Spinal Pharmacology of Nociceptive Transmission
Introduction
Acute thermal or mechanical stimuli—or chemicals released from damaged tissue—applied acutely to the skin, muscle, or viscera in the absence of prior conditioning or training evoke a constellation of well-defined behavior and characteristic changes in autonomic function that are defined as nociception. As Sherrington wrote in 1906, “Stimuli become adequate as excitants of pain when they are of such intensity as threatens damage to the skin.” The composition of the behavioral sequelae to such stimuli in the unanesthetized, intact animal varies with the state of arousal, species, and age but will typically include signs of agitation, vocalization, and coordinated efforts to escape (e.g., withdrawal of the limb) or to attenuate the magnitude of the stimulus (e.g., licking or shaking the stimulated limb). The more intense the acute stimulus, the greater the pain indices (e.g., decreased response latency or increased magnitude of responding). With frank tissue injury or inflammation, the organism will often display evidence of ongoing pain behavior even after the injuring stimulus has been removed, and the same stimulus may now elicit an enhanced magnitude of pain behavior. The state corresponding to this facilitated behavioral response is referred to as “hyperalgesia.” Pragmatically, if the hyperalgesia includes an exaggerated response produced by a frankly non-noxious stimulus (e.g., light brushing of the skin), we may further define this second component as allodynia. Our aim is to understand the pharmacology of the systems that mediate these behaviorally relevant phenomena. Such systems may be considered in terms of the overall organization of the encoding substrates.
The present chapter seeks to provide an overview of the pharmacology associated with the several components of this afferent trafficking, with an emphasis on the effects of such agents on the pain behavior of the organism.
Thus, combined study of the behavioral states induced by specific and well-defined nociceptive stimuli with specific effort to assess receptor pharmacology within terminal regions of the anatomical tracts through which this information projects allows us to define the behavioral relevance of these systems to nociceptive processing. Such focal pharmacological manipulation in the intact and unanesthetized animal is achieved through the delivery of drugs in a reliable, delimited manner to specific regions of the central nervous system (CNS). In the brain, placement of intracerebroventricular cannulae permits assessment of central action but affords little anatomic specificity of the site of action. However, stereotaxic placement of microinjection cannulae combined with small injection volumes and iontophoretic administration of agents helps define local CNS pharmacology. Spinal drug delivery using chronic catheters or acute injections has permitted examination of the pharmacology of spinal systems that alter nociceptive transmission (Yaksh and Malkmus 1999). Factors governing the degree of localization of drug action after intracerebral or intrathecal delivery have been reviewed intensively elsewhere.
Excitatory Transmitters in the Afferent Components of Nociceptive Processing
The following sections consider the pharmacology of the systems that subserve the rostral flow of information generated by small afferent input into the dorsal horn and the subsequent projections via crossed and uncrossed tracts into the brain stem and diencephalon.
Primary Afferents: Transmitters and Receptor Systems
Physiology of the First-Order Synapse
Several properties characterize the nature of the interaction between primary afferent fibers and second-order neurons.
Post-synaptic Effect
Single-unit recording has indicated that primary afferent stimulation results in powerful excitation of dorsal horn neurons. Dating from the earliest systematic studies (Hongo et al 1968), there has been no evidence that primary afferents induce monosynaptic inhibition in the dorsal horn (see, for example, reviews of dorsal horn function: Light 1992, Willis 2001). This property suggests that the putative afferent transmitters should largely be characterized by their ability to evoke excitatory post-synaptic potentials (EPSPs) in second-order dorsal horn neurons.
Multiple Neurotransmitters in a Terminal
Stimulation of nerve filaments at intensities that activate small, slowly conducting afferents typically reveals the existence of at least two populations of EPSPs that are believed to be monosynaptic: (1) fast and of brief duration and (2) delayed and of extended duration (Urban and Randic 1984, King et al 1988, Schneider and Perl 1988, Gerber and Randic 1989a, Yoshimura and Jessell 1989). Although the presence of different EPSPs on the same membrane may reflect monosynaptic input from two different families of axons and/or the presence of interneurons contributing to the slow EPSP, such multiple EPSP morphologies in fact also reflect the presence of several distinct classes of neurotransmitters released from a given terminal acting on the dorsal horn neuron, including excitatory amino acids (Jessell et al 1986; Schneider and Perl 1988; Gerber and Randic 1989, 1989), purines (Fyffe and Perl 1984), and peptides (Ryu 1988, Murase et al 1989). Release of multiple transmitters from a single terminal at a single synapse is supported by electron microscopy, which frequently shows the presence of morphologically distinct (small clear-core versus large dense-core) populations of vesicles within the same terminal bouton (see Hokfelt 1991). These differences are consistent with the broader appreciation in neurobiology that such morphologically distinct vesicles reflect the co-containment of distinct classes of releasable neurotransmitters within the same terminal (De Biasi and Rustioni 1988). Examination of the distribution of glutamate indicates, for example, that it is probably contained in small open-core vesicles whereas large dense-core vesicles are believed to contain peptides (see Hokfelt 1991).
The association of peptides with dense-core vesicles and amino acids with clear-core vesicles has practical consequence when it comes to the depolarization/secretion properties of these transmitter classes. Dense-core vesicles typically reside farther from the synaptic density than clear-core vesicles do. The intracellular Ca2+ required to couple local depolarization to vesicular release arises from voltage-dependent Ca2+ channels within the synaptic density. Thus, in general, greater depolarization (associated with a higher firing frequency as observed after tissue injury) is required for the intracellular Ca2+ concentration to reach the mobilization threshold in the vicinity of the dense-core vesicles (Lundberg 1989, Verhage 1991). This association supports the notion that peptide release is comparatively enhanced with persistent activation.
Stimulus Intensity and Afferent Release
As reviewed elsewhere in this volume, an important characteristic of the primary afferent–encoding process is that the magnitude of the generator potential and the frequency of the action potential are largely a function of peripheral stimulus intensity. At the spinal terminal, larger generator potentials lead to the progressive opening of more voltage-sensitive calcium channels that serve to mobilize vesicles for release of transmitter. Accordingly, transmitter release and post-synaptic depolarization will typically be a function of action potential frequency. Importantly, as reviewed below, coupling between afferent traffic and release can be significantly increased or decreased by local modulatory factors that regulate excitation–secretion coupling (e.g., as in opening of the voltage-sensitive calcium channel, mobilization of synaptic proteins) or terminal depolarization.
Primary Afferent Terminal Calcium Channels
Depolarization of the primary afferent terminal leads to the opening of voltage-gated calcium channels (VGCCs). A variety of VGCCs have been identified as defined by their activation characteristics, structural subunit composition, and pharmacology (Yaksh 2006). Several are present in the dorsal root ganglion (DRG) and primary afferent fiber central terminals (Zamponi et al 2009). Activation of these channels, presynaptic to the primary afferent, serves a number of critical functions: (1) they generate depolarizing membrane current at the terminal, (2) they initiate release of transmitter by promoting the activation of membrane docking proteins such as SNAP 25 and VAMP (Atlas 2010), (3) they initiate phosphorylation of membrane proteins (e.g., N-methyl-D-aspartate [NMDA] and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA] receptors, which can enhance channel efficacy; Wang et al 2010), or (4) they activate cytosolic and membrane enzymes (e.g., phospholipase A2 [PLA2]; Svensson and Yaksh 2002). Blockade of several species of calcium channels, notably those for the N-, T-, and L-type channels, potently diminishes post-synaptic depolarization. Interestingly, direct assessment of peptide release via substance P (SP) receptor internalization has shown that N- but not T- or L-type blockers (Takasusuki 2011) prevent the evoked release of SP from small nociceptive afferents (Zamponi et al 2009, Todorovic and Jevtovik-Todorovic 2011). It should be noted that post-synaptic calcium channels are also important. Post-synaptic currents, initiated by afferent input, are also reduced by N-type channel blockers, but much less so by P/Q-type and L-type channel blockers.
Afferent Transmitters and Their Receptors
The role of distinctive populations of terminals remains to be determined, but the physiological properties of coupling of the respective receptors suggest distinct mechanisms of afferent encoding. An essential characteristic of these agents is their ability to be released into the extracellular milieu following depolarization of the primary afferent terminals. Thus, in vivo, activation of C-fiber afferents elicits the release of SP (Yaksh et al 1980, Kuraishi et al 1989), calcitonin gene–related peptide (CGRP; Saria et al 1986, Morton and Hutchison 1990), vasoactive intestinal polypeptide (VIP; Yaksh et al 1982a), somatostatin (SST; Morton et al 1988), and glutamate (Skilling et al 1988). At present, analysis of laminae I and II of the dorsal horn (regions where small afferents are known to terminate, see Chapter 5) and small DRG cells (considered to be the cells of origin of small unmyelinated and finely myelinated afferent axons) has revealed the presence of a large number of possible transmitter candidates.
As noted above, multiple neurotransmitters are commonly present within any given terminal, frequently the excitatory amino acid glutamate and a peptide such as SP. These neurotransmitters are summarized in Figures 28-1 and 28-2. Given the ability of glutamate, acting through receptor-gated Na+ or Ca2+ channels, to produce rapid EPSPs and the ability of peptides to decrease K+ conductance and yield slow, long-lasting EPSPs, co-containment allows a single terminal to evoke multiphasic post-synaptic events. Distinct populations of afferent fibers can be defined on the basis of their peptide contents (Seybold 2009). For example, histochemical analysis of lumbar DRG cells has typically revealed that 50% contain CGRP and 30% contain SP; 96% of the CGRP-positive cells also showed SP immunoreactivity (Ju et al 1987a, 1987b). Populations of C fibers have been identified as peptidergic (containing, for example, SP and CGRP) and as non-peptidergic (identified by binding of the plant lectin isolectin B4 [IB4]) (Larsson 2009, Liu and Salter 2010). A significant number of large Aβ fibers (up to 20%) are also nociceptive (see Djouhri and Lawson 2004 for review), but little is known about their specific pharmacology, and thus they will be noted but not specifically considered.
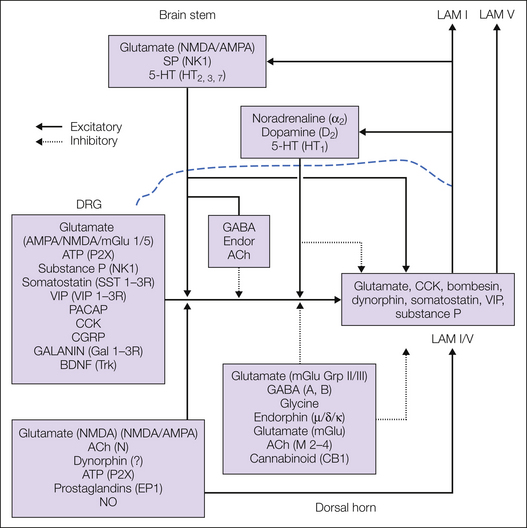
Figure 28-1 Schematic diagram indicating transmitters that are present in primary afferents and superficial (lamina I) and deep (lamina V) projection neurons.
The unifying premise is that the principal post-synaptic effect of primary afferents is monosynaptic excitation. As indicated, both classes of small afferent fibers make contact with the several families of neurons, some of which are interneurons and some of which are projection neurons. The direct primary afferent fiber drive onto the interneuron pool has not been indicated in the figure for simplicity. In either case, the principal transmitter is glutamate. Many of the afferents are also peptidergic, and a significant fraction of these axons with vanilloid receptors (TRPV1) express peptides such as substance P. When strong evidence supports a particular post-synaptic receptor, that is indicated. Some descending bulbospinal fibers (e.g., NE) produce hyperpolarization post-synaptically to elicit inhibition and depolarization presynaptically to engender primary afferent depolarization and inhibition. The latter is indicated by a solid line for differentiation (see text). Details of this schematic are discussed in the accompanying text. ACh, acetylcholine; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ATP, adenosine triphosphate; BDNF, brain-derived neurotrophic factor; CCK, cholecystokinin; CGRP, calcitonin gene–related peptide; Endor, endorphin; GABA, γ-aminobutyric acid; 5-HT, 5-hydroxytryptamine; LAM, lamina; mGlu, metabotropic glutamate; NK1, neurokinin 1; NMDA, N-methyl-D-aspartate; NE, noradrenergic; NO, nitric oxide; PACAP, pituitary adenylate cyclase–activating peptide; SP, substance P; VIP, vasoactive intestinal polypeptide.
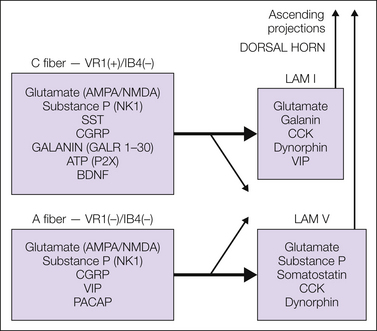
Figure 28-2 Schematic diagram indicating the dorsal horn transmitter and receptor systems that have been shown to regulate the excitability of dorsal horn input (primary afferents) and output (second-order/projection neurons).
As indicated, systems that enhance excitability of the primary afferent terminals and second-order neurons arise from intrinsic neuronal systems and from bulbospinal projections. Similarly, transmitters and receptors that reduce the excitability of afferent processing act presynaptically on both the primary afferent fibers and the second-order/projection neurons. These modulatory influences can arise from both spinally and supraspinally organized systems. Details of this schematic are discussed in the accompanying text. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ATP, adenosine triphosphate; BDNF, brain-derived neurotrophic factor; CCK, cholecystokinin; CGRP, calcitonin gene–related peptide; IB4, isolectin B4; LAM, lamina; NK1, neurokinin 1; NMDA, N-methyl-D-aspartate; PACAP, pituitary adenylate cyclase–activating peptide; SST, somatostatin; VIP, vasoactive intestinal polypeptide.
Amino Acids
Transmitter System
Glutamate is found in 65–80% of DRG and trigeminal ganglion neurons (Battaglia and Rustioni 1988, Tracey, De Biasi et al 1991). Although aspartate was at first considered to be an afferent neurotransmitter, there is no functional evidence for this and only glutamate will be considered. Many sensory neurons exhibiting glutamate immunoreactivity have small perikarya that link them to small primary afferent fibers. Electromicrographic studies using afferent transport markers have shown glutamate to be present in the dorsal horn terminals of large fractions of both myelinated and unmyelinated axons (Broman et al 1993). Specific activation of small afferents with capsaicin evokes the release of glutamate from primary afferent neurons (Jeftinija et al 1991, Sorkin et al 1993). Recovery of glutamate in microdialysates of the dorsal spinal cord in vivo is increased several-fold after the injection of noxious chemicals into the periphery (Skilling et al 1988, Sluka and Westlund 1992, Sorkin et al 1992, Malmberg and Yaksh 1995b, Marsala et al 1995), thus providing additional support for the hypothesis that glutamate is released from afferent nociceptors, although other cellular sources of excitatory amino acids are not excluded by these studies. These findings are consistent with the observation of vesicular glutamate transporters in Aβ, Aδ, and C fibers (Oliveira et al 2003, Todd et al 2003, Hughes et al 2004). Subtypes of glutamate transporters are located predominantly, perhaps exclusively, on specific cell types; for example, excitatory amino acid carrier 1 (EAAC1) is found on dorsal horn and DRG neurons and axonal terminals, whereas glutamate aspartate transporter (GLAST) and glutamate transporter-1 (GLT-1) are found on astrocytes and microglia in the spinal cord. Astrocytic transporters are thought to be important in the newly appreciated tripartite synapse, where they transport excess glutamate into astrocytes, which is then converted into glutamine by the enzyme phosphate-activated glutaminase. Glutamine is released into the synapse, where it is picked up by axon terminals and converted back into glutamate by the resident mitochondria. Under basal conditions, transporter inhibition results in increased levels of extracellular glutamate, spontaneous pain behavior, and evoked hypersensitivity. The latter two phenomena are reversed by glutamate receptor antagonists (Liaw et al 2005). Decreased dorsal horn expression of GLT-1 and GLAST is observed following partial sciatic nerve ligation (Xin et al 2009), chronic constriction injury (CCI; Ramos et al 2010), and paclitaxel neuropathy (Weng et al 2005, Wang et al 2010), thus suggesting that injury induces the loss of astrocytic transporters and the resultant glutamate-mediated excitotoxicity. Interestingly, Ramos and co-authors (2010) reported that administration of ceftriaxone, an agent that up-regulates GLT-1 expression, reverses both the loss of GLT-1 and the pain behavior seen after a variety of injury states. These data conflict with those seen after intraplantar injection of formalin or complete Freund’s adjuvant, where glutamate transporter blockade or knockdown is reported to enhance pain behavior (Niederberger et al 2003, Yaster et al 2011).
Receptors
The post-synaptic excitatory effects of spinal excitatory amino acids are reflected by their potent ability to initiate pain behavior in animals after spinal delivery. These effects are mediated by specific interactions with a variety of glutamate receptors that are broadly divided into ionotropic and metabotropic subtypes.
The ionotropic glutamate AMPA, kainate, and NMDA receptors will be considered first. Receptors in each class are constituted from multiple subunits from different gene families to form transmembrane glutamate-activated pores. Details of assembly are provided elsewhere (Mayer and Armstrong 2004). Intrathecal injections of glutamate receptor agonists have emphasized the importance of both NMDA and non-NMDA sites on dorsal horn neurons in producing powerful algogenic behavior (Aanonsen and Wilcox 1987, Sun and Larson 1991, Coderre and Melzack 1992, Malmberg and Yaksh 1992a, Kontinen and Meert 2002). Equally important is the fact that presynaptic ionotropic autoreceptors, found on primary afferent terminals, regulate the release of glutamate (see Fig. 28-3).
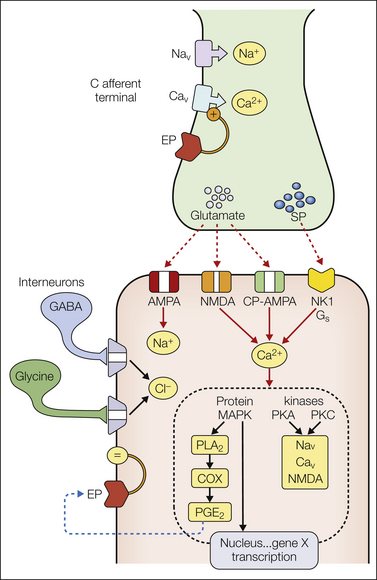
Figure 28-3 First-order synapse for a small afferent axon in the spinal dorsal horn.
As indicated, neurotransmitters are released and depolarize second-order neurons through glutamate (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]/N-methyl-D-aspartate [NMDA], ionophores depolarizing the membrane by increased cation conductance) and peptides such as substance P (interacting with metabotropic receptors; neurokinin 1, NK1). Depolarization and increased intracellular calcium activate a variety of kinases that phosphorylate target proteins. Phosphorylation of ionophores, such as sodium and calcium channels or the NMDA receptor, leads to enhanced activation. Activation of other kinases, such as mitogen-activated protein kinases (MAPKs), may immediately activate enzymes such as phospholipase A2 (PLA2), and lead to the synthesis and release of a variety of arachidonic acid products. Prostaglandins can interact with extracellular pre- and post-synaptic receptors (EPs) to increase the opening of calcium channels or post-synaptically to reduce Cl− conductance in glycine receptors otherwise activated by inhibitory interneurons. In addition, MAPKs can have direct effects on transcription and lead, for example, to increased expression of various proteins, such as cyclooxygenase-2, over a period of hours. See text for additional commentary. COX, cyclooxygenase; CP-AMPA, calcium-permeable AMPA; GABA, γ-aminobutyric acid; PGE2, prostaglandin E2; PKA, protein kinase A; PKC, protein kinase C; SP, substance P.
AMPA Receptors: Together with kainite receptors, AMPA receptors form a division of ionotropic receptors referred to as non-NMDA. Tetrameric AMPA receptors are glutamate-activated ionophores, which when activated, lead to a transient increase in the conductance of small cations (sodium) that results in depolarization. They are composed of two subunit dimers (GluA1–4) and are present in high concentration in the dorsal horn on non–primary afferent neuronal membranes and on ventral horn motor neurons and Renshaw cells (Tachibana et al 1994, Wang et al 2010). Receptor subunits have multiple phosphorylation sites that individually contribute to receptor trafficking, movement into synapses, channel conductance, and open time. Non-neuronal cells are also immunopositive for AMPA receptors. Dorsal horn AMPA receptors show a decrease after rhizotomy (Carlton et al 1998), consistent with the finding that more than one-third of putative nociceptive DRG neurons are immunopositive for AMPA receptors (Willcockson and Valtschanoff 2008). Furthermore, electrophysiological studies show activity mediated by presynaptic AMPA receptors at spinal afferent terminals (Lee et al 2002). Activation of these autoreceptors has been reported to inhibit release of glutamate (Lee et al 2002). A population of AMPA receptors are also Ca2+ permeable, a property endowed by the absence of GluA2 subunits (Hollmann 1991). Such calcium-permeable AMPA receptors are present on lamina I neurons, some of which are neurokinin 1 (NK1) receptor positive, and on outer lamina II neurons (Engelman 1999). A second population of gephyrin-coated lamina I neurons project to the midbrain and thalamus and contain GluA4 rather than GluA1 subunits (Polgár et al 2008); these neurons lack NK1 receptors (Polgár et al 2008). Other NK1-positive neurons in deeper dorsal horn laminae also lack GluR1 subunits (Todd et al 2009).
AMPA Physiology: Work with AMPA antagonists has emphasized that fast synaptic transmission between primary afferent fibers and both superficial and deep dorsal horn neurons is primarily driven by AMPA receptors (Gerber and Randic 1989a, 1989b; Yoshimura and Jessell 1990; Randic et al 1993; Yoshimura and Nishi 1993; Seagrove et al 2004); kainite and NMDA receptors contribute only a small component of the early EPSP. Accordingly, iontophoretically applied AMPA antagonists block the acute excitation in dorsal horn neurons initiated by all classes of afferent fibers. Thus, selective AMPA antagonists are effective in blocking the responses of dorsal horn neurons to acute noxious mechanical and thermal stimuli in normal animals (Dougherty et al 1992, King and Lopez-Garcia 1993). Studies of the calcium-permeable AMPA site in the spinal cord ex vivo have shown that activation of these receptors leads to increased calcium flux and serves to strengthen the synaptic transmission mediated by AMPA receptors (Gu et al 1996). Blocking spinal calcium-permeable AMPA sites with intrathecal Joro spider toxin facilitates C, but not A fiber–evoked responses of dorsal horn neurons (Stanfa et al 2000). Importantly, expression of calcium-permeable AMPA receptors on membranes of NK1-positive (Vikman et al 2008) and NK1-negative lamina I neurons (Larsson and Broman 2008) is regulated by ongoing afferent traffic and increases as a result of tissue inflammation. Afferent evoked activity, mediated via activation of either NMDA or tumor necrosis factor (TNF) receptors in the dorsal horn, enhances AMPA receptor trafficking (Choi et al 2010, Tao 2010).
AMPA-Mediated Behavior: Intrathecal injection of AMPA antagonists produces a frank block of the behavioral response to acute aversive stimuli, such as on the hot plate or tail flick test, as well as facilitated states induced by tissue injury (Pogatzki et al 2000, Nozaki-Taguchi and Yaksh 2002). Importantly, at doses that are slightly higher, hindlimb dysfunction occurs after intrathecal delivery, a finding emphasizing the effect on ventral horn function and the probable block of excitatory input from large proprioceptive afferents. Thus, although behavioral analysis suggests that AMPA antagonists alter nociceptive input, their functional profile emphasizes the broad spectrum of end points blocked after the intrathecal delivery of such antagonists. Clinical trials with AMPA receptor antagonists indicated modest anti-hyperalgesia, especially against dynamic allodynia and cold pain, and agents were generally ineffective in reversing spontaneous pain (Sang et al 1998, 2004; Gormsen et al 2009).
Animals genetically engineered to have fewer calcium-permeable AMPA receptors have reduced inflammation-induced pain behavior, whereas animals with decreased GluA2 subunits have prolonged and increased inflammatory hyperalgesia (Hartmann et al 2004). It should also be noted that intrathecal jorotoxin and philanthotoxin, blockers of the calcium-permeable AMPA site, blocked thermal injury–induced mechanical allodynia, carrageenan-evoked thermal hyperalgesia, and mechanical allodynia and had minimal effect on acute thermal escape latencies (Sorkin et al 2001, Jones and Sorkin 2004). Additionally, although AMPA receptor antagonists prevent the development of both primary and secondary hyperalgesia following surgical incision, antagonists specific to the calcium-permeable site selectively block only secondary hyperalgesia (Pogatzki et al 2003). Lack of jorotoxin blockade of primary hyperalgesia is an example of differences between calcium-permeable AMPA and NMDA receptor blockade, thus implying that the second messengers downstream of Ca2+ entry in these two systems trigger distinct second-messenger pathways (Sorkin et al 2008).
Kainate Receptors: Kainate receptors are tetramers of subunits, each with distinct physiological and pharmacological properties (Wilding and Huettner 2001). The subunits GluR5–7 can form low-affinity receptors but develop higher affinity when paired with either KA1 or KA2. When activated, kainate receptors become permeable to monovalent cations (Na+, K+), although variants are reported that are also permeable to Ca2+ (Huettner 2003). Persistent desensitization can occur at low agonist concentrations (Paternain et al 1998). Autoradiography shows dense kainate binding in laminae I and II and less dense binding in deeper laminae (Mitchell and Anderson 1991). Immunohistochemistry shows kainate subunit labeling on perikarya in laminae I–III (Yung 1998). Immunostaining also co-localizes with IB4 and cholera toxin subunit B and is significantly reduced by rhizotomy (Hwang et al 2001). Presynaptic afferent localization is confirmed by identification of kainate subunits on DRG cells labeled with IB4, vanilloid receptor 1, and P2X3 receptors, but not with SP (Lee et al 2001) (Lucifora et al 2006). Both pre- and post-synaptic kainate receptors may play a role in transmission at spinal primary afferent synapses. Presynaptically, kainate subunits are present on primary afferent terminals, where they may serve as autoreceptors (Hwang et al 2001) and increase (Lee et al 1999) or decrease (Kerchner et al 2001) release of glutamate from primary afferents.
Kainate Physiology: Kainate receptor block has revealed an AMPA/NMDA-independent slow potential that was most pronounced for stimulation intensities sufficient to activate high-threshold Aδ and C fibers (Li et al 1999). In addition, kainate receptors are found on inhibitory dorsal horn neurons, and approximately one-third of terminals in the superficial dorsal horn are positive for GABAergic markers and co-stain for kainate receptors (Lu et al 2005). Activation of these receptors may lead to increased release of γ-aminobutyric acid (GABA); paradoxically this may induce an ultimate decrease in GABA inhibition through negative feedback at GABAB autoreceptors (Kerchner et al 2001).
Kainate-Mediated Behavior: Intrathecal kainate receptor–preferring antagonists displayed antinociceptive action in the acute tail flick, hot plate, formalin, and mechanical pain threshold tests, as well as nerve injury hyperpathia (Li et al 1999).
NMDA Receptor: The NMDA receptor is a glutamate-activated calcium ionophore that is constructed from four subunits: two NR1 subunits and two from the NR2 family—the latter have a great deal more variability than the NR1 subunits (Mori and Mishina 1995). There are binding sites for glutamate and an allosteric site for glycine.
NMDA Physiology: Antagonism of the NMDA receptor has been shown to have little effect on acute post-synaptic excitation in the absence of conditioning input (Dickenson and Sullivan 1987) because of Mg2+ blockade under basal membrane voltage conditions.
NMDA-Mediated Behavior: Blockade of spinal NMDA receptors by intrathecal delivery does not alter the acute thermal or mechanical thresholds (Yaksh et al 1995). Accordingly, the details of this receptor will not be further considered here. As reviewed below, however, this receptor does play an important role in augmenting afferent-evoked excitation in the presence of conditioning stimulation.
Metabotropic Receptors: Metabotropic glutamate receptors (mGluRs) are G protein–coupled receptors that are divided into three principal groups based on their intracellular signaling cascades. In group I, mGluR1 and mGluR5 stimulate phospholipase C (PLC), thereby leading to mobilization of intracellular Ca2+, activation of protein kinase C (PKC), and phosphoinositide hydrolysis; groups II (mGluR2 and 3) and III (mGluR4 and 6–8) are negatively coupled to adenylate cyclase. At the spinal level, delivery of group I agonists enhances basal glutamate release, and group II and III agonists diminish evoked glutamate release (Kumar et al 2010). These results suggest that group I mGluRs may be pro-nociceptive by enhancing the spinal release of glutamate whereas group II and III mGluRs may be antinociceptive by suppressing the spinal release of glutamate. This supposition is strengthened by the finding that group I mGluR agonists increase phosphorylation of the spinal NMDA NR2B subunit (Guo et al 2004) and activate the mitogen-activated protein kinases (MAPKs) extracellular signal–regulated kinases 1 and 2 (ERK1 and ERK2) (Karim et al 2001). In parallel, spinal mGluR1 and mGluR5 antagonists reduce the hyperalgesia and receptor phosphorylation engendered by paw inflammation (Guo et al 2004, Montana et al 2009).
Group II agonists produce reductions in basal (Cozzi et al 1997) or stimulated (Battaglia et al 1997) glutamate levels in the caudate and striatum, respectively. In DRGs, more than half the neurons, many of them presumptive nociceptors, are positive for group II (mGluR2/3) receptors. Activation of these receptors is without effect in naïve animals but reduces both pain behavior and single-fiber activation in the sensitized state (Du et al 2008). These effects may be mediated via modulation of transient receptor potential vanilloid 1 (TRPV1) receptors and tetrodotoxin (TTX)-resistant Na+ channels. Systemic treatment with group II agonists reduces pain behavior in both nerve injury and inflammation models. This is thought to be due in great part to presynaptic inhibition of A (including Aδ) fiber input into superficial dorsal horn neurons (Gerber et al 2000b).
Group III mGluR agonists reduce release of glutamate in the nucleus accumbens (Xi et al 2003) and hippocampus (Martin et al 2007). Conversely, local application of the group I agonists dihydroxyphenylglycine (DHPG) (Moroni et al 1998) or (RS)-2-chloro-5-hydroxyphenylglycine (Pintor et al 2000) increases and local application of the group I antagonist 2-methyl-6-(phenylethynyl) pyridine (MPEP) (Thomas et al 2001) decreases glutamate levels in the parietal cortex or striatum in vivo.
Neurokinins
Transmitter System
SP was the first peptide identified as being specific for small sensory afferents and remains the best characterized. It, along with several sequence-similar peptides (e.g., neurokinin A [NKA]), are widely distributed among small IB4-negative DRG neurons whose central terminals synapse in spinal laminae I and inner II (Pan et al 2003) (see Fig. 28-2). Based on axons identified by conduction velocity, about half of C fibers and 20% of Aδ fibers contain SP (McCarthy and Lawson 1989). In addition, populations of bulbospinal-projecting neurons also contain and probably release SP (Hokfelt et al 2000). Spinal cord release of SP is secondary to direct stimulation of central C-fiber terminals by capsaicin (Jhamandas et al 1984), by acute activation of C fibers (Yaksh et al 1980, Go and Yaksh 1987), and by noxious mechanical (Oku et al 1987, Kuraishi et al 1989) and cold (Tiseo et al 1990) stimuli. Using antibody-coated microelectrodes, SP and NKA were found to be released in the superficial dorsal horn in response to noxious thermal, mechanical, and chemical stimuli (Diez Guerra et al 1988; Duggan et al 1988, 1990). Using NK1 receptor internalization as an index of synaptic activity, peripheral noxious stimuli were found to initiate a stimulation intensity–dependent release of SP (Mantyh et al 1995, Allen et al 1997, Honor et al 1999).
Receptor
Several classes of NK receptors have been identified (Almeida et al 2004). These G protein–coupled receptors stimulate PLC, thereby leading to breakdown of phosphoinositol and elevation of intracellular calcium levels. As with other G protein–coupled receptors, when this receptor is occupied, it undergoes internalization (Mantyh 2002). NK1 receptors are densely distributed on superficial dorsal horn neurons, many of which project to the brain stem (rostroventral medulla) and diencephalon (nucleus parabrachialis) (Todd 2002, Spike et al 2003) and to a lesser degree to deeper dorsal horn neurons (Stucky et al 1993). NK3 receptors are also found superficially in the dorsal horn (Ding et al 2002).
Physiology
Spinal delivery of neurokinins, particularly SP, has been shown to (1) evoke activity in nociceptive dorsal horn neurons (Salter and Henry 1991), (2) produce mild agitation (Hylden and Wilcox 1981, Seybold et al 1982), and (3) induce potent hyperalgesia (Yashpal et al 1982, Papir-Kricheli et al 1987, Malmberg and Yaksh 1992a, Hua et al 1999) in unanesthetized animals. At the several tachykinin receptors it appears that NK1 and perhaps NK2 receptors are of most importance in nociception (Fleetwood-Walker et al 1988, Laneuville et al 1988). Spinal NK1 receptor antagonists reduce the afterdischarge in dorsal horn neurons evoked by acute noxious stimulation (Radhakrishnan and Henry 1991).
Behavior
Behavioral studies in animal models have emphasized that intrathecal neurokinin antagonists fail to alter acute nociceptive behavior (e.g., hot plate test) but do diminish the hyperalgesia induced by persistent stimuli, such as in the phase 2 formalin test (Yamamoto and Yaksh 1991, Yashpal et al 1993, Hua et al 1998), carrageenan-evoked thermal hyperalgesia (Gao et al 2003), and visceral nociception (Okano et al 2002, Gaudreau and Plourde 2003). Convergent results have been reported in rats with reduced expression of NK1 protein because of intrathecal injection of antisense oligonucleotides (Hua et al 1998) and in mice lacking the NK1 receptor (Laird et al 2001). NK3-preferring antagonists depress spinal wind-up (Barbieri and Nistri 2001) and central sensitization of a spinal withdrawal reflex (Houghton et al 2000) and reduce hyperalgesia in arthritic models (Zaratin et al 2000).
Calcitonin Gene–Related Peptide
Transmitter System
CGRP-like immunoreactivity is expressed in approximately 45–70% of lumbar DRG neurons (McCarthy and Lawson 1990, Verge et al 1993). Based on identification of axonal conduction velocity, the majority of CGRP-containing neurons were classified as nociceptive (e.g., CGRP in 46% of C fibers, in 33% of Aδ fibers, and in 17% of Aβ fibers) (McCarthy and Lawson 1990). CGRP is released from the spinal terminals of primary afferent neurons by high-intensity mechanical and thermal stimuli, as well as by local injection of irritants (Morton and Hutchison 1989, Garry and Hargreaves 1992).
Receptors
The effects of CGRP are believed to be mediated by the calcitonin-like receptor, that is, a Gs-coupled seven-transmembrane–spanning receptor (Hay Conner et al 2004).
Physiology
Application of CGRP induces spinal facilitation of dorsal horn responses that were blocked by putative CGRP antagonism (Sun et al 2003). Iontophoretic application of CGRP potentiates the depolarizing effects of SP (Biella 1991).
Behavior
Intrathecal delivery of partial CGRP sequences believed to be antagonistic resulted in a reduction in the hyperalgesia induced by intradermal capsaicin (Sun et al 2003) and carrageenan (Bird et al 2006). Spinal delivery of a CGRP antagonist increased thermal escape latency with and without tissue inflammation (Yu et al 1996). In addition, CGRP antagonism diminished the writhing response induced by phenylbenzoquinone (Saxen et al 1994) and the thermal hyperalgesia and tactile allodynia otherwise observed after cord hemisection (Bennett et al 2000).
Somatostatin
Transmitter System
SST immunoreactivity is limited to populations of small cells in DRGs and small dorsal horn neurons (Tessler et al 1986, O’Brien et al 1989, Kiyama and Emson 1990, Zhang et al 1993). SST has also been identified in populations of bulbospinal-projecting cells (Krisch 1981). Early work showed that SST is released from the spinal cord by capsaicin (Gamse et al 1981). Subsequent work indicated differential release of SST in the spinal cord in response to noxious thermal but not noxious mechanical stimuli (Kuraishi et al 1985, Morton et al 1989, Tiseo et al 1990).
Receptors
SST and its analogues act through a family of G protein–coupled receptors (SST1–5) that are widely distributed in the brain and periphery. SST1, 2, and 5 inhibit the opening of voltage-sensitive calcium channels (Olias et al 2004). Binding and parallel immunohistochemistry showed SST receptor subtypes 1, 2, and 3 in laminae I–III and in the ventral horn (Segond von Banchet et al 1999). Some of this immunoreactivity is probably present on interneurons and on terminals of sensory afferents. Immunoreactivity for the SST3 receptor is also present on a large percentage of DRG neurons and motoneurons (Senaris et al 1995).
Physiology
STT has been shown to inhibit spinal dorsal horn neuronal firing in response to noxious stimuli (Randic and Miletic 1978, Sandkuhler et al 1990, Chapman and Dickenson 1992) through a decrease in post-synaptic membrane excitability by activation of inwardly rectifying K+ conductance (Kim et al 2002). Other work has emphasized a biphasic concentration-dependent activation of neurons and long-lasting depression suggesting toxicity (Delfs and Dichter 1983). After intrathecal application, SST increased the hindpaw electromyographic reflex (Wiesenfeld-Hallin 1985) and facilitated thermal nociception (Wiesenfeld-Hallin 1986).
Behavior
Considerable controversy exists regarding the effects of spinal SST and its analogues. Early work suggested that it was antinociceptive. However, other reports indicated that antinociception was observed at doses that resulted in pronounced motor dysfunction (Gaumann and Yaksh 1988, 1989; Mollenholt et al 1988; Spampinato and Ferri 1991). It is probable that important differences are related to the nature of the multiple receptors being activated by the several agonists. The spinal pharmacology of these excitatory and inhibitory receptor-mediated effects has not been fully studied to date.
Vasoactive Intestinal Polypeptide/Pituitary Adenylate Cyclase–Activating Peptide
Transmitter System
VIP and pituitary adenylate cyclase–activating peptide (PACAP) are both structurally related members of the glucagon/secretin superfamily (Dickinson and Fleetwood-Walker 1999). VIP-positive neurons are numerous in primary afferent neurons of the thoracic and, in particular, the sacral spinal nerves, as well as in cranial nerves that innervate viscera (Kuo et al 1985, Kawatani et al 1986, Yaksh et al 1988). VIP protein and mRNA expression are localized primarily in small to medium-sized DRG neurons (Nahin et al 1994, Dun et al 1996). Afferent stimulation, but not spinal capsaicin, releases VIP from the spinal cord (Yaksh et al 1982a, Takano et al 1993).
Receptors
VIP binding is concentrated in spinal laminae I and II (Yashpal et al 1991). PACAP has also been identified in small afferents, which unlike VIP, are capsaicin sensitive. Capsaicin results in the release and depletion of PACAP in the spinal cord (Zhang et al 1997). VIP and PACAP are both recognized by a family of three receptors. Cloning reveals them to be G protein–coupled, adenylate cyclase–activating receptors (Lutz et al 1993). Message for each of the three receptors is present in the spinal dorsal horn, particularly in laminae II–IV (Dickinson et al 1999).
Physiology/Behavior
Iontophoretic VIP and PACAP evoke the excitation of dorsal horn neurons (Xu and Wiesenfeld-Hallin 1991; Dickinson et al 1997, 1999). Intrathecal VIP initiates the facilitation of spinal flexor reflexes, but spinal delivery of a VIP antagonist was without effect on this reflex (Wiesenfeld-Hallin 1989). Application of PACAP or a putative PACAP agonist (maxadilan) resulted in long-lasting spinal depolarization (Xu and Wiesenfeld-Hallin 1996) and hyperalgesia (Narita et al 1996). Conversely, application of a putative PACAP antagonist was found to induce a slow depolarizing response and reduce stimulation-evoked activation in spinal cord slices. Others have reported PACAP-induced inhibition of the C fiber–evoked flexor reflex (Zhang et al 1993), block of the tail flick (Narita et al 1996), and a reduction in formalin-induced flinching (Zhang et al 1993). Accordingly, whether PACAP is nociceptive or antinociceptive is controversial and doubtless depends on the specific receptors and systems examined (Dickinson and Fleetwood-Walker 1999).
Galanin
Transmitter System
Galanin is expressed in DRGs and the spinal dorsal horn (Hokfelt et al 1987, Michener et al 1990). In the dorsal horn, galanin is primarily located in small GABAergic and enkephalinergic cells (Zhang et al 1993, Simmons et al 1995). In the DRG, neither the fiber caliber associated with galanin-positive neurons (Lawson et al 1993) nor the stimuli to which they respond have been characterized. Galanin staining density in the superficial dorsal horn decreases with C- but not with A-fiber stimulation, probably indicating release (Klein et al 1992). The physiological stimuli that evoke spinal galanin release in normal animals have not been defined. However, the peptide does not appear to be released in response to noxious thermal or mechanical stimulation (Morton and Hutchison 1989).
Receptors
Three receptors have been cloned for galanin (Gal1–3) and belong to the superfamily of G protein–coupled receptors (Branchek et al 2000). Activation of either the Gal1 or Gal3 receptor produces hyperpolarization via Gi/o, whereas Gal2 receptor activation leads to stimulation of Gq/11, thereby producing mobilization of calcium (Branchek et al 2000). All three receptor transcripts are present in the DRG and spinal cord (Waters and Krause 2000). Gal1 receptor mRNA is present in lamina II local neurons (Parker et al 1995).
Physiology
Early work indicated that intrathecal galanin facilitates the flexor reflex in response to noxious stimulation at low doses and inhibits it at higher doses (Wiesenfeld-Hallin et al 1988). It is now known that intrathecal Gal1 receptor (Gal1–29) but not Gal2 receptor (Gal2–13)–preferring agonists inhibit spinal SP release, as assessed by NK1 receptor internalization evoked by paw compression. Spinal release of prostaglandin E2 (PGE2) evoked by intrathecal SP was blocked by both Gal1 and Gal2 receptor–preferring agonists. These data were taken to support both a pre- and post-synaptic action for Gal1 receptor and a post-synaptic action for Gal2 receptor at the level of the spinal dorsal horn (Hua et al 2005).
Behavior
Intrathecal low doses of galanin produce a significant reduction in the mechanical threshold (Kerr et al 2000, Liu et al 2001), whereas higher doses are reported to produce vocalization (Cridland and Henry 1988). Based on Gal1 versus Gal2 receptor–preferring agonists, this enhanced sensitivity is believed to be mediated by the Gal2 receptor. Spinal Gal1–29 but not Gal2–11 markedly inhibited the flinching behavior induced by paw formalin, whereas both agents blocked the hyperalgesia induced by intrathecal SP (Hua et al 2004).
Adenosine Triphosphate
Transmitter System
Adenosine triphosphate (ATP) is believed to be released, in part, from primary afferent terminals (Stevens and Fields 2000, Matsuka et al 2001, Gu 2003). In culture, ATP is released from DRG axons following electrical stimulation (Stevens and Fields 2000).
Receptors
Given the multiple subunits, at least 10 functional R-homomeric and heteromeric P2X receptors have been identified (Khakh et al 2001, North 2002). P2X receptors are expressed at a variety of sites on neurons and non-neuronal cells (Kennedy et al 2003, Fields 2004). These effects are antagonized by the local application of antagonists. An important effect on the primary afferent terminal has also been postulated based on the ability of P2X agonists to initiate afferent transmitter release (see below). Current thinking points to an important role of such afferent-evoked ATP release in activating adjacent glia (Stevens and Fields 2000). Further discussion on purines in pain transmission and the results of manipulating its effects on behavior are considered below.
Brain-Derived Nerve Growth Factor
Brain-derived neurotrophic factor (BDNF) is synthesized by small DRG neurons, transported to spinal terminals (Michael et al 1997), and released via capsaicin or electrical stimulation of the dorsal roots (Lever et al 2001). Importantly, this release is maximized by high-frequency burst stimulation and diminished by NMDA receptor antagonism. The role of spinal BDNF after release is not known, although it may serve as a modulator of synaptic transmission (Snider and McMahon 1998). The complexity of its actions is suggested by the observation that although intrathecal BDNF diminishes the formalin flinching response (Siuciak et al 1995), NMDA-evoked responses are enhanced following up-regulation of BDNF in DRGs, and this enhanced excitability is reduced by BDNF-binding receptor protein (Kerr et al 1999).
Mix of Post-synaptic Effects
An important element evident from this component of the review is that the excitatory effects of primary afferent fibers are mediated by multiple transmitters (e.g., amino acids and several peptides) and by multiple receptors for a given transmitter, as with glutamate. Current evidence suggests that high-intensity afferent input initiates the concurrent release of multiple transmitters. Not surprisingly, the post-synaptic consequences are extremely complex. In this instance, concurrent spinal injection of SP and glutamate produces a significant mutual augmentation of the algogenic effect as compared with the injection of either alone (Mjellem-Joly et al 1991; see also Aanonsen and Wilcox 1987). Similar results have been noted after iontophoretic delivery of SP and glutamate onto dorsal horn neurons (Randic et al 1990, Dougherty et al 1993, Leem et al 2001). Conversely, a noxious thermal, mechanical, or subcutaneous irritant (formalin) activates a complex profile of activation of large and small afferents that serves to activate spinal c-Fos or a neuronal marker such as Zif/268. It has been shown that activation of c-Fos by thermal stimuli is reduced by an NMDA or AMPA antagonist whereas Zif/268 expression is unaltered. Following formalin application, c-Fos and Zif/268 expression was reduced by NMDA but not by AMPA antagonism alone (Rahman et al 2002). It is clear that at the level of the first synapse there is a very high degree of pharmacologically defined, behaviorally relevant encoding.
Ascending Afferent Tracts
Spinal Pharmacology of the Spinifugal Neuron
As reviewed in the preceding section, input into the dorsal horn is characterized by the concurrent release of a variety of peptides and amino acids that can each act through multiple receptors present on second-order neurons. The output function of the spinal cord is represented by activity in the projection neurons.
In brief, one may broadly consider that ascending projections arise from the superficial marginal layer (lamina I), from deeper-lying magnocellular neurons (lamina V) with dorsally projecting dendrites, and from deeper-lying cells in laminae VI/X. The ascending systems have been reviewed in detail in other portions of this text (see Chapter 12) and several systematic reviews (Willis and Westlund 1997). Consideration of the pharmacology of these cells takes the form of asking what their respective responses to locally applied agents are and what receptors are co-expressed on cells that contain retrogradely transported label injected into various supraspinal regions.
Marginal cells (in lamina I) are characterized by strong monosynaptic connections with small, often high-threshold primary afferent fibers (Craig 2000). These cells are characterized by a variety of receptors, including those for glutamate (AMPA/NMDA) and neuropeptides (e.g., NK1). Consistent with this pharmacology, marginal cells display glutamate-positive terminals with the morphology characteristic of primary afferent fibers, as well as non–primary afferent neurons. A significant proportion of these cells receive SP-positive connections suggestive of peptidergic primary afferents (Willis 2001, Todd 2002, Morris et al 2004). In addition to the excitatory input from primary afferents and from interneurons, a variety of inhibitory synaptic systems have also been identified on these marginal projection neurons (see the following section on inhibitory modulation in the dorsal horn).
Post-synaptic Effects of Projecting Neurons
As a rule, single-unit recordings suggest that the primary monosynaptic (or short-latency) effect of spinobulbar/diencephalic activity is excitation (Chung et al 1986, Sinclair et al 1991, Apkarian and Shi 1994, Ohara and Lenz 2003). Failure thus far to see evidence of monosynaptic supraspinal inhibition, of course, does not exclude such possibilities in all systems. Afferent-evoked inhibition has indeed been demonstrated in thalamic neurons, but current evidence suggests that this inhibition is mediated by local inhibitory interneurons (Zhang et al 2002). In any case, it seems certain that an important component of the direct spinifugal projection is the frequency-encoded release of excitatory transmitters (Emmers 1976).
Ascending Sensory System Transmitters
As reviewed elsewhere in this text (see Chapters 12 and 17), the intervening links between the spinal cord and higher-order (supraspinal) processing are complex. Heuristically, we may consider these links in terms of (1) the long spinifugal tracts that project and make monosynaptic connections with neurons in the brain stem (medulla, periaqueductal gray [PAG], mesencephalic reticular formation, parabrachial nucleus) and diencephalon (thalamus and hypothalamus) (Willis and Westlund 1997), (2) projections from these brain stem sites to higher diencephalic centers (e.g., “reticulothalamic”), and (3) projections from diencephalic centers to the cortex.
Spinomesencephalic and -diencephalic Projections
As reviewed above, afferent input into the dorsal horn produces an excitatory drive that is characterized by a variety of peptide and excitatory amino acid receptors. This excitation is observed in local interneurons and neurons that project from the spinal cord (see Figs. 28-1 and 28-2).
Amino Acid Projections
Glutamate has been extensively identified in neurons of the spinothalamic and its trigeminal homologue tracts, thus suggesting the probable role of this excitatory amino acid (Magnusson et al 1987, Ericson et al 1995, Persson and Broman 2004).
Peptidergic Projections
Immunohistochemical investigations examining the content of dorsal horn neurons labeled after the injection of a retrogradely transported substance into various brain stem sites have demonstrated spinal neurons containing cholecystokinin (CCK)-like immunoreactivity (LI), dynorphin 1–8, SST, bombesin, VIP, and SP projecting into the bulbar reticular formation (Standaert et al 1986, Nahin 1987, Leah et al 1988). Spinifugal cells containing CCK and dynorphin–LI labeling have been found in and around the central canal. Ascending tract cells located in lamina I and projecting into the spinomesencephalic and diencephalic pathways contain galanin, CCK, dynorphin, and VIP, whereas lamina V cells projecting in a spinoreticular component contain SST (Ju et al 1987a, 1987b; Leah et al 1988; Nahin et al 1989). SP-positive neurons or neurons containing message for preprotachykinin are sparse, but such cells projecting to the thalamus have been found in lamina I, in lamina V, and around the central canal (Battaglia and Rustioni 1992, Battaglia et al 1992, Noguchi and Ruda 1992, Nishiyama et al 1995).
Brain Stem Projection Neurons
Ascending brain stem projections are numerous and complex, and a number will be specifically considered. (1) Serotonin-containing cell bodies in the midline dorsal raphe in the mesencephalon constitute the principal source of serotonin-positive axons traveling rostrally to project throughout the diencephalon and forebrain (Arango et al 2002, Abrams et al 2004). (2) Noradrenergic fibers arising from the locus coeruleus travel rostrally and project throughout the diencephalon and forebrain (Berridge and Waterhouse 2003, Hollis et al 2004). (3) Peptidergic projections that include SP-containing fibers arise from brain stem sites projecting to the parafascicular and central medial nuclei of the thalamus (Sim and Joseph 1992). Distinct SP- and neurotensin-containing projections have also been identified from the parabrachial complex to the central nucleus of the amygdala (Block et al 1989).
Given the importance of these extraspinal terminals, the relative absence of precise information currently available on the transmitters in spinifugal pathways projecting to specific supraspinal regions is surprising. Future studies will probably provide important insight into the identity of the long-tract spinifugal systems and thus the supraspinal organization of afferent input.
Thalamocortical Projections
Though heterogeneous, the majority of thalamocortical projections appear to be excitatory (Jones 1988, 1998). Thalamic projections originating in the ventrobasal complex and projecting to layer IV of various cortical regions, including the primary somatic sensory cortex, have enriched glutamate immunoreactivity (Tsumoto 1990, Kharazia and Weinberg 1994).
Effects of Focally Injected Transmitter Agonists and Antagonists
The presence of projection neurons containing these substances gives rise to the likelihood that they may serve as neurotransmitters released into the supraspinal projection regions of these cells. Given the importance of this ascending linkage, there is surprisingly little information on the nature of the unconditioned pain behavior evoked by microinjection of these agonists into the vicinity of these terminals. In unanesthetized animals, microinjection of glutamate into the terminal region of ascending pathways, notably within the mesencephalic central gray, evokes spontaneous pain-like behavior consisting of vocalization and vigorous effort to escape. Closer examination of the pharmacology revealed the ordering of activity to be NMDA = kainate > quisqualate ≥ D-glutamate. The effects of NMDA were reversed by MK-801 and 2-amino-5-phosphonovalorate, thus emphasizing the involvement of local NMDA receptors (Jensen and Yaksh 1992). These effects are consistent with the extensive literature indicating that stimulation in the central gray can evoke signs of significant agitation (Schmitt et al 1974, Kiser et al 1978, Fardin et al 1984). Failure to observe significant pain behavior following injection of glutamate into either the thalamus or modestly so into the medulla is surprising in view of early work emphasizing that electrical stimulation in this area is able to induce prominent escape behavior (Casey 1971; see Bowsher 1976 for review of the early literature) and given that afferent-evoked excitation of thalamic cells is inhibited by both NMDA and non-NMDA antagonists (Salt 1986).
It should be emphasized that studies examining the behavioral effects arising from the direct activation of supraspinal systems must carefully consider the possibility that complex species-specific behavioral patterns, not necessarily related to pain-evoked behavior, are being activated. Many of the complex behavior patterns evoked by focal activation (e.g., within the mesencephalon) have substantial parallels with activity associated with operationally defined states of fear and anxiety in the so-called defense reaction (see Bandler et al 1991 for review). As discussed below, states of emotionality have an impact on the pain behavior evoked by a noxious stimulus. In the context of the work discussed above, this highlights the difficulty in attempting to define the link in the afferent pathways that process nociceptive information and govern the unconditioned behavior of the animal in a given environment. This subtlety will probably be an important feature of future studies on the behavioral syndromes associated with the pain state in animal models.
Modulation of the Encoding of Afferent-Evoked Activity
The perceptual processes occurring in the brain reflect the peripheral environment based on the information provided by the spinifugal pathways. This spinifugal activity reflects not only the monosynaptic excitatory input from primary afferent fibers (which transduce the physical environment) but also the composite of polysynaptic excitatory/inhibitory components activated by the afferent input. Thus, a dominant principle of the organization of this afferent input is that at all levels of the neuraxis, it is subject to pharmacological influences that increase and decrease these excitatory influences. Psychophysical studies have shown that the reported intensity of a given physical stimulus can be significantly increased or decreased by several manipulations known to alter spinal excitability and produce a state of hyper- or hypoalgesia, respectively. In the following sections, components of the spinal and supraspinal systems that underlie such regulatory contributions are considered.
Spinal Dorsal Horn Receptor Systems
Functional Properties of Dorsal Horn Encoding Endowed by Modulation of Afferent-Evoked Excitation
Several lines of evidence make it clear that the response properties of the dorsal horn neuron and, accordingly, the output carried by spinifugal projections are not simply defined by the nature of the excitatory afferent input but reflect a series of active encoding events that enhance or diminish (1) the release properties of the primary afferent terminal and (2) the excitability of the projection neurons. The presence of intervening segmental and suprasegmental interneurons linking the primary afferent input with the projecting neuron provides additional opportunities for amplification or diminution of the excitatory state of the projection neuron.
Plasticity of Dorsal Horn Systems
The complex neural linkages involving excitatory and inhibitory transmitters clearly allow considerable plasticity in the input–output relationships observed in the dorsal horn. An example of plasticity encoding is the response characteristics of a common class of spinal neurons: the wide–dynamic range (WDR) neurons that lie within the dorsal horn and receive strong convergent mono- and polysynaptic excitatory input from large (Aβ, low-threshold tactile) and small (C, high-threshold polymodal nociceptor) primary afferents (see Light 1992 for general review). The receptive field of these cells is typically complex, with dermatomal regions responding to low-threshold input overlapping or contiguous with regions in which high-intensity thermal or mechanical stimulation is effective in activating the neuron (Willis 1988). The response properties of such cells are, however, not simply defined by the nature of the afferent connectivity, but also by the influence of a number of pharmacologically distinct neuronal systems that modify the reaction of the cell to its afferent input. Two examples of the physiological response properties of these spinal neurons, which demonstrate positive and negative regulation by convergent neuronal influences, are considered below.
Neuronal Receptive Field Size
The effective receptive field of a dorsal horn cell is not invariant. Classic studies have shown that section of the lateral portion of Lissauer’s tract (an intrasegmental projection system arising in part from the substantia gelatinosa [SG]) or topical application of strychnine (a glycine receptor antagonist) increases the size of the sensory dermatome in primates (Denny-Brown et al 1973). Iontophoretic delivery of glycine and GABA antagonists similarly increases the receptive field size of dorsal horn projection neurons (Zieglgansberger and Herz 1971, Lin et al 1996). Repetitive activation of small, typically high-threshold afferent input leads to a significant increase in the size of the receptive field of a given dorsal horn neuron. In contrast, other systems may decrease the size or components of the receptive field that activate a given dorsal horn neuron. μ-Opioid agonists diminish the size of the high-threshold (C-fiber) component of the receptive field but have little or no effect on the low-threshold component (Yaksh 1978).
Neuronal Response to Afferent Input
The magnitude of the response may be altered in the absence of a change in stimulus magnitude. Thus, as noted above, repetitive activation of C fibers will lead to an augmented response to subsequent afferent input, a phenomenon referred to as “wind-up” (Mendell 1966). In addition to modifying the magnitude of response to a given noxious stimulus, local application of glycine or GABA antagonists augments dorsal horn WDR neuron responses to low-threshold (Aβ) afferent input (Khayyat et al 1975, Yokota et al 1979). Conversely, agonists of specific dorsal horn receptor classes, such as those for the μ- and δ-opioid and α2-adrenergic receptors, induce powerful suppression of the small afferent-induced excitation of these cells (see below). Furthermore, consistent with the effects of activating these specific receptor systems, considerable evidence points to a complex set of bulbospinal modulatory substrates that, by acting through these receptor systems, produce corresponding changes in dorsal horn output. Thus, brain stem stimulation can diminish the slope of the response (frequency of discharge)–versus–stimulus intensity curve of dorsal horn neurons, as well as shift the intercept of the stimulus intensity–response curve to the left, indicative of a reduction in the threshold stimulus intensity necessary to evoke activity in the cell (Gebhart et al 1983, 1984). These shifts may be modality specific, thus implicating presynaptic inhibition. Conversely, other input facilitates the response of the dorsal horn to afferent traffic (Suzuki et al 2002). These bidirectional effects on the input–output relationships of the dorsal horn mediated by spinal and supraspinally organized systems indeed form the core property of the original “gate control” formalization proposed by Melzack and Wall (1965; see also Yaksh 1999).
Importance of Spinal Plasticity to Supraspinally Mediated Functions
Understanding the systems that regulate the output function of the spinal dorsal horn has particular relevance to the pain experience. Clearly, issues related to perception, though mediated by higher-order structures, are strongly influenced by the input encoded by the spinal systems. Changes in spinal outflow typically lead to parallel alterations in the response of supraspinal target nuclei to a given stimulus (see, for example, Sherman et al 1997a, 1997b). That is to say, the nature of the experience is strongly driven by information arising from the spinal cord. Alterations in this spinal outflow modify perception of the environment.
Overview of Anatomic Elements That Modulate Dorsal Horn Input–Output Function
Regulation of the input–output function of spinal dorsal horn responses to primary afferent input has pronounced, behaviorally relevant effects on physiological function. These excitatory and inhibitory components arise from several sources: (1) locally organized segmental interneurons (see Figs. 28-1 and 28-3), (2) non-neuronal cells (see Fig. 28-4), and (3) suprasegmentally organized bulbospinal neuronal projections (see Fig. 28-5).
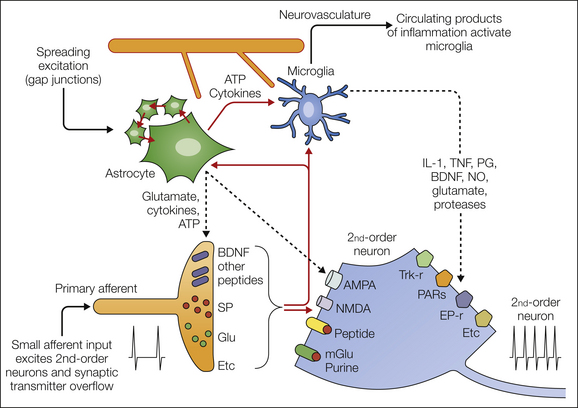
Figure 28-4 Schematic outline of currently considered mechanisms whereby non-neuronal cells might interact with dorsal horn nociceptive processing.
Primary afferent fibers release a variety of products to directly activate second-order neurons. In addition, there is overflow from the synapse, which can gain access to astrocytes, microglia, and extrasynaptic neuronal sites. Astrocytes communicate over volumes of neural tissue by calcium waves through gap junctions. They can also increase the extracellular levels of products such as adenosine triphosphate (ATP), glutamate, and a variety of cytokines. They interact reciprocally with local populations of microglia, which can be activated acutely as evidenced by the increase in phosphorylation of mitogen-activated protein kinases such as p38. Microglia can themselves be activated by neuronal products, notably the chemokine fractalkine, and in turn can release a variety of pro-inflammatory products, which by acting on eponymous receptors enhance the excitability of dorsal horn neurons. Finally, astrocytes and microglia, because of their proximity to the cerebral vasculature, can serve as sensors of circulating products and in this manner allow these products to influence neural function. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BDNF, brain-derived neurotrophic factor; IL-1, interleukin-1; mGlu, metabotropic glutamate; NMDA, N-methyl-D-aspartate; NO, nitric oxide; PARs, protease-activated receptors; PG, prostaglandin; SP, substance P; TNF, tumor necrosis factor.
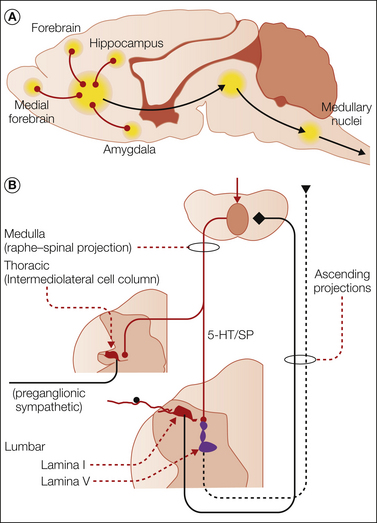
Figure 28-5 A, Convergence on bulbospinal drive arising from the forebrain, hippocampus, and amygdala through the ventral diencephalon and medial mesencephalon and into the rostroventral medulla and leading to activation of bulbospinal projections. B, Local dorsal horn organization in which descending facilitatory pathways arising from the rostroventral medulla are activated by input from lamina I dorsal horn neurons. The bulbospinal 5-hydroxytryptamine (5-HT)/substance P (SP) system projects to deep lamina V neurons and acts though 5-HT3 receptors to increase the excitability of these neurons and initiate or sustain a facilitated state of nociceptive processing. A parallel bulbospinal 5-HT pathway, which is also driven by ascending nociceptive input, has been omitted from the figure for simplicity. Both pathways are discussed in the text.
Locally Organized Segmental Interneurons
Local interneurons are heavily represented in the superficial dorsal horn, laminae I–III. Organizationally, these interneurons are GABAergic and glycinergic (inhibitory) and glutamatergic (excitatory) interneurons, with populations receiving synaptic input from primary afferents. These neurons in turn display synaptic terminations on both afferent and non-afferent terminals. The specific connectivity of these local interneurons is discussed in further detail in Chapter 5. Importantly, the organization of these local systems is functionally arranged to provide local inhibitory sculpting of local afferent-evoked excitation, particularly from large afferents (Khayyat et al 1975, Sivilotti and Woolf 1994). Conversely, the cascading organization of glutamatergic neurons provides linkages that have the ability to amplify afferent input.
Non-neuronal Cells
The spinal dorsal horn displays an abundance of astrocytes and microglia. Astrocytes derive embryologically from glial precursor cells and are classically known to contribute to CNS integrity, metabolic support, and blood–brain barrier function. This organization of pre- and post-synaptic neurons and astrocytes is commonly referred to as the tripartite synapse. Astrocytes form gap junctions with adjacent astrocytes and together form astrocytic nets over which they communicate for considerable distances via calcium waves (Scemes and Giaume 2006). Microglia are resident brain macrophages derived from circulating bone marrow–derived monocytes that enter the neuraxis at birth. These cells have been viewed largely from the perspective of immune surveillance and response to injury and infection. However, it is now appreciated that both these classes of glial cells contribute robustly to local synaptic transmission (see Fig. 28-4). Astrocytes and microglia can influence synaptic transmission by releasing a variety of active products (such as ATP, cytokines). Conversely, transmitters from primary afferents and intrinsic neurons (glutamate, ATP, SP) can overflow from the synaptic cleft to these adjacent non-neuronal cells and lead to activation of them. Neurons may activate microglia by the specific release of a membrane chemokine (fractalkine) that binds to specific microglial receptors. An added element to this role played by these non-neuronal cells is that they have the ability to surveil the circulation because of their anatomic relationship to the neurovasculature. This process is part of a complex cascade referred to broadly as “neuroinflammation.” The functional contribution of spinal non-neuronal cells to spinal nociceptive processing is supported by the observation that inhibitors of microglia/astrocyte activation can produce robust changes in pain behavior. Thus, intrathecal delivery of agents such as minocycline (a second-generation tetracycline) and pentoxifylline has been reported to block microglia activation and diminish hyperalgesic states. Similar metabolic inhibitors that block astrocyte activation (fluorocitrate) can likewise diminish hyperalgesia after nerve and tissue injury.
Suprasegmentally Organized Bulbospinal Neuronal Projections
Spinal projections originating in the brain stem and projecting into the spinal cord are characterized by being largely serotonergic and originating in the medullary midline raphe or by being noradrenergic and originating in several brain stem nuclei, including the locus coeruleus. A subpopulation of the serotonergic projections co-contain SP (Dahlstrom and Fuxe 1964, Bowker et al 1983). Neurochemical studies have shown that these neurons project into the dorsal horn and intermediolateral cell column. These bulbospinal projections may be activated by spinifugal and supraspinal linkages (see Fig. 28-5).
Spinobulbospinal
Thus, small afferent input evokes spinal release of 5-hydroxytryptamine (5-HT) and norepinephrine, indicative of the activation of bulbospinal projections (Tyce and Yaksh 1981). This linkage is mediated, in part, by spinal activation of lamina I neurons, which send projections into the caudal brain stem, particularly the caudal raphe nuclei in the rostroventral medulla (Todd et al 2000). Behavioral and electrophysiological studies have shown that the noradrenergic projections exert potent analgesic effects, as evidenced by reversal of these effects with intrathecal noradrenergic antagonists (Sagen and Proudfit 1984; see Jones 1991 for review). In contrast, the serotonergic projections have potent inhibitory or facilitatory effects on dorsal horn neurons, notably lamina V neurons, depending on the subtype of serotonin receptor involved (Willcockson et al 1984, Sorkin et al 1993), as shown by loss of this effect after destruction of the bulbospinal 5-HT pathways with the intrathecal application of a serotonin neurotoxin (5,6-dihydroxytryptamine) (Suzuki et al 2002) or by blocking one of several serotonin receptors (Zeitz et al 2002). Direct support for the functional significance of these spinobulbospinal serotonergic systems on nociception is provided by the observations that these treatments have been shown to diminish a variety of hyperpathic states associated with inflammation and nerve injury (Porreca et al 2001, Rahman et al 2006, Zhang et al 2009; but see Leong et al 2011). It should be noted that components of these descending pathways also project into the thoracic intermediolateral cell column synapsing onto preganglionic sympathetic neurons (see Fig. 28-5). These bulbospinal projections contribute to the sympathetic response initiated by spinal nociceptive input (e.g., the spinobulbospinal loop) (Ross et al 1984, Minson et al 2002).
Supraspinal–Bulbospinal
The bed nuclei from which the bulbospinal projections arise receive robust input from the rostral systems. Although space is insufficient to review this connectivity in detail, the limbic forebrain systems (septum, nucleus accumbens, hypothalamus) form a multisynaptic pathway projecting through the ventral diencephalon and mesencephalic core into the pons and rostroventral medulla to activate the bulbospinal projections described above and exert a modulatory effect on the spinal systems (see Fig. 28-1), and such linkage has been well documented (Behbehani and Fields 1979). Thus, stimulation in specific zones of the mesencephalic PAG can initiate an aversive response and produce potent effects on blood pressure (e.g., reflecting an excitatory effect on spinal preganglionic sympathetic outflow) (Blessing 2003). These linkages are particularly important because they connect regulation of spinal activation to forebrain systems known to underlie elements of anxiety and emotionality. Thus, classic conditioning studies have demonstrated that increased autonomic outflow and reflex function can result from behavioral paradigms that, for example, pair a painful stimulus with an otherwise innocuous cue (e.g., a light or brief sound). These conditioning paradigms have potent effects on forebrain function, and they are also known to activate the bulbospinal links discussed above. In short, this spinobulbospinal circuit represents a feedforward facilitatory system that can mediate a robust state of facilitated processing reflecting the role played by the supraspinal systems regulating spinal processing.
Pharmacology of Facilitatory Systems Regulating the Excitatory Efficacy of Primary Afferent Input
It is evident that a number of factors may be released that locally enhance excitability of the primary afferent terminal and accordingly alter local transmitter release in the presence of a particular afferent input. In several instances, a given system, such as that for serotonin, may indeed act by a variety of receptors to either enhance or suppress excitability. Receptors post-synaptic to the primary afferent may logically reside on the membrane of local interneurons or non-neuronal cells. These cells can release their respective products at local synapses or into the local extracellular milieu to alter local excitability.
Glutamate
Based on electrophysiology and histochemistry, glutamate is contained and released from primary afferents, spinopetal projections, and a large number of local excitatory interneurons. Extracellular levels of glutamate are also regulated by non-neuronal cells that express glutamate transporters. In the presence of various stimuli, these astrocyte pools can be released into the local milieu and contribute to local glutamatergic activity (Schousboe 2003, Fellin and Carmignoto 2004). This extracellular glutamate can then exert an effect on local activity through a variety of ionotropic and metabotropic receptors.
Glutamate Ionotropic Receptors
As reviewed above, the AMPA but not the NMDA receptor plays a pivotal role in the acute excitation initiated by release of glutamate from afferent terminals. The focus of this section is on the role of the NMDA ionophore in facilitating afferent-evoked excitation. An important component is its ability to produce significant increases in intracellular calcium. Previous comments above on Ca2+-permeable AMPA and kainate receptors should accordingly also be considered in the context of the present discussion focusing on the facilitatory effects of glutamate; however, it should be kept in mind that the subcellular machinery downstream of these various receptors differs.
NMDA Receptor: The NMDA receptor is a glutamate-activated calcium ionophore derived from a series of pore-forming and auxiliary subunits (Glu1, Glu2A through 2D, and Glu3A and 3B receptors) that determine the functional properties of native NMDA receptors (Mori and Mishina 1995). On the primary afferent, NMDA receptors are located preterminally on small primary afferents (Liu et al 1997, Li et al 2004). On non-afferent terminals, NMDA receptors are extensively distributed on both interneurons and projection neurons. NR2A and NR2B are the prevalently expressed NR2 subunits in the spinal cord (Momiyama 2000). NR2A subunits appear largely at the synapse. Conversely, NR2B subunits appear to be located extrasynaptically. It has been speculated that extrasynaptic receptors participate in the presence of high levels of extracellular glutamate.
Physiological Effects: Activation of the NMDA receptors present on small afferents initiates the release of SP, whereas antagonism of NMDA, but not AMPA receptors, has been reported to diminish release of SP from small primary afferents (Liu 1997, Marvizon et al 1997, Malcangio et al 1998). This activation by NMDA may reflect both depolarization of the terminal by the ionophore and the increased intracellular calcium that results from activation of the NMDA ionophore (see Fig. 28-3).
Direct post-synaptic activation of NMDA receptors can initiate a slow component of the afferent-evoked excitatory post-synaptic current (Popescu and Auerbach 2004). However, in the spinal dorsal horn, in the absence of conditioning stimulation the NMDA receptor fails to be functional in the presence of glutamate. This lack of activation reflects at least in part the presence of an Mg2+ ion that occupies and occludes the pore at resting membrane potential. In the presence of persistent membrane depolarization, as with frequent stimulation of C fibers, the membrane is adequately depolarized, the Mg2+ block is removed, and the channel becomes functional, with large amounts of calcium and associated currents being passed (Dickenson and Sullivan 1990). Consistent with acute activation of the AMPA ionophore and failure of the NMDA ionophore to be activated in the presence of a normal resting membrane potential, NMDA antagonists typically have little effect on acute afferent-evoked activity in dorsal horn neurons (Davies and Watkins 1983, Headley et al 1987, Sher and Mitchell 1990). In contrast, repetitive C- but not A-fiber input yields a highly augmented response to a subsequent C-fiber stimulus (Mendell and Wall 1965, Mendell 1966, Woolf and Wall 1986, Woolf and King 1987). Blockade of both AMPA and NMDA receptors will prevent the onset of wind-up and diminish the increases in receptive field size otherwise observed (Davies and Lodge 1987; Dickenson and Sullivan 1987, 1990; Woolf and Thompson 1991; Ren et al 1992).
Behavior: The behavioral effects of activating spinal NMDA systems suggest that they may play a role in facilitating the organized response of the animal to a given noxious stimulus. Thus, direct activation of spinal NMDA receptors with intrathecal agonists induces an augmented response to a noxious thermal stimulus (i.e., hyperalgesia; Moochhala and Sawynok 1984, Cridland and Henry 1986, Aanonsen and Wilcox 1987, Malmberg and Yaksh 1992b). The observation that repetitive stimulation of small afferents yields exaggerated activation of dorsal horn neurons has particular behavioral correlates. Injection of an irritant such as formalin into the paw will result in an initial burst of small afferent activity, followed by a prolonged low level of afferent discharge (Heapy et al 1987, Puig and Sorkin 1996). Single-unit recordings from dorsal horn WDR neurons display a biphasic change in activation (early, phase 1, and later, phase 2). This temporal profile observed with single-unit activity is mirrored by behavior. After intraplantar injection of formalin, the animal displays an initial transient phase of flinching and licking of the injected paw (phase 1), followed after a brief period of quiescence by a second prolonged phase of licking and flinching of the injected paw (Yaksh et al 2001b). Significantly, spinal delivery of AMPA but not NMDA antagonists diminishes the first phase, but both agents will reduce the second-phase response when assessed as single-unit activity (Chapman and Dickenson 1995) or as flinching behavior (Coderre and Melzack 1992, Chaplan et al 1997, Nishiyama et al 1999). Antagonism of allosteric enabling sites, such as the “glycine site” associated with NMDA receptors, induces similar depressive effects on dorsal horn neuronal responses facilitated by repetitive input (Dickenson and Aydar 1991). Intrathecal delivery of these agents also depresses facilitated pain states such as observed in the formalin test (Nishiyama et al 1998). This functional profile of spinal NMDA receptor antagonism, distinct from that observed with AMPA antagonism, is consistent with the lack of effect of NMDA antagonists on acute thermal and mechanical nociceptive thresholds (Yaksh et al 1995). These effects, however, indicate a prominent depressive effect on facilitated states, such as the second phase of the formalin test or the exaggerated reactivity to thermal and mechanical stimuli applied to cutaneous (Ren et al 1992a, 1992b; Yamamoto et al 1993; Hama et al 2003) or visceral (Gaudreau and Plourde 2004) tissues that are inflamed (as with carrageenan or mustard oil). Accumulating evidence implicates the importance of NMDA receptor subtypes, particularly NR2B subunit–containing receptors, in facilitated nociception (Taniguchi et al 1997, Boyce et al 1999). This subunit, as noted, possesses a restricted distribution in the superficial dorsal horn (Boyce et al 1999). It should be noted that although spinal NMDA antagonists have clearly been shown to exert anti-hyperalgesic action in many behavioral models, a discrepancy is evident between the several types of behaviors that remain at present unexplained. Thus, the primary hyperalgesia and secondary tactile allodynia observed after a cutaneous incision or a focal thermal injury to the paw is prevented by intrathecal AMPA but not by NMDA antagonists (Zahn and Brennan 1998, Zahn et al 1998, Nozaki-Taguchi and Yaksh 2002).
Metabotropic Glutamate Receptors
Receptors: Eight G protein–coupled mGlu receptors have been cloned and characterized into three groups (I–III) on the basis of their sequence homology and biochemical and pharmacological properties. Group I mGlu receptors (mGlu1/mGlu5) are coupled to a Gq-like protein and stimulate PLC. High levels of mGlu1 and mGlu5 receptor protein are found in laminae I and II of the rat dorsal horn (Vidnyanszky et al 1994, Boxall et al 1998, Berthele et al 1999, Jia et al 1999). mGlu5 receptors have also been identified in astrocytes (Balazs et al 1997).
Physiology: Activation of group I mGlu receptors produces long-term potentiation of sensory transmission in the SG region of the spinal cord (Gerber et al 2000a) mediated through enhanced release of excitatory transmitter (Park et al 2004). Intrathecal group I agonists initiate a potent thermal and mechanical hyperalgesia (Fisher and Coderre 1998). Conversely, group I mGlu receptor antagonists reduce the sustained activity of dorsal horn neurons initiated by the cutaneous application of mustard oil but have little effect on activity initiated by sustained tactile stimulation (Young et al 1994, 1995) and reduce the secondary thermal hyperalgesia produced by knee joint inflammation (Zhang et al 2002). The specific importance of the spinal mGlu1 receptor has been confirmed by knockout of spinal mGlu1 receptor protein with intrathecal antisense oligonucleotides. In these studies, deep dorsal horn neurons were strongly excited by innocuous stimuli applied to their peripheral receptive fields but displayed reductions in their sustained excitatory responses to the selective C-fiber activator mustard oil and in response to an mGlu1 receptor agonist (Young et al 1998).
Behavior: In sheep, intrathecal group I mGlu receptor antagonism diminished inflammation-induced hyperalgesia (Dolan and Nolan 2002). The probable contribution of spinal mGlu5 receptor was also supported by the ability of selective antagonism to reverse the primary mechanical hyperalgesia otherwise observed following inflammation in the absence of any change in the response to noxious mechanical or thermal stimulation and in the absence of conditioning inflammation in rats and sheep.
Serotonin
Endogenous System
Bulbospinal serotonin-containing projections arise from the midline caudal raphe nuclei and project to the spinal dorsal and ventral horns (Dahlstrom and Fuxe 1964). These cells contain and release a variety of transmitters, including serotonin and SP (Bowker et al 1983). High-intensity lumbar afferent stimulation has been shown to activate the spinobulbospinal pathways and lead to spinal release of serotonin (Tyce and Yaksh 1981). Current evidence suggests that activation of this bulbospinal circuit is dependent on local circuits (e.g., from the PAG; Behbehani and Fields 1979), as well as on spinobulbar input arising from NK1 receptor–bearing spinal marginal cells that project into the medullary brain stem (Todd et al 2000).
Receptors
At least seven major subtypes of serotonin receptors exist (Hoyer et al 1994), several of which have been identified in the spinal cord (5-HT1, 5-HT2, 5-HT3, and 5-HT4). Message for a variety of the 5-HT subtypes has also been identified in DRGs, including the 5-HT1B, 5-HT1D, 5-HT2A, 5-HT2C, 5-HT3, and 5-HT7 receptors (Pierce et al 1996). Autoradiographic, electrophysiological, and/or pharmacological studies show the presence of 5-HT1A, 5-HT1D, 5-HT2A, and 5-HT3 receptors on sensory neurons (Hamon et al 1989; Todorovic and Anderson 1990, 1992). Specifically, 5-HT1A and 5-HT1D receptors are present on unmyelinated peptidergic DRG neurons (Cardenas et al 1997, Potrebic et al 2003). 5-HT2A receptors are distributed in the spinal cord in lamina II inner and are particularly dense in lamina IX. Immunoreactive cell bodies were found to be numerous in lamina IX. Immunoreactivity is mainly post-synaptic on dendrites and cell bodies with a significant incidence of “non-synaptic” localization (Doly et al 2004). 5-HT3 receptors are expressed by small myelinated (Aδ) afferents and a small population of largely non-peptidergic C fibers (Zeitz et al 2002), as well as by dorsal horn interneurons. Immunohistochemical and autoradiographic studies have revealed that 5-HT3 receptors are located mainly in the central terminals of primary afferents and, in part, are post-synaptic on dorsal horn neurons (Kidd et al 1993, Morales et al 1998), fibers, and terminals. In situ hybridization studies have shown that DRG cells contain mRNA sequences that code for both the 5-HT3A and 5-HT3B subunits (Kia et al 1995). Most of the dorsal horn axons immunoreactive for 5-HT3A subunits are associated with capsaicin-sensitive terminals that do not bind IB4 or contain CGRP (Maxwell et al 2003). 5-HT7 receptors have been found on peptidergic small afferents and are concentrated in laminae I and II (Meuser et al 2002).
Physiological Effects
A variety of these 5-HT receptors are G protein coupled, activate PLC, increase intracellular calcium (5-HT2A–2C, 5-HT7), and stimulate cyclic adenosine monophosphate (cAMP; 5-HT4, 5-HT6, 5-HT7). Others depress cAMP (5-HT1A, 1B, 1D). Yet other 5-HT subtypes are known to be excitatory cation channels (5-HT3) (see Hoyer and Martin 1997, Barnes and Sharp 1999 for review). Given the several functionally distinct populations of receptors at the spinal level, the effects of endogenous or exogenous serotonin will clearly be complex. It is evident that a number of these receptors can indeed couple in such a manner to facilitate transmission through the dorsal horn. In some instances this effect could be mediated by a direct excitatory effect on the membrane of primary afferent fibers or projection neurons. Thus, intrathecal injection of 5-HT3 and 5-HT7, agonists that depolarize membranes, has been reported to enhance and 5-HT3 antagonists to reduce dorsal horn nociceptive responses (Meuser et al 2002, Zeitz et al 2002). Conversely, activation of raphe–spinal projections electrically or by glutamate has been reported to facilitate and to inhibit spinal nociceptive processing. The facilitatory effect is thought to be mediated by 5-HT1A receptor–mediated inhibition of spinal GABAergic neurons (Zhuo and Gebhart 1991, Zemlan et al 1994). An interesting adjunct to the role of bulbospinal serotonin projections is the possibility that they may potentiate excitability by recruiting so-called silent AMPA receptors on dorsal horn neurons to the post-synaptic membrane (Kerchner et al 1999).
Behavior
Manipulation of a variety of 5-HT receptor systems has indeed been shown to alter behavior. 5-HT3 receptor antagonism or knockdown of 5-HT3 receptor expression has no effect on acute nociception but serves to reduce facilitated states. The second-phase behavior in the formalin test is attenuated after intrathecal injection of a 5-HT3 receptor antagonist (ondansetron) (Oyama et al 1996, Zeitz et al 2002). 5-HT3 receptor antagonists reduce the mechanical hyperalgesia evoked by carrageenan (Eschalier et al 1989). Given the importance of spinal 5-HT3 receptor–mediated facilitation of dorsal horn processing and pain behavior and the apparent role of marginal cell input in driving this spinobulbospinal serotonin loop, it is important to note that destruction of NK1 receptor–bearing marginal cells by the intrathecal delivery of a neurotoxin (SP–saporin) reduces the small afferent–evoked facilitation that is otherwise observed in deep dorsal horn neurons (Suzuki et al 2002). These data jointly emphasize that the physiological and behavioral effects of bulbospinal serotonin are mediated by an effect on the spinal 5-HT3 receptor. Alternatively, spinal delivery of 5-HT3 agonists has also been shown to be antinociceptive, and the effects of PAG stimulation on spinal nociceptive processing is reversed by spinal 5-HT3 antagonism, perhaps reflecting 5-HT3–mediated excitation of GABAergic/glycinergic interneurons (Alhaider et al 1991). Spinal 5-HT7 but not 5-HT3 antagonists blocked the antinociceptive effects of rostroventral medulla application of morphine. In contrast, hyperalgesia was blocked by spinal 5-HT3 but not by 5-HT7 antagonism, thus suggesting that descending inhibitory or facilitatory pathways arising in the rostroventral medulla act at the spinal level through 5-HT7 and 5-HT3 receptors, respectively (Dogrul et al 2009).
Substance P
As reviewed previously, SP is typically released from small primary afferents. NK1 receptors are present on superficial dorsal horn neurons (e.g., marginal cells, in the SG, and to a lesser extent on the dendrites and soma of deeper-lying cell bodies; Todd 2002). Although NK1 receptor activation initiates excitation in dorsal horn neurons, NK1 antagonism has little effect on acute neuronal activation but attenuates the initiation of small afferent–evoked wind-up (De Koninck and Henry 1991, Chapman and Dickenson 1993). In that vein, pharmacological NK1 antagonism (Yamamoto and Yaksh 1991) or knockout of the NK1 receptor in transgenic animals (King et al 2000, Laird et al 2001) or with intrathecal antisense oligonucleotides (Hua et al 1998) has little effect on acute pain thresholds but moderately reduces behavioral states of hyperalgesia secondary to peripheral inflammation (e.g., intraplantar carrageenan). Similarly, blockade or knockout of the spinal NK1 receptor has little effect on the first phase but significantly diminishes the magnitude of the second-phase response to intraplantar formalin (Yamamoto and Yaksh 1991, 1992; Coderre and Melzack 1992a, 1992b; Hua et al 1998). These results suggest that, like other receptor systems post-synaptic to the primary afferent, NK1 agonists initiate a facilitated state in post-synaptic neurons.
Purine (Adenosine Triphosphate)
As reviewed above, ATP may be released from primary afferents (Matsuka et al 2001), but ATP is also released from non–primary afferent neurons and from non-neuronal cells, particularly astrocytes (Gu 2003). ATP acts as a fast excitatory neurotransmitter through a variety of ionotropic P2X (P2X1–7) and metabotropic P2Y (P2Y1, 2, 4, 6, 11, 12, 13, 14) receptors in the superficial laminae of the dorsal horn (Edwards et al 1992, Bardoni et al 1997).
Purine receptors on the central terminals of primary afferent neurons were first demonstrated by immunochemical studies using antibodies to P2X subunits (Vulchanova et al 1996, 1997). Thus, P2X3 immunoreactivity is restricted to lamina II inner and disappears after axotomy or following the destruction of IB4-positive afferent fibers with IB4–saporin (Vulchanova et al 2001). Immunostaining has also shown that P2X receptor subunits are expressed on sensory neurons that contain SP, CGRP, and SST (Petruska et al 2000). All P2X receptors are permeable to small monovalent cations; some also have significant calcium or anion permeability. Their activation by selective agonists evokes the release of a variety of neurotransmitters in different systems, including glutamate, GABA, glycine, and noradrenaline (Khakh and Henderson 1998, Boehm 1999, Hugel and Schlichter 2000, Rhee et al 2000, Deuchars et al 2001a, Gomez-Villafuertes et al 2001). Accordingly, the presence of P2X receptor subunits on the central terminals of primary afferent neurons raises the possibility that centrally released ATP (Fyffe and Perl 1984) may act on the central terminals of primary afferent neurons to either modulate or directly evoke the release of glutamate and neuropeptides. Such an effect on the release of glutamatergic primary afferent terminals in laminae I–III and V appears to be likely (Li et al 1998, Nakatsuka and Gu 2001). The Ca2+ permeability of P2X receptors makes it likely that they may, like other ligand-gated cation channels (including NMDA), contribute to the terminal release of other neurotransmitters (North 2002). These effects suggesting a role in facilitated processing are consistent with reports discussed in the preceding sections wherein intrathecal application of P2X receptor antagonists decreased inflammation-induced hyperalgesia (Zheng and Chen 2000). Non-neuronal cells express several subtypes of P2X and P2Y receptors. In microglia, ATP induces the activation of p38 or ERK1/2 MAPKs, which results in the release of TNF-α and interleukin-6 (Hide et al 2000, Inoue 2002, Inoue et al 2003). It should be noted that interpretation of purine actions is complex. Aside from the possibility of direct effects of ATP that are not mediated by “purinergic receptors” (Joo Choi et al 2003), ATP is rapidly converted to adenosine, which has its own specific receptor-mediated effects (Lao et al 2001).
Proteinases
A variety of proteases, such as thrombin and trypsin, can directly activate neuronal and non-neuronal systems through their interaction with proteinase-activated receptors (PARs). PARs (1–4) are families of guanine nucleotide binding protein–coupled receptors that when activated by serine proteases, induce an increase in intracellular calcium (cf. Russell and McDougall 2009, Garcia et al 2010). PAR1, 2, and 4 have been identified in spinal neurons and DRGs (Niclou et al 1998, Steinhoff et al 2000). Protein and message for all PARs have been demonstrated in primary rat astrocyte cultures (Wang et al 2002). Amino acid sequences acting on PAR1 or treating superfused spinal cord tissue with trypsin stimulates the release of SP (Steinhoff et al 2000). Similar intrathecal delivery results in hyperalgesia that is blocked in mice with targeted deletion of either the SP precursor or the NK1 receptor (Vergnolle et al 2001). Intrathecal delivery of PAR1 agonists will similarly produce thermal hyperalgesia and induce spinal release of prostaglandins (Koetzner et al 2004a).
Cytokines and Chemokines
Cytokines and chemokines are a structurally diverse series of secreted signaling molecules that typically act through eponymous receptors. Their role in the classic studies of immunity has shown many of them to be robust activators of a variety of cells. In general, this subject is too broad to review here in any detail. However, it has become increasingly appreciated that a number of these molecules are synthesized and released within the neuraxis and that their respective receptor sites are also found within the CNS and spinal cord on both neuronal and non-neuronal cells. As reviewed above, astrocytes and microglia have increasingly been implicated in regulating excitability of the spinal circuitry involved in facilitated states initiated by peripheral inflammation and nerve injury. Two specific examples related to the role of these cytokines in chronic pain will be cited.
Tumor Necrosis Factor
TNF is a cytokine released from astrocytes and microglia through activation of a metalloproteinase that results in proteolytic cleavage of it from the cell membrane ((McGeehan et al 1994, Zhou et al 2010). TNF binds to TNFR1 and TNFR2, both having been identified in astrocytes and microglia (Dopp et al 1997) and in neurons (Zhang et al 2010). In nerve injury and in the presence of chronic inflammation there is a prominent increase in TNF. TNF has been shown to activate DRG cells and initiate a hyperpathic state after intrathecal delivery (Leung and Cahill 2010). In the DRG, TNF serves to increase the TTX-resistant Na+ current through a p38 MAPK–mediated cascade initiated by TNFR1 (Jin and Gereau 2006). Importantly, block of TNF with the use of knockout mice or the intrathecal delivery of antibodies can produce a significant reduction in these hyperpathic states (Lee et al 2009, Zhang et al 2011).
Fractalkine
This protein, also known as CX3CL1, binds to the chemokine receptor CX3CR1. It is constitutively expressed on the neuronal membranes from whence it is released; its receptor is constitutively expressed on spinal microglia (Clark et al 2011). Intrathecal application of fractalkine results in microglia activation and a facilitated pain state. Following peripheral nerve injury and persistent inflammation, fractalkine is up-regulated. The functional contribution of fractalkine systems to nociceptive processing is supported by the observation that hyperalgesic states can be attenuated by the intrathecal delivery of a neutralizing antibody to the fractalkine receptor (Milligan et al 2008, Clark et al 2011).
Eicosanoids
One result of the increased intracellular Ca2+ that occurs in the presence of persistent dorsal horn depolarization is PLA2 activation leading to increases in intracellular arachidonic acid (see (Svensson and Yaksh 2002, Burke and Dennis 2009). A wide variety of eicosanoids are subsequently synthesized by three enzymatic pathways: (1) prostaglandins via cyclooxygenases (COXs); (2) leukotrienes, hydroxyeicosatetraenoic acids (HETEs), hepoxilins, and lipoxins via 5-, 12- and 12/15-lipoxygenases (LOXs); and (3) epoxyeicosatrienoic acids (EETs) and HETEs via cytochrome P450 (Buczynski et al 2009). Although early studies demonstrated a peripheral role of eicosanoids in nociception, it is now accepted that these systems also exert potent effects within the spinal cord dorsal horn.
Cyclooxygenase
Current work has shown that at least two constitutively expressed prostaglandin synthases called cyclooxygenases (COX-1 and COX-2) are present in spinal neurons and non-neuronal cells, such as astrocytes (O’Banion 1999). COX-1 is predominantly expressed in naïve microglia (Yermakova et al 1999), but COX-2 expression has been noted in microglia after stimulation (Bauer et al 1997). In vivo perfusion studies have shown that local depolarization (Yaksh 1982), afferent stimulation (Ramwell et al 1966; Coderre et al 1990; Malmberg and Yaksh 1995a, 1995b), or direct activation of spinal neurons with SP or NMDA results in increased extracellular levels of prostanoids in the spinal cord (Dirig and Yaksh 1999, Hua et al 1999, Yaksh et al 2001a, Svensson et al 2003) (see Fig. 28-3).
Importantly, it should be emphasized that although peripheral COX-2 is inducible (Vane et al 1998, O’Banion 1999), spinal COX-2 is constitutively expressed and appears to be engaged immediately in the presence of the appropriate stimulus (e.g., intrathecal SP/NMDA) (see Svensson and Yaksh 2002, Ghilardi et al 2004). After intracellular formation, these lipidic acids are exported to the extracellular space, where they can then exert powerful effects on adjacent neuronal elements through a family of prostaglandin receptors (DP, EP, FP, IP, and TP). These G protein–coupled receptors with seven transmembrane domains (Armstrong and Wilson 1995, Negishi et al 1995, Versteeg et al 1999) trigger intracellular signals that can be stimulatory as evidenced by activation of adenyl cyclase (Negishi et al 1995, Narumiya et al 1999) and activation of PLC (Yousufzai et al 1988, Birnbaumer et al 1990). Conversely, inhibitory effects have been demonstrated as evidenced by depression of cAMP production (Melien et al 1988, Negishi et al 1989). In situ hybridization and immunohistochemical studies have localized the receptor proteins EP1, EP2, EP3, EP4 (Kawamura et al 1997, Donaldson et al 2001), and IP (Matsumura et al 1995) to the superficial layers of spinal cord, and DP, EP1, EP3, and IP receptors have been detected on DRG neurons (Oida et al 1995, Wright et al 1999).
On the primary afferent terminal, prostaglandins will, via EP receptors, increase Ca2+ conductance through voltage-sensitive calcium channels in DRG neurons (Makhinson et al 1999) and increase secretion of primary afferent peptides such as SP (Nicol et al 1992). Similarly, block of spinal COX-2 will significantly but incompletely decrease the release of SP evoked by strong, tissue-injuring stimuli (Ghilardi et al 2004) or the release of glutamate evoked by intraplantar formalin (Malmberg and Yaksh 1995b).
Post-synaptically, activation of spinal EP2 receptors by PGE2 reduces glycinergic inhibition by phosphorylation of the α3 subunit of the glycine receptor (Harvey et al 2004). As discussed above, glycine plays an important role in regulating afferent traffic in the dorsal horn, and block of spinal glycinergic function can initiate a potent behavioral allodynia (Yaksh 1989).
Behaviorally, intrathecal prostaglandins delivered to the unanesthetized rat evoke hyperalgesia (Yaksh 1982, Taiwo and Levine 1986, Uda et al 1990), whereas spinal COX inhibitors, specifically those selective for COX-2 but not COX-1, suppress the thermal hyperalgesia induced by spinally injected SP or NMDA (Malmberg and Yaksh 1992b, Yaksh et al 2001a), the augmented flexor reflex in adjuvant-treated rats (Malmberg and Yaksh 1992a, Seybold et al 2003), and the behavioral hyperalgesia resulting from intra-abdominal acetic acid (Yaksh 1982), intraplantar formalin (Yamamoto and Nozaki-Taguchi 2002), and peripheral tissue injury (Malmberg and Yaksh 1992a, Yaksh et al 2001b, Du et al 2004). These observations suggest a role for COX products in mediating the spinal facilitated nociceptive processing leading to hyperalgesia. These results further support the assertion that COX inhibitors exert their anti-hyperalgesic effects through inhibition of spinal prostaglandin release.
Lipoxygenase
LOX catalyzes O2 insertion into polyunsaturated fatty acids. Several families of LOX enzymes have been classified according to their activity: 5-LOX, 12-LOX, and 12/15-LOX. Frequently, the several enzyme families have a number of isozymes (Buczynski et al 2009). In the presence of inflammation, there are significant increases in the expression of a variety of LOX products, including 12-HETE and Hepolillin B3 (HXB3) (Buczynski et al 2010). Importantly, intrathecal delivery of 12-LOX– but not 5- or 15-LOX–preferring inhibitors prevented increases in spinal HXB3 and the associated hyperpathia. These agents increased SP release, thus suggesting a presynaptic effect and increased intracellular calcium in DRGs through a TRPV1/TRPA1-mediated action (Gregus et al in revision).
It should be noted that the contribution by lipid mediators is only just beginning to be understood. Thus, it has been shown that spinal cord depolarization enhances the release of linoleic acid metabolites and that when examined, these substances were 12-LOX products found to be potent activators of spinal TRP receptors (Patwardhan et al 2009).
Nitric Oxide Synthase
Nicotinamide adenine dinucleotide phosphate (NADPH)-dependent nitric oxide synthase (NOS) activity is present in the superficial spinal cord and in DRGs (Hecker et al 1994, Mabuchi et al 2004). Synthesis of the second messenger nitric oxide (NO) occurs secondary to increased intracellular Ca2+. This synthesis is undertaken by a variety of constitutively expressed (neuronal [nNOS] and endothelial [eNOS]) and inducible (iNOS) isozymes. These isoforms generate NO by oxidizing a guanidino nitrogen of L-arginine, with molecular oxygen and NADPH being co-substrates (Lohse et al 1998). NO is an intercellular messenger that can diffuse in the extracellular space for distances estimated to be up to 3–400 μm. NO produces cyclic guanosine monophosphate (cGMP) through activation of guanylate cyclase. This leads to the activation of cGMP-dependent protein kinases, phosphodiesterases, and ion channels (Prast and Philippu 2001). NOS, the enzyme responsible for the synthesis of NO, has been found to occur in areas important for nociceptive processing, such as the dorsal horn (Mizukawa et al 1989, Anderson 1992) and DRG cells (diaphorase-positive type B ganglion cells) (Aimi et al 1991, Morris et al 1992). Because NO has the ability to readily penetrate cell membranes, it has been proposed as a probable candidate for a retrogradely acting messenger on presynaptic terminals (O’Dell et al 1991, Schuman and Madison 1991) (see Fig. 28-1). In the hippocampus, NO synthesis can be initiated by NMDA receptor–mediated increases in Ca2+ (Garthwaite et al 1988). The effects of NO are doubtless complex. Thus, NO can sometimes have opposite effects at low and high concentrations. Similarly, as with many transmitter systems, the effects of NO can be manifested by both excitatory and inhibitory transmitter release (Prast and Philippu 2001). Electrophysiologically, the role of spinal NOS has been accordingly complex. Thus, the prolonged discharge in WDR neurons, initiated by repetitive small afferent input, can be suppressed by inhibition of spinal NO synthesis (Haley et al 1992). It has, in fact, been demonstrated that activation of spinal NMDA initiates release of glutamate in a manner that is diminished by NOS inhibition (Sorkin 1993). Initiation of NO synthesis with nitroprusside has mixed effects. Thus, in laminae I and II neurons, the preponderant effect was inhibition and, less so, excitation. In deeper laminae (lamina X), a preponderant excitatory effect was noted (Pehl and Schmid 1997). Behaviorally, the hyperalgesia induced by activation of spinal NMDA receptors (Meller et al 1992, Malmberg and Yaksh 1993b) or by tissue injury (e.g., carrageenan or intraplantar formalin) is blocked by spinal delivery of inhibitors of NO synthesis (Malmberg and Yaksh 1993b). An important question relates to the role of the respective isoforms. Intrathecally administered, selective inhibitors of nNOS diminish carrageenan-evoked thermal hyperalgesia (Sekiguchi et al 2004). Work with selective NOS inhibitors has suggested that iNOS contributes to hyperalgesia in the later stages of carrageenan-induced inflammation whereas nNOS plays a role throughout the entire time course of the injury (Osborne and Coderre 1999). Work with nNOS and iNOS knockout mice, however, has revealed continued hyperalgesia. It was suggested that loss of one isoform could be compensated for by up-regulation of other NOS isoforms, including eNOS (Tao et al 2003, 2004).
Dynorphin
Dynorphin is expressed in laminae I, V, and VI in projection and non-projection neurons and in lamina II local circuit neurons (Ruda et al 1988, Nahin et al 1989). Spinally, dynorphin has been shown to produce motor dysfunction (Stevens and Yaksh 1986) and potent facilitation of pain behavior (Vanderah et al 1996, 2000; Laughlin et al 1997; Lai et al 2001; Kawaraguchi et al 2004). Dynorphin increases intracellular calcium (Tang et al 2000). These effects appear to reflect spinal release of glutamate (or activation of an NMDA receptor) and/or activation of a downstream cascade leading to release of prostaglandin (Koetzner et al 2004b). Thus, the allodynia and hyperalgesia produced by intrathecal dynorphin are blocked by NMDA receptor antagonism (Lai et al 2001, Laughlin et al 2001). This effect appears to be independent of an opioid effect since the actions are uniformly produced by the des-tyrosine version of the peptide. Spinal dynorphin (Dyn 1–17) in low doses is able to antagonize the effects of intrathecal opiates (Fujimoto et al 1990). This effect appears to be produced by Dyn 1–17 but not by other dynorphin analogues (Rady et al 1991) and is not mediated by a κ-opiate receptor. Again, the presence of dynorphin in spinal neurons, as well as its up-regulation following inflammation (Iadarola et al 1988), provides evidence for its possible role as an endogenous “algesic agent.”
Cholecystokinin
A number of groups have reported that CCK, particularly the octapeptide (CCK-8), may diminish the antinociceptive effects of morphine (Faris et al 1983, Wiertelak et al 1992) and reverse the inhibition of dorsal horn neurons produced by morphine (Kellstein et al 1991). Given the presence and release of CCK from the spinal cord (Yaksh et al 1982b), this peptide could serve as an endogenous opioid antagonist. Support for this hypothesis is provided by the observation that CCK antagonists (particularly of the A type) can augment the effects of morphine (O’Neill et al 1989, Kellstein et al 1991). The nature and specificity of this interaction remains to be defined (for review see Baber et al 1989). Thus, Tseng and Collins (1992) reported that intrathecal CCK would antagonize the effects of intraventricular β-endorphin.
Intracellular Processes
The excitability of neurons is governed by a variety of processes that regulate the activation of membrane and cytosolic enzymes, receptors, and channels. Two forms of post-translational protein modification that should be mentioned in conjunction with pain are ubiquitination and phosphorylation. Both processes are fast and reversible by de-ubiquitination and dephosphorylation, respectively. Frequently, phosphorylation or dephosphorylation of elements in the ubiquitination pathway renders their binding sites available for processing, and the two processes work hand in hand to induce permanent or at least long-lasting effects.
Phosphorylation
There is expansive evidence that in the presence of repetitive input, phosphorylation plays an important role in enhancing the input–output function of dorsal horn neurons. Space is inadequate to include an in-depth discussion of this topic, their targets, or the results of phosphorylation on nociceptive processing (see Mao et al 1994, Willis 2002, Obata and Noguchi 2004; see also Chapter 3 for further discussion). These enzymes play important roles in a variety of signal transduction cascades, and several examples will be noted.
Mitogen-Activated Protein Kinase: MAPKs consist of three groups: p38, ERKs, and c-Jun N-terminal kinase (JNK) (Ji et al 2009). Following tissue and nerve injury, these MAPKs can be activated to phosphorylate specific enzyme systems and initiate transcriptional regulation (see Fig. 28-3). Thus, following persistent small afferent activation (e.g., intraplantar formalin) and peripheral nerve injury, these MAPKs are activated (phosphorylated) in spinal microglia and astrocytes and thereby result in the release of algogenic mediators. A specific example is the role played by p38 MAPK, which serves to activate PLA2, an essential link in the eicosanoid cascade leading to prostaglandin synthesis and release (Schmidlin et al 2000). In addition, p38 activation serves as a transcription-activating factor that results in increased message for inducible COX-2. Inhibition of all three MAPK pathways has been shown to attenuate inflammatory and neuropathic pain in different animal models. Development of specific inhibitors of the MAPK pathways to target neurons and glial cells may lead to new therapies for pain management. Although it is well documented that the MAPK pathways can increase pain sensitivity via peripheral mechanisms, this review will focus on the central mechanisms of MAPKs, especially ERKs.
Serine/Threonine Kinases: Several families of enzymes, among them protein kinase A (PKA) and PKC, preferentially phosphorylate specific serine/threonine residues. PKA is dependent on activation of cAMP, typically by stimulatory G protein (Gs)-coupled receptors, such as the β-adrenergic and histamine H2 receptors (Purcell and Carew 2001). PKC is activated by increased intracellular calcium or diacylglycerol, secondary to the activation of phospholipases by Gq-coupled receptors, such as α1-adrenergic or 5-HT2A receptors, (Wajima et al 2000, Toselli and Taglietti 2005). Ca2+/calmodulin-dependent protein kinase II (CaM kinase II) is regulated by the Ca2+/calmodulin complex and may be activated by processes leading to increased intracellular ATP (Makhinson et al 1999, Fang et al 2002). All these enzymes are widely distributed and, as reviewed in the cited references, typically consist of multiple, differentially distributed isoforms. The role of these serine/threonine-phosphorylating enzymes has been widely studied and shown by pharmacological interventions to have profound effects on spinal sensory processing that lead to long-lasting effects on dorsal horn excitability. Several examples will be cited.
Activation of spinal kinases yields specific NMDA receptor subunit phosphorylation; for example, PKA and PKC phosphorylate the NR1, NR2A, and NR2B subunits at serine/threonine sites, and CaM kinase II phosphorylates NR2A and NR2B subunits (Omkumar et al 1996) (see Fig. 28-3). Tyrosine phosphorylation leads to increases and dephosphorylation leads to decreases in NMDA current and channel-opening probability (Wang and Salter 1994, Yu et al 1997). Thus, repetitive depolarization of the membrane will result in increased intracellular calcium through several mechanisms: (1) voltage-gated ion channels, (2) ion-gated channels (e.g., NMDA/calcium-permeable AMPA channels), and (3) G protein–coupled receptors that act to mobilize intracellular calcium (e.g., NK1 receptors). The increased calcium leads to activation of the several kinases. In addition, TrkA and TrkB receptors are present in the spinal cord, and BDNF, perhaps released from primary afferents, can induce tyrosine phosphorylation of the NMDA receptor (Di Luca et al 2001). These events can thus lead to enhanced functionality of the NMDA ionophore.
PKA has been implicated in neuronal sensitization at multiple levels of the neuraxis in multiple cell types. PKA is involved in PGE2- and CGRP-induced modulation of TTX-resistant Na+ current in DRG neurons. In addition, PKA mediates TNF-induced excitability of nociceptive DRG neurons (Zhang et al 2002). Interestingly, PKA agonists increase the production of pro-inflammatory cytokines in microglial primary cultures (Liu et al 2011). In the spinal cord, PKA agonists selectively increase the responses of spinothalamic tract neurons to innocuous but not noxious stimuli over a period of several hours (Palecek et al 1994). The hyperalgesia induced by intradermal capsaicin (Fang et al 2003), thermal injury (Jones and Sorkin 2005), and nerve injury (Gao et al 2005) was blocked or reversed by spinal treatment with PKA antagonists. Synaptic strengthening within the central nucleus of the amygdala following joint inflammation is dependent, in part, on PKA-mediated enhanced NMDA receptor function (Bird et al 2005).
PKC also plays multiple functions in sensitization of the sensory neuron in the DRG (Gold et al 1998) (Natura et al 2005); it is likely that PKC-ε is the relevant PKC isoform at this site (Khasar et al 1999). In the dorsal horn, PKC-γ is concentrated in neurons in lamina II inner but is also found in laminae I and III (Polgar et al 1999). These neurons, for the most part, do not express GABA and are thus thought to be excitatory. Many are interneurons. PKC-β II is more evenly distributed among the three superficial dorsal horn laminae. Both isoforms are up-regulated following nerve injury (Miletic et al 2000). Inhibition of spinal PKC also reduced the hyperalgesia resulting from capsaicin injection and thermal injury (Fang et al 2003, Jones and Sorkin 2005). Knockout of PKC-γ prevents the development of neuropathic pain (Malmberg et al 1997) and significantly blunts peripheral injury–induced sensitization of spinal dorsal horn WDR neurons (Martin et al 2001) without affecting acute nociceptive responses.
Ubiquitination
Mono-ubiquitination (tagging a protein with a single ubiquitin molecule) of plasma membrane receptors, predominantly G coupled-protein receptors, initiates agonist-induced receptor endocytosis, which usually culminates in recycling. In contrast, poly-ubiquitination of receptors results in endocytosis, followed by proteosome lysis. This is exemplified by the CXCR4 chemokine receptor (Marchese and Benovic 2001). δ-Opiate receptors are tagged with ubiquitin for proteolytic processing via a different pathway (Hislop et al 2009). Proteins other than membrane receptors are also targeted by ubiquitin for proteosomal lysis; an important example is the PKA regulatory subunit. Nerve injury leads to ubiquitination and proteolytic destruction of the PKA regulatory subunit (Chain et al 1999). Local administration of selective ubiquitin–proteosome antagonists blocks the CCI-induced increase in PKA activity while concurrently reversing the CCI-induced behavioral hyperalgesia (Moss et al 2002). Ossipov and colleagues (2007) identified a role for the ubiquitin–protease system in enhanced spinal release of CGRP and dynorphin, probably through actions on proteins involved in synaptic vesicle release, and others have recently shown that sphingosine-1-phosphate–induced activation of the mammalian target of rapamycin (mTor) is also dependent on E3 ubiquitin (Maeurer et al 2009).
Systems Suppressing the Excitatory Efficacy of Primary Afferent Input
Afferent traffic through the dorsal horn is subject to a variety of local circuits that limit excitatory spinifugal outflow.
Opioids
The pharmacology of opioid systems in spinal nociceptive processing is broadly considered in Chapters 30 and 31. In brief, several specific points should be emphasized in the context of the present consideration of spinal modulatory systems.
These data jointly suggest a probable segmental and suprasegmental organization of pathways through which spinal opioid receptors may be activated.
Catecholamines
Classic observations have demonstrated that increasing spinal catecholamine activity in an acutely spine-transected animal would block nociceptive flexor reflex afferent activity in a fashion reversed by phenoxybenzamine (Anden et al 1966), a finding consistent with the demonstration of catecholamine-positive cell bodies projecting from the brain stem to the spinal gray along the dorsolateral funiculus (Dahlstrom and Fuxe 1964). Currently, it is appreciated that these descending pathways represent the outflow of a broad array of brain stem and diencephalic structures that can serve to modulate spinal somatic, motor, and autonomic function by enhancing (see above sections on serotonin) and decreasing spinal afferent excitability.
Endogenous Systems
Terminals containing norepinephrine and epinephrine are present in the spinal gray and in axons that arise from neurons in the pontine A5, A6 (nucleus locus coeruleus), and A7 (subcoeruleus) cell groups (Westlund et al 1983, Rajaofetra et al 1992). Activation of these systems by direct stimulation of the bulbar catecholamine nuclei with microinjections of glutamate (Hammond et al 1985) or by activation of local input into these nuclei from the PAG (Cui et al 1999) or from ascending pathways (Tyce and Yaksh 1981) will lead to increases in spinal extracellular norepinephrine concentrations.
Adrenergic Receptors
Adrenaline and noradrenaline act through two major classes of α-adrenergic receptors: α1 and α2. There are three principal α1 receptor subtypes (Hieble et al 1995). All three subtypes have been identified in DRGs and the spinal dorsal horn, α1A, α1B, and α1D (Xie et al 2001). Three distinct subtypes of α2-adrenergic receptors are distinguished: α2A, α2B, and α2C (see Aantaa et al 1995, Bylund 1995). Species differences have been identified. An α2D receptor was found in the rat and is the rodent homologue of the human α2A receptor (MacKinnon et al 1994). Characterization of the distribution of mRNA for the three subtypes in the DRG revealed that the ordering of prevalence in a normal animal was α2C (80%), α2A (20%), and α2B adrenoceptor (rare). Importantly, with nerve injury there was a prominent increase in the number of DRG neurons expressing α2A with no change in α2C (Shi et al 2000). Distribution of the α2-subtype receptor protein is more variable. In terms of protein, more DRG cells express α2A than α2C (Stone et al 1998, Birder and Perl 1999). Thus, spinal α2A receptor protein in the spinal cord is present on the terminals of SP-containing, capsaicin-sensitive afferent fibers. In other studies, α2C receptor protein was found to be densely distributed on axons adjacent to cell bodies and the proximal dendrites of NK1 receptor–bearing lamina I cells and on distal dendrites from laminae III/IV neurons (Olave and Maxwell 2003). Importantly, these terminals were positive for glutamate transporter type 2 protein. Because this transporter is principally present in interneurons (Todd et al 2003), it was concluded that the α2C protein was present on the terminals of spinal interneurons (Olave and Maxwell 2003).
Physiological Effects: A role for both α1 and α2 subtypes in the dorsal horn in regulating nociceptive transmission appears to be likely. The α1 receptor couples to Gq/11, activates PLC, increases intracellular calcium, and activates PKC (Hague et al 2003). Pharmacological studies have suggested that these receptors may play an excitatory role in the DRG, particularly after peripheral nerve injury (Lee et al 1999; but see Xie et al 1995). On the other hand, norepinephrine, acting through an α1A receptor, increases the frequency of both GABAergic and glycinergic miniature inhibitory post-synaptic currents (mIPSCs) in SG neurons (Kawasaki et al 2003). With regard to the α2 receptor subtypes, current work has emphasized the importance of the α2 subtypes in the inhibitory regulation of dorsal horn function. These α2 adrenoceptors are G protein–coupled receptors and mediate their functions through a variety of G proteins, including Gi/o (Piascik et al 1996). When activated, these α2 receptors lead to a reduction in transmitter release by inhibition of the opening of voltage-sensitive calcium channels and membrane hyperpolarization through an increased K+ current (see North et al 1987, Maze and Tranquilli 1991). Studies in SG neurons show such an inhibition. Because the inhibitory effects occurred in the absence of a change in miniature excitatory post-synaptic current (mEPSC) amplitude, the depression of excitability is considered to be presynaptic in origin (Kawasaki et al 2003). Interestingly, it was suggested that α2 inhibition was clearly observed on both Aδ (probably non-peptidergic) and C fibers. Such effects on C fibers are consistent with reports that α2 agonists can depress the release of SP and CGRP from small primary afferents (Takano et al 1993, Supowit et al 1998) and glutamate in ex vivo models (Kamisaki et al 1993). As noted, there are multiple subclasses of α2 receptors. Based on pharmacology, it appears likely that at least some of the presynaptic effects on primary afferents reflect an action characterized by an α2A-subtype pharmacology (Kawasaki et al 2003). Evidence of the post-synaptic action of α2 agonists is provided by observations that iontophoretically applied α2-adrenergic agonists to SG neurons of the spinal cord and trigeminal nucleus produce hyperpolarization (Kawasaki et al 2003). It is interesting to note that the α2C receptor is believed to be on the terminals of glutamate-releasing interneurons that contact NK1-positive cells (Olave and Maxwell 2003). This suggests a probable role of this subclass in regulating the polysynaptic drive that leads to augmented dorsal horn output. In the spinal dorsal horn, iontophoretically activated α2 receptors lead to potent and selective inhibition of the nociceptive responses (to heat or pinch) with no effect on innocuous stimuli (Fleetwood-Walker et al 1985).
Behavior: Intrathecal delivery of noradrenaline produces potent analgesia in a variety of species, including the rat (Kuraishi 1979a 1979b; Yaksh 1979; Reddy et al 1980; Reddy and Yaksh 1980; Jensen and Yaksh 1986; Peng et al 1996), primate (Yaksh and Reddy 1981), dog (Sabbe et al 1994), and sheep (Waterman et al 1988). Work in humans has provided parallel data showing the potent spinal actions of agents such as clonidine (Eisenach et al 1996). The agonist and antagonist pharmacology of these effects clearly implicates an α2 receptor subtype (Yaksh 1985). In initial work we showed that the antagonist pharmacology of different α2-preferring agonists was distinct and proposed the importance of spinal α2C as well as spinal α2A receptors in producing antinociception, with the spinal effects on blood pressure being mediated by the former (Takano et al 1993). Subsequent work using knockout mice yielded controversial data. However, it now appears likely that only minimal differences exist between agonist-induced analgesic responses in α2A knockout and wild-type mice (Link et al 1996), whereas α2C knockout reveals the particular importance of that receptor subtype to spinal antinociception (Fairbanks et al 2002).
As noted above, early work emphasized the probable role of the noradrenergic bulbospinal pathways in regulating spinal function (Anden et al 1966). These effects were reversed by phenoxybenzamine, an adrenergic receptor antagonist (Anden et al 1966). It was demonstrated in the early 1970s that microinjection of μ-opiate agonists into the brain stem or electrical stimulation of the brain stem suppresses spinal nociceptive reflexes and produces a behaviorally defined analgesia (see Yaksh and Rudy 1978 for early review). Subsequent work demonstrated that supraspinal manipulations that block spinal reflexes or dorsal horn nociceptive neuron firing would (1) evoke the release of noradrenaline from the spinal cord (Hammond et al 1985), (2) be blocked at the spinal level by antagonism of α-noradrenergic receptors of the α2 receptor type (Kuraishi et al 1979b, Yaksh 1979, Jensen and Yaksh 1986, Peng et al 1996), and (3) be mimicked by spinal delivery of noradrenaline and other α2-adrenoceptor agonists (Kuraishi et al 1979b, Reddy et al 1980). These findings emphasized the physiological importance of bulbospinal projections and demonstrated the role played by adrenergic systems and the spinal α2 receptor in this control. Interestingly, spinal delivery of an α2non-A antagonist (prazosin) was shown to block the bulbospinal effects produced by microinjection of morphine into the PAG in a manner now believed to reflect the spinal pharmacology of an α2C receptor (Camarata and Yaksh 1985).
Dopamine
The principal emphasis with regard to catecholamines has been on the role of noradrenergic and adrenergic projections. There is additional support for the potential role of other catecholamines, including dopamine. This amine is present in descending tracts that originate in the A9 and A11 cell groups (Dahlstrom and Fuxe 1964). Activation of this pathway electrically or by iontophoretic delivery of D2 agonists inhibits nociceptive responses in dorsal horn projection neurons. Conversely, these effects were antagonized by D2 receptor antagonists (Fleetwood-Walker et al 1988). Intrathecal delivery of dopamine agonists yielded an antinociceptive action, probably mediated by a D2 action (Jensen and Yaksh 1984, Barasi and Duggal 1985).
Serotonin
As reviewed above, bulbospinal serotonin projection systems act through a variety of serotonin receptors. As indicated, one component of the serotonergic system is excitatory, initiated either by direct excitation (as with 5-HT3 or 5-HT7 activating a small afferent terminal) or indirectly by inhibition of an inhibition (disinhibition, as with 5-HT1B inhibition of local GABA release) leading to an enhanced excitatory drive. It is clear, however, that spinal serotonergic receptors can also display a counter-excitatory (inhibitory) effect. Iontophoretic delivery of 5-HT1B/D agonists suppresses dorsal horn firing (Storer and Goadsby 1997) and reduces nociceptive activation (Honda et al 2003). In other studies, 5-HT inhibited the dorsal root–evoked excitatory post-synaptic potential and the direct depolarization evoked by an NK1 agonist. Importantly, the neurons excited by NK1 agonists and inhibited by 5-HT displayed a dense 5-HT–positive plexus (Worsley et al 2005). Early work with intrathecal drug delivery reported the antinociceptive effects of spinal 5-HT. These effects were reversed by antagonists with mixed affinities but importantly included 5-HT2 receptor antagonists (Wang 1977, Yaksh and Wilson 1979). Subsequent work has shown similar effects over a broad range of models, including mechanical paw pressure, thermal escape, and the formalin test (Solomon and Gebhart 1988, Bardin et al 1997) (see Fig. 28-5).
Glutamate
As reviewed above, group I mGlu receptors have largely been reported to be facilitatory on excitatory transmission, whereas group II and III mGlu receptors have been found to largely suppress dorsal horn excitability. Group II mGlu receptors consist of mGlu2 and mGlu3, which act via negative coupling through Gi/Go proteins to adenylate cyclase to inhibit cAMP formation (for reviews see Conn and Pin 1997, Schoepp et al 1999).
Metabotropic Receptors
Group II receptors are distributed widely over presynaptic terminals in the dorsal horn (Petralia et al 1996, Lujan et al 1997, Testa et al 1998). mGlu3 receptors are expressed predominantly in the superficial dorsal horn (Ohishi et al 1993a, 1993b; Petralia et al 1996; Lujan et al 1997; Boxall et al 1998; Yung 1998; Jia et al 1999) and preferentially in axon terminals (Fagni et al 2004). Immunostaining for mGlu2/3 and mGlu7 receptors in the DRG has been reported (Ohishi et al 1995, Jia et al 1999). mGlu3 receptors are also found on glial cells (Ohishi et al 1993a, Petralia et al 1996, Boxall et al 1998, Berthele et al 1999, Jia et al 1999).
Physiology
Group II mGlu receptors negatively modulate the release of glutamate (Battaglia et al 1997) through inhibition of voltage-sensitive Ca2+ channels (Fagni et al 2000). This implies that group II mGlu receptors reduce the hyperexcitable states associated with hyperalgesia and allodynia. Groups II and III mGlu receptor agonists diminish the post-synaptic potentials evoked by primary afferent stimulation in dorsal horn neurons, thus suggesting that mGlu receptors expressed at primary afferent synapses exert a presynaptic inhibitory effect (Gerber et al 2000b). Activation of both group II and group III mGlu receptors can reverse capsaicin-induced facilitation of spinothalamic neurons (Neugebauer et al 2000). Similarly, group II mGlu receptor agonists diminish the C fiber–evoked discharges of WDR spinal dorsal horn neurons otherwise facilitated in the presence of carrageenan inflammation (Stanfa and Dickenson 1998).
Behavior
Intrathecal delivery of group II mGlu receptor agonists reversed the hyperalgesia induced by acute inflammation in rats and sheep (Fisher and Coderre 1996, Dolan and Nolan 2002).
GABA/Glycine
The majority of neurons in the dorsal and ventral horn display potent inhibition by GABA and glycine (Curtis et al 1967). Conversely, numerous inhibitory events are frequently blocked by agents such as bicuculline and strychnine, which are known to block the receptors on which these agents may act, thus indicating the role of GABAergic and glycinergic receptors, respectively (Game and Lodge 1975, Yoshimura and Nishi 1995).
GABA/Glycine System
GABA and glycine are the principal inhibitory neurotransmitters in the spinal cord. Up to 30% of neurons in laminae I and II and 45% of those in lamina III express GABA, and the majority also display glycine immunoreactivity (Todd and Sullivan 1990). It is currently thought that GABA and glycine in fact act as co-transmitters of interneurons at many synapses in the dorsal horn (Taal and Holstege 1994, Todd et al 1996, Keller et al 2001). Systematic examination has suggested that synaptic glomeruli that are presynaptic to small unmyelinated axons may display only GABA whereas glomeruli associated presynaptically with larger myelinated axons possess both neurotransmitters (Todd 1996).
Receptors
GABA and glycine both act through ligand-gated channels (GABAA and glycine receptors, respectively), whereas GABA also acts on a G protein–coupled receptor (GABAB). Details of the receptor structure and pentameric subunit composition of the GABAA ionophore (Chebib and Johnston 1999, Steiger and Russek 2004) and glycine (Breitinger and Becker 2002) are considered elsewhere. The GABAA receptor is a chloride ionophore, and when activated, Cl− moves along its concentration gradient. The normal transmembrane Cl− distribution is regulated by the cation–chloride co-transporters NKCC1 and NKCC2, which import and export Cl− ions, respectively. In the dorsal horn, the import/export activity is such that under basal conditions, intracellular Cl− is lower than extracellular Cl−; as a result, when the GABAA ionophore is activated, there is an influx of Cl− that results in a negative charge and leads to inhibition. Conversely, slightly more intracellular Cl− is present in the primary afferent terminal, and here, GABAA activation actually leads to depolarization. Though seemingly paradoxical, this modest depolarization suppresses transmitter release by inactivating calcium channels; the mechanism underlying the inhibition is referred to as primary afferent depolarization (PAD) (Rudomin 2002). Interestingly, after injury there is reduced Cl− export activity in dorsal horn neurons, which leads to accumulation of intracellular Cl− such that activation of the ionophore may now lead to an exit of Cl− (i.e., depolarization). This results in turning the GABA/glycine effect into excitation of the second-order neuron (Price et al 2005). This shift has been suggested to contribute to the hyperpathia that occurs after inflammation and nerve injury (Morales-Aza et al 2011).
GABA subunit composition defines the role of other ligands, such as benzodiazepines or neurosteroids, that can alter the effects of GABA at the GABAA ionophore (Whiting 2003). Agonist occupancy of the benzodiazepine or neurosteroid binding site enhances the activity of GABA at the GABAA receptor (Hevers and Luddens 1998). GABAA binding as well as message is present in large and small DRG cells and is found in high concentration in the superficial dorsal horn on terminals and cell bodies. Benzodiazepine subunit expression is present in DRGs and on spinal neurons (Wisden et al 1991, Bohlhalter et al 1996). The glycine ionophore, when activated, increases Cl− conductance in the post-synaptic membrane and reduces the excitability of secondary-order neurons. Glycine binding and protein are present throughout the spinal gray. It has been shown in particular, however, that receptors composed of the α3 subunit are present in high quantity in the superficial lamina II of the dorsal horn (Harvey et al 2004). The GABAB receptor is a member of the seven-transmembrane–spanning superfamily, which when coupled through Gi/o protein linkage, serves to diminish the opening of voltage-sensitive calcium channels and to hyperpolarize the membrane (Hammond 2001). GABAB binding is maximal in lamina II, with about half the binding lost after capsaicin application or rhizotomy (Price et al 1987).
Physiological Effect
Given the organizational connectivity of GABA and glycine receptors within the dorsal horn, it is not surprising that activity in dorsal horn nociceptive and non-nociceptive neurons and terminals is under powerful tonic regulation by GABAA and glycine receptors. As noted above, the principal effect of activating these receptors is to initiate inhibitory control given the transmembrane distribution of Cl− in the normal neuron. An anomalous finding is that following nerve injury, there is a change in this transmembrane gradient such that activation of GABA/glycine ionophores results in depolarization. In the following sections we will consider several examples of the effects of these inhibitory and excitatory actions.
Inhibition: Glycine functions as a fast inhibitory neurotransmitter in the superficial spinal dorsal horn (van den Pol and Gorcs 1988, Todd 1990). Local application of a glycine antagonist (strychnine) induces a powerful facilitation of WDR neuronal responses to low-threshold, otherwise innocuous mechanical stimuli (Khayyat et al 1975, Yokota et al 1979). Much later work provided confirmation of this finding by showing that with bicuculline treatment, the mechanical threshold of high-threshold flexor motor neurons is reduced and their responses to light touch or electrical stimulation of A fibers is enhanced (Sivilotti and Woolf 1994). On intercellular recording, A-fiber stimulation with bicuculline results in repetitive, long-lasting polysynaptic EPSCs following the initial fast response in superficial dorsal horn neurons (Baba et al 2003). Importantly, dorsal horn GABA-containing terminals are frequently presynaptic to the large central afferent terminal complexes and form reciprocal synapses (Barber et al 1978, Carlton and Hayes 1990). GABAergic axosomatic connections on spinothalamic cells have also been identified (Carlton et al 1992), and these receptors contribute to post-synaptic inhibition of the transmission of nociceptive information in projection neurons (Lin et al 1996). Consistent with the role played by benzodiazepines in regulating GABAA activity, midazolam increased GABAA-mediated currents in SG neurons (Kohno et al 2000). Thus, the responses of spinothalamic and deep dorsal horn neurons to low-intensity, innocuous mechanical stimuli are significantly enhanced after the intrathecal administration of bicuculline (Reeve et al 1998). In single-unit studies, the majority of SG neurons displayed IPSPs in response to a light tactile stimulus. These IPSPs were mediated by GABAA and glycine receptors, thus confirming that non-noxious mechanical stimuli activate local GABAergic or glycinergic circuitry in the dorsal horn (Narikawa et al 2000). Similarly, small afferent input was noted to drive the bicuculline-sensitive inhibition of small afferent–evoked projection neurons (Hantman et al 2004). These properties are consistent with the existence of complex functional interactions between populations of afferents serving to modulate local excitability. Consider that early work showed that iontophoretic delivery of glycine and GABA diminished the size of cutaneous receptive fields (Zieglgansberger and Herz 1971) and reduced spontaneous and afferent-evoked activity in spinothalamic tract neurons (Willcockson et al 1984). Conversely, the IPSCs elicited in SG neurons by a brush stimulus (e.g., low-threshold myelinated afferent) applied over large areas of the ipsilateral hindlimb were blocked by strychnine or bicuculline (Narikawa et al 2000). Although GABAB receptor antagonism has little evident effect on the ongoing processing of noxious or innocuous input, spinal baclofen significantly reduced the Aβ, Aδ, and C fiber–evoked responses of spinal dorsal horn neurons in a dose-related manner (Sokal and Chapman 2003).
As suggested by the above, the potent regulation by GABA- and glycine-releasing interneurons displays complex activation pharmacology. Thus, in the superficial dorsal horn, GABA/glycine-releasing interneurons have been shown to be activated by a variety of pharmacologically defined systems: (1) Muscarinic receptors on both axon terminals and somatodendritic sites serve to increase the excitability of inhibitory interneurons and enhance the release of GABA in the SG (Baba et al 1998). (2) ATP facilitates the release of glycine through P2X2 receptors in dorsal horn lamina II neurons (Ren et al 1992b, Jang et al 2001). (3) SP- and CGRP-positive boutons make presynaptic or symmetrical contact with GABAergic dendrites and soma in the superficial dorsal horn (Hiura et al 1998). (4) 5-HT through 5-HT3 and noradrenaline receptors activate GABA- and glycine-containing interneurons to cause IPSPs in the dorsal horn and/or release of GABA (Grudt et al 1995, Baba et al 2000, Kawamata et al 2003). (5) Primary afferent Aδ fibers activate glycinergic and/or GABAergic interneurons primarily through the non-NMDA receptor subclass (Yoshimura and Nishi 1995). (6) Ca2+-permeable AMPA receptors are present on a majority of GABA receptor–expressing neurons (Albuquerque et al 1999).
Excitation: After nerve injury, spinal neurons regress to a neonatal phenotype in which GABAA activation becomes excitatory. This excitatory effect is secondary to reduced activity of the membrane Cl− transporter, which changes the reversal potential for Cl− conductance. Now, increasing membrane Cl− conductance, as occurs with GABAA receptor activation, results in membrane depolarization. Under normal conditions, transmembrane [Cl−] is at equilibrium at or just below resting membrane potential. Increasing Cl− permeability by GABAA or glycine receptor (Cl− channels) yields hyperpolarization and inhibition. “Cation–Cl” co-transporters regulate Cl− gradients by exporting Cl−. The loss of dorsal horn neuronal KCC2 after nerve injury leads to increased intracellular [Cl−]i. Under these conditions, increasing Cl− permeability may lead to failure of GABAA/glycine inhibition or, in fact, turn the GABA/glycine effect into excitation of the second-order neuron (De Koninck 2007).
Behavior
Intrathecal delivery of GABAA receptor agonists has modest effects on acute escape responses (Hammond and Washington 1993) but attenuates hyperalgesia in a number of models (Malan et al 2002). With regard to benzodiazepines that interact with the GABAA site, a similar profile of effects is noted. Early work showed that intrathecal benzodiazepines reduce small afferent–evoked somatosympathetic reflexes (Niv et al 1983, Gaumann et al 1990). In acute behavioral reflex models, such as the tail flick test, there is a dose-dependent increase in latency (Allen and Yaksh 2004). Activity is also observed in models of facilitated processing, such as the formalin test (Nishiyama and Hanaoka 2003), or in models of inflammation (Kyles et al 1995). Intrathecal delivery of GABAB agonists has a moderate effect on acute nociceptive thresholds, but the maximum analgesic effects may not occur until doses are achieved that alter motor function (Wilson and Yaksh 1978, Yaksh and Reddy 1981, Aran and Hammond 1991, Malan et al 2002). In models of facilitated processing, a dose-dependent effect on hyperalgesia has been noted (Dirig and Yaksh 1995, Kaneko and Hammond 1997, Malan et al 2002).
An important aspect of GABAergic and glycinergic spinal system function is the apparent tonic role they play in regulating afferent processing that would otherwise be non-noxious in character. Transient block of spinal GABAA (bicuculline/picrotoxin) or glycine (strychnine) receptors reveals the prominent tonic role played by the GABA and glycine systems. Early work demonstrated that low-threshold tactile stimuli, typically ineffective in producing evidence of escape behavior, were able to evoke powerful pain behavior after spinal antagonism of GABA and glycine receptors (Yaksh 1989). Such observations are in concert with yet earlier studies on the activity of spinal/trigeminal single units noted above. These observations raise the likelihood that the encoding of low-threshold mechanical stimuli as innocuous depends completely on the presence of tonic activation of intrinsic glycine/GABAergic neurons that are known to exist within the spinal dorsal horn. Several lines of evidence substantiate the relevance of these dorsal horn inhibitory amino acids in regulating the behavior generated by low-threshold afferent transmission. Thus, genetic variants such as the poll Hereford calf (Gundlach et al 1988) and the spastic mouse (White and Heller 1982) have been shown to display particular sensitivity to even modest stimulation, and these models exhibit up to a 10-fold decrease in glycine binding. Second, GABAA knockout mice exhibit tactile allodynia similar to that observed with spinal GABAA receptor antagonism (Yaksh 1989, McGowan and Hammond 1993, Onaka et al 1996). The mechanical allodynia probably reflects the loss of GABAA receptors presynaptic to low-threshold Aβ (Gmelin and Zimmermann 1983) and Aδ (De Koninck and Henry 1994) primary afferent fibers. Such behavioral evidence of the effects of hypersensitivity would be consistent with loss of post-synaptic GABAA receptors on spinofugal neurons (Lin et al 1996). Third, in humans, strychnine intoxication is characterized by hypersensitivity to light touch (Arena 1970), and the role of such interneurons in the encoding of afferent input has been suggested as an important mechanism involved in the allodynia and hyperesthesia evoked following spinal cord ischemia (Hao et al 1992a, 1992b; Marsala and Yaksh 1992) and peripheral nerve injury (Yaksh et al 1992).
Adenosine
Adenosine Systems
Adenosine is generated from ATP by ectonucleotidases that dephosphorylate ATP, adenosine diphosphate (ADP), and adenosine monophosphate (AMP) (Zimmermann et al 1998). Extracellular adenosine levels can be elevated at the spinal cord level by local depolarization (K+ and capsaicin) (Sweeney et al 1989) and by a wide variety of pharmacological interventions, including the direct effects of 5-HT, noradrenaline, and opiates.
Receptors
Four receptor subtypes (A1, A2A, A2B, and A3) have been identified based on their pharmacology and cloning (Fredholm et al 2001). The three classes of receptors are all G protein coupled, with A1 and A3 typically being inhibitory (reduced cAMP) and the A2 families being excitatory (increased cAMP) (Schulte and Fredholm 2003). The spinal distribution of these adenosine receptors has been established with autoradiography and immunohistochemistry. Both approaches have shown adenosine A1 binding receptor protein in the superficial dorsal horn spinal cord (laminae I and II). This binding/protein was reduced by local kainate, but not by rhizotomy (Geiger et al 1984, Choca et al 1988, Deuchars et al 2001b), thus suggesting localization on interneurons. Adenosine A2A receptor message is present in DRGs (Kaelin-Lang et al 1998) but not in the spinal cord (Kaelin-Lang et al 1999), thus suggesting their presence on the spinal terminals of sensory afferents.
Physiological Effect
Adenosine A1 receptor activation increases K+ conductance, thereby leading to hyperpolarization and to reduced frequency of opening of voltage-sensitive calcium channels (Dunwiddie and Masino 2001). Examination of the effects of A1 receptor agonists on primary afferent terminal–releasing properties indicates inhibition of release of the primary afferent stores of CGRP, but not SP (Santicioli et al 1992, 1993; Mauborgne et al 2002). Electrophysiological studies have further shown that adenosine acts presynaptically through A1 receptors to diminish monosynaptic Aδ and C fiber–evoked depolarization in some superficial dorsal horn neurons (Lao et al 2001, Patel et al 2001).
Behavior
The intrathecal delivery of adenosine mediated by A1-type receptor pharmacology produces modest increases in acute nociceptive thresholds (Sosnowski et al 1989) but also induces significant anti-hyperalgesic action in models of post–inflammation-induced thermal hyperalgesia (Poon and Sawynok 1998), in the formalin model (Malmberg and Yaksh 1993a), and in models of neuropathic pain associated with glycine inhibition (Sosnowski and Yaksh 1989) and nerve injury (Lee and Yaksh 1996, Khandwala et al 1998, Poon and Sawynok 1998). When examined, these effects were not observed with adenosine A2–preferring agonists. Consistent with these behavioral effects, intrathecal adenosine A1 activation decreased the incidence of c-Fos–positive cells in the superficial and deep dorsal horn neurons evoked by peripheral inflammation (Sorkin et al 2003).
Important aspects of the actions of endogenous adenosine are its rapid uptake and inactivation. Accordingly, it is important to note that spinal delivery of adenosine kinase or adenosine deaminase inhibitors will of itself have significant anti-hyperalgesic effects that appear to possess an adenosine A1 antagonist pharmacology (Keil and DeLander 1992, Poon and Sawynok 1998, Lavand’homme and Eisenach 1999, McGaraughty et al 2001). These observations provide confirmation of the probable contribution of the endogenous release of adenosine to local spinal modulation.
Cannabinoids
The pharmacology of the cannabinoid (CB) system in spinal nociceptive processing is considered in Chapter 38 to which the reader is referred. In brief, the following issues related to cannabinoid actions are noteworthy in the context of CNS nociceptive encoding.
Two specific G protein–coupled cannabinoid receptors have been identified, one of which (CB1) is present on spinal neurons and the other (CB2) is present on glial cells. In the spinal cord, local delivery of CB agonists produces a significant antinociceptive effect in a variety of models.
Endogenous Agents
Identification of the presence of cannabinoid binding sites has led to discovery of endogenous agents that act on these receptors. N-Arachidonyl ethanolamide (anandamide) was identified first (Devane et al 1992), followed by a second endocannabinoid, 2-arachidonylglycerol (Mechoulam et al 1995, Sugiura et al 1995). These molecules exhibit affinity for both the CB1 and CB2 receptors (Guindon and Hohmann 2009). Local microdialysis during brain stem stimulation revealed that parameters found to be antinociceptive and antagonized by CB antagonists were associated with an increase in the release of several endocannabinoids (Guindon and Hohmann 2009). Similarly, injections of formalin into the hindpaws of rats induced the release of anandamide in the PAG (Walker et al 1999). These molecules are long-chain lipids, and there has been interest in the hypothesis that they may act at additional sites. In particular, the vanilloid (TRPV1) receptor can be activated by anandamide in a manner blocked by TRPV1 receptor antagonists (Zygmunt et al 1999).
Receptors
Based on expression and pharmacology, two cannabinoid receptor families have been identified. Although both CB1 and CB2 are negatively coupled to adenylyl cyclase, they show downstream differences. CB1 is negatively coupled through Gi/o (Felder et al 1995) to N- and P/Q-type calcium channels (Caulfield and Brown 1992, Mackie et al 1995) and positively coupled to inwardly rectifying potassium channels (Deadwyler et al 1995, Mackie et al 1995). CB2 is not coupled to either calcium Q-type or inwardly rectifying potassium channels (Felder et al 1995). In DRGs, CB1 receptors appear to be coupled primarily to N-type calcium channels (Khasabova et al 2004). The presence of cannabinoid binding in the spinal cord has been well characterized (Herkenham et al 1991). Capsaicin treatment produces a modest reduction and rhizotomy produces a small yet greater reduction in binding (Hohmann and Herkenham 1998, Hohmann et al 1999). Consistent with this proposed distribution, CB1 mRNA and protein are highly expressed in DRG cells of heterogeneous size (Sanudo-Pena et al 1999, Hohmann 2002), including TRPV1-positive cells (Ahluwalia et al 2000). CB2 receptor protein has been identified in DRG cell cultures (Ross et al 2001). These results suggest for the CB1 receptor a modest presynaptic localization in small capsaicin-sensitive and non–capsaicin-sensitive afferents, with the preponderance of post-synaptic expression in the spinal dorsal horn. In the spinal cord, CB1 message has been identified in all spinal laminae except IX (Mailleux and Vanderhaeghen 1992). Immunohistochemistry has indicated CB1 receptor protein in laminae I and II inner and lamina X (Farquhar-Smith et al 2000, Salio et al 2002). At least some dorsal horn immunoreactivity is located on GABA-positive interneurons (Salio 2002). The CB1 receptor, though constitutively expressed at a lower level in the spinal cord, displays marked up-regulation in the spinal cord and DRG after nerve injury and persistent inflammation (Hsieh et al 2011).
Physiological Effects
In culture, CB1 agonists were observed to reduce the entry of Ca2+ in DRG cultures, thus suggesting a probable effect on terminal release (Ross et al 2001). With regard to primary afferent release, in isolated DRG cell cultures, opiates but not CB1 agonists diminish release of CGRP (Khasabova et al 2004). In vitro recording suggests that CB1 activation results in inhibition of dorsal horn glutamate-mediated activation through inhibition of its release onto lamina II glycine/GABAergic interneurons (Morisset and Urban 2001). No evidence of a post-synaptic effect was noted given that AMPA-kainate–mediated post-synaptic current was unaltered. These results are consistent with the demonstrated co-localization of CB1 receptor protein with GABA-positive neurons (Jennings et al 2001). Intrathecal delivery of CB1 agonists suppressed noxious heat–evoked activity in dorsal horn WDR neurons. These results are consistent with the ability of CB1 agonists to suppress stimulus-evoked Fos expression in the lumbar spinal cord (Tsou et al 1996). Conversely, CB1 but not CB2 receptor antagonism exaggerated acute nociceptive but not non-nociceptive transmission at the level of the spinal cord, thus suggesting a tonic role of the endogenous cannabinoids at spinal CB1 receptors (Chapman 1999).
Behavioral Effects
Intrathecal and supraspinal (see below) delivery of a CB agonist produces a significant antinociceptive effect in a variety of models, including thermal escape (Yaksh 1981, Smith and Martin 1992; for an extensive review see Pertwee 2001). Current work emphasizes the clear importance of the CB1 receptor in mediating antinociception after spinal or systemic delivery. Knockout of the CB1 receptor (Ledent et al 1999, Zimmer et al 1999) or knockdown with antisense oligonucleotides (Edsall et al 1996) significantly reduces the effects of antinociceptive CB treatment. Importantly, consistent with up-regulation of the CB2 receptor after nerve injury and persistent inflammation, intrathecal CB2a agonists were effective in attenuating injury-evoked hyperalgesia (Hsieh et al 2011).
Cholinergic
Cholinergic Systems
In the spinal dorsal horn, cholinergic soma, as defined by choline acetyltransferase immunoreactivity, are present in laminae III, V, and X (Borges and Iversen 1986). A dense plexus of cholinergic terminals extends through laminae II and III (Sherriff and Henderson 1994). Pharmacological studies have suggested that the dorsal horn effects of acetylcholine are mediated through nicotinic and muscarinic receptors.
Cholinergic Receptors
Muscarinic: Five muscarinic receptors have been defined (Caulfield 1993). M1, M3, and M5 are preferentially coupled to Gq protein, whereas M2 and M4 are preferentially coupled to the Gi class (Caulfield and Birdsall 1998). In rat DRG, in situ hybridization shows M2, M3, and M4 but not M1 or M5 transcripts, with the respective signals principally localized in small to medium neurons (Tata et al 2000). M2 and M4 receptor proteins are expressed in small to medium-sized neurons, whereas M1 and M3 proteins are uniformly distributed across the neuronal population of the ganglion (Bernardini et al 1999). The M2/M3 muscarinic subtypes are highly expressed on IB4-positive neurons (Haberberger et al 1999). In the superficial dorsal horn, M2, M3, and M4 receptors but not the M1 receptor are expressed in spinal gray, with M2 binding present on a high proportion of lamina III but not lamina I neurons (Hoglund and Baghdoyan 1997, Stewart and Maxwell 2003).
Nicotinic: Nicotinic receptors are ligand-gated, action-selective channels composed of pentameric combinations of distinct subunits that yield pharmacologically distinct ionophores (Miyazawa et al 2003, Nai et al 2003). Nicotinic receptors have been identified on dorsal horn neurons, on DRG cells (Flores 2000), and of particular interest, on the terminals of vanilloid-positive/capsaicin-sensitive afferents (Roberts et al 1995, Khan et al 2003, Haberberger et al 2004).
Physiological Effects
Muscarinic: Activation of muscarinic receptors inhibits the discharge of projection neurons evoked by noxious as well as non-noxious stimuli (Chen and Pan 2004). The mechanism of this inhibition appears to be that presynaptic muscarinic activation inhibits glutamatergic excitatory input onto lamina II neurons. Interestingly, muscarinic receptor activation also excites lamina II GABAergic interneurons and increases local release of GABA (Baba et al 1998, Li et al 2002).
Nicotinic: Activation of nicotinic receptors enhances the spinal release of a variety of neurotransmitters, including glutamate (Khan et al 1996), norepinephrine (Li and Eisenach 2002), serotonin (Cordero-Erausquin and Changeux 2001), and GABA/glycine (Kiyosawa et al 2001, Cordero-Erausquin et al 2004). The effects on glutamate have been shown to enhance synaptic transmission in dorsal horn neurons, presumably mediated in part by a direct effect on primary afferent terminals (Genzen and McGehee 2003). In other work, enhanced GABAergic inhibition in the SG was initiated by nicotinic receptor activation (Takeda et al 2003). Importantly, work has suggested that the composition of the nicotinic subtypes present on inhibitory interneurons (e.g., GABA/glycine) is distinct from those expressing NK1 receptors, which are probably projection neurons (Cordero-Erausquin et al 2004).
Behavioral Effects
Muscarinic: Intrathecal delivery of muscarinic agonists (Iwamoto and Marion 1993, Honda et al 2000), as well as cholinesterase inhibitors (Naguib and Yaksh 1994, 1997; Lavand’homme and Eisenach 1999), has been reported to produce antinociception using a variety of end points. Reports have suggested that the spinal action is mediated by M2 receptors (Iwamoto and Marion 1993), but others have more recently emphasized the M3 subtype (Naguib and Yaksh 1997, Honda et al 2000).
Nicotinic: Intrathecal nicotinic agonists have been shown to produce prominent excitation followed by analgesia (Khan et al 1998, Damaj et al 2000). The mechanism of this analgesia is not well defined. As reviewed above, nicotinic receptor activation at the spinal level can indeed result in the release of a variety of modulatory transmitters, including inhibitory amino acids, norepinephrine, and serotonin, all of which could alter dorsal horn nociceptive processing. Alternately, it is appreciated that activation of nicotinic receptors can induce desensitization of the afferent terminal membrane in a manner similar to capsaicin, and such inactivation of spinal C-fiber terminals might be a component of the analgesia observed with nicotinic agonists.
Neurotensin
Neurotensin System
Neurotensin is a tridecapeptide. Neurotensin-positive terminals and cell bodies are present in small non-GABAergic interneurons in laminae I and II (Proudlock et al 1993).
Neurotensin Receptors
Neurotensin is believed to exert its effects through at least three cloned receptors (Vincent et al 1999).
Physiological Effects
Local delivery of neurotensin in the spinal dorsal horn excites spinal nociceptors (Stanzione and Zieglgansberger 1983).
Behavioral Effects
Intrathecal delivery of this peptide has been reported to yield signs of both excitation (Seybold et al 1982) and antinociceptive actions in thermal and chemical nociceptive models (Yaksh et al 1982b).
Conclusion
This chapter has sought to adhere to the organizing principle that our understanding of the pharmacology underlying pain processing requires convergence of the effect of specific agents on well-defined pain behavior and on the underlying hypothesized mechanisms. Thus, for exaggerated spinal processing or a particular receptor to be related to pain, the properties of the behavioral effects must display characteristics similar to the pharmacology of the underlying effects on cellular activity.
What has been a particularly interesting issue has been the evident importance of the role played by the spinal circuitry in the processing of afferent information. Virtually all the changes in pain states that arise from injury and inflammation co-vary with the output function of spinal projection neurons. Although it is clear that the perceptual correlates of the stimulus are defined by what happens in higher centers, it is equally clear that these systems respond to the content of the afferent input. Accordingly, the spinal action of a variety of agents is clearly sufficient to produce potent and therapeutically useful changes in pain behavior. Consequently, the progressive understanding of the biology of these processing algorithms has revealed mechanisms relevant to a variety of hyperesthetic states. Indeed, it has been revealed that the innocuous aspects of light tactile stimuli depend absolutely on the presence of effective GABA and glycinergic inhibition at the level of the first-order synapse.
Acknowledgments
Studies were supported by the National Institutes of Health (NS067459, LS; NIDA DA02110, TY NS16541, TY).
The references for this chapter can be found at www.expertconsult.com.