mechanisms, acid-base balance and hearing. Some important interactions are detailed in Drug Interactions 47-1.
| Drug | Possible effects and management |
| Antihypertensives, diuretics | Monitor blood pressure closely whenever an NSAID is used concurrently, as reduction in renal function may reduce the antihypertensive effect |
| Cyclosporin and other nephrotoxic drugs | Concurrent use with NSAIDs may result in higher plasma concentration of cyclosporin, resulting in an increased potential for nephrotoxicity. Concurrent use of nephrotoxic drugs and NSAIDs may also increase the risk for nephrotoxicity. Monitor closely during concurrent drug use |
| Lithium | NSAIDs may decrease excretion of lithium, which may result in higher plasma lithium concentration and toxicity. Monitor lithium plasma concentration and clinical symptoms |
| Methotrexate | Concurrent use of methotrexate with low to moderate doses of an NSAID may result in methotrexate toxicity due to reduced renal excretion of methotrexate. Avoid combination |
| Probenecid | May result in higher plasma concentration of the NSAIDs and increased risk of toxicity. Concurrent use with ketoprofen is not recommended. If probenecid is given with an NSAID, monitor closely, as a decrease in NSAID dosage may be indicated |
| Warfarin | May increase the risk of GI ulcers or haemorrhage. Monitor closely for signs of these effects. Warfarin may be displaced from protein-binding sites, resulting in a higher risk of bleeding episodes. Monitor international normalised ratio closely. Platelet inhibition may be dangerous in patients receiving anticoagulant or thrombolytic agents. Avoid concurrent drug administration if possible |
| Zidovudine | NSAIDs may reduce zidovudine elimination, increasing the risk of toxicity. Avoid combination or monitor for signs of toxicity, as dose reduction of zidovudine may be necessary |
Disease-Modifying antirheumatic drugs (DMARDS)
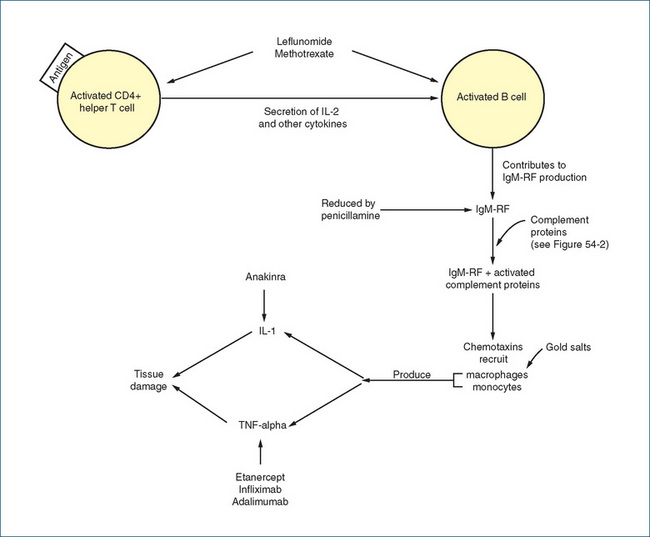
Rheumatoid arthritis affects millions of people worldwide, and women are three times more likely to have the disease than men. The onset and clinical course of rheumatoid arthritis varies and the incidence increases with age. The traditional approach for many years involved the prescribing of NSAIDs as first-line therapy but this option has been superseded, and disease-modifying antirheumatic drugs (DMARDs) are now prescribed much earlier. Use of DMARDs has substantially improved the control of rheumatoid arthritis and the long-term quality of life of the individual. In many instances, combinations of DMARDs may be used.
DMARDs comprise a group of drugs with diverse chemical structures, and their mechanisms of action in many cases are largely unknown. The main antirheumatic drugs are:
Gold salts
Gold as a pharmaceutical agent has been in use for centuries to relieve ‘the itching palm’. In more recent times, formulations in which gold is attached to sulfur (to increase solubility) have been used in the treatment of rheumatoid arthritis. Although the exact anti-inflammatory mechanism of action is unknown, these drugs appear to suppress the synovitis of the acute stage of rheumatoid disease. Proposed mechanisms of action include inhibition of sulfhydryl systems and various enzyme systems, suppression of phagocytic action of macrophages and leucocytes, and alteration of the immune response. Gold products (auranofin and aurothiomalate) are indicated for the treatment of rheumatoid arthritis; aurothiomalate is also used to treat juvenile arthritis.
The onset of action after oral administration of auranofin occurs in 3–4 months, while for parenteral aurothiomalate it is 6–8 weeks. The plasma half-life of the gold in these preparations is highly variable and may be as short as 1 week with a low dose, increasing to weeks and months with chronic therapy. In some people, gold can still be found in the liver and skin years after therapy has ceased. Auranofin is predominantly excreted in faeces, whereas aurothiomalate is mainly excreted by the kidneys, with a low percentage (10%-40%) found in faeces. These drugs are used with caution in people with inflammatory bowel disease, skin rash, diabetes or heart failure. In individuals with known gold drug hypersensitivity, blood dyscrasias, severe haematological disease, a history of bone marrow toxicity, renal or hepatic impairment, chronic skin disorders or systemic lupus erythematosus, these drugs should be avoided. Concurrent use of gold compounds with penicillamine (see below) will increase the risk of serious blood dyscrasias and/or renal toxicity.
It has long been recognised that efficacy and toxicity are positively related when it comes to the use of gold compounds. Not surprisingly, a significant number of individuals manifest adverse drug reactions, most commonly involving the skin (allergic reactions) and mucous membranes (sore, irritated tongue or gums; mouth ulcers or fungal infections). For auranofin, additional adverse drug reactions include abdominal distress or pain, gas, diarrhoea, nausea and vomiting. Those particular effects are less common with aurothiomalate, which instead may cause flushing, fainting, dizziness, palpitations and dyspnoea.
Penicillamine
Penicillamine is a chelating agent for heavy metals, such as mercury, lead, copper and iron. Knowledge of its metal-chelating properties has led to its use in Wilson’s disease (characterised by an excess of copper) and heavy-metal intoxications. After chelation by penicillamine, the metals are made more soluble so that they can be readily excreted by the kidneys. The mechanism of action of penicillamine as an antirheumatic agent is unknown, although lymphocyte function is improved and levels of IgM rheumatoid factor and immune complexes located in blood and synovial fluids are reduced. The relationship of these effects to rheumatoid arthritis is unknown.
Penicillamine is indicated for the prophylaxis and treatment of Wilson’s disease and for treating rheumatoid arthritis (especially for patients with moderate to severe arthritis who have not responded to other therapies), juvenile chronic arthritis and cystinuria. The onset of action in Wilson’s disease is 1–3 months, and in rheumatoid arthritis 2–3 months. Penicillamine is metabolised extensively in the liver, and the metabolites are excreted in both urine and faeces.
Penicillamine may impair renal and haematological function; hence its concurrent use with gold compounds or phenylbutazone may result in serious blood dyscrasias and/or renal toxicity. Also, use in patients with renal impairment or blood dyscrasias should be avoided. Adverse reactions include anorexia, diarrhoea, loss of taste senses, nausea, vomiting, abdominal pain, allergic reactions and stomatitis. Use penicillamine with caution in people with Goodpasture’s syndrome or myasthenia gravis, and avoid use in pregnancy, with the exception of patients with Wilson’s disease and certain patients with cystinuria. Women receiving penicillamine should not breastfeed.
Sulfasalazine
The use of gold salts and penicillamine is limited by their toxicity, and sulfasalazine may be chosen as a first-line drug. Sulfasalazine consists of the sulfonamide antibiotic sulfapyridine linked to the anti-inflammatory salicylate mesalazine. Sulfasalazine is poorly absorbed, and in the colon it is split by bacteria into sulfapyridine and mesalazine, which is the active component. This drug is indicated for treating rheumatoid arthritis and is also used in treating inflammatory bowel disorders (see Chapter 30).
Most adverse reactions are dose-dependent and related to the drug being a sulfonamide. Common adverse reactions include nausea, anorexia, rashes, tinnitus, dizziness and headache. Of a more serious nature are the haematological effects, which include haemolytic anaemia, agranulocytosis and thrombocytopenia. Sulfasalazine is contraindicated in people with haematological disorders or with a known sensitivity to sulfonamide derivatives. Close monitoring is required in patients with renal or hepatic impairment.
Hydroxychloroquine
The quinoline drugs, which included chloroquine and hydroxychloroquine, were originally developed as antimalarial drugs during World War II. Although primarily used as an antimalarial (see Chapter 46), hydroxychloroquine also possesses anti-inflammatory activity and is used for the treatment of mild rheumatoid arthritis. The mechanism of action is not well understood but hydroxychloroquine inhibits the release of lysosomal enzymes, PML chemotaxis, IL-1 release and the action of phospholipase A2 thus decreasing the formation of inflammatory mediators (see Figure 47-5). The onset of action is delayed; it takes several weeks to a month or more before a reduction in joint swelling is observed. Hydroxychloroquine is considered to be less effective than other DMARDs but is better tolerated, with a lower incidence of toxicity.
People commonly experience nausea, diarrhoea, abdominal cramps and anorexia. Hydroxychloroquine has a lower incidence of ocular toxicity (retinopathy) than the older drug chloroquine but can cause severe haematological reactions, including agranulocytosis, aplastic anaemia and thrombocytopenia. In people with haematological disorders hydroxychloroquine may cause further myelo-suppression and exacerbate porphyria; in addition, it may exacerbate the symptoms of myasthenia gravis and psoriasis, and is not used in pregnant women because of the risk of neurological disturbances in the fetus.
TNF-α antagonists
Tumour necrosis factor alpha (TNF-α), produced by macrophages, is a cytokine that plays an important role in normal inflammatory and immune responses. It is also a proinflammatory mediator that stimulates accumulation of neutrophils and macrophages at sites of inflammation, induces vascular adhesion molecules and matrix metalloproteinases, causes an acute phase response and stimulates macrophages to produce IL-1, which is a co-stimulator of T and B cell proliferation. TNF-α binds to two types of receptors, the type 1 TNF receptor (TFNRI) and the type 2 TNF receptor (TNFRII). It clearly plays a complex role in rheumatoid arthritis and current strategies have been to develop drugs that inhibit TNF-α production or release, to neutralise TNF-α on the cell surface or to block the TNF-α receptor or its downstream signal transduction pathway.
Adalimumab and infliximab
Adalimumab, a relatively new drug, is a recombinant human monoclonal antibody that, like infliximab, binds to TNF-α with high affinity thus impairing binding to its receptors. This results in lysis of cells expressing TNF-α receptors on their surface.
Contraindications for use are similar to those of etanercept and infliximab. It is administered SC (40 mg) once every 2 weeks. In view of the predisposition to serious infection with this drug, including tuberculosis and other opportunistic infections, signs of infection such as persistent fever should be reported immediately to the treating health professional (see Clinical Interest Box 47-4).
Clinical interest box 47-4 Infections and biological agents
Tumour necrosis factor alpha (TNF-α) is important for immune responses and host defence. Not surprisingly, the use of TNF-α antagonists predisposes patients to a range of infections, especially in the first 2 years of anti-TNF therapy, with a reduction of risk in subsequent years (Haroon & Inman 2009). The majority of infections are minor but there is concern that with the disruption of granuloma formation there is an increased risk of tuberculosis (TB). The American College of Rheumatology guidelines published in 2008 on the use of biologic agents recommended mandatory TB screening before starting therapy. In patients with a strong history suggestive of latent TB a chest X-ray and TB skin test should be performed. A positive finding would necessitate treatment of TB prior to use of anti-TNF agents. In addition, the guidelines suggest avoiding biologic agents in chronic hepatitis B and C if there is clinical evidence of liver pathology, in patients with URTI and fever, non-healed skin ulcers and life-threatening fungal infections or active viral infection with herpes zoster (Saag et al 2008; Furst et al 2008).
Infliximab is an antibody against TNF-α and comprises the antigen-binding region of the mouse antibody and the constant region of human IgG1 (Figure 47-6). It binds to soluble and membrane-bound TNF-α, preventing TNF-α from binding to its receptor and hence initiating inflammatory cell actions in chronic conditions such as rheumatoid arthritis. In addition to rheumatoid arthritis, infliximab is indicated for the treatment of Crohn’s disease and ankylosing spondylitis. Like etanercept, infliximab should be used with great caution as it may reactivate latent tuberculosis, worsen heart failure, exacerbate or induce a lupus-like syndrome and worsen multiple sclerosis. It is administered IV at a dose of 3 mg/kg, repeated every 2–6 weeks and then at 8-week intervals. Common adverse reactions include but are not limited to abdominal pain, cough, dizziness, headache, itching, fatigue and nausea.
Etanercept
Etanercept is a bioengineered fusion protein comprising two tumour necrosis factor (TNF) receptors coupled with a portion of human IgG, which binds to TNF and blocks its activity (Figure 47-6).
Etanercept has been shown to be effective when used as monotherapy in rheumatoid arthritis but has the disadvantage that it is administered subcutaneously twice a week. It is absorbed slowly, and peak plasma concentrations occur at approximately 50 hours; the half-life is in the order of 4–5 days. It is administered as 25 mg twice a week, but there is some evidence that 50 mg SC once a week may be as effective. In view of suppression of the activity of TNF-α, oral vaccines (e.g. polio vaccine) should not be administered to people receiving etanercept, and medical advice should be sought if a person is exposed to chickenpox or shingles during therapy. Contraindications to its use include hypersensitivity to etanercept and sepsis. Adverse reactions have included fatal pancytopenia and aplastic anaemia.
Cytokine modulators
Abatacept
Abatacept is an engineered drug comprising the extracellular portion of CTL-4 conjugated to the Fc portion of IgG1. It binds to CD80 and CD86 on antigen-presenting cells, which modulates a key co-stimulatory signal required for activation of T lymphocytes expressing CD28. This results in a reduction in cytokine synthesis and inflammation. It is currently indicated for use in moderate-tosevere rheumatoid arthritis with methotrexate in people who have an inadequate clinical response to methotrexate or TNF-α antagonists. Given as an IV infusion at 0, 2 and 4 weeks then at 4-week intervals, the most significant adverse effects are infusion-related reactions (e.g. headache, dizziness and hypertension usually within 1 hour of start of infusion) and infections. The latter precludes use in serious infections including active tuberculosis.
Anakinra
Multiple mediators are involved in inflammatory processes, and one such mediator is interleukin-1 (IL-1). IL-1 is produced in a variety of cells, including monocytes, macrophages and specialised cells in the synovial lining of joints. It is a proinflammatory cytokine, and suppression of its activity by antagonists that bind to the IL-1 receptor tends to reduce the inflammatory response.
Anakinra is a recombinant form of the endogenous IL-1 receptor antagonist and inhibits the action of IL-1 by competitively blocking the binding of IL-1 to IL-1 receptors on IL-1 responsive target cells. Due to its relatively short half-life (3–6 hours), it is administered daily (at the same time) by the SC route. Contraindications, adverse reactions and predisposition to infection are similar to those of the other cytokine blockers. As there are no data, the use of anakinra should be avoided in pregnancy.
Rituximab
Rituximab is a genetically engineered chimeric (mouse–human) IgG(1)-kappa monoclonal antibody that targets the CD20 antigen found on the surface of malignant and normal B lymphocytes. It depletes CD20+ B cells through a combination of complement-dependent cytotoxicity, antibody-dependent cellular cytotoxicity and induction of apopotosis. As a consequence of B-cell depletion, antibody production, cytokine networks, B-cell-mediated antigen presentation and activation of T-cells and macrophages are all affected. Rituximab causes lymphopenia in most patients, typically lasting 6 months, and a full recovery of B lymphocytes in the peripheral blood is usually seen 9–12 months after therapy, as CD20 is not expressed on haematopoietic stem cells. It is indicated for treatment of severe rheumatoid arthritis in individuals refractory to or intolerant of TNF-α antagonists. Given as an IV infusion (1 g for 2 doses 2 weeks apart) the most significant adverse effect is infusion-related reactions (e.g. headache, back pain, limb pain, heat sensations, pruritus and rash) and, rarely, anaphylaxis. The severity and frequency of infusion-related reactions can be controlled by the administration of paracetamol and an antihistamine 30–60 minutes prior to the infusion and IV methylprednisolone (100 mg) 30 minutes before the infusion (AMH 2010).
Drugs used for the treatment of gout
Gout is a disease associated with a genetically determined error of uric acid metabolism that increases its production. The hallmark of gout is hyperuricaemia, or high concentration of uric acid in the blood. It predominantly affects men in Australasian society, and the onset of gout is usually during middle age. In some people, hyperuricaemia may occur as a result of under-excretion by the kidneys. Recurrent gouty arthritis is painful, and uric acid crystal deposits can occur throughout the body, including the kidneys, which results in an inflammatory response.
Gout is characterised by defective purine metabolism and manifests itself by attacks of acute pain, swelling and tenderness of joints, such as those of the big toe, ankle, instep, knee and elbow. The amount of uric acid in the blood becomes elevated and tophi (deposits of uric acid or urates) form in the cartilage of various parts of the body. These deposits tend to grow in size. Chronic arthritis, nephritis, and premature sclerosis of blood vessels may develop if gout is uncontrolled.
Before treatment, causes of hyperuricaemia should be excluded. These include secondary hyperuricaemia as a result of increased cell breakdown in neoplastic diseases or cancer, psoriasis, Paget’s disease or renal disease. Lifestyle factors that may also cause hyperuricaemia include obesity, hypertension, excess alcohol consumption and lead exposure. Many drugs have also been reported to increase uric acid levels, including cancer chemotherapeutic drugs and thiazide diuretics. Asymptomatic hyperuricaemia in an elderly person may or may not be drug-induced and often is not treated because of potential adverse drug reactions.
Treatment goals for gout include:
Management strategies include the initial use of specific drugs for the acute attack, lifestyle modifications and drugs for preventing recurrent gout. Drugs used to treat an acute gout attack include NSAIDs, intra-articular corticosteroids and, when indicated, colchicine. NSAIDs are primarily used to treat the acute inflammation and associated pain (all appear to be equally effective) but they have no effect on the underlying metabolic problem. They are often prescribed to relieve an acute gout attack, while colchicine, because of its potentially toxic effects, is reserved for people who are not responsive to these agents or those who cannot tolerate them.
To treat chronic gouty arthritis or to prevent recurrent gout attacks, allopurinol (Drug Monograph 47-1) and probenecid are used (Figure 47-7).
Drug monograph 47-1 Allopurinol
Allopurinol decreases the production of uric acid by inhibiting xanthine oxidase, the enzyme necessary to convert hypoxanthine to xanthine, and xanthine to uric acid (Figure 47-7). It also increases the reuse of both hypoxanthine and xanthine for nucleic acid synthesis, thus resulting in a feedback inhibition of purine synthesis. The result is a decrease in uric acid concentration in both the plasma and urine that prevents or decreases urate deposits thus preventing or reducing both gouty arthritis and urate nephropathy. The reduction in urinary urate concentration prevents the formation of uric acid or calcium oxalate calculi in the kidneys.
Indications
About one million prescriptions are written in Australia each year for allopurinol. This drug is indicated for treating chronic gout, urate nephrolithiasis and acute uric acid nephropathy. It is also used for treating hyperuricaemia (due to high levels of cell breakdown) secondary to disease, chemotherapy and radiotherapy.
Pharmacokinetics
Allopurinol is well absorbed orally and about 60% of a dose is metabolised in the liver to an active metabolite, oxypurinol, which also inhibits xanthine oxidase. The plasma half-life of allopurinol is 1–2 hours while that of oxypurinol is 18–30 hours. The onset of action in reducing plasma uric acid concentration is 2–3 days, and a fall in uric acid concentration to within the normal range occurs in 1–3 weeks. A decrease in frequency of acute gout attacks may require several months of drug therapy. Approximately 20% of the drug is excreted in faeces and the remainder is excreted via the kidneys (20%–30% as unchanged drug in urine).
Warnings and contraindications
Avoid use in people with renal disease, as accumulation of the active metabolite (oxypurinol) may cause toxicity. Dosage reduction may be necessary in persons with hepatic or renal impairment. Avoid use in individuals with known allopurinol hypersensitivity.
Drug interactions
The following effects may occur when allopurinol is given with the drugs listed:
Adverse reactions
These include pruritus, allergic reaction, rash, hives, diarrhoea, abdominal distress, vomiting, alopecia, dermatitis and, rarely, bone marrow depression, liver toxicity, a hypersensitivity reaction, peripheral neuritis, renal failure and nosebleeds.
Dosage and administration
The adult dose is 100 mg daily initially, increased by 100 mg/day at monthly intervals if necessary. The maintenance dosage is 100–300 mg daily. for treatment of hyperuricaemia (from antineoplastic therapy), usually 600–800 mg orally daily is administered beginning 1–3 days before chemotherapy or radiation therapy. For maintenance therapy, adjust the dose according to plasma uric acid concentration, which is analysed about 2 days after the initiation of allopurinol and periodically thereafter.
Patients should also be advised to increase their fluid intake, if not contraindicated, to 2.5–3 L/day, to reduce their risk of forming kidney stones. In addition, a neutral or slightly alkaline urine is recommended, which can be accomplished by dietary means such as by consuming milk, fruits (except plums, prunes and cranberries) and vegetables (except for corn and lentils), and other dietary alterations as recommended.
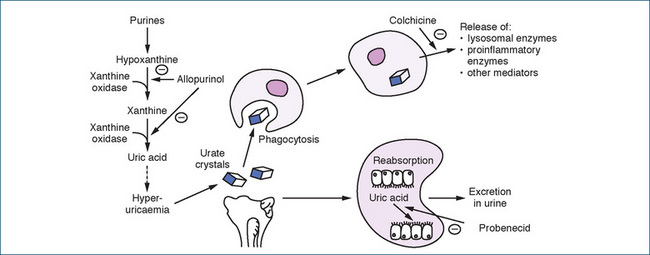
Figure 47-7 Uric acid production and the sites of action of drugs used for the treatment of hyperuricaemia. = inhibition. Reproduced with kind permission of Karen Lillywhite, Computer Assisted Learning Unit, School of Medicine, Flinders University, Adelaide, South Australia, Australia.
Colchicine
Colchicine is a plant alkaloid from the autumn crocus (Colchicum autumnale) and was introduced for the treatment of gout in 1763. It is remarkably effective during an acute attack but is of little benefit when used as a prophylactic drug. The mechanism of action of colchicine in gout is unknown, although it is reported to have anti-inflammatory effects and to inhibit neutrophil migration. It also decreases the release of a glycoprotein produced during phagocytosis of urate crystals. Additional actions include blocking the release of chemotactic factors and inflammatory mediators (Figure 47-7). These actions result in a decrease in urate deposits and inflammation, even though the drug does not affect uric acid production or excretion. Colchicine is used in low doses for the treatment of acute gout when NSAIDs or corticosteroids are inappropriate (see Clinical Interest Box 47-5).
Clinical interest box 47-5 Colchicine: NZRA consensus statement on the use of colchicine in the treatment of gout
In December 2009, the New Zealand Rheumatology Association (NZRA) stated:
…When NSAIDs are contraindicated and corticosteroids are not providing an adequate response, colchicine is an option, particularly if taken within the first 24 hours of the onset of pain. The use of two hourly dosing of colchicine to treat acute gout is no longer appropriate, especially in older patients, because of the serious adverse effects arising from large doses. The recommended dose for colchicine in the treatment of acute gout is 1.0 mg stat, followed by 0.5 mg stat six hourly, up to a maximum dose of 2.0 mg per 24 hours on the first day. An alternative regimen to consider in acute gout of less than 12 hours duration is 1.0 mg stat followed by 0.5 mg one hour later. On the subsequent days, the total dose should not exceed 1.5 mg daily. The total dose should not exceed 6 mg over four days. A prophylactic dose of colchicine may then be started after three days. Corticosteroids can be used in combination with NSAIDs or colchicine to provide further relief during acute gout.
Colchicine can also be used prophylactically in the treatment of gout with a dose ranging from 0.5 mg every other day to 0.5 mg twice daily, just short of that which will induce diarrhoea or soft stools in the patient.
Fatal and non-fatal cases of colchicine toxicity have been reported with concomitant use of P-gp and CYP3A4 inhibitors such as cyclosporin, clarithromycin, erythromycin, verapamil, diltiazem, ketoconazole, HIV protease inhibitors etc. Toxicity can also be increased by daily consumption of a litre of grapefruit juice, hepatic and renal impairment, statins, fibrates and digoxin.
Source: New Zealand Rheumatology Association Consensus Statement on the use of colchicine in the Treatment of Gout, December 2009, http://www.rheumatology.org.nz/position_statement.cfm [3 March 2010].
Colchicine is rapidly but poorly absorbed after oral administration and concentrates in white blood cells. In acute gouty arthritis, it has an onset of action within 18 hours of oral administration. The peak effect for relief of pain and inflammation is reached in 1–2 days but reduced swelling may require 3 days or more. Colchicine is partly metabolised in the liver and undergoes extensive enterohepatic recirculation, with most of the inactive metabolites eliminated in the faeces. Only 10%–20% of unchanged drug is excreted in urine. As colchicine raises plasma cyclosporin concentration, monitoring is recommended; reduction of cyclosporin dosage may be necessary.
Common adverse reactions to colchicine include diarrhoea, nausea, vomiting, abdominal pain, anorexia and, with chronic therapy, alopecia. Rare adverse reactions include hypersensitivity reactions, blood dyscrasias, neuropathy, myopathy and bone marrow suppression. Due to the inherent toxicity of colchicine, it is used with caution in people with moderate to severe liver and kidney impairment and GI disorders, and avoided in individuals with known colchicine hypersensitivity, a history of blood dyscrasias and pregnancy.
Probenecid
Probenecid is indicated for treating hyperuricaemia and chronic gouty arthritis, and as an adjunct to antibiotic therapy. It lowers the serum concentration of uric acid by competitively inhibiting the reabsorption of urate at the proximal renal tubule, thus increasing the urinary excretion of uric acid. It has no anti-inflammatory action or analgesic effect.
Probenecid was developed during the late 1940s specifically to retard the renal elimination of penicillin, as this was expensive and in short supply during World War II. It competitively inhibits the secretion of weak organic acids, such as penicillin and some of the cephalosporins, at both the proximal and distal renal tubules. The result is an increase in blood concentration and duration of action of these antibiotics. This combination is used to treat sexually transmitted diseases (e.g. gonorrhoea, acute pelvic inflammatory disease and neurosyphilis).
Probenecid is well absorbed orally and is highly bound to plasma proteins, especially to albumin. The peak uricosuric effect is reached within 30 minutes, whereas peak suppression of penicillin excretion is noted in 2 hours and lasts nearly 8 hours. Probenecid is metabolised in the liver and excreted by the kidneys. Important drug interactions are detailed in Drug Interactions 47-2.
Drug Interactions 47-2 Probenecid
| Drug | Possible effects and management |
| Aspirin or salicylates | Not recommended, because aspirin or salicylates in moderate to high doses given chronically will inhibit the effectiveness of probenecid |
| Cephalosporins, penicillins | Probenecid decreases the renal tubular secretion of penicillin and selected cephalosporins, which may result in higher plasma concentration and prolonged duration of action of the antibiotic |
| Methotrexate | Probenecid may decrease the tubular secretion of methotrexate, which may increase the risk of serious toxicity with methotrexate. Avoid combination or, if used concurrently, administer a lower dose of methotrexate and monitor closely for toxicity |
| NSAIDs: indomethacin, ketoprofen and naproxen | Probenecid decreases excretion of weak acids such as NSAIDs, which leads to higher plasma concentrations and increased potential for NSAID toxicity. The daily dose of NSAID may need to be adjusted |
| Nitrofurantoin | Probenecid may decrease the renal tubular secretion of nitrofurantoin, resulting in higher plasma concentration and possibly toxicity. This may reduce the urinary concentration and effectiveness of nitrofurantoin in urinary tract infections. Monitor effectiveness closely |
| Zidovudine | Concurrent drug administration may lead to inhibition of zidovudine metabolism and secretion, resulting in elevated plasma concentration and an increased risk of zidovudine toxicity. Avoid combination or monitor for adverse effects |
Adverse reactions to probenecid include headaches, anorexia, mild nausea or vomiting, sore gums, pain and/or blood on urination, lower back pain, frequent urge to urinate, renal stones, dermatitis and, rarely, anaphylaxis, anaemia, leucopenia and nephrotic syndrome. This drug should be avoided in people with probenecid hypersensitivity; in those with any conditions that may increase uric acid formation, such as a history of renal stones; in moderate to severe kidney function impairment; or in anyone undergoing treatment that may increase uric acid formation, such as cancer chemotherapy or radiation therapy. Also avoid use in persons with a history of blood disorders.
The adult dosage is 250 mg twice daily for 7 days, and a maintenance dose of 500 mg twice a day. The dose may be adjusted if necessary every 4 weeks up to a maximum of 2 g daily in divided doses. As an adjunct to penicillin or cephalosporin drug therapy, the dose in adults is 500 mg four times daily. For infants and children, check a current drug reference for recommended dosing schedules. In addition, patients should be instructed to maintain a high fluid intake to reduce the risk of uric acid kidney stone formation.
Immunosuppressant drugs
In Australia each year about 1000 people receive organ transplants and a range of drugs is used to prevent rejection of the transplanted kidney, liver or heart. The rejection of allogenic transplants led to the development of immunosuppressant drugs, or agents that decrease or prevent an immune response. These agents are crucial to the success of organ transplantation. A foreign substance or organ transplant in the body activates an immune response by the release of macrophages to phagocytose and process the foreign substance. In addition, IL-1 production increases, which activates helper T lymphocytes that have a surface CD3 receptor. The activated T cells stimulate production of killer, or cytotoxic, T lymphocytes, and B lymphocytes, in part by producing IL-2. T cells are necessary for cellular immunity, while B cells are responsible for humoral immunity (production of antibodies). The primary sites of action of the immunosuppressant drugs are illustrated in Figure 47-8.
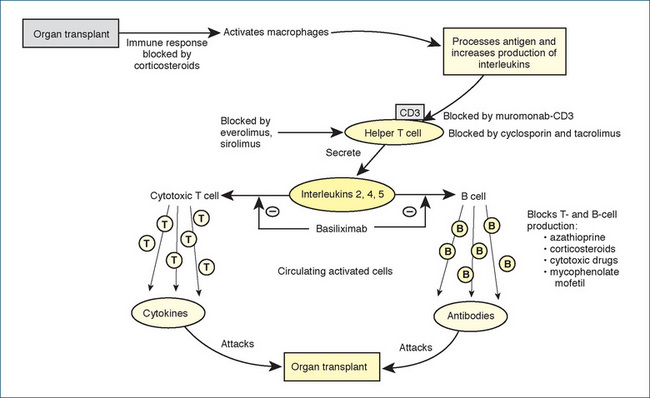
Immunodeficiency or immunosuppression may also occur from a genetic or an acquired disorder of the immune system. Acquired immune deficiency may be induced by a variety of drugs such as chemotherapeutic and immunosuppressant agents, by radiation therapy or through viral infection. Because AIDS often has devastating complications and a fatal outcome, much research interest has been directed towards developing immunomodulating or immunostimulating medications. There are several classes of immunosuppressant drugs:
Calcineurin inhibitors
Complete T-cell activation involves translocation of the nuclear factor of activated T cells (NFAT) to the nucleus, where transactivation of genes that control synthesis of cytokines such as IL-2 occurs. IL-2 stimulates T-cell proliferation and generation of cytotoxic T lymphocytes. Calcineurin is the enzyme that removes phosphate groups from NFAT, which then allows its nuclear translocation. If calcineurin is inhibited, NFAT does not enter the nucleus, gene transcription does not proceed, and the T lymphocyte does not respond to specific antigenic stimulation. The two calcineurin inhibitors in clinical use are cyclosporin and tacrolimus.
Cyclosporin
See Drug Monograph 47-2 for the pharmacology of cyclosporin.
Drug monograph 47-2 Cyclosporin
Cyclosporin is a potent immunosuppressant drug used to prevent rgan (renal, hepatic or cardiac allograft) transplant rejection and to induce or maintain remission n people with immune or inflammatory disorders. It is usually administered in combination with corticosteroids. Both calcineurin inhibitors (cyclosporin and tacrolimus) form a complex with cyclophilin that blocks the action of calcineurin in activated T cells. This prevents cytokine production and subsequent cell proliferation and differentiation. Calcineurin inhibitors do not cause significant myelosuppression or bone marrow depression.
PHARMACOKINETICS Cyclosporin is available in oral and parenteral dosage forms. After oral administration, its bioavailability is variable (about 30%), and may improve with increasing doses and chronic administration. Absorption may decrease after a liver transplant or in patients with liver impairment or GI dysfunction, such as diarrhoea or vomiting. It has a half-life of about 7 hours in children and 19 hours in adults; orally, it reaches peak plasma concentration in 3.5 hours. Cyclosporin is extensively metabolised (99.9%) in the liver by CYP3A and is subject to many drug interactions. The metabolites are eliminated principally via the faeces, with approximately 6% excreted in urine.
ADVERSE REACTIONS These are dose-related and include hirsutism, tremors, acne or oily skin, headache, leg cramps, nausea, vomiting, gingival hyperplasia (swollen, bleeding gums), seizures, nephrotoxicity, hepatotoxicity, severe hypertension and, rarely, anaphylaxis, haemolytic-uraemic syndrome, hyperkalaemia and pancreatitis. The incidence of lymphomas, skin malignancies and other lymphoproliferative-type disorders increases as the extent and duration of immunosuppression increases. Gingival hyperplasia, a common problem with the use of this drug, is generally reversible about 6 months after cyclosporin is discontinued.
DRUG INTERACTIONS Therapeutic drug monitoring plays a major role in preventing both cyclosporin toxicity and subtherapeutic dosing, both of which can be potentially catastrophic situations. The effect on the plasma cyclosporin concentration of adding, removing or changing any drug should be monitored. Multiple drug interactions have been described and the following effects may occur when cyclosporin is given with the drugs listed:
WARNINGS AND CONTRAINDICATIONS Use with caution in people with renal impairment or recent surgery. Avoid use in patients with cyclosporin hypersensitivity; recent chickenpox, herpes zoster or measles infections; and in severe liver or kidney function impairment.
DOSAGE AND ADMINISTRATION Regimens may vary in different transplant centres. The adult oral dosage is 8–15 mg/kg daily as two doses. Children may require a higher dose per kg, as they metabolise this drug rapidly. The IV dose is one-third to one-half the total daily oral dose.
Tacrolimus
Tacrolimus, in conjunction with corticosteroids, is indicated for the prophylaxis of organ (liver and kidney) rejection. It inhibits activation of T lymphocytes and is believed to bind to cyclophilin or FKBP-12 protein, forming a complex with calcineurin that prevents T-cell activation.
Oral and parenteral formulations of tacrolimus are available. Oral absorption is variable, blood concentrations are reached in 0.5–4 hours and the elimination half-life is 11–40 hours. Tacrolimus is metabolised in the liver (primarily by CYP3A) to a range of metabolites, including several active ones. Less than 1% is excreted in the urine. In view of its metabolism by CYP3A4, plasma tacrolimus concentration may be reduced by inducers and increased by inhibitors of the enzyme. Drug interactions with tacrolimus have not been so fully investigated but are similar to those of cyclosporin. The absorption of tacrolimus is reduced by aluminium hydroxide gel, resulting in decreased bioavailability. Administration of the aluminium gel should be avoided for at least 3 hours either side of dosing with tacrolimus.
Adverse reactions are numerous and commonly dose-related. They include opportunistic infections, blood disorders, diabetes, GI distress, anorexia, hyperglycaemia, hyperkalaemia, hypomagnesaemia, nephrotoxicity, neurotoxicity, headaches, insomnia, tremors, convulsions, paraesthesia, pleural effusion, peripheral oedema, pruritus, rash, cardiac disorders (e.g. cardiomyopathy and hypertension), neuropathy, muscle cramps, sweating, tinnitus, blurred vision and, rarely, anaphylaxis and hepatotoxicity.
In view of the range of adverse reactions, tacrolimus is used with caution in patients with diabetes mellitus, liver or neurological dysfunction, hepatitis B or C infection or hyperkalaemia. Its use is avoided in people with hypersensitivity to tacrolimus or polyoxyl 60 hydrogenated castor oil (the solubiliser in the IV preparation), current cancer, chickenpox, herpes zoster, infection or kidney function impairment. The last is particularly important, as tacrolimus may cause permanent renal damage.
Sirolimus derivatives
Sirolimus and everolimus
Both of these drugs act by complexing with cyclophilin of FKBP-12 in the same manner as cyclosporin and tacrolimus. Unlike the cyclosporin–FKBP-12 complex, which then inhibits calcineurin, the sirolimus/everolimus–FKBP-12 complex inhibits a kinase enzyme, which is crucial to cell cycle progression and cytokine-induced B- and T-cell proliferation. Sirolimus is one of the drugs used in drugeluting stents to reduce proliferation of smooth muscle cells and neointimal hyperplasia (refer to Clinical Interest Box 6-2). Everolimus is indicated for the prevention of kidney and heart transplant rejection, while sirolimus tends to be used in cases of renal transplant rejection.
Both drugs are administered orally, and peak blood concentrations occur within 1–2 hours. A high-fat meal decreases the peak concentration of both drugs, and it is recommended that these drugs be taken at the same time each day with the same type of food or fluid. This is important, because the plasma concentrations of these drugs are routinely monitored, and dosage adjustments may be made based on the result obtained.
Everolimus and sirolimus are metabolised by CYP3A4 and are substrates for P-glycoprotein. Not surprisingly, both are subject to many drug interactions, and concomitant administration of an inhibitor of CYP3A4 will increase plasma concentration, while administration of an inducer of CYP3A4 will decrease the plasma drug concentration.
Multiple drug interactions (see Drug Interactions 47-3) may occur with either everolimus or sirolimus. In addition, due to the involvement of CYP3A4, consumption of grapefruit should be avoided, as it may increase the risk of toxicity with either of these drugs.
Drug Interactions 47-3 Everolimus/sirolimus
| Drug | Possible effects and management |
| Cyclosporin | Increases the plasma concentration of everolimus, and plasma monitoring of everolimus concentration is necessary when adjusting cyclosporin dose |
| Erythromycin | May inhibit the metabolism of both drugs, leading to increased plasma concentrations and possible toxicity. Monitor and decrease dose if necessary |
| Itraconazole/ketoconazole | May inhibit metabolism of drugs, increasing plasma concentrations and risk of toxicity. Avoid combination or monitor plasma concentration of everolimus and sirolimus |
| Rifampicin | May increase metabolism of everolimus and sirolimus, reducing plasma drug concentration and therapeutic effect. Monitor closely if this combination is used |
The use of everolimus and sirolimus is associated with a dose-dependent rise in triglycerides and cholesterol. This may warrant either a dose reduction or treatment with a statin. Limited data are available and use should be avoided in pregnancy. Contraception is recommended during treatment and for 2–3 months after the last dose.
Common adverse reactions include hypertriglyceridaemia, hypercholesterolaemia and various haematological disorders, e.g. leucopenia, neutropenia, thrombocytopenia and raised plasma creatinine (with cyclosporin).
Cytotoxic immunosuppressant drugs
The cytotoxic immunosuppressant drugs include azathioprine, methotrexate, cyclophosphamide and 6-mercapto-purine; the last three are discussed in Chapter 42.
Azathioprine
Azathioprine is indicated as an adjunct medication to prevent rejection in renal organ transplant recipients, and for severe active rheumatoid arthritis in patients who have not responded to other therapies. The mechanism of action involves alterations in purine synthesis that primarily suppress T- and B-cell production, cell-mediated hypersensitivity and antibody production. In combination with steroids, azathioprine appears to have a steroid-conserving effect, and a lower dose of steroid may be used to treat chronic inflammatory processes when given with azathioprine.
Azathioprine is available in oral and parenteral dosage forms. When given orally, it is well absorbed from the intestinal tract. It has a half-life of 5 hours, with an onset of action of 6–8 weeks in rheumatoid arthritis and perhaps 4–8 weeks in other inflammatory disease states. It is metabolised in the liver to active metabolites (6-mercaptopurine and 6-thioinosinic acid), with further metabolism by xanthine oxidase. It is primarily excreted via the biliary system.
Adverse reactions are often observed and include anorexia, nausea, vomiting, leucopenia or infection, megaloblastic anaemia (the patient may be asymptomatic but may also have fever, chills, cough, low-back or side pain, pain on urination or increased weakness), hepatitis (infrequent), thrombocytopenia, hypersensitivity, pancreatitis, pneumonitis, sores in the mouth and on the lips and skin rash. The risk of hepatotoxicity is greater when the dosage of azathioprine exceeds 2.5 mg/kg daily.
Due to its immunosuppressant actions, azathioprine is used with caution in patients with pancreatitis or hepatic, renal or bone marrow impairment. It is also contraindicated in people with neoplastic disorders, uncontrolled infection or recent exposure to varicella virus infections.
Immunosuppressant antibodies
Antibodies directed against cell-surface antigens on T lymphocytes have been in clinical practice for decades and are widely used for preventing organ transplant rejection. Currently available immunosuppressant antibodies include antithymocyte globulins, basiliximab and muromonab-CD3.
Antithymocyte globulins
The available polyclonal antithymocyte globulins include a horse antibody directed against human thymocyte cell surface markers and a rabbit antibody raised against human T lymphoblast cell surface markers. Both of these antibodies bind to cell-surface receptors (e.g. CD2–CD4 antigens, HLA class I and II molecules) on the surface of human T lymphocytes. Once bound, these antibodies are cytotoxic and hence deplete the number of circulating lymphocytes and block lymphocyte function. Both products (horse and rabbit) contain small concentrations of antibodies that will cross-react with other blood components, and are thus not interchangeable. It is usual to perform a skin test for allergy before administration, so these globulins are contraindicated in people who manifest an allergic response to the test dose.
Antithymocyte globulins are used for the prevention and treatment of organ (kidney, heart, lung and liver) transplant rejection. They are administered IV (infusion) and the dosage is calculated on a per-kg basis. Common adverse reactions include fever, chills, dyspnoea, chest pain, hypotension, diarrhoea, nausea and vomiting and a variety of haematological complications (e.g. leucopenia and thrombocytopenia), which are all related to a cytokine release syndrome. As many people develop anti-antibodies (e.g. anti-rabbit antibodies), this limits the possibility of repeated doses. As would be expected with chronic immunosuppression, these antithymocyte globulins are associated with the development of lymphoproliferative disorders.
Basiliximab
Basiliximab is a purified monoclonal antibody immunosuppressant that is used in combination with cyclosporin and corticosteroids to prevent acute rejection of transplanted kidneys. This drug is an IL-2-receptor antagonist, binding to the IL-2 receptor complex, and thus inhibiting IL-2 binding. This inhibition results in a decreased activation of lymphocytes and an impaired immune system response to antigens. This drug is administered parenterally and has an elimination half-life of approximately 7–10 days.
Unlike the antithymocyte globulins, which produce a number of adverse reactions related to a cytokine release syndrome, basiliximab appears to be relatively free of adverse reactions other than those that would be expected to be observed in transplant patients on multiple therapies. Long-term adverse effects are unknown, but currently the drug appears not to increase the incidence of opportunistic infections or lymphoproliferative disorders (compared to baseline) in immunosuppressed patients.
Pregnancy should be prevented when using this drug and contraception continued for at least 2 months after the last dose.
Muromonab-CD3
Muromonab-CD3 is a purified mouse monoclonal antibody that reacts with CD3 receptors on the surface of T lymphocytes. It blocks the activation and functions of the T cells in response to an antigenic challenge; thus it functions as an immunosuppressant and does not cause myelosuppression.
Muromonab-CD3 is indicated for the treatment of acute renal organ transplant rejection and is usually given in combination with azathioprine, cyclosporin or corticosteroids. It is also administered to treat acute rejection (steroid-resistant) in cardiac and hepatic transplant patients. Available parenterally, it acts to reduce activated T cells within minutes of administration. It reaches steady-state plasma concentration in about 3 days and has a duration of action of about 7 days. The number of circulating CD3-positive T cells returns to baseline levels within 1 week of discontinuation of muromonab-CD3.
The most frequent adverse reactions with muromonab-CD3 occur with the first dose. The first-dose effect (cytokine release syndrome) consists of light-headedness, elevated temperature, chills, nausea, vomiting, diarrhoea, headache, dyspnoea, chest pain, tremors and trembling. These effects may be repeated to a lesser degree after the second dose but are rarely encountered with later doses. Fever and chills that occur later may be caused by infection. Anaphylaxis, hypersensitivity, encephalopathy, convulsions, cerebral oedema and aseptic meningitis syndrome are reported less often. Concurrent use with other immunosuppressant agents may increase the risk of infection and the development of lymphoproliferative diseases. A reduced dose of corticosteroids and azathioprine is recommended when muromonab-CD3 is started. (For interactions with live viral vaccines, see azathioprine above.)
Other immunosuppressants
Mycophenolate
Mycophenolate mofetil and mycophenolate sodium, used in conjunction with cyclosporin and corticosteroids, are indicated for the prophylaxis of renal transplant rejection. Both drugs are metabolised to mycophenolic acid, an active metabolite that inhibits the responses of T and B lymphocytes to mitogenic and allospecific stimulation. It also suppresses antibody formation by B cells and may inhibit the influx of leucocytes into inflammatory and graft rejection sites.
Available orally, mycophenolate is rapidly metabolised to the active metabolite mycophenolic acid and an inactive metabolite, a phenolic glucuronide. The half-life of mycophenolic acid is 18 hours, with excretion primarily as the phenolic glucuronide in urine (∼85%).
Potential interactions with other drugs used commonly in transplant patients have been investigated (see Drug Interactions 47-4).
Drug Interactions 47-4 Mycophenolate mofetil
| Drug | Possible effects and management |
| Aciclovir or ganciclovir | These drugs compete with mycophenolate mofetil for renal excretion and may increase each other’s toxicity. Avoid, or a potentially serious drug interaction may occur. If used concurrently, monitor closely |
| Antacids (magnesium and aluminium hydroxide), cholestyramine, colestipol | These drugs decrease the absorption of mycophenolate mofetil. Administer mycophenolate mofetil 1 hour before or 2 hours after antacids and bile acid sequestrants |
| Immunosuppressant agents, e.g. azathioprine, glucocorticoids, cyclophosphamide, cyclosporin | Enhanced immunosuppression. May increase the risk of developing infections and neoplasms |
Adverse reactions are dose-related and include abdominal pain, constipation or diarrhoea, nausea, vomiting, dizziness, acne, insomnia, rash, anaemia, chest pain, cough, dyspnoea, haematuria, hypertension, leucopenia, peripheral oedema, arrhythmia, arthralgia, colitis, GI bleeding, gingival hyperplasia, gingivitis, myalgia, oral moniliasis, pancreatitis, thrombocytopenia and tremors.
Mycophenolate is used with caution in patients with delayed post-transplant renal graft function, and dose reduction may be necessary. This drug should be avoided in people with mycophenolate mofetil hypersensitivity, active GI system disease or severe kidney function impairment.
Leflunomide
Leflunomide is an oral DMARD that inhibits pyrimidine synthesis, thus limiting the available pool of pyrimidine precursors needed for proliferation of cells, including T cells in the inflammatory response (Figure 47-6). It is highly protein-bound and undergoes continuous enterohepatic recirculation. In the liver, leflunomide is converted (95%) to the primary active metabolite, called A77 1726. This metabolite is excreted slowly in urine (about 30%) and faeces (about 70%) and has a long half-life of 2–3 weeks. As the active metabolite undergoes extensive enterohepatic recirculation, it may take up to 2 years for the drug concentration to decrease to an undetectable level in plasma.
Common adverse reactions with this drug include diarrhoea, nausea, rashes and alopecia. Serious reactions such as pancytopenia and skin reactions of the toxic epidermal necrolysis type have been reported. In addition, there is increasing evidence of a spectrum of severe lung toxicity including pneumonitis and pulmonary fibrosis. As clinical presentation of interstitial lung disease has occurred at varying times after commencement of leflunomide therapy, patients should be monitored closely for evidence of a decline in pulmonary function, worsening of a cough or dyspnoea. Collection of long-term safety data for leflunomide is continuing.
Immunostimulant Drugs
Advances in biotechnology have allowed the development of new agents that can either activate the body’s immune defences or modify a response to an unwanted stimulus, such as an antitumour response. These agents are called biological response modifiers; when the immune system is activated, they are commonly referred to as immunostimulants. With the advent of recombinant deoxyribonucleic acid (DNA) technology in the early 1980s, new agents were made available in larger quantities for clinical trials and investigations. Although still in its infancy, this area of study has the potential for solving some of the mysteries about disease that have eluded researchers for centuries. Research may also provide future drugs that will control the devastation, pain and suffering induced by many viral diseases (e.g. AIDS) and cancer. Common immunostimulants include vaccines, colonystimulating factors and the interferons. Interferons (alpha, beta, gamma) bind to specific cell-surface receptors that are linked through to the inner networks of the cell that control functions such as enzyme activity, cell proliferation and enhancement of immune activity.
Lymphokines (IL-1 and IL-2), interferons (primarily alpha-interferon) and granulocyte or granulocyte–macrophage colonystimulating factors (G-CSF or GM-CSF-leukine) are under investigation as immunostimulants. Lymphokines are protein substances released by sensitised lymphocytes when in contact with specific antigens. They activate macrophages to stimulate humoral and cellular immunity for the host. Interleukins have been called the chemical messengers of immune cell communication. Interleukin-2 is believed to be a T-cell growth factor that promotes the long-term survival and growth of T lymphocytes, which is necessary for the continuation of the immune response and is also involved in the rejection of transplanted organs. Aldesleukin is a recombinant variant of IL-2 that binds to the IL-2 receptor and stimulates cell proliferation and the production of interferon gamma. Although some patients have been helped with these therapies, major problems are limited effectiveness or extreme systemic toxicity. The currently available immune stimulants are listed in Table 47-4.
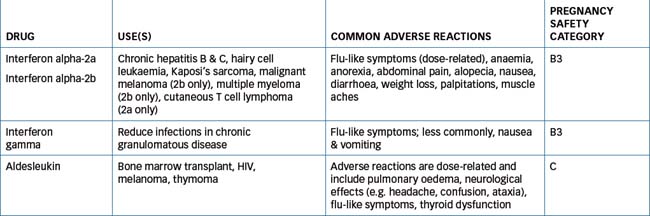
Histamine and Histamine-Receptor Antagonists (Antihistamines)
Distribution of histamine
Histamine is a chemical mediator that occurs naturally in almost all body tissues. It is present in highest concentration in the skin, lung and GI tract and, when liberated from cells, plays an early transient role in the inflammatory process.
In many tissues, the chief site of production and storage of histamine is the cytoplasmic granules of the mast cell or, in the case of blood, the basophil, which closely resembles the mast cell in function. Mast cells are small ovoid structures widely distributed in the loose connective tissue. They are especially abundant in small blood vessels and in bronchial smooth muscle, which appears to have the highest concentration of mast cells of any organ in the body. The mast cells and basophils make up the mast-cell histamine pool. A second major site of histamine production is known as the non-mast-cell pool, where the histamine is stored in the cells of the epidermis, GI mucosa and the CNS. Although histamine is present in various foods and is synthesised by intestinal flora, the amount absorbed does not contribute to the body’s stores of this amine.
Actions
The reactions mediated by histamine are attributable to binding to two distinct populations of receptors, called H1 and H2 receptors. A third histamine receptor has been identified (H3), but research is ongoing to define its role. The principal actions of histamine are listed in Table 47-5.
Table 47-5 Histamine receptor-mediating effects
| Structure | Histamine receptors | Effects |
| Vascular system | ||
| Capillary(microcirculation) | H1 and H2 | Dilation, increased permeability |
| Arteriole (smooth muscle) | H1 and H2 | Dilation |
| Smooth muscle | ||
| Bronchial, bronchiolar | H1 | Contraction |
| Gastrointestinal | H1 | Contraction |
| Exocrine glands | ||
| Gastric | H2 | Gastric acid secretion (HCl) |
| Epidermis | H1 | Triple response (flush, flare, wheal) |
| Adrenal medulla | H1 | Adrenaline and noradrenaline release |
| Central nervous system | H1 | Motion sickness |
Vascular effects
In the microcirculatory component of the cardiovascular system (arterioles, capillaries, venules), the liberation of histamine has been shown to involve both the H1 and H2 receptors. Stimulation of these receptors dilates the capillaries and venules, producing increased localised blood flow, increased capillary permeability, erythema and oedema. By activating the H1 and H2 receptors on the smooth muscles of the arterioles, histamine is also capable of eliciting a systemic response, that is, vasodilation of the arterioles, which can result in a profound fall in blood pressure.
Smooth muscle effects
Although histamine exerts a powerful relaxing effect on the smooth muscle of the arterioles, it produces a contractile action on the smooth muscles of many non-vascular organs, such as the bronchi and GI tract. In sensitised individuals, activation of the H1 receptors of the lungs can cause marked bronchial muscle contraction that often progresses to dyspnoea and airway obstruction.
Exocrine glandular effects
While histamine stimulates the gastric, salivary, pancreatic and lacrimal glands, the main effect is seen in the gastric glands. Stimulation of H2 receptors in the exocrine glands of the stomach increases production of gastric acid secretions. The high hydrochloric acid concentration is attributed to the activity of the parietal cells of the stomach and is implicated in the development of peptic ulcers (see Chapters 29 and 30).
Central nervous system effect
Histamine is known to be present throughout the tissues of the brain. Its effects seem to involve both H1 and H2 receptor mediation. The activation of H1 receptors of the semicircular canals is associated with motion sickness.
Inflammatory effects
Histamine as a chemical mediator is implicated in many pathological disorders. Although four different types of hypersensitivity responses to immunological injury exist, the type I anaphylactic reaction is the one associated with the release of histamine.
People with type I-mediated hypersensitivity develop allergies as a result of sensitisation to a foreign agent that may be ingested, inhaled or injected. An incalculable number of these allergenic agents exist. They vary widely in that different forms of allergy can develop in response to seasonal exposure to pollens, grasses and weeds, or to non-seasonal agents such as house dust, feathers, moulds and other similar substances. Hypersensitivity to a variety of foods such as shellfish or strawberries requires ingestion of the antigen. Insects such as bees or wasps and even drugs, particularly penicillin, also possess allergenic properties that may induce a severe response in hypersensitive individuals.
Type I anaphylactic hypersensitivity accounts for a substantial number of allergic disorders and involves a complex series of anomalies ranging from mild urticaria to anaphylactic shock. The mechanism of type I anaphylactic reaction involves the attachment of an antigen (Ag) to an antibody (Ab), specifically IgE, and this complex in turn becomes fixed to the mast cell. The pathological manifestations of Ag–IgE interaction are caused by mast cell degranulation, resulting in the release of histamine and other mediators responsible for producing the allergic symptoms. The type I anaphylactic reaction is responsible for various disorders, such as urticaria, atopy (allergic rhinitis and hay fever), food allergies, bronchial asthma and systemic anaphylaxis.
Urticaria
Urticaria is a vascular reaction of the skin characterised by immediate formation of a wheal and flare accompanied by severe itching. Contact with an external irritant such as drugs or foods produce the Ag–IgE-mediated response, with resultant release of histamine from the mast cells into the skin. The local vasodilation produces the red flare, and the increased permeability of the capillaries leads to tissue swelling. These swellings are often called hives; when giant hives occur, the condition is known as angioneurotic oedema. Antihistaminic drugs administered before exposure to the antigen will prevent this response.
Atopy
Atopy occurs in genetically susceptible individuals and is usually caused by seasonal pollen. This condition is manifested as an upper respiratory tract disorder known as allergic rhinitis (hay fever). After the interaction of Ag–IgE antibody on the surface of the bronchial mast cells, histamine is released, producing local vascular dilation and increased capillary permeability. This change produces a rapid fluid leakage into the tissues of the nose, resulting in swelling of the nasal linings. In certain people, antihistaminic therapy can prevent the oedematous reaction if the drug is administered before antigen exposure.
Food allergies
Food allergies involve an intestinal IgE–mast cell response to ingested antigens. If the upper GI tract is affected, vomiting results; if the lower GI tract is invaded, cramps and diarrhoea occur. This condition also has been known to produce systemic anaphylaxis after ingestion of a large amount of antigen.
Bronchial asthma
When an inhaled antigen combines with IgE, stimulation of the mast cells triggers the release of mediators in the lower respiratory tract, usually in the bronchi and bronchioles. Histamine plays a role in the early asthma response, but administration of antihistaminic drugs actually does not help to relieve bronchoconstriction because more potent chemical mediators than histamine are responsible for causing the reaction (see Chapter 28).
Systemic anaphylaxis
Systemic anaphylaxis is a generalised reaction manifested as a life-threatening systemic condition. The Ag–IgE mediator response involves the basophils of the blood and the mast cells in the connective tissue. The commonest precipitating causes of this response are drugs (particularly penicillin), insect stings (wasps and bees) and, occasionally, certain foods. The release of massive amounts of histamine into the circulation causes widespread vasodilation, resulting in a profound fall in blood pressure. The excessive dilation also allows plasma to leave the capillaries and a loss of circulatory volume ensues. When the reaction is fatal, death is usually caused not only by shock but also by laryngeal oedema. The symptoms of the latter condition include smooth muscle contraction of the bronchi and pharyngeal oedema, which usually leads to asphyxiation.
Drug allergies often develop in susceptible individuals who show no adverse effect after the first dose of a drug. A second or subsequent exposure to even a minute amount of this same antigen may elicit an exaggerated IgE response, either locally or systemically. People who exhibit such reactions are said to be allergic to the drug. The IgE-mediated response, particularly with penicillin, may occur in either the skin, producing severe urticaria, or the respiratory tract, causing bronchial asthma. Even limited contact in certain sensitised individuals can produce a fatal systemic anaphylaxis. Some of the drugs that elicit an allergic response include penicillin, chloramphenicol, streptomycin, sulfonamides and aspirin.
Antihistamines
Many histamine receptor antagonists are available over the counter and are widely used for motion sickness, vertigo and skin and allergic disorders. In general, these drugs antagonise the action of histamine at H1 receptors and are conventionally referred to as antihistamines.
With the discovery of two histamine receptors, H1 and H2, the antihistamines were divided into the H1-receptor antagonists and the H2-receptor antagonists. The H2-receptor-blocking agents, which include cimetidine and ranitidine and others, are discussed in Chapter 29. This section reviews the sedating and newer, less sedating antihistamines.
H1-receptor antagonists
The H1 antihistamines have the greatest therapeutic effect on nasal allergies. These drugs are more effective if given before histamine is released. They relieve symptoms better at the beginning of the hay fever season than during its height but fail to relieve the asthma that often accompanies hay fever. These preparations are palliative and do not immunise the individual or protect against allergic reactions. Dozens of antihistamine drugs are available. They generally differ from one another in potency, duration of action and incidence of adverse reactions, particularly sedation. In general, they fall into two categories: the older, sedating drugs and the newer, less sedating antihistamines.
Indications
The sedating antihistamines are indicated for the treatment of allergies, skin disorders, vertigo, motion sickness and nausea, and for sedation. Generally, their oral absorption pattern is good, with onset of action within 15–60 minutes for most of them. The newer, less sedating antihistamines are primarily used for allergic disorders. They have a reduced incidence of anticholinergic effects and sedation, and are generally better tolerated. Individual response to antihistamines varies, and nearly all of these drugs are recommended for short-term treatment. Antihistamines are primarily metabolised in the liver and excreted by the kidneys.
Potential interactions with other drugs are shown in Drug Interactions 47-5.
Drug Interactions 47-5 Antihistamines
| Drug | Possible effects and management |
| Alcohol, CNS depressants | Concurrent use may enhance CNS-depressant effects. If the CNS depressant also has anticholinergic adverse effects, enhanced effects may be seen. Avoid combined use |
| Anticholinergic medications, psychotropics, others | Enhanced CNS-depressant and anticholinergic adverse effects may be noted. Avoid combined use |
| Levodopa | Phenothiazine antihistamines antagonise the action of levodopa. Avoid combined use |
Adverse reactions/warnings and contraindications
The commonest adverse reactions are sedation (with oldertype antihistamines), dizziness, dry mouth, tinnitus, fatigue, headache, nausea and vomiting. The anticholinergic effect of antihistamines will exacerbate closed-angle glaucoma, pyloroduodenal obstruction, bladder neck obstruction and hyperthyroidism. Avoid using these drugs in people with specific antihistamine hypersensitivity, hypokalaemia, liver function impairment (phenothiazine-type), prostatic hypertrophy or urinary retention. The elderly may be more sensitive to adverse effects such as hypotension and dizziness; lower doses are preferable in this group.
Dosage and administration
The dosage varies for each drug and among adults, children and infants. The newer agents released are generally longeracting drugs with fewer sedative effects. Loratadine, for example, is taken once a day and has few, if any, sedative and anticholinergic effects. The available dosage forms and ADEC pregnancy categories are noted in Table 47-6.
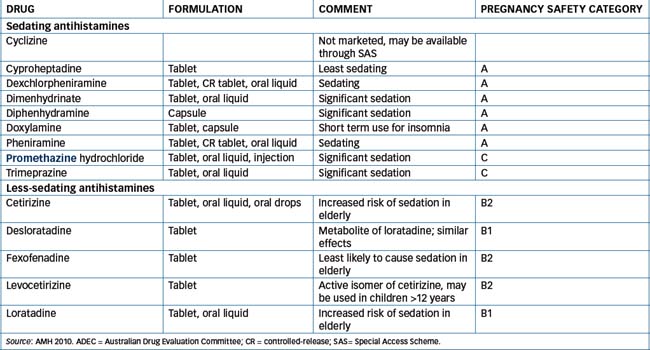
Fexofenadine, an antihistamine approved in 1996, is a metabolite of terfenadine. Manufacture of terfenadine has ceased because of the serious and potentially fatal cardiovascular drug interactions associated with its use. The likelihood of serious cardiac arrhythmias with fexofenadine is low but care should still be exercised in persons with prolonged QT interval.
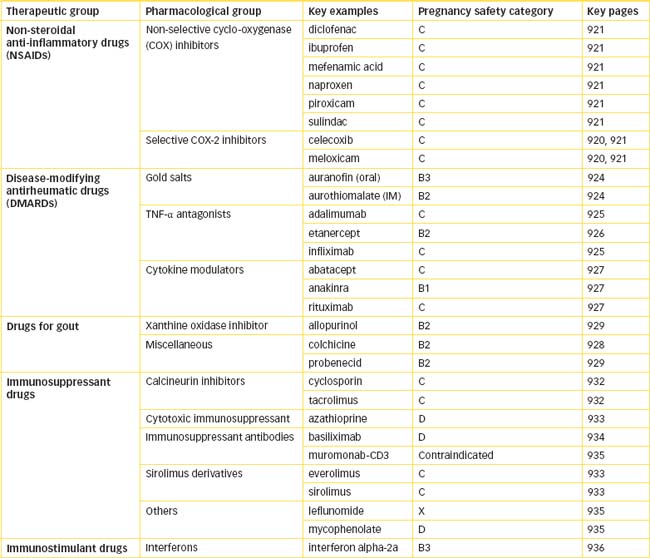
Key points
 The body’s resistance to disease is both non-specific, which offers immediate protection, and specific, i.e. an acquired specific resistance or immunity that develops more slowly. The lymphatic system, which is responsible for eliciting highly specific immune responses, comprises lymph, lymphatic vessels, lymph nodes, red bone marrow and the thymus gland, spleen and tonsils. The initial and immediate lines of defence for the body are the skin, the mucous membranes of the respiratory and gastrointestinal tracts and chemical secretions such as saliva and gastric acid. Damage to cells by bacteria or viruses, physical trauma (e.g. a cut), foreign bodies, chemical substances, surgery, radiation or electricity will elicit an inflammatory response. The four characteristic signs of inflammation are swelling (oedema), redness (erythema), pain and heat, which are accounted for by three basic events: (1) blood vessel vasodilation and increased capillary permeability, (2) cellular infiltration and (3) tissue repair. Mediators of inflammation include the complement system and local hormones such as histamine, prostaglandins and cytokines. Provocation of an immune response against specific pathogens or transplanted tissue is called specific resistance or immunity. Humoral immunity is a response effective in the extracellular environment, in contrast to cell-mediated immunity, which deals with intracellular organisms. T and B lymphocytes and the polymorphonuclear leucocytes (PMLs) are involved in the immune response, although only mononuclear T and B lymphocytes are immunocompetent cells. Antibodies are gamma globulins (a type of protein), called immunoglobulins, that are specific for particular antigens. Five classes of antibodies have been identified: IgG, IgM, IgA, IgD and IgE. The four different types of allergic or hypersensitivity reactions are type I (immediate or anaphylactic), type II (antibody-dependent), type III (complex-mediated) and type IV (delayed hypersensitivity). An individual can acquire further immune capabilities through both natural and artificial means. This type of immunity is acquired either actively or passively. Inflammatory disorders such as rheumatoid arthritis require lifelong therapy, and the choice of drugs is often weighed against the risk of adverse effects. Despite diversity in their chemical structures, all the nonsteroidal anti-inflammatory drugs (NSAIDs) possess the same therapeutic properties—analgesic, antipyretic and anti-inflammatory effects. NSAIDs also share, to varying degrees, the same adverse reactions because inhibition of prostaglandins, which accounts for all the therapeutic effects, also causes the renal and gastrointestinal toxicity. NSAIDs are indicated for the treatment of acute or chronic rheumatoid arthritis, osteoarthritis, ankylosing spondylitis and other rheumatic diseases, and mild to moderate pain. Disease-modifying antirheumatic drugs (DMARDs) comprise a group of drugs with diverse chemical structures, and their mechanisms of action in many cases are largely unknown. Use of DMARDs has substantially improved the control of rheumatoid arthritis and the long-term quality of life of the individual. In many instances, combinations of DMARDs may be used. The primary goals of therapy for gout are to end the acute attack as soon as possible, to prevent a recurrence, to prevent the formation of uric acid stones in the kidneys and to prevent or minimise the complications of sodium urate deposits in the joints. The rejection of kidney, liver and heart allergenic transplants has led to the development of immunosuppressant drugs, or agents that decrease or prevent an immune response. There are several classes of immunosuppressant drugs: the corticosteroids (discussed in Chapter 35); the calcineurin inhibitors cyclosporin and tacrolimus; the cytotoxic immunosuppressants azathioprine, methotrexate, cyclophosphamide and 6-mercaptopurine (all discussed in Chapter 42); the immunosuppressant antibodies including the antithymocyte globulins and basiliximab and muromonab-CD3; the sirolimus derivatives everolimus and sirolimus and other immunosuppressants including mycophenolate and leflunomide. Advances in biotechnology have allowed the development of new agents that can either activate the body’s immune defences or modify a response to an unwanted stimulus, such as an antitumour response. These agents are called biological response modifiers; when the immune system is activated, they are commonly referred to as immunostimulants. Common immunostimulants include vaccines, colonystimulating factors, the interferons and aldesleukin. Histamine is a natural chemical found in most body tissues and has been implicated in a range of conditions such as urticaria, atopy, food allergies, bronchial asthma and systemic anaphylaxis.
The body’s resistance to disease is both non-specific, which offers immediate protection, and specific, i.e. an acquired specific resistance or immunity that develops more slowly. The lymphatic system, which is responsible for eliciting highly specific immune responses, comprises lymph, lymphatic vessels, lymph nodes, red bone marrow and the thymus gland, spleen and tonsils. The initial and immediate lines of defence for the body are the skin, the mucous membranes of the respiratory and gastrointestinal tracts and chemical secretions such as saliva and gastric acid. Damage to cells by bacteria or viruses, physical trauma (e.g. a cut), foreign bodies, chemical substances, surgery, radiation or electricity will elicit an inflammatory response. The four characteristic signs of inflammation are swelling (oedema), redness (erythema), pain and heat, which are accounted for by three basic events: (1) blood vessel vasodilation and increased capillary permeability, (2) cellular infiltration and (3) tissue repair. Mediators of inflammation include the complement system and local hormones such as histamine, prostaglandins and cytokines. Provocation of an immune response against specific pathogens or transplanted tissue is called specific resistance or immunity. Humoral immunity is a response effective in the extracellular environment, in contrast to cell-mediated immunity, which deals with intracellular organisms. T and B lymphocytes and the polymorphonuclear leucocytes (PMLs) are involved in the immune response, although only mononuclear T and B lymphocytes are immunocompetent cells. Antibodies are gamma globulins (a type of protein), called immunoglobulins, that are specific for particular antigens. Five classes of antibodies have been identified: IgG, IgM, IgA, IgD and IgE. The four different types of allergic or hypersensitivity reactions are type I (immediate or anaphylactic), type II (antibody-dependent), type III (complex-mediated) and type IV (delayed hypersensitivity). An individual can acquire further immune capabilities through both natural and artificial means. This type of immunity is acquired either actively or passively. Inflammatory disorders such as rheumatoid arthritis require lifelong therapy, and the choice of drugs is often weighed against the risk of adverse effects. Despite diversity in their chemical structures, all the nonsteroidal anti-inflammatory drugs (NSAIDs) possess the same therapeutic properties—analgesic, antipyretic and anti-inflammatory effects. NSAIDs also share, to varying degrees, the same adverse reactions because inhibition of prostaglandins, which accounts for all the therapeutic effects, also causes the renal and gastrointestinal toxicity. NSAIDs are indicated for the treatment of acute or chronic rheumatoid arthritis, osteoarthritis, ankylosing spondylitis and other rheumatic diseases, and mild to moderate pain. Disease-modifying antirheumatic drugs (DMARDs) comprise a group of drugs with diverse chemical structures, and their mechanisms of action in many cases are largely unknown. Use of DMARDs has substantially improved the control of rheumatoid arthritis and the long-term quality of life of the individual. In many instances, combinations of DMARDs may be used. The primary goals of therapy for gout are to end the acute attack as soon as possible, to prevent a recurrence, to prevent the formation of uric acid stones in the kidneys and to prevent or minimise the complications of sodium urate deposits in the joints. The rejection of kidney, liver and heart allergenic transplants has led to the development of immunosuppressant drugs, or agents that decrease or prevent an immune response. There are several classes of immunosuppressant drugs: the corticosteroids (discussed in Chapter 35); the calcineurin inhibitors cyclosporin and tacrolimus; the cytotoxic immunosuppressants azathioprine, methotrexate, cyclophosphamide and 6-mercaptopurine (all discussed in Chapter 42); the immunosuppressant antibodies including the antithymocyte globulins and basiliximab and muromonab-CD3; the sirolimus derivatives everolimus and sirolimus and other immunosuppressants including mycophenolate and leflunomide. Advances in biotechnology have allowed the development of new agents that can either activate the body’s immune defences or modify a response to an unwanted stimulus, such as an antitumour response. These agents are called biological response modifiers; when the immune system is activated, they are commonly referred to as immunostimulants. Common immunostimulants include vaccines, colonystimulating factors, the interferons and aldesleukin. Histamine is a natural chemical found in most body tissues and has been implicated in a range of conditions such as urticaria, atopy, food allergies, bronchial asthma and systemic anaphylaxis.Review exercises
References and further reading
Australian Institute of Health and Welfare. Australia’s Health 2008: The Eleventh Biennial Health Report of the Australian Institute of Health and Welfare. Cat. No. AUS 99. Canberra:AIHW, 2008.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Bombardier C., Laine L., Reicin A., et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. New England Journal of Medicine. 2000;343:1520-1528.
Brater D.C., Harris C., Redfern J.S., Gertz B.J. Renal effects of COX-2 selective inhibitors. American Journal of Nephrology. 2001;21:1-15.
Bresalier R.S., Sandler R.S., Quan H., et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. New England Journal of Medicine. 2005;352:1092-1102.
Burke A., Smyth E., Fitzgerald G.A. Analgesic-antipyretic agents; pharmacotherapy of gout. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw–Hill, 2006. [ch 26]
European Medicines Agency Post-authorisation Evaluation of Medicines for Human Use Questions and Answers on COX-2 Inhibitors. Available: http://www.emea.europa.eu/pdfs/human/press/pr/6297805en.pdf[16 November 2009]
Furst D.E., Keystone E.C., Kirkham B., et al. Updated consensus statement on biological agents for the treatment of rheumatic disease, 2008. Annals of Rheumatic Diseases. 2008;67 (Suppl 3):iii2-iii25.
Greenberg M.E., Lai M.H., Hartel G.F., et al. Response after one dose of a monovalent influenza A (H1N1) 2009 Vaccine—Preliminary report. New England Journal of Medicine. 2009;361:1-10.
Halloran P.F. Immunosuppressive drugs for kidney transplantation. New England Journal of Medicine. 2004;351:2715-2729.
Haroon N., Inman R.D. Infectious complications of biological therapy. Current Opinion in Rheumatology. 2009;21:397-403.
Krensky A.M., Vincenti F., Bennett W.M. Immunosuppressants, tolerogens, and immunostimulants. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw–Hill, 2006. [ch 52]
Olsen N.J., Stein C.M. New drugs for arthritis. New England Journal of Medicine. 2004;350:2167-2179.
Patrono C., Baigent C. Low-dose aspirin, coxibs and other NSAIDs: a clinical mosaic emerges. Molecular Interventions. 2009;9:31-39.
Perazella M., Tray K. Selective cyclo-oxygenase-2 inhibitors: a pattern of nephrotoxicity similar to traditional nonsteroidal anti-inflammatory drugs. American Journal of Medicine. 2000;111:64-67.
Rott K.T., Agudelo C.A. Gout. Journal of the American Medical Association. 2003;289:2857-2860.
Saag K.G., Teng G.G., Patkar N.M., et al. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis and Rheumatism. 2008;59:767-784.
Silverstein F.E., Faich G., Goldstein J.L., Simon L.S., et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal antiinflammatory drugs for osteoarthritis and rheumatoid arthritis. The CLASS Study: a randomized controlled trial. Journal of the American Medical Association. 2000;284:1247-1255.
Simmons D.L., Botting R.M., Hla T. Cyclooxygenase isozymes:the biology of prostaglandin synthesis and inhibition. Pharmacological Reviews. 2004;56:387-437.
Sutaria S., Katbamna R., Underwood M. Effectiveness of interventions for the treatment of acute and prevention of recurrent gout: a systematic review. Rheumatology. 2006;45:1422-1431.
Tortora G.J., Grabowski S.R. The lymphatic system, nonspecific resistance to disease, and immunity. In Tortora G.J., Grabowski S.R., editors: Principles of Anatomy and Physiology, 9th edn, New York: John Wiley, 2000. [ch 22]