Peripheral Mechanisms of Cutaneous Nociception
Introduction
One of the vital functions of the nervous system is to provide information about the occurrence or threat of injury. The sensation of pain, by its inherent aversive nature, contributes to this function. In this chapter we consider the peripheral neural apparatus that responds to noxious (injurious or potentially injurious) stimuli and thus provides a signal to alert the organism to potential injury. Investigators have studied cutaneous sensibility by recording from single nerve fibers in different species, including humans. Stimuli are applied to the receptive field (i.e., area of the tissue responsive to the applied stimulus) of single fibers, and the characteristics of the neural response are noted. We concentrate on the skin for three reasons. First, sensory receptors in the skin have been more thoroughly studied than receptors in any other tissue. Second, the opportunity to perform correlative psychophysical studies in animals and humans allows powerful inferences to be made regarding function. Third, cutaneous pain sensation is of great clinical significance. Diseases such as post-herpetic neuralgia and others associated with small-fiber neuropathies have profound effects on cutaneous sensory function and often lead to severe pain.
Highly specialized sensory fibers, alone or in concert with other specialized fibers, provide information to the central nervous system (CNS) not only about the environment but also about the state of the organism itself. In the case of the sensory capacity of the skin, cutaneous stimuli may evoke a sense of cooling, warmth, or touch. Accordingly, certain sensory fibers are selectively sensitive to these stimuli. Warm fibers, which are predominately unmyelinated, are exquisitely sensitive to gentle warming of their punctate receptive fields. These fibers have been shown to exclusively signal the quality and intensity of the warmth sensation (Johnson et al 1979). Similarly, a subpopulation of the thinly myelinated, Aδ fibers respond selectively to gentle cooling stimuli and encode the sense of cooling (Darian-Smith et al 1973). For the sense of touch, different classes of mechanoreceptive afferent fibers are exquisitely sensitive to deformations of the skin. These low-threshold mechanoreceptors encode such features as texture and shape.
A relatively high threshold for an adequate stimulus distinguishes the remaining class of cutaneous receptors. Because these receptors respond preferentially to noxious stimuli, they are termed nociceptors (Sherrington 1906). Among the many varieties of sensory receptors, nociceptors are distinctive in that they typically respond to the multiple energy forms that produce injury (thermal, mechanical, and chemical stimuli) and provide information to the CNS regarding the location and intensity of noxious stimuli. Nociceptors may be subclassified with respect to four criteria: (1) unmyelinated C-fiber afferents (conduction velocity <2 m/sec) versus myelinated A-fiber afferents (conduction velocity >2 m/sec), (2) modalities of stimulation that evoke a response, (3) response characteristics, and (4) distinctive chemical markers (e.g., receptors expressed on the membrane). We first consider the properties of cutaneous nociceptors and then review how their function is thought to relate to the sensation of pain.
Tissue damage results in a cascade of events that lead to enhanced pain in response to natural stimuli, termed hyperalgesia. A corresponding increase in the responsiveness of nociceptors, called sensitization, occurs. The characteristics of hyperalgesia and its neurophysiological counterpart sensitization are discussed in a later section. Finally, we consider how nociceptors may play a role in accounting for the often severe pain that accompanies nervous system injury and disease.
Properties of Nociceptors in Uninjured Skin
Nature might have designed nociceptors such that each had the capacity to respond to the full complement of stimulus energy forms that pose potential risks to the organism (thermal, mechanical, and chemical). What nature has adopted instead is a mixed strategy whereby many nociceptors respond to multiple stimulus modalities (polymodal) and others have more specialized response properties. These specialized response properties probably at least in part account for different aspects of nociceptive sensory function (e.g., burning, aching, pricking, prickle, itch). As delineated later, nociceptors have distal effector functions as well, and specialization may also play a role here. The end result is that nociceptors have a complex biology and heterogeneous properties.
The receptive field of a nociceptor is often first localized by use of mechanical stimuli. Various other stimulus modalities are then applied to this receptive field. In most early studies of nociceptors, only heat and mechanical stimuli were used to study nociceptors. Therefore, the nomenclature of CMH and AMH is often used to refer to C-fiber mechano-heat–sensitive nociceptors and A-fiber mechano-heat–sensitive nociceptors, respectively. If a fiber responds to heat and mechanical stimuli, the fiber will in most cases respond to chemical stimuli as well (Davis et al 1993b). Thus, CMHs and AMHs may also be referred to as polymodal nociceptors.
The issue of whether a given nociceptor responds to a particular stimulus modality is perilous because the presumed lack of response to a given modality may in fact represent failure to apply the stimulus with sufficient intensity. The problem with the application of high-intensity stimuli is that the stimulus may alter the properties of the nociceptor in an enduring manner. A selection bias occurs: nociceptors with lower thresholds are more likely to be studied. The easiest way to find a nociceptor for electrophysiological study is to apply squeezing (mechanical) stimuli to the skin and thus identify the receptive field. This selection process identifies what are termed mechanically sensitive afferents (MSAs). In time it has become apparent that selection bias from this approach has led to oversight of an important class of nociceptors: mechanically insensitive afferents (MIAs). Because these fibers by definition have high mechanical thresholds (or are unresponsive to mechanical stimuli), finding the mechanical receptive field of these fibers is difficult. An alternative technique described by Meyer and colleagues (1991) has been to apply electrical stimuli to the skin to identify the putative receptive field. With this technique it turns out that about half of the Aδ-fiber nociceptors and 30% of the C-fiber nociceptors are MIAs, with MIAs being defined as afferents that have very high mechanical thresholds (>6 bar = 600 kPa = 60 g/mm2) or are unresponsive to mechanical stimuli (Handwerker et al 1991, Meyer et al 1991). MIAs have also been reported in the knee joint (Schaible and Schmidt 1985), viscera (Häbler et al 1988), and cornea (Tanelian 1991). As will be seen, this MIA–MSA distinction is of significance with regard to distinguishing nociceptor types. From the perspective of nomenclature, it is well to emphasize that MIAs are not defined as fibers that have no response to mechanical stimuli but rather as fibers that have a very high threshold (or no sensitivity at all) such that demonstration of a response to mechanical stimuli in electrophysiological studies is difficult.
C-Fiber Nociceptors
CMHs are commonly encountered cutaneous afferents, and activity of sufficient magnitude in these fibers is thought to evoke a burning pain sensation. The size of the receptive field appears to scale with the size of the animal. Typical values for monkey are between 15 and 20 mm2 (LaMotte and Campbell 1978), and for human they are near 100 mm2 (Schmidt et al 1997). There are often discrete areas of mechanical sensitivity (hot spots) within the receptive field, but in many fibers the areas of mechanical responsiveness tend to fuse over the region of the receptive field. Most CMHs respond to chemical stimuli (though not as well as A-fiber nociceptors; Davis et al 1993b) and can therefore be considered polymodal.
Responses to heat stimuli have been studied in considerable detail. The response of a typical CMH to a random sequence of heat stimuli ranging from 41–49°C is shown in Figure 1-1A. It can be seen that the response increases monotonically with stimulus intensity over this temperature range, which encompasses the pain threshold in humans. One ion channel involved in the transduction of heat at nerve terminals is thought to be the neuronal transient receptor potential ion channel V1 (TRPV1); activity in this channel increases with increasing temperature (Caterina et al 1997). A detailed description of the neuronal ion channels involved in stimulus transduction is presented in Chapter 2 (for review see Dubin and Papapoutian 2010). Signal transduction molecules on keratinocytes may also play a role in heat transduction by inducing the release of adenosine triphosphate (ATP), which activates purinergic receptors (P2X3 and P2Y2) on the free nerve endings (see Fig. 1-4).
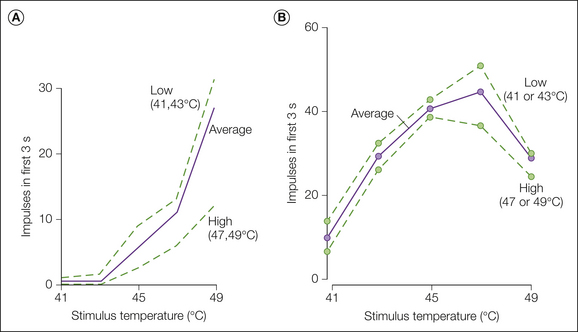
Figure 1-1 Responses of a typical C-fiber nociceptor and a warm fiber to heat stimuli.
Heat stimuli ranging from 41-49°C and lasting 3 seconds were presented at 25-second interstimulus intervals to the glabrous skin of the monkey hand. Each stimulus occurred with equal frequency and was preceded by every other stimulus an equal number of times. Within these constraints, the order of stimulus presentation was randomized. Base temperature between stimuli was 38°C. A, Monotonic stimulus–response function for a typical nociceptor. B, Non-monotonic stimulus–response function for a typical warm fiber. The solid line represents the total response to a given temperature averaged across all presentations. The dotted lines represent the stimulus–response functions obtained when the preceding temperature was of low (41 and 43°C) or high (47 and 49°C) intensity. (Reproduced with permission from LaMotte RH, Campbell JN 1978 Comparison of responses in warm and nociceptive C-fiber afferents in monkey with human judgements of thermal pain. Journal of Neurophysiology 41:509–528.)
Two types of heat response are observed following a stepped heat stimulus. Quick C (QC) fibers exhibit their peak discharge during the rising phase of the heat stimulus, whereas slow C (SC) fibers exhibit their peak discharge during the plateau phase (Fig. 1-2B). The heat thresholds (Fig. 1-2C) and mechanical thresholds of QC fibers are significantly lower than those of SC fibers, thus suggesting that they may be located more superficially in the epidermis. QC fibers respond more vigorously to pruritic stimuli than do SC fibers, which suggests that they may be important in itch sensations (Johanek et al 2008).
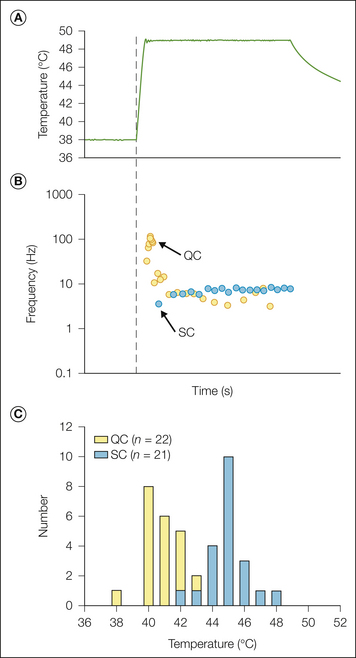
Figure 1-2 Two types of heat responses are observed in C-fiber nociceptors.
A, Stepped heat stimulus (49°C, 3 seconds) used to classify heat response. B, The quick C (QC) fiber (yellow circles) exhibits a high-frequency discharge during the rising phase of the stimulus that adapts quickly (within 1 second). The slow C (SC) fiber (blue circles) exhibits a relatively uniform discharge throughout the stimulus period. Each circle represents the time of occurrence of an action potential. C, A histogram of the heat thresholds reveals that the distributions of QC and SC fibers are almost non-overlapping. (From Johanek LM, Meyer RA, Friedman RM, et al 2008 A role for polymodal C-fiber afferents in nonhistaminergic itch. Journal of Neuroscience 28:7659–7669.)
Thermal modeling studies combined with electrophysiological analysis have indicated that (1) the heat threshold of CMHs depends on the temperature at the depth of the receptor and not the rate of increase in temperature, (2) transduction of heat stimuli (conversion of heat energy to action potentials) occurs at different skin depths for different CMHs (Tillman et al 1995b), and (3) suprathreshold responses of CMHs vary directly with the rate of increase in temperature (Tillman et al 1995a, 1995b; Yarnitsky et al 1992). The depth of the heat-responsive terminals of CMHs varies quite widely (ranging from 20–570 μm; Tillman et al 1995b). When a stepped temperature stimulus is applied to the skin, the temperature increases in the subsurface levels more slowly because of thermal inertia. The disparity in the surface temperature and the temperature at the level of the receptor varies directly with depth and indirectly with time. Given that the depth of CMH terminals varies widely, true heat thresholds are obtained when the rate of increase in temperature is very gradual or when the duration of the stimulus is very long. Although the literature reflects a wide range of heat thresholds for CMHs, when tested with these types of heat stimuli, the heat threshold of the majority of CMHs is in a remarkably narrow range of 39–41°C (Tillman et al 1995b).
The response of CMHs is also strongly influenced by the stimulus history. Both fatigue and sensitization are observed. One example of fatigue is the observation that the response to the second of two identical heat stimuli is substantially less than the response to the first stimulus. This fatigue is dependent on the time between stimuli, with full recovery taking longer than 10 minutes. A similar reduction in the intensity of pain after repeated heat stimuli is observed in human subjects (LaMotte and Campbell 1978). Fatigue is also apparent in Figure 1-1A, where the response to a given stimulus varied inversely with the intensity of the preceding stimulus. A decrease in the response to heat is also observed following mechanical stimuli applied to the receptive field or electrical stimuli applied to the nerve trunk (Peng et al 2003). This suggests that fatigue in response to a given stimulus modality can be induced by heterologous stimulation, that is, by excitation with a stimulus of a different modality. Interestingly, recovery from cross-modal or heterologous fatigue is faster than recovery from fatigue induced by a stimulus of the same modality. Presumably, this is because these heterologous stimuli do not activate and therefore do not fatigue the stimulus transduction apparatus in the same way. Alternatively, fatigue may arise from independent effects on spike initiation (from antidromic stimulation) and transduction (from natural stimulation at the receptive field). Fatigue in response to heat stimuli is also seen in vitro when small (and presumably nociceptive) dorsal root ganglion (DRG) cells are repetitively tested with heat stimuli (Greffrath et al 2002). The enhanced response, or sensitization, that may occur in CMHs after tissue injury is described below in the section on hyperalgesia.
Responses to mechanical stimuli are covered in more detail later. Suffice it here to indicate that CMHs usually display a slowly adapting response to mechanical stimuli of a given force. As noted later, MSA CMHs have a graded response to punctate stimuli, but their stimulus–response functions become saturated at levels substantially below the threshold for pain.
C-fiber MIAs are heterogeneous with regard to responses to chemical and heat stimuli, and some respond only to mechanical stimuli (but of course with a very high mechanical threshold). The sensitivity to mechanical stimuli has no obvious correlation to the heat threshold (Davis et al 1993b). In contrast to CMH afferents, some C-fiber MIAs in humans are vigorously excited when challenged with histamine or capsaicin. In addition, the activity observed in these C-fiber MIAs parallels the duration of the perception of itch (histamine) or burning pain (capsaicin) (Schmelz et al 1997, 2000b). C-fiber MIAs may therefore act as chemosensors. In addition to pronounced chemosensitivity, these fibers have some other interesting properties that could account for pain in response to tonic pressure stimuli or the neurogenic flare response (see below).
Low-threshold C-fiber mechanoreceptors that do not respond to heat have been described in the cat (Bessou and Perl 1969) and rabbit (Shea and Perl 1985). In primates, including humans, these fibers have been found in proximal areas of the body (Kumazawa and Perl 1977, Nordin 1990) and the hairy skin on the forearm (Vallbo et al 1999). These afferents are strongly activated by innocuous mechanical stimuli moved slowly across the receptive field, but they also respond to pinprick stimuli. The neuronal activity in these fibers is not critical for the perception of touch and, according to one imaging study, leads to the activation of the insular but not the sensory cortex (Olausson et al 2003). Low-threshold C-fiber mechanoreceptors are thought to mediate the sensation of “pleasant” touch and may therefore play an important role in “affiliative” behavior (Vallbo et al 1999, Wessberg et al 2003, Löken et al 2009).
Some mechano-insensitive C fibers are reported to be activated by non-noxious and noxious cold and hot stimuli. It has been hypothesized that activity in these afferents may mediate the “hot–burning” sensations caused by such stimuli. These afferents may also be involved in mediating psychophysical phenomena such as “paradoxical heat” or the thermal grill illusion (Campero et al 2009).
C-fiber afferents differ not only in their receptive features but also in their conductive properties. In fact, their conductive and receptive properties appear to correlate. When unmyelinated C-fiber afferents are activated repetitively by electrical stimuli, their conduction latency increases gradually (i.e., the conduction velocity of the afferent decreases). In addition, with increasing stimulation frequency, the amount of this activity-dependent slowing increases. Slowing in C-fiber MIAs is greater than in C-fiber MSAs (Weidner et al 1999), and mechanosensitive nociceptive afferents show more pronounced slowing than do cold-sensitive C fibers, low-threshold C fibers, or sympathetic efferent C fibers (Gee et al 1996, Serra et al 1999, Obreja et al 2010, Ringkamp et al 2010). This difference in slowing properties indicates that the ion channels responsible for conduction may be different and suggests that the ion channels responsible for spike initiation at the receptive terminal may also differ between C-fiber classes.
A-Fiber Nociceptors
A-fiber nociceptors are thought to evoke pricking pain, sharpness, and perhaps aching pain. As a general rule, A-fiber nociceptors do what C-fiber nociceptors do, but do it more robustly. They respond at higher discharge frequencies, and the discriminable information supplied to the CNS is greater (e.g., Slugg et al 2000).
Two types of A-fiber nociceptors are apparent (Dubner et al 1977, Treede et al 1998). A summary of their properties is presented in Table 1-1. Type I fibers are typically responsive to heat, mechanical, and chemical stimuli and may therefore be referred to as AMHs or polymodal nociceptors. Because the heat thresholds are high with short-duration stimuli (typically >53°C), the responsiveness of these fibers to heat has in some studies been overlooked. Consequently, these fibers have been called high-threshold mechanoreceptors (HTMs) by many investigators (e.g., Burgess and Perl 1967). Heat sensitivity in type I fibers is most likely mediated by the vanilloid receptor–like protein 1 (VRL1, renamed TRPV2) since it has a similar high threshold for activation by heat and is expressed in neurons with small myelinated axons (Caterina et al 1999). When heat thresholds are determined with long-duration temperature stimuli, however, thresholds are in the mid-40–50°C range (Treede et al 1998). Type I AMHs are seen in hairy and glabrous skin (Campbell et al 1979) and have also been described in the cat and rabbit (Fitzgerald and Lynn 1977, Roberts and Elardo 1985). The mean conduction velocity of type I AMHs in the monkey is 25 m/sec and extends as high as 55 m/sec. Thus, by conduction velocity criteria, type I AMHs fall into a category between that of Aδ and Aβ fibers. Nearly all type I AMHs are MSAs. Their receptive field size is similar to that of CMHs, but the presence of “hot spots” in response to mechanical stimuli is much more obvious.
Table 1-1
Comparison of Type I and Type II A-Fiber Nociceptors
CHARACTERISTIC |
TYPE I |
TYPE II |
| Heat threshold to short stimuli | High | Low |
| Heat threshold to long stimuli | Low | Low |
| Response to intense heat | Slowly increasing | Adapting |
| Response latency to intense heat | Long | Short |
| Peak latency to intense heat | Late | Early |
| Mechanical threshold | Most are MSAs | Most are MIAs |
| Conduction velocity | Aδ and Aβ fibers | Aδ fibers |
| Sensitization to heat injury | Yes | No |
| Location | Hairy and glabrous skin | Hairy skin |
MIAs, mechanically insensitive afferents; MSAs, mechanically sensitive afferents.
Type II A-fiber nociceptors were encountered only infrequently in early studies. It turns out that this is because the thresholds to mechanical stimuli place the majority of these fibers in the MIA category. Many have no demonstrable response to mechanical stimuli. When an unbiased electrical search stimulus is used, however, the prevalence of type I and type II A-fiber nociceptors in the hairy skin of the primate is similar. They do not occur in the glabrous skin of the hand (where type I AMHs are prevalent). Their mean conduction velocity, 15 m/sec, is also lower than that of type I AMHs. Their responses to heat resemble those observed in CMHs, and they may also be mediated by the vanilloid receptor 1 (VR1 or TRPV1). Responses to endogenous inflammatory/algesic mediators resemble those seen with type I A-fiber nociceptors (Davis et al 1993b).
Examples of the differing responses of the two types of A-fiber nociceptors to a heat stimulus are shown in Figure 1-3. Type I fibers exhibit a distinctive, gradually increasing response to heat. They sensitize to burn and chemical injury and probably play a role in the development of hyperalgesia. Type II fibers respond to heat in similar fashion to CMHs: early peak frequency and a slowly adapting response (Treede et al 1995). As noted later, type II A-fiber nociceptors are thought to signal first pain sensation in response to heat and may also contribute to pain caused by the application of capsaicin to the skin (Ringkamp et al 2001).
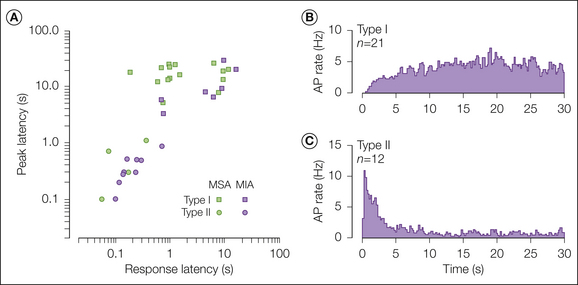
Figure 1-3 A-fiber nociceptors exhibit two types of responses to a heat stimulus.
A, Scatter plot of peak discharge latency versus response latency for mechanically insensitive afferents (MIAs; purple symbols) and mechanically sensitive afferents (MSAs; green symbols) in response to a 53°C, 30-second stimulus. Receptors that had a long peak discharge latency were considered to have a type I heat response (squares). Receptors that had a short response latency and a peak discharge near stimulus onset were considered to have a type II heat response (circles). The type II heat response was found more frequently in the MIA group (p ≤ 0.05, χ²-test). B, Average peristimulus frequency histogram (obtained with a 0.2-second bin width) of the response to the 53°C, 30-second stimulus for A-fiber nociceptors that had a type I heat response. C, Average peristimulus frequency histogram for A-fiber nociceptors that had a type II heat response. (Reproduced with permission from Treede RD, Meyer RA, Campbell JN 1998 Myelinated mechanically insensitive afferents from monkey hairy skin: heat-response properties. Journal of Neurophysiology 80:1082–1093.)
The conduction velocity of small myelinated Aδ fibers is, by definition, faster than that of unmyelinated C fibers. However, the terminal cutaneous branches of nociceptive Aδ fibers may conduct at a velocity characteristic of unmyelinated fibers (i.e., <2 m/sec) (Peng et al 1999). In addition, these unmyelinated terminals may branch off the main axon several centimeters proximal to their cutaneous receptive field.
Nociceptors Can Be Classified by Molecular Markers
The anatomical and biochemical features of nociceptive afferents have been studied extensively to correlate these features with their receptive properties. A wide range of cell markers have been used to classify nociceptive afferents and to study their peripheral and central projections. These markers include molecules expressed on the cell surface (e.g., receptors, glycoconjugates), molecules stored and released from nociceptive afferents (e.g., peptides), and enzymes. Expression of receptors for neurotrophic factors is of interest since these factors may regulate the sensitivity of nociceptive afferents in physiological and pathological states such as inflammation and neuropathy. The size of neuronal populations expressing or co-expressing different markers varies between species (Zwick et al 2002) and changes with the developmental stage (Molliver et al 1997, Guo et al 2001). Inflammation of the innervated tissue or a peripheral nerve lesion can cause substantial changes in the expression of these molecules. With the ongoing discovery of new marker molecules and the refinement of histological techniques, classification of nociceptive afferents is undergoing constant change and revision. Despite these “challenges,” however, classification of nociceptive afferents based on the expression of biochemical markers is instructive inasmuch as certain different neuronal populations are distinguishable across species. Sophisticated genetic manipulations have allowed the peripheral and central projections of defined neuronal populations to be studied in great detail. In addition, ablation experiments have been used to study the role of defined neuronal populations in animal behavior.
The cell bodies of nociceptive somatic and visceral afferents are located in DRGs. Slowly conducting Aδ and C fibers, including nociceptors, have small cell bodies (Lawson and Waddell 1991). Some of these are labeled with an antibody directed against a neurofilament protein (NF200) and are therefore thought to correspond to the somata of small myelinated Aδ afferents.
Small DRG cells are subdivided into peptidergic neurons (i.e., neurons containing peptides such as substance P [SP], calcitonin gene–related peptide [CGRP], and somatostatin [SST]) and “non-peptidergic” neurons. In the rat, about 40% of DRG cells, 50% of C fibers, and 20% of Aδ fibers are classified as peptidergic (McCarthy and Lawson 1989, Lawson et al 1996). Non-peptidergic, nociceptive neurons contain fluoride-resistant acid phosphatase (FRAP) (Silverman and Kruger 1988a), and their somata and axons bind the plant isolectin B4 (IB4) from Griffonia simplicifolia (Silverman and Kruger 1988b). It is common practice to classify neurons as “peptidergic” or “non-peptidergic” based on their binding of IB4. However, considerable co-localization of SP or CGRP and IB4 or FRAP has been reported in rats but less so in mice (Carr et al 1990, Wang et al 1994, Bergman et al 1999, Price and Flores 2007). In vivo intracellular recordings combined with immunohistochemistry have shown that cells containing SP or CGRP or cells binding IB4 are nociceptive and that non-nociceptive cells do not label with these markers (Lawson et al 1997, 2002; Gerke and Plenderleith 2001).
A group of mas-related genes (Mrgs) have been discovered that are selectively expressed in small DRG neurons and encode G protein–coupled receptors (GPCRs) (Dong et al 2001). Independently, sensory neuron–specific GPCRs (so-called sensory neuron–specific receptors [SNSRs]) in which the encoding genes were identical to some of the previously described Mrgs were identified shortly thereafter (Lembo et al 2002). For some Mrgs (MrgA–C) identified in mice, no ortholog genes exist in human or non-human primates, but closely related Mrgs (so-called MrgXs) have been identified. For other Mrgs (MrgD–G), however, ortholog genes exist in humans. Mrgs are expressed mainly in non-peptidergic, IB4-positive neurons, with some Mrgs being expressed in distinct IB4 subpopulations. In in vitro recordings, MrgD+ DRG cells showed characteristics typical of nociceptors (e.g., broad action potentials, expression of tetrodotoxin [TTX]-resistant sodium channels) (Drussor et al 2008). Receptors encoded by Mrgs respond to a variety of ligands, including β-alanine, cortistatin, peptides derived from different opioid precursors, and different RFamide peptides (Dong et al 2001, Han et al 2002, Lembo et al 2002, Robas et al 2003, Shinohara et al 2004), and they probably modulate excitability and sensitivity in this class of nociceptive afferents.
Expression of some markers appears to be related to the peripheral target tissue innervated by the neuron. Thus, almost all visceral afferents are peptidergic, but only about half the afferents projecting to the skin are (e.g., Perry and Lawson 1998) and only a small percentage of afferents projecting to muscle are labeled with IB4 (Plenderleith and Snow 1993, Ambalavanar et al 2003). MrgD-positive fibers exclusively innervate the skin, and they terminate in more superficial skin layers than do their peptidergic counterparts (Fig. 1-4) (Zylka et al 2005). Peptidergic and non-peptidergic afferents project to distinct dorsal horn laminae, with peptidergic fibers projecting mainly to lamina I and lamina II outer and IB4-binding afferents projecting preferentially to lamina II inner (e.g., Hunt and Rossi 1985, Silverman and Kruger 1988b; but see also Woodbury et al 2000).
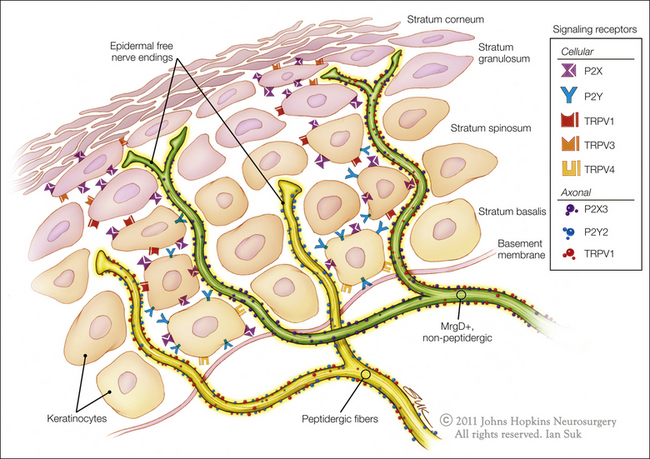
Figure 1-4 Schematic illustration of unmyelinated fiber terminations in the epidermis.
Non-peptidergic, MrgD+ neurons terminate as free nerve endings in the most superficial layers of the epidermis. Peptidergic neurons terminate in deep layers of the epidermis. Some of the signaling receptors found on keratinocytes and free nerve endings are also illustrated. (Artwork by Ian Suk, Johns Hopkins University; adapted from Dussor G, Koerber HR, Oaklander AL, et al 2009 Nucleotide signaling and cutaneous mechanisms of pain transduction. Brain Research Reviews 60:24–35.)
Although all nociceptive neurons depend on nerve growth factor (NGF) during early development, in the adult only peptidergic neurons express its receptor TrkA (tropomyosin-related kinase A) (Averill et al 1995). In contrast, most IB4-positive DRG cells do not express TrkA (Molliver et al 1995, but see also Kashiba et al 2001) but express one of the glial-derived neurotrophic factor (GDNF) family receptors (GDNFRα1–4) together with receptor tyrosine kinase Ret (Bennett et al 1998, Orozco et al 2001).
Peptidergic and non-peptidergic neurons express different receptors involved in signal transduction, and they may therefore display different sensitivity to a given stimulus. Thus the P2X3 receptor, which mediates nociceptor excitation by ATP, is primarily expressed in IB4-positive neurons (Vulchanova et al 1998). In contrast, TRPV1, which mediates responses to heat, capsaicin, and protons, is expressed in only a minority of IB4-positive cells in mice (Zwick et al 2002). In rats, however, this segregation is less obvious since about half of both IB4-positive and -negative cells express TRPV1 (Caterina et al 1997; Michael and Priestley 1999; Guo et al 1999, 2001). Species differences also exist in the co-expression of different Mrgs and their co-expression with other markers of nociceptive neurons (Zylka et al 2003).
Coupling between C-Fiber Nociceptors
Activation of one fiber by action potential activity in another is referred to as coupling. Coupling of action potential activity occurs between C fibers in the normal peripheral nerve of the monkey (Meyer et al 1985a). Coupling frequently involves conventional CMHs. Coupling is eliminated by injecting small amounts of local anesthetic at the receptive field of the CMH, thus indicating that the site of coupling is near the receptor. Collision studies indicate that the coupling is bidirectional. Sympathetic fibers do not appear to be involved in this coupling as demonstrated by experiments in which the sympathetic chain is stimulated or ablated (Meyer and Campbell 1987). The role of coupling is unknown but it may relate to the flare response or other efferent functions of nociceptors (see below). Coupling between peripheral nerve fibers is also one of the pathological changes associated with nerve injury (e.g., Blumberg and Jänig 1982, Meyer et al 1985b). In this case, coupling occurs at the site of axotomy.
Anatomical Studies of Cutaneous Nociceptors
Immunostaining for protein gene product (PGP) 9.5, a carboxy-terminal ubiquitin hydrolase, has proved particularly sensitive in identifying small-diameter afferents in the skin (Hsieh et al 1996). Vertical sections reveal that epidermal axons emerge from the superficial dermal nerve plexuses running beneath the epidermis. Schwann cells encase the axons at the dermal level, but as the axons rise into the epidermis between keratinocytes, the Schwann cell encasements are lost (Kruger et al 1981). Both clear round and large dense-core vesicles are noted at the epidermal penetration site. The vesicles are similar morphologically to vesicles present in other cells involved in hormone and neurotransmitter secretion. It is presumed that these vesicles secrete their contents into tissues on activation (see the efferent role of nociceptors below). Some of these fibers appear to innervate Langerhans cells. In small-fiber neuropathies in which patients have pain and deficits in cutaneous pain sensibility, these axonal terminals stained by PGP 9.5 are markedly decreased (Holland et al 1998).
As illustrated in Figure 1-4, free nerve endings can be traced far into the epidermal layer. These free nerve endings are probably sensory and serve the sensations of pain, temperature, and itch. The parent axons of these unmyelinated terminals are probably both myelinated and unmyelinated. Some of these free nerve endings are peptidergic and contain SP or CGRP (Gibbons et al 1987). Others are non-peptidergic and reach into the superficial layers of the epidermis.
Relationship of Nociceptor Activity to Acute Pain Sensations
Nociceptors and Pain in Response to Heat Stimuli
CMHs Signal Pain from Heat Stimuli to Glabrous Skin
We now examine the evidence that CMHs signal pain. In glabrous skin of the hand, two types of fibers, CMHs (not AMHs) and warm fibers, respond to short-duration heat stimuli (≤5 seconds) at temperatures near the pain threshold in humans (i.e., around 45°C). It is of interest, therefore, to compare how warm fibers and CMHs encode information about noxious heat stimuli. Warm fibers respond vigorously to gentle warming of the skin and are thought to signal the sensation of warmth (Johnson et al 1979). An example of the response of a warm fiber to stimuli in the noxious heat range is shown in Figure 1-1B. The response of warm fibers is not monotonic over this temperature range. In the example shown in Figure 1-1B, the total response evoked at 49°C was less than that at 45°C. Psychophysical studies in humans demonstrate that pain increases monotonically with stimulus intensities between 40 and 50°C. Because the responses of CMHs increase monotonically over this temperature range (Fig. 1-1A) and the responses of warm fibers do not (Fig. 1-1B), it follows that CMHs probably signal the sensation of heat pain to the glabrous skin of the hand (LaMotte and Campbell 1978).
Other evidence in support of a role for CMHs in pain sensation includes the following: (1) human judgments of pain in response to stimuli over the range of 41–49°C correlate well with the activity of CMH nociceptors over this range (Fig. 1-5, Meyer and Campbell 1981b); (2) selective A-fiber ischemic blocks or C-fiber (local anesthetic) blocks indicate that C-fiber function is necessary for perception of thermal pain near the pain threshold (Torebjörk and Hallin 1973); (3) the stimulus interaction effects observed in psychophysical experiments (LaMotte and Campbell 1978) are also observed in recordings from CMHs (Fig. 1-1A); (4) the latency to pain sensation on glabrous skin following stepped changes in temperature is long and consistent with input from CMHs (Campbell and LaMotte 1983); and (5) in patients with congenital insensitivity to pain, microscopic examination of peripheral nerves indicates an absence of C fibers (Bischoff 1979).
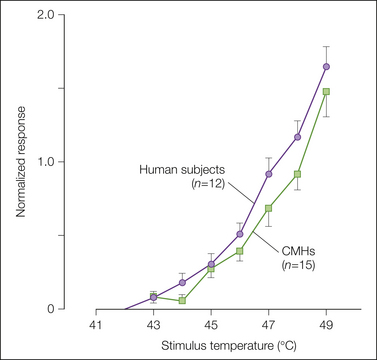
Figure 1-5 Correlation of the response of C-fiber nociceptors in the monkey with pain ratings in human subjects.
The close match between the curves supports a role for C-fiber nociceptors in heat pain sensation from glabrous skin. The first stimulus of the heat sequence was always 45°C. The remaining nine stimuli ranged from 41–49°C in 1°C increments and were presented in random order. Human judgments of pain were measured with a magnitude estimation technique: subjects assigned an arbitrary number (the modulus) to the magnitude of pain evoked by the first 45°C stimulus and judged the painfulness of all subsequent stimuli as a ratio of this modulus. The response to a given stimulus was normalized by dividing by the modulus for each human subject or by the average response to the first 45°C stimulus for the C-fiber mechano-heat–sensitive nociceptors (CMHs). (Originally published in Meyer RA, Campbell JN 1981 Peripheral neural coding of pain sensation. Johns Hopkins APL Technical Digest 2:164–171. Copyright 1981 AAAS.)
Human Microneurographic Recordings
Microneurography has been used to record from nociceptive afferents in awake humans and allows correlations between the discharge of afferents and the reported sensations of the subject. The technique involves percutaneous insertion of a microelectrode into fascicles of nerves such as the superficial radial nerve at the wrist. These studies have demonstrated that the properties of nociceptors in humans and monkeys are similar. In some experiments the microelectrode is also used to stimulate an identified, single nerve fiber in awake human subjects to evoke specific sensations. Some, however, argue that the size of the stimulating electrode is too large to stimulate individual units (Wall and McMahon 1985). Given this reservation, the following evidence from microneurographic studies in humans points to the capacity of CMH activity to evoke pain: (1) intraneural electrical stimulation of presumed single identified CMHs in humans elicits pain (Torebjörk and Ochoa 1980), (2) the heat threshold for activation of CMHs recorded in awake humans is just below the pain threshold (Van Hees and Gybels 1981), and (3) a linear relationship exists between responses of CMHs recorded in awake humans and ratings of pain over the temperature range 39–51°C (Torebjörk et al 1984).
Correlations between Psychophysical Measures of the Heat Pain Threshold and Neurophysiological Results
We noted above that the heat threshold of CMHs is dependent on temperature at the level of the receptor and is independent of the rate of change in temperature. At the same time when threshold temperature is measured at the surface of skin, CMHs have a lower threshold when the rate of increase in temperature is slow. As discussed earlier, the reason for this relates to thermal inertia.
Human pain thresholds are sometimes measured as the temperature that corresponds to the first report of pain as skin temperature is increased linearly (Marstock technique). Investigators have noted that faster rates of change in temperature lead to lower estimates of the heat pain threshold (Yarnitsky and Ochoa 1990, Tillman et al 1995a). This is the opposite of the situation with the surface temperature threshold of CMHs but fits with the finding that suprathreshold responses of CMHs vary directly with the rate of increase in temperature. Thus it is unlikely that the threshold responses of CMHs are responsible for the heat pain thresholds. Rather, it appears that nociceptors must reach a certain discharge frequency (about 0.5 impulses/sec) for pain to be perceived (Van Hees 1976, Yarnitsky et al 1992, Tillman et al 1995a).
A-Fiber Nociceptors and Heat Pain
As shown in Figure 1-6, a long-duration heat stimulus applied to the glabrous skin of the hand in human subjects evokes substantial pain for the duration of the stimulus. CMHs exhibit a prominent discharge during the early phase of the stimulus, but this response adapts within seconds to a low level. In contrast, type I AMHs are initially unresponsive but then discharge vigorously. Therefore, type I AMHs probably contribute to the pain during a sustained, high-intensity heat stimulus (Meyer and Campbell 1981a).
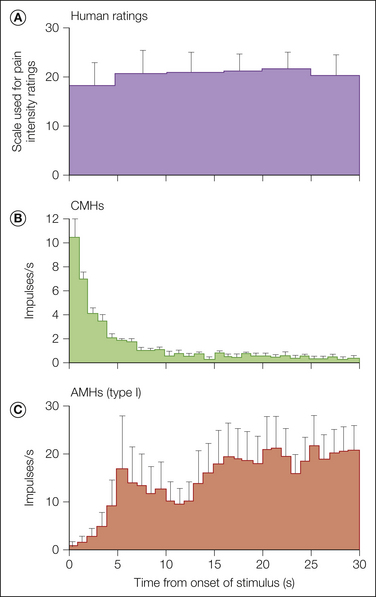
Figure 1-6 Ratings of pain by human subjects during a long-duration, intense heat stimulus (53°C, 30 seconds) applied to the glabrous skin of the hand compared with responses of C-fiber mechano-heat–sensitive nociceptors (CMHs) and type I A-fiber mechano-heat–sensitive nociceptors (AMHs).
A, Pain was intense throughout the stimulus. B, The brisk response of CMHs at the beginning of the stimulus changed to a low rate of discharge after 5 seconds. C, The response of AMHs increased during the first 5 seconds and remained high throughout the stimulus. (Reprinted with permission from Meyer RA, Campbell JN 1981 Myelinated nociceptive afferents account for the hyperalgesia that follows a burn to the hand. Science 213:1527–1529.)
In hairy skin, stepped heat stimuli evoke a double pain sensation (Lewis and Pochin 1937). The first perception is a sharp pricking sensation, and the second sensation is a burning feeling that occurs after a momentary lull during which little if anything is felt. Myelinated afferent fibers must signal the first pain since the latency of response to the first pain is too small to be carried by C fibers (Campbell and LaMotte 1983). Type II A-fiber nociceptors (see Fig. 1-3) are ideally suited to signal this first pain sensation: (1) the thermal threshold is near the threshold temperature for the first pain (Dubner et al 1977), (2) the receptor utilization time (time between onset of the stimulus and activation of the receptor) is short (Treede et al 1998), and (3) the burst of activity at the onset of the heat stimulus is consistent with the perception of a momentary pricking sensation. The absence of a first pain sensation to heat stimuli applied to the glabrous skin of the human hand correlates with the failure to find type II A-fiber nociceptors on the glabrous skin of the hand in the monkey.
The preceding discussion indicates that nociceptors may signal pain in response to heat stimuli. However, two caveats are in order: (1) This does not mean that activity in nociceptors always signals pain. It is clear that low-level discharge rates in nociceptors do not always lead to sensation (e.g., Van Hees and Gybels 1981, Cervero et al 1993). Central mechanisms, including attentional and emotional states, quite obviously play a crucial role in whether and how much nociceptor activity leads to the perception of pain. (2) It is probable that receptors other than nociceptors signal pain in certain circumstances. For example, the pain in response to light touch that occurs after certain nerve injuries or with tissue injury appears to be signaled by activity in low-threshold mechanoreceptors (see below).
Nociceptors and Pain in Response to Controlled Mechanical Stimuli
A-Fiber Nociceptors Signal Sharp Pain
A-fiber and C-fiber MSAs respond well to punctate mechanical stimuli. When a controlled-force stimulus is applied to the receptive field, the response is greatest at the onset of the stimulus and then slowly adapts. Like heat, repeated presentations of a mechanical stimulus lead to pronounced fatigue. A-fiber nociceptors recover faster from fatigue than do C-fiber nociceptors (Fig. 1-7).
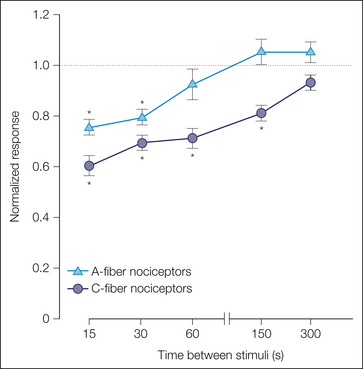
Figure 1-7 A-fiber nociceptors recover faster from fatigue than do C-fiber nociceptors.
Mechanical stimuli were presented to the receptive field of A-fiber and C-fiber nociceptors at different interstimulus intervals (with 10 minutes between stimulus pairs). The A-fiber response (triangles) recovered within 60 seconds, whereas the C-fiber response (circles) took more than 150 seconds to recover. To normalize the data, the response to the test stimulus was divided by the response to the immediately preceding conditioning stimulus. (Adapted from Slugg RM, Meyer RA, Campbell JN 2000 Response of cutaneous A- and C-fiber nociceptors in the monkey to controlled-force stimuli. Journal of Neurophysiology 83:2179–2191.)
Much has been learned about the features of a mechanical stimulus that determine the response of nociceptors to mechanical stimuli. The discharge of nociceptors increases with increased force and pressure, but these functions vary depending on probe size: the smaller the probe, the greater the response (Garell et al 1996). For cylindrical probes of different diameter, the discharges are comparable if the intensity of the stimulus is calculated according to force per length of the perimeter of the cylindrical probe. This suggests that the stress/strain maximum that occurs at the edge of the cylindrical stimulus is the critical parameter for excitation of nociceptor terminals.
For a given probe size, the response of A-fiber nociceptors increases monotonically with force, whereas the response of C-fiber nociceptors becomes saturated at higher force levels (Fig. 1-8A; Slugg et al 2000). In general, the discharge in A fibers is greater than that in C fibers.
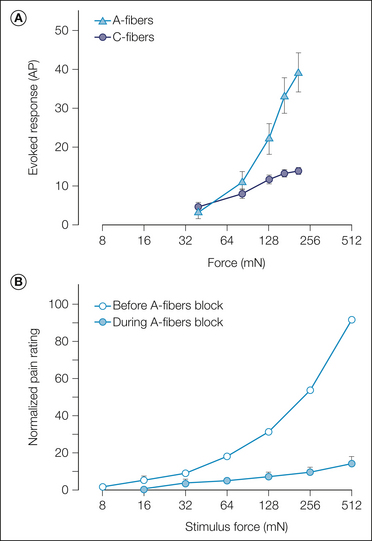
Figure 1-8 Comparison of responses of nociceptors to mechanical stimuli in the monkey with pain ratings in human subjects.
These data provide evidence that A-fiber nociceptors signal the pain reported from sharp probes. A, Average responses of A-fiber nociceptors (triangles) and C-fiber nociceptors (circles) to controlled-force stimuli. The A fibers exhibited a monotonically increasing response, whereas the response of the C fibers reached a plateau at the higher force levels (0.4-mm-diameter cylindrical probes; the total response to a stimulus 3 seconds in duration is plotted). B, Average pain ratings in response to controlled-force stimuli (open circle) increased monotonically in a manner comparable to that observed for the A-fiber nociceptors. Selective block of A-fiber function led to a significant decrease in pain ratings (filled circles). All pain ratings for a given subject were normalized by dividing by that subject’s average rating of the maximum stimulus (0.2-mm-diameter cylindrical probes, stimulus duration of 1 second). (A, Adapted from Slugg RM, Meyer RA, Campbell JN 2000 Response of cutaneous A- and C-fiber nociceptors in the monkey to controlled-force stimuli. Journal of Neurophysiology 83:2179–2191; B, adapted from Magerl W, Fuchs PN, Meyer RA, et al 2001 Roles of capsaicin-insensitive nociceptors in cutaneous pain and secondary hyperalgesia. Brain 124:1754–1764.)
The area of the receptive field that responds to mechanical stimuli also responds to heat stimuli (Treede et al 1990). However, the transducer elements that account for mechanosensitivity are probably different from those responsible for heat. For example, the heat response of nociceptors is readily sensitized by a heat injury, whereas the mechanical response is not (see below).
A-fiber nociceptors appear to be responsible for the sharp pain reported in response to punctate mechanical stimuli: (1) the reaction time to perception of pain is short, (2) the stimulus–response function of A-fiber nociceptors (Fig. 1-8A) is comparable to the pain ratings of human subjects (Fig. 1-8B) over a similar force range, and (3) the pain in response to sharp probes is dramatically reduced during selective blockade of A-fiber function (Fig. 1-8B; Magerl et al 2001).
Pretreatment of the skin with capsaicin abolishes heat pain sensitivity but does not greatly affect mechanical pain (Magerl et al 2001). This suggests that the A-fibers involved in sharp pain are capsaicin insensitive; they could be type I AMHs or HTMs.
C-Fiber MIAs Signal Pain in Response to Tonic Pressure
When long-duration mechanical stimuli are applied to human subjects, the pain increases throughout the stimulus (Adriaensen et al 1984). However, the response of MSAs to long-duration suprathreshold stimuli adapts with time. Although C-fiber MIAs are, by definition, normally insensitive to mechanical stimuli, they develop a response to prolonged mechanical stimulation (Schmidt et al 2000). In addition, the pain associated with a tonic stimulus persists through selective A-fiber blockade (Andrew and Greenspan 1999b). Thus it appears that C-fiber MIAs signal the pain associated with tonic pressure.
Nociceptors and Cold Pain Sensation
Cold pain differs from heat pain in a number of important factors: (1) the cold pain threshold (≈14°C on hairy skin; Harrison and Davis 1999) is much farther from resting skin temperature (33°C) than the heat pain threshold (about 45°C), (2) the slope of the stimulus–response function is much steeper for heat pain than for cold pain (Morin and Bushnell 1998), and (3) the lag in response between stimulus onset and pain report suggests that cold pain is subserved by deeper receptors whereas heat pain seems to be subserved by superficial receptors. Klement and Arndt (1992) demonstrated that cold pain could be evoked by cold stimuli applied within the veins of human subjects. A local anesthetic applied within the vein, but not in the overlying skin, abolished cold pain sensibility. It is therefore possible that cold pain is served, at least in part, by vascular receptors.
Just as the sensation of warmth is served by a specific set of primary afferents (predominantly C fibers), the sense of cooling is served by a specific set of primary afferents (i.e., cold fibers). Cold fibers are predominantly of the A type. They exhibit ongoing activity at room temperature, and their response increases markedly with gentle cooling. Stimuli that induce cold pain are not encoded well by these cold fibers. Although the majority of nociceptors have some response to ice stimuli applied to the skin, Simone and Kajander (1997) showed that all A-fiber nociceptors respond to cold stimuli below 0°C. C-fiber nociceptors may play a role in signaling cold pain sensation as well (LaMotte and Thalhammer 1982). A non-selective cation channel has been identified (called ANKTM1 or transient receptor potential ankyrin 1 [TRPA1]) that has an activation threshold (17.5°C) comparable to the cold pain threshold (Story et al 2003). This channel is found in a subset of nociceptive sensory neurons that are responsive to intense heat and capsaicin. However, the role of TRPA1 in mediating noxious cold is still debated.
Nociceptors and Chemically Evoked Sensations
Many chemical agents produce pain when applied to the skin. In many cases the pain from these agents probably results from tissue injury and is therefore indirect. (Chemical mediators associated with inflammation are described later.) One exception that has received a lot of attention is capsaicin. Intradermal injection of capsaicin produces intense burning pain that lasts for several minutes. When capsaicin is injected into the receptive field of C-fiber MSAs, the response is weak (relative to the heat response) and of short duration (Baumann et al 1991). In contrast, A-fiber and C-fiber MIAs exhibit a long-lasting, vigorous response to capsaicin (Schmelz et al 2000b, Ringkamp et al 2001), thus suggesting that these fibers are responsible for the pain induced by capsaicin. The pungent effects of capsaicin appear to be mediated by the TRPV1 receptor expressed on nociceptive fibers. This receptor appears to be activated by heat and protons (acid) as well.
Another chemical of interest is histamine, which produces a long-lasting itch when applied to the skin. Injection of histamine into the receptive field of C-fiber MSAs leads to a lasting response (Johanek et al 2008). Iontophoresis of histamine into the receptive field of a subpopulation of C-fiber MIAs also produces a vigorous, long-lasting response (Schmelz et al 1997), which suggests that both CMHs and C-fiber MIAs may play a role in histamine-induced itch. Histamine probably activates nociceptors via the H1 receptor located on peripheral terminals.
Because cowhage spicules produce an intense itch that is not blocked by topical antihistamines (Johanek et al 2008), and they provide a useful tool to investigate the chronic itch in patients that is resistant to antihistamine treatment. In about half of normal subjects, cowhage-induced itch is greatly attenuated during selective blockade of myelinated fibers. Although C-fiber MIAs do not respond to cowhage, QC fibers and A-fiber nociceptors respond vigorously to cowhage (Ringkamp et al 2011). The active ingredient in cowhage is the cysteine protease mucunain, which activates nociceptive terminals via protease-activated receptor 2 (PAR-2) and PAR-4 (Reddy et al 2008).
Hyperalgesia: Role of Nociceptors and Other Afferent Fibers
To understand the peripheral neural mechanisms of pain induced by noxious stimuli is to understand only one aspect of pain sensibility. There is, in fact, a dynamic plasticity that relates stimulus intensity and sensation. Of great biological importance in this regard is the phenomenon of hyperalgesia. Hyperalgesia is defined as a leftward shift of the stimulus–response function that relates the magnitude of pain to stimulus intensity. An example of this is seen in Figure 1-9A, which shows human judgments of pain induced by heat stimuli before and after a burn. It is evident that the threshold for pain is lowered and pain in response to suprathreshold stimuli is enhanced.
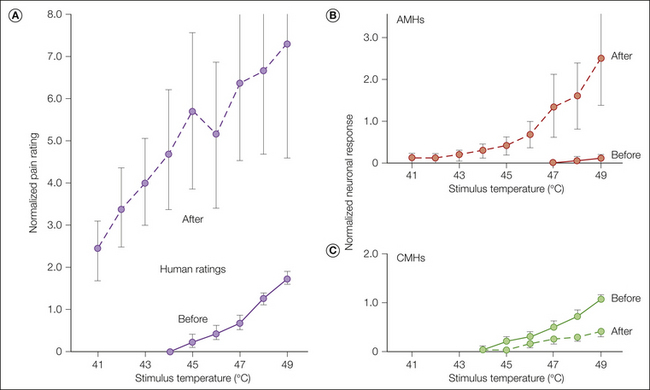
Figure 1-9 Hyperalgesia and nociceptor sensitization after a cutaneous burn injury.
Responses to heat stimuli were obtained 5 minutes before and 10 minutes after a 53°C, 30-second burn on the glabrous skin of the hand. The burn resulted in increases in the magnitude of pain (hyperalgesia) in human subjects that were matched by enhanced responses (sensitization) in type I A-fiber mechano-heat–sensitive nociceptors (AMHs) in the monkey. In contrast, C-fiber mechano-heat–sensitive nociceptors (CMHs) exhibited decreased sensitivity after the burn. A, Human judgments of pain. B, Responses of type I AMHs in the monkey. C, Responses of CMHs in the monkey. The same type of random heat sequence and normalization described in Figure 1-5 was used. Because the AMHs did not respond to the 45°C stimulus before the burn, the AMH data were normalized by dividing by the response to the first 45°C stimulus after the burn. (Reprinted with permission from Meyer RA, Campbell JN 1981 Myelinated nociceptive afferents account for the hyperalgesia that follows a burn to the hand. Science 213:1527–1529.)
Hyperalgesia is a consistent feature of somatic and visceral tissue injury and inflammation. Pharyngitis is associated with hyperalgesia in pharyngeal tissues such that merely swallowing induces pain. Micturition in the presence of a urinary tract infection is painful, again reflecting the presence of hyperalgesia. In arthritis, slight motion of the joint results in pain. A sunburn leads to pain with light touch and gentle heating.
The peripheral neural mechanisms of hyperalgesia have been studied in various tissues, including the joints, cornea, testicle, gastrointestinal tract, and bladder. Much of the theoretical work on hyperalgesia, however, has evolved from studies of the skin, and it is this work that will receive attention here.
Hyperalgesia occurs not only at the site of injury but also in the surrounding uninjured area. Hyperalgesia at the site of injury is termed primary hyperalgesia, whereas hyperalgesia in the uninjured skin surrounding the injury is termed secondary hyperalgesia (Lewis 1935). Hyperalgesia exemplifies the functional plasticity of the nervous system. As we will see, the neural mechanisms for primary and secondary hyperalgesia differ.
In discussing hyperalgesia, it is useful to consider the following variables: (1) energy form of the injury, (2) type of tissue involved, (3) energy form of the test stimulus, and (4) location of the testing relative to the area injured. These variables interact in complex ways. For example, it will be shown that nociceptors will become sensitized to mechanical stimuli (the energy form of the test stimulus), but only after certain forms of injury (i.e., injection of inflammatory mediators).
An experimental design frequently used for study of the neural mechanisms of hyperalgesia is to characterize the response properties of a given fiber, then apply a manipulation that under usual circumstances would produce hyperalgesia, and finally assess whether this manipulation has altered the response properties of the fiber in question. Cutaneous hyperalgesia has been studied after thermal injury (burn or freeze lesions), after local administration of chemicals (e.g., capsaicin, mustard oil, or menthol), after a mechanical injury to the skin (e.g., incision, crushing), and after exposure to ultraviolet radiation. The main features of the hyperalgesia that develops after these various injuries are quite similar.
As shown in Figure 1-10, the relative locations of the injury site, the test site, and the receptive field of the sensory neuron being studied dictate whether the experiment provides information regarding the mechanisms of primary or secondary hyperalgesia (Treede et al 1992). These three variables may interact in any of six ways. As shown in Figure 1-10, when the injury and the test site coincide (Fig. 1-10A and B), the study has provided a basis by which to consider the mechanism of primary hyperalgesia, whereas when the test site and the injury site diverge (Fig. 1-10C–F), the study has provided a basis by which to account for secondary hyperalgesia.
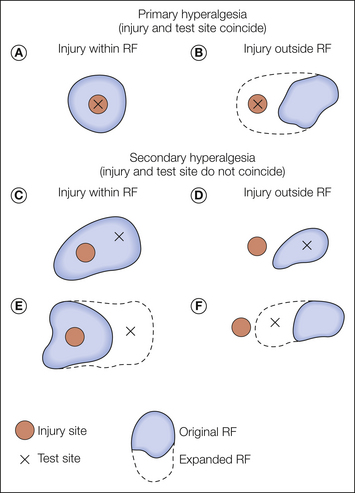
Figure 1-10 Experimental configurations for testing the neural mechanisms of primary and secondary hyperalgesia.
To study primary hyperalgesia, the site of injury (indicated by filled circles) and the site of testing (indicated by the X’s) must coincide. Alterations in the stimulus–response function from stimuli applied to the original receptive field (RF) (A) and expansion of the RF toward the injury site (B) are substrates for primary hyperalgesia. To study secondary hyperalgesia, the site of injury and the site of testing must not coincide (C and D). Sensitization of the stimulus–response function as revealed by testing within the original RF may occur following injuries within (C) or outside the RF (D). Expansion of the RF to include a test site outside the original RF may occur for injuries within (E) or outside (D) the RF. (Reprinted from Treede RD, Meyer RA, Raja SN, et al 1992 Peripheral and central mechanisms of cutaneous hyperalgesia. Progress in Neurobiology 38:397–421. Copyright 1992, with permission from Elsevier.)
When the paradigms shown in Figure 1-10A and B are used, it is found that under certain circumstances, nociceptors exhibit an increased response to the test stimulus. Thus, peripheral neural mechanisms are likely to account for at least some aspects of primary hyperalgesia. In contrast, primary afferent nociceptors do not develop an enhanced response to the test stimulus when the paradigms shown in Figure 1-10C–F are investigated. By default, therefore, the mechanism for secondary hyperalgesia must reside within the CNS.
Primary Hyperalgesia
We first consider the situation in which a burn injury is applied to the skin and the test stimulus is heat applied to the location of the burn injury. When a burn is applied to the glabrous skin of the hand, marked hyperalgesia to heat develops as shown in Figure 1-9A (Meyer and Campbell 1981a). The hyperalgesia is manifested as a leftward shift of the stimulus–response function that relates the magnitude of pain to stimulus intensity. For example, the 41°C stimulus was not painful before the burn but after the injury was as painful as the 49°C stimulus before the injury.
Peripheral Sensitization as a Mechanism for Primary Hyperalgesia to Heat Stimuli
Substantial evidence favors the concept that the primary hyperalgesia to heat stimuli that develops at the site of a burn injury is mediated by sensitization of nociceptors (Meyer and Campbell 1981a, LaMotte et al 1982). Sensitization is defined as a leftward shift of the stimulus–response function that relates the magnitude of the neural response to stimulus intensity. Sensitization is characterized by a decrease in threshold, an augmented response to suprathreshold stimuli, and ongoing spontaneous activity. These properties correspond to the properties of hyperalgesia (Table 1-2).
Table 1-2
Comparison of Characteristics of Hyperalgesia and Sensitization
HYPERALGESIA (SUBJECT RESPONSE) |
SENSITIZATION (FIBER RESPONSE) |
| Decreased pain threshold | Decreased threshold for response |
| Increased pain in response to suprathreshold stimuli | Increased response to suprathreshold stimuli |
| Spontaneous pain | Spontaneous activity |
To explain the hyperalgesia that occurs with a burn on the glabrous skin of the hand, a correlative analysis of subjective ratings of pain in humans with responses of nociceptors (CMHs and type I AMHs) in anesthetized monkeys was performed (Meyer and Campbell 1981a). Test heat stimuli were applied to the glabrous skin of the hand before and after a 53°C, 30-second burn. The burn led to prominent hyperalgesia in the human subjects (Fig. 1-9A). The CMHs showed a decreased response following the burn (Fig. 1-9C), whereas the type I AMHs were markedly sensitized (Fig. 1-9B). Thus, it is likely that for thermal injuries on the glabrous skin of the hand, AMHs, not CMHs, code for the heat hyperalgesia.
Sensitization is not a uniform property of nociceptors. Tissue type and the nature of the injury are important variables. For example, CMHs that innervate hairy skin become sensitized, whereas as described above, CMHs that innervate the glabrous skin of the hand do not become sensitized to a burn injury (Campbell and Meyer 1983). Thus, CMHs appear to play a role in accounting for hyperalgesia to heat stimuli on hairy skin (LaMotte et al 1983). These data support the conclusion that the hyperalgesia to heat stimuli that occurs at the site of an injury is due to sensitization of primary afferent nociceptors.
Hyperalgesia to Mechanical Stimuli
Distinguishing hyperalgesia to mechanical stimuli in the primary and secondary zones may be incorrect in some respects since the mechanism for hyperalgesia in the two zones may have some common elements. The mechanisms discussed in this section, however, will be limited to those applicable to the primary zone.
Different forms of mechanical hyperalgesia have been characterized. One form is evident when the skin is gently stroked with a cotton swab and is referred to as “stroking hyperalgesia,” “dynamic hyperalgesia,” or “allodynia.” The second form of hyperalgesia is evident when punctate stimuli, such as von Frey probes, are applied and, accordingly, has been termed “punctate hyperalgesia.” Hyperalgesia to tonic stimulation with a blunt probe, called “pressure hyperalgesia,” and impact hyperalgesia to shooting small bullets against the skin at a controlled velocity have also been described in the primary hyperalgesic zone (Kilo et al 1994). As discussed in the later section on secondary hyperalgesia, the mechanism for these different forms of mechanical hyperalgesia is probably different. Stroking hyperalgesia is thought to be signaled by low-threshold mechanoreceptors, whereas punctate hyperalgesia is mediated at least in part by nociceptors. Pressure hyperalgesia and impact hyperalgesia are probably mediated by sensitized C fibers. Another form of mechanical hyperalgesia termed “progressive tactile hypersensitivity,” which may contribute to the allodynia associated with inflammation, has been described (Ma and Woolf 1997).
Nociceptor Sensitization as a Mechanism for Mechanical Hyperalgesia in the Primary Zone
Primary hyperalgesia to mechanical stimuli appears to be due, at least in part, to sensitization of primary afferent nociceptors to mechanical stimuli. This sensitization is manifested in several ways.
Lowered Threshold
Thresholds to mechanical stimulation of either CMHs or AMHs recorded in primates or humans, as measured with von Frey hairs (a punctate stimulus), are not changed by heat and/or mechanical injury (e.g., Thalhammer and LaMotte 1982, Campbell et al 1988a). However, MIAs have been shown to develop mechanical sensitivity after inflammation. Figure 1-11 shows the response of an Aδ-fiber MIA to mechanical stimuli before and after exposure to a mixture of algesic inflammatory mediators (bradykinin, histamine, serotonin, and prostaglandin E1 [PGE1]). This MIA was unresponsive to the 5-bar von Frey probe initially, but a robust response to this probe developed after inflammation.
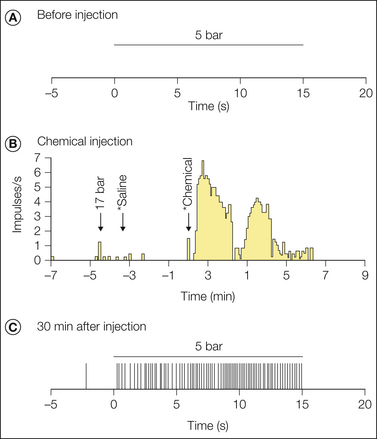
Figure 1-11 Example of sensitization to mechanical stimuli for an Aδ-fiber nociceptor following a chemical injection.
A, The fiber did not respond to the application of a 5-bar stimulus for 15 seconds to the most sensitive area within its receptive field. The initial mechanical threshold for this fiber was 10 bar, and therefore it was a mechanically insensitive afferent (MIA). B, This MIA responded vigorously to a 10-μL intradermal injection of a chemical mixture containing 10 nmol bradykinin, 0.3 nmol prostaglandin E1, 30 nmol serotonin, and 30 nmol histamine. (Each asterisk indicates the time of needle insertion; bin size = 5 seconds). C, Sensitization to mechanical stimuli was demonstrated in this fiber 30 minutes after chemical injection. The fiber now responded to application of the 5-bar stimulus. Each vertical tic corresponds to the time of occurrence of an action potential. The von Frey threshold decreased (from 10 to 4 bar), and the receptive field area increased (from 9 to 88 mm2). No response to heat was observed either before or after the injection. (Reproduced with permission from Davis KD, Meyer RA, Campbell JN 1993 Chemosensitivity and sensitization of nociceptive afferents that innervate the hairy skin of monkey. Journal of Neurophysiology 69:1071–1081.)
Increased Response to Suprathreshold Stimuli
Although inflammation does not result in a reduction in the mechanical threshold of AMHs and CMHs, responses to suprathreshold stimuli may be augmented (Cooper et al 1991). Inflammation of the rat paw results in an enhanced response to suprathreshold mechanical stimuli, spontaneous activity, and expanded receptor fields for both A- and C-fiber nociceptors (Andrew and Greenspan 1999a).
Expansion of the Receptive Field
The receptive fields of AMH fibers, as well as some CMH fibers, expand modestly into the area of an adjacent heat (Thalhammer and LaMotte 1982) or mechanical (Reeh et al 1987) injury. As a result of this expansion, heat or mechanical stimuli delivered after the injury will activate a greater number of fibers. This spatial summation would be expected to induce more pain.
Loss of Central Inhibition as a Mechanism of Mechanical Hyperalgesia in the Primary Zone
Under usual circumstances, production of pain from activation of nociceptors with mechanical stimuli is inhibited in the CNS by the concurrent activation of low-threshold mechanoreceptors (e.g., Bini et al 1984). There is evidence that injury decreases the responsiveness of low-threshold mechanoreceptors. Hyperalgesia to mechanical stimuli in the primary zone could therefore be due to injury to low-threshold mechanoreceptors, which would lead to central disinhibition of nociceptor input and thus result in enhanced pain (i.e., hyperalgesia).
Inflammatory Mediators and Nociceptors
Injury results in the local release of numerous chemicals from non-neuronal cells (e.g., fibroblasts, mast cells, neutrophils, monocytes, and platelets), as well as from the sensory terminals of primary afferent fibers that mediate or facilitate the inflammatory process. Inflammatory mediators include prostaglandins, leukotrienes, bradykinin, serotonin, histamine, SP, thromboxanes, platelet-activating factor, purines such as adenosine and ATP, protons, and free radicals (Fig. 1-12, see also Basbaum et al 2009). Cytokines, such as interleukins and tumor necrosis factor, and neurotrophins, especially NGF, are also generated during inflammation. NGF not only is necessary for the survival of nociceptors during development but may also play an important role during inflammatory processes in adult animals. Some of these agents can directly activate nociceptors, whereas others act indirectly via inflammatory cells, which in turn release algogenic agents. Other mediators lead to sensitization of the nociceptor response to natural stimuli and therefore play a role in primary hyperalgesia. The variety of chemical mediators released during inflammation can have a synergistic effect in potentiating nociceptor responses.
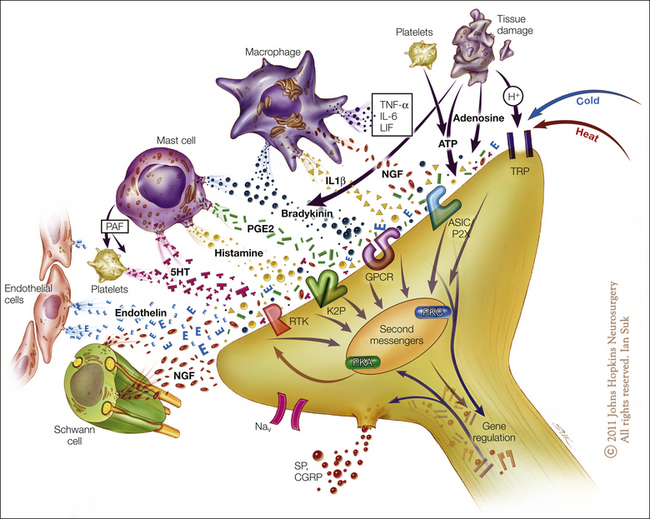
Figure 1-12 Potential mediators of peripheral sensitization after inflammation.
Tissue injury and inflammation lead to the release of numerous chemicals from non-neuronal and neuronal cells, such as mast cells, macrophages, platelets, immune and endothelial cells, Schwann cells, keratinocytes, fibroblasts, and peripheral nociceptor terminals. Mediators released include protons (H+), purines (adenosine, adenosine triphosphate), nerve growth factor (NGF), cytokines such as tumor necrosis factor (TNF-α) and interleukins (IL-1β, IL-6), leukemia inhibitory factor (LIF), prostaglandin E2 (PGE2), bradykinin, histamine, serotonin (5-HT), platelet activating factor (PAF), and endothelin. These mediators may act directly to alter the sensitivity of peripheral nociceptors or indirectly via coupling to one or more peripheral membrane-bound receptors, including transient receptor potential (TRP) channels, acid-sensitive ion channels (ASICs), purinergic (P2X) receptors, G protein–coupled receptors (GPCRs), two-pore potassium channels (K2P), and receptor tyrosine kinase (RTK). Binding of the ligands to these receptors can initiate a cascade of events that includes activation of second-messenger systems (protein kinase A [PKA] and C [PKC]) and alteration of gene regulation. (Artwork by Ian Suk, Johns Hopkins University; adapted from Woolf CJ, Costigan M 1999 Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proceedings of the National Academy of Sciences of the United States of America 96:7723–7730.)
A variety of metabotropic and ionotropic receptors, including purinergic and glutamatergic receptors, have been identified on DRG cells and on the peripheral terminals of nociceptive afferent fibers. Activation of these receptors may modulate the sensitivity of peripheral nociceptors to exogenous stimuli (Carlton and Coggeshall 1998).
Arachidonic Acid Metabolites
The prostaglandins, thromboxanes, and leukotrienes are a large family of arachidonic acid metabolites collectively known as eicosanoids. The eicosanoids are generally considered to not activate nociceptors directly but rather enhance the sensation of pain in response to natural stimuli and other endogenous chemicals by increasing the frequency of action potential firing (for reviews see Schaible et al 2002, Cunha and Ferreira 2003, Momin and McNaughton 2009). A sensitizing and direct excitatory effect of PGE2 and PGI2, however, has been demonstrated in afferents innervating joints. Prostaglandins are synthesized by the constitutive enzyme cyclooxygenase-1 (Cox-1) and by Cox-2, an enzyme induced in peripheral tissues by inflammation (Ballou et al 2000). Several prostaglandins, PGI2, PGE1, PGE2, and PGD2, are considered to play a role in inflammatory pain and hyperalgesia. Prostaglandins reduce the threshold for initiation of action potentials and increase the excitability of sensory neurons by decreasing the threshold for activation of a nociceptor-specific voltage-activated Na current, Nav1.8, and increasing intracellular cyclic adenosine monophosphate (cAMP) levels (England et al 1996, Gold et al 1996). The prostaglandin-induced increase in firing frequency may also result from an increase in the hyperpolarization-activated current (Ih), which leads to faster depolarization toward the action potential threshold, the consequence of which is a decrease in the time interval between successive action potentials (Momin and McNaughton 2009). Of the leukotrienes (metabolites of the lipoxygenase pathway), LTD4 and LTB4 have been suggested to play a role in hyperalgesia (Levine et al 1984) and in sensitization to mechanical stimuli (Martin et al 1987).
Bradykinin
Several lines of evidence suggest that bradykinin may also play a critical role in inflammatory pain and hyperalgesia (see Couture et al 2001, Meini and Maggi 2008 for reviews). Bradykinin is released on tissue injury (e.g., from plasma), is present in inflammatory exudates, and excites and sensitizes unmyelinated and myelinated nociceptors to natural stimuli (Beck and Handwerker 1974, Khan et al 1992). Administration of exogenous bradykinin produces pain and transient hyperalgesia to heat in humans (Manning et al 1991). Bradykinin acts on B1 and B2 receptors to induce nociceptor sensitization by activation of phospholipase C (PLC) and protein kinase C (PKC), production of arachidonic acids, and modulation of the TRPV1 channel (see the section on the vanilloid receptor below) (Reeh and Sauer 1997, Banik et al 2001).
Protons
The low pH levels found in inflamed tissue have led to the hypothesis that local acidosis may contribute to the pain and hyperalgesia associated with inflammation. Continuous administration of low-pH solutions in humans causes pain and hyperalgesia to mechanical stimuli (Steen and Reeh 1993). This correlates with the observation that protons selectively activate nociceptors and produce sensitization of nociceptors to mechanical stimuli. Excitation of nociceptors by protons does not undergo tachyphylaxis or adaptation, and a synergistic excitatory effect of protons and a combination of inflammatory mediators has been reported (Steen et al 1996).
A class of acid-sensing ion channels (ASICs), a subgroup of the degenerin/epithelial sodium channel (DEG/ENaC) family of proteins, has emerged as sensors of low pH (see Holzer 2009, Sluka et al 2009 for review). ASICs signal moderate decreases in extracellular pH, in contrast to TRPV1, which is activated by severe acidosis (pH values below 6). ASIC1A and ASIC3 have been identified in DRG neurons, and their expression is increased by inflammation, nerve injury, and bone cancer, thus suggesting that ASICs may play a role in mediating or modulating pain in these conditions. The observation that a non-selective ASIC inhibitor, amiloride, reduces cutaneous acid-evoked pain in humans suggests that ASICs may be a potential therapeutic target for inflammatory pain (Ugawa et al 2002).
Serotonin
Mast cells, on degranulation, release platelet-activating factor, which in turn leads to the release of serotonin (5-hydroxytryptamine [5-HT]) from platelets. Serotonin causes pain when applied to a human blister base (Richardson and Engel 1986) and can activate nociceptors (Lang et al 1990). Serotonin can also potentiate the pain induced by bradykinin and enhance the response of nociceptors to bradykinin. Additional evidence for a role of 5-HT in nociception stems from observations that 40% of lumbar DRG neurons, mostly small to medium-sized cells, are immunoreactive for the 5-HT2A receptor and many of these cells also express the TRPV1 receptor (Van Steenwinckel et al 2009).
Histamine
Release of SP from nociceptor terminals can cause the release of histamine from mast cells. Histamine can lead to a variety of responses, including vasodilatation and edema. The role of histamine in pain sensation is less clear since application of exogenous histamine to the skin produces itch and not pain sensations (Simone et al 1991a). Histamine excites polymodal visceral nociceptors, especially when applied in high concentrations (Koda et al 1996), and potentiates the responses of nociceptors to bradykinin and heat (Mizumura et al 1995). Mechanosensitive cutaneous nociceptors in rats and humans respond only weakly to histamine (Lang et al 1990), but a subpopulation of mechano-insensitive C fibers was vigorously excited by histamine (Schmelz et al 1997). Activation of histamine H3 receptors, a ligand-gated ion channel that modulates the influx of Na+, however, leads to decreased release of inflammatory peptides and reduced pain and inflammation (Cannon et al 2007).
Purines
During inflammation and tissue injury, purines such as adenosine and its mono- or polyphosphate derivatives (AMP, ADP, ATP) may be released or leak into the extracellular space and activate nociceptors (for review see Burnstock 2009). Platelets are a rich source of ATP, and aggregation of platelets or lysis of cells can lead to release of ATP. Adenosine and its phosphates have been reported to induce pain in a human blister base. Intra-arterial or intradermal injection of adenosine also causes pain, and intravenous/intracoronary infusion of adenosine induces angina-like symptoms (Sylvén et al 1986). In animals, adenosine enhances the response to formalin, presumably via the A2 receptor. Animals lacking the adenosine A2a receptor are hypoalgesic to heat stimuli (Ledent et al 1997).
A number of lines of evidence support the potential role of ATP as a peripheral mediator of pain. ATP is found at increased levels at sites of inflammation and can activate nociceptors. Psychophysical studies in humans indicate that iontophoresis of ATP into normal skin results in dose-related pain. ATP-induced pain is dependent on capsaicin-sensitive neurons; repeated topical application of capsaicin reduces the ATP-induced pain to 25% of normal. In addition, the ATP-induced pain is increased two- to three-fold when iontophoresed into skin made hyperalgesic by acute capsaicin treatment or by ultraviolet inflammation. Thus, in inflammatory conditions ATP may activate nociceptors and serve as an endogenous mediator of pain (Hamilton et al 2000). In human microneurographic studies, injection of ATP activated 60% of mechano-responsive and mechano-insensitive C-nociceptive fibers without sensitizing these fibers to mechanical or heat stimuli (Hilliges et al 2002).
Receptors for ATP have been found on primary sensory neurons both in the DRG and in the periphery. Multiple purinergic (P2) receptors have been suggested to be involved in pain signaling and modulation. ATP presumably activates nociceptive neurons in normal skin via the P2X3 receptor and the heteromeric P2X2/P2X3 receptor (Chen et al 1995, Lewis et al 1995, Cook et al 1997). Messenger RNA for most of the P2X receptors (1–6) has been found in DRG neurons. In particular, both mRNA for the P2X3 receptor and the receptor protein itself have been found in small-diameter neurons in the DRG. Local intradermal injection of agents activating P2X receptors results in dose-related pain behavior in rodents that is mediated by capsaicin-sensitive neurons (Bland-Ward and Humphrey 1997) and enhanced pain behavior in response to formalin (Sawynok and Reid, 1997). The proportion of C-fiber nociceptors responding and the magnitude of their response are increased by P2X agonists in inflamed skin. Activation of homomeric P2X3 receptors is thought to contribute to acute nociception and inflammatory pain, whereas activation of heteromeric P2X2/3 receptors appears to modulate the longer-lasting nociceptive sensitivity associated with nerve injury or chronic inflammation (Burnstock 2009).
Recently, it has been suggested that peripheral adenosine receptors may also be involved in the modulation of inflammatory pain. A1 adenosine receptors are expressed in DRG cells, and peripheral activation of these receptors results in a reduction in inflammatory hyperalgesia via interactions with the nitric oxide/cyclic guanosine monophosphate/protein kinase G intracellular signaling pathways (Lima et al 2010).
Cytokines
During inflammation, cytokines (e.g., interleukin-1β [IL-1β], tumor necrosis factor α [TNF-α], IL-6) are released by a variety of cells (e.g., macrophages, Schwann cells) and regulate the inflammatory response (see Miller et al 2009, Schaible 2010). Clinical studies have shown that TNF-α levels in synovial fluid are increased in painful joints (Shafer et al 1994). Treatment with antibodies against TNF-α has been reported to improve the symptoms accompanying rheumatoid arthritis, including pain (Elliott et al 1994). Studies in animals have demonstrated mechanical and thermal hyperalgesia after systemic or local injection of IL-1, IL-6, and TNF-α. Additionally, treatment with antiserum against TNF-α is able to inhibit or delay the onset of hyperalgesia in experimental models of inflammation (Woolf et al 1997).
Cytokines may excite nociceptors either by rapid alterations in the properties of ion channels expressed in sensory neurons; indirectly by stimulating the release of other mediators such as prostaglandins, neurotrophins, and ATP; and by longer-term changes resulting from new gene transcription. Direct excitation and sensitization of nociceptive afferent fibers to thermal and mechanical stimuli have been shown for IL-1β and TNF-α (Fukuoka et al 1994). When applied along a peripheral nerve, TNF-α induces ectopic activity in nociceptive afferent fibers (Sorkin et al 1997).
IL-6 in combination with its soluble IL-6 receptor can sensitize nociceptors to heat as evidenced by increased heat-evoked intradermal release of CGRP (Obreja et al 2002). Other cytokines, IL-1β and TNF-α, also produce transient sensitization of heat-evoked release of CGRP from nociceptors in rat skin (Oprée and Kress 2000). IL-6–deficient mice show reduced mechanical and thermal hyperalgesia following inflammation (Xu et al 1997). These studies provide evidence for a role of cytokines in inflammation-associated hyperalgesia. The sensitization of nociceptors by cytokines may be mediated by p38-induced phosphorylation of TTX-resistant sodium channels, as well as by up-regulation of TRPV1 expression and function (Jin and Gereau 2006; for review see Ma and Quirion 2007).
Excitatory Amino Acids
A number of excitatory amino acids (EAAs) and peptide receptors are present at post-synaptic sites in the dorsal horn. These receptors have been found on DRG cells and the presynaptic terminals of primary afferents and are considered to play a role in the modulation of nociceptive impulses (see Carlton 2001, Goudet et al 2009). The most studied EAA, glutamate, can act either through ligand-gated ion channels (ionotropic glutamate receptors [iGluRs]) or through G protein–coupled metabotropic receptors (mGluRs). Based on sequence homology and physiological and pharmacological properties, the mGluRs have been further divided into three groups—group I (mGluR 1 and 5), group II (mgluR 2 and 3), and group III (mGluR 4, 6, 7, and 8). iGluR, mGluR1, and mGluR5 receptors have been identified on unmyelinated axons in the skin (Bhave et al 2001, Zhou et al 2001). About 40% of lumbar DRG cells contain mGluR2/3 immunoreactivity, and a majority of these cells are IB4+ small cells.
Several lines of evidence indicate a role of peripheral mGluRs in nociception and inflammatory pain. Peripheral application of glutamate activates nociceptors, and peripheral administration of ligands binding to glutamate receptors induces pain behavior in animals. Involvement of peripheral iGluR, mGluR1, and mGluR5 in formalin-induced pain behavior and glutamate-induced thermal hyperalgesia has been demonstrated (Davidson et al 1997). Intraplantar, but not intrathecal or intracerebroventricular administration of an mGluR5 antagonist reduced inflammatory hyperalgesia. Neurons in the DRG can be double-labeled with antisera for mGluR5 and VR1, thus suggesting that mGluR5 is expressed on the peripheral terminals of nociceptive neurons and contributes to inflammatory hyperalgesia (Walker et al 2001). In particular, mGluR1 activates PLC, which leads to release of Ca2+ from intracellular stores and activation of PKC.
Endogenous sources of glutamate in the periphery include plasma, macrophages, epithelial and dendritic cells in the epidermis and dermis, and Schwann cells. In addition, peripheral processes of the primary afferents contain glutamate, and nociceptor stimulation can cause peripheral release of glutamate from the terminals of these afferents.
Peripheral mGluRs are also considered to have antinociceptive effects. Peripheral administration of group II mGluR agonists blocks PGE2-induced thermal hyperalgesia, and activation of these receptors results in depression of the responses of nociceptors sensitized by exposure to formalin or inflammatory soup (Yang and Gereau 2002, Du 2008). These observations suggest that selective group II agonists may be a therapeutic target for inflammatory pain states.
Nerve Growth Factor
NGF may contribute to inflammatory pain via direct and indirect mechanisms (for review see Pezet and McMahon 2006, Watson et al 2008). Pro-inflammatory cytokines stimulate the release of NGF from various sources, including fibroblasts, keratinocytes, Schwann cells, and inflammatory cells (lymphocytes, macrophages, and mast cells). NGF stimulates mast cells to release histamine and serotonin. NGF can also induce heat hyperalgesia by acting directly on the peripheral terminals of primary afferent fibers (Chuang et al 2001). Transgenic animals modified to overexpress NGF show hyperalgesic pain behavior (Davis et al 1993a). NGF sensitizes nociceptors and may alter the distribution of Aδ fibers such that a greater proportion of fibers have nociceptor properties (Stucky et al 1999). NGF has been implicated in the inflammation-induced changes in nociceptor response properties, such as an increase in the incidence of ongoing activity, increase in the maximum fiber following frequency, and changes in the configuration of the action potential of DRG neurons (Djourhi et al 2001). The inflammation-induced changes in nociceptive neurons are prevented by sequestration of NGF (Koltzenburg et al 1999). Cultured DRG neurons from inflamed animals exhibit spontaneous activity, and cultured DRG neurons from non-inflamed animals exhibit spontaneous activity when cultivated for 1 day with NGF (Kasai and Mizumura 2001). These studies suggest that in inflamed rats NGF may play a role in inducing spontaneous activity in DRG neurons.
NGF modulates the activity of ligand- and voltage-gated ion channels involved in nociception, such as TRPV1, P2X3, ASIC3, and Nav1.8. NGF potentiates responses of the TRPV1 receptor (see the section on the vanilloid receptors), and NGF-induced hyperalgesia is absent in TRPV1 knockout mice (Chuang et al 2001). NGF-induced hyperalgesia may be mediated via its actions on the TTX-resistant sodium channel Nav1.8. NGF-induced thermal hyperalgesia failed to develop in mice with a mutation in the Nav1.8 gene (Kerr at al 2001). Binding of NGF to TrkA stimulates the mitogen-activated protein kinase (MAPK), phosphatidyl-3′-kinase (PI3K), and PLC-γ intracellular signal transduction pathways (for details see Cheng and Ji 2008). Potential clinical therapeutic approaches being explored include humanized monoclonal antibodies to NGF or its tyrosine kinase receptor TrkA and sequestration of NGF via soluble receptor protein that binds NGF.
Other Receptors
A number of other receptor systems have been reported to play a role in the peripheral modulation of nociceptor responsiveness.
Vanilloid Receptors
The vanilloid receptor TRPV1 (also known as VR1) is present on a subpopulation of primary afferent fibers and is activated by capsaicin, heat, and protons (see Chapter 2). Following inflammation, axonal transport of TRPV1 mRNA is induced, the proportion of TRPV1-labeled unmyelinated axons in the periphery is increased by almost 100% (Carlton and Coggeshall 2001), and the sensitivity of DRG neurons and primary afferent fibers to capsaicin increases (Nicholas et al 1999, Tohda et al 2001). Certain inflammatory mediators, such as bradykinin, lower the threshold of TRPV1-mediated heat-induced currents in DRG neurons and increase the proportion of DRG cells that respond to capsaicin (Stucky et al 1998, Sugiura et al 2002). NGF also potentiates the responses of TRPV1, and NGF-induced thermal hyperalgesia is absent in TRPV1 knockout mice. These observations, along with other experiments performed in mice lacking TRPV1, indicate that this channel protein plays a critical role in inflammation-induced heat hyperalgesia (Caterina et al 2000, Davis et al 2000).
Inflammatory mediators activate or sensitize TRPV1 through a diverse array of second-messenger pathways. For example, the thermal hyperalgesia induced by bradykinin and NGF is thought to be mediated, in part, by PLC-dependent phosphorylation of TRPV1 by PKC. Activation of PLC also leads to hydrolysis of the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) and consequent reversal of TRPV1 disinhibition by that lipid (Chuang et al 2001). A PIP2 binding site that is critical for the thermal sensitivity of TRPV1 has been identified (Prescott and Julius 2003). Functional coupling between protein kinase A (PKA) and TRPV1 also appears to play an important role in inflammatory hyperalgesia (Rathee et al 2002, Distler et al 2003). Finally, some inflammatory mediators activate TRPV1 indirectly via the production of fatty acid agonists (Shin et al 2002). For instance, bradykinin, acting at B2 receptors, excites cutaneous nociceptors via production of the 12-lipoxygenase metabolite of arachidonic acid 12-hydroperoxyeicosatetraenoic acid (12-HPETE), which in turn acts as a TRPV1 agonist.
Endothelin Receptors
Endothelins are vasoactive peptides that are widely distributed in somatic and visceral tissue (for reviews see Hans et al 2009, Khodorova et al 2009). Endothelin-1 (ET-1) is synthesized and released by endothelial cells, as well as by leukocytes and macrophages, and acts via GPCRs—ETA and ETB. ETA receptors are found in a large proportion of small cells in DRGs. ETB receptors are expressed mainly in keratinocytes, DRG satellite cells, and Schwann cells and may induce the synthesis and release of PGE2. Peripheral administration of ET-1 results in hyperalgesia that is attenuated by ETA antagonists. ET-1 also potentiates the effects of other algogens such as PGE2, capsaicin, and formalin. Activation of ETA receptors on neurons results in enhanced function of TRPV1 and TTX-resistant Na channels and an increase in intracellular Ca2+ levels, which in turn activates PKC and other second-messenger systems and leads to enhanced excitability of nociceptors. Endothelins have been implicated in the pain and hyperalgesia associated with inflammation, skin incision, cancer, and sickle cell crisis. ETB receptors have been reported to mediate both pro- and antinociceptive effects. Activation of ETB receptors on keratinocytes results in the release of β-endorphins, which inhibit nociceptor activity by binding to opioid receptors on the peripheral terminals of nociceptors (Khodorova et al 2003).
Peripheral Modulators of Nociceptor Activity
GPCRs, present on the plasma membrane and terminals of nociceptive neurons, play an important role in the modulation of pain signaling. GPCRs involved in antinociceptive mechanisms include opioid, cannabinoid, SST, muscarinic acetylcholine, γ-aminobutyric acid (GABAB), mGlu, and α2-adrenergic receptors (Fig. 1-13, for review see Pan et al 2008). Most GPCR agonists that have antinociceptive action are coupled to Gi/o proteins, which modulate voltage-gated Ca2+ channels and result in a decrease in presynaptic Ca2+ entry and inhibition of neurotransmitter release. GPCRs also modulate an inwardly rectifying K+ channel, the GIRK channel, which plays a critical role in maintaining resting membrane potential and excitability.
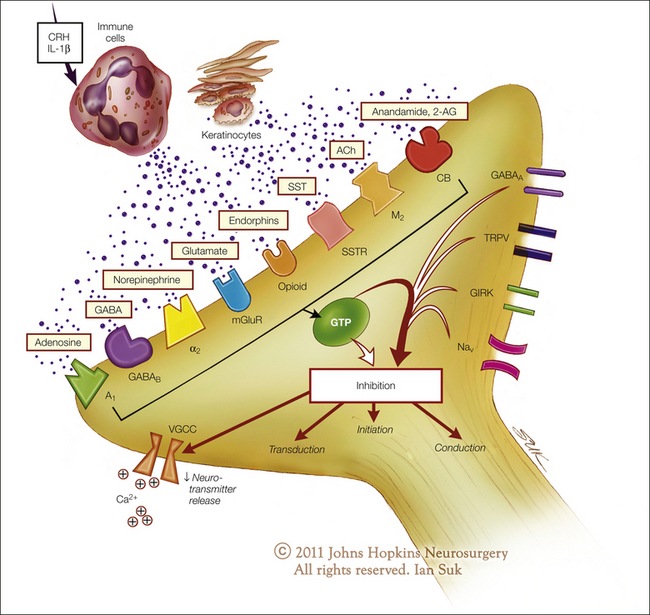
Figure 1-13 Potential peripheral modulatory mechanisms of nociceptor activity.
Several metabotropic G protein–coupled receptors (GPCRs) may play a role in inhibition of the initiation, transduction, or conduction of pain signals from peripheral nociceptive terminals. These GCPRs include opioid, cannabinoid (CB), somatostatin (SSTR), muscarinic acetylcholine (M2), γ-aminobutyric acid B (GABAB), metabotropic glutamate (mGluR), adenosine 1 (A1), and α2-adrenergic (α2) receptors. Activation of these GCPRs by their endogenous ligands leads to inhibition of voltage-gated Ca2+ channels (VGCCs), which results in a decrease in presynaptic Ca2+ entry and inhibition of neurotransmitter release. GPCRs also modulate an inwardly rectifying K+ channel, the GIRK channel, which plays an important role in maintenance of the duration and excitability of the resting membrane potential. GPCRs also regulate the function and kinetics of ion channels involved in sensory transduction, such as the transient receptor potential vanilloid (TRPV) channels and sodium channels (Nav). (Artwork by Ian Suk, Johns Hopkins University.)
The GPCRs on peripheral nociceptors are attractive potential therapeutic targets for the development of new drugs that may have some benefit, in contrast to the more traditional analgesics, which work at the level of the CNS. Drugs acting at the periphery and inhibiting the generation and signaling of nociceptive input toward the spinal cord and the brain may prevent central plastic changes such as wind-up and central sensitization. In addition, these drugs may provide analgesia without the undesirable adverse effects, such as sedation, dizziness, and cognitive dysfunction, associated with drugs acting on the CNS system (see Stein et al 2009).
Opioids
Besides their central analgesic action, morphine and other opioids produce analgesia in inflamed tissues by a peripheral mechanism (see Stein et al 2009). Opioid receptors have been demonstrated on the peripheral terminals of afferent fibers, and axonal transport of these receptors is enhanced during inflammation. Peripheral analgesia by opioids appears to be part of a physiological antinociceptive system since increased amounts of endogenous opioids have been found in inflamed tissues. Inflammatory cells such as macrophages, monocytes, and lymphocytes contain opioid peptides. Release of endogenous opioids and antinociception can be induced by IL-1β and corticotropin-releasing hormone (CRH) originating from the inflamed tissue.
An alternative mechanism for activation of endogenous opioid analgesia at the site of tissue injury has been described (see Khodorova et al 2009 for review). ET-1, a potent vasoactive peptide, is synthesized and released by epithelia after tissue injury. Although ET-1 can trigger pain by activating ETA receptors on nociceptors, it also has an analgesic effect through its actions on ETB receptors. Activation of ETB receptors on keratinocytes by ET-1 results in the release of β-endorphins and analgesia mediated via peripheral μ- and κ-opioid receptors linked to GIRKs (see Fig. 1-13).
Cannabinoids
Cannabinoids have recently emerged as a potential therapy for chronic pain. Clinical use of non-selective cannabinoids is, however, limited by their CNS actions, which lead to psychotropic effects, temporary memory impairment, and dependence. The endocannabinoid system includes the two cloned metabotropic receptors CB1 and CB2, possibly the orphan receptor GPR55, and the endogenous ligands anandamide and 2-arachidonoylglycerol. CB1 and CB2 receptors are GPCRs expressed in neural and non-neural immune cells. They are distributed at many key sites in the pain-signaling pathway, including the peripheral and central terminals of primary afferent fibers, spinal dorsal horn neurons, and the brain stem and brain. CB1 and CB2 mRNA and protein are widely expressed in the majority of DRG nociceptive neurons (Agarwal et al 2007), and their expression has been shown to be up-regulated following inflammation (Amaya et al 2006) and nerve injury (Beltramo et al 2006, Mitrirattaanakul et al 2006). Multiple lines of evidence suggest that the analgesic effects of CB1 and CB2 agonists may be mediated via their actions on nociceptive primary afferents. Cannabinoids regulate the function and kinetics of ion channels involved in sensory transduction, such as the TRP channels (e.g., TRPV1, TRPA1, TRPM8) and purinergic ion channels (P2X2, P2X2/3), as well as channels that directly affect neuronal excitability (various K+ and Ca2+ channels). Studies in animal models suggest that peripheral CB1 and CB2 receptors may be important targets in controlling the pain associated with inflammation, neuropathy, and bone cancer (see Anand et al 2009, Kress and Kuner 2009 for reviews). CB receptor agonists also enhance the analgesic effects of opioid agonists and non-steroidal anti-inflammatory drugs in experimental pain models.
Somatostatin
SST is a regulatory peptide that is widely distributed in neural and non-neural cells such as immune cells, fibroblasts, and neuroendocrine cells. Found in a subpopulation of capsaicin-sensitive peptidergic DRG neurons, SST binds to G protein–coupled membrane receptors. Activation of SST receptors opens various K+ channels and inhibits voltage-gated Ca2+ channels, which results in its anti-inflammatory and analgesic effects. SST decreases the release of peptides such as SP and CGRP from sensory nerve endings in the periphery and reduces neurogenic inflammation (for review see Pinter et al 2006). The analgesic effects of SST are thought to result from inhibition of the TRPV1 ion channel (Carlton et al 2004) and possibly via an interaction with opioid receptors. Intraplantar administration of the SST receptor agonist octreotide reduces the phase II response after formalin injection, decreases the response of CMHs to heat stimuli, and attenuates the thermal responses of nociceptors sensitized by bradykinin. Endogenous release of SST from nociceptive afferents is considered to play a modulatory role in inflammatory and neuropathic pain. Intra-articular injection of SST into the knee resulted in pain relief in patients with osteoarthritis and rheumatoid arthritis. Synthetic SST agonists may have potential as anti-inflammatory and analgesic drugs.
Cholinergic Receptors
Non-neuronally released acetylcholine, acting on peripheral cholinergic receptors, may have a modulatory role on nociception. Nicotine has a weak excitatory effect on C-fiber nociceptors and induces mild sensitization to heat, but no alterations in mechanical responsiveness. In contrast, muscarine desensitizes C nociceptors to mechanical and heat stimuli (Bernardini et al 2001). Thus, nicotinic and muscarinic receptors may have opposing effects on cutaneous nociceptors. Studies in mice with targeted deletions of the M2 receptor gene suggest that M2 receptors on cutaneous nerve endings depress the responsiveness of nociceptive fibers to noxious stimuli (see Wess et al 2003 for review). High levels of expression of M2 mRNA and considerably lower levels of M3 and M4 mRNA are detected in medium-sized and small DRG neurons in the rat (Tata et al 2000). M2, M3, and M4 muscarinic receptor subtypes may be involved in the modulation of nociceptive transduction.
γ-Aminobutyric Acid Receptors
The inhibitory neurotransmitter GABA activates both ionotropic (GABAA and GABAC) and metabotropic (GABAB) receptors. GABAA receptors have been found in DRG cells and on their central terminals in the dorsal horn. GABAA receptors have been reported to be present in 10–14% of the unmyelinated primary afferent axons in the glabrous skin of the cat (Carlton et al 1999). Behavioral studies suggest a bimodal effect of GABAA receptors on the modulation of peripheral nociceptive transmission; a low concentration of GABAA agonists attenuates and a high concentration enhances formalin-induced pain behavior. GABAB receptors are also present in primary afferents, and GABAB mRNA is expressed in DRG cells (Towers et al 2000). Degeneration of primary afferent fibers by administration of capsaicin to neonatal rats decreases GABAB receptor density by 50%, thus indicating that these receptors are localized in TRPV1-expressing nociceptive afferents (Price et al 1987). Activation of the GABAB receptor by agonists such as baclofen inhibits neuronal excitability by inhibition of N-type Ca2+ currents and potentiation of voltage-dependent K+ currents (Takeda et al 2004).
α2-Adrenoceptors
Traditionally, the analgesic effects of α2-adrenergic agonists, such as clonidine and dexmedetomidine, are thought to be secondary to their actions in the CNS (for review see Pertovaara 2006). However, peripheral α2-adrenoceptors may also be involved in modulation of nociceptor activity. Studies using selective α2-subtype knockout mice have shown that the α2A-adrenergic receptors are primarily involved in the analgesic effect of α2-adrenoceptor agonists (Stone et al 1997). Selective removal of TRPV1-expressing sensory neurons induces a large decrease in α2A- but not in α2C-adrenoceptors in the spinal dorsal horn, which suggests that α2A-adrenoceptors are located on the central terminals of primary afferent neurons whereas the α2C subtype is located primarily on spinal dorsal horn neurons (Stone et al 1998, Chen et al 2007). α2-Adrenergic agonists may inhibit the depolarization-induced Ca2+ influx and induce a GIRK current in nociceptors.
Second Messengers and Signal Transduction Pathways
As described above, inflammation is associated with the release of a host of chemical mediators (Fig. 1-12). Although some of these agents may directly activate nociceptors, most of the inflammatory mediators lead to changes in the sensory neuron rather than directly activating it. Such changes in sensory neurons include early post-translational alterations in the peripheral terminals of nociceptors (peripheral sensitization) and a delayed transcription-dependent alteration (see Woolf and Costigan 1999, Kidd and Urban 2001). Peripheral sensitization can be the result of changes in the transducer molecule (e.g., TRPV1 receptor) or in voltage-gated ion channels (e.g., sodium channels) secondary to the phosphorylation of membrane-bound proteins. Inflammation can also induce delayed and longer-lasting transcription-dependent changes in effector genes in DRG cells as a result of electrical activity and retrograde transport of specific signal molecules such as NGF. An increase in intracellular calcium induced by electrical activity activates a host of intracellular transcription factors such as the cAMP-response element–binding protein (CREB; Ji and Rupp 1997).
Considerable attention has been focused on the signal transduction mechanisms of primary afferent neurons and their alteration by inflammation. Two principal signaling pathways have been postulated to mediate inflammation-induced hyperalgesia. Inflammatory mediators such as PGE2, serotonin, and adenosine activate PKA (Gold et al 1998), whereas NGF, bradykinin, and epinephrine induce hyperalgesia in part by activating PKA but also through an ε isozyme of PKC (Khasar et al 1999). PKA and PKC sensitize nociceptors to heat by modulating the activity of TTX-resistant sodium currents. As described above, these signaling pathways also interact with the heat transducer TRPV1, which results in sensitization of the receptor to heat.
MAPKs are also reported to be involved in the transduction of extracellular stimuli (e.g., signals from extracellular growth factors such as NGF) into diverse intracellular responses and neuronal plasticity. Three subfamilies of MAPKs have been well characterized—the extracellular signal–regulated kinases (ERKs), the c-Jun amino-terminal kinases (JNKs), and the p38 enzymes. ERK is present in primary afferent neurons, is phosphorylated by nociceptive stimuli, and is thought to play a role in inflammatory hyperalgesia (Dai et al 2002). Inflammation also activates p38 in the soma of C-fiber nociceptive cells in the DRG (Ji et al 2002). Inhibiting the activation of p38 in the DRG reduces the inflammation-induced increase in TRPV1 receptors in the DRG and attenuates heat hyperalgesia. Activation of p38 in the DRG is dependent on peripheral production of NGF during inflammation. Thus, MAPKs and NGF play important regulatory roles in TRPV1 receptor expression and maintenance of heat hyperalgesia after inflammation.
Postoperative Pain and Hyperalgesia
The pain resulting from different tissue injuries may differ in its characteristics and mechanisms. Postoperative, incisional pain is a unique but common form of acute pain. Studies in rodents have characterized the primary hyperalgesia to mechanical and thermal stimuli caused by a surgical incision (Brennan et al 1996, Pogatzki and Raja 2003). Primary hyperalgesia to mechanical stimuli lasts for 2–3 days, whereas hyperalgesia to heat lasts longer—6–7 days after plantar incision. As with other types of tissue injury, secondary hyperalgesia after incision injury is present only to mechanical, not thermal, stimuli (Pogatzki et al 2000). The incision-induced primary and secondary hyperalgesia results from characteristic peripheral, spinal, and supraspinal mechanisms (Zahn and Brennan 1999, Pogatzki et al 2002). The conversion of mechanically insensitive “silent nociceptors” to mechanically responsive fibers may play an important role in the maintenance of primary mechanical hyperalgesia (Pogatzki et al 2002). Release of ATP from injured cells is considered to play an important role in the induction of mechanical allodynia after a skin incision (Tsuda et al 2001).
The incision-induced spontaneous activity in primary afferent fibers plays a critical role in maintaining wide–dynamic range neurons in the dorsal horn in a sensitized state. In contrast to the central mechanisms of hyperalgesia following other forms of cutaneous injury where N-methyl-D-aspartate (NMDA) receptors play a critical role, the hyperalgesia that results from an incision is characterized by distinct pharmacological mechanisms that are not dependent on NMDA receptors.
Role of the Sympathetic Nervous System in Inflammation
Nociceptors normally do not respond to sympathetic stimulation. In addition, sympathectomy plus depletion of catecholamine stores with reserpine has no effect on acute inflammation. In contrast, sympathectomy reduces the severity of injury in chronic adjuvant-induced arthritis (see Raja 1995, Jänig et al 1996 for reviews). Inflammation may lead to catechol sensitization of cutaneous nociceptors. Sympathetic stimulation and close arterial injection of norepinephrine (NE) also excite 35–40% of C-polymodal nociceptors in chronically inflamed rats (Sato et al 1993). This adrenergic activation of nociceptors was blocked by α2- but not by α1-adrenergic antagonists. Sympathetic efferent fibers are also thought to play a role in neurogenic inflammation.
In human skin sensitized by the topical application of capsaicin, hyperalgesia persists longer at sites where exogenous NE was administered, and this α-adrenoceptor–mediated effect was independent of the vasoconstrictor response (Drummond 1995, 1996). Additionally, local administration of an α-adrenergic antagonist reduced the spontaneous pain and hyperalgesia resulting from the intradermal injection of capsaicin (Kinnman et al 1997). However, physiological modulation of sympathetic vasoconstrictor activity by whole-body warming or cooling does not alter the intensity or spatial distribution of capsaicin-evoked spontaneous pain and mechanical hyperalgesia (Baron et al 1999). Anatomical studies indicate that SP and NMDA receptor mRNA is up-regulated in preganglionic sympathetic neurons after paw inflammation in rats (Ohtori et al 2002). These changes are postulated to possibly be evidence of a role of the sympathetic nervous system in inflammatory hyperalgesia.
Secondary Hyperalgesia
An understanding of secondary hyperalgesia is important not only with regard to understanding the neural mechanisms of acute pain but also with regard to understanding many aspects of chronic pain. In this section we consider the nature of secondary hyperalgesia and its possible peripheral and central mechanisms.
Secondary Hyperalgesia to Mechanical but Not Heat Stimuli
Primary hyperalgesia is characterized by the presence of enhanced pain in response to heat and mechanical stimuli, whereas secondary hyperalgesia is characterized by enhanced pain in response to only mechanical stimuli (e.g., Ali et al 1996). In one study in which the sensory changes that occur in the zones of primary and secondary hyperalgesia were compared (Raja et al 1984), burn injuries were induced in two locations on the glabrous skin of the hand in human subjects (Fig. 1-14). Within minutes of the injury, lightly touching the skin at the site of the two burns, as well as in a large area surrounding the burns, caused pain. The decrease in the pain threshold to von Frey hairs in the primary (injured) zone was similar to that in the area of secondary hyperalgesia (Fig. 1-14B). Marked hyperalgesia to heat was observed in the area of primary hyperalgesia (site A, the injury site, Fig. 1-14C). In the uninjured region between the two burns, however, the painfulness of the heat stimuli actually decreased (Fig. 1-14D). Notably, the area between the burns was hypo-algesic to heat while being hyperalgesic to mechanical stimuli.
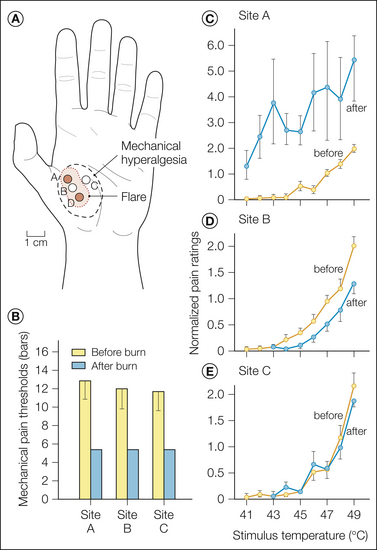
Figure 1-14 Hyperalgesia to mechanical and heat stimuli develops at the site of injury (zone of primary hyperalgesia), whereas hyperalgesia to mechanical but not heat stimuli develops in the uninjured area surrounding an injury (zone of secondary hyperalgesia).
A, Two burns (53°C, 30 seconds) were applied to the glabrous skin of the hand (sites A and D). Mechanical thresholds for pain and ratings of pain in response to heat stimuli were recorded before and after the burns at one of the injury sites (site A), in the uninjured skin between the two burns (site B), and at an adjacent site (site C). The areas of flare and mechanical hyperalgesia following the burns in one subject are also shown. In all subjects, the area of mechanical hyperalgesia was larger than the area of flare. Mechanical hyperalgesia was present even after the flare disappeared. B, Mean mechanical thresholds for pain before and after burns. The mechanical threshold for pain was significantly decreased following the burn. The mechanical hyperalgesia was of similar magnitude at each of the three test spots (A, B, C). C–E, Mean normalized ratings of the painfulness of heat stimuli (same as described in Fig. 1-5) before and after burns. C, At burn site A, all the characteristics of heat hyperalgesia (i.e., decrease in pain threshold, increased pain in response to suprathreshold stimuli, and spontaneous pain) were observed after the burns. D, In the uninjured area between the two burns (site B), pain ratings decreased after the burns. Thus, heat hypalgesia was observed. E, At site C, pain ratings before and after the burns were not significantly different. (Note that a different scale is used in C.) (Reproduced with permission from Raja SN, Campbell JN, Meyer RA 1984 Evidence for different mechanisms of primary and secondary hyperalgesia following heat injury to the glabrous skin. Brain 107:1179–1188.)
Spreading Sensitization of Nociceptors Does Not Occur
Activation of nociceptors leads to a flare response (discussed in more detail below). This response is neurogenic in the sense that it depends on intact innervation of the skin by nociceptors. The flare response extends well outside the area of initial injury. One explanation for the flare response is that it involves spreading activation of nociceptors. Activation of one nociceptor leads to the release of chemicals that activate neighboring nociceptors, which leads to further release of chemicals and activation of additional nociceptors. Lewis (1942) believed that a similar mechanism, which he termed spreading sensitization, accounted for secondary hyperalgesia. Activation and sensitization of one nociceptor lead to spread of this sensitization to another nociceptor, possibly because of the effects of a sensitizing substance released from the nociceptor initially activated. Another theoretical possibility is that coupling between nociceptors increases after injury.
Several lines of evidence indicate that spreading sensitization does not occur:
Other differences exist between flare and secondary hyperalgesia (LaMotte et al 1991):
Central Mechanisms of Secondary Hyperalgesia
If peripheral sensitization does not account for secondary hyperalgesia, the mechanisms noted in Figure 1-10C–F should be examined in the CNS. Indeed, it has been relatively easy to demonstrate enhanced responsiveness of CNS neurons to mechanical stimuli after cutaneous injury (e.g., Simone et al 1991b). Substantial evidence favors the following important tenet: the peripheral signal for pain does not reside exclusively with nociceptors. Under pathological circumstances, other receptor types, which are normally associated with the sensation of touch, acquire the capacity to evoke pain. This principle applies not only to secondary hyperalgesia but also to neuropathic pain states in general. This condition arises in part through augmentation of the responsiveness of central pain-signaling neurons to input from low-threshold mechanoreceptors, a phenomenon often termed central sensitization.
Many of the insights acquired about secondary hyperalgesia have been gained from studies with capsaicin. Investigators have been drawn to the use of capsaicin as the “injury” stimulus for several reasons:
LaMotte and colleagues performed a number of pivotal experiments to determine the relative importance of peripheral and central sensitization in secondary hyperalgesia (LaMotte et al 1991). To test whether peripheral nerve fibers are sensitized, capsaicin was administered under conditions of a proximal nerve block, and the magnitude of hyperalgesia was determined after the effects of the anesthetic dissipated. When the relevant nerve is blocked proximal to the capsaicin injection site, the CNS is spared the nociceptive input generated at the time of injection. The effects of capsaicin on the peripheral nervous system are not affected (e.g., a flare develops) since the nerve block is proximal to the area of capsaicin application. Figure 1-15 shows the results of this experiment in one subject. No hyperalgesia was present after the block had worn off. Thus, when the CNS is spared the input of nociceptors at the time of the acute insult, hyperalgesia does not develop (LaMotte et al 1991, Pedersen et al 1996).
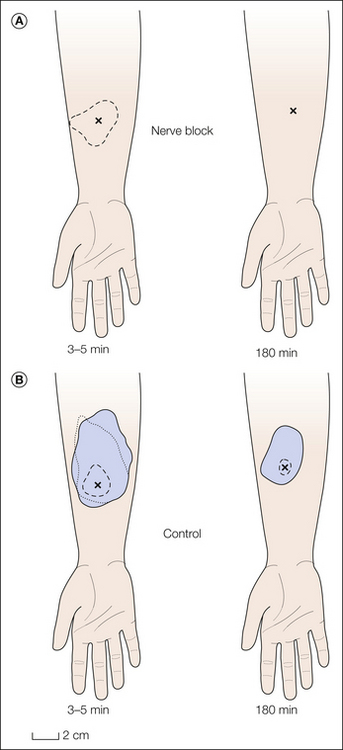
Figure 1-15 A proximal nerve block prevents the development of secondary hyperalgesia.
A, After blockade of the lateral antebrachial nerve with 1% Xylocaine, capsaicin (100 μg in 10 μL) was injected into the anesthetic skin. A flare (dashed line) developed within 5 minutes. No hyperalgesia was present 180 minutes after the capsaicin injection when the local anesthetic block had dissipated. B, On the control arm, normal flare and hyperalgesia in response to stroking (dotted line) and punctate (solid line) stimuli developed within 5 minutes. Hyperalgesia to punctate stimuli was still present 180 minutes after the capsaicin injection. (Adapted from LaMotte RH, Shain CN, Simone DA, et al 1991 Neurogenic hyperalgesia: psychophysical studies of underlying mechanisms. Journal of Neurophysiology 66:190–211.)
Additional evidence that central sensitization, not peripheral sensitization, plays a major role in secondary hyperalgesia includes the following:
Different Mechanisms for Stroking and Punctate Hyperalgesia
Two distinct forms of mechanical hyperalgesia are observed in the zone of secondary hyperalgesia: punctate hyperalgesia and stroking hyperalgesia. Hyperalgesia to blunt pressure is not observed in the secondary zone (Koltzenburg et al 1992). We will first consider stroking hyperalgesia (also called allodynia). Stroking hyperalgesia appears to be mediated by activity in low-threshold mechanoreceptors. When a pressure cuff was used to selectively block myelinated fibers, the pain in response to stroking disappeared at a time when touch sensation was lost but heat and cold sensations were still present (LaMotte et al 1991, Koltzenburg et al 1992). This is also true in patients with stroking hyperalgesia from neuropathic pain (Campbell et al 1988b). In another series of experiments, Torebjörk and colleagues (1992) performed intraneural microstimulation in awake human subjects. As shown in Figure 1-16, stimulation of primary afferent fibers normally concerned with tactile sensibility evoked pain when (but not before) secondary hyperalgesia was produced.
Figure 1-16 Microneurographic evidence that large-diameter myelinated fibers are involved in the pain observed in the zone of secondary hyperalgesia.
A, Intraneural electrical stimulation of the superficial peroneal nerve at a fixed intensity and frequency evoked a purely tactile (non-painful) sensation projected to a small area of skin on the dorsum of the foot (dark blue area). B, After intradermal injection of capsaicin (100 μg in 10 μL) adjacent to the projected zone (at the site indicated by the open circle), a zone of secondary hyperalgesia (indicated by light blue area) developed that overlapped the sensory projection field. Now, intraneural stimulation at the same intensity and frequency as in A was perceived as a tactile sensation accompanied by pain. C, When the zone of secondary hyperalgesia no longer overlapped the sensory projection field, the intraneural stimulation was again perceived as purely tactile, without any pain component. (Adapted from Torebjörk HE, Lundberg LER, LaMotte RH 1992 Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. Journal of Physiology [London] 448:765–780.)
Punctate hyperalgesia is manifested by heightened pain associated with the application of small, stiff, or sharp probes to the skin (e.g., von Frey monofilaments). Several lines of evidence indicate that punctate hyperalgesia has a different neural mechanism than stroking hyperalgesia does and is mediated by central sensitization to activity in nociceptors:
The pain in response to a controlled punctate stimulus does not vary significantly across the zone of secondary hyperalgesia but decreases precipitously at the border (Huang et al 2000). This suggests that the sensitization responsible for secondary hyperalgesia is an all-or-nothing phenomenon. In addition, subjects were able to grade the magnitude of pain from stimuli of different intensity. Interestingly, although the threshold for pain in response to punctate stimuli decreases in the zone of secondary hyperalgesia (Magerl et al 1998), the threshold for touch detection increases (Magerl and Treede 2004).
Model for Stroking Hyperalgesia
From the above we know that secondary hyperalgesia to stroking stimuli appears to be due to sensitization of central pain-signaling neurons to the input from low-threshold mechanoreceptors (Fig. 1-17). In normal skin, activity in low-threshold mechanoreceptors signals touch sensation (Fig. 1-17A). As a result of the barrage of activity in nociceptors, sensitization occurs in the CNS such that input from low-threshold mechanoreceptors gains access to the pain system (Fig. 1-17B). Now, light touching of the skin is painful. Plasticity in the response of second-order neurons in the dorsal horn appears to be a major factor that accounts for this central sensitization (see Chapter 6 for detailed discussion). However, another possibility that involves plasticity in primary afferents is that mechanoreceptors gain access to nociceptive neurons by means of a presynaptic link (Cervero et al 2003; see discussion below on primary afferent depolarization).
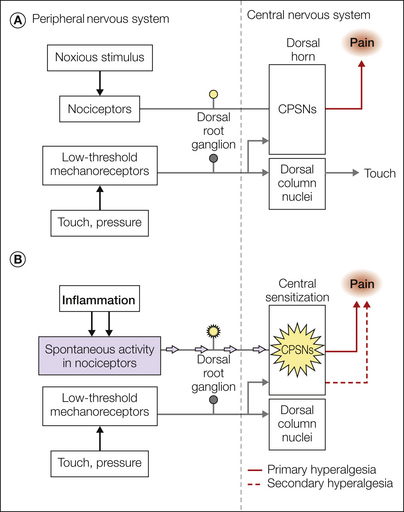
Figure 1-17 Central sensitization accounts for secondary hyperalgesia.
A, Nociceptors signal acute pain. Noxious stimuli selectively activate nociceptors that project to central pain-signaling neurons (CPSNs) in the spinal cord. The CPSNs project to higher centers, where pain is perceived. Low-threshold mechanoreceptors convey the sensation of touch. B, Injury or inflammation leads to the sensitization of primary afferent nociceptors. The enhanced responsiveness or sensitization of primary afferents accounts for primary hyperalgesia. Spontaneous activity also develops in the nociceptors and drives the development of sensitization of the CPSNs. This central sensitization involves enhanced connectivity between low-threshold mechanoreceptors and CPSNs. Now, signals from low-threshold mechanoreceptors gain access to the pain pathway, which leads to the development of secondary hyperalgesia to mechanical stimuli.
Model for Punctate Hyperalgesia
Punctate hyperalgesia appears to be mediated by central sensitization to nociceptor input. However, most nociceptors respond to heat stimuli. Why is there not hyperalgesia to heat stimuli in the secondary hyperalgesic zone? One possibility is that this central sensitization involves a mechano-specific channel. In this model, punctate hyperalgesia is mediated by mechano-specific nociceptive afferents that project via sensitized mechano-specific interneurons to central pain-signaling neurons. Support for this hypothesis comes from experiments in which the skin was pretreated with topical capsaicin to eliminate epidermal nerve fibers that are sensitive to heat (Nolano et al 1999). Such treatment led to a lack of pain in response to heat stimuli; however, secondary hyperalgesia to punctate stimuli developed after the injection of capsaicin into nearby untreated skin (Fig. 1-18; Fuchs et al 2000). Additional experiments with selective nerve fiber blocks revealed that the punctate hyperalgesia disappeared when Aδ fibers were blocked (Magerl et al 2001). Thus, punctate hyperalgesia appears to be signaled by Aδ-fiber afferents that are insensitive to capsaicin and heat.
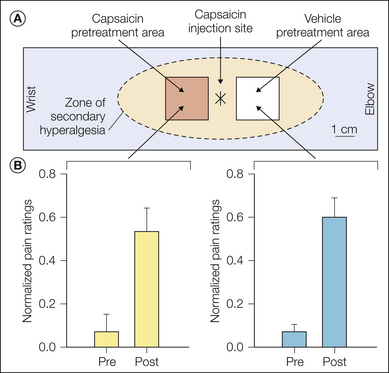
Figure 1-18 Secondary hyperalgesia to punctate mechanical stimuli occurs in skin that has been pretreated with topical capsaicin, which desensitizes unmyelinated, epidermal nerve fibers.
A, Capsaicin was applied to a 2 × 2-cm area on the volar aspect of the forearm to produce desensitization of the skin to heat stimuli. A nearby vehicle-treated area served as control. Two days later, capsaicin was injected intradermally between the two treatment areas and produced a large, symmetrical zone of secondary hyperalgesia to punctate mechanical stimuli. B, Pain ratings in response to a sharp probe increased dramatically 60 minutes after the capsaicin injection. The average pain ratings at the capsaicin pretreatment area (left panel) were not significantly different from those at the vehicle treatment area (right panel). (Adapted from Fuchs PN, Campbell JN, Meyer RA 2000 Secondary hyperalgesia persists in capsaicin desensitized skin. Pain 84:141–149.)
One well-studied form of central sensitization, termed wind-up, is characterized by a slowly increasing response of central neurons to repeated C-fiber stimulation at rates greater than 0.3 Hz (e.g., Mendell and Wall 1965). The perceptual correlate of wind-up is temporal summation (Price et al 1977). The finding that temporal summation does not change in the zone of secondary hyperalgesia argues against wind-up as a mechanism for secondary hyperalgesia (Magerl et al 1998).
Effect of Aging on Nociceptive Properties
Aging induces changes in the properties of unmyelinated nociceptive afferents (Namer et al 2009). Thus, the percentage of mechanosensitive afferents decreased whereas the percentage of mechano-insensitive afferents increased, and some fibers showed signs of sensitization, desensitization, and spontaneous activity, features previously observed in patients with neuropathic pain. In addition, changes in the conductive properties of nociceptive afferents were also observed. The mechanisms underlying these changes are unknown, but they may, for example, be due to a diminished supply of neurotrophic factors. It has been hypothesized that age-related changes could render nociceptive afferents more susceptible to neuropathy-inducing insults (Namer et al 2009).
Efferent and Trophic Functions of Nociceptors
Nociceptors, apart from signaling pain, serve regulatory and trophic functions. An efferent role for nociceptors was suggested by several investigators almost a century ago (see Lynn 1996 for historical review). Two efferent cutaneous phenomena have been considered to be dependent on the integrity of afferent nociceptive fibers and are part of the so-called neurogenic inflammation: vasodilatation, which becomes visible as a flare surrounding a site of injury, and plasma extravasation, which is manifested as a wheal at the site of injury. Several peptides have been identified in the peripheral terminals of sensory neurons, including SP and other tachykinins such as neurokinins A and K, CGRP, SST, and vasoactive intestinal polypeptide. The presence and release of SP and CGRP from capsaicin-sensitive sensory nerve endings in experimental animals, their ability to induce many of the signs of acute inflammation, including vasodilatation and plasma extravasation, and inhibition of neurogenic vasodilatation by selective neuropeptide antagonists suggest that they may be the principal mediators of neurogenic inflammation and axon reflexive flare. SP-induced vasodilatation and plasma extravasation may result from a direct effect on the vasculature or be due to release of histamine by degranulation of mast cells by SP. Differences in neurogenic inflammation and peptide release in rat and human skin have, however, been observed. Electrical stimulation results in release of CGRP and SP in rat and human skin. However, unlike rat skin, the endogenous release of peptides after strong chemical or electrical stimulation is not associated with neurogenic protein extravasation or release of mast cell mediators in human skin (see Schmelz and Petersen 2001 for review).
SP and CGRP are also reported to play a role in immunological processes (e.g., migration of leukocytes to sites of tissue injury), and they stimulate epidermal cells (e.g., keratinocytes and Langerhans cells) necessary for the maintenance and repair of skin integrity (for reviews see Maggi and Meli 1988, Holzer 1998). Other efferent actions of nociceptors that are mimicked by vasoactive neuropeptides are contraction of smooth muscles, stimulation of mucous secretion from airways, and leukocyte adhesion. Some efferent functions of nociceptors and the chemical mediators involved are shown in Figure 1-19.
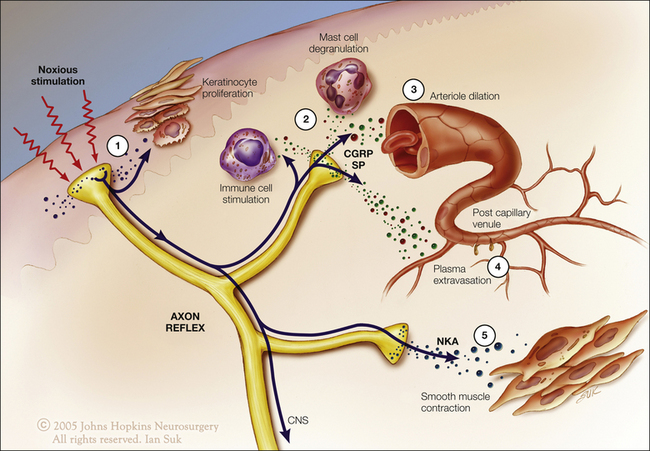
Figure 1-19 Efferent actions of nociceptors.
A noxious stimulus leads to action potentials in nociceptive fibers that propagate not only to the central nervous system but also antidromically into peripheral branches. These antidromic action potentials lead to the release of neuropeptides such as substance P, calcitonin gene–related peptide (CGRP), and neurokinin A (NKA). These substances can stimulate epidermal cells (1) and immune cells (2) or lead to vasodilatation (3), plasma extravasation (4), and smooth muscle contraction (5). (Artwork by Ian Suk, Johns Hopkins University.)
Flare is thought to be due to a peripheral axon reflex. Activation of one branch of a nociceptor by a noxious stimulus results in the antidromic invasion of action potentials into adjacent branches of the nociceptor, which in turn causes the release of vasoactive substances from terminals of the nociceptor. Capsaicin-sensitive A- and C-fiber nociceptors are thought to be involved in the flare reaction. However, the extent of the flare far exceeds the size of the receptive fields of conventional nociceptors. A possible explanation for this discrepancy is that the flare is mediated, at least in part, by a subpopulation of chemosensitive nociceptive fibers with large receptive fields. Some C-fibers with large, complex receptive fields have been reported (Meyer et al 1991, Schmelz et al 1997). Transcutaneous electrical stimulation studies in the skin of human volunteers suggest that the axon reflex flare, measured by laser Doppler imaging, was mediated via mechano-insensitive C-fiber nociceptors (Schmelz et al 2000a).
Several lines of evidence indicate that the neural substrates for vasodilatation and the perception of pain are different. (1) The magnitude of the vasodilatation induced by a noxious stimulus does not always increase with the intensity of pain (Koltzenburg and Handwerker 1994). (2) Low activity (<1 Hz) in C fibers can generate significant vasodilatation (Lynn and Shakhanbeh 1988), but in humans does not cause any conscious sensation. (3) Histamine can produce a large flare with little or no pain. Possible explanations are that different discharge patterns are needed for pain versus flare in a given fiber population or certain classes of afferents are better designed for flare than for pain and vice versa.
Similarly, pain sensations and plasma extravasation have independent mechanisms. The ability of inflammatory mediators such as bradykinin, histamine, and serotonin to induce plasma extravasation and excite nociceptors, as assessed by pain ratings and flare response, has been examined in humans. In healthy human skin, no clear relationship between nociceptor activation and plasma extravasation could be established. For example, bradykinin induced protein extravasation without pain or flare, and serotonin evoked pain and flare at concentrations that did not induce plasma extravasation. These observations suggest that the plasma extravasation induced by these mediators is mostly non-neurogenic in mechanism (Lischetzki et al 2001).
Antidromic activity involved in the effector responses can also originate from the spinal cord. A series of studies in a model of acute arthritis indicated that primary afferent input to the spinal cord activates multisynaptic central neuronal pathways, which in turn influence the development of neurogenic inflammation (for review see Sluka et al 1995). Activation of primary afferent fibers may result in depolarization of the central terminals of other afferent fibers (primary afferent depolarization [PAD]). If PAD is large enough (e.g., under peripheral inflammatory conditions), the depolarization can be sufficient to initiate action potentials at the central terminals that are conducted antidromically in the primary afferent fibers (dorsal root reflexes [DRRs]). It is postulated that the antidromic impulses (DRRs) triggered by PAD result in the release of neuropeptides in the joint from peripheral terminals of the afferents and contribute to the inflammatory process. DRRs have been recorded in C, Aδ, and Aβ fiber types in rat models of acute arthritis. The joint inflammation and the DRRs were attenuated by prior dorsal rhizotomy.
Cervero and Laird proposed that the DRRs may also explain secondary hyperalgesia (Cervero et al 2003). According to this hypothesis, the action potentials initiated in primary afferent fibers as a result of enhanced PAD propagate peripherally to produce flare and centrally to evoke pain sensation. As evidence to support this hypothesis, they reported that light stroking of the skin leads to an increase in blood flow in the zone of secondary hyperalgesia, but not in normal skin.
Nociceptive innervation of the skin has been suggested to also play a critical role in wound healing. Sensory denervation by capsaicin injection impairs cutaneous wound healing in rats (Smith and Liu 2002). Skin denervation decreases keratinocyte proliferation and leads to decreased skin thickness (Hsieh and Lin 1999). The role of cutaneous nociceptors in wound healing may be due to neuromodulatory actions of the sensory peptides SP and CGRP, which when injected at skin wound sites, promote wound healing in aged rats (Khalil and Helme 1996).
Nociceptors and Neuropathic Pain
A well-known axiom in the field of pain is that injury to the nociceptive pathways, whether it be in the peripheral nervous system or CNS, carries with it the liability that pain may result. This is paradoxical in the sense that lesions should, one would think, lead to deficits in function. The ongoing pain in patients is frequently associated with enhanced pain in response to natural stimuli, a phenomenon termed hyperalgesia. Hyperalgesia may be prominent in neuropathic conditions such as post-herpetic neuralgia, certain cases of diabetic or human immunodeficiency virus–associated neuropathy, and certain cases of traumatic nerve injury. In this section we consider the role of altered function of nociceptors in neuropathic pain.
In considering inflammatory pain it was noted earlier in this chapter that primary hyperalgesia is explained by sensitization of nociceptors whereas secondary hyperalgesia is due to central sensitization. In the case of secondary hyperalgesia, the input of low-threshold mechanoreceptors, normally concerned only with touch sensibility, leads to pain because the synaptic links with central pain-signaling cells in the dorsal horn are strengthened. A similar mechanism of central sensitization appears to also explain the allodynia seen with neuropathic pain states. This was demonstrated in human subjects by selectively blocking the neural activity in large fibers (touch fibers) with an ischemic block. When touch sensation was eliminated and the functions of other nerve fibers were still preserved, the allodynia disappeared (Campbell et al 1988b).
The relative role of central and peripheral mechanisms in neuropathic pain is not well understood and probably varies not only with the disease but also with factors such as genetic differences. In many cases, however, the abnormal input of neural activity from nociceptive afferents plays a dynamic and ongoing role in maintenance of the pain state.
Understanding of neuropathic pain involves two key concepts: (1) inappropriate activity in nociceptive fibers (injured and uninjured) and (2) central changes in sensory processing that arise from these abnormalities. To consider how these mechanisms generate heightened pain we discuss in some depth the simplest of neuropathic pain models: the sequelae of severing a nerve.
Ectopic Sensitivity Develops in Injured Fibers
When a nerve is severed, the nociceptors are also severed. The injured (transected) nociceptors could in principle function abnormally at the site of nerve transection (the neuroma). Indeed, abnormal spontaneous activity has been observed in A and C fibers originating from a neuroma (see Chapter 64). Given that a substantial proportion of C-fiber afferents are nociceptors, it is likely that this spontaneous activity is in fact occurring in nociceptive afferents. In patients with a painful neuroma and hyperalgesia, locally anesthetizing the neuroma may eliminate the pain and hyperalgesia (Gracely et al 1992). Thus, ongoing activity arising from nociceptive fibers in the neuroma contributes to the ongoing pain and hyperalgesia after nerve injury and may contribute to phantom limb pain (see Chapter 67).
Ectopic mechanical sensitivity also develops in experimental neuromas. Tapping at the site of a neuroma leads to a neural response. This may account for the observation that tapping on a neuroma is quite often found to be painful (Tinel’s sign). Superficial neuromas, which are more prone to accidental mechanical stimulation, or neuromas that are in locations associated with high mechanical stress are more likely to be painful. One strategy to alleviate neuroma pain is to resect the neuroma and move the nerve to a deep location. Since neuromas form when a nerve is cut, removing a neuroma in essence is a neuroma relocation operation. Neuroma relocation effectively relieves pain enduringly in at least some cases (Burchiel et al 1993).
The Role of the Intact Nociceptor
Several lines of evidence suggest that uninjured, intact nociceptors that share the nerve of the injured fibers play a role in neuropathic pain. Much of the evidence comes from animal models of neuropathic pain in which the L5 spinal nerve is cut and ligated (Kim and Chung 1992). This injury leads to behavioral signs of hyperalgesia to mechanical and heat stimuli applied to the ipsilateral foot. Using this model, we have learned the following: (1) Dorsal rhizotomy of the lesioned L5 root does not reverse the hyperalgesia regardless of whether this is done pre-emptively (before the L5 spinal nerve is severed) or after the lesion. Thus, interruption of input from the injured L5 spinal nerve fails to reverse the hyperalgesia in the foot, which indicates that ectopic activity from the injured nerve is not essential for the development of neuropathic pain (Li et al 2000). (2) Electrophysiological recordings from the uninjured L4 spinal nerve (the root that most overlaps the innervation territory of the L5 root) reveal abnormal spontaneous activity in C-fiber nociceptors. The spontaneous activity appears to emanate at least in part from the skin (Wu et al 2001). (3) Molecules related to pain (e.g., CGRP, brain-derived neurotrophic factor [BDNF], VR1) are up-regulated in L4 DRGs (Fukuoka et al 2000). (4) Expression of the TTX-resistant sodium channel Nav1.8 increases in the sciatic nerve (Gold et al 2003).
Additional evidence for a contribution of non-axotomized nociceptors comes from clinical studies demonstrating that distal therapies are effective in neuropathic pain states. Capsaicin causes degeneration of the cutaneous terminals of nociceptors that express TRPV1 (Nolano et al 1999). Capsaicin applied to the skin can alleviate the pain associated with nerve injuries (e.g., Robbins et al 1998). This clinical effect can be understood only by invoking a role of cutaneous nociceptors that survive the injury. Moreover, since the toxicity of capsaicin appears to be restricted to the skin, it is the cutaneous terminal of the nociceptor that must be generating pain.
Wallerian Degeneration and Neuropathic Pain
When the L5 spinal nerve is cut, the axons distal to the cut undergo wallerian degeneration. In the peripheral nerve, axons from intact nerve roots are in close proximity to degenerating axons and thus are exposed to diffusible factors released into the endoneurial space or at the nerve terminals. These factors could be derived from Schwann cells or macrophages and could affect nociceptive terminals directly or indirectly by an alteration in the cell bodies of nociceptors. As illustrated in Figure 1-20, wallerian degeneration may play a role in neuropathic pain by producing sensitization of primary afferent nociceptors and/or by leading to the development of central sensitization.
Figure 1-20 Wallerian degeneration in distal nerves may account for the development of hyperalgesia in patients with neuropathic pain.
When a nerve is cut, the nerve fibers distal to the cut undergo wallerian degeneration. Adjacent uninjured fibers are exposed to a dramatically altered endoneurial environment. The inflammatory milieu may include chemokines, cytokines, and growth factors. This may lead to the development of peripheral or central sensitization. A, Peripheral sensitization. Wallerian degeneration may lead to the sensitization of primary afferent nociceptors. Now, mechanical stimulation of the nociceptors results in an enhanced response, which accounts for the mechanical hyperalgesia. B, Central sensitization. Wallerian degeneration may lead to the development of spontaneous activity in nociceptors. This spontaneous activity may produce a state of central sensitization similar to that described for secondary hyperalgesia (see Fig. 1-17B). Now, mechanical stimulation of low-threshold mechanoreceptors activates the sensitized central pain-signaling neurons, which accounts for the mechanical hyperalgesia. CNS, central nervous system; DRG, dorsal root ganglion; PNS, peripheral nervous system.
Nociceptors and the Sympathetic Nervous System
Activity in nociceptors induces an increase in sympathetic discharge. This increased discharge is associated with the rise in blood pressure in acute pain states. Usually, the converse is not true: sympathetic activity does not affect the discharge of nociceptive neurons. In certain patients with pain, however, nociceptors acquire sensitivity to NE released by sympathetic efferents. Pain dependent on activity in the sympathetic nervous system is referred to as sympathetically maintained pain (SMP).
SMP may or may not be an important pain mechanism overall in patients, but in at least some individuals SMP may be the driving pathophysiological basis for the pain (for review see Drummond 2010). SMP in particular is noted in many cases of complex regional pain syndrome (reflex sympathetic dystrophy, causalgia). This condition is usually triggered by trauma to an extremity, with varying combinations of edema, allodynia and hyperalgesia, vasomotor and sudomotor abnormalities, and motor disturbances developing in the extremity. For SMP, procedures that interrupt the function of the sympathetic nervous system can alleviate the pain and hypersensitivity.
Sympathetically Maintained Pain Is a Receptor Disorder
Clinical studies support the concept that catechol sensitivity may develop in nociceptors after partial nerve injury. For example, intraoperative stimulation of the sympathetic chain induces pain in patients with causalgia (Walker and Nulson 1948, White and Sweet 1969). Also, physiological activation of sympathetic vasoconstrictor neurons leads to enhanced spontaneous pain and hyperalgesia in patients with SMP (Baron et al 2002). Pain is increased in the majority of patients with complex regional pain syndrome when sympathetic nervous system activity is evoked by a loud startling noise or by cooling their forehead (Drummond et al 2001). Injection of NE around stump neuromas or in the skin of patients with post-herpetic neuralgia induces an increase in spontaneous pain (Chabal et al 1992, Choi and Rowbotham 1997, Lin et al 2006). In SMP, anesthetic blockade of the sympathetic nervous system relieves the pain and hyperalgesia; intradermal injection of NE into the previously hyperalgesic area induces pain (Ali et al 2000). NE injected into normal subjects evokes little or no pain. This suggests that SMP does not arise from too much NE, but rather from the presence of adrenergic receptors in the skin that are coupled to nociceptors. Therefore, in SMP, the NE that is normally released from sympathetic terminals acquires the capacity to evoke pain.
Nerve Injury Induces Catechol Sensitization in Nociceptors
In addition to spontaneous activity, adrenergic sensitivity develops in nociceptors after nerve injury. In a primate model of an L6 spinal nerve lesion (Ali et al 1999), intact nociceptors innervating the foot exhibited spontaneous activity and a response to the α1-adrenergic agonist phenylephrine applied to the receptive field. In monkeys in which no spinal nerve lesion was applied, little or no catechol sensitivity and spontaneous activity were present. Using a somewhat different injury model, studies in rabbits have also demonstrated catechol sensitization of intact nociceptors after injury to companion nerve fibers (Sato and Perl 1991). C fibers ending in a neuroma also display adrenergic sensitivity (e.g., Häbler et al 1987). Thus adrenergic mechanisms may play a role in activating injured nociceptors as well.
α1-Adrenergic Agonists Activate Nociceptors Leading to Central Sensitization
Clinical studies support the postulate that SMP arises from expression of α-adrenergic receptors on the terminals of nociceptors. Systemic phentolamine, an α-adrenergic antagonist, relieves pain when given to patients with SMP (Raja et al 1991). Topical application of clonidine, an α2-adrenergic agonist, to the painful skin of patients with SMP relieved hyperalgesia in the painful area (Davis et al 1991). Activation of α2-adrenoceptors located on sympathetic terminals by clonidine blocks the release of NE. When phenylephrine, a selective α1-adrenergic agonist, was applied to the clonidine-treated area, pain was rekindled in patients with SMP (Davis et al 1991). Thus, clinical data, as well as primate physiological data, suggest that in SMP, release of NE from the sympathetic terminals activates nociceptors that express α1-adrenoceptors. The spontaneous activity and excitation of nociceptors by NE lead to central sensitization. Whether a change in phenotype or some other molecular change explains this nociceptor chemical sensitization is unanswered. Of some interest is the finding that the density of α1-adrenoceptors in the epidermis of hyperalgesic skin of patients with complex regional pain syndrome is increased as measured with quantitative autoradiographic techniques (Drummond et al 1996). For SMP, procedures that reduce or eliminate excitation of α1-adrenergic receptors lessen nociceptor activity and therefore lessen the hyperalgesia. Other potential central mechanisms that modulate nociception and emotional responses by adrenergic facilitation of nociceptive transmission in the dorsal horn or thalamus and/or by depletion of bulbospinal opioids have also been postulated (Drummond 2010).
Acknowledgment
We appreciate the technical assistance of T. V. Hartke and Ian Suk for contributing original artwork. This work was supported by National Institute of Health grants P01 NS 47399, NS-14447, and NS-26363.
The references for this chapter can be found at www.expertconsult.com.
References
Adriaensen H., Gybels J., Handwerker H.O., et al. Nociceptor discharges and sensations due to prolonged noxious mechanical stimulation—a paradox. Human Neurobiology. 1984;3:53–58.
Agarwal N., Pacher P., Tegeder I., et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nature Neuroscience. 2007;10:870–879.
Ali Z., Meyer R.A., Campbell J.N. Secondary hyperalgesia to mechanical but not heat stimuli following a capsaicin injection in hairy skin. Pain. 1996;68:401–411.
Ali Z., Raja S.N., Wesselmann U., et al. Intradermal injection of norepinephrine evokes pain in patients with sympathetically maintained pain. Pain. 2000;88:161–168.
Ali Z., Ringkamp M., Hartke T.V., et al. Uninjured C-fiber nociceptors develop spontaneous activity and alpha adrenergic sensitivity following L6 spinal nerve ligation in the monkey. Journal of Neurophysiology. 1999;81:455–466.
Amaya F., Shimosato G., Kawasaki Y., et al. Induction of CB1 cannabinoid receptor by inflammation in primary afferent neurons facilitates antihyperalgesic effect of peripheral CB1 agonist. Pain. 2006;124:175–183.
Ambalavanar R., Moritani M., Haines A., et al. Chemical phenotypes of muscle and cutaneous afferent neurons in the rat trigeminal ganglion. Journal of Comparative Neurology. 2003;460:167–179.
Anand P., Whiteside G., Fowler C.J., et al. Targeting CB2 receptors and the endocannabinoid system for the treatment of pain. Brain Research Reviews. 2009;60:255–266.
Andrew D., Greenspan J.D. Mechanical and heat sensitization of cutaneous nociceptors after peripheral inflammation in the rat. Journal of Neurophysiology. 1999;82:2649–2656.
Andrew D., Greenspan J.D. Peripheral coding of tonic mechanical cutaneous pain: comparison of nociceptor activity in rat and human psychophysics. Journal of Neurophysiology. 1999;82:2641–2648.
Averill S., McMahon S.B., Clary D.O., et al. Immunocytochemical localization of trkA receptors in chemically identified subgroups of adult rat sensory neurons. European Journal of Neuroscience. 1995;7:1484–1494.
Ballou L.R., Botting R.M., Goorha S., et al. Nociception in cyclooxygenase isozyme–deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10272–10276.
Banik R.K., Kozaki Y., Sato J., et al. B2 receptor–mediated enhanced bradykinin sensitivity of rat cutaneous C-fiber nociceptors during persistent inflammation. Journal of Neurophysiology. 2001;86:2727–2735.
Baron R., Schattschneider J., Binder A., et al. Relation between sympathetic vasoconstrictor activity and pain and hyperalgesia in complex regional pain syndromes: a case-control study. Lancet. 2002;359:1655–1660.
Baron R., Wasner G., Borgstedt R., et al. Effect of sympathetic activity on capsaicin-evoked pain, hyperalgesia, and vasodilatation. Neurology. 1999;52:923–932.
Basbaum A.I., Bautista D.M., Scherrer G., et al. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284.
Baumann T.K., Simone D.A., Shain C.N., et al. Neurogenic hyperalgesia: the search for the primary cutaneous afferent fibers that contribute to capsaicin-induced pain and hyperalgesia. Journal of Neurophysiology. 1991;66:212–227.
Beck P.W., Handwerker H.O. Bradykinin and serotonin effects on various types of cutaneous nerve fibers. Pflugers Archiv. 1974;347:209–222.
Beltramo M., Bernardini N., Bertorelli R., et al. CB2 receptor–mediated antihyperalgesia: possible direct involvement of neural mechanisms. European Journal of Neuroscience. 2006;23:1530–1538.
Bennett D.L., Michael G.J., Ramachandran N., et al. A distinct subgroup of small DRG cells express GDNF receptor components and GDNF is protective for these neurons after nerve injury. Journal of Neuroscience. 1998;18:3059–3072.
Bergman E., Carlsson K., Liljeborg A., et al. Neuropeptides, nitric oxide synthase and GAP-43 in B4-binding and RT97 immunoreactive primary sensory neurons: normal distribution pattern and changes after peripheral nerve transection and aging. Brain Research. 1999;832:63–83.
Bernardini N., Sauer S.K., Haberberger R., et al. Excitatory nicotinic and desensitizing muscarinic (M2) effects on C-nociceptors in isolated rat skin. Journal of Neuroscience. 2001;21:3295–3302.
Bessou P., Perl E.R. Response of cutaneous sensory units with unmyelinated fibers to noxious stimuli. Journal of Neurophysiology. 1969;32:1025–1043.
Bhave G., Karim F., Carlton S.M., et al. Peripheral group I metabotropic glutamate receptors modulate nociception in mice. Nature Neuroscience. 2001;4:417–423.
Bini G., Crucci G., Hagbarth K.E., et al. Analgesic effect of vibration and cooling on pain induced by intraneural electrical stimulation. Pain. 1984;18:239–248.
Bischoff A. Congenital insensitivity to pain with anhidrosis. A morphometric study of sural nerve and cutaneous receptors in the human prepuce. In: Bonica J.J., Liebeskind J.C., Albe-Fessard D.G., eds. Advances in pain research and therapy. New York: Raven Press; 1979:53–65.
Bland-Ward P.A., Humphrey P.P.A. Acute nociception mediated by hindpaw P2X receptor activation in the rat. British Journal of Pharmacology. 1997;122:365–371.
Blumberg H., Jänig W. Activation of fibers via experimentally produced stump neuromas of skin nerves: ephaptic transmission or retrograde sprouting? Experimental Neurology. 1982;76:468–482.
Brennan T.J., Vandermeulen E.P., Gebhart G.F. Characterization of a rat model of incisional pain. Pain. 1996;64:493–501.
Burchiel K.J., Johans T.J., Ochoa J. Painful nerve injuries: bridging the gap between basic neuroscience and neurosurgical treatment. Acta Neurochirurgica Supplementum. 1993;58:131–135.
Burgess P.R., Perl E.R. Myelinated afferent fibres responding specifically to noxious stimulation of the skin. Journal of Physiology (London). 1967;190:541–562.
Burnstock G. Purinergic receptors and pain. Current Pharmaceutical Design. 2009;15:1717–1735.
Campbell J.N., Khan A.A., Meyer R.A., et al. Responses to heat of C-fiber nociceptors in monkey are altered by injury in the receptive field but not by adjacent injury. Pain. 1988;32:327–332.
Campbell J.N., LaMotte R.H. Latency to detection of first pain. Brain Research. 1983;266:203–208.
Campbell J.N., Meyer R.A. Sensitization of unmyelinated nociceptive afferents in the monkey varies with skin type. Journal of Neurophysiology. 1983;49:98–110.
Campbell J.N., Meyer R.A., LaMotte R.H. Sensitization of myelinated nociceptive afferents that innervate monkey hand. Journal of Neurophysiology. 1979;42:1669–1679.
Campbell J.N., Raja S.N., Meyer R.A., et al. Myelinated afferents signal the hyperalgesia associated with nerve injury. Pain. 1988;32:89–94.
Campero M., Baumann T.K., Bostock H., et al. Human cutaneous C fibres activated by cooling, heating and menthol. Journal of Physiology. 587, 2009. 5633-565
Cannon K.E., Leurs R., Hough L.B. Activation of peripheral and spinal histamine H3 receptors inhibits formalin-induced inflammation and nociception, respectively. Pharmacology, Biochemistry, and Behavior. 2007;88:122–129.
Carlton S.M. Peripheral excitatory amino acids. Current Opinion in Pharmacology. 2001;1:52–56.
Carlton S.M., Coggeshall R.E. Nociceptive integration: does it have a peripheral component? Pain Forum. 1998;7:71–78.
Carlton S.M., Coggeshall R.E. Peripheral capsaicin receptors increase in the inflamed rat hindpaw: a possible mechanism for peripheral sensitization. Neuroscience Letters. 2001;310:53–56.
Carlton S.M., Zhou S., Coggeshall R.E. Peripheral GABA(A) receptors: evidence for peripheral primary afferent depolarization. Neuroscience. 1999;93:713–722.
Carlton S.M., Zhou S., Du J., et al. Somatostatin modulates the transient receptor potential vanilloid 1 (TRPV1) ion channel. Pain. 2004;110:616–627.
Carr P.A., Yamamoto T., Nagy J.I. Calcitonin gene–related peptide in primary afferent neurons of rat: co-existence with fluoride-resistant acid phosphatase and depletion by neonatal capsaicin. Neuroscience. 1990;36:751–760.
Caterina M.J., Leffler A., Malmberg A.B., et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313.
Caterina M.J., Rosen T.A., Tominaga M., et al. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature. 1999;398:436–441.
Caterina M.J., Schumacher M.A., Tominaga M., et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824.
Cervero F., Gilbert R., Hammond R.G.E., et al. Development of secondary hyperalgesia following non-painful thermal stimulation of the skin: a psychophysical study in man. Pain. 1993;54:181–189.
Cervero F., Laird J.M.A., García-Nicas E. Secondary hyperalgesia and presynaptic inhibition: an update. European Journal of Pain. 2003;7:345–351.
Cervero F., Meyer R.A., Campbell J.N. A psychophysical study of secondary hyperalgesia: evidence for increased pain to input from nociceptors. Pain. 1994;58:21–28.
Chabal C., Jacobson L., Russell L.C., et al. Pain response to perineuromal injection of normal saline, epinephrine, and lidocaine in humans. Pain. 1992;49:9–12.
Chen C.C., Sivilotti L.G., Colquhoun D., et al. A P2X purinoceptor expressed by a subset of sensory neurons. Nature. 1995:428–431.
Chen S.R., Pan H.M., Richardson T.E., et al. Potentiation of spinal alpha(2)-adrenoceptor analgesia in rats deficient in TRPV1-expressing afferent neurons. Neuropharmacology. 2007;52:1624–1630.
Cheng J.K., Ji R.R. Intracellular signaling in primary sensory neurons and persistent pain. Neurochemical Research. 2008;33:1970–1978.
Choi B., Rowbotham M.C. Effect of adrenergic receptor activation on post-herpetic neuralgia pain and sensory disturbances. Pain. 1997;69:55–63.
Chuang H., Prescott E.D., Kong H., et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962.
Cook S.P., Vulchanova L., Hargreaves K.M., et al. Distinct ATP receptors on pain-sensing and stretch-sensing neurons. Nature. 1997;387:505–508.
Cooper B., Ahlquist M., Friedman R.M., et al. Properties of high-threshold mechanoreceptors in the goat oral mucosa. II. Dynamic and static reactivity in carrageenan-inflamed mucosa. Journal of Neurophysiology. 1991;66:1280–1290.
Couture R., Harrisson M., Vianna R.M., et al. 2001 Kinin receptors in pain and inflammation. European Journal of Pharmacology. 2001;429:161–176.
Cunha F.Q., Ferreira S.H. Peripheral hyperalgesic cytokines. Advances in Experimental Medicine and Biology. 2003;521:22–39.
Dai Y., Iwata K., Fukuoka T., et al. Phosphorylation of extracellular signal–regulated kinase in primary afferent neurons by noxious stimuli and its involvement in peripheral sensitization. Journal of Neuroscience. 2002;22:7737–7745.
Darian-Smith I., Johnson K.O., Dykes R. “Cold” fiber population innervating palmar and digital skin of the monkey: responses to cooling pulses. Journal of Neurophysiology. 1973;36:325–346.
Davidson E.M., Coggeshall R.E., Carlton S.M. Peripheral NMDA and non-NMDA glutamate receptors contribute to nociceptive behaviors in the rat formalin test. Neuroreport. 1997;8:941–946.
Davis B.M., Lewin G.R., Mendell L.M., et al. Altered expression of nerve growth factor in the skin of transgenic mice leads to changes in response to mechanical stimuli. Neuroscience. 1993;56:789–792.
Davis J.B., Gray J., Gunthorpe M.J., et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187.
Davis K.D., Meyer R.A., Campbell J.N. Chemosensitivity and sensitization of nociceptive afferents that innervate the hairy skin of monkey. Journal of Neurophysiology. 1993;69:1071–1081.
Davis K.D., Treede R.-D., Raja S.N., et al. Topical application of clonidine relieves hyperalgesia in patients with sympathetically maintained pain. Pain. 1991;47:309–317.
Distler C., Rathee P.K., Lips K.S., et al. Fast Ca2+-induced potentiation of heat-activated ionic currents requires cAMP/PKA signaling and functional AKAP anchoring. Journal of Neurophysiology. 2003;89:2499–2505.
Djouhri L., Dawbarn D., Robertson A., et al. Time course and nerve growth factor dependence of inflammation-induced alterations in electrophysiological membrane properties in nociceptive primary afferent neurons. Journal of Neuroscience. 2001;21:8722–8733.
Dong X., Han S., Zylka M.J., et al. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell. 2001;106:619–632.
Drummond P.D. Noradrenaline increases hyperalgesia to heat in skin sensitized by capsaicin. Pain. 1995;60:311–315.
Drummond P.D. Independent effects of ischaemia and noradrenaline on thermal hyperalgesia in capsaicin-treated skin. Pain. 1996;67:129–133.
Drummond P.D. Sensory disturbances in complex regional pain syndrome: clinical observations, autonomic interactions, and possible mechanisms. Pain Medicine. 2010;11:1257–1266.
Drummond P.D., Finch P.M., Skipworth S., et al. Pain increases during sympathetic arousal in patients with complex regional pain syndrome. Neurology. 2001;57:1296–1303.
Drummond P.D., Skipworth S., Finch P.M. Alpha 1-adrenoceptors in normal and hyperalgesic human skin. Clinical Science (London). 1996;91:73–77.
Du J., Zhou S., Carlton S.M. Group II metabotropic glutamate receptor activation attenuates peripheral sensitization in inflammatory states. Neuroscience. 2008;154:754–766.
Dubin A.E., Patapoutian A. Nociceptors: the sensors of the pain pathway. Journal of Clinical Investigation. 2010;120:3760–3772.
Dubner R., Hu J.W. Myelinated (A-delta) nociceptive afferents innervating the monkey`s face. Journal of Dental Research. 1977;56:A167.
Dubner R., Price D.D., Beitel R.E., et al. Peripheral neural correlates of behavior in monkey and human related to sensory-discriminative aspects of pain. In: Anderson D.J., Mathews B., eds. Pain in the trigeminal region. Amsterdam: Elsevier North Holland; 1977:57–66.
Dussor G., Zylka M.J., Anderson D.J., et al. Cutaneous sensory neurons expressing the Mrgprd receptor sense extracellular ATP and are putative nociceptors. Journal of Neurophysiology. 2008;99:1581–1589.
Elliott M.J., Maini R.N., Feldmann M., et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor α (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110.
England S., Bevan S., Docherty R.J. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP–protein kinase A cascade. Journal of Physiology. 1996;495:429–440.
Fitzgerald M., Lynn B. The sensitization of high threshold mechanoreceptors with myelinated axons by repeated heating. Journal of Physiology. 1977;265:549–563.
Fuchs P.N., Campbell J.N., Meyer R.A. Secondary hyperalgesia persists in capsaicin desensitized skin. Pain. 2000;84:141–149.
Fukuoka H., Kawatani M., Hisamitsu T., et al. Cutaneous hyperalgesia induced by peripheral injection of interleukin-1β in the rat. Brain Research. 1994;657:133–140.
Fukuoka T., Tokunaga A., Kondo E., et al. The role of neighboring intact dorsal root ganglion neurons in a rat neuropathic pain model. In: Devor M., Rowbotham M., Wiesenfeld-Hallin Z., eds. Progress in pain research and management. Seattle: ISAP Press; 2000:137–146.
Garell P.C., McGillis S.L.B., Greenspan J.D. Mechanical response properties of nociceptors innervating feline hairy skin. Journal of Neurophysiology. 1996;75:1177–1189.
Garnsworthy R.K., Gully R.L., Kenins P., et al. Identification of the physical stimulus and the neural basis of fabric-evoked prickle. Journal of Neurophysiology. 1988;59:1083–1097.
Gee M.D., Lynn B., Cotsell B. Activity-dependent slowing of conduction velocity provides a method for identifying different functional classes of C-fibre in the rat saphenous nerve. Neuroscience. 1996;73:667–675.
Gerke M.B., Plenderleith M.B. Binding sites for the plant lectin Bandeiraea simplicifolia I-isolectin B(4) are expressed by nociceptive primary sensory neurones. Brain Research. 2001;911:101–104.
Gibbins I.L., Wattchow D., Coventry B. Two immunohistochemically identified populations of calcitonin gene–related peptide (CGRP)-immunoreactive axons in human skin. Brain Research. 1987;414:143–148.
Gold M.S., Levine J.D., Correa A.M. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. Journal of Neuroscience. 1998;18:10345–10355.
Gold M.S., Reichling D.B., Shuster M.J., et al. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:1108–1112.
Gold M.S., Weinreich D., Kim C.S., et al. Redistribution of Nav1.8 in uninjured axons enables neuropathic pain. Journal of Neuroscience. 2003;23:158–166.
Goudet C., Magnaghi V., Landry M., et al. Metabotropic receptors for glutamate and GABA in pain. Brain Research Reviews. 2009;60:43–56.
Gracely R.H., Lynch S.A., Bennett G.J. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain. 1992;51:175–194.
Greffrath W., Nemenov M.I., Schwarz S., et al. Inward currents in primary nociceptive neurons of the rat and pain sensations in humans elicited by infrared diode laser pulses. Pain. 2002;99:145–155.
Guo A., Simone D.A., Stone L.S., et al. Developmental shift of vanilloid receptor 1 (VR1) terminals into deeper regions of the superficial dorsal horn: correlation with a shift from TrkA to Ret expression by dorsal root ganglion neurons. European Journal of Neuroscience. 2001;14:293–304.
Guo A., Vulchanova L., Wang J., et al. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. European Journal of Neuroscience. 1999;11:946–958.
Häbler H.-J., Jänig W., Koltzenburg M. Activation of unmyelinated afferents in chronically lesioned nerves by adrenaline and excitation of sympathetic efferents in the cat. Neuroscience Letters. 1987;82:35–40.
Häbler H.-J., Jänig W., Koltzenburg M. A novel type of unmyelinated chemosensitive nociceptor in the acutely inflamed urinary bladder. Agents and Actions. 1988;25:219–221.
Hamilton S.G., Warburton J., Bhattacharjee A., et al. ATP in human skin elicits a dose-related pain response which is potentiated under conditions of hyperalgesia. Brain. 2000;123:1238–1246.
Han S.K., Dong X., Hwang J.I., et al. Orphan G protein–coupled receptors MrgA1 and MrgC11 are distinctively activated by RF-amide–related peptides through the Galpha q/11 pathway. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14740–14745.
Handwerker H.O., Kilo S., Reeh P.W. Unresponsive afferent nerve fibres in the sural nerve of the rat. Journal of Physiology. 1991;435:229–242.
Hans G., Schmidt B.L., Strichartz G. Nociceptive sensitization by endothelin-1. Brain Research Reviews. 2009;60:36–42.
Harrison J.L., Davis K.D. Cold-evoked pain varies with skin type and cooling rate: a psychophysical study in humans. Pain. 1999;83:123–135.
Hilliges M., Weidner C., Schmelz M., et al. ATP responses in human C nociceptors. Pain. 2002;98:59–68.
Holland N.R., Crawford T.O., Hauer P., et al. Small-fiber sensory neuropathies: clinical course and neuropathology of idiopathic cases. Annals of Neurology. 1998;44:47–59.
Holzer P. 1998 Neurogenic vasodilatation and plasma leakage in the skin. General Pharmacology. 1998;30:5–11.
Holzer P. Acid-sensitive ion channels and receptors. Handbook of Experimental Pharmacology. 2009;194:283–332.
Hsieh S.T., Choi S., Lin W.-M., et al. Epidermal denervation and its effects on keratinocytes and Langerhans cells. Journal of Neurocytology. 1996;25:513–524.
Hsieh S.T., Lin W.M. Modulation of keratinocyte proliferation by skin innervation. Journal of Investigative Dermatology. 1999;113:579–586.
Huang J.H., Ali Z., Travison T.G., et al. Spatial mapping of the zone of secondary hyperalgesia reveals a gradual decline of pain with distance but sharp borders. Pain. 2000;86:33–42.
Hunt S.P., Rossi J. Peptide- and non–peptide-containing unmyelinated primary afferents: the parallel processing of nociceptive information. Society of London. Series B. Biological Sciences. 1985;308:283–289.
Jänig W., Levine J.D., Michaelis M. Interactions of sympathetic and primary afferent neurons following nerve injury and tissue trauma. In: Kumazawa T., Kruger L., Mizumura K., eds. Interactions of sympathetic and primary afferent neurons following nerve injury and tissue trauma. British Vancouver: Elsevier Science; 1996:161–184.
Ji R.R., Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. Journal of Neuroscience. 1997;17:1776–1785.
Ji R.-R., Samad T.A., Jin S.X., et al. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68.
Jin X., Gereau R.W. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. Journal of Neuroscience. 2006;26:246–255.
Johanek L.M., Meyer R.A., Friedman R.M., et al. A role for polymodal C-fiber afferents in nonhistaminergic itch. Journal of Neuroscience. 2008;28:7659–7669.
Johnson K.O., Darian S.I., LaMotte C., et al. Coding of incremental changes in skin temperature by a population of warm fibers in the monkey: correlation with intensity discrimination in man. Journal of Neurophysiology. 1979;42:1332–1353.
Kasai M., Mizumura K. Increase in spontaneous action potentials and sensitivity in response to norepinephrine in dorsal root ganglion neurons of adjuvant inflamed rats. Neuroscience Research. 2001;39:109–113.
Kashiba H., Uchida Y., Senba E. Difference in binding by isolectin B4 to trkA and c-ret mRNA–expressing neurons in rat sensory ganglia. Brain Research. Molecular Brain Research. 2001;95:18–26.
Kerr B.J., Souslova V., McMahon S.B., et al. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport. 2001;12:3077–3080.
Khalil Z., Helme R. Sensory peptides as neuromodulators of wound healing in aged rats. Journals of Gerontology. Series A. Biological Sciences and Medical Sciences. 1996;51:B354–B361.
Khan A.A., Raja S.N., Manning D.C., et al. The effects of bradykinin and sequence-related analogs on the response properties of cutaneous nociceptors in monkeys. Somatosensory & Motor Research. 1992;9:97–106.
Khasar S.G., Lin Y.H., Martin A., et al. A novel nociceptor signaling pathway revealed in protein kinase C ε mutant mice. Neuron. 1999;24:253–260.
Khodorova A., Montmayeur J.P., Strichartz G. Endothelin receptors and pain. Journal of Pain. 2009;10:4–28.
Khodorova A., Navarro B., Jouaville L.S., et al. Endothelin-B receptor activation triggers an endogenous analgesic cascade at sites of peripheral injury. Nature Medicine. 2003;9:1055–1061.
Kidd B.L., Urban L.A. Mechanisms of inflammatory pain. British Journal of Anaesthesia. 2001;87:3–11.
Kilo S., Schmelz M., Koltzenburg M., et al. Different patterns of hyperalgesia induced by experimental inflammation in human skin. Brain. 1994;117:385–396.
Kim S.H., Chung J.M. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363.
Kinnman E., Nygårds E.B., Hansson P. Peripheral alpha-adrenoreceptors are involved in the development of capsaicin induced ongoing and stimulus evoked pain in humans. Pain. 1997;69:79–85.
Klede M., Handwerker H.O., Schmelz M. Central origin of secondary mechanical hyperalgesia. Journal of Neurophysiology. 2003;90:353–359.
Klement W., Arndt J.O. The role of nociceptors of cutaneous veins in the mediation of cold pain in man. Journal of Physiology (London). 1992;449:73–83.
Koda H., Minagawa M., Si-Hong L., et al. H1-receptor–mediated excitation and facilitation of the heat response by histamine in canine visceral polymodal receptors studied in vitro. Journal of Neurophysiology. 1996;76:1396–1404.
Koltzenburg M., Bennett D.L., Shelton D.L., et al. Neutralization of endogenous NGF prevents the sensitization of nociceptors supplying inflamed skin. European Journal of Neuroscience. 1999;11:1698–1704.
Koltzenburg M., Handwerker H.O. Differential ability of human cutaneous nociceptors to signal mechanical pain and to produce vasodilatation. Journal of Neuroscience. 1994;14:1756–1765.
Koltzenburg M., Lundberg L.E.R., Torebjörk H.E. Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain. 1992;51:207–219.
Koppert W., Dern S.K., Sittl R., et al. A new model of electrically evoked pain and hyperalgesia in human skin: the effects of intravenous alfentanil, S(+)-ketamine, and lidocaine. Anesthesiology. 2001;95:395–402.
Kress M., Kuner R. Mode of action of cannabinoids on nociceptive nerve endings. Experimental Brain Research. 2009;196:79–88.
Kruger L., Perl E.R., Sedivec M.J. Fine structure of myelinated mechanical nociceptor endings in cat hairy skin. Journal of Comparative Neurology. 1981;198:137–154.
Kumazawa T., Perl E.R. Primate cutaneous receptors with unmyelinated (C) fibres and their projection to the substantia gelatinosa Paris. Journal of Physiology. 1977;73:287–304.
LaMotte R.H., Campbell J.N. Comparison of responses of warm and nociceptive C-fiber afferents in monkey with human judgements of thermal pain. Journal of Neurophysiology. 1978;41:509–528.
LaMotte R.H., Shain C.N., Simone D.A., et al. Neurogenic hyperalgesia: psychophysical studies of underlying mechanisms. Journal of Neurophysiology. 1991;66:190–211.
LaMotte R.H., Thalhammer J.G. Response properties of high-threshold cutaneous cold receptors in the primate. Brain Research. 1982;244:279–287.
LaMotte R.H., Thalhammer J.G., Robinson C.J. Peripheral neural correlates of magnitude of cutaneous pain and hyperalgesia: a comparison of neural events in monkey with sensory judgements in human. Journal of Neurophysiology. 1983;50:1–26.
LaMotte R.H., Thalhammer J.G., Torebjörk H.E., et al. Peripheral neural mechanisms of cutaneous hyperalgesia following mild injury by heat. Journal of Neuroscience. 1982;2:765–781.
Lang E., Novak A., Reeh P.W., et al. Chemosensitivity of fine afferents from rat skin in vitro. Journal of Neurophysiology. 1990;63:887–901.
Lawson S.N., Crepps B.A., Perl E.R. Relationship of substance P to afferent characteristics of dorsal root ganglion neurones in guinea-pig. Journal of Physiology. 1997;505:177–191.
Lawson S.N., Crepps B., Perl E.R. Calcitonin gene–related peptide immunoreactivity and afferent receptive properties of dorsal root ganglion neurones in guinea-pigs. Journal of Physiology. 2002;540:989–1002.
Lawson S.N., McCarthy P.W., Prabhakar E. Electrophysiological properties of neurones with CGRP-like immunoreactivity in rat dorsal root ganglia. Journal of Comparative Neurology. 1996;365:355–366.
Lawson S.N., Waddell P.J. Soma neurofilament immunoreactivity is related to cell size and fibre conduction velocity in rat primary sensory neurons. Journal of Physiology. 1991;435:41–63.
Ledent C., Vaugeois J.M., Schiffmann S.N., et al. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. 1997;388:674–678.
Lembo P.M., Grazzini E., Groblewski T., et al. Proenkephalin A gene products activate a new family of sensory neuron–specific GPCRs. Nature Neuroscience. 2002;5:201–209.
Levine J.D., Lau W., Kwiat G., et al. Leukotriene B4 produces hyperalgesia that is dependent on polymorphonuclear leukocytes. Science. 1984;225:743–745.
Lewis C., Neidhart S., Holy C., et al. Coexpression of P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory neurons. Nature. 1995;377:432–435.
Lewis T. Experiments relating to cutaneous hyperalgesia and its spread through somatic fibres. Clinical Science (London). 1935;2:373–423.
Lewis T. Pain. New York: Macmillan; 1942.
Lewis T., Pochin E.E. The double pain response of the human skin to a single stimulus. Clinical Science (London). 1937;3:67–76.
Li Y., Dorsi M.J., Meyer R.A., et al. Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain. 2000;85:493–502.
Lima F.O., Souza G.R., Verri W.A., Jr., et al. Direct blockade of inflammatory hypernociception by peripheral A1 adenosine receptors: involvement of the NO/cGMP/PKG/KATP signaling pathway. Pain. 2010;151:506–515.
Lin E.E., Horasek S., Agarwal S., et al. Local administration of norepinephrine in the stump evokes dose-dependent pain in amputees. Clinical Journal of Pain. 2006;22:482–486.
Lischetzki G., Rukwied R., Handwerker H.O., et al. Nociceptor activation and protein extravasation induced by inflammatory mediators in human skin. European Journal of Pain. 2001;5:49–57.
Löken L.S., Wessberg J., Morrinson I., et al. Coding of pleasant touch by unmyelinated afferents in humans. Nature Neuroscience. 2009;12:547–548.
Lynn B. Efferent function of nociceptors. In: Belmonte C., Cervero F., eds. Neurobiology of nociceptors. Oxford: Oxford University Press; 1996:418–438.
Lynn B., Shakhanbeh J. Neurogenic inflammation in the skin of the rabbit. Agents and Actions. 1988;25:228–230.
Ma Q.P., Woolf C.J. The progressive tactile hyperalgesia induced by peripheral inflammation is nerve growth factor dependent. Neuroreport. 1997;8:807–810.
Ma W., Quirion R. Inflammatory mediators modulating the transient receptor potential vanilloid 1 receptor: therapeutic targets to treat inflammatory and neuropathic pain. Expert Opinion on Therapeutic Targets. 2007;11:307–320.
Magerl W., Fuchs P.N., Meyer R.A., et al. Roles of capsaicin-insensitive nociceptors in cutaneous pain and secondary hyperalgesia. Brain. 2001;124:1754–1764.
Magerl W., Treede R.D. Secondary tactile hypoesthesia: a novel type of pain-induced somatosensory plasticity in human subjects. Neuroscience Letters. 2004;361:136–139.
Magerl W., Wilk S.H., Treede R- D. Secondary hyperalgesia and perceptual wind-up following intradermal injection of capsaicin in humans. Pain. 1998;74:257–268.
Maggi C.A., Meli A. The sensory-efferent function of capsaicin-sensitive sensory neurons. General Pharmacology. 1988;19:1–43.
Manning D.C., Raja S.N., Meyer R.A., et al. Pain and hyperalgesia after intradermal injection of bradykinin in humans. Clinical Pharmacology and Therapeutics. 1991;50:721–729.
Martin H.A., Basbaum A.I., Kwiat G.C., et al. Leukotriene and prostaglandin sensitization of cutaneous high-threshold C- and A-delta mechanonociceptors in the hairy skin of rat hindlimbs. Neuroscience. 1987;22:651–659.
McCarthy P.W., Lawson S.N. Cell type and conduction velocity of rat primary sensory neurons with substance P–like immunoreactivity. Neuroscience. 1989;28:745–753.
Meini S., Maggi C.A. Knee osteoarthritis: a role for bradykinin? Inflammation Research. 2008;57:351–361.
Mendell L.M., Wall P. Responses of single dorsal cord cells to peripheral cutaneous unmyelinated fibres. Nature. 1965;206:97–99.
Meyer R.A., Campbell J.N. Myelinated nociceptive afferents account for the hyperalgesia that follows a burn to the hand. Science. 1981;213:1527–1529.
Meyer R.A., Campbell J.N. Peripheral neural coding of pain sensation. Johns Hopkins APL Technical Digest. 1981;2:164–171.
Meyer R.A., Campbell J.N. Coupling between unmyelinated peripheral nerve fibers does not involve sympathetic efferent fibers. Brain Research. 1987;437:181–182.
Meyer R.A., Campbell J.N., Raja S.N. Antidromic nerve stimulation in monkey does not sensitize unmyelinated nociceptors to heat. Brain Research. 1988;441:168–172.
Meyer R.A., Davis K.D., Cohen R.H., et al. Mechanically insensitive afferents (MIAs) in cutaneous nerves of monkey. Brain Research. 1991;561:252–261.
Meyer R.A., Raja S.N., Campbell J.N. Coupling of action potential activity between unmyelinated fibers in the peripheral nerve of monkey. Science. 1985;227:184–187.
Meyer R.A., Raja S.N., Campbell J.N., et al. Neural activity originating from a neuroma in the baboon. Brain Research. 1985;325:255–260.
Michael G.J., Priestley J.V. Differential expression of the mRNA for the vanilloid receptor subtype 1 in cells of the adult rat dorsal root and nodose ganglia and its downregulation by axotomy. Journal of Neuroscience. 1999;19:1844–1854.
Miller R.J., Jung H., Bhangoo S.K., et al. Cytokine and chemokine regulation of sensory neuron function. Handbook of Experimental Pharmacology. 2009;194:417–449.
Mitrirattanakul S., Ramakul N., Guerrero A.V., et al. Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain. 2006;126:102–114.
Mizumura K., Minagawa M., Koda H., et al. Influence of histamine on the bradykinin response of canine testicular polymodal receptors in vitro. Inflammation Research. 1995;44:376–378.
Molliver D.C., Radeke M.J., Feinstein S.C., et al. Presence or absence of TrkA protein distinguishes subsets of small sensory neurons with unique cytochemical characteristics and dorsal horn projections. Journal of Comparative Neurology. 1995;361:404–416.
Molliver D.C., Wright D.E., Leitner M.L., et al. IB4-binding DRG neurons switch from NGF to GDNF dependence in early postnatal life. Neuron. 1997;19:849–861.
Momin A., McNaughton P.A. Regulation of firing frequency in nociceptive neurons by pro-inflammatory mediators. Experimental Brain Research. 2009;196:45–52.
Morin C., Bushnell M.C. Temporal and qualitative properties of cold pain and heat pain: a psychophysical study. Pain. 1998;74:67–73.
Namer B., Barta B., Orstavik K., et al. Microneurographic assessment of C-fibre function in aged healthy subjects. Journal of Physiology. 2009;587:419–428.
Nicholas R.S., Winter J., Wren P., et al. Peripheral inflammation increases the capsaicin sensitivity of dorsal root ganglion neurons in a nerve growth factor–dependent manner. Neuroscience. 1999;91:1425–1433.
Nolano M., Simone D.A., Wendelschafer-Crabb G., et al. Topical capsaicin in humans: parallel loss of epidermal nerve fibers and pain sensation. Pain. 1999;81:135–145.
Nordin M. Low-threshold mechanoreceptive and nociceptive units with unmyelinated (C) fibres in the human supraorbital nerve. Journal of Physiology (London). 1990;426:229–240.
Obreja O., Ringkamp M., Namer B., et al. Patterns of activity-dependent conduction velocity changes differentiate classes of unmyelinated mechano-insensitive afferents including cold nociceptors, in pig and in human. Pain. 2010;148:59–69.
Obreja O., Schmelz M., Poole S., et al. Interleukin-6 in combination with its soluble IL-6 receptor sensitises rat skin nociceptors to heat, in vivo. Pain. 2002;96:57–62.
Ohtori S., Takahashi K., Ino H., et al. Up-regulation of substance P and NMDA receptor mRNA in dorsal horn and preganglionic sympathetic neurons during adjuvant-induced noxious stimulation in rats. Annals of Anatomy. 2002;184:71–76.
Olausson H., Lamarre Y., Backlund H. Unmyelinated tactile afferents signal touch and project to insular cortex. Nature Neuroscience. 2003;5:900–904.
Oprée A., Kress M. Involvement of the proinflammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene–related peptide release from rat skin. Journal of Neuroscience. 2000;20:6289–6293.
Orozco O.E., Walus L., Sah D.W., et al. GFRalpha3 is expressed predominantly in nociceptive sensory neurons. European Journal of Neuroscience. 2001;13:2177–2182.
Pan H.L., Wu Z.Z., Zhou H.Y., et al. Modulation of pain transmission by G-protein–coupled receptors. Pharmacology & Therapeutics. 2008;117:141–161.
Pedersen J.L., Crawford M.E., Dahl J.B., et al. Effect of preemptive nerve block on inflammation and hyperalgesia after human thermal injury. Anesthesiology. 1996;84:1020–1026.
Peng Y.B., Ringkamp M., Campbell J.N., et al. Electrophysiological assessment of the cutaneous arborization of A-delta-fiber nociceptors. Journal of Neurophysiology. 1999;82:1164–1177.
Peng Y.B., Ringkamp M., Meyer R.A., et al. Fatigue and paradoxical enhancement of heat response in C-fiber nociceptors from cross-modal excitation. Journal of Neuroscience. 2003;23:4766–4774.
Perry M.J., Lawson S.N. Differences in expression of oligosaccharides, neuropeptides, carbonic anhydrase and neurofilament in rat primary afferent neurons retrogradely labelled via skin, muscle or visceral nerves. Neuroscience. 1998;85:293–310.
Pertovaara A. Noradrenergic pain modulation. Progress in Neurobiology. 2006;80:53–83.
Pezet S., McMahon S.B. Neurotrophins: mediators and modulators of pain. Annual Review of Neuroscience. 2006;29:507–538.
Pinter E., Helyes Z., Szolcsanyi J. Inhibitory effect of somatostatin on inflammation and nociception. Pharmacology & Therapeutics. 2006;112:440–456.
Plenderleith M.B., Snow P.J. The plant lectin Bandeiraea simplicifolia I-B4 identifies a subpopulation of small diameter primary sensory neurones which innervate the skin in the rat. Neuroscience Letters. 1993;159:17–20.
Pogatzki E.M., Gebhart G.F., Brennan T.J. Characterization of Adelta- and C-fibers innervating the plantar rat hindpaw one day after an incision. Journal of Neurophysiology. 2002;87:721–731.
Pogatzki E.M., Raja S.N. A mouse model of incisional pain. Anesthesiology. 2003;99:1023–1027.
Pogatzki E.M., Zahn P.K., Brennan T.J. Effect of pretreatment with intrathecal excitatory amino acid receptor antagonists on the development of pain behavior caused by plantar incision. Anesthesiology. 2000;93:489–496.
Prescott E.D., Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300:1284–1288.
Price D.D., Hu J.W., Dubner R., et al. Peripheral suppression of first pain and central summation of second pain evoked by noxious heat pulses. Pain. 1977;3:57–68.
Price G.W., Kelly J.S., Bowery N.G. The location of GABAB receptor binding sites in mammalian spinal cord. Synapse. 1987;1:530–538.
Price T.J., Flores C.M. Critical evaluation of the colocalization between calcitonin gene–related peptide, substance P, transient receptor potential vanilloid subfamily 1 immunoreactivities, and isolectin B4 binding in primary afferent neurons of the rat and mouse. Journal of Pain. 2007;8:263–272.
Raja S.N. Role of the sympathetic nervous system in acute pain and inflammation. Annals of Medicine. 1995;27:241–246.
Raja S.N., Campbell J.N., Meyer R.A. Evidence for different mechanisms of primary and secondary hyperalgesia following heat injury to the glabrous skin. Brain. 1984;107:1179–1188.
Raja S.N., Treede R.-D., Davis K.D., et al. Systemic alpha-adrenergic blockade with phentolamine: a diagnostic test for sympathetically maintained pain. Anesthesiology. 1991;74:691–698.
Rathee P.K., Distler C., Obreja O., et al. PKA/AKAP/VR-1 module: a common link of Gs-mediated signaling to thermal hyperalgesia. Journal of Neuroscience. 2002;22:4740–4745.
Reddy V.B., Iuga A.O., Shimada S.G., et al. Cowhage-evoked itch is mediated by a novel cysteine protease: a ligand of protease-activated receptors. Journal of Neuroscience. 2008;28:4331–4335.
Reeh P.W., Bayer J., Kocher L., et al. Sensitization of nociceptive cutaneous nerve fibers from the rat tail by noxious mechanical stimulation. Experimental Brain Research. 1987;65:505–512.
Reeh P.W., Kocher L., Jung S. Does neurogenic inflammation alter the sensitivity of unmyelinated nociceptors in the rat? Brain Research. 1986;384:42–50.
Reeh P.W., Sauer S.K. Chronic aspects in peripheral nociception. In: Jensen T.S., Turner J.A., Wiesenfeld-Hallin Z., eds. Progress in pain research and management. Seattle: IASP Press; 1997:115–131.
Richardson B.P., Engel G. The pharmacology and function of 5-HT3 receptors. Trends in Neurosciences. 1986;9:424–427.
Ringkamp M., Johanek L.M., Borzan J., et al. Conduction properties distinguish unmyelinated sympathetic efferent fibers and unmyelinated primary afferent fibers in the monkey. PLoS One. 5, 2010. e9076
Ringkamp M., Peng Y.B., Wu G., et al. Capsaicin responses in heat-sensitive and heat-insensitive A-fiber nociceptors. Journal of Neuroscience. 2001;21:4460–4468.
Ringkamp M., Schepers R.J., Shimada S.G., et al. A role for nociceptive, myelinated nerve fibers in itch sensation. Journal of Neuroscience. 2011;31:14841–14849.
Robas N., Mead E., Fidock M. MrgX2 is a high potency cortistatin receptor expressed in dorsal root ganglion. Journal of Biological Chemistry. 2003;278:44400–44404.
Robbins W.R., Staats P.S., Levine J., et al. Treatment of intractable pain with topical large-dose capsaicin: preliminary report. Anesthesia and Analgesia. 1998;86:579–583.
Roberts W.J., Elardo S.M. 1985 Sympathetic activation of A-delta nociceptors. Somatosensory Research. 1985;3:33–44.
Sang C.N., Gracely R.H., Max M.B., et al. Capsaicin-evoked mechanical allodynia and hyperalgesia cross nerve territories—evidence for a central mechanism. Anesthesiology. 1996;85:491–496.
Sato J., Perl E.R. Adrenergic excitation of cutaneous pain receptors induced by peripheral nerve injury. Science. 1991;251:1608–1610.
Sato J., Suzuki S., Iseki T., et al. Adrenergic excitation of cutaneous nociceptors in chronically inflamed rats. Neuroscience Letters. 1993;164:225–228.
Sawynok J., Reid A. Peripheral adenosine 5′-triphosphate enhances nociception in the formalin test via activation of a purinergic p2x receptor. European Journal of Pharmacology. 1997;330:115–121.
Schaible H.-G., Ebersberger A., Von Banchet G.S. Mechanisms of pain in arthritis. Annals of the New York Academy of Sciences. 2002;966:343–354.
Schaible H.G., Schmidt R.F. Effects of an experimental arthritis on the sensory properties of fine articular afferent units. Journal of Neurophysiology. 1985;54:1109–1122.
Schaible H.G., Von Banchet G.S., Boettger M.K., et al. The role of proinflammatory cytokines in the generation and maintenance of joint pain. Annals of the New York Academy of Sciences. 2010;1193:60–69.
Schmelz M., Michael K., Weidner C., et al. Which nerve fibers mediate the axon reflex flare in human skin? Neuroreport. 2000;11:645–648.
Schmelz M., Petersen L.J. Neurogenic inflammation in human and rodent skin. News in Physiological Sciences. 2001;16:33–37.
Schmelz M., Schmidt R., Bickel A., et al. Specific C-receptors for itch in human skin. Journal of Neuroscience. 1997;17:8003–8008.
Schmelz M., Schmidt R., Handwerker H.O., et al. Encoding of burning pain from capsaicin-treated human skin in two categories of unmyelinated nerve fibres. Brain. 2000;123:560–571.
Schmelz M., Schmidt R., Ringkamp M., et al. Limitation of sensitization to injured parts of receptive fields in human skin C-nociceptors. Experimental Brain Research. 1996;109:141–147.
Schmidt R., Schmelz M., Ringkamp M., et al. Innervation territories of mechanically activated C nociceptor units in human skin. Journal of Neurophysiology. 1997;78:2641–2648.
Schmidt R., Schmelz M., Torebjörk H.E., et al. Mechano-insensitive nociceptors encode pain evoked by tonic pressure to human skin. Neuroscience. 2000;98:793–800.
Serra J., Campero M., Ochoa J., et al. Activity-dependent slowing of conduction differentiates functional subtypes of C fibres innervating human skin. Journal of Physiology (London). 1999;515:799–811.
Shafer D.M., Assael L., White L.B., et al. Tumor necrosis factor-α as a biochemical marker of pain and outcome in temporomandibular joints with internal derangements. Journal of Oral and Maxillofacial Surgery. 1994;52:786–791.
Shea V.K., Perl E.R. Sensory receptors with unmyelinated (C) fibers innervating the skin of the rabbit’s ear. Journal of Neurophysiology. 1985;54:491–501.
Sherrington C.S. The integrative action of the nervous system. New York: Scribner; 1906.
Shin J., Cho H., Hwang S.W., et al. Bradykinin–12-lipoxygenase–VR1 signaling pathway for inflammatory hyperalgesia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10150–10155.
Shinohara T., Harada M., Ogi K., et al. Identification of a G protein–coupled receptor specifically responsive to beta-alanine. Journal of Biological Chemistry. 2004;279:23559–23564.
Silverman J.D., Kruger L. Acid phosphatase as a selective marker for a class of small sensory ganglion cells in several mammals: spinal cord distribution, histochemical properties, and relation to fluoride-resistant acid phosphatase (FRAP) of rodents. Somatosensory Research. 1988;5:219–246.
Silverman J.D., Kruger L. Lectin and neuropeptide labeling of separate populations of dorsal root ganglion neurons and associated “nociceptor” thin axons in rat testis and cornea whole-mount preparations. Somatosensory Research. 1988;5:259–267.
Simone D.A., Alreja M., LaMotte R.H. Psychophysical studies of the itch sensation and itchy skin (“allokinesis”) produced by intracutaneous injection of histamine. Somatosensory & Motor Research. 1991;8:271–279.
Simone D.A., Baumann T.K., LaMotte R.H. Dose-dependent pain and mechanical hyperalgesia in humans after intradermal injection of capsaicin. Pain. 1989;38:99–107.
Simone D.A., Kajander K.C. Responses of cutaneous A-fiber nociceptors to noxious cold. Journal of Neurophysiology. 1997;77:2049–2060.
Simone D.A., Sorkin L.S., Oh U., et al. Neurogenic hyperalgesia: central neural correlates in responses of spinothalamic tract neurons. Journal of Neurophysiology. 1991;66:228–246.
Slugg R.M., Meyer R.A., Campbell J.N. Response of cutaneous A- and C-fiber nociceptors in the monkey to controlled-force stimuli. Journal of Neurophysiology. 2000;83:2179–2191.
Sluka K.A., Willis W.D., Westlund K.N. The role of dorsal root reflexes in neurogenic inflammation. Pain Forum. 1995;4:141–149.
Sluka K.A., Winter O.C., Wemmie J.A. Acid-sensing ion channels: a new target for pain and CNS diseases. Current Opinion in Drug Discovery & Development. 2009;12:693–704.
Smith P.G., Liu M. Impaired cutaneous wound healing after sensory denervation in developing rats: effects on cell proliferation and apoptosis. Cell and Tissue Research. 2002;307:281–291.
Sorkin L.S., Xiao W.-H., Wagner R., et al. Tumour necrosis factor-α induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81:255–262.
Steen K.H., Reeh P.W. Sustained graded pain and hyperalgesia from harmless experimental tissue acidosis in human skin. Neuroscience Letters. 1993;154:113–116.
Steen K.H., Steen A.E., Kreysel H.W., et al. Inflammatory mediators potentiate pain induced by experimental tissue acidosis. Pain. 1996;66:163–170.
Stein C., Clark J.D., Oh U., et al. Peripheral mechanisms of pain and analgesia. Brain Research Reviews. 2009;60:90–113.
Stone L.S., Broberger C., Vulchanova L., et al. Differential distribution of alpha2A and alpha2C adrenergic receptor immunoreactivity in the rat spinal cord. Journal of Neuroscience. 1998;18:5928–5937.
Stone L.S., MacMillan L.B., Kitto K.F., et al. The alpha2a adrenergic receptor subtype mediates spinal analgesia evoked by alpha2 agonists and is necessary for spinal adrenergic-opioid synergy. Journal of Neuroscience. 1997;17:7157–7165.
Story G.M., Peier A.M., Reeve A.J., et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–829.
Stucky C.L., Abrahams L.G., Seybold V.S. Bradykinin increases the proportion of neonatal rat dorsal root ganglion neurons that respond to capsaicin and protons. Neuroscience. 1998;84:1257–1265.
Stucky C.L., Koltzenburg M., Schneider M., et al. Overexpression of nerve growth factor in skin selectively affects the survival and functional properties of nociceptors. Journal of Neuroscience. 1999;19:8509–8516.
Sugiura T., Tominaga M., Katsuya H., et al. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. Journal of Neurophysiology. 2002;88:544–548.
Sylvén C., Edlund A., Brandt R., et al. Angina pectoris–like pains provoked by intravenous adenosine. British Medical Journal. 1986;293:1027–1028.
Szolcsányi J. Capsaicin, irritation, and desensitization: neurophysiological basis and future perspectives. In: Green B.G., Mason J.R., Kare M.R., eds. Chemical Senses, vol. 2: Irritation. New York: Marcel Dekker; 1990:141–169.
Takeda M., Tanimoto T., Ikeda M., et al. Activation of GABAB receptor inhibits the excitability of rat small diameter trigeminal root ganglion neurons. Neuroscience. 2004;123:491–505.
Tanelian D.L. Cholinergic activation of a population of corneal afferent nerves. Experimental Brain Research. 1991;86:414–420.
Tata A.M., Vilaro M.T., Mengod G. Muscarinic receptor subtypes expression in rat and chick dorsal root ganglia. Brain Research. Molecular Brain Research. 2000;82:1–10.
Thalhammer J.G., LaMotte R.H. Spatial properties of nociceptor sensitization following heat injury of the skin. Brain Research. 1982;231:257–265.
Thalhammer J.G., LaMotte R.H., Heat sensitization of one-half of a cutaneous nociceptor`s receptive field does not alter the sensitivity of the other half. Bonica J.J., Lindblom U., Iggo A., eds. Advances in pain research and therapy, vol. 5. New York: Raven Press; 1983:71–75.
Tillman D.B., Treede R.-D., Meyer R.A., et al. Response of C fibre nociceptors in the anaesthetized monkey to heat stimuli: correlation with pain threshold in humans. Journal of Physiology. 1995;485:767–774.
Tillman D.B., Treede R.-D., Meyer R.A., et al. Response of C fibre nociceptors in the anaesthetized monkey to heat stimuli: estimates of receptor depth and threshold. Journal of Physiology. 1995;485.3:753–765.
Tohda C., Sasaki M., Konemura T., et al. Axonal transport of VR1 capsaicin receptor mRNA in primary afferents and its participation in inflammation-induced increase in capsaicin sensitivity. Journal of Neurochemistry. 2001;76:1628–1635.
Torebjörk E., Ochoa J. Specific sensations evoked by activity in single identified sensory units in man. Acta Physiologica Scandinavica. 1980;110:445–447.
Torebjörk H.E., Hallin R.G. Perceptual changes accompanying controlled preferential blocking of A and C fibre responses in intact human skin nerves. Experimental Brain Research. 1973;16:321–332.
Torebjörk H.E., LaMotte R.H., Robinson C.J. Peripheral neural correlates of magnitude of cutaneous pain and hyperalgesia: simultaneous recordings in humans of sensory judgments of pain and evoked responses in nociceptors with C-fibers. Journal of Neurophysiology. 1984;51:325–339.
Torebjörk H.E., Lundberg L.E.R., LaMotte R.H. Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. Journal of Physiology (London). 1992;448:765–780.
Towers S., Princivalle A., Billinton A., et al. GABAB receptor protein and mRNA distribution in rat spinal cord and dorsal root ganglia. European Journal of Neuroscience. 2000;12:3201–3210.
Treede R.-D., Cole J.D. Dissociated secondary hyperalgesia in a subject with large fibre sensory neuropathy. Pain. 1993;53:169–174.
Treede R.-D., Meyer R.A., Campbell J.N. Comparison of heat and mechanical receptive fields of cutaneous C-fiber nociceptors in monkey. Journal of Neurophysiology. 1990;64:1502–1513.
Treede R.-D., Meyer R.A., Campbell J.N. Myelinated mechanically insensitive afferents from monkey hairy skin: heat-response properties. Journal of Neurophysiology. 1998;80:1082–1093.
Treede R.-D., Meyer R.A., Raja S.N., et al. Peripheral and central mechanisms of cutaneous hyperalgesia. Progress in Neurobiology. 1992;38:397–421.
Treede R.-D., Meyer R.A., Raja S.N., et al. Evidence for two different heat transduction mechanisms in nociceptive primary afferents innervating monkey skin. Journal of Physiology. 1995;483:747–758.
Tsuda M., Koizumi S., Inoue K. Role of endogenous ATP at the incision area in a rat model of postoperative pain. Neuroreport. 2001;12:1701–1704.
Ugawa S., Ueda T., Ishida Y., et al. Amiloride-blockable acid-sensing ion channels are leading acid sensors expressed in human nociceptors. Journal of Clinical Investigation. 2002;110:1185–1190.
Vallbo A.B., Olausson H., Wessberg J. Unmyelinated afferents constitute a second system coding tactile stimuli of the human hairy skin. Journal of Neurophysiology. 1999;81:2753–2763.
Van Hees J., Human C-fiber input during painful and nonpainful skin stimulation with radiant heat. Bonica J.J., Albe-Fessard D.G., eds. Advances in pain research and therapy, vol. 1. New York: Raven Press; 1976:35–40.
Van Hees J., Gybels J.C. C Nociceptor activity in human nerve during painful and nonpainful skin stimulation. Journal of Neurology, Neurosurgery, and Psychiatry. 1981;44:600–607.
Van Steenwinckel J., Noghero A., Thibault K., et al. The 5-HT2A receptor is mainly expressed in nociceptive sensory neurons in rat lumbar dorsal root ganglia. Neuroscience. 2009;161:838–846.
Vulchanova L., Riedl M.S., Shuster S.J., et al. P2X3 is expressed by DRG neurons that terminate in inner lamina II. European Journal of Neuroscience. 1998;10:3470–3478.
Walker A.E., Nulson F. Electrical stimulation of the upper thoracic portion of the sympathetic chain in man. Archives of Neurology and Psychiatry. 1948;59:559–560.
Walker K., Reeve A., Bowes M., et al. mGlu5 receptors and nociceptive function II. mGlu5 receptors functionally expressed on peripheral sensory neurones mediate inflammatory hyperalgesia. Neuropharmacology. 2001;40:10–19.
Wall P.D., McMahon S.B. Microneuronography and its relation to perceived sensation. A critical review. Pain. 1985;21:209–229.
Wang H., Rivero-Melian C., Robertson B., et al. Transganglionic transport and binding of the isolectin B4 from Griffonia simplicifolia I in rat primary sensory neurons. Neuroscience. 1994;62:539–551.
Watson J.J., Allen S.J., Dawbarn D. Targeting nerve growth factor in pain: what is the therapeutic potential? BioDrugs. 2008;22:349–359.
Weidner C., Schmelz M., Schmidt R., et al. Functional attributes discriminating mechano-insensitive and mechano-responsive C nociceptors in human skin. Journal of Neuroscience. 1999;19:10184–10190.
Wess J., Duttaroy A., Gomeza J., et al. Muscarinic receptor subtypes mediating central and peripheral antinociception studied with muscarinic receptor knockout mice: a review. Life Sciences. 2003;72:2047–2054.
Wessberg J., Olausson H., Fernström K.W., et al. Receptive field properties of unmyelinated tactile afferents in the human skin. Journal of Neurophysiology. 2003;89:1567–1575.
White J.C., Sweet W.H. Pain and the neurosurgeon: a forty year experience. Springfield, IL: Charles C Thomas; 1969.
Woodbury C.J., Ritter A.M., Koerber H.R. On the problem of lamination in the superficial dorsal horn of mammals: a reappraisal of the substantia gelatinosa in postnatal life. Journal of Comparative Neurology. 2000;417:88–102.
Woolf C.J., Allchorne A., Safieh-Garabedian B., et al. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor α. British Journal of Pharmacology. 1997;121:417–424.
Woolf C.J., Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7723–7730.
Wu G., Ringkamp M., Hartke T.V., et al. Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. Journal of Neuroscience. 21, 2001. RC140
Xu X.J., Hao J.X., Andell-Jonsson S., et al. Nociceptive responses in interleukin-6–deficient mice to peripheral inflammation and peripheral nerve section. Cytokine. 1997;9:1028–1033.
Yang D., Gereau R.W. Peripheral group II metabotropic glutamate receptors (mGluR2/3) regulate prostaglandin E2–mediated sensitization of capsaicin responses and thermal nociception. Journal of Neuroscience. 2002;22:6388–6393.
Yarnitsky D., Ochoa J.L. Studies of heat pain sensation in man: perception thresholds, rate of stimulus rise and reaction time. Pain. 1990;40:85–91.
Yarnitsky D., Simone D.A., Dotson R.M., et al. Single C nociceptor responses and psychophysical parameters of evoked pain: effect of rate of rise of heat stimuli in humans. Journal of Physiology (London). 1992;450:581–592.
Zahn P.K., Brennan T.J. Incision-induced changes in receptive field properties of rat dorsal horn neurons. Anesthesiology. 1999;91:772–785.
Zhou S., Komak S., Du J., et al. Metabotropic glutamate 1alpha receptors on peripheral primary afferent fibers: their role in nociception. Brain Research. 2001;913:18–26.
Zwick M., Davis B.M., Woodbury C.J., et al. Glial cell line–derived neurotrophic factor is a survival factor for isolectin B4–positive, but not vanilloid receptor 1–positive, neurons in the mouse. Journal of Neuroscience. 2002;22:4057–4065.
Zylka M.J., Dong X., Southwell A.L., et al. Atypical expansion in mice of the sensory neuron-specific Mrg G protein–coupled receptor family. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10043–10048.
Zylka M.J., Rice F.L., Anderson D.J. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron. 2005;45:17–25.
Basbaum A.I., Bautista D.M., Scherrer G., et al. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284.
Bessou P., Perl E.R. Response of cutaneous sensory units with unmyelinated fibers to noxious stimuli. Journal of Neurophysiology. 1969;32:1025–1043.
Burgess P.R., Perl E.R. Myelinated afferent fibres responding specifically to noxious stimulation of the skin. Journal of Physiology (London). 1967;190:541–562.
Campbell J.N., Raja S.N., Meyer R.A., et al. Myelinated afferents signal the hyperalgesia associated with nerve injury. Pain. 1988;32:89–94.
Carlton S.M. Peripheral excitatory amino acids. Current Opinion in Pharmacology. 2001;1:52–56.
Caterina M.J., Leffler A., Malmberg A.B., et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313.
Davis J.B., Gray J., Gunthorpe M.J., et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187.
Dong X., Han S., Zylka M.J., et al. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell. 2001;106:619–632.
Dubin A.E., Patapoutian A. Nociceptors: the sensors of the pain pathway. Journal of Clinical Investigation. 2010;120:3760–3772.
Dubner R., Price D.D., Beitel R.E., et al. Peripheral neural correlates of behavior in monkey and human related to sensory-discriminative aspects of pain. In: Anderson D.J., Mathews B., eds. Pain in the trigeminal region. Amsterdam: Elsevier North Holland; 1977:57–66.
Dussor G., Zylka M.J., Anderson D.J., et al. Cutaneous sensory neurons expressing the Mrgprd receptor sense extracellular ATP and are putative nociceptors. Journal of Neurophysiology. 2008;99:1581–1589.
Gracely R.H., Lynch S.A., Bennett G.J. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain. 1992;51:175–194.
Ji R.-R., Samad T.A., Jin S.X., et al. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68.
Johanek L.M., Meyer R.A., Friedman R.M., et al. A role for polymodal C-fiber afferents in nonhistaminergic itch. Journal of Neuroscience. 2008;28:7659–7669.
Kruger L., Perl E.R., Sedivec M.J. Fine structure of myelinated mechanical nociceptor endings in cat hairy skin. Journal of Comparative Neurology. 1981;198:137–154.
LaMotte R.H., Campbell J.N. Comparison of responses of warm and nociceptive C-fiber afferents in monkey with human judgements of thermal pain. Journal of Neurophysiology. 1978;41:509–528.
LaMotte R.H., Shain C.N., Simone D.A., et al. Neurogenic hyperalgesia: psychophysical studies of underlying mechanisms. Journal of Neurophysiology. 1991;66:190–211.
Lembo P.M., Grazzini E., Groblewski T., et al. Proenkephalin A gene products activate a new family of sensory neuron–specific GPCRs. Nature Neuroscience. 2002;5:201–209.
Lewis T. Pain. New York: Macmillan; 1942.
Meyer R.A., Campbell J.N. Myelinated nociceptive afferents account for the hyperalgesia that follows a burn to the hand. Science. 1981;213:1527–1529.
Miller R.J., Jung H., Bhangoo S.K., et al. Cytokine and chemokine regulation of sensory neuron function. Handbook of Experimental Pharmacology. 2009;194:417–449.
Price D.D., Hu J.W., Dubner R., et al. Peripheral suppression of first pain and central summation of second pain evoked by noxious heat pulses. Pain. 1977;3:57–68.
Raja S.N., Campbell J.N., Meyer R.A. Evidence for different mechanisms of primary and secondary hyperalgesia following heat injury to the glabrous skin. Brain. 1984;107:1179–1188.
Raja S.N., Treede R.-D., Davis K.D., et al. Systemic alpha-adrenergic blockade with phentolamine: a diagnostic test for sympathetically maintained pain. Anesthesiology. 1991;74:691–698.
Sato J., Perl E.R. Adrenergic excitation of cutaneous pain receptors induced by peripheral nerve injury. Science. 1991;251:1608–1610.
Schaible H.G., Von Banchet G.S., Boettger M.K., et al. The role of proinflammatory cytokines in the generation and maintenance of joint pain. Annals of the New York Academy of Sciences. 2010;1193:60–69.
Schmelz M., Schmidt R., Bickel A., et al. Specific C-receptors for itch in human skin. Journal of Neuroscience. 1997;17:8003–8008.
Sluka K.A., Winter O.C., Wemmie J.A. Acid-sensing ion channels: a new target for pain and CNS diseases. Current Opinion in Drug Discovery & Development. 2009;12:693–704.
Stein C., Clark J.D., Oh U., et al. Peripheral mechanisms of pain and analgesia. Brain Research Reviews. 2009;60:90–113.
Story G.M., Peier A.M., Reeve A.J., et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–829.
Torebjörk H.E., Lundberg L.E.R., LaMotte R.H. Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. Journal of Physiology (London). 1992;448:765–780.
Treede R.-D., Meyer R.A., Raja S.N., et al. Peripheral and central mechanisms of cutaneous hyperalgesia. Progress in Neurobiology. 1992;38:397–421.
Treede R.-D., Meyer R.A., Raja S.N., et al. Evidence for two different heat transduction mechanisms in nociceptive primary afferents innervating monkey skin. Journal of Physiology. 1995;483:747–758.
Weidner C., Schmelz M., Schmidt R., et al. Functional attributes discriminating mechano-insensitive and mechano-responsive C nociceptors in human skin. Journal of Neuroscience. 1999;19:10184–10190.
Woolf C.J., Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7723–7730.
Zylka M.J., Rice F.L., Anderson D.J. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron. 2005;45:17–25.