PHYSIOLOGIC BASIS OF PAIN: PERIPHERAL MECHANISMS
PHYSIOLOGIC BASIS OF PAIN: CENTRAL MECHANISMS
▪ Allodynia: a painful response to a normally nonpainful stimulus
▪ Hyperalgesia: a heightened painful response to a painful stimulus
The Anterolateral Pain Pathway System Includes the Following Tracts
The sensations of pain and temperature are subsets of somatic sensation and are carried from the periphery through small myelinated and unmyelinated nerve endings called nociceptors. Acute nociceptive pain is triggered by noxious stimuli, such as a pinprick and excessive heat, by inflammation, or by damage to peripheral nerves. This type of pain is perceived after electrophysiologic transmission along peripheral nerves into the spinal cord, and from there to the cerebral cortex along specific pain pathways. In contrast, neuropathic pain can occur in the absence of a stimulus and is characterized by reduced pain thresholds so that normally non-noxious stimuli elicit the sensation of pain. Furthermore, an unpleasant affective and emotional experience may be associated with pain, which, once again, may or may not be associated with actual tissue damage.1-3 If pain was only a function of nociception, in the strict anatomic and physiologic sense, then when the painful stimulus is removed, the perception of pain should be eliminated. However, surgical procedures designed to transect the nociceptive transmission pathways in patients with severe neuropathic pain have met with limited success and have not brought consistent or permanent pain control.4 Thus, pain is more than just the transmission of sensations from nociceptors that have been stimulated by noxious stimuli.
Pain serves as a warning signal to alert an individual of potential harm so that an appropriate response can result. That response may be a withdrawal mechanism to prevent or minimize injury, or the response may be to seek medical help if the injury warrants such attention. The behavior that an individual demonstrates in response to painful stimuli is an adapted behavior that involves learning and memory. Not all individuals respond the same way to nociceptive input because the perception of pain is subjective. As a result, pain can be difficult to evaluate and treat. To aid with this evaluation and treatment of pain, this chapter is designed to review current pain terminology and mechanisms of pain in both the peripheral nervous system (PNS) and central nervous system (CNS) to enhance the understanding of the role that clinicians play in pain management. In addition, this knowledge will allow the clinician to determine the level within the nociceptive pathways at which specific interventions are most effective in modulating pain.
The International Association for the Study of Pain (IASP) has developed a classification system for pain that includes definitions of pain terms and descriptions of pain syndromes.5-7 Terminology used to define pain has undergone alterations and additions through the past decades. Two new terms have been added by the IASP—neuropathic pain and peripheral neuropathic pain. Also, the terms sympathetically maintained pain and sympathetically independent pain have been used (Table 113-1, online). These latter two types of pain are associated with complex regional pain syndromes, types I and II.5-7 These two syndromes were formerly called reflex sympathetic dystrophy and causalgia; some authors still use these older terms.
Pain Term |
Description |
Abnormal pain |
Includes allodynia, hyperalgesia, neuropathic pain, and inflammatory pain in addition to chronic pain |
Acute pain |
Pain of rapid or sudden onset; described as being sharp and localized |
Allodynia |
Perception of pain produced by stimuli that are not normally painful, such as stroking of skin, light pinch, or movement of joint |
Analgesia |
The absence of pain in response to stimulation that would normally be painful |
Central sensitization |
Remodeling of circuitry in the dorsal horns and at supraspinal sites |
Chronic pain |
Pain of long duration with a slower, gradual onset; described as dull, diffuse, and poorly localized |
Hyperalgesia |
A heightened response to noxious (painful) stimuli |
Hypoalgesia |
Diminished pain in response to a normally painful stimulus |
Inflammatory pain |
Result of tissue injury or inflammatory disease |
Mirror allodynia |
Heightened sensitivity to nonpainful stimuli in the limb contralateral to the site of nerve injury |
Neuropathic pain |
Persistent, intense burning or electric-like pain resulting from direct injury, disease, or lesion to peripheral nerves or central neurons |
Peripheral sensitization |
Primary afferents and postsynaptic neurons in the spinal cord sensitized to innocuous stimuli |
Radicular pain |
Deep, radiating pain due to nerve root irritation, e.g., sciatica |
Recurrent pain |
Recurring or episodic; pain that accompanies a reinjury, or it may be a painful episode associated with a disease process such as rheumatoid arthritis |
Sympathetic pain |
Originating from the sympathetic nervous system: complex regional pain syndrome: type I (causalgia) and type II (reflex sympathetic dystrophy) |
For more details, see references 1, 5-7, 15, 31, 93, 94.
Pain can be defined in a temporal manner: acute, chronic, and recurrent. There are also several abnormal pain states, including allodynia, hyperalgesia, neuropathic pain, and inflammatory pain. The absence of pain, called analgesia, is the absence of pain in response to a stimulation that would normally be painful.
Pain originating in the skin is termed superficial pain. This type of pain typically follows a dermatomal pattern or the skin regions innervated by a cutaneous nerve. In contrast, deep pain is more difficult to evaluate using dermatomal maps because it arises from muscles, joints, and blood vessels as well as from compressed or inflamed neurons innervating musculoskeletal tissues.8 For example, radicular pain involving nerve root irritation is a type of deep pain. The use of myotomal and sclerotomal charts is more appropriate than dermatomal charts for diagnosing deep pain symptoms.
Acute pain is defined as pain of rapid or sudden onset (see Table 113-1, online). Acute pain serves as a strong warning of tissue injury and signals the individual to withdraw from potential harm or to seek medical care. This type of pain is usually described as being sharp and localized.9,10 Unless there is pre-existing maladaptive pain, the individual’s response to pain is appropriate in relation to the scope and degree of the stimulus.1 Therapeutic intervention for acute pain strives to reduce the source of the pain (e.g., reduction of a fracture) and to reduce discomfort through the use of pharmacologic agents and modalities. In their model for the treatment of the patient with low back pain, DeRosa and Porterfield11 describe acute pain as a close association between the noxious stimuli, tissue damage, and nociception. This definition can be carried over to other patient populations, including the patient with hand and/or upper extremity pain. They support the use of modalities for the patient with acute pain.11
Chronic pain is considered pain of long duration with a slower, gradual onset relative to acute pain.1,12,13 Chronic pain is usually described as dull, diffuse, and poorly localized1,13 (see Table 113-1, online). Often, the true cause of this type of pain is unknown or not fully understood. The patient with chronic pain may have sustained a previous injury that generated acute pain. However, on examination, evidence of a previous injury is no longer visible, suggesting that sufficient time has passed for healing to occur.1 Examples of chronic pain include chronic neck pain and chronic lower back pain. Chronic root compression is one likely inducer of these two conditions.14,15 There may also be injuries and subsequent inflammation in surrounding muscles, facet joints, and ligaments contributing to this pain, which further sensitizes (sensitization is discussed later in this chapter) the nociceptors in these tissues contributing to dysfunction and pain.16-19 Chronic pain is often associated with abnormal pain states such as allodynia (pain caused by normally nonpainful stimulus) or hyperalgesia (increased response to a painful stimulus). The behaviors associated with chronic pain may appear to be inconsistent, inappropriate, or exaggerated because of the lack of success in pain relief. The patient often becomes frustrated, angry, and depressed. The prognosis of chronic pain is unpredictable because treatment may be directed at anatomic or physiologic abnormalities that have not been clearly identified as the source of the painful stimuli.1
Recurrent pain has been characterized as pain that recurs or is episodic (see Table 113-1, online). Predisposing factors related to recurrent pain include poor physical condition, poor body mechanics, previous injury, and disease processes.1 It may be pain that accompanies a reinjury, or it may be a painful episode associated with a disease process such as rheumatoid arthritis. Reinjury may be defined as an exacerbation of a previous condition.11 The individual’s behavioral response to recurrent pain is usually appropriate. This type of pain may be best treated by limited use of modalities and an emphasis on behavior modification techniques, patient education, and general conditioning.11
Abnormal pain states include mechanical allodynia, hyperalgesia, hypoalgesia, neuropathic pain, and inflammatory pain, in addition to chronic pain described previously (see Table 113-1, online). Allodynia (also known as mechanical allodynia or mechanical hypersensitivity) and hyperalgesia are distinguishable states of hypersensitivity that have been described as the perceptual counterparts of peripheral or central sensitization (discussed subsequently).20 Mechanical allodynia is the perception of pain produced by stimuli that do not normally provoke pain, such as stroking of skin, touch, light pinch, or movement of joints.10,21 Allodynia occurs after sensitization of the skin/tissue, for example, after sunburn, inflammation, or trauma. The mechanisms of molecular signaling that lead to sensitization of the afferents are described later in this chapter. It is important to note that with allodynia, the initiating stimulus does not normally produce a painful response and is also called a non-noxious stimulus.
Animal models of nerve injury have studied the incidence of mirror allodynia, which is limb allodynia or hypersensitivity contralateral to injury. A study by Li and colleagues22 revealed that 20% of animals with unilateral nerve injury developed mirror allodynia in the contralateral limb. The inducing type of nerve injury was varied and included axotomy, complete nerve ligation, and partial nerve ligation. Whiplash injuries also result in bilateral limb allodynia, perhaps caused by an injury to cervical nerve roots located in close proximity to the CNS.14,15 This last study also found that glial activation occurs in the spinal cord after spinal root injury and that this activation is temporally linked to the presence of bilateral allodynia. This topic is discussed further with central sensitization.
Hyperalgesia is a heightened response to noxious (painful) stimuli.23,24 Hyperalgesia is generated and maintained in the periphery through chemical mediators that cause peripheral sensitization. However, with both allodynia and hyperalgesia, there may be remodeling of circuitry within the dorsal horns and in supraspinal sites that contributes to the increased pain (i.e., central sensitization, to be discussed further later).24-27 There are various forms of hyperalgesia such as heat, cold, and chemical hyperalgesia.20 When hyperalgesia occurs locally at the site of damage and proximal tissue it is referred to as primary hyperalgesia, whereas pain spreading beyond the injury site is secondary hyperalgesia.20,28 In general, primary hyperalgesia is a consequence of peripheral sensitization, whereas secondary hyperalgesia is due to central sensitization mechanisms in the spinal cord. Inducing causes of hyperalgesia include nerve compression but also muscle injury/inflammation and inflammatory mediators released by tumors, to name a few.8,29,30
Thus, both allodynia and hyperalgesia are types of hyperesthesia, which is an increased sensitivity to stimulation. The more specific terms should be used for clarity because the allodynia occurs after a normally nonpainful stimulus and hyperalgesia occurs after a normally painful stimulus, but has a greater response than normal. There are also states of reduced sensitivity to a stimulus. Hypoalgesia is a diminished response to a stimulus that normally produces pain.
Neuropathic pain is a maladaptive type of pain resulting from direct injury, disease, or lesion to peripheral nerves or central neurons31 (see Table 113-1, online). Neuropathic pain may present with, but is not limited to, a variety of neuropathies (e.g., traumatic injury or compression of one or more nerves), neuritis (inflammation of a nerve or nerves), infections, tumor, stroke, epilepsy, and neurodegenerative disorders.26,27,32 This type of pain is defined as a persistent, intense burning or electric sensation. Examples of peripheral neuropathic pain in the upper quarter include cervical radicular pain and pain associated with peripheral nerve entrapment, such as carpal tunnel syndrome.
Neuritis is a special case of neuropathy occurring as a consequence of inflammatory processes affecting a nerve ending, its axon, or the surrounding tissues.8,29,33,34 This inflammatory state can be the result of immune-mediated inflammation35 without disruption of the axon, inflammation in the peripheral target tissue that inflames the axon terminal, or inflammation within an intact axon caused by localized inflammation in the axon or surrounding tissues.29 The result of each is often hypersensitivity of the skin or deep tissues that are innervated by the involved nerve(s). Interestingly, peripheral nerve recovery after a crush injury is also hindered by the presence of chronic inflammation in the target tissues.36 Thus, the control of inflammation through the use of ice or pharmaceutical agents should be considered in the treatment of patients with neuritis.
Neuropathic pain can also be divided into sympathetically maintained pain (SMP) and sympathetically independent pain (SIP).37 The primary source of SMP is thought to be the sympathetic nervous system. Nerve transection often produces this type of pain, which is typically characterized as spontaneous pain, sustained burning pain, tactile allodynia, and cold allodynia.37 Sprouting of sympathetic fibers that interact with cut or crushed axons is hypothesized to be the cause of this type of pain.37 Sympathetic pain often has a vasomotor dysfunction, with measurable changes in infrared thermography, specifically, decreased skin temperature in the cutaneous distribution of the involved nerve.37 Both types of complex regional pain syndrome, type I (causalgia) and type II (reflex sympathetic dystrophy), are linked to this source of pain mediation.5-7 SMP can be reversed temporarily with chemical sympathetic blocks (i.e., using phentolamine to block alpha-adrenoceptors or guanethidine to prevent the release of norepinephrine),38 although surgical sympathectomy appears to be more reliable.37 Please see Chapter 115 and Chapter 116 for a more detailed discussion on the management of complex regional pain syndrome.
In summary, it is important to note that all of these types of pain are often seen in combination with one another, although they may be experienced alone as well.
Nociception is the neural response to noxious insults that result in actual tissue damage. This response is mediated by specialized sensory receptors called nociceptors. Sensations elicited by the activation of these nociceptors include prickling, burning, stinging, soreness, and aching. The various types and roles of these nociceptors are discussed in more detail.
Tissue damage is one stimulus that specifically activates the nociceptors located in skin, muscle, joints, and viscera. Nociceptors are specialized sensory organs that, compared with other sensory end-organs, are the least differentiated and exist mostly as bare nerve endings. The three main classes of nociceptors (mechanical, thermal, and polymodal) are widely distributed in skin and deeper tissues and often work in unison. Nociceptors have varied thresholds and sensitivities to external and internal stimuli with which they are able to adapt or maladapt to their microenvironment.39 For example, the term sleeping or silent nociceptors has been used to describe high-threshold nociceptors that are not normally responsive to noninjurious stimuli but do respond during inflammation.31,39
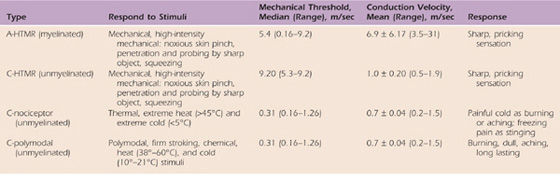
Cutaneous sensory neurons are quite diverse physiologically, anatomically, and molecularly. They can be divided into two main groups: nociceptors, which respond to noxious stimuli or to stimuli that will become noxious if prolonged, and low-threshold mechanical receptors that respond to tactile stimuli.40 Nociceptors can be divided into subgroups by myelination properties (myelinated or not, the latter called free nerve endings), stimulation modality (mechanical, chemical, thermal, or polymodal), or axon diameter (type II or IV) (Table 113-2). There is similar heterogeneity in visceral and musculoskeletal mechanical receptors and nociceptors. Because nociceptor axons carry the axon potential from the periphery into the spinal cord, nociceptors are also afferent neurons (as opposed to efferent neurons whose axons carry the axon potential from the spinal cord to targets in the periphery and are termed motor neurons).
Table 113-2 Types of Nociceptors
Nociceptor properties are from Boada and Woodbury.41,42
HTMR, high-threshold mechanoreceptors.
Mechanical nociceptors are stimulated by intensive pressure, such as a noxious skin pinch, probing the skin with sharp objects, and squeezing. The mechanical nociceptor is activated only when the amount of force is sufficient to cause tissue damage. Mechanical nociceptors are myelinated nociceptors that are also classified as A-nociceptors or A-HTMR (myelinated high-threshold mechanoreceptor).41 These same nociceptors are also known as Aδ-nociceptors, although some are also Aβ based on their axonal diameters and conduction velocities.41,42 These nerves are moderate to small in diameter (1–5 mm and thus type IV afferents), thinly to moderately myelinated, and have a broad range of conduction velocities from slow to moderately fast (see Table 113-2).41 In fact, several A-HTMR nociceptors are as fast as other types of cutaneous mechanoreceptors.41 The fastest A-HTMR are most likely Aβ-nociceptors, but most A-HTMR are Aδ-nociceptors, which have smaller diameter axons and are therefore slower. The type of pain typically perceived after activation of A-HTMR is a sharp, pricking sensation. Their firing rate increases with stimulus destructiveness. Although many think of nociceptors only as the smaller, unmyelinated C-fibers that are discussed later, these larger and faster mechanical nociceptors are the sensory neurons underlying “first pain” and therefore evolved to provide protection of the skin.41,43 Some A-nociceptors are also thermal nociceptors and respond to noxious thermal stimuli. This applies to both extreme heat (respond at more than 45°C) and extreme cold (respond at less than 5°C).44,45 These nociceptors respond in a graduated way to temperature changes, with the highest firing rates occurring with temperatures that produce actual tissue damage. Many of the C-nociceptors are also thermal receptors (see C-polymodal nociceptors).
C-nociceptors can be divided into two main groups: those that respond to mechanical stimulation but not to thermal stimuli (C-HTMR [unmyelinated high-threshold mechanoreceptor]) and those that respond to thermal as well as other noxious stimuli (C-polymodal), as shown in Table 113-2. However, mechanically insensitive cutaneous nociceptors have also been identified that respond only to heat40 and are thus called thermal receptors (see Table 113-2). The C-HTMR nociceptors are high-threshold afferents with slow conduction velocities (see Table 113-2) that respond to intensive pressure, such as a noxious skin pinch, probing the skin with sharp objects, and squeezing. In contrast, the mechanically insensitive thermal nociceptors respond to heat greater than 45°C and express a nonselective ligand-gated cation called the transient receptor potential vanilloid 1 channel receptor or TRPV1.40 In humans, specific TRPV1 antagonists or other drugs acting on this receptor could potentially be used to treat neuropathic pain associated with multiple sclerosis, chemotherapy, amputation, and osteoarthritis.46
Polymodal nociceptors are C-nociceptors that are unmyelinated and respond to several types of stimuli. They are activated by firm stroking of the skin, chemical stimuli, and high temperatures (38°–60°C) or cold temperatures (10°–21°C).42 The pain associated with activation of the C-fibers is burning, dull, aching, and long-lasting in nature. This subset of C-nociceptors is also called group IV afferents because they have unmyelinated, very small diameter axons (<1 mm) that conduct very slowly (see Table 113-2).41 C-fibers are extremely prevalent in number and make up 80% of the total fibers in cutaneous nerves.
In addition to locations in the somatic regions of the body, C-nociceptors also are located within joints, muscles, and viscera. As a component of visceral nerves, C-nociceptors are thought to transmit unpleasant sensations associated with distention of the bowel and bladder, chemical secretions, and inflammation of the visceral structures.
Another method of categorizing nociceptors includes division of the C-nociceptors into peptide or IB4 nociceptor subsets.47 The peptide subdivision expresses a variety of peptides, including substance P (SP). The peptide nociceptors terminate in lamina I and the outer portion of lamina II of the dorsal horns of the spinal cord. The IB4 subset is so named because they bind the IB4 lectin. The IB4 subdivision expresses fewer peptides than the other, expresses fluoride-resistant acid phosphatase, and terminates in the inner portion of lamina II.48 Receptor differences between these two subsets lead to differential responsiveness to chemical mediators released and contribute to varied levels of pain modulation at the site of injury.
Because nociceptors are sensory neurons, they, like other peripheral sensory neurons, are pseudounipolar in shape, with two axons branching from a main axon of the cell body. The cell body is located in the dorsal root ganglia or sensory cranial nerve ganglia, similar to other types of sensory neurons. There is then a peripherally projecting axon that carries impulses from the nociceptor terminal located in the skin, joints, or viscera. This peripheral axon transmits nociceptor information from the peripheral site centrally to the spinal cord. A centrally projecting axon projects into the spinal cord where it terminates in the dorsal horn and releases excitatory neurotransmitters, such as glutamate and SP, onto second-order projection neurons and interneurons.2,40-42,49-55 However, first, these centrally projecting nociceptive axons ascend or descend for several spinal segments in a small pathway called Lissauer’s tract (located in the posterolateral sulcus at the tip of the dorsal horn) before terminating in the dorsal horn gray matter. Both the C-nociceptor and the A-nociceptor axons synapse in the lamina of the dorsal horn. Most of the nociceptive fibers terminate in laminae I, II, and V.2,40-42,49-55 Note that lamina II is most commonly referred to as the substantia gelatinosa. However, some of these axons also extend deeper to terminate in laminae III, IV, VII, or VIII. The A-nociceptor fibers terminate primarily in laminae I, II, and V, laminae containing many wide dynamic range (WDR) neurons (see the following discussion).55 In contrast, most C-nociceptors terminate primarily in lamina II, which contains both excitatory and inhibitory interneurons that respond to both noxious and non-noxious stimuli (Fig. 113-1). Interestingly, laminae VII and VIII also receive bilateral input from polymodal nociceptors and project to neurons of the brainstem reticular formation (bilaterally); therefore, these latter laminae contribute to diffuse pain sensations.40-42,55
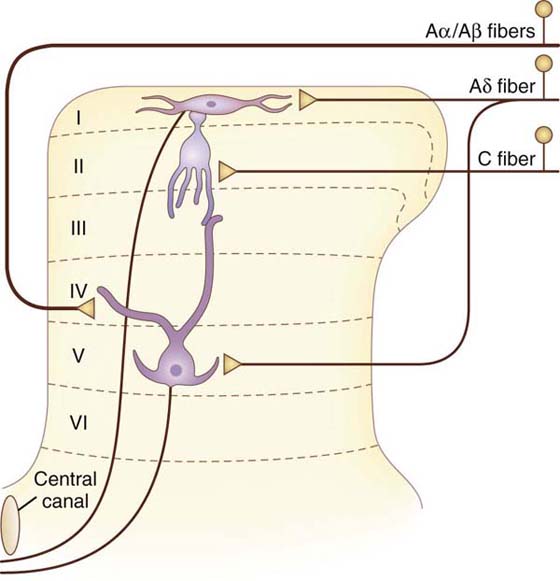
Figure 113-1 The afferent fibers of nociceptors terminate on projection neurons in the dorsal horn of the spinal cord. (Adapted from Fields HL. Pain. New York: McGraw-Hill, 1987.)
Ascending pain impulses are carried from the spinal cord via multiple pathways, including the spinothalamic, spinoreticular, spinomesencephalic, and spinohypothalamic-limbic pathways53 (Fig. 113-2). Collectively, these pathways, as well as a few others not listed here, are known as the anterolateral system. This system includes not only pain and temperature sensibilities but also crude tactile and pressure modalities from innocuous mechanoreceptors with large receptive fields.
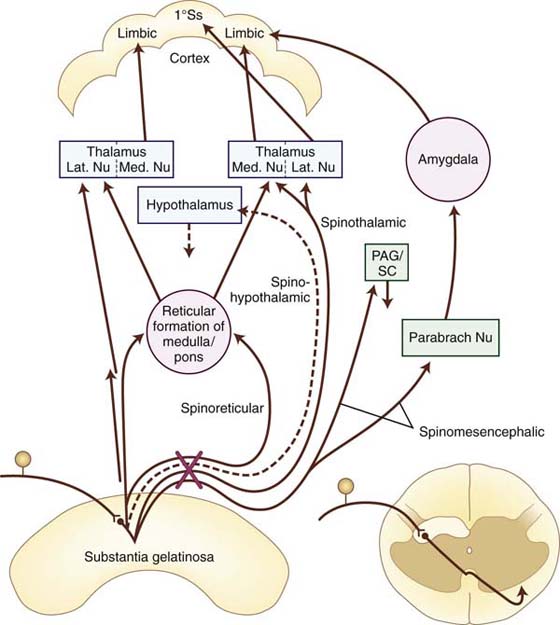
Figure 113-2 Major ascending sensory pathways of the spinal cord carrying pain-mediating fibers, the spinothalamic, spinoreticular, spinohypothalamic, and spinomesencephalic. The X indicates the decussation of indicated pathways through the anterior commissure. 1°Ss, primary somatosensory; Lat. Nu, lateral nucleus; Med. Nu, medial nucleus; PAG/SC, periaqueductal gray/superior colliculus; Parabrach Nu, parabrachial nucleus. (Adapted from Cailliet R. Neuroanatomy of pain mechanisms. In: Cailliet R, ed. Pain: Mechanisms and Management. Philadelphia: FA Davis, 1993; Basbayn AU, Jessell TM. The perception of pain. In: Kandel ER, Schwartz JH, Jessell TM, eds. Principles of Neural Science, 4th ed. New York: Elsevier, 2000.)
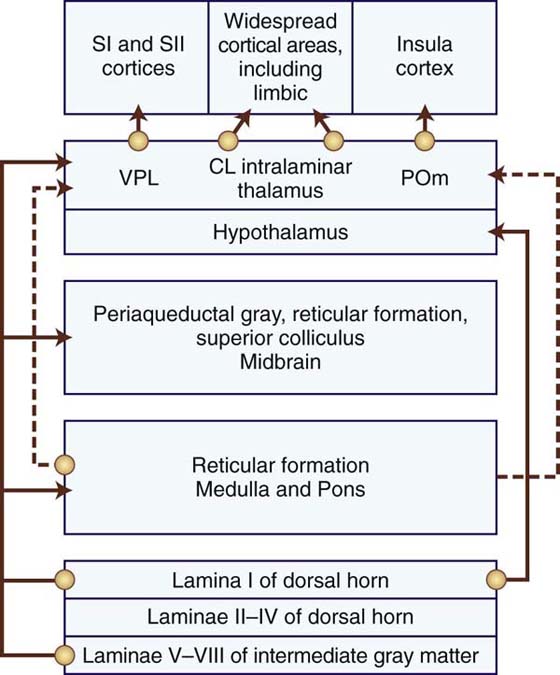
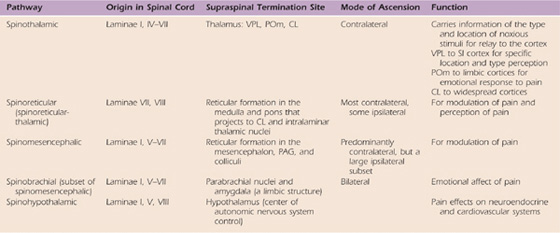
The spinothalamic tract is the primary nociceptive pathway and is also the most studied of the known pathways (Table 113-3).2,12,50,51,53,56 It consists of axons of nociceptive-specific and WDR neurons from laminae I, IV, V, VI, and VII. Although there is a small ipsilateral projection, most of the axons decussate in the ventral white commissure of the spinal cord before ascending contralaterally in the anterolateral white matter to the ventroposterolateral (VPL) nucleus, medial nucleus of posterior complex (POm), and central lateral (CL) nuclei of the thalamus. Noxious and non-noxious impulses such as pain, crude touch, and temperature are transmitted.49 The primary function of this tract is to carry the discriminatory features of type and location of the noxious stimulus via relays through the VPL nucleus to the primary somatosensory cortex (Fig. 113-3, online). However, there are also spinothalamic connections to the CL nucleus, a nucleus with projections to widespread cortical areas, and to the POm, a nucleus involved in the nondiscriminative, affective component of pain. These additional connections expand the roles of the spinothalamic tract to include the emotional response to pain and the perception of pain. The latter includes pain quality (e.g., burning, sharp, dull), cardiovascular changes (e.g., increased heart rate), and neuroendocrine effects (e.g., nausea).
Table 113-3 Components of the Anterolateral System, the Ascending Pathways of Pain Transmission
CL, centrolateral thalamic nucleus; PAG, periaqueductal gray region in the midbrain; POm, medial nucleus of the posterior thalamic complex; SI, primary somatosensory cortex; VPL, ventroposterolateral thalamic nucleus.
Figure 113-3 A schematic drawing of ascending pathways associated with pain localization and affective components of pain. The primary localization pathway for pain from the body is called the spinothalamic pathway; it ascends from the spinal cord to the ventroposterolateral (VPL) nucleus of the thalamus, which projects to the somatosensory (S) cortex areas I and II. The primary pathway involved in motivational-affective components of pain processing and perception is the spinoreticular and spinomesencephalic tracts (see text). CL, central lateral; POm, medial nucleus of the posterior complex. (Figure modified from Haines DE, ed. Fundamental Neuroscience for Basic and Clinical Application. 3rd ed. Philadelphia: Churchill Livingstone; 2006.)
The spinoreticular tract (see Table 113-3) consists primarily of fibers from nociceptive neurons in laminae VII and VIII.53 Note that a few of the neurons contributing to this tract are either inhibited or excited by innocuous cutaneous sensation. The spinoreticular tract consists primarily of axons from the cervical cord and ascends in the anterolateral quadrant of the spinal cord with the spinothalamic tract. However, not all the fibers in this tract cross the midline and ascend on the contralateral side. A small percentage of ipsilateral projections exist. Fibers of this tract project to various nuclei in the reticular formation of the medulla and pons. These reticular nuclei project to medial thalamic nuclei such as CL and other intralaminar nuclei, forming a spinoreticular-thalamic-cortical circuit. The polysynaptic nature of this circuitry allows for modulation of the pain signals through numerous brainstem and supraspinal afferents and efferents. In addition, the projections through intralaminar nuclei connect with many cortical areas including the limbic prefrontal cortex. Consequently, this pathway serves as an important route for pain perception rather than the location of the noxious stimulus.
A smaller group of nociceptive neurons located in laminae I and V to VII contribute to the spinomesencephalic tract, in which fibers project to the mesencephalic reticular formation, the periaqueductal gray (PAG) region, and superior colliculus, as well as a few collaterals to the thalamus (see Table 113-3).53 Most of the axons ascend to the contralateral midbrain, but a prominent group from the upper cervical cord ascend ipsilaterally. Axons terminating in the PAG region contribute to pain modulation via the descending pain pathways.3 The spinoparabrachial tract is a component of the spinomesencephalic tract. Fibers of this latter tract project to the parabrachial nuclei and then to the amygdala, a limbic structure serving as the center of emotion. Thus, the spinoparabrachial tract contributes to the emotional/affective component of pain.
Another pain pathway, the spinohypothalamic-limbic tract, arises from neurons in laminae I, V, and VIII.53 This tract projects to the hypothalamus, the center of supraspinal autonomic control. This pathway is one mechanism through which pain affects neuroendocrine and cardiovascular outputs (see Table 113-3).
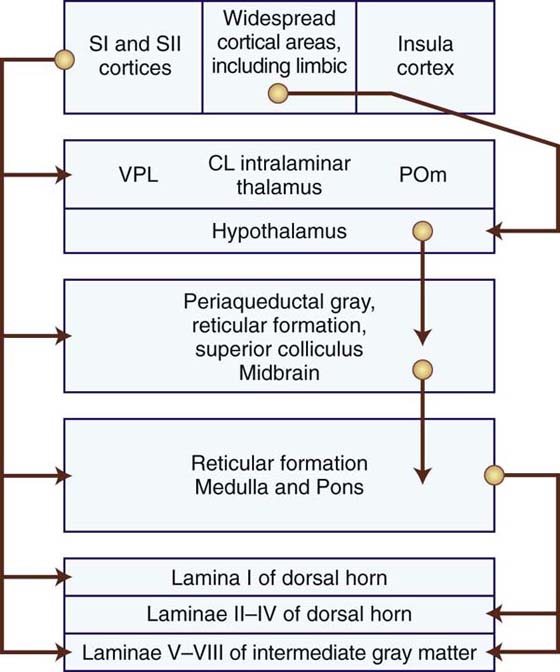
Basic research has identified and substantiated the central mechanisms of pain control. Additional research has implicated an even more potent control of nociception than the gate-control theory. Stimulation-produced analgesia was elicited in animal and human studies when direct electrical stimulation was performed in the periventricular region of the cerebral aqueduct and the third and fourth ventricles.57 Shortly after this discovery, the neural pathways that conveyed this analgesia were discovered. This descending pathway of pain control consists of excitatory connections that originate from the PAG region and synapse in the medulla within the nucleus raphe magnus51,58 (Fig. 113-4). Additional excitatory connections project from the PAG region to the nucleus reticularis paragigantocellularis and the lateral tegmental nucleus located in the medulla. Descending serotonergic neurons from the nucleus raphe magnus, as well as adrenergic neurons from the lateral tegmental nucleus, have excitatory connections on the spinal interneurons in the dorsal horns. These spinal interneurons contain endogenous opiates such as enkephalin and dynorphin, both inhibitory neuropeptides. When activated, these interneurons play a role in suppressing nociceptive activity of the incoming A- and C-nociceptors.
Figure 113-4 A schematic drawing of descending pathways associated with modulation of pain. Spinoreticular and spinomesencephalic inputs activate several descending pathways that modulate pain (see text). CL, central lateral; POm, medial nucleus of the posterior complex; S, somatosensory; VPL, ventroposterolateral. (Figure modified from Haines DE, ed. Fundamental Neuroscience for Basic and Clinical Application. 3rd ed. Philadelphia: Churchill Livingstone; 2006.).
Endogenous opiates such as enkephalins and endorphins have been discovered by several investigators.57,59,60 Cell bodies containing enkephalin and dynorphin have been located at multiple sites, including the midbrain and the spinal dorsal horns. Beta-endorphins have been located primarily within neurons of the hypothalamus that send axons to the PAG region.57 The opioid peptides within the CNS bind to specific opiate receptors. These enkephalin-containing neurons in the superficial dorsal horn inhibit the neurons that form the spinothalamic tract and, when activated, will inhibit the transmission of nociceptive information to the higher centers of the brain. The endogenous opioids also may inhibit the sensations carried by the A- and C-nociceptors through both presynaptic and postsynaptic actions on the WDR and nociceptor-specific neurons.
As discussed previously, there is an unpleasant, emotional aspect to pain, a psychological phenomenon termed secondary affect.3,61 Multiple mechanisms contribute to this affect. First, as discussed previously, parallel-serial spinal projections carrying pain information to lower brainstem and limbic structures contribute to activation of arousal, autonomic, and somatomotor systems.3,61 Second, there are parallel serial spinal projections to thalamic nuclei with both specific and widespread cortical projections. The medial thalamic nuclei, such as intralaminar and CL nuclei, project pain signals to prefrontal, insular, anterior cingulate, and posterior parietal cortices, whereas lateral thalamic nuclei, such as VPL nuclei, project predominantly to the primary and secondary somatosensory (SI and SII) cortices.3,61-63 Third, many cortical regions receive direct and/or indirect input from the numerous pain pathways.3 Fourth, research has revealed a global and bilateral integration of the brain regions mediating pain processing.64 For example, SI and SII, cortices highly involved in the localization and discrimination of pain, project to limbic cortical and subcortical regions (corticolimbic pathway), including many of the same regions that receive input directly from the ascending spinal pain pathways.3 Positron emission tomography analyses of brain regions in combination with psychophysical assessment of pain by Coghill and colleagues64 revealed that the perception of stimulus intensity involves diverse cortices such as those important in somatosensory processing (SI, SII, and posterior insular cortex), motor processing (including cerebellum, basal ganglia, supplementary motor cortex, and anterior cingulate cortex), affective and autonomic processing (anterior cingulate and insular cortices), and attentional processing so as to assign response priorities (anterior cingulate, SI, and premotor cortex).3,65 Many of these brain regions are activated bilaterally by nociceptive stimuli.64-67 Even the expectation of pain seems to have an effect on the processing of nonpainful somatosensory stimulation in many of these cortical regions.67 In summary, localization of pain, unpleasantness from the intensity of the pain, and the psychological/emotional aspects of pain are linked through numerous somatosensory, motor, and limbic cortical and subcortical centers. The involvement of the motor cortices in these global circuits and the potential for an impact on motor processing should be of interest to clinicians.
In 1965, Melzack and Wall introduced the concept that information to the projection neurons in the dorsal horn from the nociceptive afferent fibers may be modulated in some way by inputs from non-nociception afferent fibers.68-70 They proposed that processing of pain information occurred in the substantia gelatinosa of the spinal cord. Some type of “gating” mechanism involved interplay between the large primary afferents and the pain fibers (Fig. 113-5). This theory provided some rationale for many clinical observations that were previously unexplained and gave a rationale for new therapies, including transcutaneous electrical nerve stimulation of peripheral nerves and the dorsal columns of the spinal cord.71,72
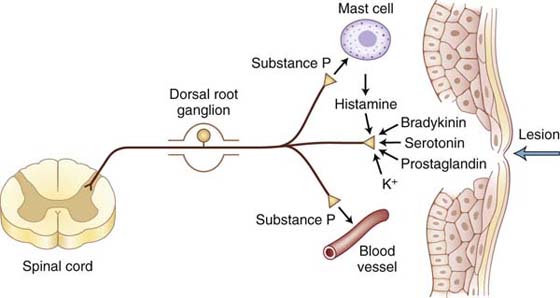
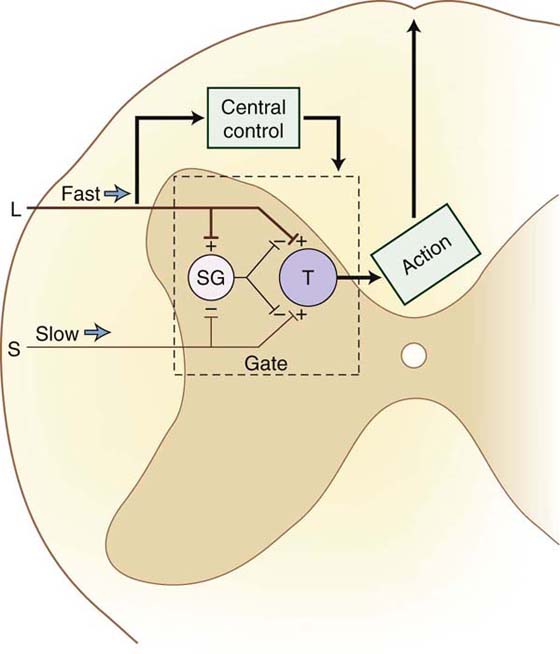
Figure 113-5 Gate-control theory of Wall and Melzack. (From Cailliet R. Pain: Mechanisms and Management. Philadelphia: FA Davis; 1993.)
The “closing of the gate” was thought to occur in lamina II of the dorsal horn. However, new evidence indicates that this gating mechanism also occurs in the dorsal root and the midbrain.73-76 The gate-control theory has undergone some refinement to account for inhibitory influences of spinal-level interneurons and the influence of descending pathways involved in pain modulation (see following discussion).73-76 The significant contribution of Melzack and Wall probably was the impetus for further investigation into pain mechanisms and pathways.68-70
The term neuroplasticity, a phenomenon that occurs in the PNS and CNS, is broadly defined as altered functioning of the nervous system in response to different environmental conditions.77 Plasticity underlies the pathophysiologic states involved in chronic pain conditions. When nociceptive pathways are suddenly activated by stimuli that are normally not painful (allodynia), peripheral sensitization is said to occur.13 Peripheral sensitization involves altered nociceptor function by which there is a reduced threshold of activation and enhanced responsiveness. As a result of peripheral sensitization, presynaptic changes of the afferent neurons increase the release of neurotransmitters. According to several investigators, this afferent barrage of excitatory transmitters into the spinal cord dorsal horn is the presynaptic component of central sensitization.78 Postsynaptic changes during central sensitization include increased excitability of spinal cord second-order neurons due to the altered receptors, second messengers, and inhibitory pathways.78
As a result of tissue damage and inflammation, a variety of chemical mediators are released into the injured site that sensitize nociceptors, thereby enhancing the sensation of nociception in response to repetitive stimuli.2,10,66,79-81 Histamine, serotonin, bradykinin, prostaglandin E2, inflammatory cytokines, norepinephrine, capsaicin, calcitonin gene–related peptide, and SP are among the chemical mediators that affect the threshold of nociceptors (Fig. 113-6). However, new mediators including proteases, growth factors, additional cytokines, and chemokines continue to be identified.39 These mediators have a variety of cellular origins. Cellular sources of inflammatory cytokines include damaged cells in the neurons or surrounding tissues, immune cells such as mast cells and macrophages found in the periphery, and spinal cord glial cells. In general, these mediators act on nociceptor terminals in peripheral tissues to decrease the stimulation threshold and increase responsiveness during inflammation.31,39 There are studies showing increased levels of tumor necrosis factor alpha at midaxonal level and tissue inflammation around an axon increased firing activity of a nociceptor axon.79,82 This increased axonal firing results in increased synaptic release of neurochemicals from the nociceptor central terminal into the spinal cord dorsal horn. Increased synaptic activity in the spinal cord after peripheral inflammation may also be a consequence of a phenotype change of the afferent sensory fibers such that low-threshold myelinated A fibers begin an abnormal inflammation-induced expression of neuropeptides involved in pain transmission.23,39 Under conditions of inflammation, non-nociceptive Aβ/δ-mechanoreceptors elicit enhanced responses to pressure and movement, and high-threshold Aδ- and C-nociceptors respond to less intense stimuli such as light pressure, and silent nociceptors become responsive to mechanical stimulation.13 The magnitude and duration of the mechanical hypersensitivity appear to be related to the severity of the nerve or tissue inflammation.83 In summary, peripheral sensitization is a function of chemically induced nociceptor hyperactivity that leads to a state of increased sensitivity to normally nonpainful stimuli (allodynia) and/or increased responsiveness to noxious stimuli (hyperalgesia) that is perceived with exaggerated or prolonged pain and may develop into chronic pain.
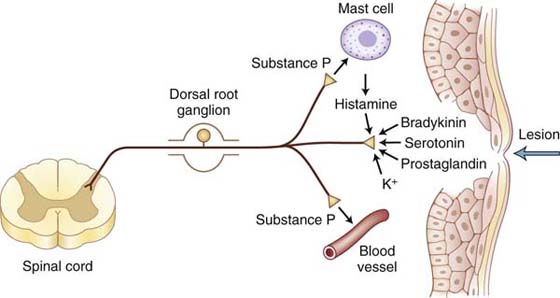
Figure 113-6 Chemical mediators can sensitize and sometimes activate the peripheral endings of nociceptors. Injury or tissue damage releases bradykinin and prostaglandins, both of which activate and sensitize nociceptors. Activation of nociceptors leads to release of substance P and other peptides. Substance P acts on mast cells in the vicinity of sensory endings to evoke degranulation and the release of histamine, which directly excites nociceptors. Substance P also produces dilation of peripheral blood vessels, and the resultant edema causes a further liberation of bradykinin.
The phenomenon of central sensitization is defined as adaptations within the CNS to external or internal stimuli that are characterized by (1) abnormal pain responses such as hyperalgesia or allodynia; (2) diverse biochemical and cellular adjustments including altered neurotransmitter, receptor, or ion channel expression; and/or (3) changes in the structure, function, and survival of neurons.84 Changes in the synaptic network involving the afferent nociceptor neurons and second-order transmission neurons are signs of persistent CNS changes that lead to sensitization.85 Two distinct forms of central sensitization have been described: activity dependent and transcription dependent.27,86 The activity-dependent form is a rapid central response induced by afferent nociceptor input that produces short-lasting synaptic changes, in turn enhancing synaptic transmission in the spinal cord.87 Such changes may be an immediate response to activity-induced excitatory facilitation and diminished inhibition.87 The transcription-dependent form refers to a slow induction of central changes in synaptic transmission distinguished by prolonged effects in which neuroplasticity may outlast the initial input or require low-threshold peripheral drive.27,84,86
Several studies found a relationship between pain behaviors such as allodynia and hyperalgesia and spinal cord inflammation using animal models of peripheral nerve injury. For example, increased expression of spinal nociceptive neuropeptides after cervical nerve root compression and transection was temporally associated with allodynia.88 In another study of peripheral nerve transection, reduced microglial and astrocytic activation in the spinal cord using pharmacologic blockade alleviated allodynia.89 In a model of neuropathic pain, inhibition of spinal cord glial activation and proinflammatory cytokines reversed allodynia after perisciatic injection of zymosan (inflammatory agent).90
The presence of mirror or bilateral allodynia (discussed briefly earlier) and extraterritorial pain provides further evidence of central sensitization.91 For instance, plasticity within the CNS is thought to underlie the spread of hyperesthesia and paresthesia symptoms from the territory of skin and muscles innervated by the median nerve to extramedian territories in carpel tunnel syndrome patients.92 Mirror or bilateral pain has been reported clinically in causalgia and reflex sympathetic dystrophy, as well as in animal models of neuropathic pain, cervical nerve root compression, and arthritis.15,91 Bilateral allodynia induced using temporary compression of cervical nerve roots correlated with glial cell activation in the spinal cord, indicating a central mechanism.15 Altered processing of afferent sensory input at the spinal level has been implicated as a probable mechanism of mirror pain.60,90 It has been proposed that spinal mediators released in the dorsal horn nerve terminal region ipsilateral to the affected peripheral nerve reach nearby nerve terminals affecting other nerves and sensory processing, in turn, producing remote and contralateral effects.91 Although evidence of central sensitization continues to be reported, the exact cellular and molecular mechanisms responsible for such neuroplasticity remain elusive. With continued research, the examination of underlying pathophysiologic mechanisms of central sensitization may lead to improved pharmacologic interventions for chronic pain.
The information provided in this chapter regarding nociception, the transmission of nociceptive input, and the mechanisms involved in pain modulation should serve as a basis for clinical decision making. Further investigations into the neuroscience of pain are required to uncover additional information regarding pain pathways and control mechanisms. The clinician must consider the source of pain mediation, the clinical assessment of pain, and the proposed mechanism of pain modulation when selecting an intervention and establishing a plan of care. The clinician should then form a hypothesis regarding the nature of the pain, the related symptoms, and the expected outcome of treatment. This is discussed in detail in the next chapter.
1. Nolan MF, Wilson MC. Patient-controlled analgesia: a method for the controlled self-administration of opioid pain medications. Phys Ther. 1995;75(5):374–379.
2. McHugh JM, McHugh WB. Pain: neuroanatomy, chemical mediators, and clinical implications. AACN Clin Issues. 2000;11(2):168–178.
3. Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science. 2000;288(5472):1769–1772.
4. Basbaum AI, Julius D. Toward better pain control. Sci Am. 2006;294(6):60–67.
5. Bruehl S, Harden RN, Galer BS, et al. External validation of IASP diagnostic criteria for complex regional pain syndrome and proposed research diagnostic criteria. International Association for the Study of Pain. Pain. 1999;81(1-2):147–154.
6. Strassels SA. After all, pain is a complex sensory and emotional experience (IASP, 1994). Clinical economics and the treatment of persistent pain. J Pain. 2006;7(11):802–803; discussion, 804-806.
7. De Laat AP. Pain in Europe. IV. Fourth Congress of the European Federation of IASP Chapters (EFIC). J Orofacial Pain. 2004;18(1):69–70.
8. Bove GM, Zaheen A, Bajwa ZH. Subjective nature of lower limb radicular pain. J Manipulative Physiol Ther. 2005;28(1):12–14.
9. Weisburg J. Pain. In: Heacox B, Andemicael-Mehreteab T, Weisberg J, eds. Physical Agents: A Comprehensive Text for Physical Therapists. Norwalk: Appleton & Lange; 1994.
10. Mogil JS, Yu L, Basbaum AI. Pain genes?: natural variation and transgenic mutants. Annu Rev Neurosci. 2000;23:777–811.
11. DeRosa CP, Porterfield JA. A physical therapy model for the treatment of low back pain. Phys Ther. 1992;72(4):261–269. discussion, 270-272.
12. Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57(1):1–164.
13. Rowbotham MC, Kidd BL, Porreca F. Role of central sensitization in chronic pain: osteoarthritis and rheumatoid arthritis compared to neuropathic pain. In: Flor H, Kalso E, Dostrovsky JO, eds. Proceedings of the 11th World Congress on Pain. Seattle: IASP Press; 2006:231–249.
14. Hubbard RD, Chen Z, Winkelstein BA. Transient cervical nerve root compression modulates pain: load thresholds for allodynia and sustained changes in spinal neuropeptide expression. J Biomech. 2008;41(3):677–685.
15. Hubbard RD, Winkelstein BA. Transient cervical nerve root compression in the rat induces bilateral forepaw allodynia and spinal glial activation: mechanical factors in painful neck injuries. Spine. 2005;30(17):1924–1932.
16. Winkelstein BA, DeLeo JA. Nerve root injury severity differentially modulates spinal glial activation in a rat lumbar radiculopathy model: considerations for persistent pain. Brain Res. 2002;956(2):294–301.
17. Siegmund GP, Brault JR, Wheeler JB. Placebo whiplash data need cautious interpretation. Int J Legal Med. 2002;116(4):251. author reply, 252.
18. Sanderson SP, Houten JK. Fracture through the C2 synchondrosis in a young child. Pediatr Neurosurg. 2002;36(5):277–278.
19. Scott S, Sanderson PL. Whiplash: a biochemical study of muscle injury. Eur Spine J. 2002;11(4):389–392.
20. Treede R, Handwerker HO, Baumgartner U, et al. Hyperalgesia and allodynia: taxonomy, assessment, and mechanims. In: Brune K, Handwerker H, eds. Hyperalgesia: Molecular Mechanisms and Clinical Implications.Vol 30. Seattle: IASP Press; 2004:3–15.
21. Decosterd I, Allchorne A, Woolf CJ. Progressive tactile hypersensitivity after a peripheral nerve crush: non-noxious mechanical stimulus-induced neuropathic pain. Pain. 2002;100(1-2):155–162.
22. Li D, Yang H, Meyerson BA, Linderoth B. Response to spinal cord stimulation in variants of the spared nerve injury pain model. Neurosci Lett. 2006;400(1-2):115–120.
23. Neumann S, Doubell TP, Leslie T, Woolf CJ. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature. 1996;384(6607):360–364.
24. Urban MO, Gebhart GF. Central mechanisms in pain. Med Clin North Am. 1999;83(3):585–596.
25. Pockett S. Spinal cord synaptic plasticity and chronic pain. Anesth Analg. 1995;80(1):173–179.
26. Woolf CJ. Pain. Neurobiol Dis. 2000;7(5):504–510.
27. Woolf CJ. Dissecting out mechanisms responsible for peripheral neuropathic pain: implications for diagnosis and therapy. Life Sci. 2004;74(21):2605–2610.
28. Costigan M, Woolf CJ. No DREAM, No pain. Closing the spinal gate. Cell. 2002;108(3):297–300.
29. Bove GM, Ransil BJ, Lin HC, Leem JG. Inflammation induces ectopic mechanical sensitivity in axons of nociceptors innervating deep tissues. J Neurophysiol. 2003;90(3):1949–1955.
30. Kehl LJ, Fairbanks CA. Experimental animal models of muscle pain and analgesia. Exerc Sport Sci Rev. 2003;31(4):188–194.
31. Costigan M, Scholz J, Samad T, Woolf CJ. Pain. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Philadelphia: Elsevier; 2006. 927-937
32. Rempel DM, Diao E. Entrapment neuropathies: pathophysiology and pathogenesis. J Electromyogr Kinesiol. 2004;14(1):71–75.
33. Clark BD, Al-Shatti TA, Barr AE, et al. Performance of a high-repetition, high-force task induces carpal tunnel syndrome in rats. J Orthop Sports Phys Ther. 2004;34(5):244–253.
34. Clark BD, Barr AE, Safadi FF, et al. Median nerve trauma in a rat model of work-related musculoskeletal disorder. J Neurotrauma. 2003;20(7):681–695.
35. Chacur M, Picolo G, Teixeira CF, Cury Y. Bradykinin is involved in hyperalgesia induced by Bothrops jararaca venom. Toxicon. 2002;40(7):1047–1051.
36. Kato N, Nemoto K, Kawaguchi M, et al. Influence of chronic inflammation in peripheral target tissue on recovery of crushed nerve injury. J Orthop Sci. 2001;6(5):419–423.
37. Han DW, Kweon TD, Kim KJ, et al. Does the tibial and sural nerve transection model represent sympathetically independent pain? Yonsei Med J. 2006;47(6):847–851.
38. Kim JM, Cole DJ, Franklin GL, Mawhinney MG. Androgen-induced norepinephrine release mediating guinea pig seminal vesicle smooth muscle proliferation: potential role of pre-synaptic alpha2-adrenoceptors. Prostate. 2003;57(1):51–56.
39. Woolf CJ, Ma Q. Nociceptors—noxious stimulus detectors. Neuron. 2007;55(3):353–364.
40. Lawson JJ, McIlwrath SL, Woodbury CJ, et al. TRPV1 unlike TRPV2 is restricted to a subset of mechanically insensitive cutaneous nociceptors responding to heat. J Pain. 2008;9(4):298–308.
41. Boada MD, Woodbury CJ. Physiological properties of mouse skin sensory neurons recorded intracellularly in vivo: temperature effects on somal membrane properties. J Neurophysiol. 2007;98(2):668–680.
42. Boada MD, Woodbury CJ. Myelinated skin sensory neurons project extensively throughout adult mouse substantia gelatinosa. J Neurosci. 2008;28(9):2006–2014.
43. Campbell JN, LaMotte RH. Latency to detection of first pain. Brain Res. 1983;266(2):203–208.
44. Willis WD. Peripheral neural mechanisms of nociception. In: Wall PD, Melzack R, eds. Textbook of Pain. New York: Churchill Livingstone; 1989.
45. Basbayn AU, Jessell TM. The perception of pain. In: Kandel ER, Schwartz JH, Jessell TM, eds. Principles of Neural Science. 4th ed New York: Elsevier; 2000.
46. Gunthorpe MJ, Szallasi A. Peripheral TRPV1 receptors as targets for drug development: new molecules and mechanisms. Curr Pharm Des. 2008;14(1):32–41.
47. Snider WD, McMahon SB. Tackling pain at the source: new ideas about nociceptors. Neuron. 1998;20(4):629–632.
48. Malmberg AB, Brandon EP, Idzerda RL, et al. Diminished inflammation and nociceptive pain with preservation of neuropathic pain in mice with a targeted mutation of the type I regulatory subunit of cAMP-dependent protein kinase. J Neurosci. 1997;17(19):7462–7470.
49. Allen BJ, Li J, Menning PM, et al. Primary afferent fibers that contribute to increased substance P receptor internalization in the spinal cord after injury. J Neurophysiol. 1999;81(3):1379–1390.
50. Baranauskas G, Nistri A. Sensitization of pain pathways in the spinal cord: cellular mechanisms. Prog Neurobiol. 1998;54(3):349–365.
51. Besson JM. The neurobiology of pain. Lancet. 1999;353(9164):1610–1615.
52. Coggeshall RE, Lekan HA, White FA, Woolf CJ. A-fiber sensory input induces neuronal cell death in the dorsal horn of the adult rat spinal cord. J Comp Neurol. 2001;435(3):276–282.
53. Willis WD, Westlund KN. Neuroanatomy of the pain system and of the pathways that modulate pain. J Clin Neurophysiol. 1997;14(1):2–31.
54. Marshall GE, Shehab SA, Spike RC, Todd AJ. Neurokinin-1 receptors on lumbar spinothalamic neurons in the rat. Neuroscience. 1996;72(1):255–263.
55. Woodbury CJ, Kullmann FA, McIlwrath SL, Koerber HR. Identity of myelinated cutaneous sensory neurons projecting to nocireceptive laminae following nerve injury in adult mice. J Comp Neurol. 2008;508(3):500–509.
56. Winkelstein BA. Mechanisms of central sensitization, neuroimmunology and injury biomechanics in persistent pain: implications for musculoskeletal disorders. J Electromyogr Kinesiol. 2004;14(1):87–93.
57. Crofford LJ, Casey KL. Central modulation of pain perception. Rheum Dis Clin North Am. 1999;25(1):1–13.
58. Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66(6):355–474.
59. Agarwal N, Pacher P, Tegeder I, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nature Neurosci. 2007;10(7):870–879.
60. Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82(4):981–1011.
61. Bushnell MC, Duncan GH, Hofbauer RK, et al. Pain perception: is there a role for primary somatosensory cortex? Proc Natl Acad Sci U S A. 1999;96(14):7705–7759.
62. Casey KL. Forebrain mechanisms of nociception and pain: analysis through imaging. Proc Natl Acad Sci U S A. 1999;96(14):7668–7674.
63. Duncan GH, Kupers RC, Marchand S, et al. Stimulation of human thalamus for pain relief: possible modulatory circuits revealed by positron emission tomography. J Neurophysiol. 1998;80(6):3326–3330.
64. Coghill RC, Sang CN, Maisog JM, Iadarola MJ. Pain intensity processing within the human brain: a bilateral, distributed mechanism. J Neurophysiol. 1999;82(4):1934–1943.
65. Creac’h C, Henry P, Caille JM, Allard M. Functional MR imaging analysis of pain-related brain activation after acute mechanical stimulation. AJNR Am J Neuroradiol. 2000;21(8):1402–1406.
66. Chapman CE. Can the use of physical modalities for pain control be rationalized by the research evidence? Can J Physiol Pharmacol. 1991;69(5):704–712.
67. Sawamoto N, Honda M, Okada T, et al. Expectation of pain enhances responses to nonpainful somatosensory stimulation in the anterior cingulate cortex and parietal operculum/posterior insula: an event-related functional magnetic resonance imaging study. J Neurosci. 2000;20(19):7438–7445.
68. Wall PD. The gate control theory of pain mechanisms. A re-examination and re-statement. Brain. 1978;101(1):1–18.
69. Nathan PW. The gate-control theory of pain. A critical review. Brain. 1976;99(1):123–158.
70. Nathan PW, Rudge P. Testing the gate-control theory of pain in man. J Neurol Neurosurg Psychiatry. 1974;37(12):1366–1372.
71. Fedorczyk J. The role of physical agents in modulating pain. J Hand Ther. 1997;10(2):110–121.
72. Rizk TE, Christopher RP, Pinals RS, et al. Adhesive capsulitis (frozen shoulder): a new approach to its management. Arch Phys Med Rehabil. 1983;64(1):29–33.
73. Dickenson AH. Gate control theory of pain stands the test of time. Br J Anaesth. 2002;88(6):755–757.
74. Davis P. Pain: opening up the gate control theory. Nurs Stand. 1993;7(45):25–27.
75. Verrill P. Does the gate theory of pain supplant all others? Br J Hosp Med. 1990;43(5):325.
76. Winnie A. The gate control theory of pain–revisited. Reg Anesth. 1989;14(5):207.
77. Coderre TJ, Katz J, Vaccarino AL, Melzack R. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain. 1993;52(3):259–285.
78. Schaible HG, Ebersberger A, Von Banchet GS. Mechanisms of pain in arthritis. Ann N Y Acad Sci. 2002;966:343–354.
79. Leem JG, Bove GM. Mid-axonal tumor necrosis factor-alpha induces ectopic activity in a subset of slowly conducting cutaneous and deep afferent neurons. J Pain. 2002;3(1):45–49.
80. Lembeck F, Gamse R. Substance P in peripheral sensory processes. Ciba Found Symp. 1982;91:35–54.
81. McCarson KE. Central and peripheral expression of neurokinin-1 and neurokinin-3 receptor and substance P–encoding messenger RNAs: peripheral regulation during formalin-induced inflammation and lack of neurokinin receptor expression in primary afferent sensory neurons. Neuroscience. 1999;93(1):361–370.
82. Kehl LJ, Hamamoto DT, Wacnik PW, et al. A cannabinoid agonist differentially attenuates deep tissue hyperalgesia in animal models of cancer and inflammatory muscle pain. Pain. 2003;103(1-2):175–186.
83. Wallas TR, Winterson BJ, Ransil BJ, Bove GM. Paw withdrawal thresholds and persistent hindlimb flexion in experimental mononeuropathies. J Pain. 2003;4(4):222–230.
84. Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288(5472):1765–1769.
85. Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26(12):696–705.
86. Dubner R. Plasticity in central nociceptive pathways. In: Merskey H, Loeser JD, Dubner R, eds. The Paths of Pain. Seattle, WA: IASP Press; 2005.
87. Scholz J, Woolf CJ. Can we conquer pain? Nature Neurosci. 2002;5(Suppl):1062–1067.
88. Rothman SM, Kreider RA, Winkelstein BA. Spinal neuropeptide responses in persistent and transient pain following cervical nerve root injury. Spine. 2005;30(22):2491–2496.
89. Sweitzer SM, Schubert P, DeLeo JA. Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Ther. 2001;297(3):1210–1217.
90. Milligan ED, Twining C, Chacur M, et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23(3):1026–1040.
91. Chacur M, Milligan ED, Gazda LS, et al. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain. 2001;94(3):231–244.
92. Zanette G, Marani S, Tamburin S. Extra-median spread of sensory symptoms in carpal tunnel syndrome suggests the presence of pain-related mechanisms. Pain. 2006;122(3):264–270.
93. Mogil JS, Chesler EJ, Wilson SG, et al. Sex differences in thermal nociception and morphine antinociception in rodents depend on genotype. Neurosci Biobehav Rev. 2000;24(3):375–389.
94. Costigan M, Woolf CJ. Pain: molecular mechanisms. J Pain. 2000;1(3 Suppl):35–44.