Chapter 5 Disorders of Potassium
Hypokalemia and Hyperkalemia
Potassium is the major intracellular cation in mammalian cells, whereas sodium is the major extracellular cation. Normally, the extracellular fluid (ECF) sodium concentration is approximately 140 mEq/L, and the ECF potassium concentration is approximately 4 mEq/L. This relationship is reversed in intracellular fluid (ICF), in which the sodium concentration is approximately 10 mEq/L and the potassium concentration is approximately 140 mEq/L. In experimental studies of dogs, control values for ICF sodium and potassium concentrations in skeletal muscle were 8.4 to 13.7 and 139 to 142 mEq/L, respectively.20,107
Total body potassium content in humans is approximately 50 to 55 mEq/kg body weight, and almost all of this potassium is readily exchangeable.6,70 In one study of potassium depletion in dogs, the control value for total exchangeable potassium as determined by 50K dilution was 47.1 mEq/kg body weight (range, 39.8 to 61.1 mEq/kg).1 In cats, total body potassium is approximately 55 mEg/kg body weight.184a As much as 95% or more of total body potassium is located within cells, with muscle containing 60% to 75% of this potassium. Muscle potassium content in normal dogs and cats is approximately 400 mEq/kg.20,107,147,191 As a solute, intracellular potassium is crucial for maintenance of normal cell volume. Intracellular potassium also is important for normal cell growth because it is required for the normal function of enzymes responsible for nucleic acid, glycogen, and protein synthesis.
The remaining 5% of the body’s potassium is located in the ECF. Maintaining the ECF potassium concentration within narrow limits is critical to avoid the life-threatening effects of hyperkalemia on cardiac conduction. In humans, the serum potassium concentration is inversely correlated with the total body deficit of potassium (Fig. 5-1). Likewise, in dogs with potassium depletion induced by dietary restriction, the muscle potassium content was strongly correlated (r = 0.87) with the serum potassium concentration.147 During translocation of potassium between ICF and ECF, however, the serum potassium concentration can change without any change in the total body potassium content. One of the most important functions of potassium in the body is its role in generation of the normal resting cell membrane potential.
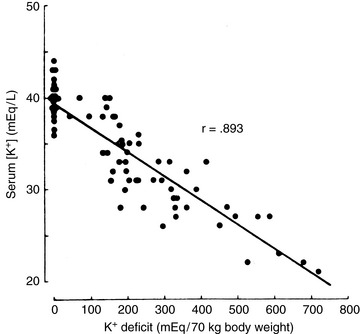
Figure 5-1 Relationship of serum potassium concentration to bodily potassium deficit. The data are derived from seven metabolic balance studies carried out on 24 human subjects depleted of potassium.
(From Sterns RH et al.: Medicine 60:339, 1981.)
The resting cell membrane potential
The normal relationship between ECF and ICF potassium concentrations is maintained by sodium, potassium-adenosinetriphosphatase (Na+, K+-ATPase) in cell membranes. This enzyme pumps sodium ions out of, and potassium ions into, the cell in a 3:2 Na/K ratio so that the intracellular concentration of potassium is much higher than its extracellular concentration. As a result, K+ ions diffuse out of the cell down their concentration gradient. However, the cell membrane is impermeable to most intracellular anions (e.g., proteins and organic phosphates). Therefore, a net negative charge develops within the cell as K+ ions diffuse out, and a net positive charge accumulates outside the cell. Consequently, a potential difference is generated across the cell membrane.
The principal extracellular cation is sodium, and it enters the cell relatively slowly down its concentration and electrical gradients, because the permeability of the cell membrane to potassium is 100-fold greater than its permeability to sodium. Diffusion of K+ ions from the cell continues until the ECF acquires sufficient positive charge to prevent further diffusion of K+ ions out of the cell. The ratio of the intracellular to extracellular concentrations of potassium ([K+]I/[K+]O) is the major determinant of the resting cell membrane potential as described by the Nernst equation:
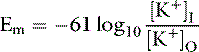
The Goldman-Hodgkin-Katz equation is a modification of the Nernst equation that allows prediction of Em based on the ionic permeability characteristics of the cell membrane to sodium and potassium and the concentrations of these ions inside and outside the cell:
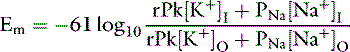
where PNa and PK are the membrane permeabilities for sodium and potassium. The term r is included in the equation to account for the effect of the electrogenic Na+, K+-ATPase pump under steady-state conditions. This term is assigned the Na/K transport ratio of 3:2 so that r = 1.5. If the membrane permeability for potassium is assigned a value of 1.0 and the cell membrane is 100 times more permeable to potassium than sodium:
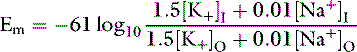
For example, using the hypothetical ECF and ICF concentrations of sodium and potassium given at the beginning of this chapter:
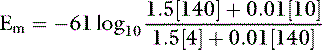
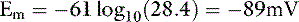
In one study of dogs with potassium deficiency, the predicted Em was −86.6 mV and the measured Em in skeletal muscle of control animals was −90.1 mV.20 The resting cell membrane potential plays a vital role in the normal function of skeletal and cardiac muscle, nerves, and transporting epithelia.
The threshold cell membrane potential
The threshold cell membrane potential is reached when sodium permeability increases to the point that sodium entry exceeds potassium exit, depolarization becomes self-perpetuating, and an action potential develops. The ability of specialized cells to develop an action potential is crucial to normal cardiac conduction, muscle contraction, and nerve impulse transmission. The excitability of a tissue is determined by the difference between the resting and threshold potentials (the smaller the difference, the greater the excitability).
Hypokalemia increases the resting potential (i.e., makes it more negative) and hyperpolarizes the cell, whereas hyperkalemia decreases the resting potential (i.e., makes it less negative) and initially makes the cell hyperexcitable (Fig. 5-2). If the resting potential decreases to less than the threshold potential, depolarization results, repolarization cannot occur, and the cell is no longer excitable. Translocation of potassium between body compartments results in a greater change in the ratio of intracellular to extracellular potassium concentrations ([K+]I/[K+]O) than does a change in total body potassium. In the former instance, the potassium concentrations of the two compartments change in opposite directions, whereas in the latter instance, they change in the same direction.

Figure 5-2 Effects of serum calcium and potassium on membrane potentials of excitable tissues. The concentration of potassium in extracellular fluids affects the resting potential, whereas calcium concentrations alter the threshold potential.
(From Leaf A, Cotran R. Renal pathophysiology. New York: Oxford University Press, 1976: 116.)
Membrane excitability also is affected by ionized calcium concentration and acid-base balance. Calcium affects the threshold potential rather than the resting potential. Ionized hypocalcemia increases membrane excitability by allowing self-perpetuating sodium permeability to be reached with a lesser degree of depolarization, whereas ionized hypercalcemia requires greater than normal depolarization for this threshold to be reached (see Fig. 5-2). Thus, hypercalcemia counteracts hyperkalemia by normalizing the difference between the resting and threshold potentials, whereas hypocalcemia exacerbates the effect of hyperkalemia on membrane excitability. This principle is the basis for treating hyperkalemia with calcium salts (see the Treatment of Hyperkalemia section). Membrane excitability is increased by alkalemia and decreased by acidemia. As a result of these factors, clinical signs are not necessarily correlated with serum potassium concentrations. Electrocardiographic findings and muscle strength reflect the functional consequences of abnormalities in serum potassium concentration.
Potassium balance
External potassium balance
External balance for potassium is maintained by matching output (primarily in urine) to input (from the diet). In the normal animal, potassium enters the body only through the gastrointestinal tract, and virtually all ingested potassium is absorbed in the stomach and small intestine. Transport of potassium in the small intestine is passive, whereas active transport (responsive to aldosterone) occurs in the colon. Colonic secretion of potassium may play an important role in extrarenal potassium homeostasis in some disease states (e.g., chronic renal failure) (Fig. 5-3).
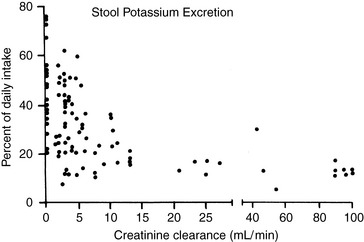
Figure 5-3 Relationship between the degree of renal insufficiency and fecal potassium excretion. Data points are compiled from three studies comprising 98 balance periods in 40 human patients. Variation in dietary protein or sodium intake did not produce consistent changes in fecal potassium excretion; thus, data points from these balance periods were included without special designation.
(From Alexander EA, Perrone RD. Regulation of extrarenal potassium metabolism. In: Maxwell MH, Kleeman CR, Narins RG, editors. Clinical disorders of fluid and electrolyte metabolism, 4th ed. New York: McGraw-Hill, 1987: 105–117, with permission of the McGraw-Hill Companies.)
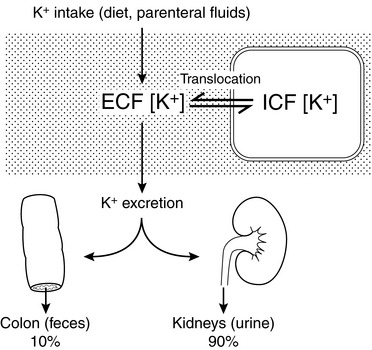
Potassium derived from the diet and endogenous cellular breakdown is removed from the body primarily by the kidneys and, to a much lesser extent, by the gastrointestinal tract. During zero balance, 90% to 95% of ingested potassium is excreted in urine, and the remaining 5% to 10% is excreted via the gastrointestinal tract. This pattern of output has been observed during control balance studies in normal dogs.12,139,152,169,182 In a study of renal handling of potassium in dogs, 90% to 98% of potassium intake was eliminated from the body by the kidneys.24
Adaptation occurs during chronic potassium loading so that the animal is protected from hyperkalemia that could occur as a result of an acute potassium load. This effect results from enhanced renal and colonic excretion of potassium, as well as from enhanced uptake of potassium by the liver and muscle, mediated by the effects of insulin and catecholamines. Potassium deprivation is associated with decreased aldosterone secretion, suppression of potassium secretion in the distal nephron, and increased reabsorption of potassium in the inner medullary collecting ducts. Skeletal muscle potassium concentration decreases, but brain and heart potassium concentrations are minimally affected during potassium depletion.20,107,178 The colon adapts to potassium deprivation by decreasing its secretion of potassium.
Internal potassium balance
Internal balance for potassium is maintained by translocation of potassium between ECF and ICF. One half to two thirds of an acute potassium load appears in the urine within the first 4 to 6 hours, and effective translocation of potassium from ECF to ICF is crucial in preventing life-threatening hyperkalemia until the kidneys have sufficient time to excrete the remainder of the potassium load. Endogenous insulin secretion and stimulation of β2-adrenergic receptors by epinephrine promote cellular uptake of potassium in the liver and muscle by increasing the activity of cell membrane Na+, K+-ATPase. The main effects of these hormones are to facilitate distribution of an acute potassium load and not to mediate minor adjustments in serum potassium concentration. The ECF concentration of potassium itself plays an important role in translocation because potassium movement into cells is facilitated by the change in chemical concentration gradient resulting from addition of potassium to ECF. The fraction of an acute potassium load taken up by the body is increased during chronic potassium depletion and decreased when total body potassium is excessive. In summary, any change in serum potassium concentration must arise from a change in intake, distribution, or excretion (Fig. 5-4).
Effect of acid-base balance on potassium distribution
The effect of acute pH changes on translocation of potassium between ICF and ECF is complex. In general, acidosis is associated with movement of potassium ions from ICF to ECF, and alkalosis is associated with movement of potassium ions from ECF to ICF. Early animal studies and observations in a small number of human patients led to the prediction that acute metabolic acidosis would be associated with a 0.6-mEq/L increment in serum potassium concentration for each 0.1-U decrement in pH. This rule of thumb has circulated widely among clinicians.31,181,188
However, a critical review of experimental studies in animals and humans demonstrated that changes in serum potassium concentration during acute acid-base disturbances were quite variable.4 The change in serum potassium concentration was greatest during acute mineral acidosis. In dogs, the increase in serum potassium concentration after administration of a mineral acid (e.g., HCl or NH4Cl) was very variable, ranging from a 0.17- to 1.67-mEq/L increment in serum potassium concentration per 0.1-U decrement in pH (mean, 0.75 mEq/L). The increment in serum potassium concentration during acute respiratory acidosis in dogs was much lower, averaging only 0.14 mEq/L per 0.1-U decrement in pH. The decrement in serum potassium concentration during metabolic alkalosis in dogs averaged 0.18 mEq/L per 0.1-U increment in pH, whereas it averaged 0.27 mEq/L per 0.1-U increment in pH during respiratory alkalosis. In another study, respiratory alkalosis induced by hyperventilation in anesthetized dogs caused a somewhat greater decrement in serum potassium concentration (0.4 mEq/L) for each 0.1-U increment in pH.136 An increase in serum potassium concentration did not occur in acute metabolic acidosis caused by organic acids (e.g., lactic acid and ketoacids).* Acute infusion of β-hydroxybutyric acid in normal dogs caused an increase in insulin in portal venous blood and hypokalemia, presumably as a result of potassium uptake by cells. Conversely, acute infusion of HCl led to increased portal vein glucagon concentration and hyperkalemia, possibly caused by potassium release from cells.5 In summary, only mineral acidosis is expected to cause any clinically relevant change in serum potassium concentration during acute acid-base disturbances.
Many factors probably contribute to the variable changes observed in serum potassium concentration during acute acid-base disturbances, including blood pH and HCO3− concentration, nature of the acid anion (mineral versus organic), osmolality, hormonal activity (e.g., catecholamines, insulin, glucagon, and aldosterone), and the metabolic and excretory roles of the liver and kidneys.4 Hyperosmolality and lack of insulin are more likely to be responsible for hyperkalemia observed in patients with diabetic ketoacidosis than is the acidosis itself.
Hyperkalemia associated with acute metabolic acidosis induced by mineral acids is transient. In a study of acute and chronic metabolic acidosis induced in dogs by administration of HCl or NH4Cl, hyperkalemia was observed after acute infusion of HCl, but hypokalemia developed after 3 to 5 days of NH4Cl administration.122 The observed hypokalemia was associated with inappropriately high urinary excretion of potassium and increased plasma aldosterone concentration.122 Similar findings have been reported in rats with chronic metabolic acidosis induced by NH4Cl. Despite a total body deficit of potassium, rats with chronic metabolic acidosis did not conserve potassium appropriately.170 This effect may be caused by a decreased filtered load of HCO3−, increased distal delivery of sodium, and increased distal tubular flow. Thus, metabolic acidosis of at least 2 to 3 days’ duration is associated with increased urinary potassium excretion and mild hypokalemia rather than hyperkalemia.79
Renal handling of potassium
The kidneys are the primary regulators of potassium balance. Potassium is filtered at the glomerulus, and approximately 70% of the filtered load is reabsorbed isosmotically with water and sodium in the proximal tubule. An additional 10% to 20% of filtered potassium is reabsorbed in the ascending limb of Henle’s loop. Finally, 10% to 20% of the filtered load is delivered to the distal nephron, where final adjustments in potassium reabsorption and secretion are made. Potassium experiences either net reabsorption or secretion in the connecting tubule, cortical collecting duct, and first portion of the outer medullary collecting duct, depending on the body’s needs. Net movement of potassium in these segments of the nephron determines urinary excretion of potassium. Potassium once again experiences reabsorption in the last portion of the outer medullary collecting duct and inner medullary collecting duct regardless of the body’s needs.
Mechanisms of renal tubular transport of potassium
The transepithelial electrical potential difference is lumen negative in the early proximal tubule, but no active transport mechanism for potassium has been discovered in this segment of the nephron. In the proximal tubule, potassium is reabsorbed along with water by solvent drag via the paracellular route. Apparently, water reabsorption increases the luminal concentration of potassium enough to overcome the unfavorable transepithelial potential difference. The transepithelial electrical potential difference becomes lumen positive in the late proximal tubule, and this facilitates reabsorption of potassium by the paracellular route. Transcellular transport of potassium in the proximal tubular cells occurs by means of potassium channels in both luminal and basolateral membranes and by a K+-Cl− cotransporter in basolateral membranes (Fig. 5-5).
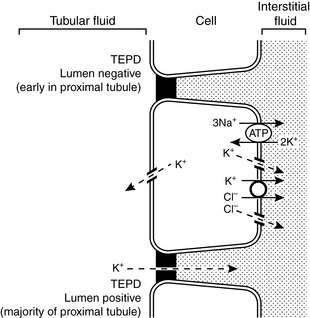
Figure 5-5 Renal tubular transport mechanisms for potassium in the proximal tubule.
TEPD, Transepithelial potential difference. (Drawing by Tim Vojt.)
In the thick ascending limb of the Henle loop, the transepithelial electrical potential difference is strongly lumen positive, and most potassium reabsorption occurs by the paracellular route. Potassium channels in the luminal membranes allow potassium to exit the cell down its concentration gradient and facilitate the electrochemical gradient for potassium reabsorption via the paracellular route. Transcellular reabsorption of potassium is facilitated by the luminal Na+-K+-2Cl− cotransporter and by potassium channels and a K+-Cl− cotransporter in the basolateral membranes (Fig. 5-6).
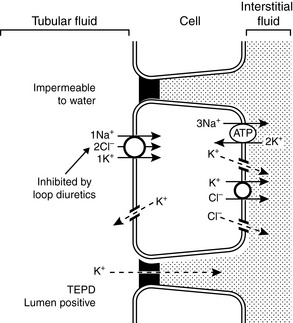
Figure 5-6 Renal tubular transport mechanisms for potassium in the thick ascending limb of Henle’s loop. TEPD, Transepithelial potential difference.
(Drawing by Tim Vojt.)
The mechanisms of renal potassium handling in the distal convoluted tubule are shown in Figure 5-7. The thiazide-sensitive Na+-Cl− cotransporter and the K+-Cl− cotransporter in the luminal membranes of these tubular cells result in secretion of potassium and reabsorption of sodium while chloride is recycled across the luminal membrane. The basolateral Na+, K+-ATPase maintains a low intracellular concentration of sodium and a high intracellular concentration of potassium that facilitate sodium reabsorption and potassium secretion across the luminal membranes.
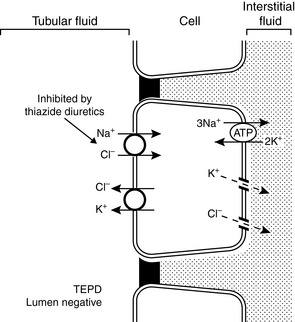
Figure 5-7 Renal tubular transport mechanisms for potassium in the distal convoluted tubule (early distal tubule).
TEPD, Transepithelial potential difference. (Drawing by Tim Vojt.)
Principal cells are found in the connecting tubule and collecting duct and are responsible for potassium secretion. The basolateral membranes of principal cells are rich in Na+, K+-ATPase, which maintains a high intracellular potassium concentration. The luminal membranes of the principal cells contain an electrogenic sodium channel (ENaC). This sodium channel is directly blocked by the diuretics amiloride and triamterene, whereas spironolactone antagonizes the effect of aldosterone on the channel. Sodium movement through this channel renders the tubular lumen negative, and the resultant increase in lumen electronegativity facilitates secretion of K+ ions through luminal K+ channels (Fig. 5-8).
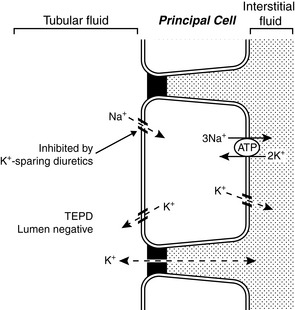
Figure 5-8 Renal tubular transport mechanisms for potassium in the principal cells of the late distal tubule and collecting duct.
TEPD, Transepithelial potential difference. (Drawing by Tim Vojt.)
There are two types of intercalated cells in the distal nephron. Type A or α intercalated cells contain H+-ATPase and H+, K+-ATPase in their luminal membranes and Cl−- HCO3− countertransporters and Cl− and K+ channels in their basolateral membranes. They also contain carbonic anhydrase. This arrangement allows the intercalated cell to secrete H+ ions and reabsorb K+ and HCO3− ions. Potassium is actively transported across the luminal membranes of type α intercalated cells by H+, K+-ATPase and then diffuses down its concentration gradient through potassium channels in the basolateral membranes (Fig. 5-9). Type A and α intercalated cells are found in the connecting tubule, cortical collecting duct, and outer medullary collecting duct. Type B and β intercalated cells are found only in the cortical collecting ducts and secrete HCO3− ions. They are able to do so because their polarity is reversed as compared with type α intercalated cells (i.e., the H+-ATPase is in the basolateral membrane, and the Cl−- HCO3− countertransporter is in the luminal membrane).
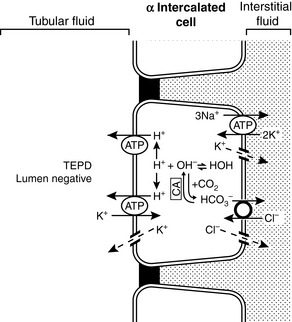
Figure 5-9 Renal tubular transport mechanisms for potassium in the α intercalated cells of the late distal tubule and collecting duct.
CA, Carbonic anhydrase; TEPD, Transepithelial potential difference. (Drawing by Tim Vojt.)
Potassium is reabsorbed from the last portion of the outer medullary collecting duct and throughout the inner medullary collecting duct. In these segments of the nephron, potassium is reabsorbed by the paracellular route despite a lumen-negative transepithelial potential difference, because reabsorption of water increases the chemical concentration gradient sufficiently to overcome the unfavorable electrical gradient.
Determinants of urinary potassium excretion
Three main factors affect potassium secretion in the distal nephron: the magnitude of the chemical concentration gradient for potassium between the tubular cells and tubular lumen, the tubular flow rate, and the transmembrane potential difference across the luminal membranes of the tubular cells. Gastrointestinal absorption of a potassium load increases the ECF concentration of potassium. This results in an increase in the number of K+ ions available for uptake at the basolateral membranes of the distal tubular cells by Na+, K+-ATPase, and the resulting increase in intracellular potassium concentration increases the chemical concentration gradient for diffusion of K+ ions out of the tubular cells across their luminal membranes.
Aldosterone is the most important hormone affecting urinary potassium excretion. Its secretion by the zona glomerulosa of the adrenal gland is stimulated directly by hyperkalemia and angiotensin II (produced in response to volume depletion), whereas adrenocorticotropic hormone (ACTH), hyponatremia, and decreased extracellular pH play permissive roles in promoting aldosterone secretion. Aldosterone release is inhibited by dopamine and atrial natriuretic factor, both of which are released in response to volume expansion.
Aldosterone increases reabsorption of Na+ and secretion of K+ and H+ ions in the distal nephron. Its primary effect is to increase the number of open Na+ channels in the luminal membranes of the principal cells. Sodium reabsorption via these luminal Na+ channels is electrogenic (i.e., it generates electronegativity in the tubular lumen). This electronegativity can be dissipated either by K+ or H+ ion secretion or by Cl− reabsorption in the distal nephron. Aldosterone increases the activity and number of Na+, K+-ATPase pumps in the basolateral membranes of the principal cells, and this effect may occur as a result of increased entry of Na+ ions across the luminal membranes. Increased Na+, K+-ATPase activity in turn increases the intracellular K+ concentration and facilitates K+ secretion across the luminal membranes. Aldosterone also increases the number of open K+ channels in the luminal membrane, thus facilitating K+ exit into tubular fluid.
Aldosterone can influence H+ secretion in two ways. It directly promotes H+ ion secretion in H+-secreting type α intercalated cells by stimulation of the H+-ATPase present in their luminal membranes. Aldosterone also promotes H+ secretion in the distal tubule by stimulating electrogenic Na+ reabsorption in principal cells and increasing lumen electronegativity, which favors enhanced H+ secretion.
An increase in distal tubular flow enhances potassium secretion by rapidly moving secreted K+ ions downstream and providing new tubular fluid from upstream in the nephron. This allows maintenance of a high chemical concentration gradient for potassium secretion and provides a “sink” for movement of K+ ions into tubular fluid. A decrease in distal tubular flow has the opposite effect and promotes dissipation of the chemical gradient for diffusion of K+ ions from principal cells into tubular fluid.
Lumen electronegativity is generated by sodium reabsorption through Na+ channels in the luminal membranes of principal cells. Normally, some of this electronegativity is dissipated by passive Cl− reabsorption. If a large concentration of a relatively nonresorbable anion (e.g., SO42−, HCO3−, penicillin) is present in distal tubular fluid, less dissipation of the electronegativity occurs, and K+ secretion is enhanced. This factor contributes to the pathophysiology of metabolic alkalosis. In this setting, there is less Cl−and more HCO3− in the distal tubular fluid, and HCO3− is relatively nonresorbable in the cortical collecting duct. This is one reason metabolic alkalosis promotes urinary K+ excretion. Amiloride is a diuretic that impairs luminal Na+ entry into principal cells by decreasing the number of open Na+ channels. This in turn reduces lumen electronegativity and impairs K+ secretion. Thus, the magnitude of distal tubular lumen electronegativity has an important effect on urinary K+ excretion.
The Na+ and Cl− concentrations of distal tubular fluid usually have little effect on K+ secretion. When the luminal Na+ concentration is very low (<25 to 35 mEq/L), however, diffusion of Na+ ions into distal tubular cells may be impaired sufficiently to produce an increase in the tubular cell transmembrane potential (making the cell interior more negative) and impeding diffusion of K+ ions from the cell into the tubular lumen.85,204,205 Extremely low luminal Cl− concentrations (<10 mEq/L) may increase net potassium secretion, possibly because some fraction of K+ reabsorption or secretion may be accomplished by K+-Cl− cotransport.198 Such a mechanism may also play a role in the pathophysiology of enhanced urinary K+ excretion during metabolic alkalosis. Antidiuretic hormone (ADH) helps minimize disruption of potassium balance during water deprivation by increasing the number of open luminal K+ channels in principal cells and facilitating potassium excretion at a time when distal tubular flow is reduced.36,69,183 Conversely, potassium excretion is not necessarily increased despite increased distal tubular flow during water diuresis because ADH is suppressed. Major factors affecting renal excretion of potassium are summarized in Figure 5-10.
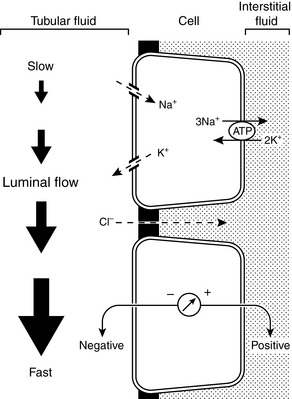
Factors influencing renal potassium excretion
Sodium Intake
High sodium intake is associated with increased urinary potassium excretion as a result of increased potassium secretion in the connecting tubule and cortical collecting duct. Increased delivery of sodium to the distal nephron results in more sodium crossing the luminal membranes of the distal tubular cells down its concentration gradient. This increased entry of Na+ ions into the tubular cells leads to increased activity of Na+, K+-ATPase in the basolateral membranes with removal of sodium to the peritubular interstitium and increased cellular uptake of potassium. This increased intracellular potassium then crosses the luminal membranes of the tubular cells and enters the tubular fluid down a favorable electrochemical gradient. Increased sodium delivery to the distal nephron also increases the distal tubular fluid flow rate, which enhances the chemical concentration gradient for potassium between the tubular cell cytoplasm and tubular fluid.
Low sodium intake is associated with decreased renal potassium excretion as a result of mechanisms opposite to those described previously. In addition, increased potassium reabsorption by type α intercalated cells occurs in the medullary collecting duct. One reason for this increased reabsorption may be increased recycling of potassium into the medullary interstitium, which may play a role in the urinary concentrating mechanism when sodium intake is restricted.
Potassium Intake
High potassium intake is associated with increased urinary potassium excretion as a result of increased tubular secretion of potassium in the connecting tubule, cortical collecting duct, and outer medullary collecting duct. This occurs because of increased numbers and activity of Na+, K+-ATPase pumps, and amplification of the basolateral membranes of principal cells, which results from an increased concentration of aldosterone. Therefore, more potassium is actively pumped into the tubular cells from the peritubular interstitium, then leaves the cells down a favorable electrochemical gradient, and enters the tubular fluid.
Low potassium intake results in decreased urinary excretion of potassium. In the presence of low potassium intake, tubular secretion by principal cells is decreased to absent in the connecting tubule, cortical collecting duct, and outer medullary collecting duct and increased reabsorption by type α intercalated cells occurs in the inner medullary collecting duct. The decrease in tubular secretion results from less potassium being available for peritubular uptake into the tubular cells by the Na+, K+-ATPase pump and a less favorable concentration gradient for potassium to leave the tubular cells and enter tubular fluid.
Dietary potassium intake also has a direct effect on the function of the luminal potassium channels in principal cells. A high potassium intake increases the activity of these channels by decreasing the phosphorylation of a specific tyrosine residue in the ROMK protein component of the channel, which in turn results in decreased removal of the channels from the luminal membranes. A low potassium diet has the opposite effect.82,201,203
Mineralocorticoids
An increased concentration of aldosterone results in increased urinary excretion of potassium as a result of increased secretion of potassium by tubular cells mainly in the cortical collecting duct. The actions of aldosterone on the principal cells result in increased uptake of potassium from the peritubular interstitium and increased movement of potassium into tubular fluid across the luminal membranes of the principal cells. A decreased transmembrane potential difference across the luminal membrane (as Na+ ions enter from tubular fluid) allows potassium to exit more easily into the tubular fluid (i.e., the interior of the cell is now less negative compared with the tubular fluid). A decreased concentration of aldosterone results in decreased urinary excretion of potassium.
Hydrogen Ion Balance
Acute mineral metabolic acidosis decreases urinary excretion of potassium. Chronic metabolic acidosis actually may increase urinary excretion of potassium. If distal tubular flow remains constant, acute (<8 hours) mineral metabolic acidosis results in decreased urinary excretion of potassium because, during metabolic acidosis caused by administration of a mineral acid, H+ ions enter cells to be buffered by intracellular proteins in exchange for K+ ions that leave cells and enter the ECF.181,188 When this ion exchange occurs across the basolateral membranes of the cells of the connecting tubule and cortical collecting ducts, the resulting decreased intracellular concentration of potassium is associated with less tubular secretion of potassium because of a less favorable chemical concentration gradient.
A critical factor determining whether acute metabolic acidosis causes this exchange of H+ and K+ ions across the cell membranes is the permeability of the anion associated with the acid. Chloride ions are relatively impermeable and cannot follow the H+ ions into the cell, whereas lactate and ketoacid anions are more permeable and can follow H+ ions into the cell so that K+ ions do not need to be exchanged with H+ ions for electroneutrality. As a result, acute mineral metabolic acidosis may be associated with H+- K+ exchange across cell membranes, but acute organic metabolic acidosis is not. Chronic (>3 days) metabolic acidosis caused by administration of a mineral acid leads to mild hypokalemia, possibly caused by stimulation of aldosterone secretion by the acidosis.79,122,170 Even in acute acidosis, a decreased filtered load of bicarbonate can reduce sodium reabsorption in the proximal tubules and increase delivery of sodium and water to the distal nephron. This increases the distal tubular flow rate and enhances urinary potassium excretion.
During alkalosis, H+ ions leave cells to titrate bicarbonate in the ECF in exchange for K+ ions that enter the cells. The increased concentration of potassium in the distal tubular cells results in increased secretion of potassium because of a more favorable chemical concentration gradient. Alkalosis also appears to directly stimulate the basolateral Na+, K+-ATPase in the principal cells of the cortical collecting duct.
Diuretics
Many clinically important diuretics (furosemide, ethacrynic acid, thiazides, and mannitol) cause increased urinary excretion of potassium and may result in depletion of body potassium stores. These diuretics increase the distal tubular delivery of sodium and the distal tubular fluid flow rate and, as a result of these effects, cause increased urinary potassium excretion for the same reason as described earlier in the discussion of the effects of high sodium intake on potassium excretion.
Normal serum concentrations
Ion-selective potentiometry and flame photometry are methods used by clinical laboratories to measure sodium and potassium concentrations in body fluids. Electrolytes in plasma are excluded from the fraction of plasma (normally about 7%) that is occupied by solids (e.g., lipids and proteins) and are confined to the aqueous phase of plasma (about 93% of total plasma volume). Flame photometry and indirect potentiometry are affected by the exclusion of electrolytes from the fraction of plasma that is occupied by solids, whereas direct potentiometry is not.194 The resulting error is usually small, but for serum sodium concentration it may be clinically relevant in patients with hyperlipemia (see Chapter 3). Potassium is present in ECF at a much lower concentration than sodium, and the effect of hyperlipemia on the measured serum potassium concentration is much less apparent.
Normal values for serum potassium concentration in dogs and cats vary slightly among laboratories but are expected to be 3.5 to 5.5 mEq/L, with an average value of approximately 4.5 mEq/L. Serum potassium concentrations exceed plasma concentrations because potassium is released from platelets during the clotting process. There is a positive correlation between platelet count and serum potassium concentration in dogs.49,155 The difference between serum and plasma potassium concentrations is most pronounced in animals with thrombocytosis.50,124,155 In one study, serum potassium concentration was greater than plasma potassium concentration by a mean of 0.63 mEq/L in dogs with normal platelet counts and by a mean of 1.55 mEq/L in dogs with thrombocytosis.155
The potassium content of erythrocytes varies in mammalian species, and hemolysis can result in hyperkalemia in species that have high red cell potassium concentrations (Table 5-1). Normal adult canine and feline red cells usually contain potassium in concentrations similar to those of plasma, and hemolysis is not associated with hyperkalemia.42,48,65,93,153 In one study, storage of canine red cells in citrate-phosphate-dextrose-adenine for 40 days resulted in an increase in plasma potassium concentration from 5 to almost 9 mEq/L despite the fact that the original intracellular potassium concentration in the red cells was only 3.8 mEq/L.153 Regardless of the underlying mechanism, this magnitude of increase in plasma potassium concentration would be unlikely to result in detectable hyperkalemia in a recipient dog transfused with blood stored in this manner.
Table 5-1 Sodium and Potassium Concentrations of Mammalian Erythrocytes
| Species | Sodium (mEq/L) | Potassium (mEq/L) |
|---|---|---|
| Human | 10-21 | 104-155 |
| Dog LK* | 93-150 | 4-11 |
| Dog HK*121 | 54 | 124 |
| Cat | 104-142 | 6-8 |
| Horse | 4-16 | 80-140 |
| Cow LK* | 72-102 | 7-37 |
| Cow HK* | 15 | 70 |
| Sheep LK* | 74-121 | 8-39 |
| Sheep HK* | 10-43 | 60-88 |
| Swine | 11-19 | 100-124 |
HK, High potassium; LK, low potassium.
* Sheep, cattle, and dogs demonstrate polymorphism with respect to their intracellular cation concentrations, depending on the level of Na+, K+ -ATPase activity in the mature red cell membranes.
The potassium concentrations of red cells from neonatal dogs are higher than those of red cells from adult dogs.42,132,145 Red cell concentrations of potassium decrease during the first weeks of life and reach normal adult concentrations by approximately 8 to 13 weeks of age. In one study, mean red cell potassium concentrations in puppies were 19.0 mEq/L at 1 day of age, 15.1 mEq/L at 5 weeks of age, and 8.7 mEq/L at 13 weeks of age.42 Reticulocytes from adult dogs also contain higher potassium concentrations than do mature red cells.120 In adult Akitas, red cell potassium concentrations may exceed 70 mEq/L, and hemolysis results in a progressive increase in plasma potassium concentration (up to 24 mEq/L) during storage of blood.48,158
Dogs may be divided genetically into two groups based on the presence or absence of Na+, K+-ATPase activity in the membranes of their mature red cells.99,121 Dogs with red cell membrane Na+, K+-ATPase activity maintain high intracellular potassium concentrations, whereas those without red cell Na+, K+-ATPase activity maintain red cell potassium concentrations similar to those of plasma. Reticulocytes from low-potassium (LK) dogs possess Na+, K+-ATPase, but it is rapidly and completely degraded by a proteolytic process during cell maturation.100,120 Reticulocytes from high-potassium (HK) dogs have twice as much Na+, K+-ATPase activity as reticulocytes from LK dogs, but in the HK dogs, degradation of the enzyme ceases early in maturation, and sufficient activity remains in the mature red cell to account for the observed high intracellular concentration of potassium.120 The HK phenotype is inherited as an autosomal recessive trait and occurs with an incidence of 26% to 38% in the Shiba and Akita breeds in Japan and 42% in the Jindo breed in Korea.76 The HK phenotype also may be seen in the Chinese shar-pei breed and can cause pseudohyperkalemia.16 Some dogs with the HK phenotype also accumulate large amounts of reduced glutathione in their erythrocytes (so-called HK/HG phenotype), which predisposes them to oxidative injury and hemolytic anemia associated with onion ingestion.209
Red cells of English springer spaniel dogs with phosphofructokinase deficiency had potassium concentrations of 19.2 to 28 mEq/L as compared with 5.1 to 7.7 mEq/L in control dogs, and hemolytic crises in affected dogs were associated with hyperkalemia.83 The higher potassium concentration in the red cells of affected dogs was attributed in part to the large number of circulating reticulocytes (7% to 26%). The same mutation has been reported to cause phosphofructokinase deficiency and hyperkalemia in whippets.80
Hemolysis in Akitas (and presumably in other HK dogs) and thrombocytosis cause what has been called pseudohyperkalemia because these effects occur in vitro. Pseudohyperkalemia also has been reported in a dog with acute lymphoblastic leukemia before chemotherapy.95 Leakage of potassium from the leukemic cells in vitro was thought to be responsible for pseudohyperkalemia in this case. Use of plasma from small blood samples collected in an excessive volume of tripotassium ethylenediaminetetraacetic acid also may result in measured hyperkalemia.
Hypokalemia
Clinical and laboratory features
Many dogs and cats with hypokalemia have no clinical signs. Muscular weakness, polyuria, polydipsia, and impaired urinary concentrating capacity are the clinical signs most likely to be recognized in dogs and cats with symptomatic hypokalemia. The pathophysiology of these clinical signs is discussed here.
The clinician should verify the abnormal serum potassium concentration with the laboratory, but measurement of potassium by flame photometry and ion-selective potentiometry is reliable, and errors are uncommon. The clinical history often provides information about the likely source of potassium loss (e.g., chronic vomiting and diuretic administration) or the possibility of translocation (e.g., insulin administration and alkalosis).
Determination of the fractional excretion of potassium (FEK) may help differentiate between renal and nonrenal sources of potassium loss. Fractional potassium excretion can be calculated and expressed as a percentage using:
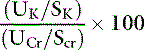
where UK is the urine concentration of K (mEq/L), SK is the serum concentration of K (mEq/L), UCr is the urine concentration of creatinine (mg/dL), and SCr is the serum concentration of creatinine (mg/dL).
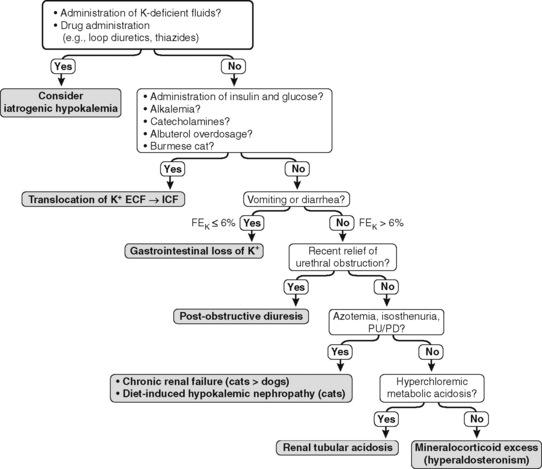
The FEK should be less than 4% for nonrenal sources of loss, and in the presence of hypokalemia, values above 4% may indicate inappropriate renal loss.60 In one study, however, FEK values for normal cats were 10.6 ± 2.1%.3 In another study of normal cats receiving a potassium-deficient diet, FEK values decreased from 10% to 12% to 3% to 6%.61 Thus, FEK values up to 6% should probably be considered normal in potassium-depleted animals with normal renal function. However, the clinical utility of FEK calculations is limited by the fact that FEK does not correlate well with 24-hour urinary excretion of potassium.3,74 The occurrence of hypokalemia in patients with metabolic alkalosis suggests vomiting of stomach contents or diuretic administration as likely causes of potassium loss. In patients with hypokalemia and metabolic acidosis, diarrhea caused by small intestinal disease, chronic renal failure, and distal renal tubular acidosis are more likely causes of potassium loss (Fig. 5-11).
The effect of aldosterone on serum potassium excretion can also be evaluated by comparing urine and serum potassium concentrations after correcting the urine potassium concentration for reabsorption of solute-free water by the kidneys. This index has been called the transtubular potassium gradient (TTKG).39,122,204,205 A value of 5.0 or higher has been said to indicate the presence of an aldosterone effect, whereas a value of 3.0 or less is expected in the absence of mineralocorticoid activity.205 Use of the TTKG is valid only when the urine osmolality is greater than 300 mOsm/kg and the urine sodium concentration is greater than 25 mEq/L. The renal TTKG may be estimated according to the equation:
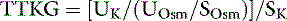
where UK is the urine potassium concentration (mEq/L), SK is the serum potassium concentration (mEq/L), UOsm is the urine osmolality (mOsm/kg), and SOsm is the serum osmolality (mOsm/kg).204,205 Values for TTKG were estimated as 3.7 ± 0.9 in normal cats and 4.2 ± 1.3 in normal dogs.52,56 Determination of TTKG was used in a dog with hypoadrenocorticism to assess the contribution of concurrent trimethoprim administration on the observed hyperkalemia.166 The causes of hypokalemia are listed in Box 5-1, and the diagnostic approach to hypokalemia is presented in Figure 5-11.
Effects of Potassium Depletion on Acid-Base Balance
Hypokalemia often is said to be associated with metabolic alkalosis, but early studies used diuretics or mineralocorticoids to induce potassium depletion. These methods probably caused disproportionate urinary loss of chloride relative to the chloride concentration of ECF, and chloride depletion presumably was the major factor responsible for the development of metabolic alkalosis (see Chapter 10).
Pure potassium depletion apparently does cause metabolic alkalosis in rats, but in dogs it leads to metabolic acidosis.20,32,77 When potassium depletion was produced during a 2- to 4-week period in dogs, and care was taken to prevent chloride depletion, metabolic acidosis developed.32,77 When potassium was restored to the diet, metabolic acidosis resolved within 5 days. The observed reduction in net acid excretion and metabolic acidosis that accompany dietary potassium depletion in the dog appear to be caused by a distal renal tubular acidification defect, which is promptly reversed by potassium repletion.77 This acidification defect is at least partially related to decreased aldosterone secretion.97
Chronic potassium depletion also appears to lead to metabolic acidosis in cats. Adult cats were fed a potassium-restricted (0.2% potassium), 32% protein diet with or without 0.8% NH4Cl.61 Serum potassium concentrations decreased from 4.3 to 4.5 mEq/L to 3.1 to 3.5 mEq/L in the NH4Cl-treated cats and to 3.6 to 3.8 mEq/L in the cats not receiving NH4Cl. Urinary FEK was appropriately decreased to 3% to 6% in both groups of cats. Potassium balance was decreased in both groups but became negative only in the NH4Cl-treated cats. Metabolic acidosis developed in both groups but was more severe in cats treated with NH4Cl. Metabolic acidosis resolved in both groups during potassium repletion.
Effects on Muscle
Muscle weakness develops when serum potassium concentration decreases to less than 3.0 mEq/L, increased creatine kinase concentration develops when serum potassium concentration decreases to less than 2.5 mEq/L, and frank rhabdomyolysis may occur when serum potassium concentration decreases to less than 2.0 mEq/L.106 Rear limb weakness may be observed in dogs and cats with hypokalemia. In cats, weakness of the neck muscles with ventroflexion of the head is commonly observed.57,59,180 Forelimb hypermetria and a broad-based hind limb stance also may be observed in hypokalemic cats. Respiratory muscle paralysis required ventilatory support in two cats with potassium depletion and was thought to be the cause of death in an experimental study of potassium depletion in dogs.59,147 Acute onset of hypokalemia and muscular weakness also have been reported in hyperthyroid cats.140 Three of the four cats in this study received fluid therapy with lactated Ringer’s solution and were treated by surgical thyroidectomy, but one cat developed hypokalemia before treatment. Serum potassium concentration was less than normal in only 5% of hyperthyroid cats in an early study,149 but in a recent study hypokalemia was present in almost 30% of hyperthyroid cats before treatment by thyroidectomy.137
The effects of progressive potassium depletion on skeletal muscle were studied in dogs and rats.20 In both species, a progressive increase in ICF sodium concentration and a progressive decrease in ICF potassium concentration were observed during potassium deficiency. In rats, hyperpolarization of the cell membrane (as predicted by the Goldman-Hodgkin-Katz equation) was detected by direct measurement at all stages of potassium depletion. In dogs, there was an initial hyperpolarization of the cell membrane (mean measured Em, −92.4 mV) during moderate potassium deficiency because [K+]O decreased proportionately more than [K+]I. There was a dramatic decrease in Em (mean measured value, −54.8 mV) at the onset of muscle weakness and paralysis in dogs with severe potassium deficiency (serum potassium concentration, 1.6 mEq/L). In rats with potassium deficiency, predicted and measured Em values were similar during both moderate and severe potassium deficiencies, and paralysis was not observed. The inability to predict resting Em in dogs with severe potassium depletion could be explained by an increase in the sodium permeability of the muscle cell membrane. This study also demonstrated the development of metabolic acidosis in dogs (pH, 7.29; HCO3−, 17.0 mEq/L) and metabolic alkalosis (pH, 7.54; HCO3−, 37.0 mEq/L) in rats with severe potassium deficiency.
Potassium is released from muscle cells during exercise, causing Vasodilatation and increased blood flow.106 This release of cellular potassium is impaired in states of potassium depletion, resulting in muscle ischemia. Muscle blood flow and potassium release increased markedly during exercise in normal but not in potassium-depleted dogs (serum potassium concentration, 2.3 mEq/L), and exercise caused rhabdomyolysis characterized by focal necrosis and inflammatory cell infiltration in potassium-depleted dogs.107 Increased creatine kinase concentrations and electromyographic abnormalities have been observed in cats with hypokalemic polymyopathy, but histopathologic lesions usually are mild or absent.59,180 In dogs with experimentally induced potassium depletion, electromyographic changes were not observed, and increased serum creatine kinase concentration and muscle histopathology were observed only in dogs that had experienced extremely rapid potassium depletion induced by administration of desoxycorticosterone acetate in addition to a potassium-deficient diet.147 Intestinal ileus has been described in human patients with potassium depletion but usually is not recognized clinically in dogs and cats.
Effects on the Cardiovascular System
Electrocardiographic changes and cardiac arrhythmias may develop, because hypokalemia delays ventricular repolarization, increases the duration of the action potential, and increases automaticity. The electrocardiographic changes associated with hypokalemia in human patients (e.g., decreased amplitude T waves, ST segment depression, and U waves) are not consistently observed in dogs and cats, but supraventricular and ventricular arrhythmias may occur. Prolongation of the QT–interval and U waves have been reported in a dog with severe hypokalemia (2.0 mEq/L) caused by chronic vomiting and in dogs with experimentally induced potassium depletion (serum potassium concentration, 2.2 mEq/L).14,86,91 In another study, development of hypokalemia in dogs over a 5-day period was associated with ST segment deviations, decreased amplitude T waves, and the appearance of U waves.68 The appearance of T waves in normal dogs is variable (e.g., positive, negative, and biphasic), and interpretation of the effects of hypokalemia on ventricular repolarization is difficult unless a baseline electrocardiogram has been obtained previously. Hypokalemia potentiates the toxic effects of digitalis on cardiac conduction and may potentiate premature contractions. Hypokalemia also renders the myocardium refractory to the effects of class I antiarrhythmic agents (e.g., lidocaine, quinidine, procainamide). Therefore, serum potassium concentration should be measured and hypokalemia should be corrected in dogs with ventricular arrhythmias unresponsive to antiarrhythmic therapy.
Effects on the Kidneys
Potassium depletion produces functional and morphologic abnormalities in the kidneys, referred to as hypokalemic nephropathy. Renal vasoconstriction leads to decreases in renal blood flow and glomerular filtration rate (GFR). Polyuria and polydipsia are observed in potassium depletion and result from impaired responsiveness of the kidneys to ADH. Defective collecting duct responsiveness to ADH is associated with decreased medullary tonicity, increased medullary blood flow, and impaired cyclic adenosine monophosphate (cAMP) generation in response to ADH. The urinary concentrating defect in potassium depletion results from decreased expression of ADH-regulated aquaporin-2 water channels in the luminal membranes of the renal epithelial cells of the cortical and medullary collecting ducts.10,125
In one study, potassium depletion in dogs over an average of 51 days led to a decrease in total exchangeable potassium from 47.1 to 35.3 mEq/kg and a decrease in serum potassium concentration from more than 4.0 mEq/L to approximately 2.5 mEq/L.1 These dogs experienced decreases in GFR, renal blood flow, and urinary concentrating capacity (UOsm after 20 hours of water deprivation) of approximately 25%. In another study, potassium depletion (serum potassium concentration, 2.1 mEq/L) in dogs had little effect on GFR but caused a 45% reduction in maximal UOsm (1902 to 1055 mOsm/kg).19 In a clinical report, a dog with chronic vomiting and hypokalemia (2.0 mEq/L) developed polyuria, polydipsia, and a urinary concentrating defect that persisted after correction of hypokalemia.86 These abnormalities were attributed to medullary washout of solute and were corrected by partial water restriction and dietary supplementation with NaCl and KCl. In yet another study, dogs subjected to potassium depletion (serum potassium concentration, 2.9 mEq/L) experienced a doubling of urine volume (596 to 1202 mL per 24 hours) and a 40% reduction in maximum urine osmolality (2006 to 1187 mOsm/kg).167
Potassium depletion increases renal ammoniagenesis and urinary net acid excretion, whereas potassium loading tends to have the opposite effect.189 In the rat, increased ammoniagenesis during potassium depletion occurs primarily via enhanced phosphate-dependent glutaminase activity and increased mitochondrial ammoniagenesis in the proximal tubular cells of the renal cortex. The decrease in ammoniagenesis during potassium loading may occur in renal tubular cells from the outer medullary region. Many experimental studies on potassium depletion and renal regulation of acid-base balance have been performed in rats. The renal response of the dog to acute acidosis is known to differ somewhat from that of the rat, and care must be taken in extrapolating data about the renal response to potassium depletion in the rat to dogs.190
Proximal renal tubular sodium reabsorption is increased during potassium depletion, possibly as a result of an increase in the activity of the proximal Na+-H+ antiporter. However, distal sodium reabsorption is decreased during potassium depletion. This presumably occurs as a result of decreased aldosterone secretion and is a direct effect of decreased ECF potassium concentration on the zona glomerulosa of the adrenal glands. Decreased distal sodium reabsorption decreases K+ and H+ ion secretion by decreasing luminal electronegativity. This decreases potassium loss in the urine but also tends to impair renal acid excretion. Thus, increased renal ammoniagenesis during potassium depletion may represent a mechanism for enhancing urinary excretion of fixed acid (as NH4+) at a time when distal H+ ion secretion is impaired. Consequently, derangements in acid-base balance are minimized.
The cytoplasmic and mitochondrial enzyme activity profile of renal tubular cells during potassium depletion is strikingly similar to that observed during chronic metabolic acidosis.189 This similarity suggests the possibility of a common effector mechanism for stimulation of renal ammoniagenesis. Intracellular pH would be a logical candidate for such an effector. As K+ ions leave cells to maintain ECF potassium concentration during potassium depletion, H+ ions enter cells and presumably lower intracellular pH. Reduced intracellular pH may in turn be the signal for increased renal ammoniagenesis from glutamine. Some studies have demonstrated reduced intracellular pH in renal tubular cells during potassium depletion, whereas others have found no change.2,177
Increased ammonia concentrations may activate the third component of complement (C3) and contribute to development of chronic tubulointerstitial disease by recruitment of immune cells.138,196 Vacuolization of proximal tubular cells is observed in human patients, whereas similar lesions are observed in the distal nephron, mainly in the medullary collecting ducts, in potassium-depleted rats. Vacuolization of proximal tubular epithelial cells has also been reported in potassium-depleted dogs.1
Specific causes of hypokalemia in dogs and cats
Hypokalemia arises from decreased intake, translocation of potassium from ECF to ICF, and excessive loss of potassium by either the gastrointestinal or urinary route. Decreased intake of potassium alone is unlikely to cause hypokalemia, but it may be a contributing factor. In chronically ill animals, for example, prolonged anorexia, loss of muscle mass, and ongoing urinary potassium losses probably combine to cause hypokalemia. A specific cause for mild hypokalemia in hospitalized dogs and cats often cannot be identified. Such hypokalemia may resolve with successful treatment of the primary disease process. Iatrogenic hypokalemia may develop when potassium-deficient fluids are administered to anorexic patients in a hospital setting. For example, lactated Ringer’s solution (potassium concentration, 4 mEq/L) is a replacement solution and does not provide sufficient potassium for maintenance needs in most animals. Solutions used for maintenance fluid therapy should contain 15 to 30 mEq/L potassium (see Chapter 14). Ingestion of certain types of clay has been associated with hypokalemia in humans because the clay can bind potassium in the gastrointestinal tract and impair its absorption, and hypokalemia has been reported in a cat after ingestion of clay cat litter containing bentonite.18,84,96
Translocation of potassium into cells may occur with alkalemia, insulin release, and catecholamine release. Alkalemia contributes to hypokalemia as K+ ions enter cells in exchange for H+ ions. Insulin promotes uptake of glucose and potassium by hepatic and skeletal muscle cells and may contribute to hypokalemia when glucose-containing fluids are administered. The stress of illness and the associated epinephrine release may also contribute to hypokalemia. Severe hypokalemia has been reported in dogs that have ingested the β2-adrenergic agonist albuterol.129,200 The mechanism of hypokalemia was presumably rapid uptake of extracellular potassium by liver and muscle cells. Hypokalemia has been associated with hypothermia, possibly as a result of potassium entry into cells.164 Mild hypokalemia was reported in 78% of dogs suffering from rattlesnake envenomation.28 Affected dogs also had transient echinocytosis that was not consistently associated with the observed hypokalemia.
A syndrome characterized by recurrent episodes of limb muscle weakness, neck ventroflexion, increased creatine kinase concentrations, and hypokalemia has been reported in related Burmese cats 4 to 12 months of age.22,103,112,127,128 This syndrome may represent an animal model of hypokalemic periodic paralysis in humans, a familial disorder characterized by episodes of sudden translocation of potassium from ECF to ICF.
Gastrointestinal loss of potassium (e.g., vomiting of stomach contents) is an important cause of hypokalemia in small animals. In one study, hypokalemia was present in 25% of dogs with gastrointestinal foreign bodies and occurred in association with hypochloremia, metabolic alkalosis, and hyponatremia.23 Chloride depletion and sodium avidity related to volume depletion contribute to perpetuation of potassium depletion and metabolic alkalosis in this setting by enhancing urinary losses of K+ and H+ ions. The effects of metabolic alkalosis on potassium balance are discussed further in Chapter 10.
Urinary loss of potassium is another important cause of hypokalemia, and hypokalemia is common in cats with chronic renal failure. Approximately 20% to 30% of cats with chronic renal failure have hypokalemia at presentation, and in one study, chronic renal disease was the most common associated disorder observed in a survey of cats with hypokalemia.54,60,64,116 Most dogs with chronic renal failure have normal serum potassium concentrations. For example, fewer than 10% of dogs with chronic renal failure caused by renal amyloidosis had hypokalemia at presentation.55 Hypokalemia also commonly occurs during the postobstructive diuresis that follows relief of urethral obstruction in cats with idiopathic lower urinary tract disease.
Renal tubular acidosis may be associated with hypokalemia (see Chapter 10). In distal (type I) renal tubular acidosis, hypokalemia is usually present before treatment, and urinary potassium losses may result in part from increased aldosterone secretion. Hypokalemia has been reported in distal renal tubular acidosis in cats.62,197,202 In proximal (type II) renal tubular acidosis, correction of acidosis requires large doses of NaHCO3, and hypokalemia usually appears during therapy. This is a result of the increased delivery of Na+ and HCO3− ions to the distal nephron. These factors enhance urinary potassium excretion by increasing distal tubular flow and lumen electronegativity (HCO3− is a relatively nonresorbable anion in the cortical collecting duct).
Finally, hypokalemic nephropathy characterized by chronic tubulointerstitial nephritis may develop in cats fed diets low in potassium and containing urinary acidifiers.30,56,57,58,60 Stimulation of aldosterone secretion by chronic metabolic acidosis and decreased gastrointestinal absorption of potassium may contribute to potassium depletion in this syndrome.61,170
Mutations in genes that encode epithelial transport proteins and channels have been associated with rare disorders of renal tubular function that cause hypokalemia in humans. One of these familial disorders, Bartter syndrome, is especially complex. It can be caused by mutations in the NKCC2 gene that codes for the luminal Na+/- K+-2Cl− cotransporter found in the thick ascending limb of Henle’s loop (type I), in the gene for the ROMK protein component of renal tubular potassium channels (type II), in the CLCNKB gene that codes for the ClC-Kb chloride channel in the basolateral membranes of tubular cells in the thick ascending limb (type III), or in the BSND gene that codes for barttin, a subunit protein of chloride channels that is required for proper insertion of the channels into the basolateral membrane (type IV).94,176 Other rare diseases can arise from a loss of function mutation in the NCCT gene for the thiazide-sensitive Na+-Cl− cotransporter in the luminal membranes of the distal convoluted tubule (Gitelman’s variant of Bartter syndrome) or a gain of function mutation in the SCNN1 gene for the luminal sodium channel (ENaC) of the principal cells of the collecting duct (Liddle syndrome). None of these rare tubular disorders has yet been recognized in veterinary medicine.
Mineralocorticoid excess is a relatively uncommon cause of urinary potassium loss and hypokalemia in dogs and cats. One report described hyperaldosteronism in a dog thought to be caused by adrenal hyperplasia of the adrenal zona glomerulosa.26 Older dogs with hyperaldosteronism as a result of aldosterone-producing adenomas or adenocarcinomas are presented for evaluation of weakness and polyuria.67,102,207 Mild to moderate hypertension may be detected, and hypokalemia, hypernatremia, mild metabolic alkalosis, dilute urine, and extremely high serum aldosterone concentrations (>3000 pmol/L; normal, 14 to 957 pmol/L) are present on laboratory evaluation. Dogs with adenomas respond well to surgical adrenalectomy, but those with adenocarcinomas experience recurrence of clinical signs if metastasis occurs. One affected dog had polyuria and hyperaldosteronism associated with a very small (2 mm) adrenal adenoma that initially was undetected by computed tomography, and the dog responded to treatment with spironolactone.159 Ultimately, the tumor was identified and removed, and the dog recovered completely. Another dog with an adrenal adenocarcinoma had clinical and laboratory features of mineralocorticoid excess, but serum aldosterone concentration was undetectable.156 Serum desoxycorticosterone concentration was measured and found to be abnormally high (288 ng/mL; normal, 16 to 46 ng/mL). After surgical removal of the tumor, serum potassium concentration normalized, but serum desoxycorticosterone concentration remained high, and the dog was treated with spironolactone.
Clinical and laboratory features of hyperaldosteronism also may be observed in dogs with hyperadrenocorticism caused by adrenal tumors that have been documented to produce a variety of steroid hormones, including aldosterone, deoxycorticosterone, and corticosterone in addition to cortisol.17,46,115,117,130
Several cats (5 to 20 years of age) with hyperaldosteronism have been reported.* Affected cats are presented for evaluation of muscle weakness (sometimes with ventroflexion of the neck), ataxia, weight loss, polyuria, polydipsia, and ocular abnormalities (e.g., mydriasis, blindness, and retinal detachments) associated with hypertension. Laboratory features consist of hypokalemia, hypernatremia, mild metabolic alkalosis, increased serum creatine kinase activity, dilute urine, extremely high serum aldosterone concentrations (usually >1000 pmol/L; normal, 194 to 388 pmol/L), and plasma renin activity that is low or at the low end of the normal reference range (0.2 to 0.5 ng/L/sec; normal, 0.2 to 1.4 ng/L/sec). In two affected cats, urinary FEK was more than 50% and consistent with inappropriate kaliuresis.75 In these two cats, chronic renal disease also was present and might have contributed to hypertension and increased FEK. Hyperaldosteronism may be associated with hyperglycemia caused by insulin resistance, and one affected cat had diabetes mellitus that persisted despite removal of the adrenal tumor.75 Aldosterone-producing adrenal tumors in cats often are 1 to 3 cm in diameter and can be visualized on abdominal ultrasonography. Cytologic evaluation of fine needle aspirates is consistent with neuroendocrine neoplasia, and histologically these tumors are adenomas or adenocarcinomas. Invasion of the caudal vena cava by the tumor can result in thromboembolism.11,75,163 Removal of adrenal tumors causing hyperaldosteronism can be successful in affected cats, but surgery carries a high risk of life-threatening intraoperative or postoperative hemorrhage.11 Medical treatment with potassium supplementation (2 to 6 mEq/day), spironolactone (2 to 4 mg/kg/day) to antagonize the effects of aldosterone, and amlodipine (0.625 to 1.25 mg/day) to control hypertension and can be used to manage affected cats. Survival times of 1 to 5 years have been reported for surgical or medical management.11 Concurrent hyperaldosteronism and hyperprogesteronism have been reported in a cat that also had clinical signs of hyperadrenocorticism and diabetes mellitus.27 See Chapter 10 for further discussion of states of mineralocorticoid excess.
Administration of loop or thiazide diuretics may cause hypokalemia as a result of increased flow rate in the distal tubules and increased secretion of aldosterone secondary to volume depletion. In one study, dogs with heart failure receiving furosemide had significantly lower mean serum potassium concentrations (mean serum potassium concentration, 3.9 mEq/L) than did normal dogs (mean serum potassium concentration, 4.4 mEq/L) or untreated dogs with arrhythmias (mean serum potassium concentration, 4.3 mEq/L).40 Of the dogs treated with furosemide, 17% had serum potassium concentrations less than 3.0 mEq/L. In another study, 10 dogs with congestive heart failure treated with captopril, furosemide, and a sodium-restricted diet did not develop significant changes in serum electrolyte concentrations.165 Penicillin derivatives may cause hypokalemia by acting as nonresorbable anions in the distal tubule and increasing secretion of potassium into tubular fluid. Amphotericin B may cause increased loss of potassium by binding to sterols in cell membranes and increasing permeability. Peritoneal dialysis can be complicated by hypokalemia if potassium-free dialysate is used for an extended time period.45
Treatment
Preparations available for parenteral use include KCl (2 mEq K+/mL) and a potassium phosphate solution containing K2HPO4 and KH2PO4 (4.36 mEq K+/mL). Potassium chloride is the additive of choice for parenteral therapy because chloride repletion is essential if vomiting or diuretic administration is the underlying cause of hypokalemia. Replacement of chloride is also essential for resolution of the metabolic alkalosis often present in such settings (see Chapter 10). When administered intravenously, KCl generally should not be infused at rates greater than 0.5 mEq/kg/hr to avoid potential adverse cardiac effects. A scale such as that shown in Table 5-2 may be used to estimate the amount of KCl to add to parenteral fluids based on serum potassium concentration.88 Infusion rates greater than 0.5 mEq/kg/hr may be required to normalize serum potassium concentration in hypokalemic patients with diabetic ketoacidosis treated with insulin. In hypokalemic human patients, potassium infusion rates up to 0.9 mEq/kg/hr were used safely in one study.89 Careful mixing of potassium chloride after addition to flexible bags of fluids is extremely important to prevent the patient from receiving a high concentration of potassium that could be life threatening. In one study, inadequate mixing of potassium chloride added to flexible bags of fluid was demonstrated to result in up to a fourfold increase in the concentration of potassium in the fluids.51 For determination of serum potassium concentration, when submitting blood samples that have been drawn from intravenous catheters in patients receiving potassium-supplemented fluids, the initial volume of blood withdrawn should be discarded, and a second sample should be submitted to the laboratory to avoid results that may be spuriously high.
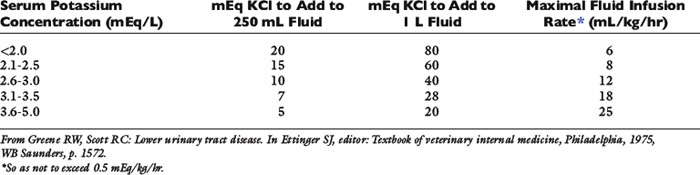
Infusion of potassium-containing fluids initially may be associated with a decrease in serum potassium concentration as a result of dilution, increased distal renal tubular flow, and cellular uptake of potassium, especially if the infused fluid also contains glucose.60 This effect may be minimized by using a fluid that does not contain glucose, administering fluids at an appropriate rate, and beginning oral potassium supplementation as soon as possible. The concentration of potassium in the infused fluid generally should not exceed 60 mEq/L, because higher concentrations of potassium may cause pain and sclerosis of peripheral veins.162 Parenteral fluids containing up to 35 mEq/L have been used safely by the subcutaneous route.72
Careful potassium supplementation is important when using insulin to treat diabetic ketoacidosis. Chronic potassium depletion is usually present in affected patients as a result of loss of muscle mass, anorexia, vomiting, and polyuria. However, serum potassium concentrations are sometimes normal or even increased because of the effects of insulin deficiency and hyperosmolality on serum potassium concentration. Because blood glucose concentration decreases with insulin treatment, marked hypokalemia may develop if supplementation is not adequate.
Potassium gluconate (e.g., Kaon and Tumil-K) is recommended for oral supplementation. In one study, orally administered KCl and KHCO3 were not palatable to cats.59 Dogs may require 2 to 44 mEq potassium per day, depending on body size.92 In cats with hypokalemic nephropathy, the initial oral dosage of potassium gluconate is 5 to 8 mEq/day divided in two or three doses, whereas the maintenance dosage can usually be reduced to 2 to 4 mEq/day.60 It is difficult to estimate the amount of potassium required to reestablish normal balance from the serum potassium concentration in a given patient because potassium is an intracellular solute. Thus, the amount of potassium required for treatment must be determined by judicious supplementation and serial measurement of serum potassium concentration during treatment and recovery.
Hyperkalemia
Hyperkalemia is uncommon if renal function and urine output are normal. Soon after ingestion of a potassium load, cellular uptake of potassium is mediated by insulin, epinephrine, and the resulting increase in ECF potassium concentration itself. Renal excretion of the potassium load then follows. Sustained, chronic hyperkalemia is almost always associated with some impairment in urinary excretion of potassium.
Clinical and laboratory features
The clinical manifestations of hyperkalemia reflect changes in cell membrane excitability and reflect the magnitude and the rapidity of onset of hyperkalemia. Muscle weakness develops with hyperkalemia, usually when serum potassium concentration exceeds 8.0 mEq/L. The electrocardiographic findings caused by hyperkalemia are often characteristic, and the electrocardiogram may be helpful in establishing a suspicion of hyperkalemia while awaiting results of the serum potassium concentration (Fig. 5-12).
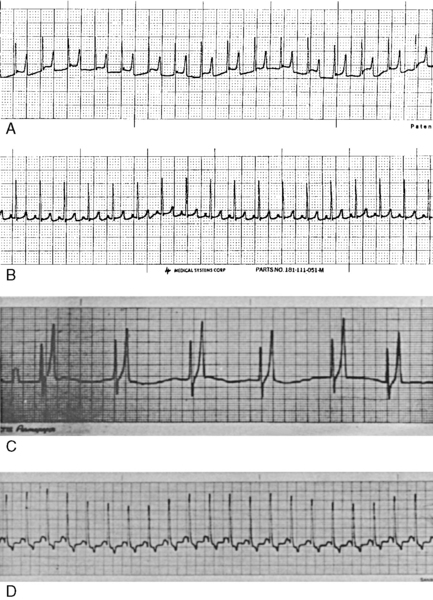
Figure 5-12 Electrocardiograms of a cat and dog with hyperkalemia. A, Electrocardiogram from an 8-year-old female domestic short-haired cat with oliguric acute renal failure and serum K+ concentration of 7.8 mEq/L. B, Electrocardiogram of the same cat after 2 mEq/kg NaHCO3 administered intravenously over 30 minutes. C, Electrocardiogram of a dog with serum K+ concentration of 9.6 mEq/L before treatment. Note tall, tented T waves and absence of P waves. D, Electrocardiogram of the same dog 15 minutes after infusion of NaHCO3.
(Parts C and D from Chew DJ, DiBartola SP. Manual of small animal nephrology and urology. New York: Churchill Livingstone, 1986: 132.)
The effects of hyperkalemia on the electrocardiogram have been studied in dogs and cats.41,43,186,187 Increased amplitude and narrowing or “tenting” of the T waves may occur with mild increases in serum potassium concentration, but these changes are inconsistent in dogs and cats. Shortening of the QT–interval may also be observed. These changes reflect abnormally rapid repolarization. Moderate hyperkalemia may result in prolongation of the PR–interval and widening of the QRS complex because of slowing of conduction through the atrioventricular system. With progression of hyperkalemia, conduction through the atrial muscle is impaired, and decreases in the amplitude and widening of the P wave are observed. In severe hyperkalemia, atrial conduction ceases, the P waves disappear, and pronounced bradycardia with a sinoventricular rhythm may be observed. In extreme hyperkalemia, the QRS complex may merge with the T wave, creating a sine wave appearance, followed by ventricular fibrillation or ventricular asystole. During progressive hyperkalemia, atrial inexcitability, depressed conduction through the specialized tissues and ventricular muscle, and the potential for reentry lead to axis deviations, widening of the QRS complex, and ventricular asystole or ventricular fibrillation. Ventricular fibrillation in hyperkalemia is most likely the result of slow intraventricular conduction and decreased duration of the refractory period. These electrocardiographic changes have also been described in cats with hyperkalemia secondary to urethral obstruction, and they represent the most life-threatening functional consequences of hyperkalemia.146 Rarely, wide-complex tachycardia without identifiable P waves may occur in cats with hyperkalemia.141 The causes of hyperkalemia are listed in Box 5-2, and the clinical approach to hyperkalemia is presented in Figure 5-13.
Box 5-2 Causes of Hyperkalemia
Translocation (ICF → ECF)
Acute mineral acidosis (e.g., HCl, NH4Cl)
Insulin deficiency (e.g., diabetic ketoacidosis)
Reperfusion of extremities after aortic thromboembolism in cats with cardiomyopathy
Hyperkalemic periodic paralysis (one case report in a pit bull)
Mild hyperkalemia after exercise in dogs with induced hypothyroidism
Infusion of lysine or arginine in total parenteral nutrition solutions
Nonspecific β-blockers (e.g., propranolol)*
Cardiac glycosides (e.g., digoxin)*
Decreased Urinary Excretion
Anuric or oliguric renal failure
Selected gastrointestinal diseases (e.g., trichuriasis, salmonellosis, perforated duodenal ulcer)
Late pregnancy in Greyhound dogs (mechanism unknown but affected dogs had gastrointestinal fluid loss)
Chylothorax with repeated pleural fluid drainage
Hyporeninemic hypoaldosteronism†
Angiotensin-converting enzyme inhibitors (e.g., enalapril)*
Angiotensin receptor blockers (e.g., losartan)*
Cyclosporin and tacrolimus*
Potassium-sparing diuretics (e.g., spironolactone, amiloride, triamterene)*
Nonsteroidal anti-inflammatory drugs*
Heparin*
Trimethoprim*
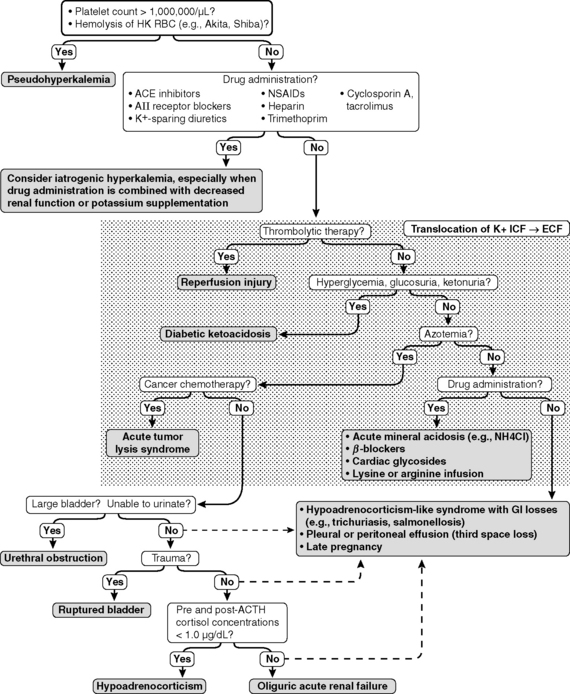
Specific causes of hyperkalemia in dogs and cats
Increased intake of potassium is unlikely to cause sustained hyperkalemia unless impaired renal excretion of potassium is present. Exceptions include iatrogenic hyperkalemia resulting from calculation errors during continuous infusion of potassium-containing fluids or administration of drugs known to predispose to hyperkalemia with concurrent potassium supplementation. Examples of the latter situation include concurrent use of nonspecific β-blockers (e.g., propranolol) or angiotensin-converting enzyme inhibitors (e.g., enalapril) with potassium supplementation (KCl used as a salt substitute contains 13.4 mEq potassium/g) during treatment of heart failure (see Chapter 21).
Translocation of potassium from ICF to ECF can cause hyperkalemia. Acute metabolic acidosis caused by mineral acids (e.g., NH4Cl and HCl) but not organic acids (e.g., lactic acid and ketoacids) causes potassium to shift out of cells in exchange for H+ ions that enter cells to be buffered. The effect of acute inorganic metabolic acidosis on serum potassium concentration in dogs is variable and was characterized by a 0.16- to 1.67-mEq/L increment in serum potassium concentration per 0.1-U decrement in pH in a review of previously published studies.4
Induction of hypothyroidism in beagles led to a 41% decrease in the Na+, K+-ATPase content of skeletal muscle, which was associated with a decrease in the mean resting plasma sodium concentration (from 148 to 142 mEq/L) and an increase in the mean resting plasma potassium concentration (from 3.7 to 4.3 mEq/L).171 Plasma potassium concentration also increased slightly after exercise in the hypothyroid dogs (up to a mean of approximately 5.0 mEq/L) but not in the euthyroid dogs presumably because decreased Na+, K+-ATPase in the hypothyroid dogs resulted in slower reuptake of potassium by muscle cells.
Insulin deficiency and hyperosmolality contribute to hyperkalemia in diabetic patients. Hyperosmolality may increase serum potassium concentration as water moves from ICF to ECF and potassium follows because of solvent drag and as a result of the increased ICF potassium concentration resulting from cellular water loss. Most diabetic patients have total body depletion of potassium caused by urinary losses, muscle mass loss, anorexia, and vomiting. A normal or low serum potassium concentration in a patient with untreated diabetic ketoacidosis indicates serious total body depletion of potassium and the need for diligent potassium supplementation. Hypokalemia at presentation is more common than hyperkalemia in diabetic dogs and cats. In one study, 43% of dogs and 83% of cats with diabetes mellitus were hypokalemic at presentation as compared with 10% of affected dogs and 8% of affected cats that were hyperkalemic.66 Other studies of cats with diabetes mellitus have confirmed that hyperkalemia is uncommon and that 44% to 70% of affected cats had hypokalemia at presentation.29,44,133,173 Hypokalemia may also develop after treatment with insulin despite potassium supplementation of fluids.118 Consequently, the clinician must pay close attention to potassium supplementation of patients with diabetic ketoacidosis.
Massive tissue breakdown may lead to transient hyperkalemia until the kidneys excrete the released potassium. Severe exercise may cause release of potassium from cells and transient hyperkalemia in humans, and this effect is less pronounced in conditioned subjects. In untrained dogs, exercise to the point of exhaustion resulted in an increase in mean serum potassium concentration from 4.4 to 6.0 mEq/L.108 Exhaustive exercise was notassociated with a significant increase in serum potassium concentration after the same dogs had been trained for 6 weeks. In racing greyhounds, no change in serum potassium concentration was observed immediately after racing, despite development of marked lactic acidosis.98 Severe rhabdomyolysis and lethal hyperkalemia (serum potassium concentrations of 10.9 to 12.6 mEq/L) occurred in three cats with hypertrophic muscular dystrophy after restraint and anesthesia.78 The sarcolemma of muscle cells in dystrophin-deficient animals is thought to be more sensitive to injury by anesthetic agents and intense activity resulting in release of intracellular contents. Life-threatening hyperkalemia developed in 16 of 46 (35%) cats with aortic thromboembolism treated with streptokinase because of reperfusion of ischemic muscle tissue and possibly decreased renal excretion associated with renal artery thrombosis.134 Acute tumor lysis syndrome complicated by renal failure and hyperkalemia has been reported in dogs and cats with lymphoma treated by radiation and chemotherapy.34,109,110,199 There is one case report of a young pit bull dog with episodic hind limb and neck weakness. Exercise or potassium challenge resulted in mild hyperkalemia.101 This case may represent an example of hyperkalemic periodic paralysis. In quarter horses, hyperkalemic periodic paralysis is an autosomal dominant trait that causes recurrent episodes of muscle fasciculations and spasm.131 It is caused by a mutation in the α subunit of the sodium channel of skeletal muscle that results in impaired channel inactivation.35,90
Decreased urinary excretion is the most important cause of hyperkalemia in small animal practice. The most common associated disorders are urethral obstruction, ruptured bladder, anuric or oliguric renal failure, and hypoadrenocorticism. The time required for development of hyperkalemia in cats after urethral obstruction is variable, but it may occur within 48 hours.71,73 After relief of obstruction, hyperkalemia resolves within 24 hours, whereas azotemia and hyperphosphatemia require 48 to 72 hours to resolve.73 After experimental bladder rupture in dogs, azotemia, hyperphosphatemia, and mild hyponatremia developed within 24 hours, whereas hyperkalemia did not develop until after 48 hours.33 In a retrospective clinical study of uroperitoneum in cats, hyperkalemia was detected in 13 of 24 (54%) animals evaluated.13
In chronic renal failure, normal potassium balance is maintained, and hyperkalemia is uncommon and develops only if oliguria occurs. This renal adaptation is accomplished by increased FEK in remnant nephrons (Fig. 5-14).24,25,179 Fecal excretion of potassium also increases during chronic progressive renal disease and represents an extrarenal adaptation to maintain potassium balance (see Fig. 5-3). However, patients with chronic renal disease have reduced ability to tolerate an acute potassium load and may require 1 to 3 days to reestablish external potassium balance when intake of potassium is abruptly increased. Dogs with experimentally induced renal disease demonstrate decreased ability to excrete a potassium load. In the first 5 hours after a potassium load, dogs with experimentally induced renal disease excreted 30% to 37% of administered potassium, whereas control dogs excreted 56% to 67%.24,25 Kaliuresis was blunted in the dogs with remnant kidneys despite exaggerated hyperkalemia and increased secretion of aldosterone, and approximately 24 hours were required for complete excretion of the potassium load. Episodes of moderate (>5.3 mEq/L) or severe (6.5 mEq/L) hyperkalemia occurred commonly in a population of dogs with naturally occurring chronic renal disease and responded well in 18 of 26 dogs managed by feeding them a potassium-reduced, home-prepared diet.184
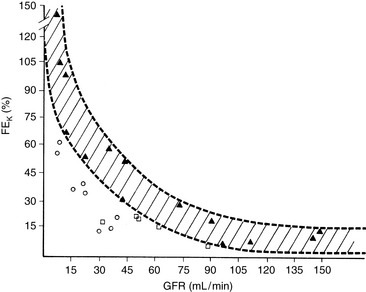
Figure 5-14 Nomogram relating fractional potassium excretion (FEK) to glomerular filtration rate (GFR). Values for patients with an intact hormonal and renal tubular secretory mechanism for potassium (closed triangles) are used to delineate the hatched area. The open squares and circles indicate patients with selective aldosterone deficiency and renal tubular secretory defects, respectively.
(From Batlle DC,Arruda JA, Kurtzman NA. Hyperkalemic distal renal tubular acidosis associated with obstructive uropathy. N Engl J Med 1981;304:373–380.)
Oliguria or anuria with hyperkalemia is more likely to occur in acute renal failure (e.g., ethylene glycol ingestion), but these findings may be observed terminally in chronic renal failure. Acute renal failure with oliguria or anuria is associated with hyperkalemia for several reasons. First, there has been insufficient time for renal adaptation to nephron loss, as occurs with chronic renal failure. Severe reductions in GFR and urine output result in inadequate distal tubular flow for effective urinary excretion of potassium. Finally, increased release of potassium from tissues during this catabolic state and acute metabolic acidosis may contribute to translocation of potassium from ICF to ECF.
Hyperkalemia, hyponatremia, and Na/K ratios less than 27:1 are usually, but not always, found in dogs and cats with hypoadrenocorticism.* In dogs with hypoadrenocorticism, hyperkalemia has been reported in 74% to 96%, hyponatremia in 56% to 100%, and Na/K ratios less than 27:1 in 85% to 100% of cases. Hyperkalemia was found in 9 of 10 cats with hypoadrenocorticism, whereas hyponatremia and Na/K ratios less than 27:1 were found in all 10 affected cats.150 Treatment is begun immediately after a presumptive diagnosis of hypoadrenocorticism is made, but conclusive diagnosis requires results of an ACTH stimulation test.
If sodium intake is sufficient to maintain normal ECF volume and distal tubular flow rate, an animal with hypoadrenocorticism may be able to maintain potassium balance. Treatment of dogs with hypoadrenocorticism with fluids alone also often decreases serum potassium concentration into the normal range. However, usually these animals are presented with anorexia and vomiting that contribute to decreased ECF volume and urine output, and without adequate endogenous mineralocorticoids, they are unable to excrete sufficient potassium to prevent frank hyperkalemia.
Electrolyte abnormalities similar to those found in dogs with hypoadrenocorticism (i.e., hyponatremia and hyperkalemia) can occur in dogs with gastrointestinal disease related to trichuriasis, salmonellosis, or perforated duodenal ulcer.53,123 Hyperkalemia in affected dogs with trichuriasis is not caused by a deficiency of aldosterone because plasma aldosterone concentrations have been found to be normal or high.87 Hyperkalemia and hyponatremia also have been observed in dogs and cats with chylous pleural and peritoneal effusions, in a dog with pleural effusion caused by a lung lobe torsion, in a dog with a neoplastic pleural effusion, in a dog with portal hypertension and peritoneal effusion associated with Bartonella henselae infection, and in cats with peritoneal effusion caused by neoplasia or feline infectious peritonitis.† The hyperkalemia observed in these situations is thought to arise from decreased renal excretion of potassium as a consequence of volume depletion (e.g., gastrointestinal fluid loss and third-space loss of fluid) and decreased distal renal tubular flow. Hyperkalemia and hyponatremia also have been reported in three female Greyhounds late in pregnancy.175 The underlying mechanism was unknown, but all of the dogs had a history of vomiting or diarrhea. Hyporeninemic hypoaldosteronism is an important cause of unexplained asymptomatic hyperkalemia in human patients, but this disorder has rarely been recognized in veterinary medicine.47 Many affected human patients have mild to moderate renal insufficiency caused by diabetic glomerulosclerosis or interstitial renal disease. Most of them have low plasma renin and aldosterone concentrations. Even in patients with normal plasma aldosterone concentrations, the concentration of this hormone must be considered abnormal in light of the hyperkalemia. Resting plasma cortisol concentrations and response to ACTH are normal. Hyperchloremic metabolic acidosis and hypertension may be observed. It is unclear whether low aldosterone concentrations are a consequence of diminished renin secretion and lack of trophic effect of angiotensin II on the zona glomerulosa of the adrenal cortex or whether there is a primary adrenal defect in aldosterone secretion. To document this syndrome in veterinary patients would require demonstration of subnormal plasma renin and aldosterone concentrations or a subnormal increase in aldosterone after volume contraction or ACTH administration. Normally, aldosterone concentrations increase in response to ACTH in the dog.207 In this study, one dog with diabetes mellitus was suspected to have hyporeninemic hypoaldosteronism based on a subnormal aldosterone response to ACTH.
Several drugs may contribute to hyperkalemia, especially when used in combination with one another, in conjunction with potassium supplementation, or in patients with renal sufficiency.148 Nonspecific β-blockers (e.g., propranolol) interfere with catecholamine-mediated uptake of potassium by liver and muscle by blocking β2-adrenergic stimulation of cell membrane Na+, K+-ATPase. Similar to digoxin, cardiac glycoside toxins found in the plant oleander (e.g., oleandrin, digitoxigenin, and Nerium) inhibit Na+, K+-ATPase and can cause hyperkalemia and arrhythmias. The deleterious effects of oleandrin are blocked by infusion of fructose-1,6-diphosphate.126 Angiotensin-converting enzyme inhibitors (e.g., enalapril) and angiotensin II receptor blockers (e.g., losartan) contribute to hyperkalemia by decreasing production of aldosterone by the adrenal glands and blunting glomerular efferent arteriolar constriction, which potentially can decrease delivery of sodium and water to the distal nephron and impair renal potassium excretion. Prostaglandins stimulate renin release, and use of nonsteroidal anti-inflammatory drugs may contribute to development of hyperkalemia. These drugs also may impair the stimulatory effect of prostaglandins on potassium channels in the luminal membranes of renal tubular cells. Heparin impairs aldosterone production by decreasing the number and affinity of angiotensin II receptors in the zona glomerulosa of the adrenal glands and may contribute to hyperkalemia in the presence of other predisposing factors.144 Potassium-sparing diuretics (e.g., spironolactone, amiloride, and triamterene) reduce urinary excretion of potassium and can cause hyperkalemia. Spironolactone competitively inhibits binding of aldosterone to its cytoplasmic receptor in the principal cells of the collecting duct. Amiloride and triamterene block sodium channels in the luminal membranes of the principal cells. Trimethoprim is similar in structure to amiloride and also inhibits sodium channels in the luminal membranes of the principal cells. Trimethoprim is most likely to cause hyperkalemia at high dosages, when urine pH is low (<6.0), and when used in patients with renal insufficiency.148 The immunosuppressive drugs cyclosporin A and tacrolimus contribute to hyperkalemia in renal transplant patients by several mechanisms, including decreased aldosterone production, inhibition of Na+, K+-ATPase, and interference with luminal potassium channels in renal tubular cells. Infusions of total parenteral nutrition solutions containing lysine and arginine may contribute to hyperkalemia because these amino acids may enter cells in exchange for potassium. In many hospitalized animals, however, the cause of mild hyperkalemia cannot be determined. In these instances, hyperkalemia often resolves with appropriate fluid therapy and treatment of the primary disease.
Treatment
Appropriate treatment is dependent on the magnitude and rapidity of onset of the hyperkalemia, as well as the underlying cause. Abnormalities of serum ionized calcium concentration and acid-base balance may aggravate the functional consequences of hyperkalemia as reflected by muscular weakness and electrocardiographic changes. Thus, if compatible electrocardiographic changes are observed, hyperkalemia should be treated regardless of its magnitude. An acute increase in serum potassium concentration to more than 6.5 mEq/L should be treated promptly. Asymptomatic animals with normal urine output and chronic hyperkalemia in the range of 5.5 to 6.5 mEq/L may not require immediate treatment, but a search for the underlying cause should be initiated.
Underlying diseases should be treated promptly (e.g., relief of urethral obstruction, establishment of urine output in patients with oliguria or anuria, and 0.9% NaCl and mineralocorticoids in patients with hypoadrenocorticism). Fluid therapy with lactated Ringer’s solution (potassium concentration, 4 mEq/L) also ameliorates hyperkalemia by improving renal perfusion and enhancing urinary excretion of potassium. However, use of a potassium-free solution (e.g., 0.9% NaCl and 0.45% NaCl) has a greater dilutional effect on the ECF potassium concentration.
Hyperkalemia may be treated by antagonizing the effects of potassium on cell membranes using calcium gluconate, by driving potassium from ECF to ICF with sodium bicarbonate or glucose (with or without concurrent insulin administration), or by removing potassium from the body with a cation exchange resin or dialysis. First, any source of intake must be discontinued (e.g., potassium-containing fluids and potassium penicillin). The clinician also should review the history to verify that the patient is not currently being treated with any drug known to contribute to hyperkalemia (e.g., nonsteroidal anti-inflammatory drugs, β-blockers, angiotensin-converting enzyme inhibitors, and potassium-sparing diuretics).
Hyperkalemia decreases the resting potential of cells. By administering calcium gluconate, the ECF concentration of calcium is increased and the threshold potential is decreased, thus normalizing the difference between the resting and threshold potential and restoring normal membrane excitability (see Fig. 5-2). Administered calcium begins to work within minutes, but its effect lasts less than 1 hour. The dosage of calcium gluconate is 2 to 10 mL of a 10% solution to be administered slowly with electrocardiographic monitoring.
Glucose works by increasing endogenous insulin release and moving potassium into cells. Its effects begin within 1 hour and last a few hours. Glucose-containing fluids (5% or 10% dextrose) or 50% dextrose (1 to 2 mL/kg) can be used for this purpose. The combination of insulin with glucose may result in greater reduction in serum potassium concentration, but there is a risk of hypoglycemia.8 Insulin (0.55 to 1.1 U/kg regular insulin added to parenteral fluids) and dextrose (2 g dextrose per unit of insulin added) have been recommended to treat hyperkalemia in cats with urethral obstruction.172
Sodium bicarbonate also works by moving K+ ions into cells as H+ ions leave cells to titrate administered HCO3− in the ECF. Bicarbonate begins to work within 1 hour, and its effects last a few hours. The usual dosage is 1 to 2 mEq/kg intravenously, and it can be repeated if necessary. In normal cats, 4 mEq/kg sodium bicarbonate given intravenously caused hypokalemia, hypernatremia, hyperosmolality, and decreased ionized calcium concentrations.37,38 If a slow sinoventricular rhythm is present and caused by hyperkalemia, atropine (0.02 to 0.04 mg/kg) may increase the firing rate of the sinus node. An experimental study in dogs demonstrated no beneficial effect of alkalinization in treating hyperkalemia in anesthetized dogs. In this study, the effects of sodium bicarbonate were similar to those of hypertonic saline.104 Sodium may have effects on cardiac muscle that account for reversal of hyperkalemic electrophysiologic changes.15 In a study of hyperkalemic human patients with end-stage renal disease, sodium bicarbonate alone did not decrease plasma potassium concentration, and it did not potentiate the potassium-lowering effects of insulin or albuterol.9
The cation exchange resin polystyrene sulfonate (Kayexalate) can be used to bind potassium and release sodium in the gastrointestinal tract. Each gram binds 1 mEq of potassium and releases 1 to 3 mEq of sodium. It can be mixed with sorbitol (to prevent constipation) and given orally or diluted in tap water and given per rectum as a retention enema using a large Foley catheter. This approach takes a few hours to work and lasts several hours. Kayexalate must be used carefully in patients with impaired ability to excrete a sodium load (e.g., those with congestive heart failure or oliguric renal failure). Intestinal necrosis also is a reported complication of polystyrene sulfonate and sorbitol given postoperatively to human patients.81,114,185
Loop or thiazide diuretics increase the distal tubular flow rate and potassium secretion and may have adjunctive value in the treatment of hyperkalemia. The β2 agonist albuterol increases cellular uptake of potassium by stimulating Na+, K+-ATPase activity and has been used to treat hyperkalemia in human patients with renal failure.7 If all of these measures fail, the clinician must consider peritoneal dialysis (see Chapter 28).45 Hemodialysis (see Chapter 29) is more efficient at removing potassium but is available only at select referral institutions. The treatment of hyperkalemia is outlined in Box 5-3.
Box 5-3 Therapeutic Considerations in the Management of Hyperkalemia*
Establish venous access and administer potassium-deficient (lactated Ringer’s) or potassium-free (0.9% NaCl, 0.45% NaCl) fluids.
Discontinue potassium intake (e.g., potassium-supplemented fluids, potassium-containing salt substitutes, potassium penicillin).
If possible, discontinue drugs that promote hyperkalemia (e.g., β-blockers, angiotensin-converting enzyme inhibitors, potassium-sparing diuretics, prostaglandin inhibitors).
Administer 1-2 mEq/kg NaHCO3 intravenously.†
Administer calcium gluconate 2-10 mL of a 10% solution slowly intravenously.
Administer 5%-10% dextrose or 1-2 mL/kg 50% dextrose intravenously. Consider 0.55-1.1 U/kg regular insulin in parenteral fluids with 2 g dextrose per unit of insulin administered.
Consider administration of sodium polystyrene sulfonate (Kayexalate) orally (20 g with 100 mL 20% sorbitol) or by retention enema (50 g in 100-200 mL tap water).‡
Consider administration of loop (furosemide, 2-4 mg/kg) or thiazide (chlorothiazide, 10-40 mg/kg; hydrochlorothiazide, 2-4 mg/kg) diuretics.
If all other measures fail, institute peritoneal dialysis.
* The therapeutic measures to be used will vary with the clinical situation (see text for discussion).
† This is the treatment most commonly used at the Ohio State University Veterinary Teaching Hospital.
† Oral administration of Kayexalate may cause nausea or vomiting. Sorbitol is added to the oral preparation to prevent constipation. Sorbitol should not be used when administering Kayexalate as a retention enema because of the possible risk of colonic necrosis.114
1 Abbrecht P.H. Effects of potassium deficiency on renal function in the dog. J Clin Invest. 1969;48:432.
2 Adam W.R., Koretsky A.P., Weiner M.W. 32P-NMR in vivo measurement of renal intracellular pH: effects of acidosis and K+ depletion in rats. Am J Physiol. 1986;251:F904.
3 Adams L.G., Polzin D.G., Osborne C.A., et al. Comparison of fractional excretion and 24-hour urinary excretion of sodium and potassium in clinically normal cats and cats with induced chronic renal failure. Am J Vet Res. 1991;52:718.
4 Adrogué H.J., Madias N.E. Changes in plasma potassium concentration during acute acid base disturbances. J Clin Invest. 1981;71:456.
5 Adrogué H.J., Chap Z., Ishida T., et al. Role of the endocrine pancreas in the kalemic response to acute metabolic acidosis in conscious dogs. J Clin Invest. 1985;75:798.
6 Alexander E.A., Perrone R.D. Regulation of extrarenal potassium metabolism. In: Maxwell M.H., Kleeman C.R., Narins R.G., editors. Clinical disorders of fluid and electrolyte metabolism. New York: McGraw-Hill; 1987:105-117.
7 Allon M., Copkney C. Albuterol and insulin for treatment of hyperkalemia in hemodialysis patients. Kidney Int. 1990;38:869.
8 Allon M., Takeshian A., Shanklin N. Effect of insulin-plus-glucose infusion with or without epinephrine on fasting hyperkalemia. Kidney Int. 1993;43:212.
9 Allon M., Shanklin N. Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol. Am J Kidney Dis. 1996;28:508.
10 Amlal H., Krane C.M., Chen Q., et al. Early polyuria and urinary concentrating defect in potassium deprivation. Am J Physiol. 2000;279:F655.
11 Ash R.A., Harvey A.M., Tasker S. Primary hyperaldosteronism in the cat: a series of 13 cases. J Feline Med Surg. 2005;7:173.
12 Atkins E.L., Schwartz W.B. Factors governing correction of the alkalosis associated with potassium deficiency: the critical role of chloride in the recovery process. J Clin Invest. 1962;41:218.
13 Aumann M., Worth L.T., Drobatz K.J. Uroperitoneum in cats: 26 cases (1986–1995). J Am Anim Hosp Assoc. 1998;34:315.
14 Bahler R.C., Rakita L. Cardiovascular function in potassium-depleted dogs. Am Heart J. 1971;81:650.
15 Ballantyne F., Davis L.D., Reynolds E.W. Cellular basis for reversal of hyperkalemic electrocardiographic changes by sodium. Am J Physiol. 1975;229:935.
16 Battison A. Apparent pseudohyperkalemia in a Chinese shar pei dog. Vet Clin Pathol. 2007;36:89.
17 Behrend E.N., Weigand C.M., Whitley E.M., et al. Corticosterone- and aldosterone-secreting adrenocortical tumor in a dog. J Am Vet Med Assoc. 2005;226:1662.
18 Bennett A., Stryjewski G. Severe hypokalemia caused by oral and rectal administration of bentonite in a pediatric patient. Pediatr Emerg Care. 2006;22:500.
19 Bennett C.M. Urine concentration and dilution in hypokalemic and hypercalcemic dogs. J Clin Invest. 1970;49:1447.
20 Bilbrey G.L., Herbin L., Carter N.W. Skeletal muscle resting membrane potential in potassium deficiency. J Clin Invest. 1973;52:3011.
21 Bissett S.A., Lamb M., Ward C.R. Hyponatremia and hyperkalemia associated with peritoneal effusion in four cats. J Am Vet Med Assoc. 2001;218:1590.
22 Blaxter A.C., Livesley P., Gruffydd-Jones T., et al. Periodic muscle weakness in Burmese kittens. Vet Rec. 1986;118:619.
23 Boag A.K., Coe R.J., Martinez T.A., et al. Acid-base and electrolyte abnormalities in dogs with gastrointestinal foreign bodies. J Vet Intern Med. 2005;19:816.
24 Bourgoignie J.J., Kaplan M., Pincus J., et al. Renal handling of potassium in dogs with chronic renal insufficiency. Kidney Int. 1981;20:482.
25 Bourgoignie J.J., Gavellas G., van Putten V., et al. Potassium-aldosterone response in dogs with chronic renal insufficiency. Miner Electrolyte Metab. 1985;11:150.
26 Breitschwerdt E.B., Meuten D.J., Greenfield C.L., et al. Idiopathic hyperaldosteronism in a dog. J Am Vet Med Assoc. 1985;187:841.
27 Briscoe K., Barrs V.R., Foster D.F., et al. Hyperaldosteronism and hyperprogesteronism in a cat. J Feline Med Surg. 2009;11:758.
28 Brown D.E., Meyer D.J., Wingfield W.E., et al. Echinocytosis associated with rattlesnake envenomation in dogs. Vet Pathol. 1994;31:654.
29 Bruskiewicz K.A., Nelson R.W., Feldman E.C., et al. Diabetic ketosis and ketoacidosis in cats: 42 cases (1980–1995). J Am Vet Med Assoc. 1997;211:188.
30 Buffington C.A., DiBartola S.P., Chew D.J., et al. Effect of low potassium commercial nonpurified diet on renal function in adult cats. J Nutr. 1991;121:S91.
31 Burnell J.M., Villamil M.F., Uyeno B.T., et al. The effect in humans of extracellular pH change on the relationship between serum potassium concentration and intracellular potassium. J Clin Invest. 1956;35:935.
32 Burnell J.M., Teubner E.J., Simpson D.P. Metabolic acidosis accompanying potassium deprivation. Am J Physiol. 1974;227:329.
33 Burrows C.F., Bovee K.C. Metabolic changes due to experimentally induced rupture of the canine urinary bladder. Am J Vet Res. 1974;35:1083.
34 Calia C.M., Hohenhaus A.E., Fox P.R., et al. Acute tumor lysis syndrome in a cat with lymphoma. J Vet Intern Med. 1996;10:409.
35 Cannon S.C., Hayward L.J., Beech J., et al. Sodium channel inactivation is impaired in equine hyperkalemic periodic paralysis. J Neurophysiol. 1995;73:1892.
36 Cassola A.C., Giebisch G., Wang W. Vasopressin increases density of apical low-conductance K channels in rat CCD. Am J Physiol. 1993;264:F502.
37 Chew D.J., Leonard M., Muir W.W. Effect of sodium bicarbonate infusions on ionized calcium and total calcium concentrations in serum of clinically normal cats. Am J Vet Res. 1989;50:145.
38 Chew D.J., Leonard M., Muir W.W. Effect of sodium bicarbonate infusion on serum osmolality, electrolyte concentrations, and blood gas tensions in cats. Am J Vet Res. 1991;52:12.
39 Choi M.J., Ziyadeh F.N. The utility of the transtubular potassium gradient in the evaluation of hyperkalemia. J Am Soc Nephrol. 2008;19:424.
40 Cobb M., Michell A.R. Plasma electrolyte concentrations in dogs receiving diuretic therapy for cardiac failure. J Small Anim Pract. 1992;33:526.
41 Cohen H.C., Gozo E.G., Pick A. The nature and type of arrhythmias in acute experimental hyperkalemia in the intact dog. Am Heart J. 1971;82:777.
42 Coulter D.B., Small L.L. Sodium and potassium concentrations of erythrocytes from perinatal, immature, and adult dogs. Cornell Vet. 1972;63:462.
43 Coulter D.B., Duncan R.J., Sander P.D. Effects of asphyxia and potassium on canine and feline electrocardiograms. Can J Comp Med. 1975;39:442.
44 Crenshaw K.L., Peterson M.E. Pretreatment clinical and laboratory evaluation of cats with diabetes mellitus: 104 cases (1992–1994). J Am Vet Med Assoc. 1996;209:943.
45 Crisp M.S., Chew D.J., DiBartola S.P., et al. Peritoneal dialysis: 27 cases (1976–1987). J Am Vet Med Assoc. 1989;195:1262.
46 Davies D.R., Foster S.F., Hopper B.J., et al. Hypokalaemic paresis, hypertension, alkalosis and adrenal-dependent hyperadrenocorticism in a dog. Aust Vet J. 2008;86:139.
47 DeFronzo R.A. Hyperkalemia and hyporeninemic hypoaldosteronism. Kidney Int. 1980;17:118.
48 Degen M. Pseudohyperkalemia in Akitas. J Am Vet Med Assoc. 1987;190:541.
49 Degen M.A. Correlation of spurious potassium elevation and platelet count in dogs. Vet Clin Pathol. 1986;15:20.
50 Degen M.A., Feldman B.F., Turrel J.M., et al. Thrombocytosis associated with a myeloproliferative disorder in a dog. J Am Vet Med Assoc. 1989;194:1457.
51 Dhein C.R., Wardrop K.J. Hyperkalemia associated with potassium chloride administration in a cat. J Am Vet Med Assoc. 1995;206:1565.
52 DiBartola S.P., Chew D.J., Jacobs G. Quantitative urinalysis including 24-hour protein excretion in the dog. J Am Anim Hosp Assoc. 1980;16:537.
53 DiBartola S.P., Johnson S.E., Davenport D.J., et al. Clinicopathologic findings resembling hypoadrenocorticism in dogs with primary gastrointestinal disease. J Am Vet Med Assoc. 1985;187:60.
54 DiBartola S.P., Rutgers H.C., Zack P.M., et al. Clinicopathologic findings associated with chronic renal disease in cats: 74 cases (1973–1984). J Am Vet Med Assoc. 1987;190:1196.
55 DiBartola S.P., Tarr M.J., Parker A.T., et al. Clinicopathologic findings in dogs with renal amyloidosis: 59 cases (1976–1986). J Am Vet Med Assoc. 1989;195:358.
56 DiBartola S.P., Buffington C.A., Chew D.J., et al. Development of chronic renal disease in cats fed a commercial diet. J Am Vet Med Assoc. 1993;202:744.
57 Dow S.W. Studies on potassium depletion in cats. In: Proceedings of the 12th Annual Kal Kan Symposium for the Treatment of Small Animal Diseases. Kal Kan Foods; 1989:61.
58 Dow S.W., Fettman M.J., LeCouteur R.S., et al. Potassium depletion in cats: renal and dietary influences. J Am Vet Med Assoc. 1987;191:1569.
59 Dow S.W., LeCouteur R.A., Fettman M.J., et al. Potassium depletion in cats: hypokalemic polymyopathy. J Am Vet Med Assoc. 1987;191:1563.
60 Dow S.W., Fettman M.J., Curtis C.R., et al. Hypokalemia in cats: 186 cases (1984–1987). J Am Vet Med Assoc. 1989;194:1604.
61 Dow S.W., Fettman M.J., Smith K.R., et al. Effects of dietary acidification and potassium depletion on acid-base balance, mineral metabolism and renal function in adult cats. J Nutr. 1990;120:569.
62 Drazner F.H. Distal renal tubular acidosis associated with chronic pyelonephritis in a cat. Calif Vet. 1980;34:15.
63 Eger C.E., Robinson W.F., Huxtable C.R.R. Primary aldosteronism (Conn’s syndrome) in a cat: a case report and review of comparative aspects. J Small Anim Pract. 1983;24:293.
64 Elliott J., Barber P.J. Feline chronic renal failure: clinical findings in 80 cases diagnosed between 1992 and 1995. J Small Anim Pract. 1998;39:78.
65 Ellory J.C., Tucker E.M. Cation transport in red blood cells. In: Agar N.S., Board P.G., editors. Red blood cells of domestic mammals. Amsterdam: Elsevier Scientific Publishing; 1983:291-314.
66 Feldman E.C., Nelson R.W. Diabetes mellitus. In: Feldman E.C., Nelson R.W., editors. Canine and feline endocrinology. 2nd ed. Philadelphia: WB Saunders; 1996:401.
67 Feldman E.C., Nelson R.W. Canine hyperadrenocorticism (Cushing’s syndrome). In: Feldman E.C., Nelson R.W., editors. Canine and feline endocrinology. 3rd ed. Philadelphia: WB Saunders; 2004:359-360.
68 Felkai F. Electrocardiographic signs in ventricular repolarization of experimentally induced hypokalemia and appearance of the U wave in dogs. Acta Vet Hung. 1985;33:221.
69 Field M.J., Stanton B.A., Giebisch G. Influence of ADH on renal potassium handling: a micropuncture and microperfusion study. Kidney Int. 1984;25:502.
70 Field M.J., Berliner R.W., Giebisch G.H. Regulation of renal potassium metabolism. In: Maxwell M.H., Kleeman G.R., Narins R.G., editors. Clinical disorders of fluid and electrolyte metabolism. New York: McGraw-Hill; 1987:119-146.
71 Finco D.R. Induced feline urethral obstruction: response of hyperkalemia to relief of obstruction and administration of parenteral fluids. J Am Anim Hosp Assoc. 1976;12:198.
72 Finco D.R. Fluid therapy. In: Kirk R.W., editor. Current veterinary therapy VI. Philadelphia: WB Saunders; 1977:8.
73 Finco D.R., Cornelius L.M. Characterization and treatment of water, electrolyte, and acid-base imbalances of induced urethral obstruction in the cat. Am J Vet Res. 1977;38:823.
74 Finco D.R., Brown S.A., Barsanti J.A., et al. Reliability of using random urine samples for spot determination of fractional excretion of electrolytes in cats. Am J Vet Res. 1997;58:1184.
75 Flood S.M., Randolph J.F., Gelzer A.R.M., et al. Primary hyperaldosteronism in two cats. J Am Anim Hosp Assoc. 1999;35:411.
76 Fujise H., Higa K., Nakayama T., et al. Incidence of dogs possessing red blood cells with high K in Japan and East Asia. J Vet Med Sci. 1997;59:495.
77 Garella S., Chang B., Kahn S.I. Alterations of hydrogen ion homeostasis in pure potassium depletion: studies in rats and dogs during the recovery phase. J Lab Clin Med. 1979;93:321.
78 Gaschen F., Gaschen L., Seiler G., et al. Lethal peracute rhabdomyolysis associated with stress and general anesthesia in three dystrophin-deficient cats. Vet Pathol. 1998;35:117.
79 Gennari F.J., Cohen J.J. Role of the kidney in potassium homeostasis: lessons from acid-base disturbances. Kidney Int. 1975;8:1.
80 Gerber K., Harvey J.W., D’Agorne S., et al. Hemolysis, myopathy, and cardiac disease associated with hereditary phosphofructokinase deficiency in two whippets. Vet Clin Pathol. 2009;38:46.
81 Gerstman B.B., Kirkman R., Platt R. Intestinal necrosis associated with postoperative orally administered sodium polystyrene sulfonate in sorbitol. Am J Kidney Dis. 1992;20:159.
82 Giebisch G., Hebert S.C., Wang W.H. New aspects of renal potassium transport. Eur J Physiol. 2003;446:289.
83 Giger U., Harvey J.W. Hemolysis caused by phosphofructokinase deficiency in English springer spaniels: seven cases (1983–1986). J Am Vet Med Assoc. 1987;191:453.
84 Gonzalez J.J., Owens W., Ungaro P.C., et al. Clay ingestion: a rare cause of hypokalemia. Ann Intern Med. 1982;97:65.
85 Good D.W., Velazquez H., Wright F.S. Luminal influences on potassium secretion: low sodium concentration. Am J Physiol. 1984;246:F609.
86 Grauer G.F., Kunze R.S. Potassium depletion nephropathy and renal medullary washout: a case report. Calif Vet. 1979;33:8.
87 Graves T., Schall W., Refsal K., et al. Basal and ACTH-stimulated plasma aldosterone concentrations are normal or increased in dogs with trichuriasis-associated pseudohypoadrenocorticism. J Am Vet Intern Med. 1994;8:287.
88 Greene R.W., Scott R.C. Lower urinary tract disease. In: Ettinger S.J., editor. Textbook of veterinary internal medicine. Philadelphia: WB Saunders; 1975:1572.
89 Hamill R.J., Robinson L.M., Wexler H.R., et al. Efficacy and safety of potassium infusion therapy in hypokalemic critically ill patients. Crit Care Med. 1991;19:694.
90 Hanna W.J., Tsushima R.G., Sah R., et al. The equine periodic paralysis Na+ channel mutation alters molecular transitions between open and inactivated states. J Physiol. 1996;497:349.
91 Hanton G., Yvon A., Provost J.P., et al. Quantitative relationship between plasma potassium levels and QT interval in beagle dogs. Lab Anim. 2007;41:204.
92 Harrison J.B., Sussman H.H., Pickering D.E. Fluid and electrolyte therapy in small animals. J Am Vet Med Assoc. 1960;137:637.
93 Harvey J.W. Erythrocyte metabolism. In: Kaneko J.J., editor. Clinical biochemistry of domestic animals. New York: Academic Press; 1989:196.
94 Hebert S.C. Bartter syndrome. Curr Opin Nephrol Hypertens. 2003;12:527.
95 Henry C.J., Lanevschi A., Marks S.L., et al. Acute lymphoblastic leukemia, hypercalcemia, and pseudohyperkalemia in a dog. J Am Vet Med Assoc. 1996;208:237.
96 Hornfeldt C.S., Westfall M.L. Suspected bentonite toxicosis in a cat from ingestion of clay cat litter. Vet Hum Toxicol. 1996;38:365.
97 Hulter H.N., Sebastian A., Sigala J.F., et al. Pathogenesis of renal hyperchloremic acidosis resulting from dietary potassium restriction in the dog: role of aldosterone. Am J Physiol. 1980;238:F79.
98 Ilkiw J.E., Davis P.E., Church D.B. Hematologic, biochemical, blood gas, and acid base values in greyhounds before and after exercise. Am J Vet Res. 1989;50:583.
99 Inaba M., Maede Y. Increase of Na+ gradient-dependent L-glutamate and L-aspartate transport in high K+ dog erythrocytes associated with high activity of (Na+, K+)-ATPase. J Biol Chem. 1984;259:312.
100 Inaba M., Maede Y. Na, K-ATPase in dog red cells: immunological identification and maturation-associated degradation by the proteolytic system. J Biol Chem. 1986;261:16099.
101 Jezyk P.F. Hyperkalemic periodic paralysis in a dog. J Am Anim Hosp Assoc. 1982;18:977.
102 Johnson K.D., Henry C.J., McCaw D.L., et al. Primary hyperaldosteronism in a dog with concurrent lymphoma. J Vet Med A Physiol Pathol Clin Med. 2006;53:467.
103 Jones B.R., Swinney G.W., Alley M.R. Hypokalemic myopathy in Burmese kittens. N Z Vet J. 1988;36:150.
104 Kaplan J.L., Braitman L.E., Dalsey W.C., et al. Alkalinization is ineffective for severe hyperkalemia in nonnephrectomized dogs. Acad Emerg Med. 1997;4:93.
105 Kitchell B.E., Fan T.M., Kordick D., et al. Peliosis hepatitis in a dog infected with Bartonella henselae. J Am Vet Med Assoc. 2000;216:519.
106 Knochel J.P. Neuromuscular manifestations of electrolyte disorders. Am J Med. 1982;72:521.
107 Knochel J.P., Schlein E.M. On the mechanism of rhabdomyolysis in potassium depletion. J Clin Invest. 1972;51:1750.
108 Knochel J.P., Blanchley J.D., Johnson J.H., et al. Muscle cell electrical hyperpolarization and reduced exercise hyperkalemia in physically conditioned dogs. J Clin Invest. 1985;75:740.
109 Laing E.J., Carter R.F. Acute tumor lysis syndrome following treatment of canine lymphoma. J Am Anim Hosp Assoc. 1988;24:691.
110 Laing E.J., Fitzpatrick P.J., Binnington A.G., et al. Half-body radiotherapy in the treatment of canine lymphoma. J Vet Intern Med. 1989;3:102.
111 Lamb W.A., Muir P. Lymphangiosarcoma associated with hyponatremia and hyperkalemia in a dog. J Small Anim Pract. 1994;35:374.
112 Lieveley P., Gruffydd-Jones T.J. Episodic collapse and weakness in cats. Vet Annu. 1989;29:261.
113 Lifton S.J., King L.G., Zerbe C.A. Glucocorticoid deficient hypoadrenocorticism in dogs: 18 cases (1986–1995). J Am Vet Med Assoc. 1996;209:2076.
114 Lillemole K.D., Romolo J.L., Hamilton S.R., et al. Intestinal necrosis due to sodium polystyrene (Kayexalate) in sorbitol enemas: clinical and experimental support for the hypothesis. Surgery. 1987;101:267-272.
115 Ling G.V., Stabenfeldt G.H., Comer K.M., et al. Canine hyperadrenocorticism: pretreatment clinical and laboratory evaluation of 117 cases. J Am Vet Med Assoc. 1979;174:1211.
116 Lulich J.P., Osborne C.A., O’Brien T.D., et al. Feline renal failure: questions, answers, questions. Compend Contin Educ Pract Vet. 1992;14:127.
117 Machida T., Uchida E., Matsuda K., et al. Aldosterone-, corticosterone- and cortisol-secreting adrenocortical carcinoma in a dog: case report. J Vet Med Sci. 2008;70:317.
118 MacIntire D.K. Treatment of diabetic ketoacidosis in dogs by continuous low-dose intravenous infusion of insulin. J Am Vet Med Assoc. 1993;202:1266.
119 MacKay A.D., Holt P.E., Sparkes A.H. Successful surgical treatment of a cat with primary aldosteronism. J Feline Med Surg. 1999;1:117.
120 Maede Y., Inaba M. (Na+, K+)-ATPase and ouabain binding in reticulocytes from dogs with high K and low K erythrocytes and their changes during maturation. J Biol Chem. 1985;260:3337.
121 Maede Y., Inaba M., Taniguchi N. Increase of Na-K ATPase activity, glutamate, and aspartate update in dog erythrocytes associated with hereditary high accumulation of GSH, glutamate, glutamine, and aspartate. Blood. 1983;61:493.
122 Magner P.O., Robinson L., Halperin R.M., et al. The plasma potassium concentration in metabolic acidosis: a re-evaluation. Am J Kidney Dis. 1988;11:220.
123 Malik R., Hunt G.B., Hinchliffe J.M., et al. Severe whipworm infection in the dog. J Small Anim Pract. 1990;31:185.
124 Mandell C.P., Goding B., Degen M.A., et al. Spurious elevation of serum potassium in two cases of thrombocythemia. Vet Clin Pathol. 1988;17:32.
125 Marples D., Frokiaer J., Dorup J., et al. Hypokalemia-induced down regulation of aquaproin-2 water channel expression in rat kidney medulla and cortex. J Clin Invest. 1996;97:1960.
126 Markov A.K., Payment M.F., Hume A.S., et al. Fructose-1,6-diphosphate in the treatment of oleander toxicity in dogs. Vet Hum Toxicol. 1999;41:9.
127 Mason K.V. A hereditary disease in Burmese cats manifested as an episodic weakness with head nodding and neck ventroflexion. J Am Anim Hosp Assoc. 1988;24:147.
128 Mason K.V. Hereditary potassium depletion in Burmese cats? J Am Anim Hosp Assoc. 1988;24:481.
129 McCown J.L., Lechner E.S., Cooke K.L. Suspected albuterol toxicosis in a dog. J Am Vet Med Assoc. 2008;232:1168.
130 Meijer J.C. Canine hyperadrenocorticism. In: Kirk R.W., editor. Current veterinary therapy VII. Philadelphia: WB Saunders; 1980:975.
131 Meyer T.S., Fedde M.R., Cox J.H., et al. Hyperkalemic periodic paralysis in horses: a review. Equine Vet J. 1999;31:362.
132 Miles P.R., Lee P. Sodium and potassium content and membrane transport properties in red blood cells from newborn puppies. J Cell Physiol. 1972;79:367.
133 Moise N.S., Reimers T.J. Insulin therapy in cats with diabetes mellitus. J Am Vet Med Assoc. 1983;182:158.
134 Moore K.E., Morris N., Dhupa N., et al. Retrospective study of streptokinase administration in 46 cats with arterial thromboembolism. J Vet Emerg Crit Care. 2000;10:245.
135 Moore L.E., Biller D.S., Smith T.A. Use of abdominal ultrasonography in the diagnosis of primary hyperaldosteronism in a cat. J Am Vet Med Assoc. 2000;217:213.
136 Muir W.W., Wagner A.E., Buchanan C. Effects of acute hyperventilation on serum potassium in the dog. Vet Surg. 1990;19:83.
137 Naan E.C., Kirpensteijn J., Kooistra H.S., et al. Results of thyroidectomy in 101 cats with hyperthyroidism. Vet Surg. 2006;35:287.
138 Nath K.A., Hostetter M.K., Hostetter T.H. Pathophysiology of chronic tubulointerstitial disease in rats: interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest. 1985;76:667.
139 Needle M.A., Kaloyanides G.J., Schwartz W.B. The effects of selective depletion of hydrochloric acid on acid base and electrolyte equilibrium. J Clin Invest. 1964;43:1836.
140 Nemzek J.A., Kruger J.M., Walshaw R., et al. Acute onset of hypokalemia and muscular weakness in four hyperthyroid cats. J Am Vet Med Assoc. 1994;205:65.
141 Norman B.C., Cote E., Barrett K.A. Wide-complex tachycardia associated with severe hyperkalemia in three cats. J Feline Med Surg. 2006;8:372.
142 Oster J.R., Perez G.O., Vaamonde C.A. Relationship between blood pH and potassium and phosphorus during acute metabolic acidosis. Am J Physiol. 1978;235:F345.
143 Oster J.R., Perez G.O., Castro A., et al. Plasma potassium response to acute metabolic acidosis induced by mineral and nonmineral acids. Miner Electrolyte Metab. 1980;4:28.
144 Oster J.R., Singer I., Fishman L.M. Heparin-induced aldosterone suppression and hyperkalemia. Am J Med. 1995;98:575.
145 Parker J.C. Dog red blood cells-adjustment in density in vivo. J Gen Physiol. 1973;61:146.
146 Parks J. Electrocardiographic abnormalities from serum electrolyte imbalance due to feline urethral obstruction. J Am Anim Hosp Assoc. 1975;11:102.
147 Patterson R.E., Haut M.J., Montgomery C.A., et al. Natural history of potassium-deficiency myopathy in the dog: role of adrenocorticosteroid in rhabdomyolysis. J Lab Clin Med. 1983;102:565.
148 Perazella M.A. Drug-induced hyperkalemia: old culprits and new offenders. Am J Med. 2000;109:307.
149 Peterson M.E., Kintzer P.P., Cavanagh P.G., et al. Feline hyperthyroidism: pretreatment clinical and laboratory evaluation of 131 cases. J Am Vet Med Assoc. 1983;183:103.
150 Peterson M.E., Greco D.S., Orth D.N. Primary hypoadrenocorticism in ten cats. J Vet Intern Med. 1989;3:55.
151 Peterson M.E., Kintzer P.P., Kass P.H. Pretreatment clinical and laboratory findings in dogs with hypoadrenocorticism-225 cases (1979–1993). J Am Vet Med Assoc. 1996;208:85.
152 Polak A., Haynie G.D., Hays R.M., et al. Effects of chronic hypercapnia on electrolyte and acid base equilibrium. J Clin Invest. 1961;40:1223.
153 Price G.S., Armstrong P.J., McLeod D.A., et al. Evaluation of citrate-phosphate-dextrose-adenine as a storage medium for packed canine erythrocytes. J Vet Intern Med. 1988;2:126.
154 Rakich P.M., Lorenz M.D. Clinical signs and laboratory abnormalities in 23 dogs with spontaneous hypoadrenocorticism. J Am Anim Hosp Assoc. 1984;20:647.
155 Reimann K.A., Knowlen G.G., Tvedten H.W. Factitious hyperkalemia in dogs with thrombocytosis: the effect of platelets on serum potassium concentration. J Vet Intern Med. 1989;3:47.
156 Reine N.J., Hohenhaus A.E., Peterson M.E., et al. Deoxycorticosterone-secreting adrenocortical carcinoma in a dog. J Vet Intern Med. 1999;13:386.
157 Renschler J.S., Dean G.A. What is your diagnosis? Abdominal mass aspirate in a cat with an increased Na:K ratio. Vet Clin Pathol. 2009;38:69.
158 Rich L.J., Berneuter D.C., Cowell R.L. Elevated serum potassium associated with delayed separation of serum from clotted blood in dogs of the Akita breed. Vet Clin Pathol. 1986;15:12.
159 Rijnberk A., Kooistra H.S., van Vonderen I.K., et al. Aldosteronoma in a dog with polyuria as the leading symptom. Domest Anim Endocrinol. 2001;20:227.
160 Rijnberk A., Voorhout G., Kooistra H.S., et al. Hyperaldosteronism in a cat with metastasized adrenocortical tumor. Vet Q. 2001;23:38.
161 Rogers W., Straus J., Chew D. Atypical hypoadrenocorticism in three dogs. J Am Vet Med Assoc. 1981;179:155.
162 Rose B.D. Hypokalemia. In: Rose B.D., editor. Clinical physiology of acid-base and electrolyte disorders. New York: McGraw-Hill; 1994:811.
163 Rose S.A., Kyles A.E., Labelle P., et al. Adrenalectomy and caval thrombectomy in a cat with primary hyperaldosteronism. J Am Anim Hosp Assoc. 2007;43:209.
164 Ross L.A., Goldstein M. Biochemical abnormalities associated with accidental hypothermia in a dog and cat. St. Louis: American College of Veterinary Internal Medicine, 1981;66.
165 Roudebush P., Allen T.A., Kuehn N.F., et al. The effect of combined therapy with captopril, furosemide, and a sodium-restricted diet on serum electrolyte concentrations and renal function in normal dogs and dogs with congestive heart failure. J Vet Intern Med. 1994;8:337.
166 Rubin S.I., Toolan L., Halperin M.L. Trimethoprim-induced exacerbation of hyperkalemia in a dog with hypoadrenocorticism. J Vet Intern Med. 1998;12:186.
167 Rutecki G.W., Cox J.W., Robertson G.W., et al. Urinary concentrating ability and antidiuretic hormone responsiveness in the potassium-depleted dog. J Lab Clin Med. 1982;100:53.
168 Sadek D., Schaer M. Atypical Addison’s disease in the dog: a retrospective survey of 14 cases. J Am Anim Hosp Assoc. 1996;32:159.
169 Sapir D.G., Levine D.Z., Schwartz W.B. The effects of chronic hypoxemia on electrolyte and acid base equilibrium: an examination of normocapneic hypoxemia and of the influence of hypoxemia on the adaptation to chronic hypercapnia. J Clin Invest. 1967;46:369.
170 Scandling J.D., Ornt D.B. Mechanism of potassium depletion during chronic metabolic acidosis in the rat. Am J Physiol. 1987;252:F122.
171 Schaafsma I.A., van Emst M.G., Kooistra H.S., et al. Exercise-induced hyperkalemia in hypothyroid dogs. Domest Anim Endocrinol. 2002;22:113.
172 Schaer M. The use of regular insulin in the treatment of hyperkalemia in cats with urethral obstruction. J Am Anim Hosp Assoc. 1975;11:106.
173 Schaer M. A clinical survey of thirty cats with diabetes mellitus. J Am Anim Hosp Assoc. 1977;13:23.
174 Schaer M., Chen C.L. A clinical survey of 48 dogs with adrenocortical hypofunction. J Am Anim Hosp Assoc. 1983;19:443.
175 Schaer M., Halling K.B., Collins K.E., et al. Combined hyponatremia and hyperkalemia mimicking acute hypoadrenocorticism in three pregnant dogs. J Am Vet Med Assoc. 2001;218:897.
176 Schlingmann K.P., Konrad M., Jeck N., et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med. 2004;350:1314.
177 Schoolwerth A.C., Culpepper R.M. Measurement of intracellular pH in suspensions of renal tubules from potassium-depleted rats. Miner Electrolyte Metab. 1990;16:191.
178 Schrock H., Kuschinsky W. Consequences of chronic potassium depletion for the ionic composition of brain, heart, skeletal muscle, and cerebrospinal fluid. Miner Electrolyte Metab. 1989;15:171.
179 Schultze R.G., Taggart D.D., Shapiro H., et al. On the adaptation in potassium excretion associated with nephron reduction in the dog. J Clin Invest. 1971;50:1061.
180 Schunk K.L. Feline polymyopathy. Proceedings of the American College of Veterinary Internal Medicine. 1984:197. Washington, DC
181 Schwartz W.B., Orning K.J., Porter R. The internal distribution of hydrogen ions with varying degrees of metabolic acidosis. J Clin Invest. 1957;36:373.
182 Schwartz W.B., Brackett N.C., Cohen J.J. The response of extracellular hydrogen ion concentration to graded degrees of chronic hypercapnia: the physiologic limits of the defense of pH. J Clin Invest. 1965;44:291.
183 Sealey J.E., Laragh J.H. A proposed cybernetic system for sodium and potassium homeostasis: coordination of aldosterone and intrarenal physical factors. Kidney Int. 1974;6:281.
184 Segev G., Fascetti A.J., Weeth L.P., et al. Correction of hyperkalemia in dogs with chronic kidney disease consuming commercial renal therapeutic diets by a potassium-reduced home-prepared diet. J Vet Intern Med. 2010;24:546.
184a Stanton C.A., Hamar D.W., Johnson D.E., et al. Bioelectrical impedance and zoometry for body composition analysis in domestic cats. Am J Vet Res. 1992;53:251.
185 Sterns R.H., Rojas M., Bernstein P., et al. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol. 2010;21:733.
186 Surawicz B. Arrhythmias and electrolyte disturbances. Bull N Y Acad Med. 1967;43:1160.
187 Surawicz B. Relationship between electrocardiogram and electrolytes. Am Heart J. 1967;73:814.
188 Swan R.C., Pitts R.F. Neutralization of infused acid by nephrectomized dogs. J Clin Invest. 1955;34:205.
189 Tannen R.L. Relationship of renal ammonia production and potassium homeostasis. Kidney Int. 1977;11:453.
190 Tannen R.L., Sastrasinh S. Response of ammonia metabolism to acute acidosis. Kidney Int. 1984;11:453.
191 Theisen S.K., DiBartola S.P., Radin M.J., et al. Muscle potassium content and potassium gluconate supplementation in normokalemic cats with naturally occurring chronic renal failure. J Vet Intern Med. 1997;11:212.
192 Thompson A.L., Scott-Moncrieff J.C., Anderson J.D. Comparison of classic hypoadrenocorticism with glucocorticoid-deficient hypoadrenocorticism in dogs: 46 cases (1985–2005). J Am Vet Med Assoc. 2007;230:1190.
193 Thompson M.D., Carr A.P. Hyponatremia and hyperkalemia associated with chylous pleural and peritoneal effusion in a cat. Can Vet J. 2002;43:610.
194 Tietz N.W., Pruden E.L., Siggaard-Andersen O. Electrolytes, blood gases, and acid-base balance. In: Tietz N.W., editor. Textbook of clinical chemistry. Philadelphia: WB Saunders; 1986:1181.
195 Tobin R.B. Varying role of extracellular electrolytes in metabolic acidosis and alkalosis. Am J Physiol. 1958;195:687.
196 Tolins J.P., Hostetter M.K., Hostetter T.H. Hypokalemic nephropathy in the rat: role of ammonia in chronic tubular injury. J Clin Invest. 1987;79:1447.
197 Torrente C., Silvestrini P., Ruiz de Gopegui R. Severe life-threatening hypokalemia in a cat with suspected distal renal tubular acidosis. J Vet Emerg Crit Care. 2010;20:250.
198 Velazquez H., Wright F.S., Good D.W., et al. Luminal influences on potassium secretion: chloride replacement with sulfate. Am J Physiol. 1982;242:F46.
199 Vickery K.R., Thamm D.H. Successful treatment of acute tumor lysis syndrome in a dog with multicentric lymphoma. J Vet Intern Med. 2007;21:1401.
200 Vite C.H., Gfeller R.W. Suspected albuterol intoxication in a dog. J Vet Emerg Crit Care. 1994;4:7.
201 Wang W.H. Regulation of renal potassium transport by dietary potassium intake. Annu Rev Physiol. 2004;66:547.
202 Watson A.D.J., Culvenor J.A., Middleton D.J., et al. Distal renal tubular acidosis in a cat with pyelonephritis. Vet Rec. 1986;119:65.
203 Welling P.A., Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol. 2009;297:F849.
204 West M.L., Bendz O., Chen C.B., et al. Development of a test to evaluate the transtubular potassium concentration gradient in the cortical collecting duct in vivo. Miner Electrolyte Metab. 1986;12:226.
205 West M.L., Marsden P.A., Richardson R.M.A., et al. New clinical approach to evaluate disorders of potassium excretion. Miner Electrolyte Metab. 1986;12:234.
206 Willard M.D., Schall W.D., McCaw D.E., et al. Canine hypoadrenocorticism: report of 37 cases and review of 39 previously reported cases. J Am Vet Med Assoc. 1982;180:59.
207 Willard M.D., Refsal K., Thacker E. Evaluation of plasma aldosterone concentrations before and after ACTH administration in clinically normal dogs and in dogs with various diseases. Am J Vet Res. 1987;48:1713.
208 Willard M.D., Fossum T.W., Torrance A., et al. Hyponatremia and hyperkalemia associated with idiopathic or experimentally induced chylothorax in four dogs. J Am Vet Med Assoc. 1991;199:353.
209 Yamato O., Hayashi M., Kasai E., et al. Reduced glutathione accelerates the oxidative damage produced by sodium n-propylthiosulfate, one of the causative agents of onion-induced hemolytic anemia in dogs. Biochim Biophys Acta. 1999;1427:175.
210 Zenger E. Persistent hyperkalemia associated with nonchylous pleural effusion in a dog. J Am Anim Hosp Assoc. 1992;28:411.