Chapter 10 Metabolic Acid-Base Disorders
Metabolic disturbances of acid-base balance are associated with many disease states, and identification of the acid-base disturbance may facilitate diagnosis of the underlying disease process. For example, observation of hypochloremic metabolic alkalosis on a serum biochemical profile of a vomiting dog may lead to recognition of gastrointestinal obstruction as the cause. The regulation of normal acid-base balance is considered in detail in Chapter 9.
Metabolic acidosis
Metabolic acidosis is characterized by a primary decrease in plasma HCO3− concentration, increased [H+], decreased pH, and a secondary, or adaptive, decrease in PCO2. In one study, metabolic acidosis was the most common acid-base disturbance in dogs and cats.61
Metabolic acidosis can be caused by loss of HCO3−-rich fluid from the body, addition of fixed acid to the body or its production by metabolism within the body, or failure of renal excretion of fixed acid. Loss of HCO3−- rich fluid usually occurs via the gastrointestinal tract (e.g., small bowel diarrhea), but it also may occur via the kidneys (e.g., carbonic anhydrase inhibitors, proximal renal tubular acidosis). The HCO3− concentration of diarrheal fluid exceeds that of plasma, whereas its Cl− concentration is lower. The loss of such fluid results in a hyperchloremic metabolic acidosis. Examples of the addition of fixed acid to the body include toxins (e.g., ethylene glycol, salicylate) and compounds used therapeutically (e.g., ammonium chloride, cationic amino acids). Examples of metabolic production of fixed acid within the body include lactic acidosis and diabetic ketoacidosis. Renal failure, hypoadrenocorticism, and distal renal tubular acidosis are examples of impaired urinary excretion of fixed acid. Small bowel diarrhea, renal failure, hypoadrenocorticism, diabetic ketoacidosis, and lactic acidosis during cardiovascular collapse are the most common causes of metabolic acidosis in small animal practice.
Body buffer response to an acute acid load
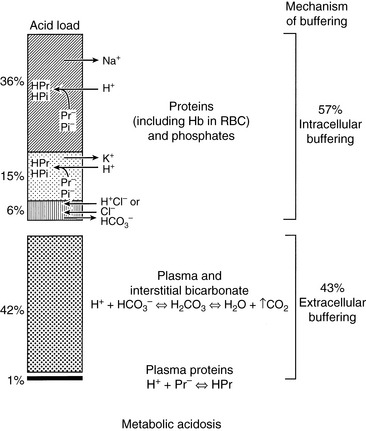
When HCl was infused acutely into nephrectomized dogs, approximately 40% of the acid was buffered by extracellular HCO3−, 10% by red cell buffers (primarily hemoglobin), and 50% by intracellular buffers of soft tissues and bone (primarily proteins and phosphates).223 In nonnephrectomized unanesthetized dogs infused intermittently with HCl, intracellular buffers contributed approximately 50% of the buffer response, regardless of the magnitude of the H+ load.210 Within a few minutes of an acute fixed acid load, administered H+ is buffered by HCO3− in plasma water. Plasma proteins and phosphates play a minor role in this acute response. Some of the administered acid enters red cells and is buffered by hemoglobin. The CO2 produced by the combination of the H+ with HCO3− ions is rapidly removed from the body by alveolar ventilation. Within 30 minutes, the acid load has been distributed to the interstitial fluid, where HCO3− again plays the dominant role in the acute buffer response. After several hours, H+ enters intracellular water in exchange for sodium and potassium ions. These hydrogen ions are buffered within cells by proteins and phosphates. In early studies,210,223 serum potassium concentration increased, but serum sodium concentration decreased after infusion of HCl. The relative roles of these buffers are depicted in Figure 10-1.
Respiratory response to an acute acid load
A fixed acid load increases [H+] and thereby stimulates peripheral and central chemoreceptors to increase alveolar ventilation. This effect begins within hours and is complete within 12 to 24 hours. In humans, there is an approximately 1.2-mm Hg reduction in PCO2 for each 1-mEq/L decrement in plasma HCO3− concentration to a minimum PCO2 of approximately 10 mm Hg.99,195 In dogs with uncomplicated metabolic acidosis induced by chronic feeding of HCl, the observed compensatory respiratory response is an approximately 0.7-mm Hg decrement in PCO2 per 1-mEq/L decrement in plasma HCO3− concentration.* In these studies, the smallest observed respiratory response was an approximately 0.5-mm Hg decrement in PCO2 per milliequivalents per liter decrement in plasma HCO3− concentration,3 and the largest response was a 1.1-mm Hg decrement in PCO2 per milliequivalents per liter decrement in plasma HCO3− concentration.66 Data are limited on the respiratory response of cats to metabolic acidosis, but there is some evidence that the cat fails to develop respiratory compensation to the same extent as observed in the dog in spontaneous236 and NH4Cl-induced metabolic acidosis.43,85,137,211,212
The classic explanation of the respiratory response to metabolic acidosis is that the increase in [H+] (decrease in pH) stimulates ventilation, and the resultant decrease in PCO2 returns the HCO3−/ PCO2 ratio and pH toward normal. This is true in acute metabolic acidosis, but the resultant secondary hypocapnia has been observed to decrease plasma HCO3− concentration further in chronic metabolic acidosis, presumably by reducing renal HCO3− reabsorption. This secondary hypocapnia contributes to 40% of the observed decrease in plasma HCO3− concentration during chronic HCl acidosis.147 Thus, chronic metabolic acidosis decreases plasma HCO3− concentration by two mechanisms: the effect of the administered HCl on body buffers and a reduction in renal HCO3− reabsorption that accompanies secondary hyperventilation. In this study, serum potassium concentration decreased during development of chronic HCl acidosis (contrary to what is typically described for acute metabolic acidosis caused by mineral acids), whereas serum sodium concentration was unchanged.147
Renal response to an acute acid load
The role of the kidneys is to excrete the fixed acid load imposed by the underlying disease process responsible for metabolic acidosis. The kidneys accomplish this task primarily by augmenting its excretion of NH4+. Titratable acidity changes little unless there is a change in the filtered load of phosphate. Chloride ions accompany the NH4+ into urine while HCO3− is regenerated and reabsorbed into extracellular fluid (ECF) to restore HCO3− that was titrated during the acute fixed acid load. Within 48 hours of a fixed acid load, approximately 25% of the added acid has been excreted in the urine, and the remainder is excreted during the next 4 days.232 The kidney can increase its NH4+ excretion as much as fivefold to tenfold during chronic metabolic acidosis.219,235,238 There is some evidence that cats do not adapt to metabolic acidosis by enhanced renal ammoniagenesis.137 The role of the kidneys in regulation of acid-base balance is discussed further in Chapter 9.
Clinical features of metabolic acidosis
The clinical signs in small animals with metabolic acidosis are more likely to be caused by the underlying disease responsible for metabolic acidosis than by the acidosis itself. In humans, respiratory compensation for metabolic acidosis leads to characteristic hyperventilation, recognized by a deep, rhythmic breathing pattern (i.e., Kussmaul respirations). Such a characteristic respiratory pattern has not been described in small animal patients, and metabolic acidosis is usually suspected by observation of a low total CO2 content on a biochemical profile and confirmed by blood gas analysis.
Severe acidosis has serious detrimental effects on cardiovascular function, including decreased cardiac output, decreased arterial blood pressure, and decreased hepatic and renal blood flow.4 Myocardial contractility is decreased when blood pH falls below 7.20.161,180 Impaired contractility may result from a decrease in myocardial intracellular pH (pHi) and displacement of calcium ions from critical binding sites on contractile proteins. Acidosis may predispose the heart to ventricular arrhythmias or ventricular fibrillation. Acidosis has a direct arterial vasodilating effect that is offset by increased release of endogenous catecholamines. However, the inotropic response to catecholamines is impaired, and this may be associated with a reduction in the number of β-adrenergic receptors.151 Acidosis has a direct vasoconstrictive effect on the venous side of the circulation, which tends to centralize blood volume and predisposes to pulmonary congestion. Acidosis shifts the oxygen-hemoglobin dissociation curve to the right, thus enhancing O2 release from hemoglobin, but this effect is offset by a decrease in red cell 2,3-diphosphoglycerate, which develops after 6 to 8 hours of acidosis and shifts the curve back to the left.161
Acidemia produces insulin resistance that impairs peripheral uptake of glucose and inhibits anaerobic glycolysis by inhibiting phosphofructokinase.7 During severe acidosis, the liver may be converted from a consumer to a producer of lactate.144 Severe acidosis also impairs the ability of the brain to regulate its volume, leading to obtundation and coma. Acute mineral acidosis causes hyperkalemia by a transcellular shifting of potassium from intracellular fluid to ECF in exchange for hydrogen ions. This effect causes a very variable change in serum potassium concentration and is not observed with organic acidosis.6 Acute reduction in blood pH causes displacement of calcium ions from negatively charged binding sites (e.g., −COO− groups) on proteins (primarily albumin) as these sites become protonated, and an increase in ionized serum calcium concentration results. Chronic metabolic acidosis leads to release of buffer (mainly calcium carbonate) from bone, and osteodystrophy and hypercalciuria result.
Diagnosis of metabolic acidosis
Metabolic acidosis is associated with several different diseases and should be considered in any severely ill patient. Often, the diagnosis is first suspected by review of the electrolyte and total CO2 results on the patient’s biochemical profile. It is confirmed by blood gas analysis. The causes of metabolic acidosis may be divided into those associated with a normal anion gap (hyperchloremic metabolic acidosis) and those associated with an increased anion gap (normochloremic metabolic acidosis) (Box 10-1).
Box 10-1 Causes of Metabolic Acidosis
Normal Anion Gap (Hyperchloremic)
Carbonic anhydrase inhibitors (e.g., acetazolamide)
Cationic amino acids (e.g., lysine, arginine, histidine)
Posthypocapnic metabolic acidosis
Dilutional acidosis (e.g., rapid administration of 0.9% saline)
Hypoadrenocorticism‡
* Patients with diabetic ketoacidosis may have some component of hyperchloremic metabolic acidosis in conjunction with increased anion gap acidosis.7,9
† The metabolic acidosis early in renal failure may be hyperchloremic and later convert to typical increased anion gap acidosis.239
† Patients with hypoadrenocorticism typically have hypochloremia caused by impaired water excretion, absence of aldosterone, impaired renal function, and lactic acidosis. These factors prevent manifestation of hyperchloremia.
The anion gap represents the difference between the commonly measured plasma cations and the commonly measured anions. This concept is discussed in detail in Chapters 9 and 12. The normal electrolyte composition of canine plasma is compared with that in normal (hyperchloremic) and increased (normochloremic) anion gap metabolic acidosis in Figure 10-2. The anion gap concept is useful in the diagnostic approach to the patient with metabolic acidosis, but it must not be taken literally. In reality, electroneutrality is maintained, and there is no actual anion gap. Normally, the anion gap is made up of the net negative charge on sulfates, phosphates, plasma proteins, and organic anions (e.g., lactate, citrate). Recent studies have shown that in normal dogs and cats, a substantial portion of the anion gap arises from the negative charge on plasma proteins. The net protein charge of plasma at p. 7.40 was calculated to be 16.0 mEq/L in dogs,60 and this value was determined to be 13.7 mEq/L in cats.155 Factors other than metabolic acidosis also may affect the value of the anion gap, and these are discussed in Chapter 12.
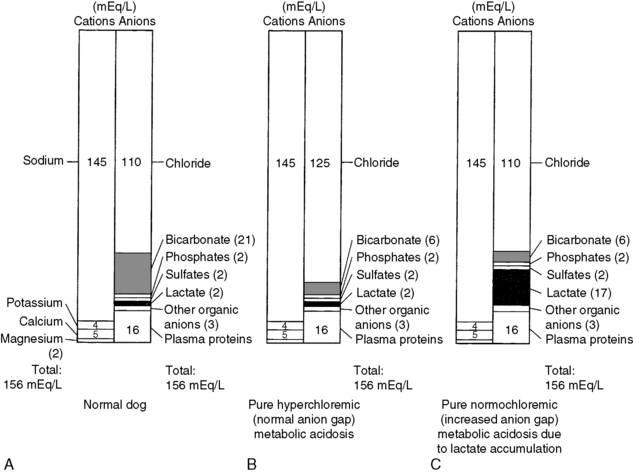
Figure 10-2 Theoretical examples of electrolyte distribution in (A) normal canine plasma, (B) a dog with pure hyperchloremic (normal anion gap) metabolic acidosis, and (C) a dog with normochloremic (increased anion gap) metabolic acidosis caused by lactate accumulation (i.e., lactic acidosis).
(Adapted from Toto RD. Metabolic acid-base disorders. In: Kokko JP, Tannen RL, editors. Fluids and electrolytes, 2nd ed. Philadelphia: WB Saunders, 1990: 324.)
When the anion gap is calculated as [(Na+ + K+) − (Cl− + HCO3−)], normal values in dogs are in the range of 12 to 25 mEq/L.4,60,191,217 Values for the anion gap may be somewhat higher in cats (17 to 31 mEq/L) than in dogs (13 to 25 mEq/L) because of some unaccounted protein and phosphate charge.60,155 In other studies, the mean anion gap for normal cats (calculated as described above) was approximately 20 mEq/L.42,45,46 If the observed metabolic acidosis is characterized by a high anion gap, it is assumed to have arisen from an acid that does not contain chloride as its anion. Examples include some inorganic acids (e.g., phosphates, sulfates) or organic acids (e.g., lactate, ketoacids, salicylate, metabolites of ethylene glycol). In this setting, titration of body buffers by the acid results in accumulation of an anion other than chloride. If the observed metabolic acidosis is characterized by a normal anion gap, there is a reciprocal increase in the plasma chloride concentration to balance the decrease in plasma HCO3− concentration. In the following discussion, the causes of metabolic acidosis have been divided into those associated with a normal anion gap and those associated with an increased anion gap.
Disorders associated with a normal anion gap
Diarrhea
The concentration of HCO3− in intestinal fluid usually is higher than that of plasma, whereas its Cl− concentration is lower. This results from the addition of alkaline pancreatic and biliary secretions to luminal contents and from secretion of HCO3− in exchange for Cl− in the ileum (Fig. 10-3 and Table 10-1). In some diseases of the small intestine, increased delivery of ileal contents to the colon may overwhelm the considerable capacity of the colon for reabsorption of fluid and electrolytes. As a result, severe acute small bowel diarrhea may cause loss of HCO3− in excess of Cl− with resultant hyperchloremic metabolic acidosis. The acidosis is not purely hyperchloremic but rather is mixed if volume depletion and impaired tissue perfusion lead to lactic acid accumulation.
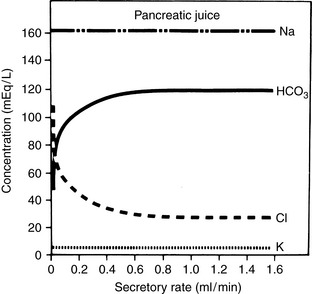
Figure 10-3 Influence of secretory rate on electrolyte composition of canine pancreatic juice. Note the inverse relationship between Cl− and HCO3− concentrations and the relatively constant concentrations of Na+ and K+.
(From Cohen JJ, Kassirer JP. Acid-base. Boston: Little, Brown, 1982: 135.)
Table 10-1 Electrolyte Composition of Luminal Fluid at the End of Individual Segments of the Gastrointestinal Tract

In one study of 134 dogs with gastroenteritis caused by parvoviral infection, only 13% had low total CO2 concentrations.121 In another study of 17 dogs with parvoviral gastroenteritis, 59% had normal pH at presentation.108 In the animals with abnormal blood gas results, alkalemia (6 of 17) was more common than acidemia (1 of 17). The majority (64%) of the dogs in this study were presented for both vomiting and diarrhea. Hypochloremia is more common than hyperchloremia in parvoviral gastroenteritis.108,121 In another study consisting of 25 puppies with parvoviral enteritis, plasma concentrations of sodium, potassium, chloride, and bicarbonate were lower than those of control dogs; however, increases in serum L-lactate concentration were uncommon, and increases in serum D-lactate concentration were not observed.169 Most dogs in this study had mild compensated metabolic acidosis.
Renal Tubular Acidosis
Renal tubular acidosis (RTA) is characterized by hyperchloremic metabolic acidosis caused by either decreased HCO3− reabsorption (proximal RTA) or defective acid excretion (distal RTA) in the presence of a normal glomerular filtration rate (GFR). RTA is uncommonly recognized in small animal practice.
Distal Renal Tubular Acidosis
In distal (classic or type 1) RTA, the urine cannot be maximally acidified because of impaired hydrogen ion secretion in the collecting ducts, and urine pH typically is above 6.0, despite moderately to markedly decreased plasma HCO3− concentration. Increased urine pH (>6.0) in the presence of acidosis is the hallmark of distal RTA. Urinary tract infection by a urease-positive organism (e.g., Proteus sp., Staphylococcus aureus) must be ruled out before considering distal RTA. Urinary net acid excretion is decreased, but bicarbonaturia usually is mild because urinary HCO3− concentration is only 1 to 3 mEq/L in the pH range of 6.0 to 6.5. Nephrolithiasis (usually calcium phosphate stones), nephrocalcinosis (resulting from alkaline urine pH and decreased urinary citrate concentration), bone demineralization (resulting from loss of bone buffer stores during chronic acidosis), and urinary potassium wasting with hypokalemia are features of distal RTA in human patients. Mutations in cytosolic carbonic anhydrase, the basolateral Cl−/HCO3− anion exchanger, and luminal H+-ATPase that affect function of the α-intercalated cells have been associated with inherited forms of distal renal tubular acidosis in humans.173 Urinary fractional excretion of HCO3− is normal (<5%) in distal RTA when plasma HCO3− concentration is increased to normal by alkali administration.
A diagnosis of distal RTA may be confirmed by an ammonium chloride tolerance test during which urine pH is monitored (using a pH meter) before and at hourly intervals for 5 hours after oral administration of 0.2 g/kg NH4Cl. Under such conditions, the urine pH of normal dogs decreased to a minimum value of 5.16 at 4 hours after administration of ammonium chloride.214 Dogs in this study also developed systemic acidosis (pH approximately 7.22 and HCO3− approximately 14 mEq/L at 2 hours after ammonium chloride administration). The amount of alkali required to correct the acidosis in human patients with distal RTA is variable but typically less than that required in proximal RTA. The required dosage of alkali in distal RTA may be as little as 1 mEq/kg/day (i.e., that required to offset daily endogenous acid production) or more than 2 to 4 mEq/kg/day. A combination of potassium and sodium citrate (depending on potassium balance) may be the preferred source of alkali.196
Proximal Renal Tubular Acidosis
In proximal (type 2) RTA, renal reabsorption of HCO3− is markedly reduced and urinary fractional excretion of HCO3− is increased (>15%) when plasma HCO3− concentration is increased to normal. Bicarbonaturia is absent and urine pH is appropriately low when metabolic acidosis is present and plasma HCO3− concentration is decreased because distal acidifying ability is intact. When plasma HCO3− concentration is decreased, the filtered load of HCO3− is reduced, and almost all of the filtered HCO3− is reabsorbed in the distal tubules, despite the presence of the proximal tubular defect. Thus, proximal RTA can be viewed as a “self-limited” disorder in which plasma HCO3− stabilizes at a lower than normal concentration after the filtered load falls sufficiently enough that distal HCO3− reabsorption can maintain plasma HCO3− at a new but lower steady-state concentration. Mutations in renal tubular transport proteins, such as the electrogenic basolateral Na+/ 3HCO3− cotransporter73 and one of the five forms of the luminal Na+/H+ antiporter, have been implicated in the pathogenesis of inherited forms of proximal renal tubular acidosis in humans.118
Other abnormalities of proximal tubular function typically accompany impaired HCO3− reabsorption in proximal RTA, and these include defects in glucose, phosphate, sodium, potassium, uric acid, and amino acid reabsorption. This combination of proximal tubular defects is known as Fanconi syndrome. Serum potassium concentration usually is normal in affected human patients at the time of diagnosis, but alkali therapy may precipitate hypokalemia and aggravate urinary potassium wasting, presumably by increasing distal delivery of sodium and HCO3−.
The diagnosis of proximal RTA is made by finding an acid urine pH (<5.5 to 6.0) in the presence of hyperchloremic metabolic acidosis and a normal GFR but an increased urine pH (>6.0) and increased urinary fractional excretion of HCO3− (>15%) after plasma HCO3− concentration has been increased to normal by alkali administration. If present, the detection of other defects in proximal tubular function (e.g., glucosuria with normal blood glucose concentration) establishes the diagnosis. Correction of metabolic acidosis by alkali therapy is more difficult in proximal RTA than in distal RTA because of the marked bicarbonaturia that occurs when plasma HCO3− concentration is increased to normal. Alkali dosages in excess of 10 mEq/kg/day may be required to correct the plasma HCO3− concentration, and such therapy may result in frank hypokalemia. Thus, potassium citrate may be the preferred source of alkali.
Multiple renal tubular reabsorptive defects resembling Fanconi syndrome have been reported in young basenji dogs.26-28,80 Clinical findings included polyuria, polydipsia, weight loss, dehydration, and weakness. Affected dogs had abnormal fractional reabsorption of glucose, bicarbonate, phosphate, sodium, potassium, and urate, and they had isolated cystinuria or generalized aminoaciduria. The renal tubular disorder in affected basenji dogs is thought to be the result of a metabolic or membrane defect affecting sodium movement or increased back leak or cell-to-lumen flux of amino acids. In one study, brush border membranes isolated from basenji dogs with Fanconi syndrome had decreased sodium-dependent glucose transport but no abnormality of cystine uptake despite the observed reabsorptive defect for cystine.157 Defective urinary concentrating ability leads to isosthenuria or hyposthenuria, and the GFR may be normal initially but decreased later in the course of the disease. Hypokalemia has also been observed late in the course of the disease.80 Death usually results from acute renal failure and papillary necrosis or acute pyelonephritis. A distinctive renal lesion is hyperchromatic karyomegaly of renal tubular cells.
Fanconi syndrome has been observed sporadically in other breeds81,143,156,182,213 and has been reported in association with administration of some drugs.16,28,160 In one case, Fanconi syndrome developed in association with primary hypoparathyroidism and resolved after treatment with calcium and calcitriol.88 Rickets in growing children and osteomalacia in adults are features of Fanconi syndrome in human patients that usually are not observed in affected dogs. However, congenital Fanconi syndrome and renal dysplasia were associated with histologic features of rickets in two Border terriers.64 The skeletal abnormalities in one of the affected dogs resolved after treatment with calcitriol and potassium phosphate. Transient Fanconi syndrome and proximal renal tubular acidosis also have been reported in a dog with high liver enzyme activities, and toxin exposure was considered as a possible explanation.115 Idiopathic transient renal tubular dysfunction also has been reported in a Labrador retriever126 and greyhound.1 Fanconi-like syndrome occurred in Australian dogs that had been fed dried chicken treats from China in 2007 and another product (not containing chicken and not from China) in 2009.87 Affected dogs had polyuria, polydipsia, glucosuria, acidosis, hypokalemia, hypophosphatemia, and azotemia. Most of them survived with conservative medical management. Finally, Fanconi syndrome has been reported in several dogs with copper storage hepatopathy, and tubular dysfunction resolved after copper chelation therapy.10,111
In one report, an 8-year-old female German shepherd had hyperchloremic metabolic acidosis, polyuria, polydipsia, isosthenuria, glucosuria with normal blood glucose concentration, and alkaline urine pH (7.46) after oral administration of NH4Cl.70 The metabolic acidosis was unresponsive to NaHCO3 administration at dosages up to 4 mEq/kg/day. This dog appeared to have distal (type 1) RTA and renal glucosuria. In another case of apparent distal RTA, a 5-year-old mixed breed dog was presented for evaluation of anorexia and was determined to have alkaline urine pH with hyperchloremic metabolic acidosis.191 In another report, an 8-year-old female German shepherd was presented for polyuria, polydipsia, weight loss, and lethargy.24 It had a normal GFR, metabolic acidosis, hyposthenuria, and intermittent glucosuria. Fractional reabsorption of sodium, glucose, and HCO3− was decreased, but reabsorption of chloride, phosphate, potassium, urate, and amino acids was normal. The dog gained weight, and its clinical signs were reversed after treatment with NaHCO3 at approximately 10 mEq/kg/ day. This dog appeared to have proximal (type 2) RTA.
Distal RTA has been reported in two cats with pyelonephritis caused by Escherichia coli.77,236 Clinical signs included polyuria, polydipsia, anorexia, lethargy, enlarged kidneys, and isosthenuria. In one cat, urine pH was 5.0 at the time pyelonephritis was first diagnosed, but distal RTA was documented at a later time by the presence of hyperchloremic metabolic acidosis, alkaline urine pH, and failure to lower urine pH after oral administration of NH4Cl.77 Findings were similar for the other cat, but hyperphosphaturia and persistent hypokalemia also were detected.236 Distal RTA and hepatic lipidosis were reported in another cat without urinary tract infection29 and in a cat with concurrent hyperaldosteronism and severe hypokalemia.228 Distal renal tubular acidosis also has been reported in association with immune-mediated hemolytic anemia in three dogs.215 Distal renal tubular acidosis is associated with some immune-mediated diseases in human patients, but not specifically immune-mediated hemolytic anemia. The clinical features of proximal (type 2) and distal (type 1) RTA are summarized in Table 10-2.
Table 10-2 Clinical Features of Proximal and Distal Renal Tubular Acidosis
| Clinical Feature | Proximal RTA | Distal RTA |
|---|---|---|
| Hypercalciuria | Yes | Yes |
| Hyperphosphaturia | Yes | Yes |
| Urinary citrate | Normal | Decreased |
| Bone disease | Less severe | More severe |
| Nephrocalcinosis | No | Yes |
| Nephrolithiasis | No | Yes (calcium phosphate) |
| Hypokalemia | Mild | Mild to severe |
| Potassium wasting | Worsened by NaHCO3 | Improved by NaHCO3 |
| Alkali required for treatment | >10 mEq/kg/day | <3 mEq/kg/day |
| Other defects of proximal tubular function* | Yes | No |
| Reduction in plasma HCO3− | Moderate | Variable (can be severe) |
| FeHCO3− at normal plasma HCO3− concentration | >15% | <5% |
| Urine pH during acidemia | <5.5 | >6.0 |
| Urine pH after Nh4cl | <5.5 | >6.0 |
Fe, fractional excretion.
* Decreased fractional reabsorption of sodium, potassium, phosphate, urate, glucose, and amino acids.
Hyporeninemic hypoaldosteronism, characterized by hyperkalemia with decreased plasma renin and aldosterone concentrations, occurs in some human patients, notably those with diabetes mellitus who also have mild to moderate renal insufficiency.65 The hyperchloremic metabolic acidosis observed in these patients has been called Type 4 RTA. This syndrome has not been characterized in veterinary medicine but should be considered in dogs and cats with hyperkalemia and mild to moderate hyperchloremic metabolic acidosis after hypoadrenocorticism has been ruled out by an adrenocorticotropic hormone (ACTH) response test. The diagnosis may be established by finding an inappropriately decreased plasma aldosterone concentration in the presence of hyperkalemia.
Carbonic Anhydrase Inhibitors
Carbonic anhydrase inhibitors, such as acetazolamide, decrease proximal tubular reabsorption of HCO3− in the kidneys by noncompetitive inhibition of luminal and cellular carbonic anhydrase. Hypokalemia is caused by increased sodium delivery to the distal nephron and its reabsorption there in exchange for potassium. As hyperchloremic metabolic acidosis develops, the filtered load of HCO3− decreases and the effect of carbonic anhydrase inhibitors on HCO3− reabsorption is limited. Acetazolamide given at 7 to 10 mg/kg three times daily causes self-limited hyperchloremic metabolic acidosis, mild to moderate hypokalemia, and mild hypocalcemia in dogs.107,201 The effects of acetazolamide were greatest after 3 days of administration, and blood chemistry results stabilized after 5 days of administration.201 Acetazolamide is used most commonly in small animal practice for the treatment of glaucoma.
Ammonium Chloride
Administration of NH4Cl is equivalent to administration of HCl because the NH4+ is converted in the liver to urea and H+. Ammonium chloride has been used commonly as a urinary acidifier in dogs and cats. A study of cats receiving 800 mg of NH4Cl per day as a powder or tablet showed that venous blood pH and HCO3− concentrations were decreased to values at the lower end of the normal range.211 A combination product supplying 580 mg each of NH4Cl and D,L-methionine had a more notable effect on venous blood pH and HCO3− concentrations than that observed with 800 mg of NH4Cl alone, but results were still within the reported normal range.212 In another study of cats, NH4Cl at 300 mg/kg/day did not significantly alter venous blood pH, PCO2, or HCO3− concentration, but 400 mg/kg/day significantly decreased blood HCO3− concentration during the course of the study.85 Ammonium chloride at a dosage of 535 mg/kg/day administered to dogs over 6 days caused hyperchloremic metabolic acidosis and was associated with hypokalemia, presumably related to increased aldosterone secretion.150 In another study of dogs, NH4Cl at 200 mg/kg/day reduced urine pH to approximately 5.0 and produced mild metabolic acidosis without change in serum potassium concentration.208
In young, growing and adult dogs, the addition of NH4Cl to the diet leads to demineralization of bone.34,125 Chronic acid feeding has also been reported to affect bone metabolism in cats. Diets containing 3% NH4Cl slowed growth of young cats, decreased blood pH and HCO3− concentrations, and lowered urine pH. Urinary calcium excretion increased in these cats, and bone demineralization was observed on histologic examination of caudal vertebrae.32 Adult cats fed 1.5% NH4Cl for 6 months developed hyperchloremic metabolic acidosis and negative balance for calcium and potassium,43 but no significant changes in trabecular bone remodeling or bone mineral density were found.44 In one study, administration of NH4Cl to cats fed a potassium-restricted diet resulted in hypokalemia, possibly by reducing gastrointestinal absorption of potassium.76 Results of these studies indicate that NH4Cl should be used with caution and blood gases should be monitored during therapy.
Infusion of Cationic Amino Acids
Metabolism of cationic amino acids (e.g., lysine, arginine, histidine) results in production of H+ as the NH4+ from these amino acids is converted to urea in the liver. For this reason, amino acid-containing fluids used in total parenteral nutrition can contribute to hyperchloremic metabolic acidosis. Other contributing factors are the presence of sulfur-containing amino acids (e.g., methionine, cysteine) in the fluid and development of hypophosphatemia during refeeding, which may reduce renal excretion of titratable acid.
Posthypocapnic Metabolic Acidosis
During compensation for chronic respiratory alkalosis, renal net acid excretion decreases with consequent reduction in plasma HCO3− and increase in plasma Cl− concentrations. When the stimulus for hyperventilation is removed and PCO2 increases, pH decreases because it requires 1 to 3 days for the kidneys to increase net acid excretion and to increase plasma HCO3− concentration. Until this occurs, a state of “posthypocapnic” metabolic acidosis exists. Recovery is spontaneous as long as sodium and phosphate are available in the diet to allow the appropriate increase in renal net acid excretion.90
Dilutional Acidosis
Dilutional acidosis refers to a decrease in plasma HCO3− concentration that occurs when extracellular volume is expanded using an alkali-free chloride-containing solution such as 0.9% NaCl. The high chloride concentration of 0.9% NaCl and the highly resorbable nature of the chloride ion in the renal tubules contribute to the decrease in plasma HCO3− concentration and the increase in Cl− concentration. Dilutional acidosis can be corrected by substitution of a solution with a lower chloride concentration (e.g., lactated Ringer’s solution, 0.45% NaCl).
Hypoadrenocorticism
Aldosterone increases renal tubular lumen negativity by enhancing sodium reabsorption in the collecting duct and secondarily increases hydrogen ion secretion. It also directly stimulates H+ secretion by increasing the activity of the luminal H+-ATPase pump in the medullary collecting duct. These effects allow urinary excretion of H+ and K+ when distal delivery of sodium is decreased. Deficiency of aldosterone in hypoadrenocorticism results in metabolic acidosis and hyperkalemia. Metabolic acidosis of variable severity is common in dogs with hypoadrenocorticism.159,190 In one study, low total CO2 concentration suggesting the presence of metabolic acidosis was found in 81 of 200 (41%) dogs with hypoadrenocorticism.190 In a study of 10 cats with hypoadrenocorticism, 3 were reported to have decreased serum total CO2 concentrations.189 Treatment of hypoadrenocorticism includes volume expansion with 0.9% NaCl and replacement of deficient mineralocorticoids and glucocorticoids.
Disorders associated with an increased anion gap
Ethylene Glycol Ingestion
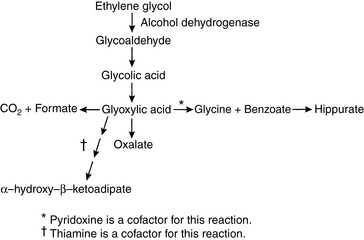
Ethylene glycol (EG) is an organic solvent (molecular mass, 62 Da) used in commercial antifreeze solutions. Ingestion of antifreeze by dogs and cats is a common cause of oliguric acute renal failure in small animal practice, and mortality exceeds 80% in affected animals.57,95,227 EG itself is not toxic, but it is converted in the liver to several metabolites that cause severe metabolic acidosis and acute renal failure (Fig. 10-4). It is rapidly absorbed from the gastrointestinal tract and is undetectable in plasma of dogs 48 hours after administration.175,205
Pathophysiology
EG is first metabolized in the liver to glycoaldehyde by alcohol dehydrogenase. Glycoaldehyde uncouples oxidative phosphorylation and may contribute to neurologic signs observed early in the course of intoxication. Subsequent steps in metabolism produce glycolic and glyoxylic acids. Glycolic acid is primarily responsible for the severe metabolic acidosis that occurs in animals poisoned by EG.50 Renal tubular injury results from glycoaldehyde, glycolic acid, and glyoxylic acids, and calcium oxalate crystals are deposited within renal tubules. The observation of these birefringent crystals in the presence of acute tubular nephrosis confirms the diagnosis of EG intoxication.
Vomiting, polydipsia, and polyuria may occur soon after ingestion of EG, but the owners of poisoned animals often do not detect these signs. Within 12 hours of ingestion, neurologic signs (e.g., lethargy, ataxia, stupor, seizures, coma) may develop. Cardiac and pulmonary manifestations (e.g., tachypnea, tachycardia) occur 12 to 24 hours after ingestion but rarely are detected in clinical cases. Oxalate crystals may be detected in the urine as early as 3 to 6 hours after ingestion of EG.68,69 Renal failure occurs in dogs as early as 24 to 48 hours after ingestion and is manifested by anorexia, lethargy, vomiting, and oliguria or anuria.97 In cats, azotemia may develop within 12 to 24 hours after ingestion of EG.68 Unfortunately, most dogs and cats with EG poisoning are presented for veterinary attention after renal failure has already developed.
A severe normochloremic (i.e., high anion gap) metabolic acidosis occurs within 3 hours of EG ingestion and persists for at least 24 hours.68,69,97,227 Serum hyperosmolality and osmolal gap peak 1 to 6 hours after ingestion and persist for 12 to 24 hours,68,69,97 but the osmolal gap may be normal in animals presented later in the course of the disease.227 Activated charcoal preparations containing propylene glycol and glycerol can increase osmolality and osmolal gap, and potentially complicate the diagnosis of EG ingestion. Measured serum osmolality peaked at 4 hours (353 mOsm/kg), osmolal gap at 6 hours (52 mOsm/kg), and serum lactate concentration at 4 hours (4.5 mmol/L) after administration of 4 g/kg of an activated charcoal preparation containing propylene glycol and glycerol.33 Results returned to baseline 24 hours after administration of the activated charcoal preparation.
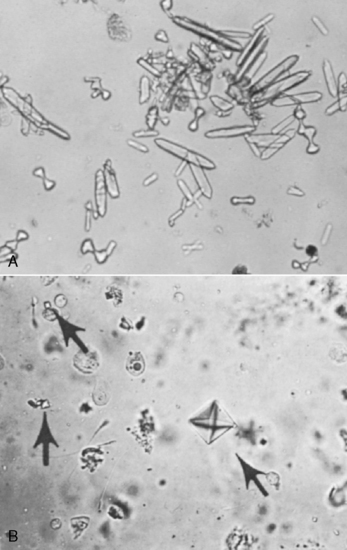
Calcium oxalate dihydrate crystals (“Maltese cross” or “envelope” forms) may be observed in the urine, but calcium oxalate monohydrate crystals (“picket fence” or “dumbbell” forms) are observed more commonly. Calcium oxalate dihydrate crystals occasionally are found in the urine of normal dogs and cats, whereas calcium oxalate monohydrate crystals rarely are seen except in animals that have ingested EG (Fig. 10-5).68,227 Crystals previously referred to as hippurates actually are calcium oxalate monohydrate crystals.134,226 Other laboratory findings include azotemia, isosthenuria, hypocalcemia, hyperphosphatemia, and hyperglycemia.227 Hyperphosphatemia observed very early in the course of EG intoxication (3 to 12 hours after ingestion) probably is the result of the high phosphorus content of rust-retardant antifreeze preparations.57,69 Hyperechogenicity of the renal cortex is observed on renal ultrasonography as early as 5 hours after ingestion of EG.2
Treatment
The response to treatment depends on the amount of EG ingested and the amount of time that elapses before treatment. In early studies, dogs that ingested less than 10 mL/kg EG were saved if treated within 2 to 4 hours of ingestion,17,175,205 and cats survived up to 6 mL/kg EG if treated within 4 hours.187 Treatment consists of inducing vomiting with apomorphine or performing gastric lavage with activated charcoal if ingestion has been recent (<8 hours before presentation). Severe hypocalcemia is corrected with calcium gluconate, and NaHCO3 is administered to combat metabolic acidosis. A NaHCO3 dosage of 1 to 2 mEq/kg may be used empirically. Calcium gluconate and NaHCO3 must not be given simultaneously because calcium carbonate crystals form, and the solution becomes turbid. Attempts to stimulate urine production with furosemide (2 to 4 mg/kg) or mannitol (1 g/kg) usually are futile.
Alcohol dehydrogenase has greater affinity for ethanol than EG. For this reason, 20% ethanol has been administered intravenously to affected dogs at a dosage of 5.5 mL/kg every 4 hours for five treatments and then every 6 hours for four additional treatments.96 Cats are treated with 20% ethanol at a dosage of 5 mL/kg every 6 hours for five treatments and then every 8 hours for four additional treatments. This treatment is unlikely to be of benefit if more than 12 to 24 hours have elapsed since ingestion of EG. Fomepizole (4-methylpyrazole) is a pharmacologic inhibitor of alcohol dehydrogenase that can be used to treat dogs with EG toxicosis.67,69 In dogs, it is superior to ethanol because it does not cause central nervous system (CNS) depression, but it must be administered within 8 hours of EG ingestion. The dosage of fomepizole used in dogs with EG intoxication is 20 mg/kg intravenously, followed by 15 mg/kg intravenously at 12 and 24 hours and 5 mg/kg intravenously at 36 hours.57,67,69 Unfortunately, fomepizole was not efficacious in EG-intoxicated cats unless administered at the same time as the EG was consumed.68 A study to investigate the difference in efficacy of fomepizole between dogs and cats found that the percentage inhibition of canine and feline alcohol dehydrogenase was similar when the concentration of fomepizole applied to feline liver homogenates was 6 times higher than that applied to canine liver homogenates.58 When cats that received lethal doses of EG were treated within 3 hours of ingestion using 125 mg/kg fomepizole followed by 31 mg/kg at 12, 24, and 36 hours, 5 of 6 survived.59 One cat developed acute renal failure but recovered. Cats treated with this high dosage of fomepizole developed mild sedation, but no biochemical evidence of toxicity was identified.
Thiamine promotes conversion of glyoxylate to glycine, and pyridoxine promotes conversion of glyoxylate to α-hydroxy-β-ketoadipate (see Fig. 10-4). These vitamins may be administered to promote alternative pathways of glyoxylate metabolism, but efficacy has not been demonstrated for such treatment. In one study, all nonazotemic dogs treated with fomepizole within 2 to 8.5 hours after EG ingestion survived, whereas only 1 of 21 azotemic dogs treated 8.5 to 38 hours after ingestion survived.57
Peritoneal dialysis or hemodialysis is necessary if the animal has anuric or oliguric renal failure at the time of presentation. Early dialysis may also be helpful to remove toxic intermediate metabolites. Despite dialysis, affected dogs may progress to end-stage renal disease and become dependent on dialysis. The prognosis for survival in adult dogs and cats with anuric or oliguric acute renal failure caused by EG intoxication is unfortunately very poor.57,227
Salicylate Intoxication
Aspirin (acetylsalicylic acid) is hydrolyzed to salicylic acid (pK’a = 3.0) in the liver. Salicylate intoxication is uncommon in small animal practice and is an example of a mixed acid-base disturbance characterized by metabolic acidosis and respiratory alkalosis. Salicylate intoxication in anesthetized, spontaneously breathing dogs resulted in a mixed respiratory alkalosis and metabolic acidosis.218 The stimulation of ventilation is caused by a direct effect of salicylate on the medullary respiratory center. Salicylate also uncouples oxidative phosphorylation in mitochondria, and the associated disturbances in carbohydrate metabolism lead to metabolic acidosis characterized by an increased anion gap associated with accumulation of lactic acid, ketoacids, and other organic acids. Salicylate usually makes a minor contribution to the observed increase in unmeasured anions.
Gastric lavage with activated charcoal should be performed if ingestion occurred less than 6 to 12 hours before admission. Administration of NaHCO3 promotes removal of salicylate from tissues and enhances its urinary excretion by the mechanism of diffusion trapping. Alkalinization of ECF and urine increases the proportion of drug present in the ionized form and thus favors diffusion of more nonionized salicylic acid from cells into ECF and urine, where it can be trapped as the poorly diffusible ionized form. An attempt should be made to maintain urine pH above 7.5 during alkaline diuresis with NaHCO3, especially if metabolic acidosis is the predominant acid-base disturbance. Alkalinization should be carried out with caution, if at all, when respiratory alkalosis is the predominant acid-base disturbance. Glucose infusion is recommended to prevent reduction in CNS glucose concentration. Hypokalemia may develop during treatment as a result of NaHCO3 administration and diuresis, and parenteral fluids should be supplemented with potassium as needed.
Metaldehyde Intoxication
Metaldehyde is a tetramer of acetaldehyde used as a snail and slug bait that can cause seizures and hyperthermia in dogs that ingest it.241 It is hydrolyzed to acetaldehyde in the stomach, which then is absorbed and metabolized to acetic acid (pK’a = 4.75). Acidemia was present in 6 of 11 intoxicated dogs in which arterial blood gas analysis was performed. Three dogs had metabolic acidosis and three had mixed acid-base disturbances that were not further characterized. Conversion of acetaldehyde to acetic acid could explain development of metabolic acidosis, and ventilatory disturbances associated with generalized seizures (either respiratory alkalosis or acidosis) could have contributed to development of mixed acid-base disorders. With supportive care, blood gas abnormalities resolve within 24 to 48 hours.
Diabetic Ketoacidosis
Pathophysiology
Overproduction of acetoacetic acid (pK’a = 3.58) and β-hydroxybutyrate (pK’a = 4.70) by the liver occurs in diabetes mellitus because of a deficiency of insulin and relative excess of glucagon. An increase in glucagon and a decrease in insulin shift the liver from its normal role in esterification of fatty acids into triglycerides to β-oxidation of fatty acids into ketoacids. At the normal pH of ECF (7.40), these organic acids are completely dissociated, and the hydrogen ions that are released titrate HCO3− and other body buffers. Acetone is formed by the nonenzymatic decarboxylation of acetoacetate and does not contribute additional fixed acid. The pathophysiology and treatment of diabetic ketoacidosis are discussed in detail in Chapter 20.
Metabolic acidosis is common in dogs and cats with diabetic ketoacidosis. In one series, mean plasma HCO3− concentration in 72 dogs with diabetic ketoacidosis was approximately 11 mEq/L at the time of diagnosis with a range of 4 to 20 mEq/L, whereas the mean HCO3− concentration in 20 affected cats was 13 mEq/L with a range of 8 to 22 mEq/L.83 In an early study of dogs with diabetes mellitus, mean plasma HCO3− concentration was 13.7 mEq/L in eight survivors (range, 9.3 to 21.0 mEq/L) and 18.1 mEq/L in five nonsurvivors (range, 13.4 to 30.2 mEq/L).138 In another study of dogs with diabetic ketoacidosis, mean arterial pH and HCO3− concentration were 7.201 (range, 6.986 to 7.395) and 11.1 mEq/L (range, 4.1 to 19.7 mEq/L) before treatment and 7.407 ± 0.053 and 18.2 ± 0.7 mEq/L 24 hours after treatment.142 Only three dogs (those with pH <7.1) received sodium bicarbonate treatment. Metabolic acidosis with median pH of 7.14 (range, 7.04 to 7.24) and HCO3− concentration of 10 mEq/L (range, 6 to 15 mEq/L) was found in 25 of 33 cats evaluated by venous blood gas analysis in a survey of cats with diabetic ketoacidosis.31 Cats with HCO3− concentrations below 14 mEq/L received bicarbonate supplementation of their fluids. In another series of diabetic cats, median total CO2 was 13 mEq/L in ketoacidotic cats and 15 mEq/L in nonketoacidotic cats.63 In a study of 116 dogs with diabetes mellitus, 43 (37%) had diabetic ketoacidosis with median venous blood pH of 7.228 (range, 6.979 to 7.374) and median bicarbonate concentration of 10.1 mEq/L (range, 4.0 to 19.3 mEq/L).78 In a study of 127 dogs with ketoacidosis, acid-base status at presentation had substantial impact on outcome.117 Nonsurvivors had lower venous pH and larger base deficits, and for each unit improvement in base deficit there was a 9% increase in likelihood of discharge from the hospital.
The nitroprusside reagent (e.g., Acetest, Bayer, Tarrytown, N.Y.) detects only ketone (−C=O) groups (e.g., acetoacetate, acetone). The concentration of β-hydroxybutyrate typically exceeds that of acetoacetate in uncontrolled diabetic ketoacidosis, and the dipstick reaction underestimates the degree of ketonuria. This problem can be overcome by adding a few drops of hydrogen peroxide to urine, which nonenzymatically converts β-hydroxybutyrate to acetoacetate.171 When insulin is administered and metabolism of ketones proceeds, there is a shift toward acetoacetate, and the dipstick reaction transiently becomes more strongly positive. This possibility should be recognized by the clinician and should not cause concern. In a study of 116 diabetic dogs (of which 88 had not previously received insulin), all ketotic and ketoacidotic dogs and 21 of 32 (66%) “nonketotic” dogs (i.e., negative urine dipstick test for ketones) had abnormally high serum β-hydroxybutyrate concentrations (>0.15 mmol/L) at presentation.78 Although not as readily available, measurement of plasma ß-hydroxybutyrate concentrations is more valuable than use of dipstick tests in the characterization of ketonemia in diabetic dogs and cats.74,242,243 The increase in unmeasured anions (as reflected in the anion gap) gives a rough estimate of the concentration of ketoanions in serum. However, this estimate is inaccurate if lactic acidosis develops because lactate also is an unmeasured anion. In one study of diabetic dogs, however, acidosis was correlated primarily with serum ketone concentration, and not with serum lactate concentration.79
To some extent, the anions of these ketoacids are excreted in urine along with sodium and potassium for electroneutrality. These organic anions are lost from the body and cannot be metabolized to HCO3− after correction of diabetic ketoacidosis with insulin therapy. Their loss thus contributes to depletion of body buffer and cation stores. Osmotic diuresis is induced by hyperglycemia and also contributes to the whole-body cation deficit. The extent of impairment in renal function may determine whether patients with diabetic ketoacidosis have an increased anion gap metabolic acidosis or hyperchloremic metabolic acidosis at the time of presentation. Patients with severe volume depletion have an increased anion gap because of retention of ketoanions, whereas those without volume depletion have hyperchloremia as a result of increased urinary excretion of the sodium and potassium salts of ketoanions and retention of chloride.5,9
Treatment
The best treatment for the acidosis of uncontrolled diabetes mellitus is fluid therapy and insulin. Insulin administration allows glucose use by skeletal muscle and adipose tissue, decreases hepatic glucose production, prevents lipolysis and ketogenesis, and permits peripheral metabolism of ketoacids. Several regimens for administration of insulin to ketoacidotic dogs and cats have been described.84 The particular protocol of insulin administration is probably less crucial to the ultimate outcome than the individualized care provided by the veterinarian during management of the diabetic animal.
Several factors may contribute to a delay in the repair of the HCO3− deficit in patients with diabetic ketoacidosis.100 Ketoacid anions that have been excreted in the urine are lost to the body and cannot be metabolized to HCO3−. After treatment with fluids and insulin, recovery may be faster in patients with a high anion gap because the retained ketoanions are metabolized, yielding HCO3−.7,9 Thus, withholding alkali may be more rational for diabetic patients with high anion gap metabolic acidosis than for those with hyperchloremic metabolic acidosis. Dilutional acidosis may occur if ECF volume (ECFV) is expanded with alkali-free solutions such as 0.9% saline. If hyperventilation persists, it may impair renal reabsorption of HCO3−, and renal acid excretion may require several days to become fully augmented.
The use of NaHCO3 to treat diabetic ketoacidosis is highly controversial, and clear benefits of its use have not been demonstrated in human patients. For example, there was no difference in recovery (based on rate of decrease of blood glucose and ketone concentrations and rate of increase of blood or cerebrospinal fluid [CSF] pH or HCO3− concentration) when NaHCO3 was or was not administered to human patients with diabetic ketoacidosis who presented with blood pH values in the range of 6.90 to 7.14.166 In another study, treatment with NaHCO3 delayed resolution of ketosis in diabetic ketoacidosis.178
There are several theoretical arguments against the use of NaHCO3 in diabetic ketoacidosis. Acidosis in the CNS may develop after NaHCO3 administration. The blood-brain barrier is permeable to CO2 but less permeable to the charged HCO3− ion. If NaHCO3 is administered, pH increases in ECF as the HCO3−/ PCO2 ratio increases, and compensatory hyperventilation decreases somewhat. As a result, PCO2 increases and CO2 diffuses into the CNS. However, bicarbonate diffusion into CNS lags behind that of CO2. During this time, the HCO3− PCO2 ratio and pH in the CNS may decrease. This has been referred to as paradoxical CNS acidosis.192 The frequency of occurrence of this complication and its clinical significance are uncertain.135
The pathophysiology of diabetic ketoacidosis also affects oxygen delivery to tissues. Chronic acidosis shifts the oxygen-hemoglobin dissociation curve to the right, thus enhancing delivery of oxygen to the tissues. Conversely, phosphorus deficiency in diabetes decreases red cell 2,3-diphosphoglycerate concentration and causes a shift of the oxygen-hemoglobin dissociation curve back to the left. Correction of acidosis with NaHCO3 shifts the curve farther to the left and potentially decreases oxygen delivery to tissues. However, administration of insulin and fluid therapy also lead to correction of the acidosis and should have a similar effect on the oxygen-hemoglobin dissociation curve.
Overzealous therapy with NaHCO3 may contribute to late development of metabolic alkalosis because insulin promotes metabolism of retained ketoacid anions to HCO3−. This excess HCO3− should be readily excreted in the urine if renal function is adequate. Other potentially detrimental effects of NaHCO3 therapy include aggravation of hyperosmolality as a consequence of the obligatory sodium load, tetany resulting from a sudden decrease in ionized serum calcium concentration, and precipitation of severe hypokalemia as extracellular potassium ions move into cells during administration of insulin and correction of acidosis. For all these reasons, NaHCO3 is not used unless severe acidosis (pH <7.1 to 7.2) is present and then only in small amounts (see the Treatment of Metabolic Acidosis section).
Uremic Acidosis
Pathophysiology
The metabolic acidosis of chronic renal failure is usually mild to moderate in severity (plasma HCO3− concentration, 12 to 15 mEq/L) and may be hyperchloremic early in the course of the disease process.239 Later in the course of the disease, the anion gap increases because of retention of phosphates, sulfates, and organic anions. Acid-base status is usually well preserved in chronic renal failure until GFR decreases to 10% to 20% of normal. In retrospective studies of small animal patients with chronic renal failure, plasma HCO3− concentrations were less than 16 mEq/L in 40% of dogs with chronic renal failure caused by amyloidosis72 and less than 15 mEq/L in 63% of cats with chronic renal failure of various causes.71 A high anion gap was observed in 43% of affected dogs (>25 mEq/L) and in 19% of affected cats (>35 mEq/L) in these studies. In acute renal failure, there has been insufficient time for the kidneys to adapt to the disease state, and the metabolic acidosis of acute renal failure is usually more severe than that observed in chronic renal failure. Complications such as sepsis and marked tissue catabolism may contribute to the severity of metabolic acidosis in acute renal failure.
Delivery of HCO3− from the proximal tubules to the distal nephron is increased in chronic renal failure.235 In dogs with experimentally induced unilateral renal disease, renal HCO3− reabsorption was not different in the diseased and control kidneys, but bicarbonaturia developed when the normal kidney was removed, and the contralateral diseased kidney was forced to function in a uremic environment.165 The osmotic diuresis characteristic of uremia may thus contribute to the increased delivery of HCO3− to the distal tubules. Increased parathyroid hormone concentration as a result of renal secondary hyperparathyroidism does not seem to have important adverse effects on HCO3− reabsorption in experimentally induced renal disease in dogs.12,206,207 The ability to lower urine pH maximally is preserved in chronic renal failure.
The main method by which the diseased kidney responds to chronic retention of fixed acid is by enhanced renal ammoniagenesis. Total ammonium excretion decreases during progressive chronic renal disease, but ammonium excretion is observed to be markedly increased when expressed per 100 mL GFR or per remnant nephron.75,202 On a per nephron basis, the diseased kidney can increase its ammonium excretion threefold to fivefold.219,235,238 This adaptive mechanism seems to be fully expended when the GFR decreases to less than 20% of normal. At this point, the diseased kidneys can no longer effectively cope with the daily fixed acid load, and a new steady state is established at a lower than normal plasma HCO3− concentration. The relatively mild decrease in plasma HCO3− concentration that is observed in chronic renal failure has been attributed to the contribution of the large reservoir of buffer (e.g., calcium carbonate) in bone. However, the capacity of the skeleton to buffer the amount of acid that accumulates in long-standing chronic renal failure has been questioned.177 The decrease in total ammonium excretion that occurs in chronic renal failure may be counterbalanced by decreased urinary excretion of organic anions (e.g., citrate, lactate, pyruvate, ketoanions).56 Metabolism of these retained organic anions would result in a net gain of HCO3− that would offset the decreased excretion of H+ in the form of NH4+.
The amount of phosphate buffer available in urine in chronic renal failure is relatively fixed and likely to be at its maximum because of hyperphosphatemia and the effects of increased plasma parathyroid hormone concentration.202,219 Furthermore, phosphorus binders and dietary phosphorus restriction are commonly used to treat chronic renal failure and may limit the amount of phosphate that can contribute to titratable acidity. When expressed on a per nephron basis, however, titratable acidity is increased in chronic renal failure.164
Treatment
Whether to treat well-compensated mild to moderate metabolic acidosis in adult patients with chronic renal failure is controversial. The potential benefits of such treatment include minimizing potential depletion of bone buffers, preventing the catabolic effects of uremic acidosis on muscle protein, preventing tubulointerstitial damage resulting from complement activation by ammonia, and improving the patient’s ability to combat a superimposed acidotic crisis (e.g., acute diarrhea).197 Thus, treatment with oral NaHCO3 at a dosage of 0.5 to 1.0 mEq/kg/day or an amount sufficient to maintain plasma HCO3− concentration at 15 mEq/L or above is reasonable if the patient can tolerate the associated sodium load. One teaspoon of baking soda contains 5 g NaHCO3 (1.3 g of which is sodium). An advantage of using calcium carbonate (e.g., Tums [GlaxoSmithKline, Brentford, UK], Os-Cal [GlaxoSmithKline]) as a phosphorus binder in chronic renal failure is that this compound can serve as both a source of alkali and a source of calcium, if small amounts of calcitriol (2 to 3 ng/kg/day) are also provided. The patient should be monitored for development of hypercalcemia when calcium carbonate and calcitriol are administered concurrently. Potassium and sodium citrate should not be used for alkali therapy in chronic renal failure patients that also are being treated with aluminum-containing phosphorus binders (e.g., aluminum hydroxide, aluminum carbonate) because citrate can increase aluminum absorption from the gastrointestinal tract in this clinical setting.162
Lactic Acidosis
Lactic acidosis is characterized by an accumulation of lactate in body fluids and a plasma lactate concentration greater than 5 mEq/L.144,183 The pK’a of lactic acid is 3.86, and it is completely dissociated at the normal pH of ECF (7.40). Lactic acidosis has been divided into two categories (Box 10-2).55,112,136 In type A (hypoxic) lactic acidosis, mitochondrial function is normal but O2 delivery to tissues is inadequate. In type B (nonhypoxic) lactic acidosis, there is adequate O2 delivery to tissues but defective mitochondrial oxidative function and abnormal carbohydrate metabolism. Inborn errors of metabolism affecting gluconeogenesis and mitochondrial oxidative function are documented to cause type B lactic acidosis in humans. Defects in mitochondrial oxidative function are called mitochondrial myopathies and are caused by hereditary defects in specific mitochondrial enzyme systems. A number of case reports suggest that similar defects occur in dogs.116,179,181,233 Pyruvate dehydrogenase deficiency is suspected to occur in clumber spaniels.109,127 This discussion focuses on type A (hypoxic) lactic acidosis.
Box 10-2 Causes of L-Lactic Acidosis*
Normal Physiology
Lactate is a metabolic end product. Its production allows regeneration of cytosolic nicotinamide adenine dinucleotide (NAD+) during anaerobic metabolism, and its ultimate fate is reoxidation back to pyruvate:
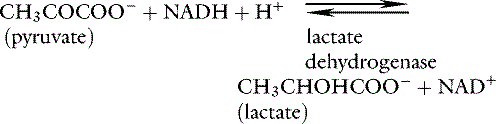
The equilibrium of this reaction is far to the right, and the normal ratio of lactate to pyruvate is 10:1. The main determinants of cytosolic lactate concentration are the concentration of pyruvate and the NADH/NAD+ ratio, both of which are affected by mitochondrial oxidative function.
Pyruvate is produced in the cytosol by anaerobic glycolysis (Embden-Meyerhof pathway). Under aerobic conditions, NADH is oxidized to NAD+ in the mitochondria and pyruvate enters the mitochondria for conversion to acetylcoenzyme A (CoA) and use in the tricarboxylic acid (Krebs) cycle, or it is converted to oxaloacetate and used for gluconeogenesis in the liver and renal cortex. Under anaerobic conditions (e.g., tissue hypoxia), oxidative pathways in the mitochondria are disrupted, and NAD+ must be replenished by reduction of pyruvate to lactate in the cytosol. Thus, lactate accumulation is the price to be paid for maintaining energy production under anaerobic conditions.
At rest, skin, red cells, brain, skeletal muscle, and gut all produce lactate. During tissue hypoxia, skeletal muscle and gut become the major producers of lactate. The liver and kidneys are the main consumers of lactate, using it for gluconeogenesis (primarily in the liver) or oxidizing it to CO2 and water. Protons are consumed when lactate is metabolized:
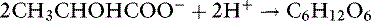
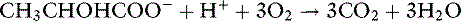
Both of these reactions require normal mitochondrial oxidative function. The protons are consumed when adenosine triphosphate (ATP) is synthesized from adenosine diphosphate (ADP) and when NADH is oxidized to NAD+ in the mitochondria.136,144 Protons are released by hydrolysis of ATP to ADP and by reduction of NAD+ to NADH, reactions that occur mainly in the cytosol. The protons do not arise from dissociation of lactic acid because the anion lactate is the predominant metabolite at normal hepatocyte pHi (pHi = 7.00 to 7.20). Thus, lactic acidosis reflects the imbalance between ATP hydrolysis and synthesis and between reduction and oxidation of NAD+. The protons produced during anaerobic glycolysis are buffered by bicarbonate and nonbicarbonate buffers. Protons are consumed and the buffers replenished when lactate is metabolized to glucose or oxidized to CO2 and water.
Pathophysiology
Lactic acidosis occurs when production of lactate by muscle and gut exceeds its use by liver and kidneys. Both pathways of lactate use depend on intact mitochondrial oxidative function, and clinical settings characterized by tissue hypoxia are the most common causes of lactic acidosis (see Box 10-2). Hepatic uptake of lactate is decreased when arterial PO2 decreases to approximately 30 mm Hg.225 Severe acidosis further impairs hepatic uptake of lactate, and the liver eventually becomes a producer rather than a consumer of lactate.140
In an experimental model of hypoxic lactic acidosis (type A) induced by ventilating dogs with 8% O2, lactate concentration was more than 5 mEq/L, pH was less than 7.2, HCO3− concentration was less than 12 mEq/L, PO2 was less than 30 mm Hg, and hepatocyte pHi was less than 7.00.11 When a similar degree of acidosis was created by infusing lactic acid into dogs with normal PO2, hepatocyte pHi remained greater than 7.00, and hepatic extraction of lactate (as a percentage of the delivered load) was approximately three times higher than that observed in the hypoxic animals. Hypoxemia reduces hepatic O2 uptake, and hepatocyte pHi decreases, presumably as a result of CO2 accumulation within cells. This study demonstrated that impaired hepatic extraction of lactate is related to decreased hepatic O2 uptake and pHi but not to arterial pH. During severe hypoxia, increased lactate production by gut and muscle and decreased hepatic extraction of lactate lead to progressive lactic acidosis. Impaired hepatic extraction of lactate and increased splanchnic production also contribute to the lactic acidosis of sepsis in dogs.48
Clinical Features
Lactic acidosis may occur in several clinical settings, especially those associated with poor perfusion and tissue hypoxia (e.g., cardiac arrest and cardiopulmonary resuscitation, shock, left ventricular failure). The clinician should strongly consider the possibility of lactic acidosis in such settings (see Box 10-2). Usually, lactic acidosis results from accumulation of the L isomer of lactate. D-Lactic acidosis, characterized by the accumulation of the D isomer, is rare but has been reported in human patients with “short-bowel syndrome” in whom gut bacteria metabolize glucose to D-lactate. Increased concentrations of D-lactate have been observed in cats fed propylene glycol45,46 and in cats with diabetic ketoacidosis, possibly as a result of hepatic ketone metabolism.47 Severe D-lactic acidosis has been documented in a cat with pancreatic insufficiency, likely as a consequence of intestinal bacterial overgrowth.184
Lactic acidosis should be suspected whenever there is an unexplained increase in unmeasured anions (i.e., an unexplained increase in the anion gap). Confirmation requires measurement of plasma lactate concentration, but this has not been performed commonly in small animal practice. Care should be taken to prevent vascular stasis when collecting venous blood for lactate determinations, and blood samples should be centrifuged immediately after collection to prevent a spurious increase in lactate concentration related to anaerobic glycolysis by red cells. Lactate concentrations in dogs have been reported in many experimental studies.* From results of these studies, normal plasma lactate concentrations in dogs are expected to be less than 2 mEq/L. Control plasma lactate concentrations in cats were 1.46 mEq/L in one study.13 In an experimental model of hemorrhagic shock in dogs, plasma lactate concentration increased from 1.5 to 5.5 mEq/L but did not completely account for the observed increases in anion gap and strong ion gap.30 Other organic anions (especially acetate and citrate) also contributed to the changes in the anion gap and strong ion gap.
Racing caused venous lactate concentrations in greyhounds to increase from 0.57 to 28.93 mEq/L, but lactate concentrations returned to 0.53 mEq/L 3 hours after exercise.119 Arterial pH decreased from 7.365 to 6.997 and returned to 7.372 3 hours after exercise, and HCO3− concentration decreased from 21.1 to 3.1 mEq/L and returned to 20.5 mEq/L 3 hours after exercise. Plasma potassium concentration does not increase in response to organic acidosis as it does in acute mineral acidosis.6 In the racing greyhounds, there was no change in plasma potassium concentration despite severe lactic acidosis.
Cardiac Arrest and Cardiopulmonary Resuscitation
Oxygen delivery to, and CO2 removal from, tissues are dependent on adequate tissue perfusion. Cardiac arrest is an extreme example of impaired tissue perfusion. During cardiopulmonary resuscitation (CPR), reduced tissue perfusion and reduced O2 delivery cause anaerobic metabolism and lactic acidosis. In dogs, lactate concentrations increased linearly during the time between cardiac arrest and the onset of CPR.38 Lactate concentrations increased progressively during closed-chest CPR in dogs39 and remained stable but did not decrease during 30 minutes of open-chest CPR.38 In this model, closed-chest CPR did not provide adequate tissue perfusion and O2 delivery to halt anaerobic metabolism.
During CPR, arterial blood gases reflect alveolar-arterial gas exchange, whereas mixed venous blood gases reflect tissue acid-base status and oxygenation.154 Respiratory alkalosis develops in arterial blood as a result of mechanical ventilation, whereas respiratory acidosis develops in venous blood because of poor tissue perfusion and impaired transport of accumulated CO2 to the lungs. In one study of human patients undergoing CPR, average arterial pH was 7.41, whereas average mixed venous pH was 7.15.237 Arterial PCO2 averaged 32 mm Hg and mixed venous PCO2 was 74 mm Hg, whereas arterial and venous HCO3− concentrations were similar.
Closed-chest CPR, initiated after 6 minutes of cardiac arrest, was studied in dogs.204 Sodium bicarbonate (2 mEq/kg) was administered after 20 minutes of cardiac arrest. Administration of NaHCO3 increased both arterial and venous pH. Before NaHCO3, arterial PCO2 was approximately 40 mm Hg, and with CPR it decreased to 20 mm Hg as a result of mechanical ventilation. After NaHCO3, arterial PCO2 increased to 30 mm Hg. Venous PCO2 was nearly 50 mm Hg, and it slowly increased during 30 minutes of cardiac arrest to 60 mm Hg in untreated dogs. Bicarbonate treatment caused venous PCO2 to increase transiently to 100 mm Hg, and it decreased to 70 mm Hg 10 minutes after NaHCO3 administration. The pH of CSF was not changed by NaHCO3 administration.
The normal arteriovenous pH gradient in dogs is 0.01 to 0.04.8,20,152 Reduced cardiac output increases arteriovenous pH and PCO2 gradients as a result of arterial hypocapnia and venous hypercapnia.8,20,154,237 The ventilation-to-perfusion ratio is increased because of decreased pulmonary blood flow, accounting for the observed arterial hypocapnia. Venous hypercapnia results from anaerobic metabolism and a greater than normal addition of CO2 to venous blood from hypoperfused tissues and diminished CO2 excretion in the lungs because of pulmonary hypoperfusion. These increases in arteriovenous pH and PCO2 gradients occur only if pulmonary ventilation continues. Respiratory arrest abolishes arteriovenous pH and PCO2 gradients.8 In summary, arterial PCO2 is not an accurate reflection of CO2 removal from tissues during CPR, and analysis of mixed venous PCO2 is recommended.8,20,152,154,237
During CPR and ventilation with 100% O2, arterial PO2 may be normal, but tissue perfusion is low (20% to 25% of normal).112 After NaHCO3 administration, additional CO2 is produced, and venous hypercapnia persists if ventilation is inadequate. Improving tissue perfusion is much more important during CPR than is NaHCO3 administration. Effective cardiac compression and adequate perfusion allow delivery of O2 to and removal of CO2 from tissues. Conversely, tissue acidosis is aggravated and pHi is decreased by NaHCO3 administration if the CO2 generated cannot be removed from the tissues by the lungs. The increase in tissue CO2 decreases pHi because CO2 diffuses more rapidly into cells than does the charged HCO3−, thereby lowering the intracellular HCO3−/PCO2 ratio. Intracellular acidosis of the myocardium leads to impaired cardiac contractility, decreased cardiac output, and aggravation of lactic acidosis. Thus, the main goals of CPR are to provide adequate tissue perfusion by effective cardiac compression and to ventilate the patient with 100% O2. In one study of short (5 minutes) and prolonged (15 minutes) cardiac arrest in dogs, NaHCO3 administration improved acidosis without a significant increase in PCO2.234 The authors concluded that NaHCO3 might be useful to reverse the acidosis of cardiac arrest if ventilation is adequate and NaHCO3 is administered in a reasonable therapeutic window.
Lymphosarcoma in Dogs
Dogs with lymphosarcoma had higher lactate concentrations than control animals, and their lactate concentrations increased significantly 30 minutes after administration of 500 mg/kg dextrose.231 Blood lactate concentrations were higher before and 1 hour after infusion of lactated Ringer’s solution in dogs with lymphosarcoma as compared with control animals.230 Blood lactate concentration returned to baseline during the second hour of the 6-hour infusion. The authors concluded that dogs with stage III or IV lymphosarcoma might have abnormal carbohydrate metabolism and a transient inability to handle lactate loads. Tumors may produce increased amounts of lactate as a result of excessive anaerobic metabolism and possibly as a result of less than normal hepatic extraction of lactate. Induction of remission with doxorubicin chemotherapy did not improve hyperlactatemia in dogs with lymphosarcoma.176
Treatment
The outcome of lactic acidosis depends on the severity and reversibility of the underlying disease process responsible for the acid-base disturbance. If treatment of lactic acidosis is to be successful, prompt diagnosis and correction of the underlying disease state are crucial. Tissue perfusion and oxygen delivery should be improved by aggressive fluid therapy to expand ECFV. Ventilation with O2 should be considered if the patient’s spontaneous ventilation is inadequate. Infections should be treated with appropriate antimicrobial agents, and cardiac output should be improved, if necessary, by administration of inotropic agents. If the underlying disease cannot be corrected, the prognosis for patients with lactic acidosis is very poor. If the underlying disease can be corrected, the accumulated lactate is metabolized, yielding an equivalent amount of HCO3−, and the acidosis is reversed.
When the pH of the patient’s blood decreases to below 7.1 to 7.2, administration of alkali is justified to prevent the detrimental effects of severe acidosis on the cardiovascular system (e.g., impaired myocardial contractility, impaired cardiovascular responsiveness to catecholamines, increased susceptibility to ventricular arrhythmias). Small doses of NaHCO3 should be administered to increase the patient’s pH to 7.2.4,112,144
Approximately 10% to 15% of administered NaHCO3 is converted immediately to CO2.112 It is essential that ventilation increase to allow removal of accumulated CO2 from the body. It is probably safe to administer NaHCO3 if the patient can reasonably be expected to increase ventilation spontaneously. If not, administration of NaHCO3 may be detrimental. In any case, NaHCO3 should be administered slowly to minimize the increase in mixed venous PCO2.
The volume of distribution (Vd) of administered HCO3− is variable, depending on the severity of the acidosis.3 Thus, there is no simple way to calculate the dosage of NaHCO3 required to increase the pH to 7.2. Volumes of distribution of 0.21 and 0.5 have been recommended for calculation of the bicarbonate space.4,112 Sodium bicarbonate should be used cautiously and only in amounts necessary to increase the pH to 7.2. It should be administered slowly over several minutes to a few hours, and at least 30 minutes should be allowed to elapse after the infusion before judging its effect.4
The use of NaHCO3 in lactic acidosis is controversial.170,220 Using the canine model of hypoxic lactic acidosis described above,11 affected dogs were left untreated, treated with 2.5 mEq/kg NaHCO3, or treated with 2.5 mEq/kg 1 M NaCl.92,93 Animals treated with bicarbonate showed a greater decrease in pH and HCO3− concentration and higher lactate concentration than the other groups. Gut lactate production was greater in dogs that received NaHCO3 than in dogs that received NaCl, and portal vein PCO2 was higher in the group that received NaHCO3. Arterial blood pressure and cardiac output declined in the untreated group and the group that received NaHCO3 but were higher in the group that received NaCl. Increased portal vein PCO2 and hepatic accumulation of lactate presumably caused hepatocyte pHi to decrease. The ability of the liver to extract lactate depends on adequate hepatic blood flow and normal hepatocyte pHi, both of which are decreased in this model. During hypoxia (PO2 <30 mm Hg), the liver is unable to increase its lactate extraction, despite an increased load delivered from the ischemic gut. The investigators concluded that use of NaHCO3 during lactic acidosis might not be effective and might even be detrimental.
Dichloroacetate (DCA) stimulates the enzyme pyruvate dehydrogenase, which converts pyruvate to acetyl CoA.62 In the canine model of hypoxic lactic acidosis described before,11 DCA was compared with NaCl.91 DCA increased pH and HCO3− concentration and maintained a constant lactate concentration, whereas NaCl treatment was associated with a decrease in pH and HCO3− concentration and an increase in lactate concentration. Hepatic lactate extraction increased with DCA, whereas liver and muscle accumulation of lactate decreased. Muscle pHi increased with DCA, but neither treatment changed arterial blood pressure or cardiac output. DCA was also studied in a cardiac arrest model in dogs.216 This study compared DCA, DCA and NaHCO3, NaHCO3, and no treatment. Bicarbonate treatment increased arterial pH, but DCA did not. DCA did not decrease lactate concentration or increase pH in either the peripheral circulation or CNS. In a canine model of hemorrhagic shock, DCA administration decreased arterial lactate concentrations but was associated with decreased cardiac stroke volume, decreased myocardial efficiency, and reduced myocardial lactate consumption.15 Thus, there are conflicting results regarding the usefulness of DCA in canine models of lactic acidosis.
Carbicarb is an equimolar mixture of Na2CO3 and NaHCO3 that limits the generation of CO2 during the buffering process:
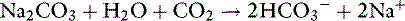
However, some of the HCO3− generated from this reaction can buffer H+ released from nonbicarbonate buffers and generate CO2 in the presence of carbonic anhydrase:
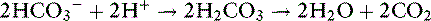
In the canine model of hypoxic lactic acidosis described earlier,11 2.5 mEq/kg Carbicarb was compared with 2.5 mEq/kg NaHCO3.21 Arterial pH increased after administration of Carbicarb but decreased after NaHCO3. Mixed venous PCO2 was unchanged after Carbicarb administration but increased after NaHCO3. Arterial lactate concentration increased after administration of NaHCO3 but stabilized after Carbicarb, whereas lactate use by the gut, muscle, and liver improved with Carbicarb but decreased after NaHCO3. Hepatocyte pHi increased after Carbicarb and decreased after NaHCO3. Arterial blood pressure decreased to a lesser extent and cardiac output stabilized with Carbicarb, whereas cardiac output decreased with NaHCO3. It was concluded that Carbicarb had a beneficial effect on myocardial contractility. Myocardial contractility may decrease after NaHCO3 administration as a result of increased venous PCO2 and decreased myocardial pHi. Decreased cardiac output follows and leads to decreased blood flow and decreased O2 delivery to gut, muscle, and liver, resulting in decreased lactate use and increased production. Carbicarb improved arterial pH without impairing myocardial contractility, presumably because it did not increase venous PCO2. This study suggests that Carbicarb is superior to NaHCO3 in the treatment of lactic acidosis in dogs.
In another study, Carbicarb was compared with sodium bicarbonate and hypertonic saline in a canine model of hemorrhagic shock.19 All dogs received identical sodium loads. Groups that received Carbicarb and sodium bicarbonate experienced similar increases in serum bicarbonate, but arterial PCO2 increased more in bicarbonate-treated dogs than in those treated with Carbicarb. Hemodynamics, oxygen delivery, and oxygen consumption improved in all three groups, and these effects were attributed to the sodium load. Carbicarb, NaHCO3, and NaCl were compared in a model of hypoxic lactic acidosis in anesthetized, mechanically ventilated dogs.193 Carbicarb increased arterial pH, base excess, and cardiac index without an increase in lactate. Bicarbonate increased PCO2, but no adverse effects of NaHCO3 on hemodynamics or pHi were detected.
A sodium-free 0.3 N solution of tromethamine (THAM) is another CO2-consuming alkalinizing agent that is capable of buffering both nonvolatile (H+) and volatile (H2CO3 derived from CO2) acid. THAM and sodium bicarbonate had similar buffering ability when evaluated in dogs with experimentally induced metabolic acidosis.163 Dogs treated with THAM did not experience the transient hypernatremia and hypercapnia that were observed in bicarbonate-treated dogs.
Treatment of metabolic acidosis
The main goal in the treatment of metabolic acidosis is prompt diagnosis and specific treatment of the underlying cause of the acid-base disorder. Correction of the underlying disease that is responsible for the patient’s metabolic acidosis may be all that is necessary (e.g., fluids and insulin in diabetic ketoacidosis). In some instances, however, the underlying disease cannot be corrected (e.g., chronic renal failure), and alkali therapy must be considered.
In general, administration of NaHCO3 should be reserved for clinical settings in which the patient’s blood pH is less than 7.1 to 7.2, and NaHCO3 should be administered only in amounts necessary to increase the pH to 7.2. Therapy with sodium bicarbonate is less likely to be harmful in animals with simple hyperchloremic metabolic acidosis (normal anion gap) because of the absence of unmeasured organic anions. In patients with normochloremic metabolic acidosis (increased anion gap), unmeasured organic anions (e.g., ketoacids, lactate) are present and can be metabolized to HCO3− during recovery. Administration of NaHCO3 in such a setting may result in late development of metabolic alkalosis. This complication should not be serious if renal function is normal because the kidneys can excrete the excess HCO3−.
Severe acidosis may lead to life-threatening cardiovascular complications (e.g., impaired cardiac contractility, impaired pressor response to catecholamines, sensitization to ventricular arrhythmias).161 Thus, if blood pH is less than 7.1 to 7.2, judicious treatment with NaHCO3 is justified. The aim of therapy should be to increase the patient’s pH to 7.2 ([H+] = 63 nEq/L), at which point the risk of life-threatening hemodynamic complications is reduced.
For example, consider a 10-kg dog with a pH of 7.000, [H+] = 100 nEq/L, [HCO3−] = 6 mEq/L, and PCO2 = 25 mm Hg. We assume that normal values are a pH of 7.387, [H+] = 41 nEq/L, [HCO3−] = 21 mEq/L, and PCO2 = 36 mm Hg and that the normal compensatory respiratory response to metabolic acidosis is a 0.7-mm Hg decrement in PCO2 per 1.0 mEq/L decrement in [HCO3−]. How much NaHCO3 must be administered to increase the dog’s pH to 7.200 ([H+] = 63 nEq/L)? This may be determined using the Henderson equation:
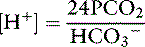
Thus, the desired [HCO3−] would be 24(25)/63 or 9.5 mEq/L if we assume that the PCO2 will not change. However, alveolar hyperventilation is likely to subside somewhat as the acidemia is partially corrected. If we assume that the PCO2 will increase to 28 mm Hg, the required [HCO3−] is 24(28)/63 or 10.7 mEq/L. Thus, we want to increase the dog’s [HCO3−] to 9.5 to 10.7 mEq/L.
We still must determine how much NaHCO3 to administer. This can be calculated using the formula:
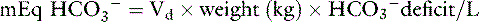
where Vd is the volume of distribution for HCO3−. However, the volume of distribution of HCO3− varies inversely with the initial HCO3− concentration and changes for at least 90 minutes after HCO3− administration to dogs.3 In this study, dogs with chronic metabolic acidosis and initial plasma HCO3− concentrations of 10 mEq/L were given 5 mEq/kg NaHCO3 and had average Vd values of 60% at 30 minutes and 76% at 90 minutes. This increase in Vd represents distribution of administered HCO3− from extracellular to intracellular sites. Bicarbonate distributes to ECF within 15 minutes and to intracellular and bone buffers within 2 to 4 hours.198 Thus, it is impossible to assign a single value for the Vd of NaHCO3 administered to dogs with metabolic acidosis. Any dosage recommendations must be considered only rough guidelines to treatment.
The dogs in this study3 had ECFVs equal to approximately 24.5% of body weight as measured by radiosulfate space. If we arbitrarily choose 0.5, a value approximately twice ECFV:
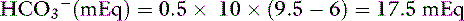
Thus, the desired amount of NaHCO3 is between 17.5 and 23.5 mEq. The NaHCO3 should be administered over the first few hours of therapy and blood gases reevaluated before making a decision about additional alkali administration. This amount of NaHCO3 represents a dose of 1.7 to 2.3 mEq/kg, and an empirical dose of 2 mEq/kg could safely have been used.
In patients with severe acidosis, any additional small reduction in plasma HCO3− concentration represents a large percentage change and can markedly increase [H+] (and reduce pH).199 For example, consider a normal dog with a pH of 7.387, [H+] = 41 nEq/L, PCO2 = 36 mm Hg, and [HCO3−] = 21 mEq/L that sustains a peracute reduction in [HCO3−] of 2 mEq/L (new [HCO3−] = 19 mEq/L) before respiratory compensation can develop. The new [H+] can be calculated from the Henderson equation as 24(36)/19 = 45 nEq/L (p. 7.347). This represents a 0.04-U change in pH and a 4-nEq/L change in [H+]. Now consider a dog with a pH of 7.102, [H+] = 79 nEq/L, PCO2 = 23 mm Hg, and [HCO3−] = 7 mEq/L that sustains a peracute reduction in [HCO3−] of 2 mEq/L (new [HCO3−] = 5 mEq/L) before respiratory compensation can develop. The dog’s new [H+] is 24(23)/5 = 110 nEq/L (p. 6.959). This represents a 0.14-U change in pH and a 31-nEq/L change in [H+]. This change in [H+] is almost eight times greater than that observed in the previous example. Thus, a small change in [HCO3−] has a much more dramatic effect on [H+] and pH when the initial [HCO3−] concentration is very low. For this reason, patients with very low plasma HCO3− concentrations and pH values less than 7.1 to 7.2 should be treated promptly with small amounts of NaHCO3 to increase their pH to the hemodynamically safe value of 7.2.
Potential complications of NaHCO3 therapy include volume overload caused by administered sodium, tetany resulting from decreased serum ionized calcium concentration caused by increased binding of calcium to plasma proteins, decreased O2 delivery to tissues because of increased affinity of hemoglobin for O2, paradoxical CNS acidosis as hyperventilation abates and CO2 diffuses into CSF, late development of alkalosis as metabolism of organic anions (e.g., ketoanions, lactate) replenishes body HCO3− stores, and hypokalemia as potassium ions enter and H+ ions exit intracellular fluid in response to alkalinization of ECF.105
Metabolic alkalosis
Metabolic alkalosis is characterized by a primary increase in plasma HCO3− concentration, decreased [H+], increased pH, and a secondary or adaptive increase in PCO2. Metabolic alkalosis was the third most common acid-base disturbance in dogs and cats in one study.61
Metabolic alkalosis can be caused by loss of chloride-rich fluid from the body via either the gastrointestinal tract or kidneys or by chronic administration of alkali. In the normal animal, renal excretion of exogenously administered alkali is very efficient, and it is difficult to create metabolic alkalosis by administration of alkali unless there is some factor preventing renal HCO3− excretion. Most cases of metabolic alkalosis in small animal practice are caused either by vomiting of stomach contents or by administration of diuretics. In a review of 962 dogs evaluated by blood gas determinations, 20 (2%) were found to be alkalemic.194 Of these 20 dogs, 13 had metabolic alkalosis and 7 had respiratory alkalosis. Of the 13 dogs with metabolic alkalosis, 10 had a history of gastrointestinal disease. In a study of 138 dogs with gastrointestinal foreign bodies, 77.5% of which were located in the stomach or duodenum, hypochloremia (51.2%), metabolic alkalosis (45.2%), hypokalemia (25%), and hyponatremia (20.5%) were commonly observed laboratory abnormalities.22
Classification of metabolic alkalosis
Patients with metabolic alkalosis may be divided into two groups.101,122,123,200,229 One group has ECFV depletion and avid renal retention of sodium and chloride. These patients respond to chloride administration and are said to have chloride-responsive metabolic alkalosis. The other group has normal or increased ECFV, and all sodium chloride ingested on a daily basis is excreted in the urine. These patients do not respond to chloride administration and are said to have chloride-resistant metabolic alkalosis.
In most instances of chloride-responsive metabolic alkalosis, the chloride concentration of the fluid lost from the body is greater than that of the ECF, so there has been a disproportionate loss of chloride. For example, the chloride concentration of gastric fluid is approximately 150 mEq/L, whereas serum chloride concentration is approximately 110 mEq/L in the dog and 120 mEq/L in the cat. Chloride-responsive metabolic alkalosis is much more common in small animal practice than is chloride-resistant metabolic alkalosis.
Development of chloride-responsive metabolic alkalosis
The pathophysiology of chloride-responsive metabolic alkalosis can be understood by considering the events associated with selective removal of gastric HCl.131,132,172 Loss of H+ from the stomach is associated, milliequivalent for milliequivalent, with an increase in the concentration of HCO3− in ECF. Plasma HCO3− concentration and the filtered load of HCO3− in the kidneys increase. Natriuresis, kaliuresis, suppression of net acid excretion with bicarbonaturia, increased urine flow rate, and renal water loss follow, but bicarbonaturia is transient and insufficient to return plasma HCO3− concentration to normal.172 These events occurred without any change in the GFR in a study of dogs made alkalotic by hemofiltration and replacement of ECF with a solution containing HCO3− as the only anion.23 It is believed that the abatement of bicarbonaturia was caused by renal sodium avidity, engendered by the volume deficit that developed as a result of the initial natriuresis and diuresis. Renal sodium avidity is thus established and contributes to perpetuation of the alkalosis and development of a potassium deficit as long as chloride intake remains deficient. These events constitute the development phase of chloride-responsive metabolic alkalosis.
Probably the most important factors in the maintenance phase of chloride-responsive metabolic alkalosis are ECFV depletion and the chloride deficit, two factors that are difficult to separate experimentally.51,98,124,174,203 Other factors that contribute to perpetuation of metabolic alkalosis are the effects of aldosterone and the potassium deficit. Aldosterone concentration is increased by ECFV depletion and results in increased distal renal Na+-H+ and Na+-K+ exchange. This results in perpetuation of alkalosis and development of a potassium deficit. Potassium depletion leads to a transcellular shift of H+ from ECF to intracellular fluid in exchange for potassium ions. When this shift occurs in renal tubular cells, it decreases pHi and enhances H+ secretion by the renal tubular cells, further aggravating the alkalosis. Hypokalemia also stimulates renal ammoniagenesis, presumably through stimulation of glutaminase via decreased pHi. The increase in renal ammonium excretion enhances renal acid excretion and contributes to increased plasma HCO3− concentration. Hypokalemia also may decrease the GFR as a consequence of glomerular hemodynamic changes and may directly impair chloride reabsorption in the distal nephron, resulting in enhanced lumen electronegativity and facilitation of H+ secretion into tubular fluid.
Response of the body to metabolic alkalosis
The body’s response to metabolic alkalosis is the reverse of its response to administration of a mineral acid such as HCl. The kidneys are more effective in excreting an alkaline load than an acid load, provided that the subject is not sodium avid and sufficient chloride is provided.
Acute Buffer Response
In an early study of the buffer response to alkali, nephrectomized dogs were given 20 mEq/kg NaHCO3 with a resultant increase in plasma HCO3− concentration to approximately 60 mEq/L.222 Of the administered HCO3−, almost one third (32%) was titrated by intracellular buffers. Of this 32%, 4% was converted to carbonic acid by H+ from lactic acid released into ECF from cells. Increased pHi enhances cellular production of lactic acid by stimulation of phosphofructokinase. Approximately 2% entered red cells in exchange for chloride (so-called chloride shift), and 26% was titrated by H+ released from intracellular proteins and phosphates while sodium and potassium ions entered cells to maintain electroneutrality. By comparison, intracellular buffers handle approximately 50% of a mineral acid load.210,223
Approximately two thirds (68%) of the HCO3− load was confined to ECF. In response to the increase in pH, plasma proteins buffered 1% of this HCO3−. That is, plasma proteins released hydrogen ions in numbers sufficient to convert 1% of the infused HCO3− to carbonic acid. The remaining 67% was retained in the ECF compartment and contributed to the observed increase in plasma HCO3− concentration. These buffer reactions are summarized in Figure 10-6.
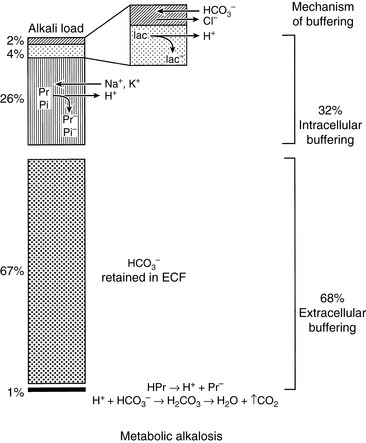
Respiratory Response to Metabolic Alkalosis
The decrease in [H+] that accompanies chronic metabolic alkalosis stimulates chemoreceptors and is responsible for the observed decrease in alveolar ventilation. Secondary or adaptive alveolar hypoventilation protects pH in the presence of increased plasma HCO3− concentration (Fig. 10-7). A review of studies of dogs with experimentally induced metabolic alkalosis suggests that for each 1.0-mEq/L increase in plasma HCO3− concentration, there is an adaptive 0.55- to 0.77-mm Hg increase in PCO2.23,40,145,146,186 This adaptive hypoventilation is associated with some degree of hypoxemia. Arterial PO2 decreased to 60 to 70 mm Hg in dogs made alkalotic by feeding a diet with a chloride deficit and administering furosemide.186

Figure 10-7 Beneficial effect of respiratory adaptation on [H+] and pH.
(From Harrington JT, Kassirer JP. Metabolic alkalosis. In: Cohen JJ, Kassirer JP, editors. Acid-base. Boston: Little, Brown, 1982: 237.)
The ventilatory response to metabolic alkalosis usually is considered to be less marked than the response to metabolic acidosis (i.e., a 0.6-mm Hg increase in PCO2 for each 1-mEq/L increase in plasma HCO3− concentration in metabolic alkalosis as compared with a 1.2-mm Hg decrease in PCO2 for each 1-mEq/L decrease in plasma HCO3− concentration in metabolic acidosis). This view has been challenged by a study of the ventilatory response of dogs to HCl acidosis and metabolic alkalosis induced by diuretics, removal of gastric acid, or mineralocorticoid administration.146 The ventilatory responses to all of these experimental acid-base disturbances were not significantly different from one another, and it was concluded that an average change of 0.74 mm Hg PCO2 can be expected for each 1.0-mEq/L change of plasma HCO3− concentration of metabolic origin. In one study, the respiratory compensation for metabolic alkalosis ranged from a 0.4- to 0.6-mm Hg increment in PCO2 for each 1-mEq/L increment in HCO3− for arterial, mixed venous, and jugular venous samples in dogs made alkalotic by the administration of furosemide.120 As a rule, a 1-mEq/L increase in plasma HCO3− concentration is expected to be associated with an adaptive 0.7-mm Hg increase in PCO2 in dogs with metabolic alkalosis.
Renal Response to Metabolic Alkalosis
In the normal animal, the kidneys rapidly and effectively excrete administered alkali. Metabolic alkalosis persists only if renal excretion of HCO3− is impaired. This may occur if GFR is decreased (i.e., decreased filtered load of HCO3−), a continued high rate of alkali administration, or some stimulus for the kidneys to retain sodium in the presence of a relative chloride deficit. In most dogs and cats with metabolic alkalosis, a combination of renal sodium avidity and diminished chloride availability is responsible for perpetuation of the alkalosis. A potassium deficit and hypokalemia develop as the kidneys increase Na+-K+ exchange in the distal nephron.
When sodium, chloride, and water are removed in proportion to their concentrations in ECF, sodium avidity develops but alkalosis does not.102 When the sodium deficit in an alkalotic animal is repaired by infusing a fluid identical in composition to the alkalotic ECF, metabolic alkalosis is corrected by selective retention of chloride.51 This occurs even when the filtered load of chloride is kept constant during the infusion of fluid.52 Thus, both sodium avidity and decreased chloride availability seem to be necessary for the perpetuation of metabolic alkalosis.
Potassium deficiency does not cause alkalosis but rather is a result of the alkalotic state. In fact, isolated potassium deficiency in dogs leads to mild metabolic acidosis.35,36 When potassium retention is prevented but sodium chloride is supplied, alkalosis is corrected despite a persisting potassium deficit.14,131,172 If potassium is supplied but chloride is not, alkalosis cannot be corrected.130 Administration of potassium chloride leads to complete correction of both alkalosis and the potassium deficit.
The renal response to hypercapnia in metabolic alkalosis was studied in normal unanesthetized dogs made alkalotic by dietary chloride restriction and administration of ethacrynic acid.145 Adaptive hypercapnia was allowed to develop and then prevented by exposure to hypoxia. During development of metabolic alkalosis, serum sodium concentration remained unchanged, but serum chloride, potassium, and phosphorus concentrations decreased, and lactate and unmeasured anion (i.e., anion gap) concentrations increased. With hypercapnia, plasma HCO3− concentration was maintained at 7.7 mEq/L above control values, whereas without hypercapnia it was maintained at 4.5 mEq/L above control values. Thus, approximately 60% of the increase in plasma HCO3− concentration was caused by the renal response to chloride and volume depletion, whereas 40% of the increase could be attributed to adaptive hypercapnia. This response appeared to be a direct effect of PCO2 on renal acid excretion and HCO3− reabsorption and was not related to any change in extracellular pH because the degree of alkalemia remained unchanged throughout the experiment. This portion of the increase in plasma HCO3− concentration (40%) may be considered maladaptive because it contributes to a higher extracellular pH. When metabolic alkalosis persists, this indiscriminate renal response to hypercapnia results in a further increase in plasma HCO3− concentration and abrogates the original beneficial effect of the increased plasma HCO3− concentration on extracellular pH.
Clinical features of metabolic alkalosis
The clinical features of dogs and cats with metabolic alkalosis are usually those of the underlying disease process. Neurologic signs have been reported in human patients with severe metabolic alkalosis and include agitation, disorientation, stupor, and coma.103 Muscle twitching and seizures may occur but have been observed rarely in dogs with severe metabolic alkalosis.
Clinical signs also may result from the accompanying potassium depletion. Signs of potassium depletion include muscle weakness of varying severity, cardiac arrhythmias, alterations in renal function (e.g., defective concentrating ability), and gastrointestinal motility disturbances (e.g., ileus). These complications are discussed in Chapter 5.
Muscle twitching may occur as a result of decreased serum ionized calcium concentration because alkalosis increases the number of negative charges on proteins, allowing more calcium ions to be bound (Fig. 10-8). Serum ionized calcium concentration decreases and may account for neuromuscular irritability by rendering the threshold potential of cells more negative (i.e., bringing the resting potential closer to the threshold potential) (Fig. 10-9). Administration of a single dose (4 mEq/kg) of sodium bicarbonate to normal cats resulted in a 10% decrease in serum ionized calcium concentration and an 8% decrease in serum total calcium concentration. These changes persisted for 3 hours, but no clinical signs were observed.41
Figure 10-8 Effects of alkalosis and acidosis on the charge of plasma proteins.
(Modified from Pitts RF. Physiology of the kidney and body fluids, 2nd ed. Chicago: Year Book Medical, 1974: 186.)
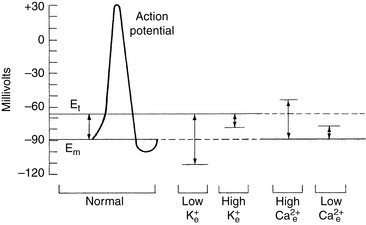
Figure 10-9 Effect of hypocalcemia on the threshold potential (Et) of cells. The height of the arrows is equal to the difference between the resting and threshold potentials and represents the excitability of the cell membrane.
(Adapted from Leaf A, Cotran R: Renal pathophysiology, New York, 1976, Oxford University Press, p. 116. IN Rose BD: Clinical physiology of acid-base and electrolyte disorders, ed. 3, New York, 1989, McGraw-Hill, p. 704.)
Metabolic alkalosis shifts the oxygen-hemoglobin dissociation curve to the left (Bohr effect) and impairs oxygen release from hemoglobin. This effect probably is not clinically relevant because an increase in red cell 2,3-diphosphoglycerate concentration occurs after 6 to 8 hours of metabolic alkalosis and results in a shift of the curve back to the right.18
Diagnosis of metabolic alkalosis
Specific clinical manifestations of metabolic alkalosis have not been reported in dogs and cats. The clinician must have a high index of suspicion for this disorder when presented with an animal having compatible clinical signs, usually chronic vomiting of stomach contents. Thus, an accurate history is the key to suspecting the diagnosis. Metabolic alkalosis also can be suspected from the results of routine serum biochemical tests. Blood gas analysis should be performed if decreased serum chloride and potassium concentrations are observed and total CO2 content is increased. Blood gas analysis allows the clinician to determine whether primary metabolic alkalosis is present and whether the magnitude of respiratory compensation is as predicted (see earlier). The concentration of unmeasured anions (i.e., anion gap) in metabolic alkalosis may increase because of loss of hydrogen ions from nonbicarbonate buffers. The increased anion gap is primarily caused by increased numbers of negative charges on proteins and partially the result of the increase in plasma protein concentration that occurs as a consequence of ECFV depletion.4
Urine pH is low during the maintenance phase of metabolic alkalosis because of enhanced distal Na+-H+ exchange and reabsorption of all filtered HCO3−. However, urine pH is alkaline during development of and recovery from metabolic alkalosis. Thus, urinary pH is of little diagnostic significance in metabolic alkalosis.
Causes of Metabolic Alkalosis
Metabolic alkalosis can be caused by continuous administration of alkali, disproportionate loss of chloride (chloride-responsive alkalosis), or excessive mineralocorticoid effect (chloride-resistant alkalosis). In some instances, the mechanism of metabolic alkalosis is unknown, and these examples are classified as miscellaneous. Most dogs with gastric dilatation-volvulus have metabolic acidosis or normal blood gas values at presentation,167,240 but, uncommonly, metabolic alkalosis and hypokalemia have been reported.129 The causes of metabolic alkalosis are listed in Box 10-3, and the pathophysiology of the major types of metabolic alkalosis is considered further here.
Chloride-Responsive Metabolic Alkalosis
Chronic vomiting of stomach contents and administration of diuretics are the most common causes of chloride-responsive metabolic alkalosis in dogs and cats.
Administration of Alkali
Acute administration of 4 mEq/kg NaHCO3 to normal unanesthetized cats resulted in mild increases in venous blood pH and HCO3− concentration lasting 180 minutes.42 A slight decrease in serum chloride concentration persisted for 30 minutes, whereas a mild increase in PCO2 persisted for 60 minutes. A solution of NaHCO3 (6.6 mEq/L) infused over 30 minutes into anesthetized dogs caused transient increases in arterial PCO2, pH, base excess, and standard bicarbonate concentration.106 Prompt renal excretion of administered NaHCO3 presumably prevented any persistent change in acid-base values in these acute studies. Renal acid excretion decreases, urine pH increases, and administered NaHCO3 is excreted within hours. There is an acute increase in carbonic acid and PCO2 as body buffers release H+ to combine with the administered HCO3−. The excess NaHCO3 is excreted in the urine, increased ventilation occurs in response to increased PCO2, and acid-base balance is restored to normal.
When alkali is administered chronically, plasma HCO3− concentration becomes a function of the daily dosage administered but returns to normal within a few days after alkali administration is discontinued. If alkali is given to subjects rendered sodium avid by previous dietary salt restriction, smaller dosages of alkali result in greater increases in plasma HCO3− concentration than are observed when higher alkali dosages are used in subjects receiving normal amounts of dietary salt.
Sources of alkali other than NaHCO3 may also contribute to metabolic alkalosis. Such organic anions include lactate that has accumulated during lactic acidosis, ketoacids in uncontrolled diabetes mellitus, and citrate in banked blood or that administered in an attempt to prevent recurrence of calcium oxalate urolithiasis. These organic anions yield HCO3− when metabolized:
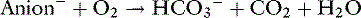
This reaction often serves to replace the HCO3− titrated during development of the acidosis (e.g., lactic acidosis, diabetic ketoacidosis). If NaHCO3 has been administered during treatment, however, metabolism of the organic anion after correction of the acidosis can result in metabolic alkalosis. If renal function is normal and volume depletion is not present, the kidneys promptly excrete the excess HCO3− and restore normal acid-base balance.
Administration of nonabsorbable alkali (e.g., aluminum hydroxide used as a phosphorus binder in patients with renal failure) usually does not cause metabolic alkalosis. Neutralization of H+ by Al(OH)3 in the stomach results in the net addition of HCO3− to ECF. Combination of Al3+ with HCO3− secreted by the pancreas produces insoluble Al2(CO3)3 in the duodenum, and there is no net increase in HCO3− ions in ECF. If, however, Al(OH)3 is administered concurrently with a cationic exchange resin (e.g., polystyrene sulfonate), the resin can bind Al3+, leaving HCO3− secreted by the pancreas to be reabsorbed in the small intestine, thus resulting in alkalinization of ECF. When renal failure is present, the kidneys have reduced capacity to excrete retained HCO3−, and metabolic alkalosis could result. This sequence of events is most likely to occur in an animal with oliguric renal failure that is treated concurrently with Al(OH)3 for hyperphosphatemia and with polystyrene sulfonate for hyperkalemia.
Gastric Fluid Loss
The H+ and Na+ concentrations of gastric fluid are inversely related to one another, whereas the K+ concentration is relatively stable (approximately 10 mEq/L). The Cl− concentration is very high (approximately 150 mEq/L) and remains remarkably constant even when hypochloremia develops. Subtracting the sum of the Na+ and K+ concentrations of gastric fluid from the Cl− concentration yields an approximation of the H+ concentration. The composition of gastric fluid is compared with that of other body fluids in Figure 10-10. When a dog or cat vomits stomach contents, water is lost along with large amounts of HCl and small amounts of potassium and sodium.

Figure 10-10 Comparison of electrolyte composition of gastric juice to other body fluids using Gamblegrams. Strong ions are crosshatched.
(From Jones NL. Blood gases and acid-base physiology, 2nd ed. New York: Thieme Medical, 1987: 133.)
The H+ produced during gastric acid secretion originates from the dissociation of carbonic acid; thus an equal number of HCO3− ions are generated in ECF. In the normal animal, gastric acid secretion does not disturb acid-base balance because the increase in ECF HCO3− concentration that accompanies parietal cell H+ secretion is balanced by pancreatic HCO3− secretion in the duodenum, and Cl− secreted into the stomach is recaptured lower in the gastrointestinal tract. When stomach contents are lost, H+ and Cl− are removed from this system, and HCO3− secreted into the duodenum by the pancreas is no longer titrated by gastric H+ but is reabsorbed farther down in the gastrointestinal tract in place of Cl−. The normal relationship between gastric and pancreatic secretions in the gastrointestinal tract is shown in Figure 10-11. Continued loss of gastric fluid can result in marked increases in plasma HCO3− concentration, and chronic vomiting of stomach contents is the most common cause of metabolic alkalosis in small animal practice.
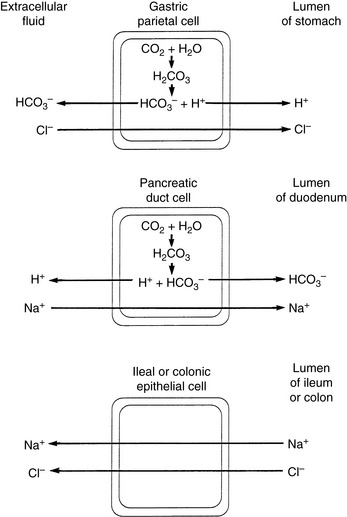
Figure 10-11 Normal relationship between gastric and pancreatic secretions in the gastrointestinal tract.
(Modified from Guyton AC. Textbook of medical physiology, 7th ed. Philadelphia: WB Saunders, 1986: 775–779.)
In studies of gastric alkalosis, experimental subjects are rendered sodium avid by feeding a low-salt diet. Gastric fluid is then continuously removed by nasogastric suction, and fluid and electrolyte losses other than HCl are quantitatively replaced.131,132,172 The effects of repeated gastric drainage over 3 days on plasma HCO3− and chloride concentrations and on potassium, sodium, and chloride balance in experimental dogs are shown in Figure 10-12. Note that the resulting metabolic alkalosis is corrected by provision of NaCl despite a progressively negative potassium balance. In the clinical setting, persistent vomiting of stomach contents leads to fluid and electrolyte losses (H+ and Cl− > Na+ and K+), and anorexia prevents adequate dietary intake of electrolytes. In patients with pyloric obstruction, gastrin secretion is enhanced, and gastric acid secretion is stimulated further. Overproduction of gastrin by a gastrin-secreting tumor may also stimulate gastric acid secretion. In one dog with gastrinoma, severe metabolic alkalosis and hypokalemia were associated with a history of chronic vomiting.221
Figure 10-12 Plasma composition and electrolyte balance in a representative study of selective HCl depletion.
(From Needle MA, Kaloyanides GJ, Schwartz WB. The effects of selective depletion of hydrochloric acid on acid-base and electrolyte equilibrium. J Clin Invest 1964;43:1839, with copyright permission of the American Society for Clinical Investigation.)
Renal avidity for sodium and defense of the ECFV occur because of ongoing fluid and electrolyte losses in the vomiting animal or intake of a low-salt diet and nasogastric suction in the experimental setting. To maintain ECFV, the kidneys must reabsorb sodium by all available mechanisms. Because of ongoing loss of gastric HCl and insufficient dietary intake of salt, there is a chloride deficit; consequently, the kidneys must reabsorb less sodium with chloride and more sodium in exchange for hydrogen and potassium ions. The latter two mechanisms contribute to perpetuation of the metabolic alkalosis and development of potassium depletion as shown in Figure 10-13. The low urine pH during the maintenance phase reflects increased distal Na+-H+ exchange in the sodium-avid state. This observation has led to the term “paradoxical aciduria” to describe the finding of low urine pH in patients with metabolic alkalosis. However, consideration of the relevant pathophysiology shows that this reduction in urine pH is the appropriate renal response under the circumstances. The extent of potassium depletion that develops is related to the severity and chronicity of the metabolic alkalosis.
Figure 10-13 Effects of chloride and potassium depletion on acid-base balance. See text for explanation.
(Drawing by Tim Vojt.)
Provision of chloride as the sodium or potassium salt allows correction of the alkalosis because the kidneys may now preferentially reabsorb sodium with chloride and rely less on Na+-K+ and Na+-H+ exchange.14,130,131,172 This allows retained HCO3− to be excreted in the urine. Urine pH increases as HCO3− is excreted, indicating a favorable response to therapy. Chloride once again appears in the urine when the alkalosis is resolved. The critical factor in resolution of this form of alkalosis is the provision of chloride as a resorbable anion. Alkalosis can be corrected without provision of sodium or potassium as long as chloride is provided. Clinically, however, alkalosis is corrected by administering some combination of NaCl and KCl.
Diuretic Administration
Diuretics cause approximately equal losses of sodium and chloride in the urine, but the concentration of chloride in ECF is less than that of sodium by approximately 35 mEq/L. Thus, these drugs may cause chloride-responsive metabolic alkalosis by a disproportionate loss of chloride in urine and creation of a relative chloride deficit in ECF. Increased renal sodium avidity is also an important factor in development of the metabolic alkalosis and potassium depletion that may occur during diuretic administration.
Loop diuretics inhibit NaCl reabsorption in the thick ascending limb of Henle’s loop by competing with chloride for the Na+-K+-2Cl− luminal carrier. This causes increased delivery of sodium to the distal nephron, where accelerated Na+-H+ and Na+-K+ exchange occurs as the kidneys attempt to retain more sodium. Increased reliance of the kidneys on these mechanisms for sodium reabsorption contributes to metabolic alkalosis and potassium depletion. These complications are less likely when thiazide diuretics are used. Thiazide diuretics inhibit NaCl transport in the distal tubule and connecting segment. They are less potent than the loop diuretics because their main effect occurs at sites in the nephron distal to those responsible for the majority of sodium reabsorption.
In response to hypokalemia, transcellular shifts of H+ from ECF into renal tubular cells may occur in exchange for K+. The resultant increase in intracellular H+ concentration facilitates renal Na+-H+ exchange and aggravates metabolic alkalosis. Stimulation of the renin-angiotensin-aldosterone system by decreased effective circulating volume also favors increased Na+-H+ and Na+-K+ exchange in the distal nephron. These latter effects are probably important in most forms of chloride-responsive metabolic alkalosis.
Many animals treated with diuretics have congestive heart failure as their primary disease process. If the treatment plan for the animal includes a low-sodium diet, renal sodium avidity is guaranteed and increases the tendency toward metabolic alkalosis and potassium depletion. Complications from diuretic therapy are unlikely if the animal is drinking water and eating a diet with adequate amounts of chloride. However, complications can develop if the animal becomes anorexic.
Posthypercapnia
Blood pH increases rapidly when PCO2 is suddenly reduced in patients with chronic hypercapnia. This has been called posthypercapnic metabolic alkalosis. In such patients, plasma HCO3− concentration has previously been increased by adaptive changes in renal HCO3− reabsorption. In response to the lowered PCO2, it takes several hours for the kidneys to decrease Na+-H+ exchange and begin to excrete the previously retained HCO3−. It may take several days for the kidneys to excrete all of the excess HCO3−, and sufficient chloride must be available during this time for reabsorption with sodium. Chloride deficiency during recovery from chronic hypercapnia plays a role in sustaining posthypercapnic metabolic alkalosis. Provision of chloride allows the alkalosis to be corrected.209 Posthypercapnic metabolic alkalosis occurs most commonly in human patients with chronic pulmonary disease who are treated by mechanical ventilation. It is important that PCO2 is decreased slowly and that adequate chloride intake is provided to prevent this complication.
Chloride-Resistant Metabolic Alkalosis
Several disorders in human medicine may cause chloride-resistant metabolic alkalosis. Of these, primary hyperaldosteronism and hyperadrenocorticism may occur in small animal practice. However, chloride-resistant metabolic alkalosis is rare in dogs and cats.
Primary Hyperaldosteronism
In primary hyperaldosteronism, increased secretion of aldosterone, usually by an adrenocortical tumor, results in sodium retention, volume expansion, hypernatremia, mild to moderate hypertension, potassium deficiency, hypokalemia, and metabolic alkalosis resistant to chloride administration. Plasma renin activity is low, but plasma aldosterone concentration is high. Affected human patients are in salt balance at an expanded ECFV and excrete ingested NaCl in the urine. Stimulation of distal nephron Na+-H+ and Na+-K+ exchange by excess mineralocorticoids is probably the most important pathophysiologic feature of primary hyperaldosteronism.
Several dogs and cats with primary hyperaldosteronism caused by aldosterone-producing adenomas or adenocarcinomas of the adrenal gland have been reported in the veterinary literature. Clinical features in affected animals included polyuria, polydipsia, weakness, hypertension, hypokalemia, hypernatremia, mild metabolic alkalosis, dilute urine, and extremely high serum aldosterone concentrations (for additional information and references see Chapter 5).
Hyperadrenocorticism
Metabolic alkalosis occurs in approximately one third of human patients with Cushing’s syndrome.104 It is more common in patients with adrenocortical carcinomas and in those with ectopic production of ACTH by nonadrenal malignancies than in those with pituitary-dependent hyperadrenocorticism. The frequency of metabolic alkalosis and serum electrolyte disturbances in dogs with hyperadrenocorticism is uncertain. Serum sodium and potassium concentrations often are normal in dogs with hyperadrenocorticism. This may reflect the fact that 80% to 85% of dogs with hyperadrenocorticism have pituitary-dependent disease. In a large group of dogs with hyperadrenocorticism, 21 of 52 (40%) dogs had increased serum sodium concentrations and 25 of 52 (48%) had decreased serum potassium concentrations.139 The relative frequency of pituitary- and adrenal-dependent disease was not reported in this study. In another study, mild hypernatremia and hypokalemia were observed occasionally in dogs with hyperadrenocorticism, and total CO2 content was increased in 33% of affected dogs.188 In another report, hypokalemia was found in only 5% of dogs with pituitary-dependent hyperadrenocorticism but in 45% of those with adrenocortical neoplasia.158 A high rate of secretion of cortisol and other corticosteroids, such as desoxycorticosterone and corticosterone in patients with adrenocortical malignancies, could be responsible for hypernatremia, hypokalemia, and metabolic alkalosis in adrenal-dependent hyperadrenocorticism.
Miscellaneous
Large doses of penicillin, ampicillin, or carbenicillin administered as a sodium salt can lead to hypokalemia and metabolic alkalosis in human patients. The drug may increase lumen electronegativity in the distal nephron by acting as a nonresorbable anion and enhancing Na+-H+ and Na+-K+ exchange. “Refeeding” alkalosis can occur in human patients when glucose is administered after prolonged fasting. The mechanism for this type of alkalosis is unknown. These types of metabolic alkalosis have not been reported in the veterinary literature.
Treatment of metabolic alkalosis
Acid-base disturbances are secondary phenomena. Diagnosis and definitive treatment of the responsible disease process are integral to the successful resolution of acid-base disorders. However, it must be remembered that alkalosis persists until chloride is replaced if vomiting of stomach contents or diuretic administration is responsible for the metabolic alkalosis. The goal of treatment in chloride-responsive metabolic alkalosis is to replace the chloride deficit while providing sufficient potassium and sodium to replace existing deficits. Definitive treatment of the underlying disease process (e.g., removal of a gastric foreign body) prevents recurrence of the metabolic alkalosis.
Patients with chronic pulmonary disease that have hypoxemia and hypercapnia are at greater risk from metabolic alkalosis than others because superimposition of metabolic alkalosis can further reduce ventilation and lead to worsening of hypoxemia. Thus, metabolic alkalosis should be treated appropriately if present and avoided if not present. Giving oxygen to patients with metabolic alkalosis should also be avoided if possible because this may impair ventilation and further aggravate hypercapnia.
Potassium without chloride (e.g., potassium phosphate) corrects neither the alkalosis nor the potassium deficit because administered potassium is excreted in the urine. A chloride salt must be given for alkalosis to be resolved and potassium retention to occur. Provision of chloride as either the sodium or potassium salt corrects chloride-responsive metabolic alkalosis. This therapy allows the kidneys to reabsorb the sodium the body requires with chloride to maintain electroneutrality. Thus, a NaCl solution (0.45% or 0.9%) with added KCl is the fluid of choice for dogs and cats with chloride-responsive metabolic alkalosis. It is best to use solutions containing NaCl and KCl because affected animals typically have been sick long enough to develop clinically relevant potassium deficits. Administering 0.9% NaCl without KCl can cause diuresis and increased urinary excretion of potassium, thus worsening any potassium deficit. As shown in Figure 10-12, provision of NaCl corrects metabolic alkalosis induced in dogs by gastric drainage, but the potassium deficit persists unless potassium is provided. A few days may be required to restore normal electrolyte and acid-base balance, but in nearly all instances, these measures are sufficient to resolve the alkalosis. In human patients with severe metabolic alkalosis or in those with severely impaired renal function, HCl or arginine HCl has been used for rapid correction of metabolic alkalosis, but there is no report of the use of these compounds in animals with metabolic alkalosis, and their use is not recommended.
H2-blocking drugs such as cimetidine, ranitidine, or famotidine may be considered as adjunctive therapy if gastric losses are ongoing because this approach reduces gastric acid secretion. For the patient with heart failure receiving loop diuretics, oral KCl administration is the best way to provide chloride without sodium and prevent further retention of fluid and aggravation of edema. Even in the presence of sodium avidity, provision of chloride lessens Na+-H+ and Na+-K+ exchange at distal nephron sites and prevents development of alkalosis when loop diuretics are used. Simultaneous use of distal blocking agents such as spironolactone, triamterene, or amiloride may also be considered. These drugs work in the principal cells of the cortical collecting tubule and impair Na+-H+ and Na+-K+ exchange by inhibiting aldosterone-sensitive sodium channels. In metabolic alkalosis caused by chronic administration of alkali, discontinuation of the source of alkali results in correction of the alkalosis over a few days, provided that renal function is normal.
Chloride-resistant metabolic alkalosis is uncommon in comparison with chloride-responsive metabolic alkalosis. When present, its successful treatment requires that the underlying disease be diagnosed and treated before alkalosis can be resolved.
1 Abraham L.A., Tyrrell D., Charles J.A. Transient renal tubulopathy in a racing Greyhound. Aust Vet J. 2006;84:398.
2 Adams W.H., Toal R.L., Walker M.A., et al. Early renal ultrasonographic findings in dogs with experimentally induced ethylene glycol nephrosis. Am J Vet Res. 1989;50:1370.
3 Adrogue H.J., Brensilver J., Cohen J., et al. Influence of steady-state alterations in acid-base equilibrium on the fate of administered bicarbonate in the dog. J Clin Invest. 1983;71:867.
4 Adrogue H.J., Brensilver J., Madias N.E. Changes in the plasma anion gap during chronic metabolic acid-base disturbances. Am J Physiol. 1978;235:F291.
5 Adrogue H.J., Eknoyan G., Suki W.K. Diabetic ketoacidosis: role of the kidney in the acid-base homeostasis re-evaluated. Kidney Int. 1984;25:591.
6 Adrogue H.J., Madias N.E. Changes in plasma potassium concentration during acute acid base disturbances. J Clin Invest. 1981;71:456.
7 Adrogue H.J., Madias N.E. Management of life-threatening acid-base disorders. N Engl J Med. 1998;338:26.
8 Adrogue H.J., Rashad M.N., Gorin A.B., et al. Arteriovenous acid-base disparity in circulatory failure: studies on mechanism. Am J Physiol. 1989;257:F1087.
9 Adrogue H.J., Wilson H., Boyd A.E., et al. Plasma acid-base patterns in diabetic ketoacidosis. N Engl J Med. 1982;307:1603.
10 Appleman E.H., Cianciolo R., Mosenco A.S., et al. Transient acquired Fanconi syndrome associated with copper storage hepatopathy in 3 dogs. J Vet Intern Med. 2008;22:1038.
11 Arieff A.I., Graf H. Pathophysiology of type A hypoxic lactic acidosis in dogs. Am J Physiol. 1987;253:E271.
12 Arruda J.A.L., Carrasquillo T., Cubria A., et al. Bicarbonate reabsorption in chronic renal failure. Kidney Int. 1976;9:481.
13 Atkins C.E., Tyler R., Greenlee P. Clinical, biochemical, acid-base, and electrolyte abnormalities in cats after hypertonic sodium phosphate enema administration. Am J Vet Res. 1985;46:980.
14 Atkins E.L., Schwartz W.B. Factors governing correction of the alkalosis associated with potassium deficiency: the critical role of chloride in the recovery process. J Clin Invest. 1962;41:218.
15 Barbee R.W., Kline J.A., Watts J.A. Depletion of lactate by dichloroacetate reduces cardiac efficiency after hemorrhagic shock. Shock. 2000;14:208-214.
16 Bark H., Perk R. Fanconi syndrome associated with amoxicillin therapy in the dog. Canine Pract. 1995;20:19.
17 Beckett S.D., Shields R.P. Treatment of acute ethylene glycol (antifreeze) poisoning in the dog. J Am Vet Med Assoc. 1971;158:472.
18 Bellingham A.J., Detter J.C., Lenfant C. Regulatory mechanisms of hemoglobin oxygen affinity in acidosis and alkalosis. J Clin Invest. 1971;50:700.
19 Benjamin J., Oropello J.M., Abalos A.M., et al. Effects of acid-base correction on hemodynamics, oxygen dynamics, and resuscitability in severe canine hemorrhagic shock. Crit Care Med. 1994;22:1616.
20 Bergman K.S., Harris B.H. Arteriovenous pH difference: a new index of perfusion. J Pediatr Surg. 1988;23:1190.
21 Bersin R.M., Arieff A.L. Improved hemodynamic function during hypoxia with Carbicarb, a new agent for the management of acidosis. Circulation. 1988;77:227.
22 Boag A.K., Coe R.J., Martinez T.A., et al. Acid-base and electrolyte abnormalities in dogs with gastrointestinal foreign bodies. J Vet Intern Med. 2005;19:816.
23 Borkan S., Northrup T.E., Cohen J.J., et al. Renal response to metabolic alkalosis induced by isovolemic hemofiltration in the dog. Kidney Int. 1987;32:322.
24 Bovee K.C. Characterization and treatment of isolated renal tubular acidosis in a dog. Washington, DC: American College of Veterinary Internal Medicine. 1984:48.
25 Bovee K.C., Joyce T., Reynolds R., et al. The Fanconi syndrome in basenji dogs: a new model for renal transport defects. Science. 1978;201:1129.
26 Bovee K.C., Joyce T., Reynolds R., et al. Spontaneous Fanconi syndrome in the dog. Metabolism. 1978;27:45.
27 Bovee K.C., Joyce T., Blazer-Yost B., et al. Characterization of renal defects in dogs with a syndrome similar to Fanconi syndrome in man. J Am Vet Med Assoc. 1979;174:1094.
28 Brown S.A., Rackich P.M., Barsanti J.A., et al. Fanconi syndrome and acute renal failure associated with gentamicin therapy in a dog. J Am Anim Hosp Assoc. 1986;22:635.
29 Brown S.A., Spyridakis L.K., Crowell W.A. Distal renal tubular acidosis and hepatic lipidosis in a cat. J Am Vet Med Assoc. 1986;189:1350.
30 Bruegger D., Kemming G.I., Jacob M., et al. Causes of metabolic acidosis in canine hemorrhagic shock: role of unmeasured ions. Crit Care. 2007;11:R130.
31 Bruskiewicz K.A., Nelson R.W., Feldman E.C., et al. Diabetic ketosis and ketoacidosis in cats: 42 cases (1980–1995). J Am Vet Med Assoc. 1997;211:188.
32 Buffington C.A., Cook N.E., Rogers Q.R., et al. The role of diet in feline struvite urolithiasis syndrome. In: Burger I.H., Rivers J.P.W., editors. Nutrition of the dog and cat. London: Cambridge University Press; 1989:357.
33 Burkitt J.M., Haskins S.C., Aldrich A., et al. Effects of oral administration of a commercial activated charcoal suspension on serum osmolality and lactate concentration in the dog. J Vet Intern Med. 2005;19:683.
34 Burnell J.M. Changes in bone sodium and carbonate in metabolic acidosis and alkalosis in the dog. J Clin Invest. 1971;50:327.
35 Burnell J.M., Dawbron J.K. Acid-base parameters in potassium depletion in the dog. Am J Physiol. 1970;218:1583.
36 Burnell J.M., Teubner E.J., Simpson D.P. Metabolic acidosis accompanying potassium deprivation. Am J Physiol. 1974;227:329.
37 Cain S.M., Dunn J.E. Transient arterial lactic acid changes in unanesthetized dogs at 21,000 feet. Am J Physiol. 1964;206:1437.
38 Carden D.L., Martin G.B., Nowak R.M., et al. Lactic acidosis as a predictor of downtime during cardiopulmonary arrest in dogs. Am J Emerg Med. 1985;3:120.
39 Carden D.L., Martin G.B., Nowak R.M., et al. Lactic acidosis during closed-chest CPR in dogs. Ann Emerg Med. 1987;16:1317.
40 Chazan J.A., Appleton F.M., London A.M., et al. Effects of chronic metabolic acid-base disturbances on the composition of cerebrospinal fluid in the dog. Clin Sci. 1969;36:345.
41 Chew D.J., Leonard M., Muir W.W. Effect of sodium bicarbonate infusions on ionized calcium and total calcium concentrations in serum of clinically normal cats. Am J Vet Res. 1989;50:145.
42 Chew D.J., Leonard M., Muir W.W. Effect of sodium bicarbonate infusion on serum osmolality, electrolyte concentrations, and blood gas tensions in cats. Am J Vet Res. 1991;52:12.
43 Ching S.V., Fettman M.J., Hamar D.W., et al. The effect of chronic dietary acidification using ammonium chloride on acid-base and mineral metabolism in the adult cat. J Nutr. 1989;119:902.
44 Ching S.V., Norrdin R.W., Fettman M.J., et al. Trabecular bone remodeling and bone mineral density in the adult cat during chronic dietary acidification with ammonium chloride. J Bone Miner Res. 1990;5:547.
45 Christopher M.M., Eckfeldt J.H., Eaton J.W. Propylene glycol ingestion causes D-lactic acidosis. Lab Invest. 1990;62:114.
46 Christopher M.M., Perman V., White J.G., et al. Propylene glycol-induced Heinz body formation and D-lactic acidosis in cats. Prog Clin Biol Res. 1989;319:69.
47 Christopher M.M., Broussard J.D., Fallin C.W., et al. Increased serum D-lactate associated with diabetic ketoacidosis. Metabolism. 1995;44:287.
48 Chrusch C., Bands C., Bose D., et al. Impaired hepatic extraction and increased splanchnic production contribute to lactic acidosis in canine sepsis. Am J Respir Crit Care Med. 2000;161:517-526.
49 Clark D.D., Chang B.S., Garella S.G., et al. Secondary hypocapnia fails to protect “whole body” intracellular pH during chronic HCl-acidosis in the dog. Kidney Int. 1983;23:336.
50 Clay K.L., Murphy R.C. On the metabolic acidosis of ethylene glycol intoxication. Toxicol Appl Pharmacol. 1977;39:39.
51 Cohen J.J. Correction of metabolic alkalosis by the kidney after isometric expansion of extracellular fluid. J Clin Invest. 1968;47:1181.
52 Cohen J.J. Selective chloride retention in repair of metabolic alkalosis without increasing filtered load. Am J Physiol. 1970;218:165.
53 Cohen J.J., Brackett N.C., Schwartz W.B. The nature of the carbon dioxide titration curve in the normal dog. J Clin Invest. 1964;43:777.
54 Cohen J.J., Madias N.E., Wolf C.J., et al. Regulation of acid-base equilibrium in chronic hypocapnia: evidence that the response of the kidney is not geared to the defense of extracellular [H+]. J Clin Invest. 1976;57:1483.
55 Cohen R.D., Woods R.A. Clinical and biochemical aspects of lactic acidosis. London: Blackwell Scientific; 1976.
56 Cohen R.M., Feldman G.M., Fernandez P.C. The balance of acid, base and charge in health and disease. Kidney Int. 1997;52:287.
57 Connally H.E., Thrall M.A., Forney S.D., et al. Safety and efficacy of 4-methylpyrazole for treatment of suspected or confirmed ethylene glycol intoxication: 107 cases (1983–1995). J Am Vet Med Assoc. 1996;209:1880.
58 Connally H.E., Hamar D.W., Thrall D.W. Inhibition of canine and feline alcohol dehydrogenase activity by fomepizole. Am J Vet Res. 2000;61:450.
59 Connally H.E., Thrall M.A., Hamar D.W. Safety and efficacy of high-dose fomepizole compared with ethanol as therapy for ethylene glycol intoxication in cats. J Vet Emerg Crit Care. 2010;20:191.
60 Constable P.D., Stämpfli H.R. Experimental determination of net protein charge and Atot and Ka of nonvolatile buffers in canine plasma. J Vet Intern Med. 2005;19:507.
61 Cornelius L.M., Rawlings C.A. Arterial blood gas and acid base values in dogs with various diseases and signs of disease. J Am Vet Med Assoc. 1981;178:992.
62 Crabb D.W., Young E.A., Harris R.A. The metabolic effects of dichloroacetate. Metab Clin Exp. 1981;30:1024.
63 Crenshaw K.L., Peterson M.E. Pretreatment clinical and laboratory evaluation of cats with diabetes mellitus: 104 cases (1992–1994). J Am Vet Med Assoc. 1996;209:943.
64 Darrigrand-Haag R.A., Center S.A., Randolph J.F., et al. Congenital Fanconi syndrome associated with renal dysplasia in 2 Border terriers. J Vet Intern Med. 1996;10:412.
65 DeFronzo R.A. Hyperkalemia and hyporeninemic hypoaldosteronism. Kidney Int. 1980;17:118.
66 DeSousa R.C., Harrington J.T., Ricanati E.S., et al. Renal regulation of acid-base equilibrium during chronic administration of mineral acid. J Clin Invest. 1974;53:465.
67 Dial S.M., Thrall M., Hamar D.W. 4-Methylpyrazole as treatment for naturally acquired ethylene glycol intoxication in dogs. J Am Vet Med Assoc. 1989;195:73.
68 Dial S.M., Thrall M.A.H., Hamar D.W. Comparison of ethanol and 4-methylpyrazole as treatments for ethylene glycol intoxication in cats. Am J Vet Res. 1994;55:1771.
69 Dial S.M., Thrall M.A.H., Hamar D.W. Efficacy of 4-methylpyrazole for treatment of ethylene glycol intoxication in dogs. Am J Vet Res. 1994;55:1762.
70 DiBartola S.P., Leonard P.O. Renal tubular acidosis in a dog. J Am Vet Med Assoc. 1982;180:70.
71 DiBartola S.P., Rutgers H.C., Zack P.M., et al. Clinicopathologic findings associated with chronic renal disease in cats: 74 cases (1973–1984). J Am Vet Med Assoc. 1987;190:1196.
72 DiBartola S.P., Tarr M.J., Parker A.T., et al. Clinicopathologic findings in dogs with renal amyloidosis: 59 cases (1976–1986). J Am Vet Med Assoc. 1989;195:358.
73 Dinour D., Chang M.H., Satoh J., et al. A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J Biol Chem. 2004;279:52238-52246.
74 Di Tommaso M.G., Aste G., Rocconi F., et al. Evaluation of a portable meter to measure ketonemia and comparison with ketonuria for the diagnosis of canine diabetic ketoacidosis. J Vet Intern Med. 2009;23:466.
75 Dorhout-Mees E.J., Machado M., Slatopolsky E., et al. The functional adaptation of the diseased kidney. III. Ammonium excretion. J Clin Invest. 1966;45:289.
76 Dow S.W., Fettman M.J., Smith K.R., et al. Effects of dietary acidification and potassium depletion on acid-base balance, mineral metabolism and renal function in adult cats. J Nutr. 1990;120:569.
77 Drazner F.H. Distal renal tubular acidosis associated with chronic pyelonephritis in a cat. Calif Vet. 1980;34:15.
78 Duarte R., Simoes D.M.N., Franchini M.L., et al. Accuracy of serum β-hydroxybutyrate measurements for the diagnosis of diabetic ketoacidosis in 116 dogs. J Vet Intern Med. 2002;16:411-417.
79 Durocher L.L., Hinchcliff K.W., DiBartola S.P., et al. Acid-base and hormonal abnormalities in dogs with naturally-occurring diabetes mellitus. J Am Vet Med Assoc. 2008;232:1310.
80 Easley J.R., Breitschwerdt E.B. Glucosuria associated with renal tubular dysfunction in three basenji dogs. J Am Vet Med Assoc. 1976;168:938.
81 Escolar E., Perezalenza D., Diaz M., et al. Canine Fanconi syndrome. J Small Anim Pract. 1993;34:567.
82 Evans G.O. Plasma lactate measurements in healthy beagles. Am J Vet Res. 1987;48:131.
83 Feldman E.C., Nelson R.W. Canine and feline endocrinology and reproduction. Philadelphia: WB. 1996:400.
84 Feldman E.C., Nelson R.W. Canine and feline endocrinology and reproduction. Philadelphia: WB Saunders. 1996:411.
85 Finco D.R., Barsanti J.A., Brown S.A. Ammonium chloride as a urinary acidifier in cats: efficacy, safety, and rationale for its use. Mod Vet Pract. 1986;67:537.
86 Fine A., Brosnan J.T., Herzberg G.R. Release of lactate by the liver in metabolic acidosis in vivo. Metabolism. 1984;33:393.
87 Fleeman L.M., Thompson M.F., Foster S.F. Outbreak of acquired proximal renal tubulopathy (Fanconi-like syndrome) in Australian dogs. Perth, Australia: Australian Veterinary Association - WA Division Conference; 2009. September
88 Freeman L.M., Breitschwerdt E.B., Keene B.W., et al. Fanconi’s syndrome in a dog with primary hypoparathyroidism. J Vet Intern Med. 1994;8:349.
89 Gennari F.J., Goldstein M.B., Schwartz W.B. The nature of the renal adaptation to chronic hypocapnia. J Clin Invest. 1972;51:1722.
90 Gougoux A., Kaehny W.D., Cohen J.J. Renal adaptation to chronic hypocapnia: dietary constraints in achieving H+ retention. Am J Physiol. 1975;229:1330.
91 Graf H., Leach W., Arieff A.I. Effects of dichloroacetate in the treatment of hypoxic lactic acidosis in dogs. J Clin Invest. 1985;76:919.
92 Graf H., Leach W., Arieff A.I. Evidence for a detrimental effect of bicarbonate therapy in hypoxic lactic acidosis. Science. 1985;227:754.
93 Graf H., Leach W., Arieff A.I. Metabolic effects of sodium bicarbonate in hypoxic lactic acidosis in dogs. Am J Physiol. 1985;249:F630.
94 Graham T.E., Barclay J.K., Wilson B.A. Skeletal muscle lactate release and glycolytic intermediates during hypercapnia. J Appl Physiol. 1986;60:568.
95 Grauer G.F., Thrall M.A. Ethylene glycol (antifreeze) poisoning in the dog and cat. J Am Anim Hosp Assoc. 1982;18:492.
96 Grauer G.F., Thrall M.A. Ethylene glycol (antifreeze) poisoning. In: Kirk R.W., editor. Current veterinary therapy IX. Philadelphia: WB Saunders; 1986:206.
97 Grauer G.F., Thrall M.A., Henre B.A., et al. Early clinicopathologic findings in dogs ingesting ethylene glycol. J Am Vet Med Assoc. 1984;45:2299.
98 Harrington J.T. Metabolic alkalosis. Kidney Int. 1984;26:88.
99 Harrington J.T., Cohen J.J. Metabolic acidosis. In: Cohen J.J., Kassirer J.P., editors. Acid-base. Boston: Little, Brown; 1982:128.
100 Harrington J.T., Cohen J.J. Metabolic acidosis. In: Cohen J.J., Kassirer J.P., editors. Acid-base. Boston: Little, Brown; 1982:157.
101 Harrington J.T., Kassirer J.P. Metabolic alkalosis. In: Cohen J.J., Kassirer J.P., editors. Acid-base. Boston: Little, Brown; 1982:227.
102 Harrington J.T., Kassirer J.P. Metabolic alkalosis. In: Cohen J.J., Kassirer J.P., editors. Acid-base. Boston: Little, Brown; 1982:232.
103 Harrington J.T., Kassirer J.P. Metabolic alkalosis. In: Cohen J.J., Kassirer J.P., editors. Acid-base. Boston: Little, Brown; 1982:240.
104 Harrington J.T., Kassirer J.P. Metabolic alkalosis. In: Cohen J.J., Kassirer J.P., editors. Acid-base. Boston: Little, Brown; 1982:280.
105 Hartsfield S.M. Sodium bicarbonate and bicarbonate precursors for treatment of metabolic acidosis. J Am Vet Med Assoc. 1981;179:914.
106 Hartsfield S.M., Thurmon J.C., Corbin J.E., et al. Effects of sodium acetate, bicarbonate and lactate on acid-base status in anaesthetized dogs. J Vet Pharmacol Ther. 1981;4(1):51-61.
107 Haskins S.C., Munger R.J., Helphrey M.G., et al. Effect of acetazolamide on blood acid-base and electrolyte values in dogs. J Am Vet Med Assoc. 1981;179:914.
108 Heald R.D., Jones B.D., Schmidt D.A. Blood gas and electrolyte concentrations in canine parvoviral enteritis. J Am Anim Hosp Assoc. 1986;22:745.
109 Herrtage M.E., Houlton J.E. Collapsing clumber spaniels. Vet Rec. 1979;105:334.
110 Hetenyi G., Paradis H., Kucharczyk J. Glucose and lactate turnover and gluconeogenesis in chronic metabolic acidosis and alkalosis in normal and diabetic dogs. Can J Physiol Pharmacol. 1988;66:140.
111 Hill T.L., Breitschwerdt E.B., Cecere T., et al. Concurrent hepatic copper toxicosis and Fanconi’s syndrome in a dog. J Vet Intern Med. 2008;22:219.
112 Hindman B.J. Sodium bicarbonate in the treatment of subtypes of acute lactic acidosis: physiologic considerations. Anesthesiology. 1990;72:1064.
113 Honer W.G., Jennings D.B. PCO2 modulation of ventilation and HCO3− buffer during chronic metabolic acidosis. Respir Physiol. 1983;54:241.
114 Hornbein T.F., Pavlin E.G. Distribution of H+ and HCO3− between CSF and blood during respiratory alkalosis in dogs. Am J Physiol. 1975;228:1149.
115 Hostutler R.A., DiBartola S.P., Eaton K.A. Transient proximal renal tubular acidosis and Fanconi syndrome in a dog. J Am Vet Med Assoc. 2004;224:1611-1614.
116 Houlton J.E., Herrtage M.E. Mitochondrial myopathy in the Sussex spaniel. Vet Rec. 1980;106:206.
117 Hume D.Z., Drobatz K.J., Hess R.S. Outcome of dogs with diabetic ketoacidosis: 127 dogs (1993–2003). J Vet Intern Med. 2006;20:547.
118 Igarashi T., Sekine T., Inatomi J., et al. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol. 2002;13:2171-2177.
119 Ilkiw J.E., Davis P.E., Church D.B. Hematologic, biochemical, blood gas, and acid base values in greyhounds before and after exercise. Am J Vet Res. 1989;50:583.
120 Ilkiw J.E., Rose R.J., Martin I.C.A. A comparison of simultaneous collected arterial, mixed venous, jugular venous and cephalic venous blood samples in the assessment of blood gas and acid base status in dogs. J Vet Intern Med. 1991;5:294.
121 Jacobs R.M., Weiser M.G., Hall R.L., et al. Clinicopathologic findings of canine parvoviral enteritis. J Am Anim Hosp Assoc. 1980;16:809.
122 Jacobson H.R. Chloride-responsive metabolic alkalosis. In: Seldin D.W., Gebisch G., editors. The regulation of acid-base balance. New York: Raven Press; 1989:431.
123 Jacobson H.R. Chloride-resistant metabolic alkalosis. In: Seldin D.W., Gebisch G., editors. The regulation of acid-base balance. New York: Raven Press; 1989:459.
124 Jacobson H.R., Seldin D.W. On the generation, maintenance, and correction of metabolic alkalosis. Am J Physiol. 1983;245:F425.
125 Jaffe H.C., Bodansky A., Chandler J.P. Ammonium chloride decalcification as modified by calcium intake: the relationship between generalized osteoporosis and osteitis fibrosa. J Exp Med. 1932;56:823.
126 Jamieson P.M., Chandler M.L. Transient renal tubulopathy in a Labrador retriever. J Small Anim Pract. 2001;42:546.
127 Jarvinen A.K., Sankari S. Lactic acidosis in a clumber spaniel. Acta Vet Scand. 1996;37:119.
128 Jennings D.B., Davidson J.S.D. Acid-base and ventilatory adaptations in conscious dogs during chronic hypercapnia. Respir Physiol. 1984;58:377.
129 Kagan K.G., Schaer M. Gastric dilatation and volvulus in a dog-a case justifying electrolyte and acid-base assessment. J Am Vet Med Assoc. 1983;182:703.
130 Kassirer J.P., Berkman P.M., Lawrenz D.R., et al. The critical role of chloride in the correction of hypokalemic alkalosis in man. Am J Med. 1965;38:172.
131 Kassirer J.P., Schwartz W.B. Correction of metabolic alkalosis in man without repair of potassium deficiency: a reevaluation of the role of potassium. Am J Med. 1966;38:19.
132 Kassirer J.P., Schwartz W.B. The response of normal man to selective depletion of hydrochloric acid. Factors in the genesis of persistent gastric alkalosis. Am J Med. 1966;40:10.
133 Kazemi H., Valenca L.M., Shannon D.C. Brain and cerebrospinal fluid lactate concentration in respiratory acidosis and alkalosis. Respir Physiol. 1969;6:178.
134 Kramer J.W., Bistline D., Sheridan P., et al. Identification of hippuric acid crystals in the urine of ethylene glycol-intoxicated dogs and cats. J Am Vet Med Assoc. 1984;184:584.
135 Kreisberg R.A. Diabetic ketoacidosis: new concepts and trends in pathogenesis and treatment. Arch Intern Med. 1978;88:681.
136 Kreisberg R.A. Pathogenesis and management of lactic acidosis. Annu Rev Med. 1984;35:181.
137 Lemieux G., Lemieux C., Duplessis S., et al. Metabolic characteristics of cat kidney: failure to adapt to metabolic acidosis. Am J Physiol. 1990;259:R277.
138 Ling G.V., Lowenstine L.J., Pulley L.T., et al. Diabetes mellitus in dogs: a review of initial evaluation, immediate and long-term management, and outcome. J Am Vet Med Assoc. 1977;170:521.
139 Ling G.V., Stabenfeldt G.H., Comer K.M., et al. Canine hyperadrenocorticism: pretreatment clinical and laboratory evaluation of 117 cases. J Am Vet Med Assoc. 1979;174:1211.
140 Lloyd M.H., Iles R.A., Simpson B.R., et al. The effect of simulated metabolic acidosis on intracellular pH and lactate metabolism in the isolated perfused rat liver. Clin Sci. 1973;45:543.
141 Lowance D.C., Garfinkel H.B., Mattern W.D., et al. The effect of chronic hypotonic volume expansion on the renal regulation of acid-base equilibrium. J Clin Invest. 1972;51:2928.
142 Macintire D.K. Treatment of diabetic ketoacidosis in dogs by continuous low-dose intravenous infusion of insulin. J Am Vet Med Assoc. 1993;202:1266.
143 MacKenzie C.P., van den Broek A. The Fanconi syndrome in a whippet. J Small Anim Pract. 1982;23:469.
144 Madias N.E. Lactic acidosis. Kidney Int. 1986;29:752.
145 Madias N.E., Adrogue H.J., Cohen J.J. Maladaptive renal response to secondary hypercapnia in chronic metabolic alkalosis. Am J Physiol. 1980;238:F283.
146 Madias N.E., Bossert W.H., Adrogue H.J. Ventilatory response to chronic metabolic acidosis and alkalosis in the dog. J Appl Physiol. 1984;56:1640.
147 Madias N.E., Schwartz W.B., Cohen J.J. The maladaptive renal response to secondary hypocapnia during chronic HCl acidosis in the dog. J Clin Invest. 1977;60:1393.
148 Madias N.E., Wolf C.J., Cohen J.J. Regulation of acid-base equilibrium in chronic hypercapnia. Kidney Int. 1985;27:538.
149 Madias N.E., Zelman S.J. The renal response to chronic mineral acid feeding: a re-examination of the role of systemic pH. Kidney Int. 1986;29:667.
150 Magner P.O., Robinson L., Halperin R.M., et al. The plasma potassium concentration in metabolic acidosis: a re-evaluation. Am J Kidney Dis. 1988;11:220.
151 Marsh J.D., Margolis T.I., Kim D. Mechanism of diminished contractile response to catecholamines during acidosis. Am J Physiol. 1988;254:H20.
152 Martin G.B., Carden D.L., Nowak R.M., et al. Comparison of central venous and arterial pH and PCO2 during open chest CPR in the canine model. Ann Emerg Med. 1985;14:529.
153 Maskrey M., Jennings D.B. Ventilation and acid-base balance in awake dogs exposed to heat and CO2. J Appl Physiol. 1985;58:549.
154 Mathias D.W., Clifford P.S., Klopfenstein H.S. Mixed venous blood gases are superior to arterial blood gases in assessing acid-base status and oxygenation during acute cardiac tamponade in dogs. J Clin Invest. 1988;82:833.
155 McCullough S.M., Constable P.D. Calculation of the total plasma concentration of nonvolatile weak acids and the effective dissociation constant of nonvolatile buffers in plasma for use in the strong ion approach to acid-base balance in cats. Am J Vet Res. 2003;64:1047-1051.
156 McEwan N.A., Macartney L. Fanconi’s syndrome in a Yorkshire terrier. J Small Anim Pract. 1987;28:737.
157 McNamara P.D., Rea C.T., Bovee K.C., et al. Cystinuria in dogs: comparison of the cystinuric component of the Fanconi syndrome in basenji dogs to isolated cystinuria. Metabolism. 1989;38:8.
158 Meijer J.C. Canine hyperadrenocorticism. In: Kirk R.W., editor. Current veterinary therapy VII. Philadelphia: WB Saunders; 1980:975.
159 Melian C., Peterson M.E. Diagnosis and treatment of naturally-occurring hypoadrenocorticism in 42 dogs. J Small Anim Pract. 1996;37:268.
160 Meyer D.J. Temporary remission of hypoglycemia in a dog with an insulinoma after treatment with streptozotocin. Am J Vet Res. 1977;38:1201.
161 Mitchell J.H., Wildenthal K., Johnson R.L. The effects of acid-base disturbances on cardiovascular and pulmonary function. Kidney Int. 1972;1:375.
162 Molitoris B.A., Froment D.H., MacKenzie T.A., et al. Citrate: a major factor in the toxicity of orally administered aluminum compounds. Kidney Int. 1989;36:949-953.
163 Moon P.E., Gabor L., Gleed R.D., et al. Acid-base, metabolic, and hemodynamic effects of sodium bicarbonate or tromethamine administration in anesthetized dogs with experimentally induced metabolic acidosis. Am J Vet Res. 1997;58:771.
164 Morrin P.A.F., Bricker N.S., Kime S.W., et al. Observations on the acidifying capacity of the experimentally diseased kidney of the dog. J Clin Invest. 1962;41:1297.
165 Morrin P.A.F., Gedney W.B., Newmark L.N., et al. Bicarbonate reabsorption in the dog with experimental renal disease. J Clin Invest. 1962;41:1303.
166 Morris L.R., Murphy M.B., Kitabchi A.E. Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med. 1986;105:836.
167 Muir W.W. Acid-base and electrolyte disturbances in dogs with gastric-dilatation volvulus. J Am Vet Med Assoc. 1982;181:229.
168 Musch T.I., Friedman D.B., Haidet G.C., et al. Arterial blood gases and acid-base status of dogs during graded dynamic exercise. J Appl Physiol. 1986;61:1914.
169 Nappert G., Dunphy E., Ruben D., et al. Determination of serum organic acids in puppies with naturally-acquired parvoviral enteritis. Can J Vet Res. 2002;66:15-18.
170 Narins R.G., Cohen J.J. Bicarbonate therapy for organic acidosis: the case for its continued use. Ann Intern Med. 1987;106:615.
171 Narins R.G., Jones E.R., Stom M.C., et al. Diagnostic strategies in disorders of fluid, electrolyte and acid base homeostasis. Am J Med. 1982;72:496.
172 Needle M.A., Kaloyandies G.J., Schwartz W.B. The effects of selective depletion of hydrochloric acid on acid base and electrolyte equilibrium. J Clin Invest. 1964;43:1836.
173 Nicoletta J.A., Schwartz G.J. Distal renal tubular acidosis. Curr Opin Pediatr. 2004;16:194-198.
174 Norris S.H., Kurtzman N.A. Does chloride play an independent role in the pathogenesis of metabolic alkalosis? Semin Nephrol. 1988;8:101.
175 Nunamaker D.M., Medway W., Berg P. Treatment of ethylene glycol poisoning in the dog. J Am Vet Med Assoc. 1971;159:310.
176 Ogilvie G.K., Vail D.M., Wheeler S.L., et al. Effects of chemotherapy and remission on carbohydrate metabolism in dogs with lymphoma. Cancer. 1992;69:233-238.
177 Oh M.S. Irrelevance of bone buffering to acid-base homeostasis in chronic metabolic acidosis. Nephron. 1991;59:7.
178 Okuda Y., Adrogue H.J., Field J.B., et al. Counterproductive effects of sodium bicarbonate in diabetic ketoacidosis. J Clin Endocrinol Metab. 1996;81:314.
179 Olby N.J., Chan K.K., Targett M.P., et al. Suspected mitochondrial myopathy in a Jack Russell terrier. J Small Anim Pract. 1997;38:213.
180 Orchard C.H., Kentish J.C. Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol. 1990;258:C967.
181 Paciello O., Maiolino P., Fatone G., et al. Mitochondrial myopathy in a German shepherd dog. Vet Pathol. 2003;40:507-511.
182 Padrid P. Fanconi syndrome in a mixed breed dog. Mod Vet Pract. 1988;69:162.
183 Pang D.S., Boysen S. Lactate in veterinary critical care: pathophysiology and management. J Am Anim Hosp Assoc. 2007;43:270.
184 Parker R.A., Cohn L.A., Wohlstadter D.R., et al. D-lactic acidosis secondary to exocrine pancreatic insufficiency in a cat. J Vet Intern Med. 2005;19:106-110.
185 Pavlin E.G., Hornbein T.F. Distribution of H+ and HCO3− between CSF and blood during respiratory acidosis. Am J Physiol. 1975;228:1145.
186 Penman R.W., Luke R.F., Jarboe T.M. Respiratory effects of hypochloremic alkalosis and potassium depletion in the dog. J Appl Physiol. 1972;33:170.
187 Penumarthy L., Oehme F.W. Treatment of ethylene glycol toxicosis in cats. Am J Vet Res. 1974;36:209.
188 Peterson M. Hyperadrenocorticism. Vet Clin North Am. 1984;14:731.
189 Peterson M.E., Greco D.S., Orth D.N. Primary hypoadrenocorticism in ten cats. J Vet Intern Med. 1989;3:55.
190 Peterson M.E., Kintzer P.P., Kass P.H. Pretreatment clinical and laboratory findings in dogs with hypoadrenocorticism-225 cases (1979–1993). J Am Vet Med Assoc. 1996;208:85.
191 Polzin D.J., Stevens J.B., Osborne C.A. Clinical evaluation of the anion gap in evaluation of acid-base disorders in dogs. Compend Contin Educ Pract Vet. 1982;4:102.
192 Posner J., Plum F. Spinal fluid pH and neurologic symptoms in systemic acidosis. N Engl J Med. 1967;277:605.
193 Rhee K.H., Toro L.O., McDonald G.G., et al. Carbicarb, sodium bicarbonate, and sodium chloride in hypoxic lactic acidosis. Chest. 1993;104:913.
194 Robinson E.P., Hardy R.M. Clinical signs, diagnosis, and treatment of alkalemia in dogs: 20 cases (1982–1984). J Am Vet Med Assoc. 1988;192:943.
195 Rose B.D. Clinical physiology of acid-base and electrolyte disorders. New York: McGraw-Hill. 1994:542.
196 Rose B.D. Clinical physiology of acid-base and electrolyte disorders. New York: McGraw-Hill. 1994:588.
197 Rose B.D. Clinical physiology of acid-base and electrolyte disorders. New York: McGraw-Hill. 1994:564.
198 Rose B.D. Clinical physiology of acid-base and electrolyte disorders. New York: McGraw-Hill. 1994:591.
199 Rose B.D. Clinical physiology of acid-base and electrolyte disorders. New York: McGraw-Hill. 1994:589.
200 Rose B.D. Clinical physiology of acid-base and electrolyte disorders. New York: McGraw-Hill. 1994:515.
201 Rose R.J., Carter J. Some physiological and biochemical effects of acetazolamide in the dog. J Vet Pharmacol Ther. 1979;2:215.
202 Sabatini S. The acidosis of chronic renal failure. Med Clin North Am. 1983;67:845.
203 Sabatini S., Kurtzman N.A. The maintenance of metabolic alkalosis: factors which decrease bicarbonate excretion. Kidney Int. 1984;25:357.
204 Sanders A.B., Otto C.W., Kern K.B., et al. Acid-base balance in a canine model of cardiac arrest. Ann Emerg Med. 1988;17:667.
205 Sanyer J.L., Oehme F.W., McGavin M.D. Systematic treatment of ethylene glycol toxicosis in dogs. Am J Vet Res. 1973;34:527.
206 Schmidt R.W., Bricker N.S., Gavellas G. Renal bicarbonate reabsorption in experimental uremia in the dog. Kidney Int. 1976;10:287.
207 Schmidt R.W., Gavellas G. Bicarbonate reabsorption in dogs with experimental renal disease: effects of proportional reduction of sodium or phosphate intake. Kidney Int. 1977;12:393.
208 Schober K.E. Investigation into intraerythrocytic and extraerythrocytic acid-base and electrolyte changes after long-term ammonium chloride administration in dogs. Am J Vet Res. 1996;57:743.
209 Schwartz W.B., Hays R.M., Pak A., et al. Effects of chronic hypercapnia on electrolyte and acid-base equilibrium. II. Recovery, with special reference to the influence of chloride intake. J Clin Invest. 1961;40:1238.
210 Schwartz W.B., Orning K.J., Porter R. The internal distribution of hydrogen ions with varying degrees of metabolic acidosis. J Clin Invest. 1957;36:373.
211 Senior D.F., Sundstrom D.A., Wolfson B.B. Effectiveness of ammonium chloride as a urinary acidifier in cats fed a popular brand of canned cat food. Feline Pract. 1986;16:24.
212 Senior D.F., Sundstrom D.A., Wolfson B.B. Testing the effects of ammonium chloride and DL-methionine on the urinary pH of cats. Vet Med. 1986;81:88.
213 Settles E.L., Schmidt D. Fanconi syndrome in a Labrador retriever. J Vet Intern Med. 1994;8:390.
214 Shaw D.H. Acute response of urine pH following ammonium chloride administration to dogs. Am J Vet Res. 1989;50:1829.
215 Shearer L.R., Boudreau A.E., Holowaychuk M.K. Distal renal tubular acidosis and immune-mediated hemolytic anemia in 3 dogs. J Vet Intern Med. 2009;23:1284.
216 Sheikh A., Fleisher G., Delgado-Paredes C., et al. Effect of dichloroacetate in the treatment of anoxic lactic acidosis in dogs. Crit Care Med. 1986;14:970.
217 Shull R.M. The value of anion gap and osmolal gap determinations in veterinary medicine. Vet Clin Pathol. 1978;7:12.
218 Silva P.R.M., Fonseca-Costa A., Zin W.A., et al. Respiratory and acid-base parameters during salicylic intoxication in dogs. Braz J Med Biol Res. 1986;19:279.
219 Simpson D.P. Control of hydrogen ion homeostasis and renal acidosis. Medicine (Baltimore). 1971;50:503.
220 Stacpoole P.W. Lactic acidosis: the case against bicarbonate therapy. Ann Intern Med. 1986;105:276.
221 Straus E., Johnson G.F., Yalow R.S. Canine Zollinger-Ellison syndrome. Gastroenterology. 1977;72:380.
222 Swan R.C., Axelrod D.R., Seip M., et al. Distribution of sodium bicarbonate infused into nephrectomized dogs. J Clin Invest. 1955;34:1795.
223 Swan R.C., Pitts R.F. Neutralization of infused acid by nephrectomized dogs. J Clin Invest. 1955;34:205.
224 Takano N. Blood lactate accumulation and its causative factors during passive hyperventilation in dogs. Jpn J Physiol. 1966;16:481.
225 Tashkin D.P., Goldstein P.J., Simmons D.H. Hepatic lactate uptake during decreased liver perfusion. Am J Physiol. 1972;223:968.
226 Thrall M.A., Grauer G.F., Mero K.N. Clinicopathologic findings in dogs and cats with ethylene glycol intoxication. J Am Vet Med Assoc. 1984;184:37.
227 Thrall M.A., Dial S.M., Winder D.R. Identification of calcium oxalate monohydrate crystals by X-ray diffraction in urine of ethylene glycol-intoxicated dogs. Vet Pathol. 1985;22:625.
228 Torrente C., Silvestrini P., Ruiz de Gopegui R. Severe life-threatening hypokalemia in a cat with suspected distal renal tubular acidosis. J Vet Emerg Crit Care. 2010;20:250.
229 Toto R.D., Alpern R.J. Metabolic acid-base disorders. In: Kokko J.P., Tannen R.L., editors. Fluids and electrolytes. Philadelphia: WB Saunders; 1996:201.
230 Vail D.M., Ogilvie G.K., Fettman M.J., et al. Exacerbation of hyperlactatemia by infusion of lactated Ringer’s solution in dogs with lymphoma. J Vet Intern Med. 1990;4:228.
231 Vail D.M., Ogilvie G.K., Wheeler S.L., et al. Alterations in carbohydrate metabolism in canine lymphoma. J Vet Intern Med. 1990;4:8.
232 Valtin H., Gennari F.J. Acid-base disorders: basic concepts and management. Boston: Little, Brown; 1987.
233 Vijayasarathy C., Giger U., Prociuk U., et al. Canine mitochondrial myopathy associated with reduced mitochondrial messenger RNA and altered cytochrome C oxidase activities in fibroblasts and skeletal muscle. Comp Biochem Physiol. 1994;109:887.
234 Vukmir R.B., Bircher N.G., Radovsky A., et al. Sodium bicarbonate may improve outcome in dogs with brief or prolonged cardiac arrest. Crit Care Med. 1995;23:515.
235 Warnock D.G. Uremic acidosis. Kidney Int. 1988;34:278.
236 Watson A.D.J., Culvenor J.A., Middleton D.J., et al. Distal renal tubular acidosis in a cat with pyelonephritis. Vet Rec. 1986;119:65.
237 Weil M.H., Rackow E.C., Trevino R., et al. Difference in acid-base state between venous and arterial blood during cardiopulmonary resuscitation. N Engl J Med. 1986;315:153.
238 Welbourne T., Weber M., Bank N. The effect of glutamine administration on urinary ammonium excretion in normal subjects and patients with renal disease. J Clin Invest. 1972;51:1852.
239 Widmer B., Gerhard R.E., Harrington J.T., et al. Serum electrolyte and acid-base composition: the influence of graded degrees of chronic renal failure. Arch Intern Med. 1979;139:1099.
240 Wingfield W.E., Twedt D.C., Moore R.W., et al. Acid-base and electrolyte values in dogs with acute gastric dilatation-volvulus. J Am Vet Med Assoc. 1982;180:1070.
241 Yas-Natan E., Segev G., Aroch I. Clinical, neurological and clinicopathological signs, treatment and outcome of metaldehyde intoxication in 18 dogs. J Small Anim Pract. 2007;48:438.
242 Zeugswetter F., Pagiz M. Ketone measurements using dipstick methodology in cats with diabetes mellitus. J Small Anim Pract. 2009;50:4.
243 Zeukswetter F., Handl S., Iben C., et al. Efficacy of plasma beta-hydroxybutyrate concentration as a marker for diabetes mellitus in acutely sick cats. J Feline Med Surg. 2010;12:300.