Chapter 3 Disorders of Sodium and Water
Hypernatremia and Hyponatremia
The volume and tonicity of body fluids are maintained within a narrow normal range by regulation of sodium and water balance. The volume of extracellular fluid (ECF) is determined by the total body sodium content, whereas the osmolality and sodium concentration of ECF are determined by water balance. The kidneys play a crucial role in these processes by balancing the excretion of salt and water with their intake and by avidly conserving them when intake is restricted (Table 3-1).
Table 3-1 Renal Regulation of Sodium and Water Balance
| Osmoregulation | Volume Regulation | |
|---|---|---|
| What is sensed | Plasma osmolality | Effective circulating volume |
| Sensors | Hypothalamic osmoreceptors | Carotid sinus |
| Aortic arch | ||
| Glomerular afferent arterioles | ||
| Cardiac atria | ||
| Large pulmonary vessels | ||
| Effectors | Vasopressin | Renin-angiotensin-aldosterone system |
| Thirst | Sympathetic nervous system | |
| Atrial natriuretic peptide | ||
| “Pressure natriuresis” | ||
| Antidiuretic hormone | ||
| What is affected | Water excretion | Urine sodium excretion |
| Water intake |
Modified from Rose BD. Clinical physiology of acid base and electrolyte disorders, 4th ed. New York: McGraw-Hill, 1994: 256, with permission of the McGraw-Hill Companies.
Terminology
Osmolality
The osmolality of a solution refers to the concentration of osmotically active particles in that solution. Osmolality is a function only of the number of particles and is not related to their molecular weight, size, shape, or charge. One mole of a nondissociating substance (e.g., glucose or urea) dissolved in 1 kg of water decreases the freezing point of the resultant solution by 1.86° C. Such a solution has an osmolality of 1 Osm/kg or 1000 mOsm/kg.
The term osmolarity refers to the number of particles of solute per liter of solution, whereas the term osmolality refers to the number of particles of solute per kilogram of solvent. When considering the physiology of body fluids, the difference between osmolality and osmolarity is negligible because body fluids typically are dilute aqueous solutions. In clinical medicine, the term osmolality is used, and the osmolality of body fluids usually is measured by freezing-point depression osmometry. A solution is said to be hyperosmotic if its osmolality is greater than that of the reference solution (often plasma) and hypoosmotic if its osmolality is less than that of the reference solution. An isosmotic solution has an osmolality identical to that of the reference solution.
The normal plasma osmolality of dogs and cats is slightly higher than that of humans and ranges from 290 to 310 mOsm/kg in dogs and from 290 to 330 mOsm/kg in cats. In one study, 20 dogs under resting conditions had plasma osmolality values of 292 to 308 mOsm/kg with a mean value of 301 mOsm/kg.67 In a study of the effects of sodium bicarbonate infusion in cats, baseline serum osmolality ranged from 290 to 330 mOsm/kg.22 Plasma osmolality can be estimated from the equation:
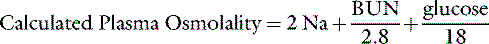
where BUN is blood urea nitrogen. In this equation, the concentrations of urea and glucose in milligrams per deciliter are converted to millimoles per liter by the conversion factors 2.8 and 18. The measured osmolality should not exceed the calculated osmolality by more than 10 mOsm/kg.42,149 If it does, an abnormal osmolal gap is said to be present. This occurs when an unmeasured solute (i.e., one not accounted for in the equation) is present in large quantity in plasma (e.g., mannitol or metabolites of ethylene glycol) or when hyperlipemia or hyperproteinemia results in pseudohyponatremia (see section on Hyponatremia with Normal Plasma Osmolality).42,50,56
Specific gravity
The term specific gravity refers to the ratio of the weight of a volume of liquid to the weight of an equal volume of distilled water. Specific gravity depends not only on the number of particles present in the solution but also on their molecular weight. The clinician can easily measure specific gravity with a hand-held refractometer. Multiplying the last two digits of the urine specific gravity (USG) by 36 gives a rough estimate of urine osmolality in dogs.71 This rule may be misleading if the urine sample contains a large amount of high-molecular-weight solute, because substances with high molecular weights have a greater effect on specific gravity than on osmolality. The effects on urine osmolality of some solutes are shown in Table 3-2.
Table 3-2 Effect of Selected Solutes on Urine Osmolality⁎
| Substance | Molecular Mass (da) | Contribution to Osmolality (mOsm/kg) |
|---|---|---|
| Albumin | 69,000 | 0.144 |
| Diatrizoate ion | 613 | 16.313 |
| Glucose | 180 | 55.555 |
⁎ 1.0 g/dL of each of the listed solutes added to distilled water would increase specific gravity by 0.010, but would have the effects on osmolality shown in the table.
Tonicity or effective osmolality
Changes in the osmolality of ECF may or may not initiate movement of water between the intracellular and extracellular compartments. A change in the concentration of permeant solutes (e.g., urea, ethanol) does not cause movement of water because these solutes are distributed equally throughout total body water (TBW). A change in the concentration of impermeant solutes (e.g., glucose, sodium) does cause movement of water because such solutes do not readily cross cell membranes. Tonicity refers to the ability of a solution to initiate water movement and is dependent on the presence of impermeant solutes in the solution.41 Thus, tonicity may be thought of as effective osmolality. A solution is hypertonic to a reference solution from which it is separated by a semipermeable membrane if its concentration of impermeant solutes is greater than that of the reference solution. A solution is hypotonic to the reference solution if its concentration of impermeant solutes is less than that of the reference solution. A solution is isotonic to the reference solution if its concentration of impermeant solutes equals that of the reference solution.
Tonicity or effective osmolality may be estimated as Posm − BUN/2.8. Consider a dog with the following laboratory values: serum sodium, 125 mEq/L; BUN, 280 mg/dL; and glucose, 90 mg/dL. This patient is hyponatremic and azotemic and has plasma hyperosmolality (calculated plasma osmolality = 355 mOsm/kg) but hypotonicity (effective plasma osmolality = 255 mOsm/kg). Clinical measurement of osmolality by freezing-point depression osmometry does not distinguish between permeant and impermeant solutes and thus does not provide direct information about the tonicity of a solution.
Diuresis
The term diuresis refers to urine flow that is greater than normal (i.e., >1 to 2 mL/kg/hr in dogs and cats). The term solute, or osmotic, diuresis refers to increased urine flow caused by excessive amounts of nonreabsorbed solute within the renal tubules (e.g., polyuria associated with diabetes mellitus, administration of mannitol). During osmotic diuresis, urine osmolality approaches plasma osmolality. The term water diuresis refers to increased urine flow caused by decreased reabsorption of solute-free water in the collecting ducts (e.g., polyuria associated with psychogenic polydipsia or diabetes insipidus). During water diuresis, urine osmolality is less than plasma osmolality.
The term isosthenuria refers to urine with an osmolality equal to that of plasma, and hyposthenuria refers to urine with an osmolality less than that of plasma. The term hypersthenuria, or baruria, refers to urine with an osmolality greater than that of plasma, but this term is rarely used and only to describe urine that is very concentrated.
Types of dehydration
Dehydration occurs when fluid loss from the body exceeds fluid intake. Dehydration may be classified according to the type of fluid lost from the body and the tonicity of the remaining body fluids. Pure water loss and loss of hypotonic fluid result in hypertonic dehydration because the tonicity of the remaining body fluids is increased. Loss of fluid with the same osmolality as that of ECF results in isotonic dehydration, because there is no osmotic stimulus for water movement and the remaining body fluids are unchanged in tonicity. Loss of hypertonic fluid or loss of isotonic fluid with water replacement results in hypotonic dehydration because the remaining body fluids become hypotonic. The types of dehydration and their relative effects on the volume and tonicity of the intracellular and extracellular compartments are shown in Figure 3-1.

Serum sodium concentration
The serum sodium concentration is an indication of the amount of sodium relative to the amount of water in the ECF and provides no direct information about total body sodium content. Patients with hyponatremia or hypernatremia may have decreased, normal, or increased total body sodium content. An increased serum sodium concentration (hypernatremia; >155 mEq/L in dogs or >162 mEq/L in cats) implies hyperosmolality, whereas a decreased serum sodium concentration (hyponatremia; <140 mEq/L in dogs or <149 mEq/L in cats) usually, but not always, implies hypoosmolality. Hyponatremia develops when the patient is unable to excrete ingested water or when urinary and insensible fluid losses have a combined osmolality greater than that of ingested or parenterally administered fluids. Hypernatremia develops when water intake has been inadequate, when the lost fluid is hypotonic to ECF, or when an excessive amount of sodium has been ingested or administered parenterally.
Normal physiology
Renal handling of sodium
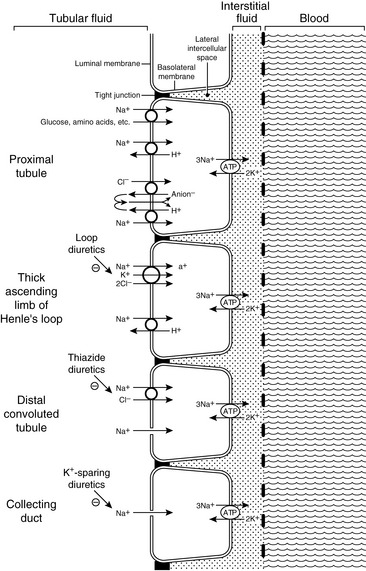
Sodium is filtered by the glomeruli and reabsorbed by the renal tubules. The metabolic energy (i.e., adenosine triphosphate [ATP]) for sodium transport in the kidneys is required by Na+, K+-adenosinetriphosphatase (Na+, K+-ATPase) in the basolateral membranes of the tubular cells. This enzyme translocates sodium from the cytoplasm of the tubular cells to the peritubular interstitium and maintains a low intracellular concentration of sodium, which promotes sodium entry into the cell at the luminal surface.
Approximately 67% of the filtered load of sodium is reabsorbed isosmotically with water in the proximal tubules. In the early proximal tubule, sodium crosses the luminal membrane by cotransport with glucose, amino acids, and phosphate and in exchange for H+ ions via the luminal Na+-H+ antiporter (during the latter process HCO3− is reabsorbed). Reabsorption of water and sodium with HCO3− and other solutes in this segment of the nephron increases the Cl− concentration in tubular fluid and facilitates Cl− reabsorption later in the proximal tubule. In the late proximal tubule, sodium is reabsorbed primarily with Cl−. In this region, the luminal Na+-H+ antiporter works in parallel with a luminal Cl−-anion− antiporter, and the net effect is NaCl reabsorption (H+ anion− is recycled back and forth across the membrane).
Approximately 25% of the filtered load of sodium is reabsorbed in the loop of Henle, primarily in the thick ascending limb. In the thin descending and ascending limbs of the Henle loop, sodium and Cl− are passively reabsorbed. In the thick ascending limb, sodium crosses the luminal membranes via the Na+-H+ antiporter and by an Na+-K+-2Cl− cotransporter.123 This Na+-K+-2Cl− cotransporter is the site of action of the loop diuretics furosemide and bumetanide. There is a strong electrochemical gradient for Na+ entry across the luminal membrane in this region (i.e., strongly lumen-positive transepithelial potential difference and high luminal sodium concentration).
Approximately 5% of the filtered load of sodium is reabsorbed in the distal convoluted tubule and connecting segment. In the early distal tubule (up to the connecting segment), sodium crosses the luminal membrane by means of an Na+-Cl− cotransporter. This cotransporter is inhibited by the thiazide diuretics.37
Approximately 3% of the filtered load of sodium is reabsorbed in the collecting ducts, and this segment of the nephron is responsible for altering sodium reabsorption in response to dietary fluctuations. In the late distal tubule (so-called connecting segment) and collecting ducts, sodium enters passively through Na+ channels in the luminal membranes of the principal cells.127,147 This movement of Na+ generates a lumen-negative transepithelial potential difference that facilitates Cl− reabsorption. The Na+ channel in the principal cells is blocked by the diuretics amiloride and triamterene. One of the main effects of aldosterone is to increase the number of open luminal Na+ channels in the cortical collecting ducts, thus altering sodium reabsorption in response to changes in dietary sodium intake. The renal tubular mechanisms for sodium reabsorption are summarized in Figure 3-2.
Renal regulation of sodium balance
ECF volume is directly dependent on body sodium content. The body is able to sense and respond to very small changes in sodium content. The adequacy of body sodium content is perceived as the fullness of the circulating blood volume. The term effective circulating volume has been used to refer to the relative fullness of the circulating portion of the extracellular compartment as perceived by the body. There are several sensors in the afferent limb of the body’s regulatory system for control of sodium balance (see Table 3-1). Low-pressure mechanoreceptors (i.e., volume receptors) in the cardiac atria and pulmonary vessels and high-pressure baroreceptors (i.e., pressure receptors) in the aortic arch and carotid sinus play a primary role in the body’s ability to sense the adequacy of the circulating volume. Within the kidneys, the juxtaglomerular apparatus responds to changes in perfusion pressure with changes in renin production and release. Less well characterized are receptors in the liver and the central nervous system that may contribute to sodium homeostasis.
The kidneys constitute the primary efferent limb of sodium control and regulate sodium balance by excreting an amount of sodium each day equal to that ingested. There are several overlapping control mechanisms for regulation of renal handling of sodium. This redundancy serves to protect against sodium imbalance should one control mechanism fail. The two points of control for sodium balance in the kidneys are glomerular filtration and tubular reabsorption. Autoregulation maintains renal blood flow and glomerular filtration rate (GFR) relatively constant despite fluctuations in systemic arterial pressure; thus, the filtered load of sodium is also kept relatively constant (see Chapter 2).
Glomerulotubular Balance
Even slight changes in GFR have the potential to have drastic effects on sodium balance if the absolute amount of sodium reabsorbed by the tubules remains constant. Consider a normal 10-kg dog in sodium balance with a serum sodium concentration of 145 mEq/L and a GFR of 4 mL/min/kg. The daily filtered load of sodium in this dog would be 57.6 L/day × 145 mEq/L = 8352 mEq/day. If the kidneys reabsorb 99.5% of the filtered load of sodium (8310 mEq/day), the amount excreted in the urine is 42 mEq/day. Consider what would happen if there was a primary (i.e., spontaneous) increase in GFR of only 1%, but the absolute amount of sodium reabsorbed remained unchanged. The filtered load of sodium would be 58.2 L/day × 145 mEq/L = 8439 mEq/day, but the amount reabsorbed would remain 8310 mEq/day. This would result in the excretion of 129 mEq/day, an amount three times that normally excreted. Under these conditions, the dog would develop negative sodium balance. Glomerulotubular balance prevents this scheme of events from occurring.
If spontaneous (primary) fluctuations in GFR occur, the absolute tubular reabsorption of filtered solutes changes in a similar direction. Thus, the fraction of the filtered load that is reabsorbed remains relatively constant despite spontaneous changes in GFR. This principle is called glomerulotubular balance, and its mechanisms are incompletely understood.
One mechanism is related to the fact that much of the sodium in the proximal tubules is reabsorbed along with several other solutes (e.g., glucose, amino acids, phosphate, and bicarbonate). A spontaneous increase in GFR increases the filtered load of all of these solutes, and their increased concentration in the proximal tubule enhances sodium reabsorption. Changes in peritubular capillary hydrostatic and oncotic pressures probably also play an important role in glomerulotubular balance. If GFR spontaneously increases without a change in renal plasma flow (RPF) (i.e., the filtration fraction increases), the blood leaving the efferent arterioles has lower hydrostatic pressure and higher oncotic pressure, thus favoring water and solute reabsorption in the proximal tubules (Fig. 3-3). Autoregulation (see Chapter 2) also contributes to glomerulotubular balance. When renal perfusion pressure is increased, afferent arteriolar constriction prevents transmission of the increased hydrostatic pressure to the glomerular capillaries and minimizes any increase in GFR and filtered solute load.
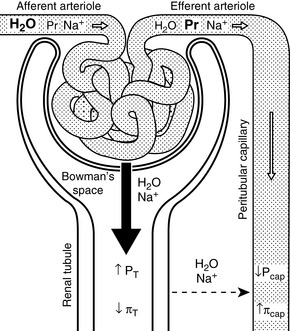
Figure 3-3 Effects of changes in Starling forces on tubular reabsorption of water and sodium. If glomerular filtration rate (GFR) increases without a change in renal plasma flow (RPF) (or if RPF decreases more than GFR as may occur in dehydration), the filtration fraction (GFR/RPF) will increase (i.e., more water and sodium will be filtered from the glomeruli into the Bowman space). This sequence of events will result in lower hydrostatic pressure (Pcap) and higher oncotic pressure (πcap) in the peritubular capillaries (downstream from the glomerular capillaries) and higher hydrostatic pressure (PT) and lower oncotic pressure (πT) in the renal tubules (downstream from the Bowman space). These changes in Starling forces will facilitate water and sodium reabsorption from the tubular fluid into the peritubular capillaries, thus minimizing loss of water and sodium in the urine.
(Drawing by Tim Vojt.)
Ingestion of a sodium load causes thirst, water consumption, and expansion of ECF volume. These events lead to a compensatory (secondary) increase in GFR by increasing hydrostatic pressure and decreasing oncotic pressure in the glomerular capillaries. Increased stretching of the afferent arterioles decreases renin secretion (and ultimately angiotensin II production). Volume expansion also causes increased atrial stretch, release of atrial natriuretic peptide, and natriuresis.
There is a paradox here. How can an increase in GFR in one situation cause an increase in the tubular reabsorption of sodium and in another situation cause a decrease in the tubular reabsorption of sodium? The answer to the paradox lies in the fundamental difference between the kidneys’ reaction to a spontaneous (primary) increase and their reaction to a compensatory (secondary) increase in GFR. Glomerulotubular balance is evoked in the former but not the latter situation.
Aldosterone
Changes in renal reabsorption of sodium in response to dietary fluctuations in sodium intake are mediated by the hormone aldosterone, which is synthesized in the zona glomerulosa of the adrenal cortex. The production and release of aldosterone are stimulated by angiotensin II, hyperkalemia, and adrenocorticotropic hormone (ACTH). Its release is inhibited by dopamine and atrial natriuretic peptide. Aldosterone increases sodium reabsorption by increasing the number and activity of open sodium channels in the luminal membranes of the principal cells in the collecting ducts.
Peritubular Capillary Factors (Starling Forces)
Increased sodium intake leads to expansion of the ECF volume and compensatory increases in both GFR and RPF (i.e., the filtration fraction remains unchanged). This increases hydrostatic pressure and decreases oncotic pressure in the peritubular capillaries, thus reducing sodium and water reabsorption in the proximal tubules. Decreased sodium intake leads to volume contraction. In this setting, RPF decreases more than GFR (i.e., the filtration fraction increases). This results in decreased hydrostatic and increased oncotic pressures in the peritubular capillaries and enhanced proximal tubular reabsorption of sodium and water (see Fig. 3-3).
Catecholamines
Catecholamine-induced vasoconstriction usually affects the efferent more than the afferent arterioles. The resultant increase in filtration fraction alters peritubular capillary hemodynamics so as to favor water and sodium reabsorption (i.e., decreased hydrostatic pressure and increased oncotic pressure). Catecholamines also directly stimulate proximal tubular sodium reabsorption through an α1-adrenergic effect and stimulate renin release from the granular cells of the juxtaglomerular apparatus through a β1-adrenergic effect. The angiotensin II ultimately produced also stimulates proximal tubular sodium reabsorption. The direct effects of catecholamines on proximal tubular sodium reabsorption are important because they offset the tendency of the increase in systemic arterial pressure to cause pressure natriuresis (see the Pressure Natriuresis section).
Angiotensin II
Decreased perfusion pressure in the afferent arterioles increases renin release from the granular cells of the juxtaglomerular apparatus and initiates the cascade of events leading to production of angiotensin II. Angiotensin II-induced vasoconstriction causes efferent more than afferent arteriolar constriction, which results in an increase in filtration fraction and changes in peritubular capillary Starling forces (decreased hydrostatic pressure and increased oncotic pressure) that facilitate proximal tubular reabsorption of sodium and water (see Fig. 3-3). Angiotensin II also directly stimulates the Na+-H+ antiporter in the proximal tubules, which facilitates sodium reabsorption and stimulates secretion of aldosterone from the adrenal gland.
Atrial Natriuretic Peptide
Atrial natriuretic peptide is one member of a family of natriuretic proteins that also includes brain natriuretic peptide (which ironically predominates in the cardiac ventricles) and C-type natriuretic peptide in the central nervous system.97 Atrial natriuretic peptide is synthesized and stored in atrial myocytes until it is released in response to atrial distention caused by volume expansion. It has a number of effects that facilitate renal excretion of sodium. Atrial natriuretic peptide causes dilation of the afferent arterioles and constriction of the efferent arterioles, leading to a primary increase in the GFR. It relaxes mesangial cells, resulting in an increase in the glomerular surface area available for filtration. Atrial natriuretic peptide also inhibits sodium reabsorption in the cortical and inner medullary collecting ducts and inhibits renin secretion, thereby decreasing production of angiotensin II and limiting the effects of angiotensin II on proximal tubular sodium reabsorption. Finally, it inhibits aldosterone secretion by adrenal zona glomerulosa.
Pressure Natriuresis
Renal sodium excretion and water excretion are markedly increased when renal arterial pressure increases even slightly without a change in the GFR. The mechanism for pressure natriuresis appears to be entirely intrarenal and does not require neural or endocrine input (i.e., it occurs in the isolated denervated kidney). The effectors of sodium balance are summarized in Table 3-3.

Regulation of water balance
The osmolality of ECF and serum sodium concentration are regulated by adjusting water balance. Osmoreceptors in the hypothalamus constitute the afferent limb (sensors) for regulation of water balance. Vasopressin (antidiuretic hormone) release is stimulated when the osmoreceptors shrink in response to plasma hyperosmolality and is inhibited when they swell in response to plasma hypoosmolality. Vasopressin (water output) and thirst (water input) constitute the efferent limb (effectors) for the regulation of water balance (see Table 3-1).
Vasopressin (Antidiuretic Hormone)
Vasopressin (antidiuretic hormone [ADH]) is a nine-amino acid peptide synthesized in neurons of the supraoptic and paraventricular nuclei in the hypothalamus (Fig. 3-4). It travels down the axons of these neurons and is released into the circulation at the level of the neurohypophysis.
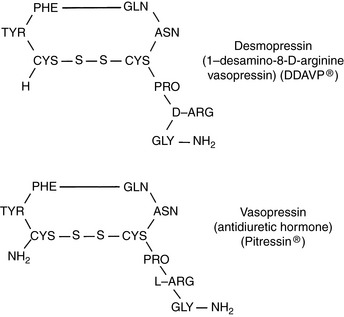
Figure 3-4 Comparison of the chemical structures of desmopressin and vasopressin. PHE, Phenylalanine; TYR, tyrosine; GLN, glutamine; ASN, asparagine; CYS, cysteine; PRO, proline; ARG, arginine; GLY, glycine.
Vasopressin increases the reabsorption of water in the collecting ducts of the kidneys and increases the permeability of the medullary collecting ducts to urea.1 Vasopressin attaches to V2 receptors on the basolateral membranes of the principal cells of the cortical and medullary collecting ducts. The hormone-receptor complex activates a guanine nucleotide regulatory protein (Gs), resulting in replacement of guanosine diphosphate (GDP) with guanosine triphosphate (GTP) and stimulation of adenyl cyclase in the cell membrane. Formation of cyclic adenosine monophosphate (cAMP) results in activation of protein kinase A, which in turn phosphorylates a specific serine residue on subunits of the tetrameric aquaporin 2 (AQP2) proteins found in membranes of subapical vesicles in the cytoplasm of the principal cells. Phosphorylation results in trafficking and insertion of AQP2 water channels into the luminal membranes of the principal cells.121,164 When vasopressin is absent or in low concentration, AQP2 channels are removed from the luminal membrane by endocytosis. Aquaporin 3 (AQP3) and 4 (AQP4) channels are found in the basolateral membranes of the principals cells and represent exit pathways for water that enters the cells via the luminal AQP2 channels. The AQP3 channel is found in the cortical and outer medullary collecting ducts, whereas AQP4 is located primarily in the inner medullary collecting ducts. In the absence of vasopressin, urine osmolality can be decreased to as low as 50 mOsm/kg by continued reabsorption of sodium without water as tubular fluid passes down the collecting ducts. The V1A receptors are located in vascular smooth muscle and cause vasoconstriction when AVP binds to them. V1B receptors are found primarily in the hypothalamus where AVP binding leads to increased secretion of corticotropin.
The effect of vasopressin on urea reabsorption may be important in the pathogenesis of medullary washout of solute in chronic polyuric states. Chronic diuresis can lead to depletion of urea from the medullary interstitium by suppression of vasopressin release and impaired urea reabsorption in the medullary collecting ducts. During antidiuresis, urea may constitute more than 40% of the medullary solute. During diuresis, however, it may constitute less than 10% of the medullary solute.17,98 The urinary concentrating mechanism is discussed in Chapter 2.
Stimuli for Vasopressin Release
The major stimulus for vasopressin release is hypertonicity of plasma reaching the osmoreceptors of the hypothalamus. The threshold for vasopressin release in humans corresponds to a plasma osmolality of 280 mOsm/kg, and similar or slightly higher threshold values have been observed in healthy experimental dogs.31,135,137 Below this osmolality, vasopressin release is suppressed, and urine is maximally diluted. One hour after oral administration of water at 40 mL/kg, normal dogs developed a mean urine osmolality of 132 mOsm/kg (range, 68 to 244 mOsm/kg).67 In humans, the release of vasopressin is maximal at a plasma osmolality of 294 mOsm/kg, and at this plasma osmolality the thirst mechanism becomes operative.137 Thus, changes in plasma osmolality as small as 1% to 2% above normal lead to maximal vasopressin release. The gain of the system is such that a 1 mOsm/kg increase in plasma osmolality leads to an almost 100 mOsm/kg increase in urine osmolality. The vasopressin system curtails water excretion, but further defense against hypertonicity requires a normal thirst mechanism and access to water. The thirst mechanism has both osmoreceptors and volume receptors. The volume receptors for the thirst mechanism are stimulated by angiotensin II and may be under control of the renin-angiotensin system.108
The next most important stimulus for vasopressin release is volume depletion sensed by baroreceptors in the left atrium, aortic sinus, and carotid sinuses. A decrease in blood volume of 5% to 10% lowers the threshold for vasopressin release and increases the sensitivity of the osmoregulatory mechanism (Fig. 3-5).59,137 Nonosmotic stimulation of vasopressin by actual or perceived volume depletion plays a major role in the generation and perpetuation of hyponatremia in states of true volume depletion and in some conditions (e.g., heart failure, liver failure, nephrotic syndrome) associated with hypervolemia (see Hypovolemic Hyponatremia and Hypervolemic Hyponatremia sections).
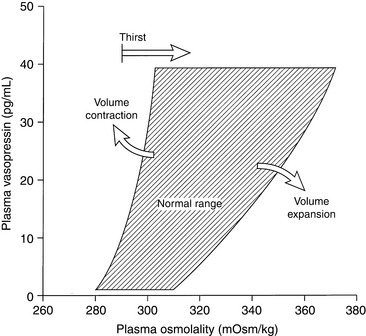
Figure 3-5 Relationship between plasma osmolality and plasma vasopressin concentration. Volume depletion lowers the threshold for vasopressin release and increases the sensitivity of the osmoregulatory system, whereas volume expansion has the opposite effect.
(Drawing by Tim Vojt.)
Other stimuli for vasopressin release include nausea, pain, and emotional anxiety. Many drugs and some electrolyte disturbances affect the release and renal action of vasopressin. The effects of some of these are depicted in Figure 3-6.
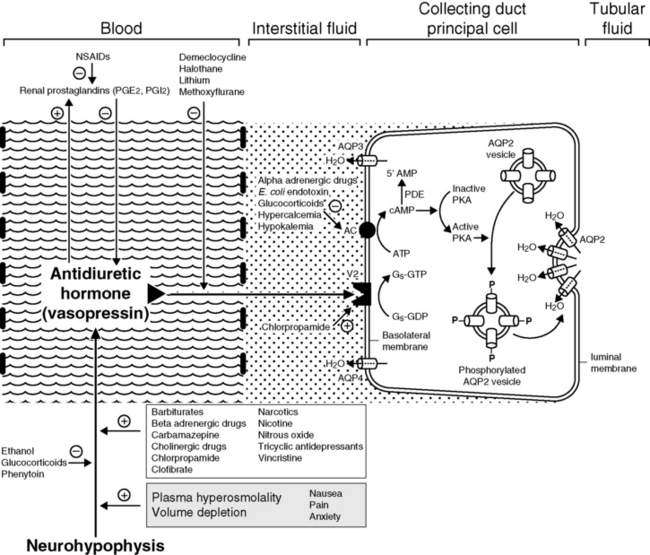
Figure 3-6 Effects of selected drugs and electrolytes on vasopressin release and action. AC, Adenyl cyclase; 5’-AMP, 5’-adenosine monophosphate; AQP, aquaporin; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; Gs, stimulatory guanine nucleotide regulatory protein; GDP, guanosine diphosphate; GTP, guanosine triphosphate; NSAIDs, nonsteroidal anti-inflammatory drugs; PDE, phosphodiesterase; PGE, prostaglandin E; PGI, prostacyclin; PKA, protein kinase A.
(Drawing by Tim Vojt.)
Role of the Kidneys in Water Balance
Three conditions must be met for the kidneys to excrete a water load normally. First, there must be adequate delivery of tubular fluid to distal diluting sites (ascending limb of Henle’s loop) where NaCl is removed without water, rendering the tubular fluid hypotonic to the medullary interstitium. Adequate distal delivery requires a normal RPF, normal GFR, and normal isosmotic reabsorption of sodium and water from the proximal tubules mediated by aquaporin 1 (AQP1) channels in the luminal and basolateral membranes of these cells. In the presence of volume depletion, RPF is usually decreased more than the GFR, and enhanced proximal tubular reabsorption of sodium and water may result from changes in postglomerular hemodynamics (see Fig. 3-3). These factors may prevent adequate distal delivery of tubular fluid for dilution.
Second, the ascending limb of Henle’s loop must function normally. That is, NaCl must be removed from this segment of the nephron without water. Loop diuretics (e.g., furosemide and ethacrynic acid) impair NaCl removal from this portion of the nephron, and some interstitial renal diseases may disrupt the normal architecture of this region, leading to impaired dilution of tubular fluid in the ascending limbs of Henle’s loop.
Last, in the absence of vasopressin, the collecting ducts must remain impermeable to water throughout their course. If any of these conditions is not met, a disorder of water excretion and a state of ECF hypotonicity and hyponatremia may result.
In the absence of vasopressin, the collecting ducts remain impermeable to water, the urine becomes maximally dilute, and polyuria develops. Hypertonicity and hypernatremia occur if the animal is unable to drink enough water to balance the tremendous loss of water in the urine. Hypertonicity and hypernatremia also may develop in states of osmotic diuresis (e.g., diabetes mellitus, mannitol administration, chronic renal failure, postobstructive diuresis). Urine osmolality approaches plasma osmolality during osmotic diuresis, and the solute responsible for the diuresis displaces sodium and other electrolytes in urine.51 Hypertonicity develops to the extent that displaced sodium remains in the ECF.
Defense Against Hypotonicity
It is crucial to the survival of the animal that the brain be protected against changes in plasma tonicity, because an increase in brain water content of more than 10% is incompatible with life.151 The fact that animals with chronic hyponatremia may have serum sodium concentrations that are 10% or more below normal attests to the brain’s ability to adapt to hypotonicity. For example, based on osmotic considerations alone, a decrease in serum sodium concentration from 145 to 132 mEq/L would correspond to an increase in intracellular water of 10%. During acute hypotonicity, water moves into the brain. The increase in hydrostatic pressure in the interstitial compartment of the brain immediately forces sodium-containing ECF into the cerebrospinal fluid. This movement of fluid out of the brain occurs within minutes and limits the change in brain water content to much less than would be anticipated based on osmotic considerations alone.151 During the first 24 hours of hypotonicity, movement of potassium out of cells also contributes substantially to the protection of the brain from an acute decrease in plasma osmolality. After 24 to 48 hours, a reduction in the cellular content of organic solutes contributes to the brain’s defense against hypotonicity. These organic osmolytes are substances that can be used by cells to maintain intracellular tonicity without having adverse effects on cellular metabolism and include amino acids (e.g., taurine, glutamate, and glutamine), methylamines (e.g., phosphocreatine), and polyols (e.g., myoinositol).6,134 The very devices that protect the brain against plasma hypotonicity predispose it to injury when hyponatremia is corrected. Solutes lost during adaptation must be recovered, and this process requires several days. If correction of hyponatremia proceeds more quickly than recovery of lost solutes can occur, a devastating complication of treatment called osmotic demyelination syndrome (myelinolysis) may occur (see Treatment of Hyponatremia section).
Clinical approach to the patient with hypernatremia
Hypernatremia is less common than hyponatremia. Intense thirst normally protects against development of hypernatremia unless water is not available or a neurologic disorder is present that either prevents access to water or interferes with recognition of thirst. All clinical conditions associated with hypernatremia reflect hyperosmolality and hypertonicity of the ECF if the solute in question is impermeant. A deficit of pure water, loss of hypotonic fluids, or gain of sodium can cause hypertonicity of the ECF and hypernatremia. The causes of hypernatremia are listed in Box 3-1, and the clinical approach to the patient with hypernatremia is outlined in Figure 3-7.
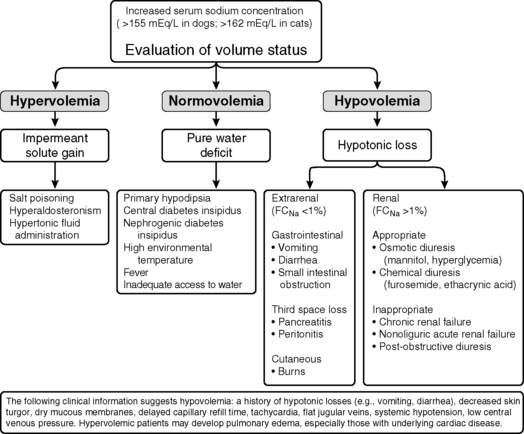
Figure 3-7 Clinical approach to the patient with hypernatremia. FCNa, Fractional clearance of sodium.
Pure water loss
When a deficit of pure water develops, the ECF becomes hypertonic in relation to the intracellular fluid (ICF), and osmotic forces cause movement of water from the intracellular to the extracellular compartment. The result is that the volume loss is shared proportionately between the extracellular and intracellular compartments. Approximately two thirds of the volume loss comes from the intracellular compartment and one third from the extracellular compartment. Plasma volume is one fourth of the ECF, and thus one twelfth of the volume loss (¼ × ⅓) is derived from the intravascular space. The oncotic pressure generated by plasma proteins favors retention of water within vessels, and the plasma compartment may not share proportionately in the volume loss.41 As a result of these factors, volume depletion is usually not a clinical feature of pure water loss. It is almost impossible for a conscious animal with an intact thirst mechanism and access to water to develop hypertonicity caused by pure water loss. Thus, hypertonicity associated with pure water loss usually implies that water intake has been defective.
Consider a normal 10-kg dog with a serum osmolality of 300 mOsm/kg. We assume that TBW is 60% of body weight, with 40% being intracellular and 20% extracellular, and that the major extracellular (i.e., NaCl) and intracellular (i.e., KCl) solutes are impermeant. The number of osmoles of solute in ECF would be 2 L × 300 mOsm/kg = 600 mOsm, and the number in ICF would be 4 L × 300 mOsm/kg = 1200 mOsm. Without access to drinking water, a loss of 1 L of pure water from ECF would cause water to move from ICF to ECF so as to equalize osmolality between the compartments according to the following equation:
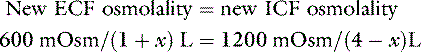
where x is the volume of water moving between compartments:
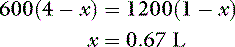
The new volumes and osmolalities are:
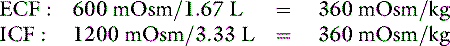
Note that the intracellular compartment has lost an amount equal to two thirds of the water deficit (0.67 L) and that the final ECF volume (1.67 L) is lower than the original volume (2 L) by an amount equal to one third of the total water deficit (0.33 L). Thus, the two compartments have shared proportionately in the water loss. These changes are depicted in Figure 3-8.
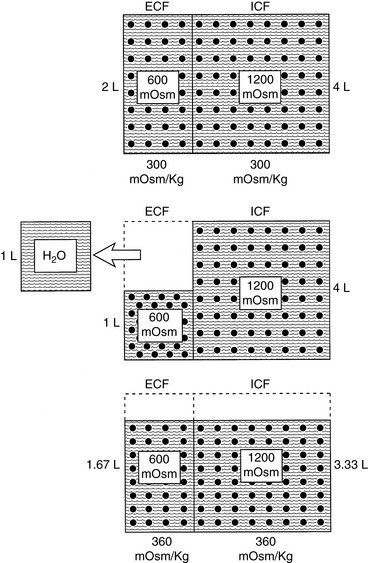
Figure 3-8 Effect of loss of 1 L of water on volume and tonicity of extracellular fluid (ECF) and intracellular fluid (ICF).
(Drawing by Tim Vojt.)
Development of a pure water deficit is uncommon in small animal medicine. The main causes of hypertonicity related to pure water deficit are hypodipsia, caused by neurologic disease, and diabetes insipidus, which represents abnormal renal loss of water. Other causes of pure water deficit include respiratory losses during exposure to high environmental temperature (e.g., panting), fever, and inadequate access to water (e.g., frozen water bowl, inattentive owner).
Rarely, chronic hypernatremia may occur in fully conscious animals that have access to water. In these cases, abnormal osmoregulation of ADH release caused by underlying hypothalamic lesions results in hypodipsia. Animals that are unable to obtain water because central nervous system disease has resulted in an altered sensorium may also be hypernatremic; but in these instances, the hypernatremia is simply a result of water deprivation. Hypodipsic hypernatremia related to defective osmoregulation of ADH has been reported in a dog with hydrocephalus and normal pituitary function.31 In normal individuals, administration of hypertonic saline increases plasma osmolality and simultaneously causes volume expansion. Osmoreceptors are stimulated by hyperosmolality but inhibited by volume expansion. Normally, the response to hyperosmolality takes precedence, and ADH secretion increases, resulting in decreased urine volume and increased urine osmolality. The affected dog experienced increased urine volume and decreased urine osmolality in response to an infusion of hypertonic saline, indicating defective osmoreceptor function as observed in human patients with hypodipsic hypernatremia. Similarly, destruction of osmoreceptors in the hypothalamus was thought to be responsible for adipsia and hypernatremia in a dog with focal granulomatous meningoencephalitis.104 Weakness and polymyopathy have been reported in a young cat with hypodipsia, hypernatremia, and hypertonicity associated with hydrocephalus and hypopituitarism, and hypernatremia, adipsia, and diabetes insipidus have been observed in a young dalmatian dog with dysplasia of the rostral diencephalon.5,34 Hypernatremia also has been reported in a dog63 and cat115 with central nervous system lymphoma.
Hypodipsia, hypernatremia, and hypertonicity caused by an abnormal thirst mechanism have been reported in young female miniature schnauzers and in a young Great Dane.27,70,76,112,159 One miniature schnauzer with hypodipsic hypernatremia had severe behavioral disturbances, and holoprosencephaly was found at necropsy.153 Another had dysgenesis of the corpus callosum and other forebrain structures.112 Grossly visible neuroanatomic abnormalities were not identified in a previous report.27 Whether a spectrum of neuroanatomic abnormalities exists in these dogs (which appear to have a form of congenital adipsic hypernatremia) is not known. Infusion of hypertonic saline has been shown to lead to an increase in urine volume and a decrease in urine osmolality compatible with defective osmoregulation of ADH.27 Clinical signs in affected dogs are associated with hypertonicity and include anorexia, lethargy, weakness, disorientation, ataxia, and seizures. Affected dogs can be managed clinically by addition of water to their food, but hypernatremia and neurologic dysfunction recur whenever water supplementation is discontinued. In a Norwegian elkhound with adipsic hypernatremia, the adipsia resolved spontaneously at 2 years of age,62 and an adipsic Labrador retriever with hypothyroidism responded to treatment with levothyroxine.81
Central or pituitary diabetes insipidus (CDI) is caused by a partial or complete lack of vasopressin production and release from the neurohypophysis.65 It may result from trauma or neoplasia or may be idiopathic in dogs and cats.⁎ Visceral larva migrans also has been reported to cause CDI in a dog.99 In one dog with hypernatremia, hypertonicity, and gastric dilation-volvulus, CDI was present and caused by neurohypophyseal atrophy secondary to a cystic craniopharyngeal duct.36 Congenital CDI is rare58,86,163 but has been reported in two sibling Afghan pups.131 Traumatic CDI may be transient in nature. Hypophysectomy for treatment of hyperadrenocorticism results in transient CDI that may take several weeks to resolve.102 Marked hypernatremia occurs in dogs in the first 24 hours after hypophysectomy and can be prevented by prophylactic treatment with desmopressin (DDAVP).64 In the month after surgery, serum sodium concentrations in control dogs were not markedly different from those observed in the DDAVP-treated group, suggesting that the dogs with untreated CDI drank sufficient water to maintain relatively normal plasma osmolality. The transient nature of CDI after hypophysectomy may result from the fact that some of the vasopressin-producing neurons from the hypothalamus terminate in the median eminence.
Animals with CDI have severe polydipsia and polyuria. Their urine typically is hyposthenuric (urine osmolality, 60 to 200 mOsm/kg), but urine osmolality may approach 400 to 500 mOsm/kg in the presence of dehydration. Variability in USG and urine osmolality values at the time of presentation in dogs and cats with diabetes insipidus presumably is related to hydration status and severity of vasopressin deficiency. In one study, dogs were classified as having complete or partial CDI based on the magnitude of increase in their USG and urine osmolality after induction of 5% dehydration.65 Dogs with complete CDI had USG values of 1.001 to 1.007 that did not change substantially after induction of 5% dehydration, whereas dogs with partial CDI had USG values of 1.002 to 1.016 that increased to 1.010 to 1.018 after induction of 5% dehydration. In both groups, there was a substantial (>50%) increase in USG 2 hours after administration of 1 to 5 U of aqueous arginine vasopressin. Affected dogs responded well to administration of DDAVP acetate (1 to 2 drops in both eyes every 12 to 24 hours), but the prognosis was dependent on the underlying cause of CDI. Many older dogs with CDI had tumors in the region of the pituitary gland and developed neurologic signs.
Increased plasma osmolality and hypernatremia may occur in dogs and cats with CDI. These results suggest that some affected dogs and cats do not obtain enough water to maintain water balance and are presented in a hypertonic state. Severe hypernatremia and neurologic dysfunction may occur if the animal cannot maintain adequate water intake.36,133 In contrast, with psychogenic polydipsia, plasma osmolality and serum sodium concentration may be lower than normal at presentation.91 Administration of vasopressin leads to an increase in urine osmolality or specific gravity in dogs and cats with CDI, but the initial response may be less than expected because of renal medullary washout of solute. In one study, USG values increased to 1.018 to 1.022 after vasopressin administration in dogs with complete CDI and to 1.018 to 1.036 in dogs with partial CDI.65
Treatment with vasopressin restores medullary hypertonicity and normal urinary concentrating ability. Historically, vasopressin tannate in oil (pitressin tannate) has been used to treat CDI in small animal practices. The dosage is 3 to 5 U for dogs or 1 to 2 U for cats given intramuscularly or subcutaneously every 24 to 72 hours as needed to control polyuria and polydipsia. To avoid the possibility of water intoxication, it is recommended that the treatment interval be determined by recurrence of polyuria. This product is no longer commercially available.
DDAVP is a structural analogue of vasopressin (see Fig. 3-4) that has a more potent antidiuretic effect than vasopressin but a minimal vasopressive effect and is relatively resistant to metabolic degradation. DDAVP is available as a nasal spray (0.1 mg/mL), injectable solution (4 µg/mL), or tablet for oral administration (0.1 and 0.2 mg). The injectable solution is much more expensive than the nasal spray, and the nasal spray has been used subcutaneously in dogs and in a cat with CDI at a dosage of 1 µg/kg without adverse effects.86,87 Polyuria and polydipsia in a cat with CDI were controlled with 1 µg/kg administered subcutaneously every 12 hours or 1.5 µg/kg administered conjunctivally every 8 hours. One drop of the nasal spray contains 1.5 to 4 µg of DDAVP, and the duration of effect varies from 8 to 24 hours.43 In humans, the bioavailability of DDAVP after oral administration was 0.1% as compared with 3% to 5% after intranasal administration, and gastrointestinal absorption was improved when it was given in a fasted state.46,136 In dogs, an antidiuretic effect was observed even after orally administered doses as low as 50 µg.160
Chlorpropamide is a sulfonylurea hypoglycemic agent that potentiates the renal tubular effects of small amounts of vasopressin and may be useful in management of animals with partial CDI. Its effect may occur by up-regulation of ADH receptors in the kidneys.35 The recommended dosage of chlorpropamide is 10 to 40 mg/kg/day orally, and hypoglycemia is a potential adverse effect. It has been useful in the management of CDI (up to 50% reduction in urine output) in some reports but not in others, possibly because some animals have partial and some have complete CDI.86,140
In the broadest sense, the term nephrogenic diabetes insipidus (NDI) may be used to describe a diverse group of disorders in which structural or functional abnormalities interfere with the ability of the kidneys to concentrate urine (Box 3-2).13,90 Congenital NDI is a rare disorder in small animal medicine.13,80,90 Affected animals are presented at a very young age for severe polyuria and polydipsia. In reported cases, urine osmolality and specific gravity have been in the hyposthenuric range. Affected animals show no response to water deprivation testing, exogenous vasopressin administration, or hypertonic saline infusion. In one case report, the plasma vasopressin concentration was markedly increased.80 Congential NDI in human patients can arise from mutations in the V2 receptor (X-linked recessive inheritance) or from mutations in the AQP2 channel (autosomal recessive inheritance). Low affinity V2 receptors were thought to be responsible for congenital NDI in a family of Siberian huskies.103
Box 3-2 Causes of Nephrogenic Diabetes Insipidus
Thiazide diuretics (chlorothiazide 20 to 40 mg/kg every 12 hours or hydrochlorothiazide 2.5 to 5.0 mg/kg twice a day) have been used to treat animals with CDI and NDI. Diuretic administration results in mild dehydration, enhanced proximal renal tubular reabsorption of sodium, decreased delivery of tubular fluid to the distal nephron, and reduced urine output. Thiazides have been reported to result in a 20% to 50% reduction in urine output in dogs with NDI and in cats with CDI.13,18,86,90,154 In other reports, thiazides were reported to be ineffective in reducing urine output in a dog and a cat with CDI.57,72 Restriction of dietary sodium and protein reduces the amount of solute that must be excreted in the urine each day and thus further reduces obligatory water loss and polyuria. A low-salt diet and hydrochlorothiazide (2 mg/kg orally twice a day) were used successfully to manage a dog with congenital NDI for 2 years.154 The dog’s water consumption decreased from an average of approximately 900 mL/kg/day to 200 mL/kg/day with treatment.
Hypotonic fluid loss
When hypotonic fluid is lost from the extracellular compartment, the osmotic stimulus for water to move from the intracellular to the extracellular compartment is less than the stimulus for water movement created by pure water loss. Thus, hypotonic losses cause a greater reduction in the ECF volume, and the animal is more likely to show clinical signs of volume depletion (e.g., tachycardia, weak pulses, and delayed capillary refill time). As the tonicity of the fluid lost increases toward the normal tonicity of ECF, the volume deficit of the extracellular compartment becomes progressively more severe (Fig. 3-9). In the case of isotonic losses, no osmotic stimulus for water movement is present. The entire loss is borne by the extracellular compartment, and hypovolemic shock may occur if the loss has been of sufficient magnitude (e.g., severe hemorrhage).
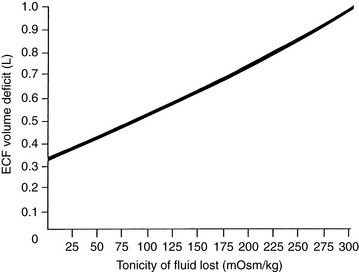
Figure 3-9 Magnitude of extracellular fluid (ECF) volume deficit caused by loss of 1 L of fluid of varying tonicity.
Consider what would occur in the previous example if our 10-kg dog had suffered a loss from the extracellular compartment of 1 L of fluid with an osmolality of 150 mOsm/kg. Such a loss would leave 450 mOsm of solute and 1 L of water in the extracellular compartment. Once again, water moves from the intracellular to the extracellular compartment until the osmolality has been equalized. Thus:
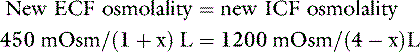
where x is the volume of water moving between compartments:
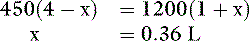
The new volumes and osmolalities are:
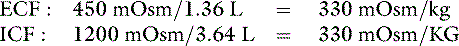
Note that the extracellular volume deficit is more severe than in the previous example of pure water loss (0.64 L vs. 0.33 L). These changes are depicted in Figure 3-10. The more closely the fluid lost approximates ECF in tonicity, the greater the volume loss from the ECF compartment.
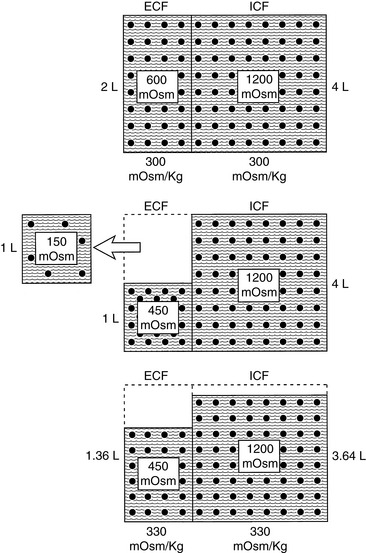
Figure 3-10 Effect of loss of 1 L of hypotonic fluid (150 mOsm/kg) on volume and tonicity of extracellular fluid (ECF) and intracellular fluid (ICF).
(Drawing by Tim Vojt.)
For simplicity, these examples are based on many assumptions that in reality may not be true. For example, TBW is not 60% of body weight in all individuals, the number of osmoles in the ECF may have been altered by electrolyte losses not detected clinically, the effects of hydrostatic forces resulting from extracellular volume depletion have not been considered, some solutes may not be strictly impermeant, and compensatory physiologic responses have not been considered. Nonetheless, such calculations are helpful in understanding the pathophysiology of hypertonic states, and they provide useful clinical approximations.
Hypotonic fluid losses are the most common type encountered in small animal medicine. They may be classified as extrarenal (e.g., gastrointestinal, third-space loss, and cutaneous) or renal. Causes of gastrointestinal losses include vomiting, diarrhea, and small intestinal obstruction; causes of third-space losses include pancreatitis and peritonitis. Cutaneous losses are usually not clinically important in dogs and cats. Eccrine sweat glands are limited to the foot pads and serve no thermoregulatory function, and burns are encountered uncommonly in small animal practice. Renal losses may result from osmotically (e.g., diabetes mellitus, mannitol) or chemically (e.g., furosemide, corticosteroids) induced diuresis or from defective urinary concentrating ability related to intrinsic renal disease (e.g., chronic renal failure, nonoliguric acute renal failure, postobstructive diuresis).
Gain of impermeant solute
Gain of impermeant solute is uncommon in small animal medicine. The addition of a sodium salt to ECF causes hypernatremia, whereas gain of an impermeant solute that does not contain sodium (e.g., glucose and mannitol) initially causes hyponatremia because water is drawn into ECF. However, hypernatremia occurs as osmotic diuresis develops because urine osmolality approaches plasma osmolality and the sodium-free solute replaces sodium in urine. The sodium displaced from urine remains in the ECF and contributes to hypernatremia.
The development of hypertonicity as a result of excessive salt ingestion is unlikely if the animal in question has an intact thirst mechanism and access to water. The addition of impermeant solute without water expands the extracellular compartment at the expense of the intracellular compartment as water moves from ICF to ECF to equalize osmolality. This volume overload may lead to pulmonary edema if the patient has underlying cardiac disease.
Consider again our example of the 10-kg dog. The addition of 200 mOsm of solute to the ECF without any water would be equivalent to ingestion of 5.85 g of sodium chloride (5.85 g NaCl = 100 mmol Na and 100 mmol Cl). The addition of this impermeant solute to ECF causes movement of water from the intracellular to extracellular compartments until osmolality has been equalized. Thus:
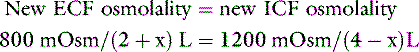
where x is the volume of water moving between compartments:
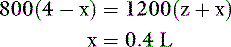
The new volumes and osmolalities are:
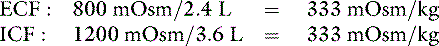
Note that ECF volume has been expanded by 0.4 L and that this volume has been derived from ICF. In the normal animal, this expansion of the extracellular compartment leads to natriuresis, and the volume deficit is repaired by ingestion of water in response to plasma hyperosmolality. These changes are depicted in Figure 3-11.
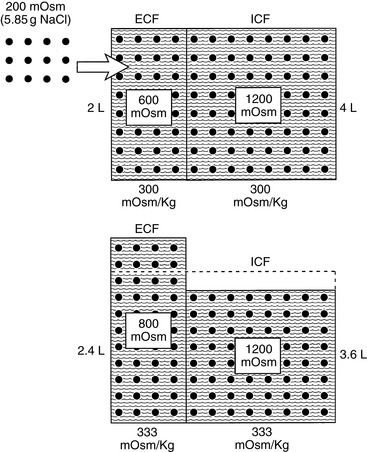
Figure 3-11 Effect of addition of 200 mOsm solute (5.85 g NaCl) on volume and tonicity of extracellular fluid (ECF) and intracellular fluid (ICF).
(Drawing by Tim Vojt.)
In one report of salt poisoning in dogs, a defective water softener resulted in delivery of drinking water containing 10% sodium chloride as compared with normal tap water containing less than 0.1%.78 The affected dogs developed progressive ataxia, seizures, prostration, and death. Their serum sodium concentrations ranged from 185 to 190 mEq/L. Histopathology showed focal areas of perivascular hemorrhage and edema in the midbrain. In another case report, presumptive salt poisoning resulted from ingestion of seawater and subsequent restriction of fresh drinking water.23 Another dog developed fatal hypernatremia after it ingested a large amount of a salt-flour mix.84 After ingestion of a salt-flour figurine, the dog began vomiting and developed polyuria and polydipsia. The owner removed the dog’s water source, and it ingested more of the salt-flour mix. Seizures, pyrexia, and sinus tachycardia developed, and the serum sodium concentration reached 211 mEq/L.
Approximately 22% of dogs that ingest paintballs (which may contain polyethylene glycol, glycerol, and sorbitol) develop hypernatremia.32 Hyperchloremia and hypokalemia also are reported. Clinical signs include vomiting, ataxia, diarrhea, and tremors. These ingredients act as osmotic laxatives, causing a shift in water from the tissues into the lumen of the bowel and resulting in hypernatremia. Warm water enemas may facilitate removal of paintball ingredients from the bowel, but activated charcoal products generally should not be used because they may contain sorbitol. Depending on the duration of onset, 5% dextrose in water (acute onset) or 0.45% NaCl (unknown onset) can be administered to gradually correct hypernatremia. Parenteral fluids can be supplemented with potassium chloride if serum potassium concentration decreases below 2.5 mEq/L.
Therapeutic administration of hyperosmolar solutions containing large amounts of sodium during cardiac resuscitation can cause hypernatremia and hypertonicity (e.g., hypertonic saline, sodium bicarbonate). For example, serum sodium concentration reached 174 mEq/L within 15 minutes after beginning infusion of 7.2% NaCl at a rate of 15 mL/kg in normal beagles.2 Sodium phosphate enemas may also result in mild hypernatremia.3 Primary hyperaldosteronism also may be associated with hypernatremia. It is rare in dogs, but several cases have been reported in cats (see Chapter 5 ). Mild hypernatremia also may occur in dogs with hyperadrenocorticism.101,128
Clinical signs of hypernatremia
The clinical signs of hypernatremia primarily are neurologic and related to osmotic movement of water out of brain cells. A rapid decrease in brain volume may cause rupture of cerebral vessels and focal hemorrhage. The severity of clinical signs is related more to the rapidity of onset of hypernatremia than to the magnitude of hypernatremia. In dogs and cats, clinical signs of hypernatremia are observed when the serum sodium concentration exceeds 170 mEq/L.66,78,84,133 If hypernatremia develops slowly, the brain has time to adapt to the hypertonic state by production of intracellular solutes (e.g., inositol and amino acids) called osmolytes or idiogenic osmoles. These substances prevent dehydration of the brain and allow patients with chronic hypernatremia to be relatively asymptomatic.
Where described in dogs and cats, clinical signs of hypernatremia and hypertonicity have included anorexia, lethargy, vomiting, muscular weakness, behavioral change, disorientation, ataxia, seizures, coma, and death.⁎ If hypotonic losses are the cause of hypernatremia, clinical signs of volume depletion (e.g., tachycardia, weak pulses, and delayed capillary refill time) may be observed on physical examination. If hypernatremia has developed as a result of a gain of sodium, signs of volume overload (e.g., pulmonary edema) may be observed, especially in patients with underlying cardiac disease. Patients with CDI or NDI typically are presented for evaluation of severe polydipsia and polyuria.
Treatment of hypernatremia
The main goals in treating patients with hypernatremia are to replace the water and electrolytes that have been lost and, if necessary, to facilitate renal excretion of excess sodium. The first priority in treatment should be to restore the ECF volume to normal. The next priority is to diagnose and treat the underlying disease responsible for the water and electrolyte deficits.
Pure water loss
Total body solute (TBS) is the product of TBW and plasma osmolality (Posm). If a patient’s fluid loss has been limited to pure water, the following relationship is true:
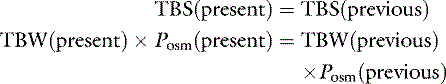
If we assume that body water (TBW) is 60% of body weight measured in kilograms (Wt) and that 2.1 × PNa is an estimate of Posm:
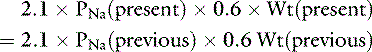
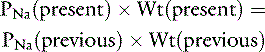
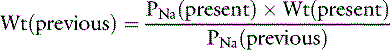
The water deficit is the difference between the previous and present body weights:
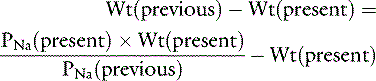
or
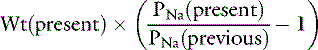
Consider a previously normal dog that has been deprived of water for several days. The dog weighs 10 kg at presentation, and its serum sodium concentration is 170 mEq/L. Assuming a previously normal serum sodium concentration of 145 mEq/L, the dog’s water deficit can be calculated:
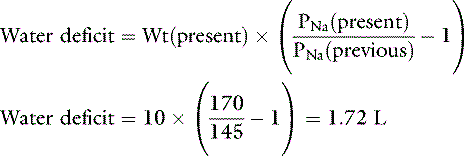
The original estimates of TBW and serum sodium concentration may be modified based on information available to the clinician at presentation. For example, if the dog’s normal serum sodium concentration is known from a previous admission, this value can be substituted in place of 145 mEq/L. If the dog’s previous normal body weight is known, the water deficit may simply be estimated as the difference between the previous and present body weights. The assumption inherent in the latter calculation is that the patient has not gained or lost tissue mass. For a short period, this is a reasonable assumption because loss of 1 kg of tissue mass requires an expenditure of approximately 1600 kcal. This caloric expenditure would require fasting for 2 to 3 days in a normal 10-kg dog with a basal energy requirement of approximately 700 kcal.
A pure water deficit can be replaced by giving 5% dextrose in water intravenously. This solution technically is only slightly hypotonic to plasma (278 mOsm/kg), but the glucose ultimately enters cells and is metabolized so that administration of 5% dextrose is equivalent to administration of water. The water deficit must be replaced and hypernatremia corrected slowly over 48 hours. The brain adapts to hypertonicity by the production of osmolytes or idiogenic osmoles that prevent cellular dehydration. Excessively rapid lowering of the serum sodium concentration may result in movement of water into brain cells and development of cerebral edema. In human patients with hypernatremia of chronic or unknown duration, correction of the serum sodium concentration at a rate of less than 10 to 12 mEq/L per 24 hours minimizes the risk of neurologic complications related to water intoxication.6,152 The animal’s serum sodium concentration should be monitored serially during replacement of the water deficit.
Hypotonic loss
As described earlier, hypotonic losses cause more severe extracellular volume contraction than do losses of pure water. As the tonicity of the fluid lost approaches the tonicity of ECF, the extracellular volume deficit becomes greater (see Fig. 3-9). As a result, signs of volume depletion are more likely with hypotonic losses, and the original replacement fluid should be isotonic so that extracellular volume repletion can proceed rapidly.
In the presence of hemorrhagic shock, whole blood, plasma, or a colloid solution is the ideal fluid to administer. The hemoglobin in whole blood improves oxygen-carrying capacity. The plasma proteins in whole blood and plasma or the dextrans in a colloid solution increase and maintain intravascular volume by increasing oncotic pressure. In many animals that have experienced severe hypotonic losses over an extended time period, replacement of the ECF volume with an isotonic crystalloid solution (e.g., 0.9% NaCl and lactated Ringer’s solution) is adequate. A volume up to four times the suspected intravascular deficit may be required because the isotonic crystalloid solution distributes rapidly throughout the ECF compartment (ECF volume is four times intravascular volume). After the extracellular volume has been expanded, hypotonic fluids (e.g., 0.45% NaCl and half-strength lactated Ringer’s solution) can be administered to provide fluids for maintenance needs and ongoing losses (see Chapter 14).
Gain of impermeant solute
The patient with an excess of sodium-containing impermeant solute in the ECF can be treated by administration of 5% dextrose intravenously. The main disadvantage of this approach is that it causes further expansion of the extracellular compartment in a patient already suffering from ECF volume expansion. In an animal with normal cardiac and renal function, this volume expansion leads to diuresis and natriuresis, and ECF volume returns to normal. In an animal with underlying cardiac disease or oliguria related to primary renal disease, this approach may lead to development of pulmonary edema. Administration of a loop diuretic (e.g., furosemide and ethacrynic acid) promotes excretion of the existing sodium load and hastens return of ECF volume to normal. As in the case of pure water deficit, it is essential that fluid administration proceeds slowly and that serum sodium concentration be lowered gradually over 48 hours to avoid neurologic complications.
Clinical approach to the patient with hyponatremia
The presence of hyponatremia usually, but not always, implies hypoosmolality. Thus, the first step in the approach to the patient with hyponatremia is to determine whether hypoosmolality of the ECF is present. This can be determined by measurement of plasma osmolality. The evaluation of hyponatremia then may be approached using the patient’s plasma osmolality as a guide. This approach is outlined in Fig. 3-12, and the causes of hyponatremia are listed in Box 3-3.
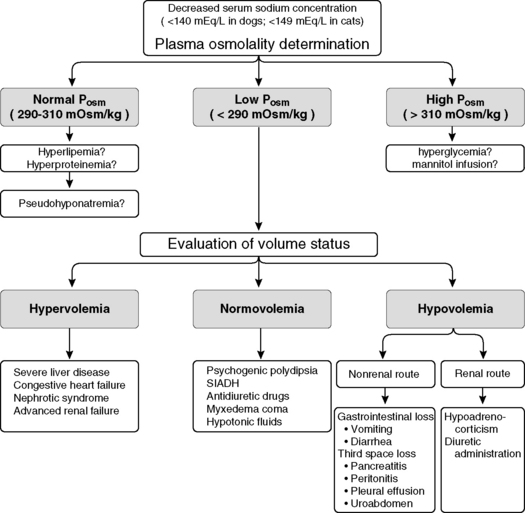
Figure 3-12 Clinical approach to the patient with hyponatremia. Posm, Plasma osmolality; SIADH, syndrome of inappropriate antidiuretic hormone.
(From DiBartola SP. Hyponatremia. Vet Clin North Am Small Anim Pract 1998;28:515–532.)
Hyponatremia with normal plasma osmolality
Sodium is present as charged particles in the aqueous phase of body fluids. Approximately 93% of plasma volume is occupied by water, and the remaining 7% consists largely of proteins and lipids. Historically, serum sodium concentration has been measured by flame photometry. Flame photometry measures the number of sodium ions in a specific volume of plasma or serum. Thus, the sodium concentration is measured as if the sodium ions were present throughout the entire sample volume, whereas actually they are active only in the aqueous phase. Normally, this error is small. In plasma or serum samples containing a large amount of lipid or protein, however, the error may be larger, and the decrease in measured serum sodium concentration could be misleading to the clinician (Fig. 3-13). When serum sodium concentration is measured by direct potentiometry using ion-selective electrodes, large amounts of lipid or protein in the sample should not affect the measured serum sodium concentration. However, if the serum sample is diluted before measurement, large amounts of lipid or protein may still affect the measured serum sodium concentration.89 Therefore, the clinician must be familiar with the laboratory method used so as to interpret serum sodium concentrations properly. The occurrence of a decreased serum sodium concentration as a result of laboratory methodology in the presence of normal plasma osmolality is called pseudohyponatremia or factitious hyponatremia. Pseudohyponatremia occurs in conditions associated with hyperlipidemia or severe hyperproteinemia.
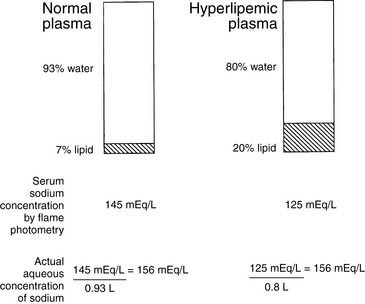
Figure 3-13 Effect of increased plasma lipids on serum sodium concentration (pseudohyponatremia or factitious hyponatremia).
(From DiBartola SP. Hyponatremia. Vet Clin North Am Small Anim Pract 1998;28:515–532.)
Plasma osmolality in patients with pseudohyponatremia is normal, because lipids and proteins are very large molecules that contribute very little to plasma osmolality. If pseudohyponatremia is present, the calculated plasma osmolality is low because of a spuriously low serum sodium concentration, whereas the measured osmolality is normal. Thus, when an abnormal osmolal gap is present and the measured osmolality is normal, pseudohyponatremia should be suspected. The diagnosis of pseudohyponatremia can be made by visual inspection of plasma for lipemia and by measurement of the total plasma protein concentration. Hyperlipemia severe enough to cause pseudohyponatremia is visible to the naked eye as lactescent plasma. Each milligram per deciliter of lipid in serum reduces the sodium concentration by 0.002 mEq/L (e.g., a serum triglyceride concentration of 1000 mg/dL would be expected to reduce the serum sodium concentration by 2 mEq/L).118 In the case of hyperproteinemia, each gram per deciliter of protein above a concentration of 8 g/dL reduces the serum sodium concentration by approximately 0.25 mEq/L (e.g., the serum sodium concentration of a patient with a serum protein concentration of 12 g/dL would be expected to be reduced by 1 mEq/L).151 At such protein concentrations, the plasma may be viscous, and this is likely to occur mainly in patients with plasma cell dyscrasias. Thus, whereas pseudohyponatremia may be intellectually interesting, it is unlikely to be of clinical relevance in most instances. Furthermore, pseudohyponatremia itself has no consequences for the health of the patient. Its importance lies in the ability of the clinician to recognize it and refrain from treating the patient for hyponatremia. Treatment should be directed at the underlying disorder causing hyperproteinemia or hyperlipidemia.
Hyponatremia with increased plasma osmolality
If an impermeant solute is added to ECF, water moves from ICF to ECF, and the osmolality of both compartments increases (see Fig. 3-11).41 If the added solute is something other than sodium, the serum sodium concentration is reduced by the translocation of water, but the plasma osmolality is higher than normal.
Hyponatremia with hyperosmolality is usually caused by hyperglycemia in diabetes mellitus, wherein each 100-mg/dL increase in glucose may decrease the serum sodium concentration by 1.6 mEq/L.82 This correction factor worked well up to a blood glucose concentration of 440 mg/dL in a study in normal humans made transiently hyperglycemic by infusion of somatostatin, but the correction factor was much greater at higher blood glucose concentrations.73 The authors concluded that an overall correction factor of a 2.4-mEq/L decrement in sodium for each 100-mg/dL increment in glucose would be preferable. In the diabetic patient, both hyperlipidemia and hyperglycemia may contribute to decreased serum sodium concentration. Administration of the osmotic diuretic mannitol also can cause hyponatremia with plasma hyperosmolality. The calculated osmolality is normal, the measured osmolality is high, and the osmolal gap is increased in the presence of mannitol, which is an unmeasured osmole. Hyperglycemia does not affect the osmolal gap because the plasma glucose concentration is part of the equation used to calculate plasma osmolality (i.e., it is a measured osmole).
Initially, TBW content is not altered in the setting of hyponatremia with hyperosmolality. Rather, there is an altered distribution of water between intracellular and extracellular compartments. However, a reduction in TBW content develops to the extent that these substances cause an osmotic diuresis.
Hyponatremia with decreased plasma osmolality
The total body sodium content and ECF volume of patients with hyponatremia and hypoosmolality may be normal, decreased, or increased, and hyponatremia may be classified according to the volume status of the patient as hypovolemic, normovolemic, and hypervolemic. In most instances, nonosmotic stimulation of antidiuretic hormone results in water retention and development of hyponatremia. Therefore, the second step in the evaluation of the patient with hyponatremia is to estimate total body sodium content and ECF volume status. This is best done by clinical assessment of the patient based on history, physical examination, and a few ancillary tests. A good history often indicates a source of fluid loss (e.g., vomiting, diarrhea, or diuretic administration), and the physical examination provides important clues to the patient’s volume status. The following physical findings should be assessed: skin turgor, moistness of the mucous membranes, capillary refill time, pulse rate and character, appearance of the jugular veins (distended or flat), and presence or absence of ascites or edema. Measurements of hematocrit and total plasma protein concentration, as well as systemic blood pressure and central venous pressure determinations, if available, further clarify the patient’s ECF volume status.
Hypovolemic Hyponatremia (Hyponatremia with Volume Depletion)
For a patient with volume depletion (hypovolemia) to develop hyponatremia, the total body deficit of sodium must exceed that of water. Hyponatremic patients with volume depletion have lost fluid by renal or nonrenal routes. Gastrointestinal losses (e.g., vomiting, diarrhea) and third-space losses, such as pleural effusion or peritoneal effusion caused by peritonitis, pancreatitis, or uroabdomen, are the most important nonrenal losses of fluid and NaCl.19,162 Gastrointestinal losses are often hypotonic in nature. The question thus arises, “If the losses are hypotonic, how does the patient become hyponatremic?” The answer follows from three physiologic events and reflects the body’s tendency to preserve volume at the expense of tonicity. First, volume depletion decreases GFR, enhances isosmotic reabsorption of sodium and water in the proximal tubules, and decreases delivery of tubular fluid to distal diluting sites. These events impair excretion of water. Second, volume depletion is a strong nonosmotic stimulus for vasopressin release, and the increased plasma vasopressin concentration further impairs water excretion. Third, the patient is thirsty because of volume depletion and continues to drink water if it is available. All of these factors have a dilutional effect on the remaining body fluids. In one study, approximately 20% of dogs with gastrointestinal foreign bodies had hyponatremia.10
Recall the previous example of the loss of 1 L of fluid with an osmolality of 150 mOsm/kg and consider what would happen if the animal in question drinks 1 L of pure water after sustaining the hypotonic loss. The added water increases the ECF volume from 1.36 to 2.36 L, and the resulting hypotonicity rapidly drives water into cells to equalize osmolality:
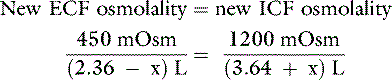
where x is the volume of water moving between compartments:
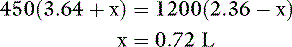
The new volumes and osmolalities are:
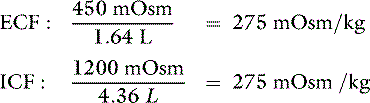
Note that in this example the intracellular compartment is expanded (4.36 L). The volume of the extracellular compartment (1.64 L) is greater than it was when the same hypotonic loss was not replaced (1.36 L) but still less than the previous normal value (2 L). Thus, hypotonic (or isotonic) losses replaced by pure water lead to expansion of the ICF space. These changes are depicted in Figure 3-14.
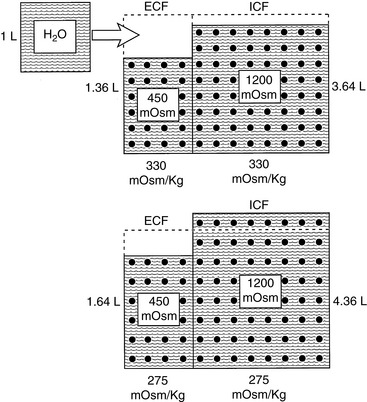
Figure 3-14 Effect of drinking 1 L of water after a loss of 1 L of hypotonic fluid (150 mOsm/kg) on volume and tonicity of extracellular fluid (ECF) and intracellular fluid (ICF).
(Drawing by Tim Vojt.)
Renal fluid and NaCl losses resulting in hyponatremia are usually caused by hypoadrenocorticism or diuretic administration. In one study, 81% of 225 dogs with hypoadrenocorticism were hyponatremic at presentation.129 Mineralocorticoid deficiency in hypoadrenocorticism results in urinary loss of NaCl and depletion of ECF volume. Volume depletion in patients with hypoadrenocorticism is a strong nonosmotic stimulus for vasopressin release and impairs water excretion.6 Hyperkalemia typically accompanies hyponatremia in hypoadrenocorticism.129,132,144,161 However, some dogs with hypoadrenocorticism have only glucocorticoid deficiency at the time of presentation and thus have normal serum potassium concentrations.100,139,156 Glucocorticoids are necessary for complete suppression of vasopressin release, and in their absence impaired water excretion and hyponatremia can occur.28 Occasionally, dogs with gastrointestinal fluid losses develop electrolyte disturbances that mimic hypoadrenocorticism.30,107 Hyponatremia associated with third-space loss of fluid has been reported with pleural effusion related to chylothorax, lung lobe torsion, and neoplasia.93,155,162,166 In these reports, hyponatremia was attributed at least in part to removal of sodium-rich fluid by thoracocentesis. However, many of these animals had evidence of volume depletion, and it is likely that nonosmotic vasopressin secretion also played a role in the development of hyponatremia. Affected dogs also had mild hyperkalemia attributed to decreased renal excretion of potassium caused by volume depletion and decreased distal renal tubular flow. Similar findings have been observed in dogs and cats with peritoneal effusion and in dogs in late pregnancy.9,85,145 The pathogenesis of hyponatremia and mild hyperkalemia in dogs with gastrointestinal losses is probably similar to that described for dogs with pleural and peritoneal effusions, but the explanation for the rare occurrence of similar electrolyte abnormalities in dogs in late pregnancy is unknown. When the cause of hyponatremia and hyperkalemia is unclear, an ACTH stimulation test should be performed to rule out hypoadrenocorticism.
Diuretics contribute to impaired water excretion and dilution of sodium in the ECF by decreased distal delivery of tubular fluid and nonosmotic stimulation of vasopressin release, which occur in response to volume depletion. Furthermore, potassium depletion caused by diuretics can contribute to hyponatremia because shifting of intracellular potassium into the extracellular compartment in exchange for sodium may occur. Hyponatremia has been associated with chronic blood loss in dogs.158 It was thought that defective urinary concentrating ability in these dogs was caused by impaired vasopressin release in response to plasma hypoosmolality and loss of NaCl from the renal medullary interstitium. Some of these dogs had hypoadrenocorticism and gastrointestinal fluid losses that might have contributed to their hyponatremia. Normal concentrating ability returned after resolution of hyponatremia.
Hypervolemic Hyponatremia (Hyponatremia with Volume Excess)
Hyponatremia may occur despite the presence of increased total body sodium and expansion of the ECF compartment in patients with ascites or edema. Some of the pathophysiologic events in these patients impair the excretion of ingested water and exert a dilutional effect on the serum sodium concentration. Hypervolemic hyponatremia is observed in three clinical conditions: congestive heart failure, severe liver disease, and nephrotic syndrome. In these disorders, there is a perception of circulating volume depletion by the body, and the regulatory mechanisms invoked result in volume expansion. This perceived volume deficit has been referred to as decreased effective circulating volume or decreased effective arterial blood volume.
Three major pathophysiologic mechanisms are operative in the pathogenesis of sodium retention and impaired water excretion in these clinical conditions. The renin-angiotensin system is activated by reduced renal perfusion and causes increased sodium retention by the kidneys. Decreased renal perfusion, decreased GFR, and increased proximal tubular reabsorption of sodium and water result in decreased delivery of tubular fluid to distal diluting sites and impairment of free water excretion. A decrease in effective arterial blood volume results in nonosmotic stimulation of vasopressin release and further impairment of water excretion. Impaired free water excretion causes dilution of retained sodium and results in hyponatremia despite the presence of increased total body sodium content and expansion of the ECF compartment. In addition, a primary intrarenal mechanism for sodium retention is thought to be operative in patients with the nephrotic syndrome.
In cirrhosis and the nephrotic syndrome, intravascular volume may be reduced as a result of decreased oncotic pressure caused by hypoalbuminemia. This volume depletion causes nonosmotic stimulation of vasopressin release and impaired water excretion. In liver failure, low effective arterial volume is caused by marked peripheral (primarily splanchnic) vasodilation.53,96,113 Reduction of cardiac output and arterial hypotension also are contributory factors. The presence of hyponatremia in patients with cirrhosis and ascites is associated with a poor prognosis for survival.109 In congestive heart failure, decreased cardiac output is sensed by baroreceptors in the carotid and aortic sinuses, resulting in nonosmotic release of vasopressin. With chronic left atrial distention, the sensitivity of baroreceptors located in this site is presumably blunted, explaining the relative lack of vasopressin suppression that would be expected in acute left atrial distention. Increased sympathetic nervous activity also occurs as well as activation of the renin-angiotensin system. The presence of hyponatremia in heart failure patients is correlated with disease severity and clinical outcome.88,125,150
The pathophysiology of sodium retention in the nephrotic syndrome is complex. In some nephrotic patients with hypervolemia, the renin-angiotensin system appears to be suppressed. This conclusion is based on decreased plasma concentrations of renin and aldosterone and suggests a primary intrarenal mechanism for sodium retention.15 The site of this intrarenal mechanism of sodium retention is not clear. In one experimental study, a distal site was implicated, whereas some investigators have suggested that alterations in filtration fraction and the glomerular ultrafiltration coefficient may be responsible.33,79
In severe liver disease, arteriovenous shunting, splanchnic venous pooling, ascites caused by portal hypertension, and decreased oncotic pressure caused by hypoalbuminemia all may lead to decreased effective circulating volume resulting in nonosmotic stimulation of vasopressin release and activation of the renin-angiotensin system.39 Sodium retention and impairment of water excretion result.
Hypervolemic hyponatremia may also be seen in advanced renal failure. Positive water balance may occur in the presence of continued polydipsia if there are an insufficient number of functional nephrons to excrete the required amount of free water. Approximately 70% of filtered water is reabsorbed isosmotically in the proximal tubules. If GFR is very low, the amount of water that can be excreted even with complete suppression of vasopressin release may be insufficient to prevent positive water balance in the presence of continued water intake. For example, consider a 10-kg dog with advanced renal failure and a GFR of 2 mL/min (approximately 5% of normal). The daily filtered load of water would be 2.88 L, and if 2.02 L (70%) is reabsorbed in the proximal tubules, the maximum volume of water that could be excreted is 860 mL. In the presence of polydipsia, it is conceivable that water intake would exceed this volume and dilutional hyponatremia would develop.
Normovolemic Hyponatremia (Hyponatremia with Normal Volume)
Normovolemic hyponatremia may occur as a result of psychogenic polydipsia, clinical conditions characterized by inappropriate secretion of vasopressin, administration of hypotonic fluids or drugs with antidiuretic effects, and myxedema coma of severe hypothyroidism. Approximately 67% of TBW is located within cells. Therefore, only 33% of the water retained in these disorders is distributed to the extracellular compartment, and only 8% is located in the plasma compartment. However, this mild volume expansion does increase GFR and decrease proximal tubular reabsorption of sodium and water, thus leading to natriuresis. If excessive water intake or inappropriate vasopressin release continues, a new steady state is achieved with a slightly expanded ECF volume and plasma hypoosmolality. Overt signs of hypervolemia are usually not present because the majority of retained water is distributed to the intracellular compartment.
Psychogenic polydipsia usually occurs in large-breed dogs. The owner may report that the dog has a nervous disposition or that polydipsia seemed to begin after some stressful event. Some hyperactive dogs placed in an exercise-restricted environment have developed psychogenic polydipsia, and some dogs with this disorder may have developed it as a learned behavior to gain attention from the owner.44 Some dogs with psychogenic polydipsia lower their water intake dramatically as a result of the stress of hospitalization, and this is sometimes a useful diagnostic observation. In one study, dogs with psychogenic polydipsia had a daily water consumption of 150 to 250 mL/kg, USG of 1.001 to 1.003, urine osmolality of 102 to 112 mOsm/kg, plasma osmolality of 285 to 295 mOsm/kg, and serum sodium concentration of 131 to 140 mEq/L.91 Hyponatremia with plasma hypoosmolality was thus documented in this study. Approximately 67% of affected dogs had a normal response to water deprivation, whereas others had some degree of medullary washout but responded to gradual water deprivation. Psychogenic polydipsia has not yet been reported in cats.
The syndrome of inappropriate ADH secretion (SIADH) refers to vasopressin release in the absence of normal osmotic or nonosmotic stimuli. This syndrome occurs in human patients and may be drug induced or associated with various types of malignancies, pulmonary diseases, and central nervous system disorders.167 Several patterns of vasopressin secretion have been observed in human patients with SIADH: erratic changes in secretion unrelated to plasma osmolality, a normal increase in vasopressin secretion in response to changes in plasma osmolality but occurring at a lower threshold (“reset osmostat”), normal vasopressin secretion when plasma osmolality is normal or increased but inability to reduce vasopressin secretion appropriately after a water load (“vasopressin leak”), and low basal vasopressin concentration that fails to increase as plasma osmolality increases, suggesting increased renal sensitivity to vasopressin or presence of another antidiuretic substance. Because not all human patients with SIADH have increased circulating concentrations of ADH, the name “syndrome of inappropriate antidiuresis” has been proposed.38
SIADH is rare in dogs. It has been reported in association with dirofilariasis, undifferentiated carcinoma, neoplasia in the region of the hypothalamus, granulomatous amebic meningoencephalitis, and hydrocephalus.12,14,52,77,148 Inappropriate vasopressin secretion may have played a role in the pathogenesis of hyponatremia in a dog with glucocorticoid deficiency.28 Idiopathic SIADH also has been characterized in dogs.47,135 Two of the dogs with idiopathic SIADH had hyponatremia, hypoosmolality, and inappropriately high vasopressin concentrations (7 to 30 pmol/L). Urine osmolality was inappropriately high (213 to 535 mOsm/kg) in one dog in the presence of plasma hypoosmolality. The threshold and sensitivity of vasopressin secretion were studied by infusion of hypertonic saline. One dog demonstrated a pattern of reset osmostat and the other a pattern consistent with vasopressin leak.135 The vasopressin receptor antagonist OPC-31260, used at a dosage of 3 mg/kg orally twice a day for a 3-year period, successfully increased urine output and decreased USG in a dog with idiopathic SIADH, but serum sodium concentration was not normalized.47
The diagnosis of SIADH must be made by excluding other causes of hyponatremia. The following criteria should be met before establishing a diagnosis of SIADH:
1. Hyponatremia with plasma hypoosmolality.
2. Inappropriately high urine osmolality in the presence of plasma hypoosmolality. (Urine osmolality is often >300 mOsm/kg in human patients with SIADH. A urine osmolality >100 mOsm/kg should be considered abnormal in a patient with hyponatremia and plasma hypoosmolality. A urine osmolality <100 mOsm/kg would normally be expected as a result of complete suppression of vasopressin release. Urine osmolality is important in distinguishing psychogenic polydipsia and SIADH. Urine is maximally diluted in psychogenic polydipsia but not in SIADH.)
3. Normal renal, adrenal, and thyroid function.
4. Presence of natriuresis despite hyponatremia and plasma hypoosmolality as a result of mild volume expansion (urine sodium concentration usually >20 mEq/L in human patients).
5. No evidence of hypovolemia, which could result in nonosmotic stimulation of vasopressin release.
6. No evidence of ascites or edema, which could result in hypervolemic hyponatremia (i.e., no evidence of severe liver disease, congestive heart failure, or nephrotic syndrome).
Impaired osmoregulation of vasopressin release was observed in 11 dogs with liver disease (7 of which had large congenital portosystemic shunts).143 Either the threshold for vasopressin release was increased or the magnitude of response decreased in these dogs, but plasma vasopressin and sodium concentrations were within the normal reference range. Affected dogs had evidence of excessive glucocorticoid secretion, and their response was similar to that previously described for dogs with spontaneous hyperadrenocorticism.8
Cerebral salt wasting occurs in critically ill human patients with intracranial injury, often subarachnoid hemorrhage, but also may be observed after neurosurgical procedures.7,26 Hyponatremia resulting from cerebral salt wasting must be differentiated from that caused by SIADH because patients with the former disorder are volume depleted and require NaCl and water replacement, whereas those with SIADH require water restriction. Atrial and brain natriuretic factors likely are responsible for the urinary loss of sodium in affected patients.110 Recognition of volume depletion depends on clinical findings such as changes in skin turgor, systemic blood pressure, central venous pressure, heart rate, and character of peripheral pulses. Hyponatremia must be corrected slowly (see Treatment of Hyponatremia section), and SIADH should be suspected if hyponatremia worsens after saline infusion. Fludrocortisone also has been used in human patients with cerebral salt wasting to facilitate sodium retention.
Severe hypothyroidism with myxedema in humans can result in hyponatremia, possibly because of decreased distal delivery of tubular fluid and nonosmotic stimulation of vasopressin release. Hyponatremia in this setting is corrected by thyroid hormone replacement. In four reported cases of myxedema coma in dogs, hyponatremia was found in two of three dogs in which the serum sodium concentration was measured.20,83
Exercise-associated hyponatremia has been reported in human athletes during prolonged exertion. It is thought to be caused by excessive consumption of water or hypotonic fluids as well as nonosmotic stimulation of vasopressin release associated with volume depletion that impairs water excretion.141 Sodium loss in sweat also may play a contributory role. Women, people of low body weight, and those taking nonsteroidal anti-inflammatory drugs are at increased risk.138 Total body exchangeable cation content (sodium and potassium) decreases during long-distance exercise in Alaskan sled dogs despite no apparent change in TBW, and consequently hyponatremia develops.74 The observed hyponatremia was mild (i.e., 139.7 mEq/L after the race as compared with 148.6 mEq/L before the race).75 Dogs do not sweat appreciably, and the mild hyponatremia was attributed to increased urinary losses of sodium in association with increased protein catabolism and excretion of large amounts of urea in urine.
Drugs that stimulate the release of vasopressin or potentiate its renal effects may lead to normovolemic hyponatremia. Nitrous oxide, barbiturates, isoproterenol, and narcotics are drugs used during anesthesia and surgery that stimulate vasopressin release from the neurohypophysis and may contribute to impaired water excretion in the postoperative period. Anxiety, stress, and pain associated with surgical procedures also may contribute to increased plasma vasopressin concentrations and result in decreased renal excretion of water. These events may result in postoperative hyponatremia, especially if the patient receives hypotonic fluids.6,38 In fact, routine use of hypotonic fluids in patients in whom water excretion is impaired by nonosmotic stimulation of vasopressin release is thought to be the main contributing factor to hospital-acquired hyponatremia in human patients and can be prevented by administration of isotonic fluids such as 0.9% NaCl.114
Chlorpropamide potentiates the action of vasopressin, possibly by inhibiting vasopressin-stimulated production of prostaglandin E2 or by up-regulating vasopressin receptors in the kidneys.35 Nonsteroidal anti-inflammatory drugs have a similar effect because of their inhibition of prostaglandin production. The antineoplastic drugs vincristine and cyclophosphamide also impair water excretion. Figure 3-6 shows the effects of various drugs on the release and action of vasopressin.
Clinical signs of hyponatremia
The clinical signs of hyponatremia are related more to the rapidity of onset than to the severity of the associated plasma hypoosmolality. In human patients, deaths and severe complications of hyponatremia were most common when the serum sodium concentration acutely decreased to less than 120 mEq/L or at a rate greater than 0.5 mEq/L/hr.25 Cerebral edema and water intoxication occur if hyponatremia develops faster than the brain’s defense mechanisms can be called into play. Reduction in plasma osmolality and influx of water into the central nervous system cause the clinical signs observed in acute hyponatremia. A 30- to 35-mOsm/kg gradient can result in translocation of water between plasma and the brain in dogs.61 Clinical signs are often absent in chronic disorders characterized by slower decreases in serum sodium concentration and plasma osmolality. During hyponatremia of chronic onset, brain volume is adjusted toward normal by loss of potassium and organic osmolytes from cells.6,134
Acute water intoxication is likely only if the patient has some underlying cause of impaired water excretion at the time a water load occurs. For example, water-loaded dogs given repositol vasopressin developed signs of acute water intoxication.69 Early signs were mild lethargy, nausea, and slight weight gain; more severe signs included vomiting, coma, and a marked increase in body weight. One dog in this study died from pulmonary and cerebral edema. Weakness, incoordination, and seizures may also result from acute water intoxication. In one clinical report, a Labrador retriever developed acute hyponatremia (125 mEq/L) and severe neurologic signs (i.e., coma) after swimming for many hours in a lake.157 The dog spontaneously underwent marked diuresis and recovered with supportive care, suggesting that it was capable of suppressing vasopressin release in response to the water load.
Treatment of hyponatremia
The two main goals of treatment in hyponatremia are to diagnose and manage the underlying disease and, if necessary, increase serum sodium concentration and plasma osmolality. Severe, symptomatic hyponatremia of acute onset (<24 to 48 hours’ duration) may result in seizures, cerebral edema, or death and requires prompt treatment. In human patients with acute hyponatremia, correction of serum sodium concentration may be required at rates up to 12 mEq/L/day.151 However, severe, symptomatic hyponatremia of rapid onset is rare in small animal practice. Because of inexperience with the management of acute hyponatremia in dogs and cats, and the known risks of overly rapid correction of hyponatremia, only use of conventional crystalloid solutions (e.g., lactated Ringer’s solution and 0.9% saline) is recommended. Use of 3% NaCl is not recommended.
Patients with chronic hyponatremia often have few or no clinical signs directly attributable to their hypoosmolality. This is probably because the brain has had sufficient time to adapt to plasma hypotonicity. In fact, treatment of chronic hyponatremia can be more dangerous than the disorder itself. In human patients, complications of treatment may occur when chronic (>48 hours’ duration) hyponatremia is corrected too rapidly (i.e., when the serum sodium concentration is increased by >10 to 12 mEq/L in 24 hours).95,151
When hyponatremia and hypoosmolality are corrected, potassium and organic osmolytes lost during adaptation must be restored to the cells of the brain. If replacement of these solutes does not keep pace with the increase in serum sodium concentration that occurs as a result of treatment, brain dehydration and injury—called osmotic demyelination or myelinolysis—may result.95,151 Experimental studies have confirmed that this syndrome is a result of a rapid and large increase in serum sodium concentration and is not a consequence of hyponatremia and hypoosmolality. Human patients with hyponatremia of more than 72 hours’ duration are more susceptible than those with hyponatremia of less than 24 hours’ duration.151 The neural lesions of myelinolysis develop several days after correction of hyponatremia and consist of myelin loss and injury to oligodendroglial cells in the pons and other sites in the brain (e.g., thalamus, subcortical white matter, and cerebellum). Lesions may take several days to develop, but on magnetic resonance imaging they are hyperintense on T2-weighted images, hypointense on T1-weighted images, and are not enhanced after gadolinium injection.95 The ability to reaccumulate organic osmolytes may vary among different regions of the brain and thus account for why some regions (e.g., midbrain) are more susceptible to osmotic demyelination.
Similar lesions have been reported in experimental dogs with hyponatremia with correction rates of 15 mEq/L/day even without overcorrection to hypernatremia.94 In veterinary medicine, myelinolysis first was reported in two dogs after correction of hyponatremia associated with trichuriasis.122 In one dog, a serum sodium concentration of 101 mEq/L had been corrected to 136 mEq/L in less than 38 hours (correction rate, >22 mEq/L/day), and in the other, a serum sodium concentration of 108 mEq/L had been corrected to 134 mEq/L in less than 38 hours (correction rate, >16 mEq/L/day). Clinical signs developed 3 to 4 days after correction of hyponatremia and consisted of lethargy, weakness, and ataxia progressing to hypermetria and quadriparesis. Lesions were detected by magnetic resonance imaging and were located in the thalamus as compared with the more typical pontine location in affected human patients. From this experience, it was recommended that dogs with asymptomatic chronic hyponatremia be treated by mild water restriction and monitoring of serum sodium concentration. Symptomatic dogs with chronic hyponatremia should be treated conservatively at correction rates of less than 10 to 12 mEq/L/day (0.5 mEq/L/hr). Serial monitoring of serum sodium concentration is necessary because the actual rate of correction may not correspond to the calculated rate of correction. Correction should be carried out with conventional crystalloid solutions (e.g., lactated Ringer’s solution and 0.9% NaCl) in a volume calculated specifically to replace the patient’s volume deficit. The clinician must remember that volume repletion in hypovolemic patients abolishes the nonosmotic stimulus for vasopressin release and allows the animal to excrete solute-free water via the kidneys. This in itself tends to correct the hyponatremia. Thus, caution should be exercised even when using conventional crystalloid fluid therapy.
Three additional cases of suspected myelinolysis in dogs with chronic hyponatremia caused by hypoadrenocorticism or trichuriasis have been reported.11,24,105 The rates of correction of hyponatremia in these dogs were 22 mEq/L on day 1 and 17 mEq/L on day 2, 32 mEq/L over 2 days, and 17 mEq/L in 9 hours.11,24,105 The neurologic signs that developed (e.g., spastic tetraparesis, loss of postural and proprioceptive responses, dysphagia, trismus, and decreased menace response) were similar to those originally described by O’Brien.122 The dogs of these reports gradually recovered over several weeks.
Water intake should be carefully restricted to a volume less than urine output in normovolemic patients with hyponatremia (e.g., psychogenic polydipsia), or drugs causing an antidiuretic effect should be discontinued if possible. Demeclocycline and lithium inhibit vasopressin release and have been used to treat SIADH in humans, but water restriction is probably the safest approach.48
In edematous patients, dietary sodium restriction and diuretic therapy should be considered. A 0.9% NaCl solution can be administered concurrently with loop diuretics (e.g., furosemide) to effect more rapid correction of hyponatremia in overhydrated symptomatic patients. The occurrence of chronic hyponatremia in patients with congestive heart failure is often a sign of advanced disease and responds poorly to treatment. Administration of furosemide and an angiotensin-converting enzyme inhibitor (e.g., enalapril) may improve stroke volume and cardiac output by reducing preload and afterload and may decrease vasopressin secretion and enhance water excretion, which in turn may facilitate resolution of hyponatremia.
Arginine vasopressin (AVP) receptor antagonists (vaptans) block either V2 receptors (lixivaptan, tolvaptan, satavaptan) or both V2 and V1A receptors (conivaptan).⁎ Based on their mechanism of action, these drugs increase free water excretion by the kidneys and effectively normalize serum sodium concentration in patients with non-osmotic release of AVP causing euvolemic (e.g., SIADH) or hypervolemic (e.g., congestive heart failure, liver failure) hyponatremia.111,117 Patients with hypovolemic hyponatremia should be treated with an infusion of 0.9% NaCl or other isotonic fluid to replace their volume deficits. The AVP receptor antagonists increase water but not solute excretion via the kidneys, and likely will have major impact on the clinical management of euvolemic and hypervolemic hyponatremia in the near future.60
Conivaptan is administered as an intravenous bolus in 5% dextrose followed by a constant rate infusion, whereas tolvaptan, lixivaptan, and satavaptan are administered orally. In humans, adverse effects of the vaptans generally are limited to thirst and dry mouth. To minimize the risk of osmotic demyelination, correction of serum sodium concentration should be limited to <8 mEq/L per 24 hours, especially when hyponatremia is chronic or of unknown duration. The pH of the conivaptan solution is low (3.0) and infusion site reactions are common.
Because they antagonize only the V2 receptor, lixivaptan and tolvaptan do not cause changes in blood pressure. Vasopressin receptor antagonists promote aquaresis and correct hyponatremia in heart failure patients, but long-term beneficial effects on patient survival have not yet been documented.21,40 Conivaptan may be especially helpful in heart failure patients as a result of its effects of the V1A receptor and potential to decrease total peripheral resistance.54 Although the combined V2/V1A receptor antagonist conivaptan might be expected to lower blood pressure, it only causes hypotension in 2.5% of treated patients, although orthostatic hypotension may be seen in 5% of treated patients.124
Conivaptan is both a substrate for and inhibitor of CYP3A4 (the 3A4 isoform of the cytochrome P-450 enzyme). Administration of conivaptan with other CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin) will markedly increase its plasma concentration, and use with other drugs metabolized by CYP3A4 (e.g., statins, midazolam, amlodipine) will increase the plasma concentrations of these drugs and potentially lead to toxicity.
Although considered primarily aquaretics, tolvaptan and lixivaptan may increase sodium excretion at higher dosages, possibly by blocking the sodium-retaining effect of AVP on the thick ascending limb of Henle’s loop. Unresolved is whether or not V2-receptor antagonists could decrease concentrations of factor VIII and von Willebrand factor, which are known to be increased by AVP.
Clinical approach to polyuria and polydipsia
Normal daily water intake and urine output in dogs and cats are influenced by the nutrient, mineral, and water content of the diet. Normal water intake should not exceed 90 mL/kg/day in dogs and 45 mL/kg/day in cats. Normal urine output ranges from 20 to 45 mL/kg/day in dogs and cats. Dogs with disorders such as psychogenic polydipsia, CDI, and NDI may have water consumption as much as five times the normal.
Dogs and cats with polyuria and polydipsia are encountered frequently in small animal practice. The causes of polyuria and polydipsia, their pathophysiologic mechanisms, and the necessary confirmatory laboratory tests are presented in the Table 3-4. The most common causes are chronic renal failure in dogs and cats, diabetes mellitus in dogs and cats, hyperadrenocorticism in dogs, and hyperthyroidism in cats. These common causes must always be ruled out before beginning an exhaustive diagnostic evaluation of the animal.
Table 3-4 Causes of Polyuria and Polydipsia in Small Animal Practice
| Disease | Mechanism of Polyuria and Polydipsia | Confirmatory Tests |
|---|---|---|
| Chronic renal disease⁎ (S) | Osmotic diuresis in remnant nephrons | ECC |
| Disruption of medullary architecture by structural disease | CBC | |
| Profile | ||
| Urinalysis | ||
| Radiography | ||
| Ultrasonography | ||
| Hyperadrenocorticism⁎ (W) | Defective ADH release and action | LDDST, HDDST |
| Psychogenic | Plasma ACTH | |
| Ultrasonography | ||
| Diabetes mellitus⁎ (S) | Osmotic diuresis caused by glucosuria | Blood glucose |
| Urinalysis | ||
| Hyperthyroidism⁎ (W) | Increased medullary blood flow and MSW | T4 |
| Psychogenic | Technetium scan | |
| Hypercalciuria | ||
| Pyometra (W) | Escherichia coli endotoxin | History |
| Immune complex glomerulonephritis | Physical | |
| CBC | ||
| Abdominal radiographs | ||
| Postobstructive diuresis (S) | Elimination of retained solutes | History |
| Defective response to ADH | Physical examination | |
| Defective sodium reabsorption | Urinalysis | |
| Hypercalcemia (W) | Defective ADH action | Serum calcium |
| Increased medullary blood flow | ||
| Impaired NaCl transport in the loop of Henle | ||
| Hypercalcemic nephropathy | ||
| Direct stimulation of thirst center | ||
| Liver disease (W) | Decreased urea synthesis with loss of medullary solute | Liver enzymes |
| Decreased metabolism of endogenous hormones | Serum bile acids | |
| (e.g., cortisol, aldosterone) | Blood ammonia | |
| Psychogenic (hepatic encephalopathy) | Liver biopsy | |
| Hypokalemia | ||
| Pyelonephritis (W) | E. coli endotoxin | Urinalysis |
| Increased renal blood flow | Urine culture | |
| MSW | CBC | |
| Renal parenchymal damage | Excretory urography | |
| Abdominal ultrasonography | ||
| Hypoadrenocorticism (W) | Renal sodium loss with MSW | Serum sodium and potassium |
| ACTH stimulation | ||
| Hypokalemia (W) | Defective ADH action | Serum potassium |
| Increased medullary blood flow and loss of medullary solute | ||
| Diuretic phase of oliguric ARF (S) | Elimination of retained solutes | History |
| Defective sodium reabsorption | CBC | |
| Profile | ||
| Urinalysis | ||
| Abdominal ultrasonography | ||
| Renal biopsy | ||
| Partial urinary tract obstruction (S) | Redistribution of renal blood flow | History |
| Defective sodium reabsorption | Physical examination | |
| Renal parenchymal damage | ||
| Drugs (W) | Various mechanisms depending on drug | History |
| Salt administration (S) | Osmotic diuresis caused by excess sodium administered | History |
| Excessive parenteral fluid administration (W) (polyuria only) | Water diuresis caused by excess water administered | History |
| Central diabetes insipidus (CDI) (W) | Congenital lack of ADH (rare) | Water deprivation test |
| Acquired lack of ADH (idiopathic, tumor, trauma) | Exogenous ADH test | |
| ADH assay | ||
| Nephrogenic diabetes insipidus (NDI) (W) | Congenital lack of renal response to ADH (very rare) | Water deprivation test |
| Acquired lack of renal response to ADH (see Table 3-5) | Exogenous ADH test | |
| ADH assay | ||
| ECC | ||
| Psychogenic polydipsia (PP) (W) | Neurobehavioral disorder (anxiety?) | Water deprivation test |
| Increased renal blood flow | Exogenous ADH test | |
| MSW | Behavioral history | |
| Renal glucosuria (S) | Solute diuresis caused by glucosuria | Blood glucose |
| Urinalysis | ||
| Primary hypoparathyroidism (W) | Unknown (psychogenic?) | Serum calcium |
| Serum phosphorus | ||
| Serum PTH | ||
| Acromegaly (W, S) | Insulin antagonism | Neuroradiography |
| Glucose intolerance | Growth hormone assay | |
| Diabetes mellitus in affected cats | ||
| Polycythemia (W) | Unknown (increased blood viscosity?) | CBC |
| Multiple myeloma (W) | Unknown (increased blood viscosity?) | Serum protein electrophoresis |
| Renal MSW (W) | Depletion of medullary interstitial solute (urea, sodium, potassium) | Gradual water deprivation |
| (3-5 days) | ||
| Hickey-Hare test |
Abbreviations: (W), water diuresis; (S), solute diuresis; ACTH, adrenocorticotropic hormone; ADH, antidiuretic hormone; ARF, acute renal failure; CBC, complete blood count; ECC, endogenous creatinine clearance; HDDST, high-dose dexamethasone suppression test; LDDST, low-dose dexamethasone suppression test; MSW, medullary washout of solute; PTH, parathyroid hormone.
⁎ Most common causes of polyuria and polydipsia.
Adapted from Bruyette DS, Nelson RW. How to approach the problems of polyuria and polydipsia. Vet Med 1986;81:112.
Determination of the specific gravity of a random urine sample from the animal is a logical starting point for evaluation of polyuria and polydipsia. If a random USG is greater than 1.030 to 1.035, the clinician should obtain additional history to rule out other disorders that may have been confused with polyuria (e.g., urinary incontinence and dysuria). If a random USG is less than 1.025 to 1.030, an initial diagnostic evaluation is warranted.
Many causes of polyuria and polydipsia can be ruled out by an initial database consisting of a complete history and physical examination, complete blood count, biochemical profile (including electrolytes), urinalysis, urine culture, and abdominal radiographs. If the animal is otherwise healthy, it is helpful to instruct the owner to quantitate and record the animal’s daily water consumption at home over a 3- to 5-day period. Determination of water intake at home prevents potential reduction in water intake precipitated by the stress of hospitalization.
With some exceptions (e.g., psychogenic polydipsia), polydipsia usually occurs as a consequence of polyuria. If polydipsia occurs without polyuria, the clinician must consider causes such as high ambient temperature (i.e., increased insensible water losses), regular prolonged exercise, water consumption to replace a previous hydration deficit, and third-space distribution of consumed water. Excessive administration of parenteral fluids causes polyuria without polydipsia. The diagnostic approach to polyuria and polydipsia is summarized in Table 3-4 and Figure 3-15.
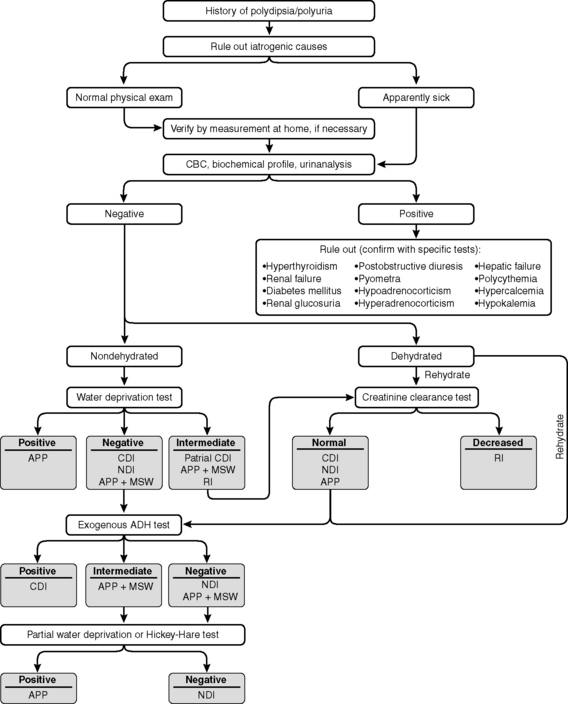
Figure 3-15 Clinical approach to the patient with polydipsia and polyuria. APP, Apparent psychogenic polydipsia; CBC, complete blood count; CDI, central diabetes insipidus; MSW, medullary solute washout; NDI, nephrogenic diabetes insipidus; RI, renal insufficiency with solute diuresis.
(From Fenner WR. Quick reference to veterinary medicine, 2nd ed. Philadelphia: JB Lippincott, 1991: 110.)
Laboratory evaluation of polyuria and polydipsia
Endogenous creatinine clearance
In chronic progressive renal disease, urinary concentrating ability is impaired after two thirds of the nephron population has become nonfunctional, whereas azotemia does not develop until three quarters of the nephrons have become nonfunctional. Thus, the main indication for determination of endogenous creatinine clearance is the clinical suspicion of renal disease in a patient with polyuria and polydipsia but normal BUN and serum creatinine concentrations. The only requirements for determination of endogenous creatinine clearance are an accurately timed collection of urine (usually 24 hours), determination of the patient’s body weight, and measurement of serum and urine creatinine concentrations. Failure to collect all urine produced results in an erroneously reduced calculated clearance value. Use of creatinine clearance as an estimate of GFR is discussed further in Chapter 2.
Water deprivation test
The water deprivation test is indicated in evaluation of animals with confirmed polydipsia and polyuria, the cause of which remains undetermined after the initial diagnostic evaluation. It is usually performed in animals with hyposthenuria (USG <1.007) that are suspected to have CDI, NDI, or psychogenic polydipsia. An animal that is dehydrated but has dilute urine has already failed the test and should not be subjected to water deprivation. In such an animal, failure to concentrate urine is probably caused by structural or functional renal dysfunction or administration of drugs that interfere with urinary concentrating ability. The water deprivation test is also contraindicated in animals that are azotemic. The test should be performed with extreme caution in animals with severe polyuria because such patients may rapidly become dehydrated during water deprivation if they have defective urinary concentrating ability.
At the beginning of the water deprivation test, the bladder must be emptied and baseline data collected (body weight, hematocrit, total plasma proteins, skin turgor, serum osmolality, urine osmolality, and USG). Water is then withheld, and these parameters are monitored every 2 to 4 hours. Urine and serum osmolalities are the best parameters to follow, but osmolality results are often not immediately available to the clinician. Thus, USG and body weight assume great importance for decision making during performance of the test. An increase in total plasma protein concentration is a relatively reliable indicator of progressive dehydration, but increases in hematocrit and changes in skin turgor are less reliable.67 Serum creatinine and BUN concentrations should not increase during a properly conducted water deprivation test.
The bladder should be emptied at the time of each urine collection. Maximal stimulation of ADH release is present after loss of 5% of body weight. The test is concluded when the patient either demonstrates adequate concentrating ability or becomes dehydrated as evidenced by loss of 5% or more of its original body weight. It is important when weighing the animal to use the same scale each time and to empty the bladder at each evaluation.
In normal dogs, dehydration becomes evident after a mean of 42 hours but occasionally may not occur until after 96 hours.67 The time required for dehydration to develop during water deprivation testing in dogs with disorders characterized by polyuria and polydipsia may be as short as a few hours or up to 12 hours. By the time dehydration is evident, normal dogs develop a USG of 1.050 to 1.076, urine osmolality of 1787 to 2791 mOsm/kg, and a urine/plasma osmolality ratio of 5.7 to 8.9.67 Normal cats developed USG values of 1.047 to 1.087 and urine osmolalities of 1581 to 2984 mOsm/kg after water deprivation of sufficient duration (approximately 40 hours) to induce 5% loss of body weight.142 Failure to achieve maximal urinary solute concentration does not localize the level of the malfunction, but a structural or functional defect may be present anywhere along the hypothalamic-pituitary-renal axis. Furthermore, animals with renal medullary solute washout may have impaired concentrating capacity regardless of the underlying cause of polyuria and polydipsia.
If there has been less than a 5% increase in urine osmolality or less than 10% change in USG for three consecutive determinations or if the animal has lost 5% or more of its original weight, 0.25 to 0.5 U/kg aqueous vasopressin (pitressin) (up to a total dose of 5 U) or 5 µg of DDAVP may be given subcutaneously and parameters of urinary concentrating ability monitored at 30, 60, and 120 minutes after ADH injection. Normal dogs and those with psychogenic polydipsia should show no additional response to ADH administration in this setting. The expected responses to water deprivation for dogs with various disorders of water balance are shown in Figure 3-16.
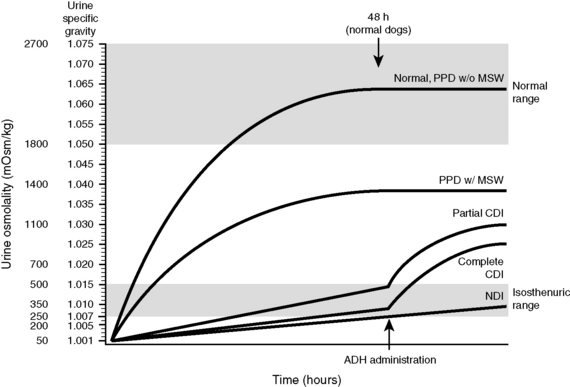
Modified water deprivation test
A modified water deprivation test has been described for the diagnosis of polyuric disorders in dogs. Water is removed from the animal’s cage and the urinary bladder emptied, after which urine osmolality or specific gravity is measured and the bladder emptied on an hourly basis. Maximal urine solute concentration is defined as occurring whenever less than a 5% increase in urine osmolality occurs on sequential determinations. This maximal concentration occurred at a mean urine osmolality of 1414 mOsm/kg in normal dogs after 24 hours of water deprivation.116 At this time, 2 to 3 U of aqueous vasopressin was administered subcutaneously and the urine osmolality determined at 1 and 2 hours after injection. Further increase in urine osmolality after administration of vasopressin should not exceed 10% in normal dogs. In this study, dogs with CDI showed an average 292% increase in urine osmolality after aqueous ADH, dogs with partial CDI an average 28% increase, and dogs with hyperadrenocorticism an average 20% increase. The time required to develop dehydration ranged from 3 to 11.5 hours in dogs with psychogenic polydipsia, complete or partial CDI, and hyperadrenocorticism.
Gradual water deprivation
Gradual water deprivation can be performed to eliminate diagnostic confusion caused by renal medullary solute washout. The owner can be instructed to restrict water consumption to 120 mL/kg/day 72 hours before, to 90 mL/kg/day 48 hours before, and to 60 mL/kg/day 24 hours before the scheduled water deprivation test. In dogs with psychogenic polydipsia, this promotes release of endogenous vasopressin, increased permeability of the inner medullary collecting ducts to urea, and restoration of the normal gradient of medullary hypertonicity. An alternative approach is to instruct the owner to reduce water consumption by approximately 10% per day over a 3- to 5-day period (but not to less than 60 mL/kg/day). This approach should be used only in animals that are otherwise healthy on initial clinical evaluation, and the owner should provide dry food ad libitum and weigh the dog daily to monitor for loss of body weight.
Hickey-hare test
In the Hickey-Hare test, water (20 mL/kg) is administered by stomach tube, an indwelling urinary catheter is placed, and urine flow (milliliters per minute) is determined.92 Hypertonic saline (2.5%) is administered intravenously at a rate of 0.25 mL/min/kg for 45 minutes. Urine volume is recorded every 15 minutes during the infusion and for 45 minutes afterward. The normal response to this procedure is a decrease in the rate of urine production caused by stimulation of ADH release by plasma hyperosmolality. It is useful in the differentiation of psychogenic polydipsia with renal medullary solute washout from NDI after negative water deprivation and exogenous ADH test results. In NDI, there should be no change or an actual increase in urine flow, whereas in psychogenic polydipsia with renal medullary solute washout, repletion of solute (e.g., NaCl) should have occurred, and the response to hypertonic saline should be normal (decreased urine volume). This test is cumbersome, is contraindicated for patients with congestive heart failure, and may lead to signs of hypernatremia in patients that cannot excrete a sodium load. It has largely been replaced by gradual water deprivation as described previously.
Exogenous antidiuretic hormone testing
The exogenous vasopressin test may be used for debilitated patients in which water deprivation is considered hazardous or to further characterize a concentrating defect detected by the routine water deprivation test. In the aqueous vasopressin test, an intravenous infusion of aqueous vasopressin (pitressin) at 10 mU/kg is given over 60 minutes. The bladder is emptied at the start of the study, and parameters of urinary concentrating ability are measured before and at 30-minute intervals for 3 hours after beginning the infusion. The bladder is emptied at each measurement. In one report, maximal response to aqueous vasopressin in water-loaded dogs usually occurred at 60 minutes (range, 30 to 90 minutes) and consisted of USG values of 1.012 to 1.033, urine osmolalities of 429 to 1437 mOsm/kg, and urine/plasma osmolality ratios of 1.5 to 5.1.68 Water should be provided ad libitum during testing, but water loading should not be performed in clinical patients.
In the repositol vasopressin test, 3 to 5 U of vasopressin tannate in oil (pitressin tannate) is given intramuscularly, and the bladder is emptied 3 to 6 hours after injection. Parameters of urinary concentrating ability are measured before and at 6, 9, 12, and 24 hours after injection. Oral water loading must be avoided because of the danger of potentially lethal water intoxication.69 Maximal response to repositol vasopressin occurred 8 to 12 hours after injection and consisted of USG values of 1.028 to 1.057, urine osmolalities of 1052 to 1850 mOsm/kg, and urine/plasma osmolality ratios of 3.9 to 6.7.69
The standard or modified water deprivation test is the preferred initial test of urinary concentrating ability because mean maximal values are usually higher with this test and results are easier to interpret (Table 3-5). Why higher values for parameters of urinary concentrating ability are achieved with this test as compared with the exogenous vasopressin tests is unknown. Possible explanations include the actions of antidiuretic substances other than ADH that may be present in hydropenic individuals, the effect of slower renal medullary blood flow in dehydrated patients, and intensification of the medullary interstitial gradient in dehydrated individuals.

1 Abramov M., Beauwens R., Cogan E. Cellular events in vasopressin action. Kidney Int. 1987;32(Suppl. 21):S56.
2 Ajito T., Suzuki K., Iwabuchi S. Effect of intravenous infusion of 7.2% hypertonic saline solution on serum electrolytes and osmotic pressure in healthy beagles. J Vet Med Sci. 1999;61:637.
3 Atkins C.E., Tyler R., Greenlee P. Clinical, biochemical, acid-base, and electrolyte abnormalities in cats after hypertonic sodium phosphate enema administration. Am J Vet Res. 1985;46:980.
4 Authement J.M., Boudrieau R.J., Kaplan P.M. Transient traumatically induced central diabetes insipidus in a dog. J Am Vet Med Assoc. 1989;194:683.
5 Bagley R.S., de Lahunta A., Randolph J.F., et al. Hypernatremia, adipsia, and diabetes insipidus in a dog with hypothalamic dysplasia. J Am Anim Hosp Assoc. 1993;29:267.
6 Bagshaw S.M., Townsend D.R., McDermid R.C. Disorders of sodium and water balance in hospitalized patients. Can J Anaesth. 2009;56:151.
7 Betjes M.G.H. Hyponatremia in acute brain disease: the cerebral salt wasting syndrome. Eur J Int Med. 2002;13:9.
8 Biewenga W.J., Rijnberk A., Mol J.A. Osmoregulation of systemic vasopressin release during long-term glucocorticoid excess: a study in dogs with hyperadrenocorticism. Acta Endocrinol. 1991;124:583.
9 Bissett S.A., Lamb M., Ward C.R. Hyponatremia and hyperkalemia associated with peritoneal effusion in four cats. J Am Vet Med Assoc. 2001;218:1590.
10 Boag A.K., Coe R.J., Martinez T.A., et al. Acid-base and electrolyte abnormalities in dogs with gastrointestinal foreign bodies. J Vet Intern Med. 2005;19:816.
11 Brady C.A., Vite C.E., Drobatz K.J. Severe neurological sequelae in a dog after treatment of hypoadrenal crisis. J Am Vet Med Assoc. 1999;215:222.
12 Breitschwerdt E.B., Root C.R. Inappropriate secretion of antidiuretic hormone in a dog. J Am Vet Med Assoc. 1979;175:181.
13 Breitschwerdt E.B., Verlander J.W., Bribernik T.N. Nephrogenic diabetes insipidus in three dogs. J Am Vet Med Assoc. 1981;179:235.
14 Brofman P.J., Knostman K.A., DiBartola S.P. Granulomatous amebic meningoencephalitis causing the syndrome of inappropriate secretion of antidiuretic hormone in a dog. J Vet Intern Med. 2003;17:230.
15 Brown E., Markandu N.D., Roulston J.E., et al. Is the renin-angiotensin-aldosterone system involved in the sodium retention of the nephrotic syndrome? Nephron. 1982;32:102.
16 Bruyette D.S., Nelson R.W. How to approach the problems of polyuria and polydipsia. Vet Med. 1986;81:112.
17 Bulger R.E. Composition of renal medullary tissue. Kidney Int. 1987;31:557.
18 Burnie A.G., Dunn J.K. A case of central diabetes insipidus in the cat: diagnosis and treatment. J Small Anim Pract. 1979;23:237.
19 Burrows C.F., Bovee K.C. Metabolic changes due to experimentally induced rupture of the canine urinary bladder. Am J Vet Res. 1974;35:1083.
20 Chastain C.B., Graham C.L., Riley M.G. Myxedema coma in two dogs. Canine Pract. 1982;9:20.
21 Chatterjee K. Hyponatremia in heart failure. J Intensive Care Med. 2009;24:347.
22 Chew D.J., Leonard M., Muir W.W. Effect of sodium bicarbonate infusion on serum osmolality, electrolyte concentrations, and blood gas tensions in cats. Am J Vet Res. 1991;52:12.
23 Chew M. Salt poisoning in a boxer bitch. Vet Rec. 1969;85:685.
24 Churcher R.K., Watson A.D.J., Eaton A. Suspected myelinolysis following rapid correction of hyponatremia in a dog. J Am Anim Hosp Assoc. 1999;35:493.
25 Cluitmans F.H., Meinders A.E. Management of severe hyponatremia: rapid or slow correction? Am J Med. 1990;88:161.
26 Coenraad M.J., Meinders A.E., Taal J.C., et al. Hyponatremia in intracranial disorders. Neth J Med. 2001;58:123.
27 Crawford M.A., Kittleson M.D., Fink G.D. Hypernatremia and adipsia in a dog. J Am Vet Med Assoc. 1984;184:818.
28 Crow S.E., Stockham S.L. Profound hyponatremia associated with glucocorticoid deficiency in a dog. J Am Anim Hosp Assoc. 1985;21:393.
29 Davenport D.J., Chew D.J., Johnson G.C. Diabetes insipidus associated with metastatic pancreatic carcinoma in a dog. J Am Vet Med Assoc. 1986;189:204.
30 DiBartola S.P., Johnson S.E., Davenport D.J., et al. Clinicopathologic findings resembling hypoadrenocorticism in dogs with primary gastrointestinal disease. J Am Vet Med Assoc. 1985;187:60.
31 DiBartola S.P., Johnson S.E., Johnson G.C., et al. Hypodipsic hypernatremia in a dog with defective osmoregulation of antidiuretic hormone. J Am Vet Med Assoc. 1994;204:922.
32 Donaldson C.W. Paintball toxicosis in dogs. Vet Med. 2003;98:995.
33 Dorhout Mees E.J. Edema formation in the nephrotic syndrome. Contrib Nephrol. 1984;43:64.
34 Dow S.W., Fettman M.J., LeCouteur R.A., et al. Hypodipsic hypernatremia and associated myopathy in a hydrocephalic cat with transient hypopituitarism. J Am Vet Med Assoc. 1987;191:212.
35 Durr J.A., Hensen J., Ehnis T., et al. Chlorpropamide upregulates antidiuretic hormone receptors and unmasks constitutive receptor signaling. Am J Physiol. 2000;278:F799.
36 Edwards D.F., Richardson D.C., Russell R.G. Hypernatremic, hypertonic dehydration in a dog with diabetes insipidus and gastric dilatation-volvulus. J Am Vet Med Assoc. 1983;182:973.
37 Ellison D.H., Velazquez H. Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol. 1987;253:F546.
38 Ellison D.H., Berl T. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064.
39 Epstein M. Derangements of renal water handling in liver disease. Gastroenterology. 1985;89:1415.
40 Farmakis D., Filippatos G., Parissis J., et al. Hyponatremia in heart failure. Heart Fail Rev. 2009;14:59.
41 Feig P.U., McCurdy D.K. The hypertonic state. N Engl J Med. 1977;297:1444.
42 Feldman B.F., Rosenberg D.P. Clinical use of anion and osmolal gaps in veterinary medicine. J Am Vet Med Assoc. 1981;178:396.
43 Feldman E.C., Nelson R.W. Water metabolism and diabetes insipidus. In: Feldman E.C., Nelson R.W., editors. Canine and feline endocrinology and reproduction. 3rd ed. Philadelphia: WB Saunders; 2004:37.
44 Feldman E.C., Nelson R.W. Water metabolism and diabetes insipidus. In: Feldman E.C., Nelson R.W., editors. Canine and feline endocrinology and reproduction. 3rd ed. Philadelphia: WB Saunders; 2004:18.
45 Ferguson D.C., Biery D.N. Diabetes insipidus and hyperadrenocorticism associated with high plasma adrenocorticotropin concentration and a hypothalamic/pituitary mass in a dog. J Am Vet Med Assoc. 1988;193:835.
46 Fjellestad-Paulsen A., Hoglund P., Lundin S., et al. Pharmacokinetics of 1-deamino-8-D-arginine vasopressin after various routes of administration in healthy volunteers. Clin Endocrinol. 1993;38:177.
47 Fleeman L.M., Irwin P.J., Phillips P.A., et al. Effects of an oral vasopressin receptor antagonist (OPC-31260) in a dog with syndrome of inappropriate secretion of antidiuretic hormone. Aust Vet J. 2000;78:825.
48 Forrest J.N., Cox M., Hong C., et al. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298:173.
49 Ghali J.K. Mechanisms, risks, and new treatment options for hyponatremia. Cardiology. 2008;111:147.
50 Gennari F.J. Serum osmolality: uses and limitations. N Engl J Med. 1984;310:102.
51 Gennari F.J., Kassirer J.P. Osmotic diuresis. N Engl J Med. 1974;291:714.
52 Giger U., Gorman N.T. Oncologic emergencies in small animals. Part II. Metabolic and endocrine emergencies. Compend Contin Educ Pract Vet. 1984;6:805.
53 Gines P., Cardenas A. The management of ascites and hyponatremia in cirrhosis. Semin Liver Dis. 2008;28:43.
54 Goldsmith S.R. Current treatments and novel pharmacologic treatments for hyponatremia in congestive heart failure. Am J Cardiol. 2005;95:14B.
55 Goldsmith S.R. Treatment options for hyponatremia in heart failure. Heart Fail Rev. 2009;14:65.
56 Grauer G.F., Grauer R.M. Veterinary clinical osmometry. Compend Contin Educ Pract Vet. 1983;5:539.
57 Green R.A., Farrow C.S. Diabetes insipidus in a cat. J Am Vet Med Assoc. 1974;164:524.
58 Greene C.E., Wong P.L., Finco D.R. Diagnosis and treatment of diabetes insipidus in two dogs using two synthetic analogs of antidiuretic hormone. J Am Anim Hosp Assoc. 1979;15:371.
59 Gross P.A., Ketteler M., Hausmann C., et al. The charted and uncharted waters of hyponatremia. Kidney Int. 1987;32(Suppl. 21):S67.
60 Gross P. Treatment of hyponatremia. Intern Med. 2008;47:885.
61 Guisado R., Arieff A.I., Massry S.G. Effects of glycerol infusions on brain water and electrolytes. Am J Physiol. 1974;227:865.
62 Hall E.F. Hypernatremia and adipsia in a dog (letter). J Am Vet Med Assoc. 1984;185:4.
63 Hanselman B., Kruth S., Poma R., et al. Hypernatremia and hyperlipidemia in a dog with central nervous system lymphosarcoma. J Vet Intern Med. 2006;20:1029.
64 Hara Y., Masuda H., Taoda T., et al. Prophylactic efficacy of desmopressin acetate for diabetes insipidus after hypophysectomy in the dog. J Vet Med Sci. 2003;65:17.
65 Harb M.F., Nelson R.W., Feldman E.C., et al. Central diabetes insipidus in dogs: 20 cases (1986-1995). J Am Vet Med Assoc. 1996;209:1884.
66 Hardy R.M. Hypernatremia. Vet Clin North Am Small Anim Pract. 1989;19:231.
67 Hardy R.M., Osborne C.A. Water deprivation test in the dog: maximal normal values. J Am Vet Med Assoc. 1979;174:479.
68 Hardy R.M., Osborne C.A. Aqueous vasopressin response test in clinically normal dogs undergoing water diuresis: technique and results. Am J Vet Res. 1987;43:1982.
69 Hardy R.M., Osborne C.A. Repositol vasopressin response test in clinically normal dogs undergoing water diuresis: technique and results. Am J Vet Res. 1991;43:1982.
70 Hawks D., Giger U., Miselis R., et al. Essential hypernatremia in a young dog. J Small Anim Pract. 1991;32:420.
71 Hendriks H.J., de Bruijne J.J., Van den Brom W.E. The clinical refractometer: a useful tool for the determination of specific gravity and osmolality of canine urine. Tijdschr Diergeneeskd. 1978;103:1065.
72 Henry W.B., Sieber S.E. Traumatic diabetes insipidus in a dog. J Am Vet Med Assoc. 1965;146:1317.
73 Hillier T.A., Abbott R.D., Barrett E.J. Hyponatremia: evaluating the correction factor for hyperglycemia. Am J Med. 1999;106:399.
74 Hinchcliff K.W., Reinhart G.A., Burr J.R., et al. Effect of racing on serum sodium and potassium concentrations and acid-base status of Alaskan sled dogs. J Am Vet Med Assoc. 1997;210:1615.
75 Hinchcliff K.W., Reinhart G.A., Burr J.R., et al. Exercise-associated hyponatremia in Alaskan sled dogs: urinary and hormonal responses. J Appl Physiol. 1997;83:824.
76 Hoskins J.D., Rothschmitt J. Hypernatremic thirst deficiency in a dog. Vet Med. 1984;79:489.
77 Houston D.M., Allen D.G., Kruth S.A., et al. Syndrome of inappropriate antidiuretic hormone secretion in a dog. Can Vet J. 1989;30:423.
78 Hughes D.E., Sokolowski J.H. Sodium chloride poisoning in the dog. Canine Pract. 1978;5:28.
79 Ichikawa I., Rennke H.G., Hoyer J.R., et al. Role for intrarenal mechanism in the impaired salt excretion of experimental nephrotic syndrome. J Clin Invest. 1983;71:91.
80 Joles J.A., Gruys E. Nephrogenic diabetes insipidus in a dog with renal medullary lesions. J Am Vet Med Assoc. 1979;174:830.
81 Kang J.H., Chang D., Lee Y.W., et al. Adipsic hypernatremia in a dog with anti-thyroid antibodies in cerebrospinal fluid and serum. J Vet Med Sci. 2006;69:751.
82 Katz M.A. Hyperglycemia-induced hyponatremia. N Engl J Med. 1973;289:843.
83 Kelly M.J., Hill J.R. Canine myxedema stupor and coma. Compend Contin Educ Pract Vet. 1984;6:1049.
84 Khanna C., Boermans H.J., Wilcock B. Fatal hypernatremia in a dog from salt ingestion. J Am Anim Hosp Assoc. 1997;33:113.
85 Kitchell B.E., Fan T.M., Kordick D., et al. Peliosis hepatitis in a dog infected with Bartonella henselae. J Am Vet Med Assoc. 2000;216:519.
86 Kraus K.H. The use of desmopressin in diagnosis and treatment of diabetes insipidus in cats. Compend Contin Educ Pract Vet. 1987;9:752.
87 Kraus K.H., Turrentine M.A., Jergens A.E., et al. Effect of desmopressin acetate on bleeding times and plasma von Willebrand factor in Doberman pinscher dogs with von Willebrand’s disease. Vet Surg. 1989;18:103.
88 Kumar S., Rubin S., Mather P.J., et al. Hyponatremia and vasopressin antagonism in congestive heart failure. Clin Cardiol. 2007;30:546.
89 Ladenson J.H., Apple F.S., Koch D.D. Misleading hyponatremia due to hyperlipemia: a method-dependent error. Ann Intern Med. 1981;95:707.
90 Lage A.L. Nephrogenic diabetes insipidus in a dog. J Am Vet Med Assoc. 1973;163:251.
91 Lage A.L. Apparent psychogenic polydipsia. In: Kirk R.W., editor. Current veterinary therapy VI. Philadelphia: WB Saunders; 1977:1098.
92 Lage A.L. Nephrogenic diabetes insipidus. In: Kirk R.W., editor. Current veterinary therapy VI. Philadelphia: WB Saunders; 1977:1102.
93 Lamb W.A., Muir P. Lymphangiosarcoma associated with hyponatremia and hyperkalemia in a dog. J Small Anim Pract. 1994;35:374.
94 Laureno R. Central pontine myelinolysis following rapid correction of hyponatremia. Ann Neurol. 1983;13:232.
95 Laureno R., Karp B.I. Myelinolysis after correction of hyponatremia. Ann Intern Med. 1997;126:57.
96 Leiva J.G., Salgado J.M., Estradas J., et al. Pathophysiology of ascites and dilutional hyponatremia: contemporary use of aquaretic agents. Ann Hepatol. 2007;6:214.
97 Levin E.R., Gardner D.G., Samson W.K. Natriuretic peptides. N Engl J Med. 1998;339:321.
98 Levitin H., Goodman A., Pigeon G., et al. Composition of the renal medulla during water diuresis. J Clin Invest. 1962;41:1145.
99 Lieberman L.L., Kircher C.H., Lein D.H. Polyuria and polydipsia associated with pituitary visceral larval migrans in a dog. J Am Anim Hosp Assoc. 1979;15:237.
100 Lifton S.J., King L.G., Zerbe C.A. Glucocorticoid deficient hypoadrenocorticism in dogs: 18 cases (1986-1995). J Am Vet Med Assoc. 1996;209:2076.
101 Ling G.V., Stabenfeldt G.H., Comer K.M., et al. Canine hyperadrenocorticism: pretreatment clinical and laboratory evaluation of 117 cases. J Am Vet Med Assoc. 1979;174:1211.
102 Lubberink A.A. Therapy for spontaneous hyperadrenocorticism. In: Kirk R.W., editor. Current veterinary therapy VII. Philadelphia: WB Saunders; 1980:979.
103 Luzius H., Jans D.A., Grunbaum E.G., et al. A low affinity vasopressin V2-receptor in inherited nephrogenic diabetes insipidus. J Recept Res. 1992;12:351.
104 MacKay B.M., Curtis N. Adipsia and hypernatraemia in a dog with focal hypothalamic granulomatous meningoencephalitis. Aust Vet J. 1999;77:14.
105 MacMillan K.L. Neurologic complications following treatment of canine hypoadrenocorticism. Can Vet J. 2003;44:490.
106 Madewell B.R., Osborne C.A., Norrdin R.A., et al. Clinicopathologic aspects of diabetes insipidus in the dog. J Am Anim Hosp Assoc. 1975;11:497.
107 Malik R., Hunt G.B., Hinchliffe J.M., et al. Severe whipworm infection in the dog. J Small Anim Pract. 1990;31:185.
108 Mann J.F.E., Johnson A.K., Ganten D., et al. Thirst and the renin angiotensin system. Kidney Int. 1987;32(Suppl. 21):S27.
109 Martin-Llahi M., Guevara M., Gines P. Hyponatremia in cirrhosis: clinical features and management. Gastroenterol Clin Biol. 2006;30:1144.
110 McGirt M.J., Blessing R., Nimjee S.M., et al. Correlation of serum brain natriuretic peptide with hyponatremia and delayed ischemic neurological deficits after subarachnoid hemorrhage. Neurosurgery. 2004;54:1369.
111 Miller M. Hyponatremia and arginine vasopressin dysregulation: mechanisms, clinical consequences, and management. J Am Geriatr Soc. 2006;54:345.
112 Miyama T.S., Iwamoto E., Umeki S., et al. Magnetic resonance imaging and clinical findings in a miniature Schnauzer with hypodipsic hypernatremia. J Vet Med Sci. 2009;71:1387.
113 Moreau R. Hyponatremia in cirrhosis: pathophysiology, prevalence, prognostic value, treatment. Acta Gastroenterol Belg. 2008;71:379.
114 Moritz M.L., Ayus J.C. Hospital-acquired hyponatremia: why are hypotonic parenteral fluids still being used? Nat Clin Pract Nephrol. 2007;3:374.
115 Morrison J.A., Fales-Williams A. Hypernatremia associated with intracranial B-cell lymphoma in a cat. Vet Clin Pathol. 2006;35:362.
116 Mulnix J.A., Rijnberk A., Hendriks H.J. Evaluation of a modified water-deprivation test for diagnosis of poly-uric disorders in dogs. J Am Vet Med Assoc. 1976;169:1327.
117 Multz A.S. Vasopressin dysregulation and hyponatremia in hospitalized patients. J Intensive Care Med. 2007;22:216.
118 Narins R.G., Jones E.R., Stom M.C., et al. Diagnostic strategies in disorders of fluid, electrolyte and acid base homeostasis. Am J Med. 1982;72:496.
119 Neer T.M., Reavis D.U. Craniopharyngioma and associated central diabetes insipidus and hypothyroidism in a dog. J Am Vet Med Assoc. 1983;182:519.
120 Nielsen L., Thompson H., Hammond G.J., et al. Central diabetes insipidus associated with primary focal B cell lymphoma in a dog. Vet Rec. 2008;162:124.
121 Neilsen S., Frokiaer J., Marples D., et al. Aquaporins in the kidney: from molecules to medicine. Physiol Rev. 2002;82:205.
122 O’Brien D.P., Kroll R.A., Johnson G.C., et al. Myelinolysis after correction of hyponatremia in two dogs. J Vet Intern Med. 1994;8:40.
123 O’Grady S.M., Palfrey H.C., Field M. Characteristics and function of Na-K-2Cl cotransport in epithelial tissues. Am J Physiol. 1987;253:C177.
124 Oh M.S. Management of hyponatremia and clinical use of vasopressin antagonists. Am J Med Sci. 2007;333:101.
125 Oren R.M. Hyponatremia in congestive heart failure. Am J Cardiol. 2005;95:2B.
126 Palm C., Pistrosch F., Hebrig K., et al. Vasopressin antagonists as aquaretic agents for the treatment of hyponatremia. Am J Med. 2006;119:S87.
127 Palmer L.G., Frindt G. Amiloride sensitive Na+ channels from the apical membrane of rat cortical collecting tubule. Proc Natl Acad Sci USA. 1986;83:2767.
128 Peterson M. Hyperadrenocorticism. Vet Clin North Am Small Anim Pract. 1984;14:731.
129 Peterson M.E., Kintzer P.P., Kass P.H. Pretreatment clinical and laboratory findings in dogs with hypoadrenocorticism:225 cases (1979-1993). J Am Vet Med Assoc. 1996;208:85.
130 Pittari J.M. Central diabetes insipidus in a cat. Feline Pract. 1996;24:18.
131 Post K., McNeill J.R.J., Clark E.G., et al. Congenital central diabetes insipidus in two sibling Afghan hound pups. J Am Vet Med Assoc. 1989;194:1086.
132 Rakich P.M., Lorenz M.D. Clinical signs and laboratory abnormalities in 23 dogs with spontaneous hypoadrenocorticism. J Am Anim Hosp Assoc. 1984;20:647.
133 Reidarson T.H., Weis D.J., Hardy R.M. Extreme hypernatremia in a dog with central diabetes insipidus: a case report. J Am Anim Hosp Assoc. 1990;26:89.
134 Reynolds R.M., Padfield P.L., Seckl J.R. Disorders of sodium balance. Br Med J. 2006;332:702.
135 Rijnberk A., Biewenga W.J., Mol J.A. Inappropriate vasopressin secretion in two dogs. Acta Endocrinol. 1988;117:59.
136 Rittig S., Jensen A.R., Jensen K.T., et al. Effect of food intake on the pharmacokinetics and antidiuretic activity of oral desmopressin (DDAVP) in hydrated normal subjects. Clin Endocrinol. 1998;48:235.
137 Robertson G.L. Thirst and vasopressin function in normal and disordered states of water balance. J Lab Clin Med. 1983;101:351.
138 Rogers I.R., Hew-Butler T. Exercise-associated hyponatremia: overzealous fluid consumption. Wilderness Environ Med. 2009;20:139.
139 Rogers W., Straus J., Chew D. A typical hypoadrenocorticism in three dogs. J Am Vet Med Assoc. 1981;179:155.
140 Rogers W.A., Valdez H., Anderson B.C., et al. Partial deficiency of antidiuretic hormone in a cat. J Am Vet Med Assoc. 1977;170:545.
141 Rosner M.H. Exercise-associated hyponatremia. Semin Nephrol. 2009;29:271.
142 Ross L.A., Finco D.R. Relationship of selected clinical renal function tests to glomerular filtration rate and renal blood flow in cats. Am J Vet Res. 1981;42:1704.
143 Rothuizen J., Biewenga W.J., Mol J.A. Chronic glucocorticoid excess and impaired osmoregulation of vasopressin release in dogs with hepatic encephalopathy. Domest Anim Endocrinol. 1995;12:13.
144 Schaer M., Chen C.L. A clinical survey of 48 dogs with adrenocortical hypofunction. J Am Anim Hosp Assoc. 1983;19:443.
145 Schaer M., Halling K.B., Collins K.E., et al. Combined hyponatremia and hyperkalemia mimicking acute hypoadrenocorticism in three pregnant dogs. J Am Vet Med Assoc. 2001;218:897.
146 Schrier R.W. Water and sodium retention in edematous disorders: role of vasopressin and aldosterone. Am J Med. 2006;119:S47.
147 Schuster V.L., Stokes J.B. Chloride transport by the cortical and outer medullary collecting duct. Am J Physiol. 1987;253:F203.
148 Shiel R.E., Pinilla M., Mooney C.T. Syndrome of inappropriate antidiuretic hormone secretion associated with congenital hydrocephalus in a dog. J Am Anim Hosp Assoc. 2009;45:249.
149 Shull R.M. The value of anion gap and osmolal gap determinations in veterinary medicine. Vet Clin Pathol. 1978;7:12.
150 Sica D.A. Hyponatremia and heart failure: treatment considerations. Congest Heart Fail. 2006;12:55.
151 Sterns R.H., Ocdol H., Schrier R.W., et al. Hyponatremia: pathophysiology, diagnosis and therapy. In: Narins R.G., editor. Maxwell and Kleeman’s clinical disorders of fluid and electrolyte metabolism. New York: McGraw-Hill; 1994:583.
152 Sterns R.H., Spital A., Clark E.C. Disorders of water balance. In: Kokko J.P., Tannen R.L., editors. Fluids and electrolytes. Philadelphia: WB Saunders; 1996:63.
153 Sullivan S.A., Harmon B.G., Purinton P.T., et al. Lobar holoprosencephaly in a miniature Schnauzer with hypodipsic hypernatremia. J Am Vet Med Assoc. 2003;223:1783.
154 Takemura N. Successful long-term treatment of congenital nephrogenic diabetes insipidus in a dog. J Small Anim Pract. 1998;39:592.
155 Thompson M.D., Carr A.P. Hyponatremia and hyperkalemia associated with chylous pleural and peritoneal effusion in a cat. Can Vet J. 2002;43:610.
156 Thompson A.L., Scott-Moncrieff J.C., Anderson J.D. Comparison of classic hypoadrenocorticism with glucocorticoid-deficient hypoadrenocorticism in dogs: 46 cases (1985-2005). J Am Vet Med Assoc. 2007;230:1190.
157 Toll J., Barr S.C., Hickford F.H. Acute water intoxication in a dog. J Vet Emerg Crit Care. 1999;9:19.
158 Tyler R.D., Qualls C.W., Heald R.D., et al. Renal concentrating ability in dehydrated hyponatremic dogs. J Am Vet Med Assoc. 1987;191:1095.
159 Van Heerden J., Geel J., Moore D.J. Hypodipsic hypernatremia in a miniature schnauzer. J S Afr Vet Assoc. 1992;63:39.
160 Vilhardt H., Bie P. Antidiuretic response in conscious dogs following peroral administration of arginine vasopressin and its analogs. Eur J Pharmacol. 1983;93:201.
161 Willard M.D., Schall W.D., McCaw D.E., et al. Canine hypoadrenocorticism: report of 37 cases and review of 39 previously reported cases. J Am Vet Med Assoc. 1982;180:59.
162 Willard M.D., Fossum T.W., Torrance A., et al. Hyponatremia and hyperkalemia associated with idiopathic or experimentally induced chylothorax in four dogs. J Am Vet Med Assoc. 1991;199:353.
163 Winterbotham J., Mason K. Congenital diabetes insipidus in a kitten. J Small Anim Pract. 1983;24:569.
164 Yamamoto T., Sasaki S. Aquaporins in the kidney: emerging new aspects. Kidney Int. 1998;54:1041.
165 Yeates K.E., Morton A.R. Vasopressin antagonists: role in the management of hyponatremia. Am J Nephrol. 2006;26:348.
166 Zenger E. Persistent hyperkalemia associated with nonchylous pleural effusion in a dog. J Am Anim Hosp Assoc. 1992;28:411.
167 Zerbe R., Stropes L., Robertson G. Vasopressin function in the syndrome of inappropriate antidiuresis. Annu Rev Med. 1980;31:315.