Representation of Pain in the Brain
Historical Perspective
The role of the brain in pain processing has long remained elusive. More than one hundred years ago, Head and Holmes (1911) observed that soldiers with extensive injuries to the cerebral cortex still perceived pain and thus concluded that the cortex played only a minimal role in pain perception. Penfield and Boldrey (1937) reached a similar conclusion when they found that patients rarely reported pain on electrical stimulation of their exposed cerebral cortex during surgery to remove epileptic foci. Although Dejerine and Roussy (1906), Marshall (1951), and others did not agree with this view and produced clinical evidence that they thought was more consistent with the idea that the cortex is involved in pain perception, until recently the dominant viewpoint remained that the cortex has a minimal role in pain perception. Nevertheless, the complex nature of the pain experience, which encompasses both sensory features and emotional and motivational components (Melzack and Casey 1968, Price 1988), suggests that the conscious appreciation of pain must include the activation and interaction of multiple brain regions. It is thus not surprising that specific lesions or focal stimulation of the cortex did not produce the experience of pain.
By the late 1980s, multiple lines of evidence suggested that several regions of the cerebral cortex could participate in pain processing. Some patients with epileptic foci involving the primary or secondary somatosensory cortices (S1 and S2, respectively) had been observed to experience painful seizures (Young and Blume 1983, Young et al 1986). In addition, lesions involving these areas in humans had on occasion been shown to reduce pain perception (White and Sweet 1969). A few single neurons responding to noxious skin stimulation had been identified in S1 and S2 of the monkey (Robinson and Burton 1980, Kenshalo et al 1988, Dong et al 1989) and in frontal cortices of the rat (Mantz et al 1988, 1990). Finally, neurosurgeons had observed that lesions involving the anterior cingulate cortex (ACC) reduced the distress associated with chronic intractable pain (Foltz and Lowell 1962, Hurt and Ballantine 1973). Nevertheless, the paucity of both animal and human evidence of involvement of the cerebral cortex in pain perception led to the continued view in medical textbooks that pain was a subcortical phenomenon.
The advent of modern human brain imaging studies in the early 1990s allowed us to begin unraveling the role of the brain in the complex experience of pain. Hemodynamic correlates of pain were first imaged in the human brain in the 1970s by Lassen and colleagues (1978) with the radioisotope xenon 133. This technique provided little spatial resolution but suggested that during pain, blood flow to the frontal lobes was increased. The first three human brain imaging studies of pain using today’s technology were published in the early 1990s by Talbot and colleagues (1991) and Jones and co-workers (1991), who used positron emission tomography (PET), and by Apkarian and associates (1992), who used single-photon emission computed tomography (SPECT). All three studies used painful cutaneous heat, and despite differences in their results, together they indicated that multiple cortical and subcortical brain areas are activated during short-duration pain induced by heat. Non-invasive human brain imaging continues to provide new insight into the human brain in health and disease at an unprecedented and unabatingly fast pace.
Defining a Pain Network in the Brain
Human brain activity can be imaged with several techniques, including PET, SPECT, functional magnetic resonance imaging (fMRI), electroencephalographic (EEG) dipole source analysis, and magnetoencephalographic (MEG) analysis. Each of these techniques has advantages and disadvantages in terms of spatial and temporal resolution, sensitivity, and cost (Box 7-1). However, all provide measures that we can use as indirect indices of neural activity. Furthermore, despite the many differences among these techniques, the results derived from each are generally congruous.
Hundreds of human brain imaging studies have now examined the cortical and subcortical brain regions involved in acute pain processing in healthy subjects. Although there are many differences in activation patterns across studies, a consistent cortical and subcortical network has emerged that includes sensory, limbic, associative, and motor areas. The regions most commonly activated are the S1, S2, ACC, insular cortex (IC), prefrontal cortex (PFC), thalamus, and cerebellum (Fig. 7-1). Pain-evoked activity in these areas is frequently observed with either PET or fMRI techniques, and the activation in these regions is consistent with anatomical studies that show probable nociceptive connectivity to these regions (Apkarian et al 2005).
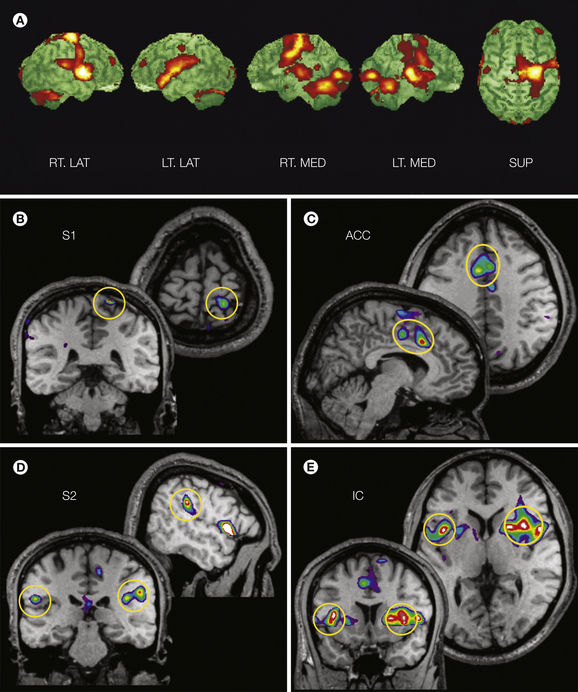
Figure 7-1 Pain-evoked activation in the human brain.
A, Surface-rendered subtraction images of activation patterns obtained with positron emission tomography scans showing right lateral (RT. LAT), left lateral (LT. LAT), right medial (RT. MED), left medial (LT. MED), and superior (SUP) views. Activation observed during repetitive innocuous warm stimuli (40°C) is subtracted from that observed during repetitive painful heat stimulation (50°C). The scans began 40 seconds after onset of the painful heat stimulation and reveal responses bilaterally in the anterior cingulate cortex (ACC), insular cortex (IC), thalamus, cerebellum, and sensorimotor cortex. B–E, Functional and anatomical magnetic resonance imaging (MRI) of four subjects exposed to repetitive 9-second noxious heat stimuli on the leg (46°C) in comparison to repetitive warm stimuli (36°C). The circled areas represent regions showing significantly greater activation during noxious heat than during the warm stimuli. These areas include the primary and secondary somatosensory cortices (S1, S2), ACC, and IC. For all images the right hemisphere is shown on the right. Responses with a statistical significance of P < 0.05 are shown superimposed in stereotactic coordinates (Talairach and Tournoux 1988) on MRI. (A From Casey KL, Morrow TJ, Lorenz J et al 2001 Temporal and spatial dynamics of human forebrain activity during heat pain: analysis by positron emission tomography. Journal of Neurophysiology 85:951–959, Fig. 4; B–E adapted from Bushnell MC, Duncan GH, Hofbauer RK et al 1999 Pain perception: is there a role for primary somatosensory cortex? Proceedings of the National Academy of Sciences of the United States of America 96:7705–7709.)
As discussed in Chapter 2 and shown in Figure 7-2, regions activated by pain in imaging studies receive either direct or indirect nociceptive input. In primates, S1 and S2 receive noxious and innocuous somatosensory input from the somatosensory thalamus (Friedman and Murray 1986, Rausell and Jones 1991, Shi and Apkarian 1995). The cingulate cortex receives input from medial thalamic nuclei that contain nociceptive neurons, including the nucleus parafascicularis and ventrocaudal part of the nucleus medialis dorsalis, as well as from lateral thalamic regions, including the ventral aspect of the ventroposterior nucleus and the ventroposterior inferior nucleus (Apkarian and Shi 1998, Craig and Dostrovsky 2001). Nociceptive input to the ACC is further suggested by the observations that painful stimuli evoke potentials over the human anterior cingulate gyrus and that single nociceptive neurons are present in the ACC of humans (Lenz et al 1998, Hutchison et al 1999), monkeys (Koyama et al 1998), and rabbits (Sikes and Vogt 1992). These data indicate a specific role for parts of ACC in pain processing that is distinct from, though probably related to, the role of the ACC in cognitive processes such as attention (Davis et al 1997, Derbyshire and Jones 1998). The IC also receives direct thalamocortical nociceptive input in the primate (Dostrovsky and Craig 1996), and nociceptive activity has been recorded from the human IC (Frot and Mauguiere 2003). A recent study using a viral anterograde transport technique provided the first direct evidence of the spinothalamic–thalamocortical pathway in monkeys (Dum et al 2009) by documenting that the pathway accesses S2, IC, and ACC more heavily than S1. Moreover, the data provide good evidence that parts of the posterior cingulate cortex receiving spinothalamic–thalamocortical input are cingulate motor areas with direct projections to the primary motor cortex. Thus the posterior cingulate regions often observed in human pain brain imaging studies potentially provide a direct route for controlling motor responses to painful stimuli. The prefrontal cortical regions are activated in a number of imaging studies of acute pain in normal subjects, and recent anatomical evidence points to massive innervation of the superficial layers of at least the dorsolateral PFC through medial thalamic neurons that respond to noxious stimuli throughout the body in rats (Monconduit and Villanueva 2005); thus this thalamocortical pathway provides the substrate for modulating higher cortical processes by nociceptive input.
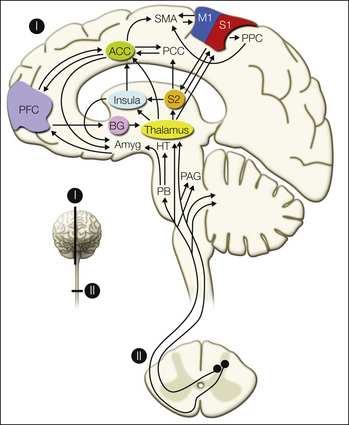
Figure 7-2 Schematic of ascending pathways, subcortical structures, and cerebral cortical structures involved in processing pain.
ACC, anterior cingulate cortex; Amyg, amygdala; BG, basal ganglia; M1, primary motor cortex; HT, hypothalamus; PAG, periaqueductal gray; PB, parabrachial nucleus of the dorsolateral pons; PCC, posterior cingulate cortex; PFC, prefrontal cortex; PPC, posterior parietal complex; S1 and S2, primary and secondary somatosensory cortical areas; SMA, supplementary motor area. (From Apkarian AV, Bushnell MC, Treede RD et al 2005 Human brain mechanisms of pain perception and regulation in health and disease. European Journal of Pain 9:463–484.)
Numerous subcortical areas have shown activation in human imaging studies. The most common subcortical pain-related activation takes place in the thalamus and cerebellum. Several nuclei in the thalamus receive nociceptive input from the dorsal horn, and the cerebellum also has reciprocal spinal connectivity (Saab and Willis 2003). The striatum, mainly the caudate putamen, is also often reported to be active in human pain imaging studies, nociceptive neurons have been described in rats and monkeys within this region (Chudler 1998), and a recent viral anterograde labeling study showed that a specific population of spinal cord lamina 5 neurons project directly to the basal ganglia (Braz et al 2005). Some human pain imaging studies also indicate activity in the nucleus accumbens and amygdala (Becerra et al 2001, Baliki et al 2010), and this activity is probably a reflection of nociceptive transmission through spino–parabrachial–amygdala projections (Bernard et al 1996). The periaqueductal gray (PAG) has also been observed to be active in human imaging studies of somatic and visceral pain, especially when the brain stem is specifically studied (Dunckley et al 2005). Thus there is evidence that multiple ascending nociceptive pathways are engaged in signaling the information integrated at the cortical level, in addition to the commonly cited spinothalamic pathways (see Chapter 12 for additional discussion).
Brain Processing of the Multidimensionality of Pain
There is now evidence from brain imaging, lesion, and electrophysiological studies that different cortical regions may be preferentially involved in different aspects of the complex experience of pain. Most evidence suggests that the somatosensory cortices are most important for the perception of sensory features, such as the location and duration of pain, whereas the limbic and paralimbic regions, such as the ACC and IC, are more important for the emotional and motivational aspects of pain. S1 and S2 contain neurons that code the spatial, temporal, and intensive aspects of innocuous and noxious somatosensory stimuli (Kenshalo and Isensee 1983, Kenshalo et al 1988, Chudler et al 1990), characteristics that could subserve the sensory-discriminative dimension of pain processing. Furthermore, clinical studies of lesions involving S1 and/or S2 show deficits in pain sensations (Greenspan et al 1999, Ploner et al 1999). Strikingly, Ploner and colleagues observed that a patient who suffered a stroke involving S1 and S2 could not localize or describe the nature of a painful stimulus but instead reported a poorly localized and ill-defined unpleasant feeling when presented with a noxious stimulus. Thus this individual appears to have pain affect without a clear sensory-discriminative component of pain sensation. Recently, Starr and co-authors (2009) reported on two patients with unilateral IC lesions regarding pain perception and brain activity. Surprisingly, both patients had higher ratings of acute thermal pain than did controls and exhibited increased S1 activity in the absence of IC activity, thus implying that disruption of nociceptive input to the IC may be compensated by increased transmission of nociceptive information to S1 and that increased acute thermal pain sensitivity results.
The ACC and IC have long been considered to be components of the limbic (emotional) part of the brain (Penfield and Boldrey 1937, MacLean 1949) and therefore potential candidates for processing the affective–motivational dimension of pain. This idea was substantiated by clinical reports that patients who had undergone cingulotomy showed attenuated emotional responses to pain (Foltz and Lowell 1962, Foltz and White 1968, Corkin and Hebben 1981). The results of imaging studies confirm these clinical reports. Rainville and co-workers (1997) demonstrated selective modulation of ACC pain-evoked activity after hypnotic suggestion of changes in pain unpleasantness and also showed a significant correlation between ACC activity and subjects’ ratings of pain unpleasantness, thus strongly suggesting involvement of the ACC in the affective dimension of the pain experience. Similarly, Tolle and colleagues (1999) used regression analysis to show that pain-evoked activation in the ACC is more related to the affective component of the pain experience, and Zubieta and associates (2001) showed that the affective component of the McGill Pain Questionnaire correlated with μ-opioid receptor activation in the ACC during sustained pain. The most compelling evidence for the role of the ACC in pain behavior comes from manipulating its activity in rats during associative learning. Concomitant activation of the ACC during conditioning produces aversive learning, whereas inhibiting ACC activity seems to block aversive learning (Johansen and Fields 2004). Therefore, at least in rodents, involvement of the ACC seems to be necessary and sufficient for noxious stimuli to produce an aversive learned behavior.
The IC has been implicated in visceral sensory and motor integration, emotional responses, and memory functions and is also consistently activated by painful stimuli. Coghill and colleagues (1999) observed a systematic relationship between the intensity of painful heat stimuli and IC activation, and Craig and associates (2000) observed a similar correlation between IC activity and the intensity of cold stimuli, thus suggesting that the IC may be involved in coding of noxious and innocuous temperature. Other data indicate that IC activity may be important for pain affect. Insular lesions are sometimes characterized by the condition of pain asymbolia, in which pain sensations appear to be normal but behavioral and physiological responses to the noxious stimulus are inappropriate (Berthier et al 1988). Recent studies continue to provide evidence that the IC may be critical and the most specific portion of the cortex in pain perception, with a posterior area encoding nociceptive stimulus properties and a more anterior region related to the subjective experience of pain. The posterior IC is the cortical area that receives the densest spinothalamic–thalamocortical nociceptive input in the cortex (Dum et al 2009). Stimulation of the human IC has been shown to provoke extremely unpleasant painful sensations (Ostrowsky et al 2002), and more recent studies indicate that these sensations are topographically organized and seem to be evoked by electrical stimulation of the IC to the exclusion of the rest of the cortex (Mazzola et al 2009) (Fig. 7-3). Although large unilateral IC lesions may enhance pain perception, focal posterior IC lesions seem to lead to central pain and specific deficits in acute pain perception (Garcia-Larrea et al 2010). Both animal and human studies implicate the IC in autonomic control (Oppenheimer et al 1996, Verberne and Owens 1998). Craig (2003a, 2003b), in integrating these findings, has stated that pain has a specific emotion that reflects the homeostatic behavioral drive and suggests that IC pain-evoked activity is central to this drive. Two separate approaches for studying the role of the IC in human pain have reached the conclusion that the anterior portion of the IC uniquely encodes the subjective magnitude of painful stimuli (Baliki et al 2009, Oshiro et al 2009). This integrated subjective perception may in fact be the signal dictating, for acute pain, the related behavioral drive.
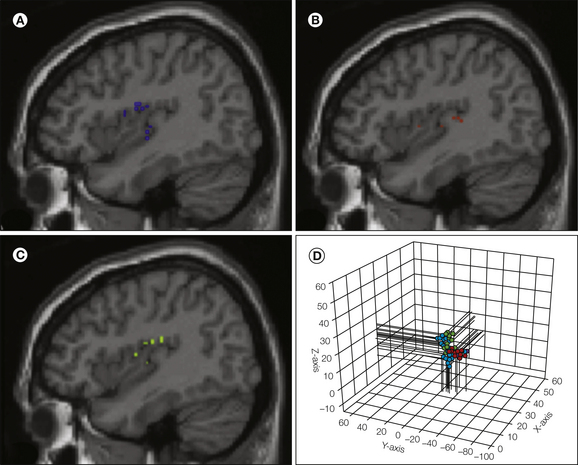
Figure 7-3 Electrical stimulation of the insula in humans evokes pain perception with somatotopic organization.
Twenty-seven sites are shown projected on a sagittal insula magnetic resonance imaging slice. Stimuli evoking pain on the face (blue symbols, panel A), lower limb (red, panel B), and upper limb (green, panel C) are shown separately. Panel D illustrates the three-dimensional relationship between all 27 sites, where the face is localized more rostral to the limb regions and the upper limb is more superior to the lower limb region. (From Mazzola L, Isnard J, Peyron R et al 2009 Somatotopic organization of pain responses to direct electrical stimulation of the human insular cortex. Pain 146:99–104.)
Prefrontal activity, mainly in the vicinity of area 10, has been observed in a number of imaging studies. However, Coghill and colleagues (1999) noted that prefrontal pain-evoked activity does not show the same systematic relationship to perceived pain intensity seen in other regions but instead exhibits the highest activity when a stimulus just becomes painful, with lower activation being associated with higher levels of pain (Fig. 7-4). Similarly, Strigo and co-workers (2003) found significantly more prefrontal activity in response to a painful cutaneous stimulus than to a painful visceral stimulus despite the visceral stimulus being perceived as more unpleasant. Thus, the prefrontal pain-evoked activity may be related to the cognitive aspects of pain perception rather than directly to pain sensation or affect. Lorenz and colleagues (2002) shed further light on the specific role of subregions of the frontal cortex in pain perception. Using capsaicin-evoked thermal allodynia, the authors compared brain activity evoked during capsaicin-produced allodynia and normal heat-induced pain of equal intensity. The contrast showed large activity in the allodynia case that included multiple frontal regions, as well as medial thalamus, nucleus accumbens, and midbrain activity. Network analysis of this activity demonstrated that dorsal frontal and orbital frontal cortical activities were antagonistic to each other, with the dorsal region limiting the activity of the orbital region and the latter acting in concert with other regions. They thus concluded that the orbital frontal–accumbens–medial thalamus network is engaged in affective perception of pain whereas the dorsal frontal cortex acts as a “top-down” controller that modulates pain and therefore limits the extent of suffering.
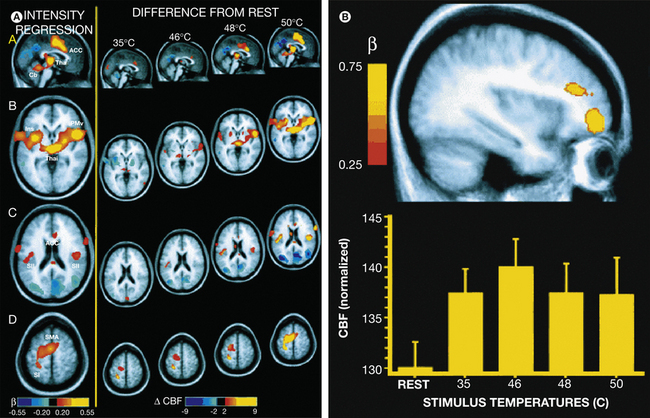
Figure 7-4 A, Multiple regression analysis reveals that activation in the thalamus, anterior cingulate cortex (ACC), insular cortex, secondary somatosensory cortex (SII), supplementary motor area (SMA), and primary somatosensory cortex (SI) is significantly related to subjects’ perceptions of pain intensity (left panel, regression coefficients [β] are color-coded such that red-yellow voxels are positively related to pain intensity whereas blue-violet voxels are inversely related to pain intensity, P < 0.001). Progressive increases in activation are evident within these areas as stimulus temperature increases (right panel, difference in cerebral blood flow [CBF] between each temperature and rest). Functional data are displayed on the averaged structural magnetic resonance imaging data of all subjects. The left side of the image corresponds to the subjects’ left hemisphere. Cb, cerebellum; Ins, insula; PMv, ventral premotor cortex; Thal, thalamus. B, Regions of the right prefrontal cortex were activated during thermal stimulation in a manner not linearly related to perceived pain intensity (top panel, P < 0.03). In the ventral focus, maximum activation was observed during stimulation approximating the pain threshold (46°C, bottom panel, means ± SE). (Adapted from Coghill RC, Sang CN, Maisog JM et al 1999 Pain intensity processing within the human brain: a bilateral, distributed mechanism. Journal of Neurophysiology 82:1934–1943.)
Another brain region activated in the majority of pain studies is the cerebellum. The cerebellum has been implicated in the control of various functions, including motor, sensory, and cognitive, and according to recent evidence, it is involved in nociceptive activities as well (Ekerot et al 1991, Gao et al 1996, Bower 1997, Saab et al 2000). Several human and animal imaging studies have reported activity in the cerebellum following painful somatic and visceral stimulation (Casey et al 1994, Derbyshire and Jones 1998, Iadarola et al 1998, Becerra et al 1999, Mertz et al 2000, Saab et al 2000). Furthermore, studies using electrical and chemical stimulation of the cerebellar cortex show that the cerebellum plays a role in the modulation of both visceral and somatic nociceptive responses (Saab et al 2001, Saab and Willis 2001). Pain-evoked cerebellar activation is present in anesthetized humans, who are not consciously aware of the pain (Hofbauer et al 2004), thus further suggesting that such activity may be more important in the regulation of afferent nociceptive activity than in the perception of pain.
How do we Distinguish Location and Quality of Pain?
Neuroimaging studies have examined brain regions activated by many types of painful stimulation, including noxious heat and cold, muscle stimulation using electric shock or hypertonic saline, topical and intradermal capsaicin, colonic distention, rectal distention, gastric distention, esophageal distention, ischemia, cutaneous electric shock, ascorbic acid, and laser heat, as well as an illusion of pain evoked by combinations of innocuous temperatures. Despite the differences in sensation, emotion, and behavioral responses provoked by these different types of pain, individuals can easily identify each as being painful. Thus there appears to be a common construct of “pain” with an underlying network of brain activity in the areas described above. Nevertheless, despite the similarities in pain experiences and similarities in neural activation patterns, each pain experience is unique. Subjects can usually differentiate noxious heat from noxious cold from noxious pressure. This ability to differentiate types of pain is particularly puzzling since there is ubiquitous convergence of information from cutaneous, visceral, and muscle tissue throughout the afferent nociceptive system (McMahon et al 1995). The convergence and the similarities in brain regions activated by different types of pain are consistent with phenomena such as referred pain but cannot explain either the ability to identify the origin of the pain or the contrasting behavioral reactions to cutaneous and visceral pain (withdrawal versus quiescence).
There is evidence from single-neuron recordings, MEG, PET, and fMRI that neural activity in the S1 cortex could underlie identification of the locus of cutaneous pain. Kenshalo and colleagues (1983, 1988) showed that S1 nociceptive neurons have discrete receptive fields such that different neurons respond to painful stimulation in different skin areas. Correspondingly, EEG, PET, and fMRI studies have shown a topographic organization of nociceptive responses in the S1 cortex similar to the organization of tactile responses (i.e., mediolateral organization of foot, hand, face, and intra-abdominal areas) (Tarkka and Treede 1993, Andersson et al 1997, DaSilva et al 2002, Strigo et al 2003, Vogel et al 2003), thus suggesting that responses in the S1 cortex may be important for pain localization. Yet it is now apparent that the IC also participates in pain localization (Mazzola et al 2009). Strigo and co-workers (2003) used fMRI to directly compare brain activation produced by esophageal distention and by cutaneous heat on the chest that were matched for pain intensity. They found that the two qualitatively different types of pain produced different primary loci of activation within the IC, S1, motor cortex, and PFC. Such local differences in responses within the “pain network” might subserve our ability to distinguish visceral and cutaneous pain, as well as the differential emotional, autonomic, and motor responses associated with these different sensations.
Laterality of Pain Representation
Many nociceptive pathways are bilateral, but the spinothalamic pathway is mainly contralateral, and its ipsilateral component becomes smaller from rats to monkeys (see Chapter 12). Generally, brain imaging studies of pain show bilateral activity in S2 and the IC and contralateral activity in S1. Activity in the ACC seems predominantly contralateral, although this is hard to quantify given its midline location. Coghill and colleagues (see Fig. 7-4) found that for low-intensity stimuli, activity that is dependent on stimulus intensity is mainly contralateral whereas activity that is not correlated with stimulus intensity is mainly right brain dominant. Still, regardless of whether the stimulus intensity is high or low, humans have little difficulty identifying the body side where a painful stimulus is applied. It is remarkable that although IC activity, as observed in human brain imaging studies, is almost always bilateral, stimulation within the IC evokes pain sensations that are mostly contralateral for body and trunk sensations and more bilateral for facial sensations (Mazzola et al 2009). It therefore seems that the IC and perhaps the S1 cortex are the best candidates for detecting the laterality of a painful stimulus since these cortical structures show laterality of activity for evoked sensations, have a somatotopic organization, and are thought to be important in untangling the sensory-discriminative aspects of pain.
Distinct Brain Responses to Nociception and to Subjective Perceived Pain
A recent study examined discrimination between stimulus localization and stimulus intensity to subdivide the brain areas related to acute pain (Oshiro et al 2009) and advance the exciting notion that pain, similar to vision and audition, may also consist of a ventral, in this case intensity-coding, stream terminating in the IC and a dorsal, spatial localization stream. A number of studies have now emphasized the need to identify brain activity relative to the subjective perception of pain (Davis et al 2002, Lorenz et al 2002, Giesecke et al 2004, Kwan et al 2005, Mazzola et al 2009). After all, the brain should reflect perception, and in fact the classic view has been that even peripheral primary afferent nociceptors show close correspondence in their stimulus–response curves to the psychophysical power function for pain. A study by Baliki and associates (2009) differentiated between perceived pain and stimulus representation for thermal painful stimuli and demonstrated that the part of the IC in contiguity with more dorsal cortex best related to the perceived subjective magnitude of acute pain; surprisingly, the same region encoded just as faithfully magnitudes for rating sizes of bars presented visually. Thus, although this part of the IC reflects subjective pain best, it is a multimodal area that might distill magnitude information for various sensory modalities. The study also demonstrated a 1:1 correspondence between activity in this region and ratings of perceived pain for each and every stimulus across all subjects, thus showing that the region responds proportionally every time that a subject reports perception of pain. Relative to subjective rating of perceived pain, IC responses could be divided into two regions—nociceptive and pain-perceptive regions—with each showing distinct anatomical connectivity as determined by diffusion tensor imaging (Baliki et al 2009; also see Craig 2009 for a review of insula function).
Temporal Sequence of Cortical Activity during Pain Perception
Most information about the temporal sequence of pain-evoked brain activation comes from EEG or MEG studies. The dual pain sensation elicited by a single brief painful stimulus that is due to the different conduction times in nociceptive A and C fibers (about a 1-second difference) is reflected in two sequential brain activations on EEG and MEG recordings from the S1, S2, and ACC (Bromm and Treede 1983, Arendt-Nielsen 1990, Bragard et al 1996, Magerl et al 1999, Opsommer et al 2001, Ploner et al 2002, Tran et al 2002, Iannetti et al 2003). EEG mapping studies (Kunde and Treede 1993, Miyazaki et al 1994), source analysis (Tarkka and Treede 1993, Valeriani et al 1996, Ploner et al 1999), and intracranial recordings (Lenz et al 1998, Frot et al 1999) show that the earliest pain-induced brain activity originates in the vicinity of S2. In contrast, tactile stimuli activate this region only after processing in S1 (Ploner et al 2000). The adjacent dorsal IC is activated slightly but significantly later than the operculum (Frot and Mauguiere 2003). These observations support the suggestion derived from anatomical studies that the S2 region and adjacent IC are primary receiving areas for nociceptive input to the brain (Apkarian and Shi 1994, Craig 2002, Dum et al 2009). Another study has shown that brief painful stimuli evoke sustained cortical activity corresponding to sustained pain perception. The time courses of activation disclosed that the first pain was particularly related to activation of S1 whereas the second pain was closely related to activation of the ACC. Both sensations were associated with S2 activation. These results are interpreted in view of the different biological functions of the first and second pain. The first pain signals threat and provides precise sensory information for immediate withdrawal, whereas the second pain attracts longer-lasting attention and motivates behavioral responses to limit injury and optimize recovery (Ploner et al 2002). An MEG study by Maihofner and colleagues (2002) differentiated between brain regions involved in cold perception from those involved in painful cold. Cold perception resulted in activity in the posterior IC with a mean peak latency of 190 msec for contralateral and 240 msec for ipsilateral insular activity. Noxious cold stimulation initially activated the IC in the same latency ranges as innocuous cold stimuli. Additionally, activations were found in the contralateral and ipsilateral S2 areas (peak latencies of 304 ± 22.7 and 310.1 ± 19.4 msec, respectively), and more variable activation was found in the cingulate cortex. Neither cold nor painful cold produced detectable activation of S1. The results suggest different processing of cold, painful cold, and touch in the human brain.
Temporal differences in brain activity for pain have also been studied by blood flow techniques. Casey and associates (2001) showed that the brain activity pattern for a repetitive thermal painful stimulus is different depending on the past history. When the brain was imaged immediately after the start of the pain, the authors observed very different brain regional responses than when the scan began after the pain had been present for 40 seconds. Perceptually, participants perceived the ongoing pain as more intense and unpleasant than the same stimulus at its onset. A number of brain regions increased in activity between early and late scans (e.g., contralateral S1, bilateral S2, parts of the IC and thalamus). Other brain regions were active only in the early stimulation scans (e.g., perigenual ACC, PFC, and anterior IC). The full significance of these differences remains unclear, although these results undoubtedly imply that the temporal dynamics of brain activity should be taken into consideration to understand the role of the brain circuitry in pain perception. More recently, fMRI has been used to examine the temporal sequence of brain activity for acute thermal pain (Baliki et al 2009; see also Apkarian et al 2011), and the authors could chart transfer of brain regional information as noxious stimuli are converted to perceived pain. The analysis showed that parts of the ACC and amygdala responded in a predictive pattern; the thalamus, basal ganglia, part of the IC, and the supplementary motor region were activated next, and these regions better related to the stimulus; and the perception-related part of the IC was activated last, together with the higher cognitive frontal and parietal regions (Fig. 7-5). Thus there seems to be a well-organized temporal sequence of brain activity that transforms noxious stimulus parameters to pain perception, even to pain relief (for more details see also Apkarian et al 2011).
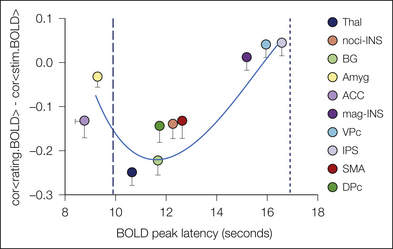
Figure 7-5 Temporal sequence of brain areas activated when participants rated the magnitude of pain perceived for a thermal painful stimulus.
For each brain region identified as activated for the task, the brain oxygenation level–dependent (BOLD) signal was compared with the shape of the stimulus and perception responses to generate a similarity value between them (y-axis). Negative values on the y-axis indicate better correspondence to the shape of the stimulus, whereas positive values indicate better correspondence to the time course of perception. The peak responses of different areas were also compared with the peak of the stimulus and perception (x-axis). Time scale on the x-axis is relative to the start of the thermal stimulus. Ten seconds corresponds to the peak of the stimulus and 18 seconds to the peak of pain perception. Thus, the mean location of any brain region within this two-dimensional space provides a temporal and shape-related assessment of its activity in relation to the thermal stimulus and perception of pain. Brain areas activated before 10 seconds must be considered predictive because they precede the stimulus peak (ACC, anterior cingulate cortex; Amyg, amygdala). Areas clustered just after the stimulus peak also show a better shape relationship with the stimulus and are thought to reflect stimulus parameters; that is, they can be considered nociceptive regions (BG, basal ganglion; DPc, dorsal premotor cortex; noci-INS, nociceptive portion of the insula; SMA, supplementary motor area; Thal, thalamus). The clusters of areas responding the latest show better similarity to pain perception and either are reflecting pain perception (mag-INS, magnitude-related portion of the insula; VPc, ventral premotor cortex, a region contiguous with the mag-INS) or are involved in task completion (i.e., magnitude estimation [IPS, inferior parietal sulcus]). (From Baliki MN, Geha PY, Apkarian AV 2009 Parsing pain perception between nociceptive representation and magnitude estimation. Journal of Neurophysiology 101:875–887.)
The Brain’s Role in Modulating Pain
In the last 30 years, much attention has been directed toward nociceptive modulation in the spinal cord involving both intrinsic mechanisms and descending control from the brain stem (Chapter 8). However, descending projections from the cerebral cortex feed into these modulatory systems, and additional pain modulation can also take place within the cortex (Fig. 7-6).
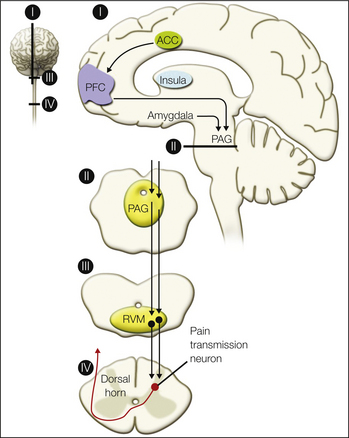
Figure 7-6 Descending pain modulatory pathways originating from the cerebral cortex.
One pathway involves descending input from the anterior cingulate cortex (ACC) to the prefrontal cortex (PFC) and then to the periaqueductal gray (PAG). Another descending pathway arrives at the PAG from the insula via the amygdala. A descending pathway from the PAG through the rostroventral medulla (RVM) to the dorsal horn of the spinal cord influences nociceptive afferent transmission. (From Schweinhardt P, Bushnell MC 2010 Pain imaging in health and disease—how far have we come? Journal of Clinical Investigation 120:3788–3797.)
Opiates in the Brain
Opiate responses in the human brain have been studied by two approaches: examination of brain metabolic function in response to pharmacological agents and direct measurement of receptors, whether static or before and after various challenges. The latter involves the use of radiolabeled pharmaceuticals introduced at tracer doses. Acquisition of data over time, as the radiotracer binds to specific receptor sites, together with appropriate kinetic models, allows quantification of receptor sites and enzyme function in human subjects with PET or SPECT. Exogenous administration of μ-opioid receptor agonist drugs has been shown to dose dependently increase metabolic activity in regions rich in μ-opioid receptors, such as the ACC, PFC, thalamus, basal ganglia, and amygdala (Firestone et al 1996, Schlaepfer et al 1998, Wagner et al 2001, Wise et al 2002, 2004, Pattinson et al 2007). The effects of the μ-opioid agonist fentanyl on regional cerebral blood flow (rCBF) after pain-related activation in the brain have also been explored. Using painful cold with or without fentanyl, Casey and colleagues (2000) showed that the increases in rCBF elicited by the stimulus were prominently reduced by the μ-opioid agonist in most regions, thus confirming an inhibitory effect of fentanyl on measures of pain-induced neuronal activity. Other studies have confirmed the effects of the μ-opioid agonist remifentanil on pain-evoked cortical activity in the ACC and IC (Petrovic et al 2002, Wise et al 2002, 2004).
Dynamic changes in the activity of the endogenous opioid system and μ-opioid receptors have also been examined by using a combination of a selective μ-opioid radiotracer, [11C]carfentanil, and models of sustained muscular or cutaneous pain in healthy subjects (Zubieta et al 2001, Bencherif et al 2002, Zubieta et al 2003, Wager et al 2007). Reductions in the in vivo availability of μ-opioid receptors, reflecting activation of this neurotransmitter system, were observed in the ACC, PFC, IC, thalamus, ventral basal ganglia (nucleus accumbens and ventral pallidum), amygdala, and PAG. Activation of certain elements of this neurotransmitter system was also linearly correlated with suppression of the sensory and affective qualities of the pain, as rated by the volunteers (Zubieta et al 2001). An area within the dorsal ACC was uniquely associated with suppression of the affective quality of pain, and this same region has been implicated in the representation of pain affect (Rainville et al 1997). These findings confirm the widespread involvement of μ-opioid receptors in regulation of the experience of pain, including not only areas involved in descending inhibition (PAG, thalamus, and amygdala) but also areas probably involved in more complex functions such as assessment of stimulus salience, as well as affective and integrative aspects of the pain experience (e.g., ventral basal ganglia, IC, ACC, and PFC). Furthermore, substantial interindividual differences have been observed in both receptor-binding levels and the magnitude of activation of this neurotransmitter system, and recent evidence suggests that the brain opiate system may be altered in some chronic pain patients (see later).
An additional contribution to variability in μ-opioid receptor binding and the capacity to activate this neurotransmitter system in response to sustained pain has been described as a function of a common polymorphism of the catechol O-methyltransferase (COMT) enzyme. Substitution of valine (Val) by methionine (Met) at codon 158 of the COMT gene is associated with a three-fold to four-fold reduction in the capacity to metabolize catecholamines (e.g., noradrenaline and dopamine). The alleles are co-dominant, which results in the lowest COMT function in Met homozygous individuals, highest in Val homozygotes, and intermediate in heterozygotes. These alterations in catecholaminergic neurotransmission result in downstream changes in the capacity to activate responses of the μ-opioid system to sustained pain, with the lowest function in Met/Met, intermediate in Met/Val, and highest in Val/Val subjects. Furthermore, there appear to be compensatory changes in μ-opioid receptor binding in opposite directions (Zubieta et al 2003). Aside from the importance of this work in understanding interindividual variations in the regulation of pain, it also describes a point of interaction between neurotransmitter systems, such as the catecholaminergic and opioidergic systems, involved in responses to stress, salient stimuli, and reward with pain regulatory mechanisms. These interactions are observed at the level of dopaminergic pathways, such as the ventral basal ganglia and anterior cortical regions, but also in areas with prominent noradrenergic innervation, such as the thalamus.
Dopamine and Pain
Although brain dopamine is best known for its role in pleasure, motivation, and motor control, there is now substantial evidence that it also plays a role in pain modulation. Animal studies involving either electrical stimulation of dopaminergic structures, such as the striatum, nucleus accumbens, or ventral tegmental area, or administration of compounds that increase synaptic levels of dopamine, such as dopamine reuptake inhibitors, show that increased dopaminergic activity attenuates nocifensive behavior in animals (Chudler and Dong 1995, Altier and Stewart 1999, Magnusson and Fisher 2000). On the other hand, deactivation of dopaminergic structures leads to hyperalgesia in animals (Saade et al 1997, Magnusson and Martin 2002, Chudler and Lu 2008). Using competitive binding PET methods and the dopamine D2/D3 radiotracer [11C]raclopride with sustained muscular pain, studies have shown that healthy individuals release dopamine in the striatum in response to pain (Scott et al 2006, Wood et al 2007b). These and other human PET studies indicate that the binding potential of [11C]raclopride in healthy subjects is related to their pain sensitivity (Hagelberg et al 2002, 2004, Pertovaara et al 2004, Martikainen et al 2005, Scott et al 2006, Wood et al 2007b). Together, this evidence suggests that one role of striatal dopamine is pain regulation. Recent evidence suggests that some chronic pain states may be associated with altered dopaminergic neurotransmission, as detailed later in this chapter.
Mechanisms Underlying Psychological Modulation of Pain
Attentional and emotional factors are known to modulate pain perception in the clinic and in the laboratory (Beydoun et al 1993, Villemure and Bushnell 2002, Roy et al 2008). Nevertheless, the mechanisms underlying such modulation have been difficult to explore in animal studies. The advent of human brain imaging provided an important new avenue for deciphering the neural basis of psychological modulation of pain. In recent years, brain imaging experiments have explored the mechanisms underlying attentional and emotional modulation of pain, as well as activity related to the expectation of pain and placebo analgesia.
Effect of Attention and Distraction on Pain-Evoked Activity in the Brain
Before brain imaging allowed the exploration of psychological variables in humans, a few studies were performed in which attentional state was manipulated in non-human primates and action potentials from single neurons were recorded. These studies revealed that for some neurons in the dorsal horn and the thalamus, the activity evoked by a noxious stimulus was enhanced when the monkey was performing a task that required attention to the noxious stimulus rather than when performing a task that required attention to be diverted to a visual stimulus (Bushnell et al 1984, Bushnell and Duncan 1989). Human brain imaging studies that use distracting tasks have revealed modulation of pain-evoked activity in the thalamus and several cortical regions, including S1, ACC, and IC (Bushnell et al 1999, Longe et al 2001, Bantick et al 2002, Brooks et al 2002, Valet et al 2004, Wiech et al 2005, Villemure and Bushnell 2009). Other regions, including PAG, parts of the ACC, and the orbitofrontal cortex (within the PFC), are activated when subjects perform distracting tasks, thus suggesting that these regions may be involved in the modulatory circuitry related to attention (Petrovic et al 2000, Frankenstein et al 2001, Tracey et al 2002, Valet et al 2004). However, many distracting tasks require increased cognitive demand, thereby possibly leading to increased arousal and/or altered emotional state of the subject. Since emotional states can also alter pain (Zelman et al 1991, De Wied and Verbaten 2001, Meagher et al 2001, Marchand and Arsenault 2002, Villemure and Bushnell 2009, Wiech and Tracey 2009, Villemure and Schweinhardt 2010), it is important to differentiate between modulatory circuits related to attention and those related to emotions.
EEG and MEG studies show that cognitive modulation of pain by attention involves early sensory processing in S2–IC (Legrain et al 2002, Nakamura et al 2002) and later processing in the ACC (Beydoun et al 1993, Kanda et al 1996, Siedenberg and Treede 1996, Garcia-Larrea et al 1997, Tiede et al 2010). Perceptual changes in pain related to attentional state appear to reflect in part a change in cortical processing and in part a decrease in ascending afferent input from the spinal cord because of activation of descending inhibitory controls. EEG signals can document this type of inhibitory control in humans (Plaghki et al 1994, Hoshiyama and Kakigi 2000, Reinert et al 2000).
Effect of Emotional State on Pain-Evoked Activity
A number of studies have now used neuroimaging to evaluate the effects of emotional state on pain processing (Phillips et al 2003, Roy et al 2009, Villemure and Bushnell 2009, Wiech and Tracey 2009, Berna et al 2010). Negative emotional states produced by looking at emotional faces, listening to unpleasant music, or smelling unpleasant odors alter pain perception, with the largest effect on pain unpleasantness rather than the sensory-discriminative components of the sensation (Villemure and Bushnell 2002, Roy et al 2008, Villemure and Bushnell 2009). Similarly, emotional state alters pain-evoked cortical activation, most commonly in regions associated with the affective component of pain processing, particularly the ACC and IC (Phillips et al 2003, Roy et al 2009, Villemure and Bushnell 2009, Berna et al 2010, Ploner et al 2011).
What Brain Circuits Are Involved in Attentional and Emotional Modulation of Pain?
As mentioned earlier, some researchers have suggested that the opiate-sensitive descending pathway from the frontal cortex to the amygdala, PAG, rostral ventral medulla, and spinal cord dorsal horn (Fields 2000) is involved in attentional modulation of pain (Tracey et al 2002, Valet et al 2004). Nevertheless, these studies have generally used tasks that may involve simultaneously altered attention, arousal, and emotional state. By separating attentional state and mood in the same study, Villemure and Bushnell (2009) found that the fronto-PAG circuitry is more likely to be involved in emotional modulation of pain whereas activation in the superior parietal lobe, which is part of Corbetta and Shulman’s proposed “top-down orienting of attention” system (Corbetta and Shulman 2002), is more important for attentional modulation (Fig. 7-7).
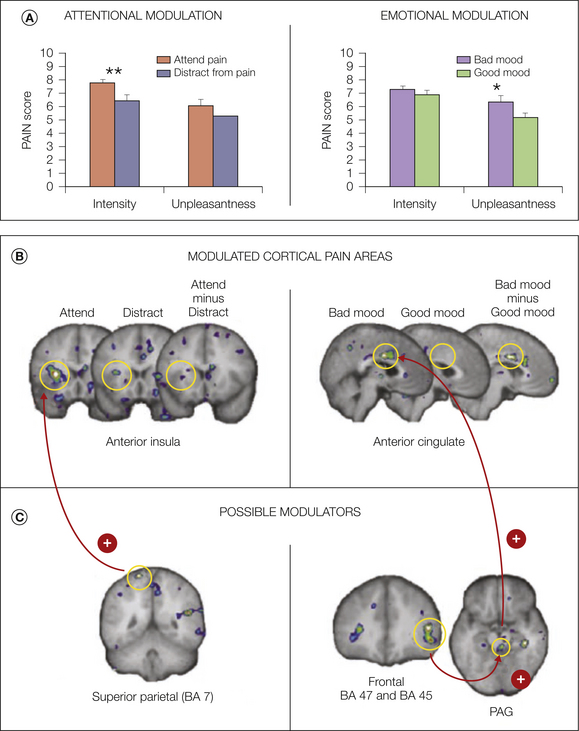
Figure 7-7 A, Attention preferentially modulates pain intensity ratings, whereas emotional state preferentially modulates pain unpleasantness. B, Cortical pain areas that showed the largest modulations in attentional and emotional conditions. Attention modulated activity in the anterior insular cortex (IC), and emotions modulated activity in the anterior cingulate cortex (ACC). C, Potential modulators of the signal in pain-processing areas. Superior posterior parietal cortex activity co-varied positively with activity in the IC when subjects attended to the pain; activity in BA 45, BA 47, and periaqueductal gray (PAG) positively co-varied with activity in the ACC when mood was deteriorated by the presence of an unpleasant odor. (From Villemure C, Schweinhardt P 2010 Supraspinal pain processing: distinct roles of emotion and attention. Neuroscientist 16:276–284, Fig. 1.)
Effect of Expectation on Pain-Evoked Activity
Imaging studies reveal that anticipation or expectation of pain can activate pain-related areas in the absence of a physical pain stimulus (Porro et al 2002, Jensen et al 2003, Yaguez et al 2005). In addition, regions thought to be involved in pain modulation, such as the PAG, PFC, and ventral striatum, are activated during a period of pain expectation, thus suggesting that such activation may modulate the impending pain-evoked activation (Beydoun et al 1993, Hsieh et al 1999, Ploghaus et al 1999, Sawamoto et al 2000, Porro et al 2002, Jensen et al 2003, Fairhurst et al 2007, Lopez-Sola et al 2010). Several studies in patients with chronic pain and/or depression show enhanced responses during anticipation of pain, which may be one factor contributing to the enhanced pain perception in these patients (Song et al 2006, Strigo et al 2008, Burgmer et al 2010).
Anticipation and Placebo Analgesia
Anticipation of a reduction in pain is a main factor contributing to the placebo effect (Benedetti et al 2005). Neuroimaging studies of expectation-related placebo analgesia show that during the anticipation period immediately preceding the application of a noxious stimulus, a cingulo–fronto–PAG descending modulatory pathway is activated, similar to that activated during emotional modulation of pain (Wager et al 2004, Eippert et al 2009a, Watson et al 2009). Furthermore, using a PET μ-opioid competitive binding assay, Zubieta and colleagues (Zubieta et al 2005, Wager et al 2007) showed that placebo treatment affected endogenous opioid activity in a number of regions, including the PAG, ACC, and lateral PFC, thus supporting the idea that activation of the cingulo–fronto–PAG pathway during the expectation of pain relief leads to a reduction in nociceptive afferent drive. This idea is further supported by a study that examined the influence of naloxone (a μ-opioid antagonist) on placebo-related brain activation (Eippert et al 2009a) and found that naloxone modulated placebo-induced responses in key structures of the descending pain control system, including the PAG and rostral ventral medulla. Furthermore, naloxone abolished the placebo-induced coupling between the rostral ACC and PAG. Functional imaging studies also provide evidence that nociceptive afferent drive at the level of the spinal cord, thalamus, and cortex is reduced during placebo analgesia (Wager et al 2004, Eippert et al 2009b, Watson et al 2009).
Brain Structure and Function in Clinical Pain
Human brain imaging techniques have afforded an exciting opportunity to examine pain processing, as well as brain structure, in clinical pain conditions. Although significant progress has been made, the study of chronic pain has been considerably more difficult than the study of acute experimental pain for a variety of reasons, including the lack of controllability and heterogeneity of patient groups. In particular, it is often difficult to determine whether brain alterations are a cause or a consequence of chronic pain because of a distinct lack of longitudinal studies. Nevertheless, it is clear that although clinical pain states fundamentally activate similar brain regions as do acute pain stimuli, differences exist that seem to reflect changes in the psychological state related to chronic pain states, as well as alterations in pain modulatory systems. In addition to changes in processing, evidence is accumulating that prolonged pain is associated with structural brain alterations. Finally, two clinical pain states, cluster headache and migraine, will be discussed in more detail because ample evidence suggests that the primary pathophysiology of these conditions is located in the brain.
Cerebral Processing of Clinical Pain
Several methods have been used to examine the processing of clinical pain. Brain activation associated with ongoing pain without experimental provocation has been investigated with CBF PET studies (Di Piero et al 1991, Hsieh et al 1995). Spontaneous fluctuations in ongoing pain have been exploited with fMRI (Apkarian et al 2001, Baliki et al 2006), and other studies have provoked pain typical for certain clinical conditions, such as dynamic mechanical allodynia and static mechanical hyperalgesia in the case of neuropathic pain (Petrovic et al 1999, Peyron et al 2004, Maihofner et al 2005, Schweinhardt et al 2006), to examine processing of clinical pain. All these different approaches have shown that brain activation associated with clinical pain states overlaps with areas that process acute pain stimuli. Nevertheless, differences are observed for clinical pain and can be grouped broadly into three categories. (1) S1 and S2 are less consistently activated (Apkarian et al 2005), perhaps particularly in the case of ongoing pain (Hsieh et al 1995), which might imply that sensory discrimination is less pronounced for ongoing pain than for brief stimuli. (2) Some brain areas exhibit increased activation (Apkarian et al 2005, Tillisch et al 2010). Baliki and colleagues (2006) used an interesting approach in analyzing patients with chronic low back pain that helped in understanding the engagement of additional forebrain areas in clinical pain: when activations present during periods of rapidly increasing pain were statistically separated from periods of sustained high-level pain, activations associated with the rapid increases in pain were similar to those observed in healthy people during acute pain. Conversely, a different brain circuit was engaged that involved the PFC and amygdala during periods of high sustained pain. The finding of more frequent PFC activation with clinical pain than with experimental pain had been shown previously in a meta-analysis (Apkarian et al 2005), and increased amygdala activation was recently corroborated in a meta-analysis of rectal distention in patients with irritable bowel syndrome (IBS) (Tillisch et al 2010) (Fig. 7-8). The same meta-analysis also found brain stem structures to more frequently be activated in IBS patients than in controls (Tillisch et al 2010) (see Fig. 7-8). In view of the importance of the PFC for higher-order processing, increased prefrontal activation is likely to reflect the more elaborated emotional and cognitive states associated with clinical pain than with experimental pain (Apkarian et al 2005). Moreover, as outlined earlier, the PFC plays an important role in pain modulation, in particular, in mediating the pain modulatory effects of psychological states. Therefore, increased prefrontal activation could also reflect increased engagement of pain facilitatory or inhibitory circuits, depending on the exact area. In the case of IBS and potentially other functional pain syndromes, increased amygdala activation might reflect a contribution of arousal and emotion circuitry to pain amplification, which could be manifested by more prominent brain stem activation (Tillisch et al 2010). (3) Thalamic activation is decreased. Chronic pain conditions are often accompanied by reduced spontaneous activity in the thalamus and an associated abnormal burst discharge (Gucer et al 1978, Hirayama et al 1989, Lenz et al 1989). PET studies show areas of reduced thalamic perfusion that spatially correspond to the electrophysiological changes (Di Piero et al 1991, Hsieh et al 1995, Iadarola et al 1995, Peyron et al 1995, Duncan et al 1998). Reversal of thalamic hypoperfusion with successful pain relief has been described in patients with cancer pain (Di Piero et al 1991) and various neuropathic pain conditions (Hsieh et al 1995, Iadarola et al 1995, Peyron et al 1995, Duncan et al 1998). In the case of neuropathic pain, decreased spontaneous activity in the thalamus is consistent with deafferentation of the somatosensory system, and it is therefore not straightforward to establish any pain-relevant pathophysiology of decreased thalamic activity. However, a clever study of a patient with Wallenberg’s syndrome allowed differentiation of deafferentation and pain (Garcia-Larrea et al 2006) and showed that thalamic hypoactivity contralateral to the pain did not merely reflect deafferentation but appeared to be related to pain pathophysiology. In addition, it is intriguing to note that thalamic hypoperfusion has been observed in patients with fibromyalgia (reviewed by Williams and Gracely 2006), which is not linked to deafferentation.
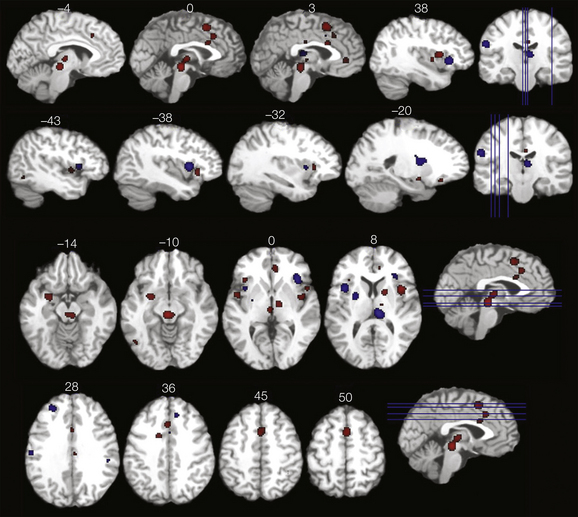
Figure 7-8 Selected axial and sagittal slices representing brain areas demonstrating greater activation in response to rectal distention in patients with irritable bowel syndrome (IBS) than in controls (red areas) and greater activation in controls than in patients with IBS (blue areas) in a meta-analysis of 18 studies. (From Tillisch K, Mayer EA, Labus JS 2010 Quantitative meta-analysis identifies brain regions activated during rectal distension in irritable bowel syndrome. Gastroenterology 140:91–100, Fig. 3.)
Cerebral Processing of Experimental Pain in Chronic Pain States
Instead of imaging brain activation associated with clinical pain, it would be more practical in many instances to use acute experimental stimuli to probe the pain-processing circuitry in patients with chronic pain. Such studies have been performed predominantly in patients with so-called functional pain syndromes, including fibromyalgia, IBS, and vulvovestibulitis. In line with the generalized hyperalgesia that is observed in many of these patients, activation of pain-processing regions is typically increased when stimuli are used that are of the same intensity as in the healthy control group (e.g., Verne et al 2003, Cook et al 2004, Giesecke et al 2004, Pukall et al 2005). Increased brain activation indicates that the nociceptive signal is amplified somewhere along the pain transmission pathways, although it does not pinpoint the mechanism of amplification. In an interesting study, Lawal and colleagues (2006) found enhanced activation even in response to subliminal esophageal stimulation in IBS patients, perhaps suggesting that at least part of the amplification is related to non-psychological factors. When the perceived intensity is equated between patients and controls rather than stimulus intensity, brain activation is not generally different in patients (Giesecke et al 2004). In conclusion, the enhanced brain activation in response to experimental pain stimuli confirms patients’ reports of increased pain sensitivity. In the next section, studies are presented that provide more specific information on how altered cerebral pain modulation might amplify the processing and experience of pain.
Evidence for Altered Supraspinal Pain Modulation in Clinical Pain
The idea that faulty supraspinal pain modulatory circuitry contributes to clinical pain has become increasingly popular in recent years, perhaps also because in many instances a mismatch between perceived pain intensity and objectifiable (peripheral) disease cannot be resolved with current diagnostic tools. Although animal data provide strong evidence for an altered balance between endogenous pain inhibition and facilitation, data in humans are still relatively scarce. As discussed in detail in Chapter 8, the central nervous system (CNS) can augment pain processing through increased facilitation or decreased inhibition. It is well known from the animal literature that pathways that modulate ascending nociceptive signals descend from the brain stem to the spinal cord (Fields and Heinricher 1985, Porreca et al 2002). Consequently, the brain stem has received much attention in human pain imaging studies in recent years. A study in healthy volunteers in whom sensitization to punctuate stimuli was achieved by capsaicin injection provided evidence that the brain stem is also involved in pain facilitation in humans (Lee et al 2008). Evidence in patients comes from a study of individuals with painful hip osteoarthritis (Gwilym et al 2009). Patients rated punctuate stimuli as “sharper” when applied to the lateral aspect of the thigh (i.e., in an area of spinal convergence with sensory innervation of the hip joint). Increased activation in response to the punctuate stimuli was observed in the ACC, dorsolateral PFC, and PAG. The magnitude of PAG activation correlated with the degree of patients’ neuropathic pain symptoms (Fig. 7-9), as measured by the PainDETECT questionnaire. Thus, this study provides evidence that brain stem mechanisms might play a role in neuropathic symptoms in a disorder that has traditionally been considered a nociceptive condition.
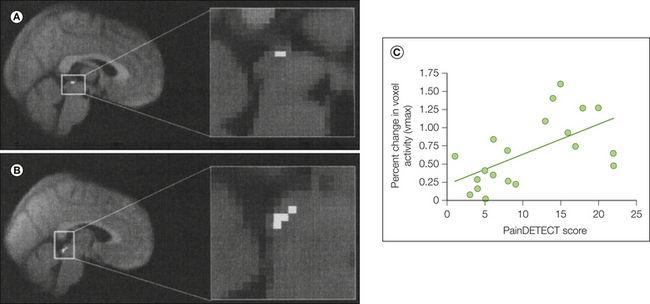
Figure 7-9 Increased activation of periaqueductal gray (PAG) in patients with osteoarthritis and neuropathic pain characteristics.
A, Increased PAG activation in response to punctuate stimuli in patients versus healthy controls (a PAG mask was used to tease out the effect). B, Increased PAG activation in response to punctuate stimuli in patients who score high on the PainDETECT questionnaire (which tests for symptoms compatible with central sensitization) versus patients who score low (analysis masked for PAG). C, Correlation between clinical manifestations of central sensitization (as shown by the total score on the PainDETECT) and PAG activation in response to punctate stimulation in patients. Activation is defined by using the percent change in the parameter estimate of the peak active voxel (vmax) within the masked region of interest of the PAG (r = 0.60, P = 0.006). (From Gwilym SE, Keltner JR, Warnaby CE et al 2009 Psychophysical and functional imaging evidence supporting the presence of central sensitization in a cohort of osteoarthritis patients. Arthritis and Rheumatism 61:1226–1234.)
Animal studies have largely concentrated on the brain stem as the site of endogenous pain modulation, but many studies in healthy volunteers have shown that the higher brain centers are important pain modulators, at least in humans, that mediate the effects of cognition, emotion, and other high-order processes on pain processing. The medial PFC appears to be an important site of supraspinal pain facilitation. Patients with rheumatoid arthritis were shown to activate the medial PFC exclusively in response to clinically relevant pain and not with disease-irrelevant pain (Schweinhardt et al 2008). The degree of activation in this region correlated with the severity of depressive symptoms in this largely not clinically depressed sample. Importantly, medial PFC hyperactivity was related to a measure of the patients’ clinical pain that partly accounted for systemic inflammation, thus indicating that the medial PFC was indeed involved in enhancing clinical pain. In keeping with a pain facilitatory role, activity in the medial PFC in patients with IBS has been shown to disrupt a functional connection between the lateral PFC and PAG (Mayer et al 2005), two areas that have been implicated in endogenous pain inhibition. (At this point, it has to be emphasized that current human brain imaging methods cannot differentiate between activation in brain stem facilitatory or inhibitory circuitries because of their close proximity. Therefore, the PAG is considered in some studies to be part of a facilitatory circuit and in others to contribute to pain inhibition, in line with the dual role that the PAG (and other brain stem structures) is known to have in pain modulation. Inferences regarding the nature of the signal can currently be based only on other variables, such as patients’ symptoms.)
A study in healthy volunteers provides another compelling demonstration of the antagonistic actions of different areas of the PFC: activation of the (dorso-) lateral PFC suppressed activity in an orbital frontal–rostral cingulate–medial thalamus network associated with the unpleasantness of capsaicin-related pain in healthy volunteers (Lorenz et al 2002, 2003).
Functional Reorganization of the Somatosensory Cortex
EEG and MEG studies in humans have advanced our understanding of cortical reorganization in phantom limb pain. Animal experiments had demonstrated that the receptive fields of neurons in S1 that respond to innocuous stimuli move to adjacent skin areas when nerve lesions or amputations interrupt their original input. This reorganization of receptive fields of deafferentiated neurons was initially thought to be a protective mechanism against the development of phantom sensations. When this prediction was tested in human amputees, however, the opposite relationship was observed: the amount of phantom limb pain was positively correlated with the amount of cortical reorganization, as determined by using innocuous somatosensory stimuli (Flor et al 1995, Montoya et al 1998, Grusser et al 2001, Karl et al 2001). This observation parallels findings that greater damage to the somatosensory pathways is associated with greater pain, such as with syringomyelia and post-herpetic neuralgia (Fields et al 1998, Hatem et al 2010, Petersen and Rowbotham 2010). Interestingly, however, the positive correlation between S1 reorganization and pain in amputees is maintained during pharmacological pain relief (Birbaumer et al 1997, Huse et al 2001), thus indicating that the extent of damage to the somatosensory afferents is not the only factor driving this relationship. Correspondingly, similar maladaptive plasticity in S1 and reversal with successful treatment have been described for non-specific chronic low back pain (Flor et al 1997) and complex regional pain syndrome (CRPS) type 1 (Maihofner et al 2003, 2004, Pleger et al 2006), conditions without detectable nerve lesions. Primate data support an effect of nociceptive afferent input on S1 processing of innocuous stimuli in the context of an intact somatosensory system and suggest modulation of the response of rapidly adapting neurons in area 3b/1 by γ-aminobutyric acid (GABA) released on activation of nociceptive neurons in area 3a as a potential mechanism (Whitsel et al 2010). In contrast, the opposite relationship—the pathophysiological contribution of S1 reorganization to pain—is still not well understood. Nevertheless, treatment approaches aimed at enhancing and correcting sensorimotor input have been shown to result in relief of pain in patients with CRPS (Pleger et al 2005, Moseley et al 2008) and amputees, in whom concomitant normalization of S1 maps relating to non-noxious input was observed (Lotze et al 1999, MacIver et al 2008).
Structural Brain Alterations in Patients with Chronic Pain
In recent years it has become clear that individuals who suffer from long-term pain have structural brain changes, first demonstrated by Apkarian and colleagues (2004) (Fig. 7-10). Most studies thus far have investigated gray matter, but more recently, alterations in white matter have also been demonstrated. Gray matter is typically observed to be decreased in important pain-processing or pain-modulating regions such as the ACC, IC, thalamus, and frontal cortex (Apkarian et al 2004, Schmidt-Wilcke et al 2005, Rocca et al 2006, Schmidt-Wilcke et al 2006, Kuchinad et al 2007, Schmidt-Wilcke et al 2008, Davis et al 2008, Geha et al 2008, Kim et al 2008, Schmitz et al 2008, Valfre et al 2008, Burgmer et al 2009, Obermann et al 2009, Rodriguez-Raecke et al 2009, Valet et al 2009, Vartiainen et al 2009, Seminowicz et al 2010), as well as in the (para-) hippocampus (Schmidt-Wilcke et al 2005, Kuchinad et al 2007, Lutz et al 2008, Valet et al 2009). Greater decreases in gray matter are observed in patients with longer pain duration (Apkarian et al 2004, Schmidt-Wilcke et al 2005, Rocca et al 2006, Kuchinad et al 2007, Geha et al 2008, Kim et al 2008, Schmitz et al 2008, Valfre et al 2008, Valet et al 2009) (see Fig. 7-10), thus suggesting that alterations in gray matter are a consequence of pain or (prolonged) nociceptive input. This concept is further supported by two additional lines of evidence. First, a longitudinal study in a rat model of neuropathic pain demonstrated prefrontal changes that occurred several months after nerve injury (Seminowicz et al 2009). Second, DaSilva and colleagues (2008) observed that the reduced thickness of the sensorimotor cortex in patients with trigeminal neuralgia was co-localized with activation related to provocation of the patients’ dynamic mechanical allodynia, thus indicating that excessive nociceptive input might lead to reductions in gray matter.
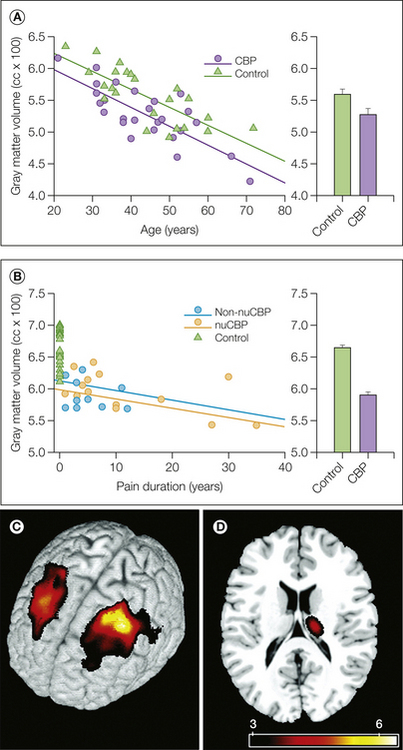
Figure 7-10 Decreased whole-brain cortical gray matter volume in patients with chronic back pain (CBP).
A and B, Skull-normalized gray matter volumes are shown for patients and matched control subjects. A, Gray matter volumes as a function of age. The difference in intercepts corresponds to an average decrease of 30 cm3 in gray matter volume in patients versus control subjects. B, Gray matter volumes as a function of pain duration after correcting for age and gender. Individual control subjects are shown at a pain duration of 0. Data for patients with a neuropathic component (nuCBP) and without a neuropathic component (non-nuCBP) are presented separately. Individual whole-brain gray matter volumes in CBP patients are all below the mean volume for controls. Group-averaged gray matter volumes (mean ± SEM) are shown in the right bar graph before (top) and after (bottom) correcting for age and gender. Lines are best linear fits for each group. C and D, Regional decreases in gray matter density in patients with CBP. Gray matter density is reduced in the left and right dorsolateral prefrontal cortex (C), as well as in the thalamus (D). Decreases were observed in the right thalamus, although the left and right sides of the body were similarly affected across patients. (From Apkarian AV, Sosa Y, Sonty S et al 2004 Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. Journal of Neuroscience 24:10410–10415.)
Several studies have now demonstrated that gray matter concentrations return to baseline levels when the pain resolves (Gwilym et al 2010, Obermann et al 2009, Rodriguez-Raecke et al 2009), a finding indicating that neuronal death is almost certainly not responsible for the observed reductions in gray matter. There is, however, evidence that changes in neuronal tissue, such as perhaps reduced dendritic or synaptic density, might contribute to the observed reductions in gray matter: several studies in patients with chronic pain have shown decreased levels of the neuronal marker N-acetylaspartate (NAA) with proton magnetic resonance spectroscopy (1H-MRS). Grachev and colleagues (2000) were the first to report decreased NAA levels in patients with chronic pain and, in fact, related this finding to potential morphometric alterations. Unfortunately, studies combining measurements of NAA and gray matter in the same patients are still largely missing. A preliminary study of 20 healthy elderly individuals with varying levels of chronic pain found pain severity to be related to lower NAA levels in the hippocampus, as well as to reduced hippocampal volumes (Zimmerman et al 2009), thus suggesting that smaller hippocampi might be partly explained by a decrease in neuronal tissue. Several additional studies have reported decreased NAA levels in various brain regions without providing information on gray matter concentrations. Two studies of fibromyalgia observed decreased hippocampal NAA levels (Emad et al 2008, Wood et al 2009), and one of them reported an inverse relationship between NAA and the severity of fibromyalgia (as measured by the fibromyalgia impact questionnaire [FIQ]) (Wood et al 2009). Decreased NAA levels have also been observed in the thalamus (Fukui et al 2006, Sorensen et al 2008) and PFC (Grachev et al 2000, 2002a, 2002b), regions that are frequently affected by decreased gray matter, as predicted by Grachev and colleagues (Grachev et al 2000) and in which altered pain processing has been demonstrated in chronic pain patients.
The functional significance of changes in gray matter is largely unknown. A few animal studies indicate that alterations in cerebral gray matter might contribute to pain itself, as well as to the sequelae of living with pain. Rapid morphological and functional changes occurred in the medial PFC after spared-nerve injury, and interestingly, an index of sensitization (N-methyl-D-aspartate [NMDA]/α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA] ratio of the synaptic current elicited by afferent fiber stimulation) correlated with the tactile thresholds in the injured paw (Metz et al 2009). In the longitudinal rat study described earlier, the occurrence of prefrontal gray matter alterations coincided with the development of anxiety-like behavior several months after induction of the pain (Seminowicz et al 2009).
Relatively recently, researchers have begun to study white matter alterations in the brains of patients with chronic pain. Geha and colleagues (2008) found decreased diffusion directionality (fractional anisotropy [FA]) in the cingulum of patients with CRPS, which could possibly indicate decreased tract myelination or reduced parallel fiber organization. Fewer white matter connections were found to originate in patients from this spot of altered diffusion than in control subjects. Furthermore, the ventral medial PFC, an area of decreased gray matter, showed an altered anatomical connectivity pattern, thus adding further evidence to impaired white matter connectivity in the patients in this study (Geha et al 2008). Some clinical significance of decreased diffusion directionality is provided by a study of patients with fibromyalgia in which a relationship was demonstrated between decreased FA in the thalamocortical tract and the degree of stiffness experienced by patients (Lutz et al 2008). Schmitz and colleagues assessed the concentration of white matter in migraine patients rather than investigating the diffusion properties of white matter. They found that patients with a high attack frequency had decreased white matter concentrations in the frontal and parietal areas (Schmitz et al 2008), thus suggesting that migraine attacks lead to white matter damage.
Neurochemical Alterations in Patients with Chronic Pain
1H-MRS can provide information on brain chemicals other than NAA, including choline-containing compounds, which indicate membrane turnover and cellular density, and glutamate/glutamine. Elevated glutamate levels, which relate to intracellular and extracellular neurotransmitter levels, have been observed in the IC during the interictal phase in migraine patients (Prescot et al 2009), as well as in patients with fibromyalgia (Harris et al 2008, 2009). Glutamate levels in the insula decreased after treatment of fibromyalgia patients, and a striking relationship was observed between decreased glutamate and improvement in clinical pain, as well as increased pressure pain thresholds (Harris et al 2008). Although more studies of this kind are needed and should preferably examine levels of GABA simultaneously, these observations provide compelling evidence that the presence of chronic pain has an underlying chemical basis in the brain.
Another methodology that can be used to study neurochemistry and, in particular, neurotransmitters, is PET. With the respective tracers, alterations in opioidergic and dopaminergic neurotransmitter systems have been observed in patients with chronic pain. Several studies have reported decreased binding of exogenously administered opioidergic tracers in key pain-processing areas in a variety of different pain conditions, including rheumatoid arthritis (Jones et al 1994), peripheral neuropathic pain (Jones et al 1999, Maarrawi et al 2007), and fibromyalgia (Harris et al 2007). Following pain reduction, normalization of binding levels was observed (Jones et al 1994). Decreased binding of the tracer can be due to either decreased receptor availability/affinity or increased endogenous opioid release. The latter explanation is supported by increased cerebrospinal fluid met-enkephalin immunoreactivity in chronic pain patients (Langemark et al 1995, Baraniuk et al 2004). Although increased release of endogenous opioids is likely to reflect engagement of endogenous antinociceptive systems, the situation is different for central neuropathic pain, in which a lateralized decrease of binding extending the anatomical damage has been observed (Maarrawi et al 2007). This finding suggests opioid receptor loss or inactivation, perhaps caused by metabolic depression and/or degeneration of opioid receptor–bearing neurons secondary to central lesions. Differences in central opioidergic systems in patients with central and peripheral neuropathic pain could explain their different sensitivities to opiate medications.
Several investigations of the dopaminergic system point to an attenuation of dopaminergic activity in some chronic pain states. This is interesting because the dopaminergic pathways seem to be important for endogenous pain modulation (see earlier in this chapter and reviews in animals [Altier and Stewart 1999] and humans [Hagelberg et al 2004]). Reductions in presynaptic dopaminergic function, as measured by the dopamine precursor [18F]fluorodopa, have been reported in idiopathic burning mouth syndrome (Jaaskelainen et al 2001), as well as in fibromyalgia (Wood et al 2007a). These data are consistent with findings of increases in D2 (but not D1) receptor binding in the basal ganglia in patients with burning mouth syndrome and atypical facial pain (Hagelberg et al 2003a, 2003b) because increased receptor availability could signify lower endogenous dopamine levels. One study in patients thus far has investigated release of dopamine in response to a pain challenge. Although healthy controls release dopamine in the striatum during tonic noxious muscle stimulation (Scott et al 2006, Wood et al 2007b), the dopamine response of fibromyalgia patients was not found to differ between painful and non-painful muscle stimulation (Wood et al 2007b).
Specific Chronic Pain Conditions
Two chronic headache syndromes are discussed for which there is evidence of specific functional and structural brain changes that are of pathophysiological relevance.
Migraine
Functional and structural imaging studies have provided evidence that migraine headaches are related to episodic dysfunction of the brain stem nuclei involved in the trigeminovascular nociceptive system. Increases in rCBF occur in the dorsal midbrain and the dorsolateral pons during migraine attacks (Weiller et al 1995, Bahra et al 2001, Afridi et al 2005a, 2005b). The persistence of increased brain stem rCBF despite relief of symptoms with sumatriptan (Weiller et al 1995) has been interpreted as brain stem dysfunction being inherent to migraine rather than being caused by the attacks. The pathophysiological relevance of these observations is emphasized by reports that electrical stimulation of the dorsal midbrain can provoke migraine-like headache when stimulated in patients implanted with electrodes for pain control (Raskin et al 1987, Veloso et al 1998). Furthermore, lesions in the dorsal midbrain have been reported to produce migraine (Haas et al 1993, Goadsby 2002a). Finally, structural abnormalities in the brain stem have been reported in migraine, such as increased PAG density (Rocca et al 2006) and perturbation of iron homeostasis (Welch et al 2001).
Migraine with Aura
In at least 20% of patients, migraine attacks are preceded by transient neurological symptoms, the most prevalent one being visual auras. Similarities between migraine visual aura and cortical spreading depression (CSD) have led to the hypothesis that CSD is responsible for the aura symptoms (Olesen et al 1981, Lauritzen 1994). This view has been greatly advanced and substantiated by brain imaging studies. CSD is characterized by a slowly propagating wave (2–6 mm/min) of sustained strong neuronal depolarization that generates transient (on the order of seconds), intense spike activity as it progresses into the tissue, followed by neural suppression that can last for minutes. The depolarization phase is associated with an increase in rCBF, whereas the phase of reduced neural activity is associated with a reduction in rCBF (Lauritzen 1994). fMRI studies show cerebrovascular changes at a rate typical for CSD in the cortex of migraineurs while experiencing a visual aura (Welch et al 1998, Hadjikhani et al 2001). These changes in fMRI signal develop first in the extrastriate cortex, contralateral to the visual changes, and then slowly migrate toward more anterior regions of the visual cortex, in agreement with retinotopic progression of the visual percept from the center of vision toward the periphery. As would be predicted if the aura symptoms were caused by CSD, Hadjikhani and colleagues (2001) showed that scintillations, which correspond to increased visual activity, were paralleled by increases in fMRI signal and that subsequent decreases in fMRI signal corresponded perceptually to scotomas. Similar results have also been obtained with MEG (Bowyer et al 2001).
Although more work needs to be done, animal studies do provide a link between brain stem dysfunction and vascular changes, which predominantly affect the occipital cortex (Lance et al 1983, Goadsby and Duckworth 1989). These results support the conceptualization of migraine as a neurovascular disorder with primary brain stem dysfunction leading to secondary changes in blood flow in the posterior circulation, which could lead to CSD in susceptible patients (Denuelle et al 2008).
Cluster Headache
Traditionally, cluster headache was thought to be a vascular condition. However, its strong circadian and seasonal rhythmicity (Goadsby 2002b) cannot be readily explained by a vascular hypothesis and led to the concept of a CNS origin for cluster headache (Strittmatter et al 1996). More specifically, dysfunction of the hypothalamic area is postulated as the primary cause of acute cluster attacks given the role that this area plays in circadian rhythm and sleep–wake cycling. Brain imaging studies lend much support to this hypothesis. PET studies have shown significant activation during nitroglycerin-induced acute cluster headache in the ipsilateral posterior hypothalamus as compared with the headache-free state (May et al 1998, 2000). Such hypothalamic activation was not observed in patients outside the bout in whom nitroglycerin did not provoke an attack. In contrast to migraine, no brain stem activation was found during the acute attack. This is remarkable because migraine and cluster headache have often been discussed as related disorders. The importance of hypothalamic activation in cluster headache is further emphasized by a study in healthy volunteers in whom experimental pain was induced by injection of capsaicin into the forehead (May et al 1998). Sensory innervation of the forehead is provided by the ophthalmic division of the trigeminal nerve (i.e., the division in which pain in cluster headache predominantly occurs). In fact, the acute attack of pain and some of the autonomic symptoms characteristic of cluster headache (conjunctival injection, lacrimation) may be regarded as manifestations of the trigeminal autonomic reflex (Goadsby and Edvinsson 1994). No hypothalamic activation was observed following injection of capsaicin into the forehead, thus highlighting the idea that the hypothalamus is primarily affected in cluster headache. The notion of hypothalamic abnormalities is further corroborated by increased gray matter volume in this region (May et al 1999), as well as by decreased levels of the neuronal marker NAA and choline, a marker of membrane turnover and cellular density (Lodi et al 2006, Wang et al 2006).
Based on evidence of the pathophysiological relevance of the posterior hypothalamus, deep brain stimulation of the border zone between the posterior hypothalamus and the ventral tegmental area has been introduced for intractable cluster headaches and yields promising results (Leone et al 2008). In light of the involvement of the trigeminal system, it is interesting that a PET study of hypothalamic electrical stimulation in patients with cluster headaches found increased rCBF in an area compatible with the trigeminal ganglion/nucleus, in addition to activation of the stimulation site (May et al 2006). Animal studies have described anatomical connections between hypothalamic nuclei and the trigeminal nucleus (Malick and Burstein 1998, Malick et al 2000).
In summary, current data suggest that migraine and cluster headache, far from being primarily vascular disorders, are conditions whose genesis is to be found in the CNS. These are excellent examples of how brain imaging has advanced our understanding of chronic pain conditions.
Conclusion
A number of pathways transmit nociceptive information from the spinal cord to the brain. Consequently, multiple regions of the brain are activated during the complex experience of pain. Cortical regions activated during pain include the limbic, paralimbic, and sensory areas, notably the ACC, IC, PFC, S1, and S2. Several lines of evidence suggest that these regions have a differential contribution to pain, with the ACC being particularly important for the emotional aspects of pain and the somatosensory and insular cortices having a prominent role in coding features such as pain location. Brain areas are involved in both opiate and non-opiate pain modulation, particularly modulation derived from psychological factors, such as attentional and emotional state. Finally, clinical pain states often activate similar brain regions as do acute pain states, but differences also exist that probably underlie disruptions in pain modulatory systems, as well as alterations in psychological state related to chronic pain states.
The references for this chapter can be found at www.expertconsult.com.
References
Afridi S.K., Giffin N.J., Kaube H., et al. A positron emission tomographic study in spontaneous migraine. Archives of Neurology. 2005;62:1270–1275.
Afridi S.K., Matharu M.S., Lee L., et al. A PET study exploring the laterality of brainstem activation in migraine using glyceryl trinitrate. Brain. 2005;128:932–939.
Altier N., Stewart J. The role of dopamine in the nucleus accumbens in analgesia. Life Sciences. 1999;65:2269–2287.
Andersson J.L., Lilja A., Hartvig P., et al. Somatotopic organization along the central sulcus, for pain localization in humans, as revealed by positron emission tomography. Experimental Brain Research. 1997;117:192–199.
Apkarian A.V., Bushnell M.C., Treede R.D., et al. Human brain mechanisms of pain perception and regulation in health and disease. European Journal of Pain. 2005;9:463–484.
Apkarian A.V., Hashmi J.A., Baliki M.N. Pain and the brain: specificity and plasticity of the brain in clinical chronic pain. Pain. 2011;152(suppl 3):S49–S64.
Apkarian A.V., Krauss B.R., Fredrickson B.E., et al. Imaging the pain of low back pain: functional magnetic resonance imaging in combination with monitoring subjective pain perception allows the study of clinical pain states. Neuroscience Letters. 2001;299:57–60.
Apkarian A.V., Shi T. Squirrel monkey lateral thalamus. I. Somatic nociresponsive neurons and their relation to spinothalamic terminals. Journal of Neuroscience. 1994;14:6779–6795.
Apkarian A.V., Shi T. Thalamocortical connections of the cingulate and insula in relation to nociceptive inputs to the cortex. In: Ayrapetyan S.N., Apkarian A.V., eds. Pain mechanisms and management. Washington, DC: IOS Press; 1998:212–220.
Apkarian A.V., Sosa Y., Sonty S., et al. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. Journal of Neuroscience. 2004;24:10410–10415.
Apkarian A.V., Stea R.A., Manglos S.H., et al. Persistent pain inhibits contralateral somatosensory cortical activity in humans. Neuroscience Letters. 1992;140:141–147.
Arendt-Nielsen L. Second pain event related potentials to argon laser stimuli: recording and quantification. Journal of Neurology, Neurosurgery, and Psychiatry. 1990;53:405–410.
Bahra A., Matharu M.S., Buchel C., et al. Brainstem activation specific to migraine headache. Lancet. 2001;357:1016–1017.
Baliki M.N., Chialvo D.R., Geha P.Y., et al. Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. Journal of Neuroscience. 2006;26:12165–12173.
Baliki M.N., Geha P.Y., Apkarian A.V. Parsing pain perception between nociceptive representation and magnitude estimation. Journal of Neurophysiology. 2009;101:875–887.
Baliki M.N., Geha P.Y., Fields H.L., et al. Predicting value of pain and analgesia: nucleus accumbens response to noxious stimuli changes in the presence of chronic pain. Neuron. 2010;66:149–160.
Bantick S.J., Wise R.G., Ploghaus A., et al. Imaging how attention modulates pain in humans using functional MRI. Brain. 2002;125:310–319.
Baraniuk J.N., Whalen G., Cunningham J., et al. Cerebrospinal fluid levels of opioid peptides in fibromyalgia and chronic low back pain. BMC Musculoskeletal Disorders. 2004;5:48.
Becerra L.R., Breiter H.C., Stojanovic M., et al. Human brain activation under controlled thermal stimulation and habituation to noxious heat: an fMRI study. Magnetic Resonance in Medicine. 1999;41:1044–1057.
Becerra L., Breiter H.C., Wise R., et al. Reward circuitry activation by noxious thermal stimuli. Neuron. 2001;32:927–946.
Bencherif B., Fuchs P.N., Sheth R., et al. Pain activation of human supraspinal opioid pathways as demonstrated by [11C]-carfentanil and positron emission tomography (PET). Pain. 2002;99:589–598.
Benedetti F., Mayberg H.S., Wager T.D., et al. Neurobiological mechanisms of the placebo effect. Journal of Neuroscience. 2005;25:10390–10402.
Berna C., Leknes S., Holmes E.A., et al. Induction of depressed mood disrupts emotion regulation neurocircuitry and enhances pain unpleasantness. Biology of Psychiatry. 2010;67:1083–1090.
Bernard J.F., Bester H., Besson J.M. Involvement of the spino-parabrachio-amygdaloid and -hypothalamic pathways in the autonomic and affective emotional aspects of pain. Progress in Brain Research. 1996;107:243–255.
Berthier M., Starkstein S., Leiguarda R. Asymbolia for pain: a sensory-limbic disconnection syndrome. Annals of Neurology. 1988;24:41–49.
Beydoun A., Morrow T.J., Shen J.F., et al. Variability of laser-evoked potentials: attention, arousal and lateralized differences. Electroencephalography and Clinical Neurophysiology. 1993;88:173–181.
Birbaumer N., Lutzenberger W., Montoya P., et al. Effects of regional anesthesia on phantom limb pain are mirrored in changes in cortical reorganization. Journal of Neuroscience. 1997;17:5503–5508.
Bower J.M. Is the cerebellum sensory for motor’s sake, or motor for sensory’s sake: the view from the whiskers of a rat? Progress in Brain Research. 1997;114:463–496.
Bowyer S.M., Aurora K.S., Moran J.E., et al. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Annals of Neurology. 2001;50:582–587.
Bragard D., Chen A.C., Plaghki L. Direct isolation of ultra-late (C-fibre) evoked brain potentials by CO2 laser stimulation of tiny cutaneous surface areas in man. Neuroscience Letters. 1996;209:81–84.
Braz J.M., Nassar M.A., Wood J.N., et al. Parallel “pain” pathways arise from subpopulations of primary afferent nociceptor. Neuron. 2005;47:787–793.
Bromm B., Treede R.D. CO2 laser radiant heat pulses activate C nociceptors in man. Pflugers Archiv. 1983;399:155–156.
Brooks J.C., Nurmikko T.J., Bimson W.E., et al. fMRI of thermal pain: effects of stimulus laterality and attention. NeuroImage. 2002;15:293–301.
Burgmer M., Gaubitz M., Konrad C., et al. Decreased gray matter volumes in the cingulo-frontal cortex and the amygdala in patients with fibromyalgia. Psychosomatic Medicine. 2009;71:566–573.
Burgmer M., Pogatzki-Zahn E., Gaubitz M., et al. Fibromyalgia unique temporal brain activation during experimental pain: a controlled fMRI study. Journal of Neural Transmission. 2010;117:123–131.
Bushnell M.C., Duncan G.H. Sensory and affective aspects of pain perception: is medial thalamus restricted to emotional issues? Experimental Brain Research. 1989;78:415–418.
Bushnell M.C., Duncan G.H., Dubner R., et al. Activity of trigeminothalamic neurons in medullary dorsal horn of awake monkeys trained in a thermal discrimination task. Journal of Neurophysiology. 1984;52:170–187.
Bushnell M.C., Duncan G.H., Hofbauer R.K., et al. Pain perception: is there a role for primary somatosensory cortex? Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7705–7709.
Casey K.L., Minoshima S., Berger K.L., et al. Positron emission tomographic analysis of cerebral structures activated specifically by repetitive noxious heat stimuli. Journal of Neurophysiology. 1994;71:802–807.
Casey K.L., Morrow T.J., Lorenz J., et al. Temporal and spatial dynamics of human forebrain activity during heat pain: analysis by positron emission tomography. Journal of Neurophysiology. 2001;85:951–959.
Casey K.L., Svensson P., Morrow T.J., et al. Selective opiate modulation of nociceptive processing in the human brain. Journal of Neurophysiology. 2000;84:525–533.
Chudler E.H. Response properties of neurons in the caudate-putamen and globus pallidus to noxious and non-noxious thermal stimulation in anesthetized rats. Brain Research. 1998;812:283–288.
Chudler E.H., Anton F., Dubner R., et al. Responses of nociceptive SI neurons in monkeys and pain sensation in humans elicited by noxious thermal stimulation: effect of interstimulus interval. Journal of Neurophysiology. 1990;63:559–569.
Chudler E.H., Dong W.K. The role of the basal ganglia in nociception and pain. Pain. 1995;60:3–38.
Chudler E.H., Lu Y. Nociceptive behavioral responses to chemical, thermal and mechanical stimulation after unilateral, intrastriatal administration of 6-hydroxydopamine. Brain Research. 2008;1213:41–47.
Coghill R.C., Sang C.N., Maisog J.M., et al. Pain intensity processing within the human brain: a bilateral, distributed mechanism. Journal of Neurophysiology. 1999;82:1934–1943.
Cook D.B., Lange G., Ciccone D.S., et al. Functional imaging of pain in patients with primary fibromyalgia. Journal of Rheumatology. 2004;31:364–378.
Corbetta M., Shulman G.L. Control of goal-directed and stimulus-driven attention in the brain. Nature Reviews. Neuroscience. 2002;3:201–215.
Corkin S., Hebben N. Subjective estimates of chronic pain before and after psychosurgery or treatment in a pain unit. Pain. 1981;11(suppl 1):S150.
Craig A., Dostrovsky J. Differential projections of thermoreceptive and nociceptive lamina I trigeminothalamic and spinothalamic neurons in the cat. Journal of Neurophysiology. 2001;86:856–870.
Craig A.D. How do you feel? Interoception: the sense of the physiological condition of the body. Nature Reviews. Neuroscience. 2002;3:655–666.
Craig A.D. Interoception: the sense of the physiological condition of the body. Current Opinion in Neurobiology. 2003;13:500–505.
Craig A.D. A new view of pain as a homeostatic emotion. Trends in Neurosciences. 2003;26:303–307.
Craig A.D. How do you feel—now? The anterior insula and human awareness. Nature Reviews. Neuroscience. 2009;10:59–70.
Craig A.D., Chen K., Bandy D., et al. Thermosensory activation of insular cortex. Nature Neuroscience. 2000;3:184–190.
DaSilva A.F., Becerra L., Makris N., et al. Somatotopic activation in the human trigeminal pain pathway. Journal of Neuroscience. 2002;22:8183–8192.
DaSilva A.F., Becerra L., Pendse G., et al. Colocalized structural and functional changes in the cortex of patients with trigeminal neuropathic pain. PLoS One. 2008;3:e3396.
Davis K.D., Pope G., Chen J., et al. Cortical thinning in IBS: implications for homeostatic, attention, and pain processing. Neurology. 2008;70:153–154.
Davis K.D., Pope G.E., Crawley A.P., et al. Neural correlates of prickle sensation: a percept-related fMRI study. Nature Neuroscience. 2002;5:1121–1122.
Davis K.D., Taylor S.J., Crawley A.P., et al. Functional MRI of pain- and attention-related activations in the human cingulate cortex. Journal of Neurophysiology. 1997;77:3370–3380.
Dejerine J., Roussy G. Le syndrome thalamique. Revue Neurologique. 1906;12:521–532.
Denuelle M., Fabre N., Payoux P., et al. Posterior cerebral hypoperfusion in migraine without aura. Cephalalgia. 2008;28:856–862.
Derbyshire S.W., Jones A.K. Cerebral responses to a continual tonic pain stimulus measured using positron emission tomography. Pain. 1998;76:127–135.
De Wied M., Verbaten M.N. Affective pictures processing, attention, and pain tolerance. Pain. 2001;90:163–172.
Di Piero V., Jones A.K., Iannotti F., et al. Chronic pain: a PET study of the central effects of percutaneous high cervical cordotomy. Pain. 1991;46:9–12.
Dong W.K., Salonen L.D., Kawakami Y., et al. Nociceptive responses of trigeminal neurons in SII-7b cortex of awake monkeys. Brain Research. 1989;484:314–324.
Dostrovsky J.O., Craig A.D. Nociceptive neurons in primate insular cortex. Society for Neuroscience Abstracts. 1996;22:111.
Dum R.P., Levinthal D.J., Strick P.L. The spinothalamic system targets motor and sensory areas in the cerebral cortex of monkeys. Journal of Neuroscience. 2009;29:14223–14235.
Duncan G.H., Kupers R.C., Marchand S., et al. Stimulation of human thalamus for pain relief: possible modulatory circuits revealed by positron emission tomography. Journal of Neurophysiology. 1998;80:3326–3330.
Dunckley P., Wise R.G., Fairhurst M., et al. A comparison of visceral and somatic pain processing in the human brainstem using functional magnetic resonance imaging. Journal of Neuroscience. 2005;25:7333–7341.
Eippert F., Bingel U., Schoell E.D., et al. Activation of the opioidergic descending pain control system underlies placebo analgesia. Neuron. 2009;63:533–543.
Eippert F., Finsterbusch J., Bingel U., et al. Direct evidence for spinal cord involvement in placebo analgesia. Science. 2009;326:404.
Ekerot C.F., Garwicz M., Schouenborg J. Topography and nociceptive receptive fields of climbing fibres projecting to the cerebellar anterior lobe in the cat. Journal of Physiology. 1991;441:257–274.
Emad Y., Ragab Y., Zeinhom F., et al. Hippocampus dysfunction may explain symptoms of fibromyalgia syndrome. A study with single-voxel magnetic resonance spectroscopy. Journal of Rheumatology. 2008;35:1371–1377.
Fairhurst M., Wiech K., Dunckley P., et al. Anticipatory brainstem activity predicts neural processing of pain in humans. Pain. 2007;128:101–110.
Fields H.L. Pain modulation: expectation, opioid analgesia and virtual pain. Progress in Brain Research. 2000;122:245–253.
Fields H.L., Heinricher M.M. Anatomy and physiology of a nociceptive modulatory system. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 1985;308:361–374.
Fields H.L., Rowbotham M., Baron R. Postherpetic neuralgia: irritable nociceptors and deafferentation. Neurobiology of Disease. 1998;5:209–227.
Firestone L.L., Gyulai F., Mintun M., et al. Human brain activity response to fentanyl imaged by positron emission tomography. Anesthesia and Analgesia. 1996;82:1247–1251.
Flor H., Braun C., Elbert T., et al. Extensive reorganization of primary somatosensory cortex in chronic back pain patients. Neuroscience Letters. 1997;224:5–8.
Flor H., Elbert T., Knecht S., et al. Phantom-limb pain as a perceptual correlate of cortical reorganization following arm amputation. Nature. 1995;375:482–484.
Foltz E.L., Lowell E.W. Pain “relief” by frontal cingulumotomy. Journal of Neurosurgery. 1962;19:89–100.
Foltz E.L., White L.E. The role of rostral cingulumotomy in “pain” relief. International Journal of Neurology. 1968;6:353–373.
Frankenstein U.N., Richter W., McIntyre M.C., et al. Distraction modulates anterior cingulate gyrus activations during the cold pressor test. NeuroImage. 2001;14:827–836.
Friedman D.P., Murray E.A. Thalamic connectivity of the second somatosensory area and neighboring somatosensory fields of the lateral sulcus of the macaque. Comparative Neurology. 1986;252:348–374.
Frot M., Mauguiere F. Dual representation of pain in the operculo-insular cortex in humans. Brain. 2003;126:438–450.
Frot M., Rambaud L., Guenot M., et al. Intracortical recordings of early pain-related CO2-laser evoked potentials in the human second somatosensory (SII) area. Clinical Neurophysiology. 1999;110:133–145.
Fukui S., Matsuno M., Inubushi T., et al. N-Acetylaspartate concentrations in the thalami of neuropathic pain patients and healthy comparison subjects measured with (1)H-MRS. Magnetic Resonance Imaging. 2006;24:75–79.
Gao J.H., Parsons L.M., Bower J.M., et al. Cerebellum implicated in sensory acquisition and discrimination rather than motor control. Science. 1996;272:545–547.
Garcia-Larrea L., Maarrawi J., Peyron R., et al. On the relation between sensory deafferentation, pain and thalamic activity in Wallenberg’s syndrome: a PET-scan study before and after motor cortex stimulation. European Journal of Pain. 2006;10:677–688.
Garcia-Larrea L., Perchet C., Creac’h C., et al. Operculo-insular pain (parasylvian pain): a distinct central pain syndrome. Brain. 2010;133:2528–2539.
Garcia-Larrea L., Peyron R., Laurent B., et al. Association and dissociation between laser-evoked potentials and pain perception. Neuroreport. 1997;8:3785–3789.
Geha P.Y., Baliki M.N., Harden R.N., et al. The brain in chronic CRPS pain: abnormal gray-white matter interactions in emotional and autonomic regions. Neuron. 2008;60:570–581.
Giesecke T., Gracely R.H., Grant M.A., et al. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis and Rheumatism. 2004;50:613–623.
Goadsby P.J. Neurovascular headache and a midbrain vascular malformation: evidence for a role of the brainstem in chronic migraine. Cephalalgia. 2002;22:107–111.
Goadsby P.J. Pathophysiology of cluster headache: a trigeminal autonomic cephalgia. Lancet Neurology. 2002;1:251–257.
Goadsby P.J., Duckworth J.W. Low frequency stimulation of the locus coeruleus reduces regional cerebral blood flow in the spinalized cat. Brain Research. 1989;476:71–77.
Goadsby P.J., Edvinsson L. Human in vivo evidence for trigeminovascular activation in cluster headache. Neuropeptide changes and effects of acute attacks therapies. Brain. 1994;117:427–434.
Grachev I.D., Fredrickson B.E., Apkarian A.V. Abnormal brain chemistry in chronic back pain: an in vivo proton magnetic resonance spectroscopy study. Pain. 2000;89:7–18.
Grachev I.D., Fredrickson B.E., Apkarian A.V. Brain chemistry reflects dual states of pain and anxiety in chronic low back pain. Journal of Neural Transmission. 2002;109:1309–1334.
Grachev I.D., Thomas P.S., Ramachandran T.S. Decreased levels of N-acetylaspartate in dorsolateral prefrontal cortex in a case of intractable severe sympathetically mediated chronic pain (complex regional pain syndrome, type I). Brain and Cognition. 2002;49:102–113.
Greenspan J.D., Lee R.R., Lenz F.A. Pain sensitivity alterations as a function of lesion location in the parasylvian cortex. Pain. 1999;81:273–282.
Grusser S.M., Winter C., Muhlnickel W., et al. The relationship of perceptual phenomena and cortical reorganization in upper extremity amputees. Neuroscience. 2001;102:263–272.
Gucer G., Niedermeyer E., Long D.M. Thalamic EEG recordings in patients with chronic pain. Journal of Neurology. 1978;219:47–61.
Gwilym S.E., Fillipini N., Douaud G., et al. Thalamic atrophy associated with painful osteoarthritis of the hip is reversible after arthroplasty; a longitudinal voxel-based-morphometric study. Arthritis and Rheumatism. 2010;62:2930–2940.
Gwilym S.E., Keltner J.R., Warnaby C.E., et al. Psychophysical and functional imaging evidence supporting the presence of central sensitization in a cohort of osteoarthritis patients. Arthritis and Rheumatism. 2009;61:1226–1234.
Haas D.C., Kent P.F., Friedman D.I. Headache caused by a single lesion of multiple sclerosis in the periaqueductal gray area. Headache. 1993;33:452–455.
Hadjikhani N., Sanchez Del Rio M., Wu O., et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4687–4692.
Hagelberg N., Forssell H., Aalto S., et al. Altered dopamine D2 receptor binding in atypical facial pain. Pain. 2003;106:43–48.
Hagelberg N., Forssell H., Rinne J.O., et al. Striatal dopamine D1 and D2 receptors in burning mouth syndrome. Pain. 2003;101:149–154.
Hagelberg N., Jaaskelainen S.K., Martikainen I.K., et al. Striatal dopamine D2 receptors in modulation of pain in humans: a review. European Journal of Pharmacology. 2004;500:187–192.
Hagelberg N., Martikainen I.K., Mansikka H., et al. Dopamine D2 receptor binding in the human brain is associated with the response to painful stimulation and pain modulatory capacity. Pain. 2002;99:273–279.
Harris R.E., Clauw D.J., Scott D.J., et al. Decreased central mu-opioid receptor availability in fibromyalgia. Journal of Neuroscience. 2007;27:10000–10006.
Harris R.E., Sundgren P.C., Craig A.D., et al. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis and Rheumatism. 2009;60:3146–3152.
Harris R.E., Sundgren P.C., Pang Y., et al. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis and Rheumatism. 2008;58:903–907.
Hatem S.M., Attal N., Ducreux D., et al. Clinical, functional and structural determinants of central pain in syringomyelia. Brain. 2010;133:3409–3422.
Head H., Holmes G. Sensory disturbances from cerebral lesions. Brain. 1911;34:102–154.
Hirayama T., Dostrovsky J.O., Gorecki J., et al. Recordings of abnormal activity in patients with deafferentation and central pain. Stereotactical and Functional Neurosurgery. 1989;52:120–126.
Hofbauer R.K., Fiset P., Plourde G., et al. Dose-dependent effects of propofol on the central processing of thermal pain. Anesthesiology. 2004;100:386–394.
Hoshiyama M., Kakigi R. After-effect of transcutaneous electrical nerve stimulation (TENS) on pain-related evoked potentials and magnetic fields in normal subjects. Clinical Neurophysiology. 2000;111:717–724.
Hsieh J.C., Belfrage M., Stone-Elander S., et al. Central representation of chronic ongoing neuropathic pain studied by positron emission tomography. Pain. 1995;63:225–236.
Hsieh J.C., Stone-Elander S., Ingvar M. Anticipatory coping of pain expressed in the human anterior cingulate cortex: a positron emission tomography study. Neuroscience Letters. 1999;262:61–64.
Hurt R.W., Ballantine H.T., Jr. Stereotactic anterior cingulate lesions for persistent pain: a report on 68 cases. Clinical Neurosurgery. 1973;21:334–351.
Huse E., Larbig W., Flor H., et al. The effect of opioids on phantom limb pain and cortical reorganization. Pain. 2001;90:47–55.
Hutchison W.D., Davis K.D., Lozano A.M., et al. Pain-related neurons in the human cingulate cortex. Nature Neuroscience. 1999;2:403–405.
Iadarola M.J., Berman K.F., Zeffiro T.A., et al. Neural activation during acute capsaicin-evoked pain and allodynia assessed with PET. Brain. 1998;121:931–947.
Iadarola M.J., Max M.B., Berman K.F., et al. Unilateral decrease in thalamic activity observed with positron emission tomography in patients with chronic neuropathic pain. Pain. 1995;63:55–64.
Iannetti G.D., Truini A., Romaniello A., et al. Evidence of a specific spinal pathway for the sense of warmth in humans. Journal of Neurophysiology. 2003;89:562–570.
Jaaskelainen S.K., Rinne J.O., Forssell H., et al. Role of the dopaminergic system in chronic pain—a fluorodopa-PET study. Pain. 2001;90:257–260.
Jensen J., McIntosh A.R., Crawley A.P., et al. Direct activation of the ventral striatum in anticipation of aversive stimuli. Neuron. 2003;40:1251–1257.
Johansen J.P., Fields H.L. Glutamatergic activation of anterior cingulate cortex produces an aversive teaching signal. Nature Neuroscience. 2004;7:398–403.
Jones A.K., Brown W.D., Friston K.J., et al. Cortical and subcortical localization of response to pain in man using positron emission tomography. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 1991;244:39–44.
Jones A.K., Cunningham V.J., Ha-Kawa S., et al. Changes in central opioid receptor binding in relation to inflammation and pain in patients with rheumatoid arthritis. British Journal of Rheumatology. 1994;33:909–916.
Jones A.K., Kitchen N.D., Watabe H., et al. Measurement of changes in opioid receptor binding in vivo during trigeminal neuralgic pain using [11C] diprenorphine and positron emission tomography. Journal of Cerebral Blood Flow and Metabolism. 1999;19:803–808.
Kanda M., Fujiwara N., Xu X., et al. Pain-related and cognitive components of somatosensory evoked potentials following CO2 laser stimulation in man. Electroencephalography and Clinical Neurophysiology. 1996;100:105–114.
Karl A., Birbaumer N., Lutzenberger W., et al. Reorganization of motor and somatosensory cortex in upper extremity amputees with phantom limb pain. Journal of Neuroscience. 2001;21:3609–3618.
Kenshalo D.R., Jr., Chudler E.H., Anton F., et al. SI nociceptive neurons participate in the encoding process by which monkeys perceive the intensity of noxious thermal stimulation. Brain Research. 1988;454:378–382.
Kenshalo D.R., Jr., Isensee O. Responses of primate SI cortical neurons to noxious stimuli. Journal of Neurophysiology. 1983;50:1479–1496.
Kim J.H., Suh S.I., Seol H.Y., et al. Regional grey matter changes in patients with migraine: a voxel-based morphometry study. Cephalalgia. 2008;28:598–604.
Koyama T., Tanaka Y.Z., Mikami A. Nociceptive neurons in the macaque anterior cingulate activate during anticipation of pain. Neuroreport. 1998;9:2663–2667.
Kuchinad A., Schweinhardt P., Seminowicz D.A., et al. Accelerated brain gray matter loss in fibromyalgia patients: premature aging of the brain? Journal of Neuroscience. 2007;27:4004–4007.
Kunde V., Treede R.D. Topography of middle-latency somatosensory evoked potentials following painful laser stimuli and non-painful electrical stimuli. Electroencephalography and Clinical Neurophysiology. 1993;88:280–289.
Kwan C.L., Diamant N.E., Pope G., et al. Abnormal forebrain activity in functional bowel disorder patients with chronic pain. Neurology. 2005;65:1268–1277.
Lance J.W., Lambert G.A., Goadsby P.J., et al. Brainstem influences on the cephalic circulation: experimental data from cat and monkey of relevance to the mechanism of migraine. Headache. 1983;23:258–265.
Langemark M., Bach F.W., Ekman R., et al. Increased cerebrospinal fluid met-enkephalin immunoreactivity in patients with chronic tension-type headache. Pain. 1995;63:103–107.
Lassen N.A., Ingvar D.H., Skinhoj E. Brain function and blood flow: changes in the amount of blood flowing in areas of the human cerebral cortex, reflecting changes in the activity of those areas, are graphically revealed with the aid of a radioactive isotope. Scientific American. 1978;139:62–71.
Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117:199–210.
Lawal A., Kern M., Sidhu H., et al. Novel evidence for hypersensitivity of visceral sensory neural circuitry in irritable bowel syndrome patients. Gastroenterology. 2006;130:26–33.
Lee M.C., Zambreanu L., Menon D.K., et al. Identifying brain activity specifically related to the maintenance and perceptual consequence of central sensitization in humans. Journal of Neuroscience. 2008;28:11642–11649.
Legrain V., Guerit J.M., Bruyer R., et al. Attentional modulation of the nociceptive processing into the human brain: selective spatial attention, probability of stimulus occurrence, and target detection effects on laser evoked potentials. Pain. 2002;99:21–39.
Lenz F.A., Kwan H.C., Dostrovsky J.O., et al. Characteristics of the bursting pattern of action potentials that occurs in the thalamus of patients with central pain. Brain Research. 1989;496:357–360.
Lenz F.A., Rios M., Chau D., et al. Painful stimuli evoke potentials recorded from the parasylvian cortex in humans. Journal of Neurophysiology. 1998;80:2077–2088.
Leone M., Proietti Cecchini A., Franzini A., et al. Lessons from 8 years’ experience of hypothalamic stimulation in cluster headache. Cephalalgia. 2008;28:787–797. discussion 798
Lodi R., Pierangeli G., Tonon C., et al. Study of hypothalamic metabolism in cluster headache by proton MR spectroscopy. Neurology. 2006;66:1264–1266.
Longe S.E., Wise R., Bantick S., et al. Counter-stimulatory effects on pain perception and processing are significantly altered by attention: an fMRI study. Neuroreport. 2001;12:2021–2025.
Lopez-Sola M., Pujol J., Hernandez-Ribas R., et al. Dynamic assessment of the right lateral frontal cortex response to painful stimulation. NeuroImage. 2010;50:1177–1187.
Lorenz J., Cross D., Minoshima S., et al. A unique representation of heat allodynia in the human brain. Neuron. 2002;35:383–393.
Lorenz J., Minoshima S., Casey K.L. Keeping pain out of mind: the role of the dorsolateral prefrontal cortex in pain modulation. Brain. 2003;126:1079–1109.
Lotze M., Grodd W., Birbaumer N., et al. Does use of a myoelectric prosthesis prevent cortical reorganization and phantom limb pain? Nature Neuroscience. 1999;2:501–502.
Lutz J., Jager L., de Quervain D., et al. White and gray matter abnormalities in the brain of patients with fibromyalgia: a diffusion-tensor and volumetric imaging study. Arthritis and Rheumatism. 2008;58:3960–3969.
Maarrawi J., Peyron R., Mertens P., et al. Differential brain opioid receptor availability in central and peripheral neuropathic pain. Pain. 2007;127:183–194.
MacIver K., Lloyd D.M., Kelly S., et al. Phantom limb pain, cortical reorganization and the therapeutic effect of mental imagery. Brain. 2008;131:2181–2191.
MacLean P.D. Psychosomatic disease and the “visceral brain.” Recent developments bearing on the Papez theory of emotion. Psychosomatic Medicine. 1949;11:338–353.
Magerl W., Ali Z., Ellrich J., et al. C- and A delta-fiber components of heat-evoked cerebral potentials in healthy human subjects. Pain. 1999;82:127–137.
Magnusson J.E., Fisher K. The involvement of dopamine in nociception: the role of D(1) and D(2) receptors in the dorsolateral striatum. Brain Research. 2000;855:260–266.
Magnusson J.E., Martin R.V. Additional evidence for the involvement of the basal ganglia in formalin-induced nociception: the role of the nucleus accumbens. Brain Research. 2002;942:128–132.
Maihofner C., Forster C., Birklein F., et al. Brain processing during mechanical hyperalgesia in complex regional pain syndrome: a functional MRI study. Pain. 2005;114:93–103.
Maihofner C., Handwerker H.O., Neundorfer B., et al. Patterns of cortical reorganization in complex regional pain syndrome. Neurology. 2003;61:1707–1715.
Maihofner C., Handwerker H.O., Neundorfer B., et al. Cortical reorganization during recovery from complex regional pain syndrome. Neurology. 2004;63:693–701.
Maihofner C., Kaltenhauser M., Neundorfer B., et al. Temporo-spatial analysis of cortical activation by phasic innocuous and noxious cold stimuli—a magnetoencephalographic study. Pain. 2002;100:281–290.
Malick A., Burstein R. Cells of origin of the trigeminohypothalamic tract in the rat. Journal of Comparative Neurology. 1998;400:125–144.
Malick A., Strassman R.M., Burstein R. Trigeminohypothalamic and reticulohypothalamic tract neurons in the upper cervical spinal cord and caudal medulla of the rat. Journal of Neurophysiology. 2000;84:2078–2112.
Mantz J., Godbout R., Tassin J.P., et al. Inhibition of spontaneous and evoked unit activity in the rat medial prefrontal cortex by mesencephalic raphe nuclei. Brain Research. 1990;524:22–30.
Mantz J., Milla C., Glowinski J., et al. Differential effects of ascending neurons containing dopamine and noradrenaline in the control of spontaneous activity and of evoked responses in the rat prefrontal cortex. Neuroscience. 1988;27:517–526.
Marchand S., Arsenault P. Odors modulate pain perception: a gender-specific effect. Physiology and Behavior. 2002;76:251–256.
Marshall J. Sensory disturbances in cortical wounds with special reference to pain. Journal of Neurology, Neurosurgery, and Psychiatry. 1951;14:187–204.
Martikainen I.K., Hagelberg N., Mansikka H., et al. Association of striatal dopamine D2/D3 receptor binding potential with pain but not tactile sensitivity or placebo analgesia. Neuroscience Letters. 2005;376:149–153.
May A., Ashburner J., Buchel C., et al. Correlation between structural and functional changes in brain in an idiopathic headache syndrome. Nature Medicine. 1999;5:836–838.
May A., Bahra A., Buchel C., et al. Hypothalamic activation in cluster headache attacks. Lancet. 1998;352:275–278.
May A., Bahra A., Buchel C., et al. PET and MRA findings in cluster headache and MRA in experimental pain. Neurology. 2000;55:1328–1335.
May A., Leone M., Boecker H., et al. Hypothalamic deep brain stimulation in positron emission tomography. Journal of Neuroscience. 2006;26:3589–3593.
Mayer E.A., Berman S., Suyenobu B., et al. Differences in brain responses to visceral pain between patients with irritable bowel syndrome and ulcerative colitis. Pain. 2005;115:398–409.
Mazzola L., Isnard J., Peyron R., et al. Somatotopic organization of pain responses to direct electrical stimulation of the human insular cortex. Pain. 2009;146:99–104.
McMahon S.B., Dmitrieva N., Koltzenburg M. Visceral pain. British Journal of Anaesthesia. 1995;75:132–144.
Meagher M.W., Arnau R.C., Rhudy J.L. Pain and emotion: effects of affective picture modulation. Psychosomatic Medicine. 2001;63:79–90.
Melzack R., Casey K.L. Sensory, motivational and central control determinants of pain: a new conceptual model. In: Kenshalo D.R., ed. The skin senses. Springfield, IL: Charles C Thomas; 1968:423–443.
Mertz H., Morgan V., Tanner G., et al. Regional cerebral activation in irritable bowel syndrome and control subjects with painful and nonpainful rectal distention. Gastroenterology. 2000;118:842–848.
Metz A.E., Yau H.J., Centeno M.V., et al. Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2423–2428.
Miyazaki M., Shibasaki H., Kanda M., et al. Generator mechanism of pain-related evoked potentials following CO2 laser stimulation of the hand: scalp topography and effect of predictive warning signal. Journal of Clinical Neurophysiology. 1994;11:242–254.
Monconduit L., Villanueva L. The lateral ventromedial thalamic nucleus spreads nociceptive signals from the whole body surface to layer I of the frontal cortex. European Journal of Neuroscience. 2005;21:3395–3402.
Montoya P., Ritter K., Huse E., et al. The cortical somatotopic map and phantom phenomena in subjects with congenital limb atrophy and traumatic amputees with phantom limb pain. European Journal of Neuroscience. 1998;10:1095–1102.
Moseley G.L., Zalucki N.M., Wiech K. Tactile discrimination, but not tactile stimulation alone, reduces chronic limb pain. Pain. 2008;137:600–608.
Nakamura Y., Paur R., Zimmermann R., et al. Attentional modulation of human pain processing in the secondary somatosensory cortex: a magnetoencephalographic study. Neuroscience Letters. 2002;328:29–32.
Obermann M., Nebel K., Schumann C., et al. Gray matter changes related to chronic posttraumatic headache. Neurology. 2009;73:978–983.
Olesen J., Larsen B., Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Annals of Neurology. 1981;9:344–352.
Oppenheimer S.M., Kedem G., Martin W.M. Left-insular cortex lesions perturb cardiac autonomic tone in humans. Clinical Autonomic Research. 1996;6:131–140.
Opsommer E., Weiss T., Plaghki L., et al. Dipole analysis of ultralate (C-fibres) evoked potentials after laser stimulation of tiny cutaneous surface areas in humans. Neuroscience Letters. 2001;298:41–44.
Oshiro Y., Quevedo A.S., McHaffie J.G., et al. Brain mechanisms supporting discrimination of sensory features of pain: a new model. Journal of Neuroscience. 2009;29:14924–14931.
Ostrowsky K., Magnin M., Ryvlin P., et al. Representation of pain and somatic sensation in the human insula: a study of responses to direct electrical cortical stimulation. Cerebral Cortex. 2002;12:376–385.
Pattinson K.T., Rogers R., Mayhew S.D., et al. Pharmacological FMRI: measuring opioid effects on the BOLD response to hypercapnia. Journal of Cerebral Blood Flow and Metabolism. 2007;27:414–423.
Penfield W., Boldrey E. Somatic motor and sensory representation in the cerebral cortex of man as studied by electrical stimulation. Brain. 1937;60:389–443.
Pertovaara A., Martikainen I.K., Hagelberg N., et al. Striatal dopamine D2/D3 receptor availability correlates with individual response characteristics to pain. European Journal of Neuroscience. 2004;20:1587–1592.
Petersen K.L., Rowbotham M.C. Natural history of sensory function after herpes zoster. Pain. 2010;150:83–92.
Petrovic P., Ingvar M., Stone-Elander S., et al. A PET activation study of dynamic mechanical allodynia in patients with mononeuropathy. Pain. 1999;83:459–470.
Petrovic P., Kalso E., Petersson K.M., et al. Placebo and opioid analgesia—imaging a shared neuronal network. Science. 2002;295:1737–1740.
Petrovic P., Petersson K.M., Ghatan P.H., et al. Pain-related cerebral activation is altered by a distracting cognitive task. Pain. 2000;85:19–30.
Peyron R., Garcia-Larrea L., Deiber M.P., et al. Electrical stimulation of precentral cortical area in the treatment of central pain: electrophysiological and PET study. Pain. 1995;62:275–286.
Peyron R., Schneider F., Faillenot I., et al. An fMRI study of cortical representation of mechanical allodynia in patients with neuropathic pain. Neurology. 2004;63:1838–1846.
Phillips M.L., Gregory L.J., Cullen S., et al. The effect of negative emotional context on neural and behavioural responses to oesophageal stimulation. Brain. 2003;126:669–684.
Plaghki L., Delisle D., Godfraind J.M. Heterotopic nociceptive conditioning stimuli and mental task modulate differently the perception and physiological correlates of short CO2 laser stimuli. Pain. 1994;57:181–192.
Pleger B., Ragert P., Schwenkreis P., et al. Patterns of cortical reorganization parallel impaired tactile discrimination and pain intensity in complex regional pain syndrome. NeuroImage. 2006;32:503–510.
Pleger B., Tegenthoff M., Ragert P., et al. Sensorimotor retuning [corrected] in complex regional pain syndrome parallels pain reduction. Annals of Neurology. 2005;57:425–429.
Ploghaus A., Tracey I., Gati J.S., et al. Dissociating pain from its anticipation in the human brain. Science. 1999;284:1979–1981.
Ploner M., Gross J., Timmermann L., et al. Cortical representation of first and second pain sensation in humans. Proceedings of the National Academy of Sciences of the United States of America. 2002;9:12444–12448.
Ploner M., Lee M.C., Wiech K., et al. Flexible cerebral connectivity patterns subserve contextual modulations of pain. Cerebral Cortex. 2011;21:719–726.
Ploner M., Schmitz F., Freund H.J., et al. Parallel activation of primary and secondary somatosensory cortices in human pain processing. Journal of Neurophysiology. 1999;81:3100–3104.
Ploner M., Schmitz F., Freund H.J., et al. Differential organization of touch and pain in human primary somatosensory cortex. Journal of Neurophysiology. 2000;83:1770–1776.
Porreca F., Ossipov M.H., Gebhart G.F. Chronic pain and medullary descending facilitation. Trends in Neurosciences. 2002;25:319–325.
Porro C.A., Baraldi P., Pagnoni G., et al. Does anticipation of pain affect cortical nociceptive systems? Journal of Neuroscience. 2002;22:3206–3214.
Prescot A., Becerra L., Pendse G., et al. Excitatory neurotransmitters in brain regions in interictal migraine patients. Molecular Pain. 2009;5:34.
Price D.D. Psychological and neural mechanisms of pain. New York: Raven Press; 1988.
Pukall C.F., Strigo I.A., Binik Y.M., et al. Neural correlates of painful genital touch in women with vulvar vestibulitis syndrome. Pain. 2005;115:118–127.
Rainville P., Duncan G.H., Price D.D., et al. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science. 1997;277:968–971.
Raskin N.H., Hosobuchi Y., Lamb S. Headache may arise from perturbation of brain. Headache. 1987;27:416–420.
Rausell E., Jones E.G. Chemically distinct compartments of the thalamic VPM nucleus in monkeys relay principal and spinal trigeminal pathways to different layers of the somatosensory cortex. Journal of Neuroscience. 1991;11:226–237.
Reinert A., Treede R., Bromm B. The pain inhibiting pain effect: an electrophysiological study in humans. Brain Research. 2000;862:103–110.
Robinson C.J., Burton H. Somatic submodality distribution within the second somatosensory (SII), 7b, retroinsular, postauditory, and granular insular cortical areas of M. fascicularis. Journal of Comparative Neurology. 1980;192:93–108.
Rocca M.A., Ceccarelli A., Falini A., et al. Brain gray matter changes in migraine patients with T2-visible lesions: a 3-T MRI study. Stroke. 2006;37:1765–1770.
Rodriguez-Raecke R., Niemeier A., Ihle K., et al. Brain gray matter decrease in chronic pain is the consequence and not the cause of pain. Journal of Neuroscience. 2009;29:13746–13750.
Roy M., Peretz I., Rainville P. Emotional valence contributes to music-induced analgesia. Pain. 2008;134:140–147.
Roy M., Piche M., Chen J.I., et al. Cerebral and spinal modulation of pain by emotions. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20900–20905.
Saab C.Y., Kawasaki M., Al Chaer E.D., et al. Cerebellar cortical stimulation increases spinal visceral nociceptive responses. Journal of Neurophysiology. 2001;85:2359–2363.
Saab C.Y., Makki A., Quast M.J., et al. Is cerebellum involved in pain? In: Devor M., Rowbotham M., Wiesenfeld-Hallen Z., eds. Proceedings of the 9th World Congress on Pain. Seattle: IASP Press, 2000. 515–511
Saab C.Y., Willis W.D. Nociceptive visceral stimulation modulates the activity of cerebellar Purkinje cells. Exp Brain Res. 2001;140:122–126.
Saab C.Y., Willis W.D. The cerebellum: organization, functions and its role in nociception. Brain Research and Brain Research Reviews. 2003;42:85–95.
Saade N.E., Atweh S.F., Bahuth N.B., et al. Augmentation of nociceptive reflexes and chronic deafferentation pain by chemical lesions of either dopaminergic terminals or midbrain dopaminergic neurons. Brain Research. 1997;751:1–12.
Sawamoto N., Honda M., Okada T., et al. Expectation of pain enhances responses to nonpainful somatosensory stimulation in the anterior cingulate cortex and parietal operculum/posterior insula: an event-related functional magnetic resonance imaging study. Journal of Neuroscience. 2000;20:7438–7445.
Schlaepfer T.E., Strain E.C., Greenberg B.D., et al. Site of opioid action in the human brain: mu and kappa agonists’ subjective and cerebral blood flow effects. American Journal of Psychiatry. 1998;155:470–473.
Schmidt-Wilcke T., Ganssbauer S., Neuner T., et al. Subtle grey matter changes between migraine patients and healthy controls. Cephalalgia. 2008;28:1–4.
Schmidt-Wilcke T., Leinisch E., Ganssbauer S., et al. Affective components and intensity of pain correlate with structural differences in gray matter in chronic back pain patients. Pain. 2006;125:89–97.
Schmidt-Wilcke T., Leinisch E., Straube A., et al. Gray matter decrease in patients with chronic tension type headache. Neurology. 2005;65:1483–1486.
Schmitz N., Admiraal-Behloul F., Arkink E.B., et al. Attack frequency and disease duration as indicators for brain damage in migraine. Headache. 2008;48:1044–1055.
Schweinhardt P., Glynn C., Brooks J., et al. An fMRI study of cerebral processing of brush-evoked allodynia in neuropathic pain patients. NeuroImage. 2006;32:256–265.
Schweinhardt P., Kalk N., Wartolowska K., et al. Investigation into the neural correlates of emotional augmentation of clinical pain. NeuroImage. 2008;40:759–766.
Scott D.J., Heitzeg M.M., Koeppe R.A., et al. Variations in the human pain stress experience mediated by ventral and dorsal basal ganglia dopamine activity. Journal of Neuroscience. 2006;26:10789–10795.
Seminowicz D.A., Labus J.S., Bueller J.A., et al. Regional gray matter density changes in brains of patients with irritable bowel syndrome. Gastroenterology. 139, 2010. 48.e2–57.e2
Seminowicz D.A., Laferriere A.L., Millecamps M., et al. MRI structural brain changes associated with sensory and emotional function in a rat model of long-term neuropathic pain. NeuroImage. 2009;47:1007–1014.
Shi T., Apkarian A.V. Morphology of thalamocortical neurons projecting to the primary somatosensory cortex and their relationship to spinothalamic terminals in the squirrel monkey. Journal of Comparative Neurology. 1995;361:1–24.
Siedenberg R., Treede R.D. Laser-evoked potentials: exogenous and endogenous components. Electroencephalography and Clinical Neurophysiology. 1996;100:240–249.
Sikes R.W., Vogt B.A. Nociceptive neurons in area 24 of rabbit cingulate cortex. Journal of Neurophysiology. 1992;68:1720–1732.
Song G.H., Venkatraman V., Ho K.Y., et al. Cortical effects of anticipation and endogenous modulation of visceral pain assessed by functional brain MRI in irritable bowel syndrome patients and healthy controls. Pain. 2006;126:79–90.
Sorensen L., Siddall P.J., Trenell M.I., et al. Differences in metabolites in pain-processing brain regions in patients with diabetes and painful neuropathy. Diabetes Care. 2008;31:980–981.
Starr C.J., Sawaki L., Wittenberg G.F., et al. Roles of the insular cortex in the modulation of pain: insights from brain lesions. Journal of Neuroscience. 2009;29:2684–2694.
Strigo I.A., Duncan G.H., Boivin M., et al. Differentiation of visceral and cutaneous pain in the human brain. Journal of Neurophysiology. 2003;89:3294–3303.
Strigo I.A., Simmons A.N., Matthews S.C., et al. Association of major depressive disorder with altered functional brain response during anticipation and processing of heat pain. Archives of General Psychiatry. 2008;65:1275–1284.
Strittmatter M., Hamann G.F., Grauer M., et al. Altered activity of the sympathetic nervous system and changes in the balance of hypophyseal, pituitary and adrenal hormones in patients with cluster headache. Neuroreport. 1996;7:1229–1234.
Talbot J.D., Marrett S., Evans A.C., et al. Multiple representations of pain in human cerebral cortex. Science. 1991;251:1355–1358.
Tarkka I.M., Treede R.D. Equivalent electrical source analysis of pain-related somatosensory evoked potentials elicited by a CO2 laser. Journal of Clinical Neurophysiology. 1993;10:513–519.
Tiede W., Magerl W., Baumgartner U., et al. Sleep restriction attenuates amplitudes and attentional modulation of pain-related evoked potentials, but augments pain ratings in healthy volunteers. Pain. 2010;148:36–42.
Tillisch K., Mayer E.A., Labus J.S. Quantitative meta-analysis identifies brain regions activated during rectal distension in irritable bowel syndrome. Gastroenterology. 2010;140:91–100.
Tolle T.R., Kaufmann T., Siessmeier T., et al. Region-specific encoding of sensory and affective components of pain in the human brain: a positron emission tomography correlation analysis. Annals of Neurology. 1999;45:40–47.
Tracey I., Ploghaus A., Gati J.S., et al. Imaging attentional modulation of pain in the periaqueductal gray in humans. Journal of Neuroscience. 2002;22:2748–2752.
Tran T.D., Inui K., Hoshiyama M., et al. Cerebral activation by the signals ascending through unmyelinated C-fibers in humans: a magnetoencephalographic study. Neuroscience. 2002;113:375–386.
Valeriani M., Restuccia D., DiLazzaro V., et al. Central nervous system modifications in patients with lesion of the anterior cruciate ligament of the knee. Brain. 1996;119:1751–1762.
Valet M., Gundel H., Sprenger T., et al. Patients with pain disorder show gray-matter loss in pain-processing structures: a voxel-based morphometric study. Psychosomatic Medicine. 2009;71:49–56.
Valet M., Sprenger T., Boecker H., et al. Distraction modulates connectivity of the cingulo-frontal cortex and the midbrain during pain—an fMRI analysis. Pain. 2004;109:399–408.
Valfre W., Rainero I., Bergui M., et al. Voxel-based morphometry reveals gray matter abnormalities in migraine. Headache. 2008;48:109–117.
Vartiainen N., Kallio-Laine K., Hlushchuk Y., et al. Changes in brain function and morphology in patients with recurring herpes simplex virus infections and chronic pain. Pain. 2009;144:200–208.
Veloso F., Kumar K., Toth C. Headache secondary to deep brain implantation. Headache. 1998;38:507–515.
Verberne A.J., Owens N.C. Cortical modulation of the cardiovascular system. Progress in Neurobiology. 1998;54:149–168.
Verne G.N., Himes N.C., Robinson M.E., et al. Central representation of visceral and cutaneous hypersensitivity in the irritable bowel syndrome. Pain. 2003;103:99–110.
Villemure C., Bushnell M.C. Cognitive modulation of pain: how do attention and emotion influence pain processing? Pain. 2002;95:195–199.
Villemure C., Bushnell M.C. Mood influences supraspinal pain processing separately from attention. Journal of Neuroscience. 2009;29:705–715.
Villemure C., Schweinhardt P. Supraspinal pain processing: distinct roles of emotion and attention. Neuroscientist. 2010;16:276–284.
Vogel H., Port J.D., Lenz F.A., et al. Dipole source analysis of laser-evoked subdural potentials recorded from parasylvian cortex in humans. Journal of Neurophysiology. 2003;89:3051–3060.
Wager T.D., Scott D.J., Zubieta J.K. Placebo effects on human mu-opioid activity during pain. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11056–11061.
Wager T.D., Rilling J.K., Smith E.E., et al. Placebo-induced changes in FMRI in the anticipation and experience of pain. Science. 2004;303:1162–1167.
Wagner K.J., Willoch F., Kochs E.F., et al. Dose-dependent regional cerebral blood flow changes during remifentanil infusion in humans: a positron emission tomography study. Anesthesiology. 2001;94:732–739.
Wang S.J., Lirng J.F., Fuh J.L., et al. Reduction in hypothalamic 1H-MRS metabolite ratios in patients with cluster headache. Journal of Neurology, Neurosurgery, and Psychiatry. 2006;77:622–625.
Watson A., El-Deredy W., Iannetti G.D., et al. Placebo conditioning and placebo analgesia modulate a common brain network during pain anticipation and perception. Pain. 2009;145:24–30.
Weiller C., May A., Limmroth V., et al. Brain stem activation in spontaneous human migraine attacks. Nature Medicine. 1995;1:658–660.
Welch K.M., Cao Y., Aurora S., et al. MRI of the occipital cortex, red nucleus, and substantia nigra during visual aura of migraine. Neurology. 1998;51:1465–1469.
Welch K.M., Nagesh V., Aurora S.K., et al. Periaqueductal gray matter dysfunction in migraine: cause or the burden of illness? Headache. 2001;41:629–637.
White J.C., Sweet W.H. Pain and the neurosurgeon: a forty-year experience. Springfield, IL: Charles C Thomas; 1969.
Whitsel B.L., Favorov O.V., Li Y., et al. Nociceptive afferent activity alters the SI RA neuron response to mechanical skin stimulation. Cerebral Cortex. 2010;20:2900–2915.
Wiech K., Seymour B., Kalisch R., et al. Modulation of pain processing in hyperalgesia by cognitive demand. NeuroImage. 2005;27:59–69.
Wiech K., Tracey I. The influence of negative emotions on pain: behavioral effects and neural mechanisms. NeuroImage. 2009;47:987–994.
Williams D.A., Gracely R.H. Biology and therapy of fibromyalgia. Functional magnetic resonance imaging findings in fibromyalgia. Arthritis Research & Therapy. 2006;8:224.
Wise R.G., Rogers R., Painter D., et al. Combining fMRI with a pharmacokinetic model to determine which brain areas activated by painful stimulation are specifically modulated by remifentanil. NeuroImage. 2002;16:999–1014.
Wise R.G., Williams P., Tracey I. Using fMRI to quantify the time dependence of remifentanil analgesia in the human brain. Neuropsychopharmacology. 2004;29:626–635.
Wood P.B., Ledbetter C.R., Glabus M.F., et al. Hippocampal metabolite abnormalities in fibromyalgia: correlation with clinical features. Journal of Pain. 2009;10:47–52.
Wood P.B., Patterson J.C., 2nd., Sunderland J.J., et al. Reduced presynaptic dopamine activity in fibromyalgia syndrome demonstrated with positron emission tomography: a pilot study. Journal of Pain. 2007;8:51–58.
Wood P.B., Schweinhardt P., Jaeger E., et al. Fibromyalgia patients show an abnormal dopamine response to pain. European Journal of Neuroscience. 2007;25:3576–3582.
Yaguez L., Coen S., Gregory L.J., et al. Brain response to visceral aversive conditioning: a functional magnetic resonance imaging study. Gastroenterology. 2005;128:1819–1829.
Young G.B., Barr H.W.K., Blume W.T. Painful epileptic seizures involving the second sensory area. Annals of Neurology. 1986;19:412.
Young G.B., Blume W.T. Painful epileptic seizures. Brain. 1983;106:537–554.
Zelman D.C., Howland E.W., Nichols S.N., et al. The effects of induced mood on laboratory pain. Pain. 1991;46:105–111.
Zimmerman M.E., Pan J.W., Hetherington H.P., et al. Hippocampal correlates of pain in healthy elderly adults: a pilot study. Neurology. 2009;73:1567–1570.
Zubieta J.K., Bueller J.A., Jackson L.R., et al. Placebo effects mediated by endogenous opioid activity on mu-opioid receptors. Journal of Neuroscience. 2005;25:7754–7762.
Zubieta J.K., Heitzeg M.M., Smith Y.R., et al. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–1243.
Zubieta J.K., Smith Y.R., Bueller J.A., et al. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–315.
Apkarian A.V., Bushnell M.C., Treede R.D., et al. Human brain mechanisms of pain perception and regulation in health and disease. European Journal of Pain. 2005;9:463–484.
Apkarian A.V., Sosa Y., Sonty S., et al. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. Journal of Neuroscience. 2004;24:10410–10415.
Arendt-Nielsen L. Second pain event related potentials to argon laser stimuli: recording and quantification. Journal of Neurology, Neurosurgery, and Psychiatry. 1990;53:405–410.
Baliki M.N., Geha P.Y., Apkarian A.V. Parsing pain perception between nociceptive representation and magnitude estimation. Journal of Neurophysiology. 2009;101:875–887.
Coghill R.C., Sang C.N., Maisog J.M., et al. Pain intensity processing within the human brain: a bilateral, distributed mechanism. Journal of Neurophysiology. 1999;82:1934–1943.
Dum R.P., Levinthal D.J., Strick P.L. The spinothalamic system targets motor and sensory areas in the cerebral cortex of monkeys. Journal of Neuroscience. 2009;29:14223–14235.
Flor H., Elbert T., Knecht S., et al. Phantom-limb pain as a perceptual correlate of cortical reorganization following arm amputation. Nature. 1995;375:482–484.
Giesecke T., Gracely R.H., Grant M.A., et al. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis and Rheumatism. 2004;50:613–623.
Harris R.E., Sundgren P.C., Craig A.D., et al. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis and Rheumatism. 2009;60:3146–3152.
Lee M.C., Zambreanu L., Menon D.K., et al. Identifying brain activity specifically related to the maintenance and perceptual consequence of central sensitization in humans. Journal of Neuroscience. 2008;28:11642–11649.
Mazzola L., Isnard J., Peyron R., et al. Somatotopic organization of pain responses to direct electrical stimulation of the human insular cortex. Pain. 2009;146:99–104.
Melzack R., Casey K.L. Sensory, motivational and central control determinants of pain: a new conceptual model. In: Kenshalo D.R., ed. The skin senses. Springfield, Ill.: Charles C Thomas; 1968:423–443.
Ploghaus A., Tracey I., Gati J.S., et al. Dissociating pain from its anticipation in the human brain. Science. 1999;284:1979–1981.
Ploner M., Schmitz F., Freund H.J., et al. Parallel activation of primary and secondary somatosensory cortices in human pain processing. Journal of Neurophysiology. 1999;81:3100–3104.
Rodriguez-Raecke R., Niemeier A., Ihle K., et al. Brain gray matter decrease in chronic pain is the consequence and not the cause of pain. Journal of Neuroscience. 2009;29:13746–13750.
Seminowicz D.A., Laferriere A.L., Millecamps M., et al. MRI structural brain changes associated with sensory and emotional function in a rat model of long-term neuropathic pain. NeuroImage. 2009;47:1007–1014.
Talbot J.D., Marrett S., Evans A.C., et al. Multiple representations of pain in human cerebral cortex. Science. 1991;251:1355–1358.
Villemure C., Bushnell M.C. Mood influences supraspinal pain processing separately from attention. Journal of Neuroscience. 2009;29:705–715.
Wager T.D., Rilling J.K., Smith E.E., et al. Placebo-induced changes in FMRI in the anticipation and experience of pain. Science. 2004;303:1162–1167.
Wood P.B., Schweinhardt P., Jaeger E., et al. Fibromyalgia patients show an abnormal dopamine response to pain. European Journal of Neuroscience. 2007;25:3576–3582.
Zubieta J.K., Bueller J.A., Jackson L.R., et al. Placebo effects mediated by endogenous opioid activity on mu-opioid receptors. Journal of Neuroscience. 2005;25:7754–7762.
Zubieta J.K., Smith Y.R., Bueller J.A., et al. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–315.