Vasodilators and antihypertensives
Vasodilators are a generic group of drugs that are primarily used in the intensive care unit (ICU) for the management of acute hypertensive states and emergencies. In addition, they have an important role in the management of myocardial ischaemia, systemic and pulmonary hypertension and cardiac failure.1
Physiology
Blood pressure is controlled by a complex physiological neurohormonal system involving all components of the cardiovascular system.2,3 Traditionally, clinical practice has focused on the arterial circulation as the major regulator of systemic pressure. The importance of venous circulation in determining mean arterial pressure and cardiac output is discussed in Chapter 90.
The role of the peripheral vasculature, including both arteriolar and venous systems, in the regulation of blood pressure may be conceptually regarded as a balance between vasodilatation and vasoconstriction3 (Fig. 91.1).
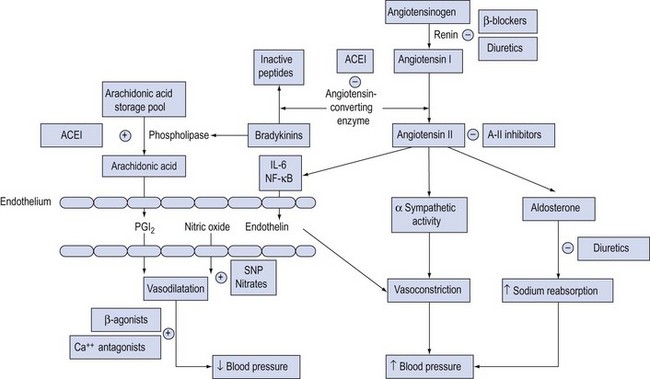
Figure 91.1 Schematic diagram of the neurohormonal factors determining vasomotor tone. Mechanism of action of vasodilators is shown by (−) for inhibition, (+) for stimulation. ACEI = angiotensin-converting enzyme inhibitors; A-II = angiotensin II; IL = interleukin; NF = nuclear factor; PGI2 = prostacyclin; SNP = sodium nitroprusside.
Calcium flux
The concentration of intracellular ionised calcium is the primary determinant of vascular smooth muscle tone: increases lead to smooth muscle contraction; decreases cause relaxation. Control of calcium influx and efflux is determined by adrenergic receptor occupation and changes in membrane potential, mediated through voltage-gated channels (see Ch. 90, Fig. 90.2).
Endothelial system
The endothelium plays a central role in blood pressure homeostasis by secreting substances such as nitric oxide, prostacyclin and endothelin.3 These substances are continuously released by the endothelium and are integral in regional autoregulation.4
Nitric oxide is synthesised from L-arginine by nitric oxide synthases and diffuses into underlying smooth muscle where it activates guanylate cyclase to increase cyclic guanosine monophosphate (cGMP) resulting in relaxation of underlying smooth muscle and vasodilatation.
Prostacyclin is synthesised via the arachidonic pathway and has a minor role in the control of vascular tone.
Endothelins are endothelium-derived vasoconstrictor peptides that are associated with increases in vascular smooth muscle intracellular calcium. They bind to endothelin receptors within the vascular smooth muscle and lead to vasoconstriction, usually in response to shear stresses, tissue hypoxia, angiotensin II and inflammatory mediators (e.g. interleukin-6 and nuclear factor-κB (NF-κB)).
Renin–angiotensin–aldosterone system
Angiotensinogen is converted by renin to form angiotensin I, which is subsequently converted to angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II has a number of effects that are responsible for blood pressure homeostasis. These include release of aldosterone, direct activation of alpha-adrenergic (α-adrenergic) receptors on vascular smooth muscle and a direct effect in the endothelium. These effects are directed at maintaining blood pressure and are integral in the stress response. Angiotensin-converting enzyme is also responsible for the inactivation of bradykinins that have predominantly vasodilatory effects, coupled to arachidonic acid synthesis and generation of prostacyclin.
Adrenergic system
The sympathetic nervous system is integrally involved with all of the above systems, regulating vascular tone at central, ganglionic and local neural levels. Adrenergic stimulation of β-receptors is associated with vasodilatation; α-receptor stimulation results in vasoconstriction. The vascular effects of the catecholamines and vasopressors are discussed in Chapter 90.
Adrenergic stimulation is the predominant system in regulating venous tone.5 This is due to endothelial differences in veins resulting in less production of nitric oxide and reduced responsiveness to angiotensin II.
Pathophysiology
Hypertensive states develop as a result of impaired or abnormal homeostatic processes, causing an imbalance between vasoconstrictive and vasodilatory effects.
Essential hypertension is the most common cause of hypertension and is due to abnormal neurohormonal regulation, particularly exaggerated effects of renin–angiotensin activity.
Secondary causes of hypertension include structural abnormalities such as aortic stenosis or renal artery stenosis; endocrine conditions such as phaeochromocytoma, Cushing's syndrome and pregnancy-induced hypertension; and central causes such as hypertensive encephalopathy or raised intracranial pressure.
Calcium channel blockers
Calcium channel blockers have numerous effects on the cardiovascular system, influencing heart rate conduction, myocardial contractility and vasomotor tone. Entry of calcium through voltage-gated calcium channels is a major determinant of arteriolar, but not venous, tone.6 Calcium channel blockers are recommended as first-step treatment for hypertension in Caucasian patients >55 years and Afro-Caribbean patients of any age.7
There are three major groups of calcium channel blockers that have different sites of action and thus different therapeutic effects: dihydropyridines (e.g. nifedipine, nimodipine, nicardipine, amlodipine, felodipine and clevidipine), phenylalkylamines (e.g. verapamil) and benzothiazepines (e.g. diltiazem).
Magnesium is a physiological calcium antagonist, and is used therapeutically as magnesium sulphate.
Nifedipine
Nifedipine is a predominant arteriolar vasodilator, with minimal effect on venous capacitance vessels and no direct depressant effect on heart rate conduction.
It may be administered intravenously, orally or sublingually, and has a rapid onset of action (2–5 minutes) and duration of action of 20–30 minutes.
Nifedipine can be used to treat angina pectoris, especially that due to coronary artery vasospasm. Peripheral vasodilatation results in decreased systemic blood pressure, often associated with sympathetic stimulation resulting in increased cardiac output and heart rate, which may counter the negative inotropic, chronotropic and dromotropic effects of nifedipine. Nevertheless, nifedipine may be associated with profound hypotension in patients with ventricular dysfunction, aortic stenosis and/or concomitant beta blockade. For this reason, the use of sublingual nifedipine as a method of treating hypertensive emergencies is no longer recommended.8
Nifedipine and related drugs may cause diuretic-resistant peripheral oedema that is due to redistribution of extracellular fluid rather than sodium and water retention.
Nimodipine
Nimodipine is a highly lipid-soluble analogue of nifedipine. High lipid solubility facilitates entrance into the central nervous system where it causes selective cerebral arterial vasodilatation.
It may be used to attenuate cerebral arterial vasospasm following aneurysmal subarachnoid haemorrhage. Improved outcomes have been demonstrated in patients with Grade 1 and 2 subarachnoid haemorrhage.9 Systemic hypotension may result from peripheral vasodilatation that may compromise cerebral blood flow in susceptible patients. Similarly, cerebral vasodilatation may increase intracranial pressure in patients with reduced intracranial elastance.
The recommended dose following aneurysmal subarachnoid haemorrhage is 60 mg orally 4-hourly, but it can be given at 30 mg 2-hourly to reduce variation in blood pressure. The use of intravenous nimodipine is not recommended owing to its profound effect on blood pressure.
Amlodipine
Amlodipine is an oral preparation that has a similar pharmacodynamic profile to nifedipine. In addition to arteriolar vasodilatory and cardiac effects, amlodipine has been shown to exert specific anti-inflammatory effects in hypertension, diabetic nephropathy and in modulating high-density lipoprotein (HDL) in patients with hypercholesterolaemia.10 These effects have seen amlodipine increasingly being used for treatment of hypertension in high-risk patients, and may have a role in stable critically ill patients with associated comorbidities.
Verapamil
The primary effect of verapamil is on the atrioventricular node and this drug is principally used as an antiarrhythmic for the treatment of supraventricular tachyarrhythmias. For this reason, concomitant therapy with beta blockers or digoxin is not recommended.
Verapamil is not as active as nifedipine in its effects on smooth muscle and it therefore causes less pronounced decrease in systemic blood pressure and is also negatively inotropic. It has a limited role as a vasodilator.11
Diltiazem
Diltiazem has a similar cardiovascular profile to verapamil, although its vasodilatory properties are intermediate between nifedipine and verapamil. Diltiazem exerts minimal cardiodepressant effects and is less likely to potentiate beta blockers.
Magnesium sulphate
Magnesium regulates intracellular calcium and potassium levels by activation of membrane pumps and competition with calcium for transmembrane channels. Physiological effects are widespread, affecting cardiovascular, central and peripheral nervous systems and the musculoskeletal junction.12
It acts as a direct arteriolar and venous vasodilator causing reductions in blood pressure. Modulation of centrally mediated and peripheral sympathetic tone results in variable effects on cardiac output and heart rate.
Consequently, it has an established role in the treatment of pre-eclampsia and eclampsia,13 perioperative management of phaeochromocytoma14 and treatment of autonomic dysfunction in tetanus.15 It has been proposed as a possible agent for the prevention of secondary ischaemia after aneurysmal subarachnoid haemorrhage; however, evidence of a benefit on outcome is still lacking.16
Direct-acting vasodilators
These drugs act directly on vascular smooth muscle and exert their effects predominantly by increasing the concentration of endothelial nitric oxide. These drugs are also known as nitrovasodilators.17
Sodium nitroprusside
Sodium nitroprusside is a non-selective vasodilator that causes relaxation of arterial and venous smooth muscle. It is compromised of a ferrous ion centre associated with five cyanide moieties and a nitrosyl group. The molecule is 44% cyanide by weight.
It is reconstituted from a powdered form. The solution is light sensitive so it requires protection from exposure to light (e.g. by wrapping administration sets in aluminium foil). Prolonged exposure to light may be associated with an increase in release of hydrogen cyanide, although this is seldom clinically significant.
When infused intravenously, sodium nitroprusside interacts with oxyhaemoglobin, dissociating immediately to form methaemoglobin while releasing free cyanide and nitric oxide. The latter is responsible for the vasodilatory effect of sodium nitroprusside.
Dosage is from 0.5 µg/kg/min to 8 µg/kg/min, but should always start at a low infusion rate and build up slowly.
Onset of action is almost immediate with a transient duration, requiring continuous intravenous infusion to maintain a therapeutic effect. Tachyphylaxis can occur and large doses should not be used if the desired therapeutic effect is not attained, as this may be associated with toxicity.
Sodium nitroprusside produces direct venous and arterial vasodilatation, resulting in a prompt decrease in systemic blood pressure. The effect on cardiac output is variable. Decreases in right atrial pressure reflect pooling of blood in the venous system, which may decrease cardiac output. This may result in reflex tachycardia that may oppose the overall reduction in blood pressure. In patients with left ventricular failure, the effect on cardiac output will depend on initial left ventricular end-diastolic pressure.
Sodium nitroprusside may potentially increase myocardial ischaemia in patients with coronary artery disease by causing an intracoronary steal of blood flow away from ischaemic areas by arteriolar vasodilatation. Secondary tachycardia may also exacerbate myocardial ischaemia.
Due to its non-selectivity, sodium nitroprusside has direct effects on most vascular beds. In the cerebral circulation, sodium nitroprusside is a cerebral vasodilator, leading to increases in cerebral blood flow and blood volume. This may be critical in patients with increased intracranial pressure. Rapid and profound reductions in mean arterial pressure produced by sodium nitroprusside may exceed the autoregulatory capacity of the brain to maintain adequate cerebral blood flow.
Sodium nitroprusside is a pulmonary vasodilator and may attenuate hypoxic pulmonary vasoconstriction, resulting in increased intrapulmonary shunting and decreased arterial oxygen tension. This phenomenon may be exacerbated by associated hypotension.
The prolonged use of large doses of sodium nitroprusside may be associated with toxicity related to the production and cyanide and, to a lesser extent, methaemoglobin.18
Free cyanide produced by the dissociation of sodium nitroprusside reacts with methaemoglobin to form cyanmethaemoglobin, or is metabolised by rhodenase in the liver and kidneys to form thiocyanate. A healthy adult can eliminate cyanide at a rate equivalent to a sodium nitroprusside infusion of 2 µg/kg per min or up to 10 µg/kg per min for 10 minutes, although there is marked inter-individual variability.
Toxicity should be of concern in patients who become resistant to sodium nitroprusside despite maximum infusion rates and who develop an unexplained lactic acidosis. In high doses, cyanide may cause seizures.
Treatment of suspected cyanide toxicity is cessation of the infusion and administration of 100% oxygen. Sodium thiosulphate (150 mg/kg) converts cyanide to thiocyanate, which is excreted renally. For severe cyanide toxicity, sodium nitrite may be infused (5 mg/kg) to produce methaemoglobin and subsequently cyanmethaemoglobin. Hydroxycobalamin, which binds cyanide to produce cyanocobalamin, may also be administered (5 g over 15 minutes, which may be repeated in severe cases).
Glyceryl trinitrate
Glyceryl trinitrate is an organic nitrate that generates nitric oxide through a different mechanism from sodium nitroprusside.
The pharmacokinetics allows glyceryl trinitrate to be given by infusion, with a longer onset and duration of action than sodium nitroprusside. The intravenous dosage can start at 5 µg/min and increase incrementally to 200 µg/min (max dose 400 µg /min). Glyceryl trinitrate may also be administered sublingually, orally or transdermally.
Tachyphylaxis is common with glyceryl trinitrate; doses should not be increased if patients no longer respond to standard doses. Glass bottles or polyethylene administration sets are required as glyceryl trinitrate is absorbed into standard polyvinylchloride sets.
The effects on the peripheral vasculature are dose-dependent, acting principally on venous capacitance vessels to produce venous pooling and decreased ventricular preload. Together with furosemide, glyceryl trinitrate is particularly useful in treating acute cardiac failure and pulmonary oedema.
Glyceryl trinitrate primarily dilates larger conductance vessels of the coronary circulation, resulting in increased coronary blood flow to ischaemic subendocardial areas, thereby relieving angina pectoris. This is in contrast to sodium nitroprusside, which may cause a coronary steal phenomenon.
Reductions in blood pressure are more dependent on blood volume than with sodium nitroprusside. Precipitous falls in blood pressure may occur in hypovolaemic patients with small doses of glyceryl trinitrate. In euvolaemic patients, reflex tachycardia is not as pronounced as with sodium nitroprusside. At higher doses, arteriolar vasodilatation occurs without significant changes in calculated systemic vascular resistance. More recently glyceryl trinitrate has been suggested as one approach to improve microcirculatory flow in septic shock, but only after adequate fluid resuscitation.19
Glyceryl trinitrate is a cerebral vasodilator and should be used with caution in patients with known or suspected raised intracranial pressure. Headache due to this mechanism is a common side-effect in conscious patients.
Isosorbide dinitrate
Isosorbide dinitrate is the most commonly administered oral nitrate for the prophylaxis of angina pectoris. It has a physiological effect that lasts up to 6 hours in doses of 60–120 mg. The mechanism of action is the same as glyceryl trinitrate. Hypotension may follow acute administration, but tolerance to this develops with chronic therapy.20
Hydralazine
Hydralazine is a potent, arterioselective, direct-acting vasodilator that acts via stimulation of cGMP and inhibition of smooth muscle myosin light chain kinase.
Following intravenous administration, 5–10 mg intravenously, hydralazine has a rapid onset of action, usually within 5–10 minutes. It can alternatively be given by continuous intravenous infusion, initially 200–300 µg/min and maintenance usually 50–150 µg/min, and may also be administered orally. The drug is partially metabolised by acetylation, for which there is marked inter-individual variability (35% of the population are slow acetylators). Although this does not have much clinical significance regarding the antihypertensive effects, it is important with respect to toxicity.20
Hydralazine causes predominantly arteriolar vasodilatation that is widespread but not uniform. It is associated with direct and reflex sympathetic activity, so that cardiac output and heart rate are increased. Prolonged use of hydralazine stimulates renin release and is associated with sodium and water retention. Consequently, hydralazine is frequently administered with beta blockers and/or diuretics.
Chronic use of hydralazine may be associated with immunological side-effects including lupus syndrome, vasculitis, haemolytic anaemia and rapidly progressive glomerulonephritis.
Alpha-adrenergic antagonists
Several groups of compounds act as alpha-adrenergic blockers with variable affinity for populations of α-receptors. Physiology and pathophysiology may influence the responsiveness of the drug receptor–effector relationship. Receptor pathobiology is discussed in Chapter 90. Consequently, there may be marked inter- and intra-individual variability in the patient's response to these drugs.
There are six main groups of α-receptor antagonists: imidazolines (e.g. phentolamine), haloalkylamines (e.g. phenoxybenzamine), prazosin, beta-adrenergic antagonists with a receptor antagonism (labetalol, carvedilol), phenothiazines (chlorpromazine) and butyrophenones (haloperidol).
Phentolamine
Phentolamine is a non-selective, competitive antagonist at α1- and α2-receptors. At low doses, phentolamine causes prejunctional inhibition of norepinephrine (noradrenaline) release (via α2-receptor inhibition). At higher doses, more complete α-receptor blockade is achieved, with enhancement of effects of beta agonists due to increased local concentration of norepinephrine produced by α2-blockade (see Ch. 90, Fig. 90.3a).
Phentolamine is administered intravenously and may be given intermittently or by infusion. Onset is rapid (within 2 minutes), with a duration of action of 10–15 minutes.
Arteriolar and venous vasodilatation reduces systemic blood pressure. Effects on cardiac output are variable, and there is modest reflex sympathetic stimulation without significant increases in heart rate.
Phenoxybenzamine
Phenoxybenzamine is a non-selective, non-competitive, α1- and α2-receptor antagonist. Blockade is also produced on histamine, serotonin and acetylcholine muscarinic receptors. Reuptake of norepinephrine is blocked, thereby potentiating the effects of beta agonists.
Phenoxybenzamine is usually administered orally, but may also be given intravenously (taking care to avoid extravasation as it is irritant to tissues). It has a long onset of action and prolonged duration of action (3–4 days). It causes a gradual reduction in systemic blood pressure, without rapid reflex sympathetic activity.
Prolonged use is associated with increased beta-adrenergic effects, predominantly increased heart rate, for which combination therapy with beta blockade is used. Phenoxybenzamine is primarily used in the management of phaeochromocytoma, either preoperatively or long term in inoperable patients. It may also be used to control autonomic hyperreflexia in patients with spinal cord transection.21
Prazosin
Prazosin is a relatively arterioselective, competitive, α1-receptor antagonist. It acts postjunctionally and therefore does not inhibit reuptake of norepinephrine. Consequently, it produces less tachycardia for a given reduction in systemic blood pressure.
It is administered orally and usually used for essential or renovascular (hyperreninaemia) hypertension. It is frequently used in combination with beta blockers and diuretics, particularly in patients with renal dysfunction.
Labetalol
Labetalol is specific competitive antagonist at α1-, β1- and β2-adrenergic receptors. Beta blockade effects predominate, with a approximate ratio of α1 : β-receptor blockade of 1 : 4–7. Labetalol has partial agonist effects on β2-receptors.
It is administered intravenously (typically 10–50 mg), has a rapid onset of action (5–10 minutes) with a duration of 2–6 hours. It may be given by infusion (usually 15–180 mg/h).
Systemic blood pressure and cardiac output are reduced by a combination of negative inotropy, arterial and venous vasodilatation. Reflex tachycardia is attenuated by beta blockade. These properties make labetalol particularly useful in controlled hypotension during anaesthesia and surgery to reduce bleeding.
Side-effects such as bronchospasm and hyperkalaemia relate predominantly to beta blockade.
Carvedilol
Carvedilol is a non-selective beta blocker with α1-antagonist activity. Most of the vasodilator activity relates to α1-antagonism, although at high concentrations it also blocks calcium entry. The ratio of α1 : β-receptor blockade is 1 : 10.
It is administered orally; no intravenous preparation is available.
Recent studies have demonstrated slowing of progression of congestive cardiac failure and improved mortality, particularly when used in conjunction with ACE inhibitors in patients with mild to moderate cardiac failure.22,23 It may also be used in patients who cannot be treated with ACE inhibitors.
Haloperidol and chlorpromazine
These drugs act as competitive α-receptor antagonists causing non-selective vasodilatation and blockade of norepinephrine reuptake.
These drugs are primarily used as major tranquillisers or antipsychotics; their effect on the peripheral vasculature should be regarded as a side-effect, rather than a specific therapeutic action.
Reduction of systemic blood pressure is variable and may be precipitous, particularly in hypovolaemic patients with high sympathetic drive. These drugs may be useful in neurogenic hypertension, and are not regarded as first-line vasodilators.
Inodilators
Many inotropic drugs have peripheral vascular effects and these are discussed in more detail in Chapter 90.
At low doses, epinephrine, norepinephrine and dopamine are predominantly beta agonists and cause both arterial and venous vasodilatation, which may cause reductions in mean arterial pressure.
Dobutamine and isoprenaline are predominantly beta agonists and may cause decreases in mean arterial pressure, particularly in hypovolaemic patients or those with increased sympathetic drive. These agents may have a role in reducing left ventricular afterload in patients with systolic heart failure.
Milrinone is a selective type III phosphodiesterase inhibitor that prevents the breakdown of cAMP within cardiac and vascular tissues. The increased cAMP levels lead to increased levels of intracellular calcium and thus increased contraction of cardiac muscle. Within vascular smooth muscle the cAMP inhibits myosin light chain kinase producing less contraction and thus vascular relaxation.
Levosimendan is a newer calcium sensitiser that leads to a greater ventricular contraction for the same intracellular calcium concentration. It also leads to vasodilatation, mediated by activation of ATP-sensitive sarcolemmal and mitochondrial potassium channels. The drug itself has a relatively short half-life, but it has a long-acting active metabolite so that haemodynamic effects may be maintained for up to 7 days.
Both milrinone and levosimendan can lead to marked hypotension, particularly if a bolus dose is given. Therefore in critically ill patients a loading dose is best avoided and any excessive vasodilation may need to be balanced by a low dose of a vasoconstrictor. Usual infusion rates of milrinone are 0.375–0.75 µg/kg/min and levosimendan 0.05–0.2 µg/kg/min.
Angiotensin-converting enzyme inhibitors
Angiotensin-converting enzyme (ACE) inhibition has become a cornerstone in the management of patients with hypertension, cardiac failure and ischaemic heart disease.24,25 They are now recommended as first-stage treatment for hypertension in Caucasian patients <55 years and patients with diabetic nephropathy.7 These drugs act by non-selective, competitive, irreversible inhibition to the angiotensin I binding site thus reducing conversion to angiotensin II.
There are a very large number of ACE inhibitors on the market with very high penetration into the ambulant patient population. These drugs are administered orally; there are no routinely used parenteral preparations. Consequently, many critically ill patients admitted to the ICU may be taking ACE inhibitors. As a general rule, ACE inhibitors are stopped in most critically ill patients until vital organ (specifically renal) function is stabilised and the patient can take them enterally. Thereafter, doses are gradually increased over time with close monitoring of renal function.
Preparations
Captopril has a short half-life and therefore may be useful for initiating treatment within the ICU. It is administered orally in increasing doses and intervals to a maximum dose of 50 mg 8-hourly. It may be administered sublingually in acute hypertension (5–25 mg), with an onset of action in 20–30 minutes, and duration of 4 hours. There are no significant differences in the cardiovascular effects between captopril and other preparations.
Enalapril is a prodrug, effective by hepatic metabolism to enalaprilat, producing a slower and more controlled action. It is administered orally in 5 mg increments to a total of 20 mg twice daily.
Lisinopril has the advantage of single daily dosing and may be useful in stable critically ill patients.
Cardiovascular effects
The cardiovascular effects of ACE inhibition are widespread, with effects that influence the peripheral vasculature, cardiac performance, and salt and water homeostasis. Consequently, ACE inhibitors are not principally regarded as vasodilators, although they have both direct and indirect effects on the peripheral vasculature.
Increased production of endothelial vasodilators such as prostacyclin and decreased production of endothelin by angiotensin result in generalised venous and arteriolar vasodilatation. This occurs in the absence of reflex sympathetic activity or changes in heart rate, due to the modulation of adrenergic stimulation. Systemic blood pressure is reduced without changes in cardiac output or heart rate.
ACE inhibition is associated with improved myocardial performance following acute myocardial infarction due to left ventricular remodelling and improvement in neurohumoral activation. These drugs have been shown to improve survival following myocardial infarction in patients with left ventricular dysfunction.26
‘First-dose hypotension’ is described in patients receiving ACE inhibitors for the first time. This may occur particularly in patients who are salt- and water-depleted, or those who develop sensitivity to the drug. Drug sensitivity may also present as a sudden decrease in renal function following commencement of the drug (see below).
Renovascular effects
ACE inhibitors may cause renal failure, particularly in patients with renovascular disease, hyperreninaemic hypertension and acute kidney injury. The renal effects of ACE inhibitors may be potentiated by diuretics, non-steroidal anti-inflammatory drugs and beta blockers. ACE inhibitors are contraindicated in patients with bilateral renal artery stenosis.
As a rule in intensive care patients, ACE inhibitors are started in suitable patients once renal function has stabilised and the patient no longer requires inotropic support. Initial doses should be low and increased as tolerated.
Side-effects and toxicity
In addition to renal dysfunction, ACE inhibitors may be associated with a number of side-effects. The most common of these is cough, which is due to the increased production of kinins.27
Severe angioneurotic oedema causing upper-airway obstruction may occur with all ACE inhibitors, although this is less common with enalapril and lisinopril. This is due to increased activation of bradykinins. ACE inhibitors are contraindicated in patients with a history of hereditary or idiopathic angioneurotic oedema28 and should be avoided in pregnancy.
Neutropenia and agranulocytosis are uncommon but potentially lethal side-effects in susceptible patients.
Angiotensin receptor blockers
These are a newer class of antihypertensive drugs that cause irreversible, selective blockade of angiotensin II at AT1 receptors.29,30
Losartan is the prototype, which has been followed by newer compounds such as irbesartan, valsartan and telmisartan. These drugs are oral preparations; there is no parenteral form.
The cardiovascular profile of angiotensin receptor blockers is similar to the ACE inhibitors, but selective blockade of angiotensin II offers several possible advantages over ACE inhibitors. These drugs are long-acting and may be given once daily; onset of action is slow, thereby avoiding first-dose hypotension; side-effects such as cough and angioneurotic oedema are less common.31 Angiotensin receptor blockers may be used as an alternative to ACE inhibitors but they should not be combined.7
Centrally acting agents
These agents modulate adrenergic stimulation at central nervous system and spinal cord level. The vasomotor centre of the medulla mainly controls sympathetic pressor influences, although other brainstem, midbrain and spinal centres have a role.
Most central responses are mediated through α2-adrenergic receptors, which modulate the release and reuptake of norepinephrine, with subsequent effects on the peripheral vasculature and cardiac function.
Clonidine
Clonidine is a centrally acting α2-agonist that stimulates inhibitory neurons in the vasomotor centre. This results in a reduction in sympathetic outflow from the central nervous system and is associated with negative inotropy and reduction in heart rate. Systemic blood pressure is reduced by this mechanism, with associated arteriolar and venous vasodilatation. Clonidine has centrally acting analgesic properties, which make it a suitable drug in patients with postoperative hypertension.
Peripherally, it stimulates prejunctional α2-receptors, thereby decreasing norepinephrine release, but may also have an effect at postjunctional α1-receptors, causing vasoconstriction. This may present as rebound hypertension following initial reduction of blood pressure, as there is variable duration of the central and peripheral effects.
Clonidine can be administered orally (50–100 µg 8-hourly) or by intravenous injection (25–150 µg) or by infusion (maximum 750 µg in 24 hours). It has a rapid onset of action (5–10 minutes) and duration of action of 20–30 minutes.
Methyldopa
Methyldopa acts as a centrally acting ‘false’ transmitter following metabolism to methylnorepinephrine and subsequent stimulation of α2-receptors leading to reduced sympathetic outflow, although the precise mechanism is not clear.
It has a limited role in hypertensive emergencies, but is useful in accelerated essential, renovascular and pregnancy-induced hypertension.
It is administered orally in doses of 250 mg to 3 g per day although side-effects (abnormal function, depression) may be minimised if the daily dose is <1 g. It may lead to a positive direct Coombs test in 20% of patients.
Other antihypertensive agents
Beta-adrenergic antagonists
Beta blockers have been used for the treatment of hypertension for over 30 years and have an increasingly important role in the management of cardiac failure.32–34 Although no longer recommended as first-step treatment of hypertension they may be used if ACE inhibitors are poorly tolerated.7
In addition to decreasing heart rate and contractility, beta blockers have other neurohumoral effects that affect vascular tone. These relate to inhibition of renin release from juxtaglomerular cells (see Fig. 91.1) and prejunctional inhibition of norepinephrine that result in reduction in vascular tone and blood pressure. A central effect of beta blockers has also been proposed.
The mode of action has been described in terms of selectivity to blockade of beta-adrenergic receptors (β1 and/or β2). While this is an appropriate pharmacological distinction, the clinical activity of these drugs is not as predictable due to mixed populations of β1- and β2-receptors in most organs and variable receptor responsiveness in physiological and pathophysiological conditions. Consequently, there is marked inter-individual variability in the response to these drugs. In high-enough doses, whether intentionally or due to toxicity, all beta blockers will cause generalised antagonism with resultant therapeutic and toxic effects.
Lipid-soluble beta blockers include propranolol and metoprolol, which are predominantly excreted by the liver; atenolol and sotalol are water-soluble and predominantly renally excreted, warranting caution with these drugs in patients with renal dysfunction.
Beta blockers may be given orally or intravenously. As there is significant first-pass metabolism, doses for oral and intravenous administration are markedly different.
Esmolol is an intravenous beta blocker that is rapidly metabolised by red cell esterases and is therefore not dependent on renal or hepatic function. Its rapid onset of action and short duration allow infusion of drug, making it a useful drug in patients with acute hypertensive states associated with tachycardia. Labetalol and carvedilol are discussed above.
Beta blockers are frequently used as adjuncts to vasodilators in the treatment of hypertensive emergencies and states, particularly where reflex tachycardia and sympathetic stimulation occur (e.g. hydralazine, nifedipine and prazosin).
Side-effects and toxicity of beta blockers include bradycardia (which may be profound), hypotension, bronchoconstriction (caution in patients who have asthma), aggravation of peripheral vascular ischaemia, hyperkalaemia and masking of the sympathetic response to hypoglycaemia.
Diuretics
As with beta blockers, diuretics have an established place in the management of hypertension. In addition to their effects on salt and water excretion and inhibition of aldosterone, direct vasodilatory effects are associated with diuretics such as furosemide and the thiazides.
These drugs have a rapid venodilatory action, which may be due to inhibition of norepinephrine-activated chloride channels on veins. Reductions of blood pressure and right atrial pressure may occur following low doses, and may present before an associated diuresis.
All diuretics should be used with caution in patients with renal dysfunction and avoided if hypovolaemia is present or suspected.
Pulmonary Vasodilators
Pulmonary vasodilators may be used in the ICU mainly to treat acute right-sided heart failure commonly after cardiac surgery/transplantation. Many of the systemic vasodilators detailed above (namely calcium channel blockers, sodium nitroprusside and nitrates) will also have vasodilatory effects within the pulmonary circulation. Specific pulmonary vasodilators may be inhaled or administered intravenously.
Nitric Oxide
As detailed above, nitric oxide (NO) is an endogenous vasodilator. When inhaled it leads to vasodilation in only the ventilated areas of the lung, and rapid combination with haemoglobin prevents systemic vasodilation. This may improve ventilation–perfusion mismatch resulting in an improved oxygenation but controlled trials have not demonstrated any beneficial effect on outcomes in severe acute lung injury and it may have an adverse effect on kidney function.35 Its use is now mainly confined to treatment of acute pulmonary hypertension post cardiac surgery.
NO is oxidated to NO2 particularly in a high-oxygen environment and therefore NO2 levels should be carefully monitored. Similarly appropriate scavenging systems must be employed.
Rebound pulmonary hypertension may occur and so NO therapy should be slowly weaned rather than abruptly stopped.
Prostacyclin
Inhaled epoprostenol or iloprost (a prostacyclin derivative) can be used as alternatives to NO. Care should be taken to avoid systemic hypotension.
Sildenafil
Sildenafil is a phosphodiesterase type 5 inhibitor used in the management of chronic pulmonary hypertension, as well as erectile dysfunction. There are reports of its use for acute pulmonary hypertension within the ICU and that it can improve haemodynamics, however, by increasing shunt fraction it might worsen oxygenation when used in acute lung injury.36
Drug selection
The clinical use of vasodilators in intensive care is different from their use in ambulatory patients. In the critically ill patient, these drugs are primarily used to control acute rises in mean arterial pressure associated with sympathetic stimulation, or as specific treatment of hypertensive emergencies.37
The ideal vasodilator is therefore one that has a rapid and predictable onset of action, allows titration to achieve a desired systemic blood pressure, does not compromise cardiac output, does not cause significant reflex tachycardia and is non-toxic.
The selection of drug to treat hypertensive states will depend on the predominant cause of hypertension and the mechanism of action in the homeostatic pathway (outlined in Fig. 91.1).
There are no large studies investigating optimum therapy in patients presenting with hypertensive emergencies. These conditions occur in a heterogeneous group of patients and drug selection is essentially determined by the underlying pathophysiology, personal preference and experience.37
Monitoring
The principles of haemodynamic monitoring in patients receiving vasoactive drugs are outlined in Chapter 90.
Patients with severe hypertension or those receiving infusions or doses of potent vasodilators such as sodium nitroprusside and glyceryl trinitrate must be monitored via an intra-arterial catheter. Non-invasive measurement devices are not rapid or accurate enough and are not recommended in patients with hypertensive emergencies.
As peripheral vasodilators have significant effects on both the arterial and venous systems, assessment of volume status is important. In the majority of patients, establishing a euvolaemic state is essential before commencing a vasodilator.
Dosages and drug administration
Vasodilators administered via infusion are delivered through a dedicated central venous catheter using infusion pumps or syringe drivers and titrated to achieve a target mean arterial pressure. Infusion lines should be free of injection portals and clearly marked with identifying labels. Common drug doses are shown in Table 91.1.
Table 91.1
Dose and infusion concentrations of commonly used vasodilators and antihypertensives in intensive care
| AGENT | INFUSION/DOSE | CAUTION |
| Sodium nitroprusside | Diluted in 5% dextrose; range 0.5–8 µg/kg/min | Cyanide toxicity (> total dose 0.5 mg/kg per 24 hours) Photodegradation Raised intracranial pressure Rebound hypotension Shunt and oxygen desaturation |
| Glyceryl trinitrate | Diluted in 5% dextrose; range 5–200 µg/min | Drug binding to polyvinylchloride Tachyphylaxis Raised intracranial pressure |
| Hydralazine | 10–20 mg i.v. bolus 20–50 mg 6–8-hourly |
Tachycardia Myocardial ischaemia |
| Phentolamine | 1–10 mg i.v. boluses 5–30 mg/h infusion |
Tachycardia |
| Phenoxybenzamine | Oral: 10 mg/day until postural hypotension i.v.: 1 mg/kg per day |
Idiosyncratic hypotension |
| Prazosin | 2–10 mg/day, 8-hourly | |
| Nifedipine | 5–10 mg oral/sublingual | Precipitous hypotension |
| Amlodipine | 5–10 mg oral/day | Caution in renal impairment |
| Captopril | 6.25–50 mg orally, 8-hourly Acute hypertension: 6.5–25 mg sublingually p.r.n. |
Caution in renovascular hypertension and renal failure Pregnancy Angioneurotic oedema |
| Enalapril | 5–20 mg/day | |
| Enalaprilat | 0.625–5 mg bolus | Caution in renal failure and hypovolaemia |
| Losartan | 25–100 mg/day | Caution in renal failure |
| Clonidine | 25–150 µg i.v. bolus | Acute, perioperative centrally mediated hypertension May cause rebound hypertension with chronic use |
| Atenolol | 1–10 mg i.v. boluses 25–100 mg oral b.d. |
Caution in poor left ventricular function, asthma Hyperkalaemia Potentiated in renal failure |
| Metoprolol | 50–100 mg 12-hourly; 5 mg i.v. bolus | As for atenolol, safe in renal failure |
| Esmolol | Loading dose 0.5 mg/kg 50–200 µg/kg/min infusion |
|
| Labetalol | 10–50 mg i.v. boluses 0.5–4 mg/min infusion |
|
| Magnesium sulphate | 40–60 mg/kg loading (or 6 g) 2–4 g/h infusion |
Maintain serum magnesium >1.5–2 mmol/L |
Specific situations
The following is a summary of the clinical uses of the above drugs in hypertensive states commonly encountered in the ICU. Specific pharmacology and physiological effects are discussed above.
Acute hypertension
The most common cause of hypertension in intensive care patients is pain or agitation, particularly in postoperative patients. It is important that patients have adequate analgesia and sedation before antihypertensives or vasodilators are used. Other common causes of hypertension include hypothermia, urinary retention, positional discomfort and omission of pre-admission antihypertensives, particularly beta blockers. The majority of instances of acute hypertension in the ICU will respond to simple measures addressing the above.38
Sustained hypertension may be treated acutely with incremental doses or infusions of short-acting drugs such as glyceryl trinitrate, sodium nitroprusside, labetalol, hydralazine, nifedipine or clonidine. Infusions of vasodilators may be required if hypertension persists, or if the patient in unable to take longer-acting oral agents such as prazosin or amlodipine. Hypertension associated with tachycardia may be treated with beta blockers.
Hypertensive encephalopathy
Hypertensive encephalopathy is defined as an acute organic brain syndrome occurring as a result of failure of cerebrovascular autoregulation. There may be differences in the degree of hypertension that cause encephalopathy and it may be the rate of increase of blood pressure that is more important than the absolute value. It may present as confusion, visual disturbances, blindness, seizures or stroke. Uncontrolled hypertension is one of the causes of the posterior reversible encephalopathy syndrome (PRES), along with pre-eclampsia, immunosuppressant drugs and sepsis. If not adequately treated, hypertensive encephalopathy may result in intracerebral haemorrhage, coma or death.39
Hypertensive encephalopathy may occur in patients with untreated or undertreated hypertension or in association with other diseases such as renal disease (e.g. glomerulonephritis, renovascular disease), thrombotic thrombocytopenic purpura, immunosuppressive therapy, collagen vascular diseases or eclampsia. Consequently, drug treatment will depend on the context in which it occurs. It is also important to rule out other causes of neurological deterioration that may also present with hypertension (e.g. stroke, intracranial haemorrhage or space-occupying lesion).
There is no evidence from randomised controlled trials to conclude one therapy is superior to another at improving outcomes. The aim of drug therapy in these patients is to reduce blood pressure in a controlled, predictable and safe way. Acutely, short-acting, titratable parenteral drugs are suitable in emergency situations. Assuming there are no absolute contraindications to beta blockers, labetalol and esmolol are ideal drugs to use. Sodium nitroprusside can be used although it may increase intracranial pressure and reduce cerebral blood flow. Phentolamine may be equally effective.8,36
Other agents that are useful in controlling severe hypertension include hydralazine, clonidine and ACE inhibitors (although these must be used cautiously in patients with associated renal dysfunction). Severe drops in blood pressure that might compromise end-organ perfusion have been reported after the use of nifedipine and therefore its use in the emergency setting is not recommended.8 Combination therapy is often required, although this should be done with caution to minimise additive effects with resultant hypotension.
Patients with hypertensive emergencies are frequently hypovolaemic due to excessive sympathetic stimulation. In the absence of left ventricular failure, judicious fluid replacement may reduce blood pressure and improve renal function, thereby minimising precipitous hypotension that may result following administration of some drugs. Diuretics are generally avoided in these conditions unless there is evidence of left ventricular failure.40
Acute stroke
Acute stroke syndromes frequently occur in the setting of severe hypertension. The reduction of mean arterial pressure must be balanced by the maintenance of adequate cerebral perfusion pressure and cerebral blood flow. Ischaemic brain is vulnerable to critical reductions in cerebral blood flow, while excessive mean arterial pressure may increase the risk of cerebral haemorrhage.41
Acutely, blood pressure should be maintained in a normal range until intracranial pathology has been identified by CT scan. Aggressive reduction in blood pressure is not recommended in patients with ischaemic stroke, whilst hypertension in patients with aneurysmal subarachnoid haemorrhage or intracranial haemorrhage may be managed by drugs outlined above.
Aortic dissection
Aortic dissection is the most dramatic and most rapidly fatal complication of severe hypertension. The aim of medical treatment is to control blood pressure and left ventricular ejection velocity to minimise propagation of the dissection. Blood pressure should be decreased as rapidly as possible to a normal or slightly hypotensive level. Titrations are usually made to achieve systolic blood pressures of 100–110 mmHg or mean arterial pressure of 55–65 mmHg. This will depend on the patient's premorbid blood pressure and the accuracy of blood pressure measurement. It is important to maintain blood pressure at levels compatible with adequate cerebral and renal perfusion.42
This is best achieved initially by use of opioid analgesia, intravenous beta blockers (e.g. esmolol, labetalol or atenolol), and possibly adding a vasodilator such as sodium nitroprusside or glyceryl trinitrate. Tachycardia must be avoided as this is a significant determinant of aortic shear force that may exacerbate the dissection. Verapamil or diltiazem are suitable alternatives for patients who have a contraindication to beta blockade.
Aortic dissection distal to the left subclavian artery is managed conservatively with antihypertensive therapy. Proximal dissections are managed surgically after acute control of blood pressure.
Acute myocardial ischaemia
Myocardial ischaemia in the absence of obstructive coronary atherosclerosis may be precipitated by severe hypertension. This occurs by increased left ventricular wall stress, reduced preload, tachycardia and increased myocardial metabolic demand. Severe ischaemia may result in acute left ventricular failure.
Intravenous glyceryl trinitrate is useful in this situation and may be used in combination with beta blockers such as esmolol, labetalol or carvedilol.
ACE inhibitors may be used in the acute situation and may be required for longer-term treatment.
Phaeochromocytoma
Tumours of the adrenal medulla secrete catecholamines that result in initial paroxysmal, then sustained, severe hypertension. They may present to the ICU as a hypertensive emergency or perioperatively for surgical ablation.43
Acute hypertensive crises associated with phaeochromocytoma are managed with incremental doses or infusions of phentolamine. Untreated patients may be significantly hypovolaemic and may require judicious volume replacement. Beta blockers should not be used in the acute setting as these will potentiate unopposed alpha-adrenergic stimulation.
Phenoxybenzamine forms the mainstay of treatment and preparation for surgery. This is commenced in 20–30 mg increments and continued until blood pressure is controlled. Excessive beta-adrenergic effects are treated with beta blockers only after sufficient alpha blockade with phenoxybenzamine.21
Magnesium sulphate is useful in the perioperative management of phaeochromocytoma. It is given by infusion at 2–4 g/hour.14
Renal failure
Renal insufficiency may be a cause or consequence of a hypertensive emergency. Patients on haemodialysis (particularly those receiving erythropoietin therapy) and renal transplant patients (especially those receiving ciclosporin or corticosteroids) commonly present with severe hypertension. In patients with new-onset renal failure accompanying severe hypertension, blood pressure must be controlled without potentiating renal dysfunction. Drugs such as calcium channel blockers, phentolamine or prazosin may preserve renal blood flow and are appropriate in these patients. ACE inhibitors and diuretics should be used with caution until renal function has stabilised or improved.
Patients in the recovery phase of acute renal failure are usually hypertensive. This is a normal physiological response and should not be treated unless there is associated myocardial or cerebral ischaemia.44
Pre-eclampsia and eclampsia
In addition to delivery of the baby and placenta, parenteral magnesium sulphate is the treatment of choice to prevent the evolution of pre-eclampsia to eclampsia (seizures and deteriorating encephalopathy13). The recommended drugs for the treatment of severe hypertension in critically ill women during pregnancy or soon after birth include labetalol, hydralazine and nifedipine.45
ACE inhibitors and angiotensin receptor blockers are contraindicated in pregnancy. This is discussed in Chapter 63.
Drug interactions
Severe rebound hypertension may result following abrupt cessation of antihypertensive treatment. Drugs associated with this discontinuation syndrome include clonidine, methyldopa, beta blockers, guanethidine and diuretics. The degree of rebound depends on the rapidity of drug withdrawal, dosage, renovascular and cardiac function. Antihypertensives should be reintroduced according to the status of the patient and the degree of hypertension managed accordingly.
Interaction with monoamine oxidase inhibitors and drugs such as indirect sympathomimetics, narcotics and tyramine-containing foods may result in a hypertensive emergency. This is best managed acutely with alpha and/or beta blockers.
References
1. Erdmann, E. The management of heart failure – an overview. Basic Res Cardiol. 2000; 95(Suppl 1):I3–17.
2. Cohn, JN. Left ventricle and arteries: structure, function, hormones, and disease. Hypertension. 2001; 37:346–349.
3. Spieker, LE, Flammer, AJ, Luscher, TF. The vascular endothelium in hypertension. Handb Exp Pharmacol. 2006; 176:249–283.
4. Egan, K, FitzGerald, GA. Eicosanoids and the vascular endothelium. Handb Exp Pharmacol. 2006; 176:189–211.
5. Magder, S, De Varennes, B. Clinical death and the measurement of stressed vascular volume. Crit Care Med. 1998; 26:1061–1064.
6. Schulman, IH, Zachariah, M, Raij, L. Calcium channel blockers, endothelial dysfunction, and combination therapy. Aging Clin Exp Res. 2005; 17:40–45.
7. National Institute for Health and Clinical Excellence. Hypertension – The Clinical Management of Primary Hypertension. Online. Available http://guidance. nice. org. uk/CG127, 2011.
8. Marik, PE, Varon, J. Hypertensive crises: challenges and management. Chest. 2007; 131:1949–1962.
9. Rinkel, GJ, Feigin, VL, Algra, A, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev. (4):2005.
10. Zanchetti, A, Julius, S, Kjeldsen, S, et al. Outcomes in subgroups of hypertensive patients treated with regimens based on valsartan and amlodipine: an analysis of findings from the VALUE trial. J Hypertens. 2006; 24:2163–2168.
11. De Cicco, M, Macor, F, Robieux, I, et al. Pharmacokinetic and pharmacodynamic effects of high-dose continuous intravenous verapamil infusion: clinical experience in the intensive care unit. Crit Care Med. 1999; 27:332–339.
12. Saris, NE, Mervaala, E, Karppanen, H, et al. Magnesium. An update on physiological, clinical and analytical aspects. Clin Chim Acta. 2000; 294:1–26.
13. Altman, D, Carroli, G, Duley, L, et al. Do women with pre-eclampsia, and their babies, benefit from magnesium sulphate? The Magpie Trial: a randomised placebo-controlled trial. Lancet. 2002; 359:1877–1890.
14. James, MF, Cronje, L. Pheochromocytoma crisis: the use of magnesium sulfate. Anesth Analg. 2004; 99:680–686.
15. Thwaites, CL, Yen, LM, Loan, HT, et al. Magnesium sulphate for treatment of severe tetanus: a randomised controlled trial. Lancet. 2006; 368:1436–1443.
16. Dorhout Mees, SM, Algra, A, Vandertop, WP, et al. Magnesium for aneurysmal subarachnoid haemorrhage (MASH-2): a randomised placebo-controlled trial. Lancet. 2012; 380(9836):44–49.
17. Vassalle, C, Domenici, C, Lubrano, V, et al. Interaction between nitric oxide and cyclooxygenase pathways in endothelial cells. J Vasc Res. 2003; 40:491–499.
18. Alaniz, C, Watts, B. Monitoring cyanide toxicity in patients receiving nitroprusside therapy. Ann Pharmacother. 2005; 39:388–389.
19. Spronk, PE, Ince, C, Gardien, MJ, et al. Nitroglycerin in septic shock after intravascular volume resuscitation. Lancet. 2002; 360:1395–1396.
20. Ferdinand, KC. Isosorbide dinitrate and hydralazine hydrochloride: a review of efficacy and safety. Expert Rev Cardiovasc Ther. 2005; 3:993–1001.
21. Prys-Roberts, C. Phaeochromocytoma – recent progress in its management. Br J Anaesth. 2000; 85:44–57.
22. Packer, M, Coats, AJ, Fowler, MB, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001; 344:1651–1658.
23. Kopecky, SL. Effect of beta blockers, particularly carvedilol, on reducing the risk of events after acute myocardial infarction. Am J Cardiol. 2006; 98:1115–1159.
24. Stone, PH. Review: ACE inhibitors reduce mortality and cardiovascular endpoints in stable coronary artery disease. ACP J Club. 2006; 145:32.
25. Remuzzi, G, Ruggenenti, P. Overview of randomised trials of ACE inhibitors. Lancet. 2006; 368:555–556.
26. Nickenig, G, Ostergren, J, Struijker-Boudier, H. Clinical evidence for the cardiovascular benefits of angiotensin receptor blockers. J Renin Angiotensin Aldosterone Syst. 2006; 7(Suppl 1):S1–S7.
27. Dicpinigaitis, PV. Angiotensin-converting enzyme inhibitor-induced cough: ACCP evidence-based clinical practice guidelines. Chest. 2006; 129:S169–S173.
28. Beltrami, L, Zingale, LC, Carugo, S, et al. Angiotensin-converting enzyme inhibitor-related angioedema: how to deal with it. Expert Opin Drug Saf. 2006; 5:643–649.
29. See, S. Angiotensin II receptor blockers for the treatment of hypertension. Expert Opin Pharmacother. 2001; 2:1795–1804.
30. Bhatia, V, Bhatia, R, Mathew, B. Angiotensin receptor blockers in congestive heart failure: evidence, concerns, and controversies. Cardiol Rev. 2005; 13:297–303.
31. Cooper, ME, Webb, RL, de Gasparo, M. Angiotensin receptor blockers and the kidney: possible advantages over ACE inhibition? Cardiovasc Drug Rev. 2001; 19:75–86.
32. Krum, H. Guidelines for management of patients with chronic heart failure in Australia. Med J Aust. 2001; 174:459–466.
33. Packer, M. Current role of beta-adrenergic blockers in the management of chronic heart failure. Am J Med. 2001; 110(Suppl 7A):S81–S94.
34. Gheorghiade, M, Eichhorn, EJ. Practical aspects of using beta-adrenergic blockade in systolic heart failure. Am J Med. 2001; 110(Suppl 7A):S68–S73.
35. Adhikari, NKJ, Burns, KEA, Friedrich, JO, et al. Effect of nitric oxide on oxygenation and mortality in acute lung injury: systematic review and meta-analysis. BMJ. 2007; 334:779–786.
36. Cornet, AD, Hosfstra, JJ, Swart, EL, et al. Sildenafil attenuates pulmonary arterial pressure but does not improve oxygenation during ARDS. Intensive Care Med. 2010; 36:758–764.
37. Moser, M, Izzo, JL, Jr., Bisognano, J. Hypertensive emergencies. J Clin Hypertens (Greenwich). 2006; 8:275–281.
38. Slama, M, Modeliar, SS. Hypertension in the intensive care unit. Curr Opin Cardiol. 2006; 21:279–287.
39. Mabie, WC. Management of acute severe hypertension and encephalopathy. Clin Obstet Gynecol. 1999; 42:519–531.
40. Vaughan, CJ, Delanty, N. Hypertensive emergencies. Lancet. 2000; 356:411–417.
41. Sokol, SI, Kapoor, JR, Foody, JM. Blood pressure reduction in the primary and secondary prevention of stroke. Curr Vasc Pharmacol. 2006; 4:155–160.
42. Ahmad, F, Cheshire, N, Hamady, M. Acute aortic syndrome: pathology and therapeutic strategies. Postgrad Med J. 2006; 82:305–312.
43. Graham, GW, Unger, BP, Coursin, DB. Perioperative management of selected endocrine disorders. Int Anesthesiol Clin. 2000; 38:31–67.
44. Palmer, BF. Impaired renal autoregulation: implications for the genesis of hypertension and hypertension-induced renal injury. Am J Med Sci. 2001; 321:388–400.
45. National Collaborating Centre for Women's and Children's Health. Hypertension in pregnancy – the management of hypertensive disorders in pregnancy. Online. Available http://guidance. nice. org. uk/CG107, 2011.