Chapter 23 Shock Syndromes
Shock is perhaps one of the most common clinical conditions that veterinarians treat. It is also one of the least well understood. Although often thought of as a disease of the cardiovascular system, shock takes place within cells and results from inadequate production of intracellular energy. This most commonly occurs because of inadequate delivery of oxygen and nutrients to tissues by an impaired cardiovascular system. It may also occur when intracellular metabolic derangements prevent appropriate cellular energy production (e.g., cyanide preventing mitochondrial energy production). Shock represents the final common pathway to death in many critical care patients in veterinary and human medicine. Veterinary patients can be presented with shock, develop shock during the diagnosis and treatment of a wide variety of medical and surgical diseases, or develop shock during the perioperative period. All of the shock syndromes in veterinary medicine can result in high morbidity and mortality if not recognized and treated immediately. Advances in our knowledge of all aspects of shock and shock syndromes and innovative noninvasive and invasive monitoring techniques have resulted in the ability to anticipate, recognize, and treat shock syndromes more effectively. Although we have learned a great deal about the pathophysiology of this devastating condition, much work remains to be completed. Clinical trials in veterinary medicine are badly needed to evaluate novel therapies for veterinary patients, and potentially document the utility of promising therapies that might then be used to treat humans.
Definition of shock
A true understanding of shock syndromes must begin with the definition of shock. Shock is not defined by tachycardia, hypotension, circulatory collapse, stupor, coma, pale mucous membranes, or dehydration. These clinical signs may be associated with shock and are easily recognized, but they are common to many other conditions. The underlying problem or inciting event for all causes of shock is a relative insufficiency of intracellular energy production. Most often this is caused by a decrease in effective blood flow and oxygen delivery to tissues that results in failure to meet the demands of the tissues. Stated differently, this form of shock is “the state in which profound and widespread reduction of effective tissue perfusion leads first to reversible and then, if prolonged, to irreversible cellular injury.77” The decrease in effective perfusion can occur by many mechanisms, either cardiac or vascular in nature.
In some cases, the inadequate cellular energy production cannot be attributed to dysfunction of the cardiovascular system, but rather to defects in oxygen loading or unloading from the red blood cell, or to defects in gas exchange in the lungs. Alternatively, mitochondrial dysfunction or intracellular metabolic problems can also be the source of inadequate cellular energy production. Regardless of the cause, inadequate production of intracellular energy initiates a complex series of events that can result in altered cellular metabolism, cellular death, organ failure, or ultimately the death of the animal.
Classification of shock syndromes
Historically, shock has been classified into various categories and causes to assist in understanding this complex disorder. Numerous classification schemes have been presented to assist in understanding the clinical syndromes of shock. Many classification schemes are aimed at simplifying a complex disorder of the cardiovascular system into isolated components either on an anatomic or functional basis.
In this chapter classification of shock syndromes will be divided into circulatory causes, hypoxemic causes, and metabolic causes. Circulatory causes can be further divided into hypovolemic causes, cardiogenic causes, and distributive causes. Table 23-1 lists examples of each classification.

Hypovolemic shock can occur by loss of intravascular volume of any etiology, including dehydration, blood loss, and third-space loss of fluids. Cardiogenic shock can occur as a result of any cardiac abnormality that causes pump failure such as heart disease, myocardial injury, cardiac tamponade, or arrhythmias. Distributive shock occurs when cardiac function and blood volume are not affected, but there is failure of the vascular tree to allow appropriate delivery (either globally or locally) and can occur through loss of neurohormonal input (e.g., sympathetic trunk transection, relative adrenal insufficiency, catecholamine imbalance), inflammatory mediator release (e.g., sepsis, endotoxemia, and anaphylaxis), or interference with blood delivery (e.g., thromboembolism, caval syndrome, gastric dilatation-volvulus (GDV) syndrome, acute portal hypertension).
Hypoxic causes of shock all have a decrease in the oxygen content of the blood in common. Anemia, as well as alterations in hemoglobin form or function (e.g., carbon monoxide toxicity, methemoglobinemia), can all cause hypoxic shock. Failure of gas exchange in the lungs can also be a significant cause of hypoxemic shock.
Metabolic causes of shock are linked together through some failure of intracellular energy production despite normal oxygen delivery to the cell. Hypoglycemia is perhaps the most common cause of this form of shock, but toxins such as cyanide could also be a cause. In addition, cytokines may play a role in mitochondrial dysfunction in patients with sepsis and contribute some part in failure of adequate intracellular energy production.
Although such categorical organization of shock syndromes is helpful to understand the complex nature of shock, it is crucial to recognize that most clinical forms of shock may encompass several classifications. The utility of anatomic or functional categories of shock is questionable when viewed from a clinical perspective, and some have argued that such a classification is misleading because clinicians may approach a one-dimensional, easy-to-understand representation of shock with a simplistic one-dimensional approach to therapy.130 Unfortunately, shock is complex; it does not begin with a common pathophysiologic event and does not necessarily end with survival after a simple universal initial treatment. Each cause of shock sets in motion a complex series of events that include neural and hormonal responses, as well as numerous inflammatory cascades.
To confuse matters further, there are several named shock syndromes that are commonly described in the veterinary literature that include an etiologic descriptor. One example is anaphylactic shock, which is a form of shock that is triggered by exposure to an offending allergen that triggers significant mast cell degranulation. Release of numerous chemokines, especially histamine, causes widespread vasodilatation that could be classified within the distributive framework discussed above. In addition, histamine causes leak of protein and fluid from within the intravascular space, which, if of sufficient magnitude would be considered hypovolemic shock. Numerous other clinical designations or descriptors of shock exist and each have multiple potential pathophysiologic classifications that might contribute to inadequate cellular energy production. Additional examples include: septic shock (can include hypoxic, hypovolemic, cardiogenic, distributive, and metabolic components) or neurogenic shock (can include distributive, cardiogenic, or metabolic components).
Perhaps the most useful feature of these schemes is to force the clinician to consider potential dysfunction of multiple components of the cardiovascular and intracellular energy production systems and thus consider therapies that support each component of dysfunction.
Additional disagreement also centers on the use of the term distributive shock. The term distributive has been used to describe various types of high-flow shock under the assumption that blood flow is not normally distributed to tissue beds. Used in this context, “distributive shock” is a theoretical designation of a type of shock syndrome that is not defined by criteria that can be easily measured in a clinical setting. Maldistribution of or heterogeneous blood flow has been documented in people in clinically accessible microscopic vascular beds (e.g., mucous membranes, sclera, liver, nail bed). Direct observations have documented the phenomenon of maldistribution of blood flow, but such measurements are not necessarily representative of all areas and are not quantitative measures of the extent of maldistribution throughout the body. Therefore maldistribution of blood flow is a physiologic concept that may be relevant to all shock states.130
The various classifications of shock syndromes in veterinary and human medicine have been created in an attempt to simplify a complex series of physiologic events. Unfortunately, laboratory research and clinical experience have not supported any one classification of the shock syndromes as being the easiest to understand or teach. The specific details surrounding the presentation of the patient with shock should rank as most important, and valuable time should not be wasted deciding which classification scheme best describes the patient.
Pathophysiology
Adequate tissue perfusion is not dependent on one simple value, rather it requires the integration of the entire cardiovascular system and is best described by effective circulating volume. Effective circulating volume (ECV) is a difficult to define term that describes the “fullness” of blood vessels. In essence adequate ECV requires an adequate blood volume delivered at an adequate pressure.11 Although tissues require the maintenance of ECV for long-term function, the cardiovascular system considers the maintenance of normal mean arterial pressure (MAP) as its number one priority.55 This is primarily because perfusion of the heart and the brain is pressure dependent; these vital organs need a minimum MAP for adequate perfusion. In terms of physics, maintaining a constant pressure is far more feasible for the cardiovascular system than maintaining a constant flow or volume.11Figure 23-1 outlines the interrelationship of the major cardiovascular parameters and how they influence MAP.
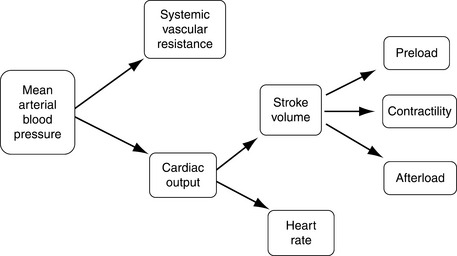
Figure 23-1 Cardiovascular parameters controlling mean arterial blood pressure. Mean arterial blood pressure is the product of cardiac output and systemic vascular resistance. Cardiac output is further dependent on both heart rate and stroke volume.
There are three main mechanisms by which decreases in ECV can occur (Table 23-2).61
1. Hypovolemia may lead to inadequate blood volume and hence inadequate preload, causing a decrease in cardiac output.
2. Cardiogenic causes in which abnormalities in cardiac function lead to inadequate cardiac output despite a normal or increased blood volume.
3. Distributive causes in which blood volume and cardiac function are normal but there are alterations in systemic vascular resistance globally or locally leading to inadequate ECV.

The cardiovascular system has several compensatory responses to defend a fall in ECV. An understanding of these responses can explain the clinical presentation of circulatory shock and the approach to therapy. Hemorrhagic shock, a form of hypovolemic shock has been modeled extensively in experimental animals and provides an ideal example for this discussion.
Hemorrhagic shock
Baroreceptor-Mediated Responses
When ongoing hemorrhage occurs in the conscious dog there is an initial normotensive period (moderate hemorrhage) followed by hypotension (severe hemorrhage).53,124 During moderate hemorrhage, the decrease in cardiac output is sensed by the high pressure baroreceptors of the carotid bodies and aortic arch. The subsequent decrease in baroreceptor afferent traffic to the vasomotor center of the brain causes sympathetic tone to increase, parasympathetic tone to decrease, and vasopressin release. This results in an increase in heart rate and systemic vascular resistance. Sympathetic mediated vasoconstriction is more prominent in precapillary arterioles and the blood vessels of the skin, skeletal muscles, and splanchnic viscera are vasoconstricted to a greater degree than the rest of the body. This is an effort to centralize blood volume and maximize perfusion of vital organs during the acute insult. Tachycardia and increases in systemic vascular resistance will contribute to the maintenance of a normal MAP in the face of volume loss (see Figure 23-1).41,53,124 This phase is also known as compensatory shock. There does appear to be some species variability in the nature of the hemodynamic responses to circulatory shock. In the dog it has been reported that there is little change in left ventricular contractility in association with the increase in sympathetic tone.60 In addition plasma epinephrine is not elevated during this normotensive hemorrhage in dogs.39
The renin-angiotensin system (RAS) is also stimulated by hemorrhage and the associated decrease in cardiac output. Renin release will be activated by the increase in sympathetic tone occurring as part of the baroreceptor response. In addition the afferent arteriole of the kidney is itself a baroreceptor and may sense the decrease in ECV directly. Plasma renin cleaves the plasma protein angiotensinogen, generating angiotensin I. Angiotensin I is rapidly converted to angiotensin II by angiotensin converting enzyme, which is present in the endothelium of the lung. Angiotensin II is one of the most potent vasoconstrictors of the body contributing to increases in systemic vascular resistance. Angiotensin II also stimulates the release of aldosterone. Both angiotensin II and aldosterone stimulate renal sodium retention in an effort to augment blood volume (see Chapter 3).11,55 As mentioned above, carotid sinus baroreceptor off-loading also stimulates the release of vasopressin (antidiuretic hormone) from the hypothalamus, which causes vasoconstriction and renal water conservation. Baroreceptor responses mediate changes on a minute-to-minute basis and are vital to surviving an acute injury or insult. The RAS responses take 10 minutes to an hour to have benefit and are more important in attempting to return the system back to the preinjury state and recovery.53
The pathophysiology above explains the six classic clinical signs of hypovolemic shock.
1. Decreased mentation due to inadequate perfusion of the brain.
2. Pale mucous membranes as a result of arteriolar constriction and decreased blood volume in the capillary beds.
3. Prolonged capillary refill time because the vasoconstricted arterioles delay the return of blood to the capillaries of the mucous membranes.
4. Tachycardia as part of the sympathetic, compensatory response.
5. Poor pulse quality due to vasoconstriction and decreased stroke volume.
6. Decreased extremity temperature compared with the core body temperature as a result of peripheral vasoconstriction.
Moderate, normotensive hemorrhage in experimental dogs occurs with the loss of less than 30% of blood volume. As the degree of hemorrhage increases there is a precipitous decrease in MAP and heart rate. In experimental dog studies, this usually occurs when following the removal or loss 30% of blood volume. This hypotensive phase is associated with central inhibition of sympathetic outflow and the animals cannot be salvaged despite administration of the shed blood and additional volume support.41,124 This is also known as decompensated or irreversible shock. Although decompensated shock is marked by bradycardia, the hypotension is independent of the bradycardia because it does not improve with atropine-induced tachycardia. The activity of the RAS continues to increase during hypotensive hemorrhage. In some species, including cats, rabbits, and rats, there is evidence that the sympathoinhibition seen with severe hemorrhage is stimulated by cardiac or cardiopulmonary receptors and transmitted via the vagus nerve.41 In contrast there is no apparent role of cardiopulmonary receptors in the generation of sympathoinhibition in dogs, the cause of the failure of sympathetic-mediated vasoconstriction in uncompensated shock in dogs is currently unknown.129 Additionally, there may be a relative vasopressin deficiency following prolonged shock states, further exacerbating the hypotension.40,78 Cardiac output and arterial blood pressure fall to zero with loss of 35% to 45% of total blood volume and is rapidly fatal.53
The clinical hallmark of decompensated shock is bradycardia and for this reason bradycardia (not due to conduction disturbances) may be a poor prognostic indicator in canine patients presenting in circulatory shock. In contrast bradycardia (or inappropriate normocardia) is not uncommon in cats having circulatory shock and does not appear to carry any prognostic significance in this species. The mechanism of bradycardia in hemodynamically unstable cats is unknown. It can occur in mild to moderate cases of shock and the heart rate generally increases as the animal is resuscitated.13,122 Hypothermia is recognized to cause bradycardia and is common in feline patients with circulatory shock, raising the possibility that body temperature could play a role in this phenomenon.107
Chemoreceptors
The chemoreceptors are specialized cells that sense decreases in oxygen, increases in carbon dioxide, and increases in hydrogen ion concentration. The peripheral chemoreceptors are found in the carotid body and aortic arch adjacent to the baroreceptors. When ECV decreases to a critical level, these cells are stimulated by the lack of oxygen and the accumulation of carbon dioxide and hydrogen ions. They transmit signals to the central vasomotor center in a manner similar to the baroreceptors, causing increases in sympathetic tone and vasopressin levels. They also stimulate the respiratory center, leading to increases in alveolar ventilation that maybe evident as tachypnea in the patient with circulatory shock. These receptors contribute to the maintenance of blood pressure in the face of severe decreases in ECV and are not thought to contribute to the regulation of MAP in the face of mild to moderate insults.11
Starling Forces
Hemorrhagic shock will also lead to alterations in Starlings forces. Following acute hemorrhage there is a sudden drop in capillary hydrostatic pressure that promotes the movement of fluid from the interstitium to the intravascular space. Interstitial fluid can replace up to 75% of the shed blood volume.10 This process is known as transcapillary refill or autotransfusion. As the protein concentration (and red blood cell concentration) of interstitial fluid is lower than that of blood, this response causes a decrease in both hematocrit and total plasma protein. In dogs and cats splenic contraction can supplement hematocrit and the end result may be a proportionally greater drop in the total protein than that of the packed cell volume following hemorrhage.22
Hypovolemic Shock
Hypovolemic shock can occur without hemorrhage; for, example severe dehydration or third-space losses can cause significant decreases in blood volume. Increased vascular permeability as may occur with severe systemic inflammation or anaphylaxis can also cause significant blood volume loss. The responses to hemorrhagic shock, as described previously, are equally applicable to other causes of hypovolemia.
Cardiogenic Shock
Cardiogenic shock causes decreases in effective circulating volume despite a normal, or frequently increased blood volume (see Table 23-2). A decrease in cardiac contractility or diastolic filling will impair stroke volume. From Figure 23-1 it can be appreciated that the compensatory responses to this abnormality will be similar to those seen in hypovolemic shock, with increases in systemic vascular resistance and tachycardia. Hence the clinical signs of cardiogenic shock will be similar to those of hypovolemic shock, namely, pale mucous membranes, prolonged capillary refill time, poor pulse quality, and differences between extremity and core temperature. Causes of cardiogenic shock include myocardial failure as may occur with dilated cardiomyopathy or end stage valvular regurgitation. Cardiogenic shock secondary to decreased diastolic filling can occur with tachyarrhythmias, hypertrophic cardiomyopathy, or pericardial tamponade.147 Patients presenting in cardiogenic shock may or may not have concurrent congestive heart failure typified by pulmonary edema, pleural effusion, or ascites. For example end-stage valvular regurgitation cases are likely to have significant morbidity associated with their congestive heart failure and are now demonstrating evidence of poor perfusion. In contrast patients with malignant tachyarrhythmias may have no evidence of congestive heart failure at the time of presentation for cardiogenic shock.
Distributive Shock
When regulation of vasomotor tone is abnormal it can cause circulatory shock despite an adequate blood volume and normal cardiac function (see Table 23-2). Global decreases in arteriolar tone will cause decreases in ECV that is sensed by the baroreceptors, as described for hemorrhagic shock. But in this scenario there is a failure of compensatory vasoconstriction in response to increases in sympathetic tone and tachycardia is the sole compensatory response. Global vasodilatation is marked by hypotension that is unresponsive to fluid administration. On physical examination these patients can have red mucous membranes, rapid capillary refill times, tachycardia, bounding pulses, and warm extremity temperatures reflecting vasodilatation.61 Some causes of distributive shock such as sepsis can cause heterogenous changes in microcirculatory vasomotor tone leading to inadequate perfusion of some tissue beds while there is normal or possibly excessive perfusion of others. If microcirculatory abnormalities occur in the absence of generalized vasodilatation the patient may have normal global hemodynamic parameters while some tissue beds are suffering from hypoperfusion and inadequate cellular energy production (also known as cryptic shock).38 This is another form of distributive shock.
Pathologic consequences of shock
Shock of any cause will result in a common pathway of cell injury and tissue damage. The pathogenesis of circulatory shock includes cellular hypoxia, inflammatory mediator generation, and free radical mediated damage.
Cellular Hypoxia
When tissue oxygen supply is inadequate, either due to global decreases in blood flow or maldistribution of blood flow, oxidative metabolism is compromised and cellular function becomes dependent on anaerobic energy production. Glycolysis, the only source of cellular energy in an anaerobic environment, is extremely inefficient with only 2 mol of adenosine triphosphate (ATP) being produced from each mole of glucose. This represents approximately 3% of the potential energy in the glucose molecules. For a short period of time this limited anaerobic energy production may prevent cell injury and death. When 1 mol of glucose is metabolized by glycolysis it produces 2 mol of pyruvate, in anaerobic conditions most of the pyruvate is then converted to lactate.54 This conversion allows glycolysis to continue as it regenerates essential NAD+ and prevents the accumulation of pyruvate. Lactate production during anaerobic metabolism occurs in conjunction with an equimolar production of hydrogen ions as a consequence of concurrent hydrolysis of ATP (Figure 23-2).120 The result is the formation of lactic acid. Given its inefficiency, anaerobic metabolism is limited in its ability to maintain normal function. In situations of acute, absolute cellular hypoxia, such as asphyxiation, anaerobic metabolism can only support life for approximately 1 minute.
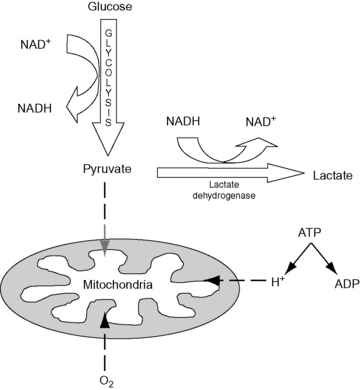
Figure 23-2 Anaerobic metabolism. Glycolysis occurs in the cytoplasm and results in the production of pyruvate, which under normal circumstances enters the mitochondria for further metabolism. In the absence of oxygen, pyruvate is converted to lactate. During anaerobic metabolism there is a concurrent accumulation of hydrogen ions from the hydrolysis of ATP; these combine with lactate to produce lactic acid.
When inadequate cellular energy metabolism occurs, cell function is compromised. Maintenance of ionic gradients across the cell membrane requires active transport systems that consume 20% to 80% of all cellular energy produced.98 Some organs are more susceptible to hypoxic injury than others. The brain and the heart are obligate, aerobic, energy-dependent organs. Neurons use the majority of cellular energy in the preservation of ionic gradients and membrane potentials while myocardial cells have a high-energy requirement to fuel contractile processes. In the face of inadequate cellular energy production, the active transport systems controlling cell volume, such as the Na-K-ATPase pump, fail. As a consequence the entry of ions such as sodium and calcium into the cell is favored. In response to the increasing intracellular osmolarity, water shifts into cells leading to cell swelling and can ultimately result in cell death.17
Increases in intracellular calcium trigger activation of calcium-dependent phospholipases and proteases that can cause cellular injury. This includes calpainlike proteases that convert xanthine dehydrogenase to xanthine oxidase.134 Without sufficient levels of xanthine dehydrogenase, intracellular hypoxanthine accumulates. These changes have important repercussions during the reperfusion period.
Free Radical Damage
Reperfusion of organs following a period of ischemia, although essential for survival, can also be a mechanism of tissue damage. When oxygen is reintroduced to cells, it is used by xanthine oxidase (which accumulates during the ischemic period) to convert hypoxanthine (also accumulated during the ischemic period) to reactive oxygen species, such as the superoxide anion and hydrogen peroxide. These products will cause direct cell injury by damaging proteins and DNA and causing lipid peroxidation. Both rises in intracellular calcium concentration and plasma membrane damage can trigger activation of phospholipase A2, leading to arachidonic acid formation and eicosanoid synthesis including thromboxane A2 and leukotrienes.148 These arachidonic acid products have many functions including pro-inflammatory, procoagulant, and vasoactive effects.
Reactive oxygen species in turn have been shown to activate the nuclear transcription factor κB (NF-κB), which causes transcription of proinflammatory mediators, including more reactive oxygen species, leukocyte adhesion molecules, and tumor necrosis factor-α.94 These processes have also been shown to damage mitochondria such that cellular energy production may remain impaired, despite adequate oxygen delivery.16,47 This abnormality has been coined “cytopathic hypoxia” and is currently considered to be a contributor to the development of multiple organ dysfunction syndrome (MODS) in various disease states.83
Inflammatory Mediators
The immune response following an insult such as hypoxic tissue injury maybe biphasic in nature with an initial proinflammatory response (systemic inflammatory response syndrome [SIRS]) mediated primarily by the innate immune system and proinflammatory cytokines. This can be followed by a compensatory antiinflammatory response syndrome (CARS), which is associated with immune suppression and the production of antiinflammatory cytokines. These immune responses have local and systemic effects that if excessive can result in MODS and death.
Cellular ischemia alters gene transcription including activation of NF-κB, which leads to cellular production of inflammatory mediators including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-2, IL-6, interferon-γ, nitric oxide synthase, and cellular adhesion molecules.10,28,94 If this inflammatory response is substantial enough it will spill over into the systemic circulation (SIRS). When regulated, these responses are important to the maintenance of an effective immune response and tissue repair. An imbalanced or overzealous inflammatory response may result in global increases in capillary permeability, vasodilatation, leukocyte activation and adhesion, procoagulant changes, and mitochondrial dysfunction.47,79,82,88 Clinically this can result in the development of both hypovolemic and distributive shock. These mechanisms are potential contributors to acquired organ dysfunction following illness or injury.88
In addition to an exuberant proinflammatory responses, an antiinflammatory response (CARS) can also occur, which has been associated with immunocompromise and in some studies it correlates to a higher mortality.151 The development of CARS is associated with increased production of antiinflammatory cytokines, such as IL-10, IL-1 receptor antagonist, and transforming growth factor-β. Leukocyte inhibition and down regulation of NF-κB have also been demonstrated.1,2 There is some discussion in the current literature that CARS occurs concomitantly with SIRS as a normal response that limits the systemic inflammatory process.2 Although there is evidence that CARS maybe associated with an enhanced susceptibility to infection, this relationship is complicated and in many respects CARS may be a protective response. Following trauma, the immune system is activated and a second immune stimulus during this period may augment the initial response (second hit phenomenon) and potentially worsen the outcome.102,150 For this reason there may be a benefit in delaying major surgery, such as fracture repair, for at least 4 days following injury.
Inadequate perfusion of the gastrointestinal tract is common in circulatory shock because sympathetic responses tend to shunt blood flow toward vital organs and cause disproportionate splanchnic hypoperfusion.11 Gastrointestinal tract hypoperfusion has been shown to be an important contributor to posttrauma multiple organ dysfunction syndrome in experimental animals and people.99,143 Ischemia and reperfusion of the gastrointestinal tract can lead to production of inflammatory mediators and activation of neutrophils. This can amplify the inflammatory response occurring following cellular ischemia subsequent to shock and lead to severe SIRS and MODS. Bacterial translocation and subsequent bacteremia can also occur following a severe circulatory shock insult and further drives the development of severe SIRS.27 In summary the immune response to injury or illness is complex and yet to be fully understood. The cellular changes that occur following an insult have a protective role but in some circumstances they can become a source of harm to the patient. As clinicians the corner stones of therapy of systemic inflammatory responses are maximizing oxygen delivery to the cells and minimizing further systemic insults. Currently there are no recommended specific therapeutic agents to modulate the immune response.
Sequelae of circulatory shock
Circulatory shock may be rapidly fatal if it leads to significant hypoperfusion of the heart and brain that cannot be adequately compensated for by cardiovascular responses and treatment is unavailable or inadequate. This can occur if the insult is severe or in association with less severe insults in patients whose ability to compensate is compromised, as may occur in anesthetized patients or in animals with comorbidities.
Circulatory shock can be fatal in the hours following insult or injury, despite aggressive resuscitative efforts, if the degree of shock was severe or prolonged enough to lead to myocardial injury and/or sympathoinhibition. This is referred to as decompensated or irreversible shock.
Animals that survive the acute episode of circulatory shock may still be at risk of developing SIRS, which, if severe, may result in MODS and possibly death in the days following the original insult or injury. The likelihood of SIRS and MODS following circulatory shock will increase with increasing severity and duration of the shock episode.28 In human medicine, other independent predictors of MODS following trauma include male gender, elderly patients, amount of red blood cell transfusions, and the persistence of an elevated blood lactate concentration 12 to 24 hours postinjury.123
There is evidence in human medicine that certain functional genetic polymorphisms can influence patient mortality.96,103 For example variants of the tumor necrosis factor gene are associated with the occurrence of sepsis and death following trauma.96 Further the cellular responses to injury show sexual dimorphism. For example, in animal experimental models and human clinical patients estrogen plays a protective role following hemorrhagic shock or sepsis.10 The role of gender and genetic predisposition in the response to injury and outcome of veterinary patients is currently unknown.
Clinical management
The initial approach to the animal with clinical evidence of cardiovascular shock should focus on resuscitation of the “ABCs,” or airway, breathing, and circulation. If the animal is not breathing at least eight times per minute or the gag reflex is absent, an endotracheal tube should be placed and positive pressure ventilation initiated, if needed. Supplemental oxygen should be supplied to all patients via mask, flow-by, or into the endotracheal tube, if applicable. If the animal has no detectable heartbeat and is pulseless, chest compressions should be performed and an electrocardiogram monitored. The administration of atropine, epinephrine, and/or vasopressin may be required to obtain return of spontaneous circulation. If the animal has received any reversible drugs (benzodiazepines, opioids, α2-agonists, etc.) before assessment, reversal of these drugs is recommended to minimize adverse cardiovascular effects of the drugs. Postoperative patients should be evaluated from a risk-benefit perspective before reversal of all opioid analgesics because rapid reversal following major surgery could lead to excessive pain. The use of an agonist-antagonist opioid drug (e.g., butorphanol) may decrease the severity of respiratory depression while allowing for continued analgesia following administration of pure μ-agonist drugs in painful patients.
Restoration of adequate circulation requires identification and control of any internal or external hemorrhage. Fluid therapy is the cornerstone of treatment for shock. Although fluid therapy is frequently contraindicated in patients with cardiogenic shock or hypervolemia, most other types of shock will be at least partially responsive to intravascular volume augmentation. Aggressive, yet judicious use of fluids will serve to increase tissue perfusion, decrease tissue hypoxemia, reduce secondary cytokine injuries, and maximize a successful outcome. A prospective study looking at people who died in the hospital after admission for treatment of injuries found that inadequate fluid resuscitation was the most common mismanagement recorded.25 Adequate intravascular volume replacement is crucial to restore perfusion to the major organs, thus reducing morbidity and mortality associated with hypovolemia. Approximately 50% of hypotensive, septic humans will have normalization of cardiovascular hemodynamics with fluid therapy alone.119
Access to the venous circulation is vital for rapid volume resuscitation. The intravenous or intraosseous routes are preferred because absorption from the subcutaneous or peritoneal space is slow and unpredictable, especially in the face of systemic vasoconstriction. Peripheral veins are preferred for the initial resuscitation efforts, but a jugular catheter may prove beneficial once the patient is more stable. The cephalic, lateral saphenous (dogs), or medial saphenous (cats) veins are most commonly used for initial placement of one or two intravenous catheters. The catheter(s) should be an over-the-needle catheter that is as large and short as possible to maximize flow rates through the catheter because the rate of flow is proportional to the radius to the fourth power and inversely proportional to the length. If venous catheter placement is not possible, either a venous cutdown or intraosseous catheter placement should be performed. Intraosseous catheter placement is further discussed in Chapter 15.
Once an intravenous catheter is placed, the clinician must decide what type and how much fluid to administer for the treatment of shock. There are basically four types of fluids that are typically used for the management of shock: crystalloids (isotonic and hypertonic), synthetic colloids, natural blood products (red blood cells, plasma, albumin), and oxygen carrying solutions. The various types and doses are listed in Table 23-3. Although the specific type of shock may help dictate the best therapeutic approach, it is important that the clinician understand the constituents of and potential side effects of each fluid type.
Table 23-3 Fluid Choices for Circulatory Support
| Fluid Type | Dose | Comments |
|---|---|---|
| Isotonic crystalloids | Dog: up to 90 mL/kg Cat: up to 60 mL/kg |
Used in animals for intravascular and interstitial volume deficits. May cause edema in animals with capillary leak or a low oncotic pressure. |
| Synthetic colloid solutions (hydroxyethyl starches) | Dog: 5-20 mL/kg Cat: 5-10 mL/kg |
Used in animals for volume replacement and oncotic support. May cause coagulopathies. |
| Human serum albumin | 2 g/kg or calculate albumin deficit (g): 10 × (desired-patient albumin) × weight (kg) × 0.3 | Used for albumin and oncotic support and volume replacement. Monitor closely for reactions. One time dose recommended. |
| Canine serum albumin | 1-2 g/kg/day | Limited safety studies thus far. Use with caution. |
| Fresh frozen plasma | 10-15 mL/kg as needed | Used to treat clotting factor deficiencies and provide albumin-containing oncotic support. |
| Frozen or cryo-poor plasma | Used to provide albumin-containing oncotic support or treatment of rodenticide toxicity. | |
| Packed red blood cells | 10-15 mL/kg to raise PCV by 10%-15% | Used to treat anemia. |
| Fresh whole blood | 20-25 mL/kg | Used to treat anemia, thrombocytopenia, clotting factor deficiencies, and provide albumin-containing oncotic support. |
Modified with permission from Silverstein DC. Daily intravenous fluid therapy. In: Silverstein DC, Hopper K, editors. Small animal critical care. St Louis: Saunders Elsevier, 2009.
Isotonic crystalloids
Isotonic crystalloids, also known as replacement fluids, are electrolyte-containing fluids with a composition similar to that of the extracellular fluid. They have a similar osmolarity as plasma and the electrolytes are small in size (i.e., sodium has a molecular weight of 23 Da compared with glucose at 180 Da). Examples include 0.9% sodium chloride, lactated Ringer’s solution, Normosol-R, and Plasmalyte 148. Although decades of investigation have not defined the ideal fluid for the treatment of shock, the initial resuscitation fluid for the treatment of patients in shock is most commonly isotonic crystalloids. A dose up to approximately one blood volume is typically used: 90 mL/kg in the dog and 50 mL/kg in the cat. Isotonic crystalloids rapidly distribute into the extracellular fluid compartment following administration, and only approximately 25% of the delivered volume remains in the intravascular space by 30 minutes postinfusion.131 Although theoretically this increase in interstitial fluid volume might predispose to interstitial edema and deranged oxygen transfer to the cells, this has not been shown in a canine hemorrhagic shock model.9 However, it is important that the veterinarian avoid overzealous use of isotonic crystalloids to prevent volume overload and interstitial edema, pulmonary edema, or cerebral edema. Patients with a low colloid osmotic pressure, pulmonary contusions, cerebral trauma, fluid nonresponsive renal disease, or cardiac disease are at highest risk for complications. In addition, substantial hemodilution of red blood cells, plasma proteins, clotting factors, and platelets can occur. Therefore, anemia, hypoproteinemia, and hypocoagulability should be anticipated following large-volume crystalloid administration.
Since it is hard to predict how a given animal will respond to a rapid fluid bolus, it is recommended that initially only one third to one half of the shock dose be given as quickly as possible (often using a pressurized fluid infusion system), followed by additional boluses as indicated by clinical parameters and repeated physical examinations. Animals with recently lacerated or ruptured blood vessels are susceptible to rebleeding following aggressive fluid therapy and a rapid increase in vascular hydrostatic pressure (and “pops the clot”). Hypotensive fluid resuscitation (to a mean arterial pressure of 60 mm Hg) may help prevent rebleeding while helping to maintain perfusion to vital organs.66
Not all isotonic fluids are created equal, as seen in Table 23-4. Isotonic saline solution (0.9% NaCl) contains a higher concentration of sodium and chloride compared with normal plasma, and will cause proportional changes (increases) in a normal animal’s electrolytes. Therefore, large amounts of 0.9% NaCl will cause a mild increase in sodium, a marked increase in chloride, and a moderate decrease in bicarbonate and potassium. The kidneys will typically compensate, if possible, by excreting the excess electrolytes and conserving potassium. Animals with hypochloremia, mild hyponatremia, or a metabolic alkalosis will often benefit from the administration of 0.9% NaCl.
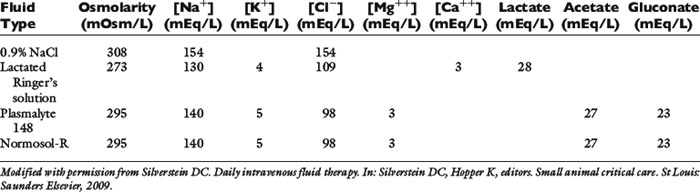
Although all isotonic crystalloids have a similar composition, there are situations when a certain fluid type might be preferable over another. Examples of specific clinical scenarios when a specific kind of isotonic crystalloid might be desirable are listed here:
1. The isotonic crystalloid of choice for animals with head trauma is 0.9% NaCl, if possible, because this fluid has the highest sodium concentration and is therefore least likely to cause a decrease in osmolarity and subsequent water movement into the brain interstitium.
2. Animals with severe hyponatremia or hypernatremia should receive crystalloid fluids that most closely match the patient’s sodium concentration during resuscitation to prevent a rapid increase or decrease in serum osmolarity and subsequent central pontine myelinolysis (often delayed in onset) or cerebral edema, respectively.
3. Animals with severe liver disease should receive nonlactate fluids. Neonates may not be able to adequately metabolize the lactate and animals with diabetic ketoacidosis could have delayed clearance of ketones following lactate administration; however, there is a lack of evidence to support these theories.
4. Patients with a hypochloremic metabolic alkalosis may benefit from 0.9% NaCl because this is the highest chloride-containing fluid.
Synthetic colloids
Synthetic colloids are polydisperse solutions with large molecules (molecular weight >20,000 Da) that do not readily sieve across the vascular membrane. Most synthetic colloidal particles are suspended within a crystalloid base solution. These fluids are hyperoncotic to the normal animal and therefore cause the movement of fluid from the extravascular to the intravascular space. Intravascular oncotic pressure is primarily regulated by albumin (69,000 Da), and the normal colloid osmotic pressure (COP) in most small animal patients is approximately 20 mm Hg. Synthetic colloids lead to an increase in blood volume that is greater than that of the infused fluid volume and also aid in the retention of this fluid in the vascular space (assuming normal capillary permeability).131 Although there is no definitive evidence to support the use of colloids over crystalloids for the treatment of shock, they may have a longer intravascular effect, require smaller volumes to achieve similar intravascular volume expansion, and prove less likely to cause interstitial edema due to their hyperoncotic characteristics. However, their use is also associated with coagulation impairment, higher costs, and possible side effects (e.g., allergic reactions or renal impairment, both primarily reported in humans).
The primary synthetic colloid solutions available contain either dextrans, gelatins, hemoglobin-based oxygen carriers (HBOCs), or hydroxyethyl starches. Dextrans are composed of naturally occurring glucose polymers, but the most commonly used and studied dextran, dextran 70, is not currently commercially available. Gelatins are made following the hydrolysis of bovine collagen and subsequent succinylation or linkage to urea. The available gelatin, oxypolygelatin, has numerous side effects and a short duration of action, making it a less desirable synthetic colloid that is unlikely to gain widespread use. HBOCs contain stroma-free, ultrapurified hemoglobin glutamers that are highly polymerized to prolong their effect in the circulation. Hydroxyethyl starches (HES) are made from a wide size range of amylopectin polymers with variable chemical modifications that influence their pharmacokinetics and metabolism. These are the most commonly used synthetic colloids and will therefore be reviewed in detail. However, the characteristics of most available synthetic colloid solutions are displayed in Table 23-5.
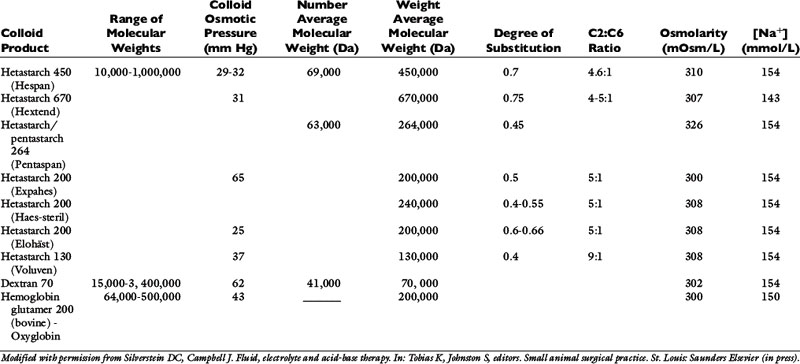
Examples of HES solutions include hetastarch, pentastarch, and tetrastarch (e.g., Voluven). HES preparations contain high polymeric glucose compounds that are manufactured by modification of the highly branched starch, amylopectin. Replacement of hydroxyl groups with hydroxyethyl groups at the C2, C3, or C6 carbon position of the constituent glucose molecules prevents rapid degradation by amylase. The ratio of substitution at the C2 versus C6 position (known as the C2:C6 ratio) also alters the half-life of the solution, with a higher ratio corresponding to a longer half-life. The degree of substitution (DS) refers to the number of hydroxyethyl groups per molecule of glucose and the higher the number of substitutions, the slower the breakdown and elimination of the molecule. However, a higher degree of substitution also means greater potential effects on coagulation.153 Hetastarch solutions have a rather high DS (0.6 to 0.7), while pentastarches and tetrastarches have a DS of 0.5 and 0.4, respectively. HES solutions are further characterized by their MW (low MW 70 kDa, medium MW 130 to 270 kDa, and high MW 450 kDa), their concentration (3%, 6%, or 10%), and their degree of substitution (0.4, 0.5, 0.6, or 0.7). It is important to note whether the MW is expressed as the number average molecular weight (MWn, most reflective of oncotic pressure) or the weight average molecular weight (MWw, exaggerated by larger particles). The MWw is determined by light scattering and is not as accurate a measure of the size of the colloid as MWn, which is the arithmetic mean of the range of molecular weights in the solution. The MWw is larger than the MWn, and as the molecular weight distribution of the colloid becomes narrower, MWw approaches and eventually equals MWn. In addition, the ability of synthetic colloids to modulate inflammation is related to their size and DS; those with a lower MW (<200 kDa) and DS (<0.4) may help to decrease capillary permeability, down-regulate the expression of adhesion molecules, inhibit neutrophil recruitment, and minimize cytokine production.46,84,145,146,159
Synthetic colloids are typically used in combination with isotonic crystalloids to maintain adequate plasma volume expansion with lower interstitial fluid volume expansion. Smaller total fluid volumes are needed and can therefore be administered more rapidly with fewer side effects. Measurements of COP are recommended with prolonged colloid administration. Animals with a colloid osmotic pressure less than 16 mm Hg may benefit from synthetic colloid administration and therapy should be adjusted to maintain values above this level. Animals with chronic hypoproteinemia may not need a COP greater than 16 mm Hg because the ratio of protein in the IV to interstitial space may not be as deranged as in those patients with acute IV losses. Total protein refractometer readings are not a valid means of monitoring colloid therapy.14
Potential side effects of synthetic colloid use are primarily related to disruption of normal coagulation. These include a decrease in factor VIII and von Willebrand factor concentrations (beyond just a dilutional effect), impairment of platelet function, and interference with the stability of fibrin clots, which makes them more susceptible to fibrinolysis.23,139,140,142,157 In addition, the time to clot formation (R), clot strength (maximum amplitude), and the speed of clot strength development (angle) using thromboelastography are all adversely affected by hydroxyethyl starches.45 The clinical manifestations of these changes are variable and depend on the status of the patient. Obviously, those patients with preexisting coagulopathies, von Willebrand disease (VWD), or moderate to severe thrombocytopenia/thrombocytopathia are at highest risk. Monitoring of the activated partial thromboplastin time (aPTT) may be helpful in assessing the adverse effects and risk level associated with the use of synthetic colloids, although there are no precise guidelines and it is difficult to predict which animals will develop clinical bleeding following administration. In general, the appropriate use of synthetic colloid solutions is deemed worth the risk, but judicious use of plasma and other blood products may also prove necessary to prevent bleeding complications, especially perioperatively. Caution should also be exercised to prevent volume overload or excessive hemodilution when large volumes of synthetic colloids are given to a patient. Additional side effects of synthetic colloids in people include renal impairment and allergic reactions, but similar problems in animals have not been documented.
Hypertonic saline
Hypertonic (7.0% to 7.5%) sodium chloride administration causes a transient osmotic shift of water from the extravascular to the IV compartment. Small volumes of approximately 4 to 6 mL/kg can be administered over 10 to 20 minutes. Rates exceeding 1 mL/kg/min may result in vagally mediated hypotension, bradycardia, and bronchoconstriction and should be avoided. Although hypertonic saline is primarily given to shift extravascular water into the IV space, there is evidence to suggest that it may also help to reduce endothelial swelling, increase cardiac contractility in normal hearts, cause mild peripheral and intestinal vasodilatation, modulate inflammation, improve intestinal edema and function, and decrease intracranial pressure.72,73,104,115,116,117,160 Hypertonic saline is especially useful for the treatment of head trauma or cardiovascular shock in animals greater than 30 kg that require large amounts of fluid for resuscitation (and time is of the essence [e.g., gastric dilatation-volvulus patients). Due to the osmotic diuresis and rapid redistribution of sodium cations that ensue following the administration of hypertonic saline, the IV volume expansion is transient (<30 minutes) and additional fluid therapy must be used.131 For example, combinations of hypertonic saline and synthetic colloid solutions have shown beneficial effects and are described below. Although 25% mannitol could also be used as a hypertonic fluid, it is less effective at increasing IV volume because the osmolarity is approximately half that of 7.5% saline. The use of 23.4% saline intravenously has been reported, but the safety margin is small and this practice is generally discouraged. However, its use for the treatment of brain injury is ongoing and research thus far looks promising.71,76,115,116, See Table 23-6 for specific characteristics of hypertonic crystalloids.
| Fluid Type | Osmolarity (mOsm/L) |
[Na+] (mEq/L) |
|---|---|---|
| 7.5% NaCl | 2400 | 1200 |
| 23.4% NaCl | 8000 | 4000 |
| 25% mannitol | 1250 |
Modified with permission from Silverstein DC, Campbell J. Fluid, electrolyte and acid-base therapy. In: Tobias K, Johnston S, editors. Small animal surgical practice. St Louis: Saunders Elsevier (in press).
Hypertonic saline administration is not without potential risks. An increase in the concentration of serum sodium and chloride will occur following administration (in addition to an increase in osmolarity). A decrease in potassium, and bicarbonate concentrations should also be anticipated. Typically, these changes are moderate and of minimal clinical importance, except in animals with preexisting electrolyte derangements or those receiving repeated doses of hypertonic saline. Hypertonic saline should not be given to dehydrated animals because these patients are interstitially volume depleted, thus limiting the effectiveness of the fluid and predisposing to further dehydration. If hypertonic solutions are administered proximal to the heart, arrhythmias might occur. If hypertonic solutions are administered in small peripheral veins, hemolysis and phlebitis can result.115
Hypertonic saline-synthetic colloid mixtures
To prolong the effect of the resuscitation fluids, a hypertonic saline/synthetic colloid mixture is often administered for resuscitation. A mixture of one part hypertonic saline (23.4% NaCl) with two parts dextran 70 or hetastarch will make a 7.5% saline mixture. There are limited veterinary studies supporting the beneficial use of this mixture for traumatic shock, endotoxic shock, hemorrhagic shock, pyometra, and gastric dilatation-volvulus in dogs.5,43,62,101,113,114,125,126,141,152,161
Hemoglobin-based oxygen carriers
Hemoglobin-based oxygen carriers (HBOC), such as Oxyglobin, have been the object of much study and interest for several years now. Although advanced scientific research is still lacking in animals, the solution contains many attractive characteristics. HBOCs improve the delivery of oxygen to the tissues by increasing both hemoglobin content and preload. However, hemoglobin concentration may not increase due to the dilutional effects of the fluid. Although Oxyglobin is the only approved HBOC for use in small animals, its future availability is uncertain.
Oxyglobin is a sterile, ultrapurified, stroma-free, polymerized bovine hemoglobin solution that is an alternative oxygen carrying solution. It is nonantigenic and therefore does not require blood typing or crossmatching before administration. Oxyglobin has an MW of 200,000 Da and a COP of 40 mm Hg. The hemoglobin concentration is 13 g/dL and it is suspended in a modified lactated Ringer’s solution with an osmolarity of 300 mOsm/L and a pH of 7.8 (see Table 23-5). The half-life ranges from 18 to 43 hours, depending on the dose administered (10 to 30 mL/kg, respectively). It can be stored at room temperature for up to 2 years, but an open bag must be used within 24 hours. It is purple in color and will cause a yellow-orange discoloration to the animal’s skin, urine, serum, sclera, and mucous membranes. Following administration of Oxyglobin, the animal’s hemoglobin must be measured to estimate oxygen carrying capacity because the hematocrit of the animal is unaffected (or diluted) by Oxyglobin. Several laboratory parameters are invalidated due to discoloration of the serum. The oxygen dissociation curve for Oxyglobin is right-shifted, so oxygen is unloaded from the red blood cells to the tissues more easily, and the nonlaminar flow of the solution increases contact between the hemoglobin polymer and the endothelium to further enhance the off-loading of oxygen. The hemoglobin may serve to perfuse areas that red blood cells cannot reach. The nitric oxide (NO) scavenging effects of Oxyglobin may be good or bad. On the one hand, the combination of reactive oxygen species and NO produces cytotoxic peroxynitrite and peroxynitrous acid that might be prevented by a NO scavenger, such as Oxyglobin. On the other hand, the vasoconstrictive effects of a nitric oxide scavenger will serve to increase afterload and decrease perfusion to many tissue beds. In pigs subjected to hemorrhagic shock with or without traumatic brain injury, HBOC-21 was able to restore cardiovascular and cerebral recovery better than hetastarch or isotonic crystalloid administration.67,69,74,86,106,118 However, Driessen et al showed that the administration of Oxyglobin to dogs with experimental hemorrhagic shock led to severe vasoconstriction and a decrease in cardiac output, despite apparent normalization of the typical physical examination parameters.34 Further studies have found that microvascular perfusion and oxygen transport are ameliorated following Oxyglobin therapy, however.32,34,35,74 Although Oxyglobin can be lifesaving in specific animals (i.e., severe, acute anemia as seen with massive blood loss or hemolytic diseases), there are some medical concerns with the safety of this product and it is not superior to natural blood products (and is more expensive). The recommended dose is 10 to 30 mL/kg (dogs), and it is important to remember that this product is a colloid and can easily contribute to IV volume overload (especially in cats). Although the product’s safety and efficacy have not been studied in cats, there are two retrospective reports of its use in this species.33,50
Blood products
The need for blood products in the shock patient is dependent on the patient’s disease process. Most previously normal patients can tolerate acute hemodilution to a hematocrit of less than 20%, although it is recommended that the hematocrit be kept above 24% in critically ill humans patients to ensure adequate oxygen delivery.155 In shock patients that are unresponsive to fluid therapy alone, the hematocrit target may be increased to greater than 30% to maximize oxygen carrying capacity. Excessive increases in hematocrit should be avoided because this will increase blood viscosity.
Most animals can tolerate an acute loss of 10% to 15% of blood volume without requiring a blood transfusion. Acute hemorrhage exceeding 20% of the blood volume often requires transfusion therapy in addition to crystalloid and colloid therapy (as discussed previously). In animals with acute blood loss requiring transfusion therapy, fresh whole blood or packed red blood cells and fresh frozen plasma should be used to stabilize clinical signs of shock, maintain the hematocrit above 24%, and keep the clotting times within the normal range. Packed red blood cells and fresh frozen plasma are administered at a dose of 10 to 15 mL/kg and fresh whole blood at a dose of 20 to 25 mL/kg (see Chapter 24 for further details on transfusion therapy).
The administration of 25% human albumin has gained recent popularity, although its potential adverse effects have also been recognized. Because it is clearly a functionally important blood component and accounts for 80% of the plasma oncotic pressure, animals with severe hypoalbuminemia may benefit from its use. Preliminary studies in dogs show that human albumin administration in dogs will increase circulating albumin concentrations, total solids, and increase COP, although the effect on mortality remains unknown.149 Current and future studies will give us more information about the use of this product. Potential risks include potentially fatal acute or delayed hypersensitivity reactions, volume overload, and coagulopathy.49,91 An increase in IgG against human albumin does occur in normal and critically ill dogs, with potentially life-threatening reactions, and therefore repeated exposures are not recommended.18,91 Canine and feline albumin are now available for purchase, although there is limited data available regarding the use of these products at this time.
Monitoring
Intensive care of the shock patient requires frequent reassessment of the animal. Several monitoring tools are useful, including physical findings (e.g., mucous membrane color, capillary refill time, pulse rate and quality, heart rate, respiratory rate), arterial blood pressure (by invasive or noninvasive means), urine output, CVP, electrocardiogram, cardiac output, PCV, TP, blood glucose, electrolytes, acid-base balance, oxygenation, and ventilation. The patient’s underlying disease process typically dictates the importance and frequency of each monitoring technique. Although oxygen transport variables (e.g., cardiac output, content of oxygen in arterial and venous blood, oxygen consumption, and oxygen delivery) provide useful information,95 most veterinary practices do not have the capability to monitor oxygen transport variables. There is increasing evidence to show that even with advanced monitoring and normalization of macrohemodynamic variables, some people, especially those with sepsis, may suffer from microcirculatory dysfunction and organ hypoperfusion.36,42,65 However, the use of current monitoring techniques are useful for assessment, prognosis, and treatment of unstable animals and are discussed below.
Physical examination
Physical examination findings are quite important in order to assess changes in cardiovascular status and indices of perfusion. Although interpretation of these findings is subjective, objective monitoring techniques can also be used to support clinical impressions. Peripheral pulse rate, quality, and synchrony with the heartbeat; respiratory rate, rhythm, and effort; mucous membrane color; and capillary refill time provide subjective information regarding the status of the cardiopulmonary system. Additional physical findings of importance include the animal’s mentation and assessment of the jugular veins.
Packed cell volume and total plasma proteins
Measurements of PCV and total plasma proteins provide essential information. The PCV indicates oxygen-carrying capacity of the blood because oxygenated hemoglobin is the source of oxygen content in arterial blood. Acute changes in blood volume may not be reflected in the PCV due to fluid shifts and potential for splenic contraction in dogs. Based on recommendations from human literature, the hemoglobin should be maintained greater than 8 g/dL (PCV >24%) to maintain oxygen delivery without undue risk of excessive transfusions.37,57 Blood or blood products should be considered when any animal is showing signs of decreased oxygen delivery (e.g., tachypnea, exercise intolerance, decreased mentation). Patients with a chronic anemia may not require blood products until the PCV is less than 15%.
Measurement of total plasma proteins provides additional valuable information. The color of the plasma can help to identify hemolysis or icterus. The refractometer reading of the total plasma protein concentration gives subjective information regarding COP in patients that have not received synthetic colloids. A decrease in the TP over time may indicate a loss or decreased production of serum proteins. With rapid drops in TP, interstitial edema is likely to result, especially in the face of crystalloid administration. The TP concentration should remain greater than 3.5 g/dL to ensure adequate intravascular oncotic pull. Colloid therapy should be strongly considered when the total plasma protein concentration is less than 3.5 g/dL. Colloid osmotic pressure can also be measured directly with an oncometer or colloid osmometer. The COP of plasma in normal patients is approximately 20 to 25 mm Hg, and values less than 16 mm Hg often are found in critically ill patients. Patients with a COP less than 16 mm Hg will likely benefit from supplemental colloid administration.
Central venous pressure
Measurement of CVP can provide valuable information about right ventricular function and intravascular volume status and is relatively easy to monitor in most veterinary practices.154 Monitoring CVP involves placement of an indwelling jugular catheter with the tip of the catheter in the thoracic cranial vena cava. If the animal has appropriate right-sided heart function, CVP provides information on filling pressures of the heart (i.e., preload). The CVP should range between 5 to 10 cmH2O in the shock patient (2 to 5 cmH2O in cats), although normal CVP values range from 0 to 10 cmH2O. Other determinants of CVP also must be considered when interpreting values, including intrathoracic pressure and venous distensibility.
Appropriate fluid resuscitation should result in an increase in CVP. However, intravascular volume expansion is temporary following crystalloid resuscitation because 75% of the fluid distributes into the extravascular space. An increase in CVP is expected following fluid therapy. However, if the CVP subsequently drops more than 3 cmH2O and the animal appears less stable following redistribution, further therapy with colloids or blood products may be indicated. Additionally, sources of intravascular volume loss or bleeding should be sought.
Arterial blood pressure
Arterial blood pressure is defined as the force that is exerted by the blood on the arterial wall. Arterial blood pressure is not cardiac output, and it should not be assumed that adequate blood pressure is synonymous with adequate cardiac output. In fact, cardiac output is a determinant of mean arterial blood pressure (i.e., mean arterial pressure = cardiac output × systemic vascular resistance). If systemic vascular resistance is increased secondary to vasoconstriction, the result is increased blood pressure. However, cardiac output can decrease during hypertension. An animal in pain can have hypertension yet lower than normal cardiac output. The animal with poor myocardial performance because of SIRS and vasoconstriction caused by pain or hypothermia can have very poor cardiac output. Therefore blood pressure monitoring should be used in addition to other monitoring techniques to provide the most accurate assessment of cardiovascular status.
Arterial blood pressure can be measured by direct or indirect methods. Direct measurement of arterial blood pressure requires a catheter placed in a peripheral artery (usually dorsal pedal or femoral), a pressure transducer, and a monitor. Accurate measurement of systolic, diastolic, and mean arterial pressures can be obtained with proper positioning of the transducer (i.e., at the level of the heart) and adequate calibration of equipment. The arterial waveform may be used to detect early deterioration of the cardiovascular system (i.e., flattening of the waveform). Placement of an arterial catheter is a challenge, especially in patients weighing less than 10 kg, and the equipment is expensive, which may deter many clinicians from measuring arterial blood pressure directly. Nonetheless it is the gold standard, becomes much easier to perform with practice, and also enables easy sampling and analysis of arterial blood.
Indirect measurement of arterial blood pressure is most feasible in clinical practice. The most important factor to remember with indirect methods is that the values obtained are not necessarily accurate, especially in smaller animals (<10 kg), severely hypothermic animals, or those with extreme vasoconstriction. However, the trend of values obtained is extremely important and should be considered more important than the actual values. Techniques for indirectly measuring blood pressure have been described elsewhere59 and are briefly reviewed below.
The two available methods of indirect arterial blood pressure monitoring are oscillometric and Doppler ultrasonic. The oscillometric method (e.g., Dinamap, GE Healthcare Systems, Waukesha, Wis.; Cardell, CAS Medical Systems Inc., Branford, Conn.) involves placement of an appropriate-sized blood pressure cuff over a peripheral artery. The mechanism of blood pressure measurement is to determine the oscillation of the artery at systolic and mean arterial pressures and convert this measurement to a numerical blood pressure. The diastolic pressure is the pressure at which the maximal oscillation has decreased by 80%. Therefore diastolic pressure measurements are least accurate. The animal should be placed in lateral recumbency to ensure that the limb is near the level of the heart. Appropriate cuff size is critical to obtain adequate readings. The width of the cuff should be approximately 40% of the circumference of the limb. A cuff that is too large results in falsely decreased values, and a cuff that is too small results in falsely increased values. The oscillometric method provides systolic, diastolic, and mean arterial pressures, as well as heart rate. The primary disadvantages of the oscillometric method include the cost of the equipment and inaccurate or unobtainable readings in animals weighing 5 to 10 kg.
The Doppler ultrasonic method uses the Doppler effect to detect movement of red blood cells past a crystal that emits Doppler waves. Each pulse of blood is converted to a sound that can easily be heard. The crystal is placed over a peripheral artery with an appropriate-sized cuff placed proximal to the crystal. A sphygmomanometer is attached to the cuff and inflated until no sound is detected. The pressure is slowly reduced until the first audible pulse is detected. Only systolic blood pressure is measured on a reliable basis, but diastolic pressure also can be obtained. The first audible pulse is the systolic blood pressure as indicated on the sphygmomanometer. The pressure continues to be slowly removed from the cuff until the audible signal changes tone. The change in tone occurs at the diastolic blood pressure. Advantages of the Doppler method include detection of an audible pulse, reasonable cost, and reliable use in very small patients.
Urine output
Urine output can be used as an indirect measurement of renal blood flow. It is easily measured by placing a urethral catheter and collection system. Urine output of less than 1 mL/kg/hr is abnormal in any animal that is volume resuscitated, especially those receiving fluid therapy, and possible causes should be explored (e.g., inadequate renal perfusion, acute renal failure, or inappropriate fluid retention).
Blood gas analysis
Arterial and venous blood gas analysis can provide valuable information about the shock patient. These values are readily obtained using a portable “point of care” blood gas analyzer. Laboratory blood gas analyzers are prohibitively expensive unless high caseload allows nearly constant usage, whereas portable blood gas analyzers can easily pay for themselves in just a few years. One disadvantage of blood gas analysis (arterial or venous) is that the results obtained represent a single moment in time, although the status of the patient may change minute by minute. The partial pressure of oxygen reflects the amount of oxygen dissolved in plasma and the saturation of hemoglobin with oxygen can be determined from the oxyhemoglobin dissociation curve.
Arterial blood gas analysis provides information regarding gas exchange in the lung and arterial acid-base balance. Arterial blood samples are most commonly collected from the femoral or dorsal pedal artery into a preheparinized syringe. The partial pressure of oxygen in arterial blood (Pao2) represents the adequacy of gas exchange in the lung.
A “mixed venous” blood gas sample must be obtained from the pulmonary artery, which requires placement of a specialized catheter. The partial pressure of oxygen in mixed venous blood (Pvo2) is a reflection of perfusion of tissues on a global basis. Normal Pvo2 values range from 35 to 45 mm Hg. Values less than 30 mm Hg indicate poor perfusion and oxygen delivery to the peripheral tissues. If a thermodilution catheter is not placed in the pulmonary artery to collect blood for Pvo2 determination, a jugular catheter placed to monitor CVP can be used to collect a venous blood sample that may approximate a true mixed venous sample.
Acid-base analysis from a blood gas sample is reviewed in Chapter 9.
Pulse oximetry
Pulse oximetry measures the saturation of hemoglobin with oxygen (Sao2) and can be monitored continuously and noninvasively. The Pao2 provides information about oxygen dissolved in plasma, whereas Sao2 provides information concerning the oxygenation of red blood cells. To be of value, pulse oximetry requires pulsatile flow of blood to the extremities (interdigital web, digit) where it is measured. Many patients with shock have decreased blood flow, especially to the extremities, which limits the effectiveness of pulse oximetry. The device may also be applied to the tongue for accurate readings, but this technique is difficult in the conscious patient. Other common areas for probe placement are the ear, axilla, vulva, and prepuce. A rectal probe may be of value in the conscious, recumbent patient.
Lactate
The clinical use of lactate measurement has gained popularity and acceptance in veterinary practice over the past 10 years due to the accessibility and reasonable cost of portable lactate analyzers. An elevated blood lactate concentration is frequently a marker of anaerobic metabolism and has been correlated with inadequate tissue oxygenation in many clinical shock syndromes.24,63,68 However, there are other clinical syndromes that can lead to hyperlactatemia, including diabetic ketoacidosis, neoplasia, drugs and toxins, hepatic insufficiency, gastrointestinal disease, or metabolic disorders. Normal reference intervals vary with the equipment used, but changes in lactate concentration in combination with other clinical indicators of shock are useful for monitoring effectiveness of therapy. Lactate concentration should decrease over time if successful cardiovascular resuscitation from shock has occurred. Occasionally, lactate concentration will increase transiently after initiation of therapy because improved perfusion results in a “washout” of waste products that did not previously enter the systemic circulation. Lactate measurement has been shown to be an effective predictor of gastric necrosis in dogs with GDV and thus serves as a useful predictor of prognosis and survival.24
Additional therapies
Various forms of shock can all lead to hypotension despite intravascular volume resuscitation, therefore necessitating the use of vasopressor and/or inotrope therapy (Table 23-7). Since both cardiac output and systemic vascular resistance affect oxygen delivery to the tissues, therapy for hypotensive patients includes maximizing cardiac function with fluid therapy and inotropic drugs and/or modifying vascular tone with vasopressor agents. The most commonly used vasopressors are exogenous catecholamines (epinephrine, norepinephrine, dopamine, and phenylephrine). Vasopressin, a nonadrenergic vasopressor agent, has also been used for the treatment of catecholamine-refractory vasodilatory shock.
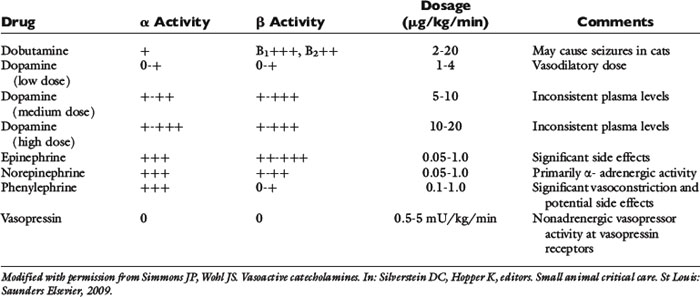
Different sympathomimetics cause various changes in the cardiovascular system, depending on the specific receptor stimulation caused by the drug.133 Conventionally, adrenergic receptor location and function involves the α1- and β2-receptors located on the vascular smooth muscle cells that lead to vasoconstriction and vasodilatation, respectively, while β1-receptors in the myocardium primarily modulate inotropic and chronotropic activity. In addition, there are dopaminergic-1 receptors in the renal, coronary, and mesenteric microvasculature that mediate vasodilatation and dopaminergic-2 receptors in the synaptic nerve terminals that inhibit the release of norepinephrine.
Dopamine has various potential actions on adrenergic and dopaminergic receptors.133 Primarily dopaminergic effects are seen at low intravenous doses (1 to 5 μg/kg/min), mainly β-adrenergic effects are seen at moderate doses (5 to 10 μg/kg/min), mixed α- and β-adrenergic effects are present at high doses (10 to 15 μg/kg/min), and primarily α-adrenergic effects are seen at very high doses (15 to 20 μg/kg/min). The actual dose response relationship is unpredictable in a given patient because it is dependent on individual variability in enzymatic dopamine inactivation, receptor down-regulation, and the degree of autonomic derangement. Dopamine can be used as a single agent therapy to provide both inotropic and pressor support in animals with vasodilatation and decreased cardiac contractility. Despite dopamine’s beneficial effects on cardiac output and blood pressure, it may have deleterious effects on renal, mesenteric, and skeletal blood flow.
Norepinephrine (NE) has mixed α- and β-adrenergic receptor agonism with preferential α-receptor activity.133 Therefore the effects on heart rate and contractility are mild, and NE is commonly used as a pressor agent in animals with normal or increased cardiac output states. The vasopressor dose of NE in humans (and extrapolated to dogs) is 0.05 to 3.3 μg/kg/min intravenously.
Epinephrine is a potent pressor with mixed α- and β-agonist activity.133 Although epinephrine is thought to have more potent β-agonist effects than NE, individual response is quite variable in patients with systemic inflammatory diseases and hypotension. Epinephrine may significantly impair splanchnic blood flow compared to other vasopressor drugs. The vasopressor dose of intravenous epinephrine is 0.01 to 0.1 μ/kg/min and for primarily β-agonist effects is 0.005 to 0.02 μg/kg/min. Epinephrine is rarely used as a sole first-line vasopressor agent due to its potential side effects, but may be necessary in critically ill animals. Phenylephrine is a pure α-agonist drug that causes profound vasoconstriction. It has been shown to cause an increase in cardiac output and blood pressure, presumably due to increased venous return to the heart and activation of α-1 receptors in the myocardium. Phenylephrine is typically used in patients that are unresponsive to other sympathomimetics, although it can be used as a sole first-line agent in vasodilated, hypotensive animals. Since phenylephrine has no β-agonist activity, it is the least arrhythmogenic of the sympathomimetic pressor drugs and is therefore desirable in animals that develop tachyarrhythmias in response to other pressor agents. The intravenous dose range is 0.5 to 3 μg/kg/min.
Dobutamine is a β-agonist with minimal α effects. It increases cardiac output, oxygen delivery, and oxygen consumption without causing vasoconstriction.133 It is therefore useful in animals with cardiac insufficiency. Dobutamine may worsen or precipitate tachyarrhythmias and may precipitate seizure activity in cats. The intravenous dose range is 1 to 5 μg/kg/min in cats and 2.5 to 20 μg/kg/min in dogs.
Vasopressin is a nonadrenergic vasopressor agent. It has both direct and indirect effects on the vascular smooth muscle via the V1 receptors and induces vasoconstriction in most vascular beds.132 In vitro, vasopressin is a more potent vasoconstrictor than phenylephrine or NE. At low doses, this drug causes vasodilatation in renal, pulmonary, mesenteric, and cerebral vasculature in an attempt to maintain perfusion to these vital organs. Low flow states secondary to hypovolemia or septic shock are associated with a biphasic response in endogenous serum vasopressin levels. There is an early increase in the release of vasopressin from the neurohypophysis in response to hypoxia, hypotension, and/or acidosis, which leads to high levels of serum vasopressin. This plays a role in the stabilization of arterial pressure and organ perfusion in the initial stages of shock. There appears to be a subsequent decrease in circulating vasopressin levels, most likely due to a depletion of hypothalamic stores. The use of vasopressin in animals in the later stages of shock, especially those that exhibit vasodilatation and are refractory to catecholamine therapy, may therefore be beneficial. The drug also enhances sensitivity to catecholamines and therefore may allow the dose of concurrent catecholamine therapy to be lowered. Experimental studies in dogs have demonstrated an increase in blood pressure and cardiac output with minimal side effects. A clinical case series using vasopressin at 0.5 to 4 mU/kg/min found an increase in blood pressure following vasopressin therapy as well. This drug will require further investigation, but may be considered in animals with catecholamine resistant vasodilatory shock, as is commonly seen in animals with SIRS, MODS, and sepsis.
Antimicrobial therapy
The Surviving Sepsis campaign of 2008 recommended administration of broad-spectrum antibacterial therapy within 1 hour of diagnosing severe sepsis or septic shock.26 Delaying administration of antibacterials or withholding their use in a septic patient increases the ability of the organisms to reproduce, spread, and induce a greater inflammatory response. Timely localization of the septic focus and procurement of infected tissue or fluid for bacterial identification and susceptibility testing is of paramount importance in the treatment of a patient with septic shock. However, sample collection may be impossible in some patients due to cardiopulmonary instability or the presence of a coagulopathy. Empiric antimicrobial therapy should be selected based on the following factors: antimicrobial characteristics (cidal versus static); most common bacterial flora in the affected tissue; ability of the antimicrobial to penetrate the infected tissue; history of recent antimicrobial use and potential for resistance; safety profile of the drug(s); and source of infection (whether nosocomial or community-acquired).
Appropriate empirical antimicrobial therapy is vital for success. One canine study found that five dogs receiving inappropriate empiric antibacterial therapy had a mortality rate of 80%.75 Broad spectrum bactericidal antimicrobial agents are typically administered to patients with septic shock via the intravenous route. Changes are made based on the results of antibacterial susceptibility testing. Possible choices of four-quadrant, empiric therapy (i.e., effective against many gram-positive and gram-negative aerobes and anaerobes) include: ampicillin (22 mg/kg IV q6 to 8h) and enrofloxacin (15 mg/kg IV q24h in dogs, 5 mg/kg IV q24h hours in cats), ampicillin and amikacin (15 mg/kg IV q24h), cefazolin (22 mg/kg IV q8h) and amikacin, ampicillin, and ceftazidime (22 mg/kg IV q8h), or clindamycin (10 mg/kg IV q8 to 12h) and enrofloxacin. Single agents such as ticarcillin/clavulanic acid (50 mg/kg IV q6h), cefoxitin (15 to 30 mg/kg IV q4 to 6h), or imipenem (5 to 10 mg/kg IV q6 to 8h, if bacterial resistance is suspected).
Gastrointestinal protectants
Stress related mucosal disease (SRMD), including both superficial and deep mucosal damage, is commonly recognized in people with critical illness such as shock.136 Hypoperfusion of the gastrointestinal tract is the most likely cause, although altered mucosal defenses, free radical damage, and increased acid production in the stomach may also contribute. Although the incidence of SRMD in small animals with shock is unknown, the primary strategy for prevention is to ensure adequate GI perfusion and employ early enteral nutrition. High-risk patients may also benefit from pharmacologic prophylaxis for SRMD. Based on the currently available evidence in human medicine, it appears that proton pump inhibitors (PPI) are superior to histamine-2 receptor antagonists (H2RA), which are superior to sucralfate in the prevention of SRMD in adult critical care patients.97,138 Drugs available include omeprazole (PPI) 0.7 to 1.0 mg/kg PO q24h, pantoprazole (PPI) 0.7 to 1.0 mg/kg IV q24h, famotidine (H2RA) 0.5 to 1.0 mg/kg IV q12 to 24h, ranitidine (H2RA) 0.5 to 4 mg/kg IV q8 to 12h, and sucralfate (protectant) 0.25 to 1 g/25kg PO q6 to 8h. Recent evidence suggests that ranitidine does not decrease acid production in dogs at clinically recommended doses.8
Controversial therapies
Glucocorticoids
Glucocorticoid therapy in shock has a long and controversial history. The initial reasoning for steroid administration to patients in shock was the mistaken belief shock was a form of an addisonian crisis. The indications and potential adverse effects of glucocorticoid therapy varies between different types of circulatory shock.
Hemorrhagic Shock
There was a great deal of interest and research into the role of glucocorticoid therapy in hemorrhagic shock in the 1960s and 1970s. Although some initial experimental studies were promising, they used models of high dose steroids administered before the episode of hemorrhage and only evaluated short-term survival.51,80 When studies were performed with a more clinically relevant design, high dose steroid therapy could not be shown to increase survival from hemorrhagic shock.30,58,80 None of these experimental studies evaluated long-term survival or the associated adverse effects of such large doses of glucocorticoids. Given the lack of scientific evidence of any benefit of glucocorticoid therapy and our current understanding of the adverse effects of high dose steroids, their routine use in patients with hemorrhagic shock, or other causes of hypovolemia, cannot be recommended.
Septic Shock
High-dose glucocorticoid therapy in septic shock has been evaluated in animal experimental studies and human clinical investigations. Several meta-analyses concluded that there was no beneficial effect associated with high-dose steroid therapy for septic shock, many of the clinical studies also suggested the possibility that this therapy could be associated with harm.21,81
More recently the syndrome of relative adrenal insufficiency (RAI) has been identified in patients with severe SIRS and sepsis. The hypothalamic-pituitary-adrenal (HPA) axis is crucial for host response to shock. Animals with inflammatory diseases leading to shock often have release of endogenous cortisol early in the course of illness. Cortisol modulates the synthesis and release of proinflammatory and antiinflammatory mediators to restrict inflammation to the infected tissues. The cause of RAI in select patients appears to be multifactorial, including factors such as vascular or ischemic damage, inflammation, and apoptosis within the HPA axis, use of drugs that modify cortisol metabolism, and/or tissue resistance to corticosteroids.108,109 RAI may occur in up to 70% of people with septic shock and is correlated with hypotension and death in these patients.7,64,85,121,127,135 Preliminary research in dogs, cats, and foals suggests that a similar phenomenon also occurs during critical illness in these species.15,20,89,90,111,112 Sepsis-induced RAI in humans is typically recognized by a basal cortisol less than 10 µg/dL or a change in cortisol (delta cortisol) of less than 9 µg/dL after administration of ACTH. Critically ill dogs with a delta cortisol less than or equal to 83 nmol/L were almost six times as likely to need vasopressor therapy in one study.89 The use of physiologic doses of hydrocortisone has been found to improve 28-day mortality and decrease the duration of vasopressor therapy in septic humans with a poor response to adrenocorticotropic hormone testing.3 However, a systematic review found no clear benefit of steroids on mortality, although subgroup analysis of low-dose steroid use suggests a beneficial drug effect on short-term mortality.4 In summary, although routine use of hydrocortisone in adults with sepsis is still controversial, its use is recommended in adult patients with septic shock that are poorly responsive to fluid and vasopressor therapy, as well as pediatric patients with suspected or proven adrenal insufficiency.4,26,87 A reported dose of hydrocortisone that has been used in dogs for the treatment of RAI is 0.5 mg/kg IV q6h.105
Anaphylaxis
Anaphylactic or anaphylactoid shock is the result of generalized mast cell and basophil degranulation resulting in the release of biochemical and vasoactive substances, such as histamine, proinflammatory cytokines, leukotrienes, and tryptase. The clinical consequences include increased vascular permeability, loss of vasomotor tone, bronchoconstriction, angioedema, and urticaria.70,156 Anaphylactic shock is typified by hypovolemic and distributive abnormalities that can be fatal. Therapy includes fluid resuscitation, antihistamine (both H1 and H2 receptor antagonists), and epinephrine administration. Glucocorticoid therapy is routinely recommended for the treatment of anaphylactic shock but there is little evidence that it is of benefit. Although not expected to have effects in the acute setting, glucocorticoids are commonly given in an effort to prevent a recurrence of clinical signs and to shorten the clinical course of the disease. To date, there are few studies evaluating the benefits of glucocorticoid therapy in anaphylaxis. One study of human patients reported that the incidence of biphasic or protracted anaphylaxis occurred despite the use of glucocorticoid therapy.137 Systemic glucocorticoids are currently recommended for the treatment of anaphylaxis in veterinary medicine.31
Hypoadrenocorticism
Hypoadrenocorticism can cause circulatory shock from the combination of hypovolemia and inadequate vasomotor tone. If significant hyperkalemia is present, the resultant bradycardia can also contribute to a decrease in cardiac output. Shock due to hypoadrenocorticism is a clear indication for glucocorticoid and mineralocorticoid therapy. Adequate fluid resuscitation and medical therapy as appropriate to lower serum potassium is also essential. Dexamethasone is the glucocorticoid therapy of choice if an ACTH stimulation test needs to be performed because it minimally interferes with endogenous cortisol measurement. A recommended dose of dexamethasone for initial stabilization of patients with hypoadrenocorticism is 0.1 to 0.2 mg/kg of dexamethasone sodium phosphate IV.44 Glucocorticoid therapy does not have to be repeated until 24 hours following this dose and at that time it maybe feasible to start oral therapy.
Bicarbonate
Metabolic acidosis is likely to be present in all patients presenting in circulatory shock, secondary to anaerobic metabolism and the subsequent accumulation of lactic acid. The treatment of lactic acidosis with bicarbonate therapy remains controversial. The rationale of treatment is predicated on the notion that having a low pH has negative effects and hence increasing pH will provide physiologic benefits. Although experimental studies have demonstrated isolated effects of acidosis, such as decreases in myocardial contractility and reduced responsiveness to catecholamines, the results of intact animal studies are less clear.48 Human clinical studies have shown that patients tolerate moderate to severe acidemia without evidence of adverse cardiovascular effects.158 Additionally acidemia may favor oxygen delivery to the tissues by causing a right shift of the oxygen-hemoglobin dissociation curve allowing for greater oxygen unloading to the tissues.56 This may be of particular benefit to the animal in shock. These contradictory findings in acidosis make it difficult to determine if restoring a normal pH is beneficial. In addition there is concern that bicarbonate therapy is itself associated with adverse effects. The few clinical trials of bicarbonate therapy for the treatment of acidemia in human medicine have had disappointing results.19,92,93,144
Bicarbonate combines with hydrogen ions in the extracellular fluid to form carbonic acid, which rapidly is convertedinto carbon dioxide and water in the presence of carbonic anhydrase. Hydrogen ions and bicarbonate cannot readily cross the cell membrane so they are somewhat trapped in the extracellular fluid space. In contrast the carbon dioxide formed as a result of bicarbonate therapy rapidly diffuses into cells and causes intracellular acidosis.12 To avoid this the patient must increase alveolar ventilation substantially to prevent elevations in blood PCO2. This is challenging for the compromised patient, such as one in circulatory shock, and paradoxical intracellular acidosis following bicarbonate therapy can occur.128
In addition the drop in ionized calcium associated with alkalinization and the hypertonicity of sodium bicarbonate can also have adverse effects. There are only two small, prospective, randomized clinical trials in human medicine evaluating the role of sodium bicarbonate in the therapy of lactic acidosis.19,92 No difference in cardiovascular performance could be identified in these studies following therapy and there was a significant drop in ionized calcium associated with therapy. Although the role of sodium bicarbonate therapy in lactic acidosis remains somewhat controversial, there is unanimous agreement that identification and treatment of the underlying cause for lactic acidosis is the first and foremost important step.6,12 Only after aggressive resuscitation efforts have been made should bicarbonate therapy be considered. The 2008 Surviving Sepsis guidelines recommend that sodium bicarbonate should not be administered to patients with a lactic acidosis unless the pH is less than 7.15.26 The role of sodium bicarbonate in patients with a pH less than 7.15 is uncertain because of the lack of studies evaluating this question. Some authors in the human literature advise against bicarbonate therapy at any pH.6,48 Sodium bicarbonate is contraindicated in patients with volume overload such as congestive heart failure or in patients with ventilatory compromise.
Case Examples
CASE 1: Hypovolemic Shock
A 2-year-old male neutered Labrador weighing 30 kg is presented collapsed. There is no history of trauma, toxin ingestion, or previous medical problems. On presentation the dog is found to be severely obtunded, has very pale mucous membranes, a capillary refill time of 4 seconds, a heart rate of 180 bpm, weak femoral pulses, and the feet are cold. The respiratory rate is 60 breaths/minute. On auscultation no murmurs or arrhythmias are heard and no abnormal breath sounds are evident.
On presentation, flow by oxygen is provided and a cephalic venous catheter is placed. Blood work collected at the time of catheter placement reveals the following:
| Parameter | Measured Value | Reference Range |
|---|---|---|
| pH | 6.911 | 7.34-7.38 |
| Pvco2 mm Hg | 45.6 | 40-46 |
| Pvo2 mm Hg | 32.7 | 49-67 |
| HCO3 mEq/L | 9.5 | 22-24 |
| SBE mEq/L | −21.2 | −2 to 0 |
| Glucose mg/dL | 133 | 65-112 |
| Lactate mmol/L | 19 | <2.5 |
| Packed cell volume % | 70 | 37-55 |
| Total protein g/dL | 3.5 | 5.4-7.1 |
A severe lactic acidosis without respiratory compensation is evident with hemoconcentration and hypoproteinemia.
Rapid administration of 2000 mL of lactated Ringer’s solution via a pressure bag and 300 mL of 6% hetastarch is provided. Following this the perfusion parameters have improved; the mucous membranes are still pale, capillary refill time is approximately 2 seconds, heart rate is 150 bpm, and the femoral pulses are assessed as being stronger. Another 1000 mL of lactated Ringer’s solution and another 200 mL of hetastarch is given rapidly. The perfusion parameters at this time are greatly improved although the heart rate remains elevated.
Following initial resuscitation, a full physical examination reveals abdominal pain. There is hemorrhagic gelatinous diarrhea evident on rectal examination. A dose of hydromorphone IV is given at this time, following which the heart rate drops to 120 bpm. A second venous blood sample is evaluated, which reveals a resolution of the lactic acidosis, PCV of 58%, and a total protein of 3.2 g/dL. An ongoing fluid plan of 1 mL/kg/hr of 6% hetastarch and 200 mL/hr of lactated Ringer’s with appropriate potassium chloride supplementation is instituted.
CASE 1 - Case Notes
This dog presented in severe circulatory shock with signs suggestive of vasoconstriction. Given the signalment, the absence of any previous history of heart disease, and no murmur or arrhythmia detected, cardiogenic shock was considered unlikely and the diagnosis of hypovolemic shock was made. The severe lactic acidosis supports the physical examination findings of severe shock. The lack of respiratory compensation is likely secondary to central nervous system depression as a result of poor brain perfusion. The very high PCV in combination with the low total protein and the acute history is classic for the syndrome known as hemorrhage gastroenteritis. This diagnosis is further supported by the finding of the bloody diarrhea on rectal examination. This disease process is characterized by rapid increase in vascular permeability of the gastrointestinal capillaries, leading to massive plasma water and protein loss into the lumen of the gastrointestinal tract. Despite its name there is minimal red blood cell loss leading to an elevation of the PCV; splenic contraction may further contribute to this increase in PCV. The consequence is hypovolemic shock and hemoconcentration in combination with hypoproteinemia due to the protein loss in the gastrointestinal tract.
This patient requires aggressive blood volume support. Isotonic crystalloid administration will aid in restoring normal blood volume but will further dilute the plasma protein concentration and the low COP will favor fluid loss to the interstitium. The addition of an artificial colloid such as hetastarch will increase COP, helping to maintain an adequate intravascular volume. This disease is not coagulopathic in nature and hence there is no indication for plasma therapy. Some infectious diseases of the gastrointestinal tract can mimic hemorrhagic gastroenteritis and fecal culture is recommended. Atypical hypoadrenocorticism can also present in this manner and should be considered. In many patients, the disease appears to be idiopathic and responds to resuscitation and general supportive care measures.
CASE 2: Cardiogenic Shock
An 8-year-old female spayed Doberman pinscher weighing 36 kg is presented collapsed. She has a history of dilated cardiomyopathy and atrial fibrillation. Her current medications include furosemide, enalapril, pimobendan, and diltiazem. On presentation the dog is obtunded, mucous membranes are pale, the capillary refill time is 2 to 3 seconds, the heart rate is 170 bpm, femoral pulse quality is decreased, and the extremity temperature feels cool. Auscultation reveals a 2/6 left-sided systolic murmur and an irregular heart rhythm, the respiratory rate is 50 breaths/min, and no crackles or wheezes are heard. Prominent jugular veins are evident and skin turgor/tent appears normal. The rectal temperature is 97.1° F.
On presentation, flow by oxygen is provided and a cephalic venous catheter is placed. A brief echocardiogram reveals biventricular enlargement with poor systolic function. Blood work collected at this time shows the following:
| Parameter | Measured Value | Reference Range |
|---|---|---|
| pH | 7.331 | 7.34-7.38 |
| Pvco2 mm Hg | 31.7 | 40-46 |
| Pvo2 mm Hg | 53.3 | 49-67 |
| HCO3 mEq/L | 16.3 | 22-24 |
| SBE mEq/L | −8.5 | −2 to 0 |
| Glucose mg/dL | 117 | 65-112 |
| Lactate mmol/L | 9.1 | <2.5 |
| Packed cell volume % | 51 | 37-55 |
| Total protein g/dL | 6.5 | 5.4-7.1 |
A lactic acidosis with appropriate respiratory compensation is evident. An electrocardiogram reveals atrial fibrillation with a ventricular rate of 170 bpm. Numerous ventricular premature contractions were also evident. Arterial blood pressure is measured with an oscillometric device; systolic pressure is 90 mm Hg, mean pressure is 64 mm Hg, and diastolic pressure is 45 mm Hg.
A diagnosis of cardiogenic shock secondary to dilated cardiomyopathy is made and a dobutamine constant rate infusion is started at an initial dose of 4 μg/kg/min. Ten minutes later there is little improvement in the perfusion parameters so the infusion rate is increased to 6 μg/kg/min. Over the next 30 minutes the dobutamine infusion rate is titrated up to a dose of 12 μg/kg/min at which time there is obvious improvement in the dogs perfusion parameters. A blood pressure measurement is repeated at this time and the systolic pressure is now 104 mm Hg, mean pressure is 75 mm Hg, and diastolic pressure is 50 mm Hg. The dog is sitting up, mucous membrane color is pale pink, capillary refill time is approximately 2 seconds, the heart rate is still 170 bpm, femoral pulse quality has improved, and body temperature is now 99.9° F.
CASE 2 - Case Notes
This dog was in circulatory shock on presentation and the physical examination findings were consistent with a vasoconstrictive form of shock. Given the signalment and history of dilated cardiomyopathy, evaluation of cardiac function is warranted in this case before considering fluid administration. The jugular vein distention is consistent with cardiogenic shock and suggests fluid therapy may not be needed at this time. The lactic acidosis is a consequence of poor tissue perfusion. A normal arterial blood pressure does not rule out the presence of shock and is consistent with compensation achieved by increased systemic vascular resistance. In this case scenario, the primary cause of shock is inadequate cardiac contractility and a parenterally administered positive inotrope such as dobutamine is indicated. Despite diltiazem therapy this dog has a rapid ventricular rate and further medical treatment to address this problem maybe warranted.
CASE 3: Septic Shock
A 5-year-old female spayed domestic short haired cat weighing 4 kg is presented collapsed. She has a history of vomiting and inappetence for the last 48 hours. There is no known history of trauma or toxin exposure; the cat has had no previous medical problems.
On presentation the cat is laterally recumbent and unresponsive, mucous membranes are very pale, capillary refill time cannot be determined, the heart rate is 140 bpm, the femoral pulses are barely palpable, and the extremities feel cold. The rectal temperature is 93.6° F. On auscultation no murmur, gallop, or arrhythmia is evident; no abnormal breath sounds are heard.
Flow by oxygen is provided and several unsuccessful attempts are made to place a peripheral venous catheter. An intraosseous catheter is placed in the proximal humerus and 100 mL of warm lactated Ringer’s solution is given rapidly. Following this therapy, a cephalic venous catheter is placed and a jugular venous blood sample is obtained for analysis.
| Parameter | Measured Value Presentation | Reference Range— Cat |
|---|---|---|
| pH | 7.045 | 7.34-7.38* |
| Pvco2 mm Hg | 50 | 38-42* |
| Pvo2 mm Hg | 34.7 | 49-67* |
| HCO3 mEq/L | 12.8 | 22-24* |
| SBE mEq/L | −12.6 | −2 to −8* |
| Glucose mg/dL | 49 | 67-168 |
| Lactate mmol/L | 6.3 | <2.5 |
| Packed cell volume % | 33 | 25-45 |
| Total protein g/dL | 6.5 | 6.0-8.6 |
Reevaluation of the patient at this time reveals pale mucous membranes, the capillary refill time still cannot be assessed, the heart rate is 160 bpm, and the femoral pulses have improved but are considered weak. The blood work reveals a mixed acid-base disorder with both a lactic acidosis and a respiratory acidosis in addition to hypoglycemia. One gram of dextrose IV is administered followed by another 60 mL of warm lactated Ringer’s solution. A repeat blood glucose measurement is now 230 mg/dL.
Following this therapy the cat is more responsive, the heart rate is 190 bpm, and femoral pulse quality has improved but does not feel normal. Active warming is started with a warm air blanket. Doppler blood pressure at this time reveals a pressure of 80 mm Hg. The cat is maintained on lactated Ringer’s with 5% dextrose at rate of 50 mL/hr. A full physical examination reveals mild dehydration, abdominal pain, and a palpable midabdominal mass. A dose of hydromorphone IV is given. Ultrasound evaluation of the abdomen reveals free abdominal fluid, a sample of which is aspirated, and microscopic evaluation reveals a septic exudate.
Intravenous, broad spectrum antimicrobials are given and reevaluation of the patient at this time shows no change in the perfusion parameters, less abdominal pain, and a temperature of 98.1° F. Doppler blood pressure at this time is 70 mm Hg. A central venous catheter is placed in the jugular vein and the central venous pressure is 2 mm Hg (2.72 cmH2O). Following 25 mL of 6% hetastarch IV, the Doppler blood pressure is 75 mm Hg and the central venous pressure is 4 mm Hg (5.44 cmH2O). Another 15 mL of 6% hetastarch is given and the blood pressure rechecked. The Doppler blood pressure remains at 75 mm Hg and the central venous pressure is now 6 mm Hg (8.16 cmH2O). A second central venous sample is collected and reveals mild improvement in the lactic acidosis and concurrent respiratory acidosis.
| Parameter | Second Sample Values | Reference Range — Cat |
|---|---|---|
| pH | 7.180 | 7.34-7.38* |
| Pvco2 mm Hg | 45.8 | 38-42* |
| Pvo2 mm Hg | 38.1 | 49-67* |
| HCO3 mEq/L | 16.9 | 22-24* |
| SBE mEq/L | −9.2 | −2 to −8* |
| Glucose mg/dL | 185 | 67-168 |
| Lactate mmol/L | 4.1 | <2.5 |
| Packed cell volume % | 26 | 25-45 |
| Total protein g/dL | 5.0 | 6.0-8.6 |
A constant rate infusion of dopamine is started at a rate of 5 μg/kg/min and over the next 30 minutes it is titrated up to rate of 10 μg/kg/min at which time the Doppler blood pressure is 100 mm Hg. The cat is now sitting up and moving around, perfusion parameters have improved with pale pink mucous membranes, capillary refill time is less than 2 seconds, heart rate is 180 bpm, and there are fair femoral pulses. The cat is now considered stable for induction of anesthesia and an exploratory laparotomy.
Case 3 - Case Notes
This cat was severely hypothermic and in circulatory shock on presentation. Venous access in such cases can be extremely challenging and intraosseous catheters can provide a rapid and reliable route of fluid and drug administration. The severe hypothermia is of concern but active warming is delayed until fluid resuscitation has been initiated to prevent peripheral vasodilatation that may occur with warming and could contribute to deterioration of the hemodynamic status of the patient. Following initial fluid therapy via the intraosseous route, peripheral perfusion improved sufficiently to allow for venous access. The initial blood work revealed a lactic acidosis that supports the diagnosis of circulatory shock. The low partial pressure of central venous oxygen also suggests inadequate oxygen delivery to the tissues. The hypoglycemia is a life-threatening abnormality that requires immediate intervention. Hypoglycemia is not an expected abnormality in association with circulatory shock and suggests the presence of concurrent disease such as severe SIRS/sepsis.
The diagnosis of septic peritonitis is confirmed by the cytology of the abdominal effusion and the administration of appropriate, parenteral antimicrobials is a priority. Hypotension, as identified by a Doppler blood pressure less than 90 mm Hg, is also a life-threatening abnormality and is consistent with severe hypovolemic shock, distributive shock with adequate blood volume, or a combination of the two. Measurement of central venous pressure is invaluable as a tool to guide fluid therapy in this scenario. Hetastarch was chosen in this case based on the likelihood that the patient will have increased capillary permeability secondary to sepsis and can be predicted to have fluid and protein loss into the abdomen. When blood pressure is not responsive to volume administration and central venous pressure suggests the patient has adequate blood volume, the diagnosis of distributive shock is made. Distributive shock requires vasopressor therapy such as dopamine, which is titrated to effect.
1 Adib-Conquy M., Adrie C., Moine P., et al. NF-kappaB expression in mononuclear cells of patients with sepsis resembles that observed in lipopolysaccharide tolerance. Am J Respir Crit Care Med. 2000;162:1877.
2 Adib-Conquy M., Cavaillon J.-M. Compensatory anti-inflammatory response syndrome. Thromb Haemost. 2009;101:36.
3 Annane D., Sebille V., Charpentier C., et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862.
4 Annane D. Adrenal insufficiency in sepsis. Curr Pharm Des. 2008;14:1882.
5 Armistead C.W.Jr, Vincent J.L., Preiser J.C., et al. Hypertonic saline solution-hetastarch for fluid resuscitation in experimental septic shock. Anesth Analg. 1989;69:714.
6 Aschner J.L., Poland R.L. Sodium bicarbonate: basically useless therapy. Pediatrics. 2008;122:831.
7 Bellissant E., Annane D. Effect of hydrocortisone on phenylephrine--mean arterial pressure dose-response relationship in septic shock. Clin Pharmacol Ther. 2000;68:293.
8 Bersenas A.M., Mathews K.A., Allen D.G., et al. Effects of ranitidine, famotidine, pantoprazole, and omeprazole on intragastric pH in dogs. Am J Vet Res. 2005;66:425.
9 Bickell W.H., Barrett S.M., Romine-Jenkins M., et al. Resuscitation of canine hemorrhagic hypotension with large-volume isotonic crystalloid: impact on lung water, venous admixture, and systemic arterial oxygen saturation. Am J Emerg Med. 1994;12:36.
10 Bird M.D., Karavitis J., Kovacs E.J. Sex differences and estrogen modulation of the cellular immune response after injury. Cell Immunol. 2008;252:57.
11 Boulpaep E.L. Regulation of arterial pressure and cardiac output. In: Boron W.F., Boulpaep E.L., editors. Medical Physiology: A Cellular and Molecular Approach. 2nd ed. Philadelphia, PA: Saunders/Elsevier; 2009:554.
12 Boyd J.H., Walley K.R. Is there a role for sodium bicarbonate in treating lactic acidosis from shock? Curr Opin Crit Care. 2008;14:379.
13 Brady C.A., Otto C.M., Van Winkle T.J., et al. Severe sepsis in cats: 29 cases (1986-1998). J Am Vet Med Assoc. 2000;217:531.
14 Bumpus S.E., Haskins S.C., Kass P.H. Effect of synthetic colloids on refractometric readings of total solids. J Vet Emerg Crit Care. 1998;8:21.
15 Burkitt J.M., Haskins S.C., Nelson R.W., et al. Relative adrenal insufficiency in dogs with sepsis. J Vet Intern Med. 2007;21:226.
16 Cairns C.B., Moore F.A., Haenel J.B., et al. Evidence for early supply independent mitochondrial dysfunction in patients developing multiple organ failure after trauma. J Trauma. 1997;42:532.
17 Chaudry I.H. Cellular mechanisms in shock and ischemia and their correction. Am J Physiol. 1983;245:R117.
18 Cohn L.A., Kerl M.E., Lenox C.E., et al. Response of healthy dogs to infusions of human serum albumin. Am J Vet Res. 2007;68:657.
19 Cooper D.J., Walley K.R., Wiggs B.R., et al. Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. A prospective, controlled clinical study. Ann Intern Med. 1990;112:492.
20 Costello M.F., Fletcher D.J., Silverstein D.C., et al. Adrenal insufficiency in feline sepsis. San Francisco: ACVECC Postgraduate Course, 2006.
21 Cronin L., Cook D.J., Carlet J., et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;23:1430.
22 Davies B.N., Withrington P.G. The actions of drugs on the smooth muscle of the capsule and blood vessels of the spleen. Pharmacol Rev. 1973;25:373.
23 de Jonge E., Levi M. Effects of different plasma substitutes on blood coagulation: a comparative review. Crit Care Med. 2001;29:1261.
24 de Papp E., Drobatz K.J., Hughes D. Plasma lactate concentration as a predictor of gastric necrosis and survival among dogs with gastric dilatation-volvulus: 102 cases (1995-1998). J Am Vet Med Assoc. 1999;215:49.
25 Deane S.A., Gaudry P.L., Woods P., et al. The management of injuries--a review of deaths in hospital. Aust N Z J Surg. 1988;58:463.
26 Dellinger R.P., Levy M.M., Carlet J.M., et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296.
27 De-Souza D.A., Greene L.J. Intestinal permeability and systemic infections in critically ill patients: effect of glutamine. Crit Care Med. 2005;33:1125.
28 Dewar D., Moore F.A., Moore E.E., et al. Postinjury multiple organ failure. Injury. 2009;40:912.
29 Di Filippo A., Ciapetti M., Prencipe D., et al. Experimentally-induced acute lung injury: the protective effect of hydroxyethyl starch. Ann Clin Lab Sci. 2006;36:345.
30 Dillon R., Hankes G.H., Nachreiner R.F., et al. Experimental hemorrhage in dogs: Effects of prednisolone sodium succinate. J Am Anim Hosp Assoc. 1978;14:673.
31 Dowling P.M. Anaphylaxis. In: Silverstein D.C., Hopper K., editors. Small Animal Critical Care Medicine. St. Louis: Saunders Elsevier; 2009:727.
32 Driessen B., Jahr J.S., Lurie F., et al. Arterial oxygenation and oxygen delivery after hemoglobin-based oxygen carrier infusion in canine hypovolemic shock: a dose-response study. Crit Care Med. 2003;31:1771.
33 Driessen B., Jahr J.S., Lurie F., et al. Effects of isovolemic resuscitation with hemoglobin-based oxygen carrier Hemoglobin glutamer-200 (bovine) on systemic and mesenteric perfusion and oxygenation in a canine model of hemorrhagic shock: a comparison with 6% hetastarch solution and shed blood. Vet Anaesth Analg. 2006;33:368.
34 Driessen B., Jahr J.S., Lurie F., et al. Inadequacy of low-volume resuscitation with hemoglobin-based oxygen carrier hemoglobin glutamer-200 (bovine) in canine hypovolemia. J Vet Pharmacol Ther. 2001;24:61.
35 Driessen B., Zarucco L., Gunther R.A., et al. Effects of low-volume hemoglobin glutamer-200 versus normal saline and arginine vasopressin resuscitation on systemic and skeletal muscle blood flow and oxygenation in a canine hemorrhagic shock model. Crit Care Med. 2007;35:2101.
36 Dubin A., Pozo M.O., Casabella C.A., et al. Increasing arterial blood pressure with norepinephrine does not improve microcirculatory blood flow: a prospective study. Crit Care. 2009;13:R92.
37 Eckardt K.U. Anemia in critical illness. Wien Klin Wochenschr. 2001;113:84.
38 Elbers P.W.G., Ince C. Mechanisms of critical illness--classifying microcirculatory flow abnormalities in distributive shock. Crit Care. 2006;10:221.
39 Engeland W.C., Dempsher D.P., Byrnes G.J., et al. The adrenal medullary response to graded hemorrhage in awake dogs. Endocrinology. 1981;109:1539.
40 Errington M.L., Rocha e Silva M. The secretion and clearance of vasopressin during the development of irreversible haemorrhagic shock. J Physiol (Lond). 1971;217:43P.
41 Evans R.G., Ventura S., Dampney R.A., et al. Neural mechanisms in the cardiovascular responses to acute central hypovolaemia. Clin Exp Pharmacol Physiol. 2001;28:479.
42 Fang X., Tang W., Sun S., et al. Comparison of buccal microcirculation between septic and hemorrhagic shock. Crit Care Med. 2006;34:S447.
43 Fantoni D.T., Auler Junior J.O., Futema F., et al. Intravenous administration of hypertonic sodium chloride solution with dextran or isotonic sodium chloride solution for treatment of septic shock secondary to pyometra in dogs. J Am Vet Med Assoc. 1999;215:1283.
44 Feldman E.C., Nelson R.W. Hypoadrenocorticism (Addison’s disease). In Feldman E.C., Nelson R.W., editors: Canine and Feline Endocrinology and Reproduction, 3rd ed, St. Louis: W.B. Saunders, 2004.
45 Felfernig M., Virmani S., Weintraud M., et al. Molecular weight of hydroxyethyl starch molecules influences coagulation profile measured by thrombelastography. J R Nav Med Serv. 2008;94:7.
46 Feng X., Ren B., Xie W., et al. Influence of hydroxyethyl starch 130/0.4 in pulmonary neutrophil recruitment and acute lung injury during polymicrobial sepsis in rats. Acta Anaesthesiol Scand. 2006;50:1081.
47 Fink M.P. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 2001;17:219.
48 Forsythe S.M., Schmidt G.A. Sodium bicarbonate for the treatment of lactic acidosis. Chest. 2000;117:260.
49 Francis A.H., Martin L.G., Haldorson G.J., et al. Adverse reactions suggestive of type III hypersensitivity in six healthy dogs given human albumin. J Am Vet Med Assoc. 2007;230:873.
50 Gibson G.R., Callan M.B., Hoffman V., et al. Use of a hemoglobin-based oxygen-carrying solution in cats: 72 cases (1998-2000). J Am Vet Med Assoc. 2002;221:96.
51 Glenn T.M., Lefer A.M. Anti-toxic action of methylprednisolone in hemorrhagic shock. Eur J Pharmacol. 1971;13:230.
52 Gunter P. Practice guidelines for blood component therapy. Anesthesiology. 1996;85:1219.
53 Guyton A.C., Hall J.E. Circulatory shock and physiology of its treatment. In: Guyton A.C., Hall J.E., editors. Textbook of Medical Physiology. 11th ed. Philadelphia: Elsevier Saunders; 2006:278.
54 Guyton A.C., Hall J.E. Metabolism of carbohydrates and formation of adenosine triphosphate. In: Guyton A.C., Hall J.E., editors. Textbook of Medical Physiology. 11th ed. Philadelphia: Elsevier Saunders; 2006:829.
55 Guyton A.C., Hall J.E. The integrated system for pressure control. In: Guyton A.C., Hall J.E., editors. Textbook of Medical Physiology. 11th ed. Philadelphia: Elsevier Saunders; 2006:216.
56 Guyton A.C., Hall J.E. Transport of oxygen and carbon dioxide in blood and tissue fluids. In Guyton A.C., Hall J.E., editors: Textbook of Medical Physiology, 11th ed, Philadelphia: Elsevier Saunders, 2006.
57 Hajjar L.A., Auler Junior J.O., Santos L., et al. Blood tranfusion in critically ill patients: state of the art. Clinics (Sao Paulo). 2007;62:507.
58 Hankes G.H., Dillon A.R., Ravis W.R. Effects of lactated ringer solution and prednisolone sodium succinate on dogs with induced hemorrhagic-shock. Am J Vet Res. 1992;53:26.
59 Henik R.A., Dolson M.K., Wenholz L.J. How to obtain a blood pressure measurement. Clin Tech Small Anim Pract. 2005;20:144.
60 Hintze T.H., Vatner S.F. Cardiac dynamics during hemorrhage. Relative unimportance of adrenergic inotropic responses. Circ Res. 1982;50:705.
61 Holmes C.L., Walley K.R. The evaluation and management of shock. Clin Chest Med. 2003;24:775.
62 Horton J.W., Walker P.B. Small-volume hypertonic saline dextran resuscitation from canine endotoxin shock. Ann Surg. 1991;214:64.
63 Hughes D. Lactate measurement: diagnostic, therapeutic and prognostic implications. In: Bonagura J.D., editor. Current Veterinary Therapy. Philadelphia: WB Saunders; 1999:112.
64 Hurcombe S.D., Toribio R.E., Slovis N., et al. Blood arginine vasopressin, adrenocorticotropin hormone, and cortisol concentrations at admission in septic and critically ill foals and their association with survival. J Vet Intern Med. 2008;22:639.
65 Ince C. Microcirculation in distress: a new resuscitation end point? Crit Care Med. 2004;32:1963.
66 Jackson K., Nolan J. The role of hypotensive resuscitation in the management of trauma. J Intensive Care Soc. 2009;10:109.
67 Johnson T., Arnaud F., Dong F., et al. Bovine polymerized hemoglobin (hemoglobin-based oxygen carrier-201) resuscitation in three swine models of hemorrhagic shock with militarily relevant delayed evacuation--effects on histopathology and organ function. Crit Care Med. 2006;34:1464.
68 Karagiannis MH, Reniker AN, Kerl ME, et al. Lactate measurement as an indicator of perfusion. Compend Contin Educ Vet 28:287
69 Katz L.M., Manning J.E., McCurdy S., et al. HBOC-201 improves survival in a swine model of hemorrhagic shock and liver injury. Resuscitation. 2002;54:77.
70 Kemp S.F., Lockey R.F. Anaphylaxis: a review of causes and mechanisms. J Allergy Clin Immunol. 2002;110:341.
71 Kerwin A.J., Schinco M.A., Tepas J.J.III, et al. The use of 23.4% hypertonic saline for the management of elevated intracranial pressure in patients with severe traumatic brain injury: a pilot study. J Trauma. 2009;67:277.
72 Kien N.D., Moore P.G., Pascual J.M., et al. Effects of hypertonic saline on regional function and blood flow in canine hearts during acute coronary occlusion. Shock. 1997;7:274.
73 Kien N.D., Reitan J.A., White D.A., et al. Cardiac contractility and blood flow distribution following resuscitation with 7.5% hypertonic saline in anesthetized dogs. Circ Shock. 1991;35:109.
74 King D.R., Cohn S.M., Proctor K.G. Resuscitation with a hemoglobin-based oxygen carrier after traumatic brain injury. J Trauma. 2005;59:553.
75 King L.G. Postoperative complications and prognostic indicators in dogs and cats with septic peritonitis: 23 cases (1989-1992). J Am Vet Med Assoc. 1994;204:407.
76 Koenig M.A., Bryan M., Lewin J.L.III, et al. Reversal of transtentorial herniation with hypertonic saline. Neurology. 2008;70:1023.
77 Kumar A., Parrillo J.E. Shock: classification, pathophysiology, and approach to management. In Parrillo J.E., Dellinger R.P., editors: Critical Care Medicine: Principles of Diagnosis and Management in the Adult, 2nd ed, Philadelphia: Mosby, 2001.
78 Landry D.W., Levin H.R., Gallant E.M., et al. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation. 1997;95:1122.
79 Landry D.W., Oliver J.A. The pathogenesis of vasodilatory shock. N Engl J Med. 2001;345:588.
80 Lefer A.M., Martin J. Mechanism of the protective effect of corticosteriods in hemorrhagic shock. Am J Physiol. 1969;216:314.
81 Lefering R., Neugebauer E.A. Steroid controversy in sepsis and septic shock: a meta-analysis. Crit Care Med. 1995;23:1294.
82 Levi M., van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26.
83 Levy R.J. Mitochondrial dysfunction, bioenergetic impairment, and metabolic down-regulation in sepsis. Shock. 2007;28:24.
84 Lv R., Zhou W., Chu C., et al. Mechanism of the effect of hydroxyethyl starch on reducing pulmonary capillary permeability in a rat model of sepsis. Ann Clin Lab Sci. 2005;35:174.
85 Manglik S., Flores E., Lubarsky L., et al. Glucocorticoid insufficiency in patients who present to the hospital with severe sepsis: a prospective clinical trial. Crit Care Med. 2003;31:1668.
86 Manning J.E., Katz L.M., Brownstein M.R., et al. Bovine hemoglobin-based oxygen carrier (HBOC-201) for resuscitation of uncontrolled, exsanguinating liver injury in swine. Carolina Resuscitation Research Group. Shock. 2000;13:152.
87 Marik P.E., Pastores S.M., Annane D., et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008;36:1937.
88 Marshall J.C. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 2001;29:S99.
89 Martin L.G., Groman R.P., Fletcher D.J., et al. Pituitary-adrenal function in dogs with acute critical illness. J Am Vet Med Assoc. 2008;233:87.
90 Martin L.G., Groman R.P. Relative adrenal insufficiency in critical illness. J Vet Emerg Crit Care. 2004;14:149.
91 Martin L.G., Luther T.Y., Alperin D.C., et al. Serum antibodies against human albumin in critically ill and healthy dogs. J Am Vet Med Assoc. 2008;232:1004.
92 Mathieu D., Neviere R., Billard V., et al. Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study. Crit Care Med. 1991;19:1352.
93 Maury E., Vassal T., Offenstadt G. Cardiac contractility during severe ketoacidosis. N Engl J Med. 1999;341:1938.
94 Meldrum D.R., Donnahoo K.K. Role of TNF in mediating renal insufficiency following cardiac surgery: evidence of a postbypass cardiorenal syndrome. J Surg Res. 1999;85:185.
95 Mellema M. Cardiac output, wedge pressure, and oxygen delivery. Vet Clin N Am Small Anim Pract. 2001;31:1175.
96 Menges T., König I.R., Hossain H., et al. Sepsis syndrome and death in trauma patients are associated with variation in the gene encoding tumor necrosis factor. Crit Care Med. 2008;36:1456.
97 Metz D.C. Preventing the gastrointestinal consequences of stress-related mucosal disease. Curr Med Res Opin. 2005;21:11.
98 Michiels C. Physiological and pathological responses to hypoxia. Am J Pathol. 2004;164:1875.
99 Moore F.A. The role of the gastrointestinal tract in postinjury multiple organ failure. Am J Surg. 1999;178:449.
100 Morales D., Madigan J., Cullinane S., et al. Reversal by vasopressin of intractable hypotension in the late phase of hemorrhagic shock. Circulation. 1999;100:226.
101 Okrasinski E.B., Krahwinkel D.J., Sanders W.L. Treatment of dogs in hemorrhagic shock by intraosseous infusion of hypertonic saline and dextran. Vet Surg. 1992;21:20.
102 Pape H.C., van Griensven M., Rice J., et al. Major secondary surgery in blunt trauma patients and perioperative cytokine liberation: determination of the clinical relevance of biochemical markers. J Trauma. 2001;50:989.
103 Pappachan J.V., Coulson T.G., Child N.J.A., et al. Mortality in adult intensive care patients with severe systemic inflammatory response syndromes is strongly associated with the hypo-immune TNF -238A polymorphism. Immunogenetics. 2009;61:657.
104 Pascual J.L., Ferri L.E., Seely A.J., et al. Hypertonic saline resuscitation of hemorrhagic shock diminishes neutrophil rolling and adherence to endothelium and reduces in vivo vascular leakage. Ann Surg. 2002;236:634.
105 Peyton J.L., Burkitt J.M. Critical illness-related corticosteroid insufficiency in a dog with septic shock. J Vet Emerg Crit Care. 2009;19:262.
106 Philbin N., Handrigan M., Rice J., et al. Resuscitation following severe, controlled hemorrhage associated with a 24 h delay to surgical intervention in swine using a hemoglobin based oxygen carrier as an oxygen bridge to definitive care. Resuscitation. 2007;74:332.
107 Polderman K.H. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37:S186.
108 Prigent H., Maxime V., Annane D. Clinical review: corticotherapy in sepsis. Crit Care. 2004;8:122.
109 Prigent H., Maxime V., Annane D. Science review: mechanisms of impaired adrenal function in sepsis and molecular actions of glucocorticoids. Crit Care. 2004;8:243.
110 Prist R., Rocha-e-Silva M., Scalabrini A., et al. A quantitative analysis of transcapillary refill in severe hemorrhagic hypotension in dogs. Shock. 1994;1:188.
111 Prittie J.E., Barton L.J., Peterson M.E., et al. Hypothalamo-pituitary-adrenal axis function in critically ill cats. Proceedings, 9th Int Vet Emerg Crit Care Symp. 2003:771.
112 Prittie J.E., Barton L.J., Peterson M.E., et al. Pituitary ACTH and adrenocortical secretion in critically ill dogs. J Am Vet Med Assoc. 2002;220:615.
113 Prough D.S., Whitley J.M., Taylor C.L., et al. Small-volume resuscitation from hemorrhagic shock in dogs: effects on systemic hemodynamics and systemic blood flow. Crit Care Med. 1991;19:364.
114 Prough D.S. Hypertonic hyperoncotic fluid resuscitation after hemorrhagic shock in dogs. Anesth Analg. 73, 1991.
115 Qureshi A.I., Suarez J.I. Use of hypertonic saline solutions in treatment of cerebral edema and intracranial hypertension. Crit Care Med. 2000;28:3301.
116 Qureshi A.I., Wilson D.A., Traystman R.J. Treatment of transtentorial herniation unresponsive to hyperventilation using hypertonic saline in dogs: effect on cerebral blood flow and metabolism. J Neurosurg Anesthesiol. 2002;14:22.
117 Radhakrishnan R.S., Radhakrishnan H.R., Xue H., et al. Hypertonic saline reverses stiffness in a Sprague-Dawley rat model of acute intestinal edema, leading to improved intestinal function. Crit Care Med. 2007;35:538.
118 Rice J., Philbin N., McGwin G., et al. Bovine polymerized hemoglobin versus Hextend resuscitation in a swine model of severe controlled hemorrhagic shock with delay to definitive care. Shock. 2006;26:302.
119 Rivers E., Nguyen B., Havstad S., et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368.
120 Robergs R.A., Ghiasvand F., Parker D. Biochemistry of exercise-induced metabolic acidosis. Am J Physiol Regul Integr Comp Physiol. 2004;287:R502.
121 Rothwell P.M., Udwadia Z.F., Lawler P.G. Cortisol response to corticotropin and survival in septic shock. Lancet. 1991;337:582.
122 Rudloff E., Kirby R. Fluid resuscitation and the trauma patient. Vet Clin North Am Small Anim Pract. 2008;38:645.
123 Sauaia A., Moore F.A., Moore E.E., et al. Early predictors of postinjury multiple organ failure. Arch Surg. 1994;129:39.
124 Schadt J.C., Ludbrook J. Hemodynamic and neurohumoral responses to acute hypovolemia in conscious mammals. Am J Physiol. 1991;260:H305.
125 Schertel E.R., Allen D.A., Muir W.W., et al. Evaluation of a hypertonic saline-dextran solution for treatment of dogs with shock induced by gastric dilatation- volvulus. J Am Vet Med Assoc. 1997;210:226.
126 Schertel E.R., Allen D.A., Muir W.W., et al. Evaluation of a hypertonic sodium chloride dextran solution for treatment of traumatic shock in dogs. J Am Vet Med Assoc. 1996;208(3):366.
127 Schroeder S., Wichers M., Klingmuller D., et al. The hypothalamic-pituitary-adrenal axis of patients with severe sepsis: altered response to corticotropin-releasing hormone. Crit Care Med. 2001;29:310.
128 Shapiro J.I. Pathogenesis of cardiac dysfunction during metabolic acidosis: therapeutic implications. Kidney Int Suppl. 1997;61:S47.
129 Shen Y.T., Knight D.R., Thomas J.X., et al. Relative roles of cardiac receptors and arterial baroreceptors during hemorrhage in conscious dogs. Circ Res. 1990;66:397.
130 Shoemaker W.C. Diagnosis and treatment of shock syndromes. In Shoemaker W.C., Ayers S., Genvick A., editors: Textbook of Critical Care, 3rd ed, Philadelphia: W.B. Saunders, 1995.
131 Silverstein D.C., Aldrich J., Haskins S., et al. Assessment of changes in blood volume in response to resuscitative fluid administration in dogs. J Vet Emerg Crit Care. 2005;15:185.
132 Silverstein D.C. Vasopressin. In: Silverstein D.C., Hopper . Small Animal Critical Care Medicine. St. Louis: Saunders Elsevier; 2009:759.
133 Simmons J.P., Wohl J.S. Vasoactive catecholamines. In: Silverstein D.C., Hopper K., editors. Small Animal Critical Care Medicine. St. Louis: Saunders Elsevier; 2009:756.
134 Sindram D., Clavien P.A. Calpain methods in hepatic ischemia-reperfusion injury. Methods Mol Biol. 2000;144:261.
135 Soni A., Pepper G.M., Wyrwinski P.M., et al. Adrenal insufficiency occurring during septic shock: incidence, outcome, and relationship to peripheral cytokine levels. Am J Med. 1995;98:266.
136 Spirt M.J. Stress-related mucosal disease: Risk factors and prophylactic therapy. Clin Ther. 2004;26:197.
137 Stark B.J., Sullivan T.J. Biphasic and protracted anaphylaxis. J Allergy Clin Immunol. 1986;78:76.
138 Stollman N., Metz D.C. Pathophysiology and prophylaxis of stress ulcer in intensive care unit patients. J Crit Care. 2005;20:35.
139 Strauss R.G., Stump D.C., Henriksen R.A., et al. Effects of hydroxyethyl starch on fibrinogen, fibrin clot formation, and fibrinolysis. Transfusion (Paris). 1985;25:230.
140 Strauss R.G., Stump D.C., Henriksen R.A. Hydroxyethyl starch accentuates von Willebrand’s disease. Transfusion (Paris). 1985;25:235.
141 Strecker U., Dick W., Madjidi A., et al. The effect of the type of colloid on the efficacy of hypertonic saline colloid mixtures in hemorrhagic shock: dextran versus hydroxyethyl starch. Resuscitation. 1993;25:41.
142 Stump D.C., Strauss R.G., Henriksen R.A., et al. Effects of hydroxyethyl starch on blood coagulation, particularly factor VIII. Transfusion (Paris). 1985;25:349.
143 Tani T., Fujino M., Hanasawa K., et al. Bacterial translocation and tumor necrosis factor-alpha gene expression in experimental hemorrhagic shock. Crit Care Med. 2000;28:3705.
144 Thorens J.B., Jolliet P., Ritz M., et al. Effects of rapid permissive hypercapnia on hemodynamics, gas exchange, and oxygen transport and consumption during mechanical ventilation for the acute respiratory distress syndrome. Intensive Care Med. 1996;22:182.
145 Tian J., Lin X., Guan R., et al. The effects of hydroxyethyl starch on lung capillary permeability in endotoxic rats and possible mechanisms. Anesth Analg. 2004;98:768.
146 Tian J., Lin X., Li Y.H., et al. Influence of hydroxyethyl starch on lipopolysaccharide-induced tissue nuclear factor kappa B activation and systemic TNF-alpha expression. Acta Anaesthesiol Scand. 2005;49:1311.
147 Topalian S., Ginsberg F., Parrillo J.E. Cardiogenic shock. Crit Care Med. 2008;36:S66.
148 Toyokuni S. Reactive oxygen species-induced molecular damage and its application in pathology. Pathol Int. 1999;49:91.
149 Trow A.V., Rozanski E.A., Delaforcade A.M., et al. Evaluation of use of human albumin in critically ill dogs: 73 cases (2003-2006). J Am Vet Med Assoc. 2008;233:607.
150 Tschoeke S.K., Hellmuth M., Hostmann A., et al. The early second hit in trauma management augments the proinflammatory immune response to multiple injuries. J Trauma. 2007;62:1396.
151 van Dissel J.T., van Langevelde P., Westendorp R.G., et al. Anti-inflammatory cytokine profile and mortality in febrile patients. Lancet. 1998;351:950.
152 Varicoda E.Y., Poli de Figueiredo L.F., Cruz R.J.Jr, et al. Blood loss after fluid resuscitation with isotonic or hypertonic saline for the initial treatment of uncontrolled hemorrhage induced by spleen rupture. J Trauma. 2003;55:112.
153 von Roten I., Madjdpour C., Frascarolo P., et al. Molar substitution and C2/C6 ratio of hydroxyethyl starch: influence on blood coagulation. Br J Anaesth. 2006;96:455.
154 Waddell L.S., Brown A.J. Hemodynamic monitoring. In: Silverstein D.C., Hopper K., editors. Small Animal Critical Care Medicine. St. Louis: Saunders Elsevier; 2009:859.
155 Weingart C., Kohn B. Clinical use of a haemoglobin-based oxygen carrying solution (Oxyglobin) in 48 cats (2002-2006). J Feline Med Surg. 2008;10:431.
156 Wiener E.S., Bajaj L. Diagnosis and emergent management of anaphylaxis in children. Adv Pediatr. 2005;52:195.
157 Wierenga J.R., Jandrey K.E., Haskins S.C., et al. In vitro comparison of the effects of two forms of hydroxyethyl starch solutions on platelet function in dogs. Am J Vet Res. 2007;68:605.
158 Wildenthal K., Mierzwiak D.S., Myers R.W., et al. Effects of acute lactic acidosis on left ventricular performance. Am J Physiol. 1968;214:1352.
159 Xie J., Lv R., Yu L., et al. Hydroxyethyl starch 130/0.4 inhibits production of plasma proinflammatory cytokines and attenuates nuclear factor-kappaB activation and toll-like receptors expression in monocytes during sepsis. J Surg Res. 2009.
160 Zakaria R., Tsakadze N.L., Garrison R.N. Hypertonic saline resuscitation improves intestinal microcirculation in a rat model of hemorrhagic shock. Surgery. 2006;140:579.
161 Zoran D.L., Jergens A.E., Riedesel D.H., et al. Evaluation of hemostatic analytes after use of hypertonic saline solution combined with colloids for resuscitation of dogs with hypovolemia. Am J Vet Res. 1992;53:1791.