Chapter 46 Antiprotozoal, Antimycobacterial and Anthelmintic Drugs
This chapter reviews the various drugs used to treat protozoal diseases such as malaria and amoebiasis, mycobacterial infections such as Mycobacterium avian complex (MAC), tuberculosis and leprosy and helminth (worm) infections. These diseases are prevalent throughout the world and their control peaks and wanes, often due to factors such as availability of drugs, patient compliance with specific drug therapies and the development of drug-resistant strains. Control of these infections is important and challenging to the health-care professional, and in some instances a global approach to eradication (as with malaria, tuberculosis and leprosy) has been adopted.
Key abbreviations
Malaria
MALARIA has been and still is a prevalent disease despite efforts to control the causative parasite and insect vector. Each year 300–500 million cases of clinical malaria occur, with a further 10,000–30,000 people contracting the disease after visiting endemic areas (Croft 2000). The majority of cases reported within Australia in recent years have been in people returning from endemic areas (AIHW 2008; Clinical Interest Box 46-1. However, in 2002, 10 local cases of malaria were diagnosed and these were the first reports of malaria in Australia since 1986. During 2005–2009 3118 cases of malaria (an average of 620/year) were notified in Australia (National Notifiable Diseases Surveillance System). Four species of the genus Plasmodium are responsible for human malaria: P. vivax, P. malariae, P. ovale and P. falciparum. P. ovale malaria, which is found in West Africa, is considered rare. P. falciparum malaria is the most lethal form of malaria and is usually resistant to chloroquine.
Clinical interest box 46-1 Malaria Notification in Australia
Over the period 2005–2009 there were 1067 notifications of malaria in Queensland, 635 in New South Wales, 52 in the ACT, 537 in Victoria, 75 in Tasmania, 143 in South Australia, 175 in the Northern Territory and 435 in Western Australia. The total number of reported cases dropped from 817 in 2005 to 430 in 2009.
Source: National Notifiable Diseases Surveillance System, Department of Health and Ageing.
Malaria is transmitted to humans by the bite of an infected female Anopheles mosquito, as well as by blood transfusion (usually P. malariae).
Life cycle of the malarial parasite
To understand the chemotherapy of malaria, it is essential to review the life cycle of the malarial parasite, the plasmodium. Figure 46-1 presents the cycle in seven basic steps. Plasmodia have two interdependent life cycles: the sexual cycle, which takes place in the mosquito, and the asexual cycle, which occurs in the human body.
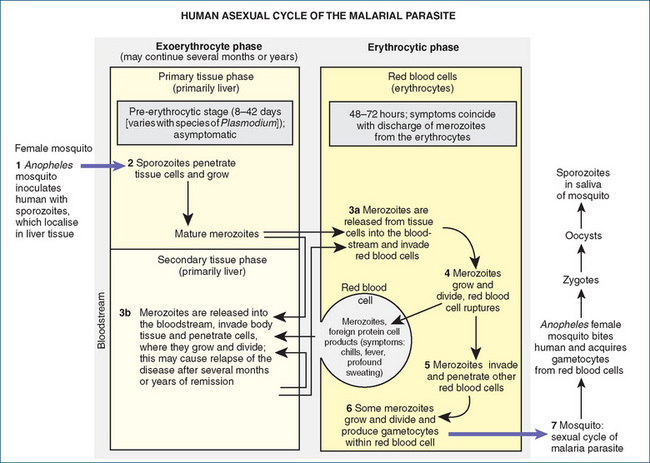
Sexual cycle
The sexual cycle is noted in step 7 of Figure 46-1. The female Anopheles mosquito becomes the carrier of the parasite by drawing blood containing male and female gametocytes (sexual forms of the parasite) from an infected person. In the stomach of the mosquito, the female gametocytes are fertilised by the male gametocytes to form zygotes, which undergo numerous cell divisions to develop into sporozoites. The formation of sporozoites in the mosquito completes the sexual cycle. Sporozoites then migrate to the salivary glands of the infected mosquito and are injected into the bloodstream of the human by the bite of the female insect (step 1, Figure 46-1).
Asexual cycle
In humans, the asexual cycle of the plasmodium consists of the exoerythrocytic phase and the erythrocytic phase.
Exoerythrocytic phase
Shortly after the introduction of the sporozoites into the circulation of the human, they leave the blood and enter fixed tissue cells (reticuloendothelial cells) of the liver, where multiplication and maturation take place (step 2). For a period of time that varies with different plasmodia (8–42 days), the individual exhibits no symptoms, no parasites are found in erythrocytes and the blood is non-infective. This phase is known as the pre-erythrocytic stage. The parasites are called primary tissue schizonts, or pre-erythrocytic forms. After this stage, the young parasites burst from the liver cells as merozoites.
Erythrocytic phase
When merozoites enter the bloodstream, they penetrate the erythrocytes and begin the erythrocytic phase of their existence (step 3a). In the case of P. vivax (but not P. falciparum), some of the merozoites invade other tissue cells to form secondary exoerythrocytic forms (step 3b). The relapses seen in P. vivax and other forms of malaria are believed to be caused by the successive formations of merozoites produced by various secondary exoerythrocytic forms of the parasite. Drugs affecting malarial parasites in the bloodstream do not always destroy those in the exoerythrocytic, or tissue, stage.
After the merozoites bore into red blood cells, they again multiply, but this time asexually, forming erythrocytic schizonts (Figure 46-2). The erythrocytic phase is completed when the parasitised red blood cells rupture, setting free many more merozoites that have formed from the schizonts. Pyrogenic substances are also liberated, causing a rapid rise in body temperature (step 4, Figure 46-1). Some of the merozoites may be destroyed in the plasma of the blood by leucocytes and other agents, but some enter other erythrocytes to repeat the cycle (step 5). The recurring chills, fever and prostration that are prominent clinical symptoms of malaria occur when the red blood cells rupture and release the young parasites with foreign protein and cell products. The erythrocytic phase lasts 48–72 hours, depending on the plasmodium involved. After a few cycles, some of the asexual forms of the malarial parasites develop into sexual forms called gametocytes (step 6). When the mosquito bites a person infected with malarial parasites and ingests the sexual forms, the cycle begins again.
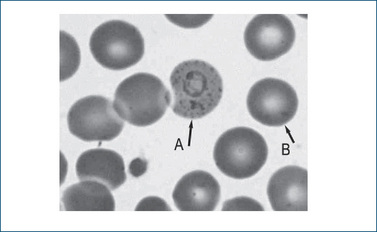
Figure 46-2 Peripheral blood film (magnification ×100) showing a normal red cell (B) and a red cell with a trophozoite of P. vivax displaying the typical signet ring form and Schüffners stippling (A). With kind permission of Ros Biebrick, Haematology, SA Pathology, Flinders Medical Centre, Adelaide, Australia.
P. vivax is the most common form of malaria; this infestation is usually mild, drug resistance is uncommon and it can easily be suppressed with antimalarial medications. The P. falciparum strain of malaria is less common but much more severe than the P. vivax form. Drug-resistant strains of P. falciparum exist, and the symptoms with this infestation occur at irregular intervals and can cause very serious complications. If untreated or if treatment is delayed, the disease may progress to irreversible cardiovascular shock and death. Although relapses are reported with P. vivax malaria, once the P. falciparum form is eliminated, no dormant forms remain in the liver; therefore no relapses are reported with P. falciparum.
People who harbour the sexual forms of plasmodia are called carriers, as it is from carriers that mosquitoes receive the parasite forms that perpetuate the disease. The asexual forms cause the clinical symptoms of malaria. Carriers should avoid giving blood because the recipient of this blood could contract malaria or become a carrier. A large number of malaria cases (some fatal) have arisen from transfusions of infected blood.
Antimalarial drugs
The emergence of drug-resistant strains of malaria, particularly that caused by P. falciparum, poses a major public health problem throughout the world. Despite the combined efforts of many countries to eradicate malaria, it remains the most devastating infectious disease in the world because of the many lives lost and the economic burden it imposes. It is essential that travellers contemplating a trip to areas of the world where malaria is endemic be aware of the need to obtain information about measures for reducing exposure to the disease. Malaria exists in New Guinea, Indonesia, Asia, Central and South America, the Middle East and many other countries. Travellers should receive malaria chemoprophylaxis before entering these areas. Current information on this can be obtained from several sources in Australasia:
The choice of a drug (schizonticide) for malaria (either prophylaxis [MP] or treatment [MT]) is based on the particular malarial strain involved and the stage of the plasmodium life cycle. Regimens include a number of drugs such as doxycycline (MP and MT) and clindamycin (MT) (refer to Chapter 44) and those discussed below.
Mefloquine (MP and MT)
Mefloquine is a blood schizonticide; it prevents the replication of asexual erythrocytic parasites but has no effect on the gametocytes of P. falciparum. Its exact mechanism of action is unknown but it is believed to inhibit protein synthesis (by binding plasmodial DNA), inhibit plasmodial haem polymerase and raise the intravascular pH of the acidic food vacuoles in the parasite. It is not effective in eliminating the exoerythrocytic or intrahepatic stages of P. vivax or P. ovale infections.
Mefloquine is indicated for the prevention and treatment of chloroquine-resistant malaria and multi-drug-resistant strains of P. falciparum, and to prevent malaria caused by P. vivax, P. ovale and P. malariae. It is a potent and fast-lacting antimalarial drug that is well absorbed orally, widely distributed in the body and reaches peak plasma concentration in 2–12 hours. It has a long elimination half-life of 13–33 days and is extensively metabolised by the liver, with <10% of the drug excreted unchanged in the urine. Most of the drug is excreted through bile in faeces.
Drug interactions with mefloquine include:
Adverse reactions are generally dose-related and occur more commonly in therapeutic than in prophylactic drug regimens. Adverse effects include nausea, vomiting, headache, dizziness, insomnia, gastric distress, diarrhoea, visual disturbances and, rarely, bradycardia and central nervous system (CNS) toxicity (depression, hallucinations, convulsions, psychosis, anxiety and confusion). Use mefloquine with caution in people with cardiac disease or hepatic impairment. Avoid use in people with epilepsy, heart block (first- or second-degree) and a history of psychiatric problems (psychosis, hallucinations, anxiety and depression). Mefloquine may impair the performance of skilled tasks for 2–3 weeks after dosing.
The usual adult dosage for prophylaxis is 250 mg once a week, commencing 2–3 weeks before departure, then on the same day of each week during and for 2 weeks after the visit to the endemic area.
Primaquine (MP and MT)
The mechanism of action of primaquine is unknown but it can bind to and alter plasmodial DNA. It is very effective in the exoerythrocytic stages of P. vivax and P. ovale malaria and against the primary phase (exoerythrocytic stage) of P. falciparum malaria. It is also effective against the sexual forms (gametocytes) of plasmodia (especially P. falciparum). Indications include the prevention of malaria relapses (radical cure) caused by P. vivax and P. ovale, and it is also effective against gametocytes of P. falciparum.
Primaquine is absorbed rapidly from the gastrointestinal tract (GIT) and reaches peak plasma concentrations within 2–3 hours. The drug undergoes extensive presystemic metabolism to the main metabolite, carboxyprimaquine, which is further metabolised to unidentified compounds. It has a half-life of about 6 hours, and the amount of unchanged drug excreted in urine is <4%.
Adverse reactions include gastric distress (stomach cramps or pain, nausea and vomiting), haemolytic anaemia (anorexia; back, leg or abdominal pain; dark urine; pale skin; weakness, fever), methaemoglobinaemia (cyanosis, dizziness, respiratory difficulty, weakness) and, rarely, leucopenia. The use of primaquine concurrently with other myelosuppressive drugs should be avoided because of the higher risk of myelosuppression. Use primaquine with caution in people with a history (personal or family) of acute haemolytic anaemia or a nicotinamide adenine dinucleotide (NADH) methaemoglobin reductase deficiency. Avoid use in people with primaquine hypersensitivity or G6PD deficiency (primaquine may cause haemolytic anaemia, especially in people deficient in G6PD) and in children under 12 months old because of increased risk of methaemoglobinaemia.
When used for prophylaxis (third-line drug) of malaria (susceptible organisms) the adult dose (base) is 30 mg daily starting 7 days before entering a malaria-endemic area and continuing for 1 week after leaving the area.
Quinine (MT)
Quinine was the first drug used to treat malaria (see Clinical Interest Box 46-2). As a schizonticidal agent, it concentrates in parasitised erythrocytes, which may be why it has selective toxicity during the erythrocytic stages of plasmodial infections. It can also bind to plasmodial DNA, thus inhibiting RNA synthesis and DNA replication.
Clinical interest box 46-2 Quinine, Gin and Tonic and Bitter Lemon for Malaria
Malaria is one of the most important infectious diseases in the world, especially in tropical areas. Epidemics (‘the ague’) have been described since the first century AD. It is due to an infection with parasites of the Plasmodium genus and involves bouts of severe fevers at regular 2- or 4-day intervals; severe damage to the brain, liver and spleen is likely if untreated. Among the early herbalist cures was St John’s wort.
The first specific effective treatment was found in the 17th century, with bark of the cinchona trees (called ‘quin’ by the Incas) found in South America. This remedy was brought from Ecuador to Europe by returning Jesuit missionaries, so the powder was called ‘Jesuit’s powder’. It quickly proved to be effective in Italy, Europe and England, and was highly prized.
The active principal was isolated in pure form in 1820 and named quinine; much effort went into procuring seeds and growing high-yield strains of the trees in Europe. Pharmacological studies on quinine led to the development of related drugs: chloroquine, mepacrine, primaquine and other aminoquinolines. Many strains of the Plasmodium parasites, however, have developed resistance to these drugs, and chloroquine-resistant malaria is now a major problem worldwide. Quinine also proved to have useful skeletal muscle-relaxant properties and has been used in the past to treat muscle spasms.
Quinine has a very bitter taste, so a tradition was instituted by the British Raj rulers in India and the Far East of putting gin into their quinine mixtures (‘tonic water’) to disguise the taste. This became a habit and nowadays tonic water (containing quinine) is taken to enhance the flavour of gin! Bitter lemon drinks also have added quinine to enhance the bitter flavours. Tonic water now contains about 83 mg quinine per litre, and bitter lemon about 44 mg quinine per litre.
Quinine sulfate is indicated in combination with other drugs (doxycycline or pyrimethamine plus sulfadoxine) for the treatment of chloroquine-resistant malaria caused by chloroquine-resistant P. falciparum.
Drug interactions with quinine include:
Adverse reactions include cinchonism (changes in colour vision or blurred vision, headache, very severe nausea and vomiting, tinnitus and transient hearing loss), GI distress (stomach pain or cramps, diarrhoea, nausea and vomiting) and, rarely, haemolytic uraemic syndrome (haemolytic anaemia, thrombocytopenia, disseminated intravascular coagulation and acute renal failure), hypoglycaemia, hypersensitivity and hepatotoxicity. Use with caution in people with a history of cardiac arrhythmias, G6PD deficiency or myasthenia gravis. Avoid use in people with quinine or quinidine hypersensitivity or a history of haemoglobinuria or optic neuritis.
The adult dosage (i.e. for persons >50 kg) for chloroquine-resistant P. falciparum malaria is 600 mg orally every 8 hours for 7 days, plus either doxycycline or clindamycin.
Combination antimalarials
A number of combination antimalarials are available. These include:
Artemether with lumefantrine (MT)
Artemether is a metabolised derivative of a natural antimalarial (artemisinin) extracted from a Chinese weed called qing hao. It acts rapidly on the asexual erythrocytic stages of P. vivax and chloroquine-sensitive/resistant and multi-drug-resistant P. falciparum. Artemether is not suitable for prophylaxis as it does not affect either primary or latent tissue-stage parasites. Lumefantrine is structurally related to quinine and is very effective against P. falciparum. The combination of lumefantrine with artemether gives effectiveness greater than that of either drug alone. The currently available drug combination is a fixed ratio of 1 part artemether to 6 parts lumefantrine. Both drugs are thought to interfere with the conversion of haem to the malaria pigment haemozoin in the food vacuole of the malarial parasite. Additionally, both inhibit nucleic acid synthesis within the malarial parasite.
Very little is known about the pharmacokinetics of this combination, due to the lack of an intravenous formulation for pharmacokinetic studies, and because large intersubject and intrasubject variability occurs in the plasma drug concentrations. Administered orally, artemether is rapidly absorbed and peak plasma concentration is reached in approximately 2 hours. In contrast, lumefantrine is highly lipophilic and absorption is slow, with peak plasma concentration reached at about 6–8 hours after dosing. Absorption is improved if the tablets are taken with food. Both drugs are extensively bound to plasma proteins (>97%), artemether being extensively metabolised to the active metabolite dihydroartemisinin, predominantly by CYP3A4/5, and lumefantrine to the active debutylated metabolite by CYP3A4. Of clinical significance is the fact that lumefantrine is an inhibitor of CYP2D6; hence drug interactions occur with other drugs (e.g. flecainide, metoprolol, clomipramine) metabolised by CYP2D6, resulting in increased plasma concentration of the coadministered drug. Relevant drug information sources should be consulted for potential drug interactions, prior to administration of artemether with lumefantrine. Although the drugs are cleared from plasma with an elimination half-life of 2 hours for artemether and a terminal half-life of 2–3 days for lumefantrine, no urinary excretion data are available.
Common adverse effects include nausea, vomiting, diarrhoea, headache, dizziness, fatigue and muscle problems (e.g. myalgia, weakness). Rarely, an increase in Q-T interval occurs and hence this combination should be avoided in persons with coexisting prolongation of the Q-T interval or in persons with a family history of sudden death. Serious birth defects have been observed in animal studies, and this combination is contraindicated in the first trimester of pregnancy.
Atovaquone with proguanil (MP and MT)
Atovaquone is a hydroxynaphthoquinone drug that is used in combination with proguanil. Together, atovaquone with proguanil causes a synergistic antimalarial effect as a result of interference with the synthesis of pyrimidines, which are required for nucleic acid synthesis. Atovaquone inhibits mitochondrial electron transport in P. falciparum. In contrast, proguanil, through its active metabolite cycloguanil, inhibits dihydrofolate reductase (like pyrimethamine) and potentiates the ability of atovaquone to collapse the mitochondrial membrane potential. The currently available drug combination is a fixed ratio of 2.5 parts atovaquone to 1 part proguanil (e.g. 250 mg atovaquone/100 mg proguanil tablet) and it is used for the treatment of uncomplicated P. falciparum malaria and for prophylaxis of P. falciparum malaria. For the latter, treatment is commenced 2–3 days prior to entering an endemic area and continued for 1 week after leaving the area. Prophylaxis may be continued for up to 3 months. Proguanil monotherapy is also indicated for malaria prophylaxis but specialist advice should be sought.
Atovaquone is poorly and erratically absorbed (affected by dose and diet), while proguanil is rapidly and extensively absorbed regardless of diet. It is recommended that the drug be taken with food or a milky drink, as dietary fat enhances absorption of atovaquone. Unlike proguanil, atovaquone is not metabolised and >90% is excreted as unchanged drug in faeces, with very little excretion via urine. Proguanil is metabolised to polar metabolites which, along with unchanged drug (˜40%), are excreted in urine. The elimination half-life of atovaquone is 2–3 days in adults (1–2 days in children), whereas the elimination half-life of proguanil is 12–15 hours.
At the doses used, adverse effects are mild (e.g. abdominal pain, nausea and vomiting) and of limited duration. The safety of the combination in human pregnancy has not been fully established, and breastfeeding is not recommended as atovaquone is excreted in breast milk.
Pyrimethamine with sulfadoxine (MT)
Pyrimethamine is an antiprotozoal agent used to treat malaria and toxoplasmosis. It binds to and inhibits the protozoal enzyme dihydrofolate reductase, thus inhibiting the conversion of dihydrofolic acid to tetrahydrofolic acid. This results in a depletion of folate, which is essential for nucleic acid synthesis and protein production. A synergistic effect is achieved in combination with sulfadoxine, which inhibits the utilisation of para-aminobenzoic acid (PABA) in the synthesis of dihydropteroic acid (see Chapter 44). Pyrimethamine with sulfadoxine is administered with oral quinine for the treatment of chloroquine-resistant P. falciparum malaria.
Pyrimethamine is orally absorbed and widely distributed in the body, although it concentrates mainly in blood cells, kidneys, liver and spleen. It reaches peak plasma concentrations in 2–6 hours and has a half-life of 35–175 hours. It is extensively metabolised in the liver to unknown metabolites and about 30% is excreted in the urine over 40 days.
Adverse reactions are usually rare but, in high doses, gastric distress (anorexia, nausea, vomiting, diarrhoea), atrophic glossitis (pain, burning or inflamed tongue, change or a loss of taste sensation) and blood dyscrasias (e.g. agranulocytosis, megaloblastic anaemia, thrombocytopenia) may be seen. When pyrimethamine is administered concurrently with other bone marrow depressants, an increase in leucopenia and/or thrombocytopenia may result. Monitor full blood counts closely. Use this combination with caution in people with liver function impairment. Avoid use in people with pyrimethamine hypersensitivity, anaemia, bone marrow suppression or a history of convulsive disorders. Pyrimethamine and sulfadoxine are both excreted in breast milk.
The adult oral dosage for the treatment of chloroquine-resistant malaria is three tablets (75 mg/1500 mg) as a single dose on the third and fourth days of oral quinine treatment.
Amoebiasis
Amoebiasis is an infection of the large intestine produced by a protozoan parasite, Entamoeba histolytica. This infestation is found worldwide but is prevalent and severe in tropical areas. Transmission is usually through ingestion of cysts (faecal to oral route) from contaminated food or water or from person-to-person contact. Poor personal hygiene can increase the spread of this parasite.
Life cycle of amoebae
This protozoan has two stages in its life cycle: the trophozoite (vegetative amoeba), which is the active, motile form; and the cyst, or inactive, drug-resistant form that appears in intestinal excretion. The trophozoite stage is capable of amoeboid motion and sexual activity. Because of its susceptibility to injury, it generally succumbs to an unfavourable environment. However, in certain circumstances, the trophozoite protects itself by entering the cystic stage. During this phase, the protozoan becomes inactive by surrounding itself with a resistant cell wall within which it can survive for a long time, even in an unsuitable environment.
The complete life cycle of the amoeba occurs in humans, the main host. It begins with ingestion of cysts that are present on hands, food or water contaminated by faeces. In the stomach, the hydrochloric acid does not destroy the swallowed cysts, which pass unharmed into the small intestine. The digestive juices penetrate the cystic walls and the trophozoites are released. The motile amoebae later pass into the colon, where they live and multiply for a time, feeding on the bacterial flora of the gut.
The presence of bacteria is essential to their survival. Finally, before excretion, the trophozoites move towards the terminal end of the bowel and again become encysted. After the cysts are eliminated in the faeces, they remain viable and infective. The cycle may begin again when the cysts appearing in faecal excretion are ingested through contamination of food or water.
The parasite causing amoebiasis replicates in three major locations: the lumen of the bowel, the intestinal mucosa and extraintestinal sites. Amoebiasis is thus classified according to its primary site of action: intestinal amoebiasis, where amoebic activity is restricted to the bowel lumen or intestinal mucosa; or extra-intestinal amoebiasis, where parasitic invasion occurs outside the intestine.
Intestinal amoebiasis
Intestinal amoebiasis may be manifested as an asymptomatic intestinal infection or a symptomatic intestinal infection that may be mild, moderate or severe. In asymptomatic intestinal amoebiasis, the action of the parasite is restricted to the lumen of the bowel. The individual is asymptomatic but becomes a carrier of the disease by passing mature cysts of the parasite in formed stools. Outside the body, the cysts can live for several weeks, surviving dry, freezing or high-temperature conditions. By this route the infection is transmitted from person to person by flies or contaminated food or water. Ordinary concentrations of chlorine used for purification do not destroy the cysts. If the carrier fails to follow any drug treatment, serious GI pathological problems eventually develop. Occasionally, mild symptoms occur, including vague abdominal pain, nausea, flatulence, fatigue and nervousness.
Symptomatic amoebiasis occurs when the trophozoites in the lumen of the bowel penetrate the mucosal lining of the colon. After they multiply and thrive on bacterial flora, a large infestation occurs, producing diarrhoea and abdominal pain. The increased loss of fluid may cause prostration. In addition, ulcerative colitis may result. This state of the disease is called intestinal amoebiasis and is usually diagnosed as mild, moderate or severe, according to the intensity of the symptoms and the extent of the disease.
Extraintestinal amoebiasis
The term extraintestinal amoebiasis means the parasites have migrated to other parts of the body, such as the liver or, occasionally, the spleen, lungs or brain. When the parasites are in the liver, necrotic foci develop because of their destructive effect on tissues. When there is liver involvement, the terms liver abscess and hepatic amoebiasis are usually used.
Amoebicidal drugs
Drugs for the treatment of amoebiasis are classified according to the site of amoebic action. Luminal amoebicides act primarily in the bowel lumen and are generally ineffective against parasites in the bowel wall or tissues. Tissue amoebicides are drugs that act primarily in the bowel wall, liver and other extraintestinal tissues. No single drug is effective for both types of amoebiasis; therefore a luminal and extraluminal (tissue) amoebicide or combination therapy is often prescribed. Drugs used for symptomatic intestinal or extraintestinal amoebiasis include metronidazole (Chapter 44) and tinidazole, and for the eradication of cysts, paromomycin.
Paromomycin
Paromomycin is both an amoebicidal and an antibacterial agent. The drug is an aminoglycoside antibiotic with antibacterial properties similar to those of neomycin. Paromomycin acts directly on intestinal amoebae and on bacteria such as Salmonella and Shigella. Because the drug is poorly absorbed from the GIT, it exerts no effect on systemic infections such as extraintestinal amoebiasis. It is indicated for the treatment of acute and chronic intestinal amoebiasis. Paromomycin is poorly absorbed from the GIT; thus most of the drug is excreted in the faeces.
Adverse reactions include nausea, diarrhoea and gastric distress. Paromomycin is an aminoglycoside, so the drug interactions, warnings and contraindications possible with aminoglycosides may also occur with paromomycin. See the discussion of aminoglycoside antibiotics in Chapter 44.
The adult dosage for treating intestinal amoebiasis is 25–35 mg/kg daily in three divided doses given with meals for 7 days. Paromomycin is available only through the Special Access Scheme (SAS).
Miscellaneous antiprotozoal drugs pentamidine
Pentamidine
Pentamidine is an aromatic diamine with a broad spectrum of activity against several species of pathogenic protozoa including Trypanosoma brucei gambiense (sleeping sickness) and the ascomycetous fungus Pneumocystis jiroveci (formerly known as Pneumocystis carinii). It is indicated for the treatment of P. jiroveci in AIDS patients and for trypanosomiasis excluding Trypanosoma brucei rhodesiense, which is refractory to pentamidine. The mechanism of action is unclear but the sites of drug action include membrane phospholipids, intracellular enzymes and DNA and RNA. The drug is administered IV as pentamidine isethionate but can also be administered via inhalation using a specialised nebuliser. It is eliminated slowly over weeks to months. Common adverse effects include nausea, vomiting, diarrhoea and a range of cardiovascular effects and abnormalities in glucose metabolism. Use as an antiprotozoal agent is under specialist advice.
Toxoplasmosis
Toxoplasmosis is caused by an intracellular parasite Toxoplasma gondii. This parasite is found worldwide in raw vegetables and the soil, and infests a variety of animals, including humans. Cats and other feline species are the natural hosts, and it is often harboured in the host with no evidence of the disease. Toxoplasmosis is contracted by ingesting cysts in inadequately cooked or raw meat, fish or vegetables or by accidentally ingesting cysts from cat faeces.
Symptomatically, the individual may experience lymphadenopathy, fever and, occasionally, a rash on the palms and soles. The most serious complication of toxoplasmosis is meningoencephalitis, which is common in HIV patients. Acute infection is treated with a combination of sulfadiazine plus pyrimethamine orally, both of which alter the folic acid cycle of the Toxoplasma organism, resulting in its death. Life-long suppressive therapy is essential to prevent relapse, and inclusion of calcium folinate reduces bone marrow suppression.
Trichomoniasis
Trichomoniasis is a disease of the vagina caused by Trichomonas vaginalis. Its characteristic presentation consists of a wet, inflamed vagina, a ‘strawberry’ cervix and a thin, yellow, frothy malodorous discharge. Usually, both sexual partners are infected by this organism, which can be identified microscopically from semen, prostatic fluid or exudate from the vagina. Infections often recur, which indicates that the protozoans persist in extravaginal foci, the male urethra or the periurethral glands and ducts of both sexes.
Metronidazole is the drug of choice for treating trichomoniasis. Treatment must be given simultaneously to both partners involved in order to affect a cure.
Mycobacterial infections
Mycobacterial infections in humans include Mycobacterium avian complex (MAC), tuberculosis and leprosy.
Mycobacterium avian complex
Mycobacterium avium complex (MAC) consists of two species: Mycobacterium avium and Mycobacterium intracellulare. M. avium accounts for the majority of MAC infections in AIDS patients while M. intracellulare is responsible for 40% of MAC infections in immunocompetent individuals. These mycobacteria are found in freshwater and saltwater worldwide and the common environmental sources of MAC include aerosolised water, household and hospital water supplies, house dust, soil, birds, farm animals and the various components that make up a cigarette.
MAC is transmitted via inhalation and oral ingestion. Following translocation across the mucosal epithelium the bacteria infect macrophages and spread in the submucosal tissue. The mycobacteria are then spread via the lymphatic system and, in immunocompromised patients (e.g. AIDS), spread to the liver, spleen, bone marrow and other sites. Specialist advice should be sought regarding drug regimens for either HIV negative or HIV positive patients. Drugs used include clarithromycin or azithromycin, amikacin or streptomycin (refer Chapter 44) and some of the drugs discussed below.
Tuberculosis
Tuberculosis (TB), a chronic granulomatous infection caused by the acid-fast bacillus Mycobacterium tuberculosis, is both a curable and a controllable infection that is transmitted from person to person. An estimated 9.2 million new cases were registered worldwide in 2006 of which 709,000 cases were HIV-positive. India, China, Indonesia, South Africa and Nigeria were ranked 1 (India) to 5 (Nigeria) for the highest number of reports. The Asian region (Southeast Asia and the Western Pacific region) accounts for 55% of the global cases (World Health Organization [WHO] 2008).
The revised WHO ‘Stop TB Strategy’ aims to have a TB-free world. The current goals are to reduce TB prevalence and death rates by 50% (relative to 1990) by 2015 and to eliminate TB as a public health problem (1 case per million population) by 2050. Within Australasia, TB remains a problem among Aboriginal people, the aged, immigrants from endemic countries and Maoris; in 2005–2009, 5584 cases were recorded by the Communicable Diseases Australia–National Notifiable Diseases Surveillance System (www.health.gov.au).
The development of drug-resistant TB is a major concern. A 2001 study estimated the incidence of resistance to at least one antituberculosis drug among new cases of TB at about 10% in Australia, 11% in New Zealand, 2% in New Caledonia, 12% in the USA and 7% in England and Wales. The highest incidences of multi-drug resistance were found in Estonia (14.1%) and Henan Province in China (10.8%). In contrast, multi-drug resistance rates were low in Australia (2%) and New Zealand (1.1%) (Espinal et al 2001).
The WHO recommends management under the Directly Observed Treatment—Short Course (DOTS) strategy as the approach to TB control and cure in 85% of all new cases. The DOTS strategy includes five steps and involves the health system in the steps to a cure:
The DOTS program needs government support and funding to achieve long-term TB control. The result of not instituting such a plan is obvious—increased numbers of cases of TB, multi-drug-resistant TB and deaths.
Pathogenesis
TB most commonly affects the lungs but other body areas can also be infected, such as bones, joints, skin, meninges or the genitourinary tract. M. tuberculosis is an aerobic bacillus that needs a highly oxygenated organ site for growth; thus the lungs, the growing ends of bones and the cerebral cortex are ideal sites. The bacilli can become inactive and be walled off by calcified and fibrous tissues, often for the lifetime of the person. If host defences break down, however, or if the individual receives an immunosuppressive drug, the bacilli may be reactivated.
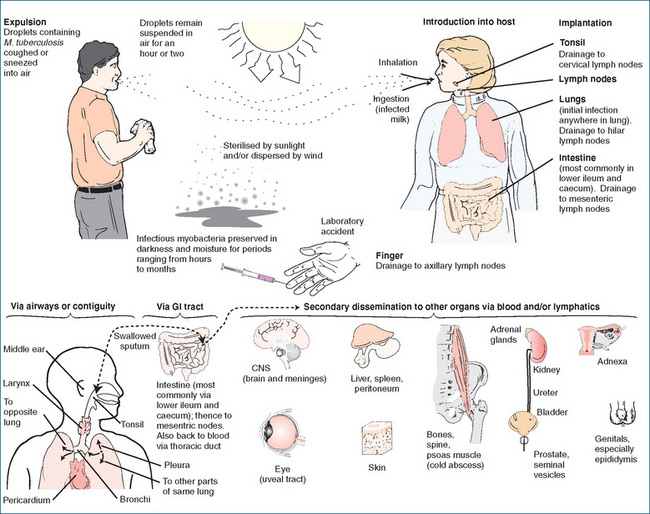
Tubercle bacilli may be transmitted via airborne droplets when an infected person coughs or sneezes, but cannot be transmitted on objects such as dishes, clothing or sheets and bedding (Figure 46-3). Persons producing sputum generally have many bacilli and are more infectious than the infected person who does not cough. Sharing an enclosed environment with an infected person creates a high risk of developing this infection (Ebert 1993).
Drug treatment regimens
Effective drug regimens are available to treat TB. Drug selection is based on the development of drug-resistant organisms and drug toxicity. General guidelines include:
Isoniazid (also known as INH) is an antimycobacterial (bactericidal) agent that affects mycobacteria in the dividing phase. Its exact mechanism of action is unknown but it is believed to inhibit mycolic acid synthesis and cause cell wall disruption in susceptible organisms. Isoniazid is indicated for the primary treatment, re-treatment and prophylaxis of TB.
Pharmacokinetics
Isoniazid is well absorbed orally and is widely distributed throughout the body. It is metabolised in the liver by acetylation, a characteristic that displays a genetic polymorphism in humans. In people who metabolise the drug quickly, often referred to as ‘fast acetylators’, peak plasma concentrations occur in 1–2 hours, while this takes 4–6 hours for those classed as ‘slow acetylators’ (refer to Chapter 7 for further details). The half-life in fast acetylators is 0.5–1.6 hours, and in slow acetylators 2–5 hours. Slow acetylators have a decrease in hepatic N-acetyltransferase, the enzyme that carries out the reaction. Excretion is primarily via the urine, partly as unchanged drug and as the inactive acetylated form.
Slow acetylators may need lower drug dosages and are more apt to develop adverse reactions, particularly peripheral neuritis. The incidence of slow acetylators is highest in Egyptian, Israeli, Scandinavian, other Caucasian and black populations, while the lowest incidence is in Eskimo, Oriental and native American populations.
Drug interactions
Various effects may occur when isoniazid is given with the drugs listed:
Alcohol. Daily use of alcohol may result in increased isoniazid metabolism and higher risk of hepatotoxicity. Monitor patients, as a drug dose adjustment may be necessary
Carbamazepine. Isoniazid may inhibit metabolism (CYP3A), resulting in increased carbamazepine plasma concentration and toxicity. Monitor plasma concentration closely and adjust dose if indicated
Disulfiram. May increase the incidence of CNS adverse effects such as ataxia, irritability, dizziness or insomnia. Monitor closely for these symptoms, as dosage reduction or even discontinuation of disulfiram may be required
Hepatotoxic drugs. May increase potential for hepatotoxicity. Avoid, or a potentially serious drug interaction may occur Ketoconazole, miconazole or rifampicin. Isoniazid with ketoconazole may decrease plasma concentration of ketoconazole; if both isoniazid and rifampicin are given with ketoconazole, the plasma levels of ketoconazole or rifampicin have been reported to be undetectable. The combination of isoniazid or rifampicin together or individually with ketoconazole or parenteral miconazole is not recommended. Rifampicin with isoniazid may increase the potential for hepatotoxicity, especially in patients with liver impairment and in fast acetylators of isoniazid. Monitor closely for hepatotoxicity, especially during the first 90 days of therapy
Phenytoin. May result in impaired phenytoin metabolism, leading to increased plasma concentration and toxicity. The phenytoin dose may need to be adjusted. Monitor plasma phenytoin concentration closely
Adverse reactions
These include gastric distress, anorexia, nausea, vomiting, weakness, hepatitis, peripheral neuritis and, rarely, blood dyscrasias, hypersensitivity and optic neuritis. Increases in plasma transaminases may occur in the first few months of treatment.
Warnings and contraindications
Use with caution in people with severe kidney disease and convulsive disorders. Avoid use in people with isoniazid, ethionamide, pyrazinamide, niacin or nicotinic acid hypersensitivity or liver function impairment, and in alcoholics.
Dosage* and administration
The adult oral dose (daily regimen) of isoniazid is 5 mg/kg (max 300 mg daily). When a twice-weekly regimen is used the dose is 15 mg/kg (max 900 mg) twice weekly. For children, the daily regimen is 10 mg/kg up to a maximum of 300 mg daily.
Additional comments
If gastric distress occurs, this drug may be taken with meals or an antacid, although aluminium antacids should be separated by at least 1 hour from isoniazid, as absorption is decreased when food or antacids are given concurrently. Coadministration of pyridoxine (25 mg) with each dose of isoniazid is recommended to reduce the risk of peripheral neuropathy in at-risk individuals.
* Dosage regimens differ slightly between various authorities.
Drug monograph 46-2 Rifampicin
Rifampicin is a broad-spectrum bactericidal antibiotic (antimycobacterial) that blocks mycobacterial RNA transcription. It is indicated for the treatment of TB and for asymptomatic carriers of Neisseria meningitidis. It is well absorbed orally and widely distributed in the body.
Pharmacokinetics
Rifampicin is lipid-soluble, so it may reach and kill intracellular and extracellular susceptible bacteria. Therapeutic plasma concentrations occur in 1.5–4 hours, and the elimination half-life is up to 5 hours. Rifampicin is metabolised in the liver to a range of metabolites, including an active metabolite (25-O-desacetylrifampicin), and excreted primarily in the faeces.
Drug interactions
Rifampicin induces hepatic cytochrome P450 enzymes, which results in increased clearance of drugs metabolised by the liver. The following drug interactions may occur when rifampicin is given with the drugs listed.
Adverse reactions
These include gastric distress; hypersensitivity; a flu-like syndrome; red–orange discolouration of urine, faeces, saliva, sputum, sweat and tears (soft contact lenses can be discoloured by rifampicin); fungal overgrowth; and, rarely, blood dyscrasias, hepatitis and interstitial nephritis.
Contraindications
Avoid use in people with liver function impairment or previous allergic reaction to rifampicin or rifabutin, and in alcoholics.
Dosage and administration
The oral dosage of rifampicin for adults and children in combination with other agents (for TB) is 10 mg/kg to a maximum of 600 mg daily, or 15 mg/kg (max 600 mg) three times a week.
Additional comments
To obtain the maximum absorption, rifampicin should be taken with a full glass of water on an empty stomach, i.e. 1 hour before or 2 hours after a meal. If gastric distress is a problem, it may be taken with food. Concurrent alcohol consumption should be avoided. Blood counts should be monitored and dental procedures deferred until blood counts are normal.
Antimycobacterial agents used for MAC and TB
Antimycobacterial agents used to treat MAC and TB are listed in alphabetical order, as follows.
Capreomycin (TB)
Capreomycin is an antimycobacterial agent with an unknown mechanism of action. It is indicated in combination therapy for the treatment of pulmonary TB after primary medications (isoniazid, rifampicin, pyrazinamide and ethambutol) fail, or when these medications cannot be used because of resistant bacilli or drug toxicity. Administered parenterally, capreomycin has a half-life of 3–6 hours and reaches peak plasma concentration in 1–2 hours after IM administration. It is excreted by the kidneys primarily unchanged.
When capreomycin is given with any of the drugs in Drug Interactions 46-1, very serious reactions may result. Avoid concurrent use if possible. Adverse reactions include nephrotoxicity, hypokalaemia, neuromuscular blockade, ototoxicity (auditory: hearing loss, ringing or buzzing noise; vestibular: ataxia, dizziness, nausea, vomiting), hypersensitivity and pain, soreness or hardness at the injection site. Use capreomycin with caution in people with dehydration. Avoid use in persons with capreomycin hypersensitivity, eighth cranial nerve damage, myasthenia gravis or Parkinson’s disease. Dosage reduction is required in people with renal impairment.
Drug interactions 46-1 Capreomycin
| Drug | Possible effects and management |
| Aminoglycosides | Increased risk of developing ototoxicity, nephrotoxicity and neuromuscular blockade. Hearing loss may progress to deafness, even after the drug is stopped. This can be a very dangerous combination. Avoid concurrent drug administration |
| Amphotericin B, cisplatin, cyclosporin, ethacrynic acid, frusemide (parenteral), paromomycin, streptomycin or vancomycin | Concurrent or even sequential use of capreomycin with any of these drugs can increase the risk of ototoxicity and nephrotoxicity. Hearing loss may occur and progress to deafness, even if drugs are stopped. Avoid if at all possible |
| Neuromuscular-blocking agents | May result in increased neuromuscular-blocking effects, causing respiratory depression or paralysis. Monitor closely, especially during surgery or in the postoperative period. If possible, avoid this combination |
Cycloserine (MAC and TB)
This is a second-line broad-spectrum antibiotic that can be bacteriostatic or bactericidal, depending on drug concentration at the infection site and on organism susceptibility. Cycloserine is an antimycobacterial agent that interferes with bacterial cell wall synthesis. In combination with other drugs, it is indicated for the treatment of TB or MAC after failure of first-line antitubercular medications.
Cycloserine is well absorbed orally and is widely distributed in body tissues and fluids. The peak plasma concentration occurs in 3–4 hours and the drug has a half-life of 10 hours. About 35% of cycloserine is metabolised, with excretion primarily via the kidneys. See Drug Interactions 46-2 for agents that interact with cycloserine and possible outcomes.
Drug interactions 46-2 Cycloserine
| Drug | Possible effects and management |
| Alcohol | In chronic alcohol abusers, cycloserine may increase the risk of seizures. Avoid or a potentially serious drug interaction may occur |
| Isoniazid | May increase CNS adverse effects such as seizures. Monitor closely, as dosage adjustments may be necessary |
Adverse reactions include headache, dose-related CNS toxicity (confusion, dizziness, sedation, irritability, restlessness, depression, tremors, nightmares, mood alterations, speech problems, anxiety and thoughts of suicide), hypersensitivity, peripheral neuropathy and convulsions. Use this drug with caution in people with severe anxiety, depression or psychosis. Avoid use in people with cycloserine hypersensitivity, a history of epilepsy and in alcoholics. Dosage reduction is required in people with renal impairment.
Ethambutol (MAC and TB)
This drug is bacteriostatic; it is believed to diffuse into the mycobacteria bacilli and suppress RNA synthesis. Ethambutol is effective only against actively dividing mycobacteria. It is indicated in combination with other drugs for the treatment and re-treatment of TB and MAC. There is an increased risk of neurotoxicity, such as optic and peripheral neuritis, when ethambutol is administered concurrently with other neurotoxic medications. Monitor closely if concurrent therapy is instituted.
Ethambutol is absorbed orally and distributed to most body tissues and fluids, with the exception of cerebrospinal fluid. High concentrations are found in the kidneys, lungs, saliva, urine and erythrocytes. Therapeutic plasma concentrations occur within 4 hours and the half-life is 3–4 hours. Ethambutol is partly metabolised (15%) in the liver, and 75% of the oral dose is excreted unchanged in urine and 10% in faeces.
Adverse reactions include gastric distress, confusion, disorientation, headache, optic neuritis, peripheral neuritis, hypersensitivity and acute gouty arthritis.
Use ethambutol with caution in people with acute gouty arthritis, as ethambutol may raise uric acid levels. Avoid use in patients with ethambutol hypersensitivity, optic neuritis or kidney function impairment.
Pyrazinamide (TB)
Pyrazinamide is an antimycobacterial agent with an unknown mechanism of action. Depending on concentration at the site of action and susceptibility of the mycobacteria, this drug can be bacteriostatic or bactericidal. It is indicated in combination with other agents for the treatment of TB. Pyrazinamide is well absorbed orally and is widely distributed in the body. The time to peak plasma concentration is 1–2 hours and the elimination half-life is 9–10 hours. Pyrazinamide is primarily metabolised in the liver and excreted by the kidneys.
Adverse reactions include arthralgia related to hyperuricaemia, pruritus, rash and, rarely, gouty arthritis and hepatotoxicity. Use this drug with caution in patients with gout and type 1 diabetes, as control may become more difficult. Avoid use in people with hypersensitivity to pyrazinamide, ethionamide, isoniazid, niacin or nicotinic acid, or severe liver disease. Use in patients with renal impairment may result in accumulation of uric acid crystals that can worsen renal impairment. This drug is only available through the SAS.
Rifabutin (TB and MAC)
Rifabutin is an antimycobacterial agent indicated for the prophylaxis of disseminated MAC in people with advanced HIV infection. It inhibits DNA-dependent RNA polymerase in susceptible Escherichia coli and Bacillus subtilis, and has in-vitro activity against many strains of M. tuberculosis. It is used for the prophylaxis and treatment of MAC and the treatment of TB if rifampicin is unsuitable.
Rifabutin is absorbed from the GIT, reaches peak plasma concentration in 2–4 hours and has a terminal half-life of 45 hours. It is highly lipophilic and crosses the blood–brain barrier; cerebrospinal fluid levels are about 50% of the corresponding plasma concentration. It is metabolised in the liver (five metabolites have been identified) and excreted primarily by the kidneys. When rifabutin is administered concurrently with zidovudine (AZT), some studies indicate that the mean zidovudine plasma concentration may decrease. Monitor closely if combination therapy is used.
Adverse reactions include nausea, vomiting, skin rash and, rarely, joint pain, a change in taste sensations, myalgia, neutropenia, pseudojaundice (yellow-tinged skin) and uveitis. Avoid use in people with rifabutin or rifampicin hypersensitivity and active TB. Dosage reduction by about 50% is required in patients with renal impairment.
Streptomycin (TB)
Streptomycin is an aminoglycoside antibiotic that is poorly absorbed from the GIT; it is therefore given intramuscularly.
It was one of the first effective agents used in the late 1940s to treat TB and it is still an important agent in managing severe TB. Like the other aminoglycosides, its major toxicities include ototoxicity and nephrotoxicity, especially when given to patients with impaired renal function or with other medications with the same toxicities. (See Chapter 44 for detailed information on the aminoglycosides.)
Leprosy
Leprosy, or Hansen’s disease, is caused by Mycobacterium leprae in humans. It was first reported in 600 BC and today the estimated number of infected people is nearly 15 million worldwide. Recent data indicate that leprosy is prevalent in many countries, including India and Brazil. By the end of 1999 10 million patients had been cured of leprosy by using multi-drug therapy (see Clinical Interest Box 46-3). In Australia during the period 2005–2009, 43 cases of leprosy were reported. Of these, 10 were from Western Australia, 5 from Victoria, 10 from New South Wales, 5 from the Northern Territory, 9 from Queensland, 3 from South Australia and 1 from Tasmania. The Australian Capital Territory was the only state free of leprosy over this period (Communicable Diseases Australia—National Notifiable Diseases Surveillance System, www.health.gov.au).
Clinical interest box 46-3 Global Alliance for Elimination of Leprosy (GAEL)
This global alliance, which comprises the leprosy endemic countries, the International Federation of Anti-Leprosy Associations, the pharmaceutical company Novartis, the WHO and the Nippon/Sasakawa Memorial Health Foundation, was formed in 1999. Its mission is to eliminate leprosy (defined as a registered prevalence of <1 case per 10,000 population) globally. The Nippon Foundation has pledged about A$50 million to implement the strategy, and Novartis is providing free multi-drug therapy for all remaining leprosy patients in the world. ‘According to official reports received during 2008 from 118 countries and territories, the global registered prevalence of leprosy at the beginning of 2008 stood at 212,802 cases, while the number of new cases detected during 2007 was 254,525 (excluding the small number of cases in Europe). The number of new cases detected globally has fallen by 11,100 cases (a 4% decrease) during 2007 compared with 2006’.
Source: Leprosy Today, www.who.int/lep [1 October 2009].
Although the precise mode of transmission is unknown, the incubation period for leprosy is a few months to decades. Large numbers of leprosy bacilli are generally shed from skin ulcers, nasal secretions, the GIT and, perhaps, biting insects.
M. leprae is a bacillus that in humans first presents as a skin lesion: a large plaque or macule that is erythematous or hypopigmented in the centre. More numerous lesions, peripheral nerve trunk involvement and the common complications of plantar ulceration of the feet, foot-drop, loss of hand function and corneal abrasions may follow.
Drug therapy can cure leprosy, stop transmission of the disease and prevent the disfigurement that results in social exclusion and stigmatisation. Since 1982, the WHO has recommended the use of multi-drug therapy comprising dapsone, rifampicin and clofazimine. This combination is bactericidal and prevents the occurrence of drug resistance. For individuals not tolerating clofazimine, an alternative regimen of monthly rifampicin, ofloxacin and minocycline for 2 years has been recommended. For individuals not tolerating rifampicin, the WHO has recommended daily administration of a combination of clofazimine, ofloxacin and minocycline for 6 months, followed by daily administration of clofazimine with either minocycline or ofloxacin for an additional 18 months (WHO 1998). These WHO guidelines are followed in Australia.
Drugs used to treat leprosy
Dapsone
Dapsone is an antibacterial (antileprosy) agent that is bacteriostatic, with an action similar to that of the sulfonamides. It may also be a dihydrofolate reductase inhibitor. Dapsone is effective against M. leprae and is indicated for the treatment of all types of leprosy and for dermatitis herpetiformis. Dapsone is absorbed orally, distributed throughout the body and found in fluids and in all body tissues. Therapeutic plasma concentrations are achieved in 2–6 hours and the half-life is around 30 hours. The drug is acetylated by N-acetyltransferase in the liver; thus slow acetylators are more apt than fast acetylators to develop higher plasma concentrations and adverse reactions. Excretion is via the kidneys.
See Drug Interactions 46-3 for drugs that may interact with dapsone and possible outcomes.
Drug interactions 46-3 Dapsone
| Drug | Possible effects and management |
|---|---|
| Didanosine (DDI) | Concurrent drug administration may reduce absorption of dapsone. Dapsone requires an acid medium for absorption, while DDI is given with a buffer to neutralise stomach acid to increase absorption. Administer dapsone a minimum of 2 hours before DDI |
| Trimethoprim, trimethoprim–sulfamethoxazole | Decreased elimination of both dapsone and trimethoprim may occur. Monitor for increased incidence of adverse effects |
Adverse reactions include hypersensitivity, haemolytic anaemia, methaemoglobinaemia and, rarely, CNS adverse effects, gastric distress, blood disorders, exfoliative dermatitis, hepatic damage, mood alterations, peripheral neuritis and the sulfone syndrome (fever, tiredness, exfoliative dermatitis, jaundice, lympadenopathy, anaemia and methaemoglobinaemia). Use dapsone with caution in people with liver function impairment. Avoid use in patients with dapsone or sulfonamide hypersensitivity, severe anaemia, severe G6PD deficiency or methaemoglobin reductase deficiency.
The adult dapsone antileprosy dosage (given in combination with other antileprosy drugs) is 100 mg orally daily. As a suppressant for dermatitis herpetiformis, the adult dosage is 50 mg orally daily initially, increased as necessary to 300 mg daily. The dosage for children as an antileprosy agent is 1–2 mg/kg orally to a maximum of 100 mg daily.
Clofazimine
The antileprosy mechanism of action of clofazimine is unknown; it has a slow bactericidal effect on M. leprae, inhibits mycobacterial growth and tends to bind preferentially to mycobacterial DNA. Oral absorption of clofazimine is variable and it is distributed primarily in fatty tissues and cells. Macrophages accumulate the drug and further distribute it throughout the body. Its half-life is about 2–3 months with chronic therapy, and peak plasma concentrations occur 1–6 hours after dosing. It is excreted primarily in faeces.
Adverse reactions include gastric distress; ichthyosis; discolouration (red and brown–black) of skin, faeces, sweat, tears and urine; a change in taste sensations; dry, burning, itching or irritated eyes; photosensitivity; and, rarely, GI bleeding, hepatitis and depression. Use clofazimine with caution in people with liver function impairment. Avoid use in patients with clofazimine hypersensitivity or a history of GI disorders.
The adult dosage in combination therapy for leprosy is 50 mg orally daily and 300 mg once monthly in multi-drug regimens. The drug is only available through the SAS.
Helminthiasis
The disease-producing helminths (worms) are classified as metazoa, or multicellular animal parasites. Unlike the protozoa, they are large organisms with complex cellular structures and feed on host tissue. They may be present in the GIT but several types also penetrate tissues and some undergo developmental changes, during which they wander extensively in the host. Because most anthelmintics used today are highly effective against specific parasites, the organism must be accurately identified before treatment is started, usually by finding the parasite ova or larvae in the faeces, urine, blood, sputum or tissues of the host.
Parasitic infestations do not necessarily cause clinical manifestations, although they may be injurious for a variety of reasons:
Worms may cause mechanical injury to the tissues and organs. Roundworms in large numbers may cause obstruction in the intestine, filariae may block lymphatic channels and cause massive oedema, and hookworms often cause extensive damage to the wall of the intestine and considerable loss of blood.
Platyhelminths (flatworms)
Cestodes
Cestodes are tapeworms, of which there are four varieties: Taenia saginata (beef tapeworm), Taenia solium (pork tapeworm), Diphyllobothrium latum (fish tapeworm) and Hymenolepis nana (dwarf tapeworm). As indicated by the common name of the worm, the parasite enters the intestine by way of improperly cooked beef, pork or fish, or from contaminated food, as in the case of the dwarf tapeworm.
The cestodes are segmented flatworms with a head, or scolex, that has hooks or suckers used to attach to tissues and a number of segments, or proglottids, which in some cases may extend for 6–9 metres in the bowel. Drugs affecting the scolex allow expulsion of the organisms from the intestine. Each of the proglottids contains both male and female reproductive units. When filled with fertilised eggs, they are expelled from the worm into the environment. On ingestion, the infected larvae develop into adults in the small intestine of the human. The larvae may travel to extraintestinal sites and enter other tissues, such as the liver, muscle and eye. The tapeworms, with the exception of the dwarf tapeworm, spend part of their life cycle in a host other than humans (pigs, fish or cattle). The dwarf tapeworm does not require any such intermediate host. The tapeworm has no digestive tract; it depends on the nutrients that are intended for the host. Subsequently, the victim suffers by eventually developing nutritional deficiency.
Trematodes
Trematodes, or flukes, are flat, non-segmented parasites with suckers that attach to and feed on host tissue. The life cycle begins with the egg, which is passed into fresh water after faecal excretion from the body of the human host. The egg containing the embryo forms into a ciliated organism, the miracidium. In the presence of water, the miracidium escapes from the egg and enters the intermediate host, the freshwater snail, which exists extensively in rice paddies and irrigation ditches. After entry, the fluke forms a cyst in the lungs of the snail. In the cyst, many organisms develop. They can penetrate other parts of the snail and grow into worms called cercariae. Eventually, the cercariae are released from the snail into the water, attaching themselves to blades of grass to encyst.
When encysted organisms in snails or fish and crabs are swallowed by humans, they develop into adult flukes in different structures of the body. The flukes are therefore classified according to the type of tissue they invade. After ingestion, the eggs of Schistosoma haematobium appear in the urinary bladder and cause inflammation of the urogenital system. This can result in chronic cystitis and haematuria. Infestations with Schistosoma japonicum and Schistosoma mansoni produce intestinal disturbance with resultant ulceration and necrosis of the rectum. S. japonicum is more concentrated in the veins of the small intestine. If the liver and spleen become infected, the disease is usually fatal. S. mansoni prefers the portal veins that drain the large intestine, particularly the sigmoid colon and rectum. Unlike the other parasites, the cercariae of S. mansoni are not ingested but burrow through the skin, especially between the toes of the human host who is standing in contaminated water. They then make their way to the portal system, where they mature into adult flukes.
Schistosomiasis (bilharziasis) is endemic to Africa, Asia, South America and Caribbean islands. The disease can be controlled largely by eliminating the intermediate host, the snail. Travellers to these areas must avoid contact with contaminated water for drinking, bathing or swimming.
Nematodes (roundworms)
Nematodes are non-segmented cylindrical worms that consist of a mouth and complete digestive tract. The adults reside in the human intestinal tract; there is no intermediate host. Two types of nematode infection exist in the human: the egg form and the larval form.
Egg infective form
Ascaris lumbricoides is a large nematode (about 30 cm in length) and is known as the ‘roundworm of humans’. The adult Ascaris usually resides in the upper end of the small intestine of the human, where it feeds on semidigested foods. The fertilised egg, when excreted with faeces, can survive in the soil for a long time. When inadvertently ingested by another host, the embryos escape from the eggs and mature into adults in the host. To prevent the disease, proper sanitary conditions and meticulous personal habits must be observed.
Infection with Enterobius vermicularis, or pinworm, is highly prevalent among children and adults. Adult pinworms reside in the large intestine but the female migrates to the anus, depositing her eggs around the skin of the anal region. This causes intense itching and can be noted especially in children. Ingestion of excreted eggs can infect an individual. Eggs that contaminate clothing, bedding, furniture and other items may be responsible for continuing the reinfection of an individual and initiating the infection of others.
Larval infective form
Necator americanus (New World) or Ancylostoma duodenale (Old World) hookworms are somewhat similar in action. They reside in the small intestine of humans. When the eggs are excreted in the faeces, the larvae hatch in the soil. The larvae can penetrate the skin of humans, particularly through the soles of the feet, producing dermatitis (ground itch). On entry into the small intestine, they develop into adult worms. During the process, they extravasate blood from the intestinal vessels and cause a profound anaemia in the victim. The presence of eggs in the faeces indicates a positive test for hookworm disease. This infection can be avoided by wearing shoes.
Trichinella spiralis is a small pork roundworm that causes trichinosis. In humans, the disease begins by ingestion of insufficiently cooked pork meat. On entry of encysted meat into the small intestine, the larvae are released from the cysts. After maturation, the females develop eggs that later form into larvae. They then migrate via the bloodstream and lymphatic system to the skeletal muscles and encyst. Encapsulation and eventually calcification of the cysts occur. Diagnosis of trichinosis is made by muscle biopsy, whereby microscopic examination reveals the presence of larvae. The disease is prevented by thoroughly cooking pork meat before eating.
Anthelmintic drugs
Anthelmintic drugs are used to rid the body of worms (helminths). Anthelmintics are among the most basic forms of chemotherapy. It has been estimated that one-third of the world’s population is infested with these parasites. The main class of anthelmintic drugs is the benzimidazoles including albendazole and mebendazole. The other anthelmintics are ivermectin, praziquantel and pyrantel.
Benzimidazoles
The benzimidazoles are vermicidal and may also be ovicidal for most helminths. They cause degeneration of a parasite’s cytoplasmic microtubules, which leads to blocking of glucose uptake in the helminth, and hence death of the parasite. They are indicated for the treatment of Trichuris (whipworm), Enterobius (pinworm), Ascaris (roundworm), Ancylostoma (common hookworm) and some tapeworms and liver flukes.
Albendazole
Albendazole is poorly absorbed and essentially remains within the GIT. Absorption is significantly increased by consumption of a fatty meal; the small amount of drug absorbed is rapidly metabolised by the liver to an active sulfoxide metabolite that is thought to be the active drug against tissue infestation. The plasma half-life of albendazole sulfoxide is 8–9 hours but, after release from cysts, low concentrations can be detected in plasma for several weeks. Elimination is principally via bile, with only small quantities detected in urine.
Adverse reactions occur with higher doses and extended dosing, and include headache, nausea, vomiting and diarrhoea. Rarely, allergic reactions, liver toxicity and haematological reactions occur.
In patients with hepatic impairment, dosage reduction may be necessary. Albendazole is contraindicated in pregnancy because of evidence of teratogenic effects in several animal species, and in lactation because it is excreted in breast milk.
Dosage varies according to the infecting species but, in general, for adults a single dose of 400 mg is given before food in the case of roundworm, threadworm or hookworm infections. Consult current drug information sources for dosing schedules relevant to other helminth species and for dosing in children.
Mebendazole
The oral absorption of mebendazole is increased if given with fatty foods. It is distributed to plasma, cyst fluid, liver, hepatic cysts and muscle tissues, with a half-life of 2.5–5.5 hours. It is metabolised in the liver and excreted primarily in faeces.
Adverse reactions are uncommon and include gastric distress, diarrhoea, nausea, vomiting, alopecia, dizziness, headache and, rarely, hypersensitivity and neutropenia. Use this drug with caution in patients with Crohn’s ileitis and ulcerative colitis. Avoid use in people with mebendazole hypersensitivity and liver function impairment.
The adult and paediatric dosage (children 2 years of age and over) is 100 mg orally as a single dose for threadworm and 100 mg twice daily for 3 days for hookworm, roundworm and whipworm. If necessary, this dosage may be repeated in 2–3 weeks.
Other anthelmintics
Praziquantel
Praziquantel is an anthelmintic agent that penetrates cell membranes and increases cell permeability in susceptible worms. This results in an increased loss of intracellular calcium, contractions and muscle paralysis of the worm. The drug also disintegrates the schistosome tegument (covering). Subsequently, phagocytes are attracted to the worm and ultimately kill it. Praziquantel is indicated for the treatment of schistosomiasis due to various blood flukes and for tapeworms but is ineffective against roundworms.
Drug monograph 46-3 Ivermectin
Ivermectin is a semisynthetic compound isolated from a fermented broth of Streptomyces avermitilis. It has a wide range of activity against helminths and is also used in veterinary medicine. It is thought to exert an effect on nematodes by stimulating the release of the inhibitory neurotransmitter γ-aminobutyric acid (GABA), which disrupts neuronal transmission. Opening of chloride channels, with subsequent increasing chloride conductance, paralyses the nematode. The sites of action in nematodes differ from those in mammals.
Indications
This drug is indicated for treating infection with Strongyloides stercoralis (a threadworm found in northern Australia) and with the immature microfilariae of Onchocerca volvulus, reducing the incidence of ‘river blindness’ by up to 80%.
Pharmacokinetics
Ivermectin is incompletely absorbed after oral administration, and peak plasma concentrations occur about 4 hours after administration. The drug is metabolised and both unchanged ivermectin and the metabolites are excreted in faeces, with less than 1% appearing in urine. The plasma half-life of ivermectin is 9–15 hours and 3 days for the metabolites.
Adverse reactions
Those commonly encountered include nausea, diarrhoea, dizziness and pruritus. Rarely, ivermectin causes tachycardia, postural hypotension, uveitis, facial and peripheral oedema or elevated liver enzymes.
Praziquantel is absorbed orally and reaches a peak plasma concentration in 1–3 hours. Half-life is 0.8–1.5 hours for praziquantel and 4–6 hours for its metabolites. It is excreted by the kidneys and is generally well tolerated.
See Drug Interactions 46-4 for a list of drugs that can interact with praziquantel, plus outcomes and management.
Drug interactions 46-4 Praziquantel
| Drug | Possible effects and management |
| Alcohol | CNS effects of praziquantel may be potentiated. Avoid this combination |
| Carbamazepine,phenytoin,dexamethasone | These drugs increase metabolism of praziquantel, reducing its effectiveness. A higher dose of praziquantel may be required |
| Cimetidine | Inhibition of metabolism of praziquantel, leading to an increased plasma concentration and effect. Use an alternative H2 antagonist |
Source: AMH 2010.
Adverse reactions include headache, light-headedness, gastric distress, sweating, rash, pruritus, dizziness, drowsiness, fever, increased sweating and GI distress, including bloody diarrhoea. Use this drug with caution in patients with severe liver disease. Avoid use in people with praziquantel hypersensitivity and ocular cysticercosis.
For intestinal tapeworms the adult dose is 10 mg/kg as a single dose. Swallow tablets whole with food and plenty of water to help disguise the bitter taste. Do not chew.
Pyrantel
Pyrantel is a depolarising neuromuscular-blocking anthelmintic agent; it causes contraction and then paralysis of the helminth’s muscles. The helminths are dislodged and then expelled from the body by peristalsis. Pyrantel is indicated for the treatment of Ascaris lumbricoides (roundworm), Enterobius vermicularis (threadworm) and hookworm, but is ineffective against whipworm. This product is poorly absorbed from the GIT. Pyrantel reaches peak plasma concentration in 1–3 hours and is primarily excreted in the faeces.
Adverse reactions include diarrhoea, vomiting, nausea, headache and abdominal cramps. Rarely, pyrantel causes anorexia, dizziness, drowsiness and an increase in liver enzymes. Exercise caution in people with dehydration or malnourishment. Avoid use in patients with pyrantel hypersensitivity.
The dosage in adults and children for threadworm and roundworm is 10 mg/kg (max 750 mg) as a single oral dose. If necessary, it may be repeated in 1 week.
Key points
 Malaria is endemic in more than 100 countries and it has been reported that, worldwide, between 300 and 500 million individuals have clinical malaria.
Malaria is endemic in more than 100 countries and it has been reported that, worldwide, between 300 and 500 million individuals have clinical malaria.
 The choice of antimalarial drugs is based on the particular malarial strain involved and the stage of the plasmodium life cycle. Amoebiasis is an infection of the large intestine produced by a protozoan parasite, Entamoeba histolytica. This infestation is found worldwide but is prevalent and severe in tropical areas. The main mycobacterial infections in humans are Mycobacterium avian complex, tuberculosis and leprosy. The World Health Organization (WHO) aims to reduce TB prevalence and death rates by 50% (relative to 1990) by 2015 and to eliminate TB as a public health problem (1 case per million population) by 2050. The WHO has recommended the Directly Observed Treatment—Short Course (DOTS) approach for the control of tuberculosis and has projected that the use of this system would result in cure of 85% of all new cases. The recommended first-line drugs for tuberculosis are isoniazid, rifampicin, pyrazinamide and ethambutol. In developing countries streptomycin is also a first-line drug. Leprosy, or Hansen’s disease, is caused by Mycobacterium leprae in humans. Today, the estimated number of infected people worldwide has fallen dramatically and many countries are now free of leprosy. Anthelmintic drugs are used to rid the body of worms (helminths). It has been estimated that one-third of the world’s population is infested with these parasites.
The choice of antimalarial drugs is based on the particular malarial strain involved and the stage of the plasmodium life cycle. Amoebiasis is an infection of the large intestine produced by a protozoan parasite, Entamoeba histolytica. This infestation is found worldwide but is prevalent and severe in tropical areas. The main mycobacterial infections in humans are Mycobacterium avian complex, tuberculosis and leprosy. The World Health Organization (WHO) aims to reduce TB prevalence and death rates by 50% (relative to 1990) by 2015 and to eliminate TB as a public health problem (1 case per million population) by 2050. The WHO has recommended the Directly Observed Treatment—Short Course (DOTS) approach for the control of tuberculosis and has projected that the use of this system would result in cure of 85% of all new cases. The recommended first-line drugs for tuberculosis are isoniazid, rifampicin, pyrazinamide and ethambutol. In developing countries streptomycin is also a first-line drug. Leprosy, or Hansen’s disease, is caused by Mycobacterium leprae in humans. Today, the estimated number of infected people worldwide has fallen dramatically and many countries are now free of leprosy. Anthelmintic drugs are used to rid the body of worms (helminths). It has been estimated that one-third of the world’s population is infested with these parasites.Review exercises
References and further reading
Antibiotic Expert Group. Therapeutic Guidelines: Antibiotic, version 13. Melbourne: Therapeutic Guidelines, 2006.
Australian Institute of Health and Welfare. Australia’s Health 2008. Cat. No. AUS 99. Canberra: AIHW, 2008.
Australian Medicines Handbook 2010. Adelaide: AMH, 2010.
Croft A. Malaria: prevention in travellers. British Medical Journal. 2000;321:154-160.
Ebert S.C. Tuberculosis. In DiPiro J.T., Talber R., et al, editors: Pharmacotherapy: A Pathophysiologic Approach, 2nd edn, Norwalk: Appleton & Lange, 1993.
Espinal M.A., Laslo A., Simonsen L., et al. Global trends in resistance to antituberculosis drugs. New England Journal of Medicine. 2001;344:1294-1303.
Petri A.W.Jr. Chemotherapy of tuberculosis, Mycobacterium avium complex disease, and leprosy. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 11th edn, New York: McGraw-Hill, 2006. [ch 47]
Skull S.A., Tallis G. Epidemiology of malaria in Victoria 1999–2000: East Timor emerges as a new source of disease. Communicable Diseases Intelligence. 2001;25:149-151.
WHO Report Global Tuberculosis Control 2008. Available: http://www.who.int/tb/publications/global_report/2008/chapter_1/en/index3.html [1 October 2009].
WHO. The Stop TB Strategy. Available: http://www.who.int/tb/strategy/en/ [1 October 2009].