Central Nervous System Mechanisms of Pain Modulation
Introduction
The relationship between a peripheral stimulus and the resulting pain experience depends on a host of variables, including behavioral context and competing sensory input. As an extreme example, traumatic injuries sustained during athletic competition or combat are often initially reported as being relatively painless, although in other circumstances similar injuries are typically extremely painful (Beecher 1959, Melzack et al 1982). The modification of neural, behavioral, and subjective pain responses by behavioral context and cognitive and emotional factors results from the action of central nervous system (CNS) networks that dynamically modulate the transmission of nociceptive messages. Interplay among various networks and the net balance between descending facilitatory and inhibitory influences allow a range of responses to a given triggering stimulus. Presumably, these pain-modulating circuits exist because the ability to suppress or enhance nocifensive reflexes and other responses normally elicited by noxious stimuli enhances survival of the individual. For example, suppression of nocifensive reflexes and pain sensation might facilitate escape and reduce distraction in the face of a threat such as a predator or aggressive conspecific. Conversely, enhanced pain in the presence of tissue injury and inflammation could promote recuperative behavior and healing. However, abnormal or sustained engagement of descending facilitation is now recognized to be a factor contributing to chronic and abnormal pain states.
Descending Modulatory Control
As early as 1911, Head and Holmes explicitly postulated modulatory influences on pain. They proposed that the thalamus is the center for the perception of pain and that the neocortex, the discriminative perception center, continuously modulates the responses of the thalamus to noxious stimuli. In their view, modulation of pain was a necessary part of the ongoing process of discriminative sensation. The first direct evidence that supraspinal sites control ascending (presumably sensory) pathways was provided by Hagbarth and Kerr (1954). Carpenter and colleagues (1965) subsequently demonstrated descending control of sensory input to the ascending pathways. However, the concept of a specific pain modulatory system was first clearly articulated by Melzack and Wall (1965) in their Gate Control Theory of Pain. Although there was limited evidence at that time for descending control of nociception, the next 10 years saw clear evidence of tonic descending inhibition of nociresponsive neurons in the dorsal horn (Fields and Basbaum 1978).
Stimulation-Produced Analgesia
The seminal observation pointing to selective pain-modulating systems was the discovery of “stimulation-produced analgesia,” which is a highly specific suppression of behavioral responses to noxious stimuli produced by electrical stimulation of the midbrain periaqueductal gray (PAG). During PAG stimulation, animals remained alert and active, and their responses to most environmental stimuli were unchanged. However, the expected responses to noxious stimuli, including orientation, vocalization, and escape, were absent. Stimulation-produced analgesia was also elicited from the rostral ventromedial medulla (RVM, Mayer and Price 1976).
A critical step was the demonstration that stimulation-produced analgesia can be induced in humans. As in animals, stimulation-produced analgesia in humans is elicited by electrical stimulation of the PAG and more rostral periventricular structures. Importantly, motor function is not affected, and patients report subjective analgesia (see reviews by Boivie and Meyerson 1982, Levy et al 2010). Although this procedure is rarely performed at present, the specificity of the analgesic effect and the fact that it is consistently elicited from discrete brain sites that are homologous in a variety of species, including humans, speak to the biological importance of pain modulation.
Discovery of the pain-modulating role of the PAG was a decisive advance in understanding brain mechanisms of pain processing. Subsequent research demonstrated that the PAG is part of a central circuit that controls nociceptive transmission at the level of the spinal cord dorsal horn via a relay in the RVM (Fig. 8-1, Fields and Heinricher 1985). In animals, simple noxious stimulus–evoked reflexes, considered to be organized primarily at the spinal level, are inhibited by stimulation of either the PAG or RVM. Furthermore, this inhibition is selective for nociceptive neurons in the spinal cord dorsal horn (Waters and Lumb 1997). In addition, lesions of the descending projections of the RVM through the dorsolateral funiculus of the spinal cord block inhibition of both nociceptive dorsal horn neurons and nocifensive reflex responses. Importantly, suppression of noxious evoked behavior is not motor inhibition since affective responses are also modulated by PAG and RVM manipulation (Borszcz et al 1996, Hirakawa et al 2000, Munn et al 2009, da Silva et al 2010a). Interest in this system was further heightened when it became clear that it is recruited by opioid analgesic drugs (Yaksh et al 1988).
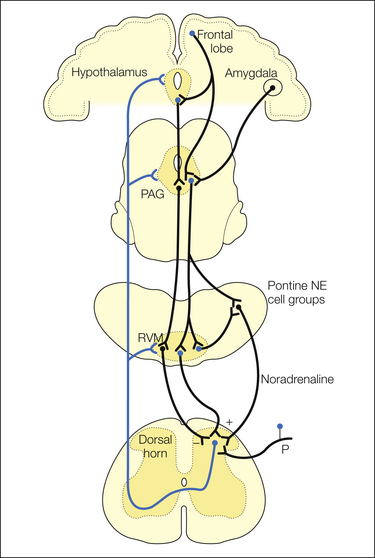
Figure 8-1 Brain stem pain-modulating pathways.
A major pain-modulating pathway has critical links in the midbrain periaqueductal gray (PAG) and rostral ventromedial medulla (RVM). The cortex, amygdala, and hypothalamus project to both the PAG and RVM (only the former are shown for clarity). The PAG controls spinal nociceptive neurons through a relay in the RVM. The RVM exerts bidirectional control over nociceptive transmission in the dorsal horn. Parallel noradrenergic pathways arising in the pontine tegmentum are also reciprocally linked with the PAG–RVM system. NE, noradrenaline; P, primary afferent.
Although other brain stem pain-modulating circuits will be mentioned (pontomedullary noradrenergic pathways, feedback via the nucleus reticularis dorsalis), this chapter emphasizes the PAG–RVM system. This focus is motivated by the demonstrated significance of this system in the actions of centrally acting analgesic drugs, its role in environmental analgesia, and evidence that it contributes to hyperalgesic states associated with inflammation, nerve injury, immune system activation, and stress.
PAG–RVM System and Facilitation of Pain Transmission
The seminal observation of stimulation-produced analgesia from the PAG and RVM and the recognition shortly thereafter that this network is recruited as part of the analgesic actions of opioids led to a widely held view of this circuit as an “analgesia system.” However, functional studies have clearly shown that brain stem modulatory systems exert bidirectional control and that pain facilitation is a major part of their function (for reviews see Porreca et al 2002, Ren and Dubner 2002, Heinricher et al 2009).
Functional studies have implicated the RVM in hyperalgesia and chronic pain triggered through both “bottom-up” and “top-down” mechanisms. Thus the RVM contributes to hyperalgesia and allodynia in inflammatory and neuropathic pain models (Porreca et al 2002, Heinricher et al 2003). Furthermore, facilitation of output from the RVM generates a tonic aversive state in rodents that can be demonstrated with place conditioning (King et al 2009). The RVM has also been implicated in deep muscle and visceral hypersensitivity and in headache-related pain (Vera-Portocarrero et al 2008, Edelmayer et al 2009, Da Silva et al 2010b, Sanoja et al 2010). It may be a factor in post-surgical hypersensitivity, although this is apparently relatively minor (Pogatzki et al 2002, Rivat et al 2009). The examples just listed illustrate that descending facilitation is engaged as part of a positive feedback loop stimulated by noxious input. Top-down facilitating modulation is also ubiquitous. The RVM is required for hyperalgesia associated with naloxone-precipitated withdrawal or prolonged opioid administration (Kaplan and Fields 1991, Vanderah et al 2001, Vera-Portocarrero et al 2011). The RVM is also engaged to produce hyperalgesia in models of mild or chronic stress (Martenson et al 2009, Imbe et al 2010, Rivat et al 2010) and as part of the sickness response triggered by systemic immune activation (Wiertelak et al 1997, Heinricher et al 2004). In these top-down examples, input from higher centers such as the amygdala and hypothalamus brings the PAG–RVM system into play to produce hyperalgesia.
Although relatively few studies have indicated a direct facilitating influence arising from the PAG, the observations outlined above indicate that the PAG–RVM system as a whole exerts true bidirectional control of nociceptive processing. Facilitation operates in parallel with descending inhibition, with differential control of input from various body regions or different modalities (Vanegas 2004, Vanegas and Schaible 2004, You et al 2010). Discovering how this system is recruited to either inhibit or facilitate nociception under different physiological conditions and in different behavioral contexts is an important challenge for future understanding of descending modulation.
Organization of the PAG–RVM System
The influence of the PAG on the dorsal horn is relayed through the RVM (see Fig. 8-1). However, both the PAG and RVM receive significant input from higher structures, thereby providing a pathway through which cognitive and emotional factors can control pain processing. The PAG–RVM system also receives somatosensory, including nociceptive, information via the spinomesencephalic and spinoreticular tracts. This system is thus positioned to integrate top-down and bottom-up influences on pain transmission.
Ascending projections from both the PAG and RVM, though not as well studied as the descending pathway, have the potential to influence cortical processing of nociceptive input (Morgan et al 1989, Munn et al 2009).
Connectivity of the PAG
The PAG receives direct projections from a number of medial prefrontal cortical areas, including the anterior cingulate, as well as from the agranular insular cortex (Beitz 1982b, An et al 1998, Floyd et al 2000). The amygdala, a forebrain structure critically involved in processing emotionally significant input, is another major source of afferents to the PAG (Aggleton 1992). Cortical afferents to the amygdala largely target its basolateral component. The basolateral amygdala then projects to the central nucleus, which in turn projects densely to the PAG (Rizvi et al 1991). The central nucleus of the amygdala also receives nociceptive input, both directly from the spinal cord (Burstein and Potrebic 1993, Gauriau and Bernard 2004) and indirectly via a large projection from dorsal horn lamina I to the parabrachial nuclei (Gauriau and Bernard 2002). The influence of the amygdala on pain is mediated through its connections with the PAG–RVM system (Helmstetter and Landeira-Fernandez 1990, Helmstetter 1992, Helmstetter and Bellgowan 1993, Helmstetter and Tershner 1994, Pavlovic et al 1996, Helmstetter et al 1998, Foo and Helmstetter 1999, McGaraughty and Heinricher 2002, McGaraughty et al 2004).
The hypothalamus and preoptic area also constitute a major source of afferents to the PAG (Bandler and Keay 1996, Rizvi et al 1996). Manipulation of various hypothalamic regions can produce analgesia (Rhodes and Liebeskind 1978, Manning et al 1994, Holden et al 2002) or hyperalgesia (Oka et al 1995, Heinricher et al 2004, Martenson et al 2009).
Brain stem input to the PAG arises from the adjacent nucleus cuneiformis, the pontomedullary reticular formation, the locus coeruleus and other catecholaminergic cell groups, and the RVM (Beitz 1982b, Herbert and Saper 1992). The PAG and adjacent nucleus cuneiformis also receive a significant projection from the dorsal horn, including spinal lamina I nociceptive neurons (Menetrey et al 1982, Keay et al 1997). The PAG can thus integrate input from the limbic forebrain and diencephalon with ascending nociceptive input from the dorsal horn (Bandler and Keay 1996).
A major output from the PAG is to the RVM. Since the PAG itself projects only minimally to the spinal cord, this connection is critical for descending pain modulation. Thus, anatomical lesions, reversible inactivation with lidocaine (lignocaine), or microinjection of excitatory amino acid receptor antagonists into the RVM abolishes the analgesia produced by stimulation of the PAG (Fields et al 1991).
The PAG also projects rostrally to the medial thalamus and orbital frontal cortex and provides a possible substrate for ascending control of nociception (Coffield et al 1992, Cameron et al 1995). PAG projections to the dorsolateral and ventrolateral pontine tegmentum and to the ventrolateral medulla are likely to be involved in other aspects of PAG function, such as autonomic regulation (Bajic and Proudfit 1999).
Connectivity of the RVM
Output from the RVM
The RVM includes the midline nucleus raphe magnus and the adjacent reticular formation ventral to the nucleus reticularis gigantocellularis. The major output of the RVM is to the spinal cord and includes serotonin-containing neurons of the B3 cell group, γ-aminobutyric acid (GABA), and several neuropeptides (see Skagerberg and Björklund 1985 for review). The spinal terminals of RVM descending axons are most dense in dorsal horn laminae I, II (the substantia gelatinosa), and V. These laminae are targets of nociceptive primary afferents and include many nociresponsive neurons (Willis and Coggleshall 1991 and see Chapter 5). Furthermore, activation of the RVM by electrical stimulation can exert both inhibitory and excitatory effects on nociceptive neurons in these laminae, including primate spinothalamic tract neurons (Willis 1988).
The details of synaptic connections of RVM terminals in the dorsal horn remain to be worked out, but there is morphological and electrophysiological evidence of a host of actions, including direct post-synaptic inhibition of projection neurons, inhibition of release of transmitters from primary afferents, excitation of inhibitory interneurons, and inhibition of excitatory interneurons (Todd 2010). Lumb and colleagues (see review by Heinricher et al 2009) demonstrated differential regulation from the brain stem of dorsal horn wide–dynamic range neurons with and without significant C-fiber input. Dorsal horn neurons without significant C-fiber input were facilitated by brain stem stimulation. Lumb and colleagues argue that facilitation of these neurons results from descending inhibition of the neurons with C-fiber evoked responses (i.e., which function as inhibitory interneurons). However, other groups have found that descending inhibition and facilitation can have distinct time courses, with facilitation preceding inhibition (You et al 2008), and as outlined below, descending facilitation and inhibition from the RVM are exerted by different RVM cell populations. These latter findings imply that descending facilitation and inhibition are parallel and at least partially independent.
Like the PAG, the RVM has ascending projections, primarily non-serotonergic (Bowker 1986). Targets include the hypothalamus, preoptic area, and central nucleus of the amygdala (Vertes 1984, Hermann et al 1996). The function or functions of these connections are unknown.
Input to the RVM
The PAG and the adjacent nucleus cuneiformis are a major source of input to the RVM. A substantial proportion of PAG afferents to spinally projecting RVM neurons are GABAergic, although non-GABAergic connections also exist (Morgan et al 2008), including serotonin and neurotensin (Beitz 1982c). A large number of enkephalin and substance P neurons are found in the PAG, but few of these neurons project directly to the RVM (Beitz 1982a). Other brain stem afferents to the RVM include the parabrachial nuclei, Kölliker–Füse and intertrigeminal region, and rostral ventrolateral medulla. The RVM also receives substantial direct projections from higher centers implicated in emotion and autonomic regulation, including the hypothalamus, preoptic area, bed nucleus of the stria terminalis, central nucleus of the amygdala, and infralimbic and prelimbic cortex (Hermann et al 1997, Murphy et al 1999, Verner et al 2008).
Direct projections from the dorsal horn to the RVM are believed to be relatively sparse but have been demonstrated (Sugiyo et al 2005). Additional possible sources of spinal information include the PAG and nucleus cuneiformis, as well as the medullary nucleus reticularis gigantocellularis, which receive direct spinomesencephalic and spinomedullary connections and project to the RVM.
Pharmacology of the PAG–RVM Pain-Modulating Network
Opioids (see also Chapter 30)
Both the PAG and the RVM support the analgesic effect of μ-opioids (Yaksh et al 1988). That is, direct focal application of morphine or selective μ-receptor agonists in either region produces antinociception comparable to that seen following systemic administration. Conversely, inactivation of or microinjection of opioid antagonists into these sites reduces the analgesic effect of systemically administered opioids. μ-Opioid receptor agonists also act directly at the output target of the PAG–RVM system, in the spinal cord dorsal horn. With cloning of the three classic opioid receptors (μ, δ, and κ), discovery of a fourth related receptor (ORL1), and generation of selective antibodies for each, it became possible to map their distributions in the CNS. Each receptor is present in both the PAG and RVM, as well as in the spinal cord dorsal horn (Mansour et al 1995, Darland et al 1998).
The effects of ligands selective for δ, κ, and ORL1 receptors are state dependent. For example, κ-opioid receptor agonists can have either an analgesic or anti-analgesic effect when microinjected into the RVM, depending on the test and the sex of the animal (Tershner et al 2000, Ackley et al 2001). δ-Opioid receptor agonists have a modest analgesic effect when microinjected into the PAG and RVM under basal conditions (see Heinricher and Fields 2003 for review) but show significant antinociceptive potency during chronic inflammation (Hurley and Hammond 2000). Injection of the ORL1 agonist nociceptin (orphanin FQ) into the RVM interferes with the antinociceptive effect of μ-opioid agonists administered systemically or microinjected into the PAG, but it also attenuates the hyperalgesia associated with acute opioid withdrawal (Heinricher et al 1997, Pan et al 2000). This apparent paradox can be understood in the context of the RVM circuitry and is explained by the fact that nociceptin inhibits the firing of both pain-inhibiting and pain-facilitating neurons in the RVM (Heinricher 2003).
Endogenous opioid peptides also play an important role in the PAG–RVM system. The contribution of endogenous opioid peptides to pain modulation was first suggested by reports that stimulation-produced analgesia in animals and humans is reduced by the narcotic antagonist naloxone. Naloxone also worsens postoperative pain in patients who have not received exogenous opioid therapy, thus establishing the relevance of endogenous opioids to common clinical situations (see Fields and Levine 1984 for review).
The different levels of the PAG–RVM circuit are linked in part through the release of endogenous opioids. Thus the antinociceptive actions of microinjected opioids (or electrical or chemical stimulation) at one site can be blocked by microinjection of opioid antagonists at a downstream site in the pathway. For example, the antinociceptive effect of a μ-opioid receptor agonist microinjected into the basolateral amygdala is reversed by injection of a μ-opioid receptor antagonist into the PAG (Tershner and Helmstetter 2000). Similarly, antinociceptive effects elicited from the PAG can be blocked by naloxone or selective μ-opioid receptor antagonists microinjected into the RVM (Kiefel et al 1993, Roychowdhury and Fields 1996). Moreover, endogenous opioid links are required for the antinociception triggered during conditioned fear, which is mediated by the PAG–RVM system (Helmstetter and Landeira-Fernandez 1990, Bellgowan and Helmstetter 1998, Foo and Helmstetter 1999).
These data demonstrate that the PAG–RVM system is linked through the release of endogenous opioids and indicate that exogenous opioids produce their effects not only by direct binding to opioid receptors in the PAG–RVM circuit but also indirectly by triggering release of endogenous opioids acting at the same receptors. This may be a critical factor in the potentiated action of exogenous opioids during inflammation (Hurley and Hammond 2001, Sykes et al 2007).
Cannabinoids
Like opioids, cannabinoids have long been used to treat pain (Walker and Huang 2002). Also like opioids, cannabinoids have considerable physical and psychological side effects that limit their use as analgesics (Malan et al 2003). Cannabinoids have significant antinociceptive efficacy in a range of animal models. The relatively few controlled clinical trials of cannabinoid analgesia in humans have yielded suggestive but mixed evidence that cannabinoid drugs have therapeutic utility. Part of the problem is that psychotropic side effects, such as mental clouding, are present in the same dose range as that for analgesia (Pertwee 2001, Iversen and Chapman 2002).
Cannabinoids again show parallels to opioids in having multiple sites of action to produce antinociception: in the periphery, spinal cord, and brain (see Hohmann 2002, Walker and Huang 2002 for reviews). Cannabinoids produce their antinociceptive effect at least in part by recruiting the PAG–RVM modulatory system. CB1 receptors are densely expressed in the PAG, and the RVM is required for the antinociceptive action of systemically administered cannabinoid agonists. Microinjection of CB1 agonists into the PAG or RVM produces antinociception, and CB1 agonists produce effects similar to morphine in identified RVM pain-modulating neurons (Meng et al 1998, Meng and Johansen 2004). Cannabinoids have negligible direct post-synaptic actions on PAG or RVM neurons but act presynaptically to block excitatory and inhibitory synaptic currents (Vaughan et al 1999, 2000). The amygdala, an important input to the PAG, also expresses CB1 receptors and is required for systemic cannabinoid antinociception. Endocannabinoids, like endogenous opioids, contribute to the antinociceptive effect of activating the PAG–RVM system (Valverde et al 2000, Hough et al 2002).
Improgan, a Novel, Non-opioid Analgesic Compound that Acts in the PAG–RVM System
Improgan is a novel, non-opioid analgesic drug that acts in both the PAG and RVM to suppress nociceptive responses in acute and chronic animal models (Nalwalk et al 2004). Improgan antinociception is blocked by the CB1 receptor antagonist rimonabant (Gehani et al 2007), thus pointing to an endocannabinoid link in its effects. By contrast, improgan antinociception is not blocked by naloxone, which indicates that this drug does not recruit endogenous opioids.
Acetylcholine
Nicotinic acetylcholine receptors in the RVM contribute to pain modulation. Nicotinic agonists microinjected into the RVM inhibit hot plate and tail flick responses to noxious heat (Iwamoto 1991), as well as the formalin response (Bannon et al 1998). Furthermore, iontophoretic (Willcockson et al 1983) or systemic (Bitner et al 1998) administration of a nicotinic agonist produces dose-related activation of RVM neurons. The pain-modulating actions of nicotinic agonists in the RVM depend on serotonergic neurons, and the nicotinic acetylcholine receptor is located predominantly on serotonergic neurons of the RVM (Bitner et al 1998). Somewhat surprisingly, although lidocaine (lignocaine) blockade of the RVM prevents the antinociceptive effects of systemic nicotine administration, it does not block the antinociceptive effects produced by ABT-564, a nicotinic cholinergic channel modulator (Decker et al 1998).
Cholecystokinin
Cholecystokinin (CCK) has both anti-opioid and pro-nociceptive actions at multiple levels of the nervous system. CCK peptides, through actions at the CCK2 receptor, act as functional antagonists to the analgesic action of opioids in behavioral studies (Crawley 1991). CCK2 agonists have cellular actions opposite those typically produced by opioids: they decrease K+ conductance (Cox et al 1995) and increase release of GABA (Miller and Lupica 1994). A corollary of the functional antagonism of opioids by CCK is that selective CCK2 antagonists can potentiate the analgesic action of morphine and endogenous opioids acting at the μ-opioid receptor and reduce some forms of opioid tolerance through action in the PAG (Zarrindast et al 1999, Tortorici et al 2003). These data indicate that endogenous CCK limits the antinociceptive action of opioids. Consistent with animal research, human studies have demonstrated that CCK antagonism enhances morphine (Price et al 1985) and placebo (Benedetti 1996) analgesia. In the RVM, CCK blocks opioid activation of physiologically identified pain-inhibiting neurons (Heinricher et al 2001a).
In addition to its anti-opioid actions, CCK exerts a pro-nociceptive effect through the PAG–RVM system. Thus, CCK receptor blockade in the RVM attenuates nociceptive hypersensitivity in a range of models (Urban et al 1996b, Kovelowski et al 2000, Ambriz-Tututi et al 2011). This pro-nociceptive action of CCK can be explained by the fact that this peptide activates putative pain-facilitating neurons in the RVM, albeit at doses higher than those required to block opioid activation of the pain-inhibiting neurons (Heinricher and Neubert 2004).
Anxiety, to which brain CCK contributes, can enhance pain sensitivity. The anti-analgesic and pro-nociceptive actions of CCK can therefore be seen as a critical link between anxiety and pain (Colloca and Benedetti 2007, Lovick 2008).
Neurotensin
As is the case with CCK, neurotensin co-distributes with opioids in pain-modulating networks. Although initial studies demonstrated an antinociceptive action of neurotensin in the PAG and RVM, more detailed dose–response studies revealed a biphasic function such that low doses microinjected into the RVM elicit a facilitatory effect on nociceptive transmission whereas higher doses inhibit nociception (Smith et al 1997). The biphasic dose–response relationship is explained by the observation that low doses of neurotensin activate the pain-facilitating neurons in this region whereas higher doses also recruit the pain-inhibiting neurons (Neubert et al 2004). These differential effects of low- and high-dose neurotensin on different RVM cell populations may be mediated by different receptors since both NTR1 and NTR2 are found within the RVM and the antinociceptive effect of neurotensin is blocked by the NTR1 antagonist (Buhler et al 2005).
The physiological significance of the pain-facilitating effect of neurotensin is confirmed by the finding that the hyperalgesic state elicited by the cutaneous inflammatory agent mustard oil is blocked by microinjection of a neurotensin receptor antagonist into the RVM (Urban et al 1996a), thus demonstrating recruitment of endogenous neurotensin in inflammation. In addition, endogenous neurotensin, like CCK, exerts an anti-opioid action (Smith et al 1997).
Substance P
Both substance P and neurokinin 1 (NK1) receptors are found in the RVM (Ljungdahl et al 1978, Nakaya et al 1994). Direct focal application of substance P into the PAG or RVM produces hypoalgesia (Malick and Goldstein 1978, Rosen et al 2004, Hamity et al 2010), and substance P at both sites is required for the analgesic effect of stimulating the lateral hypothalamus (Holden and Pizzi 2008, Holden et al 2009). Other investigators emphasize a pro-nociceptive action of this peptide in the RVM. LaGraize and colleagues (2010) reported significant hyperalgesia following RVM microinjection of substance P, and a number of groups have shown that NK1 receptor antagonists microinjected directly into the RVM attenuate hyperalgesia in inflammatory models (Pacharinsak et al 2008, Hamity et al 2010, LaGraize et al 2010). Budai and associates (2007) showed that substance P applied to individual, physiologically identified pain-facilitating neurons in the RVM enhances the neuronal response to co-applied N-methyl-D-aspartate (NMDA). The implication of these findings is that release of substance P within the RVM contributes to behavioral hypersensitivity during inflammation by increasing the excitability of RVM pain-facilitating neurons.
Further studies will clearly be needed to tease out the bimodal actions of substance P in the RVM. Linking the functional effects of substance P in the RVM to cellular mechanisms will be critical. Zhang and Hammond (2009) saw an increase in excitatory synaptic transmission when substance P was applied directly in the RVM, a finding consistent with the enhancement of NMDA-mediated activation of RVM neurons reported by Budai and co-authors (2007). Another report suggested an apparently opposing view, which is that substance P “uncouples” RVM neurons from both excitatory and inhibitory input by changing the electrical properties of the dendrites (Hahm et al 2011). One possible factor to consider in bringing these disparate findings together is that NK1 receptor expression is increased in the RVM during inflammation (LaGraize et al 2010). This increased expression might underlie a novel functional role for substance P during inflammation.
Physiology of Pain-Modulating Neurons in the PAG–RVM System
Three distinct populations of neurons in the RVM are recognized: those that discharge just before the occurrence of withdrawal from noxious heat (ON-cells), those that stop firing just before a withdrawal reflex (OFF-cells, Fig 8-2), and those that show no consistent changes in activity when withdrawal reflexes occur (NEUTRAL-cells, Fields and Heinricher 1985). A significant proportion of neurons of each class project to the spinal cord, and ON- and OFF-cells project specifically to laminae I, II, and V of the dorsal horn (Vanegas et al 1984b, Fields et al 1995). A large number of OFF-cells, NEUTRAL-cells, and at least some ON-cells are GABAergic (Winkler et al 2006). Both ON- and OFF-cells respond to manipulations of the PAG that produce behavioral analgesia (Vanegas et al 1984a).
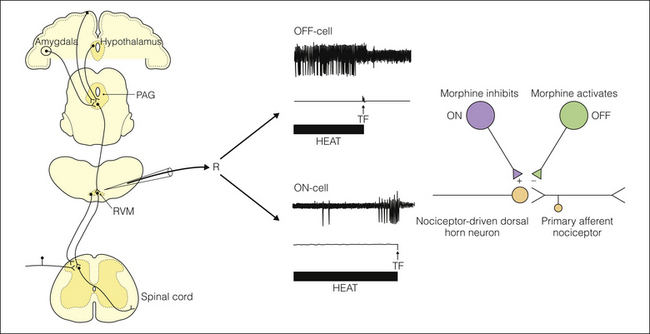
Figure 8-2 Properties of medullary pain-modulating neurons.
Left, microelectrode placement in the rostral ventromedial medulla (RVM) for single-unit extracellular recording (R). Upper right, top trace, a single oscilloscope sweep showing the OFF-cell pause occurring just before the tail flick (TF) reflex (middle trace: a force transducer showing movement) in response to noxious heat (bottom trace). Lower traces illustrate the typical ON-cell firing pattern, which is a burst beginning before the TF. The right diagram illustrates that both ON- and OFF-cells project to the spinal cord, where they exert bidirectional control over nociceptive dorsal horn neurons. As shown in Figure 8-3, ON-cells are inhibited and OFF-cells activated by morphine. PAG, periaqueductal gray.
OFF-cells exert a net inhibitory effect on nociception. Activation of OFF-cells produces behavioral antinociception (Heinricher et al 1994) and is necessary for the analgesic actions of opioids (see below). Conversely, ON-cells exert a net facilitatory effect on nociception. ON-cell activation is associated with enhanced nociception in a number of paradigms (Heinricher et al 1989, Ramirez and Vanegas 1989, Bederson et al 1990, Jinks et al 2004), and selective pharmacological activation of ON-cells produces hyperalgesia (Heinricher and Neubert 2004, Neubert et al 2004). NEUTRAL-cells remain something of an enigma, although the fact that at least a subset of NEUTRAL-cells is serotonergic (Potrebic et al 1994, Mason 1997) suggests some role for this cell class in pain modulation. Interestingly, neurons in the PAG and the adjacent dorsolateral pontine tegmentum can be divided into the same three classes of ON-, OFF-, and NEUTRAL-cells (Heinricher et al 1987, Haws et al 1989, Carlson et al 2005), which suggests that a common neural mechanism for pain modulation exists at the medullary, pontine, and midbrain levels.
The organization of the RVM suggests that the neurons of each physiological class function as a unit that exerts global control over pain transmission without topographical constraints. RVM neurons, including identified ON- and OFF-cells, project widely to the trigeminal dorsal horn and multiple spinal levels (Huisman et al 1981, Fields et al 1995). Furthermore, many RVM neurons have axons that collateralize within the RVM itself (Mason and Fields 1989), and at least in anesthetized rats, cells of the same physiological class tend to fire at the same time (Barbaro et al 1989). Thus, RVM neurons appear to function in concert to exert a broad and dynamic influence over the general responsiveness of the organism to noxious stimulation.
The inhibition of OFF-cells and activation of ON-cells associated with a nocifensive reflex can lead to general enhancement of reflex responses to subsequent stimuli delivered to any region of the body surface (Ramirez and Vanegas 1989, Foo and Mason 2003). A report that RVM neuronal responses to colorectal distention differ from those evoked by stimulation of the skin (Brink and Mason 2003) raised the possibility that PAG–RVM modulation of nociception recruited by visceral stimulation might be quite distinct from that produced by cutaneous input. However, others have since shown that ON- and OFF-cell responses evoked by colorectal distention and by intracolonic capsaicin are concordant with those evoked by cutaneous stimuli (Pinto-Ribeiro et al 2008, Sanoja et al 2010). More important, the latter authors demonstrated the functional significance of ON-cell activation in the visceral model by showing that blocking NMDA receptors in the RVM (which would be expected to reverse sensitization of the ON-cells, Xu et al 2007) prevented referred hypersensitivity in the capsaicin-treated animals. The latter finding suggests that ON-cells enhance visceral as well as cutaneous nociception. In awake unrestrained rats, RVM neurons that resemble ON-cells respond briskly to light touch and to sudden sound (Oliveras et al 1990, Leung and Mason 1999). This raises the possibility that innocuous, but possibly behaviorally significant environmental stimulation modulates nociceptive processing through the PAG–RVM system. Correlations of ON- and OFF-cell discharge with behavioral state and autonomic parameters similarly point to a potential role of these neurons in mediating the effects of a host of psychological and physiological variables on pain (Heinricher et al 2009).
Opioid Actions in the Rostral Ventromedial Medulla: ON- and OFF-Cells
Supraspinal opioid administration activates descending antinociceptive controls. How do opioids produce this effect? Morphine and other μ-opioid receptor agonists produce direct post-synaptic hyperpolarization of a subpopulation of neurons in the PAG and RVM (referred to as “secondary cells”). This hyperpolarization is mediated by increased K+ conductance (Pan et al 1990, Chieng and Christie 1994). μ-Opioid receptor agonists also act presynaptically to depress synaptic transmission. Presynaptic effects on GABAergic synaptic transmission are pronounced in the RVM, and neurons disinhibited by local μ-opioid action are referred to as “primary cells” (Pan et al 1990). (See Heinricher and Ingram 2008 for review.)
How are opioid effects at the membrane level translated into behaviorally measurable antinociception? Answering this question requires analysis at the circuit level, which is more advanced for the RVM than for the PAG. RVM OFF-cells are activated, ON-cells inhibited, and NEUTRAL-cells unaffected when morphine or μ-opioid receptor agonists are administered systemically, into the PAG or directly into the RVM (Fig. 8-3, Heinricher and Ingram 2008). Local iontophoretic application of morphine directly inhibits ON-cells but has no effect on OFF- or NEUTRAL-cells (Heinricher et al 1992). Thus, ON-cells are probably equivalent to the secondary cells that are directly hyperpolarized by μ-opioid receptor agonists, whereas the opioid activation of OFF-cells in vivo must be indirect, primarily via inhibition of GABAergic inhibitory input (Fig. 8-4).
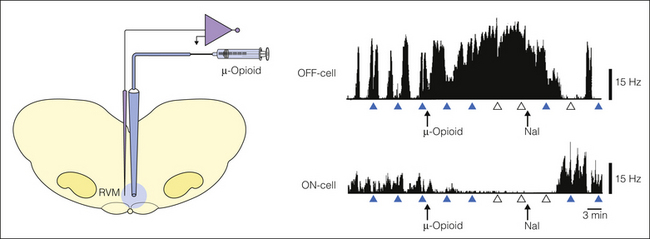
Figure 8-3 Focal application of the μ-opioid receptor agonist DAMGO into the rostral ventromedial medulla (RVM) inhibits ON-cells and activates OFF-cells. The inhibition of ON-cells is probably a direct post-synaptic effect, whereas the activation of OFF-cells must be indirect, via disinhibition (see Fig. 8-4). The opioid effect on RVM cells and nociceptive behavior is reversed by systemically administered naloxone. Arrowheads indicate times of application of heat to the tail, filled symbols indicate the presence of tail flick (TF), and open symbols indicate complete inhibition of the TF. Nal, naloxone; Hz, Hertz. (Data graphs from Heinricher MM, Morgan MM, Tortorici V, et al 1994 Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience 63:279–288.)
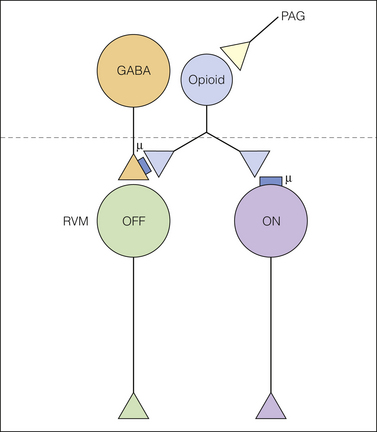
Figure 8-4 Direct and indirect effects of μ-opioids in the RVM.
Periaqueductal gray (PAG) neurons activate an opioid (presumably enkephalinergic) neuron. This endogenous opioid inhibits μ-opioid receptor–bearing ON-cells and GABAergic input to OFF-cells. Inhibition of the GABAergic input disinhibits the OFF-cell, which when activated inhibits nociceptive transmission at the level of the dorsal horn. The sources of GABAergic input to the rostral ventromedial medulla (RVM) OFF-cells and the location of the opioid neuron are not known. However, ON-cells are not the source of GABAergic input to OFF-cells because selective inhibition of RVM ON-cells does not activate RVM OFF-cells and the OFF-cell pause begins before the onset of the ON-cell burst. GABA, γ-aminobutyric acid.
Activation of OFF-cells is necessary and sufficient for the antinociceptive effects of opioids in acute pain. Selective blockade of OFF-cell activation prevents the antinociceptive effect of systemically or intraventricularly administered morphine (Heinricher et al 1997, 1999, 2001b, 2010). Inhibition of ON-cells is not by itself sufficient to account for the behavioral antinociception produced by μ-opioids under normal conditions (Heinricher et al 1999) but presumably complements and reinforces the activation of OFF-cells. However, direct opioid inhibition of RVM ON-cells may contribute significantly to antinociception in hyperalgesic states such as inflammation or opioid dependence, states characterized by increased ON-cell activity (Porreca et al 2002, Heinricher et al 2003, 2009).
As mentioned, an endogenous opioid acting on RVM neurons provides a significant link between the PAG and the RVM. Microinjecting opioid antagonists into the RVM blocks the antinociception produced by PAG manipulations, and the inhibition of RVM ON-cells by PAG morphine or bicuculline is reversed by local iontophoresis of naloxone (Pan and Fields 1996). Endogenous opioids could also act presynaptically to inhibit GABA release onto OFF-cells, again because opioid inhibition of ON-cells does not by itself produce a measurable antinociceptive effect. Figure 8-4 illustrates this opioid link in the RVM relay from the PAG to the spinal cord dorsal horn.
Insight into the role of the δ-opioid receptor in the RVM remains at an early stage, at least in comparison to our understanding of the μ-opioid receptor (see Heinricher and Fields 2003 for review). In general, δ-opioid receptor agonists produce effects that are similar to but weaker than those produced by μ-opioid receptor agonists. Microinfusion of δ-agonists into the RVM produces neuronal effects similar to those of μ-opioid receptor agonists: ON-cells are inhibited, OFF-cells are excited, and NEUTRAL-cells are unaffected. Tissue inflammation potentiates the effects mediated by the δ-opioid receptor in the RVM (Hurley and Hammond 2000), which synergizes with μ-opioid actions to enhance exogenous opioid analgesics (Hurley and Hammond 2001, Sykes et al 2007). There is also evidence that activation of the δ-opioid receptor contributes to opioid tolerance. Morphine tolerance is reduced in δ-opioid receptor–knockout mice (Zhu et al 1999). Furthermore, bivalent compounds that act as both μ-opioid receptor agonists and δ-opioid receptor antagonists show increased analgesia and reduced morphine tolerance (Daniels et al 2005).
κ-Opioid receptor actions in the RVM are superficially more complex than μ- or δ-receptor actions. Analgesia follows focal application of κ-agonists in the RVM, although some investigators report no effect. κ-Agonists in the RVM can also reduce the antinociceptive effect of morphine and interfere with stress-induced analgesia (Rossi et al 1994, Pan et al 1997, Foo and Helmstetter 2000, Ackley et al 2001). This apparent complexity is explained at the circuit level: U69593, a κ-opioid agonist, both reduces the ON-cell burst and suppresses opioid activation of OFF-cells (Meng et al 2005). The effect on ON-cells would presumably have a modest hypoalgesic effect, whereas the interference with OFF-cell activation would prevent opioid analgesia. This general schematic is confirmed by studies at the cellular level, where two inhibitory actions of κ-agonists have been demonstrated in the RVM: hyperpolarization of primary neurons (which presumably include OFF-cells) and presynaptic inhibition of glutamatergic excitation (Pan et al 1997, Ackley et al 2001, Bie and Pan 2003).
Role of Serotonin in the Descending Modulation of Nociceptive Transmission
Serotonergic neurons account for about 20% of the total RVM population (Moore 1981), and there is evidence of serotonin involvement in descending modulation of pain transmission (Le Bars 1988, Suzuki et al 2004, see also Chapter 28). Electrical stimulation of the RVM evokes the release of serotonin in spinal cord cerebrospinal fluid, and the analgesia produced by this stimulation is reduced by serotonin antagonists administered intrathecally (see Le Bars 1988 for review). Iontophoresis of serotonin inhibits the response of dorsal horn neurons to noxious stimulation (Headley et al 1978, Jordan et al 1978), and serotonin applied directly to the spinal cord inhibits nociceptive transmission (Yaksh and Wilson 1979). Furthermore, the analgesic action of systemically administered opiates can be reduced by depletion of serotonin or by neurotoxic destruction of spinal serotonin terminals.
The significance of serotonin for pain modulation has presented something of a dilemma since initial studies suggested that RVM serotonergic neurons were NEUTRAL-cells rather than ON- or OFF-cells (Mason 1997). However, a recent analysis of a large sample of RVM neurons labeled for serotonin following juxtacellular recording (Bernard et al 2010) found a substantial proportion of serotonergic neurons with ON- and OFF-cell response properties. The existence of serotonin-containing neurons with NEUTRAL-, ON-, and OFF-cell properties would, like the distribution of GABA across all three classes (Winkler et al 2006), argue against a 1:1 relationship between neurotransmitter content and physiological function.
In fact, serotonin, like the PAG–RVM system as a whole, has both anti- and pro-nociceptive roles at the level of the spinal cord. Inhibition is thought to be largely mediated via subtypes of G protein–linked 5-HT1 and 5-HT2 receptors (Millan 2002). Other evidence suggests that the inhibition is indirect and can be exerted by descending 5-HT3–mediated excitation of GABAergic inhibitory interneurons (Alhaider et al 1991). The 5-HT3 receptor is also thought to mediate the facilitatory actions of serotonin (Suzuki et al 2004). This facilitation is presumably mediated either by direct serotonin action on nociresponsive neurons that express the 5-HT3 receptor or by enhancement of release of neurotransmitters from the terminals of primary afferent nociceptors that express presynaptic 5-HT3 receptors. Suzuki and colleagues suggest that a positive feedback loop from nociceptors to the RVM and back to the dorsal horn leads to activation of spinal 5-HT3 receptors. They reported that the loss of noxious stimulus–induced facilitation of the responses of deep dorsal horn neurons following destruction of NK1 receptor–expressing lamina I neurons by neurotoxin could be mimicked by antagonism of 5-HT3 receptors at the level of the spinal cord (Suzuki et al 2002). These authors hypothesized that lamina I neurons activate descending facilitatory output from the RVM and that 5-HT3 receptors in the dorsal horn mediate this positive feedback loop. Functional studies show that intrathecal injection of the 5-HT3 antagonist ondansetron decreases pain behavior in the second phase of the formalin test (Oyama et al 1996, Zeitz et al 2002) and the hyperalgesia produced by activation of RVM ON-cells via microinjection of CCK (Dogrul et al 2009). However, Peters and colleagues (2010) failed to demonstrate an anti-allodynic effect of ondansetron following spinal nerve ligation, inconsistent with the proposal of Suzuki and colleagues (2004).
Pain-Modulating Systems in the Pontine Tegmentum: Noradrenergic Cell Groups
A second major brain stem pain-modulating system arises in the noradrenergic cell groups of the lateral pontine tegmentum (A5, A6 or locus coeruleus, and A7, see Fig. 8-1). α2-Adrenergic agents administered at the spinal level produce clinically significant analgesia (Eisenach et al 1996). Just as opioid analgesics co-opt an endogenous opioid pain-modulating system, this effect of α2-receptor agonists points to modulation of spinal nociception by endogenous noradrenergic systems. Stimulation directed at the noradrenergic cell groups produces antinociception mediated by spinal α2-adrenergic receptors (for reviews see Proudfit 1992, Pertovaara 2006). Like the PAG–RVM system, noradrenergic pathways exert bidirectional control, with antinociception mediated by spinal α2-adrenergic receptors and pro-nociception by α1 receptors (Holden et al 1999, Nuseir and Proudfit 2000).
It is not clear whether noradrenergic tone has a significant impact on acute nociceptive responsiveness under basal conditions. Thus, pharmacological blockade of spinal α1 or α2 receptors and long-term depletion of norepinephrine have been reported by several groups to have little or no effect on responses in acute pain tests (Martin et al 1999, Nuseir and Proudfit 2000, Jasmin et al 2003a). By contrast, Howorth and co-authors (2009) reported thermal hyperalgesia when a viral vector–based approach was used to reduce the excitability of pontospinal noradrenergic neurons in normal animals. The mechanical threshold was not lowered. Disparate findings also arise when considering the formalin test, with no effect, reduced responses, and enhanced responses in animals subjected to long-term depletion of norepinephrine or suppression of noradrenergic neuron excitability (Martin et al 1999, Jasmin et al 2003a, Howorth et al 2009).
Data from studies involving chronic pain models are no more consistent. There are several reports that noradrenergic depletion or antagonism facilitates responses in neuropathic pain states, which suggests that descending noradrenergic inhibitory systems are recruited following nerve injury, thus limiting the hyperalgesia that would otherwise result (Xu et al 1999, Jasmin et al 2003a). However, reducing the excitability of pontine noradrenergic neurons did not enhance either tactile or cold allodynia in animals subjected to nerve injury (Howorth et al 2009), and several other groups reported attenuation of allodynia and hyperalgesia following depletion of spinal norepinephrine (Brightwell and Taylor 2009, Martins et al 2010).
These conflicting data indicate that a simple model in which activation of pontospinal noradrenergic neurons suppresses spinal nociceptive processing will be inadequate for understanding the role of this transmitter in descending control. The fact that norepinephrine has bidirectional effects is likely to be one important complicating factor (Holden et al 1999, Nuseir and Proudfit 2000). In addition, the noradrenergic cell groups have supraspinal targets as well as descending connections, which could contribute to changes in nociception following manipulation of noradrenergic cell bodies.
It seems likely that noradrenergic pathways in the pons can be engaged independently of the PAG–RVM system. However, the PAG projects to the dorsolateral pontine area where noradrenergic neurons are found and was shown to make direct connections with A7 noradrenergic neurons (Bajic and Proudfit 1999, Bajic et al 2000). The RVM is reciprocally linked with A7 (Yeomans and Proudfit 1990, Clark and Proudfit 1991). Furthermore, although no noradrenergic neurons are present in the RVM itself, electrical stimulation of the RVM evokes the release of noradrenaline at the spinal cord (Hammond et al 1985), and the inhibition of spinal withdrawal reflexes by activation of the RVM is attenuated by noradrenergic antagonists given intrathecally (Barbaro et al 1985). Iontophoresis of α2-adrenergic antagonists onto nociceptive dorsal horn neurons blocks inhibition of these neurons by PAG stimulation (Budai et al 1998). These findings could most easily be explained by a circuit in which noradrenergic neurons are recruited by activation of the PAG or RVM. Consistent with this, RVM neurons projecting to the A7 region contain substance P, and focal application of substance P in the A7 region produces analgesia (Yeomans and Proudfit 1992). In addition, the antinociceptive effect of cholinergic stimulation in the RVM is substantially attenuated by inactivation of the A7 region (Nuseir et al 1999). Finally, spinal norepinephrine mediates the antinociceptive effect of activating RVM OFF-cells with neurotensin (Buhler et al 2008). Viewed collectively, these data indicate that the PAG–RVM system produces its antinociceptive effects at least in part by engaging the descending noradrenergic pathways. Whether this extends to pro-nociceptive output remains to be investigated.
Pontine noradrenergic systems may also mediate cortical control of spinal nociceptive transmission. Increasing GABA levels in the anterior insular cortex produces an analgesic effect that is blocked by intrathecal administration of α-adrenergic antagonists. Because this cortical region projects to the locus coeruleus, as well as the RVM, it was suggested that inhibition of insular outflow disinhibits noradrenergic neurons of the locus coeruleus (Jasmin et al 2003b). However, the possibility that the insula engages noradrenergic systems via the RVM cannot be ruled out from these data.
Plasticity of the RVM in Chronic Pain States
Molecular and Cellular Analysis of the PAG–RVM System in Chronic Pain States
The evidence that descending facilitation contributes to both inflammatory and neuropathic pain states involving cutaneous as well as deep structures prompted a search for associated changes intrinsic to RVM neurons. At the molecular and cellular level, there is evidence of striking alterations in neurotransmitters and receptors, trophic factors, and signal transduction pathways in a range of chronic pain models.
Significant effort has been focused on excitatory amino acid transmission in the RVM. During inflammation, for example, NMDA receptor subunits, including NR1, NR2A, and NR2B, are up-regulated, and there is a complex shift in the dose–response curve for exogenous NMDA microinjected into the RVM in the 24 hours following induction of peripheral inflammation with complete Freund’s adjuvant (CFA), with facilitation or inhibition of heat-evoked withdrawal occurring at different doses and time points (Guan et al 2002, Miki et al 2002). The functional significance of these changes in NMDA sensitivity is not clear since NMDA receptor blockade in the RVM had no effect on nociceptive responses in this model. However, one possibility worth considering is that NMDA receptor function is altered in neurons related to aspects of RVM function other than pain modulation, such as thermogenesis (presumably related to the stress of the chronic pain state). However, other groups have documented a necessary role for NMDA receptor activation in behavioral hypersensitivity in different pain models (Urban et al 1999, Da Silva et al 2010c). This discrepancy merits further investigation. Changes in α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptor function in the RVM during chronic pain states have also been reported. For example, phosphorylation of the GluR1 AMPA receptor subunit is increased in a chronic inflammation model (Guan et al 2004). The fact that AMPA receptor blockade in the RVM enhances sensitivity during chronic or acute inflammation, as well as in control animals (Urban et al 1999, Guan et al 2002), suggests that an AMPA receptor–mediated input drives an inhibitory RVM output. This inhibition is apparently masked during inflammation by descending facilitation from the RVM or via parallel modulatory pathways.
Brain-derived neurotrophic factor (BDNF) is broadly implicated in activity-dependent synaptic plasticity in the adult brain, but specifically in the PAG–RVM system in chronic inflammation (Guo et al 2006). Phosphorylation of the TrkB receptor for BDNF is increased in the RVM in animals treated with CFA, and down-regulation of TrkB by RNA interference attenuates hypersensitivity in this model. Administration of BDNF directly into the RVM mimics the behavioral effect of inflammation, with maximum thermal hyperalgesia occurring 6 hours after the infusion of a physiologically relevant dose of exogenous BDNF. A likely source of BDNF in the RVM is the PAG since BDNF-expressing neurons in the PAG project to the RVM and BDNF expression in the PAG is increased during inflammation produced by CFA. Further support for a BDNF projection from the PAG to the RVM comes from the fact that electrical stimulation of the PAG leads to increased phosphorylation of the TrkB receptor in the RVM.
There is also evidence of glial activation and cell loss in both the PAG and RVM in models of inflammatory and nerve injury pain (Wei et al 2008, Roberts et al 2009, Leong et al 2010, Mor et al 2010, 2011). Importantly, local glial inhibition in the RVM attenuates hypersensitivity in these models (Roberts et al 2009, Mor et al 2011).
Physiology of RVM Neurons in Chronic Pain States
Hypersensitivity measured behaviorally could reflect increased descending facilitation or decreased descending inhibition. Direct measures of the activity of the functionally distinct RVM cell classes, ON-, OFF-, and NEUTRAL-cells, are therefore required to provide a bridge between molecular mechanisms and behavioral modulation.
The RVM response to acute inflammation or to a prolonged noxious stimulus that does not produce lasting tissue damage is well defined, with a strong shift toward enhanced descending facilitation and reduced descending inhibition, as demonstrated by the prolonged activation of ON-cells and suppression of OFF-cell firing (Ramirez and Vanegas 1989, Morgan and Fields 1994, Kincaid et al 2006). Pharmacological blockade of the ON-cell response under these conditions attenuates or blocks behavioral hypersensitivity (Xu et al 2007), thus demonstrating the functional significance of this shift in the balance between ON- and OFF-cell output.
In chronic pain models, ON- and OFF-cell populations demonstrate a range of altered response patterns, which suggests that these two populations are recruited by different mechanisms under different conditions, even when the net behavioral effect is hypersensitivity. In a dural inflammation model of RVM-dependent headache-related pain, ON-cells exhibited a slowly developing and prolonged activation over a period of several hours, whereas OFF-cell firing was suppressed, but only transiently. NEUTRAL-cells did not show an immediate or delayed response to dural inflammation (Edelmayer et al 2009). In this headache model, therefore, increased facilitation is not coupled with a reduction in descending inhibition. A very different pattern was seen in animals subjected to experimental arthritis, with an increase in the firing of both ON- and OFF-cells. NEUTRAL-cells were not studied (Pinto-Ribeiro et al 2008). Although behavioral hypersensitivity was not documented in this study, it is reasonable to conclude that both descending facilitation and descending inhibition were increased, with the effects of descending inhibition presumably being masked by the facilitatory influence. In another chronic inflammation model, Miki and colleagues (1998) sampled RVM neuronal activity for periods of hours during the development of paw inflammation induced with CFA. They saw little alteration in the firing patterns of ON- or OFF-cells as inflammation progressed but indicated that some neurons classified as NEUTRAL-cells developed reflex-related changes in firing that suggested a shift to the ON- or OFF-cell group. This is an intriguing finding that should be followed up, but it should be noted that activity was compared not with the pre-CFA baseline but with earlier post-CFA time points, so it is unclear whether further changes in ON- or OFF-cell firing should have been expected. Moreover, the methods for cell classification used by Miki and co-workers may not have allowed them to distinguish ON-cells with spontaneous activity from NEUTRAL-cells (Barbaro et al 1986). In the spinal nerve ligation model of nerve injury pain, ON- and OFF-cells were sensitized following nerve injury, with lowered response thresholds for tactile stimulation (Carlson et al 2007). Ongoing discharges were not different from controls. NEUTRAL-cells were found that had properties indistinguishable from those of controls. Changes in ON- and OFF-cell response thresholds correlated with tactile allodynia in these animals.
Enhanced descending facilitation is thus coupled with a reduction in descending inhibition in acute inflammation and following nerve injury. However, the pattern of RVM recruitment is quite different in the two conditions: acute inflammation is associated with a strong shift in overall excitability of the ON- and OFF-cell populations, whereas nerve injury can be linked to a lowered response threshold. A central challenge for the future will be to link findings of neuroplasticity at the molecular and cellular level to the functionally defined circuitry of the RVM.
Physiological Function of Brain Stem Pain-Modulating Networks
Despite our extensive knowledge of the circuit properties and pharmacological responsiveness of descending modulatory networks, knowledge of their physiological function and clinical significance is tentative. Adding to the complexity of this analysis, several pain-modulating networks are operating in parallel, and understanding the net behavioral effect of their combined actions requires parsing the contribution of each under a variety of conditions. Nevertheless, there is evidence of recruitment of modulatory systems as part of counterirritation, sickness, fear, and anxiety, based on cognitive variables, including attention and expectation.
Is Pain Inhibited or Enhanced during Concurrent Noxious Stimulation at Several Regions of the Body? Multiple Bidirectional Pain-Modulating Networks
Noxious stimulation delivered to one part of the body inhibits dorsal horn nociceptive neurons in spinal segments innervating distant body parts. This phenomenon is referred to as “diffuse noxious inhibitory control” (DNIC). It has been hypothesized that DNIC functions as a contrast mechanism: noxious stimulation activates a surround inhibition that sharpens contrast between the stimulus zone and adjacent areas. This sharpened contrast would actually have a net enhancing effect on the perceived intensity of pain. However, outside the stimulated zone there would be a net analgesic effect (see Le Bars 2002 for review).
The neural basis for DNIC involves a feedback loop through a region of the caudal medulla, the subnucleus reticularis dorsalis (SRD, Villanueva et al 1996). The SRD (also referred to as the dorsal reticular nucleus) receives nociceptive input from the spinal dorsal horn and is necessary for inhibition of dorsal horn neurons by noxious stimulation of a remote body part. The PAG–RVM system had originally been hypothesized to be the efferent loop in the negative feedback circuit mediating DNIC, but lesions of the PAG or nucleus raphe magnus were ultimately found to be without effect on DNIC. SRD neurons are excited by noxious stimuli over the entire body surface. Other afferents to the SRD include the PAG and RVM, as well as the brain stem reticular formation, hypothalamus, central nucleus of the amygdala, and broad regions of the cortex, including the orbital cortex, insula, and somatosensory and motor cortex. The SRD also projects to several thalamic nuclei as part of the spinoreticular–thalamic pathway, thus suggesting a role in nociceptive transmission, as well as in descending control (see Monconduit et al 2002 for review).
In contrast to the above evidence that the SRD is a major link in a spino–bulbo–spinal inhibitory network, other data suggest that this nucleus is a source of descending facilitatory control (Lima and Almeida 2002). There are direct synaptic contacts of SRD axons on lamina I neurons, which in turn project to the SRD, thereby providing a reciprocal circuit. Lesions of the SRD result in hypoalgesia for thermal pain and for the formalin test. Formalin-evoked Fos expression is also reduced in laminae I and IV–V after lesions of SRD. Conversely, activation of SRD neurons by local microinjection of glutamate shortens tail flick latencies. These data point to the SRD as a source of descending facilitation, particularly for neurons in the superficial dorsal horn, and furthermore, that the facilitation is part of a positive feedback loop.
The recognition that multiple descending pathways modulate spinal nociceptive processing addresses the apparent inconsistency between the idea that pain inhibits pain and the extensive evidence discussed earlier that the PAG–RVM system contributes to the enhanced response in a variety of inflammation and injury models. Whether the response to a given noxious stimulus is enhanced or suppressed will depend on stimulus location and duration, the environment in which the stimulus is applied, and the behavioral state of the animal. Some nocifensor reflexes are inhibited, whereas others are facilitated by heterotopic or homotopic stimulation (Morgan and Fields 1994). In anesthetized rats, the diffuse inhibitory effect on wide–dynamic range neurons produced by noxious somatic stimuli appears to interact with the potential facilitatory action on the dorsal horn by the RVM ON-cells that are activated by the same stimulus (Hernandez et al 1994).
Importantly, a DNIC-like process in which pain sensation evoked by stimulation at one site is reduced by concurrent noxious stimulation at a second site is well documented in humans. This is suggested to be a possible basis for acupuncture and many other traditional pain therapies based on counterirritation. However, remote inhibition in humans may not involve the SRD and surround inhibition of dorsal horn neurons. Indeed, at least one imaging study suggests that the conditioned pain modulation observed in humans involves the PAG–RVM system (Wilder-Smith et al 2004). This modulation of sensory experience in humans is therefore now referred to as “conditioned pain modulation” rather than DNIC. The latter term emphasizes the sensory effect of the “conditioning” (remote) stimulus on the pain evoked by the “test” stimulus (Yarnitsky et al 2010). A host of studies now document a relative deficit in conditioned pain modulation in a diverse range of clinical pain populations (see van Wijk and Veldhuijzen 2010, Yarnitsky 2010 for reviews). Even more interesting is the suggestion that individuals vary in their inherent capacity for conditioned pain modulation and evidence that after a transient insult, prolonged pain is more likely to develop in those with less potent conditioned modulation (Yarnitsky et al 2008). Further studies will clearly be needed to fully understand the relationship between conditioned pain modulation in humans and DNIC as measured in the dorsal horn.
Hyperalgesia and the Sickness Response
The sickness response to injury or infection has classically been viewed as consisting of fever, activation of the hypothalamic–pituitary–adrenal axis, anorexia, inhibition of exploratory behavior, and increased sleep (Dantzer et al 1998, Kelley et al 2003). Nociceptive responses are also altered as part of the sickness response. Thus, systemic administration of bacterial endotoxin and other sickness-inducing agents produces a hyperalgesic state. This centrally mediated hyperalgesia has been noted on a range of pain tests, including tail flick, hot plate, and formalin tests. Watkins and colleagues (1995) proposed that this increased nociceptive sensitivity has an adaptive role in the sickness response in that it would encourage rest and recuperative behavior.
Sickness-induced hyperalgesia, like other components of the sickness response, is mediated at least in part by the cytokine interleukin-1β (IL-1β). Thus sickness induced by the administration of endotoxin is reversed by IL-1 receptor antagonist, a naturally occurring competitive antagonist of IL-1β, and mimicked by systemic administration of IL-1β (Watkins et al 1995). Importantly, direct administration of IL-1β within the brain alters nociceptive responding. Although the literature is not entirely consistent regarding the behavioral profile of intracerebroventricular IL-1β (Oka and Hori 1999), focal application of IL-1β into specific brain regions points to the preoptic region as a potential site of action (Oka et al 1995). This is of particular interest when considering brain stem modulatory systems because the medial preoptic area projects to both the PAG and RVM (Murphy et al 1999) and the RVM is required for endotoxin-induced hyperalgesia (Wiertelak et al 1997). Thus brain stem facilitatory influences, probably recruited via release of a prostanoid in the medial preoptic area (Heinricher et al 2004), appear to have a major role in the hyperalgesia observed following immune system activation by cytokines.
Defense: Threat versus Safety
In unanesthetized animals, situations that are threatening, such as an inescapable noxious stimulus, the presence of a predator, or contextual cues associated with intense or prolonged noxious stimuli, produce robust antinociceptive effects. Stress-induced analgesia can also be demonstrated in humans, thus indicating that the antinociception seen in animals is not merely hyporeflexia (Flor et al 2002, Rhudy and Meagher 2003, Yilmaz et al 2010). The behavioral antinociceptive effects involve the PAG–RVM network. For example, Watkins, Mayer, and colleagues showed that analgesia following shock to the forepaws is completely blocked by naloxone and by RVM lesions (Watkins and Mayer 1982, Watkins et al 1983). This analgesia can be conditioned to a neutral stimulus such as a light or tone contingently paired with foot shock or to the environment in which the foot shock is received. This conditioned analgesia is blocked by lesions or administration of opioid antagonists into either the PAG or RVM (Watkins et al 1982a, 1982b, Helmstetter and Landeira-Fernandez 1990, Helmstetter and Tershner 1994, Foo and Helmstetter 1999). Conditioned fear recruits the PAG–RVM system via the amygdala (Helmstetter 1992, Helmstetter and Bellgowan 1993, 1994).
It is also possible to condition an anti-analgesic effect (Watkins et al 1998). When rats exposed to inescapable shock in a defined context receive a light signal at the termination of the shock, that light becomes a safety signal. When the rats are returned to the context in which they received the shock, illuminating the light prevents the expression of conditioned fear behavior, including analgesia (Wiertelak et al 1992). Furthermore, the safety signal has a potent and generalized anti-opioid effect; it interferes with the analgesic effect of systemic or spinal morphine. Lesions of the midbrain dorsal raphe nucleus and the RVM prevent this anti-analgesic effect. Finally, spinal intrathecal injection of a CCK antagonist restores the potency of spinal morphine, which indicates that the anti-opioid effect of the safety signal is mediated by the release of CCK.
CCK also plays a role in the phenomenon of “associative opioid tolerance.” Siegel (1976) observed that there is a form of morphine tolerance that is specific to a particular context. That is, when morphine is given repeatedly and selectively (but not continuously) in a single context with salient cues, it gradually loses its antinociceptive potency. However, the same dose of morphine given in a novel context is fully effective. Loss of potency (tolerance) is therefore manifested only in the morphine-associated context. Systemic administration of a selective CCK2 receptor antagonist fully restores morphine’s analgesic potency (Mitchell et al 2000). The expression of associative morphine tolerance is correlated with an increase in Fos expression in basolateral amygdala neurons and is prevented by local microinjection of a CCK2 antagonist into the basolateral amygdala.
These experiments on conditioned analgesia, safety signal–induced anti-analgesia, and associative morphine tolerance link the modulation of pain to specific, biologically relevant environmental conditions and to well-defined neural networks and neurotransmitter systems. Indeed, pain modulation probably represents a more general behavioral function that implements action selection or decision making. That is, normal responses to a painful stimulus may conflict with other motivated behavior. Suppression of nociceptive behavior and pain sensation in the presence of a severe and immediate threat is well documented. However, less dire but nonetheless high-priority motivated behavior can also interact with pain. Feeding, for example, has long been recognized to compete with pain responses; if hunger dominates, pain responses are inhibited, and vice versa (Casey and Morrow 1983, LaGraize et al 2004). If the available food is highly palatable, the analgesic effect of anticipated feeding in rats is powerful and can be blocked by the opioid antagonist naloxone (Dum and Herz 1984). A similar competing interaction occurs when an animal has the urge to urinate. Micturition and pain behavior compete, presumably because the necessary motor patterns are mutually exclusive (Baez et al 2005). Importantly, a subset of RVM neurons are excited when a rat micturates but stop firing when micturition is inhibited and the animal instead responds to a noxious stimulus. Taken as a whole, these studies suggest that a major function of the PAG–RVM system is to implement behavioral decisions about whether to respond to a noxious stimulus.
This idea that action selection/decision making is a critical impetus for modulating pain perception is supported by studies of the ventral striatum in rodents and humans. This region, usually referred to as the nucleus accumbens (NAc), connects brain regions important for motivational processing with motor systems (Mogenson et al 1980). The NAc also has the potential to recruit the PAG–RVM system through its connections with the hypothalamus and amygdala. Although the NAc has typically been thought of as encoding signals of reward (Carlezon and Thomas 2009), human functional imaging studies have shown that the NAc is activated at short latency by noxious stimulation (Becerra et al 2001, Baliki et al 2010). Significantly, microinjection of opioids into the NAc both promotes the consumption of palatable food and suppresses responding to noxious stimulation (Will et al 2006, Gear and Levine 2011). This finding implicates the NAc in the response decision when reward-seeking and pain escape motivations compete.
The PAG–RVM System In Humans
Humans have pain-modulating circuitry homologous to the PAG–RVM system outlined in animals, a conclusion based on several lines of evidence. First, this brain stem–to–spinal cord circuitry is conserved in a variety of mammalian species, including rodents, carnivores, primates, and marsupials. Importantly, the distribution of neurotransmitters, specifically opioid peptides, in this pathway is also conserved (Emson et al 1984, Pittius et al 1984). Furthermore, the same μ-opioid receptor agonists that inhibit withdrawal and escape behavior in animals through the PAG–RVM system relieve clinically significant pain in patients. In addition, as already discussed, the idea that homologous circuitry can produce analgesia in humans is strongly supported by the observation that patients with chronic pain report relief during stimulation of the PAG region. This observation in humans represents a critical extension of animal work because it supports the idea that subjective pain relief accompanies the behavioral antinociception produced by midbrain stimulation in animals.
Functional imaging studies now document that the PAG and/or RVM is engaged in paradigms representing facilitated pain states, as well as during pharmacological and endogenous analgesia (see Bingel and Tracey 2008 for review). Analyses of placebo analgesia are particularly intriguing in showing a naloxone-sensitive functional coupling between the rostral anterior cingulate cortex and the PAG, as well as a decrease in spinal activation during placebo analgesia (Eippert et al 2009a, 2009b). There are also significant pharmacological parallels between placebo analgesia and operation of the PAG–RVM system, with attenuation by naloxone and enhancement by a CCK antagonist (Levine et al 1978, Benedetti 1996). It is also now recognized that placebo recruits descending controls to influence spinal nociceptive processing (Matre et al 2006).
Taken as a whole, these data provide the critical bridge between the human sensory experience and the pain modulatory circuitry studied so extensively in animals.
Attention
That attentional factors can reliably alter perceived pain intensity is well established (Villemure and Bushnell 2002). In a typical experimental paradigm, human subjects are trained in a task to make either a visual or a noxious thermal discrimination. Attending to the painful stimulus generally increases its perceived intensity, and distraction largely reduces it.
There is evidence that attentional modulation of pain involves descending pathways. In monkeys, the responses of some nociceptive dorsal horn neurons to a noxious thermal stimulus were found to be greater when the monkey was required to discriminate the intensity of the stimulus than when required to carry out a distracting visual task (Hayes et al 1981). Furthermore, under conditions of correct cueing of a thermal discrimination task, monkeys displayed shorter response latencies for signaling their detection of the increase in skin temperature. From this it was inferred that they perceive the stimulus as more intense (Bushnell et al 1985). These studies suggest that attentional shifts lead to a centrally generated component in the response of dorsal horn neurons to noxious thermal stimulation and that this centrally generated increment alters the perceived intensity of pain.
Although it is uncertain what neural circuits are involved in the attentional modulation of dorsal horn neurons, human functional imaging studies implicate the PAG–RVM system. Tracey and colleagues (2002) studied brain stem regions during a task in which normal human subjects rated thermal pain in the presence or absence of a visual distractor. During distraction, pain intensities were rated as significantly lower, and there was a concomitant increase in activity in the PAG. In an extension of this work, Bantick and co-workers (2002) studied subjective pain reports and forebrain functional magnetic resonance imaging signals when noxious stimuli were applied with or without a distraction task. Pain reaction times were increased and intensity ratings were predictably lowered during distraction. This was associated with activation of the rostral anterior cingulate cortex during distraction and a decrease in other areas, including the contralateral thalamus, insular cortex, and mid-cingulate region (an area activated by noxious stimuli). The authors attributed this pattern of reduced activation during distraction to reduced input via nociceptor-activated sensory pathways. These imaging results indicate that both the PAG and the anterior cingulate cortex are activated when pain intensity is reduced by distraction and point to a possible mechanism for attentional regulation of afferent pathways.
Expectations and Pain-Predictive Cues: Anterior Cingulate and Anterior Insular Cortex (See Chapter 27)
With learning, neutral contextual cues acquire the power to either increase or decrease the activity of nociceptive dorsal horn neurons in the absence of a noxious stimulus. In situations in which subjects are conditioned with specific cues that predict either painful or neutral stimuli, the cues come to have a significant effect on the pain experience (Sawamoto et al 2000, Ploghaus et al 2003). When pain-predictive cues are presented just before non-painful warm stimuli, human subjects are more likely to report the warm stimuli as being painful. In these same subjects, concomitant functional imaging demonstrates activation of the rostral anterior cingulate cortex and anterior insular cortex by cues predicting pain. Consistent with the functional imaging data, single-unit recordings in awake primates have revealed activation of a population of anterior cingulate cortex neurons specifically during pain avoidance behavior (Koyama et al 2001), and lesions of the rostral anterior cingulate cortex prevent the development of pain avoidance behavior (e.g., Johansen et al 2001). These data tie the human sensory experience to the pain modulatory circuitry studied so extensively in animals.
Non-human primate studies indicate that at least part of the expectancy-related enhancement of pain perception is due to activation of descending pathways that alter nociceptive processing in the dorsal horn. Thus, Duncan and colleagues (1987) recorded the activity of nociceptive trigeminal dorsal horn neurons, including some that project to the thalamus. The primates were trained to press a button in response to a light cue. Pressing the button initiated a trial during which the animal had to discriminate between two noxious thermal stimuli. The activity of some neurons displayed abrupt increases or decreases that were time-locked to the light cue or the button press. Importantly, these task-related changes in activity occurred before onset of the thermal stimulus. These experiments show that the activity of dorsal horn nociceptive neurons can be increased or decreased by a context-specific modulatory signal that originates in the CNS. The presence of facilitatory modulation of dorsal horn nociresponsive neurons raises the intriguing possibility that pain can be produced by a centrally originating drive of dorsal horn nociceptive neurons, without activation of primary afferent nociceptors.
Can we link the evidence implicating the rostral anterior cingulate cortex and the anterior insula in expectancy-related modulation with the changes observed in the dorsal horn? There are several possible pathways through which cortical regions activated during expectancy could reach brain stem pain-modulating nuclei. These pathways include direct projections from the anterior cingulate cortex to the PAG (An et al 1998) and direct anterior insular connections to the RVM and pontine noradrenergic cell groups (Jasmin et al 2004). There is also a dense rostral anterior cingulate cortex projection to the NAc (Knishes and Haber 1994), which in turn projects to the hypothalamus and to the basolateral amygdala. Both of the latter have connections with PAG and RVM. Further research is needed to establish that expectancy-related cortical activation modulates pain through descending influences on dorsal horn nociceptive neurons. However, as described earlier, we know that the PAG–RVM system is engaged during placebo analgesia. We also know that placebo analgesia is mediated primarily by expectancy (see Chapter 27). These two lines of evidence strongly suggest that the PAG–RVM system mediates alterations in pain as a result of expectancy.
Conclusion
The discovery of pain modulatory systems, the behavioral triggers and somatosensory stimuli that activate them, and the neurotransmitters that mediate their actions has provided us with a preliminary mechanistic understanding of the variability of pain. This greater understanding offers the promise of rationally developed treatments based on the manipulation of psychological variables, on physical methods, and on new, more selective drugs or drug combinations. These improvements in treatment will go hand in hand with progress in elucidating the neural circuits involved in pain modulation.
The references for this chapter can be found at www.expertconsult.com.
References
Ackley M.A., Hurley R.W., Virnich D.E., et al. A cellular mechanism for the antinociceptive effect of a kappa opioid receptor agonist. Pain. 2001;91:377–388.
Aggleton J.P., ed. The amygdala: neurobiological aspects of emotion, memory and mental dysfunction. New York: Wiley-Liss, 1992.
Alhaider A.A., Lei S.Z., Wilcox G.L. Spinal 5-HT3 receptor–mediated antinociception: possible release of GABA. Journal of Neuroscience. 1991;11:1881–1888.
Ambriz-Tututi M., Cruz S.L., Urquiza-Marin H., et al. Formalin-induced long-term secondary allodynia and hyperalgesia are maintained by descending facilitation. Pharmacology, Biochemistry, and Behavior. 2011;98:417–424.
An X., Bandler R., Ongur D., et al. Prefrontal cortical projections to longitudinal columns in the midbrain periaqueductal gray in macaque monkeys. Journal of Comparative Neurology 1998. 1998;401:455–479.
Baez M.A., Brink T.S., Mason P. Roles for pain modulatory cells during micturition and continence. Journal of Neuroscience. 2005;25:384–394.
Bajic D., Proudfit H.K. Projections of neurons in the periaqueductal gray to pontine and medullary catecholamine cell groups involved in the modulation of nociception. Journal of Comparative Neurology. 1999;405:359–379.
Bajic D., Proudfit H.K., Van Bockstaele E.J. Periaqueductal gray neurons monosynaptically innervate extranuclear noradrenergic dendrites in the rat pericoerulear region. Journal of Comparative Neurology. 2000;427:649–662.
Baliki M.N., Geha P.Y., Fields H.L., et al. Predicting value of pain and analgesia: nucleus accumbens response to noxious stimuli changes in the presence of chronic pain. Neuron. 2010;66:149–160.
Bandler R., Keay K.A. Columnar organization in the midbrain periaqueductal gray and the integration of emotional expression. Progress in Brain Research. 1996;107:285–300.
Bannon A.W., Decker M.W., Holladay M.W., et al. Broad-spectrum, non-opioid analgesic activity by selective modulation of neuronal nicotinic acetylcholine receptors. Science. 1998;279:77–81.
Bantick S.J., Wise R.G., Ploghaus A., et al. Imaging how attention modulates pain in humans using functional MRI. Brain. 2002;125:310–319.
Barbaro N.M., Hammond D.L., Fields H.L. Effects of intrathecally administered methysergide and yohimbine on microstimulation-produced antinociception in the rat. Brain Research. 1985;343:223–229.
Barbaro N.M., Heinricher M.M., Fields H.L. Putative pain modulating neurons in the rostral ventral medulla: reflex-related activity predicts effects of morphine. Brain Research. 1986;366:203–210.
Barbaro N.M., Heinricher M.M., Fields H.L. Putative nociceptive modulatory neurons in the rostral ventromedial medulla of the rat display highly correlated firing patterns. Somatosensory & Motor Research. 1989;6:413–425.
Becerra L., Breiter H.C., Wise R., et al. Reward circuitry activation by noxious thermal stimuli. Neuron. 2001;32:927–946.
Bederson J.B., Fields H.L., Barbaro N.M. Hyperalgesia during naloxone-precipitated withdrawal from morphine is associated with increased on-cell activity in the rostral ventromedial medulla. Somatosensory & Motor Research. 1990;7:185–203.
Beecher H.K. Relationship of significance of wound to pain experienced. Journal of the American Medical Association. 1959;161:1609–1613.
Beitz A.J. The nuclei of origin of brain stem enkephalin and substance P projections to the rodent nucleus raphe magnus. Neuroscience. 1982;7:2753–2768.
Beitz A.J. The organization of afferent projections to the midbrain periaqueductal gray of the rat. Neuroscience. 1982;7:133–159.
Beitz A.J. The sites of origin of brain stem neurotensin and serotonin projections to the rodent nucleus raphe magnus. Journal of Neuroscience. 1982;2:829–842.
Bellgowan P.S.F., Helmstetter F.J. The role of mu and kappa opioid receptors within the periaqueductal gray in the expression of conditional hypoalgesia. Brain Research. 1998;791:83–89.
Benedetti F. The opposite effects of the opiate antagonist naloxone and the cholecystokinin antagonist proglumide on placebo analgesia. Pain. 1996;64:535–543.
Bernard J., Sevoz-Couche C., Hamon M., et al. Responses of lateral paragigantocellular and raphe magnus serotonergic neurons to noxious stimuli: a comparative reappraisal using juxtacellular recording. Montreal, Canada: IASP World Congress; 2010. PW205
Bie B., Pan Z.Z. Presynaptic mechanism for anti-analgesic and anti-hyperalgesic actions of kappa-opioid receptors. Journal of Neuroscience. 2003;23:7262–7268.
Bingel U., Tracey I. Imaging CNS modulation of pain in humans. Physiology. 2008;23:371–380.
Bitner R.S., Nikkel A.L., Curzon P., et al. Role of the nucleus raphe magnus in antinociception produced by ABT-594: immediate early gene responses possibly linked to neuronal nicotinic acetylcholine receptors on serotonergic neurons. Journal of Neuroscience. 1998;18:5426–5432.
Boivie J., Meyerson B.A. A correlative anatomical and clinical study of pain suppression by deep brain stimulation. Pain. 1982;13:113–126.
Borszcz G.S., Johnson C.P., Thorp M.V. The differential contribution of spinopetal projections to increases in vocalization and motor reflex thresholds generated by the microinjection of morphine into the periaqueductal gray. Behavioral Neuroscience. 1996;110:368–388.
Bowker R.M. The relationship between descending serotonin projections and ascending projections in the nucleus raphe magnus: a double labeling study. Neuroscience Letters. 1986;70:348–353.
Brightwell J.J., Taylor B.K. Noradrenergic neurons in the locus coeruleus contribute to neuropathic pain. Neuroscience. 2009;160:174–185.
Brink T.S., Mason P. Raphe magnus neurons respond to noxious colorectal distension. Journal of Neurophysiology. 2003;89:2506–2515.
Budai D., Harasawa I., Fields H.L. Midbrain periaqueductal gray (PAG) inhibits nociceptive inputs to sacral dorsal horn nociceptive neurons through alpha2-adrenergic receptors. Journal of Neurophysiology. 1998;80:2244–2254.
Budai D., Khasabov S.G., Mantyh P.W., et al. NK-1 receptors modulate the excitability of ON cells in the rostral ventromedial medulla. Journal of Neurophysiology. 2007;97:1388–1395.
Buhler A.V., Choi J., Proudfit H.K., et al. Neurotensin activation of the NTR1 on spinally-projecting serotonergic neurons in the rostral ventromedial medulla is antinociceptive. Pain. 2005;114:285–294.
Buhler A.V., Proudfit H.K., Gebhart G.F. Neurotensin-produced antinociception in the rostral ventromedial medulla is partially mediated by spinal cord norepinephrine. Pain. 2008;135:280–290.
Burstein R., Potrebic S. Retrograde labeling of neurons in the spinal cord that project directly to the amygdala or the orbital cortex in the rat. Journal of Comparative Neurology. 1993;335:469–485.
Bushnell M.C., Duncan G.H., Dubner R., et al. Attentional influences on noxious and innocuous cutaneous heat detection in humans and monkeys. Journal of Neuroscience. 1985;5:1103–1110.
Cameron A.A., Khan I.A., Westlund K.N., et al. The efferent projections of the periaqueductal gray in the rat: a Phaseolus vulgaris-leucoagglutinin study. I. Ascending projections. Journal of Comparative Neurology. 1995;351:568–584.
Carlezon W.A., Jr., Thomas M.J. Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology. 2009;56(suppl 1):122–132.
Carlson J.D., Maire J.J., Martenson M.E., et al. Sensitization of pain-modulating neurons in the rostral ventromedial medulla after peripheral nerve injury. Journal of Neuroscience. 2007;27:13222–13231.
Carlson J.D., Selden N.R., Heinricher M.M. Nocifensive reflex-related on- and off-cells in the pedunculopontine tegmental nucleus, cuneiform nucleus, and lateral dorsal tegmental nucleus. Brain Research. 2005;1063:187–194.
Carpenter D., Engberg I., Lundberg A. Differential supraspinal control of inhibitory and excitatory actions from the FRA to ascending spinal pathways. Acta Physiolica Scandinavica. 1965;63:103–110.
Casey K.L., Morrow T.J. Nocifensive responses to cutaneous thermal stimuli in the cat: stimulus-response profiles, latencies, and afferent activity. Journal of Neurophysiology. 1983;50:1497–1515.
Chieng B., Christie M.J. Hyperpolarization by opioids acting on mu-receptors of a sub-population of rat periaqueductal gray neurones in vitro. British Journal of Pharmacology. 1994;113:121–128.
Clark F.M., Proudfit H.K. Projections of neurons in the ventromedial medulla to pontine catecholamine cell groups involved in the modulation of nociception. Brain Research. 1991;540:105–115.
Cleary D.R., Neubert M.J., Heinricher M.M. Are opioid-sensitive neurons in the rostral ventromedial medulla inhibitory interneurons? Neuroscience. 2008;151:564–571.
Coffield J.A., Bowen K.K., Miletic V. Retrograde tracing of projections between the nucleus submedius, the ventrolateral orbital cortex, and the midbrain in the rat. Journal of Comparative Neurology. 1992;321:488–499.
Colloca L., Benedetti F. Nocebo hyperalgesia: how anxiety is turned into pain. Current Opinion in Anaesthesiology. 2007;20:435–439.
Cox C.L., Huguenard J.R., Prince D.A. Cholecystokinin depolarizes rat thalamic reticular neurons by suppressing a K+ conductance. Journal of Neurophysiology. 1995;74:990–1000.
Crawley J.N. Cholecystokinin-dopamine interactions. Trends in Pharmacological Sciences. 1991;12:232–236.
Da Silva L.F., Coutinho M.R., Menescal-de-Oliveira L. Opioidergic and GABAergic mechanisms in the rostral ventromedial medulla modulate the nociceptive response of vocalization in guinea pigs. Brain Research Bulletin. 2010;82:177–183.
Da Silva L.F., DeSantana J.M., Sluka K.A. Activation of NMDA receptors in the brainstem, rostral ventromedial medulla, and nucleus reticularis gigantocellularis mediates mechanical hyperalgesia produced by repeated intramuscular injections of acidic saline in rats. Journal of Pain. 2010;11:378–387.
Da Silva L.F., Walder R.Y., Davidson B.L., et al. Changes in expression of NMDA-nr1 receptor subunits in the rostral ventromedial medulla modulate pain behaviors. Pain. 2010;151:155–161.
Daniels D.J., Lenard N.R., Etienne C.L., et al. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:19208–19213.
Dantzer R., Bluthe R.M., Laye S., et al. Cytokines and sickness behavior. Annals of the New York Academy of Sciences. 1998;840:586–590.
Darland T., Heinricher M.M., Grandy D.K. Orphanin FQ/nociceptin: a role in pain and analgesia, but so much more. Trends in Neurosciences. 1998;21:215–221.
Decker M.W., Curzon P., Holladay M.W., et al. The role of neuronal nicotinic acetylcholine receptors in antinociception: effects of ABT-594. Journal of Physiology, Paris. 1998;92:221–224.
Dogrul A., Ossipov M.H., Porreca F. Differential mediation of descending pain facilitation and inhibition by spinal 5HT-3 and 5HT-7 receptors. Brain Research. 2009;1280:52–59.
Dum J., Herz A. Endorphinergic modulation of neural reward systems indicated by behavioral changes. Pharmacology, Biochemistry, and Behavior. 1984;21:259–266.
Duncan G.H., Bushnell M.C., Bates R., et al. Task-related responses of monkey medullary dorsal horn neurons. Journal of Neurophysiology. 1987;57:289–310.
Edelmayer R.M., Vanderah T.W., Majuta L., et al. Medullary pain facilitating neurons mediate allodynia in headache-related pain. Annals of Neurology. 2009;65:184–193.
Eippert F., Bingel U., Schoell E.D., et al. Activation of the opioidergic descending pain control system underlies placebo analgesia. Neuron. 2009;63:533–543.
Eippert F., Finsterbusch J., Bingel U., et al. Direct evidence for spinal cord involvement in placebo analgesia. Science. 2009;326:404.
Eisenach J.C., De Kock M., Klimscha W. α2-Adrenergic agonists for regional anesthesia. A clinical review of clonidine (1984-1995). Anesthesiology. 1996;85:655–674.
Emson P.C., Corder R., Ratter S.J., et al. Regional distribution of pro-opiomelanocortin–derived peptides in the human brain. Neuroendocrinology. 1984;38:45–50.
Fields H.L., Basbaum A.I. Brainstem control of spinal pain-transmission neurons. Annual Review of Physiology. 1978;40:217–248.
Fields H.L., Heinricher M.M. Anatomy and physiology of a nociceptive modulatory system. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 1985;308:361–374.
Fields H.L., Heinricher M.M., Mason P. Neurotransmitters in nociceptive modulatory circuits. Annual Review of Neuroscience. 1991;14:219–245.
Fields H.L., Levine J.D. Placebo analgesia—a role for endorphins? Trends in Neurosciences. 1984;7:271–273.
Fields H.L., Malick A., Burstein R. Dorsal horn projection targets of ON and OFF cells in the rostral ventromedial medulla. Journal of Neurophysiology. 1995;74:1742–1759.
Flor H., Birbaumer N., Schulz R., et al. Pavlovian conditioning of opioid and nonopioid pain inhibitory mechanisms in humans. European Journal of Pain. 2002;6:395–402.
Floyd N.S., Price J.L., Ferry A.T., et al. Orbitomedial prefrontal cortical projections to distinct longitudinal columns of the periaqueductal gray in the rat. Journal of Comparative Neurology. 2000;422:556–578.
Foo H., Helmstetter F.J. Hypoalgesia elicited by a conditioned stimulus is blocked by a mu, but not a delta or a kappa, opioid antagonist injected into the rostral ventromedial medulla. Pain. 1999;83:427–431.
Foo H., Helmstetter F.J. Activation of kappa opioid receptors in the rostral ventromedial medulla blocks stress-induced antinociception. Neuroreport. 2000;11:3349–3352.
Foo H., Mason P. Discharge of raphe magnus ON and OFF cells is predictive of the motor facilitation evoked by repeated laser stimulation. Journal of Neuroscience. 2003;23:1933–1940.
Gauriau C., Bernard J.F. Pain pathways and parabrachial circuits in the rat. Experimental Physiology. 2002;87:251–258.
Gauriau C., Bernard J.F. A comparative reappraisal of projections from the superficial laminae of the dorsal horn in the rat: the forebrain. Journal of Comparative Neurology. 2004;468:24–56.
Gear R.W., Levine J.D. Nucleus accumbens facilitates nociception. Experimental Neurology. 2011;229:502–506.
Gehani N.C., Nalwalk J.W., Razdan R.K., et al. Significance of cannabinoid CB1 receptors in improgan antinociception. Journal of Pain. 2007;8:850–860.
Guan Y., Guo W., Robbins M.T., et al. Changes in AMPA receptor phosphorylation in the rostral ventromedial medulla after inflammatory hyperalgesia in rats. Neuroscience Letters. 2004;366:201–205.
Guan Y., Terayama R., Dubner R., et al. Plasticity in excitatory amino acid receptor–mediated descending pain modulation after inflammation. Journal of Pharmacology and Experimental Therapeutics. 2002;300:513–520.
Guo W., Robbins M.T., Wei F., et al. Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for descending pain facilitation. Journal of Neuroscience. 2006;26:126–137.
Hagbarth K.E., Kerr D.I. Central influences on spinal afferent conduction. Journal of Neurophysiology. 1954;17:295–307.
Hahm E.T., Hammond D.L., Proudfit H.K. Substance P induces the reversible formation of varicosities in the dendrites of rat brainstem neurons. Brain Research. 2011;1369:36–45.
Hamity M.V., White S.R., Hammond D.L. Effects of neurokinin-1 receptor agonism and antagonism in the rostral ventromedial medulla of rats with acute or persistent inflammatory nociception. Neuroscience. 2010;165:902–913.
Hammond D.L., Tyce G.M., Yaksh T.L. Efflux of 5-hydroxytryptamine and noradrenaline into spinal cord superfusates during stimulation of the rat medulla. Journal of Physiology. 1985;359:151–162.
Haws C.M., Williamson A.M., Fields H.L. Putative nociceptive modulatory neurons in the dorsolateral pontomesencephalic reticular formation. Brain Research. 1989;483:272–282.
Hayes R.L., Dubner R., Hoffman D.S. Neuronal activity in medullary dorsal horn of awake monkeys trained in a thermal discrimination task. II. Behavioral modulation of responses to thermal and mechanical stimuli. Journal of Neurophysiology. 1981;46:428–443.
Head H., Holmes G. Sensory disturbances from cerebral lesions. Brain. 1911;34:102–254.
Headley P.M., Duggan A.W., Griersmith B.T. Selective reduction by noradrenaline and 5-hydroxytryptamine of nociceptive responses of cat dorsal horn neurones. Brain Research. 1978;145:185–189.
Heinricher M.M. Orphanin FQ/nociceptin: from neural circuitry to behavior. Life Sciences. 2003;73:813–822.
Heinricher M.M., Barbaro N.M., Fields H.L. Putative nociceptive modulating neurons in the rostral ventromedial medulla of the rat: firing of on- and off-cells is related to nociceptive responsiveness. Somatosensory & Motor Research. 1989;6:427–439.
Heinricher M.M., Cheng Z.F., Fields H.L. Evidence for two classes of nociceptive modulating neurons in the periaqueductal gray. Journal of Neuroscience. 1987;7:271–278.
Heinricher M.M., Fields H.L. The delta opioid receptor and brain pain-modulating circuits. In: Chang K.J., Porreca F., Woods J., eds. The delta receptor. New York: Marcel Dekker; 2003:467–480.
Heinricher M.M., Ingram S.L., The brainstem and nociceptive modulation. Bushnell M.C., Basbaum A.I., eds. The senses, a comprehensive reference, Vol 5. San Diego, CA: Pain. Academic Press; 2008:593–626.
Heinricher M.M., Maire J.J., Lee D., et al. Physiological basis for inhibition of morphine and improgan antinociception by CC12, a P450 epoxygenase inhibitor. Journal of Neurophysiology. 2010;104:3222–3230.
Heinricher M.M., McGaraughty S. Analysis of excitatory amino acid transmission within the rostral ventromedial medulla: implications for circuitry. Pain. 1998;75:247–255.
Heinricher M.M., McGaraughty S., Farr D.A. The role of excitatory amino acid transmission within the rostral ventromedial medulla in the antinociceptive actions of systemically administered morphine. Pain. 1999;81:57–65.
Heinricher M.M., McGaraughty S., Grandy D.K. Circuitry underlying antiopioid actions of orphanin FQ in the rostral ventromedial medulla. Journal of Neurophysiology. 1997;78:3351–3358.
Heinricher M.M., McGaraughty S., Tortorici V. Circuitry underlying antiopioid actions of cholecystokinin within the rostral ventromedial medulla. Journal of Neurophysiology. 2001;85:280–286.
Heinricher M.M., Morgan M.M., Fields H.L. Direct and indirect actions of morphine on medullary neurons that modulate nociception. Neuroscience. 1992;48:533–543.
Heinricher M.M., Morgan M.M., Tortorici V., et al. Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience. 1994;63:279–288.
Heinricher M.M., Neubert M.J. Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. Journal of Neurophysiology. 2004;92:1982–1989.
Heinricher M.M., Neubert M.J., Martenson M.E., et al. Prostaglandin E2 in the medial preoptic area produces hyperalgesia and activates pain-modulating circuitry in the rostral ventromedial medulla. Neuroscience. 2004;128:389–398.
Heinricher M.M., Pertovaara A., Ossipov M.H., Descending modulation after injury. Dostrovsky J.O., Carr D.B., Koltzenburg M., eds. Progress in pain research and management, Vol 24. Seattle: IASP Press; 2003:251–260.
Heinricher M.M., Schouten J.C., Jobst E.E. Activation of brainstem N-methyl-D-aspartate receptors is required for the analgesic actions of morphine given systemically. Pain. 2001;92:129–138.
Heinricher M.M., Tavares I., Leith J.L., et al. Descending control of nociception: specificity, recruitment and plasticity. Brain Research Reviews. 2009;60:214–225.
Helmstetter F.J. The amygdala is essential for the expression of conditional hypoalgesia. Behavioral Neuroscience. 1992;106:518–528.
Helmstetter F.J., Bellgowan P.S. Lesions of the amygdala block conditional hypoalgesia on the tail flick test. Brain Research. 1993;612:253–257.
Helmstetter F.J., Bellgowan P.S. Effects of muscimol applied to the basolateral amygdala on acquisition and expression of contextual fear conditioning in rats. Behavioral Neuroscience. 1994;108:1005–1009.
Helmstetter F.J., Landeira-Fernandez J. Conditional hypoalgesia is attenuated by naltrexone applied to the periaqueductal gray. Brain Research. 1990;537:88–92.
Helmstetter F.J., Tershner S.A. Lesions of the periaqueductal gray and rostral ventromedial medulla disrupt antinociceptive but not cardiovascular aversive conditional responses. Journal of Neuroscience. 1994;14:7099–7108.
Helmstetter F.J., Tershner S.A., Poore L.H., et al. Antinociception following opioid stimulation of the basolateral amygdala is expressed through the periaqueductal gray and rostral ventromedial medulla. Brain Research. 1998;779:104–118.
Herbert H., Saper C.B. Organization of medullary adrenergic and noradrenergic projections to the periaqueductal gray matter in the rat. Journal of Comparative Neurology. 1992;315:34–52.
Hermann D.M., Luppi P.H., Peyron C., et al. Forebrain projections of the rostral nucleus raphe magnus shown by iontophoretic application of choleratoxin b in rats. Neuroscience Letters. 1996;216:151–154.
Hermann D.M., Luppi P.H., Peyron C., et al. Afferent projections to the rat nuclei raphe magnus, raphe pallidus and reticularis gigantocellularis pars alpha demonstrated by iontophoretic application of choleratoxin (subunit b). Journal of Chemical Neuroanatomy. 1997;13:1–21.
Hernandez N., Dmitrieva N., Vanegas H. Medullary on-cell activity during tail-flick inhibition produced by heterotopic noxious stimulation. Pain. 1994;58:393–401.
Hirakawa N., Tershner S.A., Fields H.L., et al. Bi-directional changes in affective state elicited by manipulation of medullary pain-modulatory circuitry. Neuroscience. 2000;100:861–871.
Hohmann A.G. Spinal and peripheral mechanisms of cannabinoid antinociception: behavioral, neurophysiological and neuroanatomical perspectives. Chemistry and Physics of Lipids. 2002;121:173–190.
Holden J.E., Pizzi J.A. Lateral hypothalamic–induced antinociception may be mediated by a substance P connection with the rostral ventromedial medulla. Brain Research. 2008;1214:40–49.
Holden J.E., Pizzi J.A., Jeong Y. An NK1 receptor antagonist microinjected into the periaqueductal gray blocks lateral hypothalamic–induced antinociception in rats. Neuroscience Letters. 2009;453:115–119.
Holden J.E., Schwartz E.J., Proudfit H.K. Microinjection of morphine in the A7 catecholamine cell group produces opposing effects on nociception that are mediated by alpha1- and alpha2-adrenoceptors. Neuroscience. 1999;91:979–990.
Holden J.E., Van Poppel A.Y., Thomas S. Antinociception from lateral hypothalamic stimulation may be mediated by NK1 receptors in the A7 catecholamine cell group in rat. Brain Research. 2002;953:195–204.
Hough L.B., Nalwalk J.W., Stadel R., et al. Inhibition of improgan antinociception by the cannabinoid (CB)1 antagonist N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716A): lack of obligatory role for endocannabinoids acting at CB1 receptors. Journal of Pharmacology and Experimental Therapeutics. 2002;303:314–322.
Howorth P.W., Thornton S.R., O’Brien V., et al. Retrograde viral vector–mediated inhibition of pontospinal noradrenergic neurons causes hyperalgesia in rats. Journal of Neuroscience. 2009;29:12855–12864.
Huisman A.M., Kuypers H.G., Verburgh C.A. Quantitative differences in collateralization of the descending spinal pathways from red nucleus and other brain stem cell groups in rat as demonstrated with the multiple fluorescent retrograde tracer technique. Brain Research. 1981;209:271–286.
Hurley R.W., Hammond D.L. The analgesic effects of supraspinal μ and δ opioid receptor agonists are potentiated during persistent inflammation. Journal of Neuroscience. 2000;20:1249–1259.
Hurley R.W., Hammond D.L. Contribution of endogenous enkephalins to the enhanced analgesic effects of supraspinal mu opioid receptor agonists after inflammatory injury. Journal of Neuroscience. 2001;21:2536–2545.
Imbe H., Okamoto K., Donishi T., et al. Involvement of descending facilitation from the rostral ventromedial medulla in the enhancement of formalin-evoked nocifensive behavior following repeated forced swim stress. Brain Research. 2010;1329:103–112.
Iversen L., Chapman V. Cannabinoids: a real prospect for pain relief? Current Opinion in Pharmacology. 2002;2:50–55.
Iwamoto E.T. Characterization of the antinociception induced by nicotine in the pedunculopontine tegmental nucleus and the nucleus raphe magnus. Journal of Pharmacology and Experimental Therapeutics. 1991;257:120–133.
Jasmin L., Boudah A., Ohara P.T. Long-term effects of decreased noradrenergic central nervous system innervation on pain behavior and opioid antinociception. Journal of Comparative Neurology. 2003;460:38–55.
Jasmin L., Burkey A.R., Granato A., et al. Rostral agranular insular cortex and pain areas of the central nervous system: a tract-tracing study in the rat. Journal of Comparative Neurology. 2004;468:425–440.
Jasmin L., Rabkin S.D., Granato A., et al. Analgesia and hyperalgesia from GABA-mediated modulation of the cerebral cortex. Nature. 2003;424:316–320.
Jinks S.L., Carstens E., Antognini J.F. Glutamate receptor blockade in the rostral ventromedial medulla reduces the force of multisegmental motor responses to supramaximal noxious stimuli. Neuroscience Letters. 2007;426:175–180.
Johansen J.P., Fields H.L., Manning B.H. The affective component of pain in rodents: direct evidence for a contribution of the anterior cingulate cortex. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8077–8082.
Jordan L.M., Kenshalo D.R., Jr., Martin R.F., et al. Depression of primate spinothalamic tract neurons by iontophoretic application of 5-hydroxytryptamine. Pain. 1978;5:135–142.
Kaplan H., Fields H.L. Hyperalgesia during acute opioid abstinence: evidence for a nociceptive facilitating function of the rostral ventromedial medulla. Journal of Neuroscience. 1991;11:1433–1439.
Keay K.A., Feil K., Gordon B.D., et al. Spinal afferents to functionally distinct periaqueductal gray columns in the rat: an anterograde and retrograde tracing study. Journal of Comparative Neurology. 1997;385:207–229.
Kelley K.W., Bluthe R.M., Dantzer R., et al. Cytokine-induced sickness behavior. Brain, Behavior, and Immunity. 2003;17(suppl 1):S112–S118.
Kiefel J.M., Rossi G.C., Bodnar R.J. Medullary mu and delta opioid receptors modulate mesencephalic morphine analgesia in rats. Brain Research. 1993;624:151–161.
Kincaid W., Neubert M.J., Xu M., et al. Role for medullary pain facilitating neurons in secondary thermal hyperalgesia. Journal of Neurophysiology. 2006;95:33–41.
King T., Vera-Portocarrero L., Gutierrez T., et al. Unmasking the tonic-aversive state in neuropathic pain. Nature Neuroscience. 2009;12:1364–1366.
Kovelowski C.J., Ossipov M.H., Sun H., et al. Supraspinal cholecystokinin may drive tonic descending facilitation mechanisms to maintain neuropathic pain in the rat. Pain. 2000;87:265–273.
Koyama T., Kato K., Tanaka Y.Z., et al. Anterior cingulate activity during pain-avoidance and reward tasks in monkeys. Neurosci Res. 2001;39:421–430.
Kunishio K., Haber S.N. Primate cingulostriatal projection: Limbic striatal versus sensorimotor striatal input. J Comp Neurol. 1994;350:337–356.
LaGraize S.C., Borzan J., Rinker M.M., et al. Behavioral evidence for competing motivational drives of nociception and hunger. Neuroscience Letters. 2004;372:30–34.
LaGraize S.C., Guo W., Yang K., et al. Spinal cord mechanisms mediating behavioral hyperalgesia induced by neurokinin-1 tachykinin receptor activation in the rostral ventromedial medulla. Neuroscience. 2010;171:1341–1356.
Le Bars D. Serotonin and pain. In: Osborne N.N., Hamon M., eds. Neuronal serotonin. New York: Wiley; 1988:171–226.
Le Bars D. The whole body receptive field of dorsal horn multireceptive neurones. Brain Research Reviews. 2002;40:29–44.
Leong M.L., Gu M., Speltz-Paiz R., Stahura E.I., Mottey N., Steer C.J., Wessendorf M. Neuronal loss in the rostral ventromedial medulla in a rat model of neuropathic pain. J Neuroscience. 2011;23:17028–17039.
Leung C.G., Mason P. Physiological properties of raphe magnus neurons during sleep and waking. Journal of Neurophysiology. 1999;81:584–595.
Levine J.D., Gordon N.C., Fields H.L. The mechanism of placebo analgesia. Lancet. 1978;2:654–657.
Levy R., Deer T.R., Henderson J. Intracranial neurostimulation for pain control: a review. Pain Physician. 2010;13:157–165.
Lima D., Almeida A. The medullary dorsal reticular nucleus as a pronociceptive centre of the pain control system. Progress in Neurobiology. 2002;66:81–108.
Ljungdahl A., Hokfelt T., Nilsson G. Distribution of substance P–like immunoreactivity in the central nervous system of the rat—I. Cell bodies and nerve terminals. Neuroscience. 1978;3:861–943.
Lovick T.A. Pro-nociceptive action of cholecystokinin in the periaqueductal grey: a role in neuropathic and anxiety-induced hyperalgesic states. Neuroscience and Biobehavioral Reviews. 2008;32:852–862.
Malan T.P., Jr., Ibrahim M.M., Lai J., et al. CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Current Opinion in Pharmacology. 2003;3:62–67.
Malick J.B., Goldstein J.M. Analgesic activity of substance P following intracerebral administration in rats. Life Sciences. 1978;23:835–844.
Manning B.H., Morgan M.J., Franklin K.B. Morphine analgesia in the formalin test: evidence for forebrain and midbrain sites of action. Neuroscience. 1994;63:289–294.
Mansour A., Fox C.A., Akil H., et al. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends in Neurosciences. 1995;18:22–29.
Martenson M.E., Cetas J.S., Heinricher M.M. A possible neural basis for stress-induced hyperalgesia. Pain. 2009;142:236–244.
Martin W.J., Gupta N.K., Loo C.M., et al. Differential effects of neurotoxic destruction of descending noradrenergic pathways on acute and persistent nociceptive processing. Pain. 1999;80:57–65.
Martins I., Costa-Araujo S., Fadel J., et al. Reversal of neuropathic pain by HSV-1–mediated decrease of noradrenaline in a pain facilitatory area of the brain. Pain. 2010;151:137–145.
Mason P. Physiological identification of pontomedullary serotonergic neurons in the rat. Journal of Neurophysiology. 1997;77:1087–1098.
Mason P., Fields H.L. Axonal trajectories and terminations of on- and off-cells in the cat lower brainstem. Journal of Comparative Neurology. 1989;288:185–207.
Matre D., Casey K.L., Knardahl S. Placebo-induced changes in spinal cord pain processing. Journal of Neuroscience. 2006;26:559–563.
Mayer D.J., Price D.D. Central nervous system mechanisms of analgesia. Pain. 1976;2:379–404.
McGaraughty S., Farr D.A., Heinricher M.M. Lesions of the periaqueductal gray disrupt input to the rostral ventromedial medulla following microinjections of morphine into the medial or basolateral nuclei of the amygdala. Brain Research. 2004;1009:223–227.
McGaraughty S., Heinricher M.M. Microinjection of morphine into various amygdaloid nuclei differentially affects nociceptive responsiveness and RVM neuronal activity. Pain. 2002;96:153–162.
Melzack R., Wall P.D. Pain mechanisms: a new theory. Science. 1965;150:971–979.
Melzack R., Wall P.D., Ty T.C. Acute pain in an emergency clinic: latency of onset and descriptor patterns related to different injuries. Pain. 1982;14:33–43.
Menetrey D., Chaouch A., Binder D., et al. The origin of the spinomesencephalic tract in the rat: an anatomical study using the retrograde transport of horseradish peroxidase. Journal of Comparative Neurology. 1982;206:193–207.
Meng I.D., Johansen J.P. Antinociception and modulation of rostral ventromedial medulla neuronal activity by local microinfusion of a cannabinoid receptor agonist. Neuroscience. 2004;124:685–693.
Meng I.D., Johansen J.P., Harasawa I., et al. Kappa opioids inhibit physiologically identified medullary pain modulating neurons and reduce morphine antinociception. Journal of Neurophysiology. 2005;93:1138–1144.
Meng I.D., Manning B.H., Martin W.J., et al. An analgesia circuit activated by cannabinoids. Nature. 1998;395:381–383.
Miki K., Iwata K., Tsuboi Y., et al. Responses of dorsal column nuclei neurons in rats with experimental mononeuropathy. Pain. 1998;76:407–415.
Miki K., Zhou Q.Q., Guo W., et al. Changes in gene expression and neuronal phenotype in brain stem pain modulatory circuitry after inflammation. Journal of Neurophysiology. 2002;87:750–760.
Millan M.J. Descending control of pain. Progress in Neurobiology. 2002;66:355–474.
Miller K.K., Lupica C.R. Morphine-induced excitation of pyramidal neurons is inhibited by cholecystokinin in the CA1 region of the rat hippocampal slice. Journal of Pharmacology and Experimental Therapeutics. 1994;268:753–761.
Mitchell J.M., Basbaum A.I., Fields H.L. A locus and mechanism of action for associative morphine tolerance. Nature Neuroscience. 2000;3:47–53.
Mogenson G.J., Jones D.L., Yim C.Y. From motivation to action: functional interface between the limbic system and the motor system. Progress in Neurobiology. 1980;14:69–97.
Monconduit L., Desbois C., Villanueva L. The integrative role of the rat medullary subnucleus reticularis dorsalis in nociception. European Journal of Neuroscience. 2002;16:937–944.
Moore R.Y. The anatomy of central serotonin neuron systems in the rat brain. In: Jacobs B.L., Gelperin A., eds. Serotonin neurotransmission and behavior. Cambridge, MA: MIT Press; 1981:35–71.
Mor D., Bembrick A.L., Austin P.J., et al. Anatomically specific patterns of glial activation in the periaqueductal gray of the sub-population of rats showing pain and disability following chronic constriction injury of the sciatic nerve. Neuroscience. 2010;166:1167–1184.
Morgan M.M., Fields H.L. Pronounced changes in the activity of nociceptive modulatory neurons in the rostral ventromedial medulla in response to prolonged thermal noxious stimuli. Journal of Neurophysiology. 1994;72:1161–1170.
Morgan M.M., Sohn J.H., Liebeskind J.C. Stimulation of the periaqueductal gray matter inhibits nociception at the supraspinal as well as spinal level. Brain Research. 1989;502:61–66.
Morgan M.M., Whittier K.L., Hegarty D.M., et al. Periaqueductal gray neurons project to spinally projecting GABAergic neurons in the rostral ventromedial medulla. Pain. 2008;140:376–386.
Munn E.M., Harte S.E., Lagman A., et al. Contribution of the periaqueductal gray to the suppression of pain affect produced by administration of morphine into the intralaminar thalamus of rat. Journal of Pain. 2009;10:426–435.
Murphy A.Z., Rizvi T.A., Ennis M., et al. The organization of preoptic-medullary circuits in the male rat: evidence for interconnectivity of neural structures involved in reproductive behavior, antinociception and cardiovascular regulation. Neuroscience. 1999;91:1103–1116.
Nakaya Y., Kaneko T., Shigemoto R., et al. Immunohistochemical localization of substance P receptor in the central nervous system of the adult rat. Journal of Comparative Neurology. 1994;347:249–274.
Nalwalk J.W., Svokos K., Taraschenko O., et al. Activation of brain stem nuclei by improgan, a non-opioid analgesic. Brain Research. 2004;1021:248–255.
Neubert M.J., Kincaid W., Heinricher M.M. Nociceptive facilitating neurons in the rostral ventromedial medulla. Pain. 2004;110:158–165.
Nuseir K., Heidenreich B.A., Proudfit H.K. The antinociception produced by microinjection of a cholinergic agonist in the ventromedial medulla is mediated by noradrenergic neurons in the A7 catecholamine cell group. Brain Research. 1999;822:1–7.
Nuseir K., Proudfit H.K. Bidirectional modulation of nociception by GABA neurons in the dorsolateral pontine tegmentum that tonically inhibit spinally projecting noradrenergic A7 neurons. Neuroscience. 2000;96:773–783.
Oka T., Hori T. Brain cytokines and pain. In: Watkins L.R., Maier S.F., eds. Cytokines and pain. Basel, Switzerland: Birkhauser Verlag; 1999:183–203.
Oka T., Oka K., Hosoi M., et al. The opposing effects of interleukin-1 beta microinjected into the preoptic hypothalamus and the ventromedial hypothalamus on nociceptive behavior in rats. Brain Research. 1995;700:271–278.
Oliveras J.L., Martin G., Montagne J., et al. Single unit activity at ventromedial medulla level in the awake, freely moving rat: effects of noxious heat and light tactile stimuli onto convergent neurons. Brain Research. 1990;506:19–30.
Oyama T., Ueda M., Kuraishi Y., et al. Dual effect of serotonin on formalin-induced nociception in the rat spinal cord. Neuroscience Research. 1996;25:129–135.
Pacharinsak C., Khasabov S.G., Beitz A.J., et al. NK-1 receptors in the rostral ventromedial medulla contribute to hyperalgesia produced by intraplantar injection of capsaicin. Pain. 2008;139:34–46.
Pan Z., Hirakawa N., Fields H.L. A cellular mechanism for the bidirectional pain-modulating actions of orphanin FQ/nociceptin. Neuron. 2000;26:515–522.
Pan Z.Z., Fields H.L. Endogenous opioid-mediated inhibition of putative pain-modulating neurons in rat rostral ventromedial medulla. Neuroscience. 1996;74:855–862.
Pan Z.Z., Tershner S.A., Fields H.L. Cellular mechanism for anti-analgesic action of agonists of the kappa-opioid receptor. Nature. 1997;389:382–385.
Pan Z.Z., Williams J.T., Osborne P.B. Opioid actions on single nucleus raphe magnus neurons from rat and guinea-pig in vitro. Journal of Physiology. 1990;427:519–532.
Pavlovic Z.W., Cooper M.L., Bodnar R.J. Opioid antagonists in the periaqueductal gray inhibit morphine and beta-endorphin analgesia elicited from the amygdala of rats. Brain Research. 1996;741:13–26.
Pertovaara A. Noradrenergic pain modulation. Progress in Neurobiology. 2006;80:53–83.
Pertwee R.G. Cannabinoid receptors and pain. Progress in Neurobiology. 2001;63:569–611.
Peters C.M., Hayashida K., Ewan E.E., et al. Lack of analgesic efficacy of spinal ondansetron on thermal and mechanical hypersensitivity following spinal nerve ligation in the rat. Brain Research. 2010;1352:83–93.
Pinto-Ribeiro F., Ansah O.B., Almeida A., et al. Influence of arthritis on descending modulation of nociception from the paraventricular nucleus of the hypothalamus. Brain Research. 2008;1197:63–75.
Pittius C.W., Seizinger B.R., Pasi A., et al. Distribution and characterization of opioid peptides derived from proenkephalin A in human and rat central nervous system. Brain Research. 1984;304:127–136.
Ploghaus A., Becerra L., Borras C., et al. Neural circuitry underlying pain modulation: Expectation, hypnosis, placebo. Trends Cognitive Sciences. 2003;7:197–200.
Pogatzki E.M., Urban M.O., Brennan T.J., et al. Role of the rostral medial medulla in the development of primary and secondary hyperalgesia after incision in the rat. Anesthesiology. 2002;96:1153–1160.
Porreca F., Ossipov M.H., Gebhart G.F. Chronic pain and medullary descending facilitation. Trends in Neurosciences. 2002;25:319–325.
Potrebic S.B., Fields H.L., Mason P. Serotonin immunoreactivity is contained in one physiological cell class in the rat rostral ventromedial medulla. Journal of Neuroscience. 1994;14:1655–1665.
Price D.D., von der Gruen A., Miller J., et al. Potentiation of systemic morphine analgesia in humans by proglumide, a cholecystokinin antagonist. Anesthesia and Analgesia. 1985;64:801–806.
Proudfit H.K. The behavioural pharmacology of the noradrenergic system. In: Besson J.M., Guilbaud G., eds. Towards the use of noradrenergic agonists for the treatment of pain. Amsterdam: Elsevier; 1992:119–136.
Ramirez F., Vanegas H. Tooth pulp stimulation advances both medullary off-cell pause and tail flick. Neuroscience Letters. 1989;100:153–156.
Ren K., Dubner R. Descending modulation in persistent pain: an update. Pain. 2002;100:1–6.
Rhodes D.L., Liebeskind J.C. Analgesia from rostral brain stem stimulation in the rat. Brain Research. 1978;143:521–532.
Rhudy J.L., Meagher M.W. Individual differences in the emotional reaction to shock determine whether hypoalgesia is observed. Pain Medicine. 2003;4:244–256.
Rivat C., Becker C., Blugeot A., et al. Chronic stress induces transient spinal neuroinflammation, triggering sensory hypersensitivity and long-lasting anxiety-induced hyperalgesia. Pain. 2010;150:358–368.
Rivat C., Vera-Portocarrero L.P., Ibrahim M.M., et al. Spinal NK-1 receptor–expressing neurons and descending pathways support fentanyl-induced pain hypersensitivity in a rat model of postoperative pain. European Journal of Neuroscience. 2009;29:727–737.
Rizvi T.A., Ennis M., Behbehani M.M., et al. Connections between the central nucleus of the amygdala and the midbrain periaqueductal gray: topography and reciprocity. Journal of Comparative Neurology. 1991;303:121–131.
Rizvi T.A., Murphy A.Z., Ennis M., et al. Medial preoptic area afferents to periaqueductal gray medullo-output neurons: a combined fos and tract tracing study. Journal of Neuroscience. 1996;16:333–344.
Roberts J., Ossipov M.H., Porreca F. Glial activation in the rostroventromedial medulla promotes descending facilitation to mediate inflammatory hypersensitivity. European Journal of Neuroscience. 2009;30:229–241.
Rosen A., Zhang Y.X., Lund I., et al. Substance P microinjected into the periaqueductal gray matter induces antinociception and is released following morphine administration. Brain Research. 2004;1001:87–94.
Rossi G.C., Pasternak G.W., Bodnar R.J. Mu and delta opioid synergy between the periaqueductal gray and the rostro-ventral medulla. Brain Research. 1994;665:85–93.
Roychowdhury S.M., Fields H.L. Endogenous opioids acting at a medullary μ-opioid receptor contribute to the behavioral antinociception produced by GABA antagonism in the midbrain periaqueductal gray. Neuroscience. 1996;74:863–872.
Sanoja R., Tortorici V., Fernandez C., et al. Role of RVM neurons in capsaicin-evoked visceral nociception and referred hyperalgesia. European Journal of Pain. 2010;14:120–129.
Sawamoto N., Honda M., Okada T., et al. Expectation of pain enhances responses to nonpainful somatosensory stimulation in the anterior cingulate cortex and parietal operculum/posterior insula: An event-related functional magnetic resonance imaging study. J Neuroscience. 2000;20:7438–7445.
Siegel S. Morphine analgesic tolerance: its situation specificity supports a pavlovian conditioning model. Science. 1976;193:323–325.
Skagerberg G., Björklund A. Topographic principles in the spinal projections of serotonergic and non-serotonergic brainstem neurons in the rat. Neuroscience. 1985;15:445–480.
Smith D.J., Hawranko A.A., Monroe P.J., et al. Dose-dependent pain-facilitatory and -inhibitory actions of neurotensin are revealed by SR 48692, a nonpeptide neurotensin antagonist: influence on the antinociceptive effect of morphine. Journal of Pharmacology and Experimental Therapeutics. 1997;282:899–908.
Sugiyo S., Takemura M., Dubner R., et al. Trigeminal transition zone/rostral ventromedial medulla connections and facilitation of orofacial hyperalgesia after masseter inflammation in rats. Journal of Comparative Neurology. 2005;493:510–523.
Suzuki R., Morcuende S., Webber M., et al. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nature Neuroscience. 2002;5:1319–1326.
Suzuki R., Rygh L.J., Dickenson A.H. Bad news from the brain: descending 5-HT pathways that control spinal pain processing. Trends in Pharmacological Sciences. 2004;25:613–617.
Sykes K.T., White S.R., Hurley R.W., et al. Mechanisms responsible for the enhanced antinociceptive effects of μ-opioid receptor agonists in the rostral ventromedial medulla of male rats with persistent inflammatory pain. Journal of Pharmacology and Experimental Therapeutics. 2007;322:813–821.
Tershner S.A., Helmstetter F.J. Antinociception produced by mu opioid receptor activation in the amygdala is partly dependent on activation of mu opioid and neurotensin receptors in the ventral periaqueductal gray. Brain Research. 2000;865:17–26.
Tershner S.A., Mitchell J.M., Fields H.L. Brainstem pain modulating circuitry is sexually dimorphic with respect to mu and kappa opioid receptor function. Pain. 2000;85:153–159.
Todd A.J. Neuronal circuitry for pain processing in the dorsal horn. Nature Reviews Neuroscience. 2010;11:823–836.
Tortorici V., Nogueira L., Salas R., et al. Involvement of local cholecystokinin in the tolerance induced by morphine microinjections into the periaqueductal gray of rats. Pain. 2003;102:9–16.
Tracey I., Ploghaus A., Gati J.S., et al. Imaging attentional modulation of pain in the periaqueductal gray in humans. Journal of Neuroscience. 2002;22:2748–2752.
Urban M.O., Coutinho S.V., Gebhart G.F. Involvement of excitatory amino acid receptors and nitric oxide in the rostral ventromedial medulla in modulating secondary hyperalgesia produced by mustard oil. Pain. 1999;81:45–55.
Urban M.O., Jiang M.C., Gebhart G.F. Participation of central descending nociceptive facilitatory systems in secondary hyperalgesia produced by mustard oil. Brain Research. 1996;737:83–91.
Urban M.O., Smith D.J., Gebhart G.F. Involvement of spinal cholecystokininb receptors in mediating neurotensin hyperalgesia from the medullary nucleus raphe magnus in the rat. Journal of Pharmacology and Experimental Therapeutics. 1996;278:90–96.
Valverde O., Ledent C., Beslot F., et al. Reduction of stress-induced analgesia but not of exogenous opioid effects in mice lacking CB1 receptors. European Journal of Neuroscience. 2000;12:533–539.
Vanderah T.W., Suenaga N.M., Ossipov M.H., et al. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. Journal of Neuroscience. 2001;21:279–286.
Vanegas H. To the descending pain-control system in rats, inflammation-induced primary and secondary hyperalgesia are two different things. Neuroscience Letters. 2004;361:225–228.
Vanegas H., Barbaro N.M., Fields H.L. Midbrain stimulation inhibits tail-flick only at currents sufficient to excite rostral medullary neurons. Brain Research. 1984;321:127–133.
Vanegas H., Barbaro N.M., Fields H.L. Tail-flick related activity in medullospinal neurons. Brain Research. 1984;321:135–141.
Vanegas H., Schaible H.-G. Descending control of persistent pain: inhibitory or facilitatory? Brain Research Reviews. 2004;46:295–309.
van Wijk G., Veldhuijzen D.S. Perspective on diffuse noxious inhibitory controls as a model of endogenous pain modulation in clinical pain syndromes. Journal of Pain. 2010;11:408–419.
Vaughan C.W., Connor M., Bagley E.E., et al. Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Molecular Pharmacology. 2000;57:288–295.
Vaughan C.W., McGregor I.S., Christie M.J. Cannabinoid receptor activation inhibits GABAergic neurotransmission in rostral ventromedial medulla neurons in vitro. British Journal of Pharmacology. 1999;127:935–940.
Vera-Portocarrero L.P., Ossipov M.H., King T., et al. Reversal of inflammatory and noninflammatory visceral pain by central or peripheral actions of sumatriptan. Gastroenterology. 2008;135:1369–1378.
Vera-Portocarrero L.P., Ossipov M.H., Lai J., et al. Descending facilitatory pathways from the rostroventromedial medulla mediate naloxone-precipitated withdrawal in morphine-dependent rats. Journal of Pain. 2011;12:667–676.
Verner T.A., Pilowsky P.M., Goodchild A.K. Retrograde projections to a discrete apneic site in the midline medulla oblongata of the rat. Brain Research. 2008;1208:128–136.
Vertes R.P. A lectin horseradish peroxidase study of the origin of ascending fibers in the medial forebrain bundle of the rat. The lower brainstem. Neuroscience. 1984;11:651–668.
Villanueva L., Bouhassira D., Le Bars D. The medullary subnucleus reticularis dorsalis (SRD) as a key link in both the transmission and modulation of pain signals. Pain. 1996;67:231–240.
Villemure C., Bushnell M.C. Cognitive modulation of pain: how do attention and emotion influence pain processing? Pain. 2002;95:195–199.
Walker J.M., Huang S.M. Cannabinoid analgesia. Pharmacology & Thereutics. 2002;95:127–135.
Wall P.D. The laminar organization of dorsal horn and effects of descending impulses. Journal of Physiology. 1967;188:403–423.
Waters A.J., Lumb B.M. Inhibitory effects evoked from both the lateral and ventrolateral periaqueductal grey are selective for the nociceptive responses of rat dorsal horn neurones. Brain Research. 1997;752:239–249.
Watkins L.R., Cobelli D.A., Faris P., et al. Opiate vs non-opiate footshock-induced analgesia (FSIA): the body region shocked is a critical factor. Brain Research. 1982;242:299–308.
Watkins L.R., Cobelli D.A., Mayer D.J. Classical conditioning of front paw and hind paw footshock induced analgesia (FSIA): naloxone reversibility and descending pathways. Brain Research. 1982;243:119–132.
Watkins L.R., Maier S.F., Goehler L.E. Immune activation: the role of pro-inflammatory cytokines in inflammation, illness responses and pathological pain states. Pain. 1995;63:289–302.
Watkins L.R., Mayer D.J. Organization of endogenous opiate and nonopiate pain control systems. Science. 1982;216:1185–1192.
Watkins L.R., Wiertelak E.P., McGorry M., et al. Neurocircuitry of conditioned inhibition of analgesia: effects of amygdala, dorsal raphe, ventral medullary, and spinal cord lesions on antianalgesia in the rat. Behavioral Neuroscience. 1998;112:360–378.
Watkins L.R., Young E.G., Kinscheck I.B., et al. The neural basis of footshock analgesia: the role of specific ventral medullary nuclei. Brain Research. 1983;276:305–315.
Wei F., Guo W., Zou S., et al. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. Journal of Neuroscience. 2008;28:10482–10495.
Wiertelak E.P., Maier S.F., Watkins L.R. Cholecystokinin antianalgesia: safety cues abolish morphine analgesia. Science. 1992;256:830–833.
Wiertelak E.P., Roemer B., Maier S.F., et al. Comparison of the effects of nucleus tractus solitarius and ventral medial medulla lesions on illness-induced and subcutaneous formalin-induced hyperalgesias. Brain Research. 1997;748:143–150.
Wilder-Smith C.H., Schindler D., Lovblad K., et al. Brain functional magnetic resonance imaging of rectal pain and activation of endogenous inhibitory mechanisms in irritable bowel syndrome patient subgroups and healthy controls. Gut. 2004;53:1595–1601.
Will M.J., Pratt W.E., Kelley A.E. Pharmacological characterization of high-fat feeding induced by opioid stimulation of the ventral striatum. Physiology & Behavior. 2006;89:226–234.
Willcockson W.S., Gerhart K.D., Cargill C.L., et al. Effects of biogenic amines on raphe-spinal tract cells. Journal of Pharmacology and Experimental Therapeutics. 1983;225:637–645.
Willis W.D., Jr. Anatomy and physiology of descending control of nociceptive responses of dorsal horn neurons: comprehensive review. Progress in Brain Research. 1988;77:1–29.
Willis W.D., Coggleshall R.E. Sensory mechanisms of the spinal cord. New York: Plenum Press; 1991.
Winkler C.W., Hermes S.M., Chavkin C.I., et al. Kappa opioid receptor (KOR) and GAD67 immunoreactivity are found in OFF and neutral cells in the rostral ventromedial medulla. Journal of Neurophysiology. 2006;96:3465–3473.
Xu M., Kim C.J., Neubert M.J., et al. NMDA receptor–mediated activation of medullary pro-nociceptive neurons is required for secondary thermal hyperalgesia. Pain. 2007;127:253–262.
Xu M., Kontinen V.K., Kalso E. Endogenous noradrenergic tone controls symptoms of allodynia in the spinal nerve ligation model of neuropathic pain. Eur J Pharmacol. 1999;366:41–45.
Yaksh T.L., Al-Rodhan N.R.F., Jensen T.S. Sites of action of opiates in production of analgesia. Progress in Brain Research. 1988;77:371–394.
Yaksh T.L., Wilson P.R. Spinal serotonin terminal system mediates antinociception. Journal of Pharmacology and Experimental Therapeutics. 1979;208:446–453.
Yarnitsky D. Conditioned pain modulation (the diffuse noxious inhibitory control–like effect): its relevance for acute and chronic pain states. Current Opinion in Anaesthesiology. 2010;23:611–615.
Yarnitsky D., Arendt-Nielsen L., Bouhassira D., et al. Recommendations on terminology and practice of psychophysical DNIC testing. European Journal of Pain. 2010;14:339.
Yarnitsky D., Crispel Y., Eisenberg E., et al. Prediction of chronic post-operative pain: pre-operative DNIC testing identifies patients at risk. Pain. 2008;138:22–28.
Yeomans D.C., Proudfit H.K. Projections of substance P–immunoreactive neurons located in the ventromedial medulla to the A7 noradrenergic nucleus of the rat demonstrated using retrograde tracing combined with immunocytochemistry. Brain Research. 1990;532:329–332.
Yeomans D.C., Proudfit H.K. Antinociception induced by microinjection of substance P into the A7 catecholamine cell group in the rat. Neuroscience. 1992;49:681–691.
Yilmaz P., Diers M., Diener S., et al. Brain correlates of stress-induced analgesia. Pain. 2010;151:522–529.
You H.J., Colpaert F.C., Arendt-Nielsen L. Long-lasting descending and transitory short-term spinal controls on deep spinal dorsal horn nociceptive-specific neurons in response to persistent nociception. Brain Research Bulletin. 2008;75:34–41.
You H.J., Lei J., Sui M.Y., et al. Endogenous descending modulation: spatiotemporal effect of dynamic imbalance between descending facilitation and inhibition of nociception. Journal of Physiology. 2010;588:4177–4188.
Zarrindast M.R., Nikfar S., Rezayat M. Cholecystokinin receptor mechanism(s) and morphine tolerance in mice. Pharmacology and Toxicology. 1999;84:46–50.
Zeitz K.P., Guy N., Malmberg A.B., et al. The 5-HT3 subtype of serotonin receptor contributes to nociceptive processing via a novel subset of myelinated and unmyelinated nociceptors. Journal of Neuroscience. 2002;22:1010–1019.
Zhang L., Hammond D.L. Substance P enhances excitatory synaptic transmission on spinally projecting neurons in the rostral ventromedial medulla after inflammatory injury. Journal of Neurophysiology. 2009;102:1139–1151.
Zhu Y., King M.A., Schuller A.G., et al. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron. 1999;24:243–252.
Beecher H.K. Relationship of significance of wound to pain experienced. Journal of the American Medical Association. 1959;161:1609–1613.
Bernard J., Sevoz-Couche C., Hamon M., et al. Responses of lateral paragigantocellular and raphe magnus serotonergic neurons to noxious stimuli: a comparative reappraisal using juxtacellular recording. Montreal, Canada: IASP World Congress; 2010. PW205
Boivie J., Meyerson B.A. A correlative anatomical and clinical study of pain suppression by deep brain stimulation. Pain. 1982;13:113–126.
Brightwell J.J., Taylor B.K. Noradrenergic neurons in the locus coeruleus contribute to neuropathic pain. Neuroscience. 2009;160:174–185.
Brink T.S., Mason P. Raphe magnus neurons respond to noxious colorectal distension. Journal of Neurophysiology. 2003;89:2506–2515.
Budai D., Khasabov S.G., Mantyh P.W., et al. NK-1 receptors modulate the excitability of ON cells in the rostral ventromedial medulla. Journal of Neurophysiology. 2007;97:1388–1395.
Carlson J.D., Maire J.J., Martenson M.E., et al. Sensitization of pain-modulating neurons in the rostral ventromedial medulla after peripheral nerve injury. Journal of Neuroscience. 2007;27:13222–13231.
Carlson J.D., Selden N.R., Heinricher M.M. Nocifensive reflex-related on- and off-cells in the pedunculopontine tegmental nucleus, cuneiform nucleus, and lateral dorsal tegmental nucleus. Brain Research. 2005;1063:187–194.
Carpenter D., Engberg I., Lundberg A. Differential supraspinal control of inhibitory and excitatory actions from the FRA to ascending spinal pathways. Acta Physiologica Scandinavica. 1965;63:103–110.
Da Silva L.F., Walder R.Y., Davidson B.L., et al. Changes in expression of NMDA-nr1 receptor subunits in the rostral ventromedial medulla modulate pain behaviors. Pain. 2010;151:155–161.
Dantzer R., Bluthe R.M., Laye S., et al. Cytokines and sickness behavior. Annals of the New York Academy of Sciences. 1998;840:586–590.
Edelmayer R.M., Vanderah T.W., Majuta L., et al. Medullary pain facilitating neurons mediate allodynia in headache-related pain. Annals of Neurology. 2009;65:184–193.
Fields H.L., Heinricher M.M. Anatomy and physiology of a nociceptive modulatory system. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 1985;308:361–374.
Fields H.L., Heinricher M.M., Mason P. Neurotransmitters in nociceptive modulatory circuits. Annual Review of Neuroscience. 1991;14:219–245.
Fields H.L., Levine J.D. Placebo analgesia—a role for endorphins? Trends in Neurosciences. 1984;7:271–273.
Flor H., Birbaumer N., Schulz R., et al. Pavlovian conditioning of opioid and nonopioid pain inhibitory mechanisms in humans. European Journal of Pain. 2002;6:395–402.
Guan Y., Guo W., Robbins M.T., et al. Changes in AMPA receptor phosphorylation in the rostral ventromedial medulla after inflammatory hyperalgesia in rats. Neuroscience Letters. 2004;366:201–205.
Guan Y., Terayama R., Dubner R., et al. Plasticity in excitatory amino acid receptor–mediated descending pain modulation after inflammation. Journal of Pharmacology and Experimental Therapeutics. 2002;300:513–520.
Guo W., Robbins M.T., Wei F., et al. Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for descending pain facilitation. Journal of Neuroscience. 2006;26:126–137.
Hagbarth K.E., Kerr D.I. Central influences on spinal afferent conduction. Journal of Neurophysiology. 1954;17:295–307.
Hahm E.T., Hammond D.L., Proudfit H.K. Substance P induces the reversible formation of varicosities in the dendrites of rat brainstem neurons. Brain Research. 2011;1369:36–45.
Hamity M.V., White S.R., Hammond D.L. Effects of neurokinin-1 receptor agonism and antagonism in the rostral ventromedial medulla of rats with acute or persistent inflammatory nociception. Neuroscience. 2010;165:902–913.
Haws C.M., Williamson A.M., Fields H.L. Putative nociceptive modulatory neurons in the dorsolateral pontomesencephalic reticular formation. Brain Research. 1989;483:272–282.
Head H., Holmes G. Sensory disturbances from cerebral lesions. Brain. 1911;34:102–254.
Heinricher M.M. Orphanin FQ/nociceptin: from neural circuitry to behavior. Life Sciences. 2003;73:813–822.
Heinricher M.M., Cheng Z.F., Fields H.L. Evidence for two classes of nociceptive modulating neurons in the periaqueductal gray. Journal of Neuroscience. 1987;7:271–278.
Heinricher M.M., Ingram S.L., The brainstem and nociceptive modulation. Bushnell M.C., Basbaum A.I., eds. The senses, a comprehensive reference, vol 5. San Diego, Calif: Academic Press; 2008:593–626.
Heinricher M.M., Neubert M.J. Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. Journal of Neurophysiology. 2004;92:1982–1989.
Heinricher M.M., Pertovaara A., Ossipov M.H., Descending modulation after injury. Dostrovsky J.O., Carr D.B., Koltzenburg M., eds. Progress in pain research and management, vol 24. Seattle: IASP Press; 2003:251–260.
Heinricher M.M., Tavares I., Leith J.L., et al. Descending control of nociception: specificity, recruitment and plasticity. Brain Research Reviews. 2009;60:214–225.
Hermann D.M., Luppi P.H., Peyron C., et al. Afferent projections to the rat nuclei raphe magnus, raphe pallidus and reticularis gigantocellularis pars alpha demonstrated by iontophoretic application of choleratoxin (subunit b). Journal of Chemical Neuroanatomy. 1997;13:1–21.
Hernandez N., Dmitrieva N., Vanegas H. Medullary on-cell activity during tail-flick inhibition produced by heterotopic noxious stimulation. Pain. 1994;58:393–401.
Holden J.E., Schwartz E.J., Proudfit H.K. Microinjection of morphine in the A7 catecholamine cell group produces opposing effects on nociception that are mediated by alpha1- and alpha2-adrenoceptors. Neuroscience. 1999;91:979–990.
Howorth P.W., Thornton S.R., O’Brien V., et al. Retrograde viral vector–mediated inhibition of pontospinal noradrenergic neurons causes hyperalgesia in rats. Journal of Neuroscience. 2009;29:12855–12864.
Jasmin L., Boudah A., Ohara P.T. Long-term effects of decreased noradrenergic central nervous system innervation on pain behavior and opioid antinociception. Journal of Comparative Neurology. 2003;460:38–55.
Jasmin L., Rabkin S.D., Granato A., et al. Analgesia and hyperalgesia from GABA-mediated modulation of the cerebral cortex. Nature. 2003;424:316–320.
Kelley K.W., Bluthe R.M., Dantzer R., et al. Cytokine-induced sickness behavior. Brain, Behavior, and Immunity. 2003;17(Suppl 1):S112–S118.
Le Bars D. Serotonin and pain. In: Osborne N.N., Hamon M., eds. Neuronal serotonin. New York: Wiley; 1988:171–226.
Le Bars D. The whole body receptive field of dorsal horn multireceptive neurones. Brain Research Reviews. 2002;40:29–44.
LaGraize S.C., Guo W., Yang K., et al. Spinal cord mechanisms mediating behavioral hyperalgesia induced by neurokinin-1 tachykinin receptor activation in the rostral ventromedial medulla. Neuroscience. 2010;171:1341–1356.
Leong M., Speltz-Paiz R., Mottey N., et al. Intracisternal injection of 5,7 dihydroxytryptamine (5,7-DHT) partially reverses mechanical hypersensitivity after spinal nerve ligation (SNL). Society of Neurosciences Abstracts Program. 2010:896.10.
Leung C.G., Mason P. Physiological properties of raphe magnus neurons during sleep and waking. Journal of Neurophysiology. 1999;81:584–595.
Levy R., Deer T.R., Henderson J. Intracranial neurostimulation for pain control: a review. Pain Physician. 2010;13:157–165.
Lima D., Almeida A. The medullary dorsal reticular nucleus as a pronociceptive centre of the pain control system. Progress in Neurobiology. 2002;66:81–108.
Lovick T.A. Pro-nociceptive action of cholecystokinin in the periaqueductal grey: a role in neuropathic and anxiety-induced hyperalgesic states. Neuroscience and Biobehavioral Reviews. 2008;32:852–862.
Martin W.J., Gupta N.K., Loo C.M., et al. Differential effects of neurotoxic destruction of descending noradrenergic pathways on acute and persistent nociceptive processing. Pain. 1999;80:57–65.
Martins I., Costa-Araujo S., Fadel J., et al. Reversal of neuropathic pain by HSV-1–mediated decrease of noradrenaline in a pain facilitatory area of the brain. Pain. 2010;151:137–145.
Mason P. Physiological identification of pontomedullary serotonergic neurons in the rat. Journal of Neurophysiology. 1997;77:1087–1098.
Mayer D.J., Price D.D. Central nervous system mechanisms of analgesia. Pain. 1976;2:379–404.
Melzack R., Wall P.D. Pain mechanisms: a new theory. Science. 1965;150:971–979.
Melzack R., Wall P.D., Ty T.C. Acute pain in an emergency clinic: latency of onset and descriptor patterns related to different injuries. Pain. 1982;14:33–43.
Miki K., Iwata K., Tsuboi Y., et al. Responses of dorsal column nuclei neurons in rats with experimental mononeuropathy. Pain. 1998;76:407–415.
Miki K., Zhou Q.Q., Guo W., et al. Changes in gene expression and neuronal phenotype in brain stem pain modulatory circuitry after inflammation. Journal of Neurophysiology. 2002;87:750–760.
Millan M.J. Descending control of pain. Progress in Neurobiology. 2002;66:355–474.
Monconduit L., Desbois C., Villanueva L. The integrative role of the rat medullary subnucleus reticularis dorsalis in nociception. European Journal of Neuroscience. 2002;16:937–944.
Mor D., Bembrick A.L., Austin P.J., et al. Anatomically specific patterns of glial activation in the periaqueductal gray of the sub-population of rats showing pain and disability following chronic constriction injury of the sciatic nerve. Neuroscience. 2010;166:1167–1184.
Mor D., Bembrick A.L., Austin P.J., et al. Evidence for cellular injury in the midbrain of rats following chronic constriction injury of the sciatic nerve. Journal of Chemical Neuroanatomy. 2011;41:158–169.
Morgan M.M., Fields H.L. Pronounced changes in the activity of nociceptive modulatory neurons in the rostral ventromedial medulla in response to prolonged thermal noxious stimuli. Journal of Neurophysiology. 1994;72:1161–1170.
Murphy A.Z., Rizvi T.A., Ennis M., et al. The organization of preoptic-medullary circuits in the male rat: evidence for interconnectivity of neural structures involved in reproductive behavior, antinociception and cardiovascular regulation. Neuroscience. 1999;91:1103–1116.
Nalwalk J.W., Svokos K., Taraschenko O., et al. Activation of brain stem nuclei by improgan, a non-opioid analgesic. Brain Research. 2004;1021:248–255.
Neubert M.J., Kincaid W., Heinricher M.M. Nociceptive facilitating neurons in the rostral ventromedial medulla. Pain. 2004;110:158–165.
Nuseir K., Proudfit H.K. Bidirectional modulation of nociception by GABA neurons in the dorsolateral pontine tegmentum that tonically inhibit spinally projecting noradrenergic A7 neurons. Neuroscience. 2000;96:773–783.
Oliveras J.L., Martin G., Montagne J., et al. Single unit activity at ventromedial medulla level in the awake, freely moving rat: effects of noxious heat and light tactile stimuli onto convergent neurons. Brain Research. 1990;506:19–30.
Pacharinsak C., Khasabov S.G., Beitz A.J., et al. NK-1 receptors in the rostral ventromedial medulla contribute to hyperalgesia produced by intraplantar injection of capsaicin. Pain. 2008;139:34–46.
Pertovaara A. Noradrenergic pain modulation. Progress in Neurobiology. 2006;80:53–83.
Peters C.M., Hayashida K., Ewan E.E., et al. Lack of analgesic efficacy of spinal ondansetron on thermal and mechanical hypersensitivity following spinal nerve ligation in the rat. Brain Research. 2010;1352:83–93.
Pinto-Ribeiro F., Ansah O.B., Almeida A., et al. Influence of arthritis on descending modulation of nociception from the paraventricular nucleus of the hypothalamus. Brain Research. 2008;1197:63–75.
Porreca F., Ossipov M.H., Gebhart G.F. Chronic pain and medullary descending facilitation. Trends in Neurosciences. 2002;25:319–325.
Potrebic S.B., Fields H.L., Mason P. Serotonin immunoreactivity is contained in one physiological cell class in the rat rostral ventromedial medulla. Journal of Neuroscience. 1994;14:1655–1665.
Proudfit H.K. The behavioural pharmacology of the noradrenergic system. In: Besson J.M., Guilbaud G., eds. Towards the use of noradrenergic agonists for the treatment of pain. Amsterdam: Elsevier; 1992:119–136.
Ramirez F., Vanegas H. Tooth pulp stimulation advances both medullary off-cell pause and tail flick. Neuroscience Letters. 1989;100:153–156.
Ren K., Dubner R. Descending modulation in persistent pain: an update. Pain. 2002;100:1–6.
Rhudy J.L., Meagher M.W. Individual differences in the emotional reaction to shock determine whether hypoalgesia is observed. Pain Medicine. 2003;4:244–256.
Roberts J., Ossipov M.H., Porreca F. Glial activation in the rostroventromedial medulla promotes descending facilitation to mediate inflammatory hypersensitivity. European Journal of Neuroscience. 2009;30:229–241.
Sanoja R., Tortorici V., Fernandez C., et al. Role of RVM neurons in capsaicin-evoked visceral nociception and referred hyperalgesia. European Journal of Pain. 2010;14:120–129.
Siegel S. Morphine analgesic tolerance: its situation specificity supports a pavlovian conditioning model. Science. 1976;193:323–325.
Sugiyo S., Takemura M., Dubner R., et al. Trigeminal transition zone/rostral ventromedial medulla connections and facilitation of orofacial hyperalgesia after masseter inflammation in rats. Journal of Comparative Neurology. 2005;493:510–523.
Suzuki R., Rygh L.J., Dickenson A.H. Bad news from the brain: descending 5-HT pathways that control spinal pain processing. Trends in Pharmacological Sciences. 2004;25:613–617.
Urban M.O., Coutinho S.V., Gebhart G.F. Involvement of excitatory amino acid receptors and nitric oxide in the rostral ventromedial medulla in modulating secondary hyperalgesia produced by mustard oil. Pain. 1999;81:45–55.
Vanegas H. To the descending pain-control system in rats, inflammation-induced primary and secondary hyperalgesia are two different things. Neuroscience Letters. 2004;361:225–228.
Vanegas H., Schaible H.-G. Descending control of persistent pain: inhibitory or facilitatory? Brain Research Reviews. 2004;46:295–309.
van Wijk G., Veldhuijzen D.S. Perspective on diffuse noxious inhibitory controls as a model of endogenous pain modulation in clinical pain syndromes. Journal of Pain. 2010;11:408–419.
Verner T.A., Pilowsky P.M., Goodchild A.K. Retrograde projections to a discrete apneic site in the midline medulla oblongata of the rat. Brain Research. 2008;1208:128–136.
Villanueva L., Bouhassira D., Le Bars D. The medullary subnucleus reticularis dorsalis (SRD) as a key link in both the transmission and modulation of pain signals. Pain. 1996;67:231–240.
Walker J.M., Huang S.M. Cannabinoid analgesia. Pharmacology & Thereutics. 2002;95:127–135.
Watkins L.R., Maier S.F., Goehler L.E. Immune activation: the role of pro-inflammatory cytokines in inflammation, illness responses and pathological pain states. Pain. 1995;63:289–302.
Watkins L.R., Mayer D.J. Organization of endogenous opiate and nonopiate pain control systems. Science. 1982;216:1185–1192.
Watkins L.R., Wiertelak E.P., McGorry M., et al. Neurocircuitry of conditioned inhibition of analgesia: effects of amygdala, dorsal raphe, ventral medullary, and spinal cord lesions on antianalgesia in the rat. Behavioral Neuroscience. 1998;112:360–378.
Watkins L.R., Young E.G., Kinscheck I.B., et al. The neural basis of footshock analgesia: the role of specific ventral medullary nuclei. Brain Research. 1983;276:305–315.
Wei F., Guo W., Zou S., et al. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. Journal of Neuroscience. 2008;28:10482–10495.
Willis W.D., Jr. Anatomy and physiology of descending control of nociceptive responses of dorsal horn neurons: comprehensive review. Progress in Brain Research. 1988;77:1–29.
Willis W.D., Coggleshall R.E. Sensory mechanisms of the spinal cord. New York: Plenum Press; 1991.
Winkler C.W., Hermes S.M., Chavkin C.I., et al. Kappa opioid receptor (KOR) and GAD67 immunoreactivity are found in OFF and neutral cells in the rostral ventromedial medulla. Journal of Neurophysiology. 2006;96:3465–3473.
Xu M., Kim C.J., Neubert M.J., et al. NMDA receptor–mediated activation of medullary pro-nociceptive neurons is required for secondary thermal hyperalgesia. Pain. 2007;127:253–262.
Xu M., Kontinen V.K., Kalso E. Endogenous noradrenergic tone controls symptoms of allodynia in the spinal nerve ligation model of neuropathic pain. Eur J Pharmacol. 1999;366:41–45.
Yaksh T.L., Al-Rodhan N.R.F., Jensen T.S. Sites of action of opiates in production of analgesia. Progress in Brain Research. 1988;77:371–394.
Yarnitsky D., Arendt-Nielsen L., Bouhassira D., et al. Recommendations on terminology and practice of psychophysical DNIC testing. European Journal of Pain. 2010;14:339.
Yarnitsky D., Crispel Y., Eisenberg E., et al. Prediction of chronic post-operative pain: pre-operative DNIC testing identifies patients at risk. Pain. 2008;138:22–28.
Yilmaz P., Diers M., Diener S., et al. Brain correlates of stress-induced analgesia. Pain. 2010;151:522–529.
You H.J., Colpaert F.C., Arendt-Nielsen L. Long-lasting descending and transitory short-term spinal controls on deep spinal dorsal horn nociceptive-specific neurons in response to persistent nociception. Brain Research Bulletin. 2008;75:34–41.
Zhang L., Hammond D.L. Substance P enhances excitatory synaptic transmission on spinally projecting neurons in the rostral ventromedial medulla after inflammatory injury. Journal of Neurophysiology. 2009;102:1139–1151.