Chapter 7 Pharmacogenetics
Some of the striking features associated with the use of drugs are how often the therapy fails and how often drugs cause a serious adverse reaction. It is not surprising that substantial effort is expended on trying to match the right drug with the right patient. If factors such as age, gender and disease etc are taken into consideration, the one remaining variable that can influence either efficacy or toxicity of a drug is the genetic makeup of the individual. The Human Genome Project provided enormous advances in our ability to elucidate the genetic basis of interindividual variability in drug response. Knowledge continues to grow and ∼4 million single-nucleotide polymorphisms (SNPs) in the human genome have been catalogued. Polymorphisms have been identified in drug-metabolising enzymes, drug transporters and multiple drug targets including receptors, ion channels and enzymes. For a limited number of drugs genotyping or phenotyping is one aspect of a rational prescribing framework to assist in individualising drug therapy to either avoid toxicity or optimise response. This chapter provides an overview of the field of pharmacogenetics and describes examples where pharmacogenetics has had an impact on current clinical management or where early evidence suggests a role for pharmacogenetics in the future.
Key abbreviations
G6PD glucose-6-phosphate dehydrogenase
HER human epidermal growth factor receptor
HOW often have you heard or been part of a conversation that in general revolves around the observation that one person has had a different response to a drug (either positive or negative) than another person. For example ‘I can’t take cold tablets because they make me sleepy but my best friend doesn’t have any problem’ or ‘my uncle had to stop taking one of his heart drugs because it didn’t agree with him but my other uncle takes the same drug and he is fine’. Wide interindividual variability in drug response is a feature of many drugs and this has led in more recent times to the concept of ‘individualising drug therapy’. If we start from the basic premise of choosing the right drug for the right patient at the right dose and time, hence maximising efficacy and minimising toxicity, how then do we explain dramatically different responses to an optimised dosage regimen in different patients?
Variability in response can result from pharmacodynamic and/or pharmacokinetic factors. The latter include altered absorption (of orally administered drugs), distribution, metabolism and excretion. Factors known to influence renal drug clearance include age (neonatal period and elderly), renal dysfunction and cardiac failure, and these are often taken into account when determining drug dosage. However, accounting for variability in hepatic drug metabolism is more complex. Hepatic clearance can be affected by age, diet, hormonal factors, disease states, interactions (drug–drug, drug–herb, drug–complementary/alternative medicine [CAM]) and environmental chemicals. However, of the plethora of factors that can influence hepatic drug clearance, it is now recognised that the largest interindividual variability in drug response/toxicity arises from inherited (genetic) differences that alter drug metabolism and/or alter the molecular targets of drugs including transduction mechanisms downstream of the receptor.
What is pharmacogenetics?
Clinical observations of genetic variability affecting drug response can be traced back to the 1950s with early evidence of pseudocholinesterase deficiency and adverse effects of isoniazid, a drug used at that time to treat tuberculosis. The term ‘pharmacogenetics’ was first proposed by Vogel in 1959 as the ‘study of the role of genetics in drug response’. In 2010 pharmacogenetics is now a subset of pharmacogenomics. The latter is defined as ‘the study of variations in DNA and RNA characteristics as related to drug response’ whereas pharmacogenetics is defined as ‘the study of variations in DNA sequence as related to drug response’. (Definitions for Genomic Biomarkers, Pharmacogenomics, Pharmacogenetics, Genomic Data and Sample Coding Categories ICH Topic E15 2008). Simply stated, pharmacogenetics relates to the inheritance of genes that define an individual’s variability in drug response. Studies conducted more than 40 years ago demonstrated that identical twins resembled each other in terms of how they metabolised a drug, whereas fraternal twins (developed from separate eggs) showed variations similar to the general population. This was consistent with fraternal twins having different patterns of inheritance.
Succinylcholine (suxamethonium) is a depolarising neuromuscular blocker, and prolonged succinylcholine apnoea following normal clinical dosage was first reported in 1957. This results from a low activity, in otherwise healthy people, of plasma butyrylcholinesterase (also called pseudocholinesterase), the enzyme that normally hydrolyses succinylcholine. Following the advent of molecular biology techniques in the 1980s we know that the deficiency in succinylcholine hydrolysis is inherited. It arises from multiple mutations in the butyrylcholinesterase gene, for example one of which may result in substitution of a glycine for aspartic acid at residue 70 (the Asp70Gly variant) and also a homozygous ‘silent’ variant, which has <10% of normal butyrylcholinesterase activity in plasma.
In 1956 it was reported that haemolysis after treatment with the antimalarial drug primaquine (refer to Chapter 46) occurred in patients with an inherited deficiency in erythrocyte glucose-6-phosphate dehydrogenase (G6PD). G6PD is important for balancing oxidation–reduction reactions in the cell thereby maintaining the antioxidant glutathione in its reduced state. As the erythrocyte glutathione concentration is lower in persons with G6PD deficiency, primaquine binds instead to crucial thiol groups on haemoglobin, which leads to increased erythrocyte fragility and haemolysis.
However, having more or less of an enzyme cannot be predictably good or bad as exemplified by the antituberculosis drug isoniazid (refer to Drug Monograph 46–1), which is hepatically metabolised by acetylation. Polymorphism in N-acetyltransferase divides the population into ‘rapid-acetylators’ and ‘slow-acetylators’. The rapid-acetylators metabolise a greater proportion of the drug dose and thus do not achieve a therapeutic plasma concentration, whereas the slow-acetylators may appear more sensitive to the drug and experience serious adverse effects including peripheral neuropathy. The impaired elimination of isoniazid in slow acetylators arises from the presence of two variant1 alleles in the gene encoding N-acetyltransferase-2, the enzyme that metabolises isoniazid.
These early clinical examples resulted in adverse outcomes and this led to research that focused primarily on identifying individuals with ‘genetic differences’ that placed them at risk of drug toxicity or adverse drug reactions. However, it is now evident that genetic factors can also predispose individuals to therapeutic failure. Examples of the impact of pharmacogenetics in clinical practice are detailed in later sections of this chapter.
Chromosomes, genes, DNA and protein synthesis
In humans most cells have a single nucleus, with the exception of red blood cells (no nucleus) and skeletal muscle cells, which may have multiple nuclei. The nucleus contains our hereditary material or genes (a sequence of DNA that codes for a product e.g. a protein), which control cell structure and the integrated activities of the cell. Genes are arranged along chromosomes and human somatic cells have 46 chromosomes or 23 homologous pairs, one chromosome inherited from your father and one from your mother. Each chromosome consists of an uninterrupted length of deoxyribonucleic acid (DNA) that represents multiple genes that may exist in alternative forms (alleles2). The human genome consists of 23 pairs of homologous chromosomes and the DNA sequence of each chromosome was determined in the Human Genome Project, which was completed in 2003.
The importance of DNA lies in the fact that it contains the information that codes for the synthesis of multiple proteins that ultimately determine the physical and chemical characteristics of human cells and of humans. For example, proteins are essential for cell structure; serve as hormones; regulate physiological processes; control growth and development; play a significant role in immunity as antibodies, interleukins etc; function as transport molecules (e.g. haemoglobin); are the basis of contractile elements (e.g. myosin and actin) and play a vital role as enzymes that regulate biochemical processes.
Genetic information is stored in DNA (as well as RNA) as a set of three nucleotides called a triplet comprising a combination of adenine, cytosine, guanine or thymine (Figure 7-1). Each triplet is transcribed (copied) as a complementary sequence of three ribonucleic acid (RNA) nucleotides called a codon. Each codon specifies one amino acid and a sequence of amino acids forms a protein molecule. In simplest terms protein synthesis involves transcribing the sequence of DNA into a complimentary strand of messenger RNA (mRNA). Translation of the nucleotide sequence of mRNA is accomplished by ribosomes where special transfer RNA (tRNA) molecules hold the mRNA in place and translate the mRNA into amino acids, which are then incorporated into a protein. The ribosome moves along the mRNA until it reaches a special nucleotide triplet called a stop codon. At this stage translation stops and the completed protein detaches from the tRNA. Specific sources of information should be consulted for detailed descriptions of genetics, protein synthesis etc.
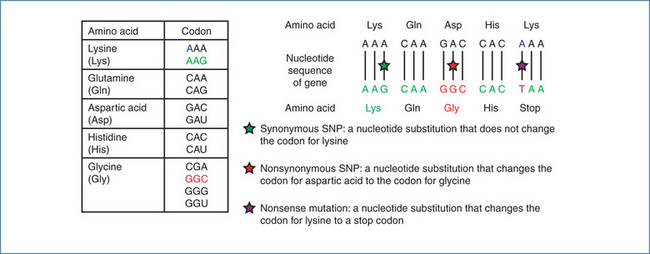
Figure 7-1 Examples of single nucleotide polymorphisms (SNPs). Most amino acids are represented by more than one codon, which is composed of three successive nucleotides. The codon specifies the amino acid and the sequence of codons in the gene controls how the cell orders the assembly of amino acids into a specific protein. The Asp-Gly substitution illustrated is the molecular basis of the deficiency in butyrylcholinesterase activity.
Variations in proteins not only occur between different species but also between individuals of the same species. This genetic variation between humans arises through mutations in genes that may occur in our own cells or has occurred in genes of our ancestors that we have inherited. Spontaneous mutations are a common occurrence in all species and occur as a result of either normal cell function or random interactions with the environment. These mutations account for evolution. However, in some instances the mutation has consequences for our health, for example a genetic disease may arise from a change in a single amino acid in a protein or a mutation that arises from an environmental exposure (e.g. radiation exposure) may result in cancer.
Genetic polymorphism
It is well recognised that there is greater variability in drug response within a population than there is in an individual administered a drug on different occasions or between identical twins. This genetic variability is thought to account for 20%–95% of the variability in drug response (Evans & McLeod 2003). Pharmacogenetic variability results from genetic polymorphism, that is, the presence of different allelic forms of a gene. Genetic polymorphism refers to a change in DNA sequence that occurs at an allele frequency of ≥1% in a population. DNA variations or mutations arise from changes in the components of DNA (i.e., the bases adenine, cytosine, guanine or thymine), which then results in a change in the nucleotide sequence. A change in the nucleotide sequence gives rise to a variant or ‘mutant’ allele compared to the reference or ‘wild-type’ allele.
Polymorphisms that arise from substitution of one nucleotide for another are called single nucleotide polymorphisms (SNPs) (Figure 7-1). If an SNP that occurs in the coding region of a gene is a nonsynonymous (missense) SNP this may result in a change in an amino acid in the encoded protein, potentially altering the structure, stability, activity and ligand-binding properties of the protein. Synonymous (silent) SNPs do not incorporate a change in an amino acid but may alter a functional activity like transcript stability. A nucleotide substitution that leads to a stop codon is called a nonsense mutation. Nucleotide insertions/deletions and frameshift mutations involving single or multiple nucleotides can also occur and these may alter gene expression and/or protein structure and function. Additionally, genes may either be duplicated, resulting in enhanced protein synthesis, or completely deleted with a consequential reduction in protein synthesis. All of these mechanisms have the potential to cause a ‘genetic difference’ in an individual and hence alter drug response.
Examples of drug-metabolising enzymes that exhibit genetic polymorphisms that influence drug response include:
Pharmacogenetic phenotypes
If a mutation in a gene results in an altered trait how does this become evident? In general if a change occurs in a single gene it gives rise to a monogenic trait that is evident from the phenotype. The phenotype refers to the trait or clinical manifestation, e.g. a person described as a ‘slow-acetylator’ or ‘fast-acetylator’ of isoniazid. For drug-metabolising enzymes the genotype–phenotype associations for a monogenic trait are shown in Table 7-1 for individuals with ‘wild-type’ (allele denoted as *1) and/or variant or ‘mutant’ (allele denoted as *2) alleles.
Table 7-1 Genotype–phenotype relationship
| DRUG METABOLISING ENZYME PHENOTYPE | RELATIVE ENZYME ACTIVITY | GENOTYPE |
| Ultrarapid-metaboliser | High | *1/*1/*1 (>2 copies of gene) |
| Extensive-metaboliser | Normal | *1/*1 (homozygous wild-type) |
| Intermediate-metaboliser | Intermediate | *1/*2 (heterozygote) |
| Poor-metaboliser | Low or absent | *2/*2 (homozygous variant) |
Although the phenotype may generally be predicted from the genotype for a trait due to a single gene (Table 7-1), the same cannot be said for the reverse. That is, a trait (phenotype) may not predict the genotype because the trait may be influenced by non-genetic factors. For example an ‘extensive-metaboliser’ with ‘normal’ enzyme activity and carrying *1/*1 alleles may be converted to a pseudo ‘poor-metaboliser’ by coadministration of an enzymeinhibiting drug even though the individual carries the *1/*1 alleles.
Ethnic and race-specific polymorphisms
Contributing to the difficulties in ‘individualising drug therapy’ is the evidence that the frequency of genetic polymorphisms differs between different ethnic groups and between different races (Clinical Interest Box 7-1). Examples include the homozygous3 Asp70Gly variant of butyryl cholinesterase, which has a frequency of 1 in 3000 persons in European and American populations while 1 in 25 are heterozygous4 carriers. In contrast the homozygous silent variant of butyrylcholinesterase occurs at a frequency of 1 in 100,000 and the heterozygotes at 1 in 60 in European and American populations (Li et al 2008). In ethnic groups such as the Vysya of India and the Alaskan Inuit, the frequency of the homozygous silent butyrylcholinesterase is 1 in 50 persons (Manoharan et al 2006).
Clinical interest Box 7-1 Polymorphism in new zealand maori
In New Zealand Maori subjects phenotyped for polymorphisms of debrisoquine (CYP2D6) and proguanil (CYP2C19), 5% were identified as poor-metabolisers of debrisoquine and 7% as poor-metabolisers of proguanil. The data for debrisoquine were similar to those reported for Caucasian populations (5%–10%) but the percentage of poor-metabolisers was higher than that found in Asian populations (0.7%–2%). For proguanil, the incidence of the poormetaboliser phenotype in Maori was higher than that for Caucasian populations but lower than the usual ranges (15%–35%) reported in Asian populations. The authors concluded that the risk of adverse drug reactions in Maori with respect to CYP2D6 and CYP2C19 would be similar to that established for Caucasian populations (Wanwimolruk et al 1995).
Similar studies were performed in South Pacific Polynesians residing in the South Island of New Zealand. The incidence of poor-metaboliser phenotypes for debrisoquine and proguanil in the South Pacific Polynesians was similar to those reported in Asian populations (Wanwimolruk et al 1998)
With regard to CYP the frequency of the CYP2D6 poor-metaboliser phenotype is 7%–10% in Caucasians and ∼1%–2% in north Asian populations (Japanese, Chinese and Koreans). CYP2D6 allele frequencies in Indigenous Australian populations from the far north of Western Australia are similar to East Asian populations (Griese et al 2001). The CYP2D6 polymorphism is of considerable clinical significance because CYP2D6 metabolises many clinically used drugs that have narrow therapeutic indices. Examples include:
Unlike CYP2D6 the poor-metaboliser phenotype of CYP2C19 is seen in 3%–5% of Caucasians and approximately 20% of north Asians. Although a limited number of drugs are metabolised by CYP2C19, pronounced pharmacodynamic effects are seen in Asians who are treated with the proton pump inhibitor omeprazole because of the higher frequency of the poor-metaboliser phenotype.
It is evident from the examples described that genetic polymorphisms frequently differ between different races and ethnic groups. Clearly in multicultural societies this creates a further layer of complexity when attempting to ‘individualise drug therapy’.
Pharmacogenetics in clinical practice
Genotyping can be performed by direct sequencing of an individual’s DNA. However, pharmacogenetic testing based on genotyping is not commonly conducted in either Australia or New Zealand. When it is performed it is generally based on phenotype using either measurement of a metabolic ratio (i.e., the metabolite:drug ratio in blood or urine) or direct measurement of the enzyme activity. The potential consequences of polymorphic drugmetabolising enzymes include:
The following sections describe some examples of clinically relevant pharmacogenetic polymorphisms.
CYP2D6
Perhexiline
Perhexiline is prescribed in Australia and New Zealand for the management of refractory angina. It has a narrow therapeutic index (plasma drug concentration range 0.15–0.6 mg/L) and exhibits dose-dependent kinetics (refer to Chapter 8), and high plasma concentrations are linked to peripheral neuropathy and potentially fatal hepatotoxicity. The normal dose range for perhexiline to maintain a therapeutic plasma concentration varies markedly, from 50–100 mg/week in poor-metabolisers to 50–600 mg/day in extensive- and ultrarapid-metabolisers. Perhexiline is metabolised by CYP2D6 and this variability in dose requirement is explained by the presence of defective CYP2D6 alleles. There is good concordance between the ratio of plasma hydroxy-perhexiline (the principal metabolite of perhexiline) and plasma perhexiline concentration and CYP2D6 genotype. Poor-metabolisers have a ratio of <0.3 (usually <0.1) and all patients commencing perhexiline therapy ‘should either be genotyped before therapy or have their plasma metabolic ratio measured within the first week of therapy’ (Miners 2007). Perhexiline therapeutic drug monitoring is offered by many clinical pathology laboratories.
Tamoxifen
Tamoxifen, commonly used to treat oestrogen responsive breast cancer, is metabolised to endoxifen, which is 100-fold more potent as a selective oestrogen receptor modulator (antagonist). CYP2D6 metabolises tamoxifen to endoxifen and variability in the plasma concentration of endoxifen has been linked to CYP2D6 genotype and phenotype (Goetz et al 2008). As would be predicted, if a patient was an extensive- or ultrarapid-metaboliser, a higher plasma concentration of endoxifen would be expected in comparison with a person who was a poor-metaboliser phenotype. Further confirmatory studies are required but one study has reported that the relapse-free survival rate is significantly lower in breast cancer patients with intermediate- or poor-metaboliser phenotype in comparison with extensive- or ultrarapid-metabolisers (Goetz et al 2007).
Opioid pain management
Current use of drugs for pain management relies heavily on evidence of clinical benefit and the absence of adverse drug reactions. Many drugs used for pain management, including codeine, methadone, morphine, oxycodone and tramadol, are metabolised by CYP enzymes, including CYP2D6. The prevalence of CYP2D6 polymorphisms in 61 ‘chronic-pain management patients’ prescribed alone or in combination oxycodone, hydrocodone, tramadol and methadone was found to be similar to the general population. Of the pain patients studied, 54% were CYP2D6 extensive-metabolisers (EM), 41% intermediatemetabolisers (IM) and 5% poor-metabolisers (PM). Although patients with the poor-metaboliser phenotype had higher steady-state drug concentrations of hydrocodone, methadone and tramadol compared to extensivemetabolisers, the differences in steady-state concentrations (PM>IM>EM) were not statistically significant. Of the study group four patients who reported adverse drug reactions (nausea and drowsiness) had impaired CYP2D6 drug metabolism based on their CYP2D6 predicted phenotype. This was a relatively small study and further large-scale prospective studies are required to establish the clinical benefit of pharmacogenetics in guiding the management of chronic pain using drugs metabolised by CYP2D6 (Jannetto & Bratanow 2009).
CYP2C9
Phenytoin
Phenytoin is an anticonvulsant that is metabolised principally by CYP2C9 (∼95%). The most common variants of CYP2C9 are CYP2C9*2 and CYP2C9*3 with frequencies of 11% and 7%, respectively, in Caucasians. The homozygous CYP2C9*3 genotype is associated with ∼ 80% reduction in metabolic drug clearance while the CYP2C9*2 genotype has only a modest effect on drug metabolism. A relationship between adverse effects (nystagmus, slurred speech and ataxia), CYP2C9 genotype (the patient was homozygous CYP2C9*3) and phenytoin dosage has been reported (Brandolese et al 2001). However, currently phenytoin genotyping is not undertaken routinely, dosing is still empirical and clinical management is supported by therapeutic drug monitoring.
Warfarin
Warfarin is a racemic drug used as an anticoagulant. S-warfarin is 3–5 times more potent as an anticoagulant that R-warfarin. Similar to phenytoin, S-warfarin is metabolised primarily by CYP2C9. The two variants CYP2C9*2 and CYP2C9*3 result in a 30%–40% reduction and almost complete loss, respectively, of S-warfarin metabolism requiring significantly lower daily dosage of warfarin to maintain a therapeutic INR in patients with either genotype (Higashi et al 2002). VKORC1 encodes the vitamin K epoxide reductase which is inhibited by warfarin, thus interfering with the carboxylation of vitamin K-dependent clotting factors. Two haplotypes5 (A and B) have been identified, with the A haplotype associated with lower levels of VKORC1 compared with the B haplotype. Individuals with the A haplotype produce a smaller amount of vitamin K epoxide reductase, requiring a lower dosage of warfarin than patients with the B haplotype. This association is independent of the CYP2C9 genotype and current evidence indicates patient age, height, CYP2C9 genotype and polymorphism of VKCOR1 account for 55% of the variability in warfarin daily dosage (Rieder et al 2005). It remains to be established whether CYP2C9 and VKCOR1 genotyping will predict warfarin dosage.
TPMT, azathioprine and 6-mercaptopurine
Variant TPMT alleles are a classic example of the synthesis of defective drug-metabolising enzymes that increase the risk of adverse drug reactions. TPMT metabolises the cytotoxic immunosuppressant drugs azathioprine (the prodrug of 6-mecaptopurine) and 6-mercaptopurine. Normally 6-mercaptopurine is ‘detoxified’ by TPMT and reduced TPMT activity is associated with a high frequency of myelosuppression (Figure 7-2). Patients who do not carry wild-type TPMT alleles have extremely low TPMT activity and almost always develop neutropenia. Indeed patients with the wild-type alleles tolerate therapy for ∼20 times longer than patients heterozygous for mutant TPMT alleles (39 versus 2 weeks). 6-Mercaptopurine and azathioprine both have a narrow therapeutic index and phenotyping provides a useful way of identifying patients potentially at risk of myelosuppression. As erythrocyte TPMT activity mirrors that of the liver, which metabolises azathioprine and 6-mercaptopurine, phenotyping is determined by measuring erythrocyte TPMT activity prior to treatment. The dose of azathioprine or 6-mercaptopurine is then adjusted and patients with the poor-metaboliser phenotype receive about 10% of the dose of those with the extensive-metaboliser phenotype. It should be noted that routine haematological monitoring is still undertaken as bone marrow suppression can occur with chronic dosing in patients with the wild-type alleles.
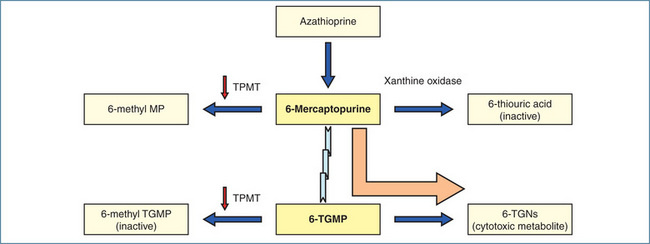
Figure 7-2 Metabolism of azathioprine and 6-mercaptopurine. 6-Mercaptopurine is either metabolised by TPMT (to 6-methyl MP) and xanthine oxidase (to 6-thiouric acid) or converted via hypoxanthine-guanine phosphoribosyltransferase to 6-thioguanine monophosphate (6-TGMP), which serves as the precursor to the pharmacologically active cytotoxic metabolites, 6-thioguanine nucleotides (TGNs). 6-TGMP is also metabolised via TPMT to the inactive 6-methyl-TGMP. In the presence of low TPMT activity (indicated by the downward arrows) metabolism occurs predominantly via 6-TGMP resulting in increased formation of 6-TGNs (indicated by the large solid arrow) and severe myelosuppression.
UGT1A1 and irinotecan
Irinotecan is a prodrug that is used in the treatment of colorectal cancer. It is metabolised to the active metabolite, 7-ethyl-10-hydroxycampto-thecan (SN-38), which functions as a topoisomerase 1 inhibitor. SN-38 is metabolised via glucuronidation, principally by UGT1A1 (refer previous section, ‘Ethnic and race-specific polymorphisms’). However, UGT1A9 also plays a minor role. Currently there are >60 known polymorphisms in the UGT1A gene and some of these have functional consequences. The UGT1A1*28 polymorphism arises from the insertion of an extra thymine–adenine repeat, making a total of seven instead of the normal six repeats, in the promoter region of the gene. Individuals who are homozygous for UGT1A1*28 have reduced UGT1A1 gene expression and the UGT1A1*28 allele is thought to contribute about 40% of the variability in enzyme activity of UGT1A1. The first report of an association between SN-38 glucuronidation and irinotecan toxicity was described in 1994 (Gupta et al 1994). Further studies of irinotecan pharmacokinetics and pharmacogenetics confirmed an association between decreased glucuronidation of SN-38, UGT1A1*28 genotype and the risk of severe diarrhoea and/or neutropenia (Iyer et al 2002; Innocenti et al 2004).
These studies ultimately led to a change in the irinotecan (Camptosar®) label in the USA in 2005. Current product information for irinotecan in the USA states: ‘Individuals who are homozygous for the UGT1A1*28 allele are at increased risk for neutropenia following initiation of Camptosar treatment. A reduced initial dose should be considered for patients known to be homozygous for the UGT1A1*28 allele. Heterozygous patients (carriers of one variant allele and one wild-type allele which results in intermediate UGT1A1 activity) may be at increased risk for neutropenia; however, clinical results have been variable and such patients have been shown to tolerate normal starting doses. When administered in combination with other agents, or as a single-agent, a reduction in the starting dose by at least one level of Camptosar should be considered for patients known to be homozygous for the UGT1A1*28 allele. However, the precise dose reduction in this patient population is not known and subsequent dose modifications should be considered based on individual patient tolerance to treatment’ (http://media.pfizer.com/files/products/uspi_camptosar.pdf [15 January 2010]). Although the USA product information alerts prescribers to the existence of UGT1A1*28 genetic variation, there is currently no recommendation that patients should be genotyped before receiving therapy.
A relationship has been established between UGT1A1*28 and neutropenia; however, the data are less convincing for diarrhoea. Some studies have found an association while others found no association. It appears that more data are needed before an association between UGT1A1*28 genotype and irinotecan-induced diarrhoea can be established. In Australasia the information on UGT1A1*28 is not stated in the product information but dosing is adjusted in the presence of diarrhoea (grades 1–4) and neutropenia (grades 2–4). Genetic testing is not routine and guidelines for dose adjustment based on genotype do not exist.
Drug molecular targets
Although genetic polymorphisms of drug-metabolising enzymes are well recognised there is increasing evidence of polymorphisms in drug targets. Of particular interest is the β2-adrenergic receptor encoded by the ADRB2 gene. To date 80 polymorphisms have been identified of which 45 are SNPs and two are insertion/deletion variants. Of these two common nonsynonymous variants (a change from glycine to arginine at amino acid position 16 [Gly16Arg] and a change from glutamine to glutamic acid at position 27 [Gln27Glu]) have functional consequences. Early studies suggested that the response to short-acting β-agonists and worsening of asthma were related to the Gly16Arg genotype; however, further studies did not support the association. Asthma is a complex disease and polymorphisms of multiple genes involved in lung function, airway responsiveness and the leukotriene pathway are currently being investigated (Lima et al 2009).
Trastuzumab (Herceptin®), a humanised monoclonal antibody, targets the extracellular domain of the human epidermal growth-factor receptor 2 (HER2) protein. HER2 is encoded by the HER2/neu gene, and plays a significant role in the proliferation of certain tumour cells. Clinically HER2 status is important in breast cancer patients as it determines the response to trastuzumab.
Similarly, mutations in the human epidermal growthfactor receptor gene (HER1) influence the response to gefitinib, which is used in the treatment of non-small-cell lung cancer. Patients carrying mutations that promote function of HER1 appear to respond better to gefitinib than those with the wild-type HER1 gene.
Abacavir, a nucleoside reverse transcriptase inhibitor (refer to Chapter 45), is used in the treatment of HIV and 5%–8% of Caucasian patients develop hypersensitivity reactions characterised by acute respiratory symptoms, rash, fever, malaise and gastrointestinal symptoms. With continued treatment the symptoms worsen and, in patients with abacavir hypersensitivity, recommencing therapy may result in life-threatening hypotension. There is a strong association between abacavir hypersensitivity and the human leucocyte antigen (HLA) allele HLA-B*5701. Screening for HLA-B*5701 decreases substantially both the incidence of abacavir hypersensitivity and early discontinuation of abacavir therapy. Polymorphic HLA alleles are also associated with the hypersensitivity reactions that occur in some patients receiving allopurinol or carbamazepine.
Polymorphisms of membrane transporters that are involved in drug transport have also been reported. The most widely studied is P-glycoprotein (P-gp, also called MDR1), which is encoded by the gene ABCB1 (refer to Chapter 6). However, results from studies investigating the activity of polymorphic variants of P-gp and responses to anticancer drugs, antihistamines, anticonvulsants, antiviral drugs, cardiac glycosides and immunosuppressants have been controversial (Giacomini & Sugiyama 2006). Further studies are ongoing to determine the clinical relevance of polymorphisms of drug transporters.
The future
Translation of pharmacogenetics into clinical practice remains intellectually challenging, in terms of assigning causation of genetic variants to a diagnostic association, and technically time-consuming, necessitating rigorous analytic validation and quality assurance/control to ensure meaningful translation to clinical practice. Additionally, the conduct of trials to establish that clinical outcomes are improved when drug therapy is individualised based on genetics is difficult because of ethical issues surrounding privacy/confidentiality and record retention, potential misuse of a person’s genetic information and health/life insurability, the problem of non-genomic confounding effects (e.g. diet, smoking, drug interactions etc) and the enor mous expense of such trials. Also the presence or absence of a particular gene or gene mutation still does not define fully the expected response to a drug because many diseases or pathophysiological conditions are multifactorial in nature.
At present there are still a number of hurdles to overcome before pharmacogenetics becomes a widespread routine clinical component of patient management. In the mean time pharmacogenetics may aid in identifying within a population those people who obtain a therapeutic benefit, those who experience an adverse effect and those who do not respond at all (Figure 7-3).
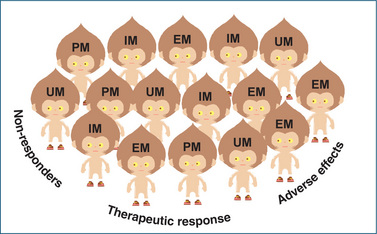
Figure 7-3 Impact of population pharmacogenetics on drug response. Cartoon of the variability in drug response (nonresponders, those who obtain a therapeutic effect or experience adverse effects) expected from a monogenetic trait in a population. EM = extensive-metaboliser, IM = intermediatemetaboliser, PM = poor-metaboliser, UM = ultrarapid-metaboliser.
Key points
 Pharmacogenomics is defined as ‘the study of variations in DNA and RNA characteristics as related to drug response’. Pharmacogenetics is defined as ‘the study of variations in DNA sequence as related to drug response’. Stated simply, pharmacogenetics relates to the inheritance of genes that define an individual’s variability in drug response. Early clinical observations identified adverse outcomes and this led to research that focused primarily on identifying individuals with ‘genetic differences’ that placed them at risk of drug toxicity or adverse drug reactions. Pharmacogenetic variability results from genetic polymorphism, that is, the presence of different allelic forms of a gene. Genetic polymorphism refers to a change in DNA sequence that occurs at an allele frequency of ≥ 1% n a population. A change in the nucleotide sequence gives rise to a variant or ‘mutant’ allele compared with the reference or ‘wild-type’ allele. Polymorphisms that arise from substitution of one nucleotide for another are called single nucleotide polymorphisms (SNPs). A nonsynonymous SNP may result in a change in an amino acid in the encoded protein, potentially altering the structure, stability, activity and ligand-binding properties of the protein. Synonymous SNPs do not incorporate a change in an amino acid but may alter a functional activity like transcript stability. Nucleotide insertions and deletions involving single or multiple nucleotides can also occur and these may alter gene expression and/or protein structure and function. Genes may also be duplicated, resulting in enhanced protein synthesis, or completely deleted with a consequential reduction in protein synthesis. If a change occurs in a single gene it gives rise to a monogenic trait that is evident from the phenotype. The phenotype refers to the clinical manifestation, e.g. a person described as a ‘slow-acetylator’ or ‘fast-acetylator’. The frequency of genetic polymorphisms differs between different ethnic groups and between different races. This contributes to the difficulties in ‘individualising drug therapy’. Genotyping can be performed by direct sequencing of an individual’s DNA. However, pharmacogenetic testing conducted in Australia and New Zealand is generally based on phenotyping rather than genotyping. The potential consequences of polymorphic drug-metabolising enzymes include: increased plasma drug concentration and duration of action, decreased plasma drug concentration and therapeutic failure, adverse drug reactions/toxicity, failure to activate a prodrug, drug metabolism via alternative pathways and exacerbation of drug interactions. Clinical examples include: CYP2D6—perhexiline, tamoxifen opioid analgesics, CYP2C9—phenytoin and warfarin, TPMT—azathioprine and 6-mercaptopurine and UGT1A1—irinotecan.
Pharmacogenomics is defined as ‘the study of variations in DNA and RNA characteristics as related to drug response’. Pharmacogenetics is defined as ‘the study of variations in DNA sequence as related to drug response’. Stated simply, pharmacogenetics relates to the inheritance of genes that define an individual’s variability in drug response. Early clinical observations identified adverse outcomes and this led to research that focused primarily on identifying individuals with ‘genetic differences’ that placed them at risk of drug toxicity or adverse drug reactions. Pharmacogenetic variability results from genetic polymorphism, that is, the presence of different allelic forms of a gene. Genetic polymorphism refers to a change in DNA sequence that occurs at an allele frequency of ≥ 1% n a population. A change in the nucleotide sequence gives rise to a variant or ‘mutant’ allele compared with the reference or ‘wild-type’ allele. Polymorphisms that arise from substitution of one nucleotide for another are called single nucleotide polymorphisms (SNPs). A nonsynonymous SNP may result in a change in an amino acid in the encoded protein, potentially altering the structure, stability, activity and ligand-binding properties of the protein. Synonymous SNPs do not incorporate a change in an amino acid but may alter a functional activity like transcript stability. Nucleotide insertions and deletions involving single or multiple nucleotides can also occur and these may alter gene expression and/or protein structure and function. Genes may also be duplicated, resulting in enhanced protein synthesis, or completely deleted with a consequential reduction in protein synthesis. If a change occurs in a single gene it gives rise to a monogenic trait that is evident from the phenotype. The phenotype refers to the clinical manifestation, e.g. a person described as a ‘slow-acetylator’ or ‘fast-acetylator’. The frequency of genetic polymorphisms differs between different ethnic groups and between different races. This contributes to the difficulties in ‘individualising drug therapy’. Genotyping can be performed by direct sequencing of an individual’s DNA. However, pharmacogenetic testing conducted in Australia and New Zealand is generally based on phenotyping rather than genotyping. The potential consequences of polymorphic drug-metabolising enzymes include: increased plasma drug concentration and duration of action, decreased plasma drug concentration and therapeutic failure, adverse drug reactions/toxicity, failure to activate a prodrug, drug metabolism via alternative pathways and exacerbation of drug interactions. Clinical examples include: CYP2D6—perhexiline, tamoxifen opioid analgesics, CYP2C9—phenytoin and warfarin, TPMT—azathioprine and 6-mercaptopurine and UGT1A1—irinotecan.References and further reading
Brandolese R., Scordo M.G., Spina E., et al. Severe phenytoin intoxication in a subject homozygous for CYP2C9*3. Clinical Pharmacology and Therapeutics. 2001;70:391-394.
Evans W.E., McLeod H.L. Pharmacogenomics—drug disposition, drug targets, and side effects. New England Journal of Medicine. 2003;348:538-549.
Evans W.E., Relling M.V. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464-468.
Definitions for Genomic Biomarkers, Pharmacogenomics, Pharmacogenetics, Genomic Data and Sample Coding Categories ICH Topic E15. International Conference on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use 2008. http://www.ema.europa.eu/pdfs/human/pharmacogenetics/19039508en.pdf [13 January 2010].
Giacomini K.M., Sugiyama Y. Membrane transporters and drug response. In Brunton L.L., Lazo J.S., Parker K.L., editors: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edn., New York: McGraw-Hill, 2006. [ch 2]
Goetz M.P., Kamal A., Ames M.M. Tamoxifen pharmacogenomics: the role of CYP2D6 as a predictor of drug response. Clinical Pharmacology and Therapeutics. 2008;83:160-166.
Goetz M.P., Knox S.K., Suman V.J., et al. The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Research Treatment. 2007;101:113-121.
Griese E.U., Ilett K.F., Kitteringham N.R., et al. Allele and genotype frequencies of polymorphic cytochromes P4502D6, 2C19 and 2E1 in Aborigines form Western Australia. Pharmacogenetics. 2001;11:69-76.
Gupta E., Lestingi T.M., Mick R., et al. Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhoea. Cancer Research. 1994;54:3723-3725.
Higashi M.K., Veenstra D.L., Kondo L.M., et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. Journal of the American Medical Association. 2002;287:1690-1698.
Innocenti F., Undevia S.D., Iyer L., et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. Journal of Clinical Oncology. 2004;22:1382-1388.
Iyer L., Das S., Janisch L., et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenetics Journal. 2002;2:43-47.
Jannetto P.J., Bratanow N.C. Utilization of pharmacogenomics and therapeutic drug monitoring for opioid pain management. Pharmacogenomics. 2009;10:1157-1167.
Li B., Duysen E.G., Carlson M., et al. The butyrylcholinesterase knockout mouse as a model for human butyrylcholinesterase deficiency. Journal of Pharmacology and Experimental Therapeutics. 2008;324:1146-1154.
Lima J.J., Blake K.V., Tantisira K.G., et al. Pharmacogenetics of asthma. Current Opinion in Pulmonary Medicine. 2009;15:57-62.
Ma M.K., Woo M.H., McLeod H.L. Genetic basis of drug metabolism. American Journal of Health-System Pharmacy. 2002;59:2061-2069.
Manoharan I., Wieseler S., Layer P., et al. Naturally occurring mutation Leu307Pro of human butyrylcholinesterase in the Vysya community of India. Pharmacogenetics and Genomics. 2006;16:461-468.
Miners J.O. Pharmacogenetics. Australian Doctor. 2007:25-29. February 9
Neitlich H.W. Increased plasma cholinesterase activity and succinylcholine resistance; a genetic variant. The Journal of Clinical Investigation. 1966;45:380-387.
Perera M., Innocenti F., Ratain M.J. Pharmacogenetic testing for uridine diphosphate glucuronosyltransferase 1A1 polymorphism: are we there yet? Pharmacotherapy. 2008;28:755-768.
Rieder M.J., Reiner A.P., Gage B.F., et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. New England Journal of Medicine. 2005;352:2285-2293.
Roden D.M., Altman R.B., Benowitz N.L., et al. Pharmacogenomics: challenges and opportunities. Annals of Internal Medicine. 2006;145:749-757.
Shin J., Kayser S.R., Langaee T.Y. Pharmacogenetics: from discovery to patient care. American Journal of Health-System Pharmacy. 2009;66:625-637.
Wanwimolruk S., Pratt E.L., Denton J.R., et al. Evidence for the polymorphic oxidation of debrisoquine and proguanil in a New Zealand Maori population. Pharmacogenetics. 1995;5:193-198.
Wanwimolruk S., Bhawan S., Coville P.F., et al. Genetic polymorphism of debrisoquine (CYP2D6) and proguanil (CYP2C19) in South Pacific Polynesian populations. European Journal of Clinical Pharmacology. 1998;54:431-435.
Weinshilboum R. Inheritance and drug response. New England Journal of Medicine. 2003;348:529-537.
Wolf C.R., Smith G., Smith R.L. Pharmacogenetics. British Medical Journal. 2000;320:987-990.
Human Genome Project Information: http://www.ornl.gov/sci/techresources/Human_Genome/home.shtml [17 February 2010]
Pharmacogenetics and Pharmacogenomics Knowledge Base (PharmGKB): www.pharmgkb.org [19 January 2010].